WO1998046597A1 - Serine protease inhibitors - Google Patents
Serine protease inhibitors Download PDFInfo
- Publication number
- WO1998046597A1 WO1998046597A1 PCT/US1998/007709 US9807709W WO9846597A1 WO 1998046597 A1 WO1998046597 A1 WO 1998046597A1 US 9807709 W US9807709 W US 9807709W WO 9846597 A1 WO9846597 A1 WO 9846597A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- hydrogen
- iii
- independently selected
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(C(C=C1)=O)N(C(C(CCC2)N2C(C(N**2)N2C(C(N)N[U])=*)=O)C=O)C1(*)O* Chemical compound *C(C(C=C1)=O)N(C(C(CCC2)N2C(C(N**2)N2C(C(N)N[U])=*)=O)C=O)C1(*)O* 0.000 description 4
- NNTUPAINNBMOFI-UHFFFAOYSA-N CC(c1ccc(C)[o]1)=C Chemical compound CC(c1ccc(C)[o]1)=C NNTUPAINNBMOFI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0806—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06026—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atom, i.e. Gly or Ala
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- Proteases are essential enzymes in the life cycle of many viruses and also play a role in various physiological processes. Many of the proteases developed by viruses are so specialized that they differ substantially from the enzymes to be found in the host, and such specialization makes the viral protease an attractive target of drug therapy. Proteases are also used by the body to regulate various processes and an unbalancing of these mechanisms leads to many disease states. Inhibition of protease, such as serine protease, is considered an ideal chemotherapy in such cases.
- Viruses suitable for serine protease inactivation include hepatitis C virus (HCV), [Love, R. A., et al.; Cell 87, 331 (1996); Kim, J. L., et al., Cell 87, 343 (1996)], human cytomeglavirus (HCMV) [ Stevens, J. T., et al, Ewr. J. BioChem. 226, 361 (1994)], various flaviviruses, e.g., yellow fever, various encephalitises [ Fraenkel- Conrat, H., et al., Virology, 2nd Ed., Prentice Hall ⁇ nglewood Cliffs, pp. 102-103 (1998)], as well as HSV-1, HSV-2, VZV, ⁇ BV, HHV-6, and HHV-7.
- HSV-1, HSV-2, VZV, ⁇ BV, HHV-6, and HHV-7 as well as HSV-1, HSV-2,
- human leukocyte elastase is a serine protease which is involved in diseases such as pulmonary emphysema, [Lungarella, G., et al., Exp. Mol. Pathol. 42, 44 (1985); Powers, J. C. Trends BioChem. Sci., I (9), 211 (1976)], arthritis [JanofF, A. et al. (eds.) Neutral Proteases in Human Polymorphonuclear Leukocytes, Urban and Schwartzenberg, Baltimore, p. 390-417, (1978); JanofF, A.
- compounds of formulas I-III are suitable, either as compounds, pharmaceutically acceptable salts or hydrates, pharmaceutical composition ingredients, whether or not as combination with other antivirals, immunomodulators, antibiotics or vaccines.
- Methods of treating infection by such viruses, and methods of preventing infection by such viruses are also disclosed.
- HSV Herpes Simplex Virus in various serotypes, e.g., HSV-1, HSV-2
- This invention relates to compounds of formula I-III, combinations thereof, or pharmaceutically acceptable salts or hydrates thereof, in the inhibition of serine protease encoded by HCV, HCMV, HSV, VZV, EBV, HHV, and flaviviruses involved with yellow fever or encephalitis.
- the compounds of the present invention are also useful for treating diseases without apparent viral etiology, including pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
- X is a single or a double bond
- X is C or N
- R 1 and R 4 are ! independently selected from the groups consisting of
- R 3 is H or (CH 2 ) n -Q, wherein n is an integer between 1 and 5 and Q is
- R 3 is absent; when either Yi or Wi are hydrogen or Yt and Wi are both hydrogens, then Zi is absent, Yi and Wi are not joined to each other and are independently selected from:
- ( ⁇ i) aryl when Yi and Wi are both not hydrogen, they are selected independently from: (i) -CH 2 -; (ii) -CHR 1 -; or (iii) -CR*R 4 -; when either Y 2 or W 2 are hydrogen or Y 2 and W 2 are both hydrogens, then Z 2 is absent, Y 2 and W are not joined to each other and are independently selected from:
- aryl when Y 2 and W 2 are both not hydrogen, they are selected independently from: (i) -CH 2 -; (ii) -CHR 1 -; or (Hi) -CR'R 4 -;
- W 3 is selected from:
- R 1 and R 4 are independently selected from the groups consisting of
- R 3 is H or (CH 2 ) consult-Q, wherein n is an integer between 1 and 5 and Q is
- Zi and Z 2 are 5 both -CH 2 -; or pharmaceutically acceptable salts or hydrates thereof.
- R 1 is independently selected from the groups consisting of
- Compound A is preferred and is defined as follows:
- the present invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising any compound of the present invention, and a pharmaceutically acceptable carrier.
- the pharmaceutical composition of the present invention is useful in the treatment of infections associated with hepatitis C and human cytomeglavirus, encephalitis, pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
- the pharmaceutical composition of the present invention is useful in the inhibition of the serine proteases of HCV, HCMV, HSV, VZV, EBV and HHV.
- the preferred compound of the present infection is compound A, shown below
- the compounds of the present invention may have asymmetric centers and occur as racemates, racemic mixtures and as individual diastereomers or enantiomers, with all isomeric forms being included in the present invention.
- alkyl is intended to include both branched- and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms (Me is methyl, Et is ethyl, Pr is propyl, Bu is butyl).
- aryl is intended to mean phenyl (Ph) or naphthyl.
- the pharmaceutically-acceptable salts of the compounds of Formulas I- III include the conventional non-toxic salts or the quaternary ammonium salts which are formed, e.g., from inorganic or organic acids or bases.
- acid addition salts include acetate, adipate, alginate, aspartate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methansulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, tosylate, and unde
- Base salts include ammonium salts, alkali metal salts such as sodium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth.
- the basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others.
- Schemes I-V for preparing the novel compounds of this invention are presented below.
- Schemes I-V are not limited by any particular substituents employed in the schemes for illustrative purposes. The examples specifically illustrate the application of the following schemes to specific compounds.
- the corresponding alpha-furfuryl amide 11 is synthesized as an intermediate.
- the asymmetric synthesis of the alpha furfuryl amide is carried out by first subjecting 2-furaldehyde 1 to enantioselective alkylation with a chiral sulfonamide-titanate complex, formed with 2, to give 3 ( in the S configuration) with typically high enantiomeric excess. See, e.g., Takahashi, H. et al., Tetrahedron Lett. 30, 7095 (1989), and Yoshioka, M. et al.,
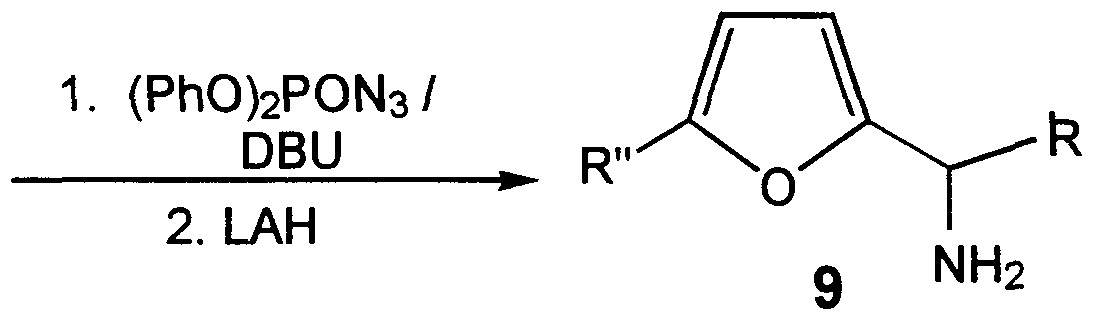
- Scheme II sets forth three racemic pathways for the synthesis of intermediate racemate 9.
- furan 7 is acylated with catalyst ZnCl 2 according to Harough, H.D. et al., J.Am.Chem.Soc. 69, 1012 (1947).
- the acylated furan 8 is then reacted with hydroxylamine to form the corresponding oxime, which in turn is reduced with lithium aluminum hydride (LAH) to give the racemic amine 9.
- LAH lithium aluminum hydride
- racemic amine 9 In a second pathway to synthesize racemic amine 9, 2-fi ⁇ raldehyde 1 is first reacted with the appropriate Grignard reagent to give the racemic alcohol 10. Conversion to the azide, followed by reduction to give racemic amine 9 is accomplished by the method of Thompson, A. et al., J.Org.Chem. 5_8, 5886 (1993).
- a third method of synthesizing racemic amine 9 involves reacting the nonenolizable aldehyde 1 with 1,1,1,3,3,3-hexamethyldisilazane (LiHMDS) to give the corresponding N-trimethylsilyl imine, which is then treated with the appropriate Grignard reagent to give 9. Tosylation of amine 9 gives 11 in Scheme III.
- Oxidative rearrangements of 11 are carried out to isolate 13. See, for example, Scheme III.
- TBHP tert-butyl hyrdoperoxide
- L-(+)-DIPT gives a mixture of 12 in the S configuration and 13 in the R configuration.
- Amide couplings used to form the compounds of this invention are typically performed by the carbodiimide method with reagents such as dicyclohexylcarbodiimide, or l-ethyl-3-(3-dimethylaminopropyl) carbodiimide.
- Other methods of forming the amide or peptide bond include, but are not limited to the synthetic routes via an acid chloride, azide, mixed anhydride or activated ester.
- solution phase amide coupling are performed, but solid-phase synthesis by classical Merrified techniques may be employed instead. The addition and removal of one or more protecting groups is also typical practice.
- the compounds of the present invention are useful in the inhibition of viral serine proteases, the prevention or treatment of infection by the human viruses encoding serine proteases, and the treatment of consequent pathological conditions such as Hepatitis C, yellow fever, viral encephalitis, herpes infection.
- the compounds of the present invention are also useful in the treatment of diseases without apparent viral etiology, including, but not limited to pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
- the compounds of the present invention may be administered orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation spray, topically, intravitreously, or rectally, in dosage unit formulations containing conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles.
- a method of treating and a pharmaceutical composition for treating infection by human viruses encoding serine proteases involves administering to a patient in need of such treatment a pharmaceutical composition comprising a pharmaceutical carrier and a therapeutically effective amount of a compound of the present invention, or a pharmaceutically acceptable salt thereof.
- compositions may be in the form of orally- administrable suspensions or tablets; nasal sprays; sterile injectable preparations, for example, as sterile injectable aqueous or oleaginous suspensions or suppositories.
- these compositions When administered orally as a suspension, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners/flavoring agents known in the art.
- these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art.
- these compositions When administered by nasal aerosol or inhalation, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- the injectable solutions or suspensions may be formulated according to known art, using suitble non-toxic, parenterally-acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitble non-toxic, parenterally-acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- compositions When rectally administered in the form of suppositories, these compositions may be prepared by mixing the drug with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquidity and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquidity and/or dissolve in the rectal cavity to release the drug.
- Dosage levels of the order of 0.02 to 5.0 or 10.0 grams-per-day are useful in the treatment or prevention of the above-indicated conditions, with oral doses two-to-five times higher.
- infection by Hepatitis C virus is effectively treated by the administration of from 10 to 50 milligrams of the compound per kilogram of body weight from one to three times per day.
- the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
- the present invention is also directed to combinations of the viral serine protease inhibitory compounds with one or more agents useful in the treatment of such viruses.
- the compounds of this invention may be effectively administered, whether at periods of pre-exposure and/or post-exposure, in combination with effective amounts of antivirals, immunomodulators, anti-infectives, or vaccines known to those of ordinary skill in the art.
- antivirals, immunomodulators, anti-infectives, or vaccines include in principle any combination with any pharmaceutical composition useful for the treatment of diseases resulting from infection by viruses encoding serine protease.
- 2-Isopropyl-2'-furyl carbinol 2-Furaldehyde (18.25 g, 0.19 mol, 1.00 eq) was dissolved in THF (35 mL) and cooled to 0°C. To this solution, isopropyl magnesium chloride (99.71 mL, 0.21 mol, 2.0 M in THF, 1.05 eq) was added dropwise. After 2 h, TLC followed the consumption of the starting material, ice-cold NH 4 CI (5%; 50 mL) was added. The reaction solution was extracted with EtOAc (150 mL), washed with water (3 x 50 mL), dried (MgSU 4 ) and concentrated to a light yellow oil.
- 2-Isopropyl-2'-furylmethylamine Following the procedure of Thompson, A.S., et al., J. Org. Chem. 58, 5886 (1993), 2-isopropyl-2-furylmethylazide (3.00 g, 18.16 mmol, 1.00 eq) was dissolved in THF (40 mL) and cooled to 0°C. To this solution, LAH (36.00 mL, 36.32 mmol, 2.00 eq) was added dropwise. After 2 h, the reaction was warmed to room temperature. The reaction was cooled to -78°C and quenched with water (50 mL).
- N-(L-Alanyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine L-Alaninyl-L-alanine acetate (1.00 g, 4.95 mmol, 1.00 eq) was dissolved in DMF (15 mL) and THF (30 mL). The reaction mixture was cooled to - 20 °C (acetone/dry ice) and N-methyl morpholine (1.09 mL, 9.91 mmol, 2.00 eq) was added, followed by slow addition of iso-butyl chloroformate (0.70 mL, 5.40 mmol, 1.09 eq).
- N-(L-Prolyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine L-Prolyl-L-alanine Acetate (0.78 g, 3.42 mmol, 1.00 eq) was dissolved in DMF (35 mL). The reaction mixture was cooled to - 20 °C (acetone/dry ice), and N-methyl morpholine (0.75 mL, 6.84 mmol, 2.00 eq) was added followed by slow addition of iso-butyl chloroformate (0.44 mL, 3.42 mmol, 1.00 eq).
- L-Prolyl - L-alaninyl-L-alanine acetate (0.72 g, 2.41 mmol, 1.00 eq) was dissolved in DMF (50 mL). The reaction mixture was cooled to - 20 °C (acetone/dry ice), and N-methyl morpholine (0.58 mL, 5.29 mmol, 2.20 eq) was added followed by slow addition of iso- butyl chloroformate (0.31 mL, 2.41 mmol, 1.00 eq).
- N-(L-alanyl-L-alanine acetate)-2-isopropyl-6-hydroxy-l ,6-dihydro-3-piperidone N-(L-alanyl-L- alanine acetate)-2-isopropyl-2'-fiuylme1hylarnine (18 mg, 0.06 mmol, 1.00 eq) was dissolved in dichloromethane (10 mL). To this solution, MCPBA (9.4 mg, 0.06 mmol, 1.00 eq) was added via four portions over 10 minutes intervals. After 10 hrs, the solvent was removed and the residue was passed through a pad of silica gel.
- N-(L-Prolyl-L-alanine acetate)-2-isopropyl-6-hydroxy- 1 , 6-dihydro-3 -piperidone N-(L- Prolyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine (35 mg, 0.10 mmol, 1.00 eq) was dissolved in dichloromethane (10 mL). To this solution, MCPBA (17 mg, 0.10 mmol, 1.00 eq) was added, After 1.2 hr, the solvent was removed and the residue was passed through a pad of silica gel. EtOAc (50 mL) was used to washed away the impurities followed by washing with acetone (25 mL).
- N- ( ⁇ -Prolyl-L-ala nyl-L-alanine acetate)-2-isopropyl-2-furylmethylamine 46 mg, 0.11 mmol, 1.00 eq
- dichloromethane 10 mL
- MCPBA 24 mg, 0.14 mmol, 1.25 eq
- porcine pancreatic elastase was performed substantially according to Powers, J.C. et al. Biochemistry 29 3108. (1990). Porcine pancreatic elastase (PPE), Suc-Ala-Ala-Ala-NA (N-succinyl-a anyl-alanyl-alanine ?- nitroanilide.), and Hepes were obtained from Sigma Chemical Co., St. Louis, MO.
- a volume of 50 ⁇ L of DMSO and a 50 ⁇ L aliquot of PPE solution (5.0 mg PPE dissolved in 5.0 mL 1 mmol HC1) were added to 0.5 mL Hepes buffer (0.1 M Hepes, 0.5 M NaCl, pH 7.5).
- a 50 ⁇ L aliquot of this solution was added to a solution of a 50 ⁇ L aliquot of substrate solution (20 mmol Suc-Ala-Ala-Ala-NA in DMSO) in 2.0 mL Hepes buffer.
- a 50 ⁇ L aliquot of inhibitor solution (20 mmole in DMSO) and a 50 ⁇ L aliquot of PPE solution were added to 0.5 mL Hepes buffer.
- Compound A was active in the assay, and showed about 20% inhibition at about 17 ⁇ M.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds such as (A) or its pharmaceutically acceptable salts or hydrates, inhibit viral and human serine proteases, and are suitable for the treatment of hepatitis C virus infection, human cytomegalovirus infection, yellow fever, viral encephalitis, pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis, herpes, and immune nephritis.
Description
TITLE OF THE INVENTION SERINE PROTEASE INHIBITORS
BACKGROUND OF THE INVENTION Proteases are essential enzymes in the life cycle of many viruses and also play a role in various physiological processes. Many of the proteases developed by viruses are so specialized that they differ substantially from the enzymes to be found in the host, and such specialization makes the viral protease an attractive target of drug therapy. Proteases are also used by the body to regulate various processes and an unbalancing of these mechanisms leads to many disease states. Inhibition of protease, such as serine protease, is considered an ideal chemotherapy in such cases.
Viruses suitable for serine protease inactivation include hepatitis C virus (HCV), [Love, R. A., et al.; Cell 87, 331 (1996); Kim, J. L., et al., Cell 87, 343 (1996)], human cytomeglavirus (HCMV) [ Stevens, J. T., et al, Ewr. J. BioChem. 226, 361 (1994)], various flaviviruses, e.g., yellow fever, various encephalitises [ Fraenkel- Conrat, H., et al., Virology, 2nd Ed., Prentice Hall Εnglewood Cliffs, pp. 102-103 (1998)], as well as HSV-1, HSV-2, VZV, ΕBV, HHV-6, and HHV-7.
In the host, human leukocyte elastase (HLΕ) is a serine protease which is involved in diseases such as pulmonary emphysema, [Lungarella, G., et al., Exp. Mol. Pathol. 42, 44 (1985); Powers, J. C. Trends BioChem. Sci., I (9), 211 (1976)], arthritis [JanofF, A. et al. (eds.) Neutral Proteases in Human Polymorphonuclear Leukocytes, Urban and Schwartzenberg, Baltimore, p. 390-417, (1978); JanofF, A. Molecular Basis of Biological Degradative Processes, Berlin, R.D., et al., (Eds.), Academic Press, New York, pp. 225-260, (1973)], pancreatitis [Geokas, M.C., et al., Lab. Invest. \9, 235 (1968)], adult respiratory distress syndrome [Burchardi, H., et al., Adv. Exp. Med. Biol. 167, 319 (1984)] and various degenerative skin disorders. The serine proteases involved with these diseases are of great interest as drug targets. Other physiological serine proteases include thrombin, which is implicated in many cardiovascular diseases [Fox, I., et al., Thromb. Haemostasis 69, 157(1993); Maffrand, J.P. Nσwv. Rev. Fr. Hemato. J4, 405 (1992)]; urokinase-type plasminogen activator (UPA), which is known to aid in the metastasis of some types of cancer [Mueller, B. M., Curr. Top. Microbiol. Immuno. 213, 65 (1996); Schmitt, M., et al., J. Obstet. Gynaecol. 2\, 151 (1995)], especially
prostate [Rabbani, S. A., et al., Int. J. Cancer 63, 840, (1995); SofF, G. A., et al., J. Clin. Invest. 96, 2593 (1995)], gastric [Herszenyi, L., et al., ActaPhysiol. Hung, 83, 213 (1995); Plebani, M., et al., Cancer 76, 367 (1995)], and breast [Duffy, M. I, et al., Enzyme Protein 49, 85, (1996); Xing, R. H., et al., Int. J. Cancer 67, 423, (1996)]; and collagenase, which plays a role in destructive corneal disease associated with rheumatoid arthritis (Riley, G. P., et al., Eye 9(6), 703 (1995)]. Serine proteases are also implicated in immune nephritis [Hruby, Z., et al., Int. Urol. Nephrol. 20, 513 (1988); Hruby, Z., et al., Nephrol. Dial. Transplant U, 32, (1996)].
Applicants have discovered new compounds useful for inhibition of serine protease as a drug target.
BRIEF DESCRIPTION OF THE INVENTION Compounds of formulas I-III, as herein defined, are disclosed. These compounds are useful in the inhibition of the serine protease encoded by HCV, HCMV, flaviviruses involved with yellow fever or encephalitis, Herpes Simplex Virus, VZV, Epstein Barr Virus, and HHV. These compounds are also suitable for the treatment of infection by such viruses, the prevention of infection by such viruses, and in the treatment of diseases resulting from infection of such viruses. The compounds of the present invention are also useful for treating diseases without apparent viral etiology, including pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
For such purposes, compounds of formulas I-III are suitable, either as compounds, pharmaceutically acceptable salts or hydrates, pharmaceutical composition ingredients, whether or not as combination with other antivirals, immunomodulators, antibiotics or vaccines. Methods of treating infection by such viruses, and methods of preventing infection by such viruses are also disclosed.
Some abbreviations that may appear in this application follow:
ABBREVIATIONS EBV Epstein Barr Virus HCV Hepatitis C Virus
HCMV Human Cytomegalovirus
HHV Human Herpes Virus
HLE Human Leukocyte Elastase
HSV Herpes Simplex Virus, in various serotypes, e.g., HSV-1, HSV-2
MCPBA 3-Chloroperoxybenzoic acid
UPA Urokinase-type Plasminogen
VZV Varicella Zoster Virus
DETAILED DESCRIPTION OF THE INVENTION
This invention relates to compounds of formula I-III, combinations thereof, or pharmaceutically acceptable salts or hydrates thereof, in the inhibition of serine protease encoded by HCV, HCMV, HSV, VZV, EBV, HHV, and flaviviruses involved with yellow fever or encephalitis. The compounds of the present invention are also useful for treating diseases without apparent viral etiology, including pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
Compounds of formula I are defined as follows:

wherein: X is a single or a double bond;
X is C or N;
R1 and R4 are ! independently selected from the groups consisting of
(i) hydrogen;
(ϋ) Cι.6 alkyl;
(iϋ) aryl;
(iv) Cι-6-OR, wherein R is H, Cι-6 alkyl or aryl;
(v) Cns-SR; and
(vi) C1-6-NR2;
R2 is
(i) OH;
(ϋ) CM alkyl;
(iϋ) O-d-6 alkyl;
(vi) aryl;

(vii) C1-6-NR2;
(i) hydrogen;
(ϋ) Cw alkyl;
(iϋ) aryl; when X is C or when X is N and X is a single bond,
R3 is H or (CH2) n-Q, wherein n is an integer between 1 and 5 and Q is
(i) OH;
(ϋ) NH2;
(iϋ) NHR;
(iv) NR2;
(v) COOH;
(vi) COOR;
(vii) SH;
(vii) S(O)R; or
(vii) SR; when X is N and X is a double bond,
R3is absent; when either Yi or Wi are hydrogen or Yt and Wi are both hydrogens, then Zi is absent, Yi and Wi are not joined to each other and are independently selected from:
(i) hydrogen; (ii) Cι.6 alkyl; or
(ϋi) aryl; when Yi and Wi are both not hydrogen, they are selected independently from: (i) -CH2-; (ii) -CHR1-; or (iii) -CR*R4 -;
when either Y2 or W2 are hydrogen or Y2 and W2 are both hydrogens, then Z2 is absent, Y2 and W are not joined to each other and are independently selected from:
(i) hydrogen; (ii) CM alkyl; or
(ϋi) aryl; when Y2 and W2 are both not hydrogen, they are selected independently from: (i) -CH2-; (ii) -CHR1-; or (Hi) -CR'R4 -;
W3 is selected from:
(i) hydrogen; (ii) Cι-6 alkyl; or (iϋ) aryl; U is selected independently from:
(i) hydrogen; (ii) -C(O)-C1-6 alkyl; (iii) -C(O)-aryl; (iv) -C(O)-O-C1-6 alkyl; (v) -C(O)-O-aryl;
(vi) -C(O)-NH-C1-6 alkyl; or (vii) -C-(O)-NH-aryl; Zi and Z are selected independently from: (i) -CH2-; (ii) -CHR1-;
(iii) -CR*R4-; or (iv) -CH2CH2-; or pharmaceutically acceptable salts or hydrates thereof.
Compounds of the formula II are defined as follows:

wherein:
R1 and R4 are independently selected from the groups consisting of
(i) hydrogen;
(ii) CM alkyl;
(iϋ) aryl;
(iv) Cι-6-OR, wherein R is H, Cι-6 alkyl or aryl;
(v) CM-SR; and
(vi) C1-6-NR2;
R2 is
(i) OH;
(ϋ) C1-6 alkyl;
(ϋi) O-Cι-6 alkyl;
(iv) aryl;
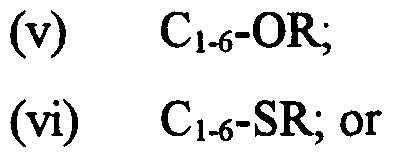
(vϋ) CM-NR2;
R3 is H or (CH2)„-Q, wherein n is an integer between 1 and 5 and Q is
0) OH;
(ϋ) NH2;
(iϋ) NHR;
(iv) NR2;
(v) COOH;
(vi) COOR;
(vϋ) SH;
(viii) S(O)R; or
(ix) SR;
R5 is
0) hydrogen;
(ϋ) C alkyl;
(iii) aryl; when either Yi or Wi are hydrogen or Yt and Wi are both hydrogens, then Zi is absent, Yi and Wi are not joined to each other and are independently selected from:
(i) hydrogen; (ii) C alkyl; or
(iii) aryl; when Yt and Wi are both not hydrogen, they are both -CH2-; when either Y2 or W2 are hydrogen or Y2 and W2 are both hydrogens, then Z2 is absent, Y2 and W2 are not joined to each other and are independently selected from:
(i) hydrogen; (ii) CM alkyl; or (iϋ) aryl; when Y2 and W2 are both not hydrogen, they are both -CH2-; W3 is:
(i) hydrogen
(ϋ) CM alkyl; or
(ϋi) aryl;
U is:
(i) hydrogen;
(ϋ) -C(O)-CM alkyl;
π
(iii) -C(O)-aryl;
(iv) -C(O)-O-CM alkyl;
(v) -C(O)-O-aryl;
(vi) -C(O)-NH-CM alkyl;
(vi) -C(O)-NH-aryl;
Zi and Z2 are 5 both -CH2-; or pharmaceutically acceptable salts or hydrates thereof.
Compounds of the formula III are defined as follows:

R1 is independently selected from the groups consisting of
(i) hydrogen
(ii) CM alkyl;
(iii) aryl;
(iv) CM-OR, wherein R is H, C alkyl or aryl;
(v) CM-SR; and

R2 is
(i) hydrogen; (ii) CM alkyl; or (ϋi) aryl;
when either Yi or Wx are hydrogen or Yi and Wi are both hydrogens, then Z\ is absent, Yi and Wi are not joined to each other and are independently selected from:
(i) hydrogen;
(ii) CM alkyl; or
(ϋi) aryl; when Yi and Wi are both not hydrogen, they are both -CH2-; W2 is:
(i) hydrogen;
(ϋ) CM alkyl; or
(ϋi) aryl;
TT *
(i) hydrogen;
(ϋ) -C(O)-CM alkyl;
(iϋ) -C(O)-aryl;
(iv) -C(O)-O-CM alkyl;
(v) -C(O)-O-aryl;
(vi) -C(O)-NH-CM alkyl; or
(vii) -C(O)-NH-aryl;
 or pharmaceutically acceptable salts or hydrates thereof.
or pharmaceutically acceptable salts or hydrates thereof.
Compound A is preferred and is defined as follows:

IV or pharmaceutically acceptable salts or hydrates thereof.
The present invention also relates to a pharmaceutical composition comprising any compound of the present invention, and a pharmaceutically acceptable carrier.
The pharmaceutical composition of the present invention is useful in the treatment of infections associated with hepatitis C and human cytomeglavirus, encephalitis, pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
The pharmaceutical composition of the present invention is useful in the inhibition of the serine proteases of HCV, HCMV, HSV, VZV, EBV and HHV.
The preferred compound of the present infection is compound A, shown below
Compound A:

The compounds of the present invention, may have asymmetric centers and occur as racemates, racemic mixtures and as individual diastereomers or enantiomers, with all isomeric forms being included in the present invention.
When any variable (e.g., R1, R2) occurs more than one time in any constituent or in formulas I-III, its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. As used herein except where noted, "alkyl" is intended to include both branched- and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms (Me is methyl, Et is ethyl, Pr is propyl, Bu is butyl). As used herein, with exceptions as noted, "aryl" is intended to mean phenyl (Ph) or naphthyl.
The pharmaceutically-acceptable salts of the compounds of Formulas I- III (in the form of water- or oil-soluble or dispersible products) include the conventional non-toxic salts or the quaternary ammonium salts which are formed, e.g., from inorganic or organic acids or bases. Examples of such acid addition salts include acetate, adipate, alginate, aspartate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methansulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, tosylate, and undecanoate. Base salts include ammonium salts, alkali metal salts such as sodium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth. Also, the basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others.
Schemes I-V for preparing the novel compounds of this invention are presented below. Schemes I-V are not limited by any particular substituents employed in the schemes for illustrative purposes. The examples specifically illustrate the application of the following schemes to specific compounds. To synthesize the end group 13 of the present invention, the corresponding alpha-furfuryl amide 11 is synthesized as an intermediate. In Scheme I, the asymmetric synthesis of the alpha furfuryl amide is carried out by first subjecting 2-furaldehyde 1 to enantioselective alkylation with a chiral sulfonamide-titanate complex, formed with 2, to give 3 ( in the S configuration) with typically high enantiomeric excess. See, e.g., Takahashi, H. et al., Tetrahedron Lett. 30, 7095 (1989), and Yoshioka, M. et al.,
Tetrahedron Lett. 30, 1657 (1989). Asymmetric conversion to the azide, followed by reduction to give amine 9 is accomplished by the method of Thompson, A. et al., J.Org.Chem. 58, 5886 (1993). Alternatively, acylated fiiran 5 is reacted with hydroxylamine to form the corresponding oxime, which in turn is reduced with lithium aluminum hydride (LAH) to give amine 6, typically in the S configuration, in the presence of either Noyori's reagent or Mosher's reagent. See, e.g., Smith, H.E., et al., J.Am.Chem.Soc. \0\, 5186 (1979); Hutchins, R.O. et al., J.Org.Chem.52, 704 (1987). Variations in Scheme I can provide either enantiomer. For example, inverting the configuration of both asymmetric carbons in 2 will give 3 in the R configuration.
SCHEME I: Asymmetric Synthesis


NH

L* = Noyori's reagent (Binal-H) Mosher's reagent (Chirald)
Scheme II sets forth three racemic pathways for the synthesis of intermediate racemate 9. In one pathway, furan 7 is acylated with catalyst ZnCl2 according to Harough, H.D. et al., J.Am.Chem.Soc. 69, 1012 (1947). The acylated furan 8 is then reacted with hydroxylamine to form the corresponding oxime, which in turn is reduced with lithium aluminum hydride (LAH) to give the racemic amine 9. See, e.g., Smith, H.E., et al., J.Am.Chem.Soc. 101, 5186 (1979). In a second pathway to synthesize racemic amine 9, 2-fiιraldehyde 1 is first reacted with the appropriate Grignard reagent to give the racemic alcohol 10. Conversion to the azide, followed by reduction to give racemic amine 9 is accomplished by the method of Thompson, A. et
al., J.Org.Chem. 5_8, 5886 (1993). A third method of synthesizing racemic amine 9 involves reacting the nonenolizable aldehyde 1 with 1,1,1,3,3,3-hexamethyldisilazane (LiHMDS) to give the corresponding N-trimethylsilyl imine, which is then treated with the appropriate Grignard reagent to give 9. Tosylation of amine 9 gives 11 in Scheme III.
SCHEME II: Racemic Pathways

0
7 8

9 NH2


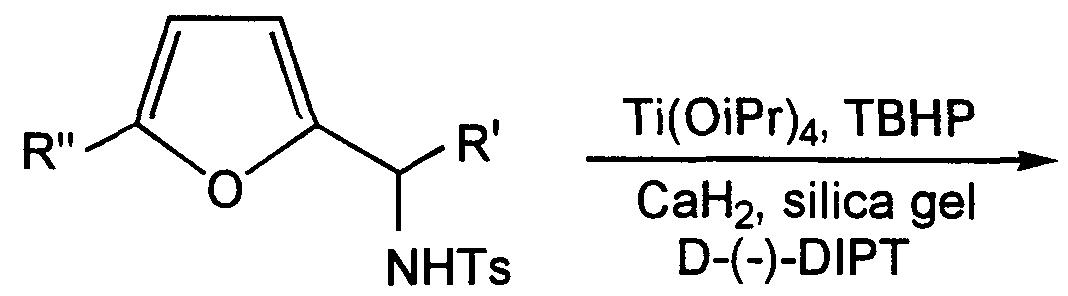
Oxidative rearrangements of 11 are carried out to isolate 13. See, for example, Scheme III. In the methods of Zhou, W.-S. et al., Tetrahedron 49, 2641 (1993), Sharpless asymmetric epόxidation of racemic 11 using tert-butyl hyrdoperoxide (TBHP) in the presence of chiral titanium-tartrate catalyst gives a mixture of 12 and 13.
Alternatively, L-(+)-DIPT gives a mixture of 12 in the S configuration and 13 in the R configuration.
SCHEME III
11

In Scheme IV, oxidation of racemic 11 with MCPBA gives 13 as a mixture of two enantiomers. See, e.g., Zhou, W.-S. et al., Tetrahedron 49, 2641 (1993). Oxidative reaarangement of tosylate 11 with the S configuration provides 13 as one enantiomer, while starting material of the R configuration gives the other enantiomer.
SCHEME IV

11 13 racemic
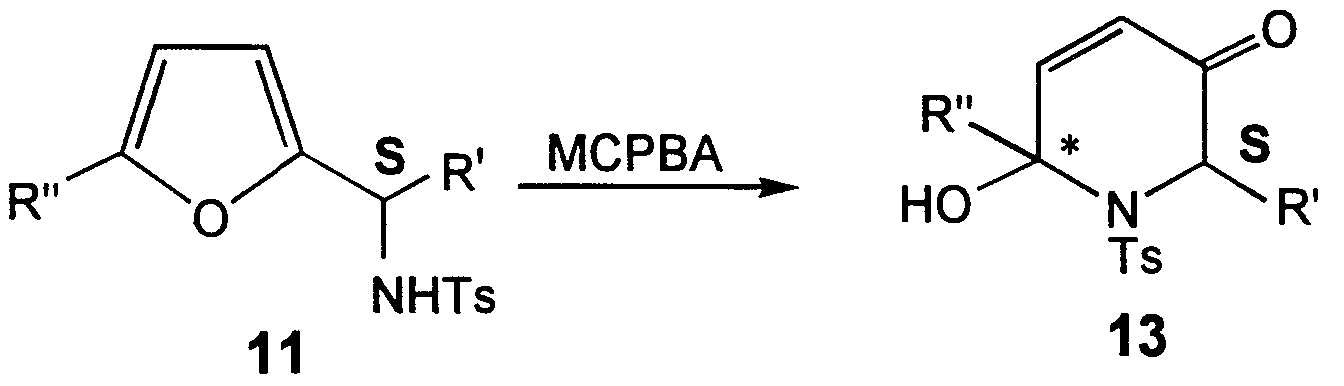

11 13
Amide couplings used to form the compounds of this invention are typically performed by the carbodiimide method with reagents such as dicyclohexylcarbodiimide, or l-ethyl-3-(3-dimethylaminopropyl) carbodiimide. Other methods of forming the amide or peptide bond include, but are not limited to the synthetic routes via an acid chloride, azide, mixed anhydride or activated ester. Typically, solution phase amide coupling are performed, but solid-phase synthesis by classical Merrified techniques may be employed instead. The addition and removal of one or more protecting groups is also typical practice.
A general synthetic approach to compounds such as A is shown in Scheme V. Grignard addition to 2-furaldehyde 1 provides racemic alcohol 10. Displacement with azide followed by LAH reduction gives racemic amine 9. Coupling
of a peptide or peptidomimetic to amine 9 via the mixed anhydride provides racemic 14, which is oxidatively rearranged using MCPBA to give compound 15 as a mixture of two enantiomers.
SCHEME V

ide idomimetic



OH
15
The compounds of the present invention are useful in the inhibition of viral serine proteases, the prevention or treatment of infection by the human viruses encoding serine proteases, and the treatment of consequent pathological conditions such as Hepatitis C, yellow fever, viral encephalitis, herpes infection. The compounds of the present invention are also useful in the treatment of diseases without apparent viral
etiology, including, but not limited to pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
For these purposes, the compounds of the present invention may be administered orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation spray, topically, intravitreously, or rectally, in dosage unit formulations containing conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles.
Thus, in accordance with the present invention there is further provided a method of treating and a pharmaceutical composition for treating infection by human viruses encoding serine proteases. The treatment involves administering to a patient in need of such treatment a pharmaceutical composition comprising a pharmaceutical carrier and a therapeutically effective amount of a compound of the present invention, or a pharmaceutically acceptable salt thereof.
These pharmaceutical compositions may be in the form of orally- administrable suspensions or tablets; nasal sprays; sterile injectable preparations, for example, as sterile injectable aqueous or oleaginous suspensions or suppositories. When administered orally as a suspension, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners/flavoring agents known in the art. As immediate release tablets, these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art. When administered by nasal aerosol or inhalation, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art. The injectable solutions or suspensions may be formulated according to known art, using suitble non-toxic, parenterally-acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution,
or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid. When rectally administered in the form of suppositories, these compositions may be prepared by mixing the drug with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquidity and/or dissolve in the rectal cavity to release the drug.
Dosage levels of the order of 0.02 to 5.0 or 10.0 grams-per-day are useful in the treatment or prevention of the above-indicated conditions, with oral doses two-to-five times higher. For example, infection by Hepatitis C virus is effectively treated by the administration of from 10 to 50 milligrams of the compound per kilogram of body weight from one to three times per day. It will be understood, however, that the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
The present invention is also directed to combinations of the viral serine protease inhibitory compounds with one or more agents useful in the treatment of such viruses. For example, the compounds of this invention may be effectively administered, whether at periods of pre-exposure and/or post-exposure, in combination with effective amounts of antivirals, immunomodulators, anti-infectives, or vaccines known to those of ordinary skill in the art. It will be understood that the scope of combinations of the compounds of this invention with antivirals, immunomodulators, anti-infectives or vaccines include in principle any combination with any pharmaceutical composition useful for the treatment of diseases resulting from infection by viruses encoding serine protease.
EXAMPLE 1

OH
2-Isopropyl-2'-furyl carbinol. 2-Furaldehyde (18.25 g, 0.19 mol, 1.00 eq) was dissolved in THF (35 mL) and cooled to 0°C. To this solution, isopropyl magnesium chloride (99.71 mL, 0.21 mol, 2.0 M in THF, 1.05 eq) was added dropwise. After 2 h, TLC followed the consumption of the starting material, ice-cold NH4CI (5%; 50 mL) was added. The reaction solution was extracted with EtOAc (150 mL), washed with water (3 x 50 mL), dried (MgSU4) and concentrated to a light yellow oil. Purification gave 2-isopropyl-2'-furyl carbinol (15.2 g, 0.11 mol, 57%). Chromatographic purification (Flash column, silica gel, 25 cm x 10 cm, 4/1; hexane/ethyl acetate); 1H NMR (CDC13, 300 MHz): δ 7.27 (d, J=1.8 Hz, 1 H) 6.24 (dd, Jι=3.0 Hz, J2=1.8 Hz, 1H) 6.13 (d, J=3.0 Hz, 1 H), 4.26 (d, J=6.9 Hz, 1H), 2.71 (s, 1 H), 2.00 (sept, J=6.6 Hz, 1 H), 0.92 (d, J=6.6 Hz, 3 H), 0.77 (d, J=6.6 Hz, 3H).
EXAMPLE 2

A.S., et al., J. Org. Chem. 58, 5886 (1993), 2-isopropyl-2'-furyl carbinol (12.50 g, 89.17 mmol, 1.00 eq) was dissolved in toluene (50 mL) and cooled to 0°C. To this
solution, DBU (16.0 mL, 107.01, 1.20 eq) was added followed by the addition of diphenyl phosphorus azide (12.93 mL, 107.01 mmol, 1.20 eq). The reaction mixture was stirred overnight and concentrated to a solid. The residue was passed through a pad of celite, washed with hexane and concentrated. Purification gave 2-isopropyl-2'- furylmethylazide (9.12 g. 55.21 mmol, 62%). Chromatographic purification (Flash column, silica gel, 25 cm x 10 cm, 19/1; hexane/ethyl acetate). 1H NMR (CDC13, 300 Mhz): δ 7.41 (d, J=1.2 Hz, 1 H), 6.37 (dd, Jr=3.0 Hz, J2=1.8 Hz, 1 H), 6.30 (d, J=3.0 Hz, 1 H), 4.11 (d, J-7.2 Hz, 1 H), 2.19 (sept, J=6.6 Hz, 1 H), 1.04 (d, J=6.6 Hz, 3 H), 0.88 (d, J=6.6 Hz, 3H).
EXAMPLE 3
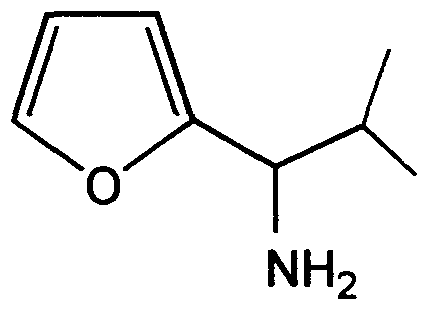
2-Isopropyl-2'-furylmethylamine. Following the procedure of Thompson, A.S., et al., J. Org. Chem. 58, 5886 (1993), 2-isopropyl-2-furylmethylazide (3.00 g, 18.16 mmol, 1.00 eq) was dissolved in THF (40 mL) and cooled to 0°C. To this solution, LAH (36.00 mL, 36.32 mmol, 2.00 eq) was added dropwise. After 2 h, the reaction was warmed to room temperature. The reaction was cooled to -78°C and quenched with water (50 mL). The reaction mixture was extracted with Et2O (3 x 100 mL), washed with water (3 x 35 mL), dried (MgSO4) and concentrated to give 2-isopropyl-2'-furylmethylamine (2.53 g, 18.16 mmol, 100%) 1H NMR (CDC13, 300 MHz): δ 7.33 (d, J=1.8 Hz 1H), 6.30 (dd, Jι=3.0 Hz, J2=1.8 Hz, 1 H), 6.11 (d, J=3.0 Hz, 1 H), 3.69 (d, J=6.2 Hz, 1 H), 2.04 (sept, J=6.6 Hz, 1 H), 1.04 (d, J=6.6 Hz, 3 H), 0.88 (d, J=6.6 Hz, 3H).
EXAMPLE 4
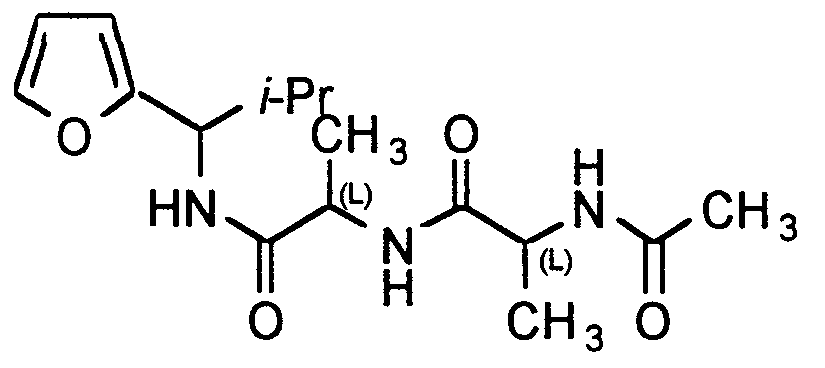
N-(L-Alanyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine. L-Alaninyl-L-alanine acetate (1.00 g, 4.95 mmol, 1.00 eq) was dissolved in DMF (15 mL) and THF (30 mL). The reaction mixture was cooled to - 20 °C (acetone/dry ice) and N-methyl morpholine (1.09 mL, 9.91 mmol, 2.00 eq) was added, followed by slow addition of iso-butyl chloroformate (0.70 mL, 5.40 mmol, 1.09 eq). The reaction mixture was stirred at - 20 °C (acetone/dry ice) for 1.5 hr, then cooled to -40 °C (acetone/dry ice), and 2-isopropyl- 2'-furylmethylamine (0.83 g, 5.93 mmol, 1.20 eq) was added. The reaction was warmed to room temperature to stand overnight. The solvents were removed under reduced pressure. Purification gave N-(L-alanyl-L-alanine acetate)-2-isopropyl-2'- furylmethylamine as a white solid (1.45 g, 4.48 mmol, 91 %). Data: TLC (silica gel, 1 :9; Methano EtOAc, Rf = 0.21); Chromatographic purification ( silica gel, 2.5 cm x 1 0 cm, 100% hexane to 50% methanol in EtOAc); 1H NMR (CD3OD, 300 MHz): 57.40- 7.38 (m, 1 H), 6.33-6.30 (m, 1 H), 6.23-6.16 (m, 1 H), 4.76-4.71 (m, 1 H), 4.41-4.26 (m, 1 H), 2.20-2.13 (m, 1 H), 1.97-1.94 (m, 3), 1.36-1.29 (m, 6H), 0.96-0.82 (m, 6H). 13C NMR (CD3OD, 100 MHz): δ 175.3,175.1, 174.4,173.4,155.6,142.9,142.9, 111.2, 107.8, 107.7, 107.6, 54.8, 54.7, 54.6, 50.9, 50.7, 50.5, 50.4, 33.2, 33.1, 22.6, 20.0,19.4, 19.3, 19.2, 18.5,18.1, 18.0, 17.7, 17.5. Anal. Calcd for Ci6H25N3O4-0.5H2O: C, 57.82; H, 7.58; N, 12.64; Found: C, 58.20; H, 7.82; N, 12.61
EXAMPLE 5

N-(L-Prolyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine. L-Prolyl-L-alanine Acetate (0.78 g, 3.42 mmol, 1.00 eq) was dissolved in DMF (35 mL). The reaction mixture was cooled to - 20 °C (acetone/dry ice), and N-methyl morpholine (0.75 mL, 6.84 mmol, 2.00 eq) was added followed by slow addition of iso-butyl chloroformate (0.44 mL, 3.42 mmol, 1.00 eq). The reaction was stirred at - 5 °C (acetone/dry ice) for 1.5 hr, then cooled to -40 °C (acetone/dry ice), and 2-isopropyl-2'-furylmethylamine (0.50 g, 3.42 mmol, 1.20 eq) was added. The reaction was warmed to room temperature to stand overnight. Work up and purification gave N-(L-prolyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine (0.82 g, 2.35 mmol, 69%). Data: TLC (silica gel, 1:1; Methanol:EtOAc, Rf = 0.40); Chromatographic purification ( silica gel, 2.5 cm x 10 cm, 100% hexane to 33 % ethanol in EtOAc); 1H NMR (CD3OD, 300 MHz): δ 7.40-7.38 (m, 1 H), 6.33- 6.30 (m, 1 H), 6.23-6.16 (m, 1 H), 4.76-4.71 (m, 1 H), 4.41- 4.26 (m, 1 H), 2.20- 2.13 (m, 1 H), 1.97-1.94 (m, 3),1.36-1.29 (m, 6H), 0.96-0.82 (m, 6H).
EXAMPLE 6

N-(L-Prolyl-L-alaninyl-L-alanine acetate)-2-isopropyl-2'-furyl methylamine. L-Prolyl - L-alaninyl-L-alanine acetate (0.72 g, 2.41 mmol, 1.00 eq) was dissolved in DMF (50 mL). The reaction mixture was cooled to - 20 °C (acetone/dry ice), and N-methyl morpholine (0.58 mL, 5.29 mmol, 2.20 eq) was added followed by slow addition of iso-
butyl chloroformate (0.31 mL, 2.41 mmol, 1.00 eq). The reaction was stirred at - 10 °C (acetone/dry ice) for 1.5 hr, then cooled to -40 °C (acetone/dry ice), and 2-isopropyl-2'- furylmethylamine (0.33 g, 2.41 mmol, 1.00 eq) was added. The reaction was warmed to room temperature to stand overnight. The solvents were removed under reduced pressure. Purification gave N-(L-prolyl-L-alaninyl-L-alanine acetate)-2-isopropyl-2'- furylmethylamine (0.83 g, 1.97 mmol, 81 %). Data: TLC (silica gel, EtOAc, Rf = 0.08); Chromatographic purification ( silica gel, 2.5 cm x 10 cm, 50 % hexane in EtOAc to 33% methanol in EtOAc); 1H NMR (CD3OD, 300 MHz): δ 7.43-7.38 (m, 1 H), 6.34- 6.16 (m, 2 H), 4.80-4.32 (m, 3H), 3.82-3.77(m, 1 H), 3.64-3.56 (m, 2H), 2.27-1.99 (m, 8H), 1.35-1.17 (m, 6H), 1.01-0.81(m, 6H). 13CNMR (CD3OD, 100 MHz): δ
174.7,173.8,173.8, 173.6, 173.4, 173.3, 173.3, 173.1, 172.9, 172.7, 155.9, 155.7, 155.5, 143.0, 142.9, 142.8, 111.2, 107.8, 107.7, 107.6, 107.5, 63.0, 62.4, 62.1, 61.8, 61.7, 61.6, 61.5, 61.4, 61.0, 60.6, 54.9, 54.8, 54.7, 54.6, 54.4, 50.5, 50.3, 48.4, 48.3, 48.0, 33.5, 33.4, 33.3, 33.1, 32.9, 32.11, 32.9, 30.8, 30.5, 30.2, 30.1, 26.1, 25.9, 25.7, 25.6, 23.6, 23.5, 22.8, 22.6, 22.6, 22.6, 20.1, 20.0,19.9, 19.6, 19.4, 19.3, 19.0. 18.6, 18.3, 18.2, 17.6, 17.2, 17.1. 17.0. 16.9. 14.6, 15.0.
EXAMPLE 7

N-(L-alanyl-L-alanine acetate)-2-isopropyl-6-hydroxy-l ,6-dihydro-3-piperidone. N-(L-alanyl-L- alanine acetate)-2-isopropyl-2'-fiuylme1hylarnine (18 mg, 0.06 mmol, 1.00 eq) was dissolved in dichloromethane (10 mL). To this solution, MCPBA (9.4 mg, 0.06 mmol, 1.00 eq) was added via four portions over 10 minutes intervals. After 10 hrs, the solvent was removed and the residue was passed through a pad of silica gel. EtOAc (50 mL) was used to washed away the impurities followed by washing with acetone (25 mL). The acetone solution was concentrated to give N-(L-alanyl-L-alanine acetate)-2-isopropyl-6-hydroxy-l,6-dihydro-3-piperidone as a white solid (1 5 mg, 0.04 mmol, 79%). 'HNMR (CD3COCD3, 300 MHz): 67.97-7.01 (m, 4H), 6.09-
6.06 (m, 1 H), 4.35-4.00 (m, 3H), 1.93-1.83 (m, 3H), 1.31-1.14 (m, 3H), 0.95-0.82 (m, 3H). HRMS MET (FAB) Calcd for Cι6H26N3O5: 340.1872. Found: 340.1871.
EXAMPLE 8

N-(L-Prolyl-L-alanine acetate)-2-isopropyl-6-hydroxy- 1 , 6-dihydro-3 -piperidone. N-(L- Prolyl-L-alanine acetate)-2-isopropyl-2'-furylmethylamine (35 mg, 0.10 mmol, 1.00 eq) was dissolved in dichloromethane (10 mL). To this solution, MCPBA (17 mg, 0.10 mmol, 1.00 eq) was added, After 1.2 hr, the solvent was removed and the residue was passed through a pad of silica gel. EtOAc (50 mL) was used to washed away the impurities followed by washing with acetone (25 mL). The acetone solution was concentrated to give a white solid, 25 mg. Chromatographic purification (silica gel, 0.5 cm x 1 cm, EtOAc to 33% acetone) gave N-(L-prolyl-L-alanine acetate)-2-isopropyl-6- hydroxy-l,6-dihydro-3-piperidone (10 mg, 0.03 mmol, 27%) * H NMR (CDC13, 300 MHz): δ 7.33-6.03 (m, 4H), 4.72-4.46 (m, 2H), 4.13-4.01 (m, 2H), 3.82-3.45 (m, 3H), 2.37-1.81 (m, 8H), 1.31-1.26 (m, 3H), 0.99-0.80 (m, 6H).
EXAMPLE 9
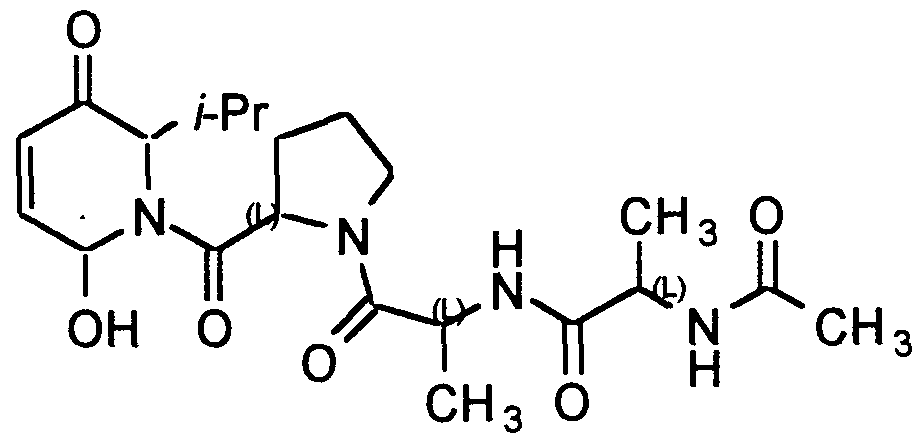
173.3,172.9,172.4,172.2,172.1, 171.9, 171.8,171.6,171.0,170.7,170.1, 155.8, 154.4, 123.0, 122.5, 108.2, 69.8, 61.8, 61.5, 60.6, 60.0, 59.5, 58.5, 58.2, 58.1, 54.0, 49.0, 48.8, 47.4, 47.1, 47.0, 46.7, 46.4, 31.9, 29.9, 29.6, 29.4, 29.3, 29.1, 28.6, 25.4, 25.2, 25.1, 24.8, 24.6, 23.3, 22.7, 21.9, 19.9, 19.3, 19.0, 18.3, 18.1, 17.9, 17.5, 16.5, 14.3. HRMS MLi+ (FAB) Calcd for C21H32N4O6Li: 443.2482. Found: 443.2493.
EXAMPLE 10
Porcine Pancreatic Elastase Assay
The assay for inhibition of porcine pancreatic elastase was performed substantially according to Powers, J.C. et al. Biochemistry 29 3108. (1990). Porcine pancreatic elastase (PPE), Suc-Ala-Ala-Ala-NA (N-succinyl-a anyl-alanyl-alanine ?- nitroanilide.), and Hepes were obtained from Sigma Chemical Co., St. Louis, MO. A volume of 50 μL of DMSO and a 50 μL aliquot of PPE solution (5.0 mg PPE dissolved in 5.0 mL 1 mmol HC1) were added to 0.5 mL Hepes buffer (0.1 M Hepes, 0.5 M NaCl, pH 7.5). A 50 μL aliquot of this solution was added to a solution of a 50 μL aliquot of substrate solution (20 mmol Suc-Ala-Ala-Ala-NA in DMSO) in 2.0 mL Hepes buffer. For tubes containing inhibitor, a 50 μL aliquot of inhibitor solution (20 mmole in DMSO) and a 50 μL aliquot of PPE solution were added to 0.5 mL Hepes buffer. Mixture was incubated for 10 minutes before a 50 μL aliquot was added to a
solution of a 50 μL aliquot of substrate solution in 2.0 mL Hepes buffer. 4-Nitroanilide hydrolysis was measured at 410 nm (ε = 8800 M-1 cm _1) using a spectrophotometer. Slopes obtained for inhibitors were compared those of the corresponding Vn's.
Compound A was active in the assay, and showed about 20% inhibition at about 17 μM.
EXAMPLE 11 Human Cytomegalovirus Protease Assay Inhibition of human cytomegalovirus protease is performed according to
Pinko, C. et al., . J.Biol.Chem 270, 23634 (1995), using either the HPLC-based peptide assay or the continuous RET fluorogenic assay.
While the foregoing specification teaches the principles of the present invention, with examples provided for the purpose of illustration, it will be understood that the practice of the invention encompasses all of the usual variations, adaptations, or modifications, as come within the scope of the following claims and its equivalents.
Claims
WHAT IS CLAIMED IS:
A compound of the formula 1

X is C or N;
R1 and R4 are ! independently selected from the groups consisting of
(i) hydrogen;
(ϋ) CM alkyl;
(ϋi) aryl;
(iv) CM-OR, wherein R is H, CM alkyl or aryl;

(vi) C1-6-NR2;
R2 is
(i) OH;
(ϋ) CM alkyl;
(iϋ) O-CM alkyl;
(vi) aryl;
(v) CMOR
 (vii) CM-NR2
(vii) CM-NR2
R5 is
(i) hydrogen;
(ϋ) C alkyl;
(iϋ) aryl; when X is C or when X is N and X is a single bond,
R3 is H or (CH2) „-Q, wh is
(i) OH;
(ϋ) NH2;
(iϋ) NHR;
(iv) NR2;
(v) COOH;
(vi) COOR;
(vϋ) SH;
(vϋ) S(O)R; or
(vϋ) SR; when X is N and X '. is a double bond,
R3 is absent; when either Yi or Wi are hydrogen or Yi and Wi are both hydrogens, then Zi is absent, Yi and Wi are not joined to each other and are independently selected from: (i) hydrogen;
(ii) CM alkyl; or
(i ) aryl; when Yi and Wt are both not hydrogen, they are selected independently from:
(i) -CH2-; (ii) -CHR1-; or
(iii) -CRJR4 -;
when either Y2 or W2 are hydrogen or Y2 and W2 are both hydrogens, then Z2 is absent, Y2 and W2 are not joined to each other and are independently selected from:
(i) hydrogen; (ii) CM alkyl; or
(i ) aryl; when Y2 and W are both not hydrogen, they are selected independently from: (i) -CH2-; (ii) -CHR1-; or (iii) -CRJR4 -;
W3 is selected from:
(i) hydrogen; (ii) CM alkyl; or (iϋ) aryl; U is selected independently from:
(i) hydrogen; (ii) -C(O)-CM alkyl; (iii) -C(O)-aryl; (iv) -C(O)-O-CM alkyl; (v) -C(O)-O-aryl;
(vi) -C(O)-NH-CM alkyl; or (vii) -C-(O)-NH-aryl; Zi and Z2 are selected independently from: (i) -CH2-; (ii) -CHR1-;
(iii) -C^R4-; or (iv) -CH2CH2-; or pharmaceutically acceptable salts or hydrates thereof.
2. A compound of the formula II
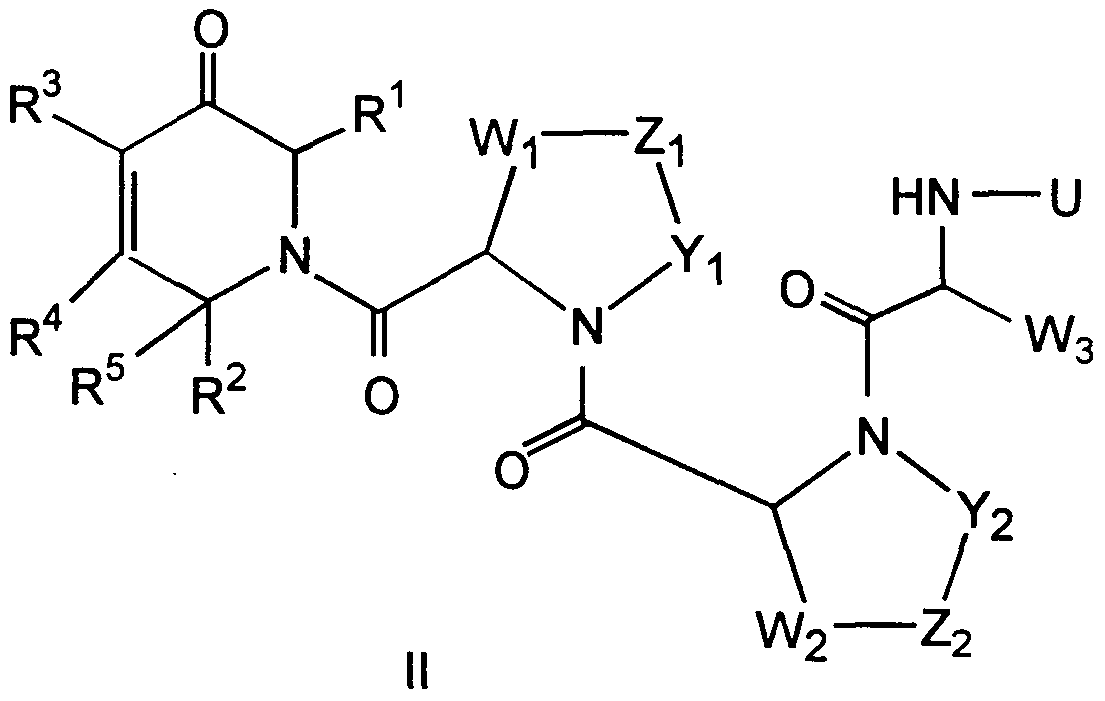
R1 and R4 are independently selected from the groups consisting of
(0 hydrogen;
(ϋ) CM alkyl;
(i ) aryl;
(iv) CM-OR, wherein R is H, CM alkyl or aryl;

(vi) CM-NR2;
R2 is
(i) OH;
(ϋ) CM alkyl;
(iϋ) O-CM alkyl;
(iv) aryl;
(v) CM-OR;
(vi) CM-SR; or
(vϋ) CM-NR2;
R3 is H or (CH2)„-Q, wherein n is an integer between 1 and 5 and C
0) OH;
(ϋ) NH2;
(iϋ) NHR;
(iv) NR2;
(v) COOH;
(vi) COOR;
(v ) SH;
(viii) S(O)R; or
(ix) SR;
R5 is
(i) hydrogen;
(ϋ) CM alkyl;
(iii) aryl; when either Yi or Wi are hydrogen or Yi and Wi are both hydrogens, then Z\ is absent, Yi and W! are not joined to each other and are independently selected from:
(i) hydrogen; (ii) CM alkyl; or
(iii) aryl; when Yi and Wi are both not hydrogen, they are both -CH2-; when either Y2 or W are hydrogen or Y2 and W2 are both hydrogens, then Z is absent, Y2 and W2 are not joined to each other and are independently selected from:
(i) hydrogen; (ii) CM alkyl; or (ϋi) aryl; when Y2 and W2 are both not hydrogen, they are both -CH2-; W3 is:
(i) hydrogen (ii) CM alkyl; or (iii) aryl; U is: (i) hydrogen;
(ii) -C(O)-CM alkyl;
(iii) -C(O)-aryl;
(iv) -C(O)-O-C1-6 alkyl;
(v) -C(O)-O-aryl;
(vi) -C(O)-NH-CM alkyl;
(vi) -C(O)-NH-aryl;
Zi and Z are s both -CH2-; or pharmaceutically acceptable salts or hydrates thereof.
3. A compound of the formula III

wherein:
R1 is independently selected from the groups consisting of
0) hydrogen
(ϋ) CM alkyl;
(iϋ) aryl;
(iv) CM-OR, wherein R is H, CM alkyl or aryl;
(v) CM-SR; and
(vi) Cι-6-NR2;
R2 is
(i) hydrogen;
(ϋ) CM alkyl; or
(iϋ) aryl;
when either Y or Wi are hydrogen or Yi and Wi are both hydrogens, then Zi is absent, Yi and Wi are not joined to each other and are independently selected from:
(i) hydrogen;
(ϋ) CM alkyl; or
(iϋ) aryl; when Yi and WΪ are both not hydrogen, they are both -CH2-;
W2 is:
(i) hydrogen;
(ϋ) CM alkyl; or
(iϋ) aryl;
U is:
(i) hydrogen;
(ϋ) -C(O)-CM alkyl;
(iii) -C(O)-aryl;
(iv) -C(O)-O-CM alkyl;
(v) -C(O)-O-aryl;
(vi) -C(O)-NH-CM alkyl; or
(vii) -C(O)-NH-aryl;
Zx is -CH2-; or pharmaceutically acceptable salts or hydrates thereof.
4. The compound IV
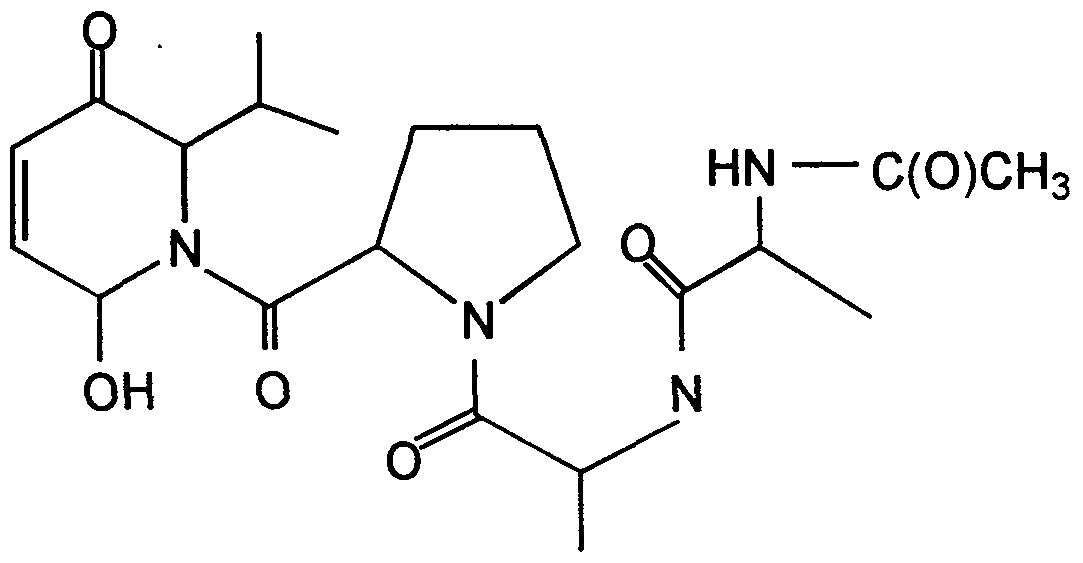
IV
or pharmaceutically acceptable salts or hydrates thereof.
5. A pharmaceutical composition comprising a compound of any of claims 1 - 4 and a pharmaceutically acceptable carrier.
6. The pharmaceutical composition of claim 5 useful in the treatment of infections i associated with hepatitis C and human cytomeglavirus, encephalitis, pulmonary emphysema, cardiovascular disease, cancer, rheumatoid arthritis and immune nephritis.
7. The pharmaceutical composition of claim 5 useful in the inhibition of the serine proteases of HCV, HCMV, HSV, VZV, EBV and HHV.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU71272/98A AU7127298A (en) | 1997-04-14 | 1998-04-14 | Serine protease inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US4364097P | 1997-04-14 | 1997-04-14 | |
| US60/043,640 | 1997-04-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998046597A1 true WO1998046597A1 (en) | 1998-10-22 |
Family
ID=21928148
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/007709 Ceased WO1998046597A1 (en) | 1997-04-14 | 1998-04-14 | Serine protease inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| AU (1) | AU7127298A (en) |
| WO (1) | WO1998046597A1 (en) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000051625A1 (en) * | 1999-03-05 | 2000-09-08 | The Trustees Of University Technology Corporation | Inhibitors of serine protease activity, methods and compositions for treatment of herpes viruses |
| US6143715A (en) * | 1997-08-11 | 2000-11-07 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptide analogues |
| WO2000051623A3 (en) * | 1999-03-05 | 2000-12-14 | Trustees Of University Technol | Inhibitors of serine protease activity, methods and compositions for treatment of nitric oxide-induced clinical conditions |
| WO2000051624A3 (en) * | 1999-03-05 | 2000-12-28 | Trustees Of University Technol | Methods and compositions useful in inhibiting apoptosis |
| WO2000052034A3 (en) * | 1999-03-05 | 2001-01-11 | Trustees Of University Technol | Inhibitors of serine protease activity, methods and compositions for treatment of viral infections |
| US6608027B1 (en) | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
| US6642204B2 (en) | 2002-02-01 | 2003-11-04 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor tri-peptides |
| US6767991B1 (en) | 1997-08-11 | 2004-07-27 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptides |
| US6869964B2 (en) | 2002-05-20 | 2005-03-22 | Bristol-Myers Squibb Company | Heterocyclicsulfonamide hepatitis C virus inhibitors |
| US6878722B2 (en) | 2002-05-20 | 2005-04-12 | Bristol-Myers Squibb Company | Substituted cycloalkyl P1′ hepatitis C virus inhibitors |
| WO2005037214A2 (en) | 2003-10-14 | 2005-04-28 | Intermune, Inc. | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of hcv replication |
| US6995174B2 (en) | 2002-05-20 | 2006-02-07 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7041698B2 (en) | 2002-05-20 | 2006-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7091184B2 (en) | 2002-02-01 | 2006-08-15 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor tri-peptides |
| US7119072B2 (en) | 2002-01-30 | 2006-10-10 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis C virus |
| US7132504B2 (en) | 2003-11-12 | 2006-11-07 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7135462B2 (en) | 2003-11-20 | 2006-11-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2007044893A2 (en) | 2005-10-11 | 2007-04-19 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis c viral replication |
| WO2007119889A1 (en) | 2006-04-18 | 2007-10-25 | Japan Tobacco Inc. | Novel piperazine compound, and use thereof as hcv polymerase inhibitor |
| US7309708B2 (en) | 2003-11-20 | 2007-12-18 | Birstol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7323447B2 (en) * | 2005-02-08 | 2008-01-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| BG65356B1 (en) * | 1999-04-06 | 2008-03-31 | Boehringer Ingelheim (Canada) Ltd. | Marcrocyclic peptides active against the hepatitis c virus,pharmaceutical compositions containing them, and methods for obtaining and use thereof |
| US7504378B2 (en) | 2002-10-25 | 2009-03-17 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7511157B2 (en) | 2004-07-20 | 2009-03-31 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor dipeptide analogs |
| US7585845B2 (en) | 2003-05-21 | 2009-09-08 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| EP2103623A2 (en) | 2005-07-25 | 2009-09-23 | Intermune, Inc. | Novel macrocyclic inhibitors of Hepatitis C virus replication |
| US7642235B2 (en) | 2003-09-22 | 2010-01-05 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| US7696242B2 (en) | 2004-07-20 | 2010-04-13 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor peptide analogs |
| US7749961B2 (en) | 2004-01-21 | 2010-07-06 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| EP2206715A1 (en) | 2004-02-24 | 2010-07-14 | Japan Tobacco, Inc. | Fused heterotetracyclic compounds and use thereof as hcv polymerase inhibitor |
| US20110046066A1 (en) * | 2008-01-11 | 2011-02-24 | Genentech, Inc. | Inhibitors of iap |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| EP2399988A2 (en) | 2006-08-11 | 2011-12-28 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Cell culture system for replication of HCV through the farnesoid X receptor (FXR) activation or inhibition and diagnostic method for HCV infection |
| US8178491B2 (en) | 2007-06-29 | 2012-05-15 | Gilead Sciences, Inc. | Antiviral compounds |
| WO2012107589A1 (en) | 2011-02-11 | 2012-08-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment and prevention of hcv infections |
| EP2520654A1 (en) | 2003-08-26 | 2012-11-07 | The Regents of the University of Colorado | Inhibitors of serine protease activity and their use in methods and compositions for treatment of bacterial infections |
| US8513186B2 (en) | 2007-06-29 | 2013-08-20 | Gilead Sciences, Inc. | Antiviral compounds |
| US8609845B2 (en) | 2004-12-20 | 2013-12-17 | Genentech, Inc. | Pyrrolidine inhibitors of IAP |
| US8835393B2 (en) | 2008-08-02 | 2014-09-16 | Genentech, Inc. | Inhibitors of IAP |
| US8907092B2 (en) | 2007-04-30 | 2014-12-09 | Genentech, Inc. | Inhibitors of IAP |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4251438A (en) * | 1979-06-14 | 1981-02-17 | The Upjohn Company | Piperazinone and piperazine polypeptides |
| US4534897A (en) * | 1980-05-27 | 1985-08-13 | The Upjohn Company | Piperazinone, piperazine, 1,4-diazepin-2-one and 1,4-diazepine intermediate compounds |
| EP0284942A2 (en) * | 1987-04-03 | 1988-10-05 | MERCK PATENT GmbH | Amino-acid derivatives |
| US5190922A (en) * | 1991-06-04 | 1993-03-02 | Abbott Laboratories | Terminally modified tri-, tetra- and pentapeptide anaphylatoxin receptor ligands |
| US5340802A (en) * | 1989-06-30 | 1994-08-23 | Abbott Laboratories | Peptide analog type-B CCK receptor ligands |
| WO1995007934A2 (en) * | 1993-09-14 | 1995-03-23 | Chiron Corporation | Il8 inhibitors |
-
1998
- 1998-04-14 WO PCT/US1998/007709 patent/WO1998046597A1/en not_active Ceased
- 1998-04-14 AU AU71272/98A patent/AU7127298A/en not_active Abandoned
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4251438A (en) * | 1979-06-14 | 1981-02-17 | The Upjohn Company | Piperazinone and piperazine polypeptides |
| US4534897A (en) * | 1980-05-27 | 1985-08-13 | The Upjohn Company | Piperazinone, piperazine, 1,4-diazepin-2-one and 1,4-diazepine intermediate compounds |
| EP0284942A2 (en) * | 1987-04-03 | 1988-10-05 | MERCK PATENT GmbH | Amino-acid derivatives |
| US5340802A (en) * | 1989-06-30 | 1994-08-23 | Abbott Laboratories | Peptide analog type-B CCK receptor ligands |
| US5190922A (en) * | 1991-06-04 | 1993-03-02 | Abbott Laboratories | Terminally modified tri-, tetra- and pentapeptide anaphylatoxin receptor ligands |
| WO1995007934A2 (en) * | 1993-09-14 | 1995-03-23 | Chiron Corporation | Il8 inhibitors |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6767991B1 (en) | 1997-08-11 | 2004-07-27 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptides |
| US6143715A (en) * | 1997-08-11 | 2000-11-07 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptide analogues |
| US8071551B2 (en) | 1999-03-05 | 2011-12-06 | BioHolding, Inc. | Methods and compositions for treating diabetes |
| WO2000051624A3 (en) * | 1999-03-05 | 2000-12-28 | Trustees Of University Technol | Methods and compositions useful in inhibiting apoptosis |
| WO2000052034A3 (en) * | 1999-03-05 | 2001-01-11 | Trustees Of University Technol | Inhibitors of serine protease activity, methods and compositions for treatment of viral infections |
| WO2000051623A3 (en) * | 1999-03-05 | 2000-12-14 | Trustees Of University Technol | Inhibitors of serine protease activity, methods and compositions for treatment of nitric oxide-induced clinical conditions |
| US7704958B1 (en) | 1999-03-05 | 2010-04-27 | Bio Holding, Inc. | Methods and compositions for inhibiting apoptosis using serine protease inhibitors |
| WO2000051625A1 (en) * | 1999-03-05 | 2000-09-08 | The Trustees Of University Technology Corporation | Inhibitors of serine protease activity, methods and compositions for treatment of herpes viruses |
| US6608027B1 (en) | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
| BG65356B1 (en) * | 1999-04-06 | 2008-03-31 | Boehringer Ingelheim (Canada) Ltd. | Marcrocyclic peptides active against the hepatitis c virus,pharmaceutical compositions containing them, and methods for obtaining and use thereof |
| US7119072B2 (en) | 2002-01-30 | 2006-10-10 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis C virus |
| US6642204B2 (en) | 2002-02-01 | 2003-11-04 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor tri-peptides |
| US7091184B2 (en) | 2002-02-01 | 2006-08-15 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor tri-peptides |
| US6869964B2 (en) | 2002-05-20 | 2005-03-22 | Bristol-Myers Squibb Company | Heterocyclicsulfonamide hepatitis C virus inhibitors |
| US7041698B2 (en) | 2002-05-20 | 2006-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US6995174B2 (en) | 2002-05-20 | 2006-02-07 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US6878722B2 (en) | 2002-05-20 | 2005-04-12 | Bristol-Myers Squibb Company | Substituted cycloalkyl P1′ hepatitis C virus inhibitors |
| US7504378B2 (en) | 2002-10-25 | 2009-03-17 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7585845B2 (en) | 2003-05-21 | 2009-09-08 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US7939667B2 (en) | 2003-05-21 | 2011-05-10 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US8067438B2 (en) | 2003-05-21 | 2011-11-29 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| EP3192872A1 (en) | 2003-08-26 | 2017-07-19 | The Regents of the University of Colorado, a body corporate | Inhibitors of serine protease activity and their use in methods and compositions for treatment of bacterial infections |
| EP2520654A1 (en) | 2003-08-26 | 2012-11-07 | The Regents of the University of Colorado | Inhibitors of serine protease activity and their use in methods and compositions for treatment of bacterial infections |
| US7642235B2 (en) | 2003-09-22 | 2010-01-05 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| EP2407470A2 (en) | 2003-10-14 | 2012-01-18 | F. Hoffmann-La Roche Ltd. | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of HCV replication |
| WO2005037214A2 (en) | 2003-10-14 | 2005-04-28 | Intermune, Inc. | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of hcv replication |
| US7132504B2 (en) | 2003-11-12 | 2006-11-07 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7309708B2 (en) | 2003-11-20 | 2007-12-18 | Birstol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7135462B2 (en) | 2003-11-20 | 2006-11-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7749961B2 (en) | 2004-01-21 | 2010-07-06 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| EP2206715A1 (en) | 2004-02-24 | 2010-07-14 | Japan Tobacco, Inc. | Fused heterotetracyclic compounds and use thereof as hcv polymerase inhibitor |
| US7696242B2 (en) | 2004-07-20 | 2010-04-13 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor peptide analogs |
| US7511157B2 (en) | 2004-07-20 | 2009-03-31 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor dipeptide analogs |
| US7767818B2 (en) | 2004-07-20 | 2010-08-03 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor dipeptide analogs |
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| US8609845B2 (en) | 2004-12-20 | 2013-12-17 | Genentech, Inc. | Pyrrolidine inhibitors of IAP |
| US9040706B2 (en) | 2004-12-20 | 2015-05-26 | Genentech, Inc. | Pyrrolidine inhibitors of IAP |
| US7323447B2 (en) * | 2005-02-08 | 2008-01-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP2305698A2 (en) | 2005-07-25 | 2011-04-06 | Intermune, Inc. | Macrocyclic inhibitors of Hepatitis C virus replication |
| EP2103623A2 (en) | 2005-07-25 | 2009-09-23 | Intermune, Inc. | Novel macrocyclic inhibitors of Hepatitis C virus replication |
| EP2305696A2 (en) | 2005-07-25 | 2011-04-06 | Intermune, Inc. | Macrocyclic inhibitors of Hepatitis C virus replication |
| EP2305697A2 (en) | 2005-07-25 | 2011-04-06 | Intermune, Inc. | Macrocyclic inhibitors of Hepatitis C virus replication |
| EP2305695A2 (en) | 2005-07-25 | 2011-04-06 | Intermune, Inc. | Macrocyclic inhibitors of Hepatitis C virus replication |
| WO2007044893A2 (en) | 2005-10-11 | 2007-04-19 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis c viral replication |
| WO2007119889A1 (en) | 2006-04-18 | 2007-10-25 | Japan Tobacco Inc. | Novel piperazine compound, and use thereof as hcv polymerase inhibitor |
| EP2399575A2 (en) | 2006-08-11 | 2011-12-28 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods, uses and compositions for treatment of an infection by a virus of the family of flaviviridae through the farnesoid X receptor (FXR) inhibition |
| EP2399988A2 (en) | 2006-08-11 | 2011-12-28 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Cell culture system for replication of HCV through the farnesoid X receptor (FXR) activation or inhibition and diagnostic method for HCV infection |
| US8907092B2 (en) | 2007-04-30 | 2014-12-09 | Genentech, Inc. | Inhibitors of IAP |
| US8513186B2 (en) | 2007-06-29 | 2013-08-20 | Gilead Sciences, Inc. | Antiviral compounds |
| US8178491B2 (en) | 2007-06-29 | 2012-05-15 | Gilead Sciences, Inc. | Antiviral compounds |
| US8809266B2 (en) | 2007-06-29 | 2014-08-19 | Gilead Sciences, Inc. | Antiviral compounds |
| US8809267B2 (en) | 2007-06-29 | 2014-08-19 | Gilead Sciences, Inc. | Antiviral compounds |
| US20110046066A1 (en) * | 2008-01-11 | 2011-02-24 | Genentech, Inc. | Inhibitors of iap |
| US8835393B2 (en) | 2008-08-02 | 2014-09-16 | Genentech, Inc. | Inhibitors of IAP |
| WO2012107589A1 (en) | 2011-02-11 | 2012-08-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment and prevention of hcv infections |
Also Published As
| Publication number | Publication date |
|---|---|
| AU7127298A (en) | 1998-11-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1998046597A1 (en) | Serine protease inhibitors | |
| EP0393457B1 (en) | Proteinase inhibitor | |
| US4935404A (en) | Phosphorus containing peptides, pharmaceutical compositions and method of treating collagenolytic conditions | |
| US5891877A (en) | Pharmaceutical compounds | |
| US5959123A (en) | 3,4-Disubstituted azetidin-2-one derivatives useful as cysteine proteinase regulators | |
| JPH10265452A (en) | Phenylsulfonamide derivative | |
| EP0769498B1 (en) | Sulfonamide derivatives with elastase inhibiting activity | |
| JPH04352757A (en) | Amino acid derivative | |
| EP0761680A2 (en) | Tetrazole compounds having Interleukin-1beta converting enzyme inhibitory activity | |
| US5061806A (en) | Phosphinic acid derivatives | |
| EP1638925A1 (en) | Protease inhibitors | |
| KR100347646B1 (en) | Matrix metalloprotease inhibitor | |
| JPH115780A (en) | Selective thrombin inhibitor | |
| KR940003297B1 (en) | 1,3,2-dioxathioene oxide derivative | |
| JP4384277B2 (en) | Substituted 6- and 7-aminotetrahydroisoquinolinecarboxylic acids | |
| KR19990028948A (en) | Piperazine Derivatives and Uses thereof | |
| CA2252086A1 (en) | 6-substituted amino-4-oxa-1-azabicyclo¬3,2,0|heptan-7-one derivatives as cysteine protease inhibitors | |
| AU7480800A (en) | Azacycloalkanone serine protease inhibitors | |
| AU3865099A (en) | Amino acid amidinohydrazones, alkoxyguanidines and aminoguanidines as protease inhibitors | |
| AU722680B2 (en) | Sulphur derivatives with a retroamide bond as endothelin-coating enzyme inhibitors | |
| JPH0581586B2 (en) | ||
| US5773428A (en) | Matrix metalloprotease inhibitors | |
| US6034095A (en) | Ketomethylene group-containing cysteine and serine protease inhibitors | |
| JPH10204054A (en) | Phenylsulfonamide derivative | |
| AU5675998A (en) | Substituted amino bicyclic-beta-lactam penam and cepham derivatives as cysteine protease inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: JP Ref document number: 1998543289 Format of ref document f/p: F |