WO1992013837A1 - A process for the preparation of 3-acylamino-4-carbamoyloxymethyl-2-azetidinone-1-sulphonic acids and intermediates for the preparation thereof - Google Patents
A process for the preparation of 3-acylamino-4-carbamoyloxymethyl-2-azetidinone-1-sulphonic acids and intermediates for the preparation thereof Download PDFInfo
- Publication number
- WO1992013837A1 WO1992013837A1 PCT/EP1992/000175 EP9200175W WO9213837A1 WO 1992013837 A1 WO1992013837 A1 WO 1992013837A1 EP 9200175 W EP9200175 W EP 9200175W WO 9213837 A1 WO9213837 A1 WO 9213837A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- carried out
- process according
- formula
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C[C@@]([C@](C*)O)C(NO*)=O Chemical compound C[C@@]([C@](C*)O)C(NO*)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/06—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D205/08—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams
- C07D205/085—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams with a nitrogen atom directly attached in position 3
Definitions
- the present invention relates to a process for the synthesis of monobacta s of formula (1)
- R represents an easily removable or pharmaceu ⁇ tically acceptable acyl residue, and of pharmaceuti ⁇ cally acceptable salts thereof, starting from (R) malic acid esters.
- R represents the acyl resi ⁇ due of O-benzylcarbonic, O-tert-butylcarbonic, phenyla- cetic, phenoxyacetic, 2-(2-a_ ⁇ ino-4-thiazolyl)-2-(Z)- (methoxyimino)acetic, 2-(2-amino-4-thiazolyl)-2-(Z)- (carboxymethoxyimino)acetic, 2-(2-amino-4-thiazolyl)-2- (Z)-(1-carboxy-l-meth 1-ethoxyimino)acetic acids.
- a 4 represents hydrogen, hydroxy or an OR1 residue, wherein R is methyl or arylalk l ⁇ group.
- object of the present invention are (3S, 4S) 3-hydrazino-4-hydroxymethyl-2-azetidinone of formula (11)
- the present invention further relates to the conversion of (11) into the well-known intermediates (3S, 4S) 3-(benzyloxycarbonylamino)-4-hydroxymethyl-2- azetidinone (13) and (3S, 4S) 3-(tert-butoxycarbonyla- mino)-4-hydroxymethyl-2-azetidinone (14), as well as to the conversion of said compounds (13) and (14) into the corresponding intermediates (3S, 4S) 3-(benzyloxycarbo- nylamino)-4-carbamoyloxymethyl-2-azetidinone (15) and (3S, 4S) 3-(tert-butoxycarbonylamino)-4-carbamoyloxyme- th l-2-azetidinone (16), the former, which is already well-known, can be converted, with well-known procedures, into monobactams (1)
- the preparation thereof requires a stereoselective (or a diastereospecific) , and enantioselective (or enantiospecific) synthesis, otherwise, in the event of a synthesis leading to racemic products, an optical resolution.
- Object of the present invention is a totally synthetic process for the preparation of the above compounds of formula (1).
- the process of the invention is carried out starting from (R) malic acid esters, which are easily obtained from L-tartaric acid.
- Scheme 1 and Scheme 2 illustrate a preferred embo ⁇ diment of the invention.
- - X is a convenient protective group, which is compati ⁇ ble with reaction conditions. Said protective group can be removed at the step wherein compound (7) is obtained. The removal of X can be carried out before, after or simultaneously the removal of OR group, in reaction conditions compatible with other functional groups present in the compound.
- Example of X are R R 5 R 6 Si or 4 R 5 R 6 SiCH 2 CH 2 OCH 2 groups, where R 4, R5 and R6 are alkyl, aryl or alkoxy groups.
- R 4, R5 and R6 are Ph-tBuSi; CAr 1Ar2Ar3, where Ar1, Ar2, Ar3 represent substituted or unsubstituted, optionally linked each other, aromatic residues (such as triphenylmethyl) ; CHiffOCH_Ar, where Ar represents a substituted or un ⁇ ubstituted aromatic residue (for example PhCH_OCH_).
- - Y is a C,-C- alkyl group, such as methyl, ethyl, n- propyl; methyl group being preferred.
- - R is a methyl group, or a CH_Ar group, where Ar is as above defined; for example benzyl group.
- - A is a tert-butoxycarbonyl or arylalkyloxycarbonyl group.
- - R is an easily removable or pharmaceutically accepta ⁇ ble acyl group, particularly the acyl residue of O-ben- zylcarbonic, O-tert-butylcarbonic, phenylacetic, phe- noxyacetic, 2-(2-amino-4-thiazolyl)-2-(Z)-(methoxyimi- no)acetic, 2-(2-amino-4-thiazolyl)-2-(Z)-(carboxyme- thoxyimino) acetic, 2-(2-amino-4-thiazolyl)-2-(Z)-(1- carboxy-1-methyl-ethoxyimino) cetic acids.
- the compounds of formula (1) can also be in the form of pharmaceutically acceptable salts, and the compound of formula (11) can also be in the form of an hydrazinium salt.
- m fact R acyl residues can be different from the ones above mentioned.
- step 1 the preparation starts from a (R) malic acid ester (2), which is converted into the diol (3) (step 1) by regioselective reduction with borane and sodium borohydride, as described by S. Saito, et al., (Chem. Lett., ' 1389 (1984)).
- reaction conditions vary according to the protective group being used.
- a dipolar aprotic solvent such as dimethylformamide or dimethyl sulfoxide
- reaction is preferably carried out in a chlorinated solvent (for example, methylene chloride) in the presence of a tertiary amine (for example diiso- propylethylamine) at a temperature ranging from 0°C to the solvent boiling temperature.
- a chlorinated solvent for example, methylene chloride
- a tertiary amine for example diiso- propylethylamine
- the protection is carried out in a halogenated solvent, such as, for example, methylene chloride, by treating with the appropriate halide, in the presence of a nitrogen base (for example, pyridine) and at a temperature ranging from 0 ⁇ C to the solvent boiling temperature.
- a nitrogen base for example, pyridine
- the protection reaction has already been described in literature (K. Prasad, et al., Tetrahedron: Asymmetry , 307 (1990)).
- Next step consists in condensing ⁇ -hydroxyesters
- a compound (4) with a di-t-butyl- or diarylalkyl azodicarboxylate Said transformation can be carried out by treating a compound (4) with at least two equivalents of a strong base, such as, for example, a lithium or sodium or potassium dialkylamide (for example, diisopropylamide) in an aprotic solvent, such as tetrahydrofurane or dimethoxyethane, at a temperature ranging from -78°C to 20°C, preferably from -40°C to 0°C, followed by the reaction with the azodicarboxylate, at a temperature ranging from -78°C to 0°C.
- a strong base such as, for example, a lithium or sodium or potassium dialkylamide (for example, diisopropylamide)
- an aprotic solvent such as tetrahydrofurane or dimethoxyethane
- Next step consists in converting the esters (5) into 0-alkylhydroxamates (6). Said conversion can be effected in two ways.
- the so obtained carboxylic acids can be isolated by extraction or by treating with an appropriate ion exchange resin and subsequent by purificating by crystallization or chromatography. Alternatively, the basic solution containing the carboxylic acid salts can be used as such for the next reaction.
- the second step consists in coupling the so obtained acids (or the salts thereof) with the appropriate O-alkylhydroxylamine (or a hydroxylammonium salt thereof).
- Said step can be carried out both starting from the carboxylic acids and starting from the crude carboxylate solution, which has previously been obtained by the above saponification.
- the coupling can be executed in an aqueous solution containing an appropriate water-soluble cosolvent, such as tetrahy- drofurane, dimethylformamide or acetonitrile, keeping pH between 4 and 7, according to the group X present, by reacting with the appropriate O-alkylhydroxylamine (or a salt thereof) (for example 1-2 equivalents), in the presence of a condensing agent, such as, for example, N,N'-dicyclohexylcarbodiimide (DCC) or l-(3- diaminopropyl)-3-ethylcarbodiimide (WSC) (1-3 equiva ⁇ lents).
- an appropriate water-soluble cosolvent such as tetrahy- drofurane, dimethylformamide or acetonitrile
- a condensing agent such as, for example, N,N'-dicyclohexylcarbodiimide (DCC) or l-
- the coupling can also be carried out activating the purified carboxylic acids by reaction with dicyclohexylcarbodiimide and N-hydroxybenzotria- zole in a dipolar aprotic solvent, such as acetoni ⁇ trile, dioxane, tetrahydrof rane or dimethylformamide and reacting the so activated adducts in the same solvent with the appropriate O-alkylhydroxylamine or a hydroxylammonium salt thereof (in the latter case also adding an equivalent amount of a tertiary amine, such as, for example, triethylamine) .
- a dipolar aprotic solvent such as acetoni ⁇ trile, dioxane, tetrahydrof rane or dimethylformamide
- the same coupling can directly be performed by using the crude alkali carboxylate solution, which has been obtained, as above described, from C 1 -C 3 alkyl ester saponification.
- the coupling can be performed by reacting with the appropriate O-alkylhydroxylamine (or a hydroxylammonium salt thereof) in the same solvent wherein saponification was carried out, optionally integrated with the addition of water or of appropriate organic cosolvents, such as dimethylformam- mide or tetrahydrofurane, in the presence of a condensing agent such as, for example, N,N'-dicy- clohexylcarbodiimide (DCC) or l-(3-diaminopropyl)-3- ethylcarbodiimide (WSC) (1-3 equivalents).
- DCC N,N'-dicy- clohexylcarbodiimide
- WSC l-(3-diaminopropyl)-3- eth
- esters (5) can be transformed into hydroxamates (6) in a single step by reacting them with the adduct which has been obtained by mixing the appropriate O-alkylhydroxylamine with trimethylaluminum in an aprotic solvent, such as, for example, tetrahy ⁇ drofurane, at a temperature ranging from -20 ⁇ C to the solvent boiling temperature (preferably from 0°C to 20°C) .
- an aprotic solvent such as, for example, tetrahy ⁇ drofurane
- Next step which consists in transforming hydroxa- mates (6) into ⁇ -lactams (7), can be performed in an appropriate organic solvent (for example tetrahydrofu ⁇ rane, acetonitrile or dimethylformamide) preferably by treating with triphenylphosphine and a dialkyl azodi ⁇ carboxylate (such as a diethyl or dii ⁇ opropyl azodicar- boxyl te) , or by treating with triphenylphosphine, carbon tetrachloride and triethylamine at a temperature ranging from 0°C to 60°C (preferably from 20°C to 30°C).
- an appropriate organic solvent for example tetrahydrofu ⁇ rane, acetonitrile or dimethylformamide
- triphenylphosphine and a dialkyl azodi ⁇ carboxylate such as a diethyl or dii ⁇ opropyl azodicar- boxyl te
- the same transformation can be carried out by converting the alcohol into an alkansul- fonyl derivative by treating, for example, with metha- nesulfonyl chloride in pyridine, followed by treatment with bases, such as sodium hydrogencarbonate or sodium carbonate in dipolar aprotic solvents, such as acetone, dioxane, etc.
- bases such as sodium hydrogencarbonate or sodium carbonate in dipolar aprotic solvents, such as acetone, dioxane, etc.
- the products (7) are purifiable by means of extraction, chromatography or crystallization.
- the conversion which is carried out with diethyl azodicarboxylate and triphenylphosphine in te- trahydrofurane at room temperature, occurs with very good yields (about 95%).
- the conversion of ⁇ -lactams (7) into compounds (10) can be accomplished in several ways. The choice of the method to
- protective group removal can be performed both before and after removing OR group (preferably before) , by treatment with a fluoride (for example tetr -n-butylammonium fluoride) in a solvent, such as tetrahydrofurane or dioxane.
- a fluoride for example tetr -n-butylammonium fluoride
- X CAr 1Ar2Ar3
- protective group removal is preferably performed before OR group removal to give the derivatives (8).
- Said unblocking can be made, for example, by treating with a strong protic acid (such as a sulfonic acid or trifluoroacetic acid) in methyl or ethyl alcohol at a temperature ranging from 0°C to 60°C, or by heating, at a temperature ranging from 20 C C to 100°C, in an acetic acid-water mixture.
- a strong protic acid such as a sulfonic acid or trifluoroacetic acid
- de- protection can be performed by hydrogenating in an appropriate solvent (for example, methyl, ethyl, n- propyl, iso-propyl alcohol or ethyl acetate) in the presence of a transition metal catalyst, such as palladium (for example, pure or supported on carbon or barium sulfate) or platinum (for example, pure or in the form of dioxide), at a pressure ranging from 1 to 10 atmospheres.
- an appropriate solvent for example, methyl, ethyl, n- propyl, iso-propyl alcohol or ethyl acetate
- a transition metal catalyst such as palladium (for example, pure or supported on carbon or barium sulfate) or platinum (for example, pure or in the form of dioxide), at a pressure ranging from 1 to 10 atmospheres.
- a transition metal catalyst such as palladium (for example, pure or supported on carbon or barium sulfate) or platinum (for example, pure or in the form of dioxide)
- Said transformation can be performed, for example, by adding a aqueous hydrochloric acid titanium trichloride solution to the substrate (9), which is dissolved in a water/alcohol system (for example water/methanol) at a pH between 3 and 10 (preferably 1 ) , maintained with buffer solutions, or by simultaneously dropping an alkali hydroxide solution.
- a water/alcohol system for example water/methanol
- good yields about 50-60%, are obtained.
- the products (8) can directly be converted into (10) in a single step, by treating them with alkali metals (for example, sodium) in liquid ammonia, optionally in the presence of organic cosolvents.
- alkali metals for example, sodium
- the compound (10) in a single step, by treating (7) with alkali metals (for example, sodium) in liquid ammonia, optionally in the presence of organic cosolvents.
- alkali metals for example, sodium
- the compound (10) can be purified by chromatography or crystallization.
- Next step consists in converting (10) into the key intermediate (11) and can be carried out by treating (10) with a strong carboxylic acid, such as, for example, trifluoroacetic or formic acid.
- a strong carboxylic acid such as, for example, trifluoroacetic or formic acid.
- a cosolvent which is compatible with reaction conditions, for example, methylene chloride, can optionally be used.
- Said reaction can be carried out with very good yields, by stirring for 1 hour a 1:1 trifluoroacetic acid: methylene chloride solution of (10), at a temperature ranging from 0°C to 25°C.
- the so obtained product (11) can be used wether as such for the next reaction, or purified by the conventional techniques (crystal- lization, ion exchange chromatography, etc.).
- the product (11) and the hydrazinium salts thereof are new, therefore they are a further object of the present invention.
- (11) can be converted into the known intermediate (12) by reacting a hydrazinium salt thereof with hydrogen in the presence of catalysts, such as platinum dioxide or Raney® Nickel, at a pressure ranging from 1 to 200 atmospheres and, depending on the used catalyst, in water, alcohol (for example, methanol or ethanol) or water-alcohol mixtures.
- catalysts such as platinum dioxide or Raney® Nickel
- (12) can be wether purified or directly reacted, as crude, with benzyloxy- carbonyl chloride or with di-t-butyl dicarbonate to give the known products (13) and (14).
- This last conversion can be performed by treating with the appro- priate acylating agent in an anhydrous solvent, such as dimethylformamide or acetonitrile and in the presence of a base, such as a tertiary amine (for example, triethylamine) ; otherwise, and preferably, in an aqueous solution kept at a pH between 8 and 10 with alkaly hydroxides (lithium, sodium or potassium) or alkali carbonates (sodium, potassium).
- a base such as a tertiary amine (for example, triethylamine)
- alkaly hydroxides lithium, sodium or potassium
- alkali carbonates sodium, potassium
- the products (13) and (14) can be transformed into the carbamates (15) and (16), the former being a well known derivative (U.S. 499,801; S. Kishimoto, et al., J. Antibiot., 36, pag. 1421 (1983)). Said conver ⁇ ion i ⁇ new, therefore it is a further object of the present invention.
- N-acyl (or N- sulfonyl) carbamate deprotection can be performed by treatment with sodium or potassium N-alkyl dithiocarbamates, while, in the case of N-sulfonylcar- bamates, by treatment with sodium sulfite.
- the above process can be alternatively carried out as far as the introduction of a ino group into 2-position of ⁇ -hydroxye ⁇ ter (4) is concerned, by electrophilic amination with other synthetic equiva ⁇ lents of NH ? + group, such as sulfonyl azides, O-substi- tuted hydroxylamines and diazonium salts.
- sulfonyl azides especially p-toluensul- fonyl azide, 2,4,6-triisopropylbenzensulfonyl azide and p-dodecylbenzensulfonyl azide, are particularly preferred.
- the following examples further illustrate the invention. EXAMPLE 1
- the suspension was diluted with brine and extracted 3 times with diethy1 ether; then the organic phase was dried over Na 2 SO. and vacuum distilled; residual pyridine was removed by azeotropic evaporation after adding 200 ml of benzene.
- the crude was purified through a 500 g SiO_ column, with a gradient eluent (8:2:0.1 to 3:7:0.1 petroleum ether/diethyl ether/triethyla ine) .
- the catalyst was filtered through a paper filter and thoroughly washed with methanol; the filtrate was sub- sequently evaporated to dryness at reduced pressure giving (9) in the form of a colourless oil, which was immediately used for the next step.
- the crude from hy- drogenation was dissolved in 8 ml of MeOH and added in a beaker containing 30 ml of phosphate buffer at pH 7. pH, which was monitored with a pH meter, was adjusted to 7 with the addition of 3N NaOH by means of a buret. 3.5 ml (8.55 mmoles) of a 30% TiCl 3 in 2N HCl solution were dropped, into the vigorously stirred solution within 15 minutes.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
A process for the preparatin of monolactams of formula (1) where R is acyl and of the pharmaceutically acceptable salts thereof, starting from (R) malic acid esters, through the new intermediate (3S, 4S) 3-hydrazino-4-hydroxymethyl azetidinone. Further, the conversion of (3S, 4S) 3-(benzyloxycarbonyl)amino-4-hydroxymethyl-2-azetidinone and (3S, 4S) 3-(tert-butoxycarbonyl)amino-4-hydroxymethyl-2-azetidinone into (3S, 4S) 3-(benzyloxycarbonyl)amino-4-(carbamoyloxy)-2-azetidinone and (3S, 4S) 3-(tert-butoxycarbonyl)amino-4-(carbamoyloxy)-2-azetidinone, respectively, is described.
Description
A PROCESS FOR THE PREPARATION OF 3-ACYLAMINO-4-CARBA- MOYLOXYMETHYL-2-AZETIDINONE-l-SULPHONIC ACIDS AND IN- TERMEDIATES FOR THE PREPARATION THEREOF
The present invention relates to a process for the synthesis of monobacta s of formula (1)

Further, the invention relates to intermediates, which are useful for the process, of the following formula
A1


and the inorganic and organic salts thereof.
The present invention further relates to the conversion of (11) into the well-known intermediates (3S, 4S) 3-(benzyloxycarbonylamino)-4-hydroxymethyl-2- azetidinone (13) and (3S, 4S) 3-(tert-butoxycarbonyla- mino)-4-hydroxymethyl-2-azetidinone (14), as well as to the conversion of said compounds (13) and (14) into the corresponding intermediates (3S, 4S) 3-(benzyloxycarbo- nylamino)-4-carbamoyloxymethyl-2-azetidinone (15) and (3S, 4S) 3-(tert-butoxycarbonylamino)-4-carbamoyloxyme- th l-2-azetidinone (16), the former, which is already well-known, can be converted, with well-known procedures, into monobactams (1)

PRIOR ART
The discovery of antimicrobial compounds, named monobactams or sulfazecins, which are characterized by a 2-azetidinonic structure bearing an acylamino group at the 3-position and a sulfonic acid group at the 1- position [R.B. Sykes et al. , Nature, 291, pag. 489 (1981); A. Imada et al., Nature, 291, pag. 590 (1981)] opened a wide line of research and many non-natural de¬ rivatives of said class have subsequently been prepared by synthetic route. Particularly, several monobactams of general formula (1) and the pharmaceutically acceptable salts thereof showed a remarkable antibiotic activity towards gram-negative bacteria, Pseudomonas aeruginosa included, as well as a consistent stability towards β-lactamases, which make them particularly interesting from a pharmacological point of view (WO
81/00103; WO 81/00183; WO 81/00252; EP-73061; US 4.572.801; 4.665.067; 4.673,739; 4.675.397; 4.782.147; 4.882.788; S. Kishimoto, et al. , J. Antibiot., 36, pag. 1421 (1983)). There is no convenient manner to obtain said monobactams through a microbiological route. Moreover,
it has been verified that only the compounds with (3S) configuration are active and that the cis derivatives (namely (4S)) are more active than the trans derivatives. Therefore, the preparation thereof requires a stereoselective (or a diastereospecific) , and enantioselective (or enantiospecific) synthesis, otherwise, in the event of a synthesis leading to racemic products, an optical resolution.
Some syntheses of the compounds of general formula (1) have been described. Said compounds are prepared starting from optically pure natural compounds, such as ascorbic acid (C.C. Wei, et al. , J. Org. Chem., 50, 3462 (1985)), or D-glyceraldehyde (A.K. Bose, et al., J. Chem. Soc, Chem. Commun., 161 (1986)), or aspartic acid (Y. Takahashi, et. al. , Chem. Pharm. Bull., 34, 2732 (1986)) or by 2+2 cycloaddition between imines and carboxylic derivatives in the presence of chiral promoters on one of the two substrates (S. Cardani et al., Tetrahedron, 5563 (1988); D.A. Evans, E.B. Sjogren, Tetrahedron Lett., 26, 3783 (1985); R.C. Thomas, Tetrahedron Lett., 5239 (1989)).
Object of the present invention is a totally synthetic process for the preparation of the above compounds of formula (1). The process of the invention is carried out starting from (R) malic acid esters, which are easily obtained from L-tartaric acid.
The compounds of formula (1) are obtained with the correct relative and absolute configuration by means of the process of the invention in a simple and indu- strially applicable way.
DETAILED DISCLOSURE OF THE INVENTION
Scheme 1 and Scheme 2 illustrate a preferred embo¬ diment of the invention.
In said Schemes: - X is a convenient protective group, which is compati¬ ble with reaction conditions. Said protective group can be removed at the step wherein compound (7) is obtained. The removal of X can be carried out before, after or simultaneously the removal of OR group, in reaction conditions compatible with other functional groups present in the compound.
Example of X are R R5R6Si or 4R5R6SiCH2CH2OCH2 groups, where R 4, R5 and R6 are alkyl, aryl or alkoxy groups. Examples of R 4, R5 and R6 are Ph-tBuSi; CAr 1Ar2Ar3, where Ar1, Ar2, Ar3 represent substituted or unsubstituted, optionally linked each other, aromatic residues (such as triphenylmethyl) ; CH„OCH_Ar, where Ar represents a substituted or unεubstituted aromatic residue (for example PhCH_OCH_). - Y is a C,-C- alkyl group, such as methyl, ethyl, n- propyl; methyl group being preferred.
- R is a methyl group, or a CH_Ar group, where Ar is as above defined; for example benzyl group.
- A is a tert-butoxycarbonyl or arylalkyloxycarbonyl group.
- R is an easily removable or pharmaceutically accepta¬ ble acyl group, particularly the acyl residue of O-ben- zylcarbonic, O-tert-butylcarbonic, phenylacetic, phe- noxyacetic, 2-(2-amino-4-thiazolyl)-2-(Z)-(methoxyimi- no)acetic, 2-(2-amino-4-thiazolyl)-2-(Z)-(carboxyme- thoxyimino) acetic, 2-(2-amino-4-thiazolyl)-2-(Z)-(1-
carboxy-1-methyl-ethoxyimino) cetic acids.
The compounds of formula (1) can also be in the form of pharmaceutically acceptable salts, and the compound of formula (11) can also be in the form of an hydrazinium salt.
SCHEME 1
OH OH
COY Step 1 HO. COY
YO2C< 2 2
(2) (3)-
1 Step 2


Step 10

(15):R-PhCH2OCO (1) (16) :R =(CH3)3COCO
As previously mentioned, Schemes 1 and 2 represent only one among the many other possible embodiments of
2 the process according to the invention, m fact R acyl residues can be different from the ones above mentioned.
Referring to Scheme 1, the preparation starts from a (R) malic acid ester (2), which is converted into the diol (3) (step 1) by regioselective reduction with borane and sodium borohydride, as described by S. Saito, et al., (Chem. Lett.,' 1389 (1984)).
Subsequently, the diol (3) can selectively be protected at the primary hydroxy group: reaction conditions vary according to the protective group being used. For R 4R5R6Si type groups, the protection is carried out by reacting the corresponding halides in a dipolar aprotic solvent, such as dimethylformamide or dimethyl sulfoxide, at a temperature ranging from 0°C to 70°C, preferably from 20°C to 50°C, in the presence of a base such as a tertiary amine, or pyridine or imidazole; particularly, in the case of X = Ph-t-BuSi, said reaction is preferably carried out in dimethylfor¬ mamide at 25°C, in the presence of imidazole, as described by G. Guanti, L. Banfi, E. Narisano, (Tetrahedron Lett., 30, 5507 (1989)). When X = R R5R6SiCH2CH2OCH2 or CH2OCH2Ar, the reaction is preferably carried out in a chlorinated solvent (for example, methylene chloride) in the presence of a tertiary amine (for example diiso- propylethylamine) at a temperature ranging from 0°C to the solvent boiling temperature. When X = CAr 1Ar2Ar3 , the protection is carried out
in a halogenated solvent, such as, for example, methylene chloride, by treating with the appropriate halide, in the presence of a nitrogen base (for example, pyridine) and at a temperature ranging from 0βC to the solvent boiling temperature. Particularly, when X = triphenylmethyl, the protection reaction has already been described in literature (K. Prasad, et al., Tetrahedron: Asymmetry , 307 (1990)).
The compounds of formula (4) are obtained with very good protection yield by using X = CAr 1Ar2Ar3 or X
= R 4R5R6Si, whenever R4, R5 and R6 are sufficiently bulky.
Next step consists in condensing β-hydroxyesters
(4) with a di-t-butyl- or diarylalkyl azodicarboxylate. Said transformation can be carried out by treating a compound (4) with at least two equivalents of a strong base, such as, for example, a lithium or sodium or potassium dialkylamide (for example, diisopropylamide) in an aprotic solvent, such as tetrahydrofurane or dimethoxyethane, at a temperature ranging from -78°C to 20°C, preferably from -40°C to 0°C, followed by the reaction with the azodicarboxylate, at a temperature ranging from -78°C to 0°C. Yields and diastereoselecti- vity depend on the kind of the protective group X and on reaction temperature. Good results are obtained using X = trityl, and carrying out enolate formation at -40°C and condensing between -40°C and 0CC. In said conditions, a clear prevalence of (2S, 3R) a ti-diaste- reoisomer, with diastereoisomeric ratio higher than 9:1, is reported and main diastereoisomer yield is about 50%. When X = Ph_t-BuSi, said condensation had
already been described (G. Guanti et al. , Tetrahedron Lett., 30, 5507 (1989)) and resulted in lower stereose- lectivity and with a slightly lower adduct yield.
The so obtained products (5) are isolated by chromatography or crystallization.
Next step consists in converting the esters (5) into 0-alkylhydroxamates (6). Said conversion can be effected in two ways. A) A two-step way. The first step consists in transfor- ming the ester group into an acid one. This can be accomplished by treating with an excess of a 0,1 to 2 N alkali hydroxide solution, such as lithium, sodium, potassium, etc, hydroxide in water, in the presence of one or more organic water-miscible cosolvent, such as methyl or ethyl alcohol, tetrahydrofurane, dioxane, dimethylformamide, acetonitrile, etc, or in an alcoholic solvent, such as methyl or ethyl alcohol, at a temperature ranging from -20°C to 60°C, preferably from 0°C to 40°C. The best results are obtained when X = CAr1Ar2Ar3 or X = R4R5R6SiCH2CH2OCH or CH2OCH2Ar. The so obtained carboxylic acids can be isolated by extraction or by treating with an appropriate ion exchange resin and subsequent by purificating by crystallization or chromatography. Alternatively, the basic solution containing the carboxylic acid salts can be used as such for the next reaction.
The second step consists in coupling the so obtained acids (or the salts thereof) with the appropriate O-alkylhydroxylamine (or a hydroxylammonium salt thereof). Said step can be carried out both starting from the carboxylic acids and starting from
the crude carboxylate solution, which has previously been obtained by the above saponification.
Starting from the carboxylic acids, the coupling can be executed in an aqueous solution containing an appropriate water-soluble cosolvent, such as tetrahy- drofurane, dimethylformamide or acetonitrile, keeping pH between 4 and 7, according to the group X present, by reacting with the appropriate O-alkylhydroxylamine (or a salt thereof) (for example 1-2 equivalents), in the presence of a condensing agent, such as, for example, N,N'-dicyclohexylcarbodiimide (DCC) or l-(3- diaminopropyl)-3-ethylcarbodiimide (WSC) (1-3 equiva¬ lents). Otherwise, the coupling can also be carried out activating the purified carboxylic acids by reaction with dicyclohexylcarbodiimide and N-hydroxybenzotria- zole in a dipolar aprotic solvent, such as acetoni¬ trile, dioxane, tetrahydrof rane or dimethylformamide and reacting the so activated adducts in the same solvent with the appropriate O-alkylhydroxylamine or a hydroxylammonium salt thereof (in the latter case also adding an equivalent amount of a tertiary amine, such as, for example, triethylamine) .
Further, the same coupling can directly be performed by using the crude alkali carboxylate solution, which has been obtained, as above described, from C1-C3 alkyl ester saponification. After acidifying to a pH between 3 and 8, the coupling can be performed by reacting with the appropriate O-alkylhydroxylamine (or a hydroxylammonium salt thereof) in the same solvent wherein saponification was carried out, optionally integrated with the addition of water or of
appropriate organic cosolvents, such as dimethylformam- mide or tetrahydrofurane, in the presence of a condensing agent such as, for example, N,N'-dicy- clohexylcarbodiimide (DCC) or l-(3-diaminopropyl)-3- ethylcarbodiimide (WSC) (1-3 equivalents). For example, when X = triphenylmethyl, the coupling is directly performed on the lithium carboxylate dissolved in a te- trahydrofurane-water mixture using O-benzylhydrox la- mine, lithium hydroxide as the base, WSC as the condensing agent. A 50-60% yield is obtained.
B) A one-step way. The esters (5) can be transformed into hydroxamates (6) in a single step by reacting them with the adduct which has been obtained by mixing the appropriate O-alkylhydroxylamine with trimethylaluminum in an aprotic solvent, such as, for example, tetrahy¬ drofurane, at a temperature ranging from -20βC to the solvent boiling temperature (preferably from 0°C to 20°C) .
Next step, which consists in transforming hydroxa- mates (6) into β-lactams (7), can be performed in an appropriate organic solvent (for example tetrahydrofu¬ rane, acetonitrile or dimethylformamide) preferably by treating with triphenylphosphine and a dialkyl azodi¬ carboxylate (such as a diethyl or diiεopropyl azodicar- boxyl te) , or by treating with triphenylphosphine, carbon tetrachloride and triethylamine at a temperature ranging from 0°C to 60°C (preferably from 20°C to 30°C). Alternatively, the same transformation can be carried out by converting the alcohol into an alkansul- fonyl derivative by treating, for example, with metha- nesulfonyl chloride in pyridine, followed by treatment
with bases, such as sodium hydrogencarbonate or sodium carbonate in dipolar aprotic solvents, such as acetone, dioxane, etc. The products (7) are purifiable by means of extraction, chromatography or crystallization. For example, when X = triphenylmethyl and R = benzyl, the conversion, which is carried out with diethyl azodicarboxylate and triphenylphosphine in te- trahydrofurane at room temperature, occurs with very good yields (about 95%). The conversion of β-lactams (7) into compounds (10) can be accomplished in several ways. The choice of the method to be used depends on the nature of the X and R groups.
In fact, in some cases it is convenient to remove the X protective group before R 0 group; in other cases it is convenient to act contrarily; finally, in some cases it is possible to remove the two groups at the same time. When X = R4R5R6Si or R4R5R6SiCH2CH2OCH2 (for example Ph2t-BuSi, Me3SiOCH2CH_OCH2) , protective group removal can be performed both before and after removing OR group (preferably before) , by treatment with a fluoride (for example tetr -n-butylammonium fluoride) in a solvent, such as tetrahydrofurane or dioxane. When
X = CAr 1Ar2Ar3, protective group removal is preferably performed before OR group removal to give the derivatives (8). Said unblocking can be made, for example, by treating with a strong protic acid (such as a sulfonic acid or trifluoroacetic acid) in methyl or ethyl alcohol at a temperature ranging from 0°C to 60°C, or by heating, at a temperature ranging from 20CC to 100°C, in an acetic acid-water mixture. When X =
CH-OCH-Ar and R = CH_Ar both groups can be removed at the same time to give the product (9) directly. When X is above (CB_2OCH2Ar) and R is methyl, then X group is removed before R group to give (8). In both cases, de- protection can be performed by hydrogenating in an appropriate solvent (for example, methyl, ethyl, n- propyl, iso-propyl alcohol or ethyl acetate) in the presence of a transition metal catalyst, such as palladium (for example, pure or supported on carbon or barium sulfate) or platinum (for example, pure or in the form of dioxide), at a pressure ranging from 1 to 10 atmospheres.
The products (8) (R = CH-Ar) can be converted into the compound (9) by hydrogenating in an appropriate solvent (for example, methyl, ethyl, n- propyl, iso-propyl alcohol or ethyl acetate) in the presence of a transition metal catalyst, such as palladium (for example, pure or supported on carbon or barium sulfate) or platinum (for example, pure or in the form of dioxide), at a pressure ranging from 1 to 10 atmospheres. For example, very high yields are obtained by operating in methyl alcohol at 1 atmosphere pressure and using 10% palladium on carbon aε catalyst. The so obtained hydroxamic acid (9) requires no further purification, but it can directly be used for the next step, which consists in reducing it to give the azetidinone (10).
Said transformation can be performed, for example, by adding a aqueous hydrochloric acid titanium trichloride solution to the substrate (9), which is dissolved in a water/alcohol system (for example
water/methanol) at a pH between 3 and 10 (preferably 1 ) , maintained with buffer solutions, or by simultaneously dropping an alkali hydroxide solution. In said conditions, good yields, about 50-60%, are obtained.
When R = methyl, the products (8) can directly be converted into (10) in a single step, by treating them with alkali metals (for example, sodium) in liquid ammonia, optionally in the presence of organic cosolvents.
Finally, when R = methyl and X = ArCH2OCH2 or = Ar 1Ar2Ar3C, (7) can also be converted directly into
(10) in a single step, by treating (7) with alkali metals (for example, sodium) in liquid ammonia, optionally in the presence of organic cosolvents. The compound (10) can be purified by chromatography or crystallization.
Next step consists in converting (10) into the key intermediate (11) and can be carried out by treating (10) with a strong carboxylic acid, such as, for example, trifluoroacetic or formic acid. A cosolvent, which is compatible with reaction conditions, for example, methylene chloride, can optionally be used. Said reaction can be carried out with very good yields, by stirring for 1 hour a 1:1 trifluoroacetic acid: methylene chloride solution of (10), at a temperature ranging from 0°C to 25°C. The so obtained product (11) can be used wether as such for the next reaction, or purified by the conventional techniques (crystal- lization, ion exchange chromatography, etc.).
The product (11) and the hydrazinium salts thereof
(for example, chloride, acetate, trifluoroacetate, formate) are new, therefore they are a further object of the present invention.
(11) can be converted into the known intermediate (12) by reacting a hydrazinium salt thereof with hydrogen in the presence of catalysts, such as platinum dioxide or Raney® Nickel, at a pressure ranging from 1 to 200 atmospheres and, depending on the used catalyst, in water, alcohol (for example, methanol or ethanol) or water-alcohol mixtures. Also (12) can be wether purified or directly reacted, as crude, with benzyloxy- carbonyl chloride or with di-t-butyl dicarbonate to give the known products (13) and (14). This last conversion can be performed by treating with the appro- priate acylating agent in an anhydrous solvent, such as dimethylformamide or acetonitrile and in the presence of a base, such as a tertiary amine (for example, triethylamine) ; otherwise, and preferably, in an aqueous solution kept at a pH between 8 and 10 with alkaly hydroxides (lithium, sodium or potassium) or alkali carbonates (sodium, potassium). As above stated the products (12), (13) and (14) are known even if they have been prepared through a different synthetic route (R.C. Thomas, Tetrahedron Lett., 5239 (1989)). The products (13) and (14) can be transformed into the carbamates (15) and (16), the former being a well known derivative (U.S. 499,801; S. Kishimoto, et al., J. Antibiot., 36, pag. 1421 (1983)). Said converεion iε new, therefore it is a further object of the present invention.
It can be performed by reacting (13) or (14) with
an acyl or sulfonyl isocyanate in an aprotic solvent, such as dimethylformamide or methylene chloride or te- trahydrof rane, followed by the resulting N-acyl (or N- sulfonyl) carbamate deprotection. In the case of N- chloracetylcarbamates, said deprotection can be performed by treatment with sodium or potassium N-alkyl dithiocarbamates, while, in the case of N-sulfonylcar- bamates, by treatment with sodium sulfite. Very good results (with overall yield of the two steps comprised between 50% and 75%) are obtained, for example, by carrying out the reaction with chloroacetyl isocyanate in dimethylformamide/methylene chloride at 0°C and by deprotecting the chloroacetyl urethane by reacting with sodium N-methyl dithiocarbamate. Compound (15) can be converted by means of well- known techniques (U.S. Patent Application 499,801; S. Kishimoto, et al., J. Antibiot., 36, pag. 1421 (1983)), into the products of general formula (1).
According to a further embodiment of the present invention, the above process can be alternatively carried out as far as the introduction of a ino group into 2-position of β-hydroxyeεter (4) is concerned, by electrophilic amination with other synthetic equiva¬ lents of NH? + group, such as sulfonyl azides, O-substi- tuted hydroxylamines and diazonium salts. According to the invention, sulfonyl azides, especially p-toluensul- fonyl azide, 2,4,6-triisopropylbenzensulfonyl azide and p-dodecylbenzensulfonyl azide, are particularly preferred. The following examples further illustrate the invention.
EXAMPLE 1
Methyl (3R) 3-hydroxy-4-(triphenylmethyl)oxybutanoate (4) (X = triphenylmethyl; Y = Me) from (3).
12.96 g (96.62 mmoles) of (3), wherein Y = Me, were dissolved in 200 ml of anhydrous methylene chloride, under nitrogen stream, and cooled to 0°C. 11.72 ml (144.93 mmoles) of pyridine and 32.32 (115.92 mmoles) of trityl chloride were added; after 15 minutes the ice bath was removed and the reaction was let to stand under stirring at r.t. for 20 hours. As the reaction resulted incomplete, 3 ml (3.71 mmoles) of pyridine and 8 g (2.8.7 mmoles) of trityl chloride were further added, and the reaction was carried out for further 3 hours. The suspension was diluted with brine and extracted 3 times with diethy1 ether; then the organic phase was dried over Na2SO. and vacuum distilled; residual pyridine was removed by azeotropic evaporation after adding 200 ml of benzene. The crude was purified through a 500 g SiO_ column, with a gradient eluent (8:2:0.1 to 3:7:0.1 petroleum ether/diethyl ether/triethyla ine) .
28.69 g (yield 79%) of product, which was crystal¬ lized from isopropyl ether/penthane to a colourless compound, were obtained. -"-H-NMR (CDC13; 200 MHz; J(Hz)): c_H 7.23-7.46 (15 H, m, trityl), 4.23 (1H, center of m, H-3), 3.68 (3H, s, OCH3), 3.17 (2H, d, J 5.4, H-4) , 2.90 (1H, d, J 4.7, OH), 2.53 e 2.57 (2H, AB portion of ABX syst., J
14.7, J^ 3.6, Jβχ 9.2, H-2).
IIRR ((cchhlloorrooffoorrmm,, ccmm"" )):: 117722!8 (ester carbonyl)
M.P. 71.8-72.6CC (iso-propyl ether/penthane)
[o(]D 13 = +5.48° (c 1.99, chloroform).
Elemental analysis for C24H240. found: C 76.54%; H 6.27%; 0 17.19%; calculated: C 76.57%; H 6.43%; 0 17.00%. EXAMPLE 2
Methyl (2S, 3R) 2-[N, '-bis-(tert-butoxycar- bonyl)hydrazino]-3-hydroxy-4-(triphenylmethyl)oxybuta- noate (5) (X = triphenylmethyl; Y = Me) from (4).
9.16 ml (65.34 mmoles) of diisopropyla ine were added, under nitrogen stream, to 80 ml of anhydrous te- trahydrofurane (THF) and the solution was cooled down to -18βC; 38.29 ml (61.26 mmoles) of n-BuLi (1.6 M hexane solution) were subsequently dropped and the solution was kept under stirring at the same temperature for 20 minutes. The reaction was then cooled to -40CC and 7.687 g (20.42 mmoles) of (4), obtained in example 1, previously dissolved in 20 ml of THF, were added. After 5 minutes, the reaction vessel was let reach 0°C and let under stirring for 30 minutes. After cooling again to -20°C, di-tert-butyl azodicarboxylate, previously dissolved in 20 ml of THF, was added and the system was kept under stirring, letting the temperature to raise till 0°C. The reaction was stopped at 0°C, by adding 7.5 ml of glacial acetic acid. After 5 minutes, the suspension was diluted with a NH.C1 saturated solution and brine and extracted with diethyl ether.
The organic phase, previously dried over Na„SO. , was concentrated under reduced pressure, to give 17.53 g of a yellow oil, which was passed through a 350 g
SiO_ chromatographic column, eluting with a 8:2:0.03 to
5:5:0.03 petroleum ether/diethyl ether/triethylamine mixture. 5.82 g (yield 48%) of (5) were obtained. 1H-NMR (DMSO-dg, 80 MHz; 130°C; J(Hz)): C-H 8.07 (IH, s broad, NH) , 7.03-7.67 (15H, m, trityl), 4.86 (IH, d, J 6.5, H-2), 4.00-4.35 (IH, m, H-3), 3.61 (3H, s, OCH_3) , 3.22 (2H, d, J 5.3, H-4) , 1.44*[9H, s, N-(Boc) ], 1.40*[9H, s, NH-(Boc) ]. IR (chloroform, cm" ): ^_ 1731 (ester carbonyl).
[o(]D 13 = +18.73° (c 2.04, chloroform).
Elemental analysis for C 3 H 2N2°8' f°un<3 c 67.04%; H
6.93%; N 4.74%; 0 21.29%; calculated: C 67.31%; H
6 . 98%; N 4 . 62%; 0 21. 1%.
* interchangeable signals
EXAMPLE 3 Benzyl (2S, 3R_) 2-[N,N'-bis-(tert-butoxycarbo- nyl)hydrazino]-3-hydroxy-4-(triphenylmethy1)oxybutane- hydroxamate (6) (X = triphenylmethyl) from (5).
2.90 g (4.78 mmoles) of ester (5), obtained in example 2, were dissolved in 20 ml of freshly distilled THF, 30 ml of distilled water were then added and the system was cooled to 0°C; 31 ml (15.3 mmoles) of 0.5 N LiOH aqueous solution were dropped within 15' . The suspension was kept under vigorous stirring at r.t. for 7 hours. The reaction was cooled to 0°C and pH was adjusted to 6 with IN HCl; 915 mg (5.74 mmoles) of 0- benzylhydroxylamine were added and pH was adjusted to 6, adding a 0.5 N LiOH aqueous solution. Finally, 1.833 g (9.56 mmoles) of WSC (l-(3-diaminopropyl)-3-ethylcar- bodiimide) were added and the system was let under stirring at r.t. for 20 hours. The aqueous phase was saturated with NaCl, then extracted with ethyl acetate.
The organic extract was dried over Na2SO. and evapora¬ ted to dryness, giving 3.56 g of crude in the form of a foam. The crude was passed through a 150 g SiO_ chroma- tographic column, eluting with a 6:4:0.03 to 4:6:0.03 petroleum ether/diethyl ethertriethy1amine mixture. 1.994 g (yield 60%) were obtained.
1H-NMR (DMSO-dg, 80 MHz, 130°C, J(Hz)): rfR 7.24-7.47 (2OH, m, trityl and benzyl aro atics), 4.79 (2H, s, OCH2Ph), 4.55 (IH, d, J 6.2, H-2) , 4.11-4.31 (IH, m, H- 3), 3.19-3.26 (IH, m, H-4) , 1.40*(9H, s, N-(Boc)), 1.37*(9H, s, NH-(BQC)) .
IR (chloroform, cm ): _> 1719, 1685, 1673 (carbonyl; hydroxamate and Boc) .
[α[]D 13 = -6.27° (c 2.41, chloroform).
* interchangeable signals
EXAMPLE 4
(3S, 4S) l-benzyloxy-3-[N,N'-bis-(tert-butoxycarbo- nyl)hydrazino3-4-( riphenylmethyl)oxymethy1-2-azetidi- none (7) (R = benzyl) from (6). 2.950 g (4.67 mmoles) of (6), obtained in example 3, were dissolved in 25 ml of anhydrous THF and added, in nitrogen stream and r.t., to 1.837 g (7.01 mmoles) of triphenylphosphine and 1.10 ml (6.99 mmoles) of diethyl azodicarboxylate. The yellow solution was let under stirring for 15 hours; the solvent was then vacuum distilled and the residue was directly passed through a 200 g Si02 chromatographic column, eluting with a 7:3 to 1:1 petroleum ether/diethyl ether mixture. 2.730 g (95% yield) of a colourless foam were obtained.
■"-H-NMR (DMSO-dg, 80 MHz, 131°C, J(Hz)): H 8.45 (IH, s
broad, NH) , 7.24-7.50 (20H, , trytil and benzyl aromatics), 4.99 (2H, s, OCH_2Ph), 4.84 (IH, d, J 5.6, H-3), 4.16 (IH, m center, H-4) , 3.45-3.62 (2H, , CH2OH), 1.37*(9H, s, N-(Boc)), 1.27*(9H, s, NH-(Boc) ) . IR (chloroform, cm"1): "_ 1783 (β-lactam carbonyl), 1722 (Boc carbonyl).
[CθD 16 = +5.07° (c 1.96, chloroform).
Elemental analysis for C4oH45N3°7' found: C 69.95%; H 6.75%; N 6.31%; 0 16.99%; calculated: C 70.67%; H 6.67%; N 6.18%; 0 16.47%.
* interchangeable signals
EXAMPLE 5
(3S, 4S) l-benzyloxy-3-[N,N' -bis-(tert-butoxycarbo- nyl)hydrazino]-4-hydroxymethyl-2-azetidinone (8) (R = benzyl) from (7).
1.045 g (1.54 mmoles) of (7) were dissolved in 20 ml of anhydrous methanol, under nitrogen stream; the solution was cooled to 0°C and 292 mg (1.54 mmoles) of p-toluensulfonic acid were added. After 5 minutes, the ice bath was removed and the system was let to stand under stirring at r.t. for 2.5 hours. Acid excess was neutralized with a NaHCO- saturated solution, then the solution was concentrated to a small volume. The residue was diluted with brine and extracted with ethyl acetate. The organic phase was dried over Na_SO. and the solvent was vacuum distilled. 1.067 g of crude were passed through a 40 g SiO_ chromatographic column, using a 1:1 to 3:7 petroleum ether/diethyl ether mixture. 458 mg (70% yield) of a white foam were obtained 1 H-NMR (DMSO-dg, 80 MHz, 129°C, J(Hz)): cTH 8.46 (IH, ε
broad, NH) , 7.28-7.40 (5H, m, benzyl aromatic), 6.91 (IH, s broad, OH), 5.00 (2H, s, OCH_2Ph) , 4.92 (IH, d, J 5.4, H-2), 4.01-4.14 (IH, m, H-3), 3.62-3.99 (2H, m,
*
CH2-OH) , 1.44 ( 9H, s , N- (Boc ) ) , 1.40 ( 9H, s , NH- (Boc ) ) . t D 15 = +7 * 65° (° 2 - 01 chloroform) .
Elemental analysis for C iH3iN3°7 ∑ found: C 57 .28%; H 6.96%; N 9.48%; O 26.28%; calculated: C 57 . 65%; H
7.14%; N 9.6%; 0 25.6%.
* interchangeable signals EXAMPLE 6
(3S, 4S) 3-[N,N*-bis-(tert-butoxycarbonyl)hydrazino]-4- hydroxymethy1-2-azetidinone (10) from (8) through in¬ termediate (9) .
175 mg of 10% Pd/carbon were added to a solution of 746 mg (1.71 mmoles) of (8), obtained in example
(5), in 20 ml of methanol. The suspension was hydroge- nated for 1 hour at r.t. and at atmospheric pressure.
The catalyst was filtered through a paper filter and thoroughly washed with methanol; the filtrate was sub- sequently evaporated to dryness at reduced pressure giving (9) in the form of a colourless oil, which was immediately used for the next step. The crude from hy- drogenation was dissolved in 8 ml of MeOH and added in a beaker containing 30 ml of phosphate buffer at pH 7. pH, which was monitored with a pH meter, was adjusted to 7 with the addition of 3N NaOH by means of a buret. 3.5 ml (8.55 mmoles) of a 30% TiCl3 in 2N HCl solution were dropped, into the vigorously stirred solution within 15 minutes. In the meantime, pH was maintained the nearest to 7 with 3N NaOH additions (about 11 ml). At the end of TiCl, additions, the system was let to
stand under stirring, at r.t., for 2 hours. The aqueous system was saturated with NaCl, pH was adjusted to 8.5 and the stirring was continued for 1 day further, in order to allow the release of the product _by Ti(III). The suspension was filtered on Celite* and the aqueous phase was extracted with ethyl acetate. The organic phase was dried over Na2S04 and the solvent was vacuum distilled.
The crude was passed through a 30 g Si02 chromato- graphic column, using a 8:2 ethyl acetate/petroleum ether mixture. 348 mg (63% yield; two steps) of a white solid, which crystallized spontaneously, were obtained. 1H-NMR (DMSO-dg, 80 MHz, 130°C, J(Hz)): Note: the spectrum gave poor resolution even at this temperature and some peaks resulted rather broadened; however, the spectrum was easier understandable when recorded in the presence of 5% D2 0' & n 4.90 (IH, m center, X part of ABCX syst., H-3), 3.52-3.79 (3H, m, ABC part of ABCX syst., H-4 + CH2OH), 1.44 (18H, s, N(Boc) + NH(Boc)). IR (chloroform, cm ) : _) 1770 (β-lactam carbonyl), 1722 (Boc carbonyl). [o(]D = +16.4° (c 1.52, methanol).
Elemental analysis for ci H 25 N 3°6' found: c 50.58%; H 7.41%; N 12.72%; 0 29.29%; calculated: C 50.75%; H 7.6%; N 12.68%; 0 28.97%.
* interchangeable signals
EXAMPLE 7
(3S, 4S) 3-hydrazino-4-hydroxymethyl-2-azetidinone (11) from (10). 95.6 mg (288.5 μmoles) of (10), obtained in example 6, were εuεpended in 1 ml of anhydrous
methylene chloride, under nitrogen stream. The suspension was cooled down to 0°C and 0.5 ml of tri- fluoroacetic acid were added and a complete dissolution was observed. After 45 minutes, the ice bath was removed and the reaction was let to stand under stirring at r.t. for 1 hour. The solvent was vacuum
_2 distilled and the residue was accurately dried at 10 mm for 24 hours, as to eliminate trifluoroacetic acid completely. The residue pale yellow oil was utilized for the following hydrogenation and for H-NMR analysis without purification.
1H-NMR (D20, 200 MHz, J(Hz)): cfH 4.64 (IH, d, J 4.6, H- 3), 3.97-4.08 (IH, m, H-4), 3.80-3.91 (2H, m, CH_2OH) .
EXAMPLE 8 (3S, 4S) 3-amino-4-hydroxymethyl-2-azetidinone (12) from (11).
The crude, obtained from example 7, was dissolved in 5 ml of water; 50 mg of t02 were added and hydroge¬ nation was carried out at r.t. and atmospheric pressure for 30 hours. The catalyst was filtered off on paper filter, thoroughly washing with water, then with methanol. The solvent was vacuum distilled and the resulting pale-yellow oil was used for next steps and for H-NMR analysis without purification. 1H-NMR (D20, 200 MHz, J(Hz)): cT 4.65 (IH, d, J 5.0, H-3), 4.07-4.12 (IH, m, H-4) , 3.92-4.00 (2H, m, CH_2OH) .
EXAMPLE 9 (3S, 4S) 3-(benzyloxycarbonylamino)-4-hydroxymethyl-2- azetidinone (13) from (12). The crude of example 8 was dissolved into 3 ml of IN NaHCO^ aqueous solution; 64 μl (403.0 μmoles) of
benzyl chloroformate were added and the reaction was let to stand under stirring at r.t. for 6 hours. The suspension was diluted with brine and extracted with ethyl acetate; the organic phase was dried over Na2SO. and vacuum distilled, giving 54 mg of crude, which was subsequently purified by means of colunm chromatography with a 95:5 ethyl acetate/petroleum ether mixture. 36.1 mg (40% yield; three steps) of a white crystalline solid were obtained. """H-NMR (chloroform-d, 200 MHz, J(Hz)): cFH 7.33 (5H, s broad, aromatic Cbz) , 6.81 (IH, s broad NH-1), 6.27 (IH, d, J 9.9, NH-(Cbz)), 5.14 (IH, dd, J 4.8 e 9.9, H- 3), 5.09 (2H, s, CH2-Ph), 3.80-3.88 (2H, m, CH_2OH) , 3.62-3.68 (2H, m, H-4 + OH). EXAMPLE 10
(3S, 4S) 3-(tert-butoxycarbonylamino)-4-hydroxymethyl- 2-azetidinone (14) from (12).
The crude of example 8 was dissolved in 2 ml of anhydrous dimethylformamide, under nitrogen stream, and 115 ml (810 μmoles) of triethylamine and 320 μl (1.35 mmoles) of di-tert-butyl dicarbonate were further added. The reaction system was let to stand for 3 days at r.t. At the end of this time, brine was added followed by extraction with ethyl acetate. The organic phase was dried over Na~ 2S04. and the solvent was vacuum distilled. 63.7 mg of crude were obtained. The subse¬ quent chromatography with a 95:5 ethyl acetate/methanol mixture gave 17.3 mg (30% yield; three steps) of a white solid. """H-NMR (DMSO-dg, 200 MHz, J(Hz)): dR 8.21 (IH, s, NH- 1), 7.31 (IH, d, J 9.8, NH-(Boc)), 4.81 (IH, dd, J 4.1
and 9.8, H-3) , 3.32-3.67 (3H, m, H-4 + CH_2OH) , 1.39
(9H, s, NH-(Boc) ) .
EXAMPLE 11
(3S, 4S) 3-(benzyloxycarbonylamino)-4-carbamoyloxyme- thyl-2-azetidinone (15) from (13).
77.4 mg (309 μmoles) of (13), obtained in example
10, were dissolved in 3.5 ml of a 6:1 methylene chloride/dimethylformamide mixture, under nitrogen stream. The solution was cooled to 0°C and 53 μl (619 μmoles) of chloroacetyl isocyanate were added; the reaction was complete after 1.5 hours. 2.39 mg (1.85 mmoles) of sodium N-methyl dithiocarbamate, previously dissolved into 2 ml of water, were added and the solution was maintained for 4 hours under vigorous stirring, until complete reaction. The aqueous phase was saturated with sodium chloride and extracted with a
85:15 chlorform/methanol mixture. The organic phase was dried over Na2-SO4. and the solvent was vacuum distilled.
The crude was purified through a chromatographic column with a 95:5 ethyl acetate/methanol mixture. 67.9 mg
(75% yield; two steps) of a white solid were obtained.
"""H-NMR (DMSO-dg, 200 MHz, J(Hz)): c5" H 8.39 (IH, s, NH-
1), 8.00 (IH, d, J 9.5, NH-(Cbz)), 7.36-7.40 (5H, m,
Cbz aromatic), 6.56 (2H, s broad, NH_2), 5.01 and 5.10 (2H, AB-system, J 12.5, CH_2-Ph), 4.96 (IH, dd, J 4.8 and 9.5, H-3), 4.10-3.93 (2H, , CH20H) , 3.80-3.90 (IH, m, H-4).
EXAMPLE 12
(3S, 4S) 3-(tert-butoxycarbonylamino)-4-carbamoyloxy- methyl-2-azetidinone (16) from (14).
The reaction was carried out under the same condi-
tions described in example 11 and led to the desired product with 63% yield.
"""H-NMR (DMSO-dg, 200 MHz, J(Hz)): cT 8.34 (IH, s, NH-
1), 7.56 (IH, d, J 9.7, NH-(Boc)), 6.55 (2H, s broad,
NH2), 4.90 (IH, dd, J 5.3 and 9.7, H-3), 3.91-4.12 (2H, m, CH2OH), 3.76-3.87 (IH, m, H-4), 1.40 (9H, s, NH-
(Boc) ) .
[0UD 18 » +56.5° (c 0.75, methanol)
Claims
(3S, 4S) azetidinones of formula

wherein: A 1, A2, A3, which are the same or different, are hydrogen or nitrogen and oxygen protective groups, and
A 4 is hydrogen, hydroxy, or OR1 group, where R1 is a methyl or an arylalkyl group; and the organic or inorganic salts thereof, as intermediates.
2. A compound according to claim 1 of formula (11)

3. A process for the preparation of (3S, 4S) 3-hydra- zino-4-hydroxymethyl-2-azetidinone (11), which consists in a) condensing (3R) 3-hydroxyesters of formula (4) OH xo.
(4)
wherein X is a protective group selected from the group consiεting of silyl, triarylmethyl or aryloxymethyl, Y is a .-C3 alkyl group, with an azodicarboxylate of formula
A - N = N - A wherein A is a tert-butoxycarboxylate or an arylalkoxy- carbonyl group; b) converting (2S, 3R) 2-N,N'-bis-(A)hydrazino-3- hydroxyesterε of formula (5)
OH
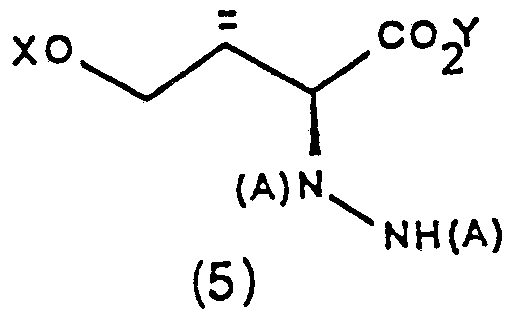
wherein X, Y and A have the above meanings, into the corresponding hydroxamates of formula (6)

(A)N
NH(A) (6)
wherein X and A have the above meanings, R is methyl or arylalkyl; c) cyclizing εaid hydroxamateε (6) into (3S, 4S) azeti¬ dinone s of formula (7) 
wherein X, A and R have the above meanings; d) removing X and R groups and reducing the resulting N-OH group to a NH group; e) removing A groups.
4. A process according to claim 3, characterized in that step a) is carried out by treating hydroxyesters (4) at a temperature ranging from -78βC to +20°C, with at least 2 equivalents of strong base in aprotic solvents and reacting the resulting enolates with an azodicarboxylate at a temperature ranging from -78°C to 0°C.
5. A process according to claim 3, characterized in that step b) is carried out by hydrolizing esters (5) with alkali hydroxydes , at a temperature ranging from 0°C to 60°C, in a system formed by water and a water- miscible solvent, or in an alcoholic system, and reacting the obtained acids with a hydroxylamine of formula NH^OR , where R has the above meanings, in the presence of condensing agents at a temperature ranging from 0°C to 40°C, in aqueous solvent.
6. A process according to claim 3, characterized in that step b) is carried out by reacting esters (5) directly with the adduct obtained from an hydroxylamine of formula NH2OR , where R has the above meanings, with trinethylaluminum in an aprotic solvent, at a tern- perature ranging from -20°C to the solvent boiling tem¬ perature.
7. A process according to claim 3, characterized in that step c) is carried out by reacting hydroxamates (6) with triphenylphosphine and a dialkyl azodicarboxy¬ late in an aprotic solvent, at a temperature ranging from 0°C to 40°C.
8. A process according to claim 3, characterized in that εtep d) , whenever X iε a protective group of ary- lalkylsilyl type, is carried out by removing, in any order X group with fluorides in a solvent selected from the group consisting in tetrahydrofurane, dioxane; and R group with hydrogenolysis on palladium or platinum, and subjecting the azetidinones (9) to the reduction of the OH group with TiCl3 in aqueous CH3OH.
9. A process according to claim 3, characterized in that step d) , whenever X is a protective group of triarylmethyl type and R is arylalkyl, is carried out by removing X wether with protic strong acids in alcoholic solvent at a temperature ranging from 0βC to 60βC, or with aqueous acetic acid at a temperature ranging from 20°C to 100°C, and subsequently removing R by hydrogenolysis on palladium or platinum, and subjecting the azetidinones (9) to the reduction of the OH group with TiCl_ in aqueous CH-OH.
10. A process according to claim 3, characterized in that step d) , whenever X is arylmethoxymethyl and R is arylalkyl, is carried out by contemporaneously removing X and R by hydrogenolysiε on palladium or platinum, and subjecting the azetidinones (9) to the reduction of the OH group with TiCl_ in aqueous CH,OH.
11. A process according to claim 3, characterized in that step d) , whenever X is a protective group of silyl type and R is methyl, is carried out by removing X by treating with fluoride in a solvent selected from the group consisting of tetrahydrofurane, dioxane and sub¬ sequently reducing the intermediates (8) directly to (10) with alkali metals in liquid ammonia.
12. A process according to claim 3, characterized in that step d) , whenever X is triarylmethyl or aryloxy- methyl and R is methyl, is carried out by reducing the intermediates (7) directly to (10) with alkali metals in liquid ammonia.
13. A process according to claim 3, characterized in that step e), whenever A is tert-butoxycarbonyl, is carried out with a strong carboxylic acid, optionally in the presence of an inert solvent, at a temperature ranging from 0βC to 25°C.
14. A process according to claim 3, characterized in that stepε d) and e), whenever A is arylalkyloxycar- bonyl, are carried out at the same time, without isolating the intermediate (10).
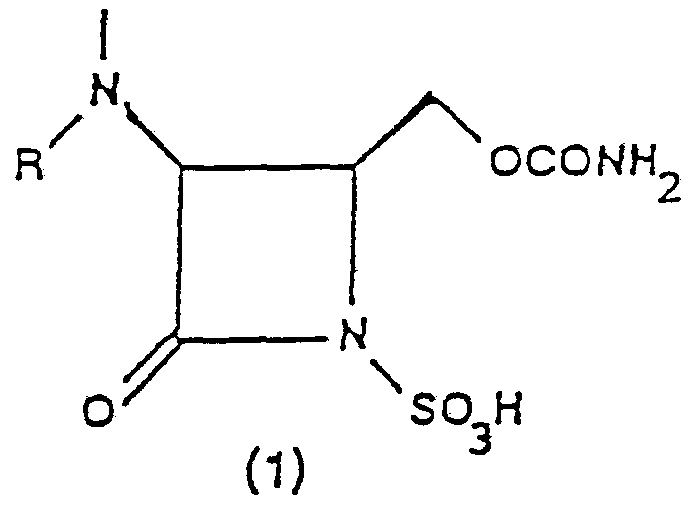
15. A process for the preparation of monobactams of formula (1)
H
where R represents an easily removable or pharmaceuti- cally acceptable acyl reεidue, conεisting in: a) converting (3S, 4S) 3-hydrazino-4-hydroxymethyl-2- azetidinone (11)

into (3S, 4S) 3-amino-4-hydroxymethyl-2-azetidinone (12)
H

b) acylating (12) to the corresponding 3-N-acylamino-2- azetidinones; c) carbamoylating to the corresponding 3-N-acylamino-2- carbamoyloxymethyl-2-azetidinoneε; d) sulfamating 3-N-acylamino-4-carbamoyloxymethyl-2- azetidinones to compounds of formula (1).
16. A process according to claim 15, characterized in that step a) is carried out by subjecting (11), or a hydrazinium salt thereof, to catalytic hydrogenation on Pt02 or Ni Raney*^, at a presεure ranging from 1 to 200 atmospheres .
17. A process according to claim 15, characterized in that step b) is carried out by acylating the amino- derivative (12) with activate derivatives of R-OH acids, where R iε as above defined.
18. A process according to claim 15, characterized in that step c), whenever R is benzyloxycarbonyl or tert- butoxycarbonyl, is carried out by treating 3-acylamino- 2-azetidinones with an acyl or sulfonyl isocyanate in aprotic solvents and by deprotecting the so obtained N- acyl or N-sulfonyl carbamates with alkali metal N- alkyldithiocarbamates or alkali sulfites, respectively.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI91A000255 | 1991-02-01 | ||
| ITMI910255A IT1244699B (en) | 1991-02-01 | 1991-02-01 | PROCESS FOR THE PREPARATION OF 3 ACYLAMINO-4- CARBAMOYLOXY-METHYL-2-AZETIDINONE-L-SULPHONIC ACIDS AND INTERMEDIATES FOR THEIR PREPARATION |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1992013837A1 true WO1992013837A1 (en) | 1992-08-20 |
Family
ID=11358378
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1992/000175 Ceased WO1992013837A1 (en) | 1991-02-01 | 1992-01-28 | A process for the preparation of 3-acylamino-4-carbamoyloxymethyl-2-azetidinone-1-sulphonic acids and intermediates for the preparation thereof |
Country Status (3)
| Country | Link |
|---|---|
| AU (1) | AU1187392A (en) |
| IT (1) | IT1244699B (en) |
| WO (1) | WO1992013837A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6982251B2 (en) | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| US7030106B2 (en) | 2001-01-26 | 2006-04-18 | Schering Corporation | Sterol absorption inhibitor compositions |
| US7053080B2 (en) | 2001-09-21 | 2006-05-30 | Schering Corporation | Methods and therapeutic combinations for the treatment of obesity using sterol absorption inhibitors |
| US7056906B2 (en) | 2001-09-21 | 2006-06-06 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| US7071181B2 (en) | 2001-01-26 | 2006-07-04 | Schering Corporation | Methods and therapeutic combinations for the treatment of diabetes using sterol absorption inhibitors |
| US7132415B2 (en) | 2001-09-21 | 2006-11-07 | Schering Corporation | Methods and therapeutic combinations for the treatment of xanthoma using sterol absorption inhibitors |
| US7192944B2 (en) | 2003-03-07 | 2007-03-20 | Schering Corp. | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7208486B2 (en) | 2003-03-07 | 2007-04-24 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7235543B2 (en) | 2003-03-07 | 2007-06-26 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7417039B2 (en) | 2001-01-26 | 2008-08-26 | Schering Corporation | Use of substituted azetidinone compounds for the treatment of sitosterolemia |
| US7459442B2 (en) | 2003-03-07 | 2008-12-02 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7560449B2 (en) | 2002-11-06 | 2009-07-14 | Schering Corporation | Methods and therapeutic combinations for the treatment of demyelination |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0093376A2 (en) * | 1982-04-30 | 1983-11-09 | Takeda Chemical Industries, Ltd. | 1-Sulfo-2-azetidinone derivatives, their production and use |
| EP0111326A1 (en) * | 1982-12-09 | 1984-06-20 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Process for the manufacture of chiral azetidinones |
| EP0411541A2 (en) * | 1989-08-02 | 1991-02-06 | Consiglio Nazionale Delle Ricerche | Process for preparing monobactames and their intermediate product |
-
1991
- 1991-02-01 IT ITMI910255A patent/IT1244699B/en active IP Right Grant
-
1992
- 1992-01-28 AU AU11873/92A patent/AU1187392A/en not_active Abandoned
- 1992-01-28 WO PCT/EP1992/000175 patent/WO1992013837A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0093376A2 (en) * | 1982-04-30 | 1983-11-09 | Takeda Chemical Industries, Ltd. | 1-Sulfo-2-azetidinone derivatives, their production and use |
| EP0111326A1 (en) * | 1982-12-09 | 1984-06-20 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Process for the manufacture of chiral azetidinones |
| EP0411541A2 (en) * | 1989-08-02 | 1991-02-06 | Consiglio Nazionale Delle Ricerche | Process for preparing monobactames and their intermediate product |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6982251B2 (en) | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| US7417039B2 (en) | 2001-01-26 | 2008-08-26 | Schering Corporation | Use of substituted azetidinone compounds for the treatment of sitosterolemia |
| US7030106B2 (en) | 2001-01-26 | 2006-04-18 | Schering Corporation | Sterol absorption inhibitor compositions |
| US7071181B2 (en) | 2001-01-26 | 2006-07-04 | Schering Corporation | Methods and therapeutic combinations for the treatment of diabetes using sterol absorption inhibitors |
| US7612058B2 (en) | 2001-01-26 | 2009-11-03 | Schering Corporation | Methods for inhibiting sterol absorption |
| US7056906B2 (en) | 2001-09-21 | 2006-06-06 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| US7132415B2 (en) | 2001-09-21 | 2006-11-07 | Schering Corporation | Methods and therapeutic combinations for the treatment of xanthoma using sterol absorption inhibitors |
| US7053080B2 (en) | 2001-09-21 | 2006-05-30 | Schering Corporation | Methods and therapeutic combinations for the treatment of obesity using sterol absorption inhibitors |
| US7560449B2 (en) | 2002-11-06 | 2009-07-14 | Schering Corporation | Methods and therapeutic combinations for the treatment of demyelination |
| US7378518B2 (en) | 2003-03-07 | 2008-05-27 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7368563B2 (en) | 2003-03-07 | 2008-05-06 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7235543B2 (en) | 2003-03-07 | 2007-06-26 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7208486B2 (en) | 2003-03-07 | 2007-04-24 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7459442B2 (en) | 2003-03-07 | 2008-12-02 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7368562B2 (en) | 2003-03-07 | 2008-05-06 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7192944B2 (en) | 2003-03-07 | 2007-03-20 | Schering Corp. | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| ITMI910255A0 (en) | 1991-02-01 |
| AU1187392A (en) | 1992-09-07 |
| ITMI910255A1 (en) | 1992-08-01 |
| IT1244699B (en) | 1994-08-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1992013837A1 (en) | A process for the preparation of 3-acylamino-4-carbamoyloxymethyl-2-azetidinone-1-sulphonic acids and intermediates for the preparation thereof | |
| CN103080088B (en) | Production method of intermediate compound for synthesizing medicament | |
| CN101160286B (en) | Process for the preparation of ccr-2 antagonist | |
| JP3639449B2 (en) | Method for producing 3-amino-pyrrolidine derivative | |
| US6087530A (en) | Process for preparing β-amino-α-hydroxy acid derivatives | |
| IE58243B1 (en) | A process for the preparation of optically-active 3-amino carboxylic acids | |
| US20070232806A1 (en) | Hydride reduction process for preparing quinolone intermediates | |
| US5179212A (en) | Process for the preparation of 3-pyrrolidinols and intermediates therefor | |
| US5475116A (en) | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones | |
| EA003022B1 (en) | Process for the synthesis of hiv protease inhibitors | |
| JP4294121B2 (en) | Process for producing pyridonecarboxylic acid derivatives and intermediates thereof | |
| EP0521686A1 (en) | Stereoselective production of hydroxyamide compounds from chiral a-amino epoxides | |
| KR20010042750A (en) | Method for producing enantiomer-free n-methyl-n-[(1s)-1-phenyl-2-((3s)-3-hydroxypyrrolidine-1-yl)ethyl]-2,2-diphenyl acetamide | |
| EP0992491A1 (en) | Processes for producing azetidine-2-carboxylic acid and intermediates thereof | |
| US5254681A (en) | Process for preparing monobactames and their intermediate product | |
| US8106230B2 (en) | Succinic acid diester derivative, process for production thereof, and use of the derivative in the production of pharmaceutical preparation | |
| EP1188744B1 (en) | Process for producing optically active 1H-3-aminopyrrolidine and derivatives thereof | |
| EP0787718A1 (en) | Process for preparing optically active 4-hydroxy-2-pyrrolidone | |
| WO2004099136A1 (en) | Process for producing pyrrolidine derivative | |
| JP3140698B2 (en) | Production method and purification method of 4-hydroxy-2-pyrrolidinone | |
| JPWO1998054132A1 (en) | Method for producing 3-(7-amidino-2-naphthyl)-2-phenylpropionic acid derivatives | |
| EP1109782A1 (en) | PROCESS FOR THE PRODUCTION OF N-PROTECTED AZETIDINE-2-CARBOXYLIC ACIDS (AzeOHs) | |
| US6911544B2 (en) | Process for the preparation of (S,S)-cis-2-phenyl-3-aminopiperidine | |
| US6617461B2 (en) | Process for preparation of optically active N-substituted azetidine-2-carboxylic acids | |
| JP3192791B2 (en) | Method for producing optically active DN-piperonyl-2-amino- (benzo [b] thiophen-3-yl) -propionylamide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR CA CS FI HU JP KP KR LK MG MN MW NO PL RO RU SD US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE BF BJ CF CG CH CI CM DE DK ES FR GA GB GN GR IT LU MC ML MR NL SE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: CA |