WO2005085212A1 - Substituted pyrimidine derivative - Google Patents
Substituted pyrimidine derivative Download PDFInfo
- Publication number
- WO2005085212A1 WO2005085212A1 PCT/JP2005/003535 JP2005003535W WO2005085212A1 WO 2005085212 A1 WO2005085212 A1 WO 2005085212A1 JP 2005003535 W JP2005003535 W JP 2005003535W WO 2005085212 A1 WO2005085212 A1 WO 2005085212A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- group
- pyrimidine
- methanol
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to a novel substituted pyrimidine derivative, and a medicament containing the same as an active ingredient, particularly a therapeutic agent for an inflammatory disease.
- Chemokines which are cell chemotactic factors, are broadly classified into two types, CXCZ chemokines and CCZ iS chemokines, depending on their structural characteristics.
- these chemokine receptors belong to the family of seven transmembrane G-protein coupled receptors, and are composed of CXC chemokine receptor and CC chemokine receptor (Pharmacological Reviews, 52, 145, 2000).
- CCR4 CC chemokine receptor 4
- Thymus and activation-regulated chemoine (I'ARC) and macrophage-derived chemokine (MDC) are specific ligands for CCR4 (CCJ chemokines) (Journal of Biological Chemistry, 272, 1503 ⁇ , 1997, Journal of Biological chemistry, 273, 1764, 1998).
- TARC was found as a T cell chemotactic factor (Journal of Biological Chemistry, 271, 21514, 1996), and MDC was discovered as a chemotactic factor for monocytes' macrophages and ⁇ cells (Journal of Experimental Medicine, 185, 1595, 1997).
- Chemokines are also known to have the characteristics of both inflammatory chemokines and homeostatic chemokines. Today, 20, 254, 1999).
- CCR4 and its ligands are involved in various diseases such as inflammatory diseases, allergic diseases, and autoimmune diseases.
- diseases such as asthma, The Journal of Clinical Investigation, 107, 1357, 2001
- atopic dermatitis Journal of Investigative Dermatology, 115, 640, 2000
- psoriasis Laboratory dermatitis
- CCR4 function modulators are expected as agents for preventing or treating these diseases and the like.
- Various drugs such as steroids are used as prophylactic or therapeutic agents for the above-mentioned inflammatory diseases, allergic diseases, autoimmune diseases, etc. There is a strong need for the development of drugs based on this.
- Patent Document 1 A gazette published after the application on which the priority of the present application is based describes that a pyrimidine derivative represented by the following general formula has an anti-inflammatory effect based on the regulation of functions of TARC and the like (Patent Document 1).
- Ar is a substituted or unsubstituted aryl
- R 1 is NR 3
- R 2 is a hydrogen atom or a substituted or unsubstituted lower alkyl
- R 3 is a substituted or unsubstituted A represents lower alkyl, substituted or unsubstituted aralkyl or substituted or unsubstituted aromatic heterocyclic aralkyl, etc.
- A represents formula (III), etc.
- m 1 is an integer of 0 to 2
- n is 0 to An integer of 4
- a 1 represents a number capable of substituting 0 force
- Q represents -NR 7
- R 6 and R 7 are the same or different and represent a hydrogen atom or a substituted or unsubstituted lower alkyl, etc. ] Or a substituted or unsubstituted alicyclic heterocyclic group.
- the application does not include compounds having aryl or heteroalkyl as R 2 or R 3 . It has been reported that a 5-cyanopyrimidine derivative represented by the following general formula has an anti-inflammatory effect based on the regulation of functions such as TARC (Patent Document 2).
- R 1 and R 3 are the same or different and each represent a hydrogen atom
- -NR 4 R 5 [R 4 and R 5 are the same or different and each represent a hydrogen atom, a substituted or unsubstituted cyclo Alkyl, substituted or unsubstituted aryl, substituted or unsubstituted aralkyl, etc.]
- R 2 represents the above formula (II), etc., where Q is a nitrogen atom or CH, and A is a short bond , Carbonyl, R, etc., R is a substituted or unsubstituted alicyclic heterocyclic group, etc., ma is an integer of 0-2, ya is 0-replaceable integer, na is an integer of 0-4.
- ya is 0-replaceable integer
- na is an integer of 0-4.
- Patent Document 3 It has been reported that a compound represented by the following general formula has a function of regulating the function of TARC or the like (Patent Document 3).
- Patent Document 4 a compound represented by the following general formula is useful as a therapeutic drug for central diseases based on GPR88 inhibition.
- R 1 is a hydrogen atom, etc.
- R 2 is a hydrogen atom, etc.
- -NR 4 [wherein, R 3 and R 4 are hydrogen atoms, etc. Or R 3 and R 4 together with an adjacent nitrogen atom form a substituted or unsubstituted heterocyclic group]
- X is 1,2,3,4-tetrazole-5-yl and the like.
- Y represents a hydrogen atom or the like
- r represents an integer of 0 to 4.
- Patent Document 1 International Publication No. 04Z074260 pamphlet
- Patent Document 2 International Publication No. 03Z082855 pamphlet
- Patent Document 3 International Publication No. 03Z104230 pamphlet
- Patent Document 4 WO 04Z054617 pamphlet
- An object of the present invention is to provide a safe and powerful preventive / therapeutic agent for inflammatory diseases, allergic diseases, autoimmune diseases and the like, which has a function of regulating CCR4 function and replaces conventional steroids. .
- the present inventors have diligently studied compounds having a CCR4 function regulating action. As a result, a pyrimidine derivative CCR4 having a saturated 6-membered heterocyclic group such as substituted piperidino at the 2-position, a substituted amino group at the 4-position, and a ring group at one of the 5- or 6-position. The present inventors have found that they are useful as function regulators, and have completed the present invention.
- the compound of the present invention is different from the compound described in Patent Document 1 in that a ring group is directly bonded to the 4-position of the pyrimidine ring via a nitrogen atom. Further, the compound of the present invention is a 5- or 6-position of the pyrimidine skeleton. And has a basic structure different from the compounds described in Patent Documents 2 and 3.
- the present invention relates to the following compounds.
- a substituted pyrimidine derivative represented by the following general formula (I) or a pharmaceutically acceptable salt thereof represented by the following general formula (I) or a pharmaceutically acceptable salt thereof.
- A: substituted !, may! /, Aryl or substituted !, may! /, Cycloalkyl,
- R 1 -R °, halogen, -OH, -OR °, halogeno lower alkyl
- R 3 — H, — R. Or— CN,
- L bond, -CO-, lower alkylene, -CO- (lower alkylene)-or-(lower alkylene) -CO-,
- R 2 same or different, -R °, halogen, -OH, -0R °, halogeno lower alkyl, -R °° -0H, -CON (R 8 ) (R 9 ), -R 00 -OR ° ,-R QQ -N (R 8 ) (R 9 ),-R QQ -CN,-R QQ -N (R 8 )-CO- R °,
- R 8 and R 9 same or different, -H or -R °,
- R 11 and R 12 same or different from each other, - 0H, -0R °, halogen, - N (R 8) ( R 9), - CN, - N (R 8) - CO- R °, - N ( R 8 ) -SO-R °, -0-CO-R °, -CO-R. Or a group consisting of -CON (R 8 ) (R 9 )
- R QQ lower alkylene
- a preferred embodiment of the present invention is a derivative shown below.
- a substituted pyrimidine derivative represented by the following general formula (II) or a pharmaceutically acceptable salt thereof represented by the following general formula (II) or a pharmaceutically acceptable salt thereof.
- A: substituted !, may! /, Aryl or substituted !, may! /, Cycloalkyl,
- R 1 -R °, halogen, -OH, -OR °, halogeno lower alkyl
- Y CR 4 or N, where X force CR 3 indicates Y, and X indicates N, Y indicates CR 4 .
- R 2 same or different, -R °, halogen, -OH, -0R °, halogeno lower alkyl, -R °° -0H, -CON (R 8 ) (R 9 ), -R 00 -OR ° ,-R QQ -N (R 8 ) (R 9 ),-R QQ -CN,-R QQ -N (R 8 )-CO- R °, -R 00 -N (R 8 ) -SO-R ° , -R °°-0-CO-R °, -R °° -CO-R. Or - R °° - CON (R 8 ) (R 9),
- R 8 and R 9 same or different, -H or -R °,
- R 5 and R 6 the same or different, the groups described in H or R 2 or R 5 and R 6 are
- R 7 -H ⁇ -R °,-R °°-OH ⁇ -CON (R 8 ) (R 9 ),-R °°-0- R °,-R °°-N (R 8 ) (R 9), - R °° - CN ⁇ -R °° - N (R 8) - CO- R. , -R. . - N (R 8) - SO - R. , -R. . -0-CO-R. , -R. . -CO-R. Or -R. . -C0N (R 8
- R QQ lower alkylene
- B is substituted, may be, pyridyl, substituted !, may be, chel, substituted, may be, furyl or substituted, may be, The derivative according to the above [5], which is a phenol.
- R 2 is halogeno lower alkyl, -R °° -OH, -R °° -OR. Or -CON (R 8
- the present invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising the substituted pyrimidine derivative represented by the above general formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the pharmaceutical composition is a CCR4 or a modulator of TARC and Z or MDC function, more preferably an inflammatory disease, an allergic disease or an autoimmune disease, particularly preferably asthma or atopic dermatitis
- the above-mentioned pharmaceutical composition which is a prophylactic or therapeutic agent for rheumatoid arthritis.
- a substituted pyrimidine derivative represented by the general formula (I) according to [1]-[10] for the manufacture of a prophylactic or therapeutic agent for an inflammatory disease, an allergic disease or an autoimmune disease.
- a pharmaceutically acceptable salt thereof, wherein the substituted pyrimidine derivative represented by the general formula (I) or the pharmaceutically acceptable salt thereof according to (1)-(10) is used in an effective amount, It is a method for preventing or treating an inflammatory disease, an allergic disease or an autoimmune disease, which is administered to a mammal.
- the substituted pyrimidine derivative of the present invention has a function of regulating the function of CCR4 or TARC and Z or MDC, and has an advantage that it has a strong effect in a model of an inflammatory disease, an allergic disease or an autoimmune disease.
- alkyl and “alkylene” mean a straight or branched hydrocarbon chain.
- “Lower alkyl” is preferably an alkyl group having 116 carbon atoms, more preferably an alkyl group having 114 carbon atoms, and still more preferably methyl and ethyl.
- “Lower alkylene” means a divalent group formed by removing one arbitrary hydrogen atom from the above “lower alkyl”, preferably alkylene having 114 carbon atoms, more preferably methylene, ethylene and It is propylene.
- Halogen refers to F, Cl, Br and I.
- Halogeno lower alkyl means preferably alkyl having 1 to 6 carbon atoms substituted with one or more halogen, more preferably C alkyl substituted with one or more F, Preferably, fluoromethyl, diflu
- Cycloalkyl is preferably cycloalkyl having 3 to 10 carbon atoms, and may be crosslinked. More preferred are cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and adamantyl.
- Aryl means an aromatic hydrocarbon group having 6 to 14 carbon atoms, and includes a phenyl group fused with “cycloalkyl”. Preferably, they are phenyl and naphthyl, and more preferably, phenyl.
- the “monocyclic or bicyclic hydrocarbon ring group” includes both monocyclic or bicyclic “cycloalkyl” and monocyclic or bicyclic “aryl”.
- the term "monocyclic heterocyclic group” refers to a monocyclic 3- to 8-membered, preferably 5- to 7-membered cyclic group containing 1 to 4 hetero atoms selected from 0, S and N forces. And includes a monocyclic heteroaryl which is an unsaturated ring, a monocyclic heterocycloalkyl which is a saturated ring, and a ring group in which the monocyclic heteroaryl is partially hydrogenated.
- the monocyclic heteroaryl preferably, pyridyl, pyrazul, pyrimidyl, pyridazyl, imidazolyl, pyrrolyl, triazolyl, tetrazolyl, chenyl, furyl, thiazolyl, pyrazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, oxaziazolyl and the like. No.
- a monocyclic heterocycloalkyl or a ring group in which a heteroaryl group is partially hydrogenated preferably, piberidyl, pyrrolidinyl, piperazinyl, azepanyl, diazepanyl, tetrahydrofuranyl, Trahydroviral, morpholinyl and the like.
- the “bicyclic heterocyclic group” is a ring group in which the above-mentioned monocyclic heterocycles are fused together or a benzene ring and a monocyclic heterocycle are condensed.
- the ring atom S or N may be oxidized to form an oxoxide-dioxide.
- an arbitrary carbon atom may be substituted with an oxo group.
- May be substituted means “unsubstituted” or “having 115 identical or different substituents”.
- the substituent in "substituted or may be aryl" and “substituted or may be cycloalkyl” is preferably halogen, lower alkyl, -OH, -0-lower alkyl, -CN , -S-lower alkyl, NO, more preferably halogen, lower alkyl, -OH
- Substituents in the “optionally substituted monocyclic or bicyclic hydrocarbon ring group” and the “optionally substituted monocyclic or bicyclic heterocyclic group” are preferably halogen, lower Alkyl, halogeno lower alkyl, -OH, -0-lower alkyl, -CN, NO,
- NH and preferably 2 is halogen, lower alkyl, or halogeno lower alkyl.
- the compound of the present invention represented by the formula (I) may have a geometric isomer or a tautomer depending on the type of a substituent. Or a mixture of the isomers.
- the compound (I) may have an asymmetric carbon atom, and an (R) -form or (S) -form optical isomer based on this may exist.
- the present invention includes all of the optical isomers as a mixture or an isolated one.
- the compound (I) also includes a pharmacologically acceptable prodrug.
- a pharmacologically acceptable prodrug is defined as the NH4 of the present invention by solvolysis or under physiological conditions. It is a compound having a group that can be converted to OH, COH, and the like. As a base to form a prodrug
- Compound (I) may form an acid addition salt or a salt with a base depending on the type of substituent.
- the strong salt is a pharmaceutically acceptable salt, specifically, an inorganic acid such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, and propion.
- Acid addition salts with organic acids such as acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, aspartic acid and glutamic acid, sodium And inorganic bases such as potassium, magnesium, calcium, and aluminum; salts with organic bases such as methylamine, ethylamine, ethanolamine, lysine, and ortin; and ammonium salts.
- the present invention also encompasses pharmaceutical compositions containing various hydrates and solvates of compound (I) and salts thereof and polymorphic substances.
- Compound (I) which is an active ingredient of the present invention, and pharmaceutically acceptable salts thereof are produced by applying various known synthetic methods, utilizing characteristics based on the basic skeleton or the type of substituent. can do.
- Such functional groups include, for example, amino groups, hydroxyl groups, carboxyl groups, and the like, and their protecting groups are described, for example, in Protective Groups in Organic Synthesis (Third Edition) by Green (TW Greene) and Utz (PGM Wuts). , 1999) ", which may be appropriately selected and used according to the reaction conditions.
- a desired compound can be obtained by introducing the protective group and performing a reaction, and then removing the protective group or converting it to a desired group as necessary.
- the prodrug of the compound (I) can be produced by introducing a specific group at the stage of a raw material or an intermediate or by carrying out a reaction using the obtained compound (I) as in the case of the above-mentioned protective group.
- the reaction is carried out by applying a method known to those skilled in the art, such as ordinary esterification, amidation, dehydration, etc. Can be performed.
- This production method is a method for producing a compound (I) of the present invention by subjecting a pyrimidine derivative (1) having a leaving group at the 2-position to a cyclic amine diversion compound (2) by ibuso substitution.
- Examples of the leaving group represented by L include a halogen, an alkylsulfiel group, an alkylsulfol
- reaction is carried out by subjecting compound (1) to a solvent inert to the reaction, in the presence or absence of a base or acid (preferably hydrogen chloride), using an equivalent or excess amount of (2) under cooling and heating under reflux. It usually takes place for one hour and five days.
- a base or acid preferably hydrogen chloride
- the solvent is not particularly limited as long as it is inert to the reaction, but, for example, aromatic hydrocarbons such as benzene, toluene, xylene, getyl ether, tetrahydrofuran (THF), 1,4-dioxane, 1,2-dimethoxy Ethers such as ethane (DME) and 1,2-dietoxetane (DEE); halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane and chloroform, methanol, ethanol, 2-propanol, butanol, etc.
- aromatic hydrocarbons such as benzene, toluene, xylene, getyl ether, tetrahydrofuran (THF), 1,4-dioxane, 1,2-dimethoxy Ethers such as ethane (DME) and 1,2-dietoxetane (DEE); halogenated hydrocarbons such as dichloromethan
- Bases include organic bases such as triethylamine, diisopropylethylamine (DIPEA), 1,8-diazabicyclo- [5.4.0] -7-indene (DBU), 2,6-lutidine, sodium carbonate, and carbonate.
- organic bases such as triethylamine, diisopropylethylamine (DIPEA), 1,8-diazabicyclo- [5.4.0] -7-indene (DBU), 2,6-lutidine, sodium carbonate, and carbonate.
- IPEA diisopropylethylamine
- DBU 1,8-diazabicyclo- [5.4.0] -7-indene
- Inorganic bases such as potassium, sodium hydride, potassium hydride, potassium tert-butoxide and the like can be mentioned.
- This production method is a method for producing a compound (la) of the present invention by subjecting a pyrimidine derivative (3) having a piperazino group (3) and a carboxylic acid conjugate (4) to an amidoni reaction.
- the reaction is carried out by using the compound (3) and the compound (4) in an equal amount or one of them in excess, and using a condensing agent (eg, dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIPC), -Ethyl-3- (3-dimethylaminopropyl) carbodiimide (WSC), 1,1-carbylbis-1H-imidazole (CDI), etc., and in some cases, further additives (eg, N-hydroxysuccinimide ( (HONSu), 1-hydroxybenzotriazole (HOBt), etc.) with stirring under cooling and heating, preferably at room temperature, usually for 1 hour to 1 day.
- a condensing agent eg, dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIPC), -Ethyl-3- (3-dimethylaminopropyl) carbodiimide (
- the active ester form of the compound (4) and the above additive may be isolated and then reacted with the compound (3).
- the solvent is not particularly limited as long as it is inert to the reaction.For example, aromatic hydrocarbons, ethers, halogenated hydrocarbons, ⁇ , ⁇ -dimethylformamide (DMF), N-methylpyrrolidone (NMP) , Dimethyl sulfoxide (DMSO) and the like. These solvents are used alone or in combination of two or more. In some cases, it is advantageous to carry out the reaction in the presence of a base such as triethylamine, DIPEA, pyridine or the like in order to make the reaction proceed smoothly.
- a base such as triethylamine, DIPEA, pyridine or the like in order to make the reaction proceed smoothly.
- the compound of the present invention having an NH group can be produced by a reduction reaction of a compound having a nitro group.
- L is -C1 or -Br
- U is -Br, -Cl, -I or -O-SO-CF
- Q is -B (OH), -B (0-
- the starting compound (1) can be produced by subjecting a pyrimidine derivative (5) to an ipamine substitution reaction with an amine compound (6), and then performing a coupling reaction using the adduct (7) and the compound (8). You. The same conditions as in the first production method can be applied to the ipso substitution reaction.
- the coupling reaction can be performed, for example, by the method described in “Chemical Experiment Course (4th edition)”, edited by The Chemical Society of Japan, Vol. 25 (1992) (Maruzen). [Formula 11]
- R 1Q represents a lower alkyl group. The same applies hereinafter.
- the compound (la) wherein L 1 is a methylsulfinyl group can be produced according to the above reaction route.
- methyl and ethyl are preferable as the lower alkyl of R 1Q.
- the coupling reaction is described in the above-mentioned Nippon Dani Gakkai, “Experimental Chemistry Course (4th edition)”, Vol. 25 (1992) (Maruzen) It can be carried out by the method described.
- the alkylation is carried out using an alkylating agent such as methyl iodide or dimethyl sulfate in a solvent such as aromatic hydrocarbons, ethers, or halogenated hydrocarbons, or a mixed solvent thereof.
- Halogenide uses a chlorinating agent such as phosphorus oxychloride, phosphorus pentachloride, or salty chloride, or a brominating agent such as phosphorus oxybromide in an equimolar excess to obtain an aromatic hydrocarbon or halogenated hydrocarbon.
- the reaction is carried out in a solvent inert to the reaction or the like or without a solvent.
- the same conditions as in the first production method can be applied to the ipso substitution reaction.
- Oxidation can be carried out by using an ordinary method of oxidation reaction of the sulfur group. For example, the method described in The Chemical Society of Japan, “Experimental Chemistry Course (4th edition)”, Vol. 23 (1992) (Maruzen) can be mentioned. . [Formula 12]
- the starting compound (3) can be produced by subjecting the pyrimidine derivative (1) to a piperazine (14) by an ipso substitution reaction.
- the same conditions as in the first production method can be applied to the ipso substitution reaction.
- the cyclic amine conjugates (2a) and (2b) can be produced by the method shown in the above formula.
- the method described in the above-mentioned “Protective Groups in Organic Synthesis (3rd edition)” for the amino group deprotection reaction and the like can be applied.
- the reductive alkylation a conventional method of reductive alkylation can be used. For example, it is described in “Experimental Chemistry Lecture (4th edition)” edited by The Chemical Society of Japan, Vol. 20 (1992) (Maruzen). Method.
- cyclic amine conjugates (2c) and (2d) can be produced by the method shown in the above formula.
- various cyclic amine compounds (2) can be prepared by, for example, alkylating a compound having a hydroxyl group with an alkylating agent (eg, alkyl halide ⁇ sulfonic acid alkyl ester) or an alkyl ether group by a Mitsunobu reaction to form a fluorinating agent.
- the compound can be converted to a compound having a phthalimido group by a Mitsunobu reaction with a fluorimido group and phthalimide, respectively.
- reaction product obtained by each of the above production methods is isolated and purified as various solvates such as a free compound, a salt thereof or a hydrate.
- the salt can be produced by subjecting it to a usual salt-forming treatment.
- Isolation and purification are performed by applying ordinary chemical operations such as extraction, concentration, evaporation, crystallization, filtration, recrystallization, and various types of chromatography.
- optical isomers can be isolated by a conventional method utilizing physical differences between the isomers.
- the optical isomers can be separated by a general optical resolution method, for example, fractional crystallization or chromatography. Further, the optical isomer is produced from an appropriate optically active starting compound.
- a vector (including the neomycin resistance gene) with the human CCR4 gene inserted downstream of the EF-1a promoter was prepared and transferred to the mouse pre B cell line B300-19 cells by the electoporation method. Transformation These cells were cultured in a medium supplemented with G418, and a single cell line that constantly and stably expresses human CCR4 was obtained by the limiting dilution method.
- Human CCR4 expressing cells were collected, washed with PBS, and suspended in Lysis Buffer (10 mM Hepes pH 7.5, 2 mM EDTA, protainase inhibitor). After placing the suspension on ice for 15 minutes, the cells were disrupted with a homogenizer and centrifuged (20000 rpm, 10 min, 4 ° C). 0 The supernatant was ultracentrifuged (22K, 30 min, 4 ° C). Thereafter, the pellet suspended in PBS was used as a membrane fraction in subsequent experiments.
- Lysis Buffer 10 mM Hepes pH 7.5, 2 mM EDTA, protainase inhibitor
- Test compounds were prepared at 20 mM Hepes pH 7.05, 100 mM NaCl, 5 mM MgCl,
- the compounds of Examples 2, 3, 6, 10, 16, 19, 20, 22, and 24 showed an inhibitory activity of 50% or more at 1 M concentration.
- the compound of Example 3 had an IC of 120 nM.
- the abdomen of Balb / c mice (6-10 weeks old, female, Nippon Chillers' Riva) was sensitized by applying 150 L of 3% oxazolone / ethanol solution (Sigma-Aldrich Japan).
- 10 L of a 1% oxazolone / ethanol solution was applied to both sides of the right ear.
- the test drug was administered 12 hours after application of the oxazolone solution (test drug administration group), and the control group received only the solvent used to dissolve the test drug.
- the inhibition rate was calculated by the following formula using the group to which the oxazolone solution was applied without sensitization as the normal group. The above test was performed on 5 animals per group.
- Inhibition rate (swelling of control group swelling of test drug administration group) xl00 / (swelling of control group-swelling of normal group)
- the compounds of Examples 2, 6, 10, 19 and 20 showed significant inhibitory activity at 30 mg / kg oral administration.
- the compound of the present invention has a strong CCR4 function regulating action, and a favorable pharmacological action was confirmed in models of inflammatory diseases and the like.
- a preparation containing one or more of compound (I) or a salt thereof as an active ingredient is prepared using a carrier, an excipient, and other additives that are usually used for formulation.
- parenteral injections such as intravenous injections, intramuscular injections, suppositories, transdermals, nasal formulations, inhalants, etc. Any of the forms may be used.
- the dose is determined as appropriate depending on the individual case, taking into account the symptoms, age, sex, etc. of the administration subject.However, for oral administration, it is usually about 0.001 mg / kg to 100 mg / kg per day for an adult. This should be given once or in 2-4 doses.
- the dose is usually in the range of 0.0001 mg / kg to 10 mg / kg per adult once or more times a day.
- the dose is usually in the range of 0.0001 mg / kg to 1 mg / kg for an adult once or more times a day.
- Tablets, powders, granules and the like are used as the solid composition for oral administration according to the present invention.
- one or more active substance (s) at least one inert excipient, such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, It is mixed with polyvinylpyrrolidone, magnesium aluminate metasilicate and the like.
- the composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as sodium carboxymethyl starch, or a solubilizer according to a conventional method.
- Tablets or pills may be coated with sugar coating or a gastric or enteric coating agent, if necessary.
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, and syrups. Agents, elixirs, etc., and generally used inert solvents such as purified water and ethanol.
- the composition may contain, in addition to the inert solvent, auxiliaries such as solubilizers, wetting agents and suspending agents, sweeteners, flavoring agents, fragrances and preservatives.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- Aqueous solvents include, for example, distilled water for injection and physiological saline.
- the non-aqueous solvent include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as ethanol, and polysorbate 80 (trade name).
- Such compositions may further include a tonicity agent, a preservative, a wetting agent, an emulsifier, a dispersant, a stabilizing agent, and a solubilizing agent. These are sterilized by, for example, filtration through a bacteria retaining filter, blending of a bactericide or irradiation. In addition, these are prepared by preparing a sterile solid composition, dissolving and suspending it in sterile water or a sterile injection solvent before use.
- Transmucosal agents such as inhalants and transnasal agents are used in solid, liquid or semi-solid form, and can be produced according to conventionally known methods.
- an excipient and as Ratatosu Ya starch furthermore, P H adjusting agent, a preservative, a surfactant, a lubricant, a stabilizing agent, a thickening agent, or the like may be added as appropriate.
- P H adjusting agent a preservative, a surfactant, a lubricant, a stabilizing agent, a thickening agent, or the like
- an appropriate inhalation or insufflation device can be used. Solutions or suspensions of the compounds, alone or as a powder in a formulated mixture, or in combination with a pharmaceutically acceptable carrier, using known devices or nebulizers, such as metered dose inhalers.
- Can be administered as A dry powder inhaler or the like can utilize a dry powder or a powder-containing capsule that can be used for single or multiple doses.
- a dry powder or a powder-containing capsule that can be used for single or multiple doses.
- it may be in the form of a pressurized aerosol spray using a suitable propellant, for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or diacid carbon, etc. .
- External preparations include ointments, plasters, creams, jellies, cataplasms, sprays, lotions, eye drops, eye ointments and the like. It contains commonly used ointment bases, lotion bases, aqueous or non-aqueous solutions, suspensions, emulsions and the like.
- ointment or lotion bases include polyethylene glycol, carboxybutyl polymer, white petrolatum, beeswax, polyoxyethylene hydrogenated castor oil, glyceryl monostearate, stearyl Anore konore, cetyl alcohol, lauromacrogol, sonolebitan sesquioleate and the like.
- N- (4-chlorophenol) -5- (4-fluorophenyl) -6-methyl-2- (methylsulfuryl) pyrimidin-4-amine was added to dichloromethane in dichloromethane under ice-cooling. Treatment with oral benzoic acid gave N- (4-chlorophenyl) -5- (4-fluorophenyl) -6-methyl-2- (methylsulfuryl) pyrimidin-4-amine. ES: 376.
- N- (4-chlorophenol) -2- (methylsulfur) -6-phenylpyrimidin-4-amine is treated with m-chlorobenzoic acid in dichloromethane at room temperature to give N There was obtained-(4-chlorophenol) -2- (methylsulfol) -6-phenylpyrimidin-4-amine.
- tert-butyl 4-oxopiperidine-1-carboxylate and (3S) -ethyl ethyl pecotate in the same manner as in Reference Example 10 to obtain tert-butyl (3S) -3- (ethoxycarbol) -1, 4′-Bipiperidine-1′-carboxylate (ES: 341) was obtained.
- the THF solution was added to a suspension of lithium hydroxide boron in THF, and the mixture was stirred under reflux with heating to give tert-butyl (3S) -3- (hydroxymethyl) -1,4′-bipiperidine-1 ′.
- -A carboxylate was obtained.
- the compound of Reference Examples 13-24 was prepared in the same manner as in Reference Example 2, the compound of Reference Examples 25-29 was prepared in the same manner as in Reference Example 3, and the compound of Reference Example 30- was prepared in the same manner as in Reference Example 4.
- the compound of Reference Example 38 and the compound of Reference Example 38 in the same manner as in Reference Example 11 was obtained by using the corresponding raw materials. Manufactured.
- Table 14 shows the structure and physical data of the compound of Reference Example (13-40).
- 6-Isopropyl-2-mercaptopyrimidin-4-ol is reacted with methane in ethanol in the presence of an aqueous sodium hydroxide solution at room temperature for 2 hours to give 6-isopropyl-2- (methylsulfuryl) pyrimidine-4- (3H )-Got on. EI: 184.
- 5-cyanouracil was reacted with a 50% aqueous solution of hydroxylamine in methanol at 70 ° C for 2.5 hours to give ⁇ '-hydroxy-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carboxyimidine. Damide was obtained. This compound was reacted with acetic anhydride in a mixed solvent of THF and DMF at 100 ° C for 1.5 days to give 5- (5-methyl-1,2,4-oxaziazol-3-yl) pyrimidine-2,4 (1H , 3H) -Zion was obtained.
- the compound of Reference Example 49-54 (the solvent was THF) and the compound of Reference Example 69 (the solvent was DEE) were used in the same manner as in the method of Reference Example 2.
- the compound of Reference Example 61 was treated in the same manner as in Reference Example 9 using the compound of 55-60 and the compound of 63-66 (the solvent was DME), and the compound of Reference Example 62 was treated in the same manner as in Reference Example 44.
- the compound of Reference Example 67 (the solvent is trifluoromethanesulfonic acid) and the compound of Reference Example 67 and the compound of Reference Example 68 in the same manner as in the method of Reference Example 4 (4 M hydrogen chloride-ethyl acetate solution instead of 1 M hydrochloric acid)
- the compound of Reference Example 70 was treated in the same manner as in Reference Example 5, and the compounds of Reference Examples 71 and 72 were treated in the same manner as in Reference Example 10.
- Example 73- The compound of Reference Example 76 was used in the same manner as in the method of Reference Example 48, and the compound of Reference Example 76 was used. Manufactured using The structures and physical data of the compounds of Reference Examples 49-76 are shown in Tables 5-8, respectively.
- N- (4-chlorophenol) -5-phenyl-2-pyrazine-1-ylpyrimidine-4-amine and (2R) -l- (t-butoxycarbol) piperidine- 1-Ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride and 1-hydroxybenzotriazole were added to a THF solution of 2-carboxylic acid and reacted at room temperature for 17 hours to give tert-butyl (2R)- 2-[(4- ⁇ 4-[(4-chlorophenyl) amino] -5-phenylpyrimidine-2-yl ⁇ piperazine-1-yl) carbol] piperidine-1-carboxylate Obtained. ES: 577.
- Example compounds shown in Tables 9 to 10 below were produced in the same manner as in Example 17 using the corresponding starting materials.
- Example 2 Using the same method as in Example 1 except that 250 mg of 2-chloro-N-cycloheptyl-5-phenylpyrimidine-4-amine was used (however, the reaction was carried out at 120 ° C. for 2 days using DEE as a solvent). ) Then, add 178 mg of (1 '-[4- (cycloheptylamino) -5-phenylpyrimidine-2-yl] -1,4, -bipiperidin-3-yl) methanol dihydrochloride to pale yellow Obtained as crystals.
- the medium was distilled off.
- the obtained residue was purified by silica gel column chromatography (form-form-methanol-28% aqueous ammonia) to give (1,- ⁇ 4-[(4-form-methyl) amino] -6-isopropyl -5-Phenylpyrimidine-2-yl ⁇ -1,4'-bipiperidin-3-yl) methanol (125 mg) was obtained.
- This compound was dissolved in 5 ml of methanol, and 0.2 ml of a 4 M hydrogen chloride-dioxane solution was heated to form a hydrochloride, and then the solvent was distilled off.
- Tables 13 to 15 show the structures of other compounds of the present invention. These are based on the above-mentioned manufacturing methods and examples.
- the compounds can be easily synthesized by using the methods described and those obvious to those skilled in the art, or variations thereof.
- the substituted pyrimidine derivative of the present invention has a function of modulating the function of CCR4 or TARC and / or MDC, and therefore has various inflammatory diseases, allergic diseases, autoimmune diseases and the like (eg, asthma, allergic rhinitis).
- Allergic conjunctivitis hay fever, dermatitis (atopic dermatitis, contact dermatitis), psoriasis, rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis Disease, insulin-dependent diabetes mellitus (IDDM), organ transplant rejection, cancer, inflammatory bowel disease (ulcerative colitis, Crohn's disease), interstitial cystitis, sepsis, pain) It is for. In particular, it is useful as a prophylactic / therapeutic agent for asthma, atopic dermatitis or rheumatoid arthritis.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
置換ピリミジン誘導体 Substituted pyrimidine derivatives
技術分野 Technical field
[0001] 本発明は、新規な置換ピリミジン誘導体、及び、それを有効成分とする医薬、特に 炎症性疾患治療剤に関する。 The present invention relates to a novel substituted pyrimidine derivative, and a medicament containing the same as an active ingredient, particularly a therapeutic agent for an inflammatory disease.
背景技術 Background art
[0002] 細胞遊走因子であるケモカインは構造的な特徴により大きく CXCZ ケモカインと CCZ iSケモカインの二種に分類される。また、これらケモカインの受容体は 7回膜貫 通 Gタンパク質共役型受容体ファミリーに属し、 CXCケモカインレセプターと CCケモ 力インレセプターから構成されている(Pharmacological Reviews, 52, 145, 2000)。 [0002] Chemokines, which are cell chemotactic factors, are broadly classified into two types, CXCZ chemokines and CCZ iS chemokines, depending on their structural characteristics. In addition, these chemokine receptors belong to the family of seven transmembrane G-protein coupled receptors, and are composed of CXC chemokine receptor and CC chemokine receptor (Pharmacological Reviews, 52, 145, 2000).
CCケモカインレセプター 4 (CCR4)は、 Tリンパ細胞及び胸腺からクローユングされ (Biochemical and Biophysical Research communications, 218, 337, 1996、 European Journal of Immunology, 26, 3021, 1996)、当初、 Th2タイプといわれる T細胞に主に 発現していると報告されていた(Journal of Experimental Medicine, 187, 875, 1998)。 し力し、その後の詳細な解析により CCR4は Thl及び Th2のェフエクタ一'メモリー T細 胞に広く存在することが示された(Journal of Immunology, 166, 103, 2001、 The Journal of Clinical Investigation, 108, 1331, 2001)。更に最近の研究では、 CCR4は ほとんどすべての皮膚指向性の T細胞(Nature, 400, 776, 1999)及び単球'マクロフ ァージ、榭状細胞、 NK細胞に存在することも明らかにされている(Arthritis & CC chemokine receptor 4 (CCR4) is clawed from T lymphocytes and thymus (Biochemical and Biophysical Research communications, 218, 337, 1996, European Journal of Immunology, 26, 3021, 1996), and is initially referred to as a Th2 type T cell. It was mainly reported to be expressed (Journal of Experimental Medicine, 187, 875, 1998). And subsequent detailed analysis indicated that CCR4 was widely present in ThE and Th2 efecta 'memory T cells (Journal of Immunology, 166, 103, 2001; The Journal of Clinical Investigation, 108 , 1331, 2001). More recent studies have also shown that CCR4 is present on almost all skin-directed T cells (Nature, 400, 776, 1999) and monocyte 'macrophages, dendritic cells, and NK cells ( Arthritis &
Rheumatism, 44, 1022, 2001)。 Rheumatism, 44, 1022, 2001).
CCケモカインで ¾>る Thymus and activation-regulated chemo ine、 I'ARC)と Macrophage- derived chemokine (MDC)は CCR4の特異的なリガンドである(Journal of Biological Chemistry, 272, 1503ο, 1997、 Journal of Biological chemistry, 273, 1764, 1998)。 TARCは T細胞遊走因子として(Journal of Biological Chemistry, 271, 21514, 1996)、また MDCは単球 'マクロファージ ·ΝΚ細胞の遊走因子として発見され (Journal of Experimental Medicine, 185, 1595, 1997)、どちらのケモカインも炎症性 ケモカインと恒常性ケモカインの特徴を併せ持つことが知られて 、る (Immunology Today, 20, 254, 1999)。 Thymus and activation-regulated chemoine (I'ARC) and macrophage-derived chemokine (MDC) are specific ligands for CCR4 (CCJ chemokines) (Journal of Biological Chemistry, 272, 1503ο, 1997, Journal of Biological chemistry, 273, 1764, 1998). TARC was found as a T cell chemotactic factor (Journal of Biological Chemistry, 271, 21514, 1996), and MDC was discovered as a chemotactic factor for monocytes' macrophages and ΝΚ cells (Journal of Experimental Medicine, 185, 1595, 1997). Chemokines are also known to have the characteristics of both inflammatory chemokines and homeostatic chemokines. Today, 20, 254, 1999).
CCR4とそのリガンドである TARC及び MDCは、炎症性疾患、アレルギー疾患、自己 免疫疾患等の様々な疾患に関与することが数多くの報告により示唆されている。例え ば、喘息(The Journal of Clinical Investigation, 107, 1357, 2001)、アトピー性皮膚炎 (Journal of Investigative Dermatology, 115, 640, 2000)、乾癬 (Laboratory Numerous reports have suggested that CCR4 and its ligands, TARC and MDC, are involved in various diseases such as inflammatory diseases, allergic diseases, and autoimmune diseases. For example, asthma (The Journal of Clinical Investigation, 107, 1357, 2001), atopic dermatitis (Journal of Investigative Dermatology, 115, 640, 2000), psoriasis (Laboratory
Investigation, 81, 335, 2001)、関節リウマチ(Arthritis & Rheumatism, 44, 2750, 2001)、炎症性腸疾患(Clinical & Experimental Immunology, 132, 332, 2003)等が挙 げられる。従って、 CCR4の機能調節剤はこれらの疾患等の予防又は治療剤として期 待される。上記炎症性疾患、アレルギー疾患、自己免疫疾患等の予防又は治療剤と しては、ステロイド剤等種々の薬剤が使用されているが、その治療効果と副作用の点 から、新たな作用機序に基づく薬剤の開発が切望されている。 Investigation, 81, 335, 2001), rheumatoid arthritis (Arthritis & Rheumatism, 44, 2750, 2001), and inflammatory bowel disease (Clinical & Experimental Immunology, 132, 332, 2003). Therefore, CCR4 function modulators are expected as agents for preventing or treating these diseases and the like. Various drugs such as steroids are used as prophylactic or therapeutic agents for the above-mentioned inflammatory diseases, allergic diseases, autoimmune diseases, etc. There is a strong need for the development of drugs based on this.
本願の優先権の基礎となる出願の後に公開された公報には、下記一般式で表され るピリミジン誘導体が TARC等の機能調節等に基づく抗炎症作用を有することが記載 されている(特許文献 1)。 A gazette published after the application on which the priority of the present application is based describes that a pyrimidine derivative represented by the following general formula has an anti-inflammatory effect based on the regulation of functions of TARC and the like (Patent Document 1).
[化 1] [Chemical 1]

(上記式 (I)中、 Arは置換もしくは非置換のァリール等、 R1は NR 3[R2は水素原子ま たは置換もしくは非置換の低級アルキルを表し、 R3は置換もしくは非置換の低級アル キル、置換もしくは非置換のァラルキルまたは置換もしくは非置換の芳香族複素環ァ ルキルを表す]等を、 Aは式 (III)等 [m1は 0から 2の整数を、 nは 0から 4の整数を、 a1は 0 力も置換可能な数を表す]を、 Qは- NR 7[R6および R7は、同一または異なって、水素 原子または置換もしくは非置換の低級アルキル等を表す]または置換もしくは非置換 の脂環式複素環基を表す。詳細は当該公報参照。 ) (In the above formula (I), Ar is a substituted or unsubstituted aryl, R 1 is NR 3 (R 2 is a hydrogen atom or a substituted or unsubstituted lower alkyl, and R 3 is a substituted or unsubstituted A represents lower alkyl, substituted or unsubstituted aralkyl or substituted or unsubstituted aromatic heterocyclic aralkyl, etc., and A represents formula (III), etc. [m 1 is an integer of 0 to 2, n is 0 to An integer of 4, a 1 represents a number capable of substituting 0 force], Q represents -NR 7 [R 6 and R 7 are the same or different and represent a hydrogen atom or a substituted or unsubstituted lower alkyl, etc. ] Or a substituted or unsubstituted alicyclic heterocyclic group.
当該出願には、 R2又は R3としてァリール又はへテロアルキルを有する化合物は含ま れない。 下記一般式で表される 5-シァノピリミジン誘導体が TARC等の機能調節等に基づく 抗炎症作用を有することが報告されている (特許文献 2)。 The application does not include compounds having aryl or heteroalkyl as R 2 or R 3 . It has been reported that a 5-cyanopyrimidine derivative represented by the following general formula has an anti-inflammatory effect based on the regulation of functions such as TARC (Patent Document 2).
[化 2] [Formula 2]

(上記式 (I)中、 R1及び R3は、同一または異なって、水素原子、 -NR4R5 [R4及び R5は、 同一または異なって、水素原子、置換もしくは非置換のシクロアルキル、置換もしくは 非置換のァリール、置換もしくは非置換のァラルキル等を表す]等を表し、 R2は上記 式 (II)等を表す。ここに、 Qは窒素原子または CH を、 Aは短結合、カルボ二ル等を 、 R は置換もしくは非置換の脂環式複素環基等を、 maは 0— 2の整数を、 yaは 0—置 換可能な整数を、 naは 0— 4の整数を表す。詳細は当該公報参照。 ) (In the above formula (I), R 1 and R 3 are the same or different and each represent a hydrogen atom, -NR 4 R 5 [R 4 and R 5 are the same or different and each represent a hydrogen atom, a substituted or unsubstituted cyclo Alkyl, substituted or unsubstituted aryl, substituted or unsubstituted aralkyl, etc.], and R 2 represents the above formula (II), etc., where Q is a nitrogen atom or CH, and A is a short bond , Carbonyl, R, etc., R is a substituted or unsubstituted alicyclic heterocyclic group, etc., ma is an integer of 0-2, ya is 0-replaceable integer, na is an integer of 0-4. For details, refer to the gazette.)
[0005] 下記一般式で示される化合物が、 TARC等の機能調節作用を有することが報告さ れている(特許文献 3)。 [0005] It has been reported that a compound represented by the following general formula has a function of regulating the function of TARC or the like (Patent Document 3).
[化 3]
![]()
![]()

(式中、 m及び ηは同一又は異なって 1一 3の整数 [但し m+nは 4以下]を、 R1は- NR4R5 [こ こで R4及び R5は H、置換していてもよいァラルキル等を示す]を、 rは 0— 4の整数を、 s は 0—置換可能な数を、 Gは窒素原子、 CH等を、 qは 0— 2の整数を、 Eは単結合、 -C(C=0)-等を、 R1Qは置換されていてもよい脂環式複素環基等を、 Aは単結合、 -0- 等を、 R3は H、置換されていてもよいアルキル等を表す。詳細は当該公報参照。 )(Where m and η are the same or different and are an integer of 13 [provided that m + n is 4 or less], R 1 is -NR 4 R 5 [where R 4 and R 5 are H, Represents an aralkyl etc.], r is an integer of 0-4, s is 0-substitutable number, G is a nitrogen atom, CH, etc., q is an integer of 0-2, E is A single bond, -C (C = 0)-, etc., R 1Q represents an optionally substituted alicyclic heterocyclic group, etc., A represents a single bond, -0-, etc., R 3 represents H, substituted Represents an alkyl or the like, which may be referred to for details.
[0006] また、下記一般式で示される化合物が、 GPR88の阻害に基づく中枢疾患の治療 薬としてを有用であることが記載されて 、る(特許文献 4)。

(式中、 Aは置換もしくは非置換のァリール、シクロアルキル等を、 R1は水素原子等を 、 R2は水素原子等を、 -NR 4 [式中、 R3及び R4は水素原子等、または R3と R4が隣接す る窒素原子と一緒になつて置換もしくは非置換の複素環基を形成する]を、 Xは 1,2,3,4-テトラゾール -5-ィル等を、 Yは水素原子などを、 rは 0— 4の整数を表す。詳 細は当該公報参照。 ) (Where A is a substituted or unsubstituted aryl, cycloalkyl, etc., R 1 is a hydrogen atom, etc., R 2 is a hydrogen atom, etc., -NR 4 [wherein, R 3 and R 4 are hydrogen atoms, etc. Or R 3 and R 4 together with an adjacent nitrogen atom form a substituted or unsubstituted heterocyclic group], and X is 1,2,3,4-tetrazole-5-yl and the like. , Y represents a hydrogen atom or the like, and r represents an integer of 0 to 4. For details, refer to the gazette.
当該公報には、 R2として環状アミノ基を有し、かつ Xとして環基を有する化合物の具 体的な開示は無い。また、 CCR4の機能調節作用及び抗炎症作用について、何ら開 示が無い。 This publication does not specifically disclose compounds having a cyclic amino group as R 2 and a cyclic group as X. In addition, there is no disclosure of a function regulating effect and an anti-inflammatory effect of CCR4.
[0007] 特許文献 1:国際公開第 04Z074260号パンフレット [0007] Patent Document 1: International Publication No. 04Z074260 pamphlet
特許文献 2:国際公開第 03Z082855号パンフレット Patent Document 2: International Publication No. 03Z082855 pamphlet
特許文献 3:国際公開第 03Z104230号パンフレット Patent Document 3: International Publication No. 03Z104230 pamphlet
特許文献 4:国際公開第 04Z054617号パンフレット Patent Document 4: WO 04Z054617 pamphlet
発明の開示 Disclosure of the invention
発明が解決しょうとする課題 Problems to be solved by the invention
[0008] 本発明の課題は、 CCR4の機能調節作用を有し、従来のステロイドに代わり炎症性 疾患、アレルギー疾患、または自己免疫疾患等の安全かつ強力な予防'治療薬を提 供することにある。 [0008] An object of the present invention is to provide a safe and powerful preventive / therapeutic agent for inflammatory diseases, allergic diseases, autoimmune diseases and the like, which has a function of regulating CCR4 function and replaces conventional steroids. .
課題を解決するための手段 Means for solving the problem
[0009] 本発明者等は、 CCR4の機能調節作用を有する化合物につき鋭意検討した。その 結果、置換ピペリジノ等の飽和 6員へテロ環基を 2位に有し、 4位に置換アミノ基を有 し、かつ、 5位または 6位の一方に環基を有するピリミジン誘導体力CCR4の機能調節 剤として有用であることを知見し、本発明を完成した。本発明化合物はピリミジン環 4 位に窒素原子を介して環基が直接結合することを特徴とする点で前記特許文献 1記 載の化合物とは構造が異なる。また、本発明化合物はピリミジン骨格の 5位又は 6位 に環基を有する構造を特徴としており、前記特許文献 2又は 3記載の化合物とは基 本構造が異なる。 [0009] The present inventors have diligently studied compounds having a CCR4 function regulating action. As a result, a pyrimidine derivative CCR4 having a saturated 6-membered heterocyclic group such as substituted piperidino at the 2-position, a substituted amino group at the 4-position, and a ring group at one of the 5- or 6-position. The present inventors have found that they are useful as function regulators, and have completed the present invention. The compound of the present invention is different from the compound described in Patent Document 1 in that a ring group is directly bonded to the 4-position of the pyrimidine ring via a nitrogen atom. Further, the compound of the present invention is a 5- or 6-position of the pyrimidine skeleton. And has a basic structure different from the compounds described in Patent Documents 2 and 3.
即ち、本発明は以下の化合物に関する。 That is, the present invention relates to the following compounds.
〔1〕下記一般式 (I)で示される置換ピリミジン誘導体又はその製薬学的に許容される 塩。 [1] A substituted pyrimidine derivative represented by the following general formula (I) or a pharmaceutically acceptable salt thereof.
[化 5] [Formula 5]

(式中の記号は以下の意味を示す。 (The symbols in the formula have the following meanings.
A:置換されて!、てもよ!/、ァリール又は置換されて!、てもよ!/、シクロアルキル、 A: substituted !, may! /, Aryl or substituted !, may! /, Cycloalkyl,
B:置換されて!ヽてもよ ヽ単環若しくは 2環式炭化水素環基、又は置換されて ヽても ょ 、単環若しくは 2環式へテロ環基、 B: A substituted or unsubstituted monocyclic or bicyclic hydrocarbon ring group, or a substituted or unsubstituted monocyclic or bicyclic heterocyclic group,
R1 :- R°、ハロゲン、 - OH、 -OR°,ハロゲノ低級アルキル、 R 1 : -R °, halogen, -OH, -OR °, halogeno lower alkyl,
X: CR3X«N, X: CR 3 X «N,
R3:— H、— R。又は— CN、 R 3 : — H, — R. Or— CN,
L:結合、 -CO-,低級アルキレン、 -CO- (低級アルキレン)-又は- (低級アルキレン )- CO-、 L: bond, -CO-, lower alkylene, -CO- (lower alkylene)-or-(lower alkylene) -CO-,
[化 6] [Formula 6]

D:単環式へテロシクロアルキル又はシクロアルキル、 D: monocyclic heterocycloalkyl or cycloalkyl,
但し、 Xが Nかつ Lが結合のとき、 Wは基 (i)であり、当該基は炭素原子で Xと結合す る、 R2:同一又は互いに異なって、 - R°、ハロゲン、 -OH, -0R°,ハロゲノ低級アルキル、 -R°°-0H, - CON(R8)(R9)、 -R00-O-R°, - RQQ- N(R8)(R9)、 - RQQ- CN、 - RQQ- N(R8)- CO- R°、Provided that when X is N and L is a bond, W is a group (i), and the group is bonded to X at a carbon atom. R 2 : same or different, -R °, halogen, -OH, -0R °, halogeno lower alkyl, -R °° -0H, -CON (R 8 ) (R 9 ), -R 00 -OR ° ,-R QQ -N (R 8 ) (R 9 ),-R QQ -CN,-R QQ -N (R 8 )-CO- R °,
R00-N(R8)-SO - R°、 - R°°- 0- CO- R°、 - R°°- CO - R。又は- R°°- CON(R8)(R9)、 R 00 -N (R 8 ) -SO-R °, -R °°-0-CO-R °, -R °° -CO-R. Or - R °° - CON (R 8 ) (R 9),
2 2 twenty two
R8及び R9 :同一又は互いに異なって、 - H又は- R°、 R 8 and R 9 : same or different, -H or -R °,
R11及び R12 :同一又は互いに異なって、 - 0H、 -0R°,ハロゲン、 - N(R8)(R9)、 - CN、 - N(R8)- CO- R°、 - N(R8)- SO - R°、 - 0- CO- R°、 -CO - R。又は- CON(R8)(R9)からなる群 R 11 and R 12: same or different from each other, - 0H, -0R °, halogen, - N (R 8) ( R 9), - CN, - N (R 8) - CO- R °, - N ( R 8 ) -SO-R °, -0-CO-R °, -CO-R. Or a group consisting of -CON (R 8 ) (R 9 )
2 2 twenty two
より選択される基で置換されて 、てもよ 、低級アルキル、 Optionally substituted with a group selected from the group consisting of lower alkyl,
R°:低級アルキル、 R °: lower alkyl,
RQQ:低級アルキレン、 R QQ : lower alkylene,
m: 0、 1、 2、 3又は 4。 ) m: 0, 1, 2, 3 or 4. )
本発明の好ましい態様としては以下に示される誘導体である。 A preferred embodiment of the present invention is a derivative shown below.
〔2〕下記一般式 (Π)で示される置換ピリミジン誘導体又はその製薬学的に許容される 塩。 [2] A substituted pyrimidine derivative represented by the following general formula (II) or a pharmaceutically acceptable salt thereof.
[化 7] [Formula 7]

(式中の記号は以下の意味を示す。 (The symbols in the formula have the following meanings.
A:置換されて!、てもよ!/、ァリール又は置換されて!、てもよ!/、シクロアルキル、 A: substituted !, may! /, Aryl or substituted !, may! /, Cycloalkyl,
B:置換されて!ヽてもよ ヽ単環若しくは 2環式炭化水素環基、又は置換されて ヽても ょ ヽ単環若しくは 2環式へテロ環基、 B: a substituted or unsubstituted monocyclic or bicyclic hydrocarbon ring group, or a substituted or unsubstituted monocyclic or bicyclic heterocyclic group,
R1 :- R°、ハロゲン、 - OH、 -OR°,ハロゲノ低級アルキル、 R 1 : -R °, halogen, -OH, -OR °, halogeno lower alkyl,
X: CR3X«N, X: CR 3 X «N,
Y: CR4又は N、但し、 X力 CR3のとき Yは Nを、 Xが Nのとき Yは CR4を示す、 Y: CR 4 or N, where X force CR 3 indicates Y, and X indicates N, Y indicates CR 4 .
Z: CR5R6、 NR7又は 0、 R2 :同一又は互いに異なって、 - R°、ハロゲン、 -OH, -0R°,ハロゲノ低級アルキル、 -R°°-0H, - CON(R8)(R9)、 -R00-O-R°, - RQQ- N(R8)(R9)、 - RQQ- CN、 - RQQ- N(R8)- CO- R°、 -R00-N(R8)-SO - R°、 - R°°- 0- CO- R°、 - R°°- CO - R。又は- R°°- CON(R8)(R9)、 Z: CR 5 R 6 , NR 7 or 0, R 2 : same or different, -R °, halogen, -OH, -0R °, halogeno lower alkyl, -R °° -0H, -CON (R 8 ) (R 9 ), -R 00 -OR ° ,-R QQ -N (R 8 ) (R 9 ),-R QQ -CN,-R QQ -N (R 8 )-CO- R °, -R 00 -N (R 8 ) -SO-R ° , -R °°-0-CO-R °, -R °° -CO-R. Or - R °° - CON (R 8 ) (R 9),
2 2 twenty two
R8及び R9 :同一又は互いに異なって、 - H又は- R°、 R 8 and R 9 : same or different, -H or -R °,
R5及び R6 :同一又は互いに異なって、 H又は R2に記載の基、或いは R5及び R6がー体 となって才キソ、 R 5 and R 6 : the same or different, the groups described in H or R 2 or R 5 and R 6 are
R7 : - Hゝ - R°、 - R°°- OHゝ - CON(R8)(R9)、 - R°°- 0- R°、 - R°°- N(R8)(R9)、 - R°°- CNゝ -R°° - N(R8)- CO- R。、 - R。。- N(R8)- SO - R。、 - R。。- 0- CO- R。、 - R。。- CO - R。又は- R。。- C0N(R8 R 7 :-H ゝ-R °,-R °°-OH ゝ-CON (R 8 ) (R 9 ),-R °°-0- R °,-R °°-N (R 8 ) (R 9), - R °° - CNゝ-R °° - N (R 8) - CO- R. , -R. . - N (R 8) - SO - R. , -R. . -0-CO-R. , -R. . -CO-R. Or -R. . -C0N (R 8
2 2 twenty two
XR9)、 XR 9 ),
R°:低級アルキル、 R °: lower alkyl,
RQQ :低級アルキレン、 R QQ : lower alkylene,
m : 0、 1、 2、 3又は 4、 m: 0, 1, 2, 3, or 4,
k: 0、 1又は 2。 ) k: 0, 1 or 2. )
前記〔2〕記載の誘導体のうち、より好ましい態様は以下に示す〔3〕一〔9〕記載の誘 導体及びそれらの塩である: Among the derivatives according to the above [2], more preferred embodiments are the derivatives according to the following [3]-[9] and salts thereof:
〔3〕 より好ましくは、 Xが CHであり、 Yが Nである前記〔2〕記載の誘導体。 [3] The derivative according to the above [2], wherein X is CH and Y is N.
〔4〕 より好ましくは、 Aが置換されて 、てもよ 、フエニルである前記〔3〕記載の誘導体 [4] More preferably, the derivative according to the above [3], wherein A is substituted or may be phenyl.
〔5〕 より好ましくは、 Bが置換されていてもよい単環式へテロァリール又は置換されて V、てもよ 、フエ-ルである前記〔4〕記載の誘導体。 [5] The derivative of the above-mentioned [4], wherein B is preferably an optionally substituted monocyclic heteroaryl or an optionally substituted V, or a phenyl.
〔6〕 より好ましくは、 Bが置換されて 、てもよ 、ピリジル、置換されて!、てもよ 、チェ- ル、置換されて 、てもよ 、フリル又は置換されて 、ても良 、フエ-ルである前記〔5〕 記載の誘導体。 [6] More preferably, B is substituted, may be, pyridyl, substituted !, may be, chel, substituted, may be, furyl or substituted, may be, The derivative according to the above [5], which is a phenol.
〔7〕 より好ましくは、 Zが CHである前記〔6〕記載の誘導体。 [7] More preferably, the derivative according to the above [6], wherein Z is CH.
2 2
〔8〕 より好ましくは、 R2がハロゲノ低級アルキル、 - R°°- OH、 - R°°- O- R。又は- CON(R8 [8] More preferably, R 2 is halogeno lower alkyl, -R °° -OH, -R °° -OR. Or -CON (R 8
)(R9)である前記〔5〕内至〔7〕記載の誘導体。 ) (R 9 ).
〔9〕 より好ましくは、 kが 1である前記〔8〕記載の誘導体。 [9] The derivative according to [8], wherein k is 1.
〔10〕 特に好ましくは以下に示す群力も選択される前記〔1〕記載の誘導体又はその 製薬学的に許容される塩: {1 '-[4- [(4-クロ口フエ-ル)ァミノ]- 5- (4-フルオロフェ -ル) ピリミジン- 2-ィル] -1 ,4'-ビピペリジン- 3-ィル }メタノール、 {1し [4- [(4-クロ口フエ-ル)ァ ミノ] -5-(4-フルオロフェ-ル )-6-メチルピリミジン- 2-ィル] -1,4しビピペリジン- 3-ィル } メタノール、 {1 '-[4- [(4-クロ口- 2-フルオロフェ -ル)ァミノ]- 5- (4-フルオロフェ -ル)ピリ ミジン- 2-ィル] -1,4しビピペリジン- 3-ィル }メタノール、(1, -{4- [(4-クロ口フエ-ル)アミ ノ]- 5-フエ-ルビリミジン- 2-ィル }- 1,4' -ビピペリジン- 3-ィル)メタノール、(1, - {4- [(4- クロ口フエ-ル)ァミノ]- 6-イソプロピル- 5-フエ-ルビリミジン- 2-ィル }- 1,4,-ビピベリジ ン -3-ィル)メタノール、 {1し[4-[(4-クロ口フエ-ル)ァミノ] -5-(2,4-ジフルオロフェ -ル) ピリミジン- 2-ィル] -1 ,4'-ビピペリジン- 3-ィル }メタノール、 {1し [4- [(4-クロ口フエ-ル)ァ ミノ] -5-(2-チェ-ル)ピリミジン- 2-ィル] -1,4しビピペリジン- 3-ィル }メタノール及び {1'- [4- [(4-クロ口- 2-フルオロフェ -ル)ァミノ]- 6- (4-フルオロフェ -ル)ピリミジン- 2-ィ ル]- 1,4-ビピペリジン- 3-ィル }メタノール。 (10) Particularly preferably, the derivative or the derivative thereof according to (1), wherein Pharmaceutically acceptable salts: {1 '-[4-[(4-chlorophenol) amino] -5- (4-fluorophenyl) pyrimidine-2-yl] -1,4' -Bipiperidin-3-yl} methanol, {1- [4-[(4-chlorophenyl) amino] -5- (4-fluorophenyl) -6-methylpyrimidine-2-yl] -1,4 ^ Bipiperidin-3-yl} methanol, {1 '-[4-[(4-chloro-2-fluorophenyl) amino] -5- (4-fluorophenyl) pyrimidine-2 -Yl] -1,4, -bipiperidin-3-yl} methanol, (1,-{4-[(4-chlorophenol) amino] -5-phenylpyrimidine-2-yl} -1,4'-Bipiperidin-3-yl) methanol, (1,-{4-[(4-chlorophenol) amino] -6-isopropyl-5-phenylpyrimidin-2-yl} -1,4, -Bipiberidin-3-yl) methanol, {1 [4-[(4-chlorophenol) amino] -5- (2,4-difluorophenyl) pyrimidine-2 -Yl] -1,4'-Bipiperidine-3- } Methanol, {1- [4-[(4-chlorophenol) amino] -5- (2-chyl) pyrimidine-2-yl] -1,4-bipiperidine-3-yl } Methanol and {1 '-[4-[(4-chloro-2-fluorophenyl) amino] -6- (4-fluorophenyl) pyrimidine-2-yl] -1,4-bipiperidine-3 -Ill} Methanol.
[0013] また、本発明は、前記一般式 (I)で示される置換ピリミジン誘導体またはその製薬学 的に許容される塩と、製薬的に許容される担体とからなる医薬組成物に関する。好ま しくは、 CCR4或いは TARC及び Z又は MDCの機能調節剤である前記医薬組成物で あり、より好ましくは炎症性疾患、アレルギー性疾患又は自己免疫性疾患、特に好ま しくは喘息、アトピー性皮膚炎又は関節リウマチの予防又は治療剤である前記医薬 組成物である。 [0013] The present invention also relates to a pharmaceutical composition comprising the substituted pyrimidine derivative represented by the above general formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier. Preferably, the pharmaceutical composition is a CCR4 or a modulator of TARC and Z or MDC function, more preferably an inflammatory disease, an allergic disease or an autoimmune disease, particularly preferably asthma or atopic dermatitis Alternatively, the above-mentioned pharmaceutical composition which is a prophylactic or therapeutic agent for rheumatoid arthritis.
また、別の態様としては、炎症性疾患、アレルギー性疾患又は自己免疫性疾患の 予防又は治療剤製造のための〔1〕一〔10〕記載の一般式 (I)で示される置換ピリミジ ン誘導体またはその製薬学的に許容される塩の使用であり、〔1〕一〔10〕記載の一般 式 (I)で示される置換ピリミジン誘導体またはその製薬学的に許容される塩の有効量 を、哺乳動物に投与することからなる炎症性疾患、アレルギー性疾患又は自己免疫 性疾患の予防又は治療法である。 In another embodiment, a substituted pyrimidine derivative represented by the general formula (I) according to [1]-[10] for the manufacture of a prophylactic or therapeutic agent for an inflammatory disease, an allergic disease or an autoimmune disease. Or a pharmaceutically acceptable salt thereof, wherein the substituted pyrimidine derivative represented by the general formula (I) or the pharmaceutically acceptable salt thereof according to (1)-(10) is used in an effective amount, It is a method for preventing or treating an inflammatory disease, an allergic disease or an autoimmune disease, which is administered to a mammal.
発明の効果 The invention's effect
[0014] 本発明の置換ピリミジン誘導体は、 CCR4或いは TARC及び Z又は MDCの機能調 節作用を有し、炎症性疾患、アレルギー疾患又は自己免疫疾患のモデルにおいて 強力な作用を示すという利点がある。 発明を実施するための最良の形態 [0014] The substituted pyrimidine derivative of the present invention has a function of regulating the function of CCR4 or TARC and Z or MDC, and has an advantage that it has a strong effect in a model of an inflammatory disease, an allergic disease or an autoimmune disease. BEST MODE FOR CARRYING OUT THE INVENTION
[0015] 以下、本発明を詳細に説明する。 Hereinafter, the present invention will be described in detail.
本明細書中、「アルキル」及び「アルキレン」とは、直鎖状又は分枝状の炭化水素鎖 を意味する。「低級アルキル」は、好ましくは炭素数 1一 6個のアルキル基であり、より 好ましくは炭素数 1一 4個のアルキル基、更に好ましくはメチル及びェチルである。「 低級アルキレン」は、上記「低級アルキル」の任意の水素原子 1個を除去してなる二 価基を意味し、好ましくは炭素数 1一 4個のアルキレンであり、より好ましくはメチレン、 エチレン及びプロピレンである。 In the present specification, “alkyl” and “alkylene” mean a straight or branched hydrocarbon chain. "Lower alkyl" is preferably an alkyl group having 116 carbon atoms, more preferably an alkyl group having 114 carbon atoms, and still more preferably methyl and ethyl. "Lower alkylene" means a divalent group formed by removing one arbitrary hydrogen atom from the above "lower alkyl", preferably alkylene having 114 carbon atoms, more preferably methylene, ethylene and It is propylene.
「ハロゲン」は、 F、 Cl、 Br及び Iを示す。「ハロゲノ低級アルキル」とは、好ましくは、 1 個以上のハロゲンで置換された炭素数 1一 6個のアルキルを意味し、より好ましくは 1 個以上の Fで置換された C アルキルであり、更に好ましくは、フルォロメチル、ジフル “Halogen” refers to F, Cl, Br and I. "Halogeno lower alkyl" means preferably alkyl having 1 to 6 carbon atoms substituted with one or more halogen, more preferably C alkyl substituted with one or more F, Preferably, fluoromethyl, diflu
1-6 1-6
ォロメチル、トリフルォロメチル及びトリフルォロェチルである。 Orthomethyl, trifluoromethyl and trifluoroethyl.
[0016] 「シクロアルキル」は、好ましくは炭素数 3— 10個のシクロアルキルであり、架橋され ていてもよい。より好ましくはシクロプロピル、シクロペンチル、シクロへキシル、シクロ ヘプチル及びァダマンチルである。「ァリール」は、炭素数 6— 14個の芳香族炭化水 素基を意味し、「シクロアルキル」と縮環したフエ二ル基を含む。好ましくはフエ-ル及 びナフチルであり、より好ましくはフエ-ルである。「単環若しくは 2環式炭化水素環基 」は、単環若しくは 2環式の「シクロアルキル」及び単環若しくは 2環式の「ァリール」の 両方を含む。 [0016] "Cycloalkyl" is preferably cycloalkyl having 3 to 10 carbon atoms, and may be crosslinked. More preferred are cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and adamantyl. “Aryl” means an aromatic hydrocarbon group having 6 to 14 carbon atoms, and includes a phenyl group fused with “cycloalkyl”. Preferably, they are phenyl and naphthyl, and more preferably, phenyl. The “monocyclic or bicyclic hydrocarbon ring group” includes both monocyclic or bicyclic “cycloalkyl” and monocyclic or bicyclic “aryl”.
[0017] 「単環式へテロ環基」とは、 0、 S及び N力 選択されるへテロ原子を 1一 4個含有す る単環 3— 8員、好ましくは 5— 7員環基であり、不飽和環である単環式へテロァリー ル、飽和環である単環式へテロシクロアルキル、及び前記単環式へテロァリールが部 分的に水素化された環基を含む。単環式へテロァリールとして好ましくは、ピリジル、 ピラジュル、ピリミジ -ル、ピリダジ -ル、イミダゾリル、ピロリル、トリァゾリル、テトラゾリ ル、チェニル、フリル、チアゾリル、ピラゾリル、イソチアゾリル、ォキサゾリル、イソォキ サゾリル、チアジアゾリル、ォキサジァゾリル等が挙げられる。単環式へテロシクロアル キル、又はへテロアリール基が部分的に水素化された環基として好ましくは、ピベリジ ル、ピロリジニル、ピペラジニル、ァゼパニル、ジァゼパニル、テトラヒドロフラニル、テ トラヒドロビラ-ル、モルホリニル等が挙げられる。 [0017] The term "monocyclic heterocyclic group" refers to a monocyclic 3- to 8-membered, preferably 5- to 7-membered cyclic group containing 1 to 4 hetero atoms selected from 0, S and N forces. And includes a monocyclic heteroaryl which is an unsaturated ring, a monocyclic heterocycloalkyl which is a saturated ring, and a ring group in which the monocyclic heteroaryl is partially hydrogenated. As the monocyclic heteroaryl, preferably, pyridyl, pyrazul, pyrimidyl, pyridazyl, imidazolyl, pyrrolyl, triazolyl, tetrazolyl, chenyl, furyl, thiazolyl, pyrazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, oxaziazolyl and the like. No. As a monocyclic heterocycloalkyl or a ring group in which a heteroaryl group is partially hydrogenated, preferably, piberidyl, pyrrolidinyl, piperazinyl, azepanyl, diazepanyl, tetrahydrofuranyl, Trahydroviral, morpholinyl and the like.
「2環式へテロ環基」は、前記の単環式へテロ環同士、又はベンゼン環と単環式へ テロ環が縮環した環基であり、好ましくは、インドリル、ベンゾフラ -ル、ベンゾチェ- ル、インダゾリル、ベンゾチアゾリル、ベンゾォキサゾリル、キノリル、イソキノリル、キナ ゾリル、キノキサリニル、ジヒドロベンゾフラニル、テトラヒドロキノリル、及びインドリニル 等が挙げられる。 The “bicyclic heterocyclic group” is a ring group in which the above-mentioned monocyclic heterocycles are fused together or a benzene ring and a monocyclic heterocycle are condensed. Preferably, indolyl, benzofuryl, benzoche , Indazolyl, benzothiazolyl, benzoxazolyl, quinolyl, isoquinolyl, quinazolyl, quinoxalinyl, dihydrobenzofuranyl, tetrahydroquinolyl, and indolinyl.
前記「単環式へテロ環基」及び「2環式へテロ環基」において、環原子である S又は N が酸ィ匕されォキシドゃジォキシドを形成してもよい。また、ヘテロシクロアルキル、及 びへテロァリールが部分的に水素化された環基においては、任意の炭素原子がォキ ソ基で置換されて 、てもよ 、。 In the “monocyclic heterocyclic group” and “bicyclic heterocyclic group”, the ring atom S or N may be oxidized to form an oxoxide-dioxide. Further, in the heterocycloalkyl and the ring group in which the heteroaryl is partially hydrogenated, an arbitrary carbon atom may be substituted with an oxo group.
[0018] 「置換されていてもよい」とは、「無置換」あるいは「同一又は異なる置換基を 1一 5個 有していること」を示す。 “May be substituted” means “unsubstituted” or “having 115 identical or different substituents”.
「置換されて 、てもよ 、ァリール」及び「置換されて 、てもよ 、シクロアルキル」にお ける置換基は、好ましくは、ハロゲン、低級アルキル、 - OH、 -0-低級アルキル、 -CN 、 -S-低級アルキル、 NOであり、更に好ましくは、ハロゲン、低級アルキル、 - OH The substituent in "substituted or may be aryl" and "substituted or may be cycloalkyl" is preferably halogen, lower alkyl, -OH, -0-lower alkyl, -CN , -S-lower alkyl, NO, more preferably halogen, lower alkyl, -OH
2 、 2,
-0-低級アルキル、 - CN、より更に好ましくは、ハロゲン、 - CNである。 -0-lower alkyl, -CN, even more preferably halogen, -CN.
「置換されて ヽてもよ ヽ単環若しくは 2環式炭化水素環基」及び「置換されて ヽても よい単環若しくは 2環式へテロ環基」における置換基は、好ましくはハロゲン、低級ァ ルキル、ハロゲノ低級アルキル、 - OH、 -0-低級アルキル、 - CN、 NO 、更 Substituents in the “optionally substituted monocyclic or bicyclic hydrocarbon ring group” and the “optionally substituted monocyclic or bicyclic heterocyclic group” are preferably halogen, lower Alkyl, halogeno lower alkyl, -OH, -0-lower alkyl, -CN, NO,
2、 NHであり 2 に好ましくは、ハロゲン、低級アルキル、ハロゲノ低級アルキルである。 2, NH, and preferably 2 is halogen, lower alkyl, or halogeno lower alkyl.
[0019] 式 (I)で示される本発明化合物(以下化合物 (I)と略す)は置換基の種類によっては 幾何異性体や互変異性体が存在する場合があるが、本発明にはこれらの異性体の 分離したもの、あるいは混合物が包含される。 [0019] The compound of the present invention represented by the formula (I) (hereinafter abbreviated as compound (I)) may have a geometric isomer or a tautomer depending on the type of a substituent. Or a mixture of the isomers.
また、化合物 (I)は不斉炭素原子を有する場合があり、これに基づく (R)体、 (S)体 の光学異性体が存在しうる。本発明はこれらの光学異性体の混合物や単離されたも のを全て包含する。 Further, the compound (I) may have an asymmetric carbon atom, and an (R) -form or (S) -form optical isomer based on this may exist. The present invention includes all of the optical isomers as a mixture or an isolated one.
更に、化合物 (I)には、薬理学的に許容されるプロドラッグも含まれる。薬理学的に 許容されるプロドラッグとは、加溶媒分解により又は生理学的条件下で本発明の NH 、 OH、 CO H等に変換できる基を有する化合物である。プロドラッグを形成する基としFurther, the compound (I) also includes a pharmacologically acceptable prodrug. A pharmacologically acceptable prodrug is defined as the NH4 of the present invention by solvolysis or under physiological conditions. It is a compound having a group that can be converted to OH, COH, and the like. As a base to form a prodrug
2 2
ては、 Prog. Med., 5, 2157-2161 (1985)や「医薬品の開発」(廣川書店、 1990年)第 7 卷 分子設計 163-198に記載の基が挙げられる。 For example, the groups described in Prog. Med., 5, 2157-2161 (1985) and “Development of Drugs” (Hirokawa Shoten, 1990), Vol. 7, Molecular Design 163-198.
[0020] 化合物 (I)は、酸付加塩又は置換基の種類によっては塩基との塩を形成する場合 もある。力かる塩としては、製薬学的に許容される塩であり、具体的には、塩酸、臭化 水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等の無機酸、ギ酸、酢酸、プロピオン酸、 シユウ酸、マロン酸、コハク酸、フマル酸、マイレン酸、乳酸、リンゴ酸、酒石酸、クェン 酸、メタンスルホン酸、エタンスルホン酸、ァスパラギン酸、グルタミン酸等の有機酸と の酸付加塩、ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウム等の無機 塩基、メチルァミン、ェチルァミン、エタノールァミン、リジン、オル-チン等の有機塩 基との塩やアンモニゥム塩等が挙げられる。 Compound (I) may form an acid addition salt or a salt with a base depending on the type of substituent. The strong salt is a pharmaceutically acceptable salt, specifically, an inorganic acid such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, and propion. Acid addition salts with organic acids such as acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, aspartic acid and glutamic acid, sodium And inorganic bases such as potassium, magnesium, calcium, and aluminum; salts with organic bases such as methylamine, ethylamine, ethanolamine, lysine, and ortin; and ammonium salts.
さらに、本発明は、化合物 (I)及びその塩の各種の水和物や溶媒和物及び結晶多 形の物質を含む医薬組成物をも包含する。 Furthermore, the present invention also encompasses pharmaceutical compositions containing various hydrates and solvates of compound (I) and salts thereof and polymorphic substances.
[0021] (製造法) [0021] (Manufacturing method)
本発明の有効成分である化合物 (I)及びその製薬学的に許容される塩は、その基 本骨格あるいは置換基の種類に基づく特徴を利用し、種々の公知の合成法を適用 して製造することができる。その際、官能基の種類によっては、当該官能基を原料乃 至中間体の段階で適当な保護基で保護、又は当該官能基に容易に転化可能な基 に置き換えておくことが製造技術上効果的な場合がある。このような官能基としては 例えばアミノ基、水酸基、カルボキシル基等であり、それらの保護基としては例えばグ リーン (T. W. Greene)及びウッツ (P. G. M. Wuts)著、「Protective Groups in Organic Synthesis (第 3版、 1999年)」に記載の保護基を挙げることができ、これらを反応条件 に応じて適宜選択して用いればよい。このような方法では、当該保護基を導入して反 応を行った後、必要に応じて保護基を除去、あるいは所望の基に転化することにより 、所望の化合物を得ることができる。 Compound (I), which is an active ingredient of the present invention, and pharmaceutically acceptable salts thereof are produced by applying various known synthetic methods, utilizing characteristics based on the basic skeleton or the type of substituent. can do. At that time, depending on the type of the functional group, it is effective in production technology to protect the functional group with an appropriate protecting group at the stage of the raw material or intermediate or to replace it with a group that can be easily converted to the functional group. May be important. Such functional groups include, for example, amino groups, hydroxyl groups, carboxyl groups, and the like, and their protecting groups are described, for example, in Protective Groups in Organic Synthesis (Third Edition) by Green (TW Greene) and Utz (PGM Wuts). , 1999) ", which may be appropriately selected and used according to the reaction conditions. In such a method, a desired compound can be obtained by introducing the protective group and performing a reaction, and then removing the protective group or converting it to a desired group as necessary.
また、化合物 (I)のプロドラッグは上記保護基と同様、原料乃至中間体の段階で特 定の基を導入、あるいは得られた化合物 (I)を用い反応を行うことで製造できる。反応 は通常のエステル化、アミド化、脱水等、当業者により公知の方法を適用することによ り行うことができる。 Further, the prodrug of the compound (I) can be produced by introducing a specific group at the stage of a raw material or an intermediate or by carrying out a reaction using the obtained compound (I) as in the case of the above-mentioned protective group. The reaction is carried out by applying a method known to those skilled in the art, such as ordinary esterification, amidation, dehydration, etc. Can be performed.
[0022] 第 1製法 [0022] First manufacturing method
[化 8] [Formula 8]

(式中 Lは脱離基を示す。以下同様。 ) (In the formula, L represents a leaving group. The same applies hereinafter.)
1 1
本製法は 2位に脱離基を有するピリミジン誘導体(1)に環状アミンィ匕合物(2)をイブ ソ置換させ、本発明化合物 (I)を製造する方法である。 This production method is a method for producing a compound (I) of the present invention by subjecting a pyrimidine derivative (1) having a leaving group at the 2-position to a cyclic amine diversion compound (2) by ibuso substitution.
Lが示す脱離基としては、ハロゲン、アルキルスルフィエル基、アルキルスルホ-ル Examples of the leaving group represented by L include a halogen, an alkylsulfiel group, an alkylsulfol
1 1
基等が挙げられる。反応は化合物(1)を反応に不活性な溶媒中、塩基又は酸 (好ま しくは塩化水素)の存在又は非存在下、当量あるいは過剰量の(2)を用いて冷却下 一加熱還流下に通常 1時間一 5日間行なわれる。溶媒としては反応に不活性であれ ば特に限定はされないが、例えばベンゼン、トルエン、キシレン等の芳香族炭化水素 類、ジェチルエーテル、テトラヒドロフラン (THF)、 1,4-ジォキサン、 1,2-ジメトキシエタ ン (DME)、 1,2-ジエトキシェタン (DEE)等のエーテル類、ジクロロメタン、 1,2-ジクロロェ タン、クロ口ホルム等のハロゲン化炭化水素類、メタノール、エタノール、 2-プロパノー ル、ブタノール等のアルコール類、 Ν,Ν-ジメチルホルムアミド (DMF)、 N-メチルピロリ ドン (NMP)、ジメチルスルホキシド (DMSO)等が挙げられる。塩基としては、トリェチル ァミン、ジイソプロピルェチルァミン (DIPEA)、 1,8-ジァザビシクロ- [5.4.0]- 7-ゥンデセ ン (DBU)、 2,6-ルチジン等の有機塩基、炭酸ナトリウム、炭酸カリウム、水素化ナトリウ ム、水素化カリウム、 tert-ブトキシカリウム等の無機塩基が挙げられる。 And the like. The reaction is carried out by subjecting compound (1) to a solvent inert to the reaction, in the presence or absence of a base or acid (preferably hydrogen chloride), using an equivalent or excess amount of (2) under cooling and heating under reflux. It usually takes place for one hour and five days. The solvent is not particularly limited as long as it is inert to the reaction, but, for example, aromatic hydrocarbons such as benzene, toluene, xylene, getyl ether, tetrahydrofuran (THF), 1,4-dioxane, 1,2-dimethoxy Ethers such as ethane (DME) and 1,2-dietoxetane (DEE); halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane and chloroform, methanol, ethanol, 2-propanol, butanol, etc. Alcohols, Ν, Ν-dimethylformamide (DMF), N-methylpyrrolidone (NMP), dimethylsulfoxide (DMSO) and the like. Bases include organic bases such as triethylamine, diisopropylethylamine (DIPEA), 1,8-diazabicyclo- [5.4.0] -7-indene (DBU), 2,6-lutidine, sodium carbonate, and carbonate. Inorganic bases such as potassium, sodium hydride, potassium hydride, potassium tert-butoxide and the like can be mentioned.
[0023] 第 2製法 [0023] Second manufacturing method
[化 9]

反応は、化合物(3)と化合物 (4)とを等量又は一方を過剰に用いて、後記の溶媒 中、縮合剤(例えば、ジシクロへキシルカルボジイミド(DCC)、ジイソプロピルカルボ ジイミド(DIPC)、 1-ェチル -3- (3-ジメチルァミノプロピル)カルボジイミド (WSC)、 1,1- カルボ-ルビス- 1H-イミダゾール (CDI)等)、場合によっては、更に添加剤(例えば、 N—ヒドロキシスクシンイミド(HONSu)、 1ーヒドロキシベンゾトリアゾール(HOBt)等)の 存在下、冷却下一加熱下、好ましくは常温下、通常 1時間一 1日間攪拌させることによ り行なうことができる。また、これらの試薬のかわりに、同等の作用を示す官能基を担 持した固相試薬(例えば PS -カルポジイミド(Argonaut Technologies社、米国)等)を使 用することも可能である。また、化合物 (4)と上記添加剤との活性エステル体をー且 単離後、化合物(3)と反応させてもよい。溶媒としては反応に不活性であれば特に限 定はされないが、例えば芳香族炭化水素類、エーテル類、ハロゲンィ匕炭化水素類、 Ν,Ν-ジメチルホルムアミド (DMF)、 N-メチルピロリドン (NMP)、ジメチルスルホキシド (DMSO)等が挙げられる。これらの溶媒は単独で、又は 2種以上混合して用いられる。 トリェチルァミン、 DIPEA、ピリジン等の塩基の存在下に反応させるのが、反応を円滑 に進行させる上で有利な場合がある。 The reaction is carried out by using the compound (3) and the compound (4) in an equal amount or one of them in excess, and using a condensing agent (eg, dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIPC), -Ethyl-3- (3-dimethylaminopropyl) carbodiimide (WSC), 1,1-carbylbis-1H-imidazole (CDI), etc., and in some cases, further additives (eg, N-hydroxysuccinimide ( (HONSu), 1-hydroxybenzotriazole (HOBt), etc.) with stirring under cooling and heating, preferably at room temperature, usually for 1 hour to 1 day. Further, instead of these reagents, it is also possible to use a solid phase reagent having a functional group having the same action (for example, PS-carpoimide (Argonaut Technologies, USA)). Alternatively, the active ester form of the compound (4) and the above additive may be isolated and then reacted with the compound (3). The solvent is not particularly limited as long as it is inert to the reaction.For example, aromatic hydrocarbons, ethers, halogenated hydrocarbons, Ν, Ν-dimethylformamide (DMF), N-methylpyrrolidone (NMP) , Dimethyl sulfoxide (DMSO) and the like. These solvents are used alone or in combination of two or more. In some cases, it is advantageous to carry out the reaction in the presence of a base such as triethylamine, DIPEA, pyridine or the like in order to make the reaction proceed smoothly.
第 3製法 Third manufacturing method
一般式 (I)における環基 A又は B、或いは基 R2上に種々の置換基を有する化合物は 、本発明化合物 (I)を原料として、当業者にとって自明である反応、又はこれらの変 法を用いることにより、容易に合成することができる。例えば以下の反応が適用できる General formula (I) in ring A or B, or a compound having various substituents on the group R 2, the invention compound (I) as a raw material, it is apparent reaction, or these variations to those skilled in the art Can be easily synthesized. For example, the following reaction can be applied
(1)アミド化及びエステルイ匕 水酸基又はアミノ基を有する本発明化合物を原料とし、カルボン酸又はそれらの反 応性誘導体を使用することにより、種々のアミドィ匕合物又はエステルイ匕合物が製造で きる。反応は、例えば日本化学会編「実験化学講座 (第 4版)」 22卷 (1992年)(丸善) 等に記載の方法を参考に実施できる。 (1) Amidation and esterification By using the compound of the present invention having a hydroxyl group or an amino group as a raw material and using a carboxylic acid or a reactive derivative thereof, various amido- or esteri-conjugated compounds can be produced. The reaction can be carried out, for example, by referring to the method described in “Experimental Chemistry Course (4th edition)”, edited by The Chemical Society of Japan, Vol. 22 (1992) (Maruzen).
(2)還元 (2) Reduction
NH基を有する本発明化合物は、ニトロ基を有する化合物の還元反応により製造で The compound of the present invention having an NH group can be produced by a reduction reaction of a compound having a nitro group.
2 2
きる。例えば日本化学会編「実験化学講座 (第 4版)」 26卷 (1992年)(丸善)等に記載 の方法で行なうことができる。 Wear. For example, the method can be carried out according to the method described in “Experimental Chemistry Lecture (4th edition)”, edited by The Chemical Society of Japan, Vol.
原料合成 Raw material synthesis
[化 10] [Formula 10]

(式中 Lは- C1又は- Brを、 Uは- Br、 - Cl、 -I又は- O- SO - CFを、 Qは- B(OH)、 - B(0-Wherein L is -C1 or -Br, U is -Br, -Cl, -I or -O-SO-CF, Q is -B (OH), -B (0-
2 2 3 2 低級アルキル)又は- SnBuを示す。以下同様。 ) 2 2 3 2 lower alkyl) or -SnBu. The same applies hereinafter. )
2 3 twenty three
原料化合物(1)はピリミジン誘導体 (5)をァミン化合物 (6)とィプソ置換反応を行な つた後、付加体 (7)と化合物(8)とを用いてカップリング反応を行うことにより製造でき る。ィプソ置換反応は前記第 1製法と同様の条件が適用できる。また、カップリング反 応は例えば日本化学会編「実験化学講座 (第 4版)」 25卷(1992年)(丸善)記載の方 法により実施することができる。 [化 11] The starting compound (1) can be produced by subjecting a pyrimidine derivative (5) to an ipamine substitution reaction with an amine compound (6), and then performing a coupling reaction using the adduct (7) and the compound (8). You. The same conditions as in the first production method can be applied to the ipso substitution reaction. The coupling reaction can be performed, for example, by the method described in “Chemical Experiment Course (4th edition)”, edited by The Chemical Society of Japan, Vol. 25 (1992) (Maruzen). [Formula 11]

(式中 R1Qは低級アルキル基を示す。以下同様。 ) (In the formula, R 1Q represents a lower alkyl group. The same applies hereinafter.)
式(1)中、 L1がメチルスルフィニル基である化合物(la)は上記の反応経路により製 造することができる。ここで、 R1Qの低級アルキルとしてはメチル及びェチルが好ましい 上記反応経路中、カップリング反応は前記の日本ィ匕学会編「実験化学講座 (第 4版) 」25卷(1992年)(丸善)記載の方法により実施することができる。アルキル化は、ヨウ ィ匕メチル、ジメチル硫酸等のアルキル化剤を用いて、芳香族炭化水素類、エーテル 類、ハロゲン化炭化水素類等の溶媒、又はそれらの混合溶媒中で行う。ハロゲンィ匕 はォキシ塩化リン、五塩化リン、塩ィ匕チォ-ル等の塩素化剤、又はォキシ臭化リン等 の臭素化剤を当量一大過剰用い、芳香族炭化水素類、ハロゲン化炭化水素類等の 反応に不活性な溶媒中又は無溶媒で実施する。ィプソ置換反応は前記第 1製法と 同様の条件を適用することができる。酸化は、スルフエ-ル基の酸化反応の常法を用 いればよぐ例えば日本化学会編「実験化学講座 (第 4版)」 23卷 (1992年)(丸善)記 載の方法が挙げられる。 [化 12]In the formula (1), the compound (la) wherein L 1 is a methylsulfinyl group can be produced according to the above reaction route. Here, methyl and ethyl are preferable as the lower alkyl of R 1Q. In the above reaction route, the coupling reaction is described in the above-mentioned Nippon Dani Gakkai, “Experimental Chemistry Course (4th edition)”, Vol. 25 (1992) (Maruzen) It can be carried out by the method described. The alkylation is carried out using an alkylating agent such as methyl iodide or dimethyl sulfate in a solvent such as aromatic hydrocarbons, ethers, or halogenated hydrocarbons, or a mixed solvent thereof. Halogenide uses a chlorinating agent such as phosphorus oxychloride, phosphorus pentachloride, or salty chloride, or a brominating agent such as phosphorus oxybromide in an equimolar excess to obtain an aromatic hydrocarbon or halogenated hydrocarbon. The reaction is carried out in a solvent inert to the reaction or the like or without a solvent. The same conditions as in the first production method can be applied to the ipso substitution reaction. Oxidation can be carried out by using an ordinary method of oxidation reaction of the sulfur group. For example, the method described in The Chemical Society of Japan, “Experimental Chemistry Course (4th edition)”, Vol. 23 (1992) (Maruzen) can be mentioned. . [Formula 12]

原料化合物(3)はピリミジン誘導体(1)をピペラジン(14)とィプソ置換反応を行なう ことにより製造できる。ィプソ置換反応は前記第 1製法と同様の条件が適用できる。 The starting compound (3) can be produced by subjecting the pyrimidine derivative (1) to a piperazine (14) by an ipso substitution reaction. The same conditions as in the first production method can be applied to the ipso substitution reaction.
[化 13][Formula 13]

(式中 Pはァミノ基の保護基を示す。以下同様。 ) (In the formula, P represents a protecting group for an amino group. The same applies hereinafter.)
環状アミンィ匕合物(2a)及び (2b)は、上記式に示す方法により製造できる。保護基 P 及び脱保護反応は、前述の「Protective Groups in Organic Synthesis (第 3版)」のァ ミノ基の脱保護反応等に記載の方法を適用することができる。また、還元的アルキル 化は、還元的アルキルィ匕の常法を用いることができ、例えば日本化学会編「実験化 学講座 (第 4版)」 20卷(1992年) (丸善)等に記載の方法が挙げられる。 The cyclic amine conjugates (2a) and (2b) can be produced by the method shown in the above formula. For the protecting group P and the deprotection reaction, the method described in the above-mentioned “Protective Groups in Organic Synthesis (3rd edition)” for the amino group deprotection reaction and the like can be applied. For the reductive alkylation, a conventional method of reductive alkylation can be used. For example, it is described in “Experimental Chemistry Lecture (4th edition)” edited by The Chemical Society of Japan, Vol. 20 (1992) (Maruzen). Method.
[0028] [化 14] [0028]

[0029] 上記各製法により得られた反応生成物は、遊離化合物、その塩あるいは水和物な ど各種の溶媒和物として単離され、精製される。塩は通常の造塩処理に付すことによ り製造できる。 [0029] The reaction product obtained by each of the above production methods is isolated and purified as various solvates such as a free compound, a salt thereof or a hydrate. The salt can be produced by subjecting it to a usual salt-forming treatment.
単離、精製は、抽出、濃縮、留去、結晶化、濾過、再結晶、各種クロマトグラフィー 等通常の化学操作を適用して行われる。 Isolation and purification are performed by applying ordinary chemical operations such as extraction, concentration, evaporation, crystallization, filtration, recrystallization, and various types of chromatography.
各種異性体は異性体間の物理ィ匕学的な差を利用して常法により単離できる。例え ば、光学異性体は一般的な光学分割法、例えば分別結晶化又はクロマトグラフィー 等により分離できる。また、光学異性体は、適当な光学活性な原料化合物より製造す ることちでさる。 Various isomers can be isolated by a conventional method utilizing physical differences between the isomers. For example, the optical isomers can be separated by a general optical resolution method, for example, fractional crystallization or chromatography. Further, the optical isomer is produced from an appropriate optically active starting compound.
[0030] 本発明化合物の薬理活性は以下の試験により確認した。 [0030] The pharmacological activity of the compound of the present invention was confirmed by the following test.
1. CCR4を介した [35S]GTP y S結合試験に対する作用 1. Effects on [ 35 S] GTP y S binding test via CCR4
(1) Human CCR4発現細胞株の取得 (1) Acquisition of human CCR4-expressing cell line
EF-1 aプロモーター下流にヒト CCR4遺伝子を挿入したベクター(ネオマイシン耐性 遺伝子含む)を作製し、マウス pre B細胞株 B300-19細胞にエレクト口ポレーシヨン法 によりトランスフエクシヨンした。これらの細胞を G418添加培地で培養し、限界希釈法 によりヒト CCR4を恒常的かつ安定に発現する単一の細胞株を取得した。 A vector (including the neomycin resistance gene) with the human CCR4 gene inserted downstream of the EF-1a promoter was prepared and transferred to the mouse pre B cell line B300-19 cells by the electoporation method. Transformation These cells were cultured in a medium supplemented with G418, and a single cell line that constantly and stably expresses human CCR4 was obtained by the limiting dilution method.
(2) Human CCR4発現細胞株膜画分の調整 (2) Preparation of membrane fraction of human CCR4-expressing cell line
ヒト CCR4発現細胞を回収し PBSで洗浄した後、 Lysis Buffer (10mM Hepes pH 7.5, 2mM EDTA, protainase inhibitor)で懸濁した。懸濁液を氷上に 15分間置いた後、ホ モジェナイザーにより細胞を破砕し遠心した(20000 rpm, 10 min, 4°C) 0さらに上清を 超遠心(22K, 30 min, 4°C)した後、ペレットを PBSに懸濁したものを膜画分として以後 の実験に用いた。 Human CCR4 expressing cells were collected, washed with PBS, and suspended in Lysis Buffer (10 mM Hepes pH 7.5, 2 mM EDTA, protainase inhibitor). After placing the suspension on ice for 15 minutes, the cells were disrupted with a homogenizer and centrifuged (20000 rpm, 10 min, 4 ° C). 0 The supernatant was ultracentrifuged (22K, 30 min, 4 ° C). Thereafter, the pellet suspended in PBS was used as a membrane fraction in subsequent experiments.
(3) GTP y S結合試験 (3) GTP y S binding test
試験化合物は、各濃度を 20 mM Hepes pH 7.05、 100 mM NaCl、 5 mM MgCl、 Test compounds were prepared at 20 mM Hepes pH 7.05, 100 mM NaCl, 5 mM MgCl,
2 2
GDP 2 μ Μ, Human MDCゝ [35S]GTP y S 150 pM、 Wheatgerm agglutinin SPA beads 1 mg及び Human CCR4発現細胞株膜画分 1 μ gを含有する反応混合液中で 1時間 30 分、室温で反応させ放射活性を測定した。 GDP 2 μ Human, Human MDC ゝ [ 35 S] GTPyS 150 pM, Wheatgerm agglutinin SPA beads 1 mg and Human CCR4 expressing cell line membrane fraction 1 μg 1 hour 30 minutes at room temperature in a reaction mixture And the radioactivity was measured.
主な実施例化合物の阻害活性値 (IC [nM])を示す。 3 shows the inhibitory activity values (IC [nM]) of the main examples.
50 50
実施例 1 : 82 Example 1: 82
実施例 2 : 110 Example 2: 110
実施例 3 : 25 Example 3: 25
実施例 6 : 72 Example 6: 72
実施例 10 : 46 Example 10: 46
実施例 16 : 28 Example 16: 28
実施例 19 : 52 Example 19: 52
実施例 20 : 85 Example 20: 85
実施例 24 : 86 Example 24: 86
この結果より、本発明化合物の強力な CCR4機能調節作用が確認された。 From these results, it was confirmed that the compound of the present invention has a strong CCR4 function regulating action.
また、比較化合物: 4-[(2,6-ジフルォロベンジル)ァミノ] -2-[(3R,5S)-3,5-ジメチル Comparative compound: 4-[(2,6-difluorobenzyl) amino] -2-[(3R, 5S) -3,5-dimethyl
-1 ,4, -ビピペリジン- 1, -ィル]ピリミジン- 5-カルボ-トリル (前記特許文献 2の化合物 4-1,4, -Bipiperidine-1, -yl] pyrimidine-5-carbo-tolyl (compound 4 of Patent Document 2 described above)
44)は 100 nMの濃度で 25%の阻害活性を示した。 44) showed 25% inhibitory activity at a concentration of 100 nM.
2.ボイデンチヤンバー法による細胞遊走活性試験 試験化合物を 106cells/well/0.2 mLとなるように前述のヒト CCR4発現細胞と懸濁し 、室温で 5分間インキュベートした。 lower well〖こはヒト MDC (3nMぺプロテック (PeproTech)社)を加え、専用フィルターを置き、試験化合物処理した細胞懸濁液を upper well〖こカ卩えて反応を開始させた。 COインキュベーターで 37°Cにて、 3時間ィ 2. Cell migration activity test by Boyden Chamber method The test compound was suspended in the above-mentioned human CCR4-expressing cells at a concentration of 10 6 cells / well / 0.2 mL, and incubated at room temperature for 5 minutes. The lower well was added with human MDC (3nM @ProproTech), a dedicated filter was placed, and the cell suspension treated with the test compound was added to the upper well to initiate the reaction. 3 hours at 37 ° C in a CO incubator
2 2
ンキュペートした後、 lower wellの細胞数を ATPLite- M (パーキンエルマ一社)により 定量化した。 After incubation, the number of cells in the lower well was quantified using ATPLite-M (PerkinElmer).
実施例 2、 3、 6、 10、 16、 19、 20、 22及び 24の化合物は、 1 M濃度で 50%以上の 阻害活性を示した。特に、実施例 3の化合物の IC は 120nMであった。 The compounds of Examples 2, 3, 6, 10, 16, 19, 20, 22, and 24 showed an inhibitory activity of 50% or more at 1 M concentration. In particular, the compound of Example 3 had an IC of 120 nM.
50 50
また、比較化合物: 4-[(2,6-ジフルォロベンジル)ァミノ] -2-[(3R,5S)-3,5-ジメチル -1,4, -ビピペリジン- 1, -ィル]ピリミジン- 5-カルボ-トリル (前記特許文献 2の化合物 4 44)は 1 μ Μ濃度で 22%の阻害活性を示した。 Comparative compound: 4-[(2,6-difluorobenzyl) amino] -2-[(3R, 5S) -3,5-dimethyl-1,4, -bipiperidine-1, -yl] pyrimidine -5-carbo-tolyl (compound 444 of Patent Document 2) showed an inhibitory activity of 22% at a concentration of 1 μΜ.
3.マウスォキサゾロン誘発接触性皮膚炎に対する作用 3.Effects on mouthoxazolone-induced contact dermatitis
Balb/cマウス(6— 10週齢、雌性、 日本チヤ一ルス'リバ一社)の腹部に 3%ォキサゾロ ン /エタノール溶液 150 L (シグマアルドリッチジャパン)を塗布により感作した。感作 後 6日目に 1%ォキサゾロン/エタノール溶液 10 Lを右耳の両面に塗布した。試験薬 物投与はォキサゾロン溶液の塗布 12時間後に実施し (試験薬物投与群)、コントロー ル群には試験薬物を溶解するのに用いた溶媒のみを投与した。右耳介の厚みは塗 布前と 20時間後にシックネスゲージ (ミツトヨ)を用いて測定し、腫れ (厚み増加分 = 20時間後測定値 塗布前測定値)を算出した。抑制率は感作せずにォキサゾロン溶 液を塗布した群をノーマル群として下式により計算した。なお、上記試験は、一群 5匹 で実施した。 The abdomen of Balb / c mice (6-10 weeks old, female, Nippon Chillers' Riva) was sensitized by applying 150 L of 3% oxazolone / ethanol solution (Sigma-Aldrich Japan). Six days after sensitization, 10 L of a 1% oxazolone / ethanol solution was applied to both sides of the right ear. The test drug was administered 12 hours after application of the oxazolone solution (test drug administration group), and the control group received only the solvent used to dissolve the test drug. The thickness of the right pinna was measured using a thickness gauge (Mitsutoyo) before and 20 hours after application, and the swelling (thickness increase = measured value 20 hours later, measured value before application) was calculated. The inhibition rate was calculated by the following formula using the group to which the oxazolone solution was applied without sensitization as the normal group. The above test was performed on 5 animals per group.
抑制率 = (コントロール群の腫れ 試験薬物投与群の腫れ ) xl00/ (コントロール群の 腫れ—ノーマル群の腫れ) Inhibition rate = (swelling of control group swelling of test drug administration group) xl00 / (swelling of control group-swelling of normal group)
実施例 2、 6、 10、 19及び 20の化合物は、 30 mg/kg経口投与で有意な抑制活性を 示した。 The compounds of Examples 2, 6, 10, 19 and 20 showed significant inhibitory activity at 30 mg / kg oral administration.
この結果より、本発明化合物の優れた抗炎症作用が確認された。 From these results, excellent anti-inflammatory action of the compound of the present invention was confirmed.
4.マウスコラーゲン誘発関節炎に対する作用 4.Effects on mouse collagen-induced arthritis
マウスコラーゲン誘発関節炎に対する作用は The Japanese Journal of Pharmacology, 88, 332 (2002)に記載の方法を用いて評価する。 Effects on mouse collagen-induced arthritis It is evaluated using the method described in Pharmacology, 88, 332 (2002).
上記の各試験例以外にも、例えば Immunology, 98, 345 (1999)に記載のマウス喘息 モデノレ、 Journal of Investigative Dermatorogy, 111, 86 (1998)に記載のォキサゾロン 誘発慢性接触性皮膚炎モデル (アトピー性皮膚炎モデル)等、抗炎症作用を評価す るために一般的に用いられる各種評価モデルにより、本発明化合物の薬理作用を確 認することができる。 In addition to the above test examples, for example, Immunology, 98, 345 (1999) mouse asthma modenole described, Journal of Investigative Dermatorogy, 111, 86 (1998) described oxazolone-induced chronic contact dermatitis model (atopic The pharmacological action of the compound of the present invention can be confirmed by various evaluation models generally used for evaluating an anti-inflammatory action, such as a dermatitis model).
以上の試験により、本発明化合物は強力な CCR4機能調節作用を有し、また炎症 性疾患等のモデルにぉ 、ても良好な薬理作用が確認された。 From the above test, the compound of the present invention has a strong CCR4 function regulating action, and a favorable pharmacological action was confirmed in models of inflammatory diseases and the like.
[0032] 化合物 (I)又はその塩の 1種又は 2種以上を有効成分として含有する製剤は通常 製剤化に用いられる担体ゃ賦形剤、その他の添加剤を用いて調製される。 [0032] A preparation containing one or more of compound (I) or a salt thereof as an active ingredient is prepared using a carrier, an excipient, and other additives that are usually used for formulation.
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、あるいは 静注、筋注等の注射剤、坐剤、経皮剤、経鼻剤あるいは吸入剤等による非経口投与 のいずれの形態であってもよい。投与量は症状、投与対象の年齢、性別等を考慮し て個々の場合に応じて適宜決定されるが、通常、経口投与の場合、成人 1日当たり 0.001 mg/kg乃至 100 mg/kg程度であり、これを 1回で、あるいは 2— 4回に分けて投 与する。また、症状によって静脈投与される場合は、通常、成人 1回当たり 0.0001 mg/kg乃至 10 mg/kgの範囲で 1日に 1回乃至複数回投与される。また、吸入の場合 は、通常、成人 1回当たり 0.0001 mg/kg乃至 1 mg/kgの範囲で 1日に 1回乃至複数回 投与される。 For oral administration, tablets, pills, capsules, granules, powders, liquids, etc., or parenteral injections, such as intravenous injections, intramuscular injections, suppositories, transdermals, nasal formulations, inhalants, etc. Any of the forms may be used. The dose is determined as appropriate depending on the individual case, taking into account the symptoms, age, sex, etc. of the administration subject.However, for oral administration, it is usually about 0.001 mg / kg to 100 mg / kg per day for an adult. This should be given once or in 2-4 doses. In the case of intravenous administration depending on the symptoms, the dose is usually in the range of 0.0001 mg / kg to 10 mg / kg per adult once or more times a day. In the case of inhalation, the dose is usually in the range of 0.0001 mg / kg to 1 mg / kg for an adult once or more times a day.
本発明による経口投与のための固体組成物としては、錠剤、散剤、顆粒剤等が用 いられる。このような固体組成物においては、一つ又はそれ以上の活性物質力 少な くとも一つの不活性な賦形剤、例えば乳糖、マン-トール、ブドウ糖、ヒドロキシプロピ ルセルロース、微結晶セルロース、デンプン、ポリビュルピロリドン、メタケイ酸アルミン 酸マグネシウム等と混合される。組成物は、常法に従って、不活性な添加剤、例えば ステアリン酸マグネシウム等の滑沢剤やカルボキシメチルスターチナトリウム等の崩壊 剤、溶解補助剤を含有していてもよい。錠剤又は丸剤は必要により糖衣又は胃溶性 若しくは腸溶性コーティング剤で被膜してもよ 、。 Tablets, powders, granules and the like are used as the solid composition for oral administration according to the present invention. In such a solid composition, one or more active substance (s) at least one inert excipient, such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, It is mixed with polyvinylpyrrolidone, magnesium aluminate metasilicate and the like. The composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as sodium carboxymethyl starch, or a solubilizer according to a conventional method. Tablets or pills may be coated with sugar coating or a gastric or enteric coating agent, if necessary.
[0033] 経口投与のための液体組成物は、薬剤的に許容される乳剤、液剤、懸濁剤、シロッ プ剤、エリキシル剤等を含み、一般的に用いられる不活性な溶剤、例えば精製水、ェ タノールを含む。この組成物は不活性な溶剤以外に可溶化剤、湿潤剤、懸濁化剤の ような補助剤、甘味剤、矯味剤、芳香剤、防腐剤を含有していてもよい。 [0033] Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, and syrups. Agents, elixirs, etc., and generally used inert solvents such as purified water and ethanol. The composition may contain, in addition to the inert solvent, auxiliaries such as solubilizers, wetting agents and suspending agents, sweeteners, flavoring agents, fragrances and preservatives.
非経口投与のための注射剤としては、無菌の水性又は非水性の液剤、懸濁剤、乳 剤を含む。水性の溶剤としては、例えば注射用蒸留水及び生理食塩水が含まれる。 非水性の溶剤としては、例えばプロピレングリコール、ポリエチレングリコール、オリー ブ油のような植物油、エタノールのようなアルコール類、ポリソルベート 80 (商品名)等 がある。このような組成物は、さらに等張化剤、防腐剤、湿潤剤、乳化剤、分散剤、安 定化剤、溶解補助剤を含んでもよい。これらは例えばバクテリア保留フィルターを通 す濾過、殺菌剤の配合又は照射によって無菌化される。また、これらは無菌の固体 組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶解、懸濁して使用す ることちでさる。 Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions. Aqueous solvents include, for example, distilled water for injection and physiological saline. Examples of the non-aqueous solvent include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as ethanol, and polysorbate 80 (trade name). Such compositions may further include a tonicity agent, a preservative, a wetting agent, an emulsifier, a dispersant, a stabilizing agent, and a solubilizing agent. These are sterilized by, for example, filtration through a bacteria retaining filter, blending of a bactericide or irradiation. In addition, these are prepared by preparing a sterile solid composition, dissolving and suspending it in sterile water or a sterile injection solvent before use.
[0034] 吸入剤や経鼻剤等の経粘膜剤は固体、液体、半固体状のものが用いられ、従来公 知の方法に従って製造することができる。例えば、ラタトースゃ澱粉のような賦形剤や 、更に、 PH調整剤、防腐剤、界面活性剤、滑沢剤、安定剤や増粘剤等が適宜添加さ れていてもよい。投与は、適当な吸入又は吹送のためのデバイスを使用することがで きる。例えば、計量投与吸入デバイス等の公知のデバイスや噴霧器を使用して、化 合物を単独で又は処方された混合物の粉末として、もしくは医薬的に許容し得る担 体と組み合わせて溶液又は懸濁液として投与することができる。乾燥粉末吸入器等 は、単回又は多数回の投与用のものであってもよぐ乾燥粉末又は粉末含有カプセ ルを利用することができる。あるいは、適当な駆出剤、例えば、クロロフルォロアルカ ン、ヒドロフルォロアルカン又は二酸ィ匕炭素等の好適な気体を使用した加圧エアゾー ルスプレー等の形態であってもよ 、。 [0034] Transmucosal agents such as inhalants and transnasal agents are used in solid, liquid or semi-solid form, and can be produced according to conventionally known methods. For example, an excipient and as Ratatosu Ya starch, furthermore, P H adjusting agent, a preservative, a surfactant, a lubricant, a stabilizing agent, a thickening agent, or the like may be added as appropriate. For administration, an appropriate inhalation or insufflation device can be used. Solutions or suspensions of the compounds, alone or as a powder in a formulated mixture, or in combination with a pharmaceutically acceptable carrier, using known devices or nebulizers, such as metered dose inhalers. Can be administered as A dry powder inhaler or the like can utilize a dry powder or a powder-containing capsule that can be used for single or multiple doses. Alternatively, it may be in the form of a pressurized aerosol spray using a suitable propellant, for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or diacid carbon, etc. .
[0035] 外用剤としては、軟膏剤、硬膏剤、クリーム剤、ゼリー剤、パップ剤、噴霧剤、ローシ ヨン剤、点眼剤、眼軟膏等を包含する。一般に用いられる軟膏基剤、ローション基剤 、水性又は非水性の液剤、懸濁剤、乳剤等を含有する。例えば、軟膏又はローション 基剤としては、ポリエチレングリコール、カルボキシビュルポリマー、白色ワセリン、サ ラシミツロウ、ポリオキシエチレン硬化ヒマシ油、モノステアリン酸グリセリン、ステアリル ァノレコーノレ、セチルアルコール、ラウロマクロゴール、セスキォレイン酸ソノレビタン等 が挙げられる。 実施例 External preparations include ointments, plasters, creams, jellies, cataplasms, sprays, lotions, eye drops, eye ointments and the like. It contains commonly used ointment bases, lotion bases, aqueous or non-aqueous solutions, suspensions, emulsions and the like. For example, ointment or lotion bases include polyethylene glycol, carboxybutyl polymer, white petrolatum, beeswax, polyoxyethylene hydrogenated castor oil, glyceryl monostearate, stearyl Anore konore, cetyl alcohol, lauromacrogol, sonolebitan sesquioleate and the like. Example
[0036] 以下、実施例に基づき本発明化合物 (I)の製法を更に詳細に説明する。本発明化 合物は下記実施例に記載の化合物に限定されるものではな 、。また原料化合物の 製法を参考例に示す。 Hereinafter, the production method of the compound (I) of the present invention will be described in more detail with reference to Examples. The compounds of the present invention are not limited to the compounds described in the following Examples. The production methods of the starting compounds are shown in Reference Examples.
また、参考例及び後記表中以下の略号を用いる。置換基の前の数字は置換位置を 示す。 The following abbreviations are used in Reference Examples and Tables below. The number before the substituent indicates the substitution position.
Ex:実施例番号、 REx:参考例番号、 Dat:物理化学的データ(F: FAB-MS(M+H)+、 FN : FAB- MS(M- H)―、 ES: ESI- MS(M+H)+、 ESN : ESI- MS(M- H)―、 EI : m- MS(M+)、 AP: APCI— MS(M+H)+、 APN:APCI— MS(M— H)―、 NMR1 : CDC1中の NMRにおける特徴 Ex: Example number, REx: Reference example number, Dat: Physicochemical data (F: FAB-MS (M + H) +, FN: FAB-MS (M-H)-, ES: ESI-MS (M + H) +, ESN: ESI-MS (M-H)-, EI: m-MS (M +), AP: APCI- MS (M + H) +, APN: APCI- MS (M-H)-, NMR1: Features in NMR in CDC1
3 Three
的なピークの δ (ppm)、 NMR2 : DMSO-d中の NMRにおける特徴的なピークの δ Δ (ppm) of typical peak, NMR2: δ of characteristic peak in NMR in DMSO-d
6 6
(ppm)、 EA:元素分析値 (%) (Cal:計算値; Fnd:実測値))、 Sal:塩及び含有溶媒 (HC1: 塩酸塩、無記載:フリー体、成分の前の数字は例えば 2HC1は 2塩酸塩を示す)、 Str: 構造式、 Syn:製造法 (数字は同様に製造した実施例番号を示す)、 Config:立体化学 (*を付記した炭素の絶対配置を示す)、 Me :メチル、 Ph:フエ-ル、 Ac :ァセチル、 Boc: tert-ブトキシカノレボニノレ。 (ppm), EA: Elemental analysis value (%) (Cal: Calculated value; Fnd: Actual measurement value)), Sal: Salt and contained solvent (HC1: Hydrochloride, No description: Free form, number before component is for example 2HC1 represents dihydrochloride), Str: Structural formula, Syn: Production method (Numbers represent the example numbers produced in the same manner), Config: Stereochemistry (Indicates the absolute configuration of carbon marked with *), Me : Methyl, Ph: Fer, Ac: Acetyl, Boc: tert-Butoxycanoleboninole.
[0037] 参考例 1 [0037] Reference Example 1
5-ブロモ -2,4-ジクロロピリミジンと 4-クロロア-リンを DIPEA存在下、ァセトニトリル中 で 3日間加熱還流して 5-ブロモ -2-クロ口- N- (4-クロ口フエ-ル)ピリミジン- 4-アミンを 得た。 ES: 318, 320。 Heat 5-bromo-2,4-dichloropyrimidine and 4-chloroaline in acetonitrile in the presence of DIPEA under reflux for 3 days to give 5-bromo-2-chloro-N- (4-chloro-methyl) Pyrimidin-4-amine was obtained. ES: 318, 320.
参考例 2 Reference example 2
5-ブロモ -2-クロ口- N- (4-クロ口フエ-ル)ピリミジン- 4-ァミンと 4-フルオロフェ -ルボ ロン酸のァセトニトリル溶液に炭酸ナトリウム水溶液、テトラキス (トリフエニルホスフィン) パラジウムを順次を加え 85°Cで加熱して 2-クロ口- N-(4-クロ口フエ-ル)- 5-(4-フルォ 口フエ-ル)ピリミジン- 4-アミンを得た。 ES: 334。 An aqueous solution of sodium carbonate and tetrakis (triphenylphosphine) palladium were added successively to an acetonitrile solution of 5-bromo-2-chloro-N- (4-chlorophenol) pyrimidine-4-amine and 4-fluorophenylboronic acid. And heated at 85 ° C. to give 2-chloro-N- (4-chlorophenol) -5- (4-fluorophenol) pyrimidin-4-amine. ES: 334.
参考例 3 Reference example 3
4_クロ口— 5_ョード _6_メチル—2_ (メチルスルフエ-ル)ピリミジンを用いて参考例 2と同 様にして、 4-クロ口- 5-(4-フルオロフェ-ル )-6-メチル -2- (メチルスルフエ-ル)ピリミジ ンを得た。 F: 269。 4 _ black port - 5 _ Yodo _ 6 _ methyl - 2 _ (Mechirusurufue - Le) pyrimidine Reference Example 2 using the same Thus, 4-chloro-5- (4-fluorophenyl) -6-methyl-2- (methylsulfur) pyrimidine was obtained. F: 269.
参考例 4 Reference example 4
4-クロ口- 5-(4-フルオロフェ-ル )-6-メチル -2- (メチルスルフエ-ル)ピリミジンと 4-ク 口ロア-リンの 2-プロパノール溶液に 1M塩酸水溶液をカ卩え、 3時間加熱還流して、 N- (4-クロ口フエ-ル)- 5- (4-フルオロフェ-ル )-6-メチル -2- (メチルスルフエ-ル)ピリ ミジン- 4-アミンを得た。 ES: 360。 A 1M aqueous hydrochloric acid solution was added to a 2-propanol solution of 4-chloro-5- (4-fluorophenyl) -6-methyl-2- (methylsulferyl) pyrimidine and 4-cyclolorine, and 3 The mixture was heated under reflux for an hour to obtain N- (4-chlorophenol) -5- (4-fluorophenyl) -6-methyl-2- (methylsulfur) pyrimidin-4-amine. ES: 360.
参考例 5 Reference Example 5
N- (4-クロ口フエ-ル)- 5- (4-フルオロフェ-ル )-6-メチル -2- (メチルスルフエ-ル)ピ リミジン- 4-アミンをジクロロメタン中、氷冷下、 m-クロ口安息香酸で処理して、 N- (4-ク ロロフエ-ル)- 5- (4-フルオロフェ-ル )-6-メチル -2- (メチルスルフィ -ル)ピリミジン -4-アミンを得た。 ES: 376。 N- (4-chlorophenol) -5- (4-fluorophenyl) -6-methyl-2- (methylsulfuryl) pyrimidin-4-amine was added to dichloromethane in dichloromethane under ice-cooling. Treatment with oral benzoic acid gave N- (4-chlorophenyl) -5- (4-fluorophenyl) -6-methyl-2- (methylsulfuryl) pyrimidin-4-amine. ES: 376.
参考例 6 Reference example 6
N- (4-クロ口フエ-ル)- 2- (メチルスルフエ-ル)- 6-フエ-ルピリミジン- 4-アミンをジク ロロメタン中、室温下、 m-クロ口安息香酸で処理して、 N- (4-クロ口フエ-ル)- 2- (メチ ルスルホ-ル)- 6-フエ-ルビリミジン- 4-アミンを得た。 F: 360。 N- (4-chlorophenol) -2- (methylsulfur) -6-phenylpyrimidin-4-amine is treated with m-chlorobenzoic acid in dichloromethane at room temperature to give N There was obtained-(4-chlorophenol) -2- (methylsulfol) -6-phenylpyrimidin-4-amine. F: 360.
参考例 7 Reference Example 7
水素化ナトリウム、 DMFの混合液に氷冷下 (4-フルオロフェ -ル)酢酸メチルおよび ギ酸メチルを滴下し、室温で 17時間攪拌した後、ヨウ化メチルをカ卩え、さらに室温で 5 時間攪拌して 2-(4-フルオロフェニル )-3-メトキシアクリル酸メチルを得た。この化合物 をエタノール中、ナトリウムメトキシド存在下、チォ尿素と反応して、 5-(4-フルオロフェ -ル) -2-チォキソ- 2,3-ジヒドロピリミジン- 4(1H)-オンを得た。 F: 223。 Methyl (4-fluorophenyl) acetate and methyl formate were added dropwise to a mixture of sodium hydride and DMF under ice-cooling, and the mixture was stirred at room temperature for 17 hours, then methyl iodide was added, and the mixture was further stirred at room temperature for 5 hours. This gave methyl 2- (4-fluorophenyl) -3-methoxyacrylate. This compound was reacted with thiourea in ethanol in the presence of sodium methoxide to give 5- (4-fluorophenyl) -2-thioxo-2,3-dihydropyrimidin-4 (1H) -one. F: 223.
参考例 8 Reference Example 8
5- (4-フルオロフェ-ル )-2-チォキソ- 2,3-ジヒドロピリミジン- 4(1H)-オンを DMF中、 ヨウ化メチルと反応して、 5-(4-フルオロフェ-ル )-2- (メチルスルフエ-ル)ピリミジン - 4(3H)-オンを得た。 ES: 237。 5- (4-Fluorophenyl) -2-thioxo-2,3-dihydropyrimidin-4 (1H) -one is reacted with methyl iodide in DMF to give 5- (4-fluorophenyl) -2 -(Methylsulfur) pyrimidin-4 (3H) -one was obtained. ES: 237.
参考例 9 Reference Example 9
5- (4-フルオロフェ-ル )-2- (メチルスルフエ-ル)ピリミジン- 4(3H)-オンをォキシ塩 ィ匕リンと 100°Cで 2時間反応して、 4-クロ口- 5-(4-フルオロフェ-ル )-2- (メチルスルフエ 二ノレ)ピリミジンを得た。 ES: 255。 5- (4-fluorophenyl) -2- (methylsulfur) pyrimidin-4 (3H) -one The reaction was carried out at 100 ° C for 2 hours with Rindo phosphorus to obtain 4-chloro-5- (4-fluorophenyl) -2- (methylsulfeninole) pyrimidine. ES: 255.
参考例 10 Reference example 10
tert-ブチル 4-ォキソピペリジン- 1-カルボキシラート、 3-ピぺリジンメタノール、 10% ノ ラジウム炭素及びメタノール混合物を、水素雰囲気下攪拌して、 tert-ブチル 3- (ヒ ドロキシメチル) -1,4'-ビピペリジン- 1'-カルボキシラート。 ES: 299。 A mixture of tert-butyl 4-oxopiperidine-1-carboxylate, 3-piperidinemethanol, 10% noradium carbon and methanol was stirred under a hydrogen atmosphere to give tert-butyl 3- (hydroxymethyl) -1, 4'-Bipiperidine-1'-carboxylate. ES: 299.
[0039] 参考例 11 [0039] Reference Example 11
tert-ブチル 3- (ヒドロキシメチル) -1,4'-ビピペリジン- 1'-カルボキシラートをメタノー ル中、 4M塩酸-酢酸ェチル溶液で処理して、 1,4'-ビピペリジン- 3-メタノール 2塩酸塩 を得た。 ES : 199。 tert-Butyl 3- (hydroxymethyl) -1,4'-bipiperidine-1'-carboxylate is treated with 4M hydrochloric acid-ethyl acetate solution in methanol to give 1,4'-bipiperidine-3-methanol 2HCl Salt was obtained. ES: 199.
参考例 12 Reference Example 12
tert-ブチル 4-ォキソピペリジン- 1-カルボキシラートと (3S)--ペコチン酸ェチルを 用いて参考例 10と同様にして tert-ブチル (3S)-3- (エトキシカルボ-ル) -1,4'-ビピ ペリジン- 1'-カルボキシラート(ES: 341)を得た。この THF溶液を水酸ィ匕ホウ素リチウ ムの THF懸濁液に加え、加熱還流下攪拌することにより、 tert-ブチル (3S)-3- (ヒドロ キシメチル) -1,4'-ビピペリジン- 1'-カルボキシラートを得た。 ES: 299。 tert-butyl 4-oxopiperidine-1-carboxylate and (3S) -ethyl ethyl pecotate in the same manner as in Reference Example 10 to obtain tert-butyl (3S) -3- (ethoxycarbol) -1, 4′-Bipiperidine-1′-carboxylate (ES: 341) was obtained. The THF solution was added to a suspension of lithium hydroxide boron in THF, and the mixture was stirred under reflux with heating to give tert-butyl (3S) -3- (hydroxymethyl) -1,4′-bipiperidine-1 ′. -A carboxylate was obtained. ES: 299.
参考例 2の方法と同様にして参考例 13— 24の化合物を、参考例 3の方法と同様に して参考例 25— 29の化合物を、参考例 4の方法と同様にして参考例 30— 37の化 合物を、参考例 5の方法と同様にして参考例 38及び 39の化合物を、参考例 11の方 法と同様にして参考例 40の化合物を、それぞれ対応する原料を使用して製造した。 参考例(13— 40)の化合物の構造及び物理ィ匕学的データを表 1一 4にそれぞれ示 す。 The compound of Reference Examples 13-24 was prepared in the same manner as in Reference Example 2, the compound of Reference Examples 25-29 was prepared in the same manner as in Reference Example 3, and the compound of Reference Example 30- was prepared in the same manner as in Reference Example 4. Using the compound of Reference Example 38 and the compound of Reference Example 38 in the same manner as in Reference Example 11, the compound of Reference Example 40 was obtained by using the corresponding raw materials. Manufactured. Table 14 shows the structure and physical data of the compound of Reference Example (13-40).
[0040] 参考例 41 [0040] Reference Example 41
ェチル 2-ピリジン- 2-ィルァセタート、無水酢酸、オルトギ酸ェチルの混合液を、 塩化亜鉛存在下 12時間加熱還流し、ェチル 3-エトキシ -2-ピリジン- 2-ィルァクリラ ートを得た。この化合物をエタノール中、ナトリウムメトキシド存在下、チォ尿素と反応 して、 5-ピリジン- 2-ィル- 2-チォキソ- 2,3-ジヒドロピリミジン- 4(1H)-オンのナトリウム塩 を得た。 ES: 206。 参考例 42 A mixed solution of ethyl 2-pyridine-2-yl acetate, acetic anhydride and ethyl ethyl formate was heated under reflux in the presence of zinc chloride for 12 hours to obtain ethyl 3-ethoxy-2-pyridine-2-yl acrylate. This compound was reacted with thiourea in ethanol in the presence of sodium methoxide to obtain the sodium salt of 5-pyridin-2-yl-2-thioxo-2,3-dihydropyrimidin-4 (1H) -one. Was. ES: 206. Reference Example 42
5-ピリジン- 2-ィル- 2-チォキソ- 2,3-ジヒドロピリミジン- 4(1H)-オンのナトリウム塩を 用いて参考例 8と同様にして 2- (メチルスルファ -ル) -5-ピリジン- 2-ィルピリミジン -4(3H)_オンを得た。この化合物のジクロロエタン溶液に、氷冷下トリエチルァミン、メ シルクロリドをカ卩えて 1時間攪拌し、 2— (メチルスルファ-ル )—5—ピリジン—2—ィルピリミジ ン -4-ィル メタンスルホナートを得た。この化合物と 4-クロロア-リンのエタノール溶 液を、メシル酸存在下 4時間加熱還流して N- (4-クロ口フエ-ル)- 2- (メチルスルファ二 ル)- 5-ピリジン- 2-ィルピリミジン- 4-アミンを得た。 ES: 329。 5-Pyridin-2-yl-2-thioxo-2,3-dihydropyrimidin-4 (1H) -one 2- (methylsulfuryl) -5-pyridine in the same manner as in Reference Example 8 using the sodium salt -2-ylpyrimidin-4 (3H) _one was obtained. Dichloroethane solution of the compound under ice-cooling Toriechiruamin, stirring the main Shirukurorido mosquito卩Ete 1 hour, 2 - pyridine-2-Irupirimiji down 4 I le methanesulfonate - (Mechirusurufa - Le) - 5 Obtained. A solution of this compound and 4-chloroaline in ethanol is heated under reflux for 4 hours in the presence of mesylic acid, and N- (4-chlorophenol) -2- (methylsulfanyl) -5-pyridine-2- The ylpyrimidin-4-amine was obtained. ES: 329.
参考例 43 Reference Example 43
6-イソプロピル- 2-メルカプトピリミジン- 4-オールをエタノール中、水酸化ナトリウム 水溶液存在下、ョードメタンと室温下 2時間反応させ、 6-イソプロピル- 2- (メチルスル ファ -ル)ピリミジン- 4- (3H)-オンを得た。 EI : 184。 6-Isopropyl-2-mercaptopyrimidin-4-ol is reacted with methane in ethanol in the presence of an aqueous sodium hydroxide solution at room temperature for 2 hours to give 6-isopropyl-2- (methylsulfuryl) pyrimidine-4- (3H )-Got on. EI: 184.
参考例 44 Reference example 44
6-イソプロピル- 2- (メチルチオ)ピリミジン- 4-(3H)-オンをクロ口ホルム中、 N-ョードコ ハク酸イミドと 70°Cで 2時間反応させ、 5-ョード -6-イソプロピル- 2- (メチルチオ)ピリミ ジン- 4- (3H)-オンを得た。 F: 311。 6-Isopropyl-2- (methylthio) pyrimidin-4- (3H) -one is reacted with N-odosuccinimide at 70 ° C for 2 hours in chloroform to give 5-Iodo-6-isopropyl-2- (Methylthio) pyrimidin-4- (3H) -one was obtained. F: 311.
参考例 45 Reference Example 45
5-シァノウラシルをメタノール中、 50%ヒドロキシルァミン水溶液と 70°Cで 2.5時間反 応させ、 Ν'-ヒドロキシ- 2,4-ジォキソ -1,2, 3,4-テトラヒドロピリミジン- 5-カルボキシイミ ダミドを得た。この化合物を THFと DMFの混合溶媒中、無水酢酸と 100°Cで 1.5日間 反応させ、 5-(5-メチル -1,2,4-ォキサジァゾール -3-ィル)ピリミジン- 2,4(1H,3H)-ジォ ンを得た。この化合物に、ォキシ塩化リン、ジェチルァ-リンをカ卩ぇ 100°Cで 3時間反 応させて、 2,4-ジクロロ- 5-(5-メチル -1,2,4-ォキサジァゾール -3-ィル)ピリミジンを得 た。 NMR1 : 9.15 (1H, s), 2.73 (3H, s)。 5-cyanouracil was reacted with a 50% aqueous solution of hydroxylamine in methanol at 70 ° C for 2.5 hours to give Ν'-hydroxy-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carboxyimidine. Damide was obtained. This compound was reacted with acetic anhydride in a mixed solvent of THF and DMF at 100 ° C for 1.5 days to give 5- (5-methyl-1,2,4-oxaziazol-3-yl) pyrimidine-2,4 (1H , 3H) -Zion was obtained. The compound was reacted with phosphorus oxychloride and getylurin at 100 ° C for 3 hours to give 2,4-dichloro-5- (5-methyl-1,2,4-oxaziazol-3-yl). L) Pyrimidine was obtained. NMR1: 9.15 (1H, s), 2.73 (3H, s).
参考例 46 Reference Example 46
tert-ブチル 4-[(3-ヒドロキシプロピル)ァミノ]ピぺリジン- 1-カルボキシラートのジク ロロェタン溶液に、酢酸、ホルムアルデヒド水溶液、トリァセトキシ水素化ホウ素ナトリ ゥムを添カ卩し、室温で 19時間反応させ、 tert-ブチル 4-[(3-ヒドロキシプロピル) (メチ ル)ァミノ]ピぺリジン- 1-カルボキシラートを得た。 F: 2730 Acetic acid, an aqueous formaldehyde solution and sodium triacetoxyborohydride were added to a solution of tert-butyl 4-[(3-hydroxypropyl) amino] piperidine-1-carboxylate in dichloroethane, and the mixture was added at room temperature for 19 hours. React with tert-butyl 4-[(3-hydroxypropyl) (methyl Ru) amino] piperidine-1-carboxylate was obtained. F: 273 0
参考例 47 Reference Example 47
tert-ブチル 4-[(3-ヒドロキシプロピル)ァミノ]ピぺリジン- 1-カルボキシラートを DMF 中、炭酸ナトリウム存在下、ヨウ化工チルと室温で 18時間反応させ、 tert-ブチル 4-[ ェチル (3-ヒドロキシプロピル)ァミノ]ピぺリジン- 1-カルボキシラートを得た。 F:287。 参考例 48 tert-Butyl 4-[(3-hydroxypropyl) amino] piperidine-1-carboxylate is reacted with iodide tyl in DMF in the presence of sodium carbonate at room temperature for 18 hours to give tert-butyl 4- [ethyl ( 3-Hydroxypropyl) amino] piperidine-1-carboxylate was obtained. F: 287. Reference Example 48
tert-ブチル 4-ォキソピペリジン- 1-カルボキシラート、 4-アミノブタン- 1-オールを 用いて参考例 10と同様の操作を行い、 tert-ブチル 4-[(4-ヒドロキシブチル)ァミノ]ピ ペリジン- 1-カルボキシラートを得た。この化合物を用いて参考例 46と同様の操作を 行!、、 tert-ブチル 4-[(4-ヒドロキシブチル) (メチル)ァミノ]ピぺリジン- 1-カルボキシラ ートを得た。この化合物を用いて参考例 11と同様の操作を行 、4- [メチル (ピペリジン -4-ィル)ァミノ]ブタノールを得た。 NMR2: 2.65 (3H, d, J = 5.2Hz), 1.89-2.05 (2H, m), 1.38-1.53 (2H, m)。 The same operation as in Reference Example 10 was performed using tert-butyl 4-oxopiperidine-1-carboxylate and 4-aminobutan-1-ol to obtain tert-butyl 4-[(4-hydroxybutyl) amino] piperidine. -1-carboxylate was obtained. The same operation as in Reference Example 46 was performed using this compound to obtain tert-butyl 4-[(4-hydroxybutyl) (methyl) amino] piperidine-1-carboxylate. The same operation as in Reference Example 11 was performed using this compound to obtain 4- [methyl (piperidin-4-yl) amino] butanol. NMR2: 2.65 (3H, d, J = 5.2 Hz), 1.89-2.05 (2H, m), 1.38-1.53 (2H, m).
[0042] 参考例 49一 76 [0042] Reference Example 49-76
参考例 1の方法と同様にして参考例 49一 54の化合物(但し、溶媒は THF)及び参 考例 69の化合物(但し、溶媒は DEE)を、参考例 2の方法と同様にして参考例 55— 6 0の化合物及び 63— 66の化合物(但し、溶媒は DME)を、参考例 9の方法と同様に して参考例 61の化合物を、参考例 44の方法と同様にして参考例 62の化合物(但し 、溶媒はトリフルォロメタンスルホン酸)を、参考例 4の方法と同様にして参考例 67の 化合物及び参考例 68の化合物(但し、 1M塩酸の代わりに 4M塩化水素-酢酸ェチル 溶液を用いた)を、参考例 5の方法と同様にして参考例 70の化合物を、参考例 10の 方法と同様にして参考例 71及び 72の化合物を、参考例 11の方法と同様にして参考 例 73— 75の化合物を、参考例 48の方法と同様にして参考例 76の化合物をそれぞ れ対応する原料を使用して製造した。参考例 49一 76の化合物の構造及び物理ィ匕 学的データを表 5— 8にそれぞれ示す。 In the same manner as in the method of Reference Example 1, the compound of Reference Example 49-54 (the solvent was THF) and the compound of Reference Example 69 (the solvent was DEE) were used in the same manner as in the method of Reference Example 2. The compound of Reference Example 61 was treated in the same manner as in Reference Example 9 using the compound of 55-60 and the compound of 63-66 (the solvent was DME), and the compound of Reference Example 62 was treated in the same manner as in Reference Example 44. The compound of Reference Example 67 (the solvent is trifluoromethanesulfonic acid) and the compound of Reference Example 67 and the compound of Reference Example 68 in the same manner as in the method of Reference Example 4 (4 M hydrogen chloride-ethyl acetate solution instead of 1 M hydrochloric acid) The compound of Reference Example 70 was treated in the same manner as in Reference Example 5, and the compounds of Reference Examples 71 and 72 were treated in the same manner as in Reference Example 10. Example 73- The compound of Reference Example 76 was used in the same manner as in the method of Reference Example 48, and the compound of Reference Example 76 was used. Manufactured using The structures and physical data of the compounds of Reference Examples 49-76 are shown in Tables 5-8, respectively.
[0043] 参考例 77 Reference Example 77
2-クロ口- N- (4-クロ口フエ-ル)- 5-フエ-ルピリミジン- 4-アミンを DEE中、ピぺラジン と 130°Cで 18時間反応させて、 N- (4-クロ口フエ-ル)- 5-フエ-ル- 2-ピぺラジン- 1-ィ ルピリミジン- 4-アミンを得た。 F: 366。 The 2-chloro-N- (4-chloropyrrole) -5-phenylpyrimidin-4-amine was reacted with piperazine in DEE at 130 ° C for 18 hours to give N- (4- Black mouth ferrule) -5-Feel-2-Pyrazine-1-y Lupyrimidin-4-amine was obtained. F: 366.
参考例 78 Reference 78
N- (4-クロ口フエ-ル)- 5-フエ-ル- 2-ピぺラジン- 1-ィルピリミジン- 4-ァミンと (2R)- l-(t-ブトキシカルボ-ル)ピぺリジン- 2-カルボン酸の THF溶液に、 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド塩酸塩、 1-ヒドロキシベンゾトリアゾール をカロえ室温下 17時間反応させて、 tert-ブチル (2R)- 2-[(4-{4-[(4-クロロフヱニル)ァ ミノ] -5-フエ-ルビリミジン- 2-ィル }ピペラジン- 1-ィル)カルボ-ル]ピぺリジン- 1-カル ボキシラートを得た。 ES: 577。 N- (4-chlorophenol) -5-phenyl-2-pyrazine-1-ylpyrimidine-4-amine and (2R) -l- (t-butoxycarbol) piperidine- 1-Ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride and 1-hydroxybenzotriazole were added to a THF solution of 2-carboxylic acid and reacted at room temperature for 17 hours to give tert-butyl (2R)- 2-[(4- {4-[(4-chlorophenyl) amino] -5-phenylpyrimidine-2-yl} piperazine-1-yl) carbol] piperidine-1-carboxylate Obtained. ES: 577.
実施例 1 Example 1
2—クロ口— N— (4—クロ口フエ-ル)— 5— (4—クロ口フエ-ル)ピリミジン— 4—ァミン 179 mgの 1,4-ジォキサン 20 ml溶液に DBU 233 mg、 1,4'-ビぺリジン- 3-ィルメタノール 2塩酸 塩 139 mgを加え、 13時間 100°Cにて加熱した。反応液を室温にし、シリカゲルを反応 溶液に加え溶媒を留去した後、残渣をシリカゲルカラムクロマトグラフィー (クロ口ホル ム-メタノール- 28%アンモニア水溶液)で精製し、 {1し [4- [(4-クロ口フエ-ル)ァミノ] -5-(4-クロ口フエ-ル)ピリミジン- 2-ィル] -1,4しビピペリジン- 3-ィル }メタノール 200 mg を薄黄色無定型結晶として得た。この化合物をメタノールに溶解し 4M塩酸/酢酸ェチ ル溶液 0.29 mlを加えた後、イソプロパノール-酢酸ェチル-メタノールより再結晶を行 つて、 {1'— [4— [(4—クロ口フエ-ル)ァミノ]— 5— (4—クロ口フエ-ル)ピリミジン— 2—ィル]—1,4'— ビピペリジン- 3-ィル }メタノール 2塩酸塩 92 mgを白色結晶として得た。 2—Black mouth—N— (4-Black mouth phenol) —5— (4—Black mouth phenol) pyrimidine—4-Amine 179 mg of 1,4-dioxane in 20 ml of DBU 233 mg, 1 , 4'-Biridine-3-ylmethanol dihydrochloride (139 mg) was added, and the mixture was heated at 100 ° C for 13 hours. The reaction solution was brought to room temperature, silica gel was added to the reaction solution, and the solvent was distilled off.The residue was purified by silica gel column chromatography (chloroform-methanol-28% aqueous ammonia solution). 4-chloro mouth phenol) -5- (4-chloro mouth pyrimidine-2-yl] -1,4 shibipiperidin-3-yl} methanol 200 mg in pale yellow amorphous crystals As obtained. This compound was dissolved in methanol, and 0.29 ml of a 4 M hydrochloric acid / ethyl acetate solution was added. The crystal was recrystallized from isopropanol-ethyl acetate-methanol to give {1 ′ — [4 — [(4-chlorophenol— Lu) amino] -5- (4-chlorophenol) pyrimidine-2-yl] -1,4'-bipiperidin-3-yl} methanol dihydrochloride (92 mg) was obtained as white crystals.
実施例 2 Example 2
2-クロ口- N- (4-クロ口フエ-ル)- 5- (4-フルオロフェ -ル)ピリミジン- 4-ァミン 236 mgを 用いて実施例 1の方法と同様に (但し、溶媒として DMF、塩基として DIPEAを用いて 80°Cで 4日間反応させた)して、 {1'-[4-[(4-クロロフヱ-ル)ァミノ] -5-(4-フルオロフェ -ル)ピリミジン- 2-ィル] -1,4'-ビピペリジン- 3-ィル }メタノール 2塩酸塩 39 mgを白色 結晶として得た。 Using 236 mg of 2-chloro-N- (4-chlorophenol) -5- (4-fluorophenol) pyrimidine-4-amine (except that DMF is used as the solvent) Then, the reaction was carried out at 80 ° C for 4 days using DIPEA as a base) to give {1 ′-[4-[(4-chlorophenyl) amino] -5- (4-fluorophenyl) pyrimidine-2 -Yl] -1,4'-Bipiperidin-3-yl} methanol dihydrochloride 39 mg was obtained as white crystals.
実施例 3 Example 3
N- (4-クロ口- 2-フルオロフェ-ル )-5- (4-フルオロフェ-ル )-2- (メチルスルフィエル) ピリミジン- 4-ァミン 450 mgを用いて実施例 1の方法と同様に(但し、溶媒として DEEを 、また、 1,4'-ビぺリジン- 3-ィルメタノール 2塩酸塩の代わりに (3S)- 1,4'-ビぺリジン -3-ィルメタノール 2塩酸塩を用いて 130°Cで 21時間反応させた)して、 N- (4-chloro-2-fluorophenyl) -5- (4-fluorophenyl) -2- (methylsulfiel) pyrimidine-4-amine (450 mg) in the same manner as in Example 1 (However, DEE is used as a solvent. The reaction was carried out at 130 ° C for 21 hours using (3S) -1,4'-viridin-3-ylmethanol dihydrochloride instead of 1,4'-viridin-3-ylmethanol dihydrochloride. Let me do)
{(3S)-1,- [4- [(4-クロ口- 2-フルオロフェ -ル)ァミノ]- 5- (4-フルオロフェ -ル)ピリミジン -2-ィル] -1,4'-ビピペリジン- 3-ィル }メタノール 2塩酸塩 217 mgを微茶褐色結晶とし て得た。 {(3S) -1,-[4-[(4-chloro-2-fluorophenyl) amino] -5- (4-fluorophenyl) pyrimidine-2-yl] -1,4'-bipiperidine -217 mg of methanol 2-hydrochloride were obtained as light brown crystals.
[0045] 実施例 4 Example 4
N- (4-クロ口フエ-ル)- 2- (メチルスルホ-ル)- 6-フエ-ルピリミジン- 4-ァミン 506 mg を用いて実施例 1の方法と同様に (但し、溶媒として 2-エトキシエタノールを、また、塩 基としてジイソプロピルェチルァミンを用いて 130°Cで 2.5日間反応させた)して (1, -{4- [(4-クロ口フエ-ル)ァミノ]- 6-フエ-ルビリミジン- 2-ィル }- 1,4,-ビピペリジン- 3-ィ ル)メタノール 2塩酸塩 87 mgを淡黄色結晶として得た。 Using 506 mg of N- (4-chlorophenol) -2- (methylsulfol) -6-phenylpyrimidine-4-amine in the same manner as in Example 1, except that the solvent The reaction was carried out with ethoxyethanol at 130 ° C for 2.5 days using diisopropylethylamine as a base) (1,-{4-[(4-chlorophenol) amino] -6 -Fervirimidine-2-yl} -1,4, -bipiperidin-3-yl) methanol dihydrochloride (87 mg) was obtained as pale yellow crystals.
実施例 5 Example 5
N- 4-クロ口- 6- (3-フルオロフェ-ル )-2- (メチルスルフエ-ル)ピリミジン- 4-ァミン 413 mgを用いて前述の参考例 5と同様の操作を行い、 N-4-クロ口- 6-(3-フルオロフェ- ル)- 2- (メチルスルフィエル)ピリミジン- 4-アミンを得た。この化合物を用いて実施例 1 と同様にして、 {1'- [4- [(4-クロ口フエ-ル)ァミノ]- 6- (3-フルオロフェ -ル)ピリミジン- 2- ィル] -1,4しビピペリジン- 3-ィル }メタノール 2塩酸塩 170 mgを淡黄色結晶として得た The same operation as in Reference Example 5 described above was performed using 413 mg of N-4-chloromouth-6- (3-fluorophenyl) -2- (methylsulfur) pyrimidine-4-amine to obtain N-4- Black mouth-6- (3-fluorophenyl) -2- (methylsulfiel) pyrimidin-4-amine was obtained. Using this compound and in the same manner as in Example 1, {1 '-[4-[(4-chlorophenol) amino] -6- (3-fluorophenyl) pyrimidine-2-yl]- 170 mg of 1,4-bipiperidin-3-yl} methanol dihydrochloride was obtained as pale yellow crystals
[0046] 実施例 6 Example 6
N- (4-クロ口フエ-ル)- 2- (メチルスルフエ-ル)- 5-フエ-ルピリミジン- 4-ァミン塩酸 塩 853 mgに飽和炭酸水素ナトリウム水溶液をカ卩え、クロ口ホルムで抽出した。有機 層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を留去し、 N-(4-ク ロロフエ-ル)- 2- (メチルスルフエ-ル)- 5-フエ-ルピリミジン- 4-ァミン 770 mgを淡黄 色油状物として得た。この化合物を用いて前述の参考例 5と同様の操作を行い、 N- (4-クロ口フエ-ル)- 2- (メチルスルフィ -ル) -5-フエ-ルピリミジン- 4-ァミン 960 mg を得た。この化合物と用いて実施例 1と同様に (但し、溶媒として DEEを用い、 130°C で 19時間反応させた)して、(1, -{4-[(4-クロ口フエニル)ァミノ] -5-フエニルピリミジン -2-ィル }-1,4,-ビピペリジン- 3-ィル)メタノール 2塩酸塩 524 mgを橙色結晶として得 た。 A saturated aqueous solution of sodium bicarbonate was added to 853 mg of N- (4-chlorophenol) -2- (methylsulfur) -5-phenylpyrimidine-4-amine hydrochloride and extracted with chloroform. did. The organic layer was washed with saturated saline, dried over anhydrous magnesium sulfate, the solvent was distilled off, and N- (4-chlorophenyl) -2- (methylsulfur) -5-phenylpyrimidine-4 was removed. 770 mg of -amine was obtained as a pale yellow oil. Using this compound, the same operation as in Reference Example 5 described above was performed to obtain N- (4-chlorophenol) -2- (methylsulfuryl) -5-phenylpyrimidine-4-amine 960 mg. Obtained. Using this compound in the same manner as in Example 1 (but using DEE as a solvent and reacting at 130 ° C. for 19 hours) to give (1,-{4-[(4-chlorophenyl) amino]] -5-Phenylpyrimidine-2-yl} -1,4, -bipiperidin-3-yl) methanol dihydrochloride (524 mg) as orange crystals It was.
実施例 7 Example 7
4-クロ口- 6- (3-メトキシフエ-ル)- 2- (メチルスルフエ-ル)ピリミジン 277 mgを用いて 前述の参考例 4と同様の操作を行!、、 N-(4-クロ口フエ-ル)- 6-(3-メトキシフエ-ル )-2- (メチルスルフエ-ル)ピリミジン- 4-ァミン 251 mgを無色結晶として得た。この化 合物を用いて前述の参考例 5と同様の操作を行 、、 N- (4-クロ口フエ-ル)- 6- (3-メト キシフエ-ル)- 2- (メチルスルフィエル)ピリミジン- 4-アミンを得た。この化合物を用い て実施例 1と同様にして、 {1し [4-[(4-クロ口フエ-ル)ァミノ]- 6-(3-メトキシフエ-ル)ピリ ミジン- 2-ィル] -1,4'-ビピペリジン- 3-ィル }メタノール 2塩酸塩 198 mgを白色結晶とし て得た。 The same operation as in Reference Example 4 described above was performed using 277 mg of 4-chloro-6- (3-methoxyphenyl) -2- (methylsulfur) pyrimidine !, N- (4-chlorophenol) 251 mg of 6- (3-methoxyphenyl) -2- (methylsulfur) pyrimidine-4-amine were obtained as colorless crystals. The same operation as in Reference Example 5 described above was performed using this compound, and N- (4-chlorophenol) -6- (3-methoxyphenyl) -2- (methylsulfiel) was obtained. Pyrimidin-4-amine was obtained. [1] [4-[(4-chlorophenol) amino] -6- (3-methoxyphenyl) pyrimidin-2-yl]- 198 mg of 1,4'-bipiperidin-3-yl} methanol dihydrochloride was obtained as white crystals.
実施例 8— 22 Example 8-22
実施例 1一 7の方法と同様にして後記表 9一 10に示す実施例化合物を、それぞれ 対応する原料を使用して製造した。 Example compounds shown in Tables 9 to 10 below were produced in the same manner as in Example 17 using the corresponding starting materials.
実施例 23 Example 23
2-クロ口- N-シクロへプチル- 5-フエ-ルビリミジン- 4-ァミン 250 mgを用いて実施 例 1の方法と同様に(但し溶媒として DEEを用い、 120°Cで 2日間反応させた)して、 {1 ' -[4- (シクロへプチルァミノ) -5-フエ-ルビリミジン- 2-ィル] -1,4,-ビピペリジン- 3-ィル) メタノール 2塩酸塩 178 mgを淡黄色結晶として得た。 Using the same method as in Example 1 except that 250 mg of 2-chloro-N-cycloheptyl-5-phenylpyrimidine-4-amine was used (however, the reaction was carried out at 120 ° C. for 2 days using DEE as a solvent). ) Then, add 178 mg of (1 '-[4- (cycloheptylamino) -5-phenylpyrimidine-2-yl] -1,4, -bipiperidin-3-yl) methanol dihydrochloride to pale yellow Obtained as crystals.
実施例 24 Example 24
N- (4-クロ口- 2-フルオロフェ-ル )-6-イソプロピル- 2- (メチルチオ)- 5-フエ-ルピリミジ ン- 4-ァミン 201 mgのクロ口ホルム (15 ml)溶液に、氷冷下 m-クロ口過安息香酸 (〉65%) 143 mgを加え、 0°C力も室温で 2時間攪拌した。反応液にチォ硫酸ナトリウム水溶液 と炭酸水素ナトリウム水溶液を加え、クロ口ホルムで抽出した。有機層を無水硫酸マグ ネシゥムで乾燥し、溶媒を留去することにより、 N_(4-クロ口フエ-ル) _6_イソプロピル -2- (メチルスルフィ -ル) -5-フエ-ルビリミジン- 4-アミンを得た。この化合物と 1.4, -ビ ピぺリジン- 3-ィルメタノール 2塩酸塩 140 mgの混合物に、 DEE 7 ml、 DBU 0.28 ml を順次カ卩え、 120°Cで 2日間攪拌した。反応液を室温まで降温し、水を加え、クロロホ ルムで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥、溶 媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (クロ口ホルム-メタノ ール -28%アンモニア水)で精製し、 (1, -{4-[(4-クロ口フエ-ル)ァミノ] -6-イソプロピル -5-フエニルピリミジン- 2-ィル }-1, 4' -ビピペリジン- 3-ィル)メタノール 125 mgを得た。 この化合物をメタノール 5 mlに溶解し、 4M塩化水素-ジォキサン溶液 0.2 mlをカロ えて塩酸塩とした後、溶媒を留去した。得られた生成物をジェチルエーテルで洗浄し て、(1, -{4- [(4-クロ口フエ-ル)ァミノ]- 6-イソプロピル- 5 -フエ-ルビリミジン- 2-ィル }-1, 4,-ビピペリジン- 3-ィル)メタノール 2塩酸塩 75 mgをベージュ色結晶として得た。 実施例 25 To a solution of 201 mg of N- (4-chloro-2-fluorophenyl) -6-isopropyl-2- (methylthio) -5-phenylpyrimidin-4-amine in 15 ml of chloroform in ice-cold form (15 ml) was added ice-cooled. 143 mg of m-chloroperbenzoic acid (> 65%) was added thereto, and the mixture was stirred at room temperature for 2 hours at 0 ° C. An aqueous solution of sodium thiosulfate and an aqueous solution of sodium hydrogen carbonate were added to the reaction solution, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off to obtain N_ (4-chlorophenol) _6_isopropyl-2- (methylsulfuryl) -5-phenylpyrimidine-4-amine Got. To a mixture of this compound and 140 mg of 1.4, -bipiperidin-3-ylmethanol dihydrochloride, 7 ml of DEE and 0.28 ml of DBU were successively added and stirred at 120 ° C for 2 days. The reaction solution was cooled to room temperature, water was added, and the mixture was extracted with chloroform. The organic layer was washed with saturated saline, dried over anhydrous magnesium sulfate, and dissolved. The medium was distilled off. The obtained residue was purified by silica gel column chromatography (form-form-methanol-28% aqueous ammonia) to give (1,-{4-[(4-form-methyl) amino] -6-isopropyl -5-Phenylpyrimidine-2-yl} -1,4'-bipiperidin-3-yl) methanol (125 mg) was obtained. This compound was dissolved in 5 ml of methanol, and 0.2 ml of a 4 M hydrogen chloride-dioxane solution was heated to form a hydrochloride, and then the solvent was distilled off. The obtained product was washed with getyl ether to give (1,-{4-[(4-chlorophenol) amino] -6-isopropyl-5-phenylpyrimidine-2-yl}- 75 mg of 1,4, -bipiperidin-3-yl) methanol dihydrochloride was obtained as beige crystals. Example 25
tert-ブチル (2R)- 2- [(4- {4- [(4-クロ口フエ-ル)ァミノ]- 5 -フエ-ルビリミジン- 2-ィル }ピペラジン- 1-ィル)カルボニル]ピぺリジン- 1-カルボキシラート 291 mgのメタノール 6 ml溶液に、 4M塩ィ匕水素-酢酸ェチル溶液 3 mlを加え、室温下 16時間攪拌した。 反応液力 溶媒を留去して得られた残渣にイソプロパノールとジイソプロピルエーテ ルの混合溶媒をカ卩えて結晶化を行 、、 N- (4-クロロフヱ-ル)- 5-フエ-ル tert-Butyl (2R)-2-[(4- {4-[(4-chlorophenol) amino] -5-phenylpyrimidine-2-yl} piperazine-1-yl) carbonyl] pi To a solution of 291 mg of lysine-1-carboxylate in 6 ml of methanol was added 3 ml of a 4M solution of hydrogen chloride-ethyl acetate, and the mixture was stirred at room temperature for 16 hours. Reaction liquid power The solvent obtained by distilling off the solvent was crystallized by adding a mixed solvent of isopropanol and diisopropyl ether to crystallize, and N- (4-chlorophenyl) -5-phenyl was added.
-2-{4-[(2R)-ピぺリジン- 2-ィルカルボ-ル]ピぺラジン- 1-ィル }ピリミジン- 4-ァミン 2 塩酸塩 242 mgを無色結晶として得た。 242 mg of 2- {4-[(2R) -piperidin-2-ylcarbol] piperazine-1-yl} pyrimidine-4-amine 2 hydrochloride was obtained as colorless crystals.
実施例 26— 39 Example 26—39
実施例 1一 25の方法と同様にして後記表 11一 12に示す実施例 26— 39の化合物 を、それぞれ対応する原料を使用して製造した。 The compounds of Examples 26 to 39 shown in Tables 11 to 12 described below were produced in the same manner as in the method of Example 125 using the corresponding raw materials.
実施例 40 Example 40
N- (4-クロ口フエ-ル)- 5-フエ-ル- 2-ピぺラジン- 1-ィルピリミジン- 4-ァミン 215 mg 及び [(2S)-l-(tert-ブトキシカルボ-ル)ピロリジン- 2-ィル]酢酸 135 mgを用いて参 考例 78の方法と同様にして、 tert-ブチル (2S)-2-[2-(4-{4-[(4-クロ口フエ-ル)ァミノ ]-5-フエニルピリミジン- 2-ィル }ピペラジン- 1-ィル) -2-ォキソェチル]ピロリジン- 1-力 ルポキシラートを得た。この化合物を用いて実施例 25と同様にして、 N-(4-クロ口フエ -ル) -5-フエ-ル- 2- {4- [(2S)-ピロリジン- 2-ィルァセチル]ピぺラジン- 1-ィル }ピリミジ ン -4-ァミン 2塩酸塩 213 mgを無色無定形固体として得た。 215 mg of N- (4-chlorophenol) -5-phenyl-2-pyrazine-1-ylpyrimidine-4-amine and ((2S) -1- (tert-butoxycarbol) pyrrolidine Tert-butyl (2S) -2- [2- (4- {4-[(4-chlorophenol) using 135 mg of -2-yl] acetic acid in the same manner as in Reference Example 78. ) Amino] -5-phenylpyrimidine-2-yl} piperazine-1-yl) -2-oxoethyl] pyrrolidine-1-force ropoxylate. In the same manner as in Example 25 using this compound, N- (4-chlorophenol) -5-phenyl-2- {4-[(2S) -pyrrolidine-2-ylacetyl] pidazine 213 mg of 1-yl} pyrimidine-4-amine dihydrochloride was obtained as a colorless amorphous solid.
実施例 1一 40の化合物の構造及び物理ィ匕学的データを表 9一 12に示す。また、表 13— 15に本発明の別の化合物の構造を示す。これらは、上記の製造法や実施例に 記載の方法及び当業者にとって自明である方法、又はこれらの変法を用いることによ り、容易に合成することができる。 The structure and physical properties of the compound of Example 140 are shown in Table 9-112. Tables 13 to 15 show the structures of other compounds of the present invention. These are based on the above-mentioned manufacturing methods and examples. The compounds can be easily synthesized by using the methods described and those obvious to those skilled in the art, or variations thereof.
[表 1] [table 1]
[0049][0049]

[0050] [表 3]

[0051] [表 4][Table 4]

[0052] [表 5] [Table 5]
n [ oo] n [oo]

SCSC00/S00Zdf/X3d εε [8挲] [eeoo] SCSC00 / S00Zdf / X3d εε [8 挲] [eeoo]

SCSC00/S00Zdf/X3d [6挲] [9S00] SCSC00 / S00Zdf / X3d [6 挲] [9S00]

SCSC00/S00idf/X3d 9ε SCSC00 / S00idf / X3d 9ε

Ex Syn Dat 1 Sal Ex Syn Dat 1 Sal
F: 512; NMR2: 7.83 (IH, s), 7.43 (IH, d, J=8.8 F: 512; NMR 2: 7.83 (IH, s), 7.43 (IH, d, J = 8.8
1 1 1 1
Hz), 1.10-1.19 (1H, m); Sal: 2HC1.0.5H2O : 496; NMR2: 8.18 (IH, s), 7.36 (2H, t, J=8.8Hz), 1.10-1.19 (1H, m ); Sal: 2HC1.0.5H 2 O: 496; NMR2: 8.18 (IH, s), 7.36 (2H, t, J = 8.8
2 2
Hz), 1.14-1.19 (IH, m); Sal: 2HC1. H20Hz), 1.14-1.19 (IH, m ); Sal:. 2HC1 H 2 0
IH, s), 7.09 (IH, d, J=7.2 IH, s), 7.09 (IH, d, J = 7.2
8 1 or F: 508; NMR2: 7.72 ( 8 1 or F: 508; NMR2: 7.72 (
Hz), 1.04-1.19 (IH, m); Sal: 2HC1.3H20 Hz), 1.04-1.19 (IH, m); Sal: 2HC1.3H 20
9 1 FxF F: 514; NMR2: 7.95 (IH, s), 7.36- 7.40 (5H, m), 9 1 F x F F: 514; NMR 2: 7.95 (IH, s), 7.36-7.40 (5H, m),
1.10-1.19(1H, m); Sal: 2HC1.3H20 1.10-1.19 (1H, m); Sal: 2HC1.3H 20
F: 514; NMR2: 7.91 (IH, s), 7.23- 7.27 (IH, m), F: 514; NMR2: 7.91 (IH, s), 7.23- 7.27 (IH, m),
10 1 10 1
2.60-2.68 (IH, m); Sal: 2HC1.1.5H20 2.60-2.68 (IH, m); Sal : 2HC1.1.5H 2 0
F: 514; NMR2: 8.18 (IH, s), 7.34 (IH, br s), F: 514; NMR 2: 8.18 (IH, s), 7.34 (IH, br s),
11 1 11 1
1.10-1.19 (IH, m): Sal: 2HC1.0.5H2O 1.10-1.19 (IH, m): Sal: 2HC1.0.5H 2 O
F: 546; NMR2: 7.92 (1H, s), 7.72- 7.83 (4H, m), F: 546; NMR2: 7.92 (1H, s), 7.72-7.83 (4H, m),
12 1 12 1
1.14-1.19 (IH, m); Sal: 2HC1. H20 1.14-1.19 (IH, m); Sal :. 2HC1 H 2 0
F: 503; NMR2: 7.98 (2H, d, 1=1.2 Hz), 7.90 (1H, F: 503; NMR2: 7.98 (2H, d, 1 = 1.2 Hz), 7.90 (1H,
13 1 NCXk s), 1.04-1.16 (IH, m); Sal: 2HC1.1.5H20 13 1 NC Xk s), 1.04-1.16 (IH, m); Sal: 2HC1.1.5H 2 0
ES: 479; NMR2: 8.04 (IH, s), 7.96 (] H, br s), ES: 479; NMR2: 8.04 (IH, s), 7.96 (] H, br s),
14 1 14 1
1.10-1.19 (IH, m); Sal: 3HC1.4H20 1.10-1.19 (IH, m); Sal: 3HC1.4H 20
F: 484; NMR2: 7.91 (IH, s), 7.79 (IH, s), F: 484; NMR2: 7.91 (IH, s), 7.79 (IH, s),
15 1 15 1
1.10-1.19 (IH, m); Sal: 2HC1.4H20 1.10-1.19 (IH, m); Sal: 2HC1.4H 20
F: 484; NMR2: 8.19 (IH. s), 7.32 (1H, s), F: 484; NMR2: 8.19 (IH. S), 7.32 (1H, s),
16 1 16 1
1.05-1.19 (IH, m); Sal: 2HC1.2H:0 1.05-1.19 (IH, m); Sal: 2HC1.2H : 0
ES: 468: NMR2: 8.05 (1H, s), 7.93 (IH. s), ES: 468: NMR2: 8.05 (1H, s), 7.93 (IH. S),
17 1 17 1
1.04-1.19 (IH, m): Sal: 2HCI.4H:0 1.04-1.19 (IH, m): Sal: 2HCI.4H : 0
F: 538; NMR2: 7.80 ( 1 H, s), 7.05 ( 1 H, s), F: 538; NMR2: 7.80 (1 H, s), 7.05 (1 H, s),
18 2 18 2
4.50-4.63 (2H, m); Sal: 2HC1.3H20 10] 4.50-4.63 (2H, m); Sal : 2HC1.3H 2 0 10]

Ex S n R1 X' Config Dat / SalEx S n R 1 X 'Config Dat / Sal
: 514; EA: Cal (C:7H30ClF2N5O.2HC1. H20) C, 53.61; H, 5.66; N, 11.58; CI, 17.58; F,: 514; EA: Cal (. C: 7 H 30 ClF 2 N 5 O.2HC1 H 2 0) C, 53.61; H, 5.66; N, 11.58; CI, 17.58; F,
3 3 -H -F S 3 3 -H -F S
6.28. Fnd: C, 53.78; H, 5.62; N, 11.76; CI, 17.84; F, 6.42.; Sal: 2HC1. H20 . 6.28 Fnd:. C, 53.78 ; H, 5.62; N, 11.76; CI, 17.84; F, 6.42 .; Sal: 2HC1 H 2 0
F: 478; NMR2: 7.74 (2H, d, J=8.8 Hz), F: 478; NMR2: 7.74 (2H, d, J = 8.8 Hz),
4 4 6-Ph -H -H S 6.67(1H, s), 2.94-3.12 (2H, m); Sal: 2HC1. 4 4 6-Ph -H -H S 6.67 (1H, s), 2.94-3.12 (2H, m); Sal: 2HC1.
2H20 2H 2 0
F: 496; NMR2: 7.56-7.61 (IH, m), 7.41 (2H, F: 496; NMR2: 7.56-7.61 (IH, m), 7.41 (2H,
5 5 -H -H RS d, J=8.8 Hz), 1.05-1.18 (lH, m); Sal: 2HC1. 5 5 -H -H RS d, J = 8.8 Hz), 1.05-1.18 (lH, m); Sal: 2HC1.
2.5H20 2.5H 2 0
F: 478; NMR2: 7.76 (1H, m), 4.51-4.66 (2H, F: 478; NMR 2: 7.76 (1H, m), 4.51-4.66 (2H,
6 6 5-Ph -H -H RS 6 6 5-Ph -H -H RS
m), 1.06-1.24 (1H, m): Sal: 2HC1.0.5H O m), 1.06-1.24 (1H, m): Sal: 2HC1.0.5H O
F: 508; NMR2: 7.74 (2H, d, J=8.8 Hz), 3.85F: 508; NMR2: 7.74 (2H, d, J = 8.8 Hz), 3.85
7 7 -H -H RS 7 7 -H -H RS
(3H, s), 1.13-1.19 (lH,m); Sal: 2HC1.2H20(3H, s), 1.13-1.19 ( lH, m); Sal: 2HC1.2H 2 0
F: 496; NMR2: 7.73 (2H, dd, J=8.8, 5.2 Hz),F: 496; NMR2: 7.73 (2H, dd, J = 8.8, 5.2 Hz),
18 ] -H -H RS 6.63(1H, s), 1.10-1.19 (IH, m); Sal: 2HC1. 18] -H -H RS 6.63 (1H, s), 1.10-1.19 (IH, m); Sal: 2HC1.
2.5H 0 2.5H 0
F: 510; NMR2: 4.58-4.78 (2H, m), 2.15 (3H, F: 510; NMR2: 4.58-4.78 (2H, m), 2.15 (3H,
19 3 6-Me -H RS 19 3 6-Me -H RS
s), 1.05-1.23 (IH, m); Sal: 2HC1.2H20s), 1.05-1.23 (IH, m); Sal: 2HC1.2H 20
F: 514; NMR2: 7.52 (2H, d, J=9.6 Hz),F: 514; NMR2: 7.52 (2H, d, J = 9.6 Hz),
20 5 -II -F RS 6.72(111, s), 1.12-1.18 (IH, m): Sal: 2HC1. 20 5 -II -F RS 6.72 (111, s), 1.12-1.18 (IH, m): Sal: 2HC1.
2.5H:0 2.5H : 0
F: 484; NMR2: 7.95(1 H, s), 7.38 (2H, d, F: 484; NMR2: 7.95 (1 H, s), 7.38 (2H, d,
21 5 6 -H -H RS J=8.8 Hz), 1.10-1.1 (1H, m); Sal: 2HC1. 21 5 6 -H -H RS J = 8.8 Hz), 1.10-1.1 (1H, m); Sal: 2HC1.
3H20 3H 2 0
F: 502: N R2: 8.04(1H, t, J=8.8 Hz).6.64 F: 502: N R2: 8.04 (1H, t, J = 8.8 Hz) .6.64
22 5 -H -F RS 22 5 -H -F RS
(lH,s), 2.63-2.65 (IH. m); Sal: 2HCI. 1.5H:0 11]
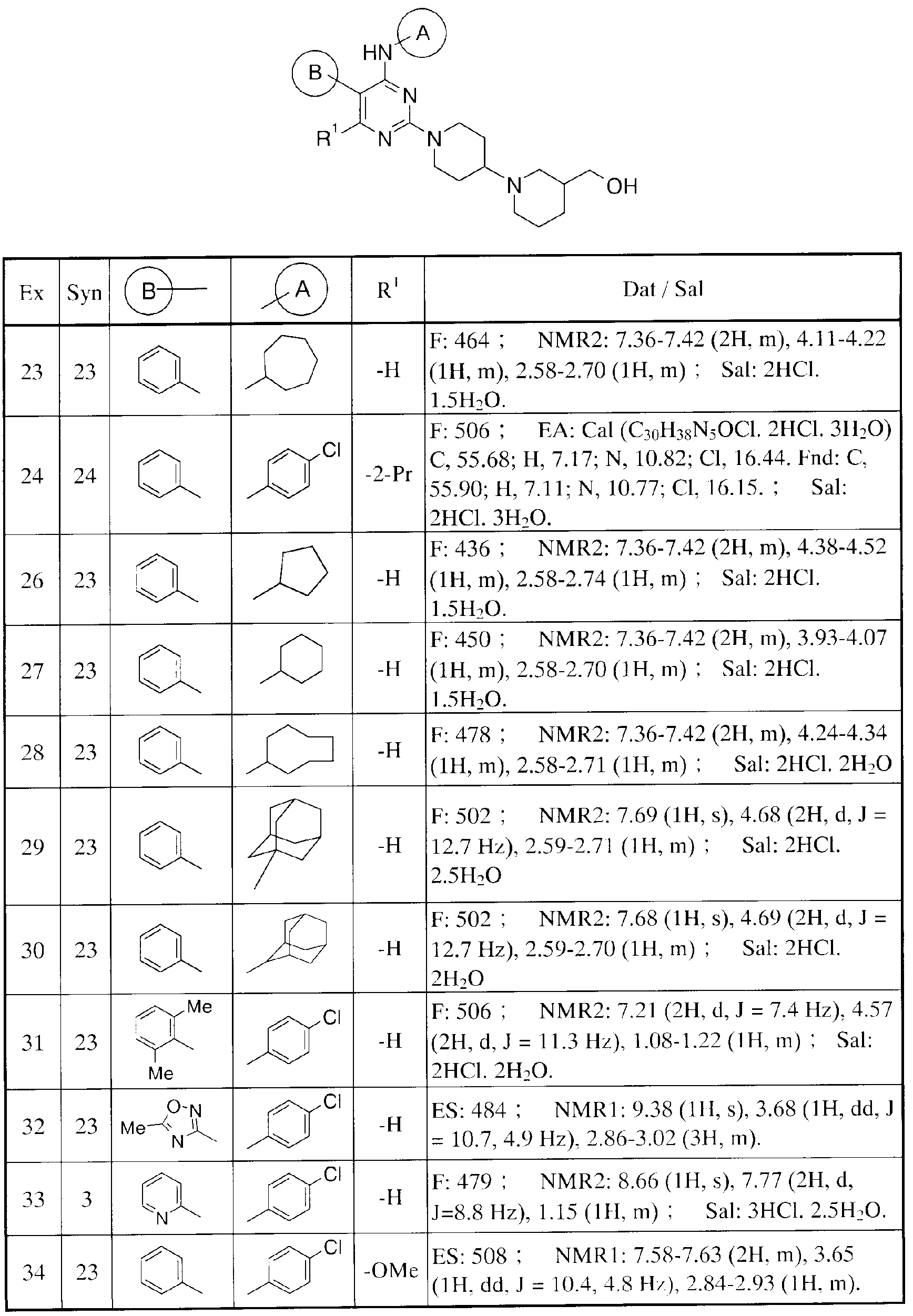
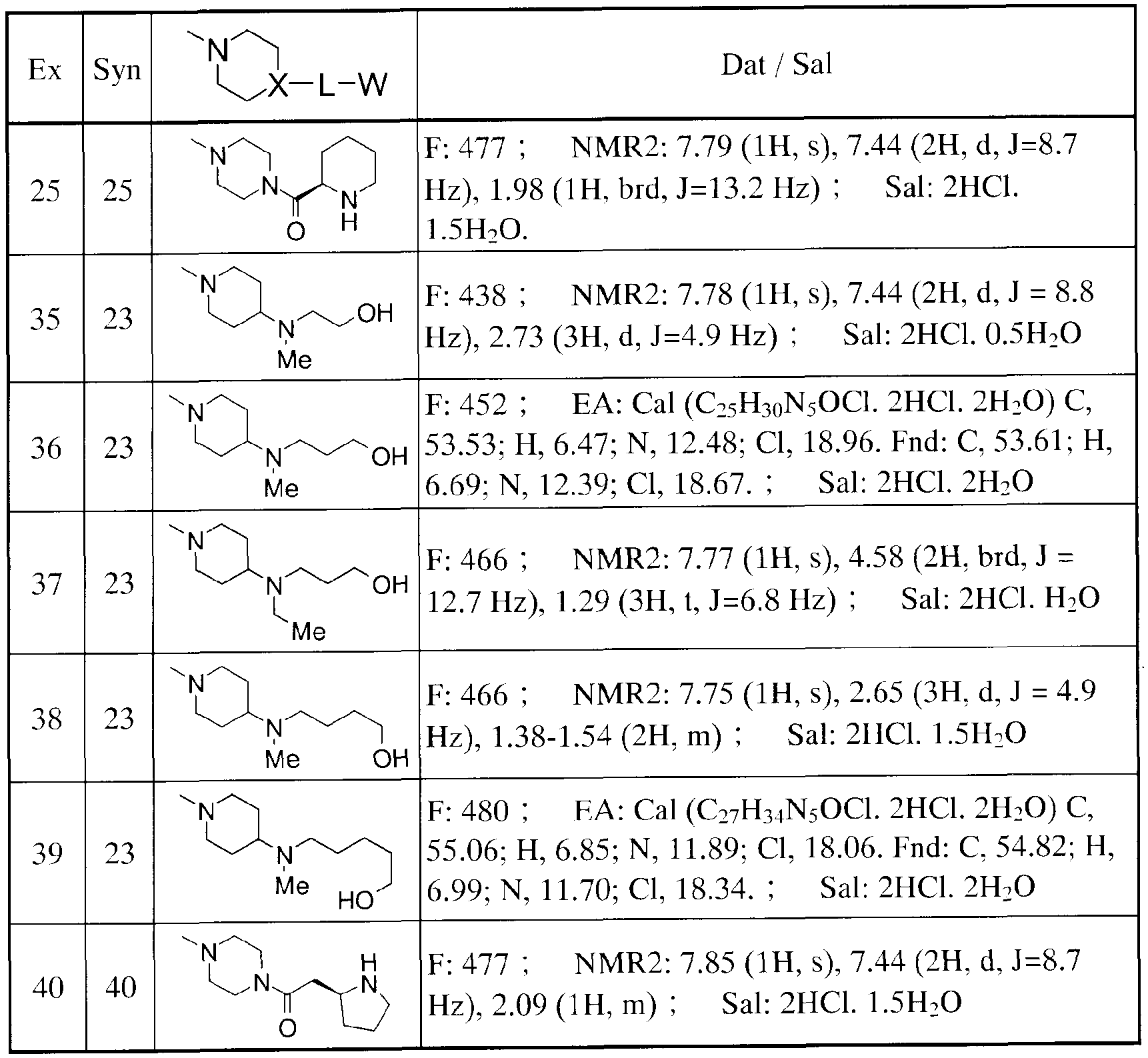
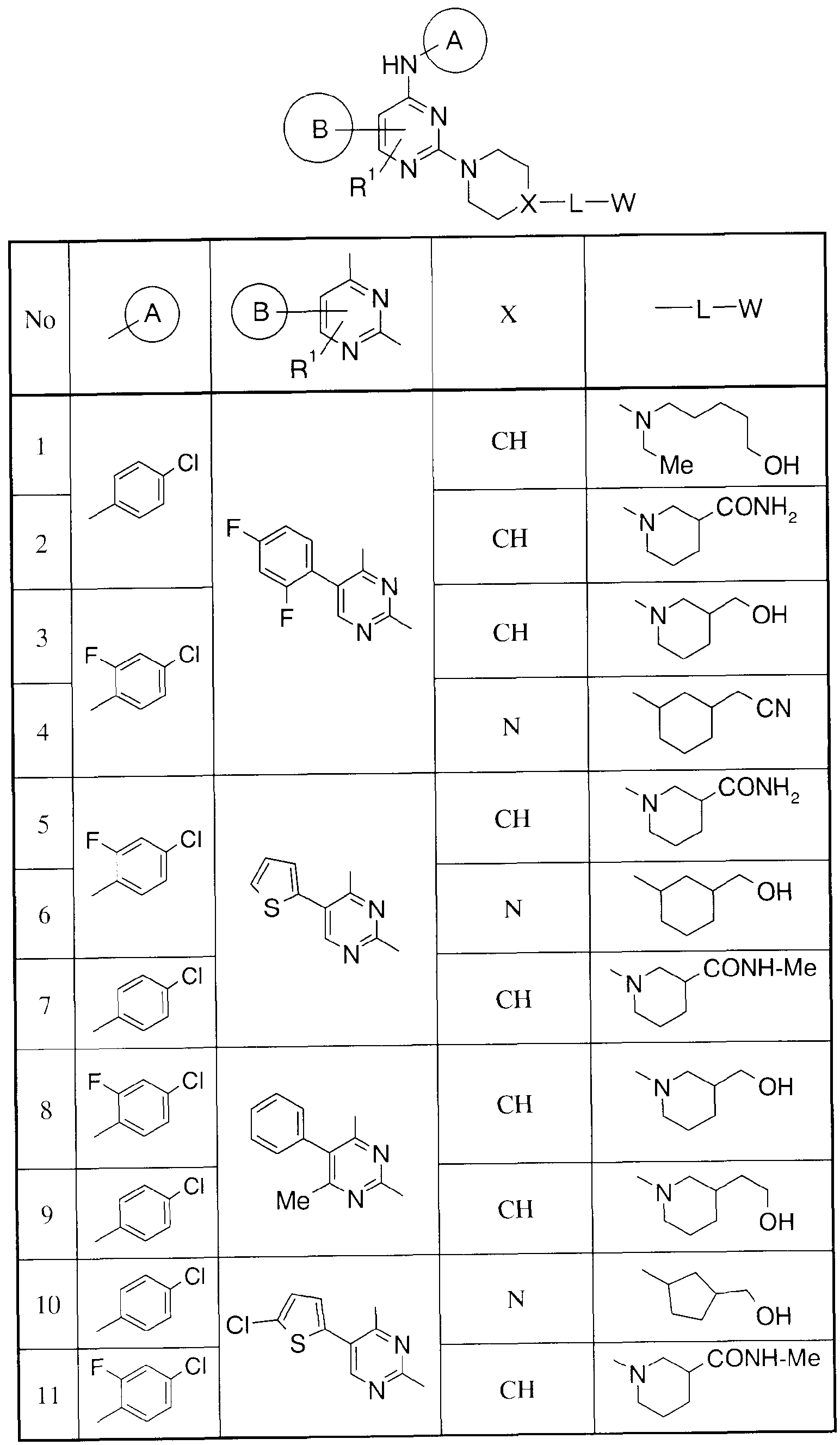
[0061] [表 14]

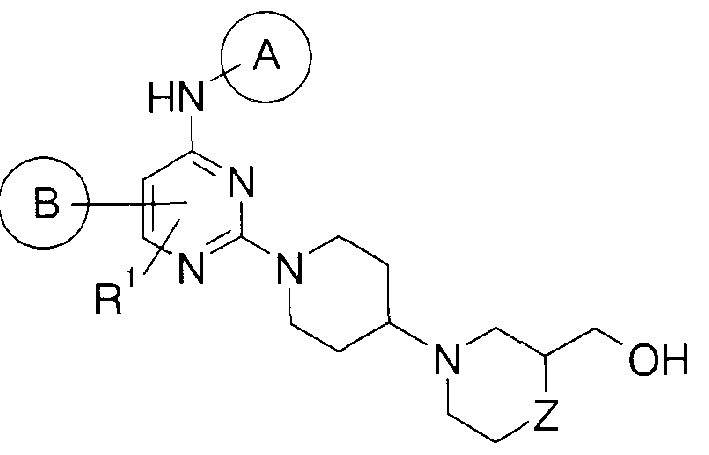

産業上の利用可能性 Industrial applicability
[0063] 本発明の置換ピリミジン誘導体は、 CCR4或いは TARC及び/又は MDCの機能調 節作用を有することから、種々の炎症性疾患、アレルギー疾患、自己免疫疾患等〔例 えば、喘息、アレルギー性鼻炎、アレルギー性結膜炎、花粉症、皮膚炎 (アトピー性 皮膚炎、接触性皮膚炎)、乾癬、関節リウマチ、全身性エリテマトーデス、多発性硬化 症、インスリン依存型糖尿病(IDDM)、臓器移植時の拒絶反応、癌、炎症性腸疾患( 潰瘍性大腸炎、クローン病)、間質性膀胱炎、敗血症、疼痛〕の予防 ·治療薬として有 用である。特に、喘息、アトピー性皮膚炎又は関節リウマチの予防 ·治療薬として有 用である。 [0063] The substituted pyrimidine derivative of the present invention has a function of modulating the function of CCR4 or TARC and / or MDC, and therefore has various inflammatory diseases, allergic diseases, autoimmune diseases and the like (eg, asthma, allergic rhinitis). , Allergic conjunctivitis, hay fever, dermatitis (atopic dermatitis, contact dermatitis), psoriasis, rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis Disease, insulin-dependent diabetes mellitus (IDDM), organ transplant rejection, cancer, inflammatory bowel disease (ulcerative colitis, Crohn's disease), interstitial cystitis, sepsis, pain) It is for. In particular, it is useful as a prophylactic / therapeutic agent for asthma, atopic dermatitis or rheumatoid arthritis.
Claims



Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004061123A JP2007217282A (en) | 2004-03-04 | 2004-03-04 | Substituted pyrimidine derivatives |
| JP2004-061123 | 2004-03-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005085212A1 true WO2005085212A1 (en) | 2005-09-15 |
Family
ID=34918042
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/003535 Ceased WO2005085212A1 (en) | 2004-03-04 | 2005-03-02 | Substituted pyrimidine derivative |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2007217282A (en) |
| WO (1) | WO2005085212A1 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007111227A1 (en) | 2006-03-24 | 2007-10-04 | Astellas Pharma Inc. | Acylaminopiperidine compound |
| WO2008146914A1 (en) * | 2007-06-01 | 2008-12-04 | Mitsubishi Tanabe Pharma Corporation | Heterocyclic compound |
| JP2008546698A (en) * | 2005-06-14 | 2008-12-25 | タイゲン バイオテクノロジー カンパニー,リミテッド | Pyrimidine compounds |
| WO2009037454A2 (en) | 2007-09-18 | 2009-03-26 | Cancer Research Technology Ltd | Cancer marker and therapeutic target |
| EP2092824A1 (en) | 2008-02-25 | 2009-08-26 | Bayer CropScience AG | Heterocyclyl pyrimidines |
| EP2120573A4 (en) * | 2007-02-12 | 2011-05-25 | Merck Sharp & Dohme | DERIVATIVES OF PIPERIDINE |
| JP2012500278A (en) * | 2008-08-20 | 2012-01-05 | シェーリング コーポレイション | Substituted pyridine derivatives and substituted pyrimidine derivatives and their use in the treatment of viral infections |
| US8193206B2 (en) | 2005-06-14 | 2012-06-05 | Taigen Biotechnology Co., Ltd. | Pyrimidine compounds |
| US8501739B2 (en) | 2005-07-04 | 2013-08-06 | High Point Pharmaceuticals, Llc | Medicaments |
| EP2805947A4 (en) * | 2012-01-16 | 2015-05-27 | Inst Pharm & Toxicology Amms | PIPERAZINYL PYRIMIDINE DERIVATIVES, PREPARATION METHOD AND USE THEREOF |
| WO2021063852A1 (en) * | 2019-09-30 | 2021-04-08 | F. Hoffmann-La Roche Ag | Substituted pyrimidine for the treatment and prophylaxis of hepatitis b virus infection |
| WO2022007869A1 (en) * | 2020-07-10 | 2022-01-13 | 浙江海正药业股份有限公司 | Pyridine or pyrimidine derivative and preparation method therefor and use thereof |
| CN116507611A (en) * | 2020-11-06 | 2023-07-28 | 佩勒梅德有限公司 | Novel capsid assembly inhibitors |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2479165B1 (en) * | 2009-09-16 | 2017-10-25 | Astellas Pharma Inc. | Glycine compound |
| WO2021143823A1 (en) * | 2020-01-16 | 2021-07-22 | 浙江海正药业股份有限公司 | Pyridine or pyrimidine derivative, and preparation method therefor and use thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5049288A (en) * | 1973-08-20 | 1975-05-01 | Thomae Gmbh Dr K | Shinkina pirimijinkagobutsuno seizohoho |
| JP2003500477A (en) * | 1999-05-31 | 2003-01-07 | エフ.ホフマン−ラ ロシュ アーゲー | 5-Phenyl-pyrimidine derivatives |
| WO2004074260A1 (en) * | 2003-02-21 | 2004-09-02 | Kyowa Hakko Kogyo Co., Ltd. | Pyrimidine derivatives |
-
2004
- 2004-03-04 JP JP2004061123A patent/JP2007217282A/en active Pending
-
2005
- 2005-03-02 WO PCT/JP2005/003535 patent/WO2005085212A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5049288A (en) * | 1973-08-20 | 1975-05-01 | Thomae Gmbh Dr K | Shinkina pirimijinkagobutsuno seizohoho |
| JP2003500477A (en) * | 1999-05-31 | 2003-01-07 | エフ.ホフマン−ラ ロシュ アーゲー | 5-Phenyl-pyrimidine derivatives |
| WO2004074260A1 (en) * | 2003-02-21 | 2004-09-02 | Kyowa Hakko Kogyo Co., Ltd. | Pyrimidine derivatives |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1890703A4 (en) * | 2005-06-14 | 2010-11-03 | Taigen Biotechnology Co Ltd | PYRIMIDINE COMPOUNDS |
| JP2008546698A (en) * | 2005-06-14 | 2008-12-25 | タイゲン バイオテクノロジー カンパニー,リミテッド | Pyrimidine compounds |
| US8193206B2 (en) | 2005-06-14 | 2012-06-05 | Taigen Biotechnology Co., Ltd. | Pyrimidine compounds |
| AU2006259525B2 (en) * | 2005-06-14 | 2012-05-24 | Gpcr Therapeutics, Inc | Pyrimidine compounds |
| US8846677B2 (en) | 2005-07-04 | 2014-09-30 | High Point Pharmaceuticals, Llc | Medicaments |
| US8501739B2 (en) | 2005-07-04 | 2013-08-06 | High Point Pharmaceuticals, Llc | Medicaments |
| US7790884B2 (en) | 2006-03-24 | 2010-09-07 | Astellas Pharma Inc. | Acylaminopiperidine compound |
| WO2007111227A1 (en) | 2006-03-24 | 2007-10-04 | Astellas Pharma Inc. | Acylaminopiperidine compound |
| EP2120573A4 (en) * | 2007-02-12 | 2011-05-25 | Merck Sharp & Dohme | DERIVATIVES OF PIPERIDINE |
| WO2008146914A1 (en) * | 2007-06-01 | 2008-12-04 | Mitsubishi Tanabe Pharma Corporation | Heterocyclic compound |
| WO2009037454A2 (en) | 2007-09-18 | 2009-03-26 | Cancer Research Technology Ltd | Cancer marker and therapeutic target |
| EP2533047A1 (en) | 2007-09-18 | 2012-12-12 | Cancer Research Technology Ltd | CCR4 as therapeutic target for cancer. |
| EP2535716A2 (en) | 2007-09-18 | 2012-12-19 | Cancer Research Technology Limited | Cancer marker and therapeutic target |
| EP2092824A1 (en) | 2008-02-25 | 2009-08-26 | Bayer CropScience AG | Heterocyclyl pyrimidines |
| JP2012500278A (en) * | 2008-08-20 | 2012-01-05 | シェーリング コーポレイション | Substituted pyridine derivatives and substituted pyrimidine derivatives and their use in the treatment of viral infections |
| EP2805947A4 (en) * | 2012-01-16 | 2015-05-27 | Inst Pharm & Toxicology Amms | PIPERAZINYL PYRIMIDINE DERIVATIVES, PREPARATION METHOD AND USE THEREOF |
| US9493453B2 (en) | 2012-01-16 | 2016-11-15 | The Institute of Pharmacology and Toxicology Academy of Military Medical Science P.L.A. China | Piperazinyl pyrimidine derivatives, preparation method and use thereof |
| WO2021063852A1 (en) * | 2019-09-30 | 2021-04-08 | F. Hoffmann-La Roche Ag | Substituted pyrimidine for the treatment and prophylaxis of hepatitis b virus infection |
| WO2022007869A1 (en) * | 2020-07-10 | 2022-01-13 | 浙江海正药业股份有限公司 | Pyridine or pyrimidine derivative and preparation method therefor and use thereof |
| CN115916765A (en) * | 2020-07-10 | 2023-04-04 | 浙江海正药业股份有限公司 | Pyridine or pyrimidine derivatives, preparation method and use thereof |
| CN116507611A (en) * | 2020-11-06 | 2023-07-28 | 佩勒梅德有限公司 | Novel capsid assembly inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007217282A (en) | 2007-08-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2005082865A1 (en) | Fused bicyclic pyrimidine derivative | |
| JP5104893B2 (en) | Diaminopyrimidine carboxamide derivatives | |
| JP5364159B2 (en) | Diaminoheterocyclic carboxamide compounds | |
| TWI465439B (en) | Triazole derivative or its salt | |
| JPWO2010058846A1 (en) | 4,6-diaminonicotinamide compound | |
| CA3037728A1 (en) | 4,6-indazole compounds and methods for ido and tdo modulation, and indications therefor | |
| AU2022217353B2 (en) | Pyridopyrimidinone derivative, preparation method therefor, and use thereof | |
| WO2012053606A1 (en) | Arylaminoheterocyclic carboxamide compound | |
| RU2741000C2 (en) | 1,4-disubstituted imidazole derivative | |
| HUE033177T2 (en) | Pyrazine is a carboxamide compound | |
| WO2011120025A1 (en) | Indazolyl-pyrimidines as kinase inhibitors | |
| WO2005085212A1 (en) | Substituted pyrimidine derivative | |
| JPWO2012036278A1 (en) | Glycine transporter inhibitor | |
| CA3142002A1 (en) | Targeted protein degradation of parp14 for use in therapy | |
| KR20120002581A (en) | Pyrimidine compounds | |
| JP5067364B2 (en) | Acylaminopiperidine compounds | |
| JP2007055940A (en) | Pyrazolopyrimidine derivatives | |
| MX2011001405A (en) | Diazepine and diazocane compounds as mc4 agonists. | |
| AU2018223982A1 (en) | 1, 4, 6-trisubstituted-2-alkyl-1H-benzo[d]imidazole derivatives as dihydroorotate oxygenase inhibitors | |
| CN114014842A (en) | A kind of heterocyclic ketone compound and its composition and its preparation method and application | |
| WO2005123697A1 (en) | Quinazoline derivatives | |
| WO2020135210A1 (en) | Substituted aryl compound and preparation method therefor and use thereof | |
| JP2006241089A (en) | A pyrrolopyrimidine derivative or a salt thereof | |
| CN102656172B (en) | 8-oxodihydropurine derivatives | |
| JP2006290791A (en) | Azole-substituted sulfonylbenzene derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: JP |