WO2004067513A1 - Antagonists for alpha-2 adrenoceptors - Google Patents
Antagonists for alpha-2 adrenoceptors Download PDFInfo
- Publication number
- WO2004067513A1 WO2004067513A1 PCT/FI2004/000038 FI2004000038W WO2004067513A1 WO 2004067513 A1 WO2004067513 A1 WO 2004067513A1 FI 2004000038 W FI2004000038 W FI 2004000038W WO 2004067513 A1 WO2004067513 A1 WO 2004067513A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- mono
- alkoxy
- quinolin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
- C07D215/42—Nitrogen atoms attached in position 4
- C07D215/44—Nitrogen atoms attached in position 4 with aryl radicals attached to said nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to new pharmacologically active derivatives of quinoline and related compounds and to their pharmaceutically acceptable salts and esters, as well as to pharmaceutical compositions containing them and to their use as alpha-2 antagonists.
- Some compounds exhibiting alpha adrenergic activity are well known in the art. It is also generally known and accepted in the art that those compounds may be used for the treatment of a wide variety of diseases and conditions of the peripheral system and the central nervous system (CNS).
- CNS central nervous system
- alpha adrenergic receptors can be divided on a pharmacological basis into alpha- 1 and alpha-2 adrenoceptors, which can both be further divided into subtypes.
- Three genetically encoded subtypes namely alpha-2 A, alpha-2B and alpha-2C adrenoceptors, have been discovered in man. Accordingly, alpha-2 adrenoceptors in humans have been subdivided into three subtypes known as alpha-2A, alpha-2B and alpha-2C adrenoceptors.
- a fourth, pharmacologically defined subtype, alpha-2D is known in rodents and in some other mammals. It represents a species homolog to the human alpha-2A adrenoceptor.
- alpha-2 adrenoceptor subtypes have distinct tissue distributions and functional roles. For instance, while alpha-2A adrenoceptors are widely expressed in various tissues, alpha- 2C adrenoceptors are concentrated in the CNS, and they appear to play a role in the modulation of specific CNS-mediated behavioural and physiological responses.
- atipamezole is a non-specific alpha-2 antagonist.
- Atipamezole has been described in, for example, EP-A-183 492 (cf. p.13, compound XV) and Haapalinna, A. et al., Naunyn- Schmiedeberg's Arch. Pharmacol. 356 (1997) 570-582.
- WO-A-99 28300 discloses substituted imidazole derivatives having agonist-like activity for alpha-2B or alpha-2B/2C adrenoceptors.
- WO 01/64645 describes derivatives of quinoline and related compounds which are stated to possess alpha-2 adrenoceptor antagonistic activity most of them being subtype selective alpha-2C adrenoceptor antagonists.
- An object of the present invention is to provide further antagonists of alpha-2 adrenoceptors that can be used for the treatment of diseases or conditions of the peripheral or central nervous system where alpha-2 antagonists are indicated to be useful.
- an object of the present invention is to provide further compounds to be used as alpha-2 antagonist agents in the treatment of mammals, including humans and animals.
- the invention also provides compounds useful as selective alpha-2C antagonist agents for the treatment of various disorders or conditions of the central nervous system where alpha-2C antagonists are indicated to be useful.
- Compounds of the invention have an improved potency and/or safety profile compared with the previously known structurally related compounds.
- Figure 1 Duration of activity of rats in the Porsolt's forced swimming test.
- the figure shows the alpha-2C-antagonistic and antidepressant-like efficacy of compounds A and D in relation to the clinically effective antidepressant fluoxetine (FLUOX).
- the present invention relates to novel alpha-2 antagonists having the general formula I:
- Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R 5 each independently being OH, halogen, ( -C ⁇ alkyl, (C 2 -C 6 )alkenyl, (C 1 -C 6 )alkoxy, halo(Ci-
- C 6 )alkyl NO , NH 2 , mono- or di(C 1 -C 6 )alkylamino, mono- or di(C 1 -C 6 )alkylamino(C ⁇ -
- Y is -CH m (R 4 )l-[CH n (R )p]v- or a single bond;
- Ri is H, ( -C f alkyl or (C 3 -C 7 )cycloalkyl; A is a benzene ring or (C 5 -C 7 )cycloalkyl;
- each R 2 is independently OH, halogen, ( -C ⁇ alkyl, (C -C 6 )alkenyl, (C 2 -C 6 )alkynyl, (C - C 7 )cycloalkyl, ( -C ⁇ alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo-(C 1 -C 6 )alkyl, NO 2 , NH 2 , mono- or di(C ⁇ -C 6 )alkylamino, amino ⁇ C ⁇ alkyl, mono- or di(C ⁇ -C 6 )al
- R 3 is H, (C 1 -C 6 )alkyl, (C 2 -C 6 )alkenyl, CN-(C ⁇ -C 6 )alkyl, (C 1 -C 6 )alkoxy-CO-(C 1 -C 6 )alkyl, (C 1 -C 6 )alkyl-CO-, NH 2 -CO-, mono- or di(C 1 -C 6 )alkylcarbamoyl, hydroxy(C 1 -C 6 )alkyl, mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl, (C 3 -C 7 )cycloalkyl, phenyl, naphthyl or benzyl, wherein the said (C 3 -C 7 )cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or substitued with 1 to 3 substituent(s) each
- Ra and Rb are independently H, OH, halogen, (C 1 -C 6 )alkyl, (C -C 6 )alkenyl, (C - C 6 )alkynyl, (d-C 6 )alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo(C 1 -C 6 )alkyl, NO 2 , NH 2 , mono- or di(C 1 -C 6 )alkylamino, mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl, amino(C 1 - C 6 )alkyl, (C ⁇ -C 6 )alkyl-S-, CN, (C 3 -C 7 )cycloalkyl, (C 3 -C 7 )cycloalkyl(C ⁇ -C 6 )alkyl, (Ci- C 6 )alkoxy-CO-,
- the moiety Q can be attached to the parent molecule in two different directions resulting in different chemical compounds.
- the invention includes within its scope both directions.
- the moiety Q can be attached to the parent molecule in two different directions resulting in different chemical compounds unless L is N and r is 0 or L is N, r is 2, and R 5 and R 5 are identical.
- the invention includes within its scope both directions.
- the compound of formula I is a compound wherein Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R 5 as defined above; Y, A, Ri-Ri and R 6 are as defined above;
- Ra and Rb are independently H, OH, halogen, (d-C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 - C 6 )alkynyl, (d-C 6 )alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo(d-C 6 )alkyl, NO , NH 2 , mono- or di(C 1 -C 6 )alkylamino, mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl, amino(C ⁇ - C 6 )alkyl, (C 1 -C 6 )alkyl-S-, CN, (C 3 -C 7 )cycloalkyl, (C 3 -C 7 )cycloalkyl(C ⁇ -C 6 )alkyl, (d- C 6 )alkoxy-CO-, (d-C
- a possible subgroup of the compound of formula I is a compound wherein
- Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R 5 as defined above;
- Y, A, R t and R 6 are as defined above; Ra and Rb are independently H, OH, halogen, (d-C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -
- C 6 )alkynyl (C ⁇ -C 6 )alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo(d-C 6 )alkyl, NO 2 , NH 2 , mono- or di(C 1 -C 6 )alkylamino, mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl, amino(C 1 - C 6 )alkyl, (d-C ⁇ alkyl-S-, CN, (C 3 -C 7 )cycloalkyl, (C 3 -C 7 )cycloalkyl(C 1 -C 6 )alkyl, (d- C 6 )alkoxy-CO-, (d-C 6 )alkyl-CO-, mono- or di(C 1 -C6)-alkylcarbamoyl, hydroxy(C 1 - C 6 )alkyl
- u is 1 or 2 and R is (d-C 6 )alkyl; or
- each R 2 is independently halogen, (d-C 6 )alkyl or (C ⁇ - C 6 )alkoxy; or
- Ra and Rb are independently H, (d-C 6 )alkyl, hydroxy(C 1 -C 6 )alkyl or phenyl; or
- Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 6 membered non-aromatic heterocyclic ring containing one heteroatom selected from N, O and S; or
- the compound is [4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-(2,3 3 7,8-tetramethyl- quinolin-4-yl)-amine, (2-methyl-3-phenyl-quinolin-4-yl)-[4-(3,4,5-trimethyl-piperazin-l- yl)-phenyl]-amine, [4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-(2-methyl-3-phenyl-quinolin- 4-yl)-amine, (7-chloro-2,3 -dimethyl-quinolin-4-yl)-[4-(3 ,4-dimethyl-piperazin- 1 -yl)- phenyl]-amine, ⁇ 4-[4-(3,4-dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl ⁇ - methanol, ⁇ 4-[4-(3,4
- Another possible subgroup of the compound of formula I is a compound wherein
- Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R 5 as defined above;
- Ra and Rb is H, OH, halogen, (C 1 -C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, (Ci- C 6 )alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo(C 1 -C 6 )alkyl, NO 2 , M ⁇ 2 , mono- or di(C ⁇ - C 6 )alkylamino, mono- or di(C ⁇ -C 6 )alkylamino(C 1 -C 6 )alkyl, amino(C ⁇ -C 6 )alkyl, (Ci- C 6 )alkyl-S- or CN and the other of Ra and Rb is (C 3 -C )cycloalkyl, (C 3 -C 7 )cycloalkyl(C ⁇ - C 6
- Ra and Rb is H or (C ⁇ -C 6 )alkyl and the other of Ra and Rb is hydroxy(C 1 - C 6 )alkyl or phenyl; or
- the compound is (3-aminomethyl-8-methyl-quinolin-4-yl)-[4-(4-methyl- piperazin- 1 -yl)-phenyl] -amine, [2,6-dimethyl-4-(4-methyl-piperazin- 1 -yl)-phenyl] -(2- phenyl-qumolm-4-yl)-amine, (2-methyl-3-phenyl-quinolin-4-yl)-[4-(4-methyl-piperazin- l-yl)-phenyl]-amine or ⁇ 8-methyl-4-[4-(4-methyl-piperazin-l-yl)-phenylamino]-quinolin- 3-yl ⁇ -methanol.
- Another possible subgroup of the compound of formula I is a compound wherein Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R 5 as defined above;
- Y, A and Ri-R 3 are as defined above; Ra and Rb are independently H, OH, halogen, (C 1 -C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 - C 6 )alkynyl, (d-C 6 )alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo(d-C 6 )alkyl, NO 2 , NH 2 , mono- or di(d-C 6 )alkylamino, mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl, amino(d- C 6 )alkyl, (d-C 6 )alkyl-S-, CN, (C 3 -C 7 )cycloalkyl, (d-C ⁇ cycloalkyKd-C ⁇ alkyl, (Ci- C 6 )alkoxy-CO-
- t is 0 or 1 and R 2 is (C ⁇ -C 6 )alkyl; or
- Ra and Rb are independently (d-C 6 )alkyl or phenyl; or
- the compound is (3-ethyl-2,8-dimethyl-quinolin-4-yl)-[4-(4-methyl- [1,4] diazepan- 1 -yl)-phenyl] -amine, [4-(4-methyl- [ 1 ,4] diazepan- 1 -yl)-phenyl] -(2-methyl- 3 -phenyl-quinolin-4-yl)-amine, (3 -ethyl-2, 8 -dimethyl-quinolin-4-yl)- [4-(3 -methyl- imidazolidin-l-yl)-phenyl] -amine, (3-ethyl-2-methyl-quinolin-4-yl)-[4-(4-methyl- [ 1,4] diazepan- l-yl)-phenyl] -amine, (2,3-dimethyl-quinolin-4-yl)-[4-(4-methyl- [ 1 ,4]diazepan
- Another possible subgroup of the compound of formula I is a compound wherein Q is
- R 5 is as defined above and r is 0 to 2;
- L is CH, CR 5 or N;
- Y, A, R1-R 4 and R 6 are as defined above;
- Ra and Rb are independently H, OH, halogen, (C 1 -C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 - C 6 )alkynyl, (d-C 6 )alkoxy, (C 1 -C 6 )alkoxy(C 1 -C 6 )alkyl, halo(d-C 6 )alkyl, NO 2 , NH 2 , mono- or di(C 1 -C 6 )alkylamino, mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl, amino(C 1 - C 6 )alkyl, (d-C 6 )alkyl-S-, CN, (C 3 -C 7 )cycloalkyl, (C 3 -C 7 )cycloalkyl(d-C 6 )alkyl, (d- C 6 )alkoxy-CO-, (C 1
- L is CH or CR 5 ;
- the compound is [6-(3,4-dimethyl-piperazin-l-yl)-pyridin-3-yl]-(2,3-dimethyl- quinolin-4-yl)-amine or ⁇ 4-[6-(3,4-dimethyl-piperazin-l-yl)-pyridin-3-ylamino]-quinolin- 3 -yl ⁇ -methanol.
- R 3 is H or (C 1 -C 6 )alkyl.
- R 3 is (C ⁇ -C 2 )alkyl.
- halo or halogen, as employed herein as such or as part of another group, refers to chlorine, bromine, fluorine or iodine.
- (CrC 6 )alkyl refers to a straight or branched chain radical having 1 to 6 carbon atoms.
- Representative examples of (C ⁇ -C 6 )alkyl include, but are not limited to, methyl, ethyl, w-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, w-pentyl, isopentyl, neopentyl, n-hexyl, and the like.
- (d-C ⁇ kenyl) refers to a straight or branched chain radical having 2 to 6 carbon atoms, and containing (a) double bond(s).
- (C 2 -C 6 )alkyny ⁇ refers to a straight or branched chain radical having 2 to 6 carbon atoms, and containing (a) triple bond(s).
- (C -C 7 )cycloalkyl refers to a saturated cyclic hydrocarbon group containing 3 to 7 carbons. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
- (C 3 -C 7 )cycloalkyl(C 1 -C 6 )alkyl refers to a "(C 3 -
- (C )cycloalkyl as defined herein, appended to the parent molecular moiety through an (d- C6)al yl group, as defined herein.
- Representative examples of (C 3 -C 7 )cycloalkyl(C ⁇ - C 6 )alkyl include, but are not limited to, cyclohexylmethyl, 1-cyclohexylethyl, 2- cyclopentylethyl, and the like.
- hydroxy refers to an - OH group.
- hydroxy(d-C 6 )alkyl refers to at least one hydroxy group, as defined herein, appended to the parent molecular moiety through an (C ⁇ -C 6 )alkyl group, as defined herein.
- Representative examples of hydroxy(d-C 6 )alkyl include, but are not limited to, hydroxymethyl, 2,2-dihydroxyethyl, 1-hydroxyethyl, 3-hydroxypropyl, 1- hydroxypropyl, 1 -methyl- 1-hydroxyethyl, 1 -methyl- 1-hydroxypropyl, and the like.
- halo(d-C 6 )alkyl refers to at least one halogen, as defined herein, appended to the parent molecular moiety through an (C 1 -C 6 )alkyl group, as defined herein.
- Representative examples of halo(C ⁇ -C 6 )alkyl include, but are not limited to, fluoromethyl, difluoromethyl, trifluoromethyl, 2-chloroethyl, 3-bromopropyl, and the like.
- cyano as employed herein as such or as part of another group, refers to a -CN group.
- s 'CN-(C ⁇ -C 6 )alky ⁇ refers to a cyano group, as defined herein, appended to the parent molecular moiety through an (d-C 6 ) lkyl group, as defined herein.
- Representative examples of CN-(C 1 -C 6 )alkyl include, but are not limited to, cyanomethyl, 1-cyanoethyl, 1-cyanopropyl, 2-cyano ⁇ ropyl, and the like.
- amino refers to a - NH 2 group.
- amino(C 1 -C 6 )alkyl refers to at least one amino group, as defined herein, appended to the parent molecular moiety through an (d-C 6 )alkyl group, as defined herein.
- amino(C ⁇ -C 6 )alkyl include, but are not limited to, aminomethyl, 2-aminoethyl, 1-aminoethyl, 2,2-diaminoethyl, 3-aminopropyl, 2-aminopropyl, 4-aminobutyl, 1 -methyl- 1-aminoethyl, and the like.
- mono- or di(C 1 -C 6 )alkylamino refers to one or two (d-C 6 )alkyl group(s), as defined herein, appended to the parent molecular moiety through an amino group, as defined herein.
- Representative examples of mono- or di(C 1 -C 6 )alkylamino include, but are not limited to methylamino, ethylamino, propylamino, butylamino, dimethylamino, diethylamino, N-ethyl-N- methylamino, and the like.
- mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl refers to a mono- or di(C 1 -C 6 )alkylamino group, as defined herein, appended to the parent molecular moiety through a (Ci -C 6 )alkyl group, as defined herein.
- mono- or di(C 1 -C 6 )alkylamino(C 1 -C 6 )alkyl include, but are not limited to, N,N- dimethylaminomethyl, N,N-diethylaminomethyl, N-methylaminoethyl, N- methylaminopropyl, N-ethyl-N-methylaminomethyl, and the like.
- (d-C ⁇ alkoxy)alkyl refers to -O-(d-C 6 )alkyl, wherein -(C 1 -C 6 )alkyl is as defined herein.
- Representative examples of (d-C 6 )alkoxy include, but are not limited to methoxy, ethoxy, propoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, and the like.
- (C ⁇ -C 6 )alkoxy(C 1 -C 6 )alkyl refers to at least one (d- C 6 )alkoxy group, as defined herein, appended to the parent molecular moiety through an (d-C 6 )alkyl group, as defined herein.
- Representative examples of (C ⁇ -C6)alkoxy(C ⁇ - C 6 )alkyl include, but are not limited to methoxymethyl, ethoxymethyl, 2-methoxyethyl, 2- ethoxyethyl, 3,3-dimethoxypropyl, 2,4-dimethoxybutyl and the like.
- mono- or di(C 1 -C 6 )-alkylcarbamoyl refers to one or two (d-C 6 )alkyl group(s), as defined herein, appended to the parent molecular moiety through a -H ⁇ CO- or - ⁇ CO- group.
- Representative examples of mono- or di(C ⁇ -C 6 )- alkylcarbamoyl include, but are not limited to N-methylcarbamoyl, N-ethylcarbamoyl, N- propylcarbamoyl, NN-dimethylcarbamoyl, NN-diethylcarbamoyl and the like.
- the compounds of formula I, as well as the pharmaceutically acceptable salts and esters thereof, are referred to below as the compounds of the invention, unless otherwise indicated.
- the invention includes within its scope all the possible stereoisomers of the compounds, including geometric isomers, e.g. Z and E isomers (cis and trans isomers), and optical isomers, e.g. diastereomers and enantiomers. Furthermore, the invention includes in its scope both the individual isomers and any mixtures thereof, e.g. racemic mixtures.
- the individual isomers may be obtained using the corresponding isomeric forms of the starting material or they may be separated after the preparation of the end compound according to conventional separation methods.
- optical isomers e.g. enantiomers
- the conventional resolution methods e.g. fractional crystallisation
- Pharmaceutically acceptable salts e.g. acid addition salts with both organic and inorganic acids are well known in the field of pharmaceuticals.
- Non-limiting examples of these salts include chlorides, bromides, sulfates, nitrates, phosphates, sulfonates, formates, tartrates, maleates, citrates, benzoates, salicylates and ascorbates.
- Pharmaceutically acceptable esters when applicable, may be prepared by known methods using pharmaceutically acceptable acids that are conventional in the field of pharmaceuticals and that retain the pharmacological properties of the free form.
- Non-limiting examples of these esters include esters of aliphatic or aromatic alcohols, e.g. methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tert-butyl esters.
- the compounds of the invention can be prepared according to methods known in the literature using suitable starting materials, for example, analogous to the methods described by B.F. Cain et al. inJ Med. Chem. 20(8) (1977) 987-996.
- A, Ra, Rb, R l5 R 2 , R 3 , R ⁇ , Q, Y, t and u are as defined above.
- Scheme 1 represents a conventional acid-catalysed coupling of the chloro-compound of formula II with a substituted aromatic amine of formula m.
- the reaction is carried out at room or elevated temperature in a suitable solvent, e.g. an alcohol such as methanol, to obtain the compound of formula I, which is then isolated from the reaction mixture in a usual manner.
- a suitable solvent e.g. an alcohol such as methanol
- the starting material ⁇ can be prepared e.g. according to scheme 2:
- Starting material IV can be prepared according to methods described in the literature (see e.g. J. Heterocyclic Chem. 34 (1997) 315-320 and references cited therein).
- starting material IV can be prepared according to scheme 3:
- Ra, Rb, R 2 and t are as defined above.
- a substituted aromatic NRi -amine HI can be prepared starting from the corresponding nitro compound V, which is reduced with an appropriate agent, e.g. hydrazine, in a suitable solvent, e.g. ethanol, and optionally alkylated with R ⁇ in a manner known in the art, when R ⁇ being (C 1 -C 6 )alkyl is desired.
- an appropriate agent e.g. hydrazine
- a suitable solvent e.g. ethanol
- X is a halogen, such as chlorine
- Y, R , R 4 and u are as described above.
- a compound VI is coupled with ⁇ ra-halogen-nitrobenzene in the presence of potassium carbonate in DMSO, to obtain a reactant V. This reaction is carried out at room or an elevated temperature.
- any starting material or intermediate can be protected, if necessary, in a manner well known in the chemical field. Any protected functionality can subsequently be deprotected in a manner known in the art.
- the compounds of the invention maybe converted, if desired, into their pharmaceutically acceptable salt or ester forms using methods well known in the art.
- Step l 7.96 g (50 mmol) of l-chloro-4-nitrobenzene, 5.01 g (50 mmol) of 2-methylpiperazine and 6.91 g (50 mmol) of potassium carbonate were mixed in 50 ml of DMSO and heated at 80°C overnight. The reaction mixture was poured into 200 ml of water and extracted with dichloromethane. The organic extract was dried over sodium sulphate and evaporated. The reaction product was purified on a silica gel column (eluent methanol: dichloromethane 1:4) to obtain 6.42 g (58 %) of 3-methyl-l-(4-nitro-phenyl)- piperazine.
- the ester (234 mg, 0.58 mmol) was dissolved in 3 ml of THF, added dropwise to a stirred suspension of 117 mg (3.1 mmol) of lithium aluminium hydride in 2 ml of THF at 0°C under an inert atmosphere.
- the reaction mixture was stirred for 30 min at 0°C and then for another lh at room temperature before it was quenched with 224 ⁇ l (12.4 mmol) of water and filtered.
- Example 12 Following the procedure outlined in Example 12, but substituting ⁇ -toluidine for aniline, ethyl(ethoxymethylene)-cyanoacetate for diethyl ethoxymethylene malonate and 1- methylpiperazine for 2-methylpiperazine, afforded 8-methyl-4-[4-(4-methyl-piperazin-l- yl)-phenylamino]-quinoline-3-carbonitrile with 18 % yield. Reduction of the nitrile group according to the method described in Example 12 afforded the title compound with 21 % yield (overall yield 4 %).
- This diamine was then cyclized by heating at 80°C for 2 h with of 118 ⁇ l of formaldehyde (2 mmol, 40 % aq. solution) in 5 ml of formic acid.
- the reaction mixture was taken up in water, made alkaline with aqueous NaOH and extracted with dichloromethane. The extract was dried over sodium sulphate, evaporated and the residue was purified on a silica gel column (eluent methanokdichloromethane, 1:9), to obtain 88 mg (37 %, overall yield 18 %) of the title compound.
- reaction mixture was stirred at 0°C for 2 h, overnight at room temperature and then refluxed for 24 h.
- the reaction mixture was evaporated and the residue purified on a silica gel column (eluent methanol: dichloromethane, 1:9), to give 0.479 g of 5, 5 -dimethyl- 1 -(4- nitro-phenyl)-piperazin-2-one as trifluoroacetic acid salt.
- the salt was taken up in water, made alkaline with sodium bicarbonate and extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated to provide 0.329 g of 5,5-dimethyl-l-(4- nitrophenyl)-piperazin-2-one (69 %).
- the piperazinone (74 mg, 0.29 mmol) was mixed with 1.5 ml of dry DMF and 16 mg (0.38 mmol) of sodium hydride (60 % in mineral oil) under argon atmosphere. 20 ⁇ l (0.33 mmol) of methyl iodide were added to the cooled (0°C) reaction mixture, which was allowed to warm up to room temperature over 2 h. The reaction mixture was then evaporated, taken up in water and extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated to yield 53 mg (70 %) of 4,5,5-trimethyl-l-(4-nitro-phenyl)-piperazin-2-one.
- step 4 substituting ethyl 2- ethylacetoacetate for ethyl 2-methylacetoacetate and aniline for 2,3-dimethylaniline, afforded 3-ethyl-2-methylquinolin-4-ol. It was dissolved in methanol and stirred overnight in a Parr hydrogenation apparatus with 20 bar H 2 at 60°C in the presence of nickel catalyst.
- Example 12 20 mg of ⁇ 4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl ⁇ -methanol (Example 12) were dissolved in 0.5 ml of dry dichloromethane and 0.3 ml of triflic anhydride were added to the solution. The reaction mixture was stirred at ambient temperature under Ar for 4 h, stripped on a rotary evaporator and dried for 1 h at high vacuum. The residue was dissolved in 2 ml of dry methanol, one drop of triethylamine was added and the solution was stirred overnight at ambient temperature under Ar.
- reaction mixture was then diluted with dichloromethane, washed 3 times with brine and dried over sodium sulfate.
- the title compound was purified from the reaction mixture on a silica gel column (gradient elution methanol: dichloromethane form 1 :9 to 1:4) to give 9.1 mg (44%).
- the compounds of the present invention show interesting pharmacological properties, namely they exhibit affinity for alpha2 adrenoceptors.
- the said pharmacological activity of the compounds of the invention is demonstrated with the pharmacological tests presented below.
- the affinity of test compounds for the three human 2 -adrenoceptor subtypes was determined in competition binding assays with the radioligand 3 H- rauwolscine.
- the biological material for the assays consisted of membranes from Shionogi SI 15 cells stably transfected with one of the three human ⁇ 2 subtypes
- Antagonist activities were determined as the ability of compounds to competitively inhibit r epinephrine-stimulated S-GTP ⁇ S binding to G proteins (Jasper et al., Biochem. Pharmacol. 55 (1998) 1035) in membranes of CHO cells stably transfected with one of the three human ⁇ 2 -adrenoceptor subtypes (Pohjanoksa et al., Eur. J. Pharmacol. 35 (1997) 53).
- Membranes (5-10 ⁇ g of protein per sample) and a minimum of 6 concentrations of test compounds were preincubated for 30 min at room temperature in 50 mM Tris, 5 mM MgCl 2 , 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 10 ⁇ M GDP, 30 ⁇ M ascorbic acid, pH 7.4 with a fixed concentration of epinephrine (5 ⁇ M for CC 2A , 15 ⁇ M for ⁇ 2B , 5 ⁇ M for ⁇ 2C ). Then trace amounts of 35 S-GTP ⁇ S (0.08 nM- 0.15 nM, specific activity 1250 Ci/mmol) were added to the incubation mixture.
- the forced swimming test (FST, Porsolt's test) is generally used in the pharmacological screening of new antidepressants.
- antidepressants increase the activity of animals compared to non-treated controls.
- Alpha-2C KO- mice i.e. mice with targeted disruption of the alpha-2C gene
- alpha-2C OE mice overexpressing the alpha-2C receptor protein
- FST was used to confirm both the alpha-2C antagonism and the antidepressant-like properties of the said compounds in vivo.
- the employed method was essentially similar as described by Porsolt et al. 1977.
- Naive rats were individually placed into glass cylinders (diameter 20 cm, height 40 cm), containing 18 cm of water maintained at 25°C. After 15 min they were removed from the cylinders and allowed to dry under warming lamp. After drying, the rats were given a subcutaneous injection of test substance or vehicle and returned to their home cages. A second injection was given 23 h after the first administration, i.e. 1 h before a second exposure to the swimming situation. In the second 5 min swimming session the duration of active behaviors for each animal was recorded.
- the figure 1 shows the in vivo activity of the compounds A and D in the FST.
- the studied alpha-2C selective compounds efficiently increased activity in the FST, as expected based on studies on transgenic mice (Scheinin, M. et al., Life Sci 68(19-20) (2001) 2277-85) and as reported with recently developed alpha-2C antagonist (WO 01/64645).
- the compounds according to the invention may be used to treat any disease or condition wherein alpha-2 antagonists are indicated to be effective.
- the compounds can also be used to reverse effects induced by alpha-2 agonists.
- the compounds of the invention may be useful in the treatment of various disorders of the central nervous system (CNS), i.e. different neurological, psychiatric and cognition disorders (such as depression, anxiety disorders, post traumatic stress disorder, schizophrenia, Parkinson's disease and other movement disorders).
- CNS central nervous system
- psychiatric and cognition disorders such as depression, anxiety disorders, post traumatic stress disorder, schizophrenia, Parkinson's disease and other movement disorders
- peripheral disorders e.g. diabetes, orthostatic hypotension, lipolytic disorders (such as obesity), Raynaud's disease or both male and female sexual dysfunctions.
- the selective alpha-2C antagonists of the present invention may be used for the treatment of various disorders or conditions of CNS-system where alpha-2C antagonists are indicated to be beneficial, for example, to alleviate the symptoms of various mental disorders propagated by stress, Parkinson's disease, depression, negative symptoms of schizophrenia, attention deficit hyperactivity disorder, post traumatic stress disorder, or anxiety disorders.
- the alpha-2C selective compounds can also be used to treat disorders and conditions associated with sensorimotor gating deficits, particularly disorders and conditions wherein the sensorimotor gating deficits result in sensory flooding and cognitive fragmentation causing dysfunction in attention and perception.
- disorders and conditions include, but are not limited to, schizophrenia, obsessive compulsive disorder, Tourette's syndrome, blepharospasm and other focal dystonias, temporal lobe epilepsy with psychosis, drug-induced psychosis (for example, psychosis caused by chronic use of dopaminergic agents) (Braff, D.L.
- the alpha-2C antagonists of the invention Due to their selectivity of action, the alpha-2C antagonists of the invention have less or no undesirable side-effects attributed to non-selective alpha-2 antagonism, such as increases in blood pressure, heart rate, salival secretions, gastrointestinal secretion, anxiety, and startle reactivity per se (Ruffolo, R.RJ. et al., Annu Rev Pharmacol Toxicol 32 (1993) 243-279).
- the compounds of the invention can be administered for example enterally, topically or " parenterally by means of any pharmaceutical formulation useful for said administration, and containing at least one active compound of fonnula I in pharmaceutically acceptable and effective amounts together with pharmaceutically acceptable diluents, carriers, and/or excipients l ⁇ iown in the art.
- the manufacture of such pharmaceutical formulations is well known in the art.
- the therapeutic dose to be given to a patient in need of treatment will vary depending on the compound being administered, the species, age and the sex of the subject being treated, the particular condition being treated, as well as the route and method of administration, and are easily detennined by person skilled in the art. Accordingly, the typical dosage for oral administration is from 5 ⁇ g/kg to 100 mg/kg per day and that for parenteral administration from 0.5 ⁇ g/kg to 10 mg/kg for an adult mammal.
- the present invention further provides a compound of the invention or an ester or salt thereof for use as alpha-2 antagonist.
- a method for the treatment of diseases or conditions where alpha-2 antagonists, e.g. alpha-2C antagonists, are indicated to be useful e.g. a method for the treatment of diseases or conditions of the central nervous system.
- a therapeutically effective amount of a compound of the invention is administered to a subject in need of such treatment.
- the use of the compounds of the invention for the manufacture of a medicament to be used for the above indications is also provided, e.g. for the manufacture of a medicament for the treatment of diseases or conditions where alpha-2 antagonists are indicated to be effective
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention provides a compound of formula (I), wherein Q, Y, A, Ra, Rb, R1 to R4, u and t are as defined in claim 1, or a pharmaceutically acceptable salt or ester thereof, useful as an alpha-2 antagonist. The compounds of formula (I) can be used for the treatment of diseases or conditions where antagonists of alpha-2 adrenoceptors are indicated to be effective.
Description
ANTAGONISTS FOR ALPHA-2 ADRENOCEPTORS
FIELD OF THE INVENTION
The present invention relates to new pharmacologically active derivatives of quinoline and related compounds and to their pharmaceutically acceptable salts and esters, as well as to pharmaceutical compositions containing them and to their use as alpha-2 antagonists.
BACKGROUND OF THE INVENTION
Some compounds exhibiting alpha adrenergic activity are well known in the art. It is also generally known and accepted in the art that those compounds may be used for the treatment of a wide variety of diseases and conditions of the peripheral system and the central nervous system (CNS).
The alpha adrenergic receptors can be divided on a pharmacological basis into alpha- 1 and alpha-2 adrenoceptors, which can both be further divided into subtypes. Three genetically encoded subtypes, namely alpha-2 A, alpha-2B and alpha-2C adrenoceptors, have been discovered in man. Accordingly, alpha-2 adrenoceptors in humans have been subdivided into three subtypes known as alpha-2A, alpha-2B and alpha-2C adrenoceptors. A fourth, pharmacologically defined subtype, alpha-2D, is known in rodents and in some other mammals. It represents a species homolog to the human alpha-2A adrenoceptor.
The alpha-2 adrenoceptor subtypes have distinct tissue distributions and functional roles. For instance, while alpha-2A adrenoceptors are widely expressed in various tissues, alpha- 2C adrenoceptors are concentrated in the CNS, and they appear to play a role in the modulation of specific CNS-mediated behavioural and physiological responses.
Compounds that are non-specific to any of the above-mentioned alpha-2 subtypes, and compounds that are specific to certain alpha-2 subtypes, are already known. For example, atipamezole is a non-specific alpha-2 antagonist. Atipamezole has been described in, for example, EP-A-183 492 (cf. p.13, compound XV) and Haapalinna, A. et al., Naunyn- Schmiedeberg's Arch. Pharmacol. 356 (1997) 570-582. WO-A-99 28300 discloses
substituted imidazole derivatives having agonist-like activity for alpha-2B or alpha-2B/2C adrenoceptors.
As to the derivatives of quinoline, WO 01/64645 describes derivatives of quinoline and related compounds which are stated to possess alpha-2 adrenoceptor antagonistic activity most of them being subtype selective alpha-2C adrenoceptor antagonists.
SUMMARY OF THE INVENTION
An object of the present invention is to provide further antagonists of alpha-2 adrenoceptors that can be used for the treatment of diseases or conditions of the peripheral or central nervous system where alpha-2 antagonists are indicated to be useful.
Accordingly, an object of the present invention is to provide further compounds to be used as alpha-2 antagonist agents in the treatment of mammals, including humans and animals.
The invention also provides compounds useful as selective alpha-2C antagonist agents for the treatment of various disorders or conditions of the central nervous system where alpha-2C antagonists are indicated to be useful.
Compounds of the invention have an improved potency and/or safety profile compared with the previously known structurally related compounds.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1: Duration of activity of rats in the Porsolt's forced swimming test. The figure shows the alpha-2C-antagonistic and antidepressant-like efficacy of compounds A and D in relation to the clinically effective antidepressant fluoxetine (FLUOX). Veh = vehicle; s.c. = subcutaneous; */?<0.05, ***p<0.001, analysed by 1-ANOVA and LSD-test.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to novel alpha-2 antagonists having the general formula I:

wherein,
Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 each independently being OH, halogen, ( -C^alkyl, (C2-C6)alkenyl, (C1-C6)alkoxy, halo(Ci-
C6)alkyl, NO , NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(Cι-
C6)alkyl, (Cι-C6)alkyl-S- or
 or Q is
or Q is

Y is -CHm(R4)l-[CHn(R )p]v- or a single bond; Ri is H, ( -Cf alkyl or (C3-C7)cycloalkyl; A is a benzene ring or (C5-C7)cycloalkyl; each R2 is independently OH, halogen, ( -C^alkyl, (C -C6)alkenyl, (C2-C6)alkynyl, (C - C7)cycloalkyl, ( -C^alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo-(C1-C6)alkyl, NO2, NH2, mono- or di(Cι-C6)alkylamino, amino^ C^alkyl, mono- or di(Cι-C6)alkylamino(Cι- C6)alkyl,
 (Cι-C6)alkyl-S-S hydroxy(C1-C6)alkyl, NH2-CO- or -CHO;
(Cι-C6)alkyl-S-S hydroxy(C1-C6)alkyl, NH2-CO- or -CHO;
R3 is H, (C1-C6)alkyl, (C2-C6)alkenyl, CN-(Cι-C6)alkyl, (C1-C6)alkoxy-CO-(C1-C6)alkyl, (C1-C6)alkyl-CO-, NH2-CO-, mono- or di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, (C3-C7)cycloalkyl, phenyl, naphthyl or benzyl, wherein the said (C3-C7)cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or
substitued with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH2, (C1-C6)alkyl, (C1-Ce)alkoxy, mono- or di(C1-C6)alkylamino or halo-(C1-C6)alkyl; each Rit is independently OH, halogen, Mϊ2, oxo, -CHO, (CrC^alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl,
 NH -CO-, mono- or di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, amino(C1-C6)alkyl, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, (C3- C7)cycloalkyl, phenyl, naphthyl or benzyl, wherein the said (C3-C7)cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or substitued with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (Ci-C6)alkyl, ( -C^alkoxy, mono- or d^C C^alkylamino or halo-(C1-C6)alkyl; or R3 and R4 or ι and ι form, together with any of the ring atom(s) to which they are attached, a condensed 5 to 7 membered carbocyclic ring or a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 or 2 substituent(s) R6 each independently being OH, oxo, (CrC^alkyl, ( -C^alkoxy, halogen or halo(d- C3)alkyl;
NH -CO-, mono- or di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, amino(C1-C6)alkyl, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, (C3- C7)cycloalkyl, phenyl, naphthyl or benzyl, wherein the said (C3-C7)cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or substitued with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (Ci-C6)alkyl, ( -C^alkoxy, mono- or d^C C^alkylamino or halo-(C1-C6)alkyl; or R3 and R4 or ι and ι form, together with any of the ring atom(s) to which they are attached, a condensed 5 to 7 membered carbocyclic ring or a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 or 2 substituent(s) R6 each independently being OH, oxo, (CrC^alkyl, ( -C^alkoxy, halogen or halo(d- C3)alkyl;
Ra and Rb are independently H, OH, halogen, (C1-C6)alkyl, (C -C6)alkenyl, (C - C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1- C6)alkyl, (Cι-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(Cι-C6)alkyl, (Ci- C6)alkoxy-CO-, (CrC^alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1- C6)alkyl, NH -CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3- C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH2, (C1-C6)alkyl, (Ci- C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and Ri form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above;
1 is 0 to 2; m is 0 to 2; n is 0 to 2; p is 0 to 2; t is 0 to 3; u is 0 to 4; and v is 0 or 1; with the provisos, that a) when Q is 1,4-phenylene, 1 is 0, m is 2, u is 0 and v is 0, then one of Ra and Rb is H, OH, halogen, (Cι-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl, (d-C^alkoxy, (Q-
C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO , MT2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1-C6)alkyl, (Cι-C6)alkyl-S- or CN and the other of Ra and Rb is (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(C1-C6)alkyl, ( - C6)alkoxy-CO-, (Cι-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1- C6)alkyl, amino(C1-C6)alkyl, (C1-C6)all:oxy(C1-C6)aιkyl, NH2-CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2, Mϊ2, (Q-C^alkyl, halo(C1-C6)alkyl, (Q-C^alkoxy or mono-or di(C1-C6)alkylamino; or Rb is H, OH, halogen, (d-C^alkyl, (C2-C6)alkenyl, (C -C6)alkynyl, (C C^alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, M32, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1-C6)alkyl, (d-C^alkyl-S-, CN, (C3- C7)cycloalkyl, (C3-C7)cycloalkyl(C1-C6)alkyl, (d-C^alkoxy-CO-, (d-C^alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1-C6)alkyl, MI2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH , (Cι-C6)alkyl, (Cι-C6)alkoxy or mono-or di(Cι-C6)alkylamino and Ra and R! form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above; b) when Q is 1,4-phenylene, 1 is 0, m is 2, u is 0, v is 0, and one of Ra and Rb is H, then the other of Ra and Rb is not ethoxy-CO-;
or a pharmaceutically acceptable salt or ester thereof.
It is evident to a person skilled in the art that, in the compounds of formula I, when Q is

Likewise, it is evident to a person skilled in the art that, in the compounds of formula I, when Q is

In one embodiment of the invention the compound of formula I is a compound wherein Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined above; Y, A, Ri-Ri and R6 are as defined above;
Ra and Rb are independently H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2- C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(d-C6)alkyl, NO , NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(Cι- C6)alkyl, (C1-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(Cι-C6)alkyl, (d- C6)alkoxy-CO-, (d-C6)alkyl-CO-, mono- or di(Cι~C6)-alkylcarbamoyl, hydroxy(C1-
C6)alkyl, NH2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3- C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH , (d-C6)alkyl, (Ci- C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and i form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms
selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R as defined above; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above;
1 is 0 to 2; m is 0 to 2 n is 0 to 2; p is 0 to 2; t is 0 to 3; u is 0 to 4; and v is O or 1; with the proviso, that when u is 0 and v is 0, then 1 is not 0.
A possible subgroup of the compound of formula I is a compound wherein
Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined above;
Y, A, R t and R6 are as defined above; Ra and Rb are independently H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2-
C6)alkynyl, (Cι-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(d-C6)alkyl, NO2, NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1- C6)alkyl, (d-C^alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(C1-C6)alkyl, (d- C6)alkoxy-CO-, (d-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1- C6)alkyl, NH2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3- C )cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH2, (d-C6)alkyl, (Ci- C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and Ri form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above;
or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above;
1 is 0 to 2; m is 0 to 2; n is 0 to 2; p is 0 to 2; t is 0 to 3; u is 0 to 4; and v is 0 or 1 ; with the provisos, that a) when u is 0 and v is 0, then 1 is not 0; b) when u is 0 and v is 1 , then 1 and p are not simultaneously 0; for example
wherein u is 1 or 2 and R is (d-C6)alkyl; or
wherein t is 0, 1 or 2 and each R2 is independently halogen, (d-C6)alkyl or (Cι- C6)alkoxy; or
wherein Ra and Rb are independently H, (d-C6)alkyl, hydroxy(C1-C6)alkyl or phenyl; or
wherein Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 6 membered non-aromatic heterocyclic ring containing one heteroatom selected from N, O and S; or
wherein the compound is [4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-(2,337,8-tetramethyl- quinolin-4-yl)-amine, (2-methyl-3-phenyl-quinolin-4-yl)-[4-(3,4,5-trimethyl-piperazin-l- yl)-phenyl]-amine, [4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-(2-methyl-3-phenyl-quinolin- 4-yl)-amine, (7-chloro-2,3 -dimethyl-quinolin-4-yl)-[4-(3 ,4-dimethyl-piperazin- 1 -yl)- phenyl]-amine, {4-[4-(3,4-dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}- methanol, {4-[4-(3,4-dimethyl-piperazin-l-yl)-phenylamino]-2-methyl-quinolin-3-yl}- methanol, (2,3-dimethyl-quinolin-4-yl)-[4-(3,3,4-trimethyl-piperazin-l-yl)-phenyl]-amine, {4- [4-(3 ,4-dimethyl-piperazin- 1 -yl)-phenylamino] -3 -methyl-quinolin-2-yl} -methanol, [4- (3 ,4-dimethyl-piperazin- 1 -yl)-phenyl] -(2-methyl- 1,2,3 ,4-tetra-hydro-
benzo[b][l,6]naphthyridin-10-yl)-amine, (2,3-dimethyl-quinolin-4-yl)-[4-(3,3,4,5,5- pentamethyl-piperazin-l-yl)-phenyl]-amine, {4-[4-(3,3,4-trimethyl-piperazin-l-yl)- phenylamino]-quinolin-3-yl} -methanol, (3-methyl-quinolin-4-yl)-[4-(3,3,4-trimethyl- piperazin- 1 -yl)-phenyl]-amine, {7-chloro-4-[4-(3 ,3 ,4-trimethyl-piperazin- 1 -yl)- phenylamino] -quinolin-3-yl} -methanol, {7-chloro-4-[4-(3 ,4-dimethyl-piperazin- 1 -yl)- phenylamino]-quinolin-3-yl} -methanol, (2,3-dimethyl-quinolin-4-yl)-[4-(3,3,4,5- tetramethyl-piperazin- 1 -yl)-phenyl] -amine, {4-[4-(3 ,4-dimethyl-piperazin- 1 -yl)- phenylamino]-7-methyl-quinolin-3-yl} -methanol, (2,3-dimethyl-quinolin-4-yι)-[4-(5- ethyl-3,3,4-trimethyl-piperazin-l-yl)-phenyl]-amine, {4-[4-(3,4-dimethyl-piperazin-l-yl)- phenylamino]-6-fluoro-quinolin-3-yl}-methanol, {6-fluoro-4-[4-(3,3,4-trimethyl- piperazin-l-yl)-phenylamino]-quinolin-3-yl}-methanol, [4-(3,4-dimethyl-piperazin-l-yl)- phenyl]-(7-methoxy-2-methyl-3-phenyl-quinolin-4-yl)-amine, [4-(3,4-dimethyl-piperazin- l-yl)-phenyl]-(2,3,5,7-tetramethyl-quinolin-4-yl)-amine, [4-(3 ,4-dimethyl-piperazin- 1-yl)- phenyl]-(2,3,7-trimethyl-quinolin-4-yl)-amine, (2,3-dimethyl-quinolin-4-yl)-[4- (octahydro-pyrido[l,2-a]pyrazin-2-yl)-phenyl]-amine, 4-[4-(3,4-dimethyl-piperazin-l-yl)- phenyl]-3,4-dihydro-lH-2-oxa-4,9-di-4-[4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-3,4- dihydro-lH-2-oxa-4,9-diaza-phenanthrene, [4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-(3- methoxymethyl-quinolin-4-yl)-amine or 1 -[4-(3-ethyl-2-methyl-quinolin-4-ylamino)- phenyl]-5,5-dimethyl-piperazin-2-one.
Another possible subgroup of the compound of formula I is a compound wherein
Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined above;
Y, A, Ri-R3 are as defined above; one of Ra and Rb is H, OH, halogen, (C1-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl, (Ci- C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, Mϊ2, mono- or di(Cι- C6)alkylamino, mono- or di(Cι-C6)alkylamino(C1-C6)alkyl, amino(Cι-C6)alkyl, (Ci- C6)alkyl-S- or CN and the other of Ra and Rb is (C3-C )cycloalkyl, (C3-C7)cycloalkyl(Cι- C6)alkyl, (C1-C6)alkoxy-CO-, (C1-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(d-C6)alkyl, amino(Cι-C6)alkyl, (C1-C6)alkoxy(C1-C6)alkyl, NH2-CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH2, (d-Ce^lkyl, halo(C1-C6)alkyl, (Cr C6)alkoxy or mono-or di(d-C6)alkylamino;
or Rb is H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl, (Cι-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1-C6)alkyl, (d-C6)alkyl-S-, CN, (C3- C7)cycloalkyl, (C3-C7)cycloalkyl(C1-C6)alkylJ (d-C6)alkoxy-CO-, (C1-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1-C6)alkyl, NH -CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (C1-C6)alkyl, (d-C6)alkoxy or mono-or di(C1-C6)alkylamino and Ra and Ri form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above; l is O; m is 2; t is 0 to 3 u is 0; and v is 0; with the proviso, that when one of Ra and Rb is H, then the other of Ra and Rb is not ethoxy-CO-; for example
wherein t is 0 or 1 and R2 is (d-C6)alkyl; or
wherein one of Ra and Rb is H or (Cι-C6)alkyl and the other of Ra and Rb is hydroxy(C1- C6)alkyl or phenyl; or
wherein the compound is (3-aminomethyl-8-methyl-quinolin-4-yl)-[4-(4-methyl- piperazin- 1 -yl)-phenyl] -amine, [2,6-dimethyl-4-(4-methyl-piperazin- 1 -yl)-phenyl] -(2- phenyl-qumolm-4-yl)-amine, (2-methyl-3-phenyl-quinolin-4-yl)-[4-(4-methyl-piperazin- l-yl)-phenyl]-amine or {8-methyl-4-[4-(4-methyl-piperazin-l-yl)-phenylamino]-quinolin- 3-yl} -methanol.
Another possible subgroup of the compound of formula I is a compound wherein Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined above;
Y, A and Ri-R3 are as defined above;
Ra and Rb are independently H, OH, halogen, (C1-C6)alkyl, (C2-C6)alkenyl, (C2- C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(d-C6)alkyl, NO2, NH2, mono- or di(d-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(d- C6)alkyl, (d-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (d-C^cycloalkyKd-C^alkyl, (Ci- C6)alkoxy-CO-, (C!-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1- C6)alkyl, Mϊ -CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C3- C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2, Mϊ2, (d-C^alkyl, (d- C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and R1 form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above; l is O; m is 2; n is 2; p is 0; t is 0 to 3; u is 0; and v is 1; for example
wherein t is 0 or 1 and R2 is (Cι-C6)alkyl; or
wherein Ra and Rb are independently (d-C6)alkyl or phenyl; or
wherein the compound is (3-ethyl-2,8-dimethyl-quinolin-4-yl)-[4-(4-methyl- [1,4] diazepan- 1 -yl)-phenyl] -amine, [4-(4-methyl- [ 1 ,4] diazepan- 1 -yl)-phenyl] -(2-methyl- 3 -phenyl-quinolin-4-yl)-amine, (3 -ethyl-2, 8 -dimethyl-quinolin-4-yl)- [4-(3 -methyl- imidazolidin-l-yl)-phenyl] -amine, (3-ethyl-2-methyl-quinolin-4-yl)-[4-(4-methyl- [ 1,4] diazepan- l-yl)-phenyl] -amine, (2,3-dimethyl-quinolin-4-yl)-[4-(4-methyl-
[ 1 ,4]diazepan- 1 -yl)-phenyl] -amine, [4-(4-methyl-[ 1 ,4]diazepan- 1 -yl)-phenyl]-(2,3,8- trimethyl-quinolin-4-yl)-amine or (3-ethyl-2,8-dimethyl-quinolin-4-yl)-(4-imidazolidin-l- yl-phenyl)-amine.
Another possible subgroup of the compound of formula I is a compound wherein Q is

Ra and Rb are independently H, OH, halogen, (C1-C6)alkyl, (C2-C6)alkenyl, (C2- C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(d-C6)alkyl, NO2, NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1- C6)alkyl, (d-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(d-C6)alkyl, (d- C6)alkoxy-CO-, (C1-C6)alkyl-CO-, mono- or di(Cι-C6)-alkylcarbamoyl, hydroxy(Cι- C6)alkyl, NH2-CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C - C )cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH2, (C1-C6)alkyl, (Ci- C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and Ri form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substiruent(s) R6 as defined above; 1 is 0 to 2; m is 0 to 2;
n is 0 to 2; p is 0 to 2; t is 0 to 3; u is 0 to 4; and v is 0 or 1; for example
wherein L is CH or CR5; or
wherein the compound is [6-(3,4-dimethyl-piperazin-l-yl)-pyridin-3-yl]-(2,3-dimethyl- quinolin-4-yl)-amine or {4-[6-(3,4-dimethyl-piperazin-l-yl)-pyridin-3-ylamino]-quinolin- 3 -yl} -methanol.
In a possible subgroup of the compound of formula I A is a benzene ring.
In another possible subgroup of the compound of formula I R3 is H or (C1-C6)alkyl.
In a further possible subgroup of the compound of formula I R3 is (Cι-C2)alkyl.
The terms employed herein have the following meanings:
The term "halo" or "halogen", as employed herein as such or as part of another group, refers to chlorine, bromine, fluorine or iodine.
The term "oxo", as employed herein, refers to an O= group.
The term "(CrC6)alkyl", as employed herein as such or as part of another group, refers to a straight or branched chain radical having 1 to 6 carbon atoms. Representative examples of (Cι-C6)alkyl include, but are not limited to, methyl, ethyl, w-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, w-pentyl, isopentyl, neopentyl, n-hexyl, and the like.
The term "(d-Cδ kenyl", as employed herein as such or as part of another group, refers to a straight or branched chain radical having 2 to 6 carbon atoms, and containing (a) double bond(s).
The term "(C2-C6)alkynyι", as employed herein as such or as part of another group, refers to a straight or branched chain radical having 2 to 6 carbon atoms, and containing (a) triple bond(s).
The term "(C -C7)cycloalkyl", as employed herein, refers to a saturated cyclic hydrocarbon group containing 3 to 7 carbons. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
The term "(C3-C7)cycloalkyl(C1-C6)alkyl", as employed herein, refers to a "(C3-
C )cycloalkyl, as defined herein, appended to the parent molecular moiety through an (d- C6)al yl group, as defined herein. Representative examples of (C3-C7)cycloalkyl(Cι- C6)alkyl include, but are not limited to, cyclohexylmethyl, 1-cyclohexylethyl, 2- cyclopentylethyl, and the like.
The term "hydroxy", as employed herein as such or as part of another group, refers to an - OH group.
The term "hydiOxy(C1-C6)alkyl", as employed herein, refers to at least one hydroxy group, as defined herein, appended to the parent molecular moiety through an (Cι-C6)alkyl group, as defined herein. Representative examples of hydroxy(d-C6)alkyl include, but are not limited to, hydroxymethyl, 2,2-dihydroxyethyl, 1-hydroxyethyl, 3-hydroxypropyl, 1- hydroxypropyl, 1 -methyl- 1-hydroxyethyl, 1 -methyl- 1-hydroxypropyl, and the like.
The term "halo(d-C6)alkyl", as employed herein, refers to at least one halogen, as defined herein, appended to the parent molecular moiety through an (C1-C6)alkyl group, as defined herein. Representative examples of halo(Cι-C6)alkyl include, but are not limited to, fluoromethyl, difluoromethyl, trifluoromethyl, 2-chloroethyl, 3-bromopropyl, and the like.
The term "cyano", as employed herein as such or as part of another group, refers to a -CN group.
The term s'CN-(Cι-C6)alkyι", as employed herein, refers to a cyano group, as defined herein, appended to the parent molecular moiety through an (d-C6) lkyl group, as defined herein. Representative examples of CN-(C1-C6)alkyl include, but are not limited to, cyanomethyl, 1-cyanoethyl, 1-cyanopropyl, 2-cyanoρropyl, and the like.
The term "amino", as employed herein as such or as part of another group, refers to a - NH2 group.
The term "amino(C1-C6)alkyl", as employed herein, refers to at least one amino group, as defined herein, appended to the parent molecular moiety through an (d-C6)alkyl group, as defined herein. Representative examples of amino(Cι-C6)alkyl include, but are not limited to, aminomethyl, 2-aminoethyl, 1-aminoethyl, 2,2-diaminoethyl, 3-aminopropyl, 2-aminopropyl, 4-aminobutyl, 1 -methyl- 1-aminoethyl, and the like.
The term "mono- or di(C1-C6)alkylamino", as employed herein as such or as part of another group, refers to one or two (d-C6)alkyl group(s), as defined herein, appended to the parent molecular moiety through an amino group, as defined herein. Representative examples of mono- or di(C1-C6)alkylamino include, but are not limited to methylamino, ethylamino, propylamino, butylamino, dimethylamino, diethylamino, N-ethyl-N- methylamino, and the like.
The term "mono- or di(C1-C6)alkylamino(C1-C6)alkyl", as employed herein, refers to a mono- or di(C1-C6)alkylamino group, as defined herein, appended to the parent molecular moiety through a (Ci -C6)alkyl group, as defined herein. Representative examples of mono- or di(C1-C6)alkylamino(C1-C6)alkyl include, but are not limited to, N,N- dimethylaminomethyl, N,N-diethylaminomethyl, N-methylaminoethyl, N- methylaminopropyl, N-ethyl-N-methylaminomethyl, and the like.
The term "(d-C^alkoxy", as employed herein as such or as part of another group, refers to -O-(d-C6)alkyl, wherein -(C1-C6)alkyl is as defined herein. Representative examples of (d-C6)alkoxy include, but are not limited to methoxy, ethoxy, propoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, and the like.
The term "(Cι-C6)alkoxy(C1-C6)alkyl", as employed herein, refers to at least one (d- C6)alkoxy group, as defined herein, appended to the parent molecular moiety through an (d-C6)alkyl group, as defined herein. Representative examples of (Cι-C6)alkoxy(Cι- C6)alkyl include, but are not limited to methoxymethyl, ethoxymethyl, 2-methoxyethyl, 2- ethoxyethyl, 3,3-dimethoxypropyl, 2,4-dimethoxybutyl and the like.
The term "mono- or di(C1-C6)-alkylcarbamoyl", as employed herein, refers to one or two (d-C6)alkyl group(s), as defined herein, appended to the parent molecular moiety through a -HΝCO- or -ΝCO- group. Representative examples of mono- or di(Cι-C6)- alkylcarbamoyl include, but are not limited to N-methylcarbamoyl, N-ethylcarbamoyl, N- propylcarbamoyl, NN-dimethylcarbamoyl, NN-diethylcarbamoyl and the like.
The compounds of formula I, as well as the pharmaceutically acceptable salts and esters thereof, are referred to below as the compounds of the invention, unless otherwise indicated.
The invention includes within its scope all the possible stereoisomers of the compounds, including geometric isomers, e.g. Z and E isomers (cis and trans isomers), and optical isomers, e.g. diastereomers and enantiomers. Furthermore, the invention includes in its scope both the individual isomers and any mixtures thereof, e.g. racemic mixtures. The individual isomers may be obtained using the corresponding isomeric forms of the starting material or they may be separated after the preparation of the end compound according to conventional separation methods. For the separation of optical isomers, e.g. enantiomers, from the mixture thereof the conventional resolution methods, e.g. fractional crystallisation, may be used.
Pharmaceutically acceptable salts, e.g. acid addition salts with both organic and inorganic acids are well known in the field of pharmaceuticals. Non-limiting examples of these salts include chlorides, bromides, sulfates, nitrates, phosphates, sulfonates, formates, tartrates, maleates, citrates, benzoates, salicylates and ascorbates. Pharmaceutically acceptable esters, when applicable, may be prepared by known methods using pharmaceutically acceptable acids that are conventional in the field of pharmaceuticals and that retain the pharmacological properties of the free form. Non-limiting examples of these esters include esters of aliphatic or aromatic alcohols, e.g. methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tert-butyl esters.
The compounds of the invention can be prepared according to methods known in the literature using suitable starting materials, for example, analogous to the methods described by B.F. Cain et al. inJ Med. Chem. 20(8) (1977) 987-996.
In general, the compounds of the invention can be prepared e.g. according to the following reaction scheme 1 :
Scheme 1

II πi

wherein A, Ra, Rb, Rl5 R2, R3, R^, Q, Y, t and u are as defined above.
Scheme 1 represents a conventional acid-catalysed coupling of the chloro-compound of formula II with a substituted aromatic amine of formula m. The reaction is carried out at room or elevated temperature in a suitable solvent, e.g. an alcohol such as methanol, to obtain the compound of formula I, which is then isolated from the reaction mixture in a usual manner.
The starting material π can be prepared e.g. according to scheme 2:
Scheme 2

wherein A, Ra, Rb, R2 and t are as defined above.
In scheme 2 compound IV is reacted with thionyl chloride in the presence of a small amount of DMF, to obtain the chlorinated reactant II. This reaction is carried out at room or an elevated temperature.
Starting material IV can be prepared according to methods described in the literature (see e.g. J. Heterocyclic Chem. 34 (1997) 315-320 and references cited therein).
For example, starting material IV can be prepared according to scheme 3:
Scheme 3

wherein Ra, Rb, R2 and t are as defined above.
In scheme 3, an aniline is reacted with an ethyl β-ketoacetate in the presence of a small amount of αr -toluenesulfonic acid in toluene, to obtain an imine, which is then cyclised by refluxing in phenyl ether. Both starting materials, aniline and ethyl β-ketoacetate, are commercially available as a variety of compounds.
Starting compounds HI can be prepared according to methods described in the literature (see e.g. Tetrahedon Lett. 38(23) (1997) 4091-4094, and J. Org. Chem. 60 (1995) 4177- 4183).
For example, a substituted aromatic NRi -amine HI can be prepared starting from the corresponding nitro compound V, which is reduced with an appropriate agent, e.g. hydrazine, in a suitable solvent, e.g. ethanol, and optionally alkylated with R\ in a manner known in the art, when R\ being (C1-C6)alkyl is desired.
Starting material V can be prepared e.g. according to scheme 4:
Scheme 4

VI V
wherein X is a halogen, such as chlorine, and Y, R , R4 and u are as described above.
hi the scheme 4 a compound VI is coupled with^αra-halogen-nitrobenzene in the presence of potassium carbonate in DMSO, to obtain a reactant V. This reaction is carried out at room or an elevated temperature.
It is obvious for a skilled person that, in the above reactions, any starting material or intermediate can be protected, if necessary, in a manner well known in the chemical field. Any protected functionality can subsequently be deprotected in a manner known in the art.
The above disclosed synthetic routes are meant to illustrate the preparation of the compounds of the invention and the preparation is by no means limited thereto, i.e. other synthetic methods that are within the general knowledge of a skilled person are also possible.
The compounds of the invention maybe converted, if desired, into their pharmaceutically acceptable salt or ester forms using methods well known in the art.
The present invention will be explained in more detail by the following examples. The examples are meant only for illustrating purposes and do not limit the scope of the invention defined in claims.
EXAMPLE 1
[4-(3,4-Dimethyl-piperasin-l-yl)-phenyl]-(253,7,8-tetramethyl-quinolin-4-yl)-amine
Step l
7.96 g (50 mmol) of l-chloro-4-nitrobenzene, 5.01 g (50 mmol) of 2-methylpiperazine and 6.91 g (50 mmol) of potassium carbonate were mixed in 50 ml of DMSO and heated at 80°C overnight. The reaction mixture was poured into 200 ml of water and extracted with dichloromethane. The organic extract was dried over sodium sulphate and evaporated. The reaction product was purified on a silica gel column (eluent methanol: dichloromethane 1:4) to obtain 6.42 g (58 %) of 3-methyl-l-(4-nitro-phenyl)- piperazine.
Step 2
6.42 g (29 mmol) of 3-methyl-l-(4-nitro-phenyl)-piperazine and 1.07 g (2.9 mmol) of tetrabutylammonium iodide were mixed with 100 ml of toluene and 25 ml of 50 % NaOH (aq.) solution at 60°C. 2.0 ml (32 mmol) of methyl iodide were added dropwise to the reaction mixture after 30 min, and heating was continued for 1.5 h. The reaction mixture was then cooled, filtered, and the precipitate was washed with toluene. The filtrate was then washed with water and the organic phase was separated. The water phase was additionally extracted with toluene and the combined organic phases were dried over sodium sulphate and evaporated to give 4.89 g (72 %) of l,2-dimethyl-4-(4-nitro-phenyl)- piperazine.
Step 3
4.85 g (21 mmol) of l,2-dimethyl-4-(4-nitro-phenyl)-piperazine was dissolved in 30 ml of ethanol, 8.0 ml (16.5 mmol) of hydrazinium hydroxide and 470 mg of 10 % palladium on activated carbon were added and the reaction mixture was refluxed for 1 h. After cooling the reaction mixture was filtered and the solvent was evaporated. The residue was dissolved in water, extracted with dichloromethane, dried over sodium sulphate and evaporated to provide 4.39 g of 4-(3 ,4-dimethyl-piperazin- l-yl)-phenylamine in a quantitative yield.
Step 4
2.45 ml (20 mmol) of 2,3-dimethylaniline, 2.85 ml (20 mmol) of ethyl 2- methylacetoacetate, 190 mg (1.0 mmol) of r -toluenesulphonic acid and 25 ml of toluene were placed into a round bottom flask equipped with a condenser and a Dean- Stark trap. The reaction mixture was refluxed overnight, cooled, and stripped on a rotary
evaporator. The resulting crude imine was dissolved in 10 ml of phenyl ether and refluxed for lh. 50 ml of diethyl ether were added to the cooled reaction mixture and the precipitate formed was filtered and washed repeatedly with diethyl ether to yield 3.11 g (77 %) of 2,3,7,8-tetramethylquinolin-4-ol.
Step 5
3.11 g (15,5 mmol) of 2,3,7,8-tetramethylquinolin-4-ol, 15 ml of thionyl chloride and 0.75 ml of NN-dimethylformamide were stirred at 80 °C for 1 h. After cooling, the reaction mixture was evaporated, the residue was dissolved in dichloromethane and poured into cold aqueous ammonia (25 %). The ammonia solution was extracted with dichloromethane, the organic phase was washed with 2M ammonia solution, dried over sodium sulphate and evaporated to obtain 3.33 g (98 %) of 4-chloro-2,3,7,8- tetramethylquinoline.
Step 6
0.214 g (1.0 mmol) of 4-chloro-2,3,7,8-tetramethylquinoline and 0.103 g (0.50 mmol) of 4-(3 ,4-dimethyl-piperazin- l-yl)-phenylamine, 0.75 ml of methanol and a few drops of concentrated hydrochloric acid were mixed and heated in a microwave oven reactor (Smith Creator™, Personal Chemistry) at 130°C for 300 s. The reaction mixture was cooled, dissolved in water, made alkaline with aqueous ΝaOH and extracted with dichloromethane. The organic extract was dried over sodium sulphate and evaporated. The crude product was purified on silica gel column (eluent methanol: dichloromethane, 1:9) to give 0.174 g (90 %) of the title compound.
1H ΝMR (CDC13, 500 MHz): δ 7.55 (1H, m), 7.15 (1H, m), 6.80 (2H, m), 6.62 (2H, m), 5.73 (1H, br s), 3.34 (2H, m), 2.89 (2H, m), 2.76 (3H, s), 2.72 (3H, s), 2.50 (2H, m), 2.45 (3H, s), 2.34 (3H, s), 2.29 (1H, m), 2.22 (3H, s), 1.13 (3H, d, J= 6.3 Hz); MS (ESf): m/z 389 (M+), 195 (M2+).
Using the same method the following compounds were prepared:
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(3-ethyl-2-methyl-quinolin-4-yl)-amine
1H ΝMR (CDC13, 500 MHz): δ 7.97 (1H, m), 7.73 (1H, m), 7.55 (1H, m), 7.26 (1H, m), 6.80 (2H, m), 6.67 (2H, m), 5.71 (1H, br s), 3.35 (2H, m), 2.88 (2H, m), 2.80 (2H, q, J=
7.7 Hz), 2.77 (3H, s), 2.45 (2H, m) 2.33 (3H, s), 2.25 (IH. m), 1.17 (3H, t, J= 7.7 Hz),
1.12 (3H, d, J= 6.2 Hz);
MS (ESf): m/z 375 (M+), 188 (M2+).
(3-Ethyl-2-methyl-quinolin-4-yl)-[4-(3,4,5-trimethyI-piperazin-l-yl)-phenyl]-amine 1H NMR (CDC13, 500 MHz): δ 7.96 (IH, m), 7.71 (IH, m), 7.55 (IH, m), 7.25 (IH, m), 6.79 (2H, m), 6.67 (2H, m), 5.70 (IH, br s), 3.33 (2H, m), 2.80 (2H, q, J= 1.6 Hz), 2.77 (3H, s), 2.52 (2H, m) 2.37 (2H, m), 2.31 (3H, s), 1.17 (3H, t, J= 1.6 Hz), 1.15 (6H, d, J= 6.2 Hz); MS (ES ): m/z 389 (M+), 195 (M2+).
(3-I§opropyl-2-methyl-quinolin-4-yl)-[4-(3,4s5-trimethyI-piperasin-l-yl)-phenyl]- amine
1H NMR (CDC13, 500 MHz): δ 7.96 (IH, m), 7.75 (IH, m), 7.56 (IH, m), 7.26 (IH, m), 6.81 (2H, m), 6.63 (2H, m), 5.73 (IH, br s), 3.63 (IH, m), 3.35 (2H, m), 2.83 (3H, s), 2.54 (2H, m), 2.40 (2H, m), 2.34 (3H, s), 1.40 (6H, d, J= 7.2 Hz), 1.17 (6H, d, J= 6.0 Hz); MS (ES ): m/z 403 (M+), 202 (M2+).
[4-(3,4-DimethyI-piperazin-l-yl)-phenyl]-(3-ethyl-2,8-dimethyl-quinolin-4-yl)-amine
1H NMR (CDC13, 500 MHz): δ 7.60 (IH, m), 7.40 (IH, m), 7.15 (IH, m), 6.78 (2H, m), 6.62 (2H, m), 5.62 (IH, br s), 3.33 (2H, m), 2.87 (2H, m), 2.79 (3H, s), 2.78 (2H, q, J= 1.1 Hz), 2.78 (3H, s), 2.46 (2H, m), 2.34 (3H, s), 2.28 (IH, m), 1.15 (3H, t, J= 7.7 Hz), 1.12 (3H, d, J= 6.3 Hz);
MS (ES ): m/z 389 (M+), 195 (M2+).
[4-(3,4-Dimethyl-piperasin-l-yl)-phenyl]-(2-phenyl-quinoIm-4-yl)-aιnme
1H NMR (CDC13, 500 MHz): δ 8.14 (IH, m), 7.94 (3H, m), 7.68 (IH, m), 7.43 (4H, m), 7.27 (IH, m), 7.20 (IH, s), 7.01 (2H, m), 3.53 (2H, m), 2.99 (2H, m), 2.63 (IH, m), 2.49 (IH, m), 2.39 (3H, s), 2.34 (IH, m), 1.19 (3H, d, J= 6.4 Hz); MS (ES ): m/z 409 (M+), 205 (M2+).
[4-(3,4-Dimethyl-piperazm-l-yl)-phenyl]-(2,35557-tetramethyl-quinoliιι-4-yl)-amme
1H MR (CDCI3, 500 MHz): δ 7.69 (IH, s), 7.00 (IH, s), 6.81 (2H, m), 6.50 (2H, m), 5.85 (IH, br s) 3.33 (2H, m), 2.87 (2H, m), 2.72 (3H, s), 2.68 (3H, s), 2.48 (2H, m), 2.44
(3H, s), 2.34 (3H, s), 2.27 (IH, m), 2.18 (3H, s), 1.12 (3H, d, J= 6.3 Hz); MS (ES ): m/z 389 (M+), 195 (M2+).
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(7-ethyl-2,3-dimethyI-qumolm-4-yI)-amme
1H NMR (CDC13, 500 MHz): δ 7.86 (IH, s), 7.67 (IH, m), 7.19 (IH, m), 6.83 (2H, m), 6.71 (2H, m), 5.97 (IH, br s) 3.38 (2H, m), 2.90 (2H, m), 2.80 (2H, q, J= 7.6 Hz), 2.72 (3H, s), 2.47 (2H, m), 2.35 (3H, s), 2.28 (IH, m), 2.21 (3H, s), 1.31 (3H, t, J= 1.6 Hz), 1.13 (3H, d, J= 6.1 Hz); MS (ESI*): m/z 389 (M ), 195 (M2+).
[4-(354-Dimethyl-piperazin-l-yl)-phenyl]-(2,35558-tetramethyl-quinolin-4-yl)-amme 1H NMR (CDCI3, 500 MHz): δ 7.31 (IH, m), 7.04 (IH, m), 6.80 (2H, m), 6.47 (2H, m), 5.77 (IH, br s) 3.32 (2H, m), 2.86 (2H, m), 2.74 (3H, s), 2.70 (6H, s), 2.45 (2H, m), 2.33 (3H, s), 2.27 (IH, m), 2.21 (3H, s), 1.12 (3H, d, J= 6.2 Hz); MS (ESI*): m/z 389 (M+), 195 (M2+).
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(2,3,8-trimethyl-quinolin-4-yl)-amine 1H NMR (CDCI3, 500 MHz): δ 7.65 (IH, m), 7.43 (IH, m), 7.22 (IH, m), 6.81 (2H, m), 6.63 (2H, m), 5.76 (IH, br s) 3.34 (2H, m), 2.87 (2H, m), 2.80 (3H, s), 2.73 (3H, s), 2.45 (2H, m), 2.33 (3H, s), 2.26 (IH, m), 2.23 (3H, s), 1.12 (3H, d, J= 6.2 Hz); MS (ESI4): m z 375 (M4), 188 (M2+).
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(6-fluoro-2,3-dimethyl-quinolin-4-yl)-amine 1H NMR (CDCI3, 500 MHz): δ 7.97 (IH, m), 7.35 (2H, m), 6.82 (2H, m), 6.64 (2H, m), 5.68 (IH, br s), 3.36 (2H, m), 2.88 (2H, m), 2.70 (3H, s), 2.47 (2H, m), 2.34 (3H, s), 2.28 (IH, m) 2.26 (3H, s), 1.13 (3H, d, J= 6.2 Hz); MS (ESI4): m/z 379 (M+), 190 (M2+).
(2,3-Dimethyl-6-nitro-quinolin-4-yl)-[4-(354-dimethyl-piperasin-l-yl)-phenyl]-amine 1H NMR (CDCI3, 500 MHz): δ 8.76 (IH, s), 8.32 (IH, m), 8.03 (IH, m), 6.85 (2H, m), 6.76 (2H, m), 6.06 (IH, br s), 3.40 (2H, m), 2.90 (2H, m), 2.74 (3H, s) 2.49 (2H, m), 2.35 (3H, s), 2.29 (IH, m), 2.24 (3H, s), 1.14 (3H, d, J= 6.2 Hz); MS (ESI4): m/z 406 (M+), 204 (M2+).
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(6-ethyl-2,3-dimethyl-quinolin-4-yl)-amine
1H NMR (CDCI3, 500 MHz): δ 8.01 (IH, m), 7.54 (IH, s), 7.46 (IH, m), 6.84 (2H, m), 6.73 (2H, m), 6.06 (IH, br s), 3.37 (2H, m), 2.91 (2H, m), 2.72 (3H, s) 2.70 (2H, q, J= 7.5 Hz), 2.50 (2H, m), 2.36 (3H, s), 2.31 (IH, m), 2.20 (3H, s), 1.19 (3H, t, J= 7.5 Hz), 1.14 (3H, d, J= 6.4 Hz); MS (ESI4): m/z 389 (M4), 195 (M2+).
EXAMPLE 2
(2-Methyl-3-phenyl-quinolin-4-yl)-[4-(3,4,5-trimethyl-piperazin-l-yl)-phenyl]-amine (Compound A)
Following the procedure outlined in Example 1, but substituting 2,6-dimethylpiperazine for 2-methylpiperazine in step 1, plus ethyl 2-phenylacetoacetate for ethyl 2- methylacetoacetate and aniline for 2,3-dimethylaniline in step 4, afforded the title compound with 21 % overall yield.
1H NMR (CDCI3, 500 MHz): δ 7.99 (IH, m), 7.63 (IH, m), 7.57 (IH, m), 7.47 (2H, m), 7.41 (IH, m), 7.28 (2H, m), 7.19 (IH, m), 6.74 (4H, m), 5.54 (IH, br s), 3.35 (2H, m), 2.51 (2H, m), 2.43 (3H, s), 2.36 (2H, m), 2.31 (3H, s), 1.15 (6H, d, J = 6.2 Hz); MS (ES ): m/z 437 (M+), 219 (M2+).
Using the same method the following compounds were prepared:
(2-Methyl-3-phenyI-quinoIin-4-yl)-[4-(4-methyl-piperazin-l-yl)-phenyl]-amine
1H NMR (CDCI3, 500 MHz): δ 8.00 (IH, m), 7.64 (IH, m), 7.58 (IH, m), 7.47 (2H, m), 7.41 (IH, m), 7.27 (2H, m), 7.20 (IH, m), 6.75 (4H, m), 5.55 (IH, br s), 3.13 (4H, m), 2.56 (4H, m), 2.44 (3H, s), 2.34 (3H, s); MS (ESI4): m/z 409 (M+), 205 (M2+)
[4-(3,4-Dimethyl-pipera22in-l-yl)-phenyl]-(7-methosy-2-methyl-3-phenyl-quinolin-4- yl)-amine MS (ESI4): m/z 453 (M+), 227 (M2+).
EXAMPLE 3
[4-(3,4-Dimethyl-piperazin-l-yl)-pheuyI]-(2-methyl-3-phenyl-quinolin-4-yl)-amine (Compound B)
Following the procedure outlined in Example 2, but substituting 2-methylpiperazine for 2,6-dimethylpiperazine, afforded the title compound.
1H NMR (CDC13, 500 MHz): δ 8.02 (IH, m), 7.60 (2H, m), 7.48 (2H, m), 7.42 (IH, m), 7.28 (2H, m), 7.20 (IH, m), 6.76 (4H, m), 5.59 (IH, br s), 3.39 (2H, m), 2.89 (2H, m), 2.50 (IH, m), 2.44 (3H, s), 2.42 (IH, m), 2.35 (3H, s), 2.25 (IH, m), 1.13 (3H, d, J = 6.3 Hz); MS MS (ESI4"): m/z 423 (M+), 212 (M2+).
EXAMPLE 4
(S)-[4-(3?4-Dimethyl-piperazin-l-yl)-phenyl]-(2-methyl-3-plιenyl-quinolin-4-yl)- amine
Following the procedure outlined in Example 3, but using chiral starting material, (S)-(+)- 2-methylpiperazine, afforded the title compound.
1H NMR (CDCI3, 500 MHz): δ 8.02 (IH, m), 7.61 (2H, m), 7.48 (2H, m), 7.42 (IH, m),
7.28 (2H, m), 7.20 (IH, m), 6.75 (4H, m), 5.59 (IH, br s), 3.39 (2H, m), 2.89 (2H, m), 2.50 (IH, m), 2.44 (3H, s), 2.42 (IH, m), 2.34 (3H, s), 2.25 (IH, m), 1.13 (3H, d, J = 6.2
Hz);
MS (ES ): m/z 423 (M4), 212 (M2+).
EXAMPLE 5
(R)-[4-(354-Dimethyl-piperazin-l-yl)-phenyl]-(2-methyl-3-pIιenyl-quinolin-4-yl)- amine
Following the procedure outlined in Example 3, but using chiral starting material, (R)-(-)- 2-methylpiperazine, afforded the title compound.
1H NMR (CDCI3, 500 MHz): δ 8.01 (IH, m), 7.61 (2H, m), 7.48 (2H, m), 7.41 (IH, m),
7.29 (2H, m), 7.20 (IH, m), 6.75 (4H, m), 5.57 (IH, br s), 3.38 (2H, m), 2.88 (2H, m), 2.49 (2H, m), 2.44 (3H,s), 2.41 (IH, m), 2.34 (3H, s), 2.25 (IH, m), 1.12 (3H, d, J = 6.3
Hz);
MS (ESI4): m/z 423 (M4), 212 (M2+).
EXAMPLE 6
[4-(3,4-DimethyI-piperazin-l-yI)-phenyI]-(2,3-dimethyl-quinoIin-4-yI)-amine
Following the procedure outlined in Example 1, but substituting aniline for 2,3- dimethylaniline in step 4, afforded the title compound with 9 % overall yield.
1H NMR (CDC13, 500 MHz): δ 7.98 (IH, m), 7.78 (IH, m), 7.57 (IH, m), 7.33 (IH, m), 6.82 (2H, m), 6.67 (2H, m), 5.82 (IH, br s), 3.36 (2H, m), 2.87 (2H, m), 2.71 (3H, s), 2.45 (2H, m), 2.33 (3H, s), 2.26 (IH, m), 2.24 (3H, s), 1.12 (3H, d, J = 6.2 Hz); MS (ESI4): m/z 361 (M+), 181 (M2+).
EXAMPLE 7
(3-Bromoquinolin-4-yl)-[4-(354-dimethylpiperazm-l-yl)-phenyI]-amine
Following the procedure outlined in Example 1, but substituting 3-bromo-4- chloroquinoline for 4-chloro-2,3,7,8-tetramethylquinoline in step 6, afforded the title compound with 42 % overall yield.
1H NMR (CDCI3, 500 MHz): δ 8.84 (IH, s), 8.00 (IH, m), 7.61 (2H, m), 7.26 (H, m), 6.90 (2H, m), 6.85 (2H, m), 6.50 (IH, br s), 3.51 (2H, m), 3.28 (2H, m), 3.01 (IH, m), 2.82 (2H, m), 2.61 (3H, m), 1.39 (3H, m);
MS (ESI4): m/z 411, 413 (M4), 206, 207 (M2+).
EXAMPLE 8
(5-Chloro-2,3-dimethylquinolin-4-yl)-[4-(3,4-dimethylpiperazin-l-yl)-phenyl]-amine
Following the procedure outlined in Example 1, but substituting 3-chloroaniline for 2,3- dimethylaniline, afforded a mixture of (5-chloro-2,3-dimethylquinolin-4-yl)-[4-(3,4- dimethylpiperazin-l-yl)-phenyl]-amine and its 7-chloro isomer. Two isomers were chromatographically separated by silica gel column (eluent methanol: dichloromethane, 1:9) to obtain the title compound with 7 % overall yield.
1H NMR (CDCI3, 500 MHz): δ 7.92 (IH, m), 7.44 (2H, m), 6.85 (2H, ), 6.63 (2H, m), 3.37 (2H, m), 2.89 (2H, m), 2.67 (3H, s), 2.47 (2H, m), 2.34 (3H, s), 2.27 (IH, m), 2.11 (3H, s), 1.13 (3H, d, J = 6.1 Hz); MS (ESI4): m/z 395 (M4), 198 (M24).
Using the same method the following compounds were prepared:
[4-(3,4-DimethyI-piperazin-l-yI)-phenyI]-(5-ethyI-2,3-dimethyl-quinoIin-4-yl)-amine
1H NMR (CDCI3, 500 MHz): δ 7.89 (IH, m), 7.51 (IH, m), 7.31 (IH, m), 6.82 (2H, m), 6.50 (2H, m), 5.84 (IH, br s) 3.34 (2H, m), 3.09 (2H, q, J= 7.5 Hz), 2.87 (2H, m), 2.70 (3H, s), 2.46 (2H, m), 2.34 (3H, s), 2.27 (IH, m), 2.22 (3H, s), 1.27 (3H, t, J= 7.5 Hz), l.l l (3H, d, J= 6.2 Hz); MS (ESI4): m/z 389 (M+), 195 (M2+).
[4-(3,4-DimethyI-piperazin-l-yl)-phenyI]-(2,3,5-trimethyl-quinoIin-4-yI)-amine
!H NMR (CDCI3, 500 MHz): δ 7.87 (IH, m), 7.45 (IH, m), 7.16 (IH, m), 6.82 (2H, m), 6.50 (2H, m), 5.83 (IH, br s), 3.33 (2H, m), 2.87 (2H, m), 2.76 (3H, s), 2.69 (3H, s), 2.46 (2H, m), 2.34 (3H, s), 2.27 (IH, m), 2.21 (3H, s), 1.12 (3H, d, J= 6.2 Hz); MS (ESf ): m/z 375 (M+), 188 (M24).
EXAMPLE 9
(7-Chloro-2,3-dimethyl-quinolin-4-yI)-[4-(3,4-dimethyI-piperazin-l-yl)-phenyl]- amine
The title compound was produced in 4% overall yield from the chromatographic separation of the corresponding 5-chloro and 7-chloro isomer mixture as described in Example 8.
1H NMR (CDCI3, 500 MHz): δ 7.97 (IH, m), 7.68 (IH, m), 7.25 (IH, m), 6.82 (2H, m), 6.67 (2H, m), 5.79 (IH, br s), 3.36 (2H, m), 2.89 (2H, m), 2.70 (3H, s), 2.48 (2H, m), 2.35 (3H, s), 2.29 (IH, m), 2.24 (3H, s), 1.13 (3H, d, J = 6.3 Hz); MS (ESI4): m/z 395 (M+), 198 (M2+).
Using the same method the following compound was prepared:
[4-(3,4-Dimethyl-pipera2in-l-yl)-phenyl]-(25357-trimethyl-quinoIin-4-yl)-amine 1H NMR (CDCI3, 500 MHz): δ 7.81 (IH, s), 7.65 (IH, m), 7.15 (IH, m), 6.83 (2H, m), 6.69 (2H, m), 5.92 (IH, br s), 3.36 (2H, m), 2.87 (2H, m), 2.70 (3H, s), 2.49 (3H, s), 2.46 (2H, m), 2.34 (3H, s), 2.27 (IH, m), 2.21 (3H, s), 1.13 (3H, d, J= 6.2 Hz); MS (ES ): m/z 375 (M4), 188 (M2+).
EXAMPLE 10
N',4'-[4-(3,4-DimethyI-piperazin-l-yl)-phenyI]-2,3-dimethyl-quinoline-4,5-diamine
Following the procedure outlined in Example 1, but substituting 3-nitroaniline for 2,3- dimethylaniline, afforded a mixture of [4-(3,4-dimethyl-piperazin-l-yι)-phenyl]-(2,3- dimethyl-7-nitroquinolin-4-yl)-amine (70 %) and [4-(3 ,4-dimethyl-piperazin- 1-yl)- phenyl]-(2,3-dimethyl-5-nitroquinolin-4-yl)-amine (30 %). This mixture was treated with hydrazinium hydroxide as described in Example 1, step 3, to obtain a mixture of N'4'-[4- (3,4-d methyl-piperazin-l-yl)-phenyl]-2,3-dimethyl-quinoline-4,5-diamine and its 7- amino isomer. Two isomers were chromatographically separated by silica gel column (eluent: triethylamine methanol: dichloromethane, 1 :9:90) to obtain the title compound with 6 % yield.
1H NMR (CDCl3/CD3OD, 500 MHz): δ 7.65 (IH, m), 7.26 (IH, m), 7.18 (2H, m), 7.03 (2H, m), 6.87 (IH, m), 3.83 (IH, m), 3.60 (5H, m), 2.94 (3H, s), 2.72 (3H, s), 2.70 (IH, m), 2.06 (3H, s), 1.57 (3H, d, J = 5.8 Hz); MS (ESI4): m/z 376 (M+), 189 (M2+).
Using the same method the following compound was prepared:
N',4'-[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-2,3-dimethyl-quinoline-4,6-diamine
MS (ESf): m/z 376 (M+), 189 (M2+).
EXAMPLE 11
Ns 54s-[4-(354-Dimethyl-piperasin-l-yl)-phenyl]-2,3-dimethyl-quinoline-4,7-diamine
The title compound was produced in 6% overall yield from the chromatographic separation of the corresponding 5-amino and 7-amino isomer mixture as described in Example 10.
1H NMR (CDCl3/CD3OD, 500 MHz): δ 7.52 (IH, m), 7.12 (IH, m), 7.00 (2H, m), 6.87 (2H, m), 6.69 (IH, m), 3.59 (2H, m), 3.50 (IH, m), 3.23 (3H, m), 2.90 (3H, s), 2.81 (IH, m), 2.66 (3H, s), 2.01 (3H, s), 1.55 (3H, m); MS (ESf): m/z 376 (M4), 189 (M24).
EXAMPLE 12
{4-[4-(3,4-DimethyI-piperazin-l-yl)-phenylamino]-quinolin-3-yl}-methanoI (Compound C)
4.5 ml (50 mmol) of aniline, 10.1 ml (50 mmol) of diethyl ethoxymethylene malonate and 30 ml of pyridine were mixed and refluxed for 3 h. The solvent was evaporated, and the remaining solid was treated with phenylether according to the method described in Example 1, step 4, to give 4-hydroxyquinoline-3-carboxylic acid ethyl ester. Following the procedure outlined in Example 1, but substituting 4-hydroxyquinoline-3-carboxylic acid ethyl ester for 2,3,7,8-tetramethylquinolin-4-ol and additionally substituting in step 6 ^-toluenesulphonic acid for concentrated hydrochloric acid and aqueous NaHCO3 for aqueous NaOH, afforded the 4-[4-(3,4-dimethylpiperazin- 1 -yl)-phenylamino]-quinoline- 3-carboxylic acid ethyl ester with 17 % overall yield. The ester (234 mg, 0.58 mmol) was dissolved in 3 ml of THF, added dropwise to a stirred suspension of 117 mg (3.1 mmol) of lithium aluminium hydride in 2 ml of THF at 0°C under an inert atmosphere. The reaction mixture was stirred for 30 min at 0°C and then for another lh at room temperature before it was quenched with 224 μl (12.4 mmol) of water and filtered. The precipitate was washed with THF, the combined filtrate was evaporated and the crude product was purified on a silica gel column (gradient elution methanol: dichloromethane from 5:95 to 1:4), to give 90 mg (yield 40 %, overall yield 7 %) of the title compound.
1H NMR (CDC13, 500 MHz): δ 8.46 (IH, s), 7.98 (IH, m), 7.70 (IH, m), 7.57 (IH, m), 7.44 (IH, br s), 7.22 (lH,m), 6.82 (4H, m), 4.78 (2H, s), 3.43 (IH, m), 3.37 (IH, m), 2.89 (2H, m), 2.52 (IH, m), 2.44 (IH, m), 2.35 (3H, s), 2.29 (IH, m), 1.14 (3H, d, J= 6.2 Hz); MS (ESf): m/z 363 (M+), 182 (M2+).
Using the same method the following compounds were prepared:
{8-Methyl-4-[4-(4-methyI-piperaain-l-yl)-phenylamino]-quinoIin-3-yl}-methanol MS (ESf): m z 363 (M+), 182 (M2+).
{4-[4-(3,4-Dimethyl-piperasin-l-yl)-phenyIamino]-7-methyl-quinolin-3-yl}-methanol
1H NMR (CDCI3, 500 MHz): δ 8.38 (IH, s), 7.74 (IH, s), 7.63 (IH, br s), 7.53 (IH, m), 7.01 (IH, m), 6.84 (4H, m), 4.77 (2H, s), 3.41 (2H, m), 2.90 (2H, m), 2.49 (2H, m), 2.45 (3H, s), 2.35 (3H, s), 2.29 (IH, m), 1.14 (3H, d, J= 6.2 Hz); MS (ESf ): m/z 377 (M+), 189 (M2+).
{4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenylamino]-6-fluoro-quinolin-3-yl}-methanol
1H NMR (CDCI3, 500 MHz): δ 8.47 (IH, s), 7.97 (IH, m), 7.31 (2H, m), 7.29 (IH, br s),
6.81 (4H, m), 4.79 (2H, s), 3.39 (2H, m), 2.89 (2H, m), 2.48 (2H, m) 2.34 (3H, s), 2.29 (IH, m), 1.13 (3H, d, J= 6.2 Hz); MS (ESf): m/z 381 (M4), 191 (M2+).
{7-ChIoro-4-[4-(3,4-dimethyI-piperazin-l-yl)-phenylamino]-quinoIin-3-yl}-methanol
1H NMR (CDC13, 500 MHz): δ 8.48 (IH, s), 7.95 (IH, m), 7.60 (IH, m), 7.41 (IH, br s), 7.13 (IH, m), 6.82 (4H, m), 4.80 (2H, s), 3.40 (2H, m), 2.89 (2H, m), 2.48 (2H, m) 2.34 (3H, s), 2.29 (IH, m), 1.14 (3H, d, J= 6.2 Hz); MS (ESf ): m/z 397 (M4), 199 (M2+).
{6,7-Dichloro-4-[4-(3s4-dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}- methanol
1H NMR (CDC13, 500 MHz): δ 8.47 (IH, s), 8.05 (IH, s), 7.72 (IH, s), 7.40 (IH, br s),
6.82 (4H, m), 4.78 (2H, s), 3.41 (2H, m), 2.87 (2H, m), 2.47 (2H, m), 2.33 (3H, s), 2.27 (lH, m), 1.13 (3H, d, J= 6.3 Hz);
MS (ESf): m/z 431 (M4), 216 (M2+).
EXAMPLE 13
(S)-{4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenyIamino]-quinoIin-3-yI}-methanol
Following the procedure outlined in Example 12, but using chiral starting material, (S)- (+)-2-methylpiperazine, afforded the title compound.
1H NMR (CDCI3, 500 MHz): δ 8.49 (IH, s), 7.99 (IH, m), 7.71 (IH, m), 7.57 (IH, m), 7.36 (IH, br s), 7.22 (lH,m), 6.82 (4H, m), 4.78 (2H, s), 3.42 (IH, m), 3.36 (IH, m), 2.88 (2H, m), 2.52 (IH, m), 2.44 (IH, m), 2.34 (3H, s), 2.28 (IH, m), 1.13 (3H, d, J= 6.2 Hz); MS (ESf): m/z 363 (M4), 182 (M2+).
EXAMPLE 14
(R)-{4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}-methanol
Following the procedure outlined in Example 12, but using chiral starting material, (R)-(- )-2-methylρiperazine, afforded the title compound.
1H NMR (CDCI3, 500 MHz): δ 8.48 (IH, s), 7.99 (IH, m), 7.72 (IH, m), 7.57 (IH, m), 7.33 (IH, br s), 7.23 (lH,m), 6.81 (4H, m), 4.78 (2H, s), 3.42 (IH, m), 3.36 (IH, m), 2.87 (2H, m), 2.51 (IH, m), 2.42 (IH, m), 2.33 (3H, s), 2.26 (IH, m), 1.12 (3H, d, J= 6.3 Hz); MS (ESf): m/z 363 (M+), 182 (M2+).
EXAMPLE 15
{4-[4-(3,4-Dimethyl-piperazin-l-yI)-phenylamino]-2-methyl-quinolin-3-yl}-methanoI
Following the procedure outlined in Example 12, but substituting 2-(l-ethoxyethylidene)- malonic acid diethyl ester (prepared according to e.g. patents US 5,041,619 and US 4,058,553) for diethyl ethoxymethylene malonate, afforded 4-[4-(3,4-dimethyl-piperazin- l-yl)-phenylamino]-2-methyl-quinoline-3-carboxylic acid ethyl ester with 26 % yield. Reduction of the ester group as described in Example 12 afforded the title compound in 62 % yield (overall yield 16 %).
1H NMR (CDCI3, 500 MHz): δ 7.94 (IH, m), 7.65 (IH, m), 7.54 (IH, m), 7.35 (IH, br s), 7.17(1H, m), 6.79 (4H, m), 4.85 (2H, s), 3.41 (IH, m), 3.35 (IH, m), 2.87 (2H, m), 2.65 (3H, s), 2.50 (IH, m), 2.42 (IH, m), 2.33, (3H, s), 2.26 (IH, m), 1.12 (3H, d, J= 6.4 Hz); MS (ESf): m/z 377 (M4), 189 (M2+).
EXAMPLE 16
(3-Aminomethyl-8-methylquinoIin-4-yl)-[4-(4-methylpiperazin-l-yl)-phenyl]-amine
Following the procedure outlined in Example 12, but substituting ø-toluidine for aniline, ethyl(ethoxymethylene)-cyanoacetate for diethyl ethoxymethylene malonate and 1- methylpiperazine for 2-methylpiperazine, afforded 8-methyl-4-[4-(4-methyl-piperazin-l- yl)-phenylamino]-quinoline-3-carbonitrile with 18 % yield. Reduction of the nitrile group according to the method described in Example 12 afforded the title compound with 21 % yield (overall yield 4 %).
MS (ESf): m/z 362 (M4), 182 (M24).
EXAMPLE 17
(3-Ethyl-2,8-dimethylquinolin-4-yl)-[4-(4-methyl[l,4]piperazin-l-yl)-phenyl]-amine
Following the procedure outlined in Example 1, but substituting homopiperazine for 2- methylpiperazine in step 1, plus ethyl 2-ethylacetoacetate for ethyl 2-methylacetoacetate and o-toluidine for 2,3-dimethylaniline in step 4, afforded the title compound with 9 % overall yield.
1H NMR (CDC13, 500 MHz): δ 7.60 (IH, m), 7.38 (IH, m), 7.13 (IH, m), 6.66 (2H, m), 6.54 (2H, m), 5.62 (IH, br s), 3.52 (2H, m), 3.40 (2H, m), 2.78 (3H, s), 2.77 (3H, s), 2.76 (2H, m), 2.71 (2H, m), 2.61 (2H, m), 2.40 (3H, s), 2.01 (2H, m), 1.16 (3H, t, J= 7.7 Hz); MS (ESf): m/z 389 (M4), 195 (M24).
Using the same method the following compound was prepared:
[4-(4-MethyI-[ls4]diazepan-l-yl)-phenyl]-(253,8-trimethyl-quinolin-4-yl)-amine
1H NMR (CDCI3, 500 MHz): δ 7.65 (IH, m), 7.41 (IH, m), 7.21 (IH, m), 6.66 (2H, m),
6.57 (2H, m), 5.74 (IH, br s), 3.51 (2H, m), 3.42 (2H, m), 2.79 (3H, s), 2.72 (3H, s), 2.69 (2H, m), 2.58 (2H, m), 2.38 (3H, s), 2.22 (3H, s), 1.99 (2H, m);
MS (ESf ): m/z 375 (M4), 188 (M2+).
EXAMPLE 18
[4-(4-MethyI-[l,4]diazepan-l-yl)-phenyI]-(2-methyl-3-phenyl-quinoIin-4-yl)-amine
Following the procedure outlined in Example 1, but substituting homopiperazine for 2- methylpiperazine in step 1, plus ethyl 2-phenylacetoacetate for ethyl 2-methylacetoacetate and aniline for 2,3-dimethylaniline in step 4, afforded the title compound with 9 % overall yield.
1H NMR (CDCI3, 500 MHz): δ 8.03 (IH, m), 7.72 (IH, m), 7.62 (IH, m), 7.55 (2H, m), 7.48 (IH, m), 7.38 (2H, m), 7.24 (IH, m)5 6.82 (2H, m), 6.61 (2H, m), 5.61 (IH, br s),
3.58 (2H, m), 3.50 (2H, m), 2.75 (2H3 m), 2.64 (2H, m), 2.49 (3H, s), 2.45 (3H, s), 2.05 (2H, m); MS (ESf): m/z 423 (M4), 212 (M2+).
EXAMPLE 19
[2,6-Dimethyl-4-(4-methyl-piperazin-l-yl)-phenyI]-(2-phenyI-quinolin-4-yl)-amine
Following the procedure outlined in Example 1, but substituting aniline for 2,3- dimethylaniline, ethyl benzoylacetate for ethyl 2-methylacetoacetate, and 4-bromo-2,6- dimethylaniline for 4-(3,4-dimethyl-piperazin-l-yl)-phenylamine, afforded 97 mg (yield 1 %) of (4-bromo-2,6-dimethyl-phenyl)-(2-phenyl-quinolin-4-yl)-amine. This amine (0.24 mmol) was dissolved in 3 ml of dry toluene and 53 μl (0.48 mmol) of 1-methylpiperazine, 54 mg (0.48 mmol) of potassium tertbutoxide, 2 mg (3 μmol) of BINAP (2,2'- bis(diphenylphosphino)-l,l'-binaphthyl) and 6 mg (6 μmol) of tris(dibenzylideneacetone)- dipalladium were added to the reaction mixture under argon atmosphere before the reaction mixture was heated at 100°C overnight. The solvent was then evaporated and the reaction mixture was purified on a silica gel column, (eluent methanokdichloromethane 1 :9), to obtain the title compound with 1 % yield.
MS (ESf): m/z 423 (M4), 212 (M2+).
EXAMPLE 20
[4-(3,4,4-TrimethylimidazoIidin-l-yl)-phenyl]-(2,3?8-trimethyl-quinolin-4-yl)amine
0.528 g (3.35 mmol) of l-chloro-4-nitrobenzene and 2.0 ml (19 mmol) of l,2-diamino-2- methylpropane were mixed and heated in a microwave oven reactor (Smith Creator™, Personal Chemistry) at 200°C for 20 min. The reaction mixture was poured into water and extracted with dichloromethane. The extract was evaporated and the reaction product was purified by silica gel column (eluent methanol'.dichloromethane, 1 :9), to obtain 0.518 g (74 %) of 2-methyl-N' , 1 ' -(4-nitro-phenyl)-propane- 1 ,2-diamine. The latter diamine was then heated for 1 h at 80 °C with 465 μl of formaldehyde (6.2 mmol, 40 % aqueous solution) in 10 ml of formic acid. The reaction mixture was taken up in water, made alkaline with aqueous NaOH and extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated. The obtained 3,4,4-trimethyl-l-(4-nitrophenyl)- imidazolidine (0.563 g, 2.4 mmol) was refluxed overnight with 10 ml of ethanol and 1.16 ml (24 mmol) of hydrazinium hydroxide in the presence of 31 mg of 10 % palladium on activated carbon. After filtration and evaporation of the solvent, the reaction product was dissolved in water, which was made alkaline with aqueous NaOH. The solution was extracted with 3% 2-propanol in dichloromethane and the organic phase was dried over sodium sulphate. Evaporation of the solvent provided 0.32 g (69 %) of N-(2-methyl-2- methylamino-propyl)-benzene-l,4-diamine. Following the procedure outlined in Example
1, but substituting o-toluidine for 2,3-dimethylaniline in steps 4-5, afforded 4-chloro- 2,3,8-trimethyl-quinoline, which was then reacted with N-(2,2-dimethyl-3-methylamino- propyl)-benzene-l,4-diamine as described in step 6, to obtain 0.228 g (95 %) of N-(2- amino-2-methyl-propyl)-N'-(2,3,8-trimethyl-quinolin-4-yl)-benzene-l,4-diamine. This diamine was then cyclized by heating at 80°C for 2 h with of 118 μl of formaldehyde (2 mmol, 40 % aq. solution) in 5 ml of formic acid. The reaction mixture was taken up in water, made alkaline with aqueous NaOH and extracted with dichloromethane. The extract was dried over sodium sulphate, evaporated and the residue was purified on a silica gel column (eluent methanokdichloromethane, 1:9), to obtain 88 mg (37 %, overall yield 18 %) of the title compound.
1H NMR (CDC13, 500 MHz): δ 7.63 (IH, m), 7.41 (IH, m), 7.20 (IH, m), 6.68 (2H, m),
6.35 (2H, m), 5.75 (lH,br s), 4.08 (2H, s), 3.17 (2H, s), 2.80 (3H, s), 2.72 (3H, s), 2.32 (3H, s), 2.21 (3H, s), 1.14 (6H, s);
MS (ESf): m/z 375 (M4), 188 (M2+).
Using the same method the following compound was prepared:
(3-Ethyl-2,8-dimethyI-quinolin-4-yl)-(4-imidazolidin-l-yl-phenyl)-amine
MS (ESf): m/z 347 (M4), 174 (M2+).
EXAMPLE 21
(3-Ethyl-2,8-dimethyl-quinolin-4-yI)-[4-(3-methyl-imidazolidin-l-yl)-phenyl]-amine
Following the procedure outlined in Example 20, but substituting ethyl 2- ethylacetoacetate for ethyl 2-methylacetoacetate, and ethane- 1,2-diamine for 1,2-diamino- 2-methylpropane, afforded the title compound with 3 % overall yield.
1H NMR (CDC13, 500 MHz): δ 7.60 (IH, m), 7.38 (IH, m), 7.12 (IH, m), 6.69 (2H, m),
6.36 (2H, m), 5.62 (IH, br s), 3.93 (2H, s), 3.37 (2H, t, J = 6.3 Hz), 2.95 (2H, t, J = 6.3 Hz), 2.78 (8H, m), 2.47 (3H, s), 1.16 (3H, t, J = 7.5 Hz);
MS (ESf): m/z 361 (M+), 181 (M2+).
EXAMPLE 22
l-[4-(2,3-DimethylquinoIin-4-ylamino)-phenyl]-4,5,5-trimethyl-piperazin-2-one
2-Methyl-N', -(4-nitrophenyl)-propane- 1,2 -diamine (0.918 g, 4.4 mmol), which was obtained as described in Example 20, was mixed with 10 ml of dry DMF and 0.228 g (5.7 mmol) of sodium hydride (60 % in mineral oil) under argon atmosphere. 584 μl (5.3 mmol) of ethyl bromoacetate were added to the cooled (0°C) reaction mixture, which was then allowed to warm up to room temperature over 2 h. The reaction mixture was then evaporated, taken up in water and extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated to give 0.929 g (72 %) of [l,l-dimethyl-2-(4- nitrophenylamino)-ethylamino] -acetic acid ethyl ester. 5 ml of trifluoroacetic acid were added dropwise to a solution of 0.567 g (1.9 mmol) of [l,l-dimethyl-2-(4-nitro- phenylamino)-ethylamino] acetic acid ethyl ester in 9 ml of dichloromethane at 0°C. The reaction mixture was stirred at 0°C for 2 h, overnight at room temperature and then refluxed for 24 h. The reaction mixture was evaporated and the residue purified on a silica gel column (eluent methanol: dichloromethane, 1:9), to give 0.479 g of 5, 5 -dimethyl- 1 -(4- nitro-phenyl)-piperazin-2-one as trifluoroacetic acid salt. The salt was taken up in water, made alkaline with sodium bicarbonate and extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated to provide 0.329 g of 5,5-dimethyl-l-(4- nitrophenyl)-piperazin-2-one (69 %). The piperazinone (74 mg, 0.29 mmol) was mixed with 1.5 ml of dry DMF and 16 mg (0.38 mmol) of sodium hydride (60 % in mineral oil) under argon atmosphere. 20 μl (0.33 mmol) of methyl iodide were added to the cooled (0°C) reaction mixture, which was allowed to warm up to room temperature over 2 h. The reaction mixture was then evaporated, taken up in water and extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated to yield 53 mg (70 %) of 4,5,5-trimethyl-l-(4-nitro-phenyl)-piperazin-2-one. The latter was treated with hydrazinium hydroxide as described in Example 1, step 3, to obtain 26 mg (56 %) of l-(4-aminophenyl)-4,5,5-trimethylpiperazin-2-one. Following the procedure outlined in Example 1, but substituting o-toluidine for 2,3-dimethylaniline in steps 4-5, afforded 4- chloro-2,3,8-frimethyl-quinoline, which was reacted with l-(4-aminophenyl)-4,5,5- trimβthylpiperazm-2-one employing the method described in step 6, providing the title compound with 93 % yield (overall yield 11 %).
MS (ESf): m z 389 (M+), 195 (M2+).
Using the same method the following compound was prepared:
l-[4-(3-Ethyl-2-methyl-quinolin-4-yIamino)-phenyl]-5,5-dimethyl-piperazin-2-one
MS (ESf): m z 389 (M4).
EXAMPLE 23
(2,3-Dimethyl-quinolin-4-yl)-[4-(3,3,4-trimethyl-piperazin-l-yl)-phenyl]-amine
28 mg (71 μmol) of l-[4-(253-Dimethyl-quinolin-4-ylamino)-phenyl]-4,5,5-trimethyl- piperazin-2-one (Example 22) were dissolved in 2 ml of dry THF and 214 μl (0.21 mmol) of IM borane-THF complex were added under argon atmosphere at room temperature. The reaction was quenched after 4 h by the addition of 3 ml of water and 3 ml of concentrated hydrochloric acid and allowed to stay for 40 min. The reaction mixture was made alkaline by adding aqueous NaOH before the water phase was extracted with dichloromethane. The extract was dried over sodium sulphate and evaporated. The crude product was purified on a silica gel column (eluent methanol: dichloromethane, 1 :9) to give 3 mg (11 %) of the title compound.
MS (ESf): m/z 375 (M4), 188 (M2+).
Using the same method the following compounds were prepared:
(2,3-Dimethyl-quinolin-4-yl)-[4-(5-ethyl-3,3,4-trimethyl-piperazin-l-yl)-phenyl]- amine
MS (ESf ): m/z 403 (M4), 202 (M2+).
(2,3-Dimethyl-quinolin-4-yl)-[4-(3?355,5-tetramethyl-piperazin-l-yl)-phenyl]-amine 1H NMR (CDC13, 500 MHz): δ 8.03 (IH, m), 7.78 (IH, m), 7.58 (IH, m), 7.33(1H, m), 6.83 (2H, m), 6.69 (2H, m), 5.92 (IH, b s), 3.38 (2H, m), 2.90 (2H, m), 2.73 (3H, s), 2.57 (IH, m), 2.46 (IH, m), 2.35 (3H, s), 2.24 (3H, s), 2.15 (IH, m), 1.73 (IH, m), 1.48 (IH, m), 0.95 (3H, m); MS (ESf): m/375 (M+), 188 (M2+).
Using the methods outlined in examples 12 and 23 the following compounds were prepared:
{7-Chloro-4-[4-(3,3,4-trimethyl-piperazin-l-yl)-phenylamino]-quinoIin-3-yl}- methanol
1H NMR (CDCI3, 500 MHz): 8.42 (IH, s), 7.93 (IH, m), 7.59 (IH, m), 7.43 (IH, b s), 7.13 (IH, m), 6.79 (4H, m), 4.77 (2H, s), 3.14 (2H, m), 2.88 (2H, m), 2.70 (2H, m), 2.30 (3H, s), 1.13 (6H, s); MS (ESf): m z 411 (M4), 206 (M2+).
{6-Fluoro-4-[4-(3,3,4-trimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}- methanol
1H NMR (CDCI3, 500 MHz): 8.44 (IH, s), 7.96 (IH, m), 7.31 (3H, m), 6.78 (4H, m), 4.78 (2H, s), 3.14 (2H, m), 2.88 (2H, m), 2.71 (2H, m), 2.30 (3H, s), 1.14 (6H, s); MS (ESf): m/z 395 (M4), 198 (M2+).
Using the methods outlined in examples 15 and 23 the following compound was prepared:
{2-Methyl-4-[4-(3,354-trimethyl-piperasin-l-yl)-phenylamino]-quinolin-3-yl}- methanol
MS (ESf): m/z 391 (M4), 196 (M2+).
EXAMPLE 24
{4-[4-(3,4-DimethyI-piperazin-l-yI)-phenyIamino]-3-methyl-quinolin-2-yl}-methanol
0.150 g (0.65 mmol) of 4-Hydroxy-3-methyl-quinoline-2-carboxylic acid ethyl ester (prepared according to e.g. J Am. Chem. Soc, vol. 68, 1964 pp. 129-132 and pp.1232- 1238) were mixed with 1 ml of phosphorus oxychloride and refluxed for 1 h. The cooled reaction mixture was evaporated and taken up in dichloromethane. The dichloromethane solution was poured into cold (0°C) aqueous ammonia and the phases were separated. The organic phase was washed with brine, dried over sodium sulphate and evaporated. The product was purified on a silica gel column (eluent methanol:dichloromethane, 1 :49) to obtain 100 mg (62 %) of 4-chloro-3-methyl-quinoline-2-carboxylic acid ethyl ester. Following the procedure outlined in Example 1, step 6, but substituting 4-chloro-3- methyl-quinoline-2-carboxylic acid ethyl ester for 4-chloro-2,3,7,8-tetramethylquinoline5 afforded 4- [4-(3 ,4-dimethyl-piperazin- 1 -yl)-phenylamino] -3 -methyl-quinoline-2- carboxylic acid ethyl ester with 21 % yield. The ester group was reduced following the method described in Example 12, affording the title compound with 54 % yield (overall yield 3 %).
1H NMR (CDCI3, 500 MHz): δ 8.04 (IH, s), 7.82 (IH, m), 7.63 (IH, m), 7.38 (IH, m), 6.83 (2H, m), 6.71 (2H, m), 5.92 (IH, br s), 4.82 (2H, s), 3.38 (2H, m), 2.90 (2H, m), 2.48 (2H, m), 2.35 (3H, s), 2.27 (IH, m), 2.12 (3H, s), 1.14 (3H, d, J= 6.1 Hz); MS (ESf): m/z 377 (M+), 189 (M24).
EXAMPLE 25
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyI]-(2-methyl-l,2,3,4-tetrahydro- benzo[b][l,6]naphthyridin-10-yl)-amine
A mixture of 1.37 g (10 mmol) of anthranilic acid, 1.15 ml (10 mmol) of l-methyl-4- piperidone and 15 ml of phosphorus oxychloride was refluxed for 3 h. After cooling the reaction mixture was evaporated and taken up in dichloromethane. The dichloromethane solution was poured into a cold (0°C) aqueous sodium bicarbonate solution and the phases were separated. The organic phase was dried over sodium sulphate and evaporated to obtain 0.49 g (22 %) of 10-chloro-2-methyl- 1,2,3, 4-tetrahydro- benzo[b][l,6]naphthyridine. Following the procedure outlined in Example 1, step 6, but substituting 10-chloro-2-methyl-l,2,3,4-tetrahydro-benzo[b][l,6]naphthyridine for 4- chloro-2,3,7,8-tetramethylquinoline, afforded the title compound with 49 % yield (overall yield 11 %).
1H NMR (DMSO-d6, 500 MHz): δ 7.92 (IH, s), 7.84 (2H, m), 7.58 (IH, m), 7.33 (IH, m), 6.79 (2H, m), 6.62 (2H, m), 3.41 (2H, m), 3.18 (IH, m), 3.08 (2H, m), 2.81 (IH, m), 2.74 (2H, m), 2.66 (IH, m), 2.32 (3H, s), 2.29 (3H, m), 2.22 (3H, s), 2.16 (IH, m), 1.03 (3H, d, J= 6.1 Hz); MS (ESf): m/z 402 (M+), 202 (M2+).
EXAMPLE 26
[6-(3,4-Dimethyl-pipera2zin-l-yl)-pyridin-3-yl]-(2 -dimethyl-quinolin-4-yl)-amine
Following the procedure outlined in Example 1, but substituting 2-chloro-5-nitropyridine for l-chloro-4-nitrobenzene in step 1, plus aniline for 2,3-dimethylaniline in step 4, afforded the title compound with 4 % overall yield.
1H NMR (CDCI3, 500 MHz): δ 8.02 (IH, s), 7.88 (IH, m), 7.76 (IH, m), 7.59 (IH, m), 7.35 (IH, m), 6.92 (IH, m), 6.56 (IH, m), 5.81 (IH, br s), 3.93 (2H, m), 3.02 (IH, m),
2.91 (IH, m), 2.72 (3H, s), 2.63 (IH, m), 2.37 (IH, m), 2.35 (3H, s), 2.25 (3H, s), 2.22 (IH, m),1.15 (3H, d, J= 6.3 Hz); MS (ESf): m/z 362 (M4), 182 (M2+).
Using the methods outlined in examples 12 and 26 the following compound was prepared:
{4-[6-(3,4-Dimethyl-piperazin-l-yl)-pyridin-3-ylamino]-quinolin-3-yl}-methanol
MS (ESf): m/z 364 (M4), 183 (M2+).
EXAMPLE 27
[4-(3,4-Dimethyl-piperazin-l-yI)-phenyl]-(3-ethyl-2-methyl-5?6,7,8-tetrahydro- quinolin-4-yl)-amine
Following the procedure outlined in Example 1, step 4, but substituting ethyl 2- ethylacetoacetate for ethyl 2-methylacetoacetate and aniline for 2,3-dimethylaniline, afforded 3-ethyl-2-methylquinolin-4-ol. It was dissolved in methanol and stirred overnight in a Parr hydrogenation apparatus with 20 bar H2 at 60°C in the presence of nickel catalyst. Thus obtained 3-ethyl-2-methyl-5,6,7,8-tetrahydroquinolin-4-ol was treated with thionyl chloride as described in Example 1, step 5, affording 4-chloro-3-ethyl-2-methyl- 5,6,7,8-tetrahydro-quinoline, which was coupled with 4-(3,4-dimethyl-piperazin-l-yl)- phenylamine by using the method described in Example 1, step 6. Finally, after silica gel purification (eluent methanohdichloromethane 1:5) the title compound was obtained with 3 % overall yield.
1H NMR (CD3OD, 500 MHz): δ 6.86 (2H, m), 6.60 (2H, m), 3.35 (IH, m), 2.91 (IH, m), 2.79 (3H, m), 2.61 (2H, q, J= 7.6 Hz), 2.47 (3H, s), 2.41 (5H, m), 2.33 (3H, s), 2.31 (IH, m), 1.78 (2H, m), 1.64 (2H, m), 1.14 (3H, d, J= 6.2 Hz) 1.02 (3H, t, J= 1.6 Hz);
MS (ESf ): m/z 379 (M+), 190 (M2+).
EXAMPLE 28
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyI]-(3-isopropyl-2-methyl-quinolin-4-yI)-amine (Compound D)
Following the procedure outlined in Example 1, but substituting ethyl 2- isopropylacetoacetate for ethyl 2-methylacetoacetate in step 1, plus aniline for 2,3- dimethylaniline in step 4, afforded the title compound with 29 % overall yield.
1H NMR (CDC13, 500 MHz): δ 7.95 (IH, m), 7.73 (IH, m), 7.54 (IH, m), 7.24 (IH, m), 6.79 (2H, m), 6.60 (2H, m), 5.71 (IH, br s), 3.61 (IH, m), 3.34 (2H, m), 2.85 (2H, m), 2.81 (3H, s), 2.45 (2H, m), 2.33 (3H, s), 2.24 (IH, m), 1.38 (6H, d, J= 7.2 Hz), 1.11 (3H, d, J= 6.2 Hz), 1.47 (2H, m); MS (ESf): m/z 389 (M+), 195 (M2+).
EXAMPLE 29
(3-Ethyl-2-methyl-quinoIin-4-yl)-[4-(4-methyl-[l,4]diasepan-l-yl)-phenyl]-amine (Compound E)
Following the procedure outlined in Example 17, but substituting aniline for ø-toluidine, afforded the title compound with 9 % overall yield.
1H NMR (CDC13, 500 MHz): δ 8.01 (IH, m), 7.80 (IH, m), 7.60 (IH, m), 7.30 (IH, m), 6.78 (2H, m), 6.64 (2H, m), 5.78 (IH, br s), 3.58 (2H, m), 3.49 (2H, m), 2.86 (2H, q, J= 7.7 Hz), 2.82 (3H, s), 2.76 (2H, m), 2.65 (2H, m), 2.44 (3H, s), 2.05 (2H, m), 1.25 (3H, t, J= 7.7 Hz); MS (ESf): m/z 375 (M4), 188 (M2+).
Using the same method the following compound was prepared:
(2,3-Dimethyl-quinoIin-4-yl)-[4-(4-methyl-[l?4]diasepan-l-yl)-phenyl]-amine
1H NMR (CDCI3, 500 MHz): δ 7.96 (IH, m), 7.78 (IH, m), 7.56 (IH, m), 7.31 (IH, m), 6.71 (2H, m)3 6.59 (2H, m), 5.82 (IH, br s), 3.52 (2H, m), 3.43 (2H, m), 2.71 (3H, s), 2.69 (2H, m), 2.57 (2H, m), 2.38 (3H, s), 2.23 (3H, s), 1.99 (2H, m); MS (ESf): m/z 361 (M+), 181 (M2+).
EXAMPLE 30
(2,3-Dimethyl-quinolin-4-yl)-[4-(3,3,4,5,5-pentamethyl-piperazin-l-yl)-phenyl]-amine
0.628 g (3.0 mmol) of 2-Methyl-N',r-(4-nitro-phenyl)-propane- 1,2-diamine (prepared as described in Example 20), 280 μl (3.6 mmol) of chloroform, 442 μl (6.0 mmol) of acetone
and 0.164 g (0.44 mmol) of tetrabutylammonium iodide were mixed and cooled down to 0°C. 1.06 ml of 50 % aqueous NaOH-solution was added dropwise to the reaction mixture to keep the temperature below 5°C. After 19 h of reaction time, 3 ml of dichloromethane was added. Phases were separated, and the organic phase was washed once with saturated NaHCO3-solution, dried over sodium sulphate and evaporated. The crude product was purified by silica gel column (eluent methanol: dichloromethane 1:9) to obtain 3,3,5,5- tetramethyl-l-(4-nitro-phenyl)-piperazin-2-one with 30 % yield. 3,3,5,5-Tetramethyl-l-(4- nitro-phenyl)-piperazin-2-one was treated with borane-THF complex employing the method described in Example 23, affording 3,3,5,5-tetramethyl-l-(4-nitro-phenyl)- piperazine with 74 % yield. Following the procedure outlined in Example 1, but substituting 3,3,5,5-tetramethyl-l-(4-nitro-phenyl)-piperazine for 3 -methyl- l-(4-nitro- phenyl)-piperazine in step 2, plus aniline for 2,3-dimethylaniline in step 4, afforded the title compound with 1 % overall yield.
1H NMR (CDC13, 500 MHz): δ 7.99 (IH, m), 7.79 (IH, m), 7.58 (IH, m), 7.34 (IH, m), 6.81 (2H, m), 6.66 (2H, m), 5.83 (IH, br s), 2.85 (2H, m), 2.72 (3H, s), 2.28 (2H, m), 2.26 (6H, s), 1.15 (12H, s); MS (ESf): m/z 403 (M+), 202 (M24).
Using the same method the following compounds were prepared:
(2,3-Dimethyl-quinolin-4-yl)-[4-(3,3,4,5-tetramethyI-piperazin-l-yl)-phenyl]-amine MS (ESf): m/z 389 (M+), 195 (M2+).
(253-Dimethyl-quinolin-4-yl)-[4-(3,3,5,5-tetramethyl-piperazin-l-yl)-phenyI]-amine
MS (ESf): m/z 389 (M4), 195 (M24).
(2,3-BimethyI-quinolin-4-yI)- [4-(l-methyl-l ,4-diaza-spiro [5.5] undec-4-yl)-phenyl] - amine Η NMR (CDCI3, 500 MHz): δ 7.98 (IH, m), 7.79 (IH, m), 7.58 (IH, m), 7.34 (IH, m), 6.81 (2H, m), 6.68 (2H, m), 5.81 (IH, b s), 3.06 (4H, m), 2.82 (2H, m), 2.72 (3H, s), 2.36 (3H, s), 2.25 (3H, s), 1.71 (6H, m), 1.58 (2H, m), 1.43 (2H, m); MS (ESf): m z 415 (M4), 208 (M2+).
[4-(3,3-Diethyl-4-methyl-piperazin-l-yl)-phenyl]-(2,3-dimethyl-quinolin-4-yl)-amine MS (ESf): m/z 403 (M+), 202 (M2+).
Using the methods outlined in examples 12 and 30 the following compound was prepared:
{4-[4-(3,3,4-Trimethyl-piperazin-l-yI)-phenyIamino]-quinolin-3-yl}-methanol
1H NMR (CDC13, 500 MHz): δ 8.46 (IH, s), 7.98 (IH, m), 7.71 (IH, m), 7.56 (IH, m), 7.46 (IH, br s), 7.22 (IH, m), 6.80 (4H, m), 4.77 (2H, s), 3.14 (2H, m), 2.89 (2H, m), 2.74 (2H, m) 2.32 (3H, s), 1.15 (6H, s,); MS (ESf): m/z 377 (M4), 189 (M24).
EXAMPLE 31
(2,3-DimethyI-quinolin-4-yI)-[4-(octahydro-pyrido[l,2-a]pyrazin-2-yl)-phenyl]-amine
0.85 ml (5.0 mmol) of triflic anhydride was added dropwise to a stirred solution of 0.804 g (4.2 mmol) of N-(2-hydroxyethyl)-phthalimide in 3 ml of dry dichloromethane under argon atmosphere at 0°C. 2,6-Lutidine (5.0 mmol) and a solution of ethyl pipecolinate (5.0 mmol) plus triethylamine (5.0 mmol) in 3 ml of dry dichloromethane were added dropwise with 10 min time intervals. The reaction mixture was stirred at room temperature for 24 h. Reaction mixture was diluted with dichloromethane and washed with acidic water. The organic phase was dried over sodium sulphate and evaporated to give crude l-[2-(l,3-dioxo-l,3-dihydro-isoindol-2-yl)-ethyl]-piperidine-2-carboxylic acid ethyl ester. The ester was cyclised by treating it with hydrazinium hydroxide in methanol at room temperature for 22 h. Thus formed hexahydro-pyrido[l,2-a]pyrazin-l-one was reduced with lithium aluminium hydride as described in Example 12, affording octahydro-pyrido[l,2-a]pyrazine. Following the procedure outlined in Example 1, but substituting octahydro-pyrido[l,2-a]pyrazine for 2-methylpiperazine in step 1, and aniline for 2,3-dimethylaniline in step 4, afforded the title compound with 1 % overall yield.
1H NMR (CDC13, 500 MHz): δ 8.01 (IH, m), 7.78 (IH, m), 7.58 (IH, m), 7.33 (IH, m), 6.82 (2H, m), 6.69 (2H, m), 5.93 (IH, br s), 3.37 (2H, m), 2.90 (3H, m), 2.72 (3H, s), 2.47 (2H, m), 2.23 (3H, s), 2.12 (IH, m), 1.80 (IH, m), 1.68 (IH, m), 1.60 (IH, m), 1.33 (2H, m), 1.14 (2H, m); MS (ESf): m/z 387 (M4), 194 (M2+).
EXAMPLE 32
4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-3,4-dihydro-lH-2-oxa-4,9-di-4-[4-(3,4- dimethyl-piperazin-l-yl)-phenyl]-3,4-dihydro-lH-2-oxa-4,9-diaza-phenanthrene
5.6 mg (15 μmol) of {4-[4-(3,4-dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}- methanol (Example 12) was dissolved in 1.5 ml of aqueous 40 % formaldehyde solution. One drop of formic acid was added and the resulting mixture was stirred at room temperature for 48 hours. The mixture was made slightly alkaline by addition of dilute NaHCO3-solution, diluted with water and extracted several times with dichloromethane. The organic phases were combined and washed additionally with brine and finally dried over sodium sulphate. Filtration and evaporation of the solvent gave crude product which was purified by silica gel column (eluent methanol: dichloromethane 1 :9) to obtain the title compound with 32% yield.
MS (ESf): m/z 375 (M+), 188 (M24).
EXAMPLE 33
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(3-methoxymethyl-quinolin-4-yl)-amine
20 mg of {4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}-methanol (Example 12) were dissolved in 0.5 ml of dry dichloromethane and 0.3 ml of triflic anhydride were added to the solution. The reaction mixture was stirred at ambient temperature under Ar for 4 h, stripped on a rotary evaporator and dried for 1 h at high vacuum. The residue was dissolved in 2 ml of dry methanol, one drop of triethylamine was added and the solution was stirred overnight at ambient temperature under Ar. The reaction mixture was then diluted with dichloromethane, washed 3 times with brine and dried over sodium sulfate. The title compound was purified from the reaction mixture on a silica gel column (gradient elution methanol: dichloromethane form 1 :9 to 1:4) to give 9.1 mg (44%).
!H NMR (CDC13, 500 MHz): δ 8.63 (IH, m), 8.05 (IH, m), 7.70 (IH, m), 7.61 (IH, m), 7.43 (IH, m), 6.86 (4H, m), 5.12 (IH, br s), 4.65 (2H, s), 3.45 (2H, m), 2.99 (2H, m), 2.65 (IH, m), 2.55 (IH, m), 2.44 (3H, s), 2.31 (IH, m), 2.04 (3H, s), 1.21 (3H, d, J= 6.3 Hz); MS (ESf ): m/z 377 (M+), 189 (M2+).
EXAMPLE 34
[4-(3,4-Dimethyl-piperazin-l-yl)-phenyl]-(3-methyl-quinolin-4-yI)-amine
Following the procedure outlined in Example 1, step 6 but substituting 4-chloro-3-methyl- quinoline (prepared according to e.g. J. Am. Chem. Soc., 68 (1964) pp. 129-132 and pp.1232-1238) for 4-chloro-2,3,7,8-tetramethylquinoline afforded the title compound with 29% yield.
1H NMR (CDC13, 500 MHz): δ 8.67 (IH, s), 8.04 (IH, m), 7.80 (IH, m), 7.59 (IH, m), 7.36 (IH, m), 6.80 (4H, m), 5.91 (IH, br s), 3.38 (2H, m), 2.89 (2H, m), 2.52 (IH, m), 2.44 (IH, m), 2.35 (3H, s), 2.29 (IH, m), 2.27 (3H, s), 1.14 (3H, d, J= 6.3 Hz); MS (ESf ): m/z 347 (M+), 174 (M2+).
Using the methods outlined in examples 34 and 23 the following compound was prepared:
(3-Methyl-quinolin-4-yl)-[4-(3,354-trimethyl-piperasin-l-yl)-phenyl]-amine
MS (ESf): m/z 361 (M4), 181 (M2+).
EXAMPLE 35
l-{4-[4-(3,4-Dimethyl-piperazin-l-yl)-phenylamino]-2-methyl-quinolin-3-yI}- ethanone
Following the procedure outlined in Example 12 but substituting 2-acetyl-3-ethoxy-but-2- enoic acid ethyl ester (prepared according to e.g. patents US 5,041,619 and US 4,058,553) for diethyl ethoxymethylene malonate and omitting the reduction step with lithium aluminium hydride, the title compound was obtained in 36% overall yield.
1H NMR (CDCI3, 500 MHz): δ 7.92 (IH, m), 7.84 (IH, b s), 7.74 (IH, m), 7.62 (IH, m), 7.24 (IH, m), 6.85 (4H, m), 3.42 (2H, m), 2.89 (2H, m), 2.72 (3H, s), 2.52 (IH, m), 2.49 (3H, s), 2.43 (IH, m), 2.35 (3H, s), 2.26 (IH, m), 1.13 (3H, d, J= 6.3 Hz); MS (ESf ): m z 389 (M+), 195 (M2+).
As already mentioned hereinbefore, the compounds of the present invention show interesting pharmacological properties, namely they exhibit affinity for alpha2 adrenoceptors. The said pharmacological activity of the compounds of the invention is demonstrated with the pharmacological tests presented below.
EXPERIMENT I: Binding affinity
The affinity of test compounds for the three human 2-adrenoceptor subtypes (α A, 2β and α2c) was determined in competition binding assays with the radioligand 3H- rauwolscine. The biological material for the assays consisted of membranes from Shionogi SI 15 cells stably transfected with one of the three human α2 subtypes
(Marjamaki et al., Biochem. Biophys. Ada 1134 (1992) 169). The membrane suspension (about 10 μg total protein per sample) and 1 nM of 3H-rauwolscine (specific activity 75- 85 Ci/mmol) were incubated with a minimum of six concentrations of the test compound in a total volume of 90 μl (50 mM KH2PO4 pH 7.5 at room temperature). Nonspecific binding was defined by 100 μM oxymetazoline and corresponded to 4-10 % of the total binding. After 30 min at room temperature, the incubation was terminated by rapid filtration (TomTec 96 harvester) through presoaked GF/B glass-fiber mats (Wallac Oy) and three washes with ice-cold 50 mM KH PO4 (pH 7.5 at room temperature). After drying, a solid scintillate (Meltex; Wallac) was melted on filter mats, and the radioactivity was measured (BetaPlate; Wallac Oy). The analysis of the experiments was carried out by nonlinear least square curve fitting. IC50s were converted to Ki's by using the equation of Cheng and Prusoff (Cheng, Y., and Prusoff, W.H., Biochem. Pharmacol. 22 (1973) 3099). The results of the experiments are reported in table 1.
Table 1. Affinities on human alpha-2 adrenoceptor subtypes

EXPERIMENT II: Antagonist activity
Antagonist activities were determined as the ability of compounds to competitively inhibit r epinephrine-stimulated S-GTPγS binding to G proteins (Jasper et al., Biochem. Pharmacol. 55 (1998) 1035) in membranes of CHO cells stably transfected with one of the three human α2-adrenoceptor subtypes (Pohjanoksa et al., Eur. J. Pharmacol. 35 (1997) 53). Membranes (5-10 μg of protein per sample) and a minimum of 6
concentrations of test compounds were preincubated for 30 min at room temperature in 50 mM Tris, 5 mM MgCl2, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 10 μM GDP, 30 μM ascorbic acid, pH 7.4 with a fixed concentration of epinephrine (5 μM for CC2A, 15 μM for α2B, 5 μM for α2C). Then trace amounts of 35S-GTPγS (0.08 nM- 0.15 nM, specific activity 1250 Ci/mmol) were added to the incubation mixture. After an additional 60 min at room temperature, the incubation was terminated by rapid vacuum filtration through glass fiber filter. Filters were washed three times with 5 ml icecold wash buffer (20 mM Tris, 5 mM MgCl2, 1 mM EDTA pH 7.4 at room temperature), dried and counted for radioactivity in a scintiallation counter. Analysis of experiments was carried out by nonlinear least square fitting. Experiments were repeated at least three times. The results of the experiments are reported in table 2.
Table 2. Antagonist potencies on human alpha-2 adrenoceptor subtypes
Compound Antag< onist potency ; nr alpha 2A alpha 2B alpha 2C
A 1 500 [3] > 3 000 [3] 7.0 ± 2.2 D > 1 000 [3] > 3 000 [3] 29 ± 5 E > 2 000 [3] > 2000 [3] 11 ± 3
EXPERIMENT Ul: Forced swimming test
The forced swimming test (FST, Porsolt's test) is generally used in the pharmacological screening of new antidepressants. In this test, antidepressants increase the activity of animals compared to non-treated controls. Alpha-2C KO- mice (i.e. mice with targeted disruption of the alpha-2C gene) appeared to be more active, and alpha-2C OE mice (overexpressing the alpha-2C receptor protein) were less active in FST (U.S. Patent No. 5,902,807 and Scheinin, M. et al., Life Sci 68(19-20) (2001) 2277-85). Therefore, FST was used to confirm both the alpha-2C antagonism and the antidepressant-like properties of the said compounds in vivo.
The employed method was essentially similar as described by Porsolt et al. 1977. Naive rats were individually placed into glass cylinders (diameter 20 cm, height 40 cm), containing 18 cm of water maintained at 25°C. After 15 min they were removed from the cylinders and allowed to dry under warming lamp. After drying, the rats were given a subcutaneous injection of test substance or vehicle and returned to their home cages. A second injection was given 23 h after the first administration, i.e. 1 h before a second
exposure to the swimming situation. In the second 5 min swimming session the duration of active behaviors for each animal was recorded.
The figure 1 shows the in vivo activity of the compounds A and D in the FST. The studied alpha-2C selective compounds efficiently increased activity in the FST, as expected based on studies on transgenic mice (Scheinin, M. et al., Life Sci 68(19-20) (2001) 2277-85) and as reported with recently developed alpha-2C antagonist (WO 01/64645).
The compounds according to the invention may be used to treat any disease or condition wherein alpha-2 antagonists are indicated to be effective. The compounds can also be used to reverse effects induced by alpha-2 agonists. Accordingly, the compounds of the invention may be useful in the treatment of various disorders of the central nervous system (CNS), i.e. different neurological, psychiatric and cognition disorders (such as depression, anxiety disorders, post traumatic stress disorder, schizophrenia, Parkinson's disease and other movement disorders). Furthermore, they may be used in the treatment of various peripheral disorders, e.g. diabetes, orthostatic hypotension, lipolytic disorders (such as obesity), Raynaud's disease or both male and female sexual dysfunctions.
The selective alpha-2C antagonists of the present invention may be used for the treatment of various disorders or conditions of CNS-system where alpha-2C antagonists are indicated to be beneficial, for example, to alleviate the symptoms of various mental disorders propagated by stress, Parkinson's disease, depression, negative symptoms of schizophrenia, attention deficit hyperactivity disorder, post traumatic stress disorder, or anxiety disorders.
In addition, the alpha-2C selective compounds can also be used to treat disorders and conditions associated with sensorimotor gating deficits, particularly disorders and conditions wherein the sensorimotor gating deficits result in sensory flooding and cognitive fragmentation causing dysfunction in attention and perception. Such disorders and conditions include, but are not limited to, schizophrenia, obsessive compulsive disorder, Tourette's syndrome, blepharospasm and other focal dystonias, temporal lobe epilepsy with psychosis, drug-induced psychosis (for example, psychosis caused by chronic use of dopaminergic agents) (Braff, D.L. et al., Psychopharmacology (Berl) 156(2-3) (2001) 234-258), Huntington's disease, Parkinson's disease, disorders caused by
fluctuation of the levels of sex hormones (such as premenstrual syndrome), and panic disorder.
Due to their selectivity of action, the alpha-2C antagonists of the invention have less or no undesirable side-effects attributed to non-selective alpha-2 antagonism, such as increases in blood pressure, heart rate, salival secretions, gastrointestinal secretion, anxiety, and startle reactivity per se (Ruffolo, R.RJ. et al., Annu Rev Pharmacol Toxicol 32 (1993) 243-279).
The compounds of the invention can be administered for example enterally, topically or " parenterally by means of any pharmaceutical formulation useful for said administration, and containing at least one active compound of fonnula I in pharmaceutically acceptable and effective amounts together with pharmaceutically acceptable diluents, carriers, and/or excipients lαiown in the art. The manufacture of such pharmaceutical formulations is well known in the art.
The therapeutic dose to be given to a patient in need of treatment will vary depending on the compound being administered, the species, age and the sex of the subject being treated, the particular condition being treated, as well as the route and method of administration, and are easily detennined by person skilled in the art. Accordingly, the typical dosage for oral administration is from 5 μg/kg to 100 mg/kg per day and that for parenteral administration from 0.5 μg/kg to 10 mg/kg for an adult mammal.
The present invention further provides a compound of the invention or an ester or salt thereof for use as alpha-2 antagonist. Furthermore, a method for the treatment of diseases or conditions where alpha-2 antagonists, e.g. alpha-2C antagonists, are indicated to be useful, e.g. a method for the treatment of diseases or conditions of the central nervous system, is provided. In such a method a therapeutically effective amount of a compound of the invention is administered to a subject in need of such treatment. The use of the compounds of the invention for the manufacture of a medicament to be used for the above indications is also provided, e.g. for the manufacture of a medicament for the treatment of diseases or conditions where alpha-2 antagonists are indicated to be effective
Those skilled in the art will appreciate that the embodiments described in this application could be modified without departing from the broad inventive concept. Those skilled in the art also understand that the invention is not limited to the particular disclosed
embodiments, but is intended to also cover modifications to the embodiments that are within the spirit and scope of the invention.
Claims
1. A compound of formula I,

Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 each independently being OH, halogen, (d-C6)alkyl, (C -C6)alkenyl, (C1-C6)alkoxy, halo(d-C6)alkyl, NO2, NH2, mono- or di(d-C6)alkylamino, mono- or di(Cι-
C6)alkylamino(C1-C6)alkyl, (C1-C6)alkyl-S- or hydroxy(C1-C6)alkyl; or Q is
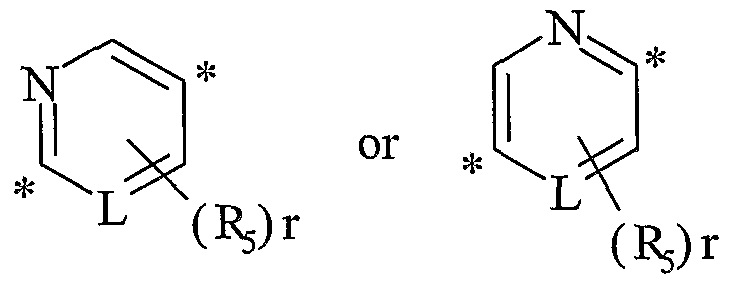
R5 is as defined above and r is 0 to 2;
L is CH, CR5 orN;
Y is -CHm(R4)l-[CHn(R4)p]v- or a single bond;
Ri is H, (Cι-C6)alkyl or (C3-C7)cycloalkyl ;
A is a benzene ring or (C5-C7)cycloalkyl; each R2 is independently OH, halogen, (Cι-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl,
(C3-C7)cycloalkyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo-(C1-C6)alkyl, NO2,
NH2, mono- or di(C1-C6)alkylamino, amino(C1-C6)alkyl, mono- or di(Cι-
C6)alkylamino(C1-C6)alkyl, (C1-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl,
(C1-C6)alkyl-S-,
 MI2-CO- or -CHO;
R3 is H, (d-C6)alkyl, (C2-C6)alkenyl, CN-(d-C6)alkyl, (d-C^alkoxy-CO-^r C6)alkyl, (d-C6)alkyl-CO-, NH2-CO-, mono- or di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, (C3-C7)cycloalkyl5 phenyl, naphthyl or benzyl, wherein the said (C -C7)cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or substitued with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (d-C6)alkyl, (d-C6)alkoxy, mono- or di(Cι- C6)alkylamino or halo-(C1-C6)alkyl; each R4 is independently OH, halogen, MI2, oxo, -CHO, (d-C6)alkyl, (C -C6)alkenyl, (C2-C6)alkynyl, CN-(C1-C6)alkyl, (d-C6)alkoxy-CO-(d-C6)alkyl, (d-C6)alkyl-CO-, MI2-CO-, mono- or di(Cι-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, amino(d-
MI2-CO- or -CHO;
R3 is H, (d-C6)alkyl, (C2-C6)alkenyl, CN-(d-C6)alkyl, (d-C^alkoxy-CO-^r C6)alkyl, (d-C6)alkyl-CO-, NH2-CO-, mono- or di(C1-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, (C3-C7)cycloalkyl5 phenyl, naphthyl or benzyl, wherein the said (C -C7)cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or substitued with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (d-C6)alkyl, (d-C6)alkoxy, mono- or di(Cι- C6)alkylamino or halo-(C1-C6)alkyl; each R4 is independently OH, halogen, MI2, oxo, -CHO, (d-C6)alkyl, (C -C6)alkenyl, (C2-C6)alkynyl, CN-(C1-C6)alkyl, (d-C6)alkoxy-CO-(d-C6)alkyl, (d-C6)alkyl-CO-, MI2-CO-, mono- or di(Cι-C6)alkylcarbamoyl, hydroxy(C1-C6)alkyl, amino(d-
C6)alkyl, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, (C3-C )cycloalkyl, phenyl, naphthyl or benzyl, wherein the said (C3-C7)cycloalkyl, phenyl, naphthyl or benzyl is unsubstituted or substitued with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (d-C6)alkyl, (d-C6)alkoxy, mono- or di(d-C6)alkylamino or halo-(C1-C6)alkyl; or R3 and Ri or Rι and R form, together with any of the ring atom(s) to which they are attached, a condensed 5 to 7 membered carbocyclic ring or a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 or 2 substituent(s) R6 each independently being OH, oxo, (d-G alkyl, (C1-C3)alkoxy, halogen or halo(C1-C3)alkyl;
Ra and Rb are independently H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2- C6)alkynyl, (d-C6)alkoxy, (Cι-C6)alkoxy(d-C6)alkyl, halo(d-C6)alkyl, NO2, NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(Cι- C6)alkyl, (C C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(d-C6)alkyl, (d-
C6)alkoxy-CO-, (Cι-C6)alkyl-CO-9 mono- or di(Cι-C6)-alkylcarbamoyl, hydroxy(Cι- C6)alkyl, NH2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (d-C6)alkyl, (d-C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and R1 form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R as defined above;
or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined above;
1 is 0 to 2; m is 0 to 2; n is 0 to 2; p is 0 to 2; t is 0 to 3; u is 0 to 4; and v is 0 or 1 ; with the provisos, that a) when Q is 1,4-phenylene, 1 is 0, m is 2, u is 0, and v is 0, then one of Ra and Rb is H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl, (d-C6)alkoxy, (d-
C6)alkoxy(Cι-C6)alkyl, halo(Cι-C6)alkyl, NO2, NH2, mono- or di(Cι- C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(d-C6)alkyl, (d-C6)alkyl-S- or CN and the other of Ra and Rb is (C3-C7)cycloalkyl, (C3- C7)cycloalkyl(d-C6)alkyl, (d-C6)alkoxy-CO-, (Cι-C6)alkyl-CO-, mono- or di(d- C6)-alkylcarbamoyl, hydroxy(C1-C6)alkyl, amino(C1-C6)alkyl, (C1-C6)alkoxy(C1-
C6)alkyl, NH2-CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH2, (C1-C6)alkyl, halo(C1-C6)alkyl, (Cι-C6)alkoxy or mono-or di(Cι - C6)alkylamino; or Rb is H, OH, halogen, (Cι-C6)alkyl, (C -C6)alkenyl, (C2-C6)alkynyl, (d- C6)alkoxy, (Cι-C6)alkoxy(Cι-C6)alkyl, halo(Cι-C6)alkyl, NO , MI2, mono- or di(CrC6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(d- C6)alkyl, (Cι-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(Cι-C6)alkyl, (d-C6)alkoxy-CO-, (d-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(d-C6)alkyl, NH2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2, NH2, (d-C6)alkyl, (d-C6)alkoxy or mono-or di(d-
C6)alkylamino and Ra and R\ form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R^ as defined above; b) when Q is 1,4-phenylene, 1 is 0, m is 2, u is 0, v is 0, and one of Ra and Rb is H, then the other of Ra and Rb is not ethoxy-CO-; or a pharmaceutically acceptable salt or ester thereof.
2. A compound according to claim 1, wherein Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined in claim 1;
Y, A, R1-R4 and R6 are as defined in claim 1;
Ra and Rb are independently H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2- C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, NH2, mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(d-
C6)alkyl, (d-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(C1-C6)alkyl, (Q- C6)alkoxy-CO-, (d-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(d- C6)alkyl, NH2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2,
NH2, (d-C6)alkyl, (d-C6)alkoxy or mono-or di(C1-C6)alkylamino; or Rb is as defined above and Ra and R1 form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1 ; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1 ;
1 is 0 to 2; m is 0 to 2 n is 0 to 2; p is 0 to 2;
t is O to 3; u is 0 to 4; and v is 0 or 1; with the proviso, that when u is 0 and v is 0, then 1 is not 0.
3. A compound according to any one of claims 1 or 2, wherein
Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined in claim 1;
Y, A, Ri-R4 and R6 are as defined in claim 1; Ra and Rb are independently H, OH, halogen, (Cι-C6)alkyl, (C2-C6)alkenyl, (C2-
C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, NH2, mono- or di(Cι-C6)alkylamino, mono- or di(C -C6)alkylamino(C1-C6)alkyl, amino(C1-
C6)alkyl, (C1-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(d-C6)alkyl, (Ci-
C6)alkoxy-CO-, (C1-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(d- C6)alkyl, NH2-CO-, -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said
(C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2,
NH2, (d-C6)alkyl, (d-C6)alkoxy or mono-or di(d-C6)alkylamino; or Rb is as defined above and Ra and R\ form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1 ; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1 ;
1 is 0 to 2; m is 0 to 2 n is 0 to 2; p is 0 to 2; t is O to 3; u is 0 to 4; and v is 0 or 1;
with the provisos, that a) when u is 0 and v is 0, then 1 is not 0; b) when u is 0 and v is 1, then 1 and p are not simultaneously 0.
4. A compound according to claim 3, wherein u is 1 or 2 and R4 is (d-C6)alkyl.
5. A compound according to any one of claims 3 or 4, wherein t is 0, 1 or 2 and each R2 is independently halogen, (d-C6)alkyl or (C1-C6)alkoxy.
6. A compound according to any one of claims 3 to 5, wherein Ra and Rb are independently H, (d-C6)alkyl, hydroxy(C1-C6)alkyl or phenyl.
7. A compound according to any one of claims 3 to 5, wherein Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 6 membered non- aromatic heterocyclic ring containing one heteroatom selected from N, O and S.
8. A compound according to any one of claims 3 to 7, wherein the compound is [4-(3,4- dimethyl-piperazin-l-yl)-phenyl]-(2,3,7,8-tetramethyl-quinolin-4-yl)-amine, (2- methyl-3-phenyl-quinolin-4-yl)-[4-(3,4,5-trimethyl-piperazin-l-yl)-phenyl]-amine, [4- (3,4-dimethyl-piperazin-l-yl)-ρhenyl]-(2-methyl-3-phenyl-quinolin-4-yl)-amine, (7- chloro-2,3-dimethyl-quinolin-4-yl)-[4-(3,4-dimethyl-piperazin-l-yl)-phenyl]-amine, {4- [4-(3 ,4-dimethyl-piperazin- 1 -yl)-phenylamino] -quinolin-3 -yl} -methanol, {4- [4- (3,4-dimethyl-piperazin-l-yl)-phenylamino]-2-methyl-quinolin-3-yl}-methanol, (2,3- dimethyl-quinolin-4-yl)-[4-(3 ,3 ,4-trimethyl-piperazin- 1 -yl)-phenyl] -amine, {4-[4-(3 ,4- dimethyl-piperazm-l-yl)-phenylamino]-3-methyl-quinolin-2-yl}-methanol, [4-(3,4- dimethyl-piperazin-l-yl)-phenyl]-(2-methyl-l,2,3,4-tetra-hydro- benzo[b] [ 1 ,6]naphthyridin- 10-yl)-amine, (2,3-dimethyl-quinolin-4-yl)-[4-(3 ,3,4,5 ,5- pentamethyl-piperazin- 1 -yl)-phenyl] -amine, {4-[4-(3 ,3 ,4-trimethyl-piperazin- 1 -yl)- phenylamino] -quinolin-3 -yl} -methanol, (3-methyl-quinolin-4-yl)-[4-(353,4-trimethyl- piperazin- 1 -yl)-phenyl]-amine, {7-chloro-4-[4-(3 ,3 ,4-trimethyl-piperazin- 1 -yl)- phenylamino] -quinolin-3 -yl} -methanol, {7-chloro-4-[4-(3,4-dimethyl-piperazin-l-yl)- phenylamino]-quinolin-3-yl}-methanol, (2,3-dimethyl-quinolin-4-yι)-[4-(3,3,4,5- tetramethyl-piperazin- 1 -yl)-phenyl]-amine, {4-[4-(3 ,4-dimethyl-piperazin- 1 -yl)- phenylamino]-7-methyl-quinolin-3-yl}-methanol, (2,3-dimethyl-quinolin-4-yl)-[4-(5-
ethyl-3 ,3 ,4-trimethyl-piperazin- 1 -yl)-phenyl]-amine, {4-[4-(3 ,4-dimethyl-piperazin- 1 - yl)-phenylamino]-6-fluoro-quinolin-3-yl}-methanol, {6-fluoro-4-[4-(3,3,4-trimethyl- piperazin- 1 -yl)-phenylamino] -quinolin-3 -yl} -methanol, [4-(3 ,4-dimethyl-piperazin- 1 - yl)-phenyl]-(7-methoxy-2-methyl-3-phenyl-quinolin-4-yl)-amine, [4-(3,4-dimethyl- piperazin-l-yl)-phenyl]-(2,3,5,7-tetramethyl-quinolin-4-yl)-amine, [4-(3,4-dimethyl- piperazin-l-yl)-phenyl]-(2,3,7-trimethyl-quinolin-4-yl)-amine, (2,3-dimethyl-quinolin- 4-yl)-[4-(octahydro-pyrido[l,2-a]pyrazin-2-yl)-phenyl]-amine, 4- [4-(3 ,4-dimethyl- piperazin- 1 -yl)-phenyl] -3 ,4-dihydro- lH-2-oxa-4,9-di-4-[4-(3 ,4-dimethyl-piperazin- 1 - yl)-phenyl]-3,4-dihydro-lH-2-oxa-4,9-diaza-phenanthrene, [4-(3,4-dimethyl- piperazin-l-yl)-phenyl]-(3-methoxymethyl-quinolin-4-yl)-amine or l-[4-(3-ethyl-2- methyl-quinolin-4-ylamino)-phenyl]-5,5-dimethyl-piperazin-2-one.
9. A compound according to claim 1, wherein
Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined in claim 1;
Y, A, Ri-R3 are as defined in claim 1; one of Ra and Rb is H, OH, halogen, (d-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl, (d-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(C1-C6)alkyl, NO2, NH2, mono- or di d-C^alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1-C6)alkyl, (d-C6)aιkyι-S- or CN and the other of Ra and Rb is (C3-C7)cycloalkyl, (C3-
C7)cycloalkyl(C1-C6)alkyl, (C1-C6)alkoxy-CO-, (C1-C6)alkyl-CO-, mono- or di(d- C6)-alkylcarbamoyl, hydroxy(C1-C6)alkyl, amino(d-C6)alkyl, (C1-C6)alkoxy(C1- C6)alkyl, NH -CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2,
NH , (Cι-Cδ)alkyl, halo(Cι-C6)alkyl, (d-C6)alkoxy or mono-or di(d-C6)alkylamino; or Rb is H, OH, halogen, (Cι-C6)alkyl, (C2-C6)alkenyl, (C2-C6)alkynyl, (Cι-C6)alkoxy, (C1-C6)alkoxy(C1-C6)alkyl, halo(Cι-C6)alkyl, NO2, MI , mono- or di(d- C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1-C6)alkyl, (Ci- C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(C1-C6)alkyl, (d-C6)alkoxy-
CO-, (d-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1-C6)alkyl, NH2-CO-5 -CHO, phenyl or 5 or 6 membered heterocycle, wherein the said (C3- C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO , NH , (d-
C6)alkyl, (C1-C6)alkoxy or mono-or di(C1-C6)alkylamino and Ra and Rt form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1 ; l is O; m is 2; t is O to 3; u is 0; and v is O; with the proviso, that when one of Ra and Rb is H, then the other of Ra and Rb is not ethoxy-CO-.
10. A compound according to claim 9, wherein t is 0 or 1 and R2 is (Cι-C6)alkyl.
11. A compound according to any one of claims 9 or 10, wherein one of Ra and Rb is H or (d-C6)alkyl and the other of Ra and Rb is hydroxy(C1-C6)alkyl or phenyl.
12. A compound according to any one of claims 9 to 11, wherein the compound is (3- aminomethyl-8-methyl-quinolin-4-yl)-[4-(4-methyl-piperazin-l-yl)-phenyl]-amine,
[2,6-dimethyl-4-(4-methyl-piperazin-l-yl)-phenyl]-(2-phenyl-quinolin-4-yl)-amine, (2-methyl-3-phenyl-quinolin-4-yl)-[4-(4-methyl-piperazin-l-yl)-phenyl]-amine or {8- methyl-4-[4-(4-methyl-piperazin-l-yl)-phenylamino]-quinolin-3-yl}-methanol.
13. A compound according to any one of claims 1 or 2, wherein
Q is 1,4-phenylene unsubstituted or substituted with 1 to 3 substituent(s) R5 as defined in claim 1 ;
Y, A and R1-R3 are as defined in claim 1;
Ra and Rb are independently H, OH, halogen, (d-C6)alkyl5 (C2-C6)alkenyl, (C - C6)alkynyl, (d-C6)alkoxy, (Cι-C6)alkoxy(C1-C6)alkyl, halo(d-C6)alkyl, NO2, NH2, mono- or di(d-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1- C6)alkyl, (d-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(d-C6)alkyl, (d- C6)alkoxy-CO-, (C1-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydroxy(C1- C6)alkyl, NH2-CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said
(C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2,
NH2, (d-C6)alkyl, (d-C6)alkoxy or mono-or di(d-C6)alkylamino; or Rb is as defined above and Ra and Ri form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1 ; l is O; m is 2; n is 2; p is 0; t is O to 3; u is 0; and v is 1.
14. A compound according to claim 13, wherein t is 0 or 1 and R is (d-Cg)aιkyl.
15. A compound according to any one of claims 13 or 14, wherein Ra and Rb are independently (d-C6)alkyl or phenyl.
16. A compound according to any one of claims 13 to 15, wherein the compound is (3- ethyl-2,8-dimethyl-quinolin-4-yl)-[4-(4-methyl-[l,4]diazepan-l-yl)-phenyl]-amine, [4- (4-methyl-[l,4]diazepan-l-yl)-phenyl]-(2-methyl-3-phenyl-quinolin-4-yl)-amine, (3- ethyl-2, 8 -dimethyl-quinolin-4-yl)- [4-(3 -methyl-imidazolidin- 1 -yl)-phenyl] -amine, (3 - ethyl-2-methyl-quinolin-4-yl)-[4-(4-methyl-[l,4]diazepan-l-yl)-phenyl]-amine, (2,3- dimethyl-quinolin-4-yl)-[4-(4-methyl-[l,4]diazepan-l-yl)-phenyl]-amine, [4-(4- methyl-[l,4]diazepan-l-yl)-phenyl]-(2,3,8-trimethyl-quinolin-4-yl)-amine or (3-ethyl- 2,8-dimethyl-quinolin-4-yl)-(4-imidazolidin-l-yl-phenyl)-amine.
7. A compound according to claim 1, wherein Q is

L is CH, CR5, orN;
Y, A, Ri-R4 and R6 are as defined in claim 1;
Ra and Rb are independently H, OH, halogen, (C1-C6)alkyl, (C2-C6)alkenyl, (C2- C6)alkynyl, (d-C6)alkoxy, (Cι.-C6)alkoxy(Cι-C6)alkyl, halo(C1-C6)alkyl, NO , NH , mono- or di(C1-C6)alkylamino, mono- or di(C1-C6)alkylamino(C1-C6)alkyl, amino(C1-
C6)alkyl, (Cι-C6)alkyl-S-, CN, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl(Cι-C6)alkyl, (d- C6)alkoxy-CO-, (d-C6)alkyl-CO-, mono- or di(C1-C6)-alkylcarbamoyl, hydiOxy(d- C6)alkyl, NH2-CO-, -CHO, phenyl, 5 or 6 membered heterocycle, wherein the said (C3-C7)cycloalkyl, phenyl or 5 or 6 membered heterocycle is unsubstituted or substituted with 1 to 3 substituent(s) each independently being OH, halogen, NO2,
NH , (d-C6)alkyl, (C1-C6)alkoxy or mono-or di(Cι-C6)alkylamino; or Rb is as defined above and Ra and R\ form, together with the atoms to which they are attached, a condensed 5 to 7 membered heterocyclic ring containing 1 or 2 heteroatoms selected from N, O and S and where the heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1; or Ra and Rb form, together with the carbon ring atoms to which they are attached, a condensed 5 to 7 membered non-aromatic carbocyclic ring or a condensed 5 to 7 membered non-aromatic heterocyclic ring containing at least one heteroatom(s) selected from N, O and S and where the carbo- or heterocyclic ring is unsubstituted or substituted with 1 to 3 substituent(s) R6 as defined in claim 1;
1 is 0 to 2; m is 0 to 2; n is 0 to 2; p is 0 to 2; t is O to 3; u is 0 to 4; and
v is 0 or 1.
18. A compound according to claim 17, wherein L is CH or CR5.
19. A compound according to any one of claims 17 or 18, wherein the compound is [6- (3,4-dimethyl-piperazin-l-yl)-pyridin-3-yl]-(2,3-dimethyl-qumolin-4-yl)-amine or {4- [6-(3,4-dimethyl-piperazin-l-yl)-pyridin-3-ylamino]-quinolin-3-yl}-methanol.
20. A compound according to any one of claims 1 to 19, wherein A is a benzene ring.
21. A compound according to any one of claims 1 to 20, wherein R3 is H or (C1-C6)alkyl.
22. A compound according to claim 21, wherein R3 is (C1-C2)alkyl.
23. A pharmaceutical composition comprising at least one compound according to any one of claims 1 to 22 and a pharmaceutically acceptable diluent, carrier and/or excipient.
24. A compound according to any one of claims 1 to 22 for use as an alpha-2 antagonist.
25. The use of a compound of formula I according to any one of claims 1 to 22 or a pharmaceutically acceptable ester or salt thereof for the manufacture of a medicament for use as an alpha-2 antagonist.
26. The use of a compound according to any one of claims 1 to 22, for the manufacture of a medicament for the treatment of a disorder of the central nervous system, diabetes, orthostatic hypotension, lipolytic disorders, Raynaud's disease or male and female sexual dysfunctions.
27. The use according to claim 26, wherein the disorder of the central nervous system is depression, anxiety disorder, post traumatic stress disorder, schizophrenia, Parkinson's disease, or another movement disorder.
28. The use of a compound according to any one of claims 1 to 22 for the manufacture of a medicament for use as a selective alpha-2C antagonist.
29. The use of a compound according to any one of claims 1 to 22 for the manufacture of a medicament for the treatment of mental disorders propagated by stress, Parkinson's disease, depression, negative symptoms of schizophrenia, attention deficit hyperactivity disorder, post traumatic stress disorder, or anxiety disorders.
30. A method for the treatment of a disease or condition where an antagonist of alpha-2 adrenoceptors is indicated to be useful, which comprises administering to a mammal in need of the treatment an effective amount of at least one compound according to any one of claims 1 to 22.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US44257003P | 2003-01-27 | 2003-01-27 | |
| US60/442,570 | 2003-01-27 | ||
| FI20030120 | 2003-01-27 | ||
| FI20030120A FI20030120A0 (en) | 2003-01-27 | 2003-01-27 | Novel alpha-2 adrenoceptor antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004067513A1 true WO2004067513A1 (en) | 2004-08-12 |
Family
ID=32826894
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FI2004/000038 Ceased WO2004067513A1 (en) | 2003-01-27 | 2004-01-27 | Antagonists for alpha-2 adrenoceptors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2004067513A1 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007071639A1 (en) * | 2005-12-21 | 2007-06-28 | Janssen Pharmaceutica N.V. | Substituted pyrazinone derivatives as alpha2c-adrenoreceptor antagonists |
| US7566786B2 (en) | 2003-05-21 | 2009-07-28 | Glaxo Group Limited | Quinoline derivatives as phosphodiesterase inhibitors |
| WO2010058060A1 (en) | 2008-11-20 | 2010-05-27 | Orion Corporation | Aryl piperazine and their use as alpha2c antagonists |
| WO2011116867A1 (en) | 2010-03-26 | 2011-09-29 | Merck Patent Gmbh | Benzonaphthyridinamines as autotaxin inhibitors |
| JP2013510103A (en) * | 2009-11-07 | 2013-03-21 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Heteroarylaminoquinolines as TGF-beta receptor kinase inhibitors |
| US8492549B2 (en) | 2007-07-20 | 2013-07-23 | Orion Corporation | 2,3-dihydrobenzo(1,4) dioxin-2-ylmethyl derivatives as alpha2C antagonists for use in the treatment of peripheric and central nervous systeme diseases |
| WO2015091414A3 (en) * | 2013-12-19 | 2015-08-20 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyl-tetrahydroquinolines and their use as alpha-2c adrenoreceptor antagonists |
| US9624199B2 (en) | 2013-12-19 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Substituted bipiperidinyl derivatives |
| US9624198B2 (en) | 2013-12-19 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyltetrahydroquinolines |
| CN106187887B (en) * | 2016-07-01 | 2018-08-14 | 上海工程技术大学 | The preparation method of 4- oxyquinoline -3- formic acid |
| WO2023038876A1 (en) * | 2021-09-07 | 2023-03-16 | Gismo Therapeutics, Inc. | Compounds and pharmaceutical compositions comprising inhibitors of amyloid peptide interactions with glycosaminoglycans, methods of treatment, and use thereof |
| US11939328B2 (en) | 2021-10-14 | 2024-03-26 | Incyte Corporation | Quinoline compounds as inhibitors of KRAS |
| CN119930515A (en) * | 2025-01-22 | 2025-05-06 | 南开大学 | 8-Substituted anilinochloroquinoline compounds and preparation method and application thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001064645A2 (en) * | 2000-03-01 | 2001-09-07 | Orion Corporation | Derivatives of quinoline as alpha-2 antagonists |
-
2004
- 2004-01-27 WO PCT/FI2004/000038 patent/WO2004067513A1/en not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001064645A2 (en) * | 2000-03-01 | 2001-09-07 | Orion Corporation | Derivatives of quinoline as alpha-2 antagonists |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7566786B2 (en) | 2003-05-21 | 2009-07-28 | Glaxo Group Limited | Quinoline derivatives as phosphodiesterase inhibitors |
| US7572915B2 (en) | 2003-05-21 | 2009-08-11 | Glaxo Group Limited | Quinoline derivatives as phosphodiesterase inhibitors |
| WO2007071639A1 (en) * | 2005-12-21 | 2007-06-28 | Janssen Pharmaceutica N.V. | Substituted pyrazinone derivatives as alpha2c-adrenoreceptor antagonists |
| US8492549B2 (en) | 2007-07-20 | 2013-07-23 | Orion Corporation | 2,3-dihydrobenzo(1,4) dioxin-2-ylmethyl derivatives as alpha2C antagonists for use in the treatment of peripheric and central nervous systeme diseases |
| US8697723B2 (en) | 2007-07-20 | 2014-04-15 | Orion Corporation | 2,3-dihydrobenzo(1,4) dioxin-2-ylmethyl derivatives as alpha2C antagonists for use in the treatment of peripheric and central nervous system diseases |
| US8809536B2 (en) | 2007-07-20 | 2014-08-19 | Orion Corporation | 2,3-dihydrobenzo[1,4] dioxin-2-ylmethyl derivatives as alpha2C antagonists for use in the treatment of peripheric and central nervous system diseases |
| WO2010058060A1 (en) | 2008-11-20 | 2010-05-27 | Orion Corporation | Aryl piperazine and their use as alpha2c antagonists |
| JP2013510103A (en) * | 2009-11-07 | 2013-03-21 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Heteroarylaminoquinolines as TGF-beta receptor kinase inhibitors |
| US8987301B2 (en) | 2009-11-07 | 2015-03-24 | Merck Patent Gmbh | Heteroarylaminoquinolines as TGF-beta receptor kinase inhibitors |
| WO2011116867A1 (en) | 2010-03-26 | 2011-09-29 | Merck Patent Gmbh | Benzonaphthyridinamines as autotaxin inhibitors |
| US9624199B2 (en) | 2013-12-19 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Substituted bipiperidinyl derivatives |
| EA032250B1 (en) * | 2013-12-19 | 2019-04-30 | Байер Фарма Акциенгезельшафт | Substituted piperidinyl-tetrahydroquinolines and use thereofas alpha-2c adrenoreceptor antagonists |
| WO2015091414A3 (en) * | 2013-12-19 | 2015-08-20 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyl-tetrahydroquinolines and their use as alpha-2c adrenoreceptor antagonists |
| US9624198B2 (en) | 2013-12-19 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyltetrahydroquinolines |
| US9944621B2 (en) | 2013-12-19 | 2018-04-17 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyl tetrahydroquinolines |
| US10961221B2 (en) | 2013-12-19 | 2021-03-30 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyl tetrahydroquinolines |
| AU2014364732B2 (en) * | 2013-12-19 | 2019-03-28 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyl-tetrahydroquinolines and their use as alpha-2C adrenoreceptor antagonists |
| CN106458978A (en) * | 2013-12-19 | 2017-02-22 | 拜耳制药股份公司 | Substituted piperidinyltetrahydroquinolines and their use as alpha-2C adrenergic receptor antagonists |
| US10323020B2 (en) | 2013-12-19 | 2019-06-18 | Bayer Pharma Aktiengesellschaft | Substituted piperidinyl tetrahydroquinolines |
| CN106458978B (en) * | 2013-12-19 | 2019-06-28 | 拜耳制药股份公司 | Substituted piperidinyl tetrahydroquinolines and their use as alpha-2C adrenergic receptor antagonists |
| TWI667234B (en) * | 2013-12-19 | 2019-08-01 | 德商拜耳製藥股份有限公司 | Substituted piperidinyltetrahydroquinolines |
| CN106187887B (en) * | 2016-07-01 | 2018-08-14 | 上海工程技术大学 | The preparation method of 4- oxyquinoline -3- formic acid |
| WO2023038876A1 (en) * | 2021-09-07 | 2023-03-16 | Gismo Therapeutics, Inc. | Compounds and pharmaceutical compositions comprising inhibitors of amyloid peptide interactions with glycosaminoglycans, methods of treatment, and use thereof |
| US11939328B2 (en) | 2021-10-14 | 2024-03-26 | Incyte Corporation | Quinoline compounds as inhibitors of KRAS |
| US12378243B2 (en) | 2021-10-14 | 2025-08-05 | Incyte Corporation | Quinoline compounds as inhibitors of KRAS |
| CN119930515A (en) * | 2025-01-22 | 2025-05-06 | 南开大学 | 8-Substituted anilinochloroquinoline compounds and preparation method and application thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU665366B2 (en) | Benzimidazolone derivatives as 5-HT1A and 5-HT2 antagonists | |
| US7875618B2 (en) | Substituted imidazo[1,5-a]quinoxalines useful as inhibitors of phosphodiesterase 10 for the treatment of neurological and other disorders | |
| EP1218352B1 (en) | Benzimidazolone derivatives having mixed serotonine and dopamine receptors affinity | |
| TW593280B (en) | CRF antagonistic quino- and quinazolines | |
| CN103717593B (en) | Regulate kinase whose compositions and method | |
| JP5419892B2 (en) | Quinazolines and related heterocyclic compounds and their therapeutic use | |
| KR20130041944A (en) | Compounds for the reduction of beta-amyloid production | |
| WO2004067513A1 (en) | Antagonists for alpha-2 adrenoceptors | |
| ZA200206956B (en) | Derivatives of quinoline as alpha-2 antagonists. | |
| CZ20012193A3 (en) | Derivatives of 2,3,4,4a-tetrahydro-1H-pyrazino(1,2-a)quinoxalin-5(6H)one, process of their preparation and their use | |
| CA2777223C (en) | 2h-pyrrol-5-amine derivatives as alpha adrenergic receptor modulators | |
| ZA200106202B (en) | Pirazino (AZA) indole derivatives. | |
| PT2235012E (en) | 4-aminopyrimidine derivatives as histamine h4 receptor antagonists | |
| JP2007512275A (en) | Pyrazolyl and imidazolyl pyrimidine | |
| AU675880B2 (en) | 1(2H-1-benzopyran-2-one-8-yl)-piperazine derivatives | |
| WO1995019968A1 (en) | Aromatic 2-amino-imidazole derivatives as alpha-2a adrenoceptor agonists | |
| SI9520094A (en) | N-substituted 3-azabicyclo (3.2.0)heptane derivatives useful as neuroleptics | |
| HRP950507A2 (en) | 2,7-SUBSTITUTED OCTAHYDRO-1H-PYRIDO(1,2-a)PYRAZINE DERIVATIVES | |
| NZ237921A (en) | Benzothienopyrazine diones; pharmaceutical compositions, methods of preparation and treatment | |
| US6593324B2 (en) | Dervatives of quinoline as alpha-2 antagonists | |
| JPWO2018168894A1 (en) | Deuterated benzimidazole compounds and their pharmaceutical uses | |
| CN120172982A (en) | Benzodihydrofuran tricyclic compound, its pharmaceutical composition and application | |
| AU690645C (en) | Aromatic 2-amino-imidazole derivatives as alpha-2A adrenoceptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 122 | Ep: pct application non-entry in european phase |