WO1998022432A1 - Novel acylamino-substituted acylanilide derivatives or pharmaceutical composition comprising the same - Google Patents
Novel acylamino-substituted acylanilide derivatives or pharmaceutical composition comprising the same Download PDFInfo
- Publication number
- WO1998022432A1 WO1998022432A1 PCT/JP1997/004174 JP9704174W WO9822432A1 WO 1998022432 A1 WO1998022432 A1 WO 1998022432A1 JP 9704174 W JP9704174 W JP 9704174W WO 9822432 A1 WO9822432 A1 WO 9822432A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- lower alkyl
- atom
- hydrogen atom
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/58—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton
- C07C255/60—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton at least one of the singly-bound nitrogen atoms being acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/03—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C311/06—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms to acyclic carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/12—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing rings
- C07C311/13—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing rings the carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
- C07C311/19—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C327/00—Thiocarboxylic acids
- C07C327/38—Amides of thiocarboxylic acids
- C07C327/40—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C327/42—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to hydrogen atoms or to acyclic carbon atoms to hydrogen atoms or to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C327/00—Thiocarboxylic acids
- C07C327/38—Amides of thiocarboxylic acids
- C07C327/48—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/36—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/34—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D309/36—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
- C07D309/38—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms one oxygen atom in position 2 or 4, e.g. pyrones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/62—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D333/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D335/00—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom
- C07D335/02—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/18—Systems containing only non-condensed rings with a ring being at least seven-membered
Definitions
- the present invention relates to a novel acylamino-substituted acylylanilide derivative, a salt thereof, and a pharmaceutical composition useful as an antiandrogen drug.
- Androgen a steroid hormone, is secreted from the testis and adrenal cortex and causes male hormones. Androgen is taken up into target cells and binds to the androgen receptor in the nucleus, and the androgen-bound receptor forms a dimer. This dimer binds to the androgen-response element on the DNA. to promote the synthesis of m RNA, by inducing protein responsible for androgen action, to express various actions in vivo (Prostate Suppl.45-51 (1996)) c androgens in disease to be ⁇ factor These include prostate cancer, benign prostatic hyperplasia, androgenesis, hirsutism, baldness, acne, seborrhea, etc. c Antiandrogens suppress the activation of androgen transcription and block the action of androgens Therefore, it is useful as a therapeutic agent for diseases in which these androgens are exacerbating factors.
- Antiandrogens are classified into compounds having a steroid skeleton similar to a substrate (steroidal antiandrogens) and compounds having a nonsteroidal skeleton (nonsteroidal antiandrogens ij).
- flucamide Japanese Patent Laid-Open No. 49-81332
- C- flutamide itself does not have an anti-androgenic effect.
- the carbon atom directly linked to the carbonyl group by metabolism ⁇ carbon It is known that the substitution of a hydroxyl group with an atom results in the formation of hydroxyflutamide, which exerts its activity, and this hydroxyl group is considered to be indispensable for the expression of antiandrogen action (J. Med. Chem 31, 954-959 (1988)).
- bicalutamide (GB 8, 221 and 421) has already been described in various countries, and GB 8, 221 and 42] shows that the acyl portion of an acylanilide derivative has aryl (or heteroaryl) sulfonyl ( Or an acylanilide derivative, such as an alkanol substituted with a sulfinyl or thio) aryl (or heteroaryl) amino.
- the compounds substantially disclosed are, like hydroxyflutamide, all compounds having a hydroxyl group at the ⁇ - carbon atom.
- carbaminoaminoacetanilide derivatives include US Pat. No. 4,532,251, and 2,6-dizino substituted with a bilazinyl carbonyl group or a substituted imidazolyl carbenyl group.
- Ricinamide has been disclosed as a fungicide. However, these compounds are
- An object of the present invention is to provide a novel acylamino-substituted acylylanilide derivative and a salt thereof, which have a strong antiandrogenic action and have less of these side effects, and further provide a drug containing the same. . Disclosure of the invention
- the present inventors have conducted intensive studies to solve these problems associated with existing antiandrogens, and surprisingly, an acylylamide derivative substituted with an acylamino group has been required for the expression of activity.
- the present invention has been found to be a compound having strong oral activity and antiandrogenic activity without having a hydroxyl group of ⁇ -carbon atom, further having few side effects, and having good oral activity. Was completed.
- the present invention relates to an acylamino-substituted acylylanilide derivative represented by the following general formula (I) or a salt thereof.
- R 1 and R 2 same or different, halogen atom, cyano, halogeno lower alkyl, nitro, carboxyl, lower alkanoyl or lower alkoxycarbonyl group
- R 3 hydrogen atom or lower alkyl group
- R 4 , R 5 , RG and R 7 identical or different, hydrogen atoms, lower alkyls which may be substituted, or heteroatoms in which R 4 and R 5 are in the form of May form a cycloalkyl group, or when n is 1, R 5 and R 6 may form a cycloalkylene group,
- a and A 2 Same or different binding or low S
- R 8 a hydrogen atom, a hydroxyl group, a lower alkoxy, or a combination of R 8 and R 5 may form a nitrogen-containing cycloalkylene group;
- R 7 and R 8 may be combined to form a nitrogen-containing cycloalkylene group:
- R 4 and R 5 represents a group other than a hydrogen atom.
- Y represented by the formula: s1 — s— or — C—N—
- R 9 lower alkyl, cycloalkyl, or aryl which may have a substituent, aralkeninole, aralkyl, or aryloxy lower alkyl, or a heteroaryl group which may be condensed with a benzene ring
- R 10 hydrogen atom or lower alkyl group
- X 2 oxygen atom or sulfur atom
- R 4 and R 5 represents a group other than a hydrogen atom: :), or an acylamino-substituted acylylanilide derivative or a salt thereof; Consists of halogen atoms, hydroxyl groups, lower alkoxy, lower alkanoyloxy, halogeno lower alkyl groups having one or more substituents of the lower alkyl group or aralkyl group of R 4 or R 5 , R "and R T A substituent selected from the group consisting of R 9 : aryl, aralkenyl, aralkyl or aryloxy lower alkyl, or benzene
- the substituent of the heteroaryl group which may be condensed with a ring is one or more of the same or different halogen atoms, hydroxyl groups, halogeno lower alkyl, lower alkyl, lower alkoxy, halogeno lower
- n is 0 and R 4 or R 5 is the same or different and is a hydrogen atom, or one or more of the same or different substituents is a hydroxyl group, lower alkoxy, lower alkanoyloxy, halogeno lower alkyl group.
- the present invention also relates to a pharmaceutical composition containing an acylamino-substituted acylylanilide derivative or a pharmaceutically acceptable salt thereof as an active ingredient, in particular, a pharmaceutical composition which is an antiandrogen, especially for prostate cancer and prostatic hyperplasia.
- a pharmaceutical composition which is an agent for preventing or treating masculosis, hirsutism, baldness, acne, and seborrhea.
- lower means a straight or branched carbon chain having 1 to 6 carbon atoms.
- An aryl group which may have a substituent, an aralkenyl group, an aralkyl group, a heteroaryl group which may be condensed with a benzene ring, or an aryloxy lower alkyl group has 1 to 3 substituents on the ring.
- halogen atom a halogeno lower alkyl group, a lower alkyl group, a lower alkoxy group, a halogeno lower alkoxy group, a cyano group, a nitro group, a lower alkyl group, a hydroxyl group, a phenyl group, a monoalkyl group.
- the substituent of “substituted or substituted, lower alkyl group or aralkyl group” may be one or more identical or different substituents.
- the group is selected from the group consisting of hydroxyl, lower alkoxy, lower alkanoyloxy, and halogeno lower alkyl:
- “Lower alkyl group” means a straight or branched lower alkyl group having 1 to 6 carbon atoms, for example, methyl, ethyl, n-propyl, isoflurane, n-butyl, isobutyl, sec-butyl, ten-butyl. —Butyl, n-pentyl, n-hexyl, etc. ⁇ 3 lower alkyl groups are preferred ⁇ ,
- lower alkylene group refers to a linear or branched lower alkylene group having 1 to 6 carbon atoms, such as methylene, ethylene, propylene, isobutylpyrene, butylene, pentamethylene, hexamethylene and the like. Preferred are lower alkylene groups having 1 to 3 carbon atoms, and more preferably methylene.
- Alkyl group means "aryl-1 lower alkylene-1”.
- Alkyloxy group means "aryl-1 lower alkylene-1-”.
- Alukeniru group means "Ariru one lower alkenylene one", preferably C 6 - a 10 Ariru one C. _ 6 alkenyl group, phenylene Rueparu, Fueniruburo Bae alkenyl, naphth / Reeteyuru, Nafuchirupuro Bae alkenyl such as Is mentioned.
- Aryloxy lower alkyl group means “aryl-O-lower alkylene-”.
- “Lower alkanoyloxy noisy Rua amino group” of the "lower Arukiru - C (two O) - NR 1 1 -” means, R '' represents a water atom or a lower Arukiru group: -:
- Halogen atom includes, for example, fluorine, chlorine, bromine or iodine atom
- Halogeno-lower alkyl group of the lower Arukiru groups of the C, "6 Arukiru group i this the Bruno nuclear ⁇ - is 3 or obtained by replacing, triflumizole Ruo b preferably methyl ::
- Halogeno lower alkoxy group means “halogeno lower alkyl mono-”
- Acyl group means a broadly defined acyl group, meaning carbonyl and sulfonyl derivatives:
- “Cycloalkyl group” means a 3- to 8-membered saturated hydrocarbon ring, preferably 3 to (member)
- Cycloalkylene group is a bond of the above cycloalkyl
- “Mono or di-lower alkylamino group” means an amino group in which the lower alkyl group is substituted by 1 or 2 groups.
- R 4 and R 5 gar body and turned by cycloalkyl group which may contain a hetero atom the cycloalkyl containing carbon atom bonded to R 4 and R 5 and them as ring atoms —
- the ring contains one heteroatom selected from nitrogen, oxygen, and sulfur.
- hetero atom for example, a sulfur atom is substituted with one or two oxo groups and
- heteroatom is preferably an oxygen atom, the concrete include Okisaniru.
- And n is a R 5 and R 6 gar body case 1, to form a cycloalkylene group" and includes a carbon atom bonded to R 5 and R 6 and its those as ring atoms It means forming the above cycloalkylene.
- Heteroaryl group optionally condensed with a benzene ring means 5 or (containing a heteroaryl group or benzene containing 1 to 3 heteroatoms selected from nitrogen, oxygen or sulfur atoms.
- a heterocyclic aryl fused with a ring, and the heteroaryl includes bilol, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, triazole, thiophene, thiopyran, furan, pyran, dioxolan, thiazole, Isothiazole, thiadiazole, Examples include azine, oxazole, isoxazole, oxaziazole, furazane, dioxazole, oxazine, oxazineazine, dioxazine, and the like.
- nitrogen-containing heteroaryl fused to a benzene ring examples include indole, isoindole, quinoline, isoquinoline, benzothiophene, and benzothiazolone.
- benzofuran, benzofurazan and the like preferably, pyridine, p : limidine, indole, quinoline, thiophene, furan and the like ::
- R 8 and R together form a nitrogen-containing cycloalkylene group or "When n is 1, R 7 and R 8 together form a nitrogen-containing cycloalkylene group” are defined as ring atoms R 8 and the 5- to 7-membered nitrogen-containing cycloalkane, or a ring atoms containing a carbon atom to which nitrogen atom and R 5 being substituted is substituted is substituted with R 8 les, Ru nitrogen atom and R 7 There substituting means to form a 4 to 7-membered nitrogen-containing cycloalkane containing Les, Ru carbon atoms and a 2, specifically pyrrole, piperidine, Kisahidoro Azepin the like to 2, Pyrrole or Pyridine is preferred ⁇ :
- the compound having a tertiary amine may be oxidized with respect to the amine, and includes all oxidized derivatives thereof.
- the compound (I) of the present invention has a tautomer based on an amide bond .
- the compound (I) may have one or more asymmetric carbon atoms.
- isomers such as (S) isomers, racemic forms, diastereomers, etc. :: Also, depending on the type of substituent, there may be a double bond, (Z) form, (E) form
- the compound having a ring may have cis-trans.
- the present invention includes all separated or mixed forms of these isomers.
- the compound of the present invention forms a salt.
- the acid addition salts with inorganic or organic acids there have is a salt with an inorganic or organic bases
- Le Shi pharmaceutically acceptable salt is preferred ⁇ : is a salt thereof , Specifically, mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid or phosphoric acid, or formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid, Addition salts with organic acids such as lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid or toluenesulfonic acid, or acidic amino acids such as aspartic acid or glutamic acid, sodium, Inorganic bases such as potassium, magnesium, calcium, aluminum, and lithium; organic bases such as methylamine, ethylamine
- the quaternary ammonium salt is specifically an ammonium salt obtained by reacting with a lower alkyl halide, a lower alkyl triflate, a lower alkyl tosylate or a benzyl halide, and preferably a salt with a methinoyl or benzyl chloride.
- the compound of the present invention can form a hydrate, a solvate with ethanol or the like, or a crystalline polymorph.
- the present invention includes all of these hydrates, solvates or crystalline polymorphs separated or mixed compounds :
- the compound (I) of the present invention can be produced by applying various production methods: The typical production methods are described below.
- This production method comprises the steps of amidating a substituted ⁇ -salt represented by the general formula (II) with a carboxylic acid or a reactive derivative or a thio-acid or a reactive derivative thereof represented by the general formula (in) to protect the compound.
- This is a method for producing the compound (I) of the present invention by removing the protecting group when having a group.
- Examples of the reactive derivative of the compound (III) include ordinary esters such as methyl ester, ethyl ester, isobutyl ester, and tert-butyl ester of carboxylic acid; acid chlorides such as acid chloride and acid bromide; acid azide; Active esters, symmetrical acid anhydrides, alkyl carbonate halides, etc., obtained by reacting with phenolic compounds such as 4-dinitrophenol and N-hydroxyamines such as I-hydroxysuccinimide and I-hydroxybenzotriazole Reaction with an organic acid-based mixed acid anhydride, diphenylphosphoryl chloride, N-methylmorpholine obtained by reacting with halocarbonic acid alkyl ester or bivaloyl halide, or triphenylphosphoric acid
- Active esters of organic origin include:
- the reaction varies depending on the reactive derivative used and the condensing agent, but is usually halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform, aromatic hydrocarbons such as benzene, toluene and xylene, polyester, and the like.
- Inert organics such as ethers such as tetrahydrofuran, esters such as ethyl acetate, acetonitrile, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone and dimethyl sulfoxide.
- the reaction is performed in a solvent, under cooling, under cooling to room temperature, or at room temperature to heating.
- oxygen, sulfur, nitrogen, etc. present in the molecule be bonded to a protecting group, and such a protecting group is described in Greene and Wuts, "Protective Groups in Organic.” And the like.
- the protecting groups described in “Synthesis” 2nd edition can be used, and these can be used and divided appropriately according to the reaction conditions.
- This production method comprises the steps of: (a) adding a substituted amine represented by the present compound (IV) or a salt thereof to a carboxylic acid or a reactive derivative thereof, a sulfonic acid or a reactive derivative thereof, thiocarboxylic acid or a reactive derivative thereof To produce the compound (1) of the present invention by using the same reaction conditions as in the first production method:
- the reaction varies depending on the reactive derivative used and the condensing agent, but is usually halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform, aromatic hydrocarbons such as benzene, toluene, xylene, ether, tetrahydrofuran, etc.
- halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform
- aromatic hydrocarbons such as benzene, toluene, xylene, ether, tetrahydrofuran, etc.
- organic solvent inert to the reaction of ethers such as ethers, esters such as ethyl acetate, and N, N-dimethylformamide N, N-dimethylacetamide, dimethyl sulfoxide, etc.
- the compound (IV) of the present invention was used in excess, or N-methylmorpholine, trimethylamine, triethylamine, N, N-dimethylaniline, pyridine, 4- (N, N-dimethylamino) pyridine, bicholine,
- a base such as lutidine
- Pyridine etc. can be used as a solvent.
- the amide group represented by the compound (la) of the present invention is converted to a thioamide group to produce the compound (lb) of the present invention.
- This reaction can be prepared by any of the known chemical methods for synthesizing thioamide derivatives from amide derivatives such as dipentapentasulfide and Lawesson's reagent:
- the reaction is usually carried out with halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as ether and tetrahydrofuran, esters such as ethyl acetate and pyridine.
- halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform
- aromatic hydrocarbons such as benzene, toluene and xylene
- ethers such as ether and tetrahydrofuran
- esters such as ethyl acetate and pyridine.
- organic solvent inert to the reaction Rejected performed under cooling to room temperature, or at room temperature to heating
- hydrolysis, hydrogenation, ureidation, etc. are also carried out by ordinary methods.
- the compound of the present invention thus produced can be isolated and purified as a salt, a hydrate, a solvate, or a polymorphic substance of the compound of the present invention as it is free.
- Salt can also be produced by subjecting it to a conventional salt formation reaction.
- the starting compound of the above-mentioned production method may contain a novel substance, it may be produced by applying the production method described in Reference Example, a method analogous to the production method, or a modified method arbitrarily practicable by those skilled in the art. it can. Isolation and purification are performed by applying ordinary chemical procedures such as extraction, concentration, distillation, crystallization, filtration, recrystallization, and various types of chromatography:
- various isomers can be separated by utilizing the difference in physicochemical properties between isomers. For example, by optical isomers selecting appropriate raw materials, or by racemic resolution method of racemic compounds, the availability of the c industry that can be force S lead to stereochemically pure isomers
- the compound of the present invention has a strong anti-androgenic effect by suppressing the transcriptional activation by androgen, and is a compound having few side effects such as central action and agonist action.
- the compound of the present invention is useful as a therapeutic or prophylactic agent for diseases in which androgen is involved as an adverse factor, for example, prostate cancer, prostatic hypertrophy, virilization, hirsutism, baldness, acne, seborrhea and the like. is there.
- the usefulness of the compound of the present invention has been confirmed by the following test methods! RU
- CHO cells were seeded on a 100-mm-diameter cell culture dish with 10 6 IX cells, and 12 to 18 hours later, a human androgen receptor expression plasmid co-precipitated with calcium phosphate, MMTV-LTR
- the luciferase jelly boater plasmid (including the neomycin resistance gene) was added and transfection was performed. After 15 hours, the medium was removed, the cells were diluted in several steps, and replated, and the medium was fed with GENETICIN® (neomycin) to a final concentration of 500 ⁇ g / ml .: About 1 week later, the cells were selected by neomycin.
- the isolated cells were detached, and cells expressing the human androgen receptor expression gene, MMTV-luciferase revoter-gene, constitutively expressing and isolated by the limiting dilution method (CHO / MMTV stable transformant:
- SV40 reporter gene stable transformant was obtained in the same manner as described above. However, the SV40 reporter plasmid and the neomycin resistance gene expression plasmid were simultaneously transfected (CHO / SV40 stable transformant; ⁇
- CHO / MMTV stable transformant cells and CHO / SV40 stable transformant cells were seeded at 1 ⁇ 10 4 cells / well in a luminobure 1 for% well cell culture, and the compound of the present invention was added 6 to 8 hours later. 18 minutes after compound addition, add 20 ⁇ l of a solution containing 1% Triton-X and 10% glycerol to lyse the cells, add 100 ⁇ l of luciferase substrate solution containing 0.47 mM luciferin, and use a luminometer. The luminescence was measured and these were taken as the luciferase activities obtained by MMTV-LTR transcriptional activation by the human androgen receptor and non-specific SV40 flow motor transcriptional activation:
- the transcription activation effect of the compound of the present invention was calculated by the following formula as a ratio to the transcription activity induced by InM DHT.
- CHO / MMTV stable transformant cells and CHO / SV40 stable transformant cells were seeded at 1 ⁇ 10 4 cells each in a luminoblate for% well cell culture, and after 6 to 8 hours, simultaneously with DHT (final concentration 0.3 nM).
- the compound of the present invention was added. Eighteen hours after the addition of the compound, the cells were lysed by adding 20 ⁇ l of a solution containing 1% Triton-X and 10% glycerol, and 100 ⁇ l of a luciferase substrate solution containing 0.47 mM luciferin was added, followed by luminescence using a luminometer. Measure the amount Luciferase activity obtained by MMTV-LTR transcriptional activation by human androgen receptor and non-specific SV40 promoter transcriptional activation:
- the inhibitory effect on transcriptional activation by the compound of the present invention was calculated by the following formula as the inhibition rate against the transcriptional activity induced by 0.3 nM DHT.
- Inhibition rate (%) 100 (1'- ⁇ ') / ( ⁇ - ⁇ )
- the IC 50 was determined from the concentration of the compound of the present invention at which the inhibition rate calculated by the above method was 50%.
- the activity of the compound of the present invention determined by the above a) and b) is shown below:
- Three-week-old male Wistar rats were simultaneously administered testosterone propionate and the compound of the present invention once a day for 5 consecutive days from 72 hours after castration. Six hours after the final administration, the wet weight of the ventral prostate was measured, and the inhibitory effect of the compound of the present invention on the increase in prostate weight by testosterone propionate was examined.
- Testosterone propionate was dissolved in cottonseed oil containing 5% ethanol and subcutaneously administered at a dose of 0.5 mg / kg body weight of the rat : the compound of the present invention was orally administered in a 0.5% methylcellulose solution ::
- the prostate weight increase inhibitory effect of the compound of the present invention was as follows: test group in which testosterone flobionate and the compound of the present invention were administered together, test group in which testosterone propionate alone was administered, and control group, and no testosterone propionate and the compound of the present invention.
- Androgen is a compound useful as a therapeutic agent for the prevention and treatment of diseases associated with exacerbation factors
- Preparations containing one or more of the compound of the present invention or a salt thereof as an active ingredient are prepared using carriers, excipients and other additives that are usually used for preparation.
- the drug can be administered orally in the form of tablets, pills, capsules, granules, powders, liquids, etc., or injections such as intravenous and intramuscular injections, and parenteral administrations such as suppositories and transdermals.
- l OOOmg about 1 to 100 mg, preferably 0.1 to 100 mg per adult for parenteral administration per day (). 1 to 100 mg, preferably about 0.001 to 50 mg, divided into 1 dose or 2 to 4 doses
- the one or more active substances include at least one inert diluent such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone, metasilicate.
- inert diluent such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone, metasilicate.
- the composition is formulated according to the usual practice with additives other than inert diluents, such as lubricants such as magnesium stearate and disintegrants such as calcium cellulose dalcholate.
- Tablets or pills may contain sucrose, gelatin, hydroxypropylcellulose, hydroxypro as required, including stabilizers such as lactose and solubilizing agents such as glutamic acid or aspartic acid.
- -Sugar coatings or gastric-soluble coatings such as ⁇ may be coated with a film of enteric substance:
- Liquid compositions for oral administration include pharmaceutically acceptable emulsifiers, solutions, suspensions, tablets, elixirs and the like, and are generally inert diluents such as purified water. Contains: In addition to the inert diluent, this composition may contain adjuvants such as wetting agents, suspending agents, sweetening agents, flavoring agents, flavoring agents, preservatives, :
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions ij, aqueous solutions including suspensions and emulsions, and suspensions include, for example, distilled water for injection and physiological saline. Contains saline.
- Non-aqueous solutions and suspensions for example blanking: b propylene glycol, Boriechiren glycolate Ichiru, vegetable oils such as Oribu oil, alcohols such as ethanol, Borisorubeto 80.
- compositions may contain adjuvants such as preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), and solubilizing agents (eg, glutamic acid, aspartic acid).
- adjuvants such as preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), and solubilizing agents (eg, glutamic acid, aspartic acid).
- Reference Example 1 52 2- (4-Trifluoromethylbenzoylamino) 2-methylpropanoate Methyl Reference Example 1-62 2- (4-Funolei Lof :: Nylsulfonylamino 2-Methylpropanoate Reference Example 1-72 2- (4-Nitrophenylsulfonylamino) methyl 2-methylpropanoate Reference Example 1-82--(4-Methoxyphenylsulfonylamino) 2-methylpropanoate Reference Example 1-93 -(4-Funolelov: Nylsulfonylamino) methyl methyl lobanoate
- REFERENCE EXAMPLE 1 15 1 (4-FUNORESHILOF ;: Nylsulfonylamino) cyclo 'methyl hexylcarboxylate Reference Example 1-16 1- (4-Fluorobe, zolylamino) cyclohexylcarboxy
- Reference Example 2-13 1- (4-Fluorophenylsulfonylamino) cyclopropylfuronic acid Reference Example 2—14— (4-1: boric acid Reference Example 2—15 1— (4-fluorophenylsulfonylamino) cyclopentylcarboxylic acid Reference Example 2—16 1— (4-funo ⁇ - Reference Example 2—17 (4-Fluorophenylsulfonylamino) cyclohexylcarboxylic acid g Reference Example 2-18 (4: no) cyclohexylcarboxylic acid
- Reference example 4 2 2 N-dioxy carbonylamino-N (4-cyano 3- -trifluoromethylphenyl) methylpropananilide
- reaction mixture was diluted with 50 ml of ethyl acetate, washed twice with 50 ml of distilled water, and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the residue was subjected to silica gel column chromatography to obtain 0.63 g of the title compound from a fraction eluted with ethyl acetate (3: 7).
- Reference Example 1 The following Reference Example was synthesized in the same manner as 1-1.
- Example 2 16 It was synthesized in Example 2 16 in the same manner as in Example 1:
- the reaction solution was diluted with 50 ml of ethyl acetate, washed with 5 () ml of 1N hydrochloric acid, 50 ml of saturated aqueous sodium hydrogen carbonate and 50 ml of distilled water, and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the residue was subjected to silica gel column chromatography and dissolved in ethyl acetate-hexane (17: 3). A crude crystal was obtained from the outlet. The crude crystals were recrystallized from ethyl acetate, and Example] 8-21 was synthesized in the same manner as in Example 3 of the title compound as colorless crystals.
- Examples 23 and 24 were synthesized in the same manner as in Example 1.
- Examples 26 to 33 were synthesized in the same manner as in Example 25-Examples 34 to 64 were synthesized in the same manner as Example 1.
- Phthanolamide 5 Dissolve Og in dichloromethane. Then, 6.37 ml of ethyl and chloroformate were added, and the mixture was stirred for 1 hour and further stirred at room temperature for 6 hours. The solvent was distilled off under reduced pressure, a mixed solvent of benzene and hexane was added, and the precipitated crystals were separated by filtration. Was distilled off to give 2-cyanobenzoic anhydride (58 mg).
- Examples 69-78 were synthesized in the same manner as in Example 68.
- Example 80 to 87 were synthesized in the same manner as in Example 79.
- Example 88
- Example 9 Examples 9] and 92 were synthesized as in Example 90:
- Example 94 was synthesized in the same manner as in Example 93.
- the organic layer was washed with saturated aqueous sodium bicarbonate solution (3 Oml), water (3 Oml), saturated aqueous citric acid solution (3) ml ⁇ 2, and dried over sodium sulfate.
- the solvent was distilled off under reduced pressure.
- the crystals were recrystallized from diisopropyl ether-ethyl acetate, and recrystallized from acetic acid acetate / lehi-fu / ro-lo'sole-hexane, to give 1 mg of the title compound as colorless crystals.
- Example 1 (X) —1 () 4 was synthesized in the same manner as in Example 25.
- Example 1 () 5-107 was synthesized in the same manner as in Example 1.
- Example 108 was synthesized in the same manner as in Example 25.
- Example K 9-11 11 was synthesized in the same manner as in Example 1.
- Examples 123 to 129 were synthesized in the same manner as in Example 1.
- Example 131 was synthesized in the same manner as in Example 130. : The structures and physical properties of these examples are shown in the table.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
明細書 Specification
新規ァシルァミノ置換ァシルァニリド誘導体又はその医薬組成物 技術分野 Novel acylamino-substituted acylylanilide derivatives or pharmaceutical compositions thereof
本発明は,抗アンドロゲン薬として有用な,新規ァシルァミノ置換ァシルァニリド誘導体及 びその塩並びに医薬組成物に関する 背景技術 The present invention relates to a novel acylamino-substituted acylylanilide derivative, a salt thereof, and a pharmaceutical composition useful as an antiandrogen drug.
ステロイドホルモンの一種であるアンドロゲンは精巣や副腎皮質から分泌され,男性ホル モン作用を引き起こす。アンドロゲンは,標的細胞内に取り込まれて核内のアンドロゲン受 容体に結合し,アンドロゲンが結合した該受容体は二量体を形成する この二量体は DNA 上のアンドロゲン一レスポンス エレメントに結合して m RNAの合成を促進し,アンドロゲ ン作用を司る蛋白を誘導することにより,生体内で種々の作用を発現させる(Prostate Suppl.45-51(1996)) cアンドロゲンが增悪因子となる疾患には,前立腺癌,前立腺肥大症, 男性化症,多毛症,禿頭症,ざ瘡,脂漏等が挙げられる c抗アンドロゲン剤は,アンドロゲン の転写活性化を抑制し,アンドロゲンの作用を遮断することから,これらのアンドロゲンが増 悪因子となる疾患の治療剤として有用である。 Androgen, a steroid hormone, is secreted from the testis and adrenal cortex and causes male hormones. Androgen is taken up into target cells and binds to the androgen receptor in the nucleus, and the androgen-bound receptor forms a dimer. This dimer binds to the androgen-response element on the DNA. to promote the synthesis of m RNA, by inducing protein responsible for androgen action, to express various actions in vivo (Prostate Suppl.45-51 (1996)) c androgens in disease to be增悪factor These include prostate cancer, benign prostatic hyperplasia, androgenesis, hirsutism, baldness, acne, seborrhea, etc. c Antiandrogens suppress the activation of androgen transcription and block the action of androgens Therefore, it is useful as a therapeutic agent for diseases in which these androgens are exacerbating factors.
抗アンドロゲン剤は,基質類似のステロイド骨格を有する化合物(ステロイド系抗アンド口 ゲン斉 と,非ステロイド骨格を有する化合物(非ステロイド系抗アンドロゲン斉 ij)に分類され ている。 Antiandrogens are classified into compounds having a steroid skeleton similar to a substrate (steroidal antiandrogens) and compounds having a nonsteroidal skeleton (nonsteroidal antiandrogens ij).
非ステロイド系抗アンドロゲン剤としては,ァシルァニリド誘導体であるフルタミド(特開昭 4 9— 81332)が知られている cフルタミド自体には抗アンドロゲン作用はなぐ代謝によって カルボニル基に直結する炭素原子( α炭素原子)に水酸基が置換することによりハイドロキ シフルタミドとなり,活性発現することが知られており,この水酸基が抗アンドロゲン作用の発 現に不可欠なものであると考えられている(J. Med. Chem 31, 954-959 (1988))。また,ビカル タミド(GB 8, 221 , 421)も既に諸タ国にて上巿されており, GB 8, 221 , 42 ]にはァシ ルァニリド誘導体のァシル部分がァリール(又はへテロアリール)スルホニル(又はスルフィ ニル若しくはチォ)ゃァリール(又はへテロアリール)ァミノで置換されたアルカノィル等であ るァシルァニリド誘導体がクレームされている。し力 ながら,実質的に開示された化合物は, ハイドロキシフルタミドと同様に,全て α炭素原子に水酸基を有する化合物である- 一方,カルボ ルアミノアセトァニリド誘導体としては, US 4, 532, 251に,ビラジニル カルボ二ル基又は置換イミダゾリルカルポ二ル基で置換された 2 , 6一ジノ As a nonsteroidal anti-androgen, flucamide (Japanese Patent Laid-Open No. 49-81332), which is an acylanilide derivative, is known. C- flutamide itself does not have an anti-androgenic effect. The carbon atom directly linked to the carbonyl group by metabolism ( α carbon It is known that the substitution of a hydroxyl group with an atom results in the formation of hydroxyflutamide, which exerts its activity, and this hydroxyl group is considered to be indispensable for the expression of antiandrogen action (J. Med. Chem 31, 954-959 (1988)). In addition, bicalutamide (GB 8, 221 and 421) has already been described in various countries, and GB 8, 221 and 42] shows that the acyl portion of an acylanilide derivative has aryl (or heteroaryl) sulfonyl ( Or an acylanilide derivative, such as an alkanol substituted with a sulfinyl or thio) aryl (or heteroaryl) amino. However, the compounds substantially disclosed are, like hydroxyflutamide, all compounds having a hydroxyl group at the α- carbon atom. On the other hand, carbaminoaminoacetanilide derivatives include US Pat. No. 4,532,251, and 2,6-dizino substituted with a bilazinyl carbonyl group or a substituted imidazolyl carbenyl group.
リシンアミドが殺菌剤として開示されてレ、る。しかしながら,これらの化合物が抗 Ricinamide has been disclosed as a fungicide. However, these compounds are
作用を有することは開示も示唆もされてレ、なレ、:;

いる =即ち,薬物の中枢への作用によると考えられる女性化乳房,乳房痛(Semm. Oncol. 18 (5 Suppl 6) 13-18 (1991), J. Med. Chem. 3 1 954-959 (1988))や長期使用によるァゴニス I、 作用の発現(J. Urol. 153 (3 part 2) 1070-1072 (1995))等である 特に,前立腺癌の治療に おいては,アンドロゲン作用を完全に遮断する必要がある為,ァゴニスト作用の発現は治療 上大きな問題となる。 Ile words, gynecomastia, which is attributed to the action of the drug in the central, mastalgia (Semm. Oncol. 18 (5 Suppl 6) 13-18 (1991), J. Med. Chem. 3 1 954-959 ( 1988)) and agonism I after long-term use, and the onset of action (J. Urol. 153 (3 part 2) 1070-1072 (1995)). Because of the need to block, the development of agonist action is a major therapeutic problem.
本発明の目的は,強力な抗アンドロゲン作用を有し,これらの副作用が少ない,新規ァシ ルァミノ置換ァシルァニリド誘導体及びその塩を提供すること,更にはこれらを含有する医 薬を提供することである。 発明の開示 An object of the present invention is to provide a novel acylamino-substituted acylylanilide derivative and a salt thereof, which have a strong antiandrogenic action and have less of these side effects, and further provide a drug containing the same. . Disclosure of the invention
本発明者らは,既存の抗アンドロゲン剤に付随するこれらの問題点を解決するべく鋭意研 究を行ったところ,意外にも,ァシルァミノ基が置換したァシルァニリド誘導体が,従来,活 性発現に必要であると考えられてレ、た α炭素原子の水酸基を有さなくとも,強レ、抗アンド口 ゲン作用を示し,更に副作用が少なく, 良好な経口活性を有する化合物である事を見出し 本発明を完成させるに至った。 The present inventors have conducted intensive studies to solve these problems associated with existing antiandrogens, and surprisingly, an acylylamide derivative substituted with an acylamino group has been required for the expression of activity. The present invention has been found to be a compound having strong oral activity and antiandrogenic activity without having a hydroxyl group of α-carbon atom, further having few side effects, and having good oral activity. Was completed.
即ち,本発明は,下記一般式( I )で示されるァシルァミノ置換ァシルァニリド誘導体又は その塩に関する。 That is, the present invention relates to an acylamino-substituted acylylanilide derivative represented by the following general formula (I) or a salt thereof.

(式中の記号は以下の意味を有する。 (The symbols in the formula have the following meanings.
R1及び R2 :同一又は異なってハロゲン原子,シァノ,ハロゲノ低級アルキル,ニトロ,カルボ キシル,低級ァルカノィル又は低級ァルコキシカルポ二ル基 R3 :水素原子又は低級アルキル基 R 1 and R 2 : same or different, halogen atom, cyano, halogeno lower alkyl, nitro, carboxyl, lower alkanoyl or lower alkoxycarbonyl group R 3 : hydrogen atom or lower alkyl group
11 : 0又は1 11: 0 or 1
R4, R5, RG及び R7 :同一又は異なって水素原子,置換基を有していても良い低級アルキル 或レ、は, R4と R5がー体となってヘテロ原子を含んでいても良いシクロアルキル基を形成し てもよく,又は, nが 1のとき R5と R6がー体となって,シクロアルキレン基を形成してもよレ、 A,及び A2:同一又は異なって結合又は低 S R 4 , R 5 , RG and R 7 : identical or different, hydrogen atoms, lower alkyls which may be substituted, or heteroatoms in which R 4 and R 5 are in the form of May form a cycloalkyl group, or when n is 1, R 5 and R 6 may form a cycloalkylene group, A and A 2 : Same or different binding or low S
R8 :水素原子,水酸基,低級アルコキシ, 或いは R8と R5がー体となって含窒素シクロアルキレン基を形成してもよく, R 8 : a hydrogen atom, a hydroxyl group, a lower alkoxy, or a combination of R 8 and R 5 may form a nitrogen-containing cycloalkylene group;
き R7と R8がー体となって含窒素シクロアルキレン基を形成してもよい:: And R 7 and R 8 may be combined to form a nitrogen-containing cycloalkylene group:
Z :ァシル基 Z: acetyl group
X,:酸素原子又は硫黄原子 X ,: oxygen atom or sulfur atom
但し, Zがへテロァリールカルボニル基の場合は, R4と R5の少なくとも一方は水素原子以 外の基を示す。 ) However, when Z is a heteroarylcarbonyl group, at least one of R 4 and R 5 represents a group other than a hydrogen atom. )
:, Zが Y— R9 (式中の記号は以下の意味を有する。 :, Z is Y—R 9 (The symbols in the formula have the following meanings.
Y : 式 一 s一 — s— 又は — C-N— で示さ; 基 Y: represented by the formula: s1 — s— or — C—N—
X 2 , (0) m J R '。 X 2 , ( 0) m JR '.
R9 :低級アルキル,シクロアルキル,又は置換基を有していてもよいァリール,ァラルケ二ノレ, ァラルキル、若しくはァリールォキシ低級アルキル,或いは,ベンゼン環と縮合してもよ いへテロアリール基 R 9 : lower alkyl, cycloalkyl, or aryl which may have a substituent, aralkeninole, aralkyl, or aryloxy lower alkyl, or a heteroaryl group which may be condensed with a benzene ring
R10 :水素原子又は低級アルキル基 R 10 : hydrogen atom or lower alkyl group
X2 :酸素原子又は硫黄原子 X 2 : oxygen atom or sulfur atom
m : 0又は 1, 2 m: 0 or 1, 2
但し, Yがカルボニル基であり, R9がへテロアリール基の場合は, R4と R5の少なくとも一方 は水素原子以外の基を示す:: )であるァシルァミノ置換ァシルァニリド誘導体又はその塩; 更に好ましくは, R4又は R5, R "及び RTの低級アルキル基若しくはァラルキル基の置換 基力 1以上の同一又は異なった,ハロゲン原子,水酸基,低級アルコキシ,低級アルカノ ィルォキシ,ハロゲノ低級アルキル基からなる群より選択される置换基であり, R9 :のァリー ル,ァラルケニル,ァラルキル若しくはァリールォキシ低級アルキル基,若しくは,ベンゼン 環と縮合してもよいへテロアリール基の置換基が, 1又はそれ以上の同一又は異なった,ハ ロゲン原子,水酸基,ハロゲノ低級アルキル,低級アルキル,低級アルコキシ,ハロゲノ低 級アルコキシ,シァノ,ニトロ,低級アルカノィルォキシ,フエニル,モノ若しくはジ低級アル キルアミノ基,カルボキシル基,低級アルコキシカルボニル基,モノ若しくはジ低級アルキル ァミノカルボニル,低級アルカノィルァミノ及びォキソ基からなる群より選択される置換基で あるァシルァミノ置換ァシルァニリド誘導体又はその塩; However, when Y is a carbonyl group and R 9 is a heteroaryl group, at least one of R 4 and R 5 represents a group other than a hydrogen atom: :), or an acylamino-substituted acylylanilide derivative or a salt thereof; Consists of halogen atoms, hydroxyl groups, lower alkoxy, lower alkanoyloxy, halogeno lower alkyl groups having one or more substituents of the lower alkyl group or aralkyl group of R 4 or R 5 , R "and R T A substituent selected from the group consisting of R 9 : aryl, aralkenyl, aralkyl or aryloxy lower alkyl, or benzene The substituent of the heteroaryl group which may be condensed with a ring is one or more of the same or different halogen atoms, hydroxyl groups, halogeno lower alkyl, lower alkyl, lower alkoxy, halogeno lower alkoxy, cyano, nitro, A substituent selected from the group consisting of lower alkanoyloxy, phenyl, mono- or di-lower alkylamino, carboxyl, lower alkoxycarbonyl, mono- or di-lower alkylaminocarbonyl, lower alkanoylamino and oxo; An acylamino-substituted acylylanilide derivative or a salt thereof,
より好ましくは, nが 0であり, R4又は R5が同一又は異なって水素原子,又は 1以上の同一 又は異なった置換基が,水酸基,低級アルコキシ,低級アルカノィルォキシ,ハロゲノ低級 アルキル基からなる群より選択される置換基を有していても良い低級アルキル若しくはァラ ルキル基であるァシルァミノ置換ァシルァニリド誘導体又はその塩; More preferably, n is 0 and R 4 or R 5 is the same or different and is a hydrogen atom, or one or more of the same or different substituents is a hydroxyl group, lower alkoxy, lower alkanoyloxy, halogeno lower alkyl group. An acylamino-substituted acylylanilide derivative or a salt thereof, which is a lower alkyl or aralkyl group which may have a substituent selected from the group consisting of:
最も好ましくは,以下よりなる群の化合物又はその塩から選択される化合物: Most preferably, a compound selected from the group consisting of: or a salt thereof:
N— (4ーシァノー 3—トリフルォロメチルフエニル)力ルバモイル]一 1ーメチルェチ N— (4-cyano 3-trifluoromethylphenyl) lvamoyl] -1-1-methylethyl
-4一フルォ口べンズアミド; -4-Fluoro-benzamide;
N— 一 [ (3 , 4—ジシァノフエ二ノレ)力ルバモイル]— 1—メチルェチル }—4一フルォロ N—one [(3,4—dicyanophenoline) rubamoyl] — 1—methylethyl} —4
N— { 1— [ (3—クロ口一 4一シァノフエニル)カノレノくモイル」― 1—メチ /レエチル i— 4ーフ ルォ口べンズアミド; N— {1 — [(3-chloro-1-41-cyanophenyl) canolenoc-moyl ”—1-methyl / reethyl i—4-fluorobenzamide;
N- { 1 - [ (4ーシァノー 3—トリフルォロメチルフエニル)力ルバモイル]— 1ーメチルェチ ル}一 2, 4, 6—トリフルォロベンズアミド; N- {1-[(4-cyano 3-trifluoromethylphenyl) rubamoyl]-1-methylethyl}-1, 2, 4, 6-trifluorobenzamide;
4—クロロー N— { 1一 [ (4—シァノー 3—トリフルォロメチルフエニル)力ルバモイル]— 1一 メチルェチル }ベンズァミド; 4-Chloro-N— {1-[(4-Cyanol 3-trifluoromethylphenyl) -lvamoyl] — 1-methylethyl} benzamide;
でめる;; Out;
また,本発明は,ァシルァミノ置換ァシルァニリド誘導体又はその製薬学的に許容される 塩を有効成分とする医薬組成物,殊に抗アンドロゲン剤である医薬組成物に関し,なかでも, 前立腺癌,前立腺肥大症,男性化症,多毛症,禿頭症,ざ瘡,脂漏の予防又は治療剤であ る医薬組成物に関する。 The present invention also relates to a pharmaceutical composition containing an acylamino-substituted acylylanilide derivative or a pharmaceutically acceptable salt thereof as an active ingredient, in particular, a pharmaceutical composition which is an antiandrogen, especially for prostate cancer and prostatic hyperplasia. The present invention relates to a pharmaceutical composition which is an agent for preventing or treating masculosis, hirsutism, baldness, acne, and seborrhea.
一般式 (I)で示される化合物について更に説明すると,次の通りである。 The compound represented by the general formula (I) will be further described as follows.
本明細書の一般式の定義において,特に断らない限り「低級」なる用語は炭素数が 1乃至 6個の直鎖又は分枝状の炭素鎖を意味する。 置換基を有していても良いァリール基,ァラルケニル基,ァラルキル基,ベンゼン環と縮合 しても良いヘテロァリール基,若しくはァリ一ルォキシ低級アルキル基は,環上に 1乃至3個 の置換基を有していてもよく,好ましくは,ハロゲン原子,ハロゲノ低級アルキル基,低級ァ ルキル基,低級アルコキシ基,ハロゲノ低級アルコキシ基,シァノ基,ニトロ基,低級アル力 ノィルォキシ基,水酸基,フエニル基,モノ若しくはジ低級アルキルアミノ基,モノ若しくはジ 低級アルキルァミノ力ルポ二ル基,低級アル力ノィルァミノ基又はォキソ基である:: In the definition of the general formula in the present specification, unless otherwise specified, the term "lower" means a straight or branched carbon chain having 1 to 6 carbon atoms. An aryl group which may have a substituent, an aralkenyl group, an aralkyl group, a heteroaryl group which may be condensed with a benzene ring, or an aryloxy lower alkyl group has 1 to 3 substituents on the ring. It may have a halogen atom, a halogeno lower alkyl group, a lower alkyl group, a lower alkoxy group, a halogeno lower alkoxy group, a cyano group, a nitro group, a lower alkyl group, a hydroxyl group, a phenyl group, a monoalkyl group. Or a di-lower alkylamino group, a mono- or di-lower alkylamino group, a lower alkylamino group or an oxo group:
R4, R5, R6及び R7におレ、て「置換基を有してレ、ても良レ、低級アルキル基又はァラルキル 基」の置換基は, 1以上の同一又は異なった置換基が,水酸基,低級アルコキシ基,低級ァ ルカノィルォキシ基,ハロゲノ低級アルキル基からなる群より選択される:: In R 4 , R 5 , R 6 and R 7 , the substituent of “substituted or substituted, lower alkyl group or aralkyl group” may be one or more identical or different substituents. The group is selected from the group consisting of hydroxyl, lower alkoxy, lower alkanoyloxy, and halogeno lower alkyl:
「低級アルキル基」は直鎖状又は分枝状の炭素数 1〜6の低級アルキル基を示し,例え ばメチル, ェチル, n—プロピル,イソフロヒ レ, n—ブチル,イソブチル, sec—ブチル, ten —ブチル, n—ペンチル, n—へキシルなどが挙げられ,炭素数!〜 3の低級アルキル基が 好ましレ \, “Lower alkyl group” means a straight or branched lower alkyl group having 1 to 6 carbon atoms, for example, methyl, ethyl, n-propyl, isoflurane, n-butyl, isobutyl, sec-butyl, ten-butyl. —Butyl, n-pentyl, n-hexyl, etc. ~ 3 lower alkyl groups are preferred \,
「低級アルキレン基」は直鎖状又は分枝状の炭素数 1〜6の低級アルキレン基を示し,例 えばメチレン, エチレン, フ Pピレン, イソフ口ピレン, ブチレン, ペンタメチレン, へキサメチ レンなどが挙げられ,炭素数 1〜3の低級アルキレン基が好ましく,更に好ましくはメチレン である:: The term "lower alkylene group" refers to a linear or branched lower alkylene group having 1 to 6 carbon atoms, such as methylene, ethylene, propylene, isobutylpyrene, butylene, pentamethylene, hexamethylene and the like. Preferred are lower alkylene groups having 1 to 3 carbon atoms, and more preferably methylene.
「ァリール基」は炭素数 6〜12の芳香族炭素水素基が好ましく,例えばフユニル, ひーナ フチル, β—ナフチルなどがあげられる 2更には,炭素数 6〜1()のものが好ましい = "Ariru group" is preferably an aromatic carbon hydrogen group having 6 to 12 carbon atoms, for example Fuyuniru, Hina Fuchiru, 2 further including β- naphthyl is preferably from 6 to 1 carbon atoms () =
「ァラルキル基」は「ァリール一低級アルキレン一」を意味する。 "Aralkyl group" means "aryl-1 lower alkylene-1".
「ァラルキルォキシ基」は「ァリール一低級アルキレン一 Ο—」を意味する。 . "Aralkyloxy group" means "aryl-1 lower alkylene-1-". .
「ァラルケニル基」は「ァリール一低級アルケニレン一」を意味し,好ましくは C6— 10ァリール 一 C. _6アルケニル基であり,フエ二ルェテニル,フエニルブロぺニル,ナフチ /レエテュル, ナフチルプロぺニルなどが挙げられる。 "Ararukeniru group" means "Ariru one lower alkenylene one", preferably C 6 - a 10 Ariru one C. _ 6 alkenyl group, phenylene Rueteniru, Fueniruburo Bae alkenyl, naphth / Reeteyuru, Nafuchirupuro Bae alkenyl such as Is mentioned.
「ァリールォキシ低級アルキル基」は「ァリール—O—低級アルキレン—」を意味する。 "Aryloxy lower alkyl group" means "aryl-O-lower alkylene-".
「低級アルコキシ基」は「低級アルキル— o—」を意味する: "Lower alkoxy" means "lower alkyl-o-":
「低級ァルコキシカルポ二ル基」は「低級ァルキル— O— C ( = o )—」を意味する:: 「低級アルカノィル基」は「低級アルキル一 C ( =〇)—」を意味する- “Lower alkoxycarbonyl” means “lower alkyl—O—C (= o) —” :: “Lower alkanoyl” means “lower alkyl-C (= 〇) —” —
「低級アルカノィルォキシ基」は「低級アルキル一 c ( = o)— o—」を意味する。 「低級アルカノィルァミノ基」は「低級ァルキル— C (二 O)— NR1 1―」を意味し, R ' 'は水 素原子又は低級ァルキル基を示す :·: “Lower alkanoyloxy group” means “lower alkyl-1 c (= o) —o—”. "Lower alkanoyloxy Noi Rua amino group" of the "lower Arukiru - C (two O) - NR 1 1 -" means, R '' represents a water atom or a lower Arukiru group: -:
「ハロゲン原子」としては例えば,フッ素,塩素,臭素又はヨウ素原子など "Halogen atom" includes, for example, fluorine, chlorine, bromine or iodine atom
「ハロゲノ低級アルキル基」の低級ァルキル基は上記の C ,„ 6ァルキル基 iこ上記ノ 原子力^— 3個置換したものであり,トリフルォロメチルが好ましい:: "Halogeno-lower alkyl group" of the lower Arukiru groups of the C, "6 Arukiru group i this the Bruno nuclear ^ - is 3 or obtained by replacing, triflumizole Ruo b preferably methyl ::
「ハロゲノ低級アルコキシ基」は「ハロゲノ低級アルキル一〇—」を意味する “Halogeno lower alkoxy group” means “halogeno lower alkyl mono-”
「ァシル基」は,広義のァシル基を意味し,カルボニル誘導体及びスルホニル誘導体を意 味する: "Acyl group" means a broadly defined acyl group, meaning carbonyl and sulfonyl derivatives:
「シクロアルキル基」は 3〜8員飽和炭化水素環を意味し,好ましくは 3〜(う員' “Cycloalkyl group” means a 3- to 8-membered saturated hydrocarbon ring, preferably 3 to (member)
「シクロアルキレン基」は上記のシクロアルキルの結合手が "Cycloalkylene group" is a bond of the above cycloalkyl
「モノ若しくはジ低級ァルキルァミノ基」とは,上記低級アルキル基が 1又は 2置換したァミ ノ基を意味する。 “Mono or di-lower alkylamino group” means an amino group in which the lower alkyl group is substituted by 1 or 2 groups.
「モノ若しくはジ低級アルキルアミノカルボニル基」とは, 「モノ若しくはジ低級アルキルアミ ノー C ( = O)—」を意味する: “Mono or di-lower alkylaminocarbonyl group” means “mono or di-lower alkylamino C (= O) —”:
「R4と R5がー体となってヘテロ原子を含んでいてもよいシクロアルキル基」を形成するとは, 環原子として R4と R5及びそれらと結合している炭素原子を含む上記シクロ— And forming a "R 4 and R 5 gar body and turned by cycloalkyl group which may contain a hetero atom", the cycloalkyl containing carbon atom bonded to R 4 and R 5 and them as ring atoms —
環上に窒素原子,酸素原子,硫黄原子から選択されるへテロ原子 1つを含んでレ The ring contains one heteroatom selected from nitrogen, oxygen, and sulfur.
また,該ヘテロ原子,例えば,硫黄原子は 1又は 2個のォキソ基で置換されてレ In addition, the hetero atom, for example, a sulfur atom is substituted with one or two oxo groups and
素原子は低級アルキル基で置換されてレ、てもよレ \;ヘテロ原子は酸素原子が好ましく,具 体的にはォキサニルが挙げられる。 Atom Les substituted by a lower alkyl group, yo Re \ be; heteroatom is preferably an oxygen atom, the concrete include Okisaniru.
「nが 1のとき R5と R6がー体となって,シクロアルキレン基を形成する」とは, R5と R6及びそ れらと結合している炭素原子と を環原子として含む上記シクロアルキレンを形成すること を意味する。 "And n is a R 5 and R 6 gar body case 1, to form a cycloalkylene group" and includes a carbon atom bonded to R 5 and R 6 and its those as ring atoms It means forming the above cycloalkylene.
「ベンゼン環と縮合していても良いヘテロァリール基」とは,窒素原子,酸素原子又は硫 黄原子から選択されるへテロ原子 1乃至 3個を含む 5又は (う員へテロァリール基,又はベン ゼン環と縮合した 2環系へテロァリール基を意味し,該ヘテロァリールとしては,ビロール,ィ ミダゾール,ピラゾール,ピリジン,ピラジン,ピリミジン,ピリダジン,トリァゾール,チォフェン, チォピラン,フラン,ピラン,ジォキソラン,チアゾ一ル,イソチアゾール,チアジアゾール,チ ァジン,ォキサゾール,イソキサゾール,ォキサジァゾ一ル, フラザン, ジォキサゾール, ォ キサジン,ォキサジァジン,ジォキサジン等が挙げられ,ベンゼン環と縮合した含窒素へテ ロアリールとしてはインドール,イソインドール,キノリン,イソキノリン,ベンゾチォフェン,ベ ンゾチアゾ一ル,ベンゾフラン,ベンゾフラザン等が挙げられる::好ましくは,ピリジン,ピ :リミ ジン,インドール, キノリン,チォフェン, フラン等である:: “Heteroaryl group optionally condensed with a benzene ring” means 5 or (containing a heteroaryl group or benzene containing 1 to 3 heteroatoms selected from nitrogen, oxygen or sulfur atoms. A heterocyclic aryl fused with a ring, and the heteroaryl includes bilol, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, triazole, thiophene, thiopyran, furan, pyran, dioxolan, thiazole, Isothiazole, thiadiazole, Examples include azine, oxazole, isoxazole, oxaziazole, furazane, dioxazole, oxazine, oxazineazine, dioxazine, and the like. Examples of nitrogen-containing heteroaryl fused to a benzene ring include indole, isoindole, quinoline, isoquinoline, benzothiophene, and benzothiazolone. And benzofuran, benzofurazan and the like:: preferably, pyridine, p : limidine, indole, quinoline, thiophene, furan and the like ::
「ヘテロァリールカルボニル基」とは, 「ヘテロァリ一ルー c (=o)—」を意味し,ヘテロァリ 一ルとは,上記の 5又は (う員へテロアリールを意味する:: “Heteroarylcarbonyl group” means “heteroaryl c (= o) —”, and heteroaryl means the above 5 or ( membered heteroaryl):
「R8と が一体となって含窒素シクロアルキレン基を形成する」又は「nが 1のとき R7と R8が 一体となって含窒素シクロアルキレン基を形成する」とは,環原子として R8が置換している窒 素原子と R5が置換している炭素原子を含む 5〜7員含窒素シクロアルカン,又は環原子とし て R8が置換してレ、る窒素原子と R7が置換してレ、る炭素原子及び A2を含む 4〜7員含窒素 シクロアルカンを形成することを意味し,具体的にはピロール,ピぺリジン, 2—へキサヒドロ ァゼピン等が挙げられ,ピロール又はピぺリジンが好ましレ \: "R 8 and R together form a nitrogen-containing cycloalkylene group" or "When n is 1, R 7 and R 8 together form a nitrogen-containing cycloalkylene group" are defined as ring atoms R 8 and the 5- to 7-membered nitrogen-containing cycloalkane, or a ring atoms containing a carbon atom to which nitrogen atom and R 5 being substituted is substituted is substituted with R 8 les, Ru nitrogen atom and R 7 There substituting means to form a 4 to 7-membered nitrogen-containing cycloalkane containing Les, Ru carbon atoms and a 2, specifically pyrrole, piperidine, Kisahidoro Azepin the like to 2, Pyrrole or Pyridine is preferred \:
本発明化合物において三級アミンを有する化合物は当該ァミンがォキシド化されていて もよく,それらのォキシド化誘導体をすベて包含するものである。 In the compound of the present invention, the compound having a tertiary amine may be oxidized with respect to the amine, and includes all oxidized derivatives thereof.
本発明化合物(I)は,アミド結合に基づく互変異性体が存在する::置換基の種類によって は, 1個乃至複数個の不斉炭素原子を有する場合もあり,これに基づく(R)体, (S)体等の 光学異性体,ラセミ体,ジァステレオマ一等が存在す ::また,置換基の種類によっては, 二重結合を有する場合もあり, (Z)体, (E)体等の幾何異性体が存在すな.更に環を有す る化合物ではシス—トランスが存在することがある。本発明は,これらの異性体の分離され たものあるいは混合物を全て包含する。 The compound (I) of the present invention has a tautomer based on an amide bond .: Depending on the type of the substituent, the compound (I) may have one or more asymmetric carbon atoms. And isomers such as (S) isomers, racemic forms, diastereomers, etc. :: Also, depending on the type of substituent, there may be a double bond, (Z) form, (E) form The compound having a ring may have cis-trans. The present invention includes all separated or mixed forms of these isomers.
本発明化合物は塩を形成する。具体的には,無機酸若しくは有機酸との酸付加塩,ある いは無機若しくは有機塩基との塩であり,製薬学的に許容しうる塩が好ましレ \:これらの塩と しては,具体的には塩酸,臭化水素酸,ヨウ化水素酸,硫酸,硝酸若しくは燐酸等の鉱酸, 又はギ酸,酢酸,プロピオン酸,シユウ酸,マロン酸,コハク酸,フマル酸,マレイン酸,乳酸, リンゴ酸,酒石酸,クェン酸,メタンスルホン酸,エタンスルホン酸,ベンゼンスルホン酸若し くは,トルエンスルホン酸等の有機酸,又はァスパラギン酸若しくはグルタミン酸などの酸性 アミノ酸との付加塩,ナトリウム,カリウム,マグネシウム,カルシウム,アルミニウム, リチウム など無機塩基,メチルァミン,ェチルァミン,エタノールァミンなどの有機塩基,リジン,オル 二チンなどの塩基性アミノ酸との塩等を挙げることが出来る。更に 4級アンモニゥム塩である こともできる。 4級アンモニゥム塩は,具体的には低級アルキルハライド,低級アルキルトリフ ラート,低級アルキルトシラート又はべンジルハライド等と反応させて得られるアンモニゥム 塩であり,好ましくはメチノレョ一ジド又はべンジルクロリド等との塩である The compound of the present invention forms a salt. Specifically, the acid addition salts with inorganic or organic acids, there have is a salt with an inorganic or organic bases, Le Shi pharmaceutically acceptable salt is preferred \: is a salt thereof , Specifically, mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid or phosphoric acid, or formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid, Addition salts with organic acids such as lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid or toluenesulfonic acid, or acidic amino acids such as aspartic acid or glutamic acid, sodium, Inorganic bases such as potassium, magnesium, calcium, aluminum, and lithium; organic bases such as methylamine, ethylamine, and ethanolamine; lysine; Examples thereof include salts with basic amino acids such as ditin. It can also be a quaternary ammonium salt. The quaternary ammonium salt is specifically an ammonium salt obtained by reacting with a lower alkyl halide, a lower alkyl triflate, a lower alkyl tosylate or a benzyl halide, and preferably a salt with a methinoyl or benzyl chloride. Is
更に,本発明化合物は水和物,エタノール等との溶媒和物や結晶多形を形成することが できる。本発明は,これらの水和物,溶媒和物又は結晶多形の分離されたものあるいは混 合化合物を全て包含する: Further, the compound of the present invention can form a hydrate, a solvate with ethanol or the like, or a crystalline polymorph. The present invention includes all of these hydrates, solvates or crystalline polymorphs separated or mixed compounds :
(製造法) (Manufacturing method)
本発明化合物(I)は,種々の製造法を適用して製造することができる::以下にその代表的 な製造法について説明する The compound (I) of the present invention can be produced by applying various production methods: The typical production methods are described below.
第一製法 First manufacturing method

(II) (l) (II) (l)
(式中の記号は,前述と同様である ) (The symbols in the formula are the same as above.)
本製造法は,一般式(II)で示される置換ァ: 塩と,一般式(in)で示される カルボン酸又はその反応性誘導体又はチォ: 酸,又はその反応性誘導体とをアミド 化し,保護基を有するときは保護基を除去する事により本発明化合物(I)を製造する方法で ある。 This production method comprises the steps of amidating a substituted α-salt represented by the general formula (II) with a carboxylic acid or a reactive derivative or a thio-acid or a reactive derivative thereof represented by the general formula (in) to protect the compound. This is a method for producing the compound (I) of the present invention by removing the protecting group when having a group.
化合物(III)の反応性誘導体としては,カルボン酸のメチルエステル,ェチルエステル,ィ ソブチルエステル, tert—ブチルエステルなどの通常のエステル,酸クロリド,酸ブロミドの 如き酸ノヽライド,酸アジド, 2, 4ージニトロフエノールなどのフエノール系化合物や Iーヒドロ キシスクシンイミド, I—ヒドロキシベンゾトリアゾールなどの N—ヒドロキシァミン系化合物等と 反応させて得られる活性エステル,対称型酸無水物,アルキル炭酸ハライドなどのハロカル ボン酸アルキルエステルやビバロイルハライドなどと反応させて得られる有機酸系混合酸無 水物や塩化ジフエニルホスホリル, N—メチルモルホリンとを反応させ, 又はトリフエニルホス Examples of the reactive derivative of the compound (III) include ordinary esters such as methyl ester, ethyl ester, isobutyl ester, and tert-butyl ester of carboxylic acid; acid chlorides such as acid chloride and acid bromide; acid azide; Active esters, symmetrical acid anhydrides, alkyl carbonate halides, etc., obtained by reacting with phenolic compounds such as 4-dinitrophenol and N-hydroxyamines such as I-hydroxysuccinimide and I-hydroxybenzotriazole Reaction with an organic acid-based mixed acid anhydride, diphenylphosphoryl chloride, N-methylmorpholine obtained by reacting with halocarbonic acid alkyl ester or bivaloyl halide, or triphenylphosphoric acid
)有機憐化合物と N—ブロモスクシンイミド等の活性化剤の組み合わせで得られ る有機憐系の活性エステルが挙げられ : ) Obtained by combining an organic compound with an activator such as N-bromosuccinimide. Active esters of organic origin include:
またカルボン酸を遊離酸で反応させるとき, 又は活性 レを単離せずに反応させる 時など,ジシクロへキシルカルポジイミド,カルボニルジイミダ 一ノレ,ジフエ二/レホスホリ /レ アジド,ジェチルホスホリルシアニドや]ーェチルー 3― (3一 メチルァミノプロピル)カルボ When reacting a carboxylic acid with a free acid, or when reacting without isolating the activity, dicyclohexylcarposimide, carbonyldiimidone, diphene / lephosphoryl / azide, getylphosphoryl cyanide, ] -Ethyl-3- (3-methylaminopropyl) carbo
:ド塩酸塩,チォニルクロリド,ォキザリルクロリド,五塩化燐,三塩化燐,ォキシ塩化燐, : Hydrochloride, thionyl chloride, oxalyl chloride, phosphorus pentachloride, phosphorus trichloride, phosphorus oxychloride,
-ルー 1—ィルォキシ卜リス(ジメチルァミノ)フォスフォニゥムへキサフルオロフ ォスフエー卜,無水トリフルォロ酢酸,無水酢酸,ピ :バロイルク口リド,メタンスルホニルクロリド ゃトシルクロリド等の縮合剤を使用するのが好適である :: 特に本発明におレ、ては酸クロリド 又は,燐酸系の混合酸無水物を用いる方法が有利である: - Lou 1- Iruokishi Kisafuruorofu Osufue Bok to Bok squirrel (Jimechiruamino) Fosufoniumu, anhydride Torifuruoro acetic acid, acetic anhydride, Pi: Baroiruku port chloride, to use a condensing agent such as methanesulfonyl chloride Ya tosyl chloride are preferred: in particular In the present invention, a method using an acid chloride or a mixed acid anhydride of phosphoric acid is advantageous:
反応は使用する反応性誘導体や縮合剤などによっても異なるが,通常ジクロロメタン,ジ クロロェタン,クロ口ホルムなどのハロゲン化炭化水素類,ベンゼン, トルエン,キシレン等の 芳香族炭化水素類,ヱ一テル,テ卜ラヒドロフラン等のエーテル類,酢酸ェチルエステル等 のエステル類,ァセトニトリル, N, N—ジメチルホルムアミド, N, N—ジメチルァセトアミド, N—メチルー 2—ピロリドンやジメチルスルホキシド等の反応に不活性な有機溶媒中,冷却 下,冷却下乃至室温下,又は室温乃至加熱下に行われる。 The reaction varies depending on the reactive derivative used and the condensing agent, but is usually halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform, aromatic hydrocarbons such as benzene, toluene and xylene, polyester, and the like. Inert organics such as ethers such as tetrahydrofuran, esters such as ethyl acetate, acetonitrile, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone and dimethyl sulfoxide. The reaction is performed in a solvent, under cooling, under cooling to room temperature, or at room temperature to heating.
尚,反応に際して,置換ァニリン(Π)を過剰に用いたり, N—メチルモルホ 11ソ 1、" ァミン, トリェチルァミン, N, N—ジメチルァニリン,ピリジン, 4一(N, N - ピリジン,ピコリン,ルチジンなどの塩基の存在下に反応させるのが,反応を円滑に進行させ る上で有利な場合がある。ピリジンなどは溶媒とすることもできる:: In the reaction, an excess of substituted aniline (Π) may be used, or N-methylmorpho 11so 1, "amine, triethylamine, N, N-dimethylaniline, pyridine, 4- (N, N-pyridine, picoline, lutidine) It is sometimes advantageous to carry out the reaction in the presence of a base such as pyridine, etc. Pyridine etc. can be used as a solvent ::
この際分子内に存在する酸素原子,硫黄原子,窒素原子等は保護基と結合してレ、ること が望ましい場合があり,このような保護基としては Greene及び Wuts著, 「Protective Groups in Organic Synthesis」第 2版に記載の保護基等を挙げることができ,これらを反応条件に応 じて適宜使レ、分けることができる。 In this case, it may be desirable that oxygen, sulfur, nitrogen, etc. present in the molecule be bonded to a protecting group, and such a protecting group is described in Greene and Wuts, "Protective Groups in Organic." And the like. The protecting groups described in “Synthesis” 2nd edition can be used, and these can be used and divided appropriately according to the reaction conditions.
第二製法 Second manufacturing method

(式中の記号は,前述と同様である。 ) 本製造法は,本発明化合物(IV)で示される置換アミン又はその塩と, - れるカルボン酸,又はその反応性誘導体,スルホン酸,又はその反応性誘導体,チォカル ボン酸, 又はその反応性誘導体とをアミド化し,本発明化合物(1)を製造する方法であり, 第一製法と同様の反応条件が使用可能である:: (The symbols in the formula are the same as above.) This production method comprises the steps of: (a) adding a substituted amine represented by the present compound (IV) or a salt thereof to a carboxylic acid or a reactive derivative thereof, a sulfonic acid or a reactive derivative thereof, thiocarboxylic acid or a reactive derivative thereof To produce the compound (1) of the present invention by using the same reaction conditions as in the first production method:
更にウレァ又はチォゥレア誘導体を合成する際には,上記の他にイソシアン酸エステル, 又はイソチォシアン酸エステル誘導体との縮合反応を用いるのが好適である.. Further, when synthesizing urea or thiourea derivatives, it is preferable to use a condensation reaction with an isocyanate ester or an isothiocyanate derivative in addition to the above.
反応は使用する反応性誘導体や縮合剤などによっても異なるが,通常ジクロロメタン,ジ クロロェタン, クロ口ホルムなどのハロゲン化炭化水素類,ベンゼン, トルエン,キシレン等の 芳香族炭化水素類,エーテル,テトラヒドロフラン等のヱ一テル類,酢酸ヱチルエステル等 のエステル類, N, N—ジメチルホルムアミドゃ N, N—ジメチルァセ卜アミドゃジメチルスル ホキシド等の反応に不活性な有機溶媒中,冷却下,冷却下乃至室温下,又は室温乃至加 熱下に行われる:. The reaction varies depending on the reactive derivative used and the condensing agent, but is usually halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform, aromatic hydrocarbons such as benzene, toluene, xylene, ether, tetrahydrofuran, etc. In an organic solvent inert to the reaction of ethers such as ethers, esters such as ethyl acetate, and N, N-dimethylformamide N, N-dimethylacetamide, dimethyl sulfoxide, etc., under cooling, under cooling to room temperature, Or at room temperature or under heating:
尚,反応に際して,本発明化合物(IV)を過剰に用いたり, N—メチルモルホリン,トリメチ ルァミン, トリェチルァミン, N, N—ジメチルァニリン,ピリジン, 4一(N, N—ジメチルァミノ) ピリジン,ビコリン,ルチジンなどの塩基の存在下に反応させるのが,反応を円滑に進行させ る上で有利な場合がある::ピリジンなどは溶媒とすることもできる:: In the reaction, the compound (IV) of the present invention was used in excess, or N-methylmorpholine, trimethylamine, triethylamine, N, N-dimethylaniline, pyridine, 4- (N, N-dimethylamino) pyridine, bicholine, The reaction in the presence of a base such as lutidine may be advantageous for the smooth progress of the reaction. :: Pyridine etc. can be used as a solvent.
第三製法 Third manufacturing method

(la) (lb) (la) (lb)
(式中の記号は,前述と同様である。 ) (The symbols in the formula are the same as above.)
本製造法は,本発明化合物(la)で示されるアミド基をチオアミド基へと変換し,本発明化 合物(lb)を製造する方法である。 In this production method, the amide group represented by the compound (la) of the present invention is converted to a thioamide group to produce the compound (lb) of the present invention.
本反応は五硫化ニ憐, Lawesson試薬等アミド誘導体からチオアミド誘導体を合成する 公知である任意の化学的方法により製造可能である: This reaction can be prepared by any of the known chemical methods for synthesizing thioamide derivatives from amide derivatives such as dipentapentasulfide and Lawesson's reagent:
反応は通常ジクロロメタン,ジクロロエタン,クロ口ホルムなどのハロゲン化炭化水素類,ベ ンゼン, トルエン,キシレン等の芳香族炭化水素類,エーテル,テトラヒドロフラン等のエーテ ル類,酢酸ェチルエステル等のエステル類,ピリジン等の反応に不活性な有機溶媒中,冷 却下,冷却下乃至室温下, 又は室温乃至加熱下に行われる The reaction is usually carried out with halogenated hydrocarbons such as dichloromethane, dichloroethane and chloroform, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as ether and tetrahydrofuran, esters such as ethyl acetate and pyridine. In an organic solvent inert to the reaction Rejected, performed under cooling to room temperature, or at room temperature to heating
また分子内に複数個のアミド基又はゥレアが存在する場合,反応条件等の調節により任 意の部位又は複数個のアミド基をチオアミド基に,ウレァをチォゥレアに変換する事が可能 である:: When there are multiple amide groups or ureas in the molecule, it is possible to convert any site or multiple amide groups to thioamide groups and urea to thiourea by adjusting the reaction conditions, etc .:
その他,加水分解,水素化,ウレイド化等も常法により行われる In addition, hydrolysis, hydrogenation, ureidation, etc. are also carried out by ordinary methods.
このようにして製造された本発明化合物は,遊離のまま,その塩,その水和物,その溶媒 和物,あるいは結晶多形の物質として単離精製される::本発明化合物(I)の塩は,常法の 造塩反応に付すことにより製造することもできる The compound of the present invention thus produced can be isolated and purified as a salt, a hydrate, a solvate, or a polymorphic substance of the compound of the present invention as it is free. Salt can also be produced by subjecting it to a conventional salt formation reaction.
上記製法の原料化合物中には、新規な物質も含まれてレ、るが、参考例記載の製法やそ の製法に準ずる方法,或いは当業者が任意に実施可能な変法を適用して製造できる。 単離精製は,抽出,濃縮,留去,結晶化,濾過,再結晶,各種クロマトグラフィー等の通 常の化学操作を適用して行われる:: Although the starting compound of the above-mentioned production method may contain a novel substance, it may be produced by applying the production method described in Reference Example, a method analogous to the production method, or a modified method arbitrarily practicable by those skilled in the art. it can. Isolation and purification are performed by applying ordinary chemical procedures such as extraction, concentration, distillation, crystallization, filtration, recrystallization, and various types of chromatography:
各種の異性体は,適当な原料化合物を選択することにより,あるレ、は異性体間の物理化 学的性質の差を利用して分離することができる。例えば,光学異性体は適当な原料を選択 することにより,あるいはラセミ化合物のラセミ分割法により,立体化学的に純粋な異性体に 導くこと力 Sできる c 産業上の利用可能性 By selecting appropriate starting compounds, various isomers can be separated by utilizing the difference in physicochemical properties between isomers. For example, by optical isomers selecting appropriate raw materials, or by racemic resolution method of racemic compounds, the availability of the c industry that can be force S lead to stereochemically pure isomers
本発明化合物はアンドロゲンによる転写活性化を抑制することにより,強力な抗アンド口 ゲン作用を有し, 中枢作用,ァゴニスト作用等の副作用の少ない化合物である。 The compound of the present invention has a strong anti-androgenic effect by suppressing the transcriptional activation by androgen, and is a compound having few side effects such as central action and agonist action.
従って,本発明化合物はアンドロゲンが增悪因子として関与する疾患,例えば,前立腺 癌,前立腺肥大症,男性化症,多毛症,禿頭症,ざ瘡,脂漏等の治療又は予防剤として有 用である。 本発明化合物の有用性は,下記の試験方法により確認されて!、る。 Therefore, the compound of the present invention is useful as a therapeutic or prophylactic agent for diseases in which androgen is involved as an adverse factor, for example, prostate cancer, prostatic hypertrophy, virilization, hirsutism, baldness, acne, seborrhea and the like. is there. The usefulness of the compound of the present invention has been confirmed by the following test methods! RU
1 . ヒトアンドロゲン受容体に対する転写活性化作用 1. Transcriptional activation of human androgen receptor
ヒト アンドロゲン受容体発現遺伝子, MMTVレボーター遺伝子安定形質転換体およびHuman androgen receptor expression gene, MMTV reporter gene stable transformant and
SV40レボーター遺伝子安定形質転換体の取得 Obtaining SV40 reporter gene stable transformant
CHO細胞を,直径 100 mmの細胞培養用ディッシュに I X 106 個播き, 12〜18時間後 に,リン酸カルシウムと共沈殿させたヒト アンドロゲン受容体発現プラスミド, MMTV-LTR ルシフェラ一ゼレボータープラスミド(ネオマイシン耐性遺伝子も含む)を加えトランスフエ クシヨンを行った。 15時間後に培地を除き,細胞を数段階に希釈し播き直し,培地に GENETICIN® (ネオマイシン)を終濃度 500 μ g/mlとなるようにカロえた,:約 1週間後,ネオ マイシンによって選択された細胞を剥がし,限界希釈法によりヒ卜 アンドロゲン受容体発 現遺伝子, MMTV-ルシフェラーゼレボータ -遺伝子を恒常的に発現する細胞を単離取 得した (CHO/MMTV安定形質転換体 : CHO cells were seeded on a 100-mm-diameter cell culture dish with 10 6 IX cells, and 12 to 18 hours later, a human androgen receptor expression plasmid co-precipitated with calcium phosphate, MMTV-LTR The luciferase jelly boater plasmid (including the neomycin resistance gene) was added and transfection was performed. After 15 hours, the medium was removed, the cells were diluted in several steps, and replated, and the medium was fed with GENETICIN® (neomycin) to a final concentration of 500 μg / ml .: About 1 week later, the cells were selected by neomycin. The isolated cells were detached, and cells expressing the human androgen receptor expression gene, MMTV-luciferase revoter-gene, constitutively expressing and isolated by the limiting dilution method (CHO / MMTV stable transformant:
上記と同様にして SV40レボーター遺伝子安定形質転換体を取得した。ただし, SV40 レボータープラスミドとネオマイシン耐性遺伝子発現プラスミドを同時にトランスフヱクトした (CHO/SV40安定形質転換体;^ An SV40 reporter gene stable transformant was obtained in the same manner as described above. However, the SV40 reporter plasmid and the neomycin resistance gene expression plasmid were simultaneously transfected (CHO / SV40 stable transformant; ^
a)ヒト アンドロゲン受容体に対する転写活性化作用の評価 (agonist作用) a) Evaluation of transcriptional activation on human androgen receptor (agonist effect)
CHO/MMTV安定形質転換体細胞および CHO/SV40安定形質転換体細胞を,それぞ れ%well細胞培養用ルミノブレ一 1、に 1 X 104個播き, 6〜8時間後に本発明化合物を添 カロした =化合物添加 18間後に 1% トリトン- Xおよび 10% グリセロールを含む溶液 20 μ 1を加え細胞を溶かし, 0.47mM ルシフェリンを含むルシフェラ一ゼ基質液 100 μ 1を加 え,ルミノメーターを用いて発光量を測定し,これらをヒト アンドロゲン受容体による MMTV-LTR転写活性化および,非特異的な SV40フロモーター転写活性化により得ら れるルシフェラーゼの活性とした:: CHO / MMTV stable transformant cells and CHO / SV40 stable transformant cells were seeded at 1 × 10 4 cells / well in a luminobure 1 for% well cell culture, and the compound of the present invention was added 6 to 8 hours later. 18 minutes after compound addition, add 20 μl of a solution containing 1% Triton-X and 10% glycerol to lyse the cells, add 100 μl of luciferase substrate solution containing 0.47 mM luciferin, and use a luminometer. The luminescence was measured and these were taken as the luciferase activities obtained by MMTV-LTR transcriptional activation by the human androgen receptor and non-specific SV40 flow motor transcriptional activation:
本発明化合物による転写活性化作用を InM DHTにより誘導される転写活性に対する 比率として以下の式により算出した。 The transcription activation effect of the compound of the present invention was calculated by the following formula as a ratio to the transcription activity induced by InM DHT.
誘導率(%)=100( -8)/(1-8) Induction ratio (%) = 100 (-8) / (1-8)
I: InM DHTを添加した場合の (MMTVルシフェラ-セ"活性)/ (SV40ルシフェラ セ'活性) B:無処置での (MMTVルシフェラ—セ'活性)/ (SV40ルシフェラ—セ"活性) I: (MMTV Luciferase activity) / (SV40 Luciferase activity) when InM DHT is added B: Untreated (MMTV Luciferase activity) / (SV40 Luciferase activity)
X:本発明化合物を添加した場合の (MMTVルシフェラ セ'活性)/ (SV40ルシフェラ セ"活性) b)ヒト アンドロゲン受容体に対する転写活性化抑制作用の評価(antagonist作用) X: (MMTV luciferase 'activity) / (SV40 luciferase' activity) when the compound of the present invention is added b) Evaluation of transcriptional activation inhibitory effect on human androgen receptor (antagonist effect)
CHO/MMTV安定形質転換体細胞および CHO/SV40安定形質転換体細胞を,それぞ れ%well細胞培養用ルミノブレートに 1 X 104個播き, 6〜8時間後に DHT (最終濃度 0.3nM)と同時に本発明化合物を添加した。化合物添加 18間後に 1% トリトン- Xおよび 10% グリセ口一ルを含む溶液 20 μ 1を加え細胞を溶かし, 0.47mM ルシフェリンを含む ルシフェラーゼ基質液 100 μ 1を加え,ルミノメ一ターを用いて発光量を測定し,これらを ヒト アンドロゲン受容体による MMTV-LTR転写活性化および,非特異的な SV40ブロ モ—ター転写活性化により得られるルシフェラーゼの活性とした:: CHO / MMTV stable transformant cells and CHO / SV40 stable transformant cells were seeded at 1 × 10 4 cells each in a luminoblate for% well cell culture, and after 6 to 8 hours, simultaneously with DHT (final concentration 0.3 nM). The compound of the present invention was added. Eighteen hours after the addition of the compound, the cells were lysed by adding 20 μl of a solution containing 1% Triton-X and 10% glycerol, and 100 μl of a luciferase substrate solution containing 0.47 mM luciferin was added, followed by luminescence using a luminometer. Measure the amount Luciferase activity obtained by MMTV-LTR transcriptional activation by human androgen receptor and non-specific SV40 promoter transcriptional activation:
本発明化合物による転写活性化抑制作用を 0.3nM DHTにより誘導される転写活性に 対する阻害率として以下の式により算出した。 The inhibitory effect on transcriptional activation by the compound of the present invention was calculated by the following formula as the inhibition rate against the transcriptional activity induced by 0.3 nM DHT.
阻害率(%)=100(1'- Χ')/(Γ-Β) Inhibition rate (%) = 100 (1'-Χ ') / (Γ-Β)
Γ:0.3ηΜ DHTのみ添加した場合の (MMTVルシフェラ—セ"活性)/ (SV40ルシフェラ—セ"活性) B:無処置での (MMTVルシフェラーセ"活性)/ (SV40ルシフェラーセ"活性) Γ: 0.3ηΜ When only DHT is added (MMTV luciferase “activity”) / (SV40 luciferase “activity”) B: Untreated (MMTV luciferase “activity” / (SV40 luciferase ”activity)
X' :本発明化合物と 0.3nM DHTを同時に添加した場合の (MMTVルシフェラ セ'活 X ': (MMTV luciferase' activity when simultaneously adding the compound of the present invention and 0.3 nM DHT
性)/ (SV40ルシフェラ—セ"活性) Sex) / (SV40 Luciferase-active)
上記の方法で算出した阻害率が 50%となる本発明化合物の濃度から IC50を求めた。 上記 a)及び b)により求められた本発明化合物の活性を以下に示す:: The IC 50 was determined from the concentration of the compound of the present invention at which the inhibition rate calculated by the above method was 50%. The activity of the compound of the present invention determined by the above a) and b) is shown below:
表 1 table 1

2.幼若去勢ラットのテストステロン誘導前立腺重量増加に対する抑制作用 2. Inhibitory effect on testosterone-induced increase in prostate weight in young castrated rats
3週令の雄性 Wistarラットを去勢後 72時間よりブロピオン酸テストステロンおよび本発明 化合物を同時に 1日 1回 5日間連続投与した。最終投与 6時間後,腹側前立腺の湿重量を 測定し,プロピオン酸テストステロンによる前立腺重量増加に対する本発明化合物の抑制 作用を検討した。 Three-week-old male Wistar rats were simultaneously administered testosterone propionate and the compound of the present invention once a day for 5 consecutive days from 72 hours after castration. Six hours after the final administration, the wet weight of the ventral prostate was measured, and the inhibitory effect of the compound of the present invention on the increase in prostate weight by testosterone propionate was examined.
プロピオン酸テストステロンは 5% エタノールを含む綿実油に溶解しラット体重 lKgあたり 0.5mgを皮下投与した :;本発明化合物は 0.5% メチルセルロース溶液に懸濁し経口投与し た:: Testosterone propionate was dissolved in cottonseed oil containing 5% ethanol and subcutaneously administered at a dose of 0.5 mg / kg body weight of the rat : the compound of the present invention was orally administered in a 0.5% methylcellulose solution ::
本発明化合物の前立腺重量増加抑制作用はフロビオン酸テストステロンおよび本発明化 合物をともに投与した群を試験群,プロピオン酸テストステロンのみを投与した群を対照群, プロピオン酸テストステロンおよび本発明化合物ともに投与しない群を無処置群として、以下 の計算式により算出した。 抑制率(%)=100(8^)/(8 ) The prostate weight increase inhibitory effect of the compound of the present invention was as follows: test group in which testosterone flobionate and the compound of the present invention were administered together, test group in which testosterone propionate alone was administered, and control group, and no testosterone propionate and the compound of the present invention. The group was set as an untreated group and calculated by the following formula. Suppression rate (%) = 100 (8 ^) / (8)
A:試験群の腹側前立腺湿重量 A: Wet weight of ventral prostate in test group
B:対照群の腹側前立腺湿重量 B: Wet weight of ventral prostate in control group
C:無処置群の腹側前立腺湿重量 C: Ventral prostate wet weight in the untreated group
上記の試験法により求められた本発明化合物の活性を以下に示す The activity of the compound of the present invention determined by the above test method is shown below.
表 2

これらの試験により,本発明化合物は純粋な抗アンドロゲン作用を有し,アンドロゲンの作 用を強く抑制することが確認された また, 中枢移行性が低レ、ことも確認されており,副作用 が少なく,アンドロゲンが増悪因子として関与する疾患の予防'治療薬として有用な化合物 である = From these tests, it was confirmed that the compound of the present invention had a pure antiandrogen action and strongly inhibited the action of androgen. In addition, it was also confirmed that the compound had a low central nervous system, and that there were few side effects. , Androgen is a compound useful as a therapeutic agent for the prevention and treatment of diseases associated with exacerbation factors =
本発明化合物又はその塩の 1種又は 2種以上を有効成分として含有する製剤は,通常製 剤化に用いられる担体ゃ賦形剤,その他の添加剤を用いて調製される。 Preparations containing one or more of the compound of the present invention or a salt thereof as an active ingredient are prepared using carriers, excipients and other additives that are usually used for preparation.
投与は錠剤,丸剤,カプセル剤,顆粒剤,散剤,液剤等による経口投与,あるいは静注, 筋注等の注射剤,坐剤,経皮等による非経口投与のいずれの形態であっても良い ::投与 量は症状,投与対象の年令,性別等を考慮して個々の場合に応じて適宜決定される力 通 常経口投与の場合成人 1日当り (). 0 1〜: l OOOmg程度,好ましくは 0. l ~ 100mg,非経口 投与の場合成人 1日当り (). 1 - l OOmg,好ましくは 0. 001〜50mg程度であり,これを 1回 で,あるいは 2〜4回に分けて投与する c The drug can be administered orally in the form of tablets, pills, capsules, granules, powders, liquids, etc., or injections such as intravenous and intramuscular injections, and parenteral administrations such as suppositories and transdermals. good:: dose symptoms, age of the administration subject, taking into account the sex and the like when an adult per day force through normal oral administration is appropriately determined on a case-by-case basis () 0 1~:. l OOOmg about 1 to 100 mg, preferably 0.1 to 100 mg per adult for parenteral administration per day (). 1 to 100 mg, preferably about 0.001 to 50 mg, divided into 1 dose or 2 to 4 doses Dosing c
本発明による経口投与のための固体組成物としては,錠剤,散剤,顆粒剤等が用いられ る。このような固体組成物においては,一つ又はそれ以上の活性物質が,少なくとも一つの 不活性な希釈剤,例えば乳糖,マンニトール,ブドウ糖,ヒドロキシプロピルセルロース,微 結晶セルロース,デンプン,ボリビニルピロリドン,メタケイ酸,アルミン酸マグネシウムと混合 される:組成物は,常法に従って,不活性な希釈剤以外の添加剤,例えばステアリン酸マグ ネシゥムのような潤滑剤や繊維素ダルコール酸カルシウムのような崩壊剤,ラクト一スのよう な安定化剤,グルタミン酸又はァスパラギン酸のような溶解補助剤を含有してレ、ても良レ \; 錠剤又は丸剤は必要によりショ糖,ゼラチン,ヒドロキシプロピルセルロース,ヒドロキシプロ -ト等の糖衣又は胃溶性あるレ、は腸溶性物質のフィルムで被 膜しても良い: As the solid composition for oral administration according to the present invention, tablets, powders, granules and the like are used. In such solid compositions, the one or more active substances include at least one inert diluent such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone, metasilicate. Mixed with acid, magnesium aluminate: The composition is formulated according to the usual practice with additives other than inert diluents, such as lubricants such as magnesium stearate and disintegrants such as calcium cellulose dalcholate. Tablets or pills may contain sucrose, gelatin, hydroxypropylcellulose, hydroxypro as required, including stabilizers such as lactose and solubilizing agents such as glutamic acid or aspartic acid. -Sugar coatings or gastric-soluble coatings such as ト may be coated with a film of enteric substance:
経口投与のための液体組成物は,薬剤的に許容される乳濁剤,溶液剤,懸濁剤, プ剤,エリキシル剤等を含み,一般的に用いられる不活性な希釈剤,例えば精製水, ールを含む::この組成物は不活性な希釈剤以外に湿潤剤,懸濁剤のような補助剤,甘味剤, 風味剤,芳香剤,防腐剤を含有していても良い,:: Liquid compositions for oral administration include pharmaceutically acceptable emulsifiers, solutions, suspensions, tablets, elixirs and the like, and are generally inert diluents such as purified water. Contains: In addition to the inert diluent, this composition may contain adjuvants such as wetting agents, suspending agents, sweetening agents, flavoring agents, flavoring agents, preservatives, :
非経口投与のための注射剤としては,無菌の水性又は非水性の溶液斉 ij,懸濁剤,乳濁 剤を包含する 水性の溶液剤,懸濁剤としては,例えば注射用蒸留水及び生理食塩水が 含まれる。非水溶性の溶液剤,懸濁剤としては,例えばブ :ロピレングリコール,ボリエチレン グリコ一ル,ォリーブ油のような植物油,エタノールのようなアルコール類,ボリソルベート 80 等がある-このような組成物はさらに防腐剤,湿潤剤,乳化剤,分散剤,安定化剤(例えば, ラクトース),溶解補助剤(例えば,グルタミン酸,ァスバラギン酸)のような補助剤を含んでい ても良い。これらは例えばバクテリア保留フィルターを通す濾過,殺菌剤の配合又は照射に よって無菌化される。また,これらは無菌の固体組成物を製造し,使用前に無菌水又は無 菌の注射用溶媒に溶解して使用することもできる: 発明を実施するための最良の形態 Injections for parenteral administration include sterile aqueous or non-aqueous solutions ij, aqueous solutions including suspensions and emulsions, and suspensions include, for example, distilled water for injection and physiological saline. Contains saline. Non-aqueous solutions and suspensions, for example blanking: b propylene glycol, Boriechiren glycolate Ichiru, vegetable oils such as Oribu oil, alcohols such as ethanol, Borisorubeto 80. - Such compositions In addition, it may contain adjuvants such as preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), and solubilizing agents (eg, glutamic acid, aspartic acid). These are sterilized by, for example, filtration through a bacteria retaining filter, blending of a bactericide or irradiation. They can also be used in the production of sterile solid compositions which are dissolved in sterile water or sterile injectable solvents before use.
以下に実施例を掲記し,本発明を更に詳細に説明する,:本発明は,これらの実施例に何 ら制限されるものではなレ、::尚,実施例で用いられる原料化合物の製造方法を参考例とし て説明する The following examples are provided to describe the present invention in more detail. The present invention is not limited to these examples. :: Production of the starting compounds used in the examples The method is explained as a reference example
参考例:! 一 1 Reference example:!
2 - (4 -フルオロフェニルスルホニルァミノ)ブタン酸メチル Methyl 2- (4-fluorophenylsulfonylamino) butanoate
2—ァミノ ブタン酸メチル塩酸塩 1 · 54gをクロ口ホルム 15mlに溶解し,氷冷下,トリェチ ルァミン 2. 23g, p—フルオロフェニルスルホニルクロリド 1. 95gを順次滴下した後,ァルゴ ン雰囲気下,室温で 4時間攪拌した。,反応液をクロ口ホルム 35mlで希釈し, 1 N塩酸 50ml, 飽和重曹水 5()mlで洗浄後,無水硫酸マグネシウムで乾燥した:減圧下に溶媒を留去した 後,残渣にジイソプロピルエーテル l ()mlを加え,析出した結晶を酢酸ェチルより再結晶し , 無色結晶の表題化合物 2. 73gを得た。 Dissolve 1.54 g of methyl 2-aminobutanoate hydrochloride in 15 ml of chloroform and add dropwise 23.23 g of triethylamine and 1.95 g of p-fluorophenylsulfonyl chloride under ice-cooling. The mixture was stirred at room temperature for 4 hours. The reaction mixture was diluted with 35 ml of chloroform, washed with 50 ml of 1N hydrochloric acid and 5 () ml of saturated aqueous sodium hydrogen carbonate, and dried over anhydrous magnesium sulfate. After distilling off the solvent under reduced pressure, diisopropyl ether was added to the residue. () ml was added, and the precipitated crystals were recrystallized from ethyl acetate to give 2.73 g of the title compound as colorless crystals.
参考例 1一 1と同様に以下の参考例を合成した。 参考例 化合物名 The following Reference Example was synthesized in the same manner as Reference Example 11-11. Reference example Compound name
参考例 1 2 リ (4—フノレ才「一 く-ノゾィルァミノ) 2 メチルプロバン酸メチル Reference Example 1 2 Li (4-Funore “Iku-Nozylamino”) 2 Methyl methyl propanoate
参考例 1 - 3 2— (2 メトキシ ゾィルァミノ)一■ 2 メチルプロパン酸メチル Reference Example 1-3-2 (2-Methoxyziramino) methyl 2-methylpropanoate
参考例 1 - 4 2— (4-シアノ^ ン ノ"ィルァミノ) 2 - メチルプロパン酸メチル Reference Example 1-42- (4-cyano ^ non "ylamino) 2-methylpropanoate
参考例 1 5 2— (4—トリフルォロメチルベンゾィルァミノ) 2 メチルプ口パン酸メチル 参考例 1 - 6 2— (4-フノレ才ロフ:: ニルスルホニルァミノ 2—メチルプロバン酸メチル 参考例 1 - 7 2— (4-ニトロフエ二ルスルホニルァミノ) 2 メチルプロパン酸メチル 参考例 1 - 8 2 - - (4-メトキシフェニルスルホニルァミノ) 2—メチルプロパン酸メチル 参考例 1 - 9 3 - (4—フノレオロフ: ニルスルホニルァミノ)ブ'ロバン酸メチル Reference Example 1 52 2- (4-Trifluoromethylbenzoylamino) 2-methylpropanoate Methyl Reference Example 1-62 2- (4-Funolei Lof :: Nylsulfonylamino 2-Methylpropanoate Reference Example 1-72 2- (4-Nitrophenylsulfonylamino) methyl 2-methylpropanoate Reference Example 1-82--(4-Methoxyphenylsulfonylamino) 2-methylpropanoate Reference Example 1-93 -(4-Funolelov: Nylsulfonylamino) methyl methyl lobanoate
参考例 1 - 10 3— (4一フルオロフ-: ニルスルホニルァミノ)ブタン酸メチル Reference Example 1-10 Methyl 3- (4-fluoro-: nylsulfonylamino) butanoate
参考例 1 - 1 1 1一 (4—フル才ロフ: 二ルスルホニルアミノ)シク 'プロピルカルボン酸メチル ■ 参考例 1 - 12 1― (4—フ /レ才 ノゾィルァミノ)シクし'プ Jピルカルボン酉 メチル REFERENCE EXAMPLE 1-1 1 1 1 (4-FURULOF: Nisulfonylamino) cyclo 'Methyl propylcarboxylate' ■ REFERENCE 1-1 Methyl
参考例 1 13 :1 一 (4 -フル才口フ ニルスルホニルァミノ)シ'クし'ペンチルカルボン酸メチル 参考例 1 - 14 1 (4—フルォロベ: ゾィルアミノ)シク口ペンチルカルボン酸メチル REFERENCE EXAMPLE 1 13: 1 Methyl (4-fluorophenylsulfonylamino) cyclopentylcarboxylate Reference Example 1-14-1 (4-Fluorobe: zylamino) methyl pentylcarboxylate
参考例 1 15 1 (4—フノレ才ロフ;: ニルスルホニルァミノ)シク 'へキシルカルボン酸メチル 参考例 1 - 16 1一 (4—フルォロベ、 ゾィルァミノ)シクロへキシルカルボン酉¾メチル REFERENCE EXAMPLE 1 15 1 (4-FUNORESHILOF ;: Nylsulfonylamino) cyclo 'methyl hexylcarboxylate Reference Example 1-16 1- (4-Fluorobe, zolylamino) cyclohexylcarboxy
参考例 1 - 17 4一 (4—フノレオロ^ : ゾィルァミノ)テ卜ラヒド πピラン—4 カルボン酸メチル 参考例 1 - 18 3- (4—フルォロ く:ノゾィルァミノ)—2.2,3 卜リメチルブタン酸ェチル 参考例 2— 1 Reference Example 1-17 4- (4-Funoleolo ^: Zyrylamino) tetrahydro πpyran-4 Methyl carboxylate Reference Example 1-18 3- (4-Fluoro-nozylamino) -2.2,3 Ethyl trimethylbutanoate Reference Example twenty one
2— (4—フノレオロフェニノレスルホニルァ: 2— (4—Funoleolopheninolesulfonyla:
2—(4—フルオロフェニルスルホニルァミノ)ブタン酸メチル 2. 73gをメタノール 4()mlに 溶解し,氷冷下, 1 N水酸化ナトリウム水溶液 2()mlを滴下後,室温で 8時間攪拌した::減圧 下にメタノールを留去した後,氷冷下, 1 N塩酸 2()mlを滴下して pH2とし,酢酸ェチル 50m 1で 4回抽出した c有機層を無水硫酸マグネシウムで乾燥し,減圧下に溶媒を留去した後, 残渣にジイソブロピルェ一テル 20mlを加え,析出した結晶を酢酸ェチルより再結晶し,無 色結晶の表題化合物 1 . 5:3gを得た:: Dissolve 2.73 g of methyl 2- (4-fluorophenylsulfonylamino) butanoate in 4 () ml of methanol, add 2 () ml of 1N aqueous sodium hydroxide solution under ice-cooling, and stir at room temperature for 8 hours. After the methanol was distilled off under reduced pressure, 2 () ml of 1N hydrochloric acid was added dropwise under ice cooling to pH2, and the organic layer c extracted four times with 50 ml of ethyl acetate was dried over anhydrous magnesium sulfate. After distilling off the solvent under reduced pressure, 20 ml of diisopropyl ether was added to the residue, and the precipitated crystals were recrystallized from ethyl acetate to obtain 1.5: 3 g of the title compound as colorless crystals ::
参考例 2— 1と同様に,以下の参考例を合成した。 The following Reference Example was synthesized in the same manner as Reference Example 2-1.
参考例 化合物名 Reference example Compound name
参考例 2— 2 2— [ (4- フ.ルオロフェニルスルホニル)メチノレァミノ]プロパン酸 Reference Example 2—2 2 — [(4-Fluorophenylsulfonyl) methinoleamino] propanoic acid
参考例 2— 3 2— (4—フルォロベンゾィルァミノ)—2—メチルプロパン酸 Reference Example 2—3 2— (4-Fluorobenzoylamino) -2-methylpropanoic acid
参考例 2— 4 2— (2 -メトキシベンゾィルァミノ)一 2—メチルプロパン酸 Reference Example 2—42- (2-Methoxybenzoylamino) -1-methylpropanoic acid
参考例 2— 5 2— (4-シァノベンゾィルァミノ) 2 メチルプロパン酸 Reference Example 2—5 2— (4-cyanobenzoylamino) 2 methylpropanoic acid
参考例 2— 6 2— (4—トリフルォロメチルベンゾィルァミノ)—2—メチルプ ン酸 Reference Example 2—62— (4-Trifluoromethylbenzoylamino) -2-methylbutyric acid
参考例 2— 7 2— (4—フルオロフェニルスルホニルァミノ) 2 - -メチルプ 'バン酸 Reference Example 2— 7 2— (4-Fluorophenylsulfonylamino) 2--methylp'banic acid
参考例 2— 8 2— (4—ニトロフエニルスルホニルァミノ) 2 メチルプ ン酉 ¾ Reference Example 2—8 2— (4-Nitrophenylsulfonylamino) 2-methylpyrrole ¾
参考例 2— 9 2— (4-メトキシフエニルスルホニルァミノ) 2 メチルプロパン酸 Reference Example 2-92- (4-methoxyphenylsulfonylamino) 2-methylpropanoic acid
参考例 2— 10 2— (N - - ンジノレォキシ 4ーフルォ t くンズアミド)一 2—メチ /レフ。ロノ、ン酸 参考例 2— 1 1 3— (4—フルオロフェニルスルホニルァミノ)プロパン酸 Reference Example 2—10 2— (N--Ninoleoxy 4-Fluoro-Kunsamide) One 2-Methyl / Ref. Lono, acid Reference Example 2-1 13- (4-Fluorophenylsulfonylamino) propanoic acid
参考例 2— 12 3- (4—フルオロフェニルスルホニルァミノ)ブタン酸 Reference Example 2—12 3- (4-Fluorophenylsulfonylamino) butanoic acid
参考例 2— 13 1— (4—フルオロフェニルスルホニルァミノ)シクロプロピルフ ルボン酸 参考例 2— 14 —(4一: ボン酸 参考例 2— 15 1一(4—フルオロフェニルスルホニルァミノ)シクロペンチルカルボン酸 参考例 2— 16 1— (4ーフノ π- 参考例 2— 17 1一(4—フルオロフェニルスルホニルァミノ)シクロへキシルカルボン g 参考例 2— 18 (4 :ノ)シク へキシルカルボン酸 Reference Example 2-13 1- (4-Fluorophenylsulfonylamino) cyclopropylfuronic acid Reference Example 2—14— (4-1: boric acid Reference Example 2—15 1— (4-fluorophenylsulfonylamino) cyclopentylcarboxylic acid Reference Example 2—16 1— (4-funo π- Reference Example 2—17 (4-Fluorophenylsulfonylamino) cyclohexylcarboxylic acid g Reference Example 2-18 (4: no) cyclohexylcarboxylic acid
参考例 2— 19 4 - (4 フル 4—カルボン酸 Reference Example 2—194- (4 full 4-carboxylic acid
参考例 2— 20 3—(4- 2.2.3 -トリメチルブタン酸 参考例 3 Reference Example 2—20 3 -— (4-2.2.3-trimethylbutanoic acid Reference Example 3
2— [ (4—フルォロフエ-ルスルホニル)メチルァミノ]フ :ロバン酸メチル 2 — [(4-Fluoroferylsulfonyl) methylamino] f : methyl rovanate
2—(4—フルオロフェニルスルホニルァミノ)フ πパン酸メチル 500mgを N, N—ジメチル ホルムアミド 5mlに溶解した後,炭酸カリウム 320mgを加え,室温で: 10分間攪拌した 反応 液を氷冷し,ヨウ化メチル 330mgを加え,アルゴン雰囲気下,室温で 12時間攪拌した 反 応液を酢酸ェチル 5()mlで希釈し,蒸留水 50mlで洗浄した後,無水硫酸マグネシウムで乾 燥した =減圧下に溶媒を留去した後,得られた粗結晶を酢酸ェチルから再結晶し,無色結 晶の表題化合物 470mgを得た。 After dissolving 500 mg of methyl 2- (4-fluorophenylsulfonylamino) pitanate in 5 ml of N, N-dimethylformamide, 320 mg of potassium carbonate was added, and the mixture was stirred at room temperature for 10 minutes. of methyl iodide was added 330 m g, under an argon atmosphere, the reaction solution was stirred at room temperature for 12 hours then diluted with acetic acid Echiru 5 () ml, washed with distilled water 50 ml, = vacuo was drying over anhydrous magnesium sulfate After evaporating the solvent, the obtained crude crystals were recrystallized from ethyl acetate to obtain 470 mg of the title compound as colorless crystals.
参考例 4一 1 Reference Example 41
2— ^ ンジルォキシカルボニルアミノー N— (4—シァノ一3—トリフルォロメチルフエニル) プロノヽンアミド 2— ^ benzyloxycarbonylamino-N— (4-cyano-13-trifluoromethylphenyl) prononamide
2 べンジルォキシカルボニルァミノプロバン酸 6. と N, N—ジメチルァセ卜アミド 70 mlの混合液を, 20 3に冷却した後,チォニルクロリド 3. 93gを滴下した =アルゴン雰囲 気下,同温度で 1時間攪拌した後, 4一アミノー2—トリフルォロメチルベンゾニトリル 5. 58g を少量ずつ加え,更に 3時間攪拌した。反応液を酢酸ェチル 2()()mlで希釈後,飽和重曹 水 200mlで洗浄し,更に蒸留水 20()mlで 2回洗浄した後,無水硫酸マグネシウムで乾燥し た。減圧下に溶媒を留去した後,残渣をシリカゲルカラムクロマトグラフィーに付し,酢酸ェ チル キサン(7: 3)溶出部より粗結晶を得た::この粗結晶を酢酸ェチルから再結晶し, 無色結晶の表題化合物 8. 17gを得た。 参考例 4一 1と同様に以下の参考例を合成した。 2 base Nji Ruo alkoxycarbonyl § amino Pro vans acid 6. and N, N-a Jimechiruase Bok amide 70 ml mixture, after cooling to 20 3, = argon Kiri囲gas under dropwise Chionirukurorido 3. 93 g, same After stirring at the temperature for 1 hour, 5.58 g of 4- amino- 2 -trifluoromethylbenzonitrile was added little by little, and the mixture was further stirred for 3 hours. The reaction solution was diluted with 2 () () ml of ethyl acetate, washed with 200 ml of saturated aqueous sodium bicarbonate, twice with 20 () ml of distilled water, and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the residue was subjected to silica gel column chromatography to obtain crude crystals from a fraction eluted with ethyl acetate ( 7 : 3) :: These crude crystals were recrystallized from ethyl acetate. 8.17 g of the title compound was obtained as colorless crystals. The following Reference Example was synthesized in the same manner as Reference Example 411.
参考例 化合物名 Reference example Compound name
参考例 4 2 2 ンジルォキシ力ルボニルアミノー N (4—シァノ 3 - -トリフルォロメチル フエニル)メチルプロパンァニリド Reference example 4 2 2 N-dioxy carbonylamino-N (4-cyano 3- -trifluoromethylphenyl) methylpropananilide
参考例 4 3 2―ベンジルォキシカルボニルアミノー N:—(4—シァノ 3 トリフルォロメチル フエニル) 3—メトキシプロパンアミド 参考例 4 4 3—べンジルォキシカルボニルアミノー N—(4—シァノー 3—トリフルォロメチル フエ二ノレ) 2, 2—ジメチノレフロノ、0ンアミド Reference Example 4 3 2-benzyl O butoxycarbonylamino over N: - (4-Shiano 3 triflate Ruo B methyl phenyl) 3-methoxy propanamide Reference Example 4 4 3 base Nji Ruo alkoxycarbonylamino over N-(4-Shiano 3 triflate Ruo Russia methyl phenylene Honoré) 2, 2-Jimechinorefurono, 0 N'amido
参考例 4 5 2—ァリルォキシカルボニルアミノー N— (3, 4—ジシァノフエ二ル)一 3—メチル 参考例 4 6 1 ベンジルォキシカルボニルァミノ -- N—(4—シァノ 3 --トリフルォロメチル フエニル)シクロブチルカルボキサミド 参考例 5 Reference Example 4 52 2-Aryloxycarbonylamino-N— (3,4-dicyanophenyl) -1-methyl Reference Example 4 61 Benzyloxycarbonylamino-N— (4-cyano 3- Trifluoromethylphenyl) cyclobutylcarboxamide Reference Example 5
2—(N—ベンジルォキシカルボニルァミノ) - 2 - ロノ ン酸 2- (N-benzyloxycarbonylamino) -2-rononic acid
1 N水酸化ナトリウム水溶液 56m]に,氷冷下, 2—了 2—メチルプロバン酸 5 · 79gを 加え,続いて,ベンジルォキシカルボニルクロリド 12. 酸化ナトリウム水溶液 75 mlをそれぞれ 4回に分けて滴下し,室温で 3時間攒桦した。反応溶液 エ で 3回洗った後, pH2になるまで I N塩酸を加え, 3()()mlの酢酸ェチルで 3回抽出した。有 機層を蒸留水で洗い,無水硫酸マグネシウムで乾燥し,減圧下に溶媒を留去した後,残渣 を酢酸ェチル—へキサン混合溶媒により再結晶し,無色結晶の表題化合物 6. 95gを得た 参考例 6 To a 1 N aqueous sodium hydroxide solution 56 m], under ice-cooling, add 5-79 g of 2-methylpropanoic acid, followed by benzyloxycarbonyl chloride. The mixture was added dropwise and left at room temperature for 3 hours. After washing with the reaction solution three times, IN hydrochloric acid was added until the pH reached 2, and the mixture was extracted three times with 3 () () ml of ethyl acetate. The organic layer was washed with distilled water, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was recrystallized from a mixed solvent of ethyl acetate-hexane to give 6.95 g of the title compound as colorless crystals. Reference Example 6
4 'ーシァノー 3 '—トリフルォロメチルトリフルォロアセ卜ァニリド 4'-cyano 3'-trifluoromethyl trifluoroacetanilide
4—アミノー 2 トリフルォロメチルベンゾニトリル 5. 0()gをクロ口ホルム 20mlに溶解し,無 水トリフルォロ酢酸 5. 3 l mlを加え室温にて 30分攪拌した 析出した結晶を濾取し, クロ口 ホルムで洗浄し表題化合物 3. 93gを得た、: 5.0 () g of 4-amino-2 trifluoromethylbenzonitrile was dissolved in 20 ml of chloroform, and 5.3 ml of anhydrous trifluoroacetic acid was added. The mixture was stirred at room temperature for 30 minutes, and the precipitated crystals were collected by filtration. After washing with chloroform, 3.93 g of the title compound was obtained:
参考例 7 Reference Example 7
4一メチルアミノー 2—トリフルォロメチルベンゾニトリル 4-Monomethylamino-2-trifluoromethylbenzonitrile
水素化ナトリウム 0. 22gを N, N—ジメチルホルムアミド 10mlに懸濁し,上記 4 ' —シァノ 一 3 ' —トリフルォロメチルトリフルォロアセトァニリド 1 . 41 gを加え氷冷下 30分攪拌した::反 応溶液にヨウ化メチル 0. 62mlを加え 4時間 60eCで攪拌後,反応溶液に水冷下飽和炭酸 カリウム水溶液 10mlを加え同温度で 1時間攪拌した。反応液を酢酸ェチル 50mlで希釈後, 蒸'留水 50mlで 2回洗浄し,無水硫酸マグネシウムで乾燥した。減圧下に溶媒を留去した後, 残渣をシリカゲルカラムクロマトグラフィーに付し,酢酸ェチル キサン(3: 7)溶出部より 表題化合物 0. 63gを得た。 Suspension of sodium hydride 0. 22 g N, N- dimethylformamide 10 ml, the 4 '- Shiano one 3' - stirred triflate Ruo b methyltriflate Ruo B acetonate § oxanilide 1 41 g was added under ice-cooling for 30 minutes. :: after stirring at reaction solution methyl iodide 0. 62 ml was added 4 hours 60 e C, water cooling under a saturated aqueous solution of potassium carbonate 10ml was added and stirred for 1 hour at the same temperature to the reaction solution. The reaction mixture was diluted with 50 ml of ethyl acetate, washed twice with 50 ml of distilled water, and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the residue was subjected to silica gel column chromatography to obtain 0.63 g of the title compound from a fraction eluted with ethyl acetate (3: 7).
参考例 8 Reference Example 8
2— (ベンジルォキシァミノ)一2—メチルプロパン酸ェチル a—ブロモイソ酪酸ェチル 1 · 95gおよび Ο—ベンジルヒドロキシ/レアミン塩酸塩 1 . 6gの N, N—ジメチルホルムアミド溶液 2()mlに無水炭酸カリウム 3. 3gをカロえ, 12():Cで 1 ()時問 攪拌した::反応溶液を減圧下濃縮し,残渣に水を加え,酢酸ェチルで抽出した。有機層を (). 5N塩酸,飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗滌した後,無水硫酸ナ トリウムで乾燥した。溶媒を減圧下に留去し,残渣をシリカゲルカラムクロマトグラフィーにて 精製し,へキサン一酢酸ェチル(1 (): 1 )溶出部より油状物として表題化合物 0. 3 を得た, 参考例 9 2- (benzyloxyamino) -1-ethyl methyl propanoate 1-95 g of a-bromoisobutyrate ethyl acetate and 1.6 g of Ο-benzylhydroxy / reamine hydrochloride in 2 () ml of N, N-dimethylformamide solution, calorie 3.3 g of anhydrous potassium carbonate. The mixture was stirred under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with (). 5N hydrochloric acid, a saturated aqueous solution of sodium hydrogencarbonate and saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography, and the title compound 0.3 was obtained as an oil from the hexane monoethyl acetate (1 (): 1) eluted portion, Reference Example 9
2— (N—ベンジルォキシー 4—フルォロベンズアミド)—2—メチルプロバン酸ェチル 2— (ベンジルォキシァミノ)一2—メチルブロバン酸ェチル 0.85 gのピリジン溶液 10 m 二 4一フルォロベンゾイルクロリド 0.68 gを加え, 10時間加熱還流した::反応溶液を減圧下濃 縮し,残渣に I N塩酸を加え,酢酸ェチルで抽出した ::有機層を水洗後,無水硫酸マグネシ ゥムで乾燥した。溶媒を減圧下留去し,得られた結晶を石油エーテルで洗滌し,表題化合 物 0.95 gを得た。 2- (N-benzyloxy-4-fluorobenzamide) -2-ethyl ethyl propylate 2- (benzyloxyamino) -1-ethyl ethyl ethyl methyl bromide 0.85 g pyridine solution 10 m 2 4-fluorobenzoyl chloride 0.68 g was added, the :: reaction solution was heated to reflux for 10 hours under reduced pressure Shimono reduced, the iN hydrochloric acid was added to the residue, and extracted with acetic acid Echiru:: the organic layer was washed with water and dried over anhydrous sulfate magnesium © beam . The solvent was distilled off under reduced pressure, and the obtained crystals were washed with petroleum ether to obtain 0.95 g of the title compound.
参考例 10— 1 Reference Example 10-1
2—アミノー N— (4—シァノ一3 - ロメチルフヱニル)フロバンアミド 2-amino-N- (4-cyano-3-lomethylphenyl) furovanamide
参考例 4一 1で合成した 2 - ニルァミノ一N— (4—シァノー 3—トリ チノレフエ-ル): , 2—ジクロロェタン 5mUこ溶解し,水冷 下,ジメチルスルフイド 79()mg,三フッ化ホウ素ジェチルエーテル錯体 6()()mgを順; アルゴン雰囲気下,室温で 13時間攪拌した後,飽和塩化アンモニゥム水溶液 10mlを加え た c更に室温で 30分攪拌した後,氷冷下, pH I Oになるまで 1 N水酸化ナトリウム水溶液を 加え,酢酸ェチル 50mlで 3回抽出し,有機層を無水硫酸マグネシウムで乾燥した =減圧下 に溶媒を留去した後,残渣にメタノールを加え,析出した結晶をメタノールより再結晶し,無 色結晶の表題化合物 189mgを得た。 Reference Example 4 2-Nilamino-N- (4-cyano-3-tritinolerefle) synthesized in 1-11: 5 mU of, 2-dichloroethane, dissolved in water, under cooling with water, 79 () mg of dimethylsulfide, trifluoride boron Jefferies chill ether complex 6 () () mg forward; under an argon atmosphere, was stirred at room temperature for 13 hours, the mixture was stirred for 30 minutes at c further at room temperature was added a saturated chloride Anmoniumu solution 10 ml, under ice-cooling, pH IO the 1 N aqueous sodium hydroxide until added, extracted three times with acetic Echiru 50 ml, the solvent was distilled off = vacuo was dried over anhydrous magnesium sulfate and the organic layer, the residue methanol was added to the precipitated crystals Was recrystallized from methanol to obtain 189 mg of the title compound as colorless crystals.
(別法) (Alternative)
2—アミノー 2—メチルプロピオン酸 277nigを N, N—ジメチルァセトァミド 3 mlに懸濁し、 — 10 Cにてチォニルクロリド 0. 206mlを加え同温にて 1時間攪拌した。次いで 4—ァミノ一 2—トリフルォロメチルベンズニ卜リルを加え、室温まで徐々に昇温しながら 2時間攪拌した 反応溶液に水を加え、酢酸ェチルで洗浄後、 1規定水酸化ナトリウム水溶液でアルカリ性に した。遊離した油状物を酢酸ュチルで抽出し、無水硫酸ナトリウムにて乾燥した 溶媒を減 圧下濃縮し、得られた残留物をシリカゲルカラムクロマトグラフィーにて精製し、クロ口ホルム 一メタノール(97 : 3)溶出部より表題化合物 4()()mgを無色油状物として得た 更に酢酸ェ チルーへキサンより再結晶を行 、表題化合物を得た ::本化合物の諸物性値は上記で得ら れた化合物と完全に一致した:: 277 nig of 2-amino-2-methylpropionic acid was suspended in 3 ml of N, N-dimethylacetamide, 0.26 ml of thionyl chloride was added at -10 C, and the mixture was stirred at the same temperature for 1 hour. Then, 4-amino-1-2-trifluoromethylbenznitrile was added, and the mixture was stirred for 2 hours while gradually warming to room temperature. Water was added to the reaction solution, washed with ethyl acetate, and then washed with a 1N aqueous sodium hydroxide solution. It was made alkaline. The released oil was extracted with butyl acetate, and the solvent dried over anhydrous sodium sulfate was reduced. The filtrate was concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography, and 4 () () mg of the title compound was obtained as a colorless oil from a fraction eluted with chloroform-methanol (97: 3). row recrystallized from hexane Chiru to give the title compound:: physical properties of this compound were in complete agreement with the compound obtained et the above:
参考例 10— 1と同様に以下の参考例を合成した The following Reference Example was synthesized as in Reference Example 10-1.
参考例 10— 2 Reference Example 10-2
2—アミノー N— (4—シァノー 3—トリフルォロメチルフエニル)一2—メチルブロバンアミド 参考例 10— 3 2-amino-N- (4-cyano 3-trifluoromethylphenyl) -1-methylbrovanamide Reference Example 10-3
N—(4—シァノー 3—トリフルォロメチルフエニル)一 2—メチルフロリンアミド塩酸塩 参考例 1 1一 1 N- (4-cyano 3-trifluoromethylphenyl) -1-methylfurolinamide hydrochloride Reference Example 1 1 1 1
2—ァミノ一 N— (13 , 4ージシァノフエニル)一 2—メチルフロバンアミド 2-amino-1 N- (13,4-dicyanophenyl) -1-2-methylfurovanamide
(1) 2—ベンジルォキシカルボニルァミノ一 2—メチルプロバン酸 30gと N, N—ジメチルァセ トアミド 130mlの混合液を, 一 2() Cに冷却した後,チォニノレクロリド K). 2mlを滴下した:.:ァ ルゴン雰囲気下, 同温度で 1時間攪拌した後, 4—ァミノフタロニトリル 18. 2gと N, N—ジメ チルァセトアミド 70mlの混合液を滴下し,同温度で 7時問,更に 0¾で 18時間攪拌した: :反 応液を酢酸ェチルで希釈後,飽和重曹水で洗净した: 更に, 1N塩酸,飽和塩化ナトリウム 水溶液で洗浄した後,無水硫酸マグネシウムで乾燥した。減圧下に溶媒を留去し,粗結晶 を得た (1) A mixture of 30 g of 2-benzyloxycarbonylamino-2-methylpropanoic acid and 130 ml of N, N-dimethylacetamide was cooled to 12 () C, and 2 ml of thionino rechloride K) was added dropwise. After stirring for 1 hour at the same temperature in an argon atmosphere, a mixed solution of 18.2 g of 4-aminophthalonitrile and 70 ml of N, N-dimethylacetoamide was added dropwise at the same temperature for 7 hours. was stirred for 18 h at 0¾:: after diluting the reaction solution with acetic acid Echiru and lavage with a saturated sodium bicarbonate solution: in addition, 1N hydrochloric acid, washed with saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain a crude crystal.
(2)得られた粗結晶をジクロロメタン 100mlに溶解し,氷冷下ジメチルスルフイド 25ml, 三フ ッ化ホウ素ジェチルェ一テル錯体 15mlを順次加え,室温で 3日間攪拌した後,更に,ジメ チノレスルフイド 25ml,三フッ化ホウ素ジェチルエーテル錯体 15mlを加え,室温で 1日間攪 拌した =反応液に 1N塩酸を加え,攪拌した後分液し,水層をクロ口ホルムで洗浄した。水層 力 )H10になるまで 1N水酸化ナトリウムを加えた後,酢酸ェチルにより抽出した =減圧下に 溶媒を留去し,得られた粗結晶を酢酸ェチルとへキサンの混合溶媒より再結晶し,表題化 合物 4. 7gを得た = (2) The obtained crude crystals were dissolved in 100 ml of dichloromethane, 25 ml of dimethyl sulfide and 15 ml of boron trifluoride-ethylester complex were sequentially added under ice cooling, and the mixture was stirred at room temperature for 3 days. 25 ml, boron trifluoride GETS chill etherate 15ml was added, 1N hydrochloric acid was added to the = reaction was 1 day stirred at room temperature, and separated after stirring, the aqueous layer was washed with black port Holm. After addition of 1N sodium hydroxide until the aqueous layer strength) H10, and extracted with acetic acid Echiru = the solvent was distilled off under reduced pressure and the resulting crude crystals were recrystallized from a mixed solvent of hexane and acetic acid Echiru 4.7 g of the title compound were obtained =
参考例 1 1一 1と同様に以下の参考例を合成した Reference Example 1 The following Reference Example was synthesized in the same manner as 1-1.
参考例 11一 2 Reference Example 11 1 2
2—アミノー 2—(3, 4—ジシァノフエ二ルカルバモイル)ェチル アセテート - 3—トリフルォロメチルフエ二ル)一 3—メ卜キシフロバンアミド ド 2. Ogをァセトニトリル 30mlに溶解し,氷冷下, ヨウ化トリメチル シラン 1. 4mlを加え,同温で 1時間攪拌した 反応液にメタノール lml,水 30mlを順次カロ え,ジェチルエーテルで洗浄した。水層が pH9になるまで飽和重曹水を加えた後,酢酸ェ チルで抽出し,無水硫酸マグネシウムで乾燥した。減圧下に溶媒を留去し,得られた粗結 晶を酢酸ェチルとへキサンの混合溶媒により再結晶し,表題化合物]. , ()gを得た:: 参考例 12— 1と同様に以下の参考例を合成した。 . 2-amino-2- (3,4-dicyanophenylcarbamoyl) ethyl acetate -Dissolve Og in 30 ml of acetonitrile, add 1.4 ml of trimethylsilane iodide under ice-cooling, and add 1 ml of the same temperature at the same temperature for 1 hour. The stirred reaction solution was weighed sequentially with 1 ml of methanol and 30 ml of water, and washed with getyl ether. After adding saturated aqueous sodium bicarbonate until the aqueous layer reached pH 9, the mixture was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the obtained crude crystal was recrystallized from a mixed solvent of ethyl acetate and hexane to give the title compound]., () G: similar to Reference Example 12-1 The following reference examples were synthesized. .
参考例 12— 2 Reference Example 12-2
1—ァミノ一 N— (4—シァノ 3—トリフルォロ' 口 1—Amino N— (4—Siano 3—Trifluolo 'mouth
参考例 12— 3 Reference Example 12-3
3—ァミノ一 N— (4—シァノ 3—トリフルォロ -2, 2 - アミド 3—Amino N— (4—Cyano 3—Trifluoro-2,2-amide
参考例 13 Reference Example 13
2 ァミノ一(3, 4 ジシァノフエニル) - 3 - 参考例 10で合成した 2—ァリルォキシカルボニルアミノー N— (3 , 4 - —3 メチルブタンアミド 1. 88g及びぎ酸 530mgのテトラヒドロフラン溶液 2-Amino (3,4-dicyanophenyl) -3--3-Synthesis of 2-aryloxycarbonylamino-N- (3,4--3-methylbutanamide 1.88 g in Example 10 and 530 mg of formic acid in tetrahydrofuran
ェニルフォスフィンパラジウム 1. 66gを加えアルゴン気流下 10時間加熱還流した 反応混 合物を減圧下濃縮し得られた残留物を 1規定塩酸及び酢酸ェチルに溶解し水層を分離し た。この水層を 1規定水酸化ナトリウム水溶液にて塩基性にし,遊離した油状物を酢酸ェチ ルで抽出後,無水硫酸ナトリウムにて乾燥した =溶媒を減圧下濃縮し,得られた残留物をシ リカゲルカラムクロマトグラフィーにて精製し,クロ口ホルム メタノール(95 : 5, V/V)溶出 部より表題化合物 435mgを無色油状物として得た。 After adding 1.66 g of phenylphosphine palladium and heating and refluxing for 10 hours in an argon stream, the reaction mixture was concentrated under reduced pressure, and the obtained residue was dissolved in 1N hydrochloric acid and ethyl acetate, and the aqueous layer was separated. The aqueous layer was basified with 1 N sodium hydroxide solution and extracted the liberated oil in acetic E Ji Le, a = the solvent was dried over anhydrous sodium sulfate and concentrated under reduced pressure, the resulting residue The residue was purified by silica gel column chromatography, and 435 mg of the title compound was obtained as a colorless oil from a fraction eluted with chloroform-methanol (95: 5, V / V).
参考例 14 Reference Example 14
1—ベンジルォキシカルポ二ルー N— (4—シァノ 3—トリフルォロメチルフエニル)ー2 —メチルプロリンアミド 1-Benzyloxycarbonyl N- (4-cyano 3-trifluoromethylphenyl) -2-methylprolinamide
1一べンジルォキシカルボ二ルー 2 メチル一プロリン 3. 2gを 1, 2 ジクロロェタン 10ml に溶解し,氷冷下,ォキサリルクロリド 470mgを滴下した後,触媒量の N, N ジメチルホル ムアミドを加え,同温で 1時間攪拌した。減圧下に溶媒を留去した後, 1 , 2—ジクロロェタン 20mlを加え再度留去した::得られた残渣を N, N ジメチルァセトアミド 5mlに溶解し,氷 冷下, 4ーァミノ一 2 トリフルォロメチルベンゾニトリル 3 , 2gを少量ずつ加えた後,室温で 6時間攪拌した 反応液を酢酸ヱチル 50mlで希釈した後, 1 N塩酸 50ml,飽和重曹水 50 ml,蒸留水 50mlでそれぞれ洗浄し,無水硫酸マグネシウムで乾燥した。減圧下に溶媒を 留去した後,クロ口ホルム一へキサンから再結晶し,表題化合物 5. 64gを得た (1) Dissolve 3.2 g of 1-benzyloxycarbonyl-2-methyl-1-proline in 10 ml of 1,2-dichloroethane, add 470 mg of oxalyl chloride dropwise under ice-cooling, and add a catalytic amount of N, N dimethylformate. Muamide was added, and the mixture was stirred at the same temperature for 1 hour. After distilling off the solvent under reduced pressure, 1,2-dichloroethane (20 ml) was added and the residue was distilled off again: The resulting residue was dissolved in N, N dimethylacetamide (5 ml), and the mixture was dissolved in ice and cooled with ice to give 4-amino-12-trifluoro. After adding 3 and 2 g of o-methylbenzonitrile in small portions, the mixture was stirred at room temperature for 6 hours. The reaction solution was diluted with 50 ml of dimethyl acetate, and washed with 50 ml of 1N hydrochloric acid, 50 ml of saturated sodium bicarbonate solution and 50 ml of distilled water. , And dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the residue was recrystallized from chloroform-hexane to obtain 5.64 g of the title compound.
参考例 15— 1 - [4一(4—フルォロベンゾィル)アミド]シクロへキシルカルボン酸 Reference Example 15-1- [4- (4-fluorobenzoyl) amido] cyclohexylcarboxylic acid
4 アミノシクロへキシルカルボン酸(cis及び Lmn.sの混合物) 1 . 43gを 1規定水酸化ナ卜 リウム水溶液 l ()mlに溶解し、トリェチルァミン 1. () lgを力 Πえた::次いで 4 フルォロベンゾィ ノレクロリド 1. 59gのテトラヒドロフラン溶液(5ml)を氷冷下滴下し、室温にて 2時間攪拌した:. 反応溶液を減圧にて濃縮し、エーテルで洗浄後、濃塩酸を用いて酸性にし、酢酸ェチルで 抽出し、乾燥後濃縮し表題化合物 2. 41gを得た。 4 1.43 g of aminocyclohexyl carboxylic acid (mixture of cis and Lmn.s) was dissolved in l () ml of 1N aqueous sodium hydroxide solution, and triethylamine 1. () lg was added:: then 4 A solution of 1.59 g of tetrafluorofuran (5 ml) was added dropwise under ice-cooling, and the mixture was stirred at room temperature for 2 hours. The reaction solution was concentrated under reduced pressure, washed with ether, acidified with conc. Extracted with ethyl, dried and concentrated to obtain 2.41 g of the title compound.
参考例 15— 1と同様にして以下の参考例を合成した:: The following Reference Example was synthesized as in Reference Example 15-1:
参考例 1 5— 2 Reference example 15-2
1一(4 フルォ口べンゾィル)ァゼチジン一: 3—カルボン酸 1- (4-fluorobenzoyl) azetidine-1: 3-carboxylic acid
これらの参考例の物性値を表に示す。 The physical properties of these Reference Examples are shown in the table.
なお,表中の記号は以下の意味を有する: The symbols in the table have the following meanings:
Ref.Ex. :参考例番号 AcOEt:酢酸ェチル Ref.Ex .: Reference example number AcOEt: Ethyl acetate
DATA :物理化学的性状 Hex :へキサン DATA: Physical and chemical properties Hex: Hexane
NMR :核磁気共鳴スぺクトノレ EtOH :エタノール NMR: nuclear magnetic resonance spectrum EtOH: ethanol
(特に明記しない限り, DMSO-d6, (Et)20:ジェチルエーテル (Unless otherwise stated, DMSO-d 6, (Et ) 2 0: Jefferies chill ether
TMS内部標準で測定) l,2-diCl-Et : l , 2—ジクロロェ mp : ¾ L, 2-diCl-Et: l, 2-dichloroe mp: 測定
表 3 Table 3
Ref.Ex DATA Ref.Ex DATA
1 -1 NMR: ό :0.77(3H,t,J=7.3Hz),1 .40-1 .66(2H,m),3.40-3.64(1 H,m),3.68(3H,s), 7.28-7.47 1 -1 NMR: ό: 0.77 (3H, t, J = 7.3 Hz), 1.40-1.66 (2H, m), 3.40-3.64 (1 H, m), 3.68 (3H, s), 7.28 -7.47
(2H,m),7.77-7.92(2H,m),8.15(1 H,d,J=8.8Hz) (2H, m), 7.77-7.92 (2H, m), 8.15 (1H, d, J = 8.8Hz)
1 -2 NMR(CDCI3,TMS internal standard) 1 -2 NMR (CDCI 3 , TMS internal standard)
5 :1 .68(6H,s),3.79(3H,s),6.77(1 H,br),7.00-7.19(2H,m),7.71 -7.87(2H,m) 5: 1.68 (6H, s), 3.79 (3H, s), 6.77 (1H, br), 7.00-7.19 (2H, m), 7.71 -7.87 (2H, m)
1 -3 NMR(CDCI3>TMS internal standard) 1 -3 NMR (CDCI 3> TMS internal standard)
b :1 .65(6H,s),3.62(3H,s),4.00(3H,s),6.95-7.45(3H,m), 8.20-8.49(2H,m)

PLlP0/L6d£/lDd OAV
N—(4 - -ル)一 2— [ (メチルスルホニル)ァミノ]フ口 参考例 1 ()一 1で合成した 2—ァミノ一 N—(4一シァノ一 3—卜リフルォロメチルフエニル) プロバンアミド 25()mgをクロ口ホルム 5mlに溶解し,氷冷下, トリェチルァミン 1 10mg,メタン スルホニルクロリド 12()mgを加え,アルゴン雰囲気下,室温で 3時間攪拌した =反応液をク ロロホルム 50mlで希釈後, 1 N塩酸 50ml ,飽和食塩水 5()mlで洗净し,無水硫酸マグネシ ゥムで乾燥した::減圧下に溶媒を留去したのち,残渣を酢酸ェチルより再結晶し,無色結品 の表題化合物 114mgを得た c: N- (4- -L) -1-2-((methylsulfonyl) amino] mouth Reference Example 1 2-amino-1-N- (4-cyano-3-3-trifluoromethylphenyl synthesized in () -11 ) Puroban'amido 25 () was dissolved mg to black port Holm 5 ml under ice-cooling, Toryechiruamin 1 10 m g, methanesulfonyl chloride 12 () mg was added, under argon, = reaction click was stirred for 3 hours at room temperature After diluting with 50 ml of roloform, washing with 50 ml of 1N hydrochloric acid and 5 () ml of saturated saline, and drying with anhydrous magnesium sulfate: The solvent was distilled off under reduced pressure, and the residue was recrystallized from ethyl acetate. This gave 114 mg of the title compound as a colorless product c:
実施例 1と同様にして実施例 2— 16を合成した :: It was synthesized in Example 2 16 in the same manner as in Example 1:
実施例 17 Example 17
N—(4ーシァノー 3—トリフルォロメチルフエニル)一 2— { [ (4—フルオロフェニゾレ)スルホ ニル]ァミノ)ブチルアミド N- (4-cyano 3-trifluoromethylphenyl) -1-2-[[(4-fluorophenizole) sulfonyl] amino] butyramide
2—(4—フルオロフェニルスルホニルァミノ)ブタン酸 500mgをテトラヒドロフラン 5mlに溶 解し,氷冷下,ォキザリルクロリド 470mgを滴下した後,触媒量の N, N—ジメチルホルムァ ミドを加え,同温度で 1時間攪拌した。減圧下に溶媒を留去した後,テトラヒドロフラン 20ml を加え再度留去した。得られた残渣を N, N—ジメチルホルムアミド 5mlに溶解し,氷冷下, 4一アミノー 2—トリフルォロメチルベンゾニトリル 580mgを少量ずつ加えた後,室温で 6時 間攪拌した。反応液を酢酸ェチル 50mlで希釈した後, 1 N塩酸 5()ml,飽和重曹水 50ml , 蒸留水 50mlでそれぞれ洗浄し,無水硫酸マグネシウムで乾燥した。減圧下に溶媒を留去 した後,残渣をシリカゲルカラムクロマトグラフィーに付し,酢酸ェチルーへキサン(17 : 3)溶 出部より粗結晶を得た。この粗結晶を酢酸ェチルから再結晶し,無色結晶の表題化合物 3 実施例 ] 7と同様にして実施例 ] 8— 21を合成した。 Dissolve 500 mg of 2- (4-fluorophenylsulfonylamino) butanoic acid in 5 ml of tetrahydrofuran, add 470 mg of oxalyl chloride dropwise under ice cooling, and add a catalytic amount of N, N-dimethylformamide. The mixture was stirred at the same temperature for 1 hour. After distilling off the solvent under reduced pressure, tetrahydrofuran (20 ml) was added and distilled off again. The obtained residue was dissolved in 5 ml of N, N-dimethylformamide, and 580 mg of 4-amino-2-trifluoromethylbenzonitrile was added little by little under ice-cooling, followed by stirring at room temperature for 6 hours. The reaction solution was diluted with 50 ml of ethyl acetate, washed with 5 () ml of 1N hydrochloric acid, 50 ml of saturated aqueous sodium hydrogen carbonate and 50 ml of distilled water, and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the residue was subjected to silica gel column chromatography and dissolved in ethyl acetate-hexane (17: 3). A crude crystal was obtained from the outlet. The crude crystals were recrystallized from ethyl acetate, and Example] 8-21 was synthesized in the same manner as in Example 3 of the title compound as colorless crystals.
実施例 22 Example 22
N—(4—シァノー 3—トリフルォロメチルフエニル)一2— (4一フルオロフェ: ニル]ァミノ)プロバンチオアミド N- (4-cyano 3-trifluoromethylphenyl) 1-2- (4-fluorophenyl) amino] propanthioamide
N—(4—シァノー 3—トリフルォロメチルフエニル)一 2— { [ (4—フルオロフェ: -ノレ]アミノ}プロパンアミド 600mgをトルエン l ()mlに溶解し, Lawesson試薬 29()mgを力 1:1 えた:. アルゴン雰囲気下, 3日間加熱還流した後,反応液を室温まで冷却し,減圧卜-に溶 媒を留去した。残渣をシリカゲルカラムクロマトグラフィーに付し,酢酸ェチル キサン (4 : 1 )溶出部より粗結晶を得た。この粗結晶を酢酸ェチルから再結晶し,無色結晶の表題 化合物 339mgを得た。 Dissolve 600 mg of N- (4-cyano 3-trifluoromethylphenyl) -1-{[(4-fluorophene: -norre] amino} propanamide in l () ml of toluene and add 29 () mg of Lawesson's reagent. After heating under reflux in an argon atmosphere for 3 days, the reaction solution was cooled to room temperature, the solvent was distilled off under reduced pressure, and the residue was subjected to silica gel column chromatography to give ethyl ethyl acetate. (4: 1) Crude crystals were obtained from the eluted portion, and the crude crystals were recrystallized from ethyl acetate to give 339 mg of the title compound as colorless crystals.
実施例 1と同様にして実施例 23 , 24を合成した Examples 23 and 24 were synthesized in the same manner as in Example 1.
実施例 25 Example 25
N— { 1— [ (4—シァノー 3—トリフルォロメチルフエニル)力ルバモイル]ー1 —メチルェチ ル} 一 4一フルォロベンズアミド N— {1 — [(4-cyano 3-trifluoromethylphenyl) -rubamoyl] -1—methylethyl} 1-41-fluorobenzamide
2— (4—フルォ口べンゾィルァミノ)一 2—メチルプロバン酸 500mgをジクロロメタン 30ml に溶解し,氷冷下,トリフエ二ノレホスフィン 933mg, N—ブロモスクシンイミド 633mgを順次 少量ずつ加えた後,アルゴン雰囲気下,同温度で 2時間攪拌した::更に 4一アミノー 2—トリ フルォロメチルベンゾニトリル 872nigを少量ずつ加え,室温で 3時間攪拌した。減圧下に溶 媒を留去した後,残渣をシリカゲルカラムクロマトグラフィーに付し,酢酸ェチル キサン ( 17: 3)溶出部より粗結晶を得た。この粗結晶を酢酸ェチルから再結晶し,無色結晶の表 題化合物 156mgを得た。 Dissolve 500 mg of 2- (4-fluorobenzoylamino) -1-methylpropanoic acid in 30 ml of dichloromethane, add 933 mg of triphenylinolephosphine and 633 mg of N-bromosuccinimide in small portions under ice-cooling, and then add argon. The mixture was stirred at the same temperature for 2 hours .: Further, 872-nig of 4-amino-2-trifluoromethylbenzonitrile was added little by little, and the mixture was stirred at room temperature for 3 hours. After evaporating the solvent under reduced pressure, the residue was subjected to silica gel column chromatography to obtain crude crystals from a fraction eluted with ethyl acetate (17: 3). The crude crystals were recrystallized from ethyl acetate to give 156 mg of the title compound as colorless crystals.
実施例 25と同様にして実施例 26— 33を合成した- 実施例 1と同様にして実施例 34— 64を合成した。 Examples 26 to 33 were synthesized in the same manner as in Example 25-Examples 34 to 64 were synthesized in the same manner as Example 1.
実施例 65 Example 65
N— { 1一 [ (4ーシァノー 3—トリフルォロメチルフヱニル)力ルバモイル] 一 1ーメチルェチ ル } 一 2—シァノベンズアミド N— {1-([4-cyano 3-trifluoromethylphenyl) -lvamoyl] 1-1-methylethyl} 1-2-cyanobenzamide
( 1 ) フタノレアミド酉 5. Ogのジクロロメタン溶 ί夜 80mlに,氷冷下, トリェチノレアミン 8. 27ml 及びクロロギ酸ェチル 6. 37mlを加え 1時間攪拌した後,室温で更に 6時間攪拌した::減圧 下,溶媒を留去し,ベンゼン一へキサン混合溶媒を加え析出した結晶を濾別した,滤液を 留去して, 2—シァノ安息香酸無水物 (う 58mgを得た: (1) Phthanolamide 5. Dissolve Og in dichloromethane. Then, 6.37 ml of ethyl and chloroformate were added, and the mixture was stirred for 1 hour and further stirred at room temperature for 6 hours. The solvent was distilled off under reduced pressure, a mixed solvent of benzene and hexane was added, and the precipitated crystals were separated by filtration. Was distilled off to give 2-cyanobenzoic anhydride (58 mg).
(2)参考例 10— 2で合成した 2—アミノー N— (4—シァノ一 3—トリフルォロメチルフヱ二ル) — 2—メチルフロバンアミド 2()0mgのジクロロメタン溶液 1 Omlに上記 2 -シァノ安息香酸無 水物 323mgを加え,室温で 9時間攪拌した。反応溶液に飽和重曹水を加え攪拌した後, 酢酸ェチルにより抽出した。有機層を無水硫酸マグネシウムにより乾燥し,減圧下,溶媒を 留去した 残渣をシリカゲルカラムクロマトグラフィーにて精製し,酢酸ェチル一へキサン (1 : 1 )溶出部より粗結晶を得た:.この粗結晶をメタノール一酢酸ェチル混合溶媒により再結 晶し,表題化合物 19()mgを得た Ω (2) Reference Example 10-2 2-amino-N- (4-cyano-13-trifluoromethylphenyl) -2-methylfrovanamide 2 () 0 mg dichloromethane solution in 1 Oml 323 mg of 2-cyanobenzoic anhydride was added, and the mixture was stirred at room temperature for 9 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction solution, and the mixture was stirred and extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography to obtain crude crystals from a fraction eluted with ethyl acetate / hexane (1: 1). the crude crystals were recrystallization with methanol monoacetate Echiru mixed solvent, to give the title compound 19 () m g Ω
実施例 66 Example 66
Ν—(4ーシァノー 3—トリフルォロメチルフエニル)一2— [2— (4—フルオロフェニル)ァ セチルァミノ]ー2—メチルプロバンアミド Ν— (4-cyano 3-trifluoromethylphenyl) 1-2- [2- (4-fluorophenyl) a cetylamino] -2-methylpropanamide
(1 ) 4—フルオロフェニル酢酸 l()2mgのジクロロメタン溶液 5mlに,氷冷下,ォキザリルク ロリド 0. 057ml及び触媒量の N , N—ジメチルホルムアミドを加え,室温で 2時問攪拌した 後,減圧下,溶媒を濃縮乾固して, 4—フルオロフェニルァセチルクロ' (1) To 5 ml of a dichloromethane solution of 2 mg of 4-fluorophenylacetic acid l () , add 0.057 ml of oxalyl chloride and a catalytic amount of N, N-dimethylformamide under ice-cooling, stir at room temperature for 2 hours, and reduce the pressure. The solvent is concentrated to dryness, and 4-fluorophenylacetylchloro '
(2)参考例 10— 2で合成した 2—アミノー N—(4ーシァノー 3— (2) 2-amino-N- (4-cyano 3-) synthesized in Reference Example 10-2
ル)一 2—メチルァロバンアミド 150mgのジクロロメタン溶液 5mlにビ'リジン 2mlを加えた後, 氷冷下上記 4一フルオロフェニルァセチルクロリドのジクロロメタン溶液 5mlを加え, 同温で 1. 5時問攪拌した後,室温で 1 · 5時間攪拌した:.:反応溶液に酢酸ェチルを加え,これを飽和 重曹水,続いて,飽和食塩水で洗浄後,無水硫酸マグネシウムで有機層を乾燥し,減圧下, 溶媒を留去して粗結晶を得た。この粗結晶を酢酸ェチルから再結晶し,表題化合物 98mg を得た。 B) Add 2 ml of bi'lysine to 5 ml of a dichloromethane solution of 150 mg of 1-methylmethylarovanamide, add 5 ml of the above-mentioned dichloromethane solution of 4-fluorophenylacetyl chloride under ice-cooling, and incubate at the same temperature for 1.5 hours. After stirring, the mixture was stirred at room temperature for 1.5 hours. :: Ethyl acetate was added to the reaction solution, and the mixture was washed with saturated aqueous sodium hydrogen carbonate and saturated brine, dried over anhydrous magnesium sulfate, and dried under reduced pressure. The solvent was distilled off to obtain crude crystals. The crude crystals were recrystallized from ethyl acetate to give 98 mg of the title compound.
実施例 67 Example 67
N— { 1— [ (4—シァノー 3—トリフルォロメチルフエニル)力ルバモイル] 一 1ーメチルェチ ル } 一 2—ヒドロキシベンズアミド N— {1 — [(4-Cyanol 3-trifluoromethylphenyl) potamyl] -1-1-methylethyl} -1-hydroxybenzamide
実施例 44で合成した 2—( U — [ (4—シァノ一 3—トリフルォロメチルフエニル)カルバモ ィル] 一 1一メチルェチル }力ルバモイル)フエニルアセテート 150mgをメタノール l mlに溶 解し, 1N水酸化ナトリウム水溶液 3mlを加えた後,室温で 2時間攪拌した。減圧下にメタノ 一ルを留去した後, 1 N塩酸を加えることにより粗結晶を得た::この粗結晶を酢酸ェチルと へキサンの混合溶媒により再結晶し,表題化合物 l()2mgを得た Dissolve 150 mg of 2-((U — [( 4 -cyano-3-trifluoromethylphenyl) carbamoyl] -111-methylethyl) potassium phenylacetate synthesized in Example 44 in 1 ml of methanol. After adding 3 ml of 1N aqueous sodium hydroxide solution, the mixture was stirred at room temperature for 2 hours. Methano under reduced pressure After distilling off the solvent, 1N hydrochloric acid was added to obtain crude crystals :: The crude crystals were recrystallized with a mixed solvent of ethyl acetate and hexane to obtain 2 mg of the title compound l ().
実施例 68 Example 68
N— U— [ (4—シァノ一3—トリフルォロメチルフエニル)力ルバモイル]一:! N—U — [(4-cyan-1-3-trifluoromethylphenyl) rubamoyl]
ル }一 8—キノリンカルボキサミド 1} 8-quinolinecarboxamide
(1 ) 8—キノリンカゾ 溶液 5mlに,氷冷下,ォキザリルクロリ ド (). 114ml及び触媒量の N, N ルホルムアミドを加え,氷冷下, 30分攪拌した後, 減圧下,溶媒を濃縮乾固して, 8—キノリンカルボユルクロリド塩酸塩を得た:: (1) To 5 ml of 8-quinoline azo solution, add oxalyl chloride (). 114 ml and a catalytic amount of N, N-formamide under ice-cooling, stir under ice-cooling for 30 minutes, and concentrate the solvent under reduced pressure to dryness. This gave 8-quinolinecarboyl chloride hydrochloride:
(2)参考例 10— 2で合成した 2—アミノー N— (4—シァノ一3—トリフルォロメチルフエ二 ル)一 2—メチルプロバンアミド 30()mgのジクロロメタン溶液 5mlに,氷冷下,上記 8—キノリ ン力ルボニルクロリド塩酸塩のジクロロメタン溶液 5 ml及び卜リェチルァミン 0. 364 mlを加え, 室温で 2時間攪拌した =反応溶液に酢酸ェチルを加え,これを飽和重曹水,続いて,水で 洗浄後,無水硫酸マグネシウムで有機層を乾燥し,減圧下,溶媒を留去した後,残澄をシリ 力ゲルカラムクロマトグラフィーに付し,酢酸ェチルーへキサン(1: 1)溶出部より粗結晶を得 た。この粗結晶を酢酸ェチルとへキサンとの混合溶媒より再結晶し,表題化合物 155mgを 得た (2) Reference Example 10-2 2-amino-N- (4-cyano-13-trifluoromethylphenyl) -1-methylpropanamide 30 () mg 30 mL dichloromethane solution in 5 ml under ice-cooling added dichloromethane 5 ml and Bok Ryechiruamin 0. 364 ml of 8-halfhearted down force Rubonirukurorido hydrochloride, acetate Echiru added to the stirred = reaction solution at room temperature for 2 hours, which saturated aqueous sodium bicarbonate solution, followed by After washing with water, the organic layer is dried over anhydrous magnesium sulfate, the solvent is distilled off under reduced pressure, and the residue is subjected to silica gel column chromatography. From the ethyl acetate-hexane (1: 1) elution part A crude crystal was obtained. The crude crystals were recrystallized from a mixed solvent of ethyl acetate and hexane to obtain 155 mg of the title compound.
実施例 68と同様にして実施例 69— 78を合成した, Examples 69-78 were synthesized in the same manner as in Example 68.
実施例 79 Example 79
N— { 1一 [ (4一シァノ一3—トリフルォロメチルフエ二ノレ)力ルバモイル] — 1一メチルェチ -4ービリジンカルボキサミド N— {1-[[4- (1-cyano-13-trifluoromethylpheninole) rubamoyl] — 1-methylethyl-4-viridinecarboxamide
ニコチン酸 0. 15gとジクロロメタン 10mlの混合液に,氷冷下, N—ヒドロキシベンゾトリ ァゾール 0. 18g, 1— (3—ジメチルアミノフロピル)ー3—ェチルカルボジイミド塩酸塩 0, 2 5gを順次加えた =同温で, 2時問攪拌した後, 2—ァミノ一 N—(4一シァノ— :3—トリフルォ ロメチルフエニル)一 2—メチルプロパンアミド 0. 30gをカロえ,室温で 6時間,更にテトラヒドロ フラン 5mlを加え同温で 65時間攪拌した =反応液に飽和重曹水を加え,酢酸ェチルで抽 出し,飽和塩化ナトリウム水溶液で洗浄した後,無水硫酸マグネシウムで乾燥した =減圧下 に溶媒を留去し,得られた粗結晶を酢酸ェチルとへキサンの混合溶媒より再結晶し,表題 化合物 0. 26gを得た。 To a mixture of 0.15 g of nicotinic acid and 10 ml of dichloromethane was added 0.18 g of N-hydroxybenzotriazole and 0.25 g of 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride under ice-cooling. sequentially added and = at the same temperature, 2 more hours after stirring, 2-Amino one N-(4 one Shiano - 3- Torifuruo Romechirufueniru) one 2-methylpropanamide 0. 30 g of Karoe, 6 hours at room temperature, further tetrahydro furan 5ml was added a saturated aqueous sodium hydrogen carbonate was added to the = reaction was stirred 65 hours at the same temperature, out extraction with acetic Echiru, washed with saturated aqueous sodium chloride solution, the solvent = vacuo drying over anhydrous magnesium sulfate The crude crystals were recrystallized from a mixed solvent of ethyl acetate and hexane to obtain 0.26 g of the title compound.
実施例 79と同様にして実施例 80― 87を合成した- 実施例 88 Examples 80 to 87 were synthesized in the same manner as in Example 79. Example 88
2— [ (4—シァノー 3—トリフルォロメチルフエ二ノレ)力ルバモイル]— 2—(2—フルォ口べ ンズアミド)ェチル アセテート 2 — [(4-Cyanol 3-Trifluoromethylphenol) Lubamoyl] —2— (2-Fluorobenzoamide) ethyl acetate
(1 ) 3—ァセトキシー 2—べンジルォキシカルボニルァミノプロバン酸 3. 9gを N, N―ジメチ ルァセトァミド 3 Oml (こ溶解し, - 2 ();Cに冷却した後, チォニルクロリド 1. 1 mlを滴下した:. アルゴン雰囲気下, で 1時間攪袢した後, 4一アミノー 3—トリフルォロメチルベンゾニトリ ル 2. 6gを少量ずつ加え, 同温度で 2時問攪拌した::反応液を酢酸ェチルで希釈後,飽和 重曹水で洗浄した。更に, 0. 1N塩酸,飽和塩化ナ卜リウム水溶液で洗浄した後,無水硫酸 マグネシウムで乾燥した。減圧下に溶媒を留去し,粗結晶を得た:: (1) 3.9 g of 3-acetoxy-2-benzyloxycarbonylaminopropanoic acid was dissolved in N, N-dimethylacetoamide 3 Oml (this was dissolved and cooled to -2 () ; C, then thionyl chloride 1.1 ml) After stirring under an argon atmosphere for 1 hour, 2.6 g of 4-amino-3-trifluoromethylbenzonitrile was added little by little, and the mixture was stirred at the same temperature for 2 hours. Was diluted with ethyl acetate, washed with saturated aqueous sodium bicarbonate, further washed with 0.1N hydrochloric acid and a saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. Got ::
( 2 ) 得られた粗結晶をジクロロメタン 100mlに溶解し,水冷下,ジメチルスルフイド 10ml, 三フッ化ホウ素ジヱチルエーテル錯体 8mlを順次加え,室温で 4時間攪拌した.反応液に 飽和重曹水を加え,酢酸ェチルで抽出した後,飽和塩化ナトリウム水溶液で洗浄した:.無 水硫酸マグネシウムで乾燥した後,減圧下に溶媒を留去し,粗結晶を得た:: (2) The obtained crude crystals were dissolved in dichloromethane (100 ml), dimethylsulfide (10 ml) and boron trifluoride diethyl ether complex (8 ml) were sequentially added under water cooling, and the mixture was stirred at room temperature for 4 hours. , Extracted with ethyl acetate, and washed with saturated aqueous sodium chloride solution. After drying over anhydrous magnesium sulfate, the solvent was distilled off under reduced pressure to obtain a crude crystal.
( 3 ) 得られた粗結晶をジクロロメタン l f5mlに溶解し,水冷下, 2—フルォロベンゾイルク口 リド 0. 56ml,トリェチルァミン ()· (06mlを順次加え,室温で 2時問攪拌した。反応液に飽和 重曹水を加え,酢酸ェチルで抽出した後, 0. I N塩酸,飽和塩化ナトリウム水溶液で洗浄し た c無水硫酸マグネシウムで乾燥した後,減圧下に溶媒を留去し,粗結晶を得た::得られた 粗結晶を酢酸ェチルとへキサンの混合溶媒により再結晶し,表題化合物 1. 25gを得た:: 実施例 89 (3) The obtained crude crystals were dissolved in 5 ml of dichloromethane, and 0.56 ml of 2-fluorobenzoyl chloride and 06 ml of triethylamine (). (06 ml) were sequentially added under cooling with water, followed by stirring at room temperature for 2 hours. to the reaction solution, saturated aqueous sodium bicarbonate was added, followed by extraction with acetic acid Echiru, 0. iN hydrochloric acid, dried with c anhydrous magnesium sulfate was washed with saturated aqueous sodium chloride solution, the solvent was distilled off under reduced pressure, the crude crystals The obtained crude crystal was recrystallized from a mixed solvent of ethyl acetate and hexane to obtain 1.25 g of the title compound.
Ν—{ 1— [ (4—シァノー 3—トリフルォロメチルフエニル)力ルバモイル] 一 2—ヒドロキシェ チル }一 2—フルォロベンズアミド Ν— {1— [(4-cyano 3-trifluoromethylphenyl) -lubamoyl] -1-2-hydroxyethyl} -1-2-fluorobenzamide
2— [ (4一シァノ一3—トリフルォロメチルフエニル)力ルバモイル] 一 2—(2—フルォ口べ ンズアミド)ェチル アセテート 0. 50gをメタノ一ノレ 5mlに溶解し,氷冷下,飽和炭酸カリウム 水溶液 2mlを加え,同温で 1時間,更に室温で 1時間攪拌した c反応液に IN塩酸を加え PH2とし,析出した結晶を廬取した: ;得られた粗結晶を水で洗浄した後,酢酸ヱチル—へ キサンから再結晶し,表題化合物 ϋ. 26gを得た:: 2— [(4-Cyan-3-trifluoromethylphenyl) potassium] Dissolve 0.50 g of 2- (2-fluorobenzoamide) ethyl acetate in 5 ml of methanol, and saturate under ice-cooling. added aqueous potassium carbonate 2 ml, 1 hour at the same temperature further and PH2 adding iN hydrochloric acid c reaction solution was stirred for 1 hour at room temperature, the precipitated crystals were Iorito:; the resulting crude crystals were washed with water After that, recrystallization from dimethyl acetate-hexane gave 26 g of the title compound ::
実施例 90 Example 90
Ν- { 1 - [ (4ーシァノー 3—トリフルォロメチルフヱニル)力ルバモイル]— 1—メチルェチ ル} 一 4—メトキシベンズアミド 2—アミノー N— (4—シァノー :3—トリフルォロメチルフエニル)一2—メチルフ :ロハンアミド 4 3()mgをテ卜ラヒドロフラン 5mlに溶解し,氷冷下, 卜リエチルァミン 26() μ I , 4—メ卜キシベン ゾイルクロリド 310mgを順次加え,氷冷下で 40分間攪拌した後,氷を加えた::更に室温で 5 分攪拌した後,減圧下に溶媒を留去し,残渣を酢酸ェチル 30mlと I N塩酸水溶液とに分配 し,有機層を飽和炭酸水素ナトリウム水溶液 15ml, / l 5ml X ,飽和食塩水 15mlで洗浄 し,硫酸ナトリウムで乾燥した c減圧下に溶媒を留去した後,残渣をシリカゲルカラムクロマ卜 グラフィ一(酢酸ェチル:へキサン = 1: 1 )にて精製し,得られた油状物質をジイソフ口ピル エーテル一酢酸ェチルにより結晶化し,得られた結晶を酢酸ェチルより再結晶し,無色結 晶の表題化合物 326mgを得た:: Ν- {1-[(4-cyano 3-trifluoromethylphenyl) rubamoyl] — 1-methylethyl} 1-4-methoxybenzamide Dissolve 43 () mg of 2-amino-N- (4-cyano: 3-trifluoromethylphenyl) -1-methylfuro : rohanamide in 5 ml of tetrahydrofuran, and cool with ice-cooling, triethylamine 26 () μI. 310 mg of 4,4-methoxybenzoyl chloride were sequentially added, and the mixture was stirred for 40 minutes under ice-cooling, and then ice was added. partitioned between Echiru 30ml and iN aqueous hydrochloric acid, the organic layer with saturated aqueous sodium bicarbonate solution 15 ml, / l 5 ml X, washed with brine 15 ml, the solvent was distilled off under c vacuum drying over sodium sulfate, The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 1), and the resulting oil was crystallized from diisobutylpyruether monoethyl acetate, and the obtained crystals were recrystallized from ethyl acetate. Crystallized and colorless crystal of the title compound 326 mg Got ::
実施例 90と同様にして実施例 9 ] , 92を合成した:: Examples 9] and 92 were synthesized as in Example 90:
実施例 93 Example 93
4ーブロモー 2—クロロー N— { 1一 [ (4一シァノー 3—トリフルォロメチルフエニル)力ルバ モイル]— 1—メチルェチルレヾンズアミド 4-Bromo-2-chloro-N— {1-([4-Cyanol 3-trifluoromethylphenyl) potassium] — 1-Methylethylbenzoylamide
4一ブロモー 2一クロ口安息香酸 52()mgをテトラヒドロフラン 1 ()mlに溶解し,氷冷下ォキザ リノレクロリド 230 μ 1, Ν, Ν—ジメチルホルムアミド 1滴を順次加え,室温にて 1時間攪拌した:: 反応液を減圧下に溶媒を留去し,残渣に 1 , 2—ジクロロェタンを加え再度留去した。残渣 をテトラヒドロフラン 4mlに溶解し,氷冷下 2—アミノー N—(4—シァノー: 3—トリフルォロメチ ノレフエニル)一 2—メチルプロノ、ンアミド 540mg,トリェチレアミン 3 10 μ 1を川頁次力 [1え, 5()分 間攪拌した。反応液に氷を加え, 2時間 30分間攪拌し,減圧下に溶媒を留去した,残渣を 酢酸ェチル 5(3mlと 1 N塩酸水溶液 30mlとに分配し,有機層を飽和炭酸水素ナトリウム水 溶液 30ml,飽和食塩水 30ml X 2で洗浄し,硫酸ナトリウムで乾燥した :;減圧下に溶媒を留 去した後,残渣を酢酸ェチルーエーテルより再結晶し,無色結晶の表題化合物 572mgを 得た = Dissolve 52 () mg of 4-bromo-2-monobenzoic acid in 1 ( ) ml of tetrahydrofuran, add 230μl of oxazinololechloride under ice-cooling and add 1 drop of ザ, Ν, Ν-dimethylformamide sequentially, and stir at room temperature for 1 hour. The solvent was distilled off from the reaction solution under reduced pressure, and 1,2-dichloroethane was added to the residue, followed by distillation again. The residue was dissolved in 4 ml of tetrahydrofuran, and 540 mg of 2-amino-N- (4-cyano: 3-trifluoromethylenolephenyl) -l-methylprono, amide and 10 μl of triethylamine were added under ice-cooling. ) The mixture was stirred for minutes. Ice was added to the reaction mixture, and the mixture was stirred for 2 hours and 30 minutes. The solvent was distilled off under reduced pressure. The residue was partitioned between ethyl acetate 5 (3 ml and 1N aqueous hydrochloric acid 30 ml), and the organic layer was diluted with saturated aqueous sodium hydrogen carbonate solution. 30 ml, washed with brine 30 ml X 2, and dried over sodium sulfate:; after distilling off the solvent under reduced pressure, the residue was recrystallized from acetic E Chiru ether to give the title compound 572M g of colorless crystals =
実施例 93と同様にして実施例 94を合成した。 Example 94 was synthesized in the same manner as in Example 93.
実施例 95 Example 95
N— ΙΊ一 [ (4—シァノ一3—トリフルォロメチルフエニル)力ルバモイル] 一 1ーメチルェチ ル ' } 一 4—ヒドロキシベンズァミド N-ΙΊ-[[(4-cyano-13-trifluoromethylphenyl) -l-bamoyl] -1-1-methylethyl '}-1-hydroxybenzamide
N— { 1一 [ (4一シァノ一3—トリフルォロメチルフエ二ノレ)力ルバモイル]― 1—メチルェチ -4ーメトキシベンズアミド 340mgをメチレンクロリド 10mlに溶解し、一 78CCにて 1M三 臭化ホウ素塩化メチレン溶液 3. 5mlを加え、室温にて 40時間攪袢した。反応液を氷冷し、 水 1 Omlを加え 5分攪拌し、クロ口ホルム 6 Omlで抽出した。有機層を飽和炭酸水素ナ卜リゥム 水溶液 3 Oml、水 3 Oml、飽和クェン酸水溶液 3 ()ml X 2で洗浄し、硫酸ナトリウムで乾燥した,. 減圧下 (こ溶媒を留去し、残渣をジィイソフロピルェ一テル―酢酸ェチルょり結晶化し、酢酸 チ /レーエ一-フー/レ- ロヒ'ソレエ- 、キサンより再結晶し,無色結晶の表題 化合物 1 mgを得た。 N— {11-[(4-Cyan-13-trifluoromethylpheninole) rubamoyl] —340 mg of 1-methyleth-4-methoxybenzamide is dissolved in 10 ml of methylene chloride, and the mixture is dissolved at 78 C C with 1 M 3.5 ml of a boron methylene chloride solution was added, and the mixture was stirred at room temperature for 40 hours. The reaction solution was ice-cooled, 1 Oml of water was added, the mixture was stirred for 5 minutes, and extracted with 6 Oml of chloroform. The organic layer was washed with saturated aqueous sodium bicarbonate solution (3 Oml), water (3 Oml), saturated aqueous citric acid solution (3) ml × 2, and dried over sodium sulfate. The solvent was distilled off under reduced pressure. The crystals were recrystallized from diisopropyl ether-ethyl acetate, and recrystallized from acetic acid acetate / lehi-fu / ro-lo'sole-hexane, to give 1 mg of the title compound as colorless crystals.
実施例 96 Example 96
N一ベンジルォキシ一 N— { 1一 [ (4 - ■3 N-benzyloxy N- {1-i [(4-■ 3
ド' De '
2— (4一フルオロー N—ゾくンジルォキシ- :ド)ー2 - 、ン酸し). 7gにジク ロロメタン 20mlを加え, 一 1 0 1 5::じで攪拌下, 五塩化リン 0. ΑΪ 2- (4-N-fluoro-N-zincsiloxy-: de) -2- -acid). Add 7 ml of dichloromethane and 20 ml of dichloromethane, and stir the mixture with 0.15 to 1 : 1.
10 Cで 1時間攪拌後 ,同温度で 4一アミノー 2—トリフルォロメチル' 小リル 0. 59gを 加え,さらに室温で 1時間攪拌した,:反応溶液をクロ口ホルムで希釈後,水洗し,無水硫酸 マグネシウムで乾燥した。溶媒を減圧下に留去し,残渣をシリカゲルカラムクロマトグラフィ 一にて精製した::へキサン一酢酸ェチル(2: 1 )溶出部より得られた結晶をジイソフ ビルェ —テルで洗滌して表題化合物 0. (う l gを得た. After stirring at 10 C for 1 hour, 0.59 g of 4-amino-2-trifluoromethyl 'small lyl was added at the same temperature, and the mixture was further stirred at room temperature for 1 hour. The reaction solution was diluted with chloroform and washed with water. , And dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography 1. The crystals obtained from the elution with hexane monoethyl acetate (2: 1) were washed with diisofurvir ether to give the title compound 0. (U got lg.
実施例 97 Example 97
N— { 1— [ (4ーシァノー 3—トリフルォロメチルフエニル)力ルバモイル]― 1ーメチルェチ ル}一 4一フルオロー N—ヒドロキシベンズアミド N— {1— [(4-cyano 3-trifluoromethylphenyl) rubamoyl] — 1-methylethyl} -1 4-fluoro-N-hydroxybenzamide
N—ベンジルォキシー N— { 1一 [ (4ーシァノー 3—トリフルォロメチルフエニル)カルバモ ィル] 一 1ーメチルェチル)一 4一フルォ口べンズアミド 0. 3gおよびぎ酸アンモニゥム 0. 1 5g のエタノール 10mlの懸濁溶液に l()。/oバラジウム炭素 0. 05gをカロえ,室温で 30分間攪拌 した cバラジゥム炭素を濾去後,水を加え,酢酸ェチルで抽出した。有機層を無水硫酸マグ ネシゥムで乾燥後,溶媒を減圧下に留去し,残渣をシリカゲルカラムクロマトグラフィーにて 精製した =へキサン—酢酸ェチル(1: 1 )溶出部より得られた結晶を酢酸ェチルおよびへキ サンの混合溶媒より再結晶し,表題化合物 0 , ]_ 8gを得た。 N-benzyloxy N— {1-[(4-cyano-3-trifluoromethylphenyl) carbamoyl] -1-methylethyl) -14-fluorobenzamide 0.3 g and ammonium formate 0.15 g ethanol 10 ml L () in the suspension solution. / o a palladium Karoe carbon 0. 05G, after filtering off the c Barajiumu carbon was stirred at room temperature for 30 minutes, water was added and extracted with acetic acid Echiru. The organic layer was dried over anhydrous magnesium sulfate, the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography = hexane-ethyl acetate (1: 1). Recrystallization from a mixed solvent of ethyl and hexane gave the title compound 0,] _ 8 g.
実施例 98 Example 98
N - (4一シァノ一3—トリフルォロメチルフエニル)一 2—メチルー 2— (3—フエ二, N- (4-Icyan-3-trifluoromethylphenyl) -2-methyl-2- (3-phenyl,
'ミド 参考例 10— 2で合成した 2 ァミノ一 N— (4 シァノ 3 トリフルォロメチルフエニル) —2 メチルブロバンアミド: 300mgのジクロロメタン溶液 10mlに,氷冷下,フエ二ルイソシァ ナ一ト 659mgを加え,室温で 4. 5時間攪拌した。反応溶液に,飽和重曹水,続いて I N塩 酸を加え,析出した粗結晶を濾取した cこの粗結晶を酢酸ュチルより再結晶し,表題化合 物 205mgを得た 2 'Mid To a 10 ml dichloromethane solution of 300 mg of 2-amino-N- (4-cyano-3 trifluoromethylphenyl) -2methylbrovanamide synthesized in Reference Example 10-2, was added 659 mg of phenylisocyanate under ice-cooling. The mixture was stirred at room temperature for 4.5 hours. To the reaction solution, saturated aqueous sodium bicarbonate solution, followed by addition of IN hydrochloric acid, collected by filtration was c crude crystals were recrystallized from acetic Yuchiru the precipitated crude crystals, to give the title compound 205 mg 2
実施例 99 Example 99
N - (4—シァノ 3 トリフルォロメチルフエニル) 2 メチル 2— (チォベンズアミド) 参考例 10— 2で合成した 2 アミノー N— (4—シァノー 3 - —2 メチルブロバンアミド 400mgをピリジン 5mlに溶解し, (チォベンゾィルチオ)酢酸 34 4mg,トリェチルァミン 226mgを加え,室温で 1 10時問攪拌した::反応液を 2N硫酸水溶液 に注ぎ,酢酸ェチルで抽出した。有機層を飽和重曹水で洗浄後,無水硫酸マグネシウムで 乾燥した。減圧下に溶媒を留去した後,残渣をシリカゲルカラムクロマトグラフィーに付し, 酢酸ェチル—へキサン : 1 )溶出部より粗結晶を得た ::この粗結晶を酢酸ェチルとへキサ ンの混合溶媒により再結晶し,表題化合物 6()mgを得た。 N- (4-cyano 3 trifluoromethylphenyl) 2-methyl 2- (thiobenzamide) 400 mg of 2 amino-N- (4-cyano 3--2-methyl bronamide synthesized in Reference Example 10-2 in 5 ml of pyridine After dissolving, 344 mg of (thiobenzoylthio) acetic acid and 226 mg of triethylamine were added, and the mixture was stirred at room temperature for 110 hours .: The reaction solution was poured into a 2N aqueous solution of sulfuric acid, and extracted with ethyl acetate. after washing, the solvent was distilled off under reduced pressure and then dried over anhydrous magnesium sulfate, the residue was subjected to silica gel column chromatography, acetic Echiru - hexane:. 1) was obtained from the eluate crude crystals: the crude The crystals were recrystallized from a mixed solvent of ethyl acetate and hexane to obtain 6 () mg of the title compound.
実施例 25と同様にして実施例 1 (X)— 1 ()4を合成し†:. Example 1 (X) —1 () 4 was synthesized in the same manner as in Example 25.
実施例 1と同様にして実施例 1 ()5― 107を合成した Example 1 () 5-107 was synthesized in the same manner as in Example 1.
実施例 25と同様にして実施例 108を合成した。 Example 108 was synthesized in the same manner as in Example 25.
実施例 1と同様にして実施例 K)9— 1 1 1を合成した。 Example K) 9-11 11 was synthesized in the same manner as in Example 1.
実施例 25と同様にして実施例 1 12— 122を合成した = Example 1 12-122 was synthesized in the same manner as in Example 25 =
実施例 1と同様にして実施例 123— 129を合成した。 Examples 123 to 129 were synthesized in the same manner as in Example 1.
実施例 130 Example 130
N - (4ーシァノー 3—トリフルォロメチルベンゾィル) [4 4 フルォロベンゾィル)アミ 参考例 15— 1で合成した [4一(4 フルォ口べンゾィル)アミド]シクロへキシルカルボン酸 を用いて、参考例 14と同様の手法を用いて反応及び後処理を行った。得られた粗抽出物 をシリカゲルカラムクロマトグラフィーにより精製し、酉乍酸ェチル キサン(1: 1 )溶出部より 単一の化合物(A)を得た。更に溶出を続け他の異性体 (B)を得た。 N- (4-cyano 3-trifluoromethylbenzoyl) [4 4 fluorobenzoyl) amide [4- (4-fluorobenzoyl) amido] cyclohexylcarboxy synthesized in Reference Example 15-1 The reaction and post-treatment were carried out in the same manner as in Reference Example 14 using an acid. The obtained crude extract was purified by silica gel column chromatography, and a single compound (A) was obtained from a portion eluted with ethyl ethyl oxane (1: 1). Elution was continued to obtain another isomer (B).
実施例 130と同様にして実施例 131を合成した。 :れらの実施例の構造及び物性値を表に示す Example 131 was synthesized in the same manner as in Example 130. : The structures and physical properties of these examples are shown in the table.
R 4 R 5 R6 R7 R 4 R 5 R 6 R 7
:で,表中の Aは一般式(I)における 部分に相当する:: , And A in the table corresponds to the part in the general formula (I) ::
![]()
![]()
Ex. :実施例番号 MS :質量分析値 Ex .: Example number MS: Mass spectrometry value
Me :メチル (i— pr)20:ジイソフロピルエMe: methyl (i- p r) 2 0: Jiisofuropirue
Et :ェチル MeOH :メタノーノレ Et: ethyl MeOH: methanol
Ac :ァセチル i-PrOH:イソフロバノール Ac: acetyl i-PrOH: isoflavanol
Benzyl -CH- 表 4 Benzyl -CH- Table 4


VL\VQIL6i£llDA zzm OAX VL \ VQIL6i £ llDA zzm OAX

9拏


PLlPQ/L6dr/lDd z zz OAX


8ε一 8ε


^ 0/ム 6U7i:)d zera/86 OAV ^ 0 / m 6U7i:) d zera / 86 OAV




6拏

ra/86 OAV

ON ON
H 〇 H 〇
\人 z V Nに \ People z v n
H H
L L拏 L L Halla


o

PLlP0/L6dT/ Dd てひ z OAV PLlP0 / L6dT / Dd Tehi z OAV
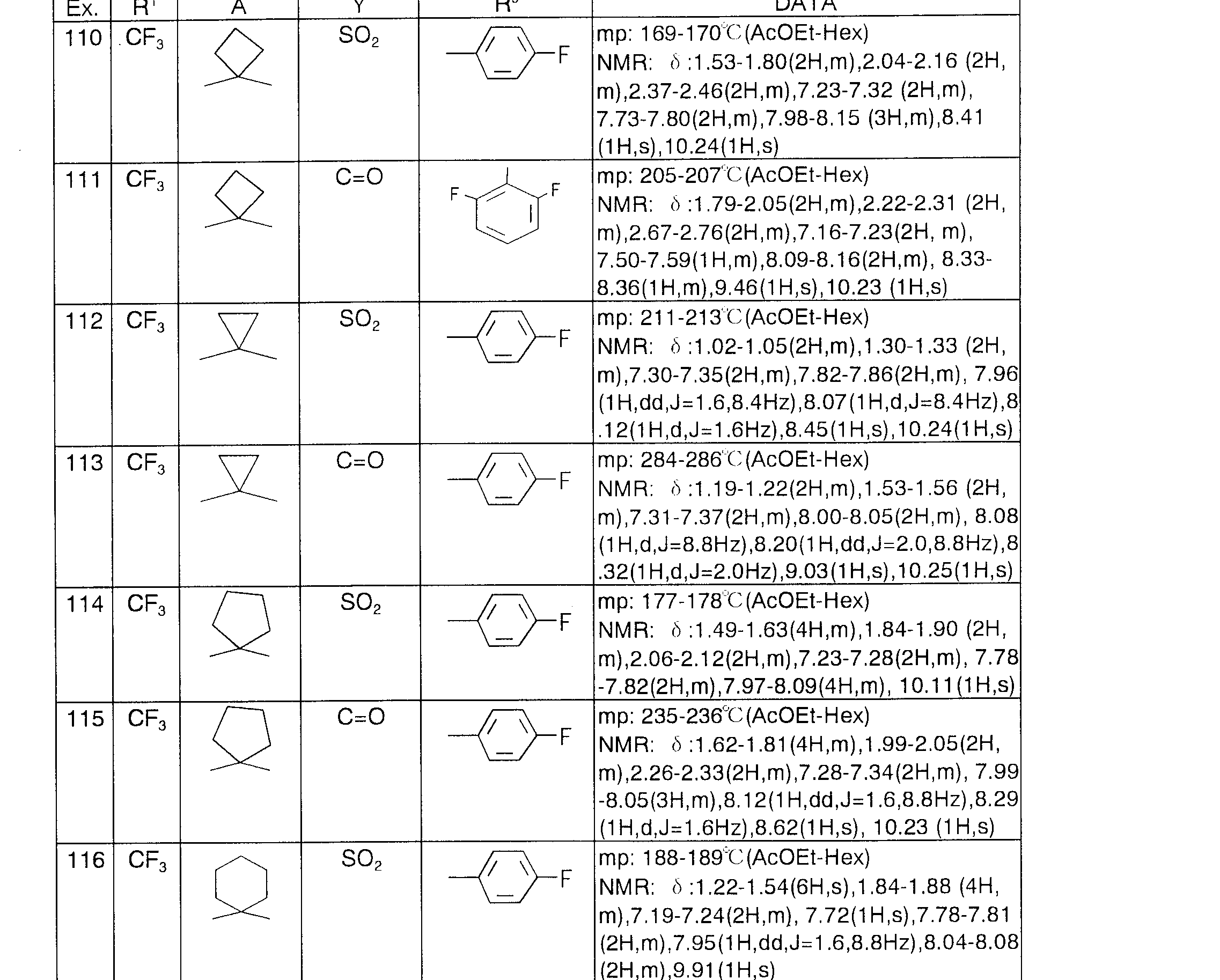



ここで,表中の記号は以下の意味を示す c Here, the symbols in the table have the following meanings c
Com. :化合物番号 表 1 4 Com .: Compound number Table 14
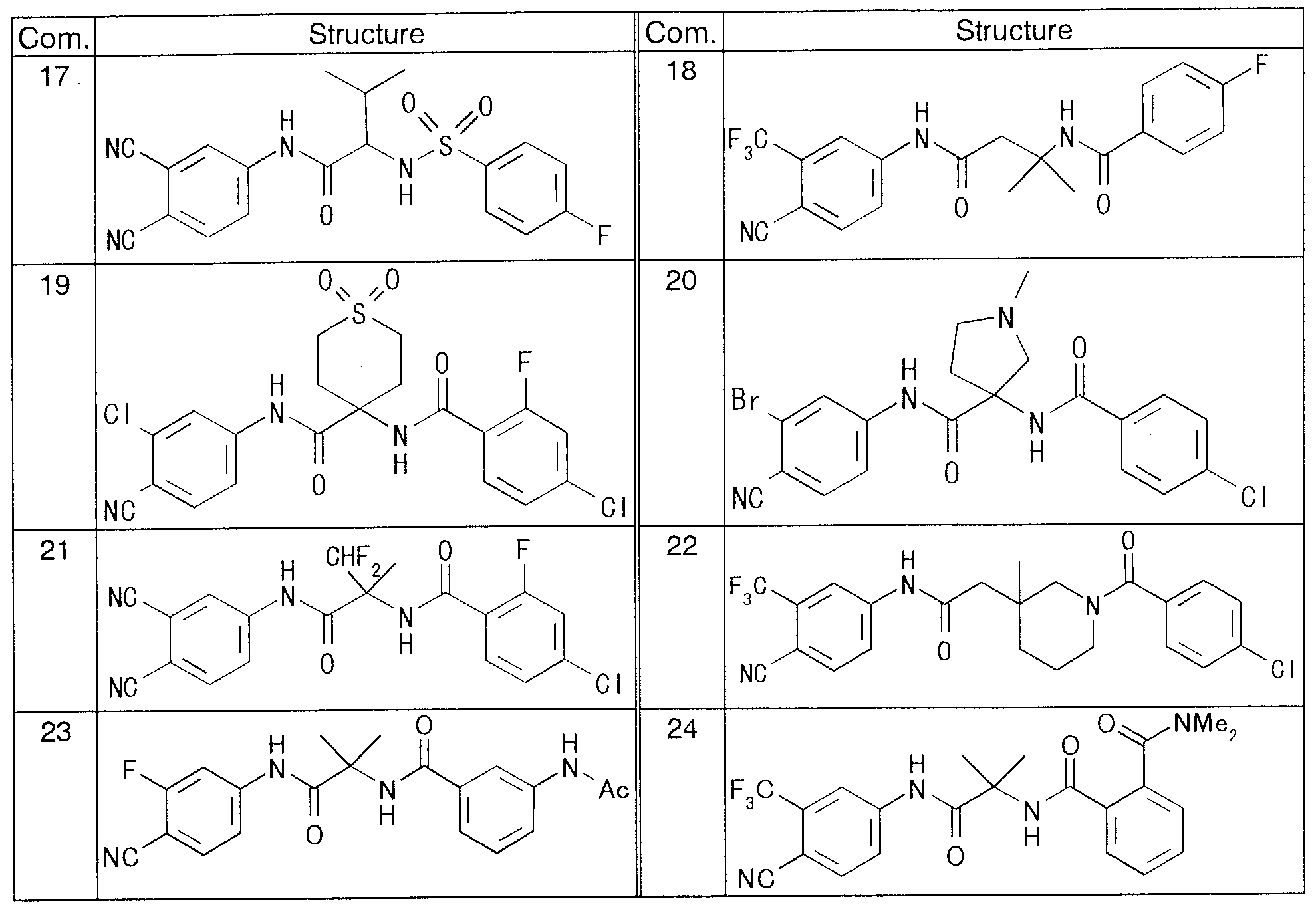
Claims
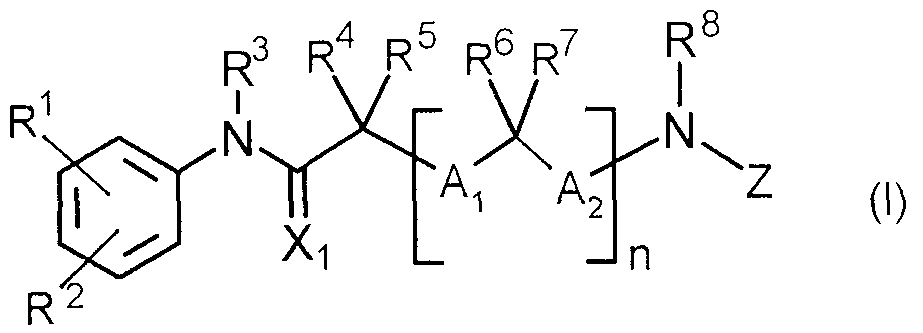
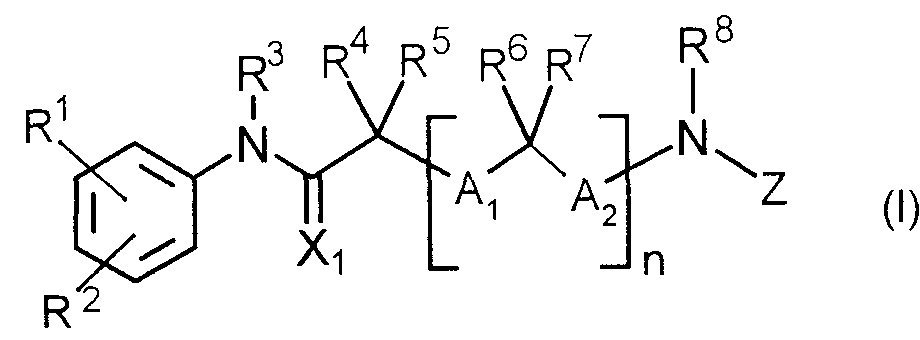
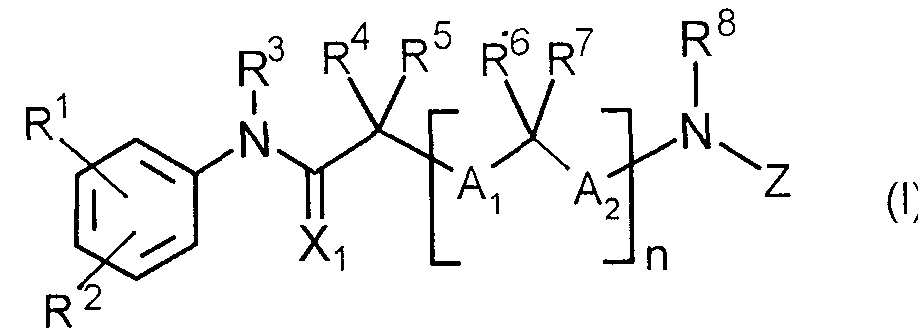
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU49664/97A AU4966497A (en) | 1996-11-18 | 1997-11-17 | Novel acylamino-substituted acylanilide derivatives or pharmaceutical composition comprising the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP8/306192 | 1996-11-18 | ||
| JP30619296 | 1996-11-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998022432A1 true WO1998022432A1 (en) | 1998-05-28 |
Family
ID=17954123
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1997/004174 Ceased WO1998022432A1 (en) | 1996-11-18 | 1997-11-17 | Novel acylamino-substituted acylanilide derivatives or pharmaceutical composition comprising the same |
Country Status (2)
| Country | Link |
|---|---|
| AU (1) | AU4966497A (en) |
| WO (1) | WO1998022432A1 (en) |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999062490A1 (en) * | 1998-06-03 | 1999-12-09 | Gpi Nil Holdings, Inc. | Small molecule sulfonamide hair growth compositions and uses |
| WO2000018361A3 (en) * | 1998-09-30 | 2000-05-25 | Procter & Gamble | Method of treating hair loss using sulfonamides |
| WO2000018358A3 (en) * | 1998-09-30 | 2000-07-27 | Procter & Gamble | Method of treating hair loss using ketoamides |
| WO2000046181A1 (en) * | 1999-02-03 | 2000-08-10 | Schering Aktiengesellschaft | Carboxylic acid derivatives, process for their preparation and their use as rotamase enzyme activity inhibitors |
| US6284779B1 (en) | 1999-02-03 | 2001-09-04 | Schering Aktiiengesellschaft | Heteroaromatic compounds |
| US6300341B1 (en) | 1998-09-30 | 2001-10-09 | The Procter & Gamble Co. | 2-substituted heterocyclic sulfonamides |
| US6307049B1 (en) | 1998-09-30 | 2001-10-23 | The Procter & Gamble Co. | Heterocyclic 2-substituted ketoamides |
| US6943187B2 (en) | 1997-06-04 | 2005-09-13 | Gpi Nil Holdings, Inc. | Pyrrolidine derivative hair growth compositions and uses |
| WO2005090282A1 (en) * | 2004-03-12 | 2005-09-29 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| US7078424B2 (en) | 1998-06-03 | 2006-07-18 | Gpi Nil Holdings, Inc. | N-linked sulfonamides of N-heterocyclic carboxylic acids or carboxylic acid isosteres |
| JP2006522037A (en) * | 2003-04-03 | 2006-09-28 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | Process for producing 1-N- (phenyl) -2-N- (phenyl) pyrrolidine-1,2-dicarboxamide derivative and 1- (phenylcarbamoyl) pyrrolidine-2-carboxylic acid derivative as an intermediate |
| US7153883B2 (en) | 1998-06-03 | 2006-12-26 | Gpi Nil Holdings Inc. | Carboxylic acids and carboxylic acid isosteres of N-heterocyclic compounds |
| JP2007500245A (en) * | 2003-06-10 | 2007-01-11 | スミスクライン ビーチャム コーポレーション | Compound |
| WO2006055184A3 (en) * | 2004-11-16 | 2007-04-05 | Janssen Pharmaceutica Nv | Novel heterocycle derivatives useful as selective androgen receptor modulators (sarms) |
| US7235576B1 (en) | 2001-01-12 | 2007-06-26 | Bayer Pharmaceuticals Corporation | Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US7329670B1 (en) | 1997-12-22 | 2008-02-12 | Bayer Pharmaceuticals Corporation | Inhibition of RAF kinase using aryl and heteroaryl substituted heterocyclic ureas |
| US7351834B1 (en) | 1999-01-13 | 2008-04-01 | Bayer Pharmaceuticals Corporation | ω-Carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US7371763B2 (en) | 2001-04-20 | 2008-05-13 | Bayer Pharmaceuticals Corporation | Inhibition of raf kinase using quinolyl, isoquinolyl or pyridyl ureas |
| US7517880B2 (en) | 1997-12-22 | 2009-04-14 | Bayer Pharmaceuticals Corporation | Inhibition of p38 kinase using symmetrical and unsymmetrical diphenyl ureas |
| US7528255B2 (en) | 1999-01-13 | 2009-05-05 | Bayer Pharmaceuticals Corporation | Hydroxy, ω-carboxyaryl substituted diphenyl ureas and dirivatives thereof as raf kinase inhibitors |
| WO2010101247A1 (en) | 2009-03-05 | 2010-09-10 | 塩野義製薬株式会社 | Cyclohexane derivative having npy y5 receptor antagonism |
| US7838541B2 (en) | 2002-02-11 | 2010-11-23 | Bayer Healthcare, Llc | Aryl ureas with angiogenesis inhibiting activity |
| US7897623B2 (en) | 1999-01-13 | 2011-03-01 | Bayer Healthcare Llc | ω-carboxyl aryl substituted diphenyl ureas as p38 kinase inhibitors |
| EP2392561A1 (en) * | 2002-07-05 | 2011-12-07 | Intrexon Corporation | Alpha-acylaminoketone ligands for modulating the expression of exogenous genes via an ecdysone receptor complex |
| US8124630B2 (en) | 1999-01-13 | 2012-02-28 | Bayer Healthcare Llc | ω-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| WO2013020440A1 (en) * | 2011-08-09 | 2013-02-14 | 上海医药集团股份有限公司 | Amide derivate, and preparation method therefor, pharmaceutical composition and use thereof |
| US8637553B2 (en) | 2003-07-23 | 2014-01-28 | Bayer Healthcare Llc | Fluoro substituted omega-carboxyaryl diphenyl urea for the treatment and prevention of diseases and conditions |
| US8796250B2 (en) | 2003-05-20 | 2014-08-05 | Bayer Healthcare Llc | Diaryl ureas for diseases mediated by PDGFR |
| US9181188B2 (en) | 2002-02-11 | 2015-11-10 | Bayer Healthcare Llc | Aryl ureas as kinase inhibitors |
| WO2016079521A1 (en) * | 2014-11-20 | 2016-05-26 | University College Cardiff Consultants Limited | Androgen receptor modulators and their use as anti-cancer agents |
| CN107072205A (en) * | 2014-09-10 | 2017-08-18 | Epizyme股份有限公司 | SMYD inhibitors |
| CN108558760A (en) * | 2018-05-29 | 2018-09-21 | 杨国宏 | A kind of aromatic amides and its preparation method and application |
| WO2018235966A1 (en) * | 2017-06-21 | 2018-12-27 | 第一三共株式会社 | Ep300/crebbp inhibitor |
| WO2019025153A1 (en) | 2017-07-31 | 2019-02-07 | Bayer Cropscience Aktiengesellschaft | USE OF SUBSTITUTED N-SULFONYL-N'-ARYLDIAMINOALKANES AND N-SULFONYL-N'-HETEROARYL DIAMINOALKANES OR THEIR SALTS TO INCREASE STRESSTOLERANCE IN PLANTS |
| CN109438297A (en) * | 2018-12-03 | 2019-03-08 | 昆明积大制药股份有限公司 | Androgen receptor antagonists, preparation method and its application |
| CN109593061A (en) * | 2018-12-03 | 2019-04-09 | 昆明积大制药股份有限公司 | Androgen receptor antagonists, preparation method and its application |
| CN109651243A (en) * | 2019-01-18 | 2019-04-19 | 西安交通大学 | A kind of class peptides and its preparation method and application containing serine |
| CN110179821A (en) * | 2019-05-17 | 2019-08-30 | 嘉兴市爵拓科技有限公司 | The composition and method and its application for promoting hair growth and/or color development to thicken |
| CN111196772A (en) * | 2018-11-16 | 2020-05-26 | 沈阳化工研究院有限公司 | Aroylamino isobutyryl derivative and application thereof |
| WO2020248327A1 (en) * | 2019-06-11 | 2020-12-17 | 江南大学 | Fatty acid synthase inhibitor and use thereof |
| WO2022007903A1 (en) * | 2020-07-09 | 2022-01-13 | 四川海思科制药有限公司 | Compound capable of inhibiting and degrading androgen receptors, and pharmaceutical compositions and pharmaceutical uses thereof |
| WO2022096542A1 (en) | 2020-11-06 | 2022-05-12 | Boehringer Ingelheim International Gmbh | 2-[thiophen-2-yl)formamido]-n-(phenyl)-2-methylpropanamide derivatives and the use thereof as medicament |
| WO2023016518A1 (en) * | 2021-08-11 | 2023-02-16 | 四川海思科制药有限公司 | Heterocyclic derivative, and composition and pharmaceutical use thereof |
| US12319651B2 (en) | 2019-04-19 | 2025-06-03 | Ligand Pharmaceuticals Incorporated | Crystalline forms and methods of producing crystalline forms of a compound |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4981332A (en) * | 1972-11-24 | 1974-08-06 | ||
| US4532251A (en) * | 1982-12-03 | 1985-07-30 | Chevron Research Company | N-substituted-N',N'-disubstituted glycinamide fungicides |
-
1997
- 1997-11-17 WO PCT/JP1997/004174 patent/WO1998022432A1/en not_active Ceased
- 1997-11-17 AU AU49664/97A patent/AU4966497A/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4981332A (en) * | 1972-11-24 | 1974-08-06 | ||
| US4532251A (en) * | 1982-12-03 | 1985-07-30 | Chevron Research Company | N-substituted-N',N'-disubstituted glycinamide fungicides |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, Vol. 101, 1984, Abstract No. 6764, VIEWEG H., "Synthesis of alpha-(Arylsulfonylamino)-Omega-Phenylalkyl carboxylic Acid 3- and -4-Amidinoanilides"; & PHARMAZIE, 1983, 38(12), 818-20. * |
Cited By (78)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6943187B2 (en) | 1997-06-04 | 2005-09-13 | Gpi Nil Holdings, Inc. | Pyrrolidine derivative hair growth compositions and uses |
| US7329670B1 (en) | 1997-12-22 | 2008-02-12 | Bayer Pharmaceuticals Corporation | Inhibition of RAF kinase using aryl and heteroaryl substituted heterocyclic ureas |
| US7625915B2 (en) | 1997-12-22 | 2009-12-01 | Bayer Healthcare Llc | Inhibition of RAF kinase using aryl and heteroaryl substituted heterocyclic ureas |
| US7517880B2 (en) | 1997-12-22 | 2009-04-14 | Bayer Pharmaceuticals Corporation | Inhibition of p38 kinase using symmetrical and unsymmetrical diphenyl ureas |
| WO1999062490A1 (en) * | 1998-06-03 | 1999-12-09 | Gpi Nil Holdings, Inc. | Small molecule sulfonamide hair growth compositions and uses |
| US7153883B2 (en) | 1998-06-03 | 2006-12-26 | Gpi Nil Holdings Inc. | Carboxylic acids and carboxylic acid isosteres of N-heterocyclic compounds |
| US7078424B2 (en) | 1998-06-03 | 2006-07-18 | Gpi Nil Holdings, Inc. | N-linked sulfonamides of N-heterocyclic carboxylic acids or carboxylic acid isosteres |
| AU760663B2 (en) * | 1998-06-03 | 2003-05-22 | Gpi Nil Holdings, Inc. | Small molecule sulfonamide hair growth compositions and uses |
| US6300341B1 (en) | 1998-09-30 | 2001-10-09 | The Procter & Gamble Co. | 2-substituted heterocyclic sulfonamides |
| US6307049B1 (en) | 1998-09-30 | 2001-10-23 | The Procter & Gamble Co. | Heterocyclic 2-substituted ketoamides |
| WO2000018358A3 (en) * | 1998-09-30 | 2000-07-27 | Procter & Gamble | Method of treating hair loss using ketoamides |
| WO2000018361A3 (en) * | 1998-09-30 | 2000-05-25 | Procter & Gamble | Method of treating hair loss using sulfonamides |
| US8124630B2 (en) | 1999-01-13 | 2012-02-28 | Bayer Healthcare Llc | ω-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US8841330B2 (en) | 1999-01-13 | 2014-09-23 | Bayer Healthcare Llc | Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US7897623B2 (en) | 1999-01-13 | 2011-03-01 | Bayer Healthcare Llc | ω-carboxyl aryl substituted diphenyl ureas as p38 kinase inhibitors |
| US7351834B1 (en) | 1999-01-13 | 2008-04-01 | Bayer Pharmaceuticals Corporation | ω-Carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US7528255B2 (en) | 1999-01-13 | 2009-05-05 | Bayer Pharmaceuticals Corporation | Hydroxy, ω-carboxyaryl substituted diphenyl ureas and dirivatives thereof as raf kinase inhibitors |
| US6284779B1 (en) | 1999-02-03 | 2001-09-04 | Schering Aktiiengesellschaft | Heteroaromatic compounds |
| WO2000046181A1 (en) * | 1999-02-03 | 2000-08-10 | Schering Aktiengesellschaft | Carboxylic acid derivatives, process for their preparation and their use as rotamase enzyme activity inhibitors |
| US7235576B1 (en) | 2001-01-12 | 2007-06-26 | Bayer Pharmaceuticals Corporation | Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US7371763B2 (en) | 2001-04-20 | 2008-05-13 | Bayer Pharmaceuticals Corporation | Inhibition of raf kinase using quinolyl, isoquinolyl or pyridyl ureas |
| US8618141B2 (en) | 2002-02-11 | 2013-12-31 | Bayer Healthcare Llc | Aryl ureas with angiogenesis inhibiting activity |
| US9181188B2 (en) | 2002-02-11 | 2015-11-10 | Bayer Healthcare Llc | Aryl ureas as kinase inhibitors |
| US7838541B2 (en) | 2002-02-11 | 2010-11-23 | Bayer Healthcare, Llc | Aryl ureas with angiogenesis inhibiting activity |
| US8242147B2 (en) | 2002-02-11 | 2012-08-14 | Bayer Healthcare Llc | Aryl ureas with angiogenisis inhibiting activity |
| US8129355B2 (en) | 2002-07-05 | 2012-03-06 | Intrexon Corporation | Ketone ligands for modulating the expression of exogenous genes via an ecdysone receptor complex |
| US9802936B2 (en) | 2002-07-05 | 2017-10-31 | Intrexon Corporation | Ketone ligands for modulating the expression of exogenous genes via an ecdysone receptor complex |
| EP2392561A1 (en) * | 2002-07-05 | 2011-12-07 | Intrexon Corporation | Alpha-acylaminoketone ligands for modulating the expression of exogenous genes via an ecdysone receptor complex |
| JP2006522037A (en) * | 2003-04-03 | 2006-09-28 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | Process for producing 1-N- (phenyl) -2-N- (phenyl) pyrrolidine-1,2-dicarboxamide derivative and 1- (phenylcarbamoyl) pyrrolidine-2-carboxylic acid derivative as an intermediate |
| US8796250B2 (en) | 2003-05-20 | 2014-08-05 | Bayer Healthcare Llc | Diaryl ureas for diseases mediated by PDGFR |
| JP2007500245A (en) * | 2003-06-10 | 2007-01-11 | スミスクライン ビーチャム コーポレーション | Compound |
| US8637553B2 (en) | 2003-07-23 | 2014-01-28 | Bayer Healthcare Llc | Fluoro substituted omega-carboxyaryl diphenyl urea for the treatment and prevention of diseases and conditions |
| WO2005090282A1 (en) * | 2004-03-12 | 2005-09-29 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| US9359285B2 (en) | 2004-03-12 | 2016-06-07 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| US8865918B2 (en) | 2004-03-12 | 2014-10-21 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| US8519158B2 (en) | 2004-03-12 | 2013-08-27 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| JP2008520665A (en) * | 2004-11-16 | 2008-06-19 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Novel heterocyclic derivatives useful as selective androgen receptor modulators (SARMs) |
| US7465809B2 (en) | 2004-11-16 | 2008-12-16 | Janssen Pharmaceutica N.V. | Heterocycle derivatives useful as selective androgen receptor modulators (SARMs) |
| WO2006055184A3 (en) * | 2004-11-16 | 2007-04-05 | Janssen Pharmaceutica Nv | Novel heterocycle derivatives useful as selective androgen receptor modulators (sarms) |
| US8088811B2 (en) | 2004-11-16 | 2012-01-03 | Janssen Pharmaceutica N.V. | Heterocycle derivatives useful as selective androgen receptor modulators (SARMs) |
| CN104803916B (en) * | 2004-11-16 | 2017-08-11 | 詹森药业有限公司 | Hete rocyclic derivatives as SARM (SARMS) |
| US7781473B2 (en) | 2004-11-16 | 2010-08-24 | J & J Pharmaceutical Research & Development, LLC. | Heterocycle derivatives useful as selective androgen receptor modulators (SARMS) |
| TWI483725B (en) * | 2004-11-16 | 2015-05-11 | Janssen Pharmaceutica Nv | Novel heterocycle derivatives useful as selective androgen receptor modulators (sarms) |
| CN104803916A (en) * | 2004-11-16 | 2015-07-29 | 詹森药业有限公司 | Novel heterocycle derivatives useful as selective androgen receptor modulators (sarms) |
| JP5685181B2 (en) * | 2009-03-05 | 2015-03-18 | 塩野義製薬株式会社 | Cyclohexane derivative having NPYY5 receptor antagonistic action |
| US8653125B2 (en) | 2009-03-05 | 2014-02-18 | Shionogi Co., Ltd. | Cyclohexane derivative having NPY Y5 receptor antagonism |
| WO2010101247A1 (en) | 2009-03-05 | 2010-09-10 | 塩野義製薬株式会社 | Cyclohexane derivative having npy y5 receptor antagonism |
| WO2013020440A1 (en) * | 2011-08-09 | 2013-02-14 | 上海医药集团股份有限公司 | Amide derivate, and preparation method therefor, pharmaceutical composition and use thereof |
| CN107072205A (en) * | 2014-09-10 | 2017-08-18 | Epizyme股份有限公司 | SMYD inhibitors |
| WO2016079521A1 (en) * | 2014-11-20 | 2016-05-26 | University College Cardiff Consultants Limited | Androgen receptor modulators and their use as anti-cancer agents |
| JPWO2018235966A1 (en) * | 2017-06-21 | 2020-04-23 | 第一三共株式会社 | EP300 / CREBBP inhibitor |
| WO2018235966A1 (en) * | 2017-06-21 | 2018-12-27 | 第一三共株式会社 | Ep300/crebbp inhibitor |
| JP7134173B2 (en) | 2017-06-21 | 2022-09-09 | 第一三共株式会社 | EP300/CREBBP inhibitor |
| US11274100B2 (en) | 2017-06-21 | 2022-03-15 | Daiichi Sankyo Company, Limited | EP300/CREBBP inhibitor |
| WO2019025153A1 (en) | 2017-07-31 | 2019-02-07 | Bayer Cropscience Aktiengesellschaft | USE OF SUBSTITUTED N-SULFONYL-N'-ARYLDIAMINOALKANES AND N-SULFONYL-N'-HETEROARYL DIAMINOALKANES OR THEIR SALTS TO INCREASE STRESSTOLERANCE IN PLANTS |
| CN108558760A (en) * | 2018-05-29 | 2018-09-21 | 杨国宏 | A kind of aromatic amides and its preparation method and application |
| CN108558760B (en) * | 2018-05-29 | 2021-12-03 | 杨国宏 | Aromatic amide compounds, preparation method and application thereof |
| CN111196772B (en) * | 2018-11-16 | 2022-09-30 | 沈阳化工研究院有限公司 | Aroylamino isobutyryl derivative and application thereof |
| CN111196772A (en) * | 2018-11-16 | 2020-05-26 | 沈阳化工研究院有限公司 | Aroylamino isobutyryl derivative and application thereof |
| CN109593061A (en) * | 2018-12-03 | 2019-04-09 | 昆明积大制药股份有限公司 | Androgen receptor antagonists, preparation method and its application |
| CN109438297B (en) * | 2018-12-03 | 2021-05-14 | 昆明积大制药股份有限公司 | Androgen receptor antagonist, preparation method and application thereof |
| CN109593061B (en) * | 2018-12-03 | 2021-09-14 | 昆明积大制药股份有限公司 | Androgen receptor antagonist, preparation method and application thereof |
| CN109438297A (en) * | 2018-12-03 | 2019-03-08 | 昆明积大制药股份有限公司 | Androgen receptor antagonists, preparation method and its application |
| CN109651243B (en) * | 2019-01-18 | 2020-05-22 | 西安交通大学 | A kind of peptoid compound containing serine and its preparation method and application |
| CN109651243A (en) * | 2019-01-18 | 2019-04-19 | 西安交通大学 | A kind of class peptides and its preparation method and application containing serine |
| US12319651B2 (en) | 2019-04-19 | 2025-06-03 | Ligand Pharmaceuticals Incorporated | Crystalline forms and methods of producing crystalline forms of a compound |
| CN110179821A (en) * | 2019-05-17 | 2019-08-30 | 嘉兴市爵拓科技有限公司 | The composition and method and its application for promoting hair growth and/or color development to thicken |
| JP2021530439A (en) * | 2019-06-11 | 2021-11-11 | 江南大学Jiangnan University | Fatty acid synthase inhibitor and its application |
| JP7256828B2 (en) | 2019-06-11 | 2023-04-12 | 江南大学 | Fatty acid synthase inhibitor and its application |
| US11911353B2 (en) | 2019-06-11 | 2024-02-27 | Jiangnan University | Fatty acid synthase inhibitor and application thereof |
| WO2020248327A1 (en) * | 2019-06-11 | 2020-12-17 | 江南大学 | Fatty acid synthase inhibitor and use thereof |
| WO2022007903A1 (en) * | 2020-07-09 | 2022-01-13 | 四川海思科制药有限公司 | Compound capable of inhibiting and degrading androgen receptors, and pharmaceutical compositions and pharmaceutical uses thereof |
| CN115697992A (en) * | 2020-07-09 | 2023-02-03 | 四川海思科制药有限公司 | Compound capable of inhibiting and degrading androgen receptor, pharmaceutical composition and pharmaceutical application thereof |
| CN115697992B (en) * | 2020-07-09 | 2025-09-19 | 西藏海思科制药有限公司 | Compound capable of inhibiting and reducing Jie Xiong hormone receptor, pharmaceutical composition and pharmaceutical application thereof |
| WO2022096542A1 (en) | 2020-11-06 | 2022-05-12 | Boehringer Ingelheim International Gmbh | 2-[thiophen-2-yl)formamido]-n-(phenyl)-2-methylpropanamide derivatives and the use thereof as medicament |
| CN116406266A (en) * | 2020-11-06 | 2023-07-07 | 勃林格殷格翰国际有限公司 | 2-[(thiophen-2-yl)carboxamido]-N-(phenyl)-2-methylpropionamide derivatives and their use as medicines |
| JP2023547529A (en) * | 2020-11-06 | 2023-11-10 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 2-[(thiophen-2-yl)formamide]-N-(phenyl)-2-methylpropanamide derivatives and their use as pharmaceuticals |
| WO2023016518A1 (en) * | 2021-08-11 | 2023-02-16 | 四川海思科制药有限公司 | Heterocyclic derivative, and composition and pharmaceutical use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU4966497A (en) | 1998-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1998022432A1 (en) | Novel acylamino-substituted acylanilide derivatives or pharmaceutical composition comprising the same | |
| KR100660309B1 (en) | Cyanophenyl derivative | |
| JP2001261657A (en) | Cyanophenyl derivative | |
| JP4449746B2 (en) | N-phenyl- (2R, 5S) dimethylpiperazine derivative | |
| US5773530A (en) | Pyrazolopyridine adenosine antagonists | |
| CN1187367C (en) | 3 alpha-hydroxy-3 beta-methoxymethyl-21-heterocycle substd. steroids with anesthetic activity | |
| JP2002088073A (en) | Anti-androgenic agent | |
| WO2006028226A1 (en) | Novel imidazolidine derivative and use thereof | |
| EP0454511A1 (en) | N-substituted heterocycle derivatives, their preparation, compositions containing them | |
| WO1993022298A1 (en) | Oxazolidine derivative and pharmaceutically acceptable salt thereof | |
| JP2006513201A (en) | 1,5-Diaryl-pyrrole-3-carboxamide derivatives and their use as cannabinoid receptor modulators | |
| JP2011509260A (en) | TRPA1 antagonist | |
| JP2004067629A (en) | Mitochondria function-activating agent and new benzimidazole derivative | |
| WO2007129745A1 (en) | Heteroarylamide lower carboxylic acid derivative | |
| KR20110067029A (en) | Novel Imidazolidine Compounds As Androgen Receptor Modulators | |
| JP2010524907A (en) | Triazolopyridinecarboxamide derivatives, their preparation and their therapeutic use | |
| KR20110034020A (en) | Alkyl thiazole carbamate derivatives, their preparation, and their use as FAH enzyme inhibitors | |
| KR20190137860A (en) | Liver X-Receptor (LXR) Modulators | |
| HUE030696T2 (en) | Process for the manufacture of dabigatran etexilate and intermediates thereof | |
| JP2025121901A (en) | Crystalline forms of farnesoid X receptor agonists | |
| JP2707936B2 (en) | β-oxo-β-benzenepropanethioamide derivative | |
| WO2009092284A1 (en) | Benzocycloheptene derivatives, preparing processes and pharmaceutical uses thereof | |
| JP5606440B2 (en) | Method for producing thiabenzoazulene propionic acid derivatives | |
| JP2014532744A (en) | Carboxamide and urea derivatives based on substituted pyrazolyls bearing phenyl moieties substituted with SO2-containing groups as vanilloid receptor ligands | |
| FR2816942A1 (en) | Benzimidazole derivatives having poly(ADP-ribose) polymerase inhibiting activity for treatment of cardiac, cerebral, inflammatory, immunological and other disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU AZ BA BB BG BR BY CA CN CU CZ EE GE GH HU ID IL IS JP KE KG KR KZ LC LK LR LS LT LV MD MG MK MN MW MX NO NZ PL RO RU SD SG SI SK SL TJ TM TR TT UA UG US UZ VN YU AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG ZW AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: CA |