THERAPEUTIC AGENTS USEFUL FOR TREATING PAIN This application claims the benefit of U.S. provisional application no. 60/528,581, filed December 9, 2003, the disclosure of the provisional application being incorporated by reference herein in its entirety.
1. Field of the Invention The present invention relates to Piperazine Compounds, compositions comprising an effective amount of Piperazine Compound and methods for treating or preventing a Condition such as pain, comprising administering to an animal in need thereof an effective amount of a Piperazine Compound.
2. Background of the Invention Pain is the most common symptom for which patients seek medical advice and treatment. Pain can be acute or chronic. While acute pain is usually self-limited, chronic pain persists for 3 months or longer and can lead to significant changes in a patient's personality, lifestyle, functional ability and overall quality of life (K.M. Foley, Pain, in Cecil Textbook of Medicine 100-107 (J.C. Bennett and F. Plum eds., 20th ed. 1996)). Chronic pain can be classified as either nociceptive or neuropathic. Nociceptive pain includes tissue injury- induced pain and inflammatory pain such as that associated with arthritis. Neuropathic pain is caused by damage to the peripheral or central nervous system and is maintained by aberrant somatosensory processing. There is a large body of evidence relating activity at both Group I mGluRs (mGluRl and mGluR5) (M.E. Fundytus, CNS Drugs 15:29-58 (2001)) and vanilloid receptors (VR1) (V. Di Marzo et al, Current Opinion in Neurobiology 12:372-379 (2002)) to pain processing. Inhibiting mGluRl or mGluR5 reduces pain, as shown by in vivo treatment with antibodies selective for either mGluRl or mGluR5, where neuropathic pain in rats was attenuated (M.E. Fundytus et al. , NeuroReport 9:731-735 (1998)). It has also been shown that antisense oligonucleotide knockdown of mGluRl alleviates both neuropathic and inflammatory pain (M.E. Fundytus et al, Brit. J. of Pharmacol. 132:354-367 (2001); M.E. Fundytus et al, Pharmacol, Biochem. & Behavior 73:401-410 (2002)). Small molecule antagonists for mGluR5-attenuated pain in in vivo animal models are disclosed
in, e.g., K. Walker et al, Neuropharmacology 40:1-9 (2000) and A. Dogrul et al, Neuroscience Let. 292:115-118 (2000)). Nociceptive pain has been traditionally managed by administering non-opioid analgesics, such as acetylsalicylic acid, choline magnesium trisalicylate, acetaminophen, ibuprofen, fenoprofen, diflusinal, and naproxen; or opioid analgesics, including morphine, hydromorphone, methadone, levorphanol, fentanyl, oxycodone, and oxymorphone. Id. In addition to the above-listed treatments, neuropathic pain, which can be difficult to treat, has also been treated with anti-epileptics (e.g., gabapentin, carbamazepine, valproic acid, topiramate, phenytoin), NMDA antagonists (e.g., ketamine, dextromethorphan), topical lidocaine (for post-herpetic neuralgia), and tricyclic antidepressants (e.g., fluoxetine, sertraline and amitriptyline). Ul is uncontrollable urination, generally caused by bladder-detrusor-muscle instability. Ul affects people of all ages and levels of physical health, both in health care settings and in the community at large. Physiologic bladder contraction results in large part from acetylcholine-induced stimulation of post-ganglionic muscarinic-receptor sites on bladder smooth muscle. Treatments for Ul include the administration of drugs having bladder-relaxant properties, which help to control bladder-detrusor-muscle overactivity. For example, anticholinergics such as propantheline bromide and glycopyrrolate, and combinations of smooth-muscle relaxants such as a combination of racemic oxybutynin and dicyclomine or an anticholinergic, have been used to treat Ul (See, e.g. , A.J. Wein, Urol Clin. N. Am. 22:557-577 (1995); Levin et al, J. Urol 128:396-398 (1982); Cooke et al, S. Afr. Med. J. 63:3 (1983); R.K. Mirakhur et al, Anaesthesia 38:1195-1204 (1983)). These drugs are not effective, however, in all patients having uninhibited bladder contractions. None of the existing commercial drug treatments for Ul has achieved complete success in all classes of Ul patients, nor has treatment occurred without significant adverse side effects. For example, drowsiness, dry mouth, constipation, blurred vision, headaches, tachycardia, and cardiac arrhythmia, which are related to the anticholinergic activity of traditional anti-UI drugs, can occur frequently and adversely affect patient compliance. Yet despite the prevalence of unwanted anticholinergic effects in many patients, anticholinergic drugs are currently prescribed for patients having Ul. The Merck Manual of Medical Information 631-634 (R. Berkow ed., 1997).
Certain pharmaceutical agents have been administered for treating addiction. U.S. Patent No. 5,556,838 to Mayer et al. discloses the use of nontoxic NMDA-blocking agents co-administered with an addictive substance to prevent the development of tolerance or withdrawal symptoms. U.S. Patent No. 5,574,052 to Rose et al. discloses co-administration of an addictive substance with an antagonist to partially block the pharmacological effects of the addictive substance. U.S. Patent No. 5,075,341 to Mendelson et al. discloses the use of a mixed opiate agonist/antagonist to treat cocaine and opiate addiction. U.S. Patent No. 5,232,934 to Downs discloses administration of 3- phenoxypyridine to treat addiction. U.S. Patents No. 5,039,680 and 5,198,459 to Imperato et al. disclose using a serotonin antagonist to treat chemical addiction. U.S.
Patent No. 5,556,837 to Nestler et al. discloses infusing BDNF or NT-4 growth factors to inhibit or reverse neurological adaptive changes that correlate with behavioral changes in an addicted individual. U.S. Patent. No. 5,762,925 to Sagan discloses implanting encapsulated adrenal medullary cells into an animal's central nervous system to inhibit the development of opioid tolerance. U.S. Patent No. 6,204,284 to Beer et al. discloses racemic (±)-l-(3,4-dichlorophenyl)-3-azabicyclo[3.1.0]hexane for use in the prevention or relief of a withdrawal syndrome resulting from addiction to drugs and for the treatment of chemical dependencies. Glutamate release is enhanced during opioid withdrawal (K. Jhamandas et al, J. of Neuroscience 16:2758-2766 (1996)). Recent evidence suggests a role for Group I mGluRs in opioid tolerance and dependence. An interaction between opioids and mGluRs was demonstrated when it was shown that an antagonist at Group I mGluRs significantly attenuated withdrawal symptoms in opioid- dependent rats (M.E. Fundytus et al, Brit. J. of Pharmacol 113:1215-1220 (1994)). More recent results show that antisense oligonucleotide knockdown of mGluRl reduces protein kinase C activity (M.E. Fundytus et al, Brit. J. of Pharmacol. 132:354-367 (2001)), which may be associated in the development of opioid tolerance and dependence (See also M.E. Fundytus, CNS Drugs 15:29-58, (2001)). Very recently, it has been shown that antisense oligonucleotide knockdown of mGluRl attenuates the development of opioid tolerance (R.N. Sharif et al, Brit. J. of Pharmacol. 136:865-872 (2002)). Selective antagonists of the mGluR5 receptor have also been shown to exert anti-dependence activity in vivo (C. Chiamulera et al, Nature Neuroscience 4:873-874 (2001)).
Without treatment, Parkinson's disease progresses to a rigid akinetic state in which patients are incapable of caring for themselves. Death frequently results from complications of immobility, including aspiration pneumonia or pulmonary embolism. Drugs commonly used for the treatment of Parkinson's disease include carbidopa/levodopa, pergolide, bromocriptine, selegiline, amantadine, and trihexyphenidyl hydrochloride. There remains, however, a need for drugs useful for the treatment of Parkinson's disease and having an improved therapeutic profile. Currently, benzodiazepines are the most commonly used anti-anxiety agents for generalized anxiety disorder. Benzodiazepines, however, carry the risk of producing impairment of cognition and skilled motor functions, particularly in the elderly, which can result in confusion, delerium, and falls with fractures. Sedatives are also commonly prescribed for treating anxiety. The azapirones, such as buspirone, are also used to treat moderate anxiety. The azapirones, however, are less useful for treating severe anxiety accompanied with panic attacks. Antagonists of the mGluR5 receptor have also been shown to exert anxiolytic and anti-depressant activity in in vivo animal models (E.
Tatarczynska et al, Brit. J. of Pharmacol. 132(7): 1423-1430 (2001) and P.J.M. Will et al, Trends in Pharmacological Sciences 22(7):331-37 (2001)). Examples of drugs for treating a seizure and epilepsy include carbamazepine, ethosuximide, gabapentin, lamotrigine, phenobarbital, phenytoin, primidone, valproic acid, trimethadione, benzodiazepines, γ-vinyl GABA, acetazolamide, and felbamate. Anti-seizure drugs, however, can have side effects such as drowsiness; hyperactivity; hallucinations; inability to concentrate; central and peripheral nervous system toxicity, such as nystagmus, ataxia, diplopia, and vertigo; gingival hyperplasia; gastrointestinal disturbances such as nausea, vomiting, epigastric pain, and anorexia; endocrine effects such as inhibition of antidiuretic hormone, hyperglycemia, glycosuria, osteomalacia; and hypersensitivity such as scarlatiniform rash, morbilliform rash, Stevens- ohnson syndrome, systemic lupus erythematosus, and hepatic necrosis; and hematological reactions such as red-cell aplasia, agranulocytosis, thrombocytopenia, aplastic anemia, and megaloblastic anemia. Ηie Merck Manual of Medical Information 345-350 (R. Berkow ed., 1997). Symptoms of strokes vary depending on what part of the brain is affected. Symptoms include loss of or abnormal sensations in an arm or leg or one side of the
body, weakness or paralysis of an arm or leg or one side of the body, partial loss of vison or hearing, double vision, dizziness, slurred speech, difficulty in thinking of the appropriate word or saying it, inability to recognize parts of the body, unusual movements, loss of bladder control, imbalance, and falling, and fainting. The symptoms can be permanent and can be associated with coma or stupor. Examples of drugs for treating strokes include anticoagulants such as heparin, drugs that break up clots such as streptokinase or tissue plasminogen activator, and drugs that reduce swelling such as mannitol or corticosteroids. Tlie Merck Manual of Medical Information 352-355 (R. Berkow ed., 1997). Pruritus is an unpleasant sensation that prompts scratching. Conventionally, pruritus is treated by phototherapy with ultraviolet B or PUVA or with therapeutic agents such as naltrexone, nalmefene, danazol, and tricyclic antidepressants. Selective antagonists of the metabotropic glutamate receptor 5 ("mGluR5") have been shown to exert analgesic activity in in vivo animal models (K. Walker et al., europharmacology 40: 1 -9 (2000) and A. Dogrul et al, Neuroscience Let. , 292(2) : 115- 118 (2000)). Selective antagonists of the mGluR5 receptor have also been shown to exert anti- Parkinson activity in vivo (K. J. Ossowska et al, Neuropharmacology 41(4):413-20 (2001) and P.J.M. Will et al, Trends in Pharmacological Sciences 22(7):331-37 (2001)). Selective antagonists of the mGluR5 receptor have also been shown to exert anti- dependence activity in vivo (C. Chiamulera et al, Nature Neuroscience 4(9):873-74 (2001)). International Publication No. WO 99/37304 by Rohne-Poulenc Rorer Pharmaceuticals, Inc. discloses oxoazaheterocyclic compounds useful for inhibiting factor Xa. International Publication No. WO 98/28980 by Merck and Co., Inc. discloses substituted bicyclic compounds useful for inhibiting farnesyl transferase. International Publication No. WO 96/32384 and U.S. Patent No. 5,786,355 to Taiho Pharmaceutical Co., Ltd. each disclose 4,6-diarylpyrimidine compounds useful as neovascular inhibitors.
International Publication No. WO 02/102381 by Vertex Pharmaceuticals, Inc. discloses acyclic piperazine and piperidine compounds useful for treating neuronal damage. International Publication No. WO 02/14314, International Publication No. 02/20012, U.S. Patent Application Publication No. US 2003/0069240 and U.S. Patent Application Publication No. US 2003/0069240 by Ortho McNeil Pharmaceutical, Inc. each disclose substituted pyrazoles useful for treating allergic conditions. E.V. Tarasov et al, Synlett. (5):625-626 (2000) discloses the preparation of bicyclic compounds by a thermally assisted intramolecular hetero Diels-Alder cycloaddition reaction of an acetylene-tethered pyrimidine that require reaction periods of extended duration, e.g., 2-6 days at 180°C. There remains, however, a clear need in the art for new drugs useful for treating or preventing pain, Ul, an addictive disorder, Parkinson's disease, parkinsonism, anxiety, epilepsy, stroke, a seizure, a pruritic condition, psychosis, a cognitive disorder, a memory deficit, restricted brain function, Huntington's chorea, ALS, dementia, retinopathy, a muscle spasm, a migraine, vomiting, dyskinesia or depression in an animal. Citation of any reference in Section 2 of this application is not to be construed as an admission that such reference is prior art to the present application.
3. Summary of the Invention The present invention encompasses compounds of Formula (I):
(0 and pharmaceutically acceptable salts thereof, where: X is -O-, -C(R (R
2)-, -N(H , -N -GOalkyl)-,
Y and Z are, independently, -C(O)- or -C(R (R2)-; each Rt and R2 is independently: (a) -H, or (b) linear or branched -(CrC6)alkyl, ~(C3-C8)cycloalkyl, phenyl or naphthyl, each of which is unsubstituted or substituted with one or more R9 groups; m is 0, 1 or 2; R3 and R4 are independently -CH2-, -CH(CH3)- or -C(O)-; each R5 is independently: (a) -halo, -CN, -OH, -NO2, -O(C1-C6)alkyl or -NH2; or (b) -(Cι-Cιo)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3- C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C10)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C1 )tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups; n is 0, 1 or 2; A is -C(O)-, -C(S)-, -CH2-, -CH(C C4 alkyl)-, -C(Q-C4 alkyl)(C1-C4 alkyl)-, -C(Rπ)(R12)- or -CH(phenyl)-, the phenyl being unsubstituted or substituted with one or more R13 groups; R6 is: (a) -(CrC10)a_kyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3-C8)cycloalkyl, -(C8-C1 )bicycloalkyl, -(C8-C1 )tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C1 )tricycloalkenyl, -(3- to 7-membered)heterocycle or -(7- to 10-membered)bicycloheterocycle, each of which is unsubstituted or substituted with one or more R13 groups, or (b) -phenyl, -naphthyl, -(C14)aryl or -(5- to 10-membered)heteroaryl, each of which is unsubstituted or substituted with one or more R15 groups;
each R7 is independently -(Ci-C6)alkyl, -(C -C6)alkenyl, -(C2-C6)alkynyl, -(C3- C8)cycloalkyl -(C5-C8)cycloalkenyl, -phenyl, -(3- to 5-membered)heterocycle, -C(halo)3, -CH(halo)2, -CH2(halo), -CN, -OH, -halo, -N3, -NO2, -N(R8)2, -CH=NR8, -NR8OH, -OR8, -C(O)R8, -C(O)OR8, -OC(O)R8, -OC(O)OR8, -SR8, -S(O)R8 or -S(O)2R8; each R8 is independently -H, -(C1-C6)alkyl, -(C2-C6)alkenyl, -(C2-C6)alkynyl,
-(C
3-C
8)cycloalkyl, -(C
5-C
8)cycloalkenyl, -phenyl, -(3- to 5-membered)heterocycle, -C(halo)
3, -CH(halo)
2 or -CH
2(halo); each R
9 is independently -(C
1-C
6)alkyl, -(C
2-C
6)alkenyl, -(C
2-C
6)alkynyl, -(C
3- C
8)cycloalkyl, -(C
5-Cs)cycloalkenyl, -phenyl or -(3- to 5~membered)heterocycle; Rio is independently -H or -( -C^alkyl; Rn and R
12 are taken together with the carbon to which they are attached to form a ~(C
3-C
8)cycloalkyl group; each R
13 is independently -CN, -OH, -halo, -N
3, -NO
2, -N(R
14)
2, -CH=NR
14, -NR
14OH, -OR
14, -C(O)R
14, -C(O)OR
14, -OC(O)R
14, -OC(O)OR
14, -SR
1 , -S(O)R
14 or -S(O)
2R
14; each R
1 is independently -H, -(C
1-C
6)alkyl, -(C
2-C
6)alkenyl, -(C
2-C
6)alkynyl, -(C
3-C
8)cycloalkyl, -(C
5-C
8)cycloalkenyl, -phenyl, -(3- to 5-membered)heterocycle, -C(halo)
3 or -CH(halo)
2; and each R
15 is independently -(C
1-C
6)alkyl, -O(C
1-C
6)alkyl, -(C
2-C
6)alkenyl, -(C
2-C
6)alkynyl, -(C
3-C
8)cycloalkyl, -(C
5-C
8)cycloalkenyl, -phenyl, -(3- to 5- membered)heterocycle, -C(halo)
3, -OC(halo)
3, -CH(halo)
2, -OCH(halo)
2, -CN, -OH, -halo, -N
3, -NO
2, -N(R
14)
2, -CH=NR
14, -NR
14OH, -OR
1 , -C(O)R
14, -C(O)OR
14, -OC(O)R
14, -OC(O)OR
14, -SR
14, -S(O)R
14 or -S(O)
2R
14. A compound of formula (I) or a pharmaceutically acceptable salt thereof (a "Piperazine Compound") is useful for treating or preventing pain, Ul, an addictive disorder, Parkinson's disease, parkinsonism, anxiety, epilepsy, stroke, a seizure, a pruritic condition, psychosis, a cognitive disorder, a memory deficit, restricted brain function, Huntington's chorea, ALS, dementia, retinopathy, a muscle spasm, a migraine, vomiting, dyskinesia or depression (each being a "Condition") in an animal. The invention further relates to a synthetic intermediate that is useful for making a Piperazine Compound. The invention further relates to a method for making a compound of formula A':
comprising the step of irradiating a compound of formula BB:
with microwave radiation under conditions that are sufficient to make a compound of formula A', where X, Y, Z, Ri, R2, R5, R7 through R9 and m are as defined above for the Piperazine Compounds of formula (I), n' is 0 or 1, Q is I, Br, Cl or F, and W is -( - C10)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3-C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C1 )tricycloalkyl, -(Cs-C1o)cycloalkenyl, -(C8-C14)bicycloalkenyl, -(C8- C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C1 )aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups, or W is H. The invention also relates to compositions comprising an effective amount of a Piperazine Compound and a pharmaceutically acceptable carrier or excipient. The compositions are useful for treating or preventing a Condition in an animal. The invention also relates to methods for treating a Condition, comprising administering to an animal in need thereof an effective amount of a Piperazine Compound. The invention also relates to methods for preventing a Condition, comprising administering to an animal in need thereof an effective amount of a Piperazine Compound.
The invention still further relates to methods for inhibiting mGluR5 function in a cell, comprising contacting a cell capable of expressing mGluR5 with an effective amount of a Piperazine Compound. The invention still further relates to methods for inhibiting mGluRl function in a cell, comprising contacting a cell capable of expressing mGluRl with an effective amount of a Piperazine Compound. The invention still further relates to a method for preparing a composition, comprising the step of admixing a Piperazine Compound and a pharmaceutically acceptable carrier or excipient. The invention still further relates to a kit comprising a container containing an effective amount of a Piperazine Compound. The kit may further comprise printed instructions for using the Piperazine Compound to treat or prevent any of the aforementioned Conditions. The present invention may be understood more fully by reference to the following detailed description and illustrative examples, which are intended to exemplify non-limiting embodiments of the invention.
4. Detailed Description of the Invention
4.1 Piperazine Compounds of Formula (I) The present invention encompasses Piperazine Compounds of Formula (I):
(I) where Rι through R15, A, Y, X, Z, m and n are as defined above for the Piperazine Compounds of formula (I). In the Piperazine Compounds of Formula (I), each R5, when present, can be bonded to either of the available carbons of the pyridine ring, i.e., as indicated in the figure above, the carbon atom at the 5-position or the carbon atom at the 6-position. In one embodiment, the Piperazine Compound has only one R5 group (i.e., n = 1), and that
R5 group is attached to the 6-position carbon atom of the pyridine ring. In another embodiment, the Piperazine Compound has only one R5 group, and that R5 group is attached to the 5-position carbon atom of the pyridine ring. In another embodiment, the Piperazine Compound has only one R5 group, that R5 group is attached to the 6-position carbon atom of the pyridine ring, and the 6-position R5 group is selected from: (a) -halo, -CN, -OH, -NO2, -O(C1-C6)alkyl or -NH2, or (b) -(Cι-Cιo)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3- C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(Cs- c cycloalkenyl, -(C8- C1 )bicycloalkenyl, -(C8-C1 )tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C1 )aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups. In another embodiment, the Piperazine Compound has only one R5 group, that R5 group is attached to the 5-position carbon atom of the pyridine ring, and the 5-position R5 group is selected from -(C1-C1o)alkyl, -(C2-C1o)alkenyl, -(C2- o)alkynyl, -(C3- C8)cycloalkyl, -(C8-C1 )bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8- C1 )bicycloalkenyl, -(C8-C1 )tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups. In another embodiment, n = 2, an R5 group is attached to the 6-position carbon atom of the pyridine ring, the 6-position R5 group is selected from: (a) -halo, -CN, -OH, -NO2, -O(d-C6)alkyl or -NH2, or (b) -(Cι-C10)alkyl, -(C2-C10)alkenyl, ~(C2-C10)alkynyl, -(C3-
C8)cycloalkyl, -(C8-C1 )bicycloalkyl, -(C8-C14)tricycloalkyl, -(Cs-C1o)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C1 )aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups, the other R5 group is attached to the 5-position carbon atom of the pyridine ring, and the 5-position R5 group is selected from -(Cι-Cιo)alkyl, -(C2-C!o)alkenyl, -(C2-C1o)alkynyl,
-(C3~C8)cycloa_kyl, -(C8-C14)bicycloalkyl, -(C8-Cι4)tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8-C14)bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-mernbered)heterocycle, -(7- to 10-membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups. In another embodiment, R is phenyl. In another embodiment, R6 is benzo(l,3)dioxolyl. In another embodiment, X is -N(H)- or -N((Cι-C4)alkyl)-. In another embodiment, X is O. In another embodiment, n is 0. In another embodiment, m is 0. In another embodiment, m is 1. In another embodiment, R3 and 1^ are each -CH2-. In another embodiment, A is -C(O)-. In another embodiment, Z is -C(O)-. In another embodiment, Y is -C(O)-. In another embodiment, Y is -C(CH3)2-. In another embodiment, A is -C(O)- and n is 0. In another embodiment, A is -C(O)-; n is 0; and R3 and R4 are independently -CH2- or -CH(CH3)-. In another embodiment, A is -C(O)-; n is 0; and R3 and R4 are each -CH2-. In another embodiment, A is -C(O)-; n is 2; and R3 and R4 are independently -CH2- or -CH(CH3)-. In another embodiment, A is -C(O)-; n is 2; and R3 and R4 are each -CH2-. In another embodiment, A is -C(O)-; m is 0; Z is -C(O)-; and X is -CH2-. In another embodiment, A is -C(O)-; m is 0; Z is -C(O)-; and X is O. In another embodiment, A is -C(O)-; m is 0; Y and Z are both -C(O)-; and X is -CH2-. In another embodiment, A is -C(O)-; m is 0; Y and Z are both -C(O)-; and X is O. hi another embodiment, A is -C(O)-; m is 0; Y is -C(Ri)(R2)-; Z is -C(O)-; and X is -CH2-.
In another embodiment, A is -C(O)-; m is 0; Y is -C(R1)(R2)-; Z is -C(O)-; and X is O. In another embodiment, R3 is -CH(CH3)- and the R3 carbon atom to which the methyl group is attached has the (R) configuration. In another embodiment, R3 is -CH(CH3)- and the R3 carbon atom to which the methyl group is attached has the (S) configuration. In another embodiment, R4 is -CH(CH3)- and the R4 carbon atom to which the methyl group is attached has the (R) configuration. In another embodiment, R is -CH(CH3)- and the R4 carbon atom to which the methyl group is attached has the (S) configuration. In another embodiment, R3 and I are each -CH(CH3)- and the R3 and R4 carbon atoms to which each methyl group is attached both have the (R) configuration. In another embodiment, R3 and 1^. are each -CH(CH3)- and the R3 and R4 carbon atoms to which each methyl group is attached both have the (S) configuration. In another embodiment, R3 and I are each -CH(CH3)- and one of the R3 and R4 carbon atoms to which a methyl group is attached has the (S) configuration and the other has the (R) configuration. In another embodiment, R3 and R are each -CH(CH3)- and the R3 carbon atom to which a methyl group is attached has the (S) configuration and the R4 carbon atom to which a methyl group is attached has the (R) configuration. In another embodiment, both R3 and R4 are each -CH(CH )- and the R4 carbon atom to which a methyl group is attached has the (S) configuration and the R3 carbon atom to which a methyl group is attached has the (R) configuration. In one embodiment, X is -C(Rι)(R2)- and one of R and R2 is hydrogen and the other is linear or branched -(C1-C6)alkyl, -(C3-C8)cycloalkyl, phenyl or naphthyl, each of which is unsubstituted or substituted with one or more R9 groups, where the carbon atom to which Ri and R2 are attached has the (R) configuration. In another embodiment, X is -C(R (R2)- and one of Rι and R2 is hydrogen and the other is linear or branched -(Cr C6)alkyl, -(C3-C8)cycloalkyl, phenyl or naphthyl, each of which is unsubstituted or substituted with one or more R9 groups, where the carbon atom to which R and R2 are attached has the (S) configuration. In one embodiment, Y is -C(R_)(R2)- and one of Ri and R2 is hydrogen and the other is linear or branched -(C1-C6)alkyl, -(C3-C8)cycloalkyl, phenyl or naphthyl, each of
which is unsubstituted or substituted with one or more R groups, where the carbon atom to which Ri and R2 are attached has the (R) configuration. In another embodiment, Y is -C(R (R2)- and one of RΪ and R2 is hydrogen and the other is linear or branched -(Q- C6)alkyl, -(C3-C8)cycloalkyl, phenyl or naphthyl, each of which is unsubstituted or substituted with one or more R groups, where the carbon atom to which R and R2 are attached has the (S) configuration. In one embodiment, Z is -C(R_)(R2)- and one of Rt and R2 is hydrogen and the other is linear or branched -(C1-C6)alkyl, -(C3-C8)cycloalkyl, phenyl or naphthyl, each of which is unsubstituted or substituted with one or more R groups, where the carbon atom to which Ri and R2 are attached has the (R) configuration. In another embodiment, Z is -C(R1)(R2)- and one of Rι and R2 is hydrogen and the other is linear or branched -(Ci- C6)alkyl, -(C3-C8)cycloalkyl, phenyl or naphthyl, each of which is unsubstituted or substituted with one or more R9 groups, where the carbon atom to which R\ and R2 are attached has the (S) configuration. In one embodiment, A is -CH(Cι-C4 alkyl)- and the carbon atom of A to which the C1-C4 alkyl group is attached has the (R) configuration. In another embodiment, A is -CH(C1-C4 alkyl)- and the carbon atom of A to which the C1-C4 alkyl group is attached has the (S) configuration. In one embodiment, A is -CH(phenyl)- and the carbon atom of A to which the phenyl group is attached has the (R) configuration. In another embodiment, A is
-CH(phenyl)- and the carbon atom of A to which the phenyl group is attached has the (S) configuration.
4.2 Piperazine Compounds of Formula (II) In one embodiment, the Piperazine Compounds are those where m = 0, Y =
-C(Rι)(R
2)-, and Z = -C(O)- and are of Formula (II):
(II) where A, X, Ri through R15 and n are as defined above for the Piperazine Compounds of formula (I). In the Piperazine Compounds of Formula (II), each R5, when present, can be bonded to either of the available carbons of the pyridine ring. In one embodiment, the Piperazine Compound has only one R5 group (i.e., n = 1), and that R5 group is attached to the 6-position carbon atom of the pyridine ring. In another embodiment, the Piperazine Compound has only one R5 group, and that R5 group is attached to the 5-position carbon atom of the pyridine ring. In another embodiment, the Piperazine Compound has only one R5 group, that R5 group is attached to the 6-position carbon atom of the pyridine ring, and the 6-position R5 group is selected from: (a) -halo, -CN, -OH, -NO2, -O(C1-C6)alkyl or -NH2, or (b) -(C1-C10)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3-
C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C1 )tricycloalkyl, -(C5-C10)cycloalkenyl, -(C8~ C1 )bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups. In another embodiment, the Piperazine Compound has only one R5 group, that R5 group is attached to the 5-position carbon atom of the pyridine ring, and the 5-position R5 group is selected from -(C1-C1o)alkyl, -(C2-Cio)alkenyl, -(C2-C1o)alkynyl, -(C3- C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C10)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C )aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups.
In another embodiment, n = 2, an R5 group is attached to the 6-position carbon atom of the pyridine ring, the 6-position R5 group is selected from: (a) -halo, -CN, -OH, -NO , -O(C1-C6)alkyl or -NH2, or (b) -(C1-C10)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3- C8)cycloalkyl, -(C8-C1 )bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C10)cycloalkenyl, -(C8- C1 )bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups, the other R5 group is attached to the 5-position carbon atom of the pyridine ring, and the 5-position R5 group is selected from -(C1-C1o)alkyl, -(C2-C10)alkenyl, -(C2-Cio)alkynyl, -(C3-C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C10)cycloalkenyl, -(C8-C14)bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10-membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups. In another embodiment, R6 is phenyl. In another embodiment, R6 is benzo(l,3)dioxolyl. In another embodiment, X is -N(H)- or -N^ -G alkyl)-. In another embodiment, n is 0. In another embodiment, R3 and R4 are each -CH2-. In another embodiment, A is -C(O)~. In another embodiment, A is -C(O)- and n is 0. In another embodiment, A is -C(O)-; n is 0; and R and R4 are independently -CH2- or -CH(CH3)-. In another embodiment, A is -C(O)-; n is 0; and R3 and R are each -CH2-. In another embodiment, A is -C(O)-; n is 2; and R3 and R4 are independently -CH2- or -CH(CH3)-. In another embodiment, A is -C(O)-; n is 2; and R3 and R are each -CH2-. In another embodiment, A is -C(O)- and X is -CH2-. In another embodiment, A is -C(O)- and X is O.
4.3 Piperazine Compounds of Formula (III) In anther embodiment, the Piperazine Compounds are those where m = 0, Y = -C(O)- and Z = -C(O)- and are of Formula (III):
(HI) where A, X, Ri through R
15 and n are as defined above for the Piperazine Compounds of formula (I). In the Piperazine Compounds of Formula (III), each R
5, when present, can be bonded to either of the available carbons of the pyridine ring. In one embodiment, the Piperazine Compound has only one R
5 group (i.e., n = 1), and that R
5 group is attached to the 6-position carbon atom of the pyridine ring. In another embodiment, the Piperazine
Compound has only one R5 group, and that R5 group is attached to the 5-position carbon atom of the pyridine ring. In another embodiment, the Piperazine Compound has only one R5 group, that R5 group is attached to the 6-position carbon atom of the pyridine ring, and the 6-position
R5 group is selected from: (a) -halo, -CN, -OH, -NO2, -O(CrC6)alkyl or -NH2, or . (b) -(C_-C10)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3- C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups. In another embodiment, the Piperazine Compound has only one R5 group, that R5 group is attached to the 5-position carbon atom of the pyridine ring, and the 5-position R5 group is selected from -(C1-C1o)alkyl, -(C2-C1o)alkenyl, -(C2-C1o)alkynyl, -(C3- C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10-
membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups. In another embodiment, n = 2, an R5 group is attached to the 6-position carbon atom of the pyridine ring, the 6-position R5 group is selected from: (a) -halo, -CN, -OH, -NO2, -O(C C6)alkyl or -NH2, or (b) -(C1-C10)alkyl, -(C2-C10)alkenyl, -(C2-C10)alkynyl, -(C3- C8)cycloalkyl, -(C8-C1 )bicycloalkyl, -(C8-C1 )tricycloalkyl, -(C5-Cio)cycloalkenyl, -(C8- C14)bicycloalkenyl, -(C8-C1 )tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups, the other R5 group is attached to the 5-position carbon atom of the pyridine ring, and the 5-position R5 group is selected from -(C1-C1o)alkyl, -(C2-C_o)alkenyl, -(C2-C1o)alkynyl, -(C3-C8)cycloalkyl, -(C8-C14)bicycloalkyl, -(C8-C14)tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8-C14)bicycloalkenyl, -(C8-Cl4)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10-membered)bicycloheterocycle, -phenyl, -naphthyl, -(C1 )aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R groups. In another embodiment, R6 is phenyl. In another embodiment, R6 is benzo(l,3)dioxolyl. In another embodiment, X is -N(H)- or -N((C1-C4)alkyl)-. In another embodiment, n is 0. In another embodiment, R3 and R4. are each -CH2-. In another embodiment, A is -C(O)-. In another embodiment, A is -C(O)- and n is 0. In another embodiment, A is -C(O)-; n is 0; and R3 and R4 are independently -CH2- or -CH(CH3)-. In another embodiment, A is -C(O)-; n is 0; and R3 and R4 are each -CH2-. In another embodiment, A is -C(O)-; n is 2; and R3 and R4 are independently -CH2- or -CH(CH3)-. In another embodiment, A is -C(O)-; n is 2; and R3 and R4 are each -CH2-. In another embodiment, A is -C(O)- and X is -CH2-.
In another embodiment, A is -C(O)- and X is O. Illustrative Piperazine Compounds of formula (I) are listed below in Tables 1-8.
Ta le 1
(IV)
8L£lt0/t 0ZSΛ/lDd t7rS9S0/S00Z OΛV
The designations (a), (b) and (c) in connection with each of the compounds of Table 1 have the following meaning: (a) means that R
a and R are -H; (b) means that R
a is -H and Rb is -OCH
3; and (c) means that R
a and R
b taken together form -O-CH
2-O-.
Table 2
(V)
The designations (a), (b) and (c) in connection with each of the compounds of Table 2 have the following meaning: (a) means that R
a and Rb are -H; (b) means that R
a is -H and Rb is -OCH
3; and (c) means that R
a and Rb taken together form -O-CH
2-O-.
Table 3
(VI)
The designations (a), (b) and (c) in connection with each of the compounds of Table 3 have the following meaning: (a) means that R
a and Rb are -H; (b) means that R
a is -H and R
b is -OCH
3; and (c) means that R
a and R taken together form -O-CH
2-O-.
Table 4
(VH)
The designations (a), (b) and (c) in connection with each of the compounds of Table 5 have the following meaning: (a) means that R
a and R are -H; (b) means that R
a is -H and R is -OCH
3; and (c) means that R
a and R
b taken together form -O-CH
2-O-.
ς
9
3i eχ _.εϊtO/ OOZSfl/I3d tZ£9£0/£00Z OΛV
The designations (a), (b) and (c) in connection with each of the compounds of Table 6 have the following meaning: (a) means that R
a and R
b are -H; (b) means that R
a is -H and R is -OCH
3; and (c) means that R
a and R
b taken together form -O-CH
2-O-.
Table 7
(X)
The designations (a), (b) and (c) in connection with each of the compounds of Table 7 have the following meaning: (a) means that R
a and Rb are -H; (b) means that R
a is -H and R
b is -OCH
3; and (c) means that R
a and R
b taken together form -O-CH
2-O-.
Table 8
(XI)
The designations (a), (b) and (c) in connection with each of the compounds of Table 8 have the following meaning: (a) means that R
a and R are -H; (b) means that R
a is -H and R is -OCH
3; and (c) means that R
a and R
b taken together form -O-CH -O-.
4.4 Definitions As used herein, the terms used above having following meaning: "-(C
1-C
1o)alkyl" means a saturated straight chain or branched non-cyclic hydrocarbon having from 1 to 10 carbon atoms. Representative saturated straight chain -(Cι-Cιo)alkyls include -methyl, -ethyl, -n-propyl, -n-butyl, -n-pentyl, -n-hexyl, -n-heptyl, -n-octyl, -n-nonyl, and -n-decyl. Representative saturated branched -( - C!o)alkyls include - so-propyl, -sec-butyl, -iso-butyl, -tert-butyl, -tso-pentyl, -2-methylbutyl, -3-methylbutyl, -2,2-dimethylbutyl, -2,3-dimethylbutyl, -2-methylpentyl, -3-methylpentyl, -4-methylpentyl, -2-methylhexyl, -3-methylhexyl, -4-methylhexyl, -5-methylhexyl, -2,3-dimethylbutyl, -2,3-dimethylpentyl, -2,4-dimethylpentyl, -2,3-dimethylhexyl, -2,4-dimethylhexyl, -2,5-dimethylhexyl, -2,2-dimethylpentyl, -2,2-dimethylhexyl, -3,3-dimethylpentyl, -3,3-dimethylhexyl, -4,4-dimethylhexyl, -2-ethylpentyl, -3-ethylpentyl, -2-ethylhexyl, -3-ethylhexyl, -4-ethylhexyl, -2-methyl- 2-ethylpentyl, -2-methyl-3-ethylpentyl, -2-methyl-4-ethylpentyl, -2-methyl-2-ethylhexyl, -2-methyl-3-ethylhexyl, -2-methyl-4-ethylhexyl, -2,2-diethylpentyl, -3,3-diethylhexyl, -2,2-diethylhexyl, -3,3-diethylhexyl and the like. "-(C
1-C
6)alkyl" means a saturated straight chain or branched non-cyclic hydrocarbon having from 1 to 6 carbon atoms. Representative saturated straight chain -(CrC^alkyls include -methyl, -ethyl, -n-propyl, -n-butyl, -n-pentyl, and -n-hexyl. Representative saturated branched -(C
1-C
6)alkyls include -.so-propyl, -.sec-butyl, -iso-butyl, -tert-butyl, -zso-pentyl, -2-methylbutyl, -3-methylbutyl, -2,2-dimethylbutyl, -2,3-dimethylbutyl, -2-methylpentyl, -3-methylpentyl, -4-methylpentyl and the like. "-(d-C^alkyl" means a saturated straight chain or branched non-cyclic hydrocarbon having from 1 to 4 carbon atoms. Representative saturated straight chain -( -G alkyls include -methyl, -ethyl, -n-propyl, and -n-butyl. Representative saturated branched -( -G^alkyls include -.so-propyl, -sec-butyl, -iso-butyl, and -tert-butyl. "-(C
2-C
1o)alkenyl" means a straight chain or branched non-cyclic hydrocarbon having from 2 to 10 carbon atoms and including at least one carbon-carbon double bond. Representative straight chain and branched (C
2-C
1o)alkyls include 1-ρentenyl, -2-pentenyl, -3 -methyl- 1-butenyl, -2-methyl-2-butenyl, -2,3-dimethyl-2-butenyl, -1-hexenyl, -2-hexenyl, -3-hexenyl, -1-heptenyl, -2-heρtenyl, -3-heptenyl, -1-octenyl,
-2-octenyl, -3-octenyl, -1-nonenyl, -2-nonenyl, -3-nonenyl, -1-decenyl, -2-decenyl, -3-decenyl and the like. "-(C
2-C
6)alkenyl" means a straight chain or branched non-cyclic hydrocarbon having from 2 to 6 carbon atoms and including at least one carbon-carbon double bond. Representative straight chain and branched (C
2-C6)alkenyls include -vinyl, -allyl, -1-butenyl, -2-butenyl, -iso-butylenyl, -1-pentenyl, -2-pentenyl, -3 -methyl- 1-butenyl, -2-methyl-2-butenyl, -2,3-dimethyl-2-butenyl, -1-hexenyl, -2-hexenyl, -3-hexenyl and the like. "-(C
2-C
1o)alkynyl" means a straight chain or branched non-cyclic hydrocarbon having from 2 to 10 carbon atoms and including at least one carbon-carbon triple bond. Representative straight chain and branched -(C
2-C
1o)alkynyls include -acetylenyl, -propynyl, -1-butynyl, -2-butynyl, -1-pentynyl, -2-pentynyl, -3-methyl-l-butynyl, -4-pentynyl, -1-hexynyl, -2-hexynyl, -5-hexynyl, -1-heptynyl, -2-heptynyl, -6-heptynyl, -1-octynyl, -2-octynyl, -7-octynyl, -1-nonynyl, -2-nonynyl, -8-nonynyl, -1-decynyl, -2-decynyl, -9-decynyl and the like. "-(C
2-C
6)alkynyl" means a straight chain or branched non-cyclic hydrocarbon having from 2 to 6 carbon atoms and including at least one carbon-carbon triple bond. Representative straight chain and branched (C
2-C
6)alkynyls include -acetylenyl, -propynyl, -1-butynyl, -2-butynyl, -1-pentynyl, -2-pentynyl, -3-methyl-l-butynyl, -4-pentynyl, -1-hexynyl, -2-hexynyl, -5-hexynyl and the like. "-(C
3-C
8)cycloalkyl" means a saturated cyclic hydrocarbon having from 3 to 8 carbon atoms. Representative (C
3-C
8)cycloalkyls include -cyclopropyl, -cyclobutyl, -cyclopentyl, -cyclohexyl, -cycloheptyl, and -cyclooctyl. "-(C
8-C
1 )bicycloalkyl" means a bi-cyclic hydrocarbon ring system having from 8 to 14 carbon atoms and at least one saturated cyclic alkyl ring. Representative -(C
8- C
14)bicycloalkyls include -indanyl, -1,2,3,4-tetrahydronaphthyl, -5,6,7,8- tetrahydronaphthyl, -perhydronaphthyl and the like. "-(C
8-C
1 )tricycloalkyl" means a tri-cyclic hydrocarbon ring system having from 8 to 14 carbon atoms and at least one saturated cyclic alkyl ring. Representative -(C
8- C
14)tricycloalkyls include -pyrenyl, -1,2,3,4-tetrahydroanthracenyl,
-perhydroanthracenyl, -aceanthreneyl, -1,2,3,4-tetrahydropenanthrenyl, -5,6,7,8- tetrahydrophenanthrenyl, -perhydrophenanthrenyl and the like.
"-(C5-C1o)cycloalkenyl" means a cyclic non-aromatic hydrocarbon having at least one carbon-carbon double bond in the cyclic system and from 5 to 10 carbon atoms. Representative (C5-C_o)cycloalkenyls include -cyclopentenyl, -cyclopentadienyl, -cyclohexenyl, -cyclohexadienyl, -cycloheptenyl, -cycloheptadienyl, -cycloheptatrienyl, -cyclooctenyl, -cyclooctadienyl, -cyclooctatrienyl, -cyclooctatetraenyl, -cyclononenyl, -cyclononadienyl, -cyclodecenyl, -cyclodecadienyl and the like. "-(C5-C8)cycloalkenyl" means a cyclic non-aromatic hydrocarbon having at least one carbon-carbon double bond in the cyclic system and from 5 to 8 carbon atoms. Representative (C5-C8)cycloalkenyls include -cyclopentenyl, -cyclopentadienyl, -cyclohexenyl, -cyclohexadienyl, -cycloheptenyl, -cycloheptadienyl, -cycloheptatrienyl, -cyclooctenyl, -cyclooctadienyl, -cyclooctatrienyl, -cyclooctatetraenyl and the like. "-(C8-C14)bicycloalkenyl" means a bi-cyclic hydrocarbon ring system having at least one carbon-carbon double bond in each ring and from 8 to 14 carbon atoms. Representative -(Cg-C1 )bicycloalkenyls include -indenyl, -pentalenyl, -naphthalenyl, -azulenyl, -heptalenyl, -1,2,7,8-tetrahydronaphthalenyl and the like. "-(C8-C1 )tricycloalkenyι" means a tri-cyclic hydrocarbon ring system having at least one carbon-carbon double bond in each ring and from 8 to 14 carbon atoms. Representative -(C8-Cι )tricycloalkenyls include -anthracenyl, -phenanthrenyl, -phenalenyl, -acenaphthalenyl, -αs-indacenyl, -s-indacenyl and the like. "-(5- to 10-membered)heteroary_" means an aromatic heterocycle ring of 5 to 10 members, including both mono- and bicyclic ring systems, where at least one carbon atom of one or both of the rings is replaced with a heteroatom independently selected from nitrogen, oxygen, and sulfur. In one embodiment, one of the -(5- to 10- membered)heteroaryl's rings contain at least one carbon atom. In another embodiment, both of the -(5- to 10-membered)heteroaryl's rings contain at least one carbon atom. Representative (5- to 10-membered)heteroaryls include pyridyl, furyl, benzofuranyl, benzo(l,3)dioxolyl, thiophenyl, benzothiophenyl, quinolinyl, pyrrolyl, indolyl, oxazolyl, benzoxazolyl, imidazolyl, benzimidazolyl, thiazolyl, benzothiazolyl, isoxazolyl, pyrazolyl, isothiazolyl, pyridazinyl, pyrimidyl, pyrazinyl, triazinyl, cinnolinyl, phthalazinyl, quinazolinyl and the like. "-(3- to 7-rnembered)heterocycle" or "-(3- to 7-membered)heterocyclo" means a 3- to 7-membered monocyclic heterocyclic ring which is either saturated, unsaturated,
non-aromatic or aromatic. A 3- or a 4-membered heterocycle can contain up to 3 heteroatoms, a 5-membered heterocycle can contain up to 4 heteroatoms, a 6-membered heterocycle can contain up to 6 heteroatoms, and a 7-membered heterocycle can contain up to 7 heteroatoms. Each heteroatom is independently selected from nitrogen, which can be quatemized; oxygen; and sulfur, including sulfoxide and sulfone. The -(3- to 7- membered)heterocycle can be attached via any heteroatom or carbon atom. Representative -(3- to 7-membered)heterocycles include pyridyl, furyl, thiophenyl, pyrrolyl, oxazolyl, imidazolyl, thiazolyl, isoxazolyl, pyrazolyl, isothiazolyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, morpholinyl, pyrrolidinonyl, pyrrolidinyl, piperidinyl, piperazinyl, hydantoinyl, valerolactamyl, oxiranyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyrindinyl, tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl and the like. "-(3- to 5-membered)heterocycle" or "-(3- to 5-membered)heterocyclo" means a 3- to 5-membered monocyclic heterocyclic ring which is either saturated, unsaturated, non-aromatic or aromatic. A 3- or 4-membered heterocycle can contain up to 3 heteroatoms and a 5-membered heterocycle can contain up to 4 heteroatoms. Each heteroatom is independently selected from nitrogen, which can be quatemized; oxygen; and sulfur, including sulfoxide and sulfone. The -(3- to 5-membered)heterocycle can be attached via any heteroatom or carbon atom. Representative -(3- to 5- membered)heterocycles include furyl, thiophenyl, pyrrolyl, oxazolyl, imidazolyl, thiazolyl, isoxazolyl, pyrazolyl, isothiazolyl, triazinyl, pyrrolidinonyl, pyrrolidinyl, hydantoinyl, oxiranyl, oxetanyl, tetrahydrofuranyl, tetrahydrothiophenyl and the like. "-(7- to 10-membered)bicycloheterocycle" or "-(7- to 10- membered)bicycloheterocyclo" means a 7- to 10-membered bicyclic, heterocyclic ring having a saturated, unsaturated, non-aromatic or aromatic group. A -(7- to 10- membered)bicycloheterocycle contains from 1 to 4 heteroatoms independently selected from nitrogen, which can be quatemized; oxygen; and sulfur, including sulfoxide and sulfone. The (7- to 10-membered)bicycloheterocycle can be attached via any heteroatom or carbon atom. Representative -(7- to 10-membered)bicycloheterocycles include -quinolinyl, -isoquinolinyl, -chromonyl, -coumarinyl, -indolyl, -indolizinyl, -benzo[b]furanyl, -benzo[b]thiophenyl, -indazolyl, -purinyl, -4H-quinolizinyl,
-isoquinolyl, -quinolyl, -phthalazinyl, -naphthyridinyl, -carbazolyl, - ?-carbolinyl, -benzo(l,3)dioxolyl and the like. A benzo(l,3)dioxolyl group has the structure:
"-(C
1 )aryl" means a 14-membered aromatic carbocyclic moiety such as anthryl or phenanthryl. "-CH (halo)" means a methyl group where one of the hydrogens of the methyl group has been replaced with a halogen. Representative -CH
2(halo) groups include -CH
2F, -CH
2C1, -CH
2Br and -CH
2I. "-CH(halo)
2" means a methyl group where two of the hydrogens of the methyl group have been replaced with a halogen. Representative -CH(halo)
2 groups include -CHF
2, -CHC1
2, -CHBr
2, -CHBrCl, -CHC1I and -CHI
2. "-C(halo)
3" means a methyl group where each of the hydrogens of the methyl group has been replaced with a halogen. Representative -C(halo)
3 groups include -CF
3, -CF
2C1, -CC1
3, -CBr
3, -CFBr
2 and -CI
3. "-Halogen" or "-halo" means -F, -Cl, -Br or -I. The phrase "pharmaceutically acceptable salt," as used herein, is any pharmaceutically acceptable salt that can be prepared from a Piperazine Compound, including a salt formed from an acid and a basic functional group, such as a nitrogen, of one of the Piperazine Compounds. Illustrative salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate (i.e., l,l'-methylene- bis-(2-hydroxy-3-naphthoate)) salts. The term "pharmaceutically acceptable salt" also refers to a salt prepared from a Piperazine Compound having an acidic functional group, such as a carboxylic acid functional group, and a pharmaceutically acceptable inorganic or organic base. Suitable bases include, but are not limited to, hydroxides of alkali metals such as sodium, potassium, and lithium; hydroxides of alkaline earth metal such as calcium and magnesium; hydroxides of other metals, such as aluminum and zinc;
ammonia and organic amines, such as unsubstituted or hydroxy-substituted mono-, di- or trialkylamines; dicyclohexylamine; tributyl amine; pyridine; N-methyl-N-ethylamine; diethylamine; triethylamine; mono-, bis- or tris-(2-hydroxy-lower alkyl amines), such as mono-, bis- or tris-(2-hydroxyethyl)amine, 2-hydroxy-tert-butylamine or tris-(hydroxymethyl)methylamine, N,N-di-lower alkyl-N-(hydroxy lower alkyl)-amines, such as N,N-dimethyl-N-(2-hydroxyethyl)amine or tri-(2-hydroxyethyl)amine; N-methyl-D-glucamine; and amino acids such as arginine, lysine and the like. An "animal" is defined herein to include any animal (e.g., human, cow, horse, sheep, pig, chicken, turkey, quail, cat, dog, mouse, rat, rabbit or guinea pig), in one embodiment a mammal such as a non-primate and a primate (e.g. , monkey or human), and in another embodiment a human. In certain embodiments, the human is an infant, child, adolescent or adult. The phrase "effective amount" when used in connection with a Piperazine Compound means an amount effective for: (a) treating or preventing a Condition; or (b) inhibiting mGluR5 or mGluRl function in a cell. The phrase "effective amount" when used in connection with another therapeutic agent means an amount for providing the therapeutic effect of the other therapeutic agent. As used herein, the term "adjunctive" is used interchangeably with "in combination" or "combination." Such terms are also used where a Piperazine
Compound and at last one therapeutic agent affects the treatment or prevention of a Condition, the same Condition in one embodiment, different Conditions in another embodiment. As used herein, the term "adjunctively administered" refers to the administration of at last one therapeutic agent in addition to a Piperazine Compound, either simultaneously with the same or at intervals prior to, during, or following administration of the a Piperazine Compound to achieve the desired therapeutic or prophylactic effect. When a first group is "substituted with one or more" second groups, each of one or more of the first group's hydrogen atoms is replaced with a second group. In one embodiment, a first group is substituted with up to three second groups. In another embodiment, a first group is substituted with one or two second groups.
In another embodiment, a first group is substituted with only one second group. The term "Ul" means urinary incontinence. The term "ALS" means amyotrophic lateral sclerosis. The phrases "treatment of," "treating" and the like include the amelioration or cessation a Condition or a symptom thereof. In one embodiment, treating includes inhibiting, for example, decreasing the overall frequency of episodes of a Condition or a symptom thereof. The phrases "prevention of," "preventing" and the like include the avoidance of the onset of a Condition or a symptom thereof.
4.5 Methods for Making the Piperazine Compounds The Piperazine Compounds can be made using conventional organic syntheses and/or by the following illustrative methods. The illustrative methods disclose one or more synthetic intermediates that are useful for making the Piperazine Compounds. All of the synthetic intermediates are also encompassed by the present invention. Certain Piperazine Compounds can be prepared according to Scheme 1.
Scheme 1
where R3, R4, R5, R6, R , R8, R13, R1 , R15, A, X, Y, Z, m and n are as defined above for the Piperazine Compounds of formula (I) and Q is I, Br, Cl or F. For example, a solution of a compound of formula A, e.g., where Q is Cl, an HC1 salt of a compound of formula B and an excess of triethylamine ("TEA," about 3-5 equivalents) in 5 mL of acetonitrile is heated in a sealed reaction vessel (e.g., a 20 mL vial) in an oil bath to about 90°C for about 12 hours. The reaction mixture is
concentrated, e.g., on rotary evaporator, and the residue is purified, e.g., via preparative thin-layer chromatography ("TLC"), to yield a Piperazine Compound. A general procedure for reacting a 2-halo-pyridine with piperazine is provided in EJ. Jacobsen et al, J. Med. Chem. 1145-1151 (1990). Piperazine Compounds can be obtained by reacting a diaminoalkyl compound of formula D with a (C1-C1o)alkyliodide, or a (C2-Cιo)alkenyliodide or (C2- C1o)alkynyliodide in which the iodine atom is bonded to an sp3 carbon atom, at low temperature, e.g., about 0°C to about -78°C, in the presence of a strong base, e.g., lithium di-.so-propylamide ("LDA"), optionally in hexamethylphosphoramide ("HMPA") as shown below in Scheme 2, e.g., for a (C1-C1o)alkyliodide reactant.
Scheme 2
where -(C1-C1o)alkyl, R3, R , R5, R , R8) A, X, Y, Z, m and n are as defined above for the
Piperazine Compounds of formula (I). A general procedure for reacting a terminal acetylene with an alkyl iodide is provided in G.M. Strunz et al, Can. J. Chem. 419-432 (1996). Piperazine Compounds where R6 contains an sp or sp carbon atom bonded to the acetylene group can be obtained by reacting a compound of formula A with an aryl iodide, or with a (C2-C1o)alkenyliodide or (C2-C1o)alkynyliodide in which the iodine atom is bonded to an sp or sp2 carbon atom, at room temperature, e.g., about 25°C, in ethyl acetate ("EtOAc") in the presence of Pd(Ph3P)2(OAc)2, Cul and TEA, as shown
below in Scheme 3, e.g., for an aryl iodide reagent containing an R^ substituent.
Scheme 3
where R3, R , R5, R , R8, R15, A, X, Y, Z, m and n are as defined above for the Piperazine Compounds of formula (I). A general procedure for reacting a terminal acetylene with an aryl iodide is provided in L.A. Hay et al, J. Org. Chem. 5050-5058 (1998). A compound of formula D', i.e., a compound of formula D where A is -C(O)-, can be prepared by reacting a compound of formula G with propynoic acid in the presence of 1-hydroxybenzotriazolehydrate ("HOBT") and 1,3-di-iso-propylcarbodiimide ("DIC") as shown in Scheme 4.
Scheme 4
where R3, R4, R5, R7, R8, X, Y, Z, m and n are as defined above for the Piperazine Compounds of formula (I). A general procedure for reacting a carboxylic acid with an amine is provided in F.M. Martin et al, Bioorg. Med. Chem. Lett. 2887-2892 (1999). A compound of formula D' can also be prepared by reacting a compound of formula G with propynoic acid chloride in the presence of a tertiary amine, such as TEA, as shown in Scheme 5.
Scheme 5
where R3, R4, R5, R7, R8, X, Y, Z, m and n are as defined above for the Piperazine Compounds of formula (I). A general procedure for reacting an acid chloride with an amine is provided in
T.R. Herrin et al, J. Med. Chem. 1216-1223 (1975).
A protected, e.g., Boc-protected, compound of formula G can be prepared by reacting a 2-halo-substituted piperadine-containing compound of formula A with a protected, e.g., Boc-protected, compound of formula I in the presence of TEA, e.g., in chloroform at a temperature of about 50°C, as shown in Scheme 6.
Scheme 6
where R3, R4, R5, R7, R8, X, Y, Z, m and n are as defined above for the Piperazine Compounds of formula (I) and Q is I, Br, Cl or F. A general procedure for reacting a 2-halo-piperidine with piperazine is provided in E. J. Jacobsen et al, J. Med. Chem. 1145-1151 (1990). Methods for using a protecting group are described below. An N-Boc group can be removed, e.g., by reacting the Boc- protected compound in 2N HCl/diethylether for about 2 hours at about 25 °C. Compounds of formula AC (i.e., unprotected compounds of formula G where m is 0, Y is -C(O)-, X is O and Z is -CH(R -) and AD (i.e., unprotected compounds of formula G where m is 0, Y is -CH(R -, X is O and Z is -C(O)-) can be prepared from a compound of formula AB as shown below in Scheme 7.
where Rls R3, R4, R5, R , R8, R9 and n are as defined above for the Piperazine Compounds of formula (I) and L is H or a protecting group. Methods for using a protecting group are described below. Selective reduction of an anhydride with sodium borohydride can be performed to yield a compound of formula AC where n is 0 and/or AD where n is 0 directly by methods known in the art, e.g., by adapting the procedures provided in Can. J. Chem., 64(6): 1031-1035 (1986) or J. Med. Chem., 34(9):2726-2735 (1991). Alternately, selective partial reduction of an anhydride with an aluminum hydride or sodium borohydride can be performed by methods known in the art, e.g., by adapting the procedures provided in Australian J. Chem., 42(6):787-795 (1989) or Tetrahedron, 44(10):2903-2912 (1988). Selective partial reduction is followed by Grignard addition, e.g., with Ri-MgCl, to yield a compound of formula AC and/or AD. Grignard addition
can be performed by methods known in the art, e.g., by adapting the procedures provided in Hecheng Huaxue, l(3):255-257 (1993) or Tetrahedron, 54:7033-7044 (1998). A protected, e.g., Boc-protected, compound of formula B, e.g., a compound of formula J or J' where A is -C(O)-or -C(C1-C alkyl)(C1-C4 alkyl)-, respectively, can be prepared by reacting a protected, e.g., Boc-protected, compound of formula I' with R6-substituted propynoic acid or R6-substituted 3-chloro-3-dialkyl-prop-l-yne as shown in Scheme 8.
Scheme 8
where R3, R , R6, R13, R14 and R15 are as defined above for the Piperazine Compounds of formula (I). A general procedure for reacting a carboxylic acid with an amine is provided in F.M. Martin et al, Bioorg. Med. Chem. Lett. 2887-2892 (1999) and a general procedure for reacting a halogenated propyne with an amine is provided in International Publication No. WO 03/004480. Methods for using a protecting group are described below.
Certain bicyclic compounds of formula A', i.e., compounds of formula A where n is 0 or 1, can be prepared as shown in Scheme 9 from a compound of formula BB, which is, in turn, prepared from a compound of formula BA, by performing a thermally assisted intramolecular Diels-Alder reaction.
Scheme 9
where R1? R2, R5, R7, R8, R9, X, Y, Z and m are as defined above for the Piperazine Compounds of formula (I), n' is 0 or 1, Q is I, Br, Cl or F, and W is -(C1-C1o)alkyl, -(C2- C10)alkenyl, -(C2-C10)alkynyl, -(C3-C8)cycloalkyl, -(C8-C1 )bicycloalkyl, -(C8- C14)tricycloalkyl, -(C5-C1o)cycloalkenyl, -(C8-C14)bicycloalkenyl, -(C8- C14)tricycloalkenyl, -(3- to 7-membered)heterocycle, -(7- to 10- membered)bicycloheterocycle, -phenyl, -naphthyl, -(C14)aryl or -(5- to 10- membered)heteroaryl, each of which is unsubstituted or substituted with one or more R7 groups, or W is H. For example, a solution of a compound of formula BB, e. g. , from about 0.5- 1.5
M in nitrobenzene, can be irradiated with microwave radiation to form a compound of formula A'. A compound of formula BB is irradiated under conditions that are sufficient to make a compound of formula A'. In one embodiment, the microwave irradiation frequency is from about 1 GHz to about 10 GHz. In another embodiment, the microwave irradiation frequency is from about 2 GHz to about 10 GHz. hi another embodiment, the microwave irradiation frequency is about 2.45 GHz. In one embodiment, a microwave-emitting apparatus operating at a frequency of about 2.45
GHz and which adjusts its microwave emissions such that a desired temperature is achieved, such as a MICROS YNTH Microwave Labstation (Milestone Microwave Laboratory System Inc., Monroe, CT), is used. The microwave irradiation causes the
solution, which can be contained in a non-metallic container, to heat from a temperature of about 25°C to a reaction temperature of from about 250°C to about 300°C. In another embodiment, the reaction temperature is from about 260°C to about 290°C. In another embodiment, the reaction temperature is from about 265°C to about 280°C. hi one embodiment, the heating occurs over a period of about 2 minutes or less. In another embodiment, the heating occurs over a period of about 1.5 minutes or less. In another embodiment, the heating occurs over a period of about 1 minute or less. Following the heating, the solution can be held at the reaction temperature with continued microwave irradiation for, in one embodiment, from about 0.5 to about 60 minutes, in another embodiment from about 0.5 to about 3 minutes, in another embodiment from about 1 to about 2 minutes, in another embodiment from about 30 to about 60 minutes, and in another embodiment from about 30 to about 50 minutes. Thereafter, irradiation can be stopped and the solution can be allowed to cool to about 25°C. Appropriate reaction conditions can be established by monitoring the formation of the compound of formula A' through routine experimentation, e.g., using TLC. A compound of formula A where n is 2 can be prepared from a compound of formula A', where n' is 0 or 1, by methods known in the art. Representative compounds of formula A and/or A', e.g., compounds of formula M, Q, T, V and Y', can be prepared by the following Schemes 10-14.
Scheme 10
(
Scheme 12
Scheme 13
In each of Schemes 10-14 above, Ri, R2, R5, R7, R8, R9, R_o, m and n are as defined above for the Piperazine Compounds of formula (I); n' is 0 or 1; each R' is independently
-H, -(C
rC
4)alkyl,
and W is as defined above for the compound of formula BB
A general procedure for Scheme 10 is provided in Tetrahedron Lett., 40(30):5483-5486 (1999). The compound of formula O can be prepared from the compound of formula N by methods known in the art, e.g. , by adapting the procedure provided in J. Med. Chem., 45:3639-3648 (2002). Compounds of formulas I, K and N are commercially available or can be prepared by methods known in the art. Piperazine Compounds where m and n are each 0, Y is -CH2-, X is -N(R)- (where R is H or (C1-C )alkyl) and Z is -C(O)- can be made by reacting a compound of formula U' (i.e. , a compound of formula U where m is 0) with a compound of formula B as shown below in Scheme 15.
Scheme 15
where A, R3 through R8 and Rπ through R15 are as defined above for the Piperazine Compounds of formula (I); n' is 0 or 1; and W is, independently, as defined above for the compound of formula BB. The reaction of 1 equivalent of U' with 1 equivalent of B can be conducted at a temperature of about 0°C. The reaction of 1 equivalent of U' with 2 equivalents of B can be conducted at a temperature of about 25°C. If any group of the compounds depicted in Schemes 1-15 above contain a -hydroxyl or -amino group, that -hydroxyl and/or -amino group can be protected using a suitable protecting group, using methods known to those skilled in the art, before the compound is reacted. Suitable protecting groups for a hydroxyl group include, but are not limited to, methyl ether, methoxymethyl ether, methoxythiomethyl ether, 2-methoxyethoxymethyl ether, bis(2-chloroethoxy)ethyl ether, tetrahydropyranyl ether, tetrahydrothiopyranyl ether, 4-methoxytetrahydropyranyl ether,
methoxytetrahydrothiopyranyl ether, tetrahydrofuranyl ether, tetrahydrothiofuranyl ether, 1-ethoxyethyl ether, 1 -methyl- 1-methoxyethyl ether, 2-(phenylselenyl ether), tert-butyl ether, allyl ether, benzyl ether, o-nitrobenzyl ether, triphenylmethyl ether, o-napthyldiphenylmethyl ether, /7-methoxydiphenylmethyl ether, 9-(9-phenyl-10-oxo)anthryl ether (tritylone), trimethylsilyl ether, .so-propyldimethylsilyl ether, tert-butyldimethylsilyl ether, tert-butyldiphenylsilyl ether, tribenzylsilyl ether, tri-wo-propylsilyl ether, formate ester, acetate ester, trichloroacetate ester, phenoxyacetate ester, wo-butyrate ester, pivaloate ester, adamantoate ester, benzoate ester, 2,4,6-trimethyl (mesitoate) ester, methyl carbonate, 2,2,2-trichlorocarbonate, allyl carbonate, p-nitrophenyl carbonate, benzyl carbonate, p-nitrobenzyl carbonate,
S-benzylthiocarbonate, N-phenylcarbamate, nitrate ester, and 2,4-dinitrophenylsulfenate ester (see, e.g., T.W. Greene et al., Protective Groups in Organic Synthesis, 17-200 (3d ed. 1999)). Suitable protecting groups for an amino group include, but are not limited to, l,l-dimethyl-2,2,2-trichloroethyl carbamate, 1 -methyl- l-(4-biphenylyl)ethyl carbamate, 2-trimethylsilylethyl carbamate, 9-fluorenylmethyl carbamate, and tert-butyl carbamate (T.W. Greene et al., Protective Groups in Organic Synthesis, 494-653 (3d ed. 1999)). Certain Piperazine Compounds may have asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms. A Piperazine Compound can be in the form of an optical isomer or a diastereomer. Accordingly, the invention encompasses Piperazine Compounds and their uses as described herein in the form of their optical isomers, diasteriomers and mixtures thereof, including a racemic mixture. Optical isomers of the Piperazine Compounds can be obtained by known techniques such as chiral chromatography or formation of diastereomeric salts from an optically active acid or base. In addition, one or more hydrogen, carbon or other atoms of a Piperazine
Compound can be replaced by an isotope of the hydrogen, carbon or other atoms. Such compounds, which are encompassed by the present invention, are useful as research and diagnostic tools in metabolism pharmacokinetic studies and in binding assays. 4.6 Therapeutic Uses of the Piperazine Compounds In accordance with the invention, the Piperazine Compounds are administered to an animal in need of treatment or prevention of a Condition.
In one embodiment, an effective amount of a Piperazine Compound can be used to treat or prevent any condition treatable or preventable by inhibiting mGluR5. Examples of conditions that are treatable or preventable by inhibiting mGluR5 include, but are not limited to, pain, an addictive disorder, Parkinson's disease, parkinsonism, anxiety, a pruritic condition, and psychosis. hi another embodiment, an effective amount of a Piperazine Compound can be used to treat or prevent any condition treatable or preventable by inhibiting mGluRl. Examples of conditions that are treatable or preventable by inhibiting mGluRl include, but are not limited to, pain, Ul, an addictive disorder, Parkinson's disease, parkinsonism, anxiety, epilepsy, a seizure, stroke, a pruritic condition, psychosis, a cognitive disorder, a memory deficit, restricted brain function, Huntington's chorea, ALS, dementia, retinopathy, a muscle spasm, a migraine, vomiting, dyskinesia and depression. The Piperazine Compounds can be used to treat or prevent acute or chronic pain. Examples of pain treatable or preventable using the Piperazine Compounds include, but are not limited to, cancer pain, labor pain, pain from physical trauma, myocardial infarction pain, pancreatic pain, colic pain, post-operative pain, headache pain, muscle pain, arthritic pain, neuropathic pain, and pain associated with a periodontal disease, including gingivitis and periodontitis. The Piperazine Compounds can also be used for treating or preventing pain associated with inflammation or with an inflammatory disease in an animal. Such pain can arise when there is an inflammation of the body tissue, and which can be a local inflammatory response and/or a systemic inflammation. For example, the Piperazine Compounds can be used to treat or prevent pain associated with inflammatory diseases including, but not limited to: organ transplant rejection; reoxygenation injury resulting from organ transplantation (See Grupp et al. , J. Mol. Cell Cardiol 31 :297-303 (1999)) including, but not limited to, transplantation of the heart, lung, liver, or kidney; chronic inflammatory diseases of the joints, including arthritis, rheumatoid arthritis, osteoarthritis and bone diseases associated with increased bone resorption; inflammatory lung diseases, such as asthma, adult respiratory distress syndrome, and chronic obstructive airway disease; inflammatory diseases of the eye, including corneal dystrophy, trachoma, onchocerciasis, uveitis, sympathetic ophthalmitis and endophthalmitis; chronic inflammatory diseases of the gum, including gingivitis and periodontitis; tuberculosis;
leprosy; inflammatory diseases of the kidney, including uremic complications, glomerulonephritis and nephrosis; inflammatory diseases of the skin, including sclerodermatitis, psoriasis and eczema; inflammatory diseases of the central nervous system, including chronic demyelinating diseases of the nervous system, multiple sclerosis, AEDS-related neurodegeneration and Alzheimer s disease, infectious meningitis, encephalomyelitis, Parkinson's disease, Huntington's disease, amyotrophic lateral sclerosis and viral or autoimmune encephalitis; autoimmune diseases, including Type I and Type II diabetes mellitus; diabetic complications, including, but not limited to, diabetic cataract, glaucoma, retinopathy, nephropathy (such as microaluminuria and progressive diabetic nephropathy), polyneuropathy, mononeuropathies, autonomic neuropathy, gangrene of the feet, atherosclerotic coronary arterial disease, peripheral arterial disease, nonketotic hyperglycemic-hyperosmolar coma, foot ulcers, joint problems, and a skin or mucous membrane complication (such as an infection, a shin spot, a candidal infection or necrobiosis lipoidica diabeticorum); immune-complex vasculitis, and systemic lupus erythematosus (SLE); inflammatory diseases of the heart, such as cardiomyopathy, ischemic heart disease hypercholesterolemia, and atherosclerosis; as well as various other diseases that can have significant inflammatory components, including preeclampsia, chronic liver failure, brain and spinal cord trauma, and cancer. The Piperazine Compounds can also be used for treating or preventing pain associated with inflammatory disease that can, for example, be a systemic inflammation of the body, exemplified by gram-positive or gram negative shock, hemorrhagic or anaphylactic shock, or shock induced by cancer chemotherapy in response to pro- inflammatory cytokines, e.g., shock associated with pro-inflammatory cytokines. Such shock can be induced, e.g., by a chemotherapeutic agent that is administered as a treatment for cancer. The Piperazine Compounds can be used to treat or prevent UL Examples of Ul treatable or preventable using the Piperazine Compounds include, but are not limited to, urge incontinence, stress incontinence, overflow incontinence, neurogenic incontinence, and total incontinence. The Piperazine Compounds can be used to treat or prevent an addictive disorder, including but not limited to, an eating disorder, an impulse-control disorder, an alcohol- related disorder, a nicotine-related disorder, an amphetamine-related disorder, a
cannabis-related disorder, a cocaine-related disorder, an hallucinogen-related disorder, an inhalant-related disorders, and an opioid-related disorder, all of which are further sub- classified as listed below. Eating disorders include, but are not limited to, Bulimia Nervosa, Nonpurging Type; Bulimia Nervosa, Purging Type; Anorexia; and Eating Disorder not otherwise specified (NOS). Impulse control disorders include, but are not limited to, Intermittent Explosive Disorder, Kleptomania, Pyromania, Pathological Gambling, Trichotillomania, and Impulse Control Disorder not otherwise specified (NOS). Alcohol-related disorders include, but are not limited to, Alcohol-Induced
Psychotic Disorder with delusions, Alcohol Abuse, Alcohol Intoxication, Alcohol Withdrawal, Alcohol Intoxication Delirium, Alcohol Withdrawal Delirium, Alcohol- Induced Persisting Dementia, Alcohol- Induced Persisting Amnestic Disorder, Alcohol Dependence, Alcohol-Induced Psychotic Disorder with hallucinations, Alcohol-Induced Mood Disorder, Alcohol- Induced Anxiety Disorder, Alcohol- iduced Sexual Dysfunction, Alcohol-Induced Sleep Disorder and Alcohol-Related Disorder not otherwise specified (NOS). Nicotine-related disorders include, but are not limited to, Nicotine Dependence, Nicotine Withdrawal, and Nicotine-Related Disorder not otherwise specified (NOS). Amphetamine-related disorders include, but are not limited to, Amphetamine
Dependence, Amphetamine Abuse, Amphetamine Intoxication, Amphetamine Withdrawal, Amphetamine Intoxication Delirium, Amphetamine-Induced Psychotic Disorder with delusions, Amphetamine-Induced Psychotic Disorders with hallucinations, Amphetamine-Induced Mood Disorder, Amphetamine-Induced Anxiety Disorder, Amphetamine-Induced Sexual Dysfunction, Amphetamine-Induced Sleep Disorder, and Amphetamine Related Disorder not otherwise specified (NOS). Cannabis-related disorders include, but are not limited to, Cannabis Dependence, Cannabis Abuse, Cannabis Intoxication, Cannabis Intoxication Delirium, Cannabis- Induced Psychotic Disorder with delusions, Cannabis-Induced Psychotic Disorder with hallucinations, Cannabis-Induced Anxiety Disorder, and Cannabis Related Disorder not otherwise specified (NOS).
Cocaine-related disorders include, but are not limited to, Cocaine Dependence, Cocaine Abuse, Cocaine Intoxication, Cocaine Withdrawal, Cocaine Intoxication Delirium, Cocaine-Induced Psychotic Disorder with delusions, Cocaine-Induced Psychotic Disorders with hallucinations, Cocaine-Induced Mood Disorder, Cocaine- Induced Anxiety Disorder, Cocaine-Induced Sexual Dysfunction, Cocaine-Induced Sleep Disorder, and Cocaine Related Disorder not otherwise specified (NOS). Hallucinogen-related disorders include, but are not limited to, Hallucinogen Dependence, Hallucinogen Abuse, Hallucinogen Intoxication, Hallucinogen Withdrawal, Hallucinogen Intoxication Delirium, Hallucinogen-Induced Psychotic Disorder with delusions, Hallucinogen-Induced Psychotic Disorders with hallucinations, Hallucinogen- Induced Mood Disorder, Hallucinogen-Induced Anxiety Disorder, Hallucinogen-Induced Sexual Dysfunction, Hallucinogen-Induced Sleep Disorder, Hallucinogen Persisting Perception Disorder (Flashbacks), and Hallucinogen Related Disorder not otherwise specified (NOS). Inhalant-related disorders include, but are not limited to, Inhalant Dependence,
Inhalant Abuse, Inhalant Intoxication, Inhalant Intoxication Delirium, Inhalant-Induced Psychotic Disorder with delusions, Inhalant-Induced Psychotic Disorder with hallucinations, Inhalant-Induced Anxiety Disorder, and Inhalant Related Disorder not otherwise specified (NOS). Opioid-related disorders include, but are not limited to, Opioid Dependence,
Opioid Abuse, Opioid Intoxication, Opioid Intoxication Delirium, Opioid-Induced Psychotic Disorder with delusions, Opioid-Induced Psychotic Disorder with hallucinations, Opioid-Induced Anxiety Disorder, Opioid Withdrawal, and Opioid Related Disorder not otherwise specified (NOS). The Piperazine Compounds can be used to treat or prevent Parkinson's disease and parkinsonism and the symptoms associated with Parkinson's disease and parkinsonism, including but not limited to, bradykinesia, muscular rigidity, resting tremor, and impairment of postural balance. The Piperazine Compounds can be used to treat or prevent generalized anxiety or severe anxiety and the symptoms associated with anxiety, including but not limited to, restlessness, tension, tachycardia, dyspnea, depression including chronic "neurotic"
depression, panic disorder, agoraphobia and other specific phobias, eating disorders, and personality disorders. The Piperazine Compounds can be used to treat or prevent epilepsy, including but not limited to, partial epilepsy, generalized epilepsy, and the symptoms associated with epilepsy, including but not limited to, simple partial seizures, jacksonian seizures, complex partial (psychomotor) seizures, convulsive seizures (grand mal or tonic-clonic seizures), petit mal (absence) seizures, and status epilepticus. The Piperazine Compounds can be used to treat or prevent a seizure, including but not limited to, infantile spasms, febrile seizures, and epileptic seizures. The Piperazine Compounds can be used to treat or prevent strokes, including but not limited to, ischemic strokes and hemorrhagic strokes. The Piperazine Compounds can be used to treat or prevent a pruritic condition, including but not limited to, pruritus caused by dry skin, scabies, dermatitis, herpetiformis, atopic dermatitis, pruritus vulvae et ani, malaria, insect bites, pediculosis, contact dermatitis, drug reactions, urticaria, urticarial eruptions of pregnancy, psoriasis, lichen planus, lichen simplex chronicus, exfoliative dermatitis, folliculitis, bullous pemphigoid, and fiberglass dermatitis. The Piperazine Compounds can be used to treat or prevent psychosis, including but not limited to, schizophrenia, including paranoid schizophrenia, hebephrenic or disorganized schizophrenia, catatonic schizophrenia, undifferentiated schizophrenia, negative or deficit subtype schizophrenia, and non-deficit schizophrenia; a delusional disorder, including erotomanic subtype delusional disorder, grandiose subtype delusional disorder, jealous subtype delusional disorder, persecutory subtype delusional disorder, and somatic subtype delusional disorder; and brief psychosis. The Piperazine Compounds can be used to treat or prevent a cognitive disorder, including but not limited to, delirium and dementia such as multi-infarct dementia, dementia pugilistica, dementia caused by AIDS, and dementia caused by Alzheimer's disease. The Piperazine Compounds can be used to treat or prevent a memory deficiency, including but not limited to, dissociative amnesia and dissociative fugue. The Piperazine Compounds can be used to treat or prevent restricted brain function, including but not limited to, that caused by surgery or an organ transplant,
restricted blood supply to the brain, a spinal cord injury, a head injury, hypoxia, cardiac arrest, or hypoglycemia. The Piperazine Compounds can be used to treat or prevent Huntington's chorea. The Piperazine Compounds can be used to treat or prevent ALS. The Piperazine Compounds can be used to treat or prevent retinopathy, including but not limited to, arteriosclerotic retinopathy, diabetic arteriosclerotic retinopathy, hypertensive retinopathy, non-proliferative retinopathy, and proliferative retinopathy. The Piperazine Compounds can be used to treat or prevent a muscle spasm. The Piperazine Compounds can be used to treat or prevent a migraine. The Piperazine Compounds can be used to treat (e. g. , inhibit) or prevent vomiting, including but not limited to, nausea vomiting, dry vomiting (retching), and regurgitation. The Piperazine Compounds can be used to treat or prevent dyskinesia, including but not limited to, tardive dyskinesia and biliary dyskinesia. The Piperazine Compounds can be used to treat or prevent depression, including but not limited to, major depression and bipolar disorder. Without wishing to be bound by theory, Applicants believe that the Piperazine Compounds are antagonists for mGluR5. The invention relates to methods for inhibiting mGluR5 function in a cell comprising contacting a cell capable of expressing mGluR5 with an effective amount of a Piperazine Compound. This method can be used in vitro, for example, as an assay to select cells that express mGluR5 and, accordingly, are useful as part of an assay to select compounds useful for treating or preventing pain, an addictive disorder, Parkinson's disease, parkinsonism, anxiety, a pruritic condition, psychosis or schizophrenia. The method is also useful for inhibiting mGluR5 function in a cell in vivo, in an animal, in a human in one embodiment, by contacting a cell, in an animal, with an effective amount of a Piperazine Compound. In one embodiment, the method is useful for treating or preventing pain in an animal in need thereof. In another embodiment, the method is useful for treating or preventing an addictive disorder in an animal in need thereof. In another embodiment, the method is useful for treating or preventing Parkinson's disease in an animal in need thereof, another embodiment, the method is useful for treating or preventing parkinsonism in an animal in need thereof. In another embodiment, the
method is useful for treating or preventing anxiety in an animal in need thereof. In another embodiment, the method is useful for treating or preventing a pruritic condition in an animal in need thereof. In another embodiment, the method is useful for treating or preventing psychosis in an animal in need thereof. In another embodiment, the method is useful for treating or preventing schizophrenia in an animal in need thereof. Examples of cells capable of expressing mGluR5 are neuronal and glial cells of the central nervous system, particularly the brain, especially in the nucleus accumbens. Methods for assaying cells that express mGluR5 are known in the art. Without wishing to be bound by theory, Applicants believe that the Piperazine Compounds are antagonists for mGluRl. The invention also relates to methods for inhibiting mGluRl function in a cell comprising contacting a cell capable of expressing mGluRl with an effective amount of a Piperazine Compound. This method can be used in vitro, for example, as an assay to select cells that express mGluRl and, accordingly, are useful as part of an assay to select compounds useful for treating or preventing a Condition. The method is also useful for inhibiting mGluRl function in a cell in vivo, in an animal, a human in one embodiment, by contacting a cell, in an animal, with an effective amount of a Piperazine Compound. In one embodiment, the method is useful for treating or preventing pain in an animal in need thereof. In another embodiment, the method is useful for treating or preventing Ul in an animal in need thereof. In another embodiment, the method is useful for treating or preventing an addictive disorder in an animal in need thereof. In another embodiment, the method is useful for treating or preventing Parkinson's disease in an animal in need thereof. In another embodiment, the method is useful for treating or preventing parkinsonism in an animal in need thereof, hi another embodiment, the method is useful for treating or preventing anxiety in an animal in need thereof. In another embodiment, the method is useful for treating or preventing epilepsy in an animal in need thereof. In another embodiment, the method is useful for treating or preventing a seizure in an animal in need thereof. In another embodiment, the method is useful for treating or preventing stroke in an animal in need thereof. In another embodiment, the method is useful for treating or preventing a pruritic condition in an animal in need thereof. In another embodiment, the method is useful for treating or preventing psychosis in an animal in need thereof. In another embodiment, the method is useful for treating or
preventing a cognitive disorder in an animal in need thereof. In another embodiment, the method is useful for treating or preventing a memory deficit in an animal in need thereof. In another embodiment, the method is useful for treating or preventing restricted brain function in an animal in need thereof. In another embodiment, the method is useful for treating or preventing Huntington's chorea in an animal in need thereof. In another embodiment, the method is useful for treating or preventing ALS in an animal in need thereof. In another embodiment, the method is useful for treating or preventing dementia in an animal in need thereof. In another embodiment, the method is useful for treating or preventing retinopathy in an animal in need thereof. In another embodiment, the method is useful for treating or preventing a muscle spasm in an animal in need thereof. In another embodiment, the method is useful for treating or preventing a migraine in an animal in need thereof. In another embodiment, the method is useful for treating or preventing vomiting in an animal in need thereof, i another embodiment, the method is useful for treating or preventing dyskinesia in an animal in need thereof. In another embodiment, the method is useful for treating or preventing depression in an animal in need thereof. Examples of cells capable of expressing mGluRl include, but are not limited to, cerebellar Purkinje neuron cells, Purkinje cell bodies (punctate), cells of spine(s) of the cerebellum; neurons and neurophil cells of olfactory-bulb glomeruli; cells of the superficial layer of the cerebral cortex; hippocampus cells; thalamus cells; superior colliculus cells; and spinal trigeminal nucleus cells. Methods for assaying cells that express mGluRl are known in the art.
4.7 Therapeutic Prophylactic Administration and Compositions of the Invention Due to their activity, the Piperazine Compounds are advantageously useful in veterinary and human medicine. As described above, the Piperazine Compounds are useful for treating or preventing a Condition in an animal in need thereof. When administered to an animal, the Piperazine Compounds can be administered as a component of a composition that comprises a pharmaceutically acceptable carrier or excipient. The present compositions, which comprise a Piperazine Compound, can be administered orally. The Piperazine Compounds can also be administered by any other
convenient route, for example, by infusion or bolus injection, by absorption through epithelial or mucocutaneous linings (e.g., oral, rectal, or intestinal mucosa) and can be adjunctively administered with another therapeutical agent. Administration can be systemic or local. Various delivery systems are known, e.g., encapsulation in liposomes, microparticles, microcapsules or capsules, and can be used to administer the Piperazine
Compound. Methods of administration include, but are not limited to, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, epidural, oral, sublingual, intracerebral, intravaginal, transdermal, rectal, by inhalation, or topical, particularly to the ears, nose, eyes, or skin. The mode of administration can be at the discretion of the practitioner. In specific embodiments, it can be desirable to administer the Piperazine
Compounds locally. This can be achieved, for example, and not by way of limitation, by local infusion during surgery, topical application, e.g., in conjunction with a wound dressing after surgery, by injection, by means of a catheter, by means of a suppository or enema, or by means of an implant, said implant being of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers. In certain embodiments, it can be desirable to introduce the Piperazine
Compounds into the central nervous system or gastrointestinal tract by any suitable route, including intraventricular, intrathecal, and epidural injection, and enema.
Intraventricular injection can be facilitated by an intraventricular catheter, for example, attached to a reservoir, such as an Ommaya reservoir. Pulmonary administration can also be employed, e.g., by use of an inhaler or nebulizer, and formulation with an aerosolizing agent, or via perfusion in a fluorocarbon or synthetic pulmonary surfactant. In certain embodiments, the Piperazine Compounds can be formulated as a suppository, with traditional binders and excipients such as triglycerides. In another embodiment, the Piperazine Compounds can be delivered in a vesicle, in particular a liposome (See Langer, Science 249: 1527-1533 (1990) and Treat et al, Liposomes in the Therapy of Infectious Disease and Cancer 317-327 and 353-365
(1989).
In yet another embodiment, the Piperazine Compounds can be delivered in a controlled-release system or sustained-release system (See, e.g., Goodson, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)). Other controlled- or sustained-release systems discussed in the review by Langer, Science 249:1527-1533 (1990) can be used. In one embodiment, a pump can be used (Langer, Science
249:1527-1533 (1990); Sefton, CRC Crit. Ref Biomed. Eng. 14:201 (1987); Buchwald et al, Surgery 88:507 (1980); and Saudek et al, N. Engl J. Med. 321:574 (1989)). In another embodiment, polymeric materials can be used (See Medical Applications of Controlled Release (Langer and Wise eds., 1974); Controlled Drug Bioavailability, Drug Product Design and Performance (Smolen and Ball eds., 1984); Ranger and Peppas, J. Macromol Sci. Rev. Macromol Chem. 23:61 (1983); Levy et al, Science 228:190 (1985); During et al., Ann. Neurol 25:351 (1989); and Howard et al, J. Neurosurg. 71:105 (1989)). In yet another embodiment, a controlled- or sustained-release system can be placed in proximity of a target of the Piperazine Compounds, e.g., the spinal column, brain, or gastrointestinal tract, thus requiring only a fraction of the systemic dose. The present compositions can optionally comprise a suitable amount of a pharmaceutically acceptable excipient so as to provide the form for proper administration to the animal. Such pharmaceutical excipients can be liquids, such as water and oils, including those of petroleum, animal, vegetable, or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like. The pharmaceutical excipient can be saline, gum acacia, gelatin, starch paste, talc, keratin, colloidal silica, urea and the like. In addition, auxiliary, stabilizing, thickening, lubricating, and coloring agents can be used. In one embodiment, the pharmaceutically acceptable excipients are sterile when administered to an animal. Water, and in one embodiment physiological saline, is a particularly useful excipient when the Piperazine Compound is administered intravenously. Saline solutions and aqueous dextrose and glycerol solutions can also be employed as liquid excipients, particularly for injectable solutions. Suitable pharmaceutical excipients also include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol,
propylene, glycol, water, ethanol and the like. The present compositions, if desired, can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents. The present compositions can take the form of solutions, suspensions, emulsions, tablets, pills, pellets, capsules, capsules containing liquids, powders, sustained-release formulations, suppositories, aerosols, sprays, suspensions, or any other form suitable for use. In one embodiment, the composition is in the form of a capsule (See, e.g., U.S. Patent No. 5,698,155). Other examples of suitable pharmaceutical excipients are described in Remington's Pharmaceutical Sciences 1447-1676 (Alfonso R. Gennaro ed., 19th ed. 1995), incorporated herein by reference. In one embodiment, the Piperazine Compounds are formulated in accordance with routine procedures as a composition adapted for oral administration to human beings. Compositions for oral delivery can be in the form of tablets, lozenges, aqueous or oily suspensions, granules, powders, emulsions, capsules, syrups, or elixirs, for example. Orally administered compositions can contain one or more agents, for example, sweetening agents such as fructose, aspartame or saccharin; flavoring agents such as peppermint, oil of wintergreen, or cherry; coloring agents; and preserving agents, to provide a pharmaceutically palatable preparation. Moreover, where in tablet or pill form, the compositions can be coated to delay disintegration and absorption in the gastrointestinal tract thereby providing a sustained action over an extended period of time. Selectively permeable membranes surrounding an osmotically active driving compound are also suitable for orally administered compositions. In these latter platforms, fluid from the environment surrounding the capsule is imbibed by the driving compound, which swells to displace the agent or agent composition through an aperture. These delivery platforms can provide an essentially zero order delivery profile as opposed to the spiked profiles of immediate release formulations. A time-delay material such as glycerol monostearate or glycerol stearate can also be used. Oral compositions can include standard excipients such as mannitol, lactose, starch, magnesium stearate, sodium saccharin, cellulose, and magnesium carbonate. In one embodiment, the excipients are of pharmaceutical grade. In another embodiment, the Piperazine Compounds can be formulated for intravenous administration. Typically, compositions for intravenous administration comprise sterile isotonic aqueous buffer. Where necessary, the compositions can also
include a solubilizing agent. Compositions for intravenous administration can optionally include a local anesthetic such as lidocaine to lessen pain at the site of the injection. The ingredients can be supplied either separately or mixed together in unit dosage form, for example, as a dry lyophilized powder or water free concentrate in a hermetically sealed container such as an ampoule or sachette indicating the quantity of active agent. Where the Piperazine Compounds are to be administered by infusion, they can be dispensed, for example, with an infusion bottle containing sterile pharmaceutical grade water or saline. Where the Piperazine Compounds are administered by injection, an ampoule of sterile water for injection or saline can be provided so that the ingredients can be mixed prior to administration. The Piperazine Compounds can be administered by controlled-release or sustained-release means or by delivery devices that are known to those skilled in the art. Examples include, but are not limited to, those described in U.S. Patent Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; 4,008,719; 5,674,533; 5,059,595; 5,591,767; 5,120,548; 5,073,543; 5,639,476; 5,354,556; and 5,733,566, each of which is incorporated herein by reference. Such dosage forms can be used to provide controlled- or sustained-release of one or more active ingredients using, for example, hydroxypropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof to provide the desired release profile in varying proportions. Suitable controlled- or sustained-release formulations known to those in the art, including those described herein, can be readily selected for use with the active ingredients of the invention. The invention thus encompasses single unit dosage forms suitable for oral administration such as, but not limited to, tablets, capsules, gelcaps, and caplets that are adapted for controlled- or sustained-release. Controlled- or sustained-release pharmaceutical compositions can have a common goal of improving drug therapy over that achieved by their non-controlled or non-sustained-release counterparts. In one embodiment, a controlled- or sustained- release composition comprises a minimal amount of a Piperazine Compound to treat or prevent the condition in a minimal amount of time. Advantages of controlled- or sustained-release compositions include extended activity of the drug, reduced dosage frequency, and increased patient compliance. In addition, controlled- or sustained-
release compositions can favorably affect the time of onset of action or other characteristics, such as blood levels of the Piperazine Compound, and can thus reduce the occurrence of adverse side effects. Controlled- or sustained-release compositions can initially release an amount of a Piperazine Compound that promptly produces the desired therapeutic or prophylactic effect, and gradually and continually release other amounts of the Piperazine Compound to maintain this level of therapeutic or prophylactic effect over an extended period of time. To maintain a constant level of the Piperazine Compound in the body, the Piperazine Compound can be released from the dosage form at a rate that will replace the amount of Piperazine Compound being metabolized and excreted from the body. Controlled- or sustained-release of an active ingredient can be stimulated by various conditions, including but not limited to, changes in pH, changes in temperature, concentration or availability of enzymes, concentration or availability of water, or other physiological conditions or compounds. The amount of the Piperazine Compound that is effective in the treatment or prevention of a condition can be determined by standard clinical techniques. In addition, in vitro or in vivo assays can optionally be employed to help identify optimal dosage ranges. The precise dose to be employed will also depend on the route of administration, and the seriousness of the Condition and can be decided according to the judgment of a practitioner and/or each animal's circumstances. Suitable effective dosage amounts, however, range from about 0.01 mg/kg of body weight to about 2500 mg/kg of body weight, although they are typically about 100 mg/kg of body weight or less. In one embodiment, the effective dosage amount ranges from about 0.01 mg/kg of body weight to about 100 mg/kg of body weight of a Piperazine Compound, in another embodiment, from about 0.02 mg/kg of body weight to about 50 mg/kg of body weight, and in another embodiment, from about 0.025 mg/kg of body weight to about 20 mg/kg of body weight. In one embodiment, an effective dosage amount is administered about every 24 hours until the Condition is abated. In another embodiment, an effective dosage amount is administered about every 12 hours until the Condition is abated. In another embodiment, an effective dosage amount is administered about every 8 hours until the Condition is abated. In another embodiment, an effective dosage amount is administered about every 6 hours until the Condition is abated. In another embodiment, an effective dosage
amount is administered about every 4 hours until the Condition is abated. The effective dosage amounts described herein refer to total amounts administered; that is, if more than one Piperazine Compound is administered, the effective dosage amounts correspond to the total amount administered. Where a cell capable of expressing mGluR5 or mGluRl is contacted with a
Piperazine Compound in vitro, the amount effective for inhibiting the mGluR5 or mGluRl receptor function in a cell will typically range from about 0.01 μg/L to about 5 mg/L, in one embodiment, from about 0.01 μg/L to about 2.5 mg/L, in another embodiment, from about 0.01 μg/L to about 0.5 mg/L, and in another embodiment, from about 0.01 μg/L to about 0.25 mg/L of a solution or suspension of a pharmaceutically acceptable carrier or excipient. In one embodiment, the volume of solution or suspension comprising the Piperazine Compound is from about 0.01 μL to about 1 mL. In another embodiment, the volume of solution or suspension is about 200 μL. Where a cell capable of expressing mGluR5 or mGluRl is contacted with a Piperazine Compound in vivo, the amount effective for inhibiting the receptor function in a cell will typically range from about 0.01 mg/kg of body weight to about 2500 mg/kg of body weight, although it typically ranges from about 100 mg/kg of body weight or less. In one embodiment, the effective dosage amount ranges from about 0.01 mg/kg of body weight to about 100 mg/kg of body weight of a Piperazine Compound, in another embodiment, from about 0.02 mg/kg of body weight to about 50 mg/kg of body weight and in another embodiment, from about 0.025 mg/kg of body weight to about 20 mg/kg of body weight. In one embodiment, an effective dosage amount is administered about every 24 h. In another embodiment, an effective dosage amount is administered about every 12 h. In another embodiment, an effective dosage amount is administered about every 8 h. In another embodiment, an effective dosage amount is administered about every 6 h. In another embodiment, an effective dosage amount is administered about every 4 h. The Piperazine Compounds can be assayed in vitro or in vivo for the desired therapeutic or prophylactic activity prior to use in humans. Animal model systems can be used to demonstrate safety and efficacy in humans. The present methods for treating or preventing a Condition in an animal in need thereof can further comprise adjunctively administering another therapeutic agent to the
animal being administered a Piperazine Compound. In one embodiment, the other therapeutic agent is adjunctively administered in an effective amount. The present methods for inhibiting mGluR5 function in a cell capable of expressing mGluR5 can further comprise contacting the cell with an effective amount of another therapeutic agent. The present methods for inhibiting mGluRl function in a cell capable of expressing mGluRl can further comprise contacting the cell with an effective amount of another therapeutic agent. Effective amounts of the other therapeutic agents are known to those skilled in the art. However, it is within the skilled artisan's purview to determine the other therapeutic agent's optimal effective- amount range. In one embodiment of the invention, where another therapeutic agent is adjunctively administered to an animal, the effective amount of the Piperazine Compound is less than its effective amount would be where the other therapeutic agent is not administered. In another embodiment of the invention, where another therapeutic agent is adjunctively administered to an animal, the effective amount of the other therapeutic agent is less than its effective amount would be where the Piperazine Compound is not administered, hi these cases, without being bound by theory, it is believed that the Piperazine Compound(s) and the other therapeutic agent(s) act synergistically to treat or prevent a Condition. The other therapeutic agent can be, but is not limited to, an opioid agonist, a non- opioid analgesic, a non-steroidal anti-inflammatory agent, an antimigraine agent, a Cox- II inhibitor, an antiemetic, a β-adrenergic blocker, an anticonvulsant, an antidepressant, a Ca -channel blocker, an anticancer agent, an agent for treating or preventing one or more Conditions, and mixtures thereof. Examples of useful opioid agonists include, but are not limited to, alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, buprenorphine, butorphanol, clonitazene, codeine, desomorphine, dextromoramide, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmorphine, etonitazene fentanyl, heroin, hydrocodone, hydromorphone, hydroxypethidine, isomethadone, ketobemidone, levorphanol, levophenacylmorphan, lofentanil, meperidine, meptazinol, metazocine,
methadone, metopon, morphine, myrophine, nalbuphine, narceine, nicomorphine, norlevorphanol, normethadone, nalorphine, normorphine, norpipanone, opium, oxycodone, oxymorphone, papaveretum, pentazocine, phenadoxone, phenomorphan, phenazocine, phenoperidine, piminodine, piritramide, proheptazine, promedol, properidine, propiram, propoxyphene, sufentanil, tilidine, tramadol, pharmaceutically acceptable salts thereof, and mixtures thereof. In certain embodiments, the opioid agonist is selected from codeine, hydromorphone, hydrocodone, oxycodone, dihydrocodeine, dihydromorphine, morphine, tramadol, oxymorphone, pharmaceutically acceptable salts thereof, and mixtures thereof. Examples of useful non-opioid analgesics include non-steroidal anti- inflammatory agents, such as aspirin, ibuprofen, diclofenac, naproxen, benoxaprofen, flurbiprofen, fenoprofen, flubufen, ketoprofen, indoprofen, piroprofen, carprofen, oxaprozin, pramoprofen, muroprofen, trioxaprofen, suprofen, aminoprofen, tiaprofenic acid, fluprofen, bucloxic acid, indomethacin, sulindac, tolmetin, zomepirac, tiopinac, zidometacin, acemetacin, fentiazac, clidanac, oxpinac, mefenamic acid, meclofenamic acid, flufenamic acid, niflumic acid, tolfenamic acid, diflurisal, flufenisal, piroxicam, sudoxicam, isoxicam, and pharmaceutically acceptable salts thereof, and mixtures thereof. Other suitable non-opioid analgesics include the following, non-limiting, chemical classes of analgesic, antipyretic, non-steroidal anti-inflammatory drugs: salicylic acid derivatives, including aspirin, sodium salicylate, choline magnesium trisalicylate, salsalate, diflunisal, salicylsalicylic acid, sulfasalazine, and olsalazin; para- aminophennol derivatives including acetaminophen and phenacetin; indole and indene acetic acids, including indomethacin, sulindac, and etodolac; heteroaryl acetic acids, including tolmetin, diclofenac, and ketorolac; anthranilic acids (fenamates), including mefenamic acid and meclofenamic acid; enolic acids, including oxicams (piroxicam, tenoxicam), and pyrazolidinediones (phenylbutazone, oxyphenthartazone); and alkanones, including nabumetone. For a more detailed description of the NSAIDs, see Paul A. Insel, Analgesic-Antipyretic and Anti-inflammatory Agents and Drugs Employed in the Treatment of Gout, in Goodman & Gilman's The Pharmacological Basis of Therapeutics 617-57 (Perry B. Molinhoff and Raymond W. Ruddon eds., 9th ed 1996) and Glen R. Hanson, Analgesic, Antipyretic and Anti-Inflammatory Drugs in Remington:
The Science and Practice of Pharmacy Vol II 1196-1221 (A.R. Gennaro ed. 19th ed. 1995) which are hereby incorporated by reference in their entireties. Examples of useful Cox-II inhibitors and 5-lipoxygenase inhibitors, as well as combinations thereof, are described in U.S. Patent No. 6,136,839, which is hereby incorporated by reference in its entirety. Examples of useful Cox-II inhibitors include, but are not limited to, rofecoxib and celecoxib. Examples of useful antimigraine agents include, but are not limited to, alpiropride, bromocriptine, dihydroergotamine, dolasetron, ergocornine, ergocorninine, ergocryptine, ergonovine, ergot, ergotamine, flumedroxone acetate, fonazine, ketanserin, lisuride, lomerizine, methylergonovine, methysergide, metoprolol, naratriptan, oxetorone, pizotyline, propranolol, risperidone, rizatriptan, sumatriptan, timolol, trazodone, zolmitriptan, and mixtures thereof. The other therapeutic agent can alternatively be an agent useful for reducing any potential side effect of a Piperazine Compounds. For example, the other therapeutic agent can be an antiemetic agent. Examples of useful antiemetic agents include, but are not limited to, metoclopromide, domperidone, prochlorperazine, promethazine, chlorpromazine, trimethobenzamide, odansetron, granisetron, hydroxyzine, acetylleucine monoethanolamine, alizapride, azasetron, benzquinamide, bietanautine, bromopride, buclizine, clebopride, cyclizine, dimenhydrinate, diphenidol, dolasetron, meclizine, methallatal, metopimazine, nabilone, oxyperndyl, pipamazine, scopolamine, sulpiride, tetrahydrocannabinol, thiethylperazine, thioproperazine, tropisetron, and mixtures thereof. Examples of useful β-adrenergic blockers include, but are not limited to, acebutolol, alprenolol, amosulabol, arotinolol, atenolol, befunolol, betaxolol, bevantolol, bisoprolol, bopindolol, bucumolol, bufetolol, bufuralol, bunitrolol, bupranolol, butidrine hydrochloride, butofilolol, carazolol, carteolol, carvedilol, celiprolol, cetamolol, cloranolol, dilevalol, epanolol, esmolol, indenolol, labetalol, levobunolol, mepindolol, metipranolol, metoprolol, moprolol, nadolol, nadoxolol, nebivalol, nifenalol, nipradilol, oxprenolol, penbutolol, pindolol, practolol, pronethalol, propranolol, sotalol, sulfinalol, talinolol, tertatolol, tilisolol, timolol, toliprolol, and xibenolol. Examples of useful anticonvulsants include, but are not limited to, acetylpheneturide, albutoin, aloxidone, aminoglutethimide, 4-amino-3-hydroxybutyric
acid, atrolactamide, beclamide, buramate, calcium bromide, carbamazepine, cinromide, clomethiazole, clonazepam, decimemide, diethadione, dimethadione, doxenitroin, eterobarb, ethadione, ethosuximide, ethotoin, felbamate, fluoresone, gabapentin, 5- hydroxytryptophan, lamotrigine, magnesium bromide, magnesium sulfate, mephenytoin, mephobarbital, metharbital, methetoin, methsuximide, 5-methyl-5-(3-phenanthryl)- hydantoin, 3-mefhyl-5-phenylhydantoin, narcobarbital, nimetazepam, nitrazepam, oxcarbazepine, paramethadione, phenacemide, phenetharbital, pheneturide, phenobarbital, phensuximide, phenylmethylbarbituric acid, phenytoin, phethenylate sodium, potassium bromide, pregabaline, primidone, progabide, sodium bromide, solanum, strontium bromide, suclofenide, sulthiame, tetrantoin, tiagabine, topiramate, trimethadione, valproic acid, valpromide, vigabatrin, and zonisamide. Examples of useful antidepressants include, but are not limited to, binedaline, caroxazone, citalopram, (S)-citalopram, dimethazan, fencamine, indalpine, indeloxazine hydrocholoride, nefopam, nomifensine, oxitriptan, oxypertine, paroxetine, sertraline, thiazesim, trazodone, benmoxine, iproclozide, iproniazid, isocarboxazid, nialamide, octamoxin, phenelzine, cotinine, rolicyprine, rolipram, maprotiline, metralindole, mianserin, mirtazepine, adinazolam, amitriptyline, amitriptylinoxide, amoxapine, butriptyline, clomipramine, demexiptiline, desipramine, dibenzepin, dimetacrine, dothiepin, doxepin, fluacizine, imipramine, imipramine N-oxide, iprindole, lofepramine, melitracen, metapramine, nortriptyline, noxiptilin, opipramol, pizotyline, propizepine, protriptyline, quinupramine, tianeptine, trimipramine, adrafinil, benactyzine, bupropion, butacetin, dioxadrol, duloxetine, etoperidone, febarbamate, femoxetine, fenpentadiol, fluoxetine, fluvoxamine, hematoporphyrin, hypericin, levophacetoperane, medifoxamine, milnacipran, minaprine, moclobemide, nefazodone, oxaflozane, piberaline, prolintane, pyrisuccideanol, ritanserin, roxindole, rubidium chloride, sulpiride, tandospirone, thozalinone, tofenacin, toloxatone, tranylcypromine, L-tryptophan, venlafaxine, viloxazine, and zimelidine. 94- Examples of useful Ca -channel blockers include, but are not limited to, bepridil, clentiazem, diltiazem, fendiline, gallopamil, mibefradil, prenylamine, semotiadil, terodiline, verapamil, amlodipine, aranidipine, barnidipine, benidipine, cilnidipine, efonidipine, elgodipine, felodipine, isradipine, lacidipine, lercanidipine, manidipine,
nicardipine, nifedipine, nilvadipine, nimodipine, nisoldipine, nitrendipine, cinnarizine, flunarizine, lidoflazine, lo erizine, bencyclane, etafenone, fantofarone, and perhexiline. Examples of useful anticancer agents include, but are not limited to, acivicin, aclarubicin, acodazole hydrochloride, acronine, adozelesin, aldesleukin, altretamine, ambomycin, ametantrone acetate, aminoglutethimide, amsacrine, anastrozole, anthramycin, asparaginase, asperlin, azacitidine, azetepa, azotomycin, batimastat, benzodepa, bicalutamide, bisantrene hydrochloride, bisnafide dimesylate, bizelesin, bleomycin sulfate, brequinar sodium, bropirimine, busulfan, cactinomycin, calusterone, caracemide, carbetimer, carboplatin, carmustine, carubicin hydrochloride, carzelesin, cedefingol, chlorambucil, cirolemycin, cisplatin, cladribine, crisnatol mesylate, cyclophosphamide, cytarabine, dacarbazine, dactinomycin, daunorubicin hydrochloride, decitabine, dexormaplatin, dezaguanine, dezaguanine mesylate, diaziquone, docetaxel, doxorubicin, doxorubicin hydrochloride, droloxifene, droloxifene citrate, dromostanolone propionate, duazomycin, edatrexate, eflornithine hydrochloride, elsamitrucin, enloplatin, enpromate, epipropidine, epirubicin hydrochloride, erbulozole, esorubicin hydrochloride, estramustine, estramustine phosphate sodium, etanidazole, etoposide, etoposide phosphate, etoprine, fadrozole hydrochloride, fazarabine, fenretinide, floxuridine, fludarabine phosphate, fluorouracil, flurocitabine, fosquidone, fostriecin sodium, gemcitabine, gemcitabine hydrochloride, hydroxyurea, idarubicin hydrochloride, ifosfamide, ilmofosine, interleukin II (including recombinant interleukin II or rIL2), interferon alpha-2a, interferon alpha-2b, interferon alpha-nl, interferon alpha- n3, interferon beta-I a, interferon gamma-I b, iproplatin, irinotecan hydrochloride, lanreotide acetate, letrozole, leuprolide acetate, liarozole hydrochloride, lometrexol sodium, lomustine, losoxantrone hydrochloride, masoprocol, maytansine, mechlorethamine hydrochloride, megestrol acetate, melengestrol acetate, melphalan, menogaril, mercaptopurine, methotrexate, methotrexate sodium, metoprine, meturedepa, mitindomide, mitocarcin, mitocromin, mitogillin, mitomalcin, mitomycin, mitosper, mitotane, mitoxantrone hydrochloride, mycophenolic acid, nocodazole, nogalamycin, ormaplatin, oxisuran, paclitaxel, pegaspargase, peliomycin, pentamustine, peplomycin sulfate, perfosfamide, pipobroman, piposulfan, piroxantrone hydrochloride, plicamycin, plomestane, porfimer sodium, porfiromycin, prednimustine, procarbazine hydrochloride, puromycin, puromycin hydrochloride, pyrazofurin, riboprine, rogletimide, safingol,
safingol hydrochloride, semustine, simtrazene, sparfosate sodium, sparsomycin, spirogermanium hydrochloride, spiromustine, spiroplatin, streptonigrin, streptozotocin, sulofenur, talisomycin, tecogalan sodium, tegafur, teloxantrone hydrochloride, temoporfin, teniposide, teroxirone, testolactone, thiamiprine, thioguanine, thiotepa, tiazofurin, tirapazamine, toremifene citrate, trestolone acetate, triciribine phosphate, trimetrexate, trimetrexate glucuronate, triptorelin, tubulozole hydrochloride, uracil mustard, uredepa, vapreotide, verteporfin, vinblastine sulfate, vincristine sulfate, vindesine, vindesine sulfate, vinepidine sulfate, vinglycinate sulfate, vinleurosine sulfate, vinorelbine tartrate, vinrosidine sulfate, vinzolidine sulfate, vorozole, zeniplatin, zinostatin, zorubicin hydrochloride. Examples of other anti-cancer drugs include, but are not limited to, 20-epi-l,25 dihydroxyvitamin D3; 5-ethynyluracil; abiraterone; aclarubicin; acylfulvene; adecypenol; adozelesin; aldesleukin; ALL-TK antagonists; altretamine; ambamustine; amidox; amifostine; aminolevulinic acid; amrubicin; amsacrine; anagrelide; anastrozole; andrographolide; angiogenesis inhibitors; antagonist D; antagonist G; antarelix; anti- dorsalizing morphogenetic protein- 1; antiandrogen; antiestrogen; antineoplaston; antisense oligonucleotides; aphidicolin glycinate; apoptosis gene modulators; apoptosis regulators; apurinic acid; ara-CDP-DL-PTBA; arginine deaminase; asulacrine; atamestane; atrimustine; axinastatin 1; axinastatin 2; axinastatin 3; azasetron; azatoxin; azatyrosine; baccatin III derivatives; balanol; batimastat; BCR/ABL antagonists; benzochlorins; benzoylstaurosporine; beta lactam derivatives; beta-alethine; betaclamycin B; betulinic acid; bFGF inhibitor; bicalutamide; bisantrene; bisaziridinylspermine; bisnafide; bistratene A; bizelesin; breflate; bropirimine; budotitane; buthionine sulfoximine; calcipotriol; calphostin C; camptothecin derivatives; canarypox IL-2; capecitabine; carboxamide-amino-triazole; carboxyamidotriazole;
CaRest M3; CARN 700; cartilage derived inhibitor; carzelesin; casein kinase inhibitors
(ICOS); castanospermine; cecropin B; cetrorelix; chloroquinoxaline sulfonamide; cicaprost; cis-porphyrin; cladribine; clomifene analogues; clotrimazole; collismycin A; collismycin B; combretastatin A4; combretastatin analogue; conagenin; crambescidin 816; crisnatol; cryptophycin 8; cryptophycin A derivatives; curacin A; cyclopentanthraquinones; cycloplatam; cypemycin; cytarabine ocfosfate; cytolytic factor; cytostatin; dacliximab; decitabine; dehydrodidemnin B; deslorelin; dexamethasone;
dexifosf amide; dexrazoxane; dexverapamil; diaziquone; didemnin B; didox; diethylnorspermine; dihydro-5-azacytidine; 9-dihydrotaxol; dioxamycin; diphenyl spiromustine; docetaxel; docosanol; dolasetron; doxifluridine; droloxifene; dronabinol; duocarmycin SA; ebselen; ecomustine; edelfosine; edrecolomab; eflornithine; elemene; emitefur; epirubicin; epristeride; estramustine analogue; estrogen agonists; estrogen antagonists; etanidazole; etoposide phosphate; exemestane; fadrozole; fazarabine; fenretinide; filgrastim; finasteride; flavopiridol; flezelastine; fluasterone; fludarabine; fluorodaunorunicin hydrochloride; forfenimex; formestane; fostriecin; fotemustine; gadolinium texaphyrin; gallium nitrate; galocitabine; ganirelix; gelatinase inhibitors; gemcitabine; glutathione inhibitors; hepsulfam; heregulin; hexamethylene bisacetamide; hypericin; ibandronic acid; idarubicin; idoxifene; idramantone; ilmofosine; ilomastat; imidazoacridones; imiquimod; immunostimulant peptides; insulin-like growth factor- 1 receptor inhibitor; interferon agonists; interferons; interleukins; iobenguane; iododoxorubicin; 4-ipomeanol; iroplact; irsogladine; isobengazole; isohomohalicondrin B; itasetron; jasplakinolide; kahalalide F; lamellarin-N triacetate; lanreotide; leinamycin; lenograstim; lentinan sulfate; leptolstatin; letrozole; leukemia inhibiting factor; leukocyte alpha interferon; leuprolide+estrogen+progesterone; leuprorelin; levamisole; liarozole; linear polyamine analogue; lipophilic disaccharide peptide; lipophilic platinum compounds; lissoclinamide 7; lobaplatin; lombricine; lometrexol; lonidamine; losoxantrone; lovastatin; loxoribine; lurtotecan; lutetium texaphyrin; lysofylline; lytic peptides; maitansine; mannostatin A; marimastat; masoprocol; maspin; matrilysin inhibitors; matrix metalloproteinase inhibitors; menogaril; merbarone; meterelin; methioninase; metoclopramide; MIF inhibitor; mifepristone; miltefosine; mirimostim; mismatched double stranded RNA; mitoguazone; mitolactol; mitomycin analogues; mitonafide; mitotoxin fibroblast growth factor-saporin; mitoxantrone; mofarotene; molgramostim; monoclonal antibody, human chorionic gonadotrophin; monophosphoryl lipid A+myobacterium cell wall sk; mopidamol; multiple drug resistance gene inhibitor; multiple tumor suppressor 1 -based therapy; mustard anticancer agent; mycaperoxide B; mycobacterial cell wall extract; myriaporone; N-acetyldinaline; N-substituted benzamides; nafarelin; nagrestip; naloxone+pentazocine; napavin; naphterpin; nartograstim; nedaplatin; nemorubicin; neridronic acid; neutral endopeptidase; nilutamide; nisamycin; nitric oxide modulators; nitroxide antioxidant; nitrullyn; O6-
benzylguanine; octreotide; okicenone; oligonucleotides; onapristone; odansetron; oracin; oral cytokine inducer; ormaplatin; osaterone; oxaliplatin; oxaunomycin; paclitaxel; paclitaxel analogues; paclitaxel derivatives; palauamine; palmitoylrhizoxin; pamidronic acid; panaxytriol; panomifene; parabactin; pazelliptine; pegaspargase; peldesine; pentosan polysulfate sodium; pentostatin; pentrozole; perflubron; perfosfamide; perillyl alcohol; phenazinomycin; phenylacetate; phosphatase inhibitors; picibanil; pilocarpine hydrochloride; pirarubicin; piritrexim; placetin A; placetin B; plasminogen activator inhibitor; platinum complex; platinum compounds; platinum-triamine complex; porfimer sodium; porfiromycin; prednisone; propyl bis-acridone; prostaglandin J2; proteasome inhibitors; protein A-based immune modulator; protein kinase C inhibitor; protein kinase C inhibitors, microalgal; protein tyrosine phosphatase inhibitors; purine nucleoside phosphorylase inhibitors; purpurins; pyrazoloacridine; pyridoxylated hemoglobin polyoxyethylene conjugate; raf antagonists; raltitrexed; ramosetron; ras farnesyl protein transferase inhibitors; ras inhibitors; ras-GAP inhibitor; retelliptine demethylated; rhenium Re 186 etidronate; rhizoxin; ribozymes; RII retinamide; rogletimide; rohitukine; romurtide; roquinimex; rubiginone Bl; ruboxyl; safingol; saintopin; SarCNU; sarcophytol A; sargramostim; Sdi 1 mimetics; semustine; senescence derived inhibitor 1; signal transduction inhibitors; signal transduction modulators; single chain antigen binding protein; sizofiran; sobuzoxane; sodium borocaptate; sodium phenylacetate; solverol; somatomedin binding protein; sonermin; sparfosic acid; spicamycin D; spiromustine; splenopentin; spongistatin 1; squalamine; stem cell inhibitor; stem-cell division inhibitors; stipiamide; stromelysin inhibitors; sulfinosine; superactive vasoactive intestinal peptide antagonist; suradista; suramin; swainsonine; syntlietic glycosaminoglycans; tallimustine; tamoxifen methiodide; tauromustine; tazarotene; tecogalan sodium; tegafur; tellurapyrylium; telomerase inhibitors; temoporfin; temozolomide; teniposide; tetrachlorodecaoxide; tetrazomine; thaliblastine; thiocoraline; thrombopoietin; thrombopoietin mimetic; thymalfasin; thymopoietin receptor agonist; thymotrinan; thyroid stimulating hormone; tin ethyl etiopurpurin; tirapazamine; titanocene bichloride; topsentin; toremifene; totipotent stem cell factor; translation inhibitors; tretinoin; triacetyluridine; triciribine; trimetrexate; triptorelin; tropisetron; turosteride; tyrosine kinase inhibitors; tyrphostins; UBC inhibitors; ubenimex; urogenital sinus-derived growth inhibitory factor; urokinase receptor antagonists; vapreotide;
variolin B; vector system, erythrocyte gene therapy; velaresol; veramine; verdins; verteporfin; vinorelbine; vinxaltine; vitaxin; vorozole; zanoterone; zeniplatin; zilascorb; and zinostatin stimalamer. Examples of useful therapeutic agents for treating or preventing Ul include, but are not limited to, propantheline, imipramine, hyoscyamine, oxybutynin, and dicyclomine. Examples of useful therapeutic agents for treating or preventing an addictive disorder include, but are not limited to, methadone, desipramine, amantadine, fluoxetine, buprenorphine, an opiate agonist, 3-phenoxypyridine, levomethadyl acetate hydrochloride, and serotonin antagonists. Examples of useful therapeutic agents for treating or preventing Parkinson's disease and parkinsonism include, but are not limited to, carbidopa/levodopa, pergolide, bromocriptine, ropinirole, pramipexole, entacapone, tolcapone, selegiline, amantadine, and trihexyphenidyl hydrochloride. Examples of useful therapeutic agents for treating or preventing anxiety include, but are not limited to, benzodiazepines, such as alprazolam, brotizolam, chlordiazepoxide, clobazam, clonazepam, clorazepate, demoxepam, diazepam, estazolam, flumazenil, flurazepam, halazepam, lorazepam, midazolam, nitrazepam, nordazepam, oxazepam, prazepam, quazepam, temazepam, and triazolam; non- benzodiazepine agents, such as buspirone, gepirone, ipsapirone, tiospirone, zolpicone, zolpidem, and zaleplon; tranquilizers, such as barbituates, e.g., amobarbital, aprobarbital, butabarbital, butalbital, mephobarbital, methohexital, pentobarbital, phenobarbital, secobarbital, and thiopental; and propanediol carbamates, such as meprobamate and tybamate. Examples of useful therapeutic agents for treating or preventing epilepsy include, but are not limited to, carbamazepine, ethosuximide, gabapentin, lamotrigine, phenobarbital, phenytoin, primidone, valproic acid, trimethadione, benzodiazepines, gabapentin, lamotrigine, γ-vinyl GABA, acetazolamide, and felbamate. Examples of useful therapeutic agents for treating or preventing a seizure include, but are not limited to, carbamazepine, ethosuximide, gabapentin, lamotrigine, phenobarbital, phenytoin, primidone, valproic acid, trimethadione, benzodiazepines, gabapentin, lamotrigine, γ-vinyl GABA, acetazolamide, and felbamate.
Examples of useful therapeutic agents for treating or preventing stroke include, but are not limited to, anticoagulants such as heparin, agents that break up clots such as streptokinase or tissue plasminogen activator, agents that reduce swelling such as mannitol or corticosteroids, and acetylsalicylic acid. Examples of useful therapeutic agents for treating or preventing a pruritic condition include, but are not limited to, naltrexone; nalmefene; danazol; tricyclics such as amitriptyline, imipramine, and doxepin; antidepressants such as those given below; menthol; camphor; phenol; pramoxine; capsaicin; tar; steroids; and antihistamines. Examples of useful therapeutic agents for treating or preventing psychosis include, but are not limited to, phenothiazines such as chlorpromazine hydrochloride, mesoridazine besylate, and thoridazine hydrochloride; thioxanthenes such as chloroprothixene and thiothixene hydrochloride; clozapine; risperidone; olanzapine; quetiapine; quetiapine fumarate; haloperidol; haloperidol decanoate; loxapine succinate; molindone hydrochloride; pimozide; and ziprasidone. Examples of useful therapeutic agents for treating or preventing Huntington's chorea include, but are not limited to, haloperidol and pimozide. Examples of useful therapeutic agents for treating or preventing ALS include, but are not limited to, baclofen, neurotrophic factors, riluzole, tizanidine, benzodiazepines such as clonazepan and dantrolene. Examples of useful therapeutic agents for treating or preventing cognitive disorders include, but are not limited to, agents for treating or preventing dementia such as tacrine; donepezil; ibuprofen; antipsychotic drugs such as thioridazine and haloperidol; and antidepressant drugs such as those given below. Examples of useful therapeutic agents for treating or preventing a migraine include, but are not limited to, sumatriptan; methysergide; ergotamine; caffeine; and beta-blockers such as propranolol, verapamil, and divalproex. Examples of useful therapeutic agents for treating (e.g., inhibiting) or preventing vomiting include, but are not limited to, 5-HT3 receptor antagonists such as odansetron, dolasetron, granisetron, and tropisetron; dopamine receptor antagonists such as prochlorperazine, thiethylperazine, chlorpromazine, metoclopramide, and domperidone; glucocorticoids such as dexamethasone; and benzodiazepines such as lorazepam and alprazolam.
Examples of useful therapeutic agents for treating or preventing dyskinesia include, but are not limited to, reserpine and tetrabenazine. Examples of useful therapeutic agents for treating or preventing depression include, but are not limited to, tricyclic antidepressants such as amitryptyline, amoxapine, bupropion, clomipramine, desipramine, doxepin, imipramine, maprotiline, nefazadone, nortriptyline, protriptyline, trazodone, trimipramine, and venlafaxine; selective serotonin reuptake inhibitors such as fluoxetine, fluvoxamine, paroxetine, citalopram, (S)-citalopram and setraline; monoamine oxidase inhibitors such as isocarboxazid, pargyline, phenelzine, and tranylcypromine; and psychostimulants such as dextroamphetamine and methylphenidate. A Piperazine Compound and the other therapeutic agent can act additively or, in one embodiment, synergistically. In one embodiment, a Piperazine Compound is adjunctively administered with another therapeutic agent; for example, a composition comprising an effective amount of a Piperazine Compound, an effective amount of another therapeutic agent can be administered. Alternatively, a composition comprising an effective amount of a Piperazine Compound and a different composition comprising an effective amount of another therapeutic agent can be concurrently administered. In another embodiment, an effective amount of a Piperazine Compound is administered prior or subsequent to administration of an effective amount of another therapeutic agent. In this embodiment, the Piperazine Compound is administered while the other therapeutic agent exerts its therapeutic effect, or the other therapeutic agent is administered while the Piperazine Compound exerts its therapeutic effect for treating or preventing a Condition. A composition of the invention is prepared by a method comprising admixing a Piperazine Compound and pharmaceutically acceptable salt and a pharmaceutically acceptable carrier or excipient. Admixing can be accomplished using methods known for admixing a compound (or salt) and a pharmaceutically acceptable carrier or excipient. In one embodiment the composition is prepared such that the Piperazine Compound is present in the composition in an effective amount.
4.8 Kits The invention encompasses kits that can simplify the administration of a Piperazine Compound to an animal. A typical kit of the invention comprises a unit dosage form of a Piperazine Compound. In one embodiment, the unit dosage form is a container, which can be sterile, containing an effective amount of a Piperazine Compound and a pharmaceutically acceptable carrier or excipient. The kit can further comprise a label or printed instructions instructing the use of the Piperazine Compound to treat a Condition. The kit can also further comprise a unit dosage form of another therapeutic agent, for example, a second container containing an effective amount of the other therapeutic agent and a pharmaceutically acceptable carrier or excipient. hi another embodiment, the kit comprises a container containing an effective amount of a Piperazine Compound, an effective amount of another therapeutic agent and a pharmaceutically acceptable carrier or excipient. Examples of other therapeutic agents include, but are not limited to, those listed above. Kits of the invention can further comprise a device that is useful for administering the unit dosage forms. Examples of such a device include but are not limited to a syringe, a drip bag, a patch, an inhaler, and an enema bag. The following examples are set forth to assist in understanding the invention and should not be construed as specifically limiting the invention described and claimed herein. The examples disclose one or more synthetic intermediates that are useful for making the Piperazine Compounds. All of the synthetic intermediates are also encompassed by the present invention. Such variations of the invention, including the substitution of all equivalents now known or later developed, which would be within the purview of those skilled in the art, and changes in formulation or changes in experimental design, are to be considered to fall within the scope of the invention incorporated herein.
5. Examples Examples 1-12 relate to the synthesis of illustrative Piperazine Compounds. 5.1 Example 1; Synthesis of Compound 1009(a) Compound 1009(a) was prepared according to the following scheme.
The conversion of commercially available Compound 4 to Compound 6 was adapted from a procedure provided in J. Med. Chem., 45:3639-3648 (2002).
A mixture of propargyl alcohol (available from Aldrich Chemical Co.,
Milwaukee, WI; 160 μl, 2.7 mmol), TEA (721 mL, 5 mmol) in 5 mL dichloromethane ("DCM") was added dropwise via pipette to a solution of Compound 6 (520 mg, 2.48 mmol) in 30 mL of DCM at about 0°C. After the addition was complete, the resulting solution was warmed over a period of about 20-30 minutes to about 25°C and stirred at about 25°C for about 12 hours. The solution mixture was then washed with an aqueous solution of saturated NFLCl. The aqueous portion was extracted with 50 mL of DCM. The combined organic portion was dried with Na
2SO , filtered and concentrated under reduced pressure with a rotary evaporator. The resulting residue was purified using flash chromatography (silica gel, 35-60 μm particle size (230-400 mesh) with a 1:4 EtOAc:hexane eluent system) to provide 490 mg of Compound 7. The structure of Compound 7 was confirmed by 1H NMR and mass spectrometry. Compound 7: 1H NMR(500 MHz, CDC1
3): δ (ppm): 8.86(s, 1H); 5.01(0, 7 = 2.8Hz, 2H); 2.62(t, 7 = 2.8Hz, 1H). MS: m/z 230.9 (M).
A solution of Compound 7 (490 mg, 2.13 mmol) in 12 mL of nitrobenzene was irradiated at about 2.45 GHz using a MICROSYNTH Microwave Labstation (Milestone Microwave Laboratory System Inc., Monroe, CT) for about 1 minute, during which time the reaction temperature rose from about 25°C to about 265°C. Microwave irradiation continued for about 2 minutes, during which time the reaction temperature was maintained at about 265°C. The irradiation was stopped, and the solution cooled to about 25°C. The progress of the reaction was monitored using TLC (2.5% methanol/DCM). After TLC revealed the formation of Compound 14, the reaction mixture was loaded onto a silica gel column of the type described above, and DCM was used to remove the nitrobenzene. The residue was purified using flash chromatography on a silica gel column of the type described above in this example and eluted with gradient elution from 2:1 EtOAc:hexane to EtOAc to provide 334.8 mg of Compound 14 (93% yield). The structure of Compound 14 was confirmed by 1H NMR and mass spectrometry. Compound 14: 1H NMR(CDC13): δ (ppm): 8.67(d, 7 = 5.0 Hz, 1H); 7.47(dt, 7 = 1.0, 5.0 Hz, 1H); 5.35(d, 7 = 0.5 Hz, 2H). MS: m/z 170 (MH).
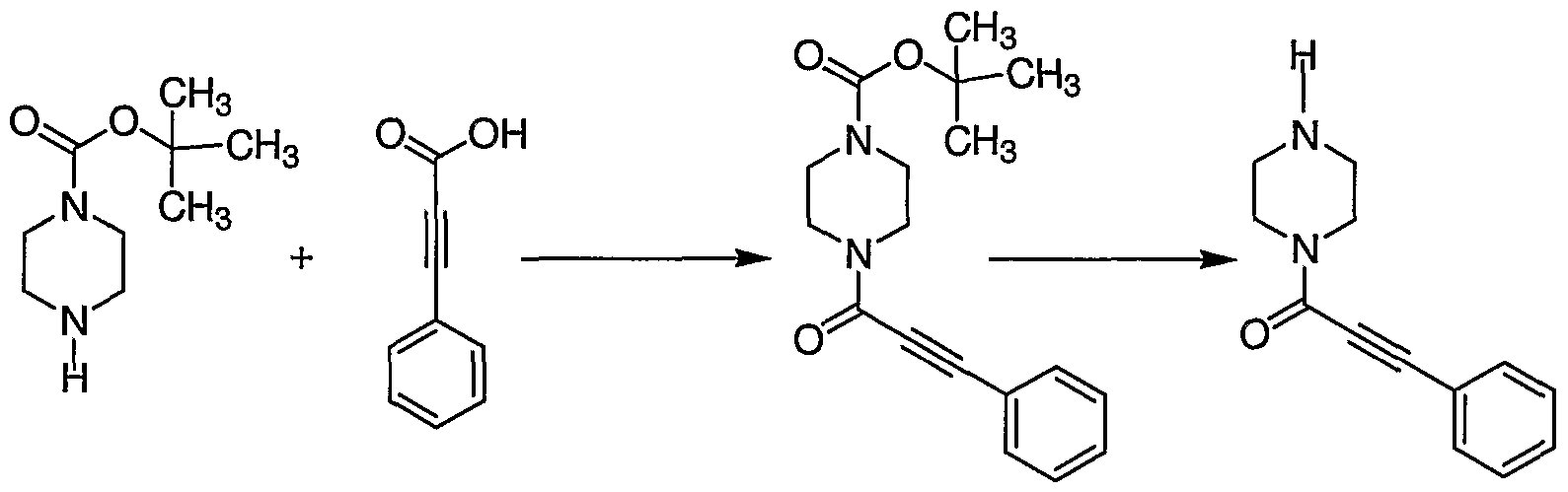
21 22 23
To a solution of t-butyl-piperizine-1-carboxylate (Compound 21; 2.8 g, 15 mmol) in 200 mL tetrahydrofuran ("THF") was added HOBT (2.03 g, 15 mmol) and l-(3- dimemylammoniumpropyl)-3-ethylcarbodimidehydrochloride (2.88 g, 15 mmol). The resulting mixture was stirred at about 25°C for about 30 minutes, and to it was added a solution of phenyl-propynoic acid (Aldrich Chemical, 2.2 g, 15 mmol) in 20 mL of THF at about 25°C. After addition was complete, the resulting mixture was stirred at about 25°C for about 2 hours. After evaporation under reduced pressure with a rotary evaporator, the residue was purified using flash chromatography on a silica gel column of the type described above in this example, eluting with 1:1 EtOAc:hexane to provide Compound 22 as an 80-90 wt.% pure product. Compound 22 was added to 30 mL of a solution of 20 wt.% trifluoroacetic acid ("TFA") in DCM. After about 2 hours at about 25°C, the mixture was concentrated under reduced pressure with a rotary evaporator. The residue was diluted with 150 mL of chloroform, and the resultant chloroform solution was dried with K
2CO
3, filtered and evaporated. The resulting residue was purified using flash chromatography on a silica gel column of the type described above in this example, eluting with 1:4 methanolxhloroform to provide 2.3 g of Compound 23 (74% yield). The structure of Compound 23 was confirmed by 1H NMR and mass spectrometry. Compound 23: 1H NMR(CDC1
3, OMSO-d6): δ (ppm): 7.60(m, 2H), 7.46(m,
3H); 3.88(m, 2H); 3.67(m, 2H); 2.95(m, 2H); 2.87(m, 2H). MS: m/z 215.3 (MH).
A solution of Compound 14 (63 mg, 0.37 mmol), the HC1 salt of Compound 23 (94 mg, 0.39 mmol) and excess TEA (about 4 equivalents) in 5 mL of acetonitrile was
placed into a sealed 20 mL vial and heated in an oil bath held at about 90°C for about 12 hours. The reaction mixture was cooled to about 25°C and concentrated on a rotary evaporator. The resultant residue was purified using preparative TLC (2.5% methanol/DCM) to provide 43 mg of Compound 1009(a) (33% yield). The structure of Compound 1009(a) was confirmed by 1H NMR and mass spectrometry. Compound 1009(a): 1H NMR(CDC13): δ (ppm): 8.36(d, 7 = 4.9 Hz, 1H); 7.57 (m, 2H); 7.40(m, 3H); 6.82(d, 7 = 4.9 Hz, 1H); 4.04(m, 2H); 3.88(m, 4H); 3.81(m, 2H). MS: m z 348.1 (MH).
5.2 Example 2: Synthesis of Compound 1441(a) Compound 1441(a) was prepared according to the following scheme.
Compound 8 was prepared from Compound 6 using a procedure that was analogous to that used to prepare Compound 7 except that 3-butyn-l-ol (Aldrich Chemical) was used in place of propargyl alcohol. Compound 15 was prepared by irradiating Compound 8 with microwave irradiation using a procedure that was analogous to that used to prepare Compound 14 except that, after the 1 minute warming period, the duration of microwave irradiation was about 45 minutes at about 265°C (31% yield). The structure of Compound 15 was confirmed by 1H NMR and mass spectrometry. Compound 15: 1H NMR(CDC13): δ (ppm): 8.48(d, 7 = 4.8Hz, 1H); 7.20(dt, 7 = 1.2, 4.8Hz, 1H); 4.52(t, 7 = 5.0Hz, 2H); 3.10(m, 2H). MS: m/z 184.1 (MH).
Compound 1441(a) was prepared from Compound 23 using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compound 15 was used in place of Compound 14 (48% yield). The structure of Compound 1441(a) was confirmed by 1H NMR and mass spectrometry. Compound 1441(a): 1H NMR(CDC13): δ(ppm): 8.23(d, 7 = 4.8 Hz, IH); 7.56(m, 2H); 7.40(m, 3H); 6.62(d, 7 = 4.8 Hz, IH); 4.47(t, 7 = 5.8 Hz, 2H); 4.02(m, 2H); 3.87(m, 2H); 3.58(m, 4H); 2.99(t, 7= 5.8 Hz, 2H). MS: m/z 362.3 (MH).
5.3 Example 3: Synthesis of Compound 1011(a) Compound 1011(a) was prepared according to the following scheme.
1 2 3
The procedure used to prepare Compound 3 from commercially available Compound 1 was adapted from a published procedure provided in Tetrahedron Lett., 40(30):5483-5486 (1999). 2.5 M n-BuLi in hexane (16 mL, 40 mmol) was added dropwise via syringe to a solution of 2,2,6,6-tetramethylpiperidine (5.1 mL, 30 mmol) in 50 mL THF at about
-78°C. After the addition was complete, the resultant mixture was stirred at about -78°C for about 30 min and Compound 1 (1.57 g, 10 mmol) in THF (10 mL) was added. The
resultant mixture was stirred at about -78°C for about 3 hours and then quenched by the addition of acetone. After being warmed to about 25°C, the reaction mixture was concentrated under reduced pressure with a rotary evaporator. The resultant residue was purified using flash chromatography on a silica gel column of the type described previously in Example 1 with gradient elution from 1:9 methano DCM to 1:4 methanohDCM to provide a mixture containing Compound 2. The structure of Compound 2 was confirmed by 1H NMR and mass spectrometry. Compound 2: 1H NMR(CDC13): δ (ppm): 8.64(d, 7 = 5.2 Hz, IH); 7.35(d, 7 = 5.2 Hz, IH); 1.69(s, 6H). MS: m/z 198 (MH). The above mixture containing Compound 2 and a catalytic amount, about 10-20 mg, of p-toluenesulfonic acid was heated to reflux in chloroform. After conversion was complete, as evidenced by liquid chromatography-mass spectrometry ("LCMS"), the reaction mixture was concentrated under reduced pressure using a rotary evaporator. The resultant residue was purified using flash chromatography on a silica gel column of the type described previously in Example 1 with gradient elution from 1:9 EtOAc:hexane to 1:1 EtOAc:hexane to provide 180 mg of Compound 3 (9% yield from Compound 1). The structure of Compound 3 was confirmed by 1H NMR and mass spectrometry. Compound 3: 1H NMR(CDC13): δ (ppm): 8.64(d, 7 = 5.2 Hz, IH); 7.35(d, 7 = 5.2 Hz, IH); 1.70(s, 6H). MS: m/z 198 (MH), 220 (M+Na).
Compound 1011(a) was prepared from Compound 23 using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compound 3 was used in place of Compound 14 (37% yield).
The structure of Compound 1011(a) was confirmed by 1H NMR and mass spectrometry. Compound 1011(a): 1H NMR(CDC13): δ (ppm): 8.37(d, 7 = 4.8 Hz, IH); 7.60(m, 2H); 7.42(m, 3H); 6.75(d, 7 = 4.8 HZ, IH); 4.06(m, 2H); 3.91(m, 4H); 3.84(m, 2H); 1.64(s, 6H). MS: m/z 376.2 (MH).
5.4 Example 4: Synthesis of Compound 1009(b) Compound 1009(b) was prepared according to the following scheme.
27 48 n-Butyl lithium in hexane (2.5 M, 8 mL) was added dropwise to l-ethynyl-4- methoxybenzene (Compound 46, Aldrich Chemical; 2.0 g, 15 mmol) in THF (20 mL) under nitrogen over about a 10 minute period at about -78°C. The mixture was stirred for about 1 hour at about -78°C after which dry CO
2 was passed through the reaction mixture for about 3 hours. The resulting solution was poured into water and washed with EtOAc. The aqueous layer was evaporated to provide crude Compound 47, which was subsequently dissolved in methanol and filtered. The filtrate was concentrated to provide 2.5 g of Compound 47 as a white solid (purity 90 wt.%; 95% yield). The structure of Compound 47 was confirmed by 1H NMR and mass spectrometry. Compound 47: 1H NMR(CDC1
3): δ ppm: 7.60(d, 7 = 9.2 Hz, 2H), 6.95 (d, 7 =
9.2 Hz, 2H), 3.90 (s, 3H). MS: m z 177 (MH).
Compound 48 was prepared from Compound 21 using a procedure that was analogous to that used to prepare Compound 22 except that 4-methoxy phenyl-propynoic acid (Compound 47) was used in place of phenyl-propynoic acid (55% yield). The structure of Compound 48 was confirmed by 1H NMR and mass spectrometry. Compound 48: 1H NMR(CDC13): δ ppm: 7.62(d, 7 = 9.2 Hz, 2H), 6.88 (d, 7 = 9.2 Hz, 2H), 3.92(s, 3H), 3.86(m, 2H), 3.70(m, 2H), 3.6 (m, 2H), 3.52(m, 2H), 1.47(m, 9H). MS: m/z 345 (MH). Compound 27 was prepared using a procedure that was analogous to that used to prepare Compound 23 except that Compound 48 was used in place of Compound 22. The structure of Compound 27 was confirmed by 1H NMR and mass spectrometry. Compound 27: 1H NMR(CDC13): δ ppm: 7.55(d, 7= 9.2 Hz, 2H), 6.85 (d, 7 = 9.2 Hz, 2H), 3.84(m, 2H), 3.76(s, 3H), 3.70(m, 2H), 2.95(m, 2H), 2.85(m, 2H). MS: m/z 245 (MH).
Compound 1009(b) was prepared from Compound 14 using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compound 27 was used in place of Compound 23 (56% yield). The structure of Compound 1009(b) was confirmed by 1H NMR and mass spectrometry. Compound 1009(b): 1H NMR(CDC13): δ (ppm): 8.33(d, 7 = 4.8 Hζ, IH), 7.51 (d, 7= 9.2 Hz, 2H), 6.89(d, 7= 9.2 Hz, 2H), 6.81(d, 7 = 4.8 Hz, IH), 5.22(s, 2H),
4.01(m, 2H), 3.88(m, 4H), 3.84(s, 3H), 3.78(m, 2H). MS: m/z 378 (MH).
5.5 Example 5: Synthesis of Compound 1009(c) Compound 1009(c) was prepared according to the following scheme.
40 41 42 43
A mixture of Compound 40 (Aldrich Chemical, 2.3 g, 32 mmol), Compound 41 (Aldrich Chemical, 5 g, 26 mmol), HOBT (4 g, 30 mmol), and DCM (200 mL) under argon in a 500 mL-flask was cooled with water and to the mixture was added DIC (4 g, 33 mmol) over about 1 hour. The resulting mixture was stirred at about 25 °C for about 10 hours. The resultant, filtered solid was removed through filtration and the filtrate was washed with water (100 mL) and aqueous 2N NaOH (40 mL). The organic layer was collected and concentrated under reduced pressure to provide crude Compound 42 (6 g) as brown solid. A mixture of crude Compound 42 (1.5 g, 6 mmol), Compound 43 (available from Oakwood Products Inc., West Columbia, SC; 2 g, 8 mmol), EtOAc (100 mL), TEA (2 mL), (Ph3P)2PdCl2 (0.2 g, 0.25 mmol), and Cul (200 mg, 1 mmol) in a 250 mL-flask was flushed with argon three times. The reaction mixture was heated to about 50°C under argon for about 6 hours. The reaction mixture was concentrated and purified using flash chromatography on a silica gel column of the type described previously in
Example 1 eluting with a mixed solvent of 3:7 EtOAc:hexane to provide 2 g of Compound 44 as a yellow solid (83% yield). The structure of Compound 44 was confirmed by 1H NMR. Compound 44: 1H NMR (CDC13): δ(ρρm): 7.11 (dd, 7 = 1.5, 8.1Hz, IH), 6.97 (d, 7= 1.3Hz, IH), 6.8 (d, 7 = 8.1Hz =, IH), 6.02 (s, 2H), 3.76-3.79 (m, 2H), 3.64-3.67 (m, 2H), 3.51-3.53 (m, 2H), 3.44-3.46 (m, 2H), 1.47 (s, 9H). Compound 44 was dissolved in 1,4-dioxane (100 mL) and cooled with ice- water. A solution of HCl (4N in 1,4-dioxane from Aldrich Chemical, 10 mL) was added over about 1 hour. The reaction mixture was heated to about 40-60°C for 1 hour. The solvent was removed to provide 1.5 g of the HCl salt of Compound 29 as a white solid (purity greater than 95 wt.%), which was used subsequently without purification. The structure of Compound 29 was confirmed by 1H NMR. Compound 29: 1H NMR (CD3OD): δ(ppm): 7.22 (dd, 7= 1.7, 8.1Hz, IH), 7.1 (d, 7= 1.3 Hz, IH), 6.91 (d, 7= 8.3Hz, IH), 6.06 (s, 2H), 4.13-4.16 (m, 2H), 3.91-3.94 (m, 2H), 3.36-3.39 (m, 2H), 3.28-3.31 (m, 2H).
Compound 1009(c) was prepared from Compound 14 using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compound 29 was used in place of Compound 23 (63% yield). The structure of Compound 1009(c) was confirmed by 1H NMR and mass spectrometry. Compound 1009(c): 1H NMR(CDC13): δ (ppm): 8.37(d, 7= 5.0Hz, IH), 7.14 (dd, 7= 1.7, 8.1 Hz, IH), 7.02 (dd, 7 = 0.5, 1.5Hz, IH), 6.82-6.85 (m, 2H), 6.04 (s, 2H), 5.24 (d, 7= 0.65Hz, 2H), 4.01-4.06 (m, 2H), 3.86-3.91 (m, 4H), 3.8-3.83 (m, 2H).
LCMS: m/z 392 (MH).
5.6 Example 6: Synthesis of Compound 865(a) Compound 865(a) was prepared according to the following scheme.
Compound 9 was prepared from Compound 6 using a procedure that was analogous to that used to prepare Compound 7 except that the reaction temperature was about -78°C and 2-propynyl amine (Aldrich Chemical) was used in place of propargyl alcohol. The reaction was complete within one hour, as evidenced by LCMS, and quenched at about -78°C by addition of saturated aqueous NH C1. The structure of Compound 9 was confirmed by 1H NMR and mass spectrometry. Compound 9: 1H NMR(CDC13): δ(ppm): 8.83(s, IH); 6.04(bs, IH, NH); 4.30(dd, 7= 2.8, 5.6Hζ, 2H); 2.35(t, 7= 2.8Hz, IH). MS: m/z 251.1 (M+Na-H).
The HCl salt of Compound 23 (52 mg, 0.24 mmol) was added to a solution of Compound 9 (50 mg, 0.22 mmol) and TEA (66 mg, 0.66 mmol) in 10 mL of DCM at about 25°C. The resulting mixture was stirred at about 25°C for about 12 hours to provide 88 mg of Compound 36 (98% yield). The structure of Compound 36 was confirmed by 1H NMR and mass spectrometry. Compound 36: 1H NMR(CDC13): Compound 36: δ (ppm): 8.40(s, IH); 7.57(m, 2H); 7.41(m, 3H); 6.34(bs, IH, NH); 4.25(dd, 7 = 2.4, 5.6 Hz, 2H); 3.94(m, 2H); 3.84(m, 2H); 3.78(m, 4H); 2.31(t, 7 = 2.8 Hz, IH). MS: m/z 408 (MH). Compound 865(a) was prepared by irradiating Compound 36 with microwave irradiation using a procedure that was analogous to that used to prepare Compound 14 except that the temperature was about 280°C at the end of the 1 minute warming period and the duration of microwave irradiation was about 1.5 minutes at about 280°C. The resulting mixture was poured onto a prepacked silica gel column of the type described previously in Example 1. Nitrobenzene was removed using DCM eluent, and a mixture of Compounds 865(a) and 16 were eluted using flash chromatography on the silica gel column with gradient elution from 2.5% methanol/DCM to 5% methanol/DCM.
Purification by preparative TLC (2.5 mL methanol/0.5 mL TFA/97 mL DCM) provided 17 mg of Compound 865(a) as a greater than 97 wt.% pure product (23% yield from Compound 9). The structure of Compound 865(a) was confirmed by 1H NMR and mass spectrometry. Compound 865(a): !H NMR(CDC13): δ(ppm): 8.18(d, 7= 5.0 Hz, lH); 7.53(m, 2H); 7.37(m, 3H); 6.95(d, 7= 5.0 Hz, IH); 4.3 l(s, 2H); 3.96(m, 2H); 3.75(m, 2H); 3.67(m, 2H); 3.59(m,2H). MS: m/z 347.1 (MH). 5.7 Example 7: Synthesis of Compound 937(a) Compound 937(a) was prepared according to the following scheme.
Compound 11 was prepared from Compound 6 using a procedure that was analogous to that used to prepare Compound 9 except that N-methyl-2-propynyl-amine (Aldrich Chemical) was used in place of 2-propynyl amine. The structure of Compound 11 was confirmed by 1H NMR and mass spectrometry. Two conformers of Compound 11 were observed in the former. Compound 11: 1H NMR(CDC13): δ(ppm): 8.83(s, 1.4H); 4.44a (d, 7= 2.8Hζ, 2H); 3.94b (d, 7 = 2.8Hz, 0.8H); 3.26b (s, 1.2H); 3.00a (s, 3H); 2.37b (t, 7 = 2.8Hz, 0.4H); 2.34a (t, 7 = 2.8Hz, IH). MS: m/z 244.1 (MH).
Compound 37 was prepared from Compound 23 using a procedure that was analogous to that used to prepare Compound 36 except that Compound 11 was used in place of Compound 9. The structure of Compound 37 was confirmed by 1H NMR and mass spectrometry. Two conformers of Compound 37 were observed in the former. Compound 37: 1H NMR(CDC13): δ (ppm): 8.44(s, 1.4H); 7.58(m, 3H); 7.43(m, 4.8H); 4.92(d, 7= 2.5, 18 Hz, IH); 4.17(dd, 7= 2.0, 18 Hz, 0.5H); 4.0-3.6(m, 14.5H); 3.22(s, 1.4H); 3.09(s, 3H); 2.42(t, 7 = 2.5 Hz, 0.4H); 2.34(t, 7= 2.5 Hz, IH). MS: m/z 422.2 (MH). Compound 937(a) was prepared by irradiating Compound 37 with microwave irradiation using a procedure that was analogous to that used to prepare Compound 14 (about 8.7% and 64% yield of Compounds 937(a) and 17, respectively). The resulting mixture was poured onto a prepacked silica gel column of the type described previously in Example 1. Nitrobenzene was removed using DCM eluent, and a mixture of
Compounds 937(a) and 17 were eluted using flash chromatography on the silica gel column with gradient elution from 2.5% methanol/DCM to 5% methanol/DCM. Purification by preparative TLC (2.5% methanol/DCM) provided Compound 937(a) (8% yield from Compound 11).
The structure of Compound 937(a) was confirmed by 1H NMR and mass spectrometry. Compound 937(a): 1H NMR(CDC13): δ (ρpm): 8.28(d, 7= 5.0 Hz, IH); 7.56(m, 2H); 7.39(m, 3H); 6.87(d, 7 = 5.0 Hz, IH); 4.33(s, 2H); 4.06(m, 2H); 3.89(m, 2H); 3.82(m, 2H); 3.73(m, 2H); 3.15(s, 3H). MS: m/z 361.2 (MH).
5.8 Example 8: Synthesis of Compound 1753(a) Compound 1753(a) was prepared according to the following scheme.
Compounds 17 and 18 were each prepared (isolated in 69% and 24% yields, respectively) by irradiating Compound 12 with microwave irradiation using a procedure that was analogous to that used to prepare Compound 14. The structures of Compounds 17 and 18 were confirmed by 1H NMR and mass spectrometry. Compound 17: 1H NMR(CDC1
3): δ (ppm): 8.51 (d, 7 = 4.8Hz, IH); 7.37(dt, 7
= 0.4, 4.8 Hz, IH); 4.40(s, 2H); 3.20(s, 3H). MS: 183.(MH); 205(M+Na). Compound 18: 1H NMR(CDC13): δ(ppm): 8.83(d, 7 = 4.8 Hz, lH); 7.73(d, 7 = 4.8Hz, IH); 3.23(s, 3H). MS: m/z 197.0 (MH).
18 23 1753(a)
Compound 1753(a) was prepared from Compound 23 using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compound 18 was used in place of Compound 14 (37% yield). The structure of Compound 1753(a) was confirmed by 1H NMR and mass spectrometry. Compound 1753(a): 1H NMR(CDC1
3): δ (ppm): 8.57(d, 7= 4.6 Hz, IH); 7.58(m, 2H); 7.40(m, 3H); 7.17(d, 7 = 4.6 Hz, IH); 4.03((m, 2H); 3.93(m, 2H); 3.87(s, 4H); 3.16(s, 3H). MS: m/z 375.2 (MH).
5.9 Example 9: Synthesis of Compound 1753(c) Compound 1753(c) was prepared according to the following scheme.
Compound 1753(c) was prepared using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compounds 18 and 29 were used in place of Compounds 14 and 23, respectively (94% yield). The structure of Compound 1753(c) was confirmed by 1H NMR and mass spectrometry. Compound 1753(c): 1H NMR(CDC13): δ (ppm): 8.57(d, 7 = 4.5 Hz, IH); 7.17(d, 7 = 4.5 Hz, IH); 7.13(dd, 7= 1.5, 8.2 Hz, IH); 7.00(m, IH); 6.81(d, 7= 8.2 Hz, IH); 6.03(s, 2H); 4.01(m, 2H); 3.93(m, 2H); 3.86(s, 4H). MS: m/z 419.1 (MH).
5.10 Example 10: Synthesis of Compound 1753(b) Compound 1753(b) was prepared according to the following scheme.
18 27 1753(b)
Compound 1753(b) was prepared using a procedure that was analogous to that used in Example 1 to prepare Compound 1009(a) except that Compounds 18 and 27 were used in place of Compounds 14 and 23, respectively (72% yield). The structure of Compound 1753(b) was confirmed by 1H NMR and mass spectrometry. Compound 1753(b): 1H NMR(CDC13): δ (ppm): 8.57(d, 7 = 4.4 Hz, IH), 7.51 (d, 7 = 8.8 Hz, 2H), 7.17(d, 7 = 4.4 Hz, IH), 6.89(d, 7 = 8.8 Hz, 2H), 4.02(m, 2H), 3.93(m, 2H), 3.87(m, 4H), 3.85(s, 3H), 3.16(s, 3H). MS: m/z 405 (MH).
5.11 Example 11: Synthesis of Compound 867(a) Compound 867(a) was prepared according to the following scheme.
Compound 13 was prepared from Compound 6 using a procedure that was analogous to that used to prepare Compound 9 except that N,N-dimethyl-prop-2- ynylamine (Aldrich Chemical) was used in place of 2-propynyl amine. The structure of Compound 13 was confirmed by 1H NMR and mass spectrometry.
Compound 13: 1H NMR(CDC13): δ (ppm): 8.80(s, IH); 6.04(bs, IH); 2.45(s, IH); 1.80(s, 6H). MS; 280(M+Na).
Compound 38 was prepared from Compound 23 using a procedure that was analogous to that used to prepare Compound 36 except that Compound 13 was used in place of Compound 9. The structure of Compound 38 was confirmed by 1H NMR and mass spectrometry. Two conformers of Compound 38 were observed in the former. Compound 38: 1H NMR(CDC13): δ (ppm): 8.80(s, 0.4H); 8.38(s, IH); 7.54(m, 3.1H); 7.41(m, 5.6H); 3.93(m, 3.1H); 3.90(m, 3.1H); 3.79(s,5.6H); 2.45(s, 0.4H); 2.43(s, IH); 1.81(s, 1.4H); 1.76(s, 6H). MS: m/z 436.2 (MH). Compound 867(a) was prepared by irradiating Compound 38 with microwave irradiation using a procedure that was analogous to that used to prepare Compound 14. The resulting mixture was poured onto a prepacked silica gel column of the type described previously in Example 1. Nitrobenzene was washed off with DCM eluent and a mixture of Compounds 867(a) and 19 was purified using flash chromatography on the silica gel column with gradient elution from 2.5% methanol DCM to 5%
methanol/DCM. Purification by preparative TLC (2.5% methanol DCM) provided Compound 867(a) (4% yield from Compound 13). The structure of Compound 867(a) was confirmed by 1H NMR and mass spectrometry. Compound 867(a): 1H NMR(CDC13): δ (ppm): 8.32(d, 7 = 5.0 Hz, IH); 7.56(m,
2H); 7.39(m, 3H); 6.80(d, 7= 5.0 Hz, IH); 4.04(m, 2H); 3.88(m, 2H); 3.84(m, 2H); 3.76(m, 2H); 1.58(s, 6H). MS: m/z 375.1 (MH).
5.12 Example 12: Compound 1777(a) Compound 1777(a) is prepared:
5.13 Example 13: Binding of an Illustrative Piperazine Compound to rnGluRS The following assay demonstrates that Compound 1441(a), an illustrative
Piperazine Compound, binds to mGluR5. Cell Cultures: Primary glial cultures were prepared from cortices of Sprague- Dawley 18 days old embryos. The cortices were dissected and then dissociated by trituration. The resulting cell homogenate was plated onto poly-D-lysine precoated T175 flasks (BIOCOAT, commercially available from Becton Dickinson and Company Inc. of Franklin Lakes, NJ) in Dulbecco's Modified Eagle's Medium ("DMEM," pH 7.4), buffered with 25 mM HEPES, and supplemented with 15% fetal calf serum ("FCS," commercially available from Hyclone Laboratories Inc. of Omaha, NE ), and incubated at 37°C and 5% CO2. After 24 hours, FCS supplementation was reduced to 10%. On day six, oligodendrocytes and microglia were removed by strongly tapping the sides of the flasks. One day following this purification step, secondary astrocytes cultures were established by subplating onto 96 poly-D-lysine precoated T175 flasks (BIOCOAT) at a density of 65,000 cells/well in DMEM and 10% FCS. After 24 hours, the astrocytes
were washed with serum free medium and then cultured in DMEM, without glutamate, supplemented with 0.5% FCS, 20 mM HEPES, 10 ng/mL epidermal growth factor ("EGF"), 1 mM sodium pyruvate, and IX penicillin/streptomycin at pH 7.5 for 3 to 5 days at 37°C and 5% CO2. The procedure allows the expression of the mGluR5 receptor by astrocytes, as demonstrated by S. Miller et al, J. Neuroscience 15(9):6103-6109 (1995). Assay Protocol: After 3-5 days incubation with EGF, the astrocytes were washed with 127 mM NaCl, 5 mM KC1, 2 mM MgCl2, 700 mM NaH2PO4, 2 mM CaCl2, 5 mM NaHCO3, 8 mM HEPES, 10 mM Glucose at pH 7.4 ("Assay Buffer") and loaded with the dye FLUO-4 (commercially available from Molecular Probes Inc. of Eugene, OR) using 0.1 mL of Assay Buffer containing FLUO-4 (3 mM final). After 90 minutes of dye loading, the cells were then washed twice with 0.2 mL Assay Buffer and resuspended in 0.1 mL of Assay Buffer. The plates containing the astrocytes were then transferred to a Fluorometric Imaging Plate reader ("FLEPR") (commercially available from Molecular Devices Corporation of Sunnyvale, CA) for the assessment of calcium mobilization flux in the presence of glutamate and in the presence or absence of antagonist. After monitoring fluorescence for 15 seconds to establish a baseline, dimethylsulfoxide ("DMSO") solutions containing various concentrations of the Piperazine Compounds diluted in Assay Buffer (0.05 mL of 4X dilutions for competition curves) were added to the cell plate and fluorescence was monitored for 2 minutes. 0.05 mL of a 4X glutamate solution (agonist) was then added to each well to provide a final glutamate concentration in each well of 10 mM. Plate fluorescence was then monitored for an additional 60 seconds after agonist addition. The final DMSO concentration in the assay was 1.0%. In each experiment, fluorescence was monitored as a function of time and the data analyzed using Microsoft Excel and GraphPad Prism. Dose-response curves were fit using a non-linear regression to determine the IC50 value. Compound 1441(a) (see Example 2) showed an IC50 value of 80.4 ± 22.4 μM (mean of 5 experiments).
5.14 Example 14: Binding of a Piperazine Compound to mGluR5 Alternatively, the following assay can be used to demonstrate that Piperazine
Compounds bind to and modulate the activity of mGluR5.
40,000 CHO-rat mGluR5 cells/well are plated into 96 well plate (Costar 3409, Black, clear bottom, 96 well, tissue culture treated) for an about 16 hour incubation in Dulbecco's Modified Eagle's Medium (DMEM, pH 7.4) and supplemented with glutamine, 10% FBS, 1% Pen Strep, and 500μg/mL Geneticin. CHO-rat mGluR5 cells are washed and treated with Optimem medium and incubated for 1-4 hours prior to loading cells. Cell plates are then washed with loading buffer (127 mM NaCl, 5 mM KC1, 2 mM MgCl2, 700 μM Na H2PO4, 2 mM CaCl2, 5 mM NaHCO3, 8 mM HEPES, and 10 mM glucose, pH 7.4) and then incubated with 3μM Fluo 4 (commercially available from Molecular probes Inc. of Eugene, OR) in 0.1 mL of loading buffer. After 90 minutes of dye loading, the cells are then washed twice with 0.2 mL loading buffer and resuspended in 0.1 mL loading buffer. The plates containing the CHO-rat mGluR5 cells are then transferred to a FLIPR for the assessment of calcium mobilization flux in the presence of glutamate and in the presence or absence of test compounds. After monitoring fluorescence for 15 seconds to establish a baseline, DMSO solutions containing various concentrations of the test compound diluted in loading buffer (0.05 mL of 4X dilutions for the competition curves) are added to the cell plate and fluorescence is monitored for 2 minutes. 0.05 mL of 4X glutamate solution (agonist) is then added to each well to provide a final glutamate concentration in each well of 10 μM. Plate fluorescence is then monitored for an additional 60 seconds after agonist addition. The final DMSO concentration in the assay is 1.0%. i each experiment, fluorescence is monitored as a function of time and the data analyzed using Microsoft Excel and GraphPad Prism. Dose-response curves are fit using a non-linear regression to determine the IC50 value. In each experiment, each data point is determined two times.
5.15 Example 15: In Vivo Assays for Treatment or Prevention of Pain The following assays can be used to demonstrate that Piperazine Compounds are useful for treating or preventing pain. Test Animals: Each experiment uses rats weighing between 200-260 g at the start of the experiment. The rats are group-housed and have free access to food and water at all times except prior to oral administration of a Piperazine Compound when food is removed for 16 hours before dosing. A control group acts as a comparison to rats
treated with a Piperazine Compound. The control group is administered the carrier for the Piperazine Compound. The volume of carrier administered to the control group is the same as the volume of carrier and Piperazine Compound administered to the test group. Acute Pain: To assess the actions of the Piperazine Compounds for the treatment or prevention of acute pain the rat tail flick test can be used. Rats are gently restrained by hand and the tail exposed to a focused beam of radiant heat at a point 5 cm from the tip using a tail flick unit (Model 7360, commercially available from Ugo Basile of Italy). Tail flick latencies are defined as the interval between the onset of the thermal stimulus and the flick of the tail. Animals not responding within 20 seconds are removed from the tail flick unit and assigned a withdrawal latency of 20 seconds. Tail flick latencies are measured immediately before (pre-treatment) and 1, 3, and 5 hours following administration of a Piperazine Compound. Data are expressed as tail flick latency(s) and the percentage of the maximal possible effect (% MPE), i.e., 20 seconds, is calculated as follows:
[ (post administration latency) - (pre-administration latency) ]
% MPE = X 100 (20 s pre-administration latency)
The rat tail flick test is described in F.E. D'Amour et al, "A Method for Determining Loss of Pain Sensation," 7. Pharmacol. Exp. Tlier. 72:74-79 (1941). Acute pain can also be assessed by measuring the animal's response to noxious mechanical stimuli by determining the paw withdrawal threshold (PWT), as described below. Inflammatory Pain: To assess the actions of the Piperazine Compounds for the treatment or prevention of inflammatory pain the Freund's complete adjuvant ("FCA") model of inflammatory pain is used. FCA-induced inflammation of the rat hind paw is associated with the development of persistent inflammatory mechanical hyperalgesia and provides reliable prediction of the anti-hyperalgesic action of clinically useful analgesic drugs (L. Bartho et al, "Involvement of Capsaicin-sensitive Neurones in Hyperalgesia
and Enhanced Opioid Antinociception in Inflammation," Naunyn-Schmiedeberg's Archives of Pharmacol. 342:666-670 (1990)). The left hind paw of each animal is administered a 50 μL intraplantar injection of 50% FCA. 24 hour post injection, the animal is assessed for response to noxious mechanical stimuli by determining the PWT, as described below. Rats are then administered a single injection of 1, 3, 10 or 30 mg kg of either a Piperazine Compound; 30 mg/kg of a control selected from Celebrex, indomethacin or naproxen; or carrier. Responses to noxious mechanical stimuli are then determined 1, 3, 5 and 24 hours post administration. Percentage reversal of hyperalgesia for each animal is defined as:
[ (post administration PWT) - (pre-administration PWT) ]
% Reversal = X 100 [ (baseline PWT) - (pre-administration PWT) ]
Neuropathic Pain: To assess the actions of the Piperazine Compounds for the treatment or prevention of neuropathic pain either the Seltzer model or the Chung model can be used. i the Seltzer model, the partial sciatic nerve ligation model of neuropathic pain is used to produce neuropathic hyperalgesia in rats (Z. Seltzer et al, "A Novel Behavioral Model of Neuropathic Pain Disorders Produced in Rats by Partial Sciatic Nerve Injury," Pain 43:205-218 (1990)). Partial ligation of the left sciatic nerve is performed under isoflurane/O2 inhalation anaesthesia. Following induction of anesthesia, the left thigh of the rat is shaved and the sciatic nerve exposed at high thigh level through a small incision and is carefully cleared of surrounding connective tissues at a site near the trocanther just distal to the point at which the posterior biceps semitendinosus nerve branches off of the common sciatic nerve. A 7-0 silk suture is inserted into the nerve with a 3/8 curved, reversed-cutting mini-needle and tightly ligated so that the dorsal 1/3 to lΛ of the nerve thickness is held within the ligature. The wound is closed with a single muscle suture (4-0 nylon (Vicryl)) and vetbond tissue glue. Following surgery, the wound area is dusted with antibiotic powder. Sham-treated rats undergo an identical surgical procedure except that the sciatic nerve is not manipulated.
Following surgery, animals are weighed and placed on a warm pad until they recover
from anesthesia. Animals are then returned to their home cages until behavioral testing begins. The animal is assessed for response to noxious mechanical stimuli by determining PWT, as described below, prior to surgery (baseline), then immediately prior to and 1, 3 and 5 hours after drug administration for the left rear paw of the animal. Percentage reversal of neuropathic hyperalgesia is defined as:
[ (post administration PWT) - (pre-administration PWT) ]
% Reversal = X 100 [ (baseline PWT) - (pre-administration PWT) ]
In the Chung model, the spinal nerve ligation model of neuropathic pain is used to produce mechanical hyperalgesia, thermal hyperalgesia and tactile allodynia in rats. Surgery is performed under isoflurane/O2 inhalation anaesthesia. Following induction of anaesthesia a 3 cm incision is made and the left paraspinal muscles are separated from the spinous process at the L4 - S2 levels. The L6 transverse process is carefully removed with a pair of small rongeurs to identify visually the L4 - L6 spinal nerves. The left L5 (or L5 and L6) spinal nerve(s) is isolated and tightly ligated with silk thread. A complete hemostasis is confirmed and the wound is sutured using non-absorbable sutures, such as nylon sutures or stainless steel staples. Sham-treated rats undergo an identical surgical procedure except that the spinal nerve(s) is not manipulated. Following surgery animals are weighed, administered a subcutaneous (s.c.) injection of saline or ringers lactate, the wound area is dusted with antibiotic powder and they are kept on a warm pad until they recover from the anesthesia. Animals are then returned to their home cages until behavioral testing begins. The animals are assessed for response to noxious mechanical stimuli by determining PWT, as described below, prior to surgery (baseline), then immediately prior to and 1, 3 and 5 hours after being administered a Piperazine Compound for the left rear paw of the animal. The animal can also be assessed for response to noxious thermal stimuli or for tactile allodynia, as described below. The Chung model for neuropathic pain is described in S.H. Kim, "An Experimental Model for Peripheral Neuropathy Produced by Segmental Spinal Nerve Ligation in the Rat," Pα 50(3):355-363 (1992).
Response to Mechanical Stimuli as an Assessment of Mechanical Hyperalgesia: The paw pressure assay can be used to assess mechanical hyperalgesia. For this assay, hind paw withdrawal thresholds (PWT) to a noxious mechanical stimulus are determined using an analgesymeter (Model 7200, commercially available from Ugo Basile of Italy) as described in C. Stein, "Unilateral Inflammation of the Hindpaw in Rats as a Model of Prolonged Noxious Stimulation: Alterations in Behavior and Nociceptive Thresholds," Pharmacol. Biochem. and Behavior 3 451-455 (1988). The maximum weight that can be applied to the hind paw is set at 250 g and the end point is taken as complete withdrawal of the paw. PWT is determined once for each rat at each time point and only the affected (ipsilateral) paw is tested. Response to Thermal Stimuli as an Assessment of Thermal Hyperalgesia: The plantar test can be used to assess thermal hyperalgesia. For this test, hind paw withdrawal latencies to a noxious thermal stimulus are determined using a plantar test apparatus (commercially available from Ugo Basile of Italy) following the technique described by K. Hargreaves et al, "A New and Sensitive Method for Measuring Thermal Nociception in Cutaneous Hyperalgesia," Pain 32(l):77-88 (1988). The maximum exposure time is set at 32 seconds to avoid tissue damage and any directed paw withdrawal from the heat source is taken as the end point. Three latencies are determined at each time point and averaged. Only the affected (ipsilateral) paw is tested. Assessment of Tactile Allodynia: To assess tactile allodynia, rats are placed in clear, plexiglass compartments with a wire mesh floor and allowed to habituate for a period of at least 15 minutes. After habituation, a series of von Frey monofilaments are presented to the plantar surface of the left (operated) foot of each rat. The series of von Frey monofilaments consists of six monofilaments of increasing diameter, with the smallest diameter fiber presented first. Five trials are conducted with each filament with each trial separated by approximately 2 minutes. Each presentation lasts for a period of 4-8 seconds or until a nociceptive withdrawal behavior is observed. Flinching, paw withdrawal or licking of the paw are considered nociceptive behavioral responses. 5.16 Example 16: In Vivo Assays for Treatment or Prevention of Anxiety The following assays can be used to demonstrate that Piperazine Compounds are useful for treating or preventing anxiety. The elevated plus maze test or the shock-probe
burying test can be used to assess the anxiolytic activity of Piperazine Compounds in rats or mice. The Elevated Plus Maze Test: The elevated plus maze consists of a platform with 4 arms, two open and two closed (50 x 10 x 50 cm enclosed with an open roof). Rats (or mice) are placed in the center of the platform, at the crossroad of the 4 arms, facing one of the closed arms. Time spent in the open arms vs the closed arms and number of open arm entries during the testing period are recorded. This test is conducted prior to drug administration and again after drug administration. Test results are expressed as the mean time spent in open arms and the mean number of entries into open arms. Known anxiolytic drugs increase both the time spent in open arms and number of open arm entries. The elevated plus maze test is described in D. Treit, "Animal Models for the Study of Anti-anxiety Agents: A Review," Neuroscience & Biobehavioral Reviews 9(2):203-222 (1985). The Shock-Probe Burying Test: For the shock-probe burying test the testing apparatus consists of a plexiglass box measuring 40 x 30 x 40 cm, evenly covered with approximately 5 cm of bedding material (odor absorbent kitty litter) with a small hole in one end through which a shock probe (6.5 cm long and 0.5 cm in diameter) is inserted. The plexiglass shock probe is helically wrapped with two copper wires through which an electric current is administered. The current is set at 2 niA. Rats are habituated to the testing apparatus for 30 min on 4 consecutive days without the shock probe in the box. On test day, rats are placed in one corner of the test chamber following drug administration. The probe is not electrified until the rat touches it with its snout or fore paws, at which point the rat receives a brief 2 mA shock. The 15 min testing period begins once the rat receives its first shock and the probe remains electrified for the remainder of the testing period. The shock elicits burying behavior by the rat.
Following the first shock, the duration of time the rat spends spraying bedding material toward or over the probe with its snout or fore paws (burying behavior) is measured as well as the number of contact-induced shocks the rat receives from the probe. Known anxiolytic drugs reduce the amount of burying behavior. In addition, an index of the rat's reactivity to each shock is scored on a 4 point scale. The total time spent immobile during the 15 min testing period is used as an index of general activity. The shock-probe
burying test is described in D. Treit, 1985, supra.
5.17 Example 17: In Vivo Assays for Treatment or Prevention of an Addictive Disorder The conditioned place preference test or drug self-administration test can be used to assess the ability of Piperazine Compounds to attenuate the rewarding properties of known drugs of abuse. The Conditioned Place Preference Test: The apparatus for the conditioned place preference test consists of two large compartments (45 x 45 x 30 cm) made of wood with a plexiglass front wall. These two large compartments are distinctly different. Doors at the back of each large compartment lead to a smaller box (36 x 18 x 20 cm) box made of wood, painted grey, with a ceiling of wire mesh. The two large compartments differ in terms of shading (white vs black), level of illumination (the plexiglass door of the white compartment is covered with aluminum foil except for a window of 7 x 7 cm), texture (the white compartment has a 3 cm thick floor board (40 x 40 cm) with nine equally spaced 5 cm diameter holes and the black has a wire mesh floor), and olfactory cues (saline in the white compartment and 1 mL of 10% acetic acid in the black compartment). On habituation and testing days, the doors to the small box remain open, giving the rat free access to both large compartments. The first session that a rat is placed in the apparatus is a habituation session and entrances to the smaller grey compartment remain open giving the rat free access to both large compartments. During habituation, rats generally show no preference for either compartment. Following habituation, rats are given 6 conditioning sessions. Rats are divided into 4 groups: carrier pre-treatment + carrier (control group), Piperazine Compound pre-treatment + carrier, carrier pre-treatment + morphine, Piperazine Compound pre-treatment + morphine. During each conditioning session the rat is injected with one of the drug combinations and confined to one compartment for 30 min. On the following day, the rat receives a carrier + carrier treatment and is confined to the other large compartment. Each rat receives three conditioning sessions consisting of 3 drug combination-compartment and 3 carrier-compartment pairings. The order of injections and the drug/compartment pairings are counterbalanced within groups. On the test day, rats are injected prior to testing (30 min to 1 hour) with either morphine or
carrier and the rat is placed in the apparatus, the doors to the grey compartment remain open and the rat is allowed to explore the entire apparatus for 20 min. The time spent in each compartment is recorded. Known drugs of abuse increase the time spent in the drug-paired compartment during the testing session. If the Piperazine Compound blocks or reduces the acquisition of morphine conditioned place preference (reward), there will be no difference in time spent in each side in rats pre-treated with a Piperazine Compound and the group will not be different from the group of rats that was given carrier + carrier in both compartments. Data will be analyzed as time spent in each compartment (drug combination-paired vs carrier-paired). Generally, the experiment is repeated with a minimum of 3 doses of a Piperazine Compound. The Drug Self- Administration Test: The apparatus for the drug self- administration test is a standard commercially available operant conditioning chamber. Before drug trials begin rats are trained to press a lever for a food reward. After stable lever pressing behavior is acquired, rats are tested for acquisition of lever pressing for drug reward. Rats are implanted with chronically indwelling jugular catheters for i.v. administration of compounds and are allowed to recover for 7 days before training begins. Experimental sessions are conducted daily for 5 days in 3 hour sessions. Rats are trained to self- administer a known drug of abuse, such as morphine. Rats are then presented with two levers, an "active" lever and an "inactive" lever. Pressing of the active lever results in drug infusion on a fixed ratio 1 (FR1) schedule (i.e., one lever press gives an infusion) followed by a 20 second time out period (signaled by illumination of a light above the levers). Pressing of the inactive lever results in infusion of excipient. Training continues until the total number of morphine infusions stabilizes to within ± 10% per session. Trained rats are then used to evaluate the effect of Piperazine Compounds pre-treatment on drug self-administration. On test day, rats are pre-treated with a Piperazine Compound or excipient and then are allowed to self- administer drug as usual. If the Piperazine Compound blocks or reduces the rewarding effects of morphine, rats pre-treated with the Piperazine Compound will show a lower rate of responding compared to their previous rate of responding and compared to excipient pre-treated rats. Data is analyzed as the change in number of drug infusions per testing session (number of infusions during test session - number of infusions during
training session).
5.18 Example 18: Functional Assay for Characterizing mGluRl Antagonistic Properties Functional assays for the characterization of mGluRl antagonistic properties are known in the art. For example, the following procedure can be used. A CHO-rat mGluRl cell line is generated using cDNA encoding rat mGluRl receptor (M. Masu and S. Nakanishi, Nature 349:760-765 (1991)). The cDNA encoding rat mGluRla receptor can be obtained from, e.g., Prof. S. Nakanishi (Kyoto, Japan). 40,000 CHO-rat mGluRl cells/well are plated into a COSTAR 3409, black, clear bottom, 96 well, tissue culture treated plate (commercially available from Fisher Scientific of Chicago, IL) and are incubated in Dulbecco's Modified Eagle's Medium (DMEM, pH 7.4) supplemented with glutamine, 10% FBS, 1% Pen/Strep, and 500 μg/mL Geneticin for about 12 hours. The CHO-rat mGluRl cells are then washed and treated with OPTIMEM medium (commercially available from Invitrogen, Carlsbad, CA) and incubated for a time period ranging from 1 to 4 hours prior to loading the cells with the dye FLUO-4. After incubation, the cell plates are washed with loading buffer (127 mM NaCl, 5 mM KC1, 2 mM MgCl2, 700 μM, NaH2PO4, 2 mM CaCl2, 5 mMNaHCO3, 8 mM HEPES, and 10 mM glucose, pH 7.4) and incubated with 3 μM FLUO-4 in 0.1 mL loading buffer for 90 min. The cells are then washed twice with 0.2 mL loading buffer, resuspended in 0.1 mL of loading buffer, and transferred to a FLIPR for measurement of calcium mobilization flux in the presence of glutamate and in the presence or absence of a Piperazine Compound. To measure calcium mobilization flux, fluorescence is monitored for about 15 s to establish a baseline and DMSO solutions containing various concentrations of a Piperazine Compound ranging from about 50 μM to about 0.8 nM diluted in loading buffer (0.05 mL of a 4X dilution) are added to the cell plate and fluorescence is monitored for about 2 min. 0.05 mL of a 4X glutamate solution (agonist) is then added to each well to provide a final glutamate concentration in each well of 10 μM and fluorescence is monitored for about one additional min. The final DMSO concentration in the assay is 1%. In each experiment fluorescence is monitored as a function of time
and the data is analyzed using a non-linear regression to determine the IC50 value. In each experiment each data point is determined twice. The present invention is not to be limited in scope by the specific embodiments disclosed in the examples which are intended as illustrations of a few aspects of the invention and any embodiments that are functionally equivalent are within the scope of this invention. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art and are intended to fall within the scope of the appended claims. A number of references have been cited, the entire disclosures of which are incorporated herein by reference.





























































































































