RU2793125C2 - Antibody-drug conjugate having an acid self-stabilizing binding site - Google Patents
Antibody-drug conjugate having an acid self-stabilizing binding site Download PDFInfo
- Publication number
- RU2793125C2 RU2793125C2 RU2020137091A RU2020137091A RU2793125C2 RU 2793125 C2 RU2793125 C2 RU 2793125C2 RU 2020137091 A RU2020137091 A RU 2020137091A RU 2020137091 A RU2020137091 A RU 2020137091A RU 2793125 C2 RU2793125 C2 RU 2793125C2
- Authority
- RU
- Russia
- Prior art keywords
- antibody
- drug
- group
- pharmaceutically acceptable
- drug conjugate
- Prior art date
Links
- 239000002253 acid Substances 0.000 title claims abstract description 24
- 230000027455 binding Effects 0.000 title claims abstract description 12
- 229940049595 antibody-drug conjugate Drugs 0.000 title claims description 99
- 239000000611 antibody drug conjugate Substances 0.000 title claims description 75
- 239000003814 drug Substances 0.000 claims abstract description 101
- 229940079593 drug Drugs 0.000 claims abstract description 99
- 150000001875 compounds Chemical class 0.000 claims abstract description 36
- 230000002378 acidificating effect Effects 0.000 claims abstract description 20
- 206010028980 Neoplasm Diseases 0.000 claims description 27
- 150000003839 salts Chemical class 0.000 claims description 24
- 150000001413 amino acids Chemical group 0.000 claims description 19
- 239000000427 antigen Substances 0.000 claims description 19
- 102000036639 antigens Human genes 0.000 claims description 19
- 108091007433 antigens Proteins 0.000 claims description 19
- 235000018102 proteins Nutrition 0.000 claims description 18
- 102000004169 proteins and genes Human genes 0.000 claims description 18
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical group O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 claims description 18
- 108090000623 proteins and genes Proteins 0.000 claims description 17
- 229940024606 amino acid Drugs 0.000 claims description 16
- 235000001014 amino acid Nutrition 0.000 claims description 16
- 108010038807 Oligopeptides Proteins 0.000 claims description 11
- 102000015636 Oligopeptides Human genes 0.000 claims description 11
- 230000008685 targeting Effects 0.000 claims description 11
- 125000003277 amino group Chemical group 0.000 claims description 8
- 201000011510 cancer Diseases 0.000 claims description 8
- -1 phosphorous ester Chemical class 0.000 claims description 8
- 125000006850 spacer group Chemical group 0.000 claims description 7
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 6
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 6
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 6
- 229940127089 cytotoxic agent Drugs 0.000 claims description 6
- 239000002254 cytotoxic agent Substances 0.000 claims description 6
- 239000012634 fragment Substances 0.000 claims description 6
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 5
- 125000001118 alkylidene group Chemical group 0.000 claims description 5
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical class C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 5
- 208000023275 Autoimmune disease Diseases 0.000 claims description 4
- 108010001857 Cell Surface Receptors Proteins 0.000 claims description 4
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 4
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 4
- 229960003767 alanine Drugs 0.000 claims description 4
- 108010027164 Amanitins Proteins 0.000 claims description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims description 3
- 229930126263 Maytansine Natural products 0.000 claims description 3
- 108091005804 Peptidases Proteins 0.000 claims description 3
- 239000004365 Protease Substances 0.000 claims description 3
- CIORWBWIBBPXCG-JZTFPUPKSA-N amanitin Chemical compound O=C1N[C@@H](CC(N)=O)C(=O)N2CC(O)C[C@H]2C(=O)N[C@@H](C(C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H](C(C)CC)C(=O)NCC(=O)N[C@H]1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-JZTFPUPKSA-N 0.000 claims description 3
- 125000000539 amino acid group Chemical group 0.000 claims description 3
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 3
- 229940124599 anti-inflammatory drug Drugs 0.000 claims description 3
- 125000003968 arylidene group Chemical group [H]C(c)=* 0.000 claims description 3
- 229960005261 aspartic acid Drugs 0.000 claims description 3
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 claims description 3
- 238000002360 preparation method Methods 0.000 claims description 3
- 229960001153 serine Drugs 0.000 claims description 3
- 229960004441 tyrosine Drugs 0.000 claims description 3
- 229960004295 valine Drugs 0.000 claims description 3
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 2
- 239000004471 Glycine Substances 0.000 claims description 2
- 206010061218 Inflammation Diseases 0.000 claims description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 claims description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 claims description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 2
- 239000004473 Threonine Substances 0.000 claims description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 2
- 235000004279 alanine Nutrition 0.000 claims description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 2
- 229960001230 asparagine Drugs 0.000 claims description 2
- 235000009582 asparagine Nutrition 0.000 claims description 2
- 235000003704 aspartic acid Nutrition 0.000 claims description 2
- 108010044540 auristatin Proteins 0.000 claims description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 claims description 2
- 235000010290 biphenyl Nutrition 0.000 claims description 2
- 239000004305 biphenyl Substances 0.000 claims description 2
- 125000006267 biphenyl group Chemical group 0.000 claims description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 2
- 235000018417 cysteine Nutrition 0.000 claims description 2
- 235000013922 glutamic acid Nutrition 0.000 claims description 2
- 239000004220 glutamic acid Substances 0.000 claims description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 2
- 208000026278 immune system disease Diseases 0.000 claims description 2
- 230000004054 inflammatory process Effects 0.000 claims description 2
- 229960000310 isoleucine Drugs 0.000 claims description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 2
- 229930182817 methionine Natural products 0.000 claims description 2
- 125000001624 naphthyl group Chemical group 0.000 claims description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 2
- 229960005190 phenylalanine Drugs 0.000 claims description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 2
- 125000000542 sulfonic acid group Chemical group 0.000 claims description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 2
- 239000004474 valine Substances 0.000 claims description 2
- AXKGIPZJYUNAIW-UHFFFAOYSA-N (4-aminophenyl)methanol Chemical compound NC1=CC=C(CO)C=C1 AXKGIPZJYUNAIW-UHFFFAOYSA-N 0.000 claims 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 claims 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 1
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 1
- 102000006240 membrane receptors Human genes 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 10
- 239000003153 chemical reaction reagent Substances 0.000 abstract description 7
- 239000000126 substance Substances 0.000 abstract description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 180
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 147
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 114
- 238000006243 chemical reaction Methods 0.000 description 108
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 90
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 48
- 125000005647 linker group Chemical group 0.000 description 44
- 239000000047 product Substances 0.000 description 44
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 43
- 239000000243 solution Substances 0.000 description 42
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 39
- 239000002904 solvent Substances 0.000 description 39
- 230000002829 reductive effect Effects 0.000 description 34
- 210000002381 plasma Anatomy 0.000 description 27
- 108091022873 acetoacetate decarboxylase Proteins 0.000 description 24
- 210000004027 cell Anatomy 0.000 description 24
- 238000011068 loading method Methods 0.000 description 24
- 238000004809 thin layer chromatography Methods 0.000 description 24
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 23
- 239000000562 conjugate Substances 0.000 description 23
- 238000001514 detection method Methods 0.000 description 22
- 238000004128 high performance liquid chromatography Methods 0.000 description 22
- 230000014759 maintenance of location Effects 0.000 description 21
- 239000003480 eluent Substances 0.000 description 19
- 238000000926 separation method Methods 0.000 description 18
- 238000010898 silica gel chromatography Methods 0.000 description 18
- 238000003756 stirring Methods 0.000 description 18
- 238000000034 method Methods 0.000 description 17
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 238000004458 analytical method Methods 0.000 description 15
- 238000000746 purification Methods 0.000 description 15
- 125000003396 thiol group Chemical group [H]S* 0.000 description 15
- 108090000765 processed proteins & peptides Proteins 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 238000007429 general method Methods 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 11
- 238000001727 in vivo Methods 0.000 description 11
- 238000003998 size exclusion chromatography high performance liquid chromatography Methods 0.000 description 11
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 10
- 239000003446 ligand Substances 0.000 description 10
- 229910052717 sulfur Inorganic materials 0.000 description 10
- 125000004434 sulfur atom Chemical group 0.000 description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 8
- 239000007791 liquid phase Substances 0.000 description 8
- 102000004196 processed proteins & peptides Human genes 0.000 description 8
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- NLMBVBUNULOTNS-HOKPPMCLSA-N [4-[[(2s)-5-(carbamoylamino)-2-[[(2s)-2-[6-(2,5-dioxopyrrol-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl n-[(2s)-1-[[(2s)-1-[[(3r,4s,5s)-1-[(2s)-2-[(1r,2r)-3-[[(1s,2r)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-o Chemical compound C1([C@H](O)[C@@H](C)NC(=O)[C@H](C)[C@@H](OC)[C@@H]2CCCN2C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC=2C=CC(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN3C(C=CC3=O)=O)C(C)C)=CC=2)C(C)C)OC)=CC=CC=C1 NLMBVBUNULOTNS-HOKPPMCLSA-N 0.000 description 7
- 229910001873 dinitrogen Inorganic materials 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 238000004007 reversed phase HPLC Methods 0.000 description 7
- 229960002317 succinimide Drugs 0.000 description 7
- 102000009027 Albumins Human genes 0.000 description 6
- 108010088751 Albumins Proteins 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- 229960000583 acetic acid Drugs 0.000 description 6
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 6
- 239000012362 glacial acetic acid Substances 0.000 description 6
- 238000012544 monitoring process Methods 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 108010024636 Glutathione Proteins 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 230000002776 aggregation Effects 0.000 description 5
- 238000004220 aggregation Methods 0.000 description 5
- 229960003180 glutathione Drugs 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 238000000108 ultra-filtration Methods 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229940126142 compound 16 Drugs 0.000 description 4
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 125000001072 heteroaryl group Chemical group 0.000 description 4
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000000178 monomer Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 150000003568 thioethers Chemical class 0.000 description 4
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 3
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 3
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 3
- 102000000844 Cell Surface Receptors Human genes 0.000 description 3
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 229940125807 compound 37 Drugs 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 229940127121 immunoconjugate Drugs 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 125000001041 indolyl group Chemical group 0.000 description 3
- 230000003211 malignant effect Effects 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 239000008363 phosphate buffer Substances 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 229940126586 small molecule drug Drugs 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 2
- ZYZCALPXKGUGJI-DDVDASKDSA-M (e,3r,5s)-7-[3-(4-fluorophenyl)-2-phenyl-5-propan-2-ylimidazol-4-yl]-3,5-dihydroxyhept-6-enoate Chemical compound C=1C=C(F)C=CC=1N1C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C(C)C)N=C1C1=CC=CC=C1 ZYZCALPXKGUGJI-DDVDASKDSA-M 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 2
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 2
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 2
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 2
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 description 2
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- 229940127007 Compound 39 Drugs 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 2
- 102000008072 Lymphokines Human genes 0.000 description 2
- 108010074338 Lymphokines Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 2
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940125846 compound 25 Drugs 0.000 description 2
- 229940125851 compound 27 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125878 compound 36 Drugs 0.000 description 2
- 229940126540 compound 41 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 2
- 229960005501 duocarmycin Drugs 0.000 description 2
- 229930184221 duocarmycin Natural products 0.000 description 2
- 229940049906 glutamate Drugs 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 2
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000007142 ring opening reaction Methods 0.000 description 2
- 229960001860 salicylate Drugs 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 230000003381 solubilizing effect Effects 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 231100001274 therapeutic index Toxicity 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- NBKZGRPRTQELKX-UHFFFAOYSA-N (2-methylpropan-2-yl)oxymethanone Chemical compound CC(C)(C)O[C]=O NBKZGRPRTQELKX-UHFFFAOYSA-N 0.000 description 1
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- ALBODLTZUXKBGZ-JUUVMNCLSA-N (2s)-2-amino-3-phenylpropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CC1=CC=CC=C1 ALBODLTZUXKBGZ-JUUVMNCLSA-N 0.000 description 1
- WJKGPJRAGHSOLM-LBPRGKRZSA-N (2s)-3-[(2-methylpropan-2-yl)oxycarbonylamino]-2-(phenylmethoxycarbonylamino)propanoic acid Chemical compound CC(C)(C)OC(=O)NC[C@@H](C(O)=O)NC(=O)OCC1=CC=CC=C1 WJKGPJRAGHSOLM-LBPRGKRZSA-N 0.000 description 1
- FOXRXVSTFGNURG-VIFPVBQESA-N (2s)-3-amino-2-(phenylmethoxycarbonylamino)propanoic acid Chemical compound NC[C@@H](C(O)=O)NC(=O)OCC1=CC=CC=C1 FOXRXVSTFGNURG-VIFPVBQESA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- HAWSQZCWOQZXHI-FQEVSTJZSA-N 10-Hydroxycamptothecin Chemical compound C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-FQEVSTJZSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 1
- YCQVNMKLANFADW-UHFFFAOYSA-N 2-bromoethyl diethyl phosphate Chemical compound CCOP(=O)(OCC)OCCBr YCQVNMKLANFADW-UHFFFAOYSA-N 0.000 description 1
- HEMGYNNCNNODNX-UHFFFAOYSA-N 3,4-diaminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1N HEMGYNNCNNODNX-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ZZNAYFWAXZJITH-UHFFFAOYSA-N 4-amino-3-nitrobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1[N+]([O-])=O ZZNAYFWAXZJITH-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- WGZYVHFXLPDNDL-UHFFFAOYSA-N 4-benzoyl-2-phenylpyrazole-3-carbonitrile Chemical compound C=1C=CC=CC=1C(=O)C(=C1C#N)C=NN1C1=CC=CC=C1 WGZYVHFXLPDNDL-UHFFFAOYSA-N 0.000 description 1
- MSNVESLISHTIRS-UHFFFAOYSA-N 9h-pyrrolo[2,1-c][1,4]benzodiazepine Chemical class N1=C2C=CC=CC2=CN2CC=CC2=C1 MSNVESLISHTIRS-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 description 1
- 108010032595 Antibody Binding Sites Proteins 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 229930182565 Australin Natural products 0.000 description 1
- 238000011725 BALB/c mouse Methods 0.000 description 1
- 102100021257 Beta-secretase 1 Human genes 0.000 description 1
- 101710150192 Beta-secretase 1 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 101100505076 Caenorhabditis elegans gly-2 gene Proteins 0.000 description 1
- 101100533230 Caenorhabditis elegans ser-2 gene Proteins 0.000 description 1
- 102000004225 Cathepsin B Human genes 0.000 description 1
- 108090000712 Cathepsin B Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- CKLJMWTZIZZHCS-UHFFFAOYSA-N D-OH-Asp Natural products OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 1
- SBJKKFFYIZUCET-JLAZNSOCSA-N Dehydro-L-ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(=O)C1=O SBJKKFFYIZUCET-JLAZNSOCSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- KRHAHEQEKNJCSD-UHFFFAOYSA-N Dihydroasparagusic acid Natural products OC(=O)C(CS)CS KRHAHEQEKNJCSD-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 108091006020 Fc-tagged proteins Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VPZXBVLAVMBEQI-VKHMYHEASA-N Glycyl-alanine Chemical compound OC(=O)[C@H](C)NC(=O)CN VPZXBVLAVMBEQI-VKHMYHEASA-N 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- QNAYBMKLOCPYGJ-UWTATZPHSA-N L-Alanine Natural products C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N L-Aspartic acid Natural products OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- FADYJNXDPBKVCA-UHFFFAOYSA-N L-Phenylalanyl-L-lysin Natural products NCCCCC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FADYJNXDPBKVCA-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-M L-tartrate(1-) Chemical compound OC(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-M 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000012515 MabSelect SuRe Substances 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 101000797092 Mesorhizobium japonicum (strain LMG 29417 / CECT 9101 / MAFF 303099) Probable acetoacetate decarboxylase 3 Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 238000006957 Michael reaction Methods 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 101100293261 Mus musculus Naa15 gene Proteins 0.000 description 1
- 108010079364 N-glycylalanine Proteins 0.000 description 1
- 101150082943 NAT1 gene Proteins 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108091005461 Nucleic proteins Chemical group 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- 241000555745 Sciuridae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101150006914 TRP1 gene Proteins 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- LVTKHGUGBGNBPL-UHFFFAOYSA-N Trp-P-1 Chemical compound N1C2=CC=CC=C2C2=C1C(C)=C(N)N=C2C LVTKHGUGBGNBPL-UHFFFAOYSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003200 antithyroid agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 150000004646 arylidenes Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- 230000000981 bystander Effects 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 230000009134 cell regulation Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 150000004294 cyclic thioethers Chemical class 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- JYGAZEJXUVDYHI-UHFFFAOYSA-N dihydroartemisininic acid Natural products C1CC(C)=CC2C(C(C)C(O)=O)CCC(C)C21 JYGAZEJXUVDYHI-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- OFDNQWIFNXBECV-VFSYNPLYSA-N dolastatin 10 Chemical class CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-VFSYNPLYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002171 ethylene diamines Chemical class 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000003168 generic drug Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- VPZXBVLAVMBEQI-UHFFFAOYSA-N glycyl-DL-alpha-alanine Natural products OC(=O)C(C)NC(=O)CN VPZXBVLAVMBEQI-UHFFFAOYSA-N 0.000 description 1
- 230000036433 growing body Effects 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 125000004474 heteroalkylene group Chemical group 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 238000010829 isocratic elution Methods 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical class OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000002132 lysosomal effect Effects 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 150000007523 nucleic acids Chemical group 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical class C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 238000012430 stability testing Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 230000008467 tissue growth Effects 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 230000005760 tumorsuppression Effects 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Images






Abstract
Description
Область техникиTechnical field
Настоящее изобретение относится к применению конъюгата антитело-лекарственное средство для лечения злокачественной опухоли или других заболеваний и, в частности, к применению специального гидрофильного кислотного стабилизированного участка соединения при получении конъюгата антитело-лекарственное средство для повышения стабильности лекарственного средства в плазме при одновременном значительном улучшении фармакокинетики (PK).The present invention relates to the use of an antibody-drug conjugate for the treatment of cancer or other diseases and, in particular, to the use of a specific hydrophilic acidic stabilized compound site in the preparation of an antibody-drug conjugate to increase drug stability in plasma while significantly improving pharmacokinetics ( P.K.).
Уровень техникиState of the art
Конъюгаты антитело-лекарственное средство (ADC) представляют собой класс новых направленных лекарственных средств, которые обычно состоят из трех компонентов: антитело или антитело-подобный лиганд, низкомолекулярное лекарственное средство и линкер, который связывает антитело с лекарственным средством. В конъюгатах антитело-лекарственное средство используется специфическое распознавание определенных антигенов антителами для транспортировки молекул лекарственного средства к месту вблизи клеток-мишеней и высвобождение их эффективно для обеспечения терапевтического эффекта. В августе 2011 года Управление США по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) одобрило новый ADC от Seattle Genetics, AdecteisTM, для лечения лимфомы Ходжкина, а также анапластической крупноклеточной лимфомы (ALCL), и клиническое применение продемонстрировало безопасность и эффективность этого класса лекарственных средств.Antibody-drug conjugates (ADCs) are a class of novel targeted drugs that typically consist of three components: an antibody or antibody-like ligand, a small molecule drug, and a linker that binds the antibody to the drug. Antibody-drug conjugates use the specific recognition of certain antigens by antibodies to transport drug molecules to a location near target cells and release them efficiently to provide a therapeutic effect. In August 2011, the US Food and Drug Administration (FDA) approved a new ADC from Seattle Genetics, Adecteis TM , for the treatment of Hodgkin's lymphoma as well as anaplastic large cell lymphoma (ALCL), and clinical use has demonstrated the safety and efficacy of this drug class.
Для конъюгатов антитело-лекарственное средство в целях обеспечения эффективного присоединения молекулы лекарственного средства к антителу, конъюгаты, в настоящее время входящие на стадию клинических испытаний, как правило, связаны через линкеры по остаткам лизина на поверхности лиганда или с остатками цистеина (полученными посредством частичного восстановления межцепочечных дисульфидных связей) в шарнирной области антитела. Однако присутствие большого количества остатков лизина (более 80) на поверхности антитела и неселективная природа присоединения приводит к неопределенности в отношении количества и присоединений и участков, что в свою очередь приводит к отсутствию однородности конъюгатов антитело-лекарственное средство. Присоединение к сульфгидрилу может эффективно снижать проблему плохого качественного состава продукта, и малеинимид, выступающий в качестве линкера, может быстро и в высокой степени селективно образовывать простые тиоэфирные продукты с сульфгидрильными группами антител в мягких условиях.For antibody-drug conjugates, in order to ensure efficient attachment of a drug molecule to an antibody, the conjugates currently in clinical trials are typically linked via linkers to lysine residues on the surface of the ligand or to cysteine residues (obtained by partial reduction of interstrand disulfide bonds) in the hinge region of the antibody. However, the presence of a large number of lysine residues (greater than 80) on the surface of the antibody and the non-selective nature of the attachment leads to uncertainty regarding the number of both attachments and sites, which in turn leads to a lack of uniformity in antibody-drug conjugates. Attachment to sulfhydryl can effectively reduce the problem of poor product quality, and maleimide acting as a linker can quickly and highly selectively form thioether products with sulfhydryl groups of antibodies under mild conditions.

Однако растущее количество исследований показало что присоединение сукцинимида к сульфгидрилу является обратимым процессом (обратная реакция Майкла), и, когда продукт присоединения попадает в плазму, можно отчетливо наблюдать обмен продукта присоединения с сульфгидрильной группой альбумина, что приводит к высвобождению молекулы лекарственного средства, вследствие присутствия большого количества белков, содержащих свободные сульфгидрильные группы, в плазме. Высвобождение молекулы лекарственного средства с одной стороны вызывает токсические побочные эффекты, в то время как с другой стороны снижает эффективность конъюгата антитело-лекарственное средство. (См. Shen, et al. "Conjugation site modulate the vivo stability and therapeutic activity of antibody-drug conjugates" Nature Biotech (2012) 30:184-189; Baldwin & Kiick, Bioconj. Chem 2011, 22, 1946-1953; Alley, et al. Bioconjugate Chem. 2008, 19, 759-765.)However, a growing body of research has shown that the addition of succinimide to sulfhydryl is a reversible process (reverse Michael reaction), and when the addition product enters the plasma, the exchange of the addition product with the sulfhydryl group of albumin can be clearly observed, resulting in the release of the drug molecule, due to the presence of a large the amount of proteins containing free sulfhydryl groups in plasma. The release of the drug molecule on the one hand causes toxic side effects, while on the other hand reduces the effectiveness of the antibody-drug conjugate. (See Shen, et al . "Conjugation site modulate the vivo stability and therapeutic activity of antibody-drug conjugates" Nature Biotech (2012) 30:184-189; Baldwin & Kiick, Bioconj. Chem 2011, 22, 1946-1953; Alley, et al . Bioconjugate Chem. 2008, 19, 759-765.)
Многие исследователи провели опубликованные испытания с целью повышения стабильности конъюгатов антитело-лекарственное средство в плазме и снижения обратной реакции Майкла на сульфгидрильной группе. Было описано, что циклический простой тиоэфирный продукт присоединения, образованный малеинимидом и сульфгидрилом, может претерпевать гидролиз в водной среде с образованием простого тиоэфирного продукта с открытым кольцом. В отличие от циклических сульфидных продуктов присоединения сульфидные продукты с открытым кольцом остаются стабильными в плазме и более не обмениваются с другими сульфгидрильными группами. Таким образом, обмена сульфгидрильными группами можно эффективно избежать, если конъюгат антитело-лекарственное средство конвертируется в структуру с открытым кольцом до его попадания в организм.Many investigators have conducted published trials to improve the stability of antibody-drug conjugates in plasma and reduce the back reaction of Michael on the sulfhydryl group. It has been described that the cyclic thioether addition product formed by maleimide and sulfhydryl can undergo hydrolysis in an aqueous medium to form an open ring thioether product. Unlike cyclic sulfide addition products, open ring sulfide products remain stable in plasma and are no longer exchanged with other sulfhydryl groups. Thus, the exchange of sulfhydryl groups can be effectively avoided if the antibody-drug conjugate is converted to an open ring structure prior to its entry into the body.

На скорость реакции присоединения с открытием кольца влияет множество факторов, включая pH и температуру реакции, а также структуру, подвергаемую реакции, и, вследствие чувствительности антител к pH и температуре, быстрое открытие кольца при pH и температуре, которые может переносить антитело, является наилучшим выбором, осуществимым посредством оптимизации структуры линкера. В патенте WO2016025752 описан способ, в котором сильная группа, притягивающая электроны, вносится снаружи сукцинимидного кольца, где указанный способ использует поглощение электронов для снижения обмена простыми тиоэфирными продуктами присоединения с альбумином; однако следствием присутствия указанной притягивающей электроны группы определенно является то, что связь сукцинимида и лекарственного средства трудно сохранить в естественных условиях и сукцинимид, несущий сильную притягивающую электроны группу, является в высокой степени чувствительным к гидролизу кольца, после чего он более не способен претерпевать дальнейшее присоединение к простому тиоэфиру.The rate of the ring-opening addition reaction is influenced by many factors, including the pH and temperature of the reaction, as well as the structure undergoing the reaction, and due to the pH and temperature sensitivity of antibodies, rapid ring opening at the pH and temperature that the antibody can tolerate is the best choice. , feasible by optimizing the structure of the linker. WO2016025752 describes a method in which a strong electron attracting group is introduced on the outside of the succinimide ring, wherein said method uses electron absorption to reduce the exchange of thioether addition products with albumin; however, a consequence of the presence of said electron-attracting group is definitely that the bond between succinimide and the drug is difficult to maintain in vivo and the succinimide bearing a strong electron-attracting group is highly susceptible to ring hydrolysis, after which it is no longer able to undergo further attachment to a simple thioether.

В патенте Seattle Genetics (США) № US20130309256 описан линкер, который вносит основную группу и притягивающую электроны группу рядом с сукцинимидом, где скорость обмена продукта присоединения с альбумином может быть эффективно снижена вследствие эффекта основной группы, значительно повышая стабильность в плазме. Как известно, pH плазмы человека является слабо основным и, хотя включение основных групп может способствовать открытию сукцинимидного кольца, повышение гидрофобности всей молекулы становится более трудным. Это явление убедительно подтверждено в последующем патенте Seattle Genetics WO2015057699. После включения стабилизированного участка присоединения, несущего основную группу, в молекулу-мишень, авторы настоящего изобретения открыли, что вся молекула конъюгата антитело-лекарственное средство все еще быстро деградировала у мышей с более высокой нагрузкой лекарственным средством вследствие гидрофобности (см. патент № WO2015057699, стр.225), и для улучшения описанной выше ситуации авторы изобретения были вынуждены внести комплексную структуру полиэтиленгликоля в боковую цепь молекулы для улучшения агрегации молекулы (см. патент № WO2015057699).Seattle Genetics (US) Patent No. US20130309256 describes a linker that introduces a main group and an electron attracting group near succinimide, where the rate of exchange of the adduct with albumin can be effectively reduced due to the effect of the main group, greatly increasing plasma stability. Human plasma pH is known to be weakly basic, and although the inclusion of basic groups may facilitate the opening of the succinimide ring, increasing the hydrophobicity of the entire molecule becomes more difficult. This phenomenon is convincingly confirmed in the subsequent Seattle Genetics patent WO2015057699. After incorporating a stabilized attachment site bearing a basic group into the target molecule, the present inventors discovered that the entire antibody-drug conjugate molecule was still rapidly degraded in mice with higher drug loading due to hydrophobicity (see Patent No. WO2015057699, page .225), and in order to improve the situation described above, the inventors were forced to introduce a complex structure of polyethylene glycol into the side chain of the molecule to improve the aggregation of the molecule (see patent No. WO2015057699).
В дополнение к снижению обмена с альбумином и повышению стабильности в плазме, другим важным фактором, который должен учитываться при конструировании конъюгата антитело-лекарственное средство, является количество лекарственного средства, которое может быть доставлено на нацеливающий агент (т.е. количество цитотоксического средства, присоединенное к каждому нацеливающему агенту (например, каждое антитело)), которое называют нагрузкой лекарственным средством. Было предположено, что более высокая нагрузка лекарственным средством обеспечивает лучшую эффективность, чем более низкая нагрузка лекарственным средством (например, нагрузка 8 элементами должна демонстрировать лучшую эффективность in vivo и in vitro, чем нагрузка 4 элементами). Обоснованием этой теории является то, что конъюгаты с более высокой нагрузкой лекарственным средством будут доставлять больше лекарственного средства (цитотоксического средства) к клеткам-мишеням. Результаты, полученные in vitro, также подтвердили, что конъюгаты с более высокой нагрузкой лекарственным средством демонстрируют более высокую активность против клеточных линий vitro. Однако определенные последующие испытания показали, что эту гипотезу еще аналогично предстоит подтвердить in vivo в моделях на животных. В литературе описано, что конъюгаты с нагрузкой 4 элементами и 8 элементами лекарственного средства ауристатина имеют неожиданно сходную активность в модели на мышах, и не наблюдали более высокой эффективности при нагрузке 8 элементами лекарственного средства. См. Hamblett, et al., Clinical Cancer Res. 10: 7063-70 (2004). Hamblett et al. выявили причины вышеупомянутого экспериментального явления и далее описали, что в моделях на животных ADC с более высокой нагрузкой выводятся из кровотока быстрее. Это более быстрое выведение указывает на тенденцию к тому, что конъюгаты с более высокой нагрузкой имеют более нестабильную PK по сравнению с конъюгатами с более низкой нагрузкой. Кроме того, конъюгаты с более высокой нагрузкой демонстрируют более низкую MTD у мышей и, таким образом, имеют более узкий терапевтический индекс. Напротив, было описано, что ADC с нагрузкой, равной двум, в искусственно модифицированном участке на моноклональном антителе, имеет те же или лучшие свойства PK и терапевтические индексы, чем некоторые из ADC с нагрузкой четырьмя элементами. См. отчеты, опубликованные Junutula, et al., Clinical Cancer Res. 16: 4769 (2010).In addition to reducing albumin turnover and improving plasma stability, another important factor to consider when designing an antibody-drug conjugate is the amount of drug that can be delivered to the targeting agent (i.e., the amount of cytotoxic agent attached to each targeting agent (for example, each antibody)), which is called the drug load. It has been suggested that higher drug loading provides better efficacy than lower drug loading (eg, 8 element loading should show better in vivo and in vitro efficacy than 4 element loading). The rationale for this theory is that conjugates with a higher drug load will deliver more drug (cytotoxic agent) to the target cells. The in vitro results also confirmed that the higher drug loading conjugates showed higher activity against in vitro cell lines. However, certain subsequent trials have shown that this hypothesis is likewise yet to be confirmed in vivo in animal models. The literature describes that the 4-unit and 8-unit drug loading conjugates of auristatin have unexpectedly similar activity in a mouse model, and no higher efficacy was observed with the 8-unit drug loading. See Hamblett, et al ., Clinical Cancer Res. 10:7063-70 (2004). Hamblett et al. identified the causes of the aforementioned experimental phenomenon and further described that in animal models, higher loading ADCs are cleared from the circulation more rapidly. This faster clearance indicates a tendency for higher loading conjugates to have a more unstable PK compared to lower loading conjugates. In addition, higher loading conjugates show lower MTD in mice and thus have a narrower therapeutic index. In contrast, ADCs with a loading of two in an artificially modified region on a monoclonal antibody have been described to have the same or better PK properties and therapeutic indices than some of the ADCs with a loading of four elements. See reports published by Junutula, et al ., Clinical Cancer Res. 16:4769 (2010).
Увеличение нагрузки лекарственным средством теоретически увеличивает количество лекарственного средства, которое несет одно антитело к клетке-мишени, однако вследствие гидрофобности лекарственного средства, гидрофобность ADC будет возрастать по мере увеличения количества лекарственного средства, вызывая агрегацию ADC в организме и снижение терапевтического индекса. Альтернативные способы преодоления тенденции к демонстрации ADC с более высокой нагрузкой менее желательных свойств PK включают добавление солюбилизирующих групп к структуре ADC. Например, к линкеру можно добавлять полимер полиэтиленгликоля или другой растворимый в воде полимер (например, область между участками связывания лекарственного средства и антитела) для преодоления тенденции ADC к агрегации. Например, несмотря на тот факт, что Seattle Genetics разработали возможность стабилизировать участки присоединения посредством включения основных групп, все еще существует необходимость в улучшении PK молекул лекарственного средства при высоких нагрузках путем, например, внесения боковой цепи ПЭГ в конъюгат участок присоединения-лекарственное средство. Однако добавление солюбилизирующих групп повышает сложность процесса получения, используемого для таких конъюгатов.Increasing the drug load theoretically increases the amount of drug that a single antibody carries to the target cell, however, due to the hydrophobicity of the drug, the hydrophobicity of the ADC will increase as the amount of drug increases, causing the ADC to aggregate in the body and reduce the therapeutic index. Alternative ways to overcome the tendency to show ADCs with a higher load of less desirable PK properties include adding solubilizing groups to the structure of the ADCs. For example, a polyethylene glycol polymer or other water-soluble polymer (eg, the region between the drug and antibody binding sites) can be added to the linker to overcome the tendency of the ADC to aggregate. For example, despite the fact that Seattle Genetics has developed the ability to stabilize attachment sites by incorporating major groups, there is still a need to improve the PK of drug molecules at high loadings by, for example, introducing a PEG side chain into the attachment site-drug conjugate. However, the addition of solubilizing groups increases the complexity of the preparation process used for such conjugates.
Таким образом, в области новых ADC, используемых для смягчения вредоносных эффектов высокого соотношения лекарственного средства и антитела (DAR), срочно необходим новый подход, который может одновременно преодолеть повышение стабильности в плазме, вызываемое обменом сукцинимидом между сульфгидрилами, также одновременно улучшая фармакокинетические свойства конъюгатов антитело-лекарственное средство с высоким DAR. Кислотный стабилизированный участок присоединения в соответствии с настоящим изобретением поразительно удовлетворяет обоим из приведенных выше требований.Thus, in the field of novel ADCs used to mitigate the deleterious effects of high drug-to-antibody ratio (DAR), a new approach is urgently needed that can simultaneously overcome the increase in plasma stability caused by succinimide exchange between sulfhydryls while also simultaneously improving the pharmacokinetic properties of antibody conjugates. -drug with high DAR. The acid stabilized attachment site of the present invention surprisingly satisfies both of the above requirements.
Сущность изобретенияThe essence of the invention
Настоящее изобретение направлено на предоставления специального гидрофильного кислотного стабилизированного конъюгата участок присоединения-лекарственное средство. При проведении экспериментов авторы настоящего изобретения неожиданно обнаружили, что, в отличие от внесения основных групп, описанных в US20130309256, внесения кислотной аминокислоты или олигопептидного элемента снижает сульфгидрилный обмен конъюгата сукцинимида и антитела при сохранении высокой степени стабильности в плазме крови. Также было экспериментально продемонстрировано, что сконструированный конъюгат демонстрирует сходный уровень гидрофобности с неконъюгированным нацеливающим реагентом вследствие включения одной или нескольких аминокислот, таким образом, сохраняя фармакокинетические (PK) свойства, сходные со свойствами неконъюгированного нацеливающего реагента in vivo. Таким образом, присутствие кислотного стабилизированного участка присоединения вносит вклад в эффективность лекарственного средства через два описанных выше каскада, которые вместе повышают стабильность лекарственного средства в плазме и улучшают фармакокинетику лекарственного средства (PK).The present invention is directed to providing a specific hydrophilic acid stabilized site-drug conjugate. Through experiments, the present inventors unexpectedly found that, in contrast to the introduction of the main groups described in US20130309256, the introduction of an acidic amino acid or an oligopeptide element reduces the sulfhydryl exchange of the succinimide-antibody conjugate while maintaining a high degree of stability in blood plasma. It has also been experimentally demonstrated that the engineered conjugate exhibits a similar level of hydrophobicity to the unconjugated targeting reagent due to the incorporation of one or more amino acids, thus maintaining pharmacokinetic (PK) properties similar to those of the unconjugated targeting reagent in vivo . Thus, the presence of an acid stabilized attachment site contributes to drug efficacy through the two cascades described above, which together increase drug plasma stability and improve drug pharmacokinetics (PK).
Введение кислотного стабилизированного участка соединения также позволяет конъюгату иметь более высокую нагрузку лекарственным средством (т.е. более высокое количество гидрофильных участков присоединения лекарственного средства на нацеливающий реагент) по сравнению с конъюгатами с более низкой нагрузкой лекарственным средством при сохранении желаемых свойств PK и демонстрации такой же или лучшей активности in vivo. (Например, конъюгат с нагрузкой 4 элементами или 8 элементами может иметь те же или лучшие свойства PK, чем его аналоги с нагрузкой 2 элементами 4 элементами, соответственно; такие конъюгаты с нагрузкой 4 элементами или 8 элементами могут иметь те же или лучшие свойства PK, чем их аналоги с нагрузкой 2 элементами или 4 элементами, соответственно).The introduction of an acidic stabilized junction site also allows the conjugate to have a higher drug loading (i.e., a higher number of hydrophilic drug attachment sites per targeting reagent) compared to lower drug loading conjugates while maintaining the desired PK properties and exhibiting the same or better in vivo activity. (For example, a conjugate loaded with 4 elements or 8 elements may have the same or better PK properties than its counterparts loaded with 2 elements with 4 elements, respectively; such conjugates loaded with 4 elements or 8 elements may have the same or better PK properties, than their counterparts with a load of 2 elements or 4 elements, respectively).
В частности, настоящее изобретение относится конъюгату антитело-лекарственное средство или его фармацевтически приемлемой соли, как показано в формуле I:In particular, the present invention relates to an antibody-drug conjugate, or a pharmaceutically acceptable salt thereof, as shown in formula I:

где:Where:
L соответствует антителу, фрагменту антитела или белку;L corresponds to an antibody, antibody fragment, or protein;
M соответствует сукцинимиду или гидролизованному сукцинимиду;M is succinimide or hydrolysed succinimide;
Ac соответствует фрагменту, состоящему из аминогруппы или кислотной группы, или олигопептиду, состоящему из множества аминокислот, где аминочасть присоединена к показанному кольцу;Ac corresponds to a fragment consisting of an amino group or an acid group, or an oligopeptide consisting of many amino acids, where the amino part is attached to the ring shown;
D соответствует лекарственной части;D corresponds to the medicinal part;
A соответствует линкерной части;A corresponds to the linker part;
Кольцо указывает на каркас, соответствующий замещенному или незамещенному C1-8 алкилидену, C1-8 гетероалкилидену, C6-10 арилидену или C4-10 гетероарилидену.The ring indicates a framework corresponding to a substituted or unsubstituted C 1-8 alkylidene, C 1-8 heteroalkylidene, C 6-10 arylidene, or C 4-10 heteroarylidene.
m соответствует целому числу от 1 до 20, и n соответствует 1 или 2.m corresponds to an integer from 1 to 20, and n corresponds to 1 or 2.
Предпочтительно, указанное антитело соответствует антителу к рецептору клеточной поверхности или ассоциированному с опухолью антигену.Preferably, said antibody corresponds to an antibody to a cell surface receptor or a tumor associated antigen.
Предпочтительно, Ac соответствует аминокислотному элементу или кислотному олигопептиду, кислотная группа Ac выбрана из одного или нескольких из группы, включающей группу карбоновой кислоты, фосфорной кислоты, фосфористой кислоты или сульфоновой кислоты.Preferably, Ac corresponds to an amino acid element or an acid oligopeptide, the acid group Ac is selected from one or more of the group consisting of a carboxylic acid, phosphoric acid, phosphorous acid or sulfonic acid group.
Более предпочтительно, Ac предпочтительно соответствует природной аминокислоте или неприродной аминокислоте, имеющей изоэлектрическую точку 7,0 или менее, или олигопептиду, состоящему из них.More preferably, Ac preferably corresponds to a natural amino acid or a non-natural amino acid having an isoelectric point of 7.0 or less, or an oligopeptide consisting of them.
В некоторых предпочтительных примерах олигопептидный элемент предпочтительно состоит из 2-5 аминокислот.In some preferred examples, the oligopeptide element preferably consists of 2-5 amino acids.
В некоторых предпочтительных примерах A соответствует расщепляемому линкеру или не расщепляемому линкеру.In some preferred examples, A corresponds to a cleavable linker or a non-cleavable linker.
В некоторых предпочтительных примерах указанное лекарственное средство соответствует цитотоксическому лекарственному средству, лекарственному средству для лечения аутоиммунного заболевания и противовоспалительному лекарственному средству.In some preferred examples, said drug corresponds to a cytotoxic drug, an autoimmune disease drug, and an anti-inflammatory drug.
Более предпочтительно, лекарственное средство D выбрано из группы, включающей лекарственные средства на основе майтанзина, лекарственные средства на основе аустралина, лекарственные средства на основе бензодипиррола, лекарственные средства на основе пирролозодиазола, аманитин и его производные и соединения камптотецина.More preferably, drug D is selected from the group consisting of maytansine drugs, australin drugs, benzodipyrrole drugs, pyrrolosodiazole drugs, amanitin and its derivatives, and camptothecin compounds.
В некоторых предпочтительных примерах A имеет следующую формулу:In some preferred examples, A has the following formula:
![]()
![]()
где C соответствует необязательному удлиняемому элементу на конце, E соответствует необязательному расщепляющемуся элементу, F соответствует спейсерному элементу и подстрочные e и f соответствуют 0 или 1. Волнистая линия указывает на точку соединения между кислотным самостабилизрованным участком присоединения и лекарственным элементом.where C corresponds to an optional extendable element at the end, E corresponds to an optional splitting element, F corresponds to a spacer element, and the subscripts e and f correspond to 0 or 1. The wavy line indicates the connection point between the acidic self-stabilized attachment site and the drug element.
В некоторых предпочтительных примерах кольцо соответствует группе C1-8 алкилена или группе C1-8 гетероалкилена.In some preferred examples, the ring corresponds to a C 1-8 alkylene group or a C 1-8 heteroalkylene group.
Более предпочтительно, кольцо соответствует группе C1-3 алкилена;More preferably, the ring corresponds to a C 1-3 alkylene group;
В некоторых предпочтительных примерах Ac выбран из группы, включающей (D/L) глицин, (D/L) аланин, (D/L) лейцин, (D/L) изолейцин, (D/L) валин, (D/L) фенилаланин, (D/L) пролин, (D/L) триптофан, (D/L) серин, (D/L) тирозин, (D/L) цистеин, (D/L) метионин, (D/L) аспарагин, (D/L) глутамин, (D/L) треонин, (D/L) аспарагиновую кислоту, (D/L) глутаминовую кислоту, или соединения следующих структурных формул:In some preferred examples, Ac is selected from the group consisting of (D/L) glycine, (D/L) alanine, (D/L) leucine, (D/L) isoleucine, (D/L) valine, (D/L) phenylalanine, (D/L) proline, (D/L) tryptophan, (D/L) serine, (D/L) tyrosine, (D/L) cysteine, (D/L) methionine, (D/L) asparagine , (D/L) glutamine, (D/L) threonine, (D/L) aspartic acid, (D/L) glutamic acid, or compounds of the following structural formulas:


где волнистая линия указывает на участок присоединения к каркасу и R соответствует произвольному линкерному фрагменту между аминогруппой и фосфатной группой.where the wavy line indicates the site of attachment to the frame and R corresponds to an arbitrary linker fragment between the amino group and the phosphate group.
Другой аспект настоящего изобретения относится к соединению участок присоединения-лекарственное средство, имеющему следующую структуру:Another aspect of the present invention relates to a site of attachment-drug having the following structure:

Ac соответствует аминокислотному элементу с изоэлектрической точкой менее 7;Ac corresponds to an amino acid element with an isoelectric point of less than 7;
D соответствует части в виде лекарственного средства;D corresponds to the drug part;
A соответствует линкерной части;A corresponds to the linker part;
q соответствует целому числу в диапазоне от 1 до 8, и n соответствует 1 или 2.q corresponds to an integer in the
Настоящее изобретение относится конъюгату антитело-лекарственное средство или его фармацевтически приемлемой соли, а также к фармацевтической композиции с фармацевтически приемлемым разбавителем, носителем или эксципиентом.The present invention relates to an antibody-drug conjugate or a pharmaceutically acceptable salt thereof, as well as to a pharmaceutical composition with a pharmaceutically acceptable diluent, carrier or excipient.
Конъюгат антитело-лекарственное средство или его фармацевтически приемлемую соль используют для получения лекарственных средств для лечения злокачественной опухоли, иммунного заболевания и воспаления.The antibody-drug conjugate, or a pharmaceutically acceptable salt thereof, is used in the manufacture of medicaments for the treatment of cancer, immune disease, and inflammation.
Описание чертежейDescription of drawings
На фиг.1 показаны результаты анализа MS-TOF, полученные для соединения 11.Figure 1 shows the results of the MS-TOF analysis obtained for
На фиг.2A представлены результаты анализа SEC-ВЭЖХ, полученные для H-11.On figa presents the results of the analysis of SEC-HPLC obtained for H-11.
Фигура 2B представлены результаты анализа SEC-ВЭЖХ, полученные для H-20.Figure 2B presents the results of the SEC-HPLC analysis obtained for H-20.
Фигура 2C представлены результаты анализа SEC-ВЭЖХ, полученные для H-29.Figure 2C presents the results of the SEC-HPLC analysis obtained for H-29.
Фигура 2D представлены результаты анализа SEC-ВЭЖХ, полученные для H-39.Figure 2D presents the results of the SEC-HPLC analysis obtained for H-39.
Фигура 2E представлены результаты анализа SEC-ВЭЖХ, полученные для H-41.Figure 2E presents the results of the SEC-HPLC analysis obtained for H-41.
Фигура 2F представлены результаты анализа SEC-ВЭЖХ, полученные для H-43.Figure 2F presents the results of the SEC-HPLC analysis obtained for H-43.
Фигура 2G представлены результаты анализа SEC-ВЭЖХ, полученные для MC-VC-PAB-MMAE.Figure 2G shows the results of the SEC-HPLC analysis obtained for MC-VC-PAB-MMAE.
Фигура 2H представлены результаты анализа SEC-ВЭЖХ, полученные для DPR-VC-PAB-MMAE.Figure 2H shows the results of the SEC-HPLC analysis obtained for DPR-VC-PAB-MMAE.
На фиг.3 представлены результаты испытания стабильности in vivo для неконъюгированных антител, ADC с кислотными стабилизированными участками присоединения и контрольных ADC.Figure 3 shows the results of in vivo stability testing for unconjugated antibodies, ADCs with acid-stabilized attachment sites, and control ADCs.
На фиг.4 представлены результаты хроматографии HIC для ADC с кислотными стабилизированными участками присоединения и контрольного ADC.Figure 4 shows the results of HIC chromatography for an ADC with acid stabilized attachment sites and a control ADC.
На фиг.5 представлены результаты эффективности in vivo для ADC с кислотными стабилизированными участками присоединения и контрольного ADC.FIG. 5 shows in vivo performance results for ADC with acid-stabilized attachment sites and control ADC.
На фиг.6 представлен график, демонстрирующий результаты эксперимента, полученные для примера 56.Figure 6 is a graph showing the results of the experiment obtained for example 56.
Конкретные варианты осуществленияSpecific Embodiments
После широкой и тщательной научно-исследовательской разработки авторы настоящего изобретения неожиданно обнаружили, что конъюгаты антитело-лекарственное средство, имеющие кислотный стабилизированный участок присоединения, демонстрируют лучшую стабильность в плазме и являются более гидрофильными, чем общепринятые конъюгаты антитело-лекарственное средство, и что конъюгированные структуры могут выдерживать более высокую нагрузку при сохранении желаемых фармакокинетических свойств структур при низкой нагрузке. Более высокая нагрузка лекарственным средством приводит к лучшей активности и терапевтической эффективности.After extensive and careful research and development, the present inventors unexpectedly found that antibody-drug conjugates having an acidic stabilized attachment site exhibit better plasma stability and are more hydrophilic than conventional antibody-drug conjugates, and that the conjugate structures can withstand a higher load while maintaining the desired pharmacokinetic properties of the structures at low load. Higher drug loading results in better potency and therapeutic efficacy.
В частности, конъюгат антитело-лекарственное средство, имеющий кислотный стабилизированный участок присоединения, предусматриваемый настоящим изобретением, имеет малеинимидный элемент, который при связывании с сульфгидрильной группой антитела и с участием стабилизирующей кислотной группы гидролизует кольцо, и полученная структура не претерпевает обмен с другими сульфгидрил-содержащими макромолекулами в плазме, таким образом, препятствуя высвобождению молекулы лекарственного средства.In particular, the antibody-drug conjugate having an acidic stabilized attachment site provided by the present invention has a maleimide element, which, when bound to the sulfhydryl group of the antibody and with the participation of the stabilizing acid group, hydrolyzes the ring, and the resulting structure does not undergo exchange with other sulfhydryl-containing macromolecules in plasma, thus preventing the release of the drug molecule.

В то же время, присутствие одной или нескольких гидрофильных кислотных групп приводит к образованию структуры с открытым кольцом, которая является более гидрофильной, чем конъюгат антитело-лекарственное средство, тем самым предотвращая такие проблемы, как снижение активности вследствие повышения нагрузки лекарственным средством.At the same time, the presence of one or more hydrophilic acid groups results in an open ring structure that is more hydrophilic than the antibody-drug conjugate, thereby preventing problems such as decreased activity due to increased drug loading.
Полученный конъюгат антитело-лекарственное средство можно использовать для того, чтобы обеспечить достижение лекарственным средством популяции клеток-мишеней, такой как группа опухолевых клеток. Конъюгат антитело-лекарственное средство специфически связывается с поверхностными белками клеток, и образовавшиеся конъюгаты случайным образом подвергаются эндоцитозу. В клетке лекарственное средство высвобождается в форме активного лекарственное вещества, обеспечивая один или несколько эффектов. Антитела включают химерные антитела, гуманизированные антитела, антитела человека; фрагменты антитела, которые могут связываться с антигенами; слитые белки Fc антител; или белки. Пригодные лекарственные средства представляют собой высокоактивные лекарственные средства, включая, но не ограничиваясь ими, майтанзиноиды, ауристатины, калихеамицины, доксорубицины, антибиотики на основе бензодипирролов (дуокармицины и CC-1065), димеры пирролобензодиазепина (PBDS), гоитрогены, камптотецин и производные, такие как SN38, иринотекан и икситекан.The resulting antibody-drug conjugate can be used to ensure that the drug reaches a target cell population, such as a group of tumor cells. The antibody-drug conjugate binds specifically to cell surface proteins and the resulting conjugates are randomly endocytosed. In the cell, the drug is released in the form of the active drug, producing one or more effects. Antibodies include chimeric antibodies, humanized antibodies, human antibodies; antibody fragments that can bind to antigens; antibody Fc fusion proteins; or squirrels. Suitable drugs are highly potent drugs including, but not limited to, maytansinoids, auristatins, calicheamicins, doxorubicins, benzodipyrrole antibiotics (duocarmycins and CC-1065), pyrrolobenzodiazepine dimers (PBDS), goitrogens, camptothecin, and derivatives such as SN38, irinotecan and ixitecan.
Подробное описание патента.Detailed description of the patent.
Сокращенные обозначения и определенияAbbreviations and definitions
Если нет иных указаний, приведенные ниже термины и выражения, используемые в настоящем описании, имеют следующие значения. Когда в настоящем описании используется торговое наименование, если контекст не указывает на иное, торговое наименование включает состав продукта, дженерическое лекарственное средство и активный фармацевтический ингредиент указанного продукта с торговым наименованием.Unless otherwise indicated, the following terms and expressions used in the present description have the following meanings. When a trade name is used in this specification, unless the context indicates otherwise, the trade name includes the formulation of the product, the generic drug, and the active pharmaceutical ingredient of the trade name of said product.
Термин "алкилен" относится к двухвалентной неразветвленной насыщенной углеводородной группе с 1-20 атомами углерода, включая группы в диапазоне от 1 до 10 атомов углерода. Примеры алкиленовых групп включают, но не ограничиваются ими, метилен (-CH2-), этилен (-CH2-CH2-), н-пропилен, н-бутилен, н-пентилен, н-гексилен и т.д. Если нет иных указаний, термин "арил" относится к полиненасыщенной, как правило, ароматической гидроксильной группе, которая может быть моноциклической или конденсированной, или к ковалентно связанному полицикличекому кольцу (с количеством колец вплоть до трех). Термин "гетероарил" относится к арильной группе (или кольцу), содержащей 1-5 гетероатомов, выбранных из группы, включающей N, O и S, где атомы азота и серы необязательно могут быть окисленными, и атомы азота необязательно могут подвергаться четвертичному аммонированию. Гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом. Неограничивающие примеры арильных групп включают: фенил, нафтил и дифенил, в то время как неограничивающие примеры гетероарильных групп включают: пиридил, пиридазинил, пиразинил, пиримидинил (пиримидинил), триазинил, хинолинил, хиноксалинил, хиназолинил, мисолинил, фталазинил, бензотриазинил, пуринил, бензимидазолил, бензопиразолил, бензотриазолил, бензотриазолил, изобензофуранил, изоиндолил, индолил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиримидинил, имидазопиридин, бензотиазолил, бензофуранил, бензотиенил, индолил, хинолинил, изохинолинил, изотиазолил, пиразолил, индолил, тиридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фуранил, тиенил и т.д. В случае описания как "замещенные", заместители ароматической и гетероароматической систем, описанные выше, выбраны из приемлемых заместителей, как указано ниже.The term "alkylene" refers to a divalent straight chain saturated hydrocarbon group with 1-20 carbon atoms, including groups in the range from 1 to 10 carbon atoms. Examples of alkylene groups include, but are not limited to, methylene (-CH2-), ethylene (-CH2-CH2-), n-propylene, n-butylene, n-pentylene, n-hexylene, etc. Unless otherwise indicated, the term "aryl" refers to a polyunsaturated, typically aromatic, hydroxyl group, which may be monocyclic or fused, or a covalently linked polycyclic ring (up to three rings). The term "heteroaryl" refers to an aryl group (or ring) containing 1-5 heteroatoms selected from the group consisting of N, O, and S, where the nitrogen and sulfur atoms may optionally be oxidized, and the nitrogen atoms may optionally undergo quaternary ammonization. The heteroaryl group may be attached to the rest of the molecule via a heteroatom. Non-limiting examples of aryl groups include: phenyl, naphthyl, and diphenyl, while non-limiting examples of heteroaryl groups include: pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl (pyrimidinyl), triazinyl, quinolinyl, quinoxalinyl, quinazolinyl, misolinyl, phthalazinyl, benzotriazinyl, purinyl, benzimidazolyl , benzopyrazolyl, benzotriazolyl, benzotriazolyl, isobenzofuranyl, isoindolyl, indolyl, benzotriazinyl, thienopyridinyl, thienopyrimidinyl, pyrimidinyl, imidazopyridine, benzothiazolyl, benzofuranyl, benzothienyl, indolyl, quinolinyl, isoquinolinyl, isothiazolyl, pyrazolyl, indolyl, trizolyl, thyridinyl, , isoxazolyl, thiadiazolyl, pyrrolyl, thiazolyl, furanyl, thienyl, etc. When described as "substituted", the substituents on the aromatic and heteroaromatic systems described above are selected from the acceptable substituents as indicated below.
Как указано в настоящем патенте, группа арилидена относится к наличию двух ковалентно связанных структур в вышеупомянутой арильной структуре в орто-, мета- или пара-положении.As stated in this patent, the arylidene group refers to the presence of two covalently linked structures in the above aryl structure in the ortho, meta or para position.
Если в настоящем описании нет иных указаний, заместители, состоящие из углеводородных групп (включая группы, обычно указываемые как алкилиден, алкенил, алкинил и циклоалкил), могут соответствуют множеству различных групп, выбранных из следующей группы: -галоген, -OR', -NR'R", -SR', -SiR'R "R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -CN и -NO2, с количеством заместителей в диапазоне от 0 до (2m'+1), где m' представляет собой общее количество атомов углерода в группе. Каждый из R', R" и R"' независимо соответствует атому водорода, незамещенному C1-8 алкилу, незамещенному арилу, арилу, замещенному 1-3 атомами галогена, незамещенному C1-8 алкилу, C1-8 алкокси или C1-8 тиоалкокси, или незамещенной арил-C1-4 алкильной группе. Когда R' и R" связаны с одним и тем же атомом азота, они могут образовывать кольца из 3, 4, 5, 6 или 7 элементов с указанным атомом азота. Например, -NR'R" включает 1-пирролидинил и 4-морфолинил.Unless otherwise indicated herein, substituents consisting of hydrocarbon groups (including groups commonly referred to as alkylidene, alkenyl, alkynyl, and cycloalkyl) may correspond to a variety of different groups selected from the following group: -halogen, -OR', -NR 'R', -SR', -SiR'R "R"', -OC(O)R', -C(O)R', -CO 2 R', -CONR'R", -OC(O) NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O) 2 R', -NH-C(NH 2 )=NH , -NR'C(NH 2 )=NH, -NH-C(NH 2 )=NR', -S(O)R', -S(O) 2 R', -S(O) 2 NR'R ", -NR'S(O) 2 R", -CN and -NO 2 , with a number of substituents in the range from 0 to (2m'+1), where m' represents the total number of carbon atoms in the group. Each of R', R" and R"' independently correspond to a hydrogen atom, an unsubstituted C 1-8 alkyl, an unsubstituted aryl, an aryl substituted with 1-3 halogen atoms, an unsubstituted C 1-8 alkyl, a C 1-8 alkoxy, or a C 1-8 thioalkoxy, or an unsubstituted aryl-C 1-4 alkyl group. When R' and R" are bonded to the same nitrogen atom, they can form rings of 3, 4, 5, 6 or 7 elements with said nitrogen atom. For example, -NR'R" includes 1-pyrrolidinyl and 4-morpholinyl.
Как используют в рамках изобретения, "производное" соединения относится к веществу, которое имеет химическую структуру, сходную со структурой соответствующего соединения, но которое также содержит по меньшей мере одну химическую группу, которая не присутствует в соединении и/или лишена по меньшей мере одной химической группы, которая присутствует в соединении. Соединение, с которым сравнивают производное, называют "исходным" соединением. Как правило, "производное" можно получать из исходного соединения посредством одной или нескольких стадий химических реакций.As used herein, a "derivative" of a compound refers to a substance that has a chemical structure similar to that of the corresponding compound, but which also contains at least one chemical group that is not present in the compound and/or lacks at least one chemical group that is present in the compound. The compound to which the derivative is compared is referred to as the "parent" compound. Typically, a "derivative" can be obtained from the parent compound through one or more chemical reaction steps.
Антитела, фрагменты антител и белкиAntibodies, antibody fragments and proteins
В вариантах осуществления настоящего изобретения антитела, фрагменты антител и белковые элементы соответствуют нацеливающим средствам, которые специфически связываются с элементом-мишенью. Антитела, как описано в контексте настоящего изобретения, способны специфически связываться с клеточными компонентами или с другими представляющими интерес молекулами-мишенями. В некоторых аспектах элемент антитела доставляет элемент лекарственного средства к конкретной популяции клеток-мишеней, с которыми взаимодействует элемент лиганда. Лиганды могут включать, но не ограничиваются ими, белки, полипептиды и пептиды, а также не белки, такие как сахара. Подходящие элементы лигандов включают, например, антитела, такие как полноразмерные (полные) антитела, а также их антигенсвязывающие фрагменты. В вариантах осуществления, где элемент лиганда представляет собой не антительный нацеливающий реагент, он может соответствовать пептиду или полипептиду, или не белковой молекуле. Примеры таких нацеливающих реагентов включают интерфероны, лимфокины, гормоны, факторы роста и колониестимулирующие факторы, витамины, молекулы транспорта питательных веществ, или любую другую связывающуюся с клеткой молекулу или вещество. В некоторых вариантах осуществления линкер ковалентно связан с атомом серы на лиганде. В некоторых аспектах атом серы представляет собой атом серы остатка цистеина, который образует межцепочечную дисульфидную связь антитела. В другом аспекте атом серы представляет собой атом серы остатка цистеина, который внесен в элемент лиганда и который образует межцепочечную дисульфидную связь антитела. В другом аспекте атом серы представляет собой атом серы остатка цистеина, который внесен в элемент лиганда (например, посредством сайт-направленного мутагенеза или химической реакции). В других аспектах атом серы, с которым связан линкер, выбран из атома серы остатка цистеина, который образует межцепочечную дисульфидную связь антитела, или фронтального остатка цистеина, который внесен в элемент лиганда (например, посредством сайт-направленного мутагенеза или химической реакции). В некоторых вариантах осуществления используют систему нумерации в соответствии с индексом EU, определяемую согласно Kabat (Kabat E.A., et al., (1991) Sequences of proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242).In embodiments of the present invention, the antibodies, antibody fragments, and protein elements correspond to targeting agents that specifically bind to the target element. Antibodies, as described in the context of the present invention, are capable of specifically binding to cellular components or other target molecules of interest. In some aspects, the antibody element delivers the drug element to a specific population of target cells with which the ligand element interacts. Ligands may include, but are not limited to, proteins, polypeptides, and peptides, as well as non-proteins such as sugars. Suitable ligand elements include, for example, antibodies, such as full-length (full) antibodies, as well as antigen-binding fragments thereof. In embodiments where the ligand element is a non-antibody targeting reagent, it may correspond to a peptide or polypeptide, or non-protein molecule. Examples of such targeting reagents include interferons, lymphokines, hormones, growth and colony stimulating factors, vitamins, nutrient transport molecules, or any other cell-binding molecule or substance. In some embodiments, the linker is covalently bonded to a sulfur atom on the ligand. In some aspects, the sulfur atom is a sulfur atom of a cysteine residue that forms an interchain disulfide bond of an antibody. In another aspect, the sulfur atom is a sulfur atom of a cysteine residue which is introduced into the ligand element and which forms an interchain disulfide bond of the antibody. In another aspect, the sulfur atom is a sulfur atom of a cysteine residue that has been introduced into the ligand element (eg, via site-directed mutagenesis or a chemical reaction). In other aspects, the sulfur atom to which the linker is bound is selected from the sulfur atom of a cysteine residue that forms an interchain disulfide bond of an antibody, or a frontal cysteine residue that is introduced into the ligand element (eg, via site-directed mutagenesis or a chemical reaction). In some embodiments, a numbering system according to the EU index is used, as defined by Kabat (Kabat EA, et al ., (1991) Sequences of proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242).
Как используют в настоящем патенте, "антитело" или "элемент антитела" включает, в пределах его объема, любую часть структуры антитела. Этот элемент может связываться, реактивно ассоциировать с или образовывать комплекс с рецептором, антигеном или другим элементом рецептора-мишени, принадлежащем клеточной популяции. Антитело может представлять собой любой белок или подобную белку молекулу, которые связывают, образуют комплекс или реагируют с частью популяции клеток, на которую направлено лечение или биологическая модификация.As used in this patent, "antibody" or "antibody element" includes, within its scope, any part of the antibody structure. This element can bind, reactively associate with, or complex with a receptor, antigen, or other target receptor element belonging to a cell population. An antibody can be any protein or protein-like molecule that binds, complexes with, or reacts with the part of the cell population that is being treated or biologically modified.
Антитела, используемые в рамках настоящего патента, включают поликлональные антитела и моноклональные антитела, где указанные поликлональные антитела представляют собой гетерогенные группы молекул антител, происходящие из сыворотки иммунного животного. Моноклональные антитела включают, но не ограничиваются ими, моноклональные антитела мыши и человека, гуманизированные моноклональные антитела или химерные моноклональные антитела, и антитела, происходящие из других видов. Моноклональные антитела человека можно получать любым из способов, известных в данной области (например, Teng, et al. 1983, proc. Nat1. Acad. Sci. USA. 80: 7308-7312; Olsson, et al. 1982, Methan, Enzymol 92: 3-16).The antibodies used herein include polyclonal antibodies and monoclonal antibodies, wherein said polyclonal antibodies are heterogeneous groups of antibody molecules derived from the serum of an immune animal. Monoclonal antibodies include, but are not limited to, murine and human monoclonal antibodies, humanized monoclonal antibodies or chimeric monoclonal antibodies, and antibodies derived from other species. Human monoclonal antibodies can be prepared by any of the methods known in the art (e.g., Teng, et al . 1983, proc. Nat1. Acad. Sci. USA. 80: 7308-7312; Olsson, et al . 1982, Methan, Enzymol 92 : 3-16).
Антитело, которое содержится в конъюгате антитело-лекарственное средство, описанном в настоящем патенте, предпочтительно должно сохранять способность связывать антиген, которую оно имело в его первоначальном диком состоянии. Таким образом, антитело, как описано в контексте настоящего изобретения, способно, предпочтительно исключительно, связываться с соответствующим антигеном. Антигены в рамках настоящего изобретения включают, например, ассоциированные с опухолью антигены (TAA), рецепторные белки клеточной поверхности и другие молекулы клеточной поверхности, регуляторы выживания клеток, регуляторы пролиферации клеток, молекулы, ассоциированные с ростом и дифференцировкой тканей (когда известно или спрогнозировано, что они являются функциональными), лимфокины, цитокины, молекулы, вовлеченные в регуляцию циркуляции клеток, молекулы, вовлеченные ангиогенез, и молекулы, ассоциированные с ангиогенезом (когда известно или спрогнозировано, что они являются функциональными). Ассоциированные с опухолью факторы могут соответствовать факторам кластеров дифференцировки (например, белкам CD).The antibody contained in the antibody-drug conjugate described in this patent should preferably retain the antigen-binding ability it had in its original wild state. Thus, an antibody, as described in the context of the present invention, is capable, preferably exclusively, of binding to the corresponding antigen. Antigens within the scope of the present invention include, for example, tumor associated antigens (TAA), cell surface receptor proteins and other cell surface molecules, cell survival regulators, cell proliferation regulators, molecules associated with tissue growth and differentiation (when known or predicted to be they are functional), lymphokines, cytokines, molecules involved in the regulation of cell circulation, molecules involved in angiogenesis, and molecules associated with angiogenesis (when known or predicted to be functional). Tumor-associated factors may correspond to factors of differentiation clusters (eg, CD proteins).
Антитела, описанные в настоящем описании для применения в конъюгате антитело-лекарственное средство, включают, но не ограничиваются ими, антитела, нацеленные на рецепторы клеточной поверхности, и антитела, нацеленные на ассоциированные с опухолью антигены. Такие ассоциированные с опухолью антигены хорошо известны специалистам в данной области и могут быть получены с использованием способов получения антител и информации, которая хорошо известна специалистам в данной области. Для разработки эффективных мишеней клеточного уровня, которые можно использовать для диагностики и лечения злокачественной опухоли, исследователи предприняли попытку идентифицировать трансмембранные или другие ассоциированные с опухолью пептиды. Эти мишени специфически экспрессируются на поверхности одной или нескольких злокачественных клеток с малой экспрессией или отсутствием экспрессии на поверхности одной или нескольких незлокачественных клеток. Как правило, такие ассоциированные с опухолью полипептиды сверхэкспрессируются в большей степени на поверхности злокачественных клеток по сравнению с поверхностью незлокачественных клеток. Идентификация таких ассоциированных с опухолью факторов имеет потенциал к значительному повышению специфичности терапии злокачественной опухоли на основе антител.Antibodies described herein for use in an antibody-drug conjugate include, but are not limited to, antibodies that target cell surface receptors and antibodies that target tumor-associated antigens. Such tumor-associated antigens are well known to those skilled in the art and can be generated using antibody production methods and information that are well known to those of skill in the art. To develop effective cellular level targets that can be used to diagnose and treat cancer, researchers have attempted to identify transmembrane or other tumor-associated peptides. These targets are specifically expressed on the surface of one or more cancer cells with little or no expression on the surface of one or more non-cancerous cells. Typically, such tumor-associated polypeptides are overexpressed to a greater extent on the surface of malignant cells compared to the surface of non-malignant cells. The identification of such tumor-associated factors has the potential to significantly increase the specificity of antibody-based cancer therapy.
Белки или пептиды, используемые в контексте настоящего патента для получения конъюгата антитело-лекарственное средство, могут представлять собой любой произвольный пептид или белок, который обладает аффинностью в отношении эпитопа или соответствующего рецептора, и они не обязательно должны принадлежать к семейству иммуноглобулинов. Эти пептиды можно выделять способами, сходными со способами, используемыми для фагового дисплея антител (Szardenings, J Recept Signal Transduct Res. 2003: 23(4): 307-49). Применение пептидов из таких случайных пептидных библиотек может быть сходным с применением антител и фрагментов антител. Пептидные или белковые связывающие молекулы могут быть связаны или соединены с макромолекулой или другим материалом, включая, но не ограничиваясь ими, альбумин, полимеры, липосомы, наночастицы или дендримеры, при условии, что указанное связывание позволяет пептиду или белку сохранять его специфичность связывания антигена.The proteins or peptides used in the context of this patent to prepare an antibody-drug conjugate can be any arbitrary peptide or protein that has affinity for an epitope or corresponding receptor, and they do not have to belong to the immunoglobulin family. These peptides can be isolated by methods similar to those used for antibody phage display (Szardenings, J Recept Signal Transduct Res. 2003: 23(4): 307-49). The use of peptides from such random peptide libraries may be similar to the use of antibodies and antibody fragments. Peptide or protein binding molecules may be linked or linked to a macromolecule or other material, including, but not limited to, albumin, polymers, liposomes, nanoparticles, or dendrimers, provided that said binding allows the peptide or protein to retain its antigen-binding specificity.
Ассоциированные с опухолью антигены включают антигены, которые хорошо известны специалистам в данной области. Последовательности нуклеиновых кислот и белков, соответствующие ассоциированным с опухолью антигенам, могут быть найдены в общедоступных базах данных, таких как Genbank. Ассоциированные с опухолью антигены, предназначенные для нацеливания посредством антител, включают все варианты аминокислотной последовательности и гомологи, обладающие по меньшей мере 70%, 80%, 85%, 90% или 95% гомологией с последовательностью, указанной в доступной литературе, или имеющие биологические свойства и характеристики, которые идентичны последовательности ассоциированного с опухолью антигена, указанной в литературе.Tumor-associated antigens include antigens that are well known to those skilled in the art. Nucleic acid and protein sequences corresponding to tumor-associated antigens can be found in public databases such as Genbank. Tumor-associated antigens intended for antibody targeting include all amino acid sequence variants and homologues that share at least 70%, 80%, 85%, 90%, or 95% homology with a sequence reported in the available literature or have biological properties. and characteristics that are identical to the tumor-associated antigen sequence reported in the literature.
Термин "ингибирование" или "ингибирование посредством" относится к снижению или полному устранению поддающегося определению количества.The term "inhibition" or "inhibition by" refers to the reduction or complete elimination of a detectable amount.
Термин "злокачественная опухоль" относится к физиологическому состоянию или заболеванию, характеризующемуся нерегулируемым клеточным ростом. "Опухоль" включает злокачественные клетки.The term "cancer" refers to a physiological condition or disease characterized by unregulated cell growth. "Tumor" includes malignant cells.
Термин "аутоиммунное заболевание" относится к заболеванию или нарушению в результате нацеливания на собственные ткани или белки индивидуума.The term "autoimmune disease" refers to a disease or disorder resulting from targeting an individual's own tissues or proteins.
Как используют в рамках изобретения, выражение "фармацевтически приемлемая соль" относится к фармацевтически приемлемой органической или неорганической соли соединения (например, лекарственного средства, конъюгата лекарственное средство-участок соединения или конъюгата лиганд-участок соединения-лекарственное средство). Указанное соединение может содержать по меньшей мере одну амино или карбоксильную группу и, таким образом, может образовывать соль аддукта с соответствующей кислотой или основанием. Иллюстративные примеры солей включают, но не ограничиваются ими: сульфат, трифторацетат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, гидросульфат, фосфат, кислый фосфат, соли изоникотиновой кислоты, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, водород тартрат, аскорбат, салицилат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, соли калия, соли натрия и т.д. Кроме того, фармацевтически приемлемые соли имеют более одного заряженного атома в их структуре. Существуют примеры, когда множество заряженных атомов, которые являются частью фармацевтически приемлемой соли, могут быть уравновешены во многих случаях. Например, фармацевтически приемлемая соль может иметь один или несколько заряженных атомов и/или один или несколько уравновешивающих атомов.As used herein, the expression "pharmaceutically acceptable salt" refers to a pharmaceutically acceptable organic or inorganic salt of a compound (eg, a drug, a drug-site conjugate of a compound, or a ligand-site of a compound-drug). Said compound may contain at least one amino or carboxyl group and thus may form an adduct salt with an appropriate acid or base. Illustrative examples of salts include, but are not limited to: sulfate, trifluoroacetate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, hydrogen sulfate, phosphate, acid phosphate, isonicotinic acid salts, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, hydrogen tartrate, ascorbate, salicylate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, potassium salts, sodium salts, etc. In addition, pharmaceutically acceptable salts have more than one charged atom in their structure. There are instances where a plurality of charged atoms that are part of a pharmaceutically acceptable salt can be balanced in many cases. For example, a pharmaceutically acceptable salt may have one or more charged atoms and/or one or more balancing atoms.
Предпочтительные лекарственные средства относятся к: цитотоксическим лекарственным средствам, используемым в терапии злокачественной опухоли, включая, но не ограничиваясь ими, майтанзин или майтанзиноиды, аналоги доластатина 10, кахимицины, доксорубицины, антибиотики на основе бензодипиррола (дуокармицины, CC-1065 и т.д.), пирроло[2,1-c][1,4]бензодиазепины (PBD) или димеры PBD и их производные, аманитин или его производные и соединения камптотецина, включая камптотецин, гидроксикамптотецин, SN-38, иктикам, иринотекан и т.д.Preferred drugs include: cytotoxic drugs used in cancer therapy, including but not limited to maytansine or maytansinoids,
С другой стороны, пригодные лекарственные средства не ограничиваются упомянутыми выше категориями, а скорее включают все лекарственные средства, которые можно использовать в конъюгате антитело-лекарственное средство.On the other hand, suitable drugs are not limited to the categories mentioned above, but rather include all drugs that can be used in the antibody-drug conjugate.
В некоторых вариантах осуществления изобретение также включает радиоизотопы, присоединенные через кислотный самостабилизированный участок присоединения. Они включают, но не ограничиваются ими: 3H, 11C, 18F, 32P, 35S, 64Cu, 68Ga, 86Y, 99Tc, 111In, 123I, 125I, 131I, 133Xe, 177Lu, 211At и 213Bi.In some embodiments, the invention also includes radioisotopes attached via an acidic self-stabilized attachment site. These include, but are not limited to: 3 H, 11 C, 18 F, 32 P, 35 S, 64 Cu, 68 Ga, 86 Y, 99 Tc, 111 In, 123 I, 125 I, 131 I, 133 Xe, 177Lu , 211At and 213Bi .
Исходя из механизма высвобождения лекарственного средства в клетку, как используют в рамках изобретения, "линкеры" или "линкеры конъюгата антитело-лекарственное средство" могут быть разделены на две категории: не расщепляющиеся линкеры и расщепляющиеся линкеры.Based on the mechanism of drug release into the cell, as used herein, "linkers" or "antibody-drug conjugate linkers" can be divided into two categories: non-cleavable linkers and cleavable linkers.
Для конъюгатов антитело-лекарственное средство, содержащих не расщепляющийся линкер, механизм высвобождения лекарственного средства является следующим: после связывания конъюгата с антигеном и эндоцитоза антитело ферментативно отщепляется в лизосоме с высвобождением активной молекулы, состоящей из низкомолекулярного лекарственного средства, линкера и аминокислотного остатка антитела. Конечное изменение молекулярной структуры лекарственного средства не снижает его цитотоксичность, однако, поскольку активная молекула заряжена (вследствие аминокислотных остатков), она не способна проникать в соседние клетки. Таким образом, указанное активное лекарственное вещество не будет способно уничтожить соседние с опухолью клетки, которые не экспрессируют антиген-мишень (антиген-негативные клетки) (известно как "эффект свидетеля") (Ducry, et al. 2010, Bioconjugate Chem. 21: 5-13).For antibody-drug conjugates containing a non-cleavable linker, the drug release mechanism is as follows: after binding of the conjugate to the antigen and endocytosis, the antibody is enzymatically cleaved in the lysosome to release the active molecule, consisting of the small molecule drug, the linker, and the amino acid residue of the antibody. The final change in the molecular structure of the drug does not reduce its cytotoxicity, however, since the active molecule is charged (due to amino acid residues), it is not able to penetrate into neighboring cells. Thus, said active drug will not be able to kill cells adjacent to the tumor that do not express the target antigen (antigen-negative cells) (known as the "bystander effect") (Ducry, et al . 2010, Bioconjugate Chem. 21:5 -13).
Расщепляющийся линкер, в соответствии с названием, может разрушаться и высвобождать активное лекарственное вещество (само низкомолекулярное лекарственное средство) в клетке-мишени. Расщепляющиеся линкеры могут быть разделены на два основных класса: химически нестабильные линкеры и ферментативно нестабильные линкеры.A cleavable linker, as the name suggests, can break down and release the active drug (the small molecule drug itself) in the target cell. Cleavable linkers can be divided into two main classes: chemically unstable linkers and enzymatically unstable linkers.
Химически нестабильные линкеры могут селективно разрушаться вследствие различий в свойствах плазмы и цитоплазмы. Такие свойства включают pH, концентрацию глутатиона и т.д.Chemically unstable linkers can be selectively degraded due to differences in plasma and cytoplasmic properties. Such properties include pH, glutathione concentration, etc.
В некоторых вариантах осуществления настоящего патента линкер соответствует pH-чувствительному линкеру, также обычно называемому чувствительным к кислоте линкером. Такие линкеры являются относительно стабильными в нейтральной среде крови (pH 7,3-7,5), но гидролизуются в слабокислой среде эндосом (pH 5,0-6,5) и лизосом (pH 4,5-5,0). В большинстве конъюгатов антитело-лекарственное средство первого поколения используются эти типы линкеров, причем их примеры включают гидразоны, карбонаты, ацетали и кетали. Вследствие ограниченной стабильности в плазме чувствительных к кислотам линкеров, конъюгаты антитело-лекарственное средство на основе таких линкеров, как правило, имеют короткое время полужизни (2-3 суток). Это короткое время полужизни в некоторой степени ограничивает применение pH-чувствительных линкеров в последнем поколении конъюгатов антитело-лекарственное средство.In some embodiments of the present patent, the linker is a pH sensitive linker, also commonly referred to as an acid sensitive linker. Such linkers are relatively stable in the neutral blood medium (pH 7.3-7.5), but are hydrolyzed in the slightly acidic environment of endosomes (pH 5.0-6.5) and lysosomes (pH 4.5-5.0). Most first generation antibody-drug conjugates use these types of linkers, examples of which include hydrazones, carbonates, acetals, and ketals. Due to the limited plasma stability of acid-sensitive linkers, antibody-drug conjugates based on such linkers typically have a short half-life (2-3 days). This short half-life somewhat limits the use of pH-sensitive linkers in the latest generation of antibody-drug conjugates.
Глутатион-чувствительные линкеры также известны как дисульфидные линкеры. Соответствующий механизм высвобождения лекарственного средства основан на различии между высокой внутриклеточной концентрацией глутатиона (миллимолярный диапазон) и относительно низкой концентрацией глутатиона (микромолярный диапазон), встречающейся в крови. Это особенно справедливо для опухолевых клеток, где низкое содержание кислорода приводит к увеличению активности редуктазы, и, таким образом, к более высоким уровням глутатиона. Дисульфидные связи являются термодинамически стабильными и, таким образом, демонстрируют высокую стабильность в плазме.Glutathione sensitive linkers are also known as disulfide linkers. The corresponding drug release mechanism is based on the difference between high intracellular glutathione concentration (millimolar range) and relatively low glutathione concentration (micromolar range) found in the blood. This is especially true in tumor cells, where low oxygen leads to increased reductase activity, and thus higher levels of glutathione. The disulfide bonds are thermodynamically stable and thus exhibit high plasma stability.
Фермент-нестабильные линкеры, такие как пептидные линкеры, позволяют лучший контроль высвобождения лекарственного средства. Пептидные линкеры могут эффективно расщепляться лизосомальными протеазами, такими как катепсин B или фибронектин (повышенные уровни этих ферментов встречаются в некоторых опухолевых тканях). Полагают, что эта пептидная связь является в высокой степени стабильной в плазме вследствие неподходящего внеклеточного pH, а также присутствия сывороточных ингибиторов протеаз, которые приводят к тому, что протеазы обычно неактивны. Учитывая высокую стабильность в плазме, а также благоприятную селективность и эффективность внутриклеточного разрушения, фермент-нестабильные линкеры широко используются в качестве расщепляющихся линкеров для конъюгатов антитело-лекарственное средство. Типичные фермент-нестабильные линкеры включают Val-Cit(vc), Phe-Lys и другие.Enzyme-unstable linkers, such as peptide linkers, allow better control of drug release. Peptide linkers can be efficiently cleaved by lysosomal proteases such as cathepsin B or fibronectin (elevated levels of these enzymes are found in some tumor tissues). This peptide bond is believed to be highly stable in plasma due to unsuitable extracellular pH as well as the presence of serum protease inhibitors which result in proteases being normally inactive. Given the high stability in plasma, as well as the favorable selectivity and efficiency of intracellular destruction, enzyme-unstable linkers are widely used as cleavable linkers for antibody-drug conjugates. Typical enzyme-unstable linkers include Val-Cit(vc), Phe-Lys, and others.
Спейсерный элемент обычно находится между расщепляющимся линкером и активным лекарственным средством, или сам по себе является частью расщепляемого линкера. Механизм действия спейсерного элемента является следующим: когда расщепляемый линкер расщепляется в подходящих условиях, спейсерный элемент претерпевает структурную перестройку, таким образом, высвобождая активное лекарственное вещество, с которым он связан. Обычно такие спейсерные элемент включают п-аминобензиловые спирты (PAB) и β-глюкурониды, замещенные или незамещенные этилендиамины и т.д.The spacer element is usually located between the cleavable linker and the active drug, or is itself part of the cleavable linker. The mechanism of action of the spacer element is as follows: when the cleavable linker is cleaved under suitable conditions, the spacer element undergoes structural rearrangement, thus releasing the active drug to which it is associated. Typically, such spacer elements include p-aminobenzyl alcohols (PAB) and β-glucuronides, substituted or unsubstituted ethylenediamines, and the like.
В настоящем патенте могут использоваться следующие сокращенные обозначения, которые следует понимать как имеющие следующие фиксированные значения: Boc: трет-бутоксикарбонил; DCC: дициклогексилкарбодиимид; DCM: дихлорметан; DIPEA: диизопропилкарбодиимид; DMF: N, N-диметилформамид; DMAP: 4-(N, N-диметиламино)пиридин; HATU: (1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиния 3-оксида гексафторфосфат; ВЭЖХ: высокоэффективная жидкостная хроматография; ПЭГ: полиэтиленгликоль; TFA: трифторуксусная кислота; THF: тетрагидрофуран; PBS: фосфатный буферный раствор (pH 7,0-7,5).The following abbreviations may be used in this patent and should be understood as having the following fixed meanings: Boc: tert-butoxycarbonyl; DCC: dicyclohexylcarbodiimide; DCM: dichloromethane; DIPEA: diisopropylcarbodiimide; DMF: N , N -dimethylformamide; DMAP: 4-( N,N -dimethylamino)pyridine; HATU: (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; HPLC: high performance liquid chromatography; PEG: polyethylene glycol; TFA: trifluoroacetic acid; THF: tetrahydrofuran PBS: phosphate buffer solution (pH 7.0-7.5).
Фармацевтически приемлемые эксципиенты включают любой носитель, разбавитель, адъювант или эксципиент, такие как консерванты и антиоксиданты, наполнители, разрыхлители, смачивающее вещества, эмульгаторы, суспендирующие вещества, растворители, диспергенты, покрывающие агенты, противомикробные и противогрибковые средства, замедляющие всасывание средства и т.д. Применение таких сред и веществ с фармацевтически активными веществами хорошо известно в данной области. Кроме случаев несовместимости между какой-либо общепринятой средой или реагентом с активным ингредиентом, их применение в терапевтических композициях также предусматривается. В качестве подходящей формы терапевтической комбинации также в указанную композицию могут быть включены дополнительные активные ингредиенты.Pharmaceutically acceptable excipients include any carrier, diluent, adjuvant or excipient such as preservatives and antioxidants, bulking agents, disintegrants, wetting agents, emulsifiers, suspending agents, solvents, dispersants, coating agents, antimicrobial and antifungal agents, retarding agents, etc. . The use of such media and substances with pharmaceutically active substances is well known in the art. Except in cases of incompatibility between any conventional medium or reagent with the active ingredient, their use in therapeutic compositions is also contemplated. As a suitable form of therapeutic combination, additional active ingredients may also be included in said composition.
Основными преимуществами настоящего изобретения являются следующие:The main advantages of the present invention are the following:
1. Конъюгат антитело-лекарственное средство, имеющий кислотный стабилизированный участок присоединения, предусматриваемый настоящим изобретением, может существенно снижать скорость обмена с сульфгидрильными группами альбумина in vivo, значительно повышая стабильность в плазме.1. An antibody-drug conjugate having an acidic stabilized attachment site provided by the present invention can significantly reduce the rate of exchange with albumin sulfhydryl groups in vivo , significantly increasing plasma stability.
2. Вследствие включения кислотной группы, кислотный стабилизированный участок присоединения, предусматриваемый настоящим изобретением, демонстрируют лучшую растворимость в воде на молекулярном уровне, эффективно улучшая свойства PK соответствующего конъюгата антитело-лекарственное средство, который демонстрирует лучшую эффективность in vivo.2. Due to the incorporation of an acidic group, the acidic stabilized attachment site provided by the present invention exhibits better water solubility at the molecular level, effectively improving the PK properties of the corresponding antibody-drug conjugate, which exhibits better in vivo efficacy.
3. Конъюгат антитело-лекарственное средство, имеющий кислотный стабилизированный участок присоединения, предусматриваемый настоящим изобретением, не только обеспечивает увеличенную стабильность плазме, он также улучшает свойства PK конечного конъюгата, и эти преимущества способны полностью удовлетворять потребности в более высокой нагрузке лекарственным средством, снижая агрегацию конъюгата антитело-лекарственное средство при высокой нагрузке лекарственным средством, и также одновременно достигая лучшей эффективности по сравнению с более низкой нагрузкой лекарственным средством.3. The antibody-drug conjugate having an acid stabilized attachment site provided by the present invention not only provides increased plasma stability, it also improves the PK properties of the final conjugate, and these advantages can fully satisfy the need for higher drug loading by reducing conjugate aggregation antibody-drug at high drug loading, while also achieving better efficacy compared to lower drug loading.
В следующем разделе изобретение описано более подробно применительно к конкретным примерам, и следует понимать, что указанные примеры используются только для иллюстрации изобретения и не предназначены для ограничения объема изобретения. Способы тестирования, для которых не указаны конкретные условия в примерах ниже, в основном проводили в обычных условиях или в условиях, рекомендованных изготовителем. Если нет иных указаний, все проценты, доли, соотношения или части приведены в расчете на массу. Если не определено иначе, подразумевается, что все профессиональные и научные термины, используемые в приведенном ниже тексте, имеют значения, в основном понятные специалистам в данной области. Кроме того, в способах по настоящему изобретению можно использовать любые способы и материалы, сходные или эквивалентные описанным способам и материалам. Предпочтительные способы и материалы, описанные в тексте ниже, приведены только для целей демонстрации.In the following section, the invention is described in more detail with reference to specific examples, and it should be understood that these examples are used only to illustrate the invention and are not intended to limit the scope of the invention. Test methods for which specific conditions are not indicated in the examples below were generally carried out under normal conditions or under the conditions recommended by the manufacturer. Unless otherwise indicated, all percentages, parts, ratios or parts are by weight. Unless otherwise specified, all professional and scientific terms used in the text below are intended to have the meanings generally understood by those skilled in the art. In addition, any methods and materials similar or equivalent to the described methods and materials can be used in the methods of the present invention. The preferred methods and materials described in the text below are for demonstration purposes only.
Общие методики, используемые в приведенных ниже вариантах осуществления изобретения, являются следующими:The general techniques used in the following embodiments of the invention are as follows:
Общая методика AGeneral procedure A
После предварительной очистки в молекулах антител с долей мономера более 95% проводили замену на фосфатный буфер, содержавший EDTA в концентрации 10 мг/мл, с использованием центрифужной пробирки для ультрафильтрации. Добавляли TCEP в молярном количестве, в 10 раз превышающем количество антитела, и полученной реакции позволяли протекать в течение восьми часов. Дисульфидные связи между цепями антитела раскрывались, и проводили определение количества свободных сульфгидрильных групп с использованием способа Эллмана для установления того, все ли дисульфидные связи раскрылись. Далее добавляли груз в молярном количестве, в 10 раз превышающем количество антитела, и полученной реакции позволяли протекать в течение восьми часов. После завершения реакции использовали центрифужную пробирку для ультрафильтрации с пороговым значением молекулярной массы 30 кДа для замены раствора на PBS, и не связавшийся груз удаляли.After preliminary purification, antibodies with a monomer fraction of more than 95% were replaced with a phosphate buffer containing EDTA at a concentration of 10 mg/ml using an ultrafiltration centrifuge tube. TCEP was added in a molar amount of 10 times the amount of antibody, and the resulting reaction was allowed to proceed for eight hours. Disulfide bonds between antibody chains were opened and free sulfhydryl groups were quantified using the Ellman method to determine if all disulfide bonds were opened. Next, a load was added in a molar amount of 10 times the amount of antibody, and the resulting reaction was allowed to proceed for eight hours. After completion of the reaction, an ultrafiltration centrifuge tube with a cut-off molecular weight of 30 kDa was used to replace the solution with PBS, and the unbound load was removed.
Общая методика BGeneral Method B
Исследование фармакокинетикиPharmacokinetic study
Способ ELISA для выявления антител в сыворотке: нанесение антитела (2 мкг/мл) проводили при 4°C в течение ночи, а затем проводили промывание посредством PBST, которое повторяли три раза, и блокирование 1% BSA+PBST при 37°C в течение одного часа; образцы сыворотки инкубировали и промывали PBST три раза; антитела для детекции (моноклональное антитело или поликлональное антитело против Fc [меченное HRP]) инкубировали при 37°C в течение одного часа, промывали три раза PBST и проявляли TMB, после чего использовали 2 M H2SO4 для завершения реакции и проводили измерения с использованием устройства для считывания микропланшетов.ELISA method for detecting antibodies in serum: application of antibody (2 μg/ml) was carried out at 4°C overnight, and then washed with PBST, which was repeated three times, and blocking with 1% BSA + PBST at 37°C for one hour; serum samples were incubated and washed with PBST three times; detection antibodies (anti-Fc monoclonal antibody or polyclonal antibody [HRP-labeled]) were incubated at 37°C for one hour, washed three times with PBST and developed with TMB, after which 2 MH 2 SO 4 was used to complete the reaction and measured using microplate readers.
Общая методика CGeneral Method C
Анализ с использованием хроматографии гидрофобного взаимодействия (HIC)Analysis using hydrophobic interaction chromatography (HIC)
Анализ ADC проводили с использованием хроматографии гидрофобного взаимодействия (HIC). Элюирование проводили с использованием 0-100% подвижной фазы B (MPB), где подвижная фаза A (MPA) состояла из 1,5 M сульфата аммония и 0,025 M фосфата натрия, и MPB состояла из 0,025 M фосфата натрия и 25% изопропанола. Объем загрузки образца составлял приблизительно 20 мкг и градиентное элюирование проводили в течение 15 минут. Для детекции использовали УФ 280 нм, и, чем более сильным был образец, транспортирующий воду, тем позднее был пик.ADC analysis was performed using hydrophobic interaction chromatography (HIC). Elution was performed using 0-100% mobile phase B (MPB), where mobile phase A (MPA) consisted of 1.5 M ammonium sulfate and 0.025 M sodium phosphate, and MPB consisted of 0.025 M sodium phosphate and 25% isopropanol. The sample loading volume was approximately 20 μg and the gradient elution was carried out for 15 minutes. UV 280 nm was used for detection, and the stronger the water-transporting sample was, the later the peak was.
Общая методика DGeneral procedure D
Испытания стабильности в плазмеPlasma stability tests
Фиксированное количество образца ADC добавляли к плазме человека, из которой уже был удален IgG человека, причем каждую пробирку с ADC получали в трех экземплярах; далее проводили инкубацию на водяной бане при 37°C в течение 0 часов и 72 часов, после чего образец ADC удаляли и в каждую пробирку добавляли 100 мкл белка A (MabSelect SuReTM LX партия:#10221479GE, промытого PBS); затем позволяли происходить адсорбции при встряхивании на вертикальном смесителе в течение двух часов, а затем проводили промывание и элюирование с получением инкубированного ADC; затем стабильность каждого инкубированного образца ADC в плазме определяли посредством ОФ-ВЭЖХ.A fixed amount of ADC sample was added to human plasma from which human IgG had already been removed, with each ADC tube obtained in triplicate; followed by incubation in a water bath at 37°C for 0 hours and 72 hours, after which the ADC sample was removed and 100 μl of protein A (MabSelect SuReTM LX batch: #10221479GE, washed with PBS) was added to each tube; then allowed to occur adsorption with shaking on a vertical mixer for two hours, and then carried out washing and elution to obtain incubated ADC; then the stability of each incubated ADC sample in plasma was determined by RP-HPLC.
Общая методика E: Сайт-специфическое присоединение ADCGeneral Method E: Site-Specific Attachment of ADC
После предварительной очистки для молекул антител с долей мономера более 95% проводили замену буфера на фосфатный буфер, содержавший EDTA в концентрации 10 мг/мл, с использованием центрифужной пробирки для ультрафильтрации. Добавляли TCEP в молярном количестве, в 10 раз превышающем количество антитела, и полученной реакции позволяли протекать в течение двух часов. С использованием центрифужной пробирки для ультрафильтрации для замены раствора на фосфатный буфер при pH 6,5, добавляли DHAA в молярном количестве, в 10 раз превышающем количество антитела, и конечной реакции позволяли протекать в течение двух часов. Далее добавляли груз в молярном количестве, в 3 раза превышающем количество антитела, и полученной реакции позволяли протекать в течение четырех часов. После завершения реакции центрифужную пробирку для ультрафильтрации с пороговым значением молекулярной массы 30 кДа использовали для замены раствора на PBS и не связанный груз удаляли с получением ADC с сайт-специфическим присоединением (DAR=2).After preliminary purification, for antibody molecules with a monomer fraction of more than 95%, the buffer was exchanged for phosphate buffer containing EDTA at a concentration of 10 mg/ml using an ultrafiltration centrifuge tube. TCEP was added in a molar amount of 10 times the amount of antibody, and the resulting reaction was allowed to proceed for two hours. Using an ultrafiltration centrifuge tube to replace the solution with phosphate buffer at pH 6.5, DHAA was added at a molar amount of 10 times the amount of antibody, and the final reaction was allowed to proceed for two hours. Next, a load was added in a molar amount of 3 times the amount of antibody, and the resulting reaction was allowed to proceed for four hours. After completion of the reaction, an ultrafiltration centrifuge tube with a cut-off molecular weight of 30 kDa was used to replace the solution with PBS and the unbound cargo was removed to give ADC with site-specific attachment (DAR=2).
Пример 1Example 1
Получение соединения 1 Get
![]()
![]()
50 г (S)-N-бензилоксикарбонил-N'-трет-бутоксикарбонил-2,3-диаминопропионовой кислоты растворяли в 500 мл дихлорметана, после чего добавляли 50 мл трифторуксусной кислоты и реакционный раствор перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционный раствор концентрировали до сухого состояния при пониженном давлении, после чего добавляли этилацетат для растворения полученного твердого вещества с последующим добавлением гексана и перемешиванием для преципитации твердых веществ, которые фильтровали и сушили с получением твердого продукта массой 21 г. LC-MS m/z (ES+): 239,1 (M+H)+ 50 g of ( S ) -N -benzyloxycarbonyl- N' -tert-butoxycarbonyl-2,3-diaminopropionic acid was dissolved in 500 ml of dichloromethane, after which 50 ml of trifluoroacetic acid was added, and the reaction solution was stirred at room temperature overnight. After completion of the reaction, the reaction solution was concentrated to dryness under reduced pressure, after which ethyl acetate was added to dissolve the resulting solid, followed by the addition of hexane and stirring to precipitate the solids, which were filtered and dried to give a solid product weighing 21 g. LC-MS m/ z(ES + ): 239.1(M+H) +
Пример 2Example 2
Получение соединения 2 Get
![]()
![]()
200 мл бензилового спирта добавляли в реакционную колбу и медленно капельно добавляли тионилхлорид (6,69 мл, 92,4 ммоль) на водяной бане; после завершения капельного добавления проводили перемешивание в течение одного часа с последующим периодическим добавлением соединения 1 (20 г, 84 ммоль) и после завершения добавления смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции бензиловый спирт отгоняли посредством масляного насоса при пониженном давлении, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=150:1) с получением 22 г продукта. LC-MS m/z (ES+): 329,1 (M+H)+.200 ml of benzyl alcohol was added to the reaction flask and thionyl chloride (6.69 ml, 92.4 mmol) was slowly added dropwise in a water bath; after completion of the drop addition, stirring was carried out for one hour, followed by periodic addition of compound 1 (20 g, 84 mmol) and after completion of the addition, the mixture was stirred at room temperature overnight. After completion of the reaction, benzyl alcohol was distilled off by an oil pump under reduced pressure, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=150:1) to obtain 22 g of a product. LC-MS m/z (ES + ): 329.1 (M+H) + .
Пример 3Example 3
Получение соединения 3 Get
![]()
![]()
Соединение 2 (10 г, 30,4 ммоль) растворяли в 150 мл ацетонитрила, добавляли диизопропилэтиламин (DIEA, 4,72 мл, 36,58 ммоль) и капельно добавляли трет-бутил-бромацетат (4,75 г, 24,4 ммоль) на ледяной бане; после капельного добавления реакции позволяли протекать в течение 30 минут, а затем реакционную смесь помещали в условия комнатной температуры для дальнейшей реакции, после чего проводили мониторинг посредством TLC (проявитель: DCM: MeOH=10:1). После завершения реакции раствор концентрировали при пониженном давлении и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=100:1) с получением 5,9 г продукта. LC-MS m/z (ES+): 443,3 (M+H)+.Compound 2 (10 g, 30.4 mmol) was dissolved in 150 ml of acetonitrile, diisopropylethylamine (DIEA, 4.72 ml, 36.58 mmol) was added and tert-butyl bromoacetate (4.75 g, 24.4 mmol) was added dropwise ) in an ice bath; after the dropwise addition, the reaction was allowed to proceed for 30 minutes, and then the reaction mixture was placed under room temperature conditions for further reaction, after which it was monitored by TLC (developer: DCM: MeOH=10:1). After completion of the reaction, the solution was concentrated under reduced pressure, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=100:1) to obtain 5.9 g of a product. LC-MS m/z (ES + ): 443.3 (M+H) + .
Пример 4Example 4
Получение соединения 4 Get Compound 4
![]()
![]()
Соединение 3 (5,9 г, 13,3 ммоль), 30 мл 1,4-диоксана и 30 мл воды добавляли в реакционную колбу, а затем добавляли DIEA (3,3 мл, 20 ммоль) и капельно добавляли Boc-ангидрид (14,5 г, 66,7 ммоль) при комнатной температуре; полученный реакционный раствор был желтым, и реакции позволяли протекать при комнатной температуре в течение приблизительно 3-4 часов после капельного добавления, после чего проводили мониторинг посредством TLC (проявитель: DCM:MeOH=30:1). После завершения реакции диоксан удаляли посредством концентрирования при пониженном давлении и добавляли DCM для проведения экстракции с последующим концентрированием при пониженном давлении с получением неочищенного желтого маслянистого продукта; его затем подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=300:1) с получением 6,6 г продукта. LC-MS m/z (ES+): 443,3 (M+H)+-Boc, 543,2 (M+H)+.Compound 3 (5.9 g, 13.3 mmol), 30 ml of 1,4-dioxane and 30 ml of water were added to the reaction flask, and then DIEA (3.3 ml, 20 mmol) was added and Boc anhydride ( 14.5 g, 66.7 mmol) at room temperature; the resulting reaction solution was yellow, and the reaction was allowed to proceed at room temperature for about 3-4 hours after the dropwise addition, after which it was monitored by TLC (developer: DCM:MeOH=30:1). After completion of the reaction, dioxane was removed by concentration under reduced pressure, and DCM was added to carry out extraction, followed by concentration under reduced pressure to give a crude yellow oily product; it was then subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=300:1) to obtain 6.6 g of a product. LC-MS m/z (ES + ): 443.3 (M+H) + -Boc, 543.2 (M+H) + .
Пример 5Example 5
Получение соединения 5 Get
![]()
![]()
6,6 г соединения 4 растворяли в 50 мл метанола, добавляли 1,32 г 5% Pd/C и проводили замену газообразного водорода от двух до трех раз пока смесь реагировала при комнатной температуре, и проводили мониторинг посредством TLC (проявитель: DCM:MeOH=5:1). После завершения реакции реакционный раствор фильтровали и концентрировали при пониженном давлении при 40°C с получением 4,4 г продукта, который использовали непосредственно в следующей реакции.6.6 g of compound 4 was dissolved in 50 ml of methanol, 1.32 g of 5% Pd/C was added, and hydrogen gas was changed two to three times while the mixture was reacting at room temperature, and monitored by TLC (developer: DCM:MeOH =5:1). After completion of the reaction, the reaction solution was filtered and concentrated under reduced pressure at 40° C. to obtain 4.4 g of a product, which was used directly in the next reaction.
Пример 6Example 6
Получение соединения 6 Getting Compound 6

Соединение 5 (4,4 г, 13,8 ммоль) растворяли в 40 мл ледяной уксусной кислоты, добавляли малеиновый ангидрид (2,71 г, 27,6 ммоль) и реакции позволяли протекать при перемешивании при комнатной температуре. Мониторинг реакции проводили посредством TLC (DCM:MeOH=3:1). После завершения реакции реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали с использованием жидкой фазы ВЭЖХ.Compound 5 (4.4 g, 13.8 mmol) was dissolved in 40 ml of glacial acetic acid, maleic anhydride (2.71 g, 27.6 mmol) was added and the reaction was allowed to proceed with stirring at room temperature. The reaction was monitored by TLC (DCM:MeOH=3:1). After completion of the reaction, the reaction solution was concentrated under reduced pressure, and the resulting residue was purified using HPLC liquid phase.
(Колонка: YMC-C18, 100 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=215 нм)(Column: YMC-C18, 100 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в вводе Solvent A: 0.2% TFA in the input
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: от 0 до 10 мин, 90%A, от 10 до 25 мин, от 90 до 45%A, от 25 до 55 мин, от 45 до 40%AGradient: 0 to 10 min, 90%A, 10 to 25 min, 90 to 45%A, 25 to 55 min, 45 to 40%A
Фракцию с временем удержания 43 мин собирали и лиофилизировали с получением 1,51 г белого твердого вещества. LC-MS m/z (ES+): 317,9 (M+H)+-Boc, 417,9 (M+H)+.The 43 min retention time fraction was collected and lyophilized to give 1.51 g of a white solid. LC-MS m/z (ES + ): 317.9 (M+H) + -Boc, 417.9 (M+H) + .
Пример 7Example 7
Получение соединения 7 Obtaining Compound 7

Соединение 6 (1,2 г, 2,88 ммоль) растворяли в 25 мл сухого толуола и 2,5 мл раствора DMA, и добавляли триэтиламин (1,2 мл, 8,65 ммоль) (можно добавлять несколько сухих молекулярных сит), проводили замену газообразного азота и смесь нагревали до 120°C, в то время как проводили мониторинг реакции посредством TLC (проявитель: DCM:MeOH=3:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол от 20:1 до 10:1) с получением 700 мг продукта. LC-MS m/z (ES+): 299,2 (M+H)+-Boc, 399,3 (M+H)+.Compound 6 (1.2 g, 2.88 mmol) was dissolved in 25 ml dry toluene and 2.5 ml DMA solution and triethylamine (1.2 ml, 8.65 mmol) was added (a few dry molecular sieves may be added), a nitrogen gas exchange was performed, and the mixture was heated to 120° C. while the reaction was monitored by TLC (developer: DCM:MeOH=3:1). After completion of the reaction, the solution was concentrated under reduced pressure using an oil pump, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol 20:1 to 10:1) to obtain 700 mg of a product. LC-MS m/z (ES + ): 299.2 (M+H) + -Boc, 399.3 (M+H) + .
Пример 8Example 8
Получение соединения 8 Getting

700 мг (1,7 ммоль) соединения 7 растворяли в 3,5 мл DMF, после чего добавляли 531 мг (2,11 ммоль) EEDQ и 733 мг (1,93 ммоль) Val-Cit-PABOH и реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM: MeOH=5:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол от 50:1 до 20:1) с получением 520 мг продукта. LC-MS m/z (ES+): 660,3 (M+H)+-Boc, 760,4 (M+H)+.700 mg (1.7 mmol) of compound 7 was dissolved in 3.5 ml of DMF, after which 531 mg (2.11 mmol) of EEDQ and 733 mg (1.93 mmol) of Val-Cit-PABOH were added and the reaction was allowed to proceed at room temperature. temperature. Monitored by TLC (developer: DCM: MeOH=5:1). After completion of the reaction, the solution was concentrated under reduced pressure using an oil pump at 45°C, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol 50:1 to 20:1) to obtain 520 mg of product. LC-MS m/z (ES + ): 660.3 (M+H) + -Boc, 760.4 (M+H) + .
Пример 9Example 9
Получение соединения 9 Getting

520 мг (0,685 ммоль) соединения 8 и 1,04 г (3,42 ммоль) NCP последовательно добавляли в реакционную колбу, добавляли 10 мл DMF для растворения твердых веществ и добавляли 0,33 мл (2,05 ммоль) DIEA, после чего реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM:MeOH=5:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=30:1) с получением 530 мг продукта. LC-MS m/z (ES+): 825,3 (M+H)+-Boc, 925,2 (M+H)+.520 mg (0.685 mmol) of
Пример 10Example 10
Получение соединения 10 Get

530 мг (0,57 ммоль) соединения 9 и 13,5 мг (0,1 моль) HOBT растворяли в 5 мл DMF, добавляли 0,17 мл (1,04 ммоль) DIEA и после активации при комнатной температуре в течение 1 ч, добавляли 373 мг (0,52 ммоль) MMAE, и раствору позволяли реагировать в течение ночи при комнатной температуре. Мониторинг посредством ВЭЖХ показал, что исходный материал MMAE полностью вступал в реакцию. Очистку проводили с использованием жидкой фазы ВЭЖХ.530 mg (0.57 mmol) of
(Колонка: YMC-C18, 50 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 50 мл/мин, детекция при λ=205 нм)(Column: YMC-C18, 50 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 90%A, 10-25 мин, 90-45%A, 25-55 мин, 45-40%AGradient: 0-10 min, 90%A, 10-25 min, 90-45%A, 25-55 min, 45-40%A
Фракцию с временем удержания 34 мин собирали и лиофилизировали с получением 200 мг продукта; LC-MS m/z (ES+): 1503,2 (M+H)+.The 34 min retention time fraction was collected and lyophilized to give 200 mg of product; LC-MS m/z (ES + ): 1503.2 (M+H) + .
Пример 11Example 11
Получение соединения 11 Getting

70 мг соединения 10 растворяли в 10 мл сухого дихлорметана, добавляли 4 мл трифторуксусной кислоты и реакции позволяли протекать при комнатной температуре в течение одного часа, а затем проводили очистку посредством ВЭЖХ.70 mg of
(Колонка: YMC-C18, 30 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=205 нм)(Column: YMC-C18, 30 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 95%A, 10-30 мин, 95-80%A, 30-50 мин, 80-50%A.Gradient: 0-10 min, 95%A, 10-30 min, 95-80%A, 30-50 min, 80-50%A.
Фракцию с временем удержания 33 мин собирали и лиофилизировали с получением 34 мг продукта; LC-MS m/z (ES+): 1347,8 (M+H)+.The 33 min retention time fraction was collected and lyophilized to give 34 mg of product; LC-MS m/z (ES + ): 1347.8 (M+H) + .
Пример 12Example 12
Получение соединения 12 Getting Compound 12

Ди-трет-бутил-L-глутамат (8 г, 30,8 ммоль) растворяли в 150 мл ацетонитрила, добавляли DIEA (6 мл, 37 ммоль), и капельно добавляли бензиловый эфир 2-бром-N-(бензилоксикарбонил)-L-аланина (10,2 г, 26,1 ммоль) на ледяной бане; после капельного добавления реакции позволяли протекать в течение 40 минут реакционный раствор помещали в условия комнатной температуры и позволяли ему реагировать при одновременном проведении мониторинга посредством TLC (проявитель: DCM:MeOH=10:1). После завершения реакции раствор концентрировали при пониженном давлении, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=100:1) с получением 8,1 г продукта. LC-MS m/z (ES+): 571,2 (M+H)+.Di-tert - butyl -L -glutamate (8 g, 30.8 mmol) was dissolved in 150 ml of acetonitrile, DIEA (6 ml, 37 mmol) was added and 2-bromo- N- (benzyloxycarbonyl) -L benzyl ester was added dropwise -alanine (10.2 g, 26.1 mmol) in an ice bath; after dropwise addition, the reaction was allowed to proceed for 40 minutes, the reaction solution was placed at room temperature and allowed to react while monitoring by TLC (developer: DCM:MeOH=10:1). After completion of the reaction, the solution was concentrated under reduced pressure, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=100:1) to obtain 8.1 g of a product. LC-MS m/z (ES + ): 571.2 (M+H) + .
Пример 13Example 13
Получение соединения 13 Obtaining Compound 13

8,1 г соединения 12 (14,2 ммоль), 50 мл 1,4-диоксана и 50 мл воды добавляли в реакционную колбу, а затем добавляли 3,5 мл DIEA (21,3 ммоль) и капельно добавляли 14,5 г Boc-ангидрида (66,7 ммоль) при комнатной температуре; полученный реакционный раствор был желтым, и реакции позволяли протекать до завершения при комнатной температуре, и в это время проводили мониторинг посредством TLC (проявитель: DCM: MeOH=30:1). После завершения реакции диоксан удаляли посредством концентрирования при пониженном давлении и добавляли DCM для проведения экстракции с последующим концентрированием при пониженном давлении с получением неочищенного желтого маслянистого продукта; затем его подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=300:1) с получением 8,55 г продукта. LC-MS m/z (ES+): 471,1 (M+H)+-Boc, 571,3 (M+H)+.8.1 g of compound 12 (14.2 mmol), 50 ml of 1,4-dioxane and 50 ml of water were added to the reaction flask, and then 3.5 ml of DIEA (21.3 mmol) were added and 14.5 g of Boc anhydride (66.7 mmol) at room temperature; the resulting reaction solution was yellow, and the reaction was allowed to proceed to completion at room temperature, and at this time was monitored by TLC (developer: DCM: MeOH=30:1). After completion of the reaction, dioxane was removed by concentration under reduced pressure, and DCM was added to carry out extraction, followed by concentration under reduced pressure to give a crude yellow oily product; then, it was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=300:1) to obtain 8.55 g of a product. LC-MS m/z (ES + ): 471.1 (M+H) + -Boc, 571.3 (M+H) + .
Пример 14Example 14
Получение соединения 14 Getting Compound 14

8,5 г соединения 13 растворяли в 70 мл метанола, добавляли 1,7 г 5% Pd/C и проводили замену газообразного водорода от двух до трех раз, в то время как смесь подвергали реакции при комнатной температуре и проводили мониторинг посредством TLC (проявитель: DCM:MeOH=5:1). После завершения реакции реакционный раствор фильтровали и концентрировали при пониженном давлении при 40°C с получением 5 г продукта, который непосредственно использовали в следующей реакции.8.5 g of compound 13 was dissolved in 70 ml of methanol, 1.7 g of 5% Pd/C was added and the hydrogen gas was replaced two to three times while the mixture was reacted at room temperature and monitored by TLC (developer : DCM:MeOH=5:1). After completion of the reaction, the reaction solution was filtered and concentrated under reduced pressure at 40°C to obtain 5 g of a product, which was directly used in the next reaction.
Пример 15Example 15
Получение соединения 15 Getting

5 г (11,2 ммоль) соединения 14 растворяли в 40 мл ледяной уксусной кислоты, добавляли 2,19 г (22,4 ммоль) малеинового ангидрида и реакции позволяли протекать при перемешивании при комнатной температуре. Проводили мониторинг реакции посредством TLC (DCM: MeOH=3:1). После завершения реакции реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали с использованием жидкой фазы ВЭЖХ.5 g (11.2 mmol) of compound 14 was dissolved in 40 ml of glacial acetic acid, 2.19 g (22.4 mmol) of maleic anhydride was added and the reaction was allowed to proceed with stirring at room temperature. The reaction was monitored by TLC (DCM: MeOH=3:1). After completion of the reaction, the reaction solution was concentrated under reduced pressure, and the resulting residue was purified using HPLC liquid phase.
(Колонка: YMC-C18, 100 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=215 нм)(Column: YMC-C18, 100 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 90%A, 10-25 мин, 90-45%A, 25-55 мин, 45-40%A.Gradient: 0-10 min, 90%A, 10-25 min, 90-45%A, 25-55 min, 45-40%A.
Фракцию с временем удержания 48 мин собирали и лиофилизировали с получением 2,3 г продукта. LC-MS m/z (ES+): 445,2 (M+H)+-Boc, 545,3 (M+H)+.The 48 min retention time fraction was collected and lyophilized to give 2.3 g of product. LC-MS m/z (ES + ): 445.2 (M+H) + -Boc, 545.3 (M+H) + .
Пример 16Example 16
Получение соединения 16 Obtaining Compound 16

2 г соединения 15 (2,88 ммоль) растворяли в 25 мл сухого толуола и 2,5 мл раствора DMA и добавляли 1,58 мл триэтиламина (11,4 ммоль), проводили замену газообразного азота и смесь нагревали до 120°C в то время как проводили мониторинг реакции посредством TLC (проявитель: DCM: MeOH=3:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол от 20:1 до 10:1) с получением 1,19 г продукта; LC-MS m/z (ES+): 427,1 (M+H)+-Boc, 527,3 (M+H)+.2 g of compound 15 (2.88 mmol) was dissolved in 25 ml of dry toluene and 2.5 ml of DMA solution and 1.58 ml of triethylamine (11.4 mmol) was added, nitrogen gas was replaced and the mixture was heated to 120°C at that time. while the reaction was monitored by TLC (developer: DCM: MeOH=3:1). After completion of the reaction, the solution was concentrated under reduced pressure using an oil pump, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol 20:1 to 10:1) to obtain 1.19 g of a product; LC-MS m/z (ES + ): 427.1 (M+H) + -Boc, 527.3 (M+H) + .
Пример 17Example 17
Получение соединения 17 Obtaining Compound 17

1,19 г (1,7 ммоль) соединения 16 растворяли в 3,5 мл DMF, после чего добавляли 531 мг (2,11 ммоль) EEDQ и 733 мг (1,93 ммоль) Val-Cit-PABOH и реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM:MeOH=5:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол от 50:1 до 25:1) с получением 753 мг продукта. LC-MS m/z (ES+): 788,5 (M+H)+-Boc, 888,4 (M+H)+.1.19 g (1.7 mmol) of compound 16 was dissolved in 3.5 ml of DMF, after which 531 mg (2.11 mmol) of EEDQ and 733 mg (1.93 mmol) of Val-Cit-PABOH were added and the reaction was allowed to proceed at room temperature. Monitored by TLC (developer: DCM:MeOH=5:1). After completion of the reaction, the solution was concentrated under reduced pressure using an oil pump at 45°C, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol 50:1 to 25:1) to obtain 753 mg of product. LC-MS m/z (ES + ): 788.5 (M+H) + -Boc, 888.4 (M+H) + .
Пример 18Example 18
Получение соединения 18 Getting Compound 18

753 мг (0,849 ммоль) соединения 17 и 1,28 г (4,24 ммоль) NCP последовательно добавляли в реакционную колбу, добавляли 15 мл DMF для растворения твердых веществ и добавляли 0,35 мл (2,55 ммоль) DIEA, после чего реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM:MeOH=5:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=25:1) с получением 762 мг продукта. LC-MS m/z (ES+): 1053,6 (M+H)+.753 mg (0.849 mmol) of compound 17 and 1.28 g (4.24 mmol) of NCP were sequentially added to the reaction flask, 15 ml of DMF was added to dissolve the solids, and 0.35 ml (2.55 mmol) of DIEA was added, after which the reaction was allowed to proceed at room temperature. Monitored by TLC (developer: DCM:MeOH=5:1). After completion of the reaction, the solution was concentrated under reduced pressure using an oil pump at 45°C, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=25:1) to obtain 762 mg of a product. LC-MS m/z (ES + ): 1053.6 (M+H) + .
Пример 19Example 19
Получение соединения 19 Getting Compound 19

762 мг (0,71 ммоль) соединения 18 и 13,5 мг (0,1 моль) HOBT растворяли в 8 мл DMF, добавляли 0,23 мл (1,42 ммоль) DIEA и после активации при комнатной температуре в течение 1 ч добавляли 463 мг (0,645 ммоль) MMAE и раствору позволяли реагировать в течение ночи при комнатной температуре. Мониторинг посредством ВЭЖХ показал, что исходный материал MMAE полностью вступал в реакцию. Проводили очистку с использованием жидкой фазы ВЭЖХ.762 mg (0.71 mmol) of compound 18 and 13.5 mg (0.1 mol) of HOBT were dissolved in 8 ml of DMF, 0.23 ml (1.42 mmol) of DIEA were added and after activation at room temperature for 1 h 463 mg (0.645 mmol) MMAE was added and the solution was allowed to react overnight at room temperature. Monitoring by HPLC showed that the MMAE starting material was fully reacted. Purification was carried out using liquid phase HPLC.
(Колонка: YMC-C18, 100 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=215 нм)(Column: YMC-C18, 100 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 90%A, 10-25 мин, 90-45%A, 25-55 мин, 45-40%AGradient: 0-10 min, 90%A, 10-25 min, 90-45%A, 25-55 min, 45-40%A
Фракцию с временем удержания 30 мин собирали и лиофилизировали с получением 300 мг продукта. LC-MS m/z (ES+): 1631,6 (M+H)+.The 30 min retention time fraction was collected and lyophilized to give 300 mg of product. LC-MS m/z (ES + ): 1631.6 (M+H) + .
Пример 20Example 20
Получение соединения 20 Getting

100 мг соединения 19 растворяли в 10 мл сухого дихлорметана, добавляли 4 мл трифторуксусной кислоты и реакции позволяли протекать при комнатной температуре в течение одного часа, а затем проводили очистку посредством ВЭЖХ.100 mg of compound 19 was dissolved in 10 ml of dry dichloromethane, 4 ml of trifluoroacetic acid was added and the reaction was allowed to proceed at room temperature for one hour, and then purification was carried out by HPLC.
(Колонка: YMC-C18, 30 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=215 нм)(Column: YMC-C18, 30 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 95%A, 10-25 мин, 95-80%A, 25-55 мин, 80-55%AGradient: 0-10 min, 95%A, 10-25 min, 95-80%A, 25-55 min, 80-55%A
Фракцию с временем удержания 30 мин собирали и лиофилизировали с получением 45 мг продукта. LC-MS m/z (ES+): 1419,1 (M+H)+.The 30 min retention time fraction was collected and lyophilized to give 45 mg of product. LC-MS m/z (ES + ): 1419.1 (M+H) + .
Примеры 21-29Examples 21-29
Синтез соединений 21-29 Synthesis of compounds 21 - 29

Пример 21Example 21
Получение соединения 21 Getting connection 21
![]()
![]()
15 г 3-нитро-4-аминобензойную кислоту растворяли в 100 мл метанола, добавляли 3 г 5% Pd/C и проводили гидрогенизацию при атмосферном давлении в течение пяти часов, а затем проводили фильтрацию; полученный остаток на фильтре промывали два раза метанолом, далее концентрировали до сухого состояния при комнатной температуре в высоком вакууме и непосредственно использовали в следующей реакции без дальнейшей очистки.15 g of 3-nitro-4-aminobenzoic acid was dissolved in 100 ml of methanol, 3 g of 5% Pd/C was added, and hydrogenation was carried out at atmospheric pressure for five hours, and then filtration was carried out; the resulting filter residue was washed twice with methanol, then concentrated to dryness at room temperature under high vacuum, and directly used in the next reaction without further purification.
Пример 22Example 22
Получение соединения 22 Getting connection 22

3,4-диаминобензойную кислоту (10 г, 65,7 ммоль) растворяли в 100 мл DMF, добавляли порошковый карбонат калия (13,6 г, 98,55 моль) и йодид калия (2,2 г, 1,31 ммоль), и капельно добавляли трет-бромуксусную кислоту в газообразном азоте, после чего капельно добавляли бутиловый эфир (12,8 г, 65,7 моль), в то время как реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM: MeOH=10:1). После завершения реакции добавляли воду, проводили экстракцию этилацетатом, и экстракт комбинировали с органической фазой; после этого проводили промывание насыщенным рассолом, а затем сушку безводным сульфатом натрия и фильтрацию, и раствор концентрировали при пониженном давлении, и полученный остаток подвергали разделению с использованием очистки посредством колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=от 150:1 до 100:1) с получением 3,46 г продукта. LC-MS m/z (ES+): 267,4 (M+H)+; 1H-ЯМР: 7,51-7,53, дд, 1H: 7,44-7,45, д, 1H: 6,66-6,69, д, 1H; 4,69, с, 2H: 1,49, с, 9H.3,4-diaminobenzoic acid (10 g, 65.7 mmol) was dissolved in 100 ml DMF, powdered potassium carbonate (13.6 g, 98.55 mol) and potassium iodide (2.2 g, 1.31 mmol) were added , and tert-bromoacetic acid in nitrogen gas was added dropwise, after which butyl ether (12.8 g, 65.7 mol) was added dropwise while the reaction was allowed to proceed at room temperature. Monitored by TLC (developer: DCM: MeOH=10:1). After completion of the reaction, water was added, extraction was carried out with ethyl acetate, and the extract was combined with the organic phase; thereafter, washing with saturated saline followed by drying with anhydrous sodium sulfate and filtration was carried out, and the solution was concentrated under reduced pressure, and the resulting residue was subjected to separation using purification by silica gel column chromatography (eluent: dichloromethane:methanol=150:1 to 100: 1) to obtain 3.46 g of product. LC-MS m/z (ES + ): 267.4 (M+H) + ; 1 H-NMR: 7.51-7.53, dd, 1H: 7.44-7.45, d, 1H: 6.66-6.69, d, 1H; 4.69, s, 2H: 1.49, s, 9H.
Пример 23Example 23
Получение соединения 23 Getting Compound 23

3 г (11,2 ммоль) соединения 22, 15 мл ледяной уксусной кислоты и малеиновый ангидрид (0,55 г, 5,6 ммоль) добавляли в реакционную колбу, и полученной реакционной смеси позволяли протекать при комнатной температуре и проводили мониторинг посредством ВЭЖХ. После завершения реакции проводили концентрирование при пониженном давлении для удаления ледяной уксусной кислоты, и проводили очистку. (Колонка: YMC-C18, 100 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=214 нм)3 g (11.2 mmol) of
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-5 мин 80%A, 5-35 мин, 80-65%A, 35-45 мин, 65-60%A, фракцию, демонстрирующую время удержания 37-42 мин, собирали и лиофилизировали с получением 2 г продукта. LC-MS m/z (ES+): 365,4 (M+H)+, 309,3 (M+H)+-tBu. 1H-ЯМР: 10,48, с, 1H; 7,41-7,44, дд, 1H; 7,35-7,36, д, 1H; 6,92, с, 2H; 6,70-6,72, д, 1H; 4,68, с, 2H; 1,42, с, 9H.Gradient: 0-5
Пример 24Example 24
Получение соединения 24 Getting connection 24

Соединение 23 (2 г, 5,49 ммоль) растворяли в 30 мл THF, и добавляли Boc-ангидрид (1,44 г, 6,59 ммоль), DMAP (1,32 г, 10,9 ммоль) и DIEA (1,8 мл, 10,98 ммоль) и реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM: MeOH=10:1). После завершения реакции раствор подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=от 100:1 до 20:1) с получением 2,2 г продукта. LC-MS m/z (ES+); 365,4(M+H)+-Boc 1H-ЯМР : 10,50, с, 1H; 7,46-7,48, дд, 1H; 7,41-7,42, д, 1H; 6,95, с, 2H; 6,73-6,75, д, 1H; 4,69, с, 2H; 1,48, с, 9H; 1,40, с, 9H.Compound 23 (2 g, 5.49 mmol) was dissolved in 30 ml THF and Boc anhydride (1.44 g, 6.59 mmol), DMAP (1.32 g, 10.9 mmol) and DIEA (1 .8 ml, 10.98 mmol) and the reaction was allowed to proceed at room temperature. Monitored by TLC (developer: DCM: MeOH=10:1). After completion of the reaction, the solution was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=100:1 to 20:1) to obtain 2.2 g of a product. LC-MS m/z (ES + ); 365.4(M+H) + -Boc 1 H-NMR: 10.50, s, 1H; 7.46-7.48, dd, 1H; 7.41-7.42, d, 1H; 6.95, s, 2H; 6.73-6.75, d, 1H; 4.69, s, 2H; 1.48, s, 9H; 1.40, s, 9H.
Пример 25Example 25
Получение соединения 25 Getting

2 г (4,31 ммоль) соединения 24 растворяли в 15 мл толуола и добавляли 2 мл DMA, триэтиламина (1,2 мл, 8,62 ммоль) и результирующей реакции позволяли протекать при 120°C. Проводили мониторинг реакции посредством TLC (DCM:MeOH=10:1). После завершения реакции раствор концентрировали при пониженном давлении, и полученный остаток подвергали разделению с использованием очистки посредством колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол от 100:1 до 50:1) с получением 1,63 г продукта; LC-MS m/z (ES+): 347,4 (M+H)+-Boc 1H-ЯМР : 7,50-7,51, дд, 1H; 7,43-7,44, д, 1H; 6,97, с, 2H; 6,75-6,77, д, 1H; 4,67, с, 2H; 1,48, с, 9H; 1,40, с, 9H.2 g (4.31 mmol) of compound 24 was dissolved in 15 ml of toluene and 2 ml of DMA, triethylamine (1.2 ml, 8.62 mmol) was added and the resulting reaction was allowed to proceed at 120°C. The reaction was monitored by TLC (DCM:MeOH=10:1). After completion of the reaction, the solution was concentrated under reduced pressure, and the resulting residue was subjected to separation using purification by silica gel column chromatography (eluent: dichloromethane:methanol 100:1 to 50:1) to obtain 1.63 g of a product; LC-MS m/z (ES + ): 347.4 (M+H) + -Boc 1 H-NMR: 7.50-7.51, dd, 1H; 7.43-7.44, d, 1H; 6.97, s, 2H; 6.75-6.77, d, 1H; 4.67, s, 2H; 1.48, s, 9H; 1.40, s, 9H.
Пример 26Example 26
Получение соединения 26 Getting Compound 26

1,6 г (3,58 ммоль) соединения 25 растворяли в 10 мл DMF, после чего добавляли 1,77 г (7,17 ммоль) EEDQ и 2,98 г (7,89 ммоль) Val-Cit-PABOH, и реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM:MeOH=10:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол от 100:1 до 25:1) с получением 2,1 г продукта. LC-MS m/z (ES+): 709,8 (M+H)+-Boc, 809,7 (M+H)+.1.6 g (3.58 mmol) of
Пример 27Example 27
Получение соединения 27 Getting Compound 27

2 г (2,47 ммоль) соединения 26 и 3,71 г (12,3 ммоль) NCP последовательно добавляли в реакционную колбу, добавляли 20 мл DMF для растворения твердых веществ и добавляли 1,2 мл (7,41 ммоль) DIEA, после чего реакции позволяли протекать при комнатной температуре. Проводили мониторинг посредством TLC (проявитель: DCM: MeOH=15:1). После завершения реакции раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=100:1) с получением 2,1 г продукта. LC-MS m/z (ES+): 973,3 (M+H)+.2 g (2.47 mmol) of compound 26 and 3.71 g (12.3 mmol) of NCP were sequentially added to the reaction flask, 20 ml of DMF was added to dissolve the solids, and 1.2 ml (7.41 mmol) of DIEA was added, after which the reaction was allowed to proceed at room temperature. Monitored by TLC (developer: DCM: MeOH=15:1). After completion of the reaction, the solution was concentrated under reduced pressure using an oil pump at 45°C, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=100:1) to obtain 2.1 g of a product. LC-MS m/z (ES + ): 973.3 (M+H) + .
Пример 28Example 28
Получение соединения 28 Getting connection 28

1 г (1 ммоль) соединения 27 и 13,5 мг (0,1 моль) HOBT растворяли в 8 мл DMF, добавляли 0,33 мл (2 ммоль) DIEA, и после активации при комнатной температуре в течение 1 ч добавляли 646 мг (0,9 ммоль) MMAE, и раствору позволяли реагировать в течение ночи при комнатной температуре. Мониторинг посредством ВЭЖХ показал, что исходный материал MMAE полностью вступал в реакцию. Очистку проводили с использованием жидкой фазы ВЭЖХ.1 g (1 mmol) of compound 27 and 13.5 mg (0.1 mol) of HOBT were dissolved in 8 ml of DMF, 0.33 ml (2 mmol) of DIEA was added, and after activation at room temperature for 1 h, 646 mg of (0.9 mmol) MMAE and the solution was allowed to react overnight at room temperature. Monitoring by HPLC showed that the MMAE starting material was fully reacted. Purification was carried out using liquid phase HPLC.
(Колонка: YMC-C18, 100 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=214 нм)(Column: YMC-C18, 100 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 90%A, 10-25 мин, 90-45%A, 25-55 мин, 45-40%A.Gradient: 0-10 min, 90%A, 10-25 min, 90-45%A, 25-55 min, 45-40%A.
Фракцию с временем удержания 30 мин собирали и лиофилизировали с получением 560 мг продукта. LC-MS m/z (ES+): 1551,7 (M+H)+.The 30 min retention time fraction was collected and lyophilized to give 560 mg of product. LC-MS m/z (ES + ): 1551.7 (M+H) + .
Пример 29Example 29
Получение соединения 29 Getting

100 мг соединения 28 растворяли в 10 мл сухого дихлорметана, добавляли 4 мл трифторуксусной кислоты и реакции позволяли протекать при комнатной температуре в течение двух часов, а затем проводили очистку посредством ВЭЖХ.100 mg of compound 28 was dissolved in 10 ml of dry dichloromethane, 4 ml of trifluoroacetic acid was added and the reaction was allowed to proceed at room temperature for two hours, and then purification was carried out by HPLC.
(Колонка: YMC-C18, 30 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=214 нм)(Column: YMC-C18, 30 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 95%A, 10-25 мин, 95-80%A, 25-55 мин, 80-55%AGradient: 0-10 min, 95%A, 10-25 min, 95-80%A, 25-55 min, 80-55%A
Фракцию с временем удержания 30 мин собирали и лиофилизировали с получением 45 мг продукта. LC-MS m/z (ES+): 1395,7 (M+H)+.The 30 min retention time fraction was collected and lyophilized to give 45 mg of product. LC-MS m/z (ES + ): 1395.7 (M+H) + .
Примеры 30-39Examples 30-39
Синтез соединений 30-39Synthesis of compounds 30-39

Пример 30Example 30
Получение соединения 30 Getting

С использованием 500-мл круглодонной колбы, карбонат калия (24,7 г, 179,24 ммоль) растворяли в 220 мл чистой воды, после чего добавляли (S)-3-амино-2-(бензилоксикарбониламино)пропионовую кислоту (16 г, 67,16 ммоль) и смесь перемешивали практически до полного растворения; далее добавляли 2-бромэтилдиэтилфосфат (10,98 г, 44,81 ммоль) и после завершения добавления смесь помещали на масляную баню при 80°C и перемешивали в течение шести часов до завершения реакции. Реакционную жидкость очищали с использованием жидкой фазы ВЭЖХ. (Колонка: YMC-C18, 100 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=214 нм)Using a 500 ml round bottom flask, potassium carbonate (24.7 g, 179.24 mmol) was dissolved in 220 ml of pure water, after which ( S )-3-amino-2-(benzyloxycarbonylamino) propionic acid (16 g, 67.16 mmol) and the mixture was stirred almost until complete dissolution; further, 2-bromoethyl diethyl phosphate (10.98 g, 44.81 mmol) was added and after completion of the addition, the mixture was placed in an oil bath at 80° C. and stirred for six hours until the reaction was complete. The reaction liquid was purified using HPLC liquid phase. (Column: YMC-C18, 100 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 5-15%B, 10-15 мин, 15%B, 15-25 мин, 15-20%B, 25-50 мин, 20-30%B, 50-55 мин, 30-50%B, 55-65 мин, 50-90%B. Фракцию с временем удержания 42-49 мин собирали и лиофилизировали с получением 8,38 г бесцветного маслянистого продукта с выходом 46,5%. Данные 1H-ЯМР (CDCl3, 400 МГц): 7,39-7,28 (м, 5H), 6,54-6,41 (м, 1H), 5,12-5,01 (м, 2H), 4,63-4,53 (м, 1H), 4,16-4,03 (м,4H), 3,48-3,36 (м,2H), 3,35-3,25 (м, 2H), 2,35-2,23 (м,2H), 1,29 (t, J=6,8Hz, 6H); LCMS[M+H]+ m/z 403,3 (вычислено для C17H27N2O7P, 402,16).Gradient: 0-10 min, 5-15%B, 10-15 min, 15%B, 15-25 min, 15-20%B, 25-50 min, 20-30%B, 50-55 min, 30 -50%B, 55-65 min, 50-90%B. The fraction with a retention time of 42-49 minutes was collected and lyophilized to obtain 8.38 g of a colorless oily product with a yield of 46.5%. 1 H-NMR data (CDCl 3 , 400 MHz): 7.39-7.28 (m, 5H), 6.54-6.41 (m, 1H), 5.12-5.01 (m, 2H ), 4.63-4.53 (m, 1H), 4.16-4.03 (m, 4H), 3.48-3.36 (m, 2H), 3.35-3.25 (m , 2H), 2.35-2.23 (m, 2H), 1.29 (t, J=6.8Hz, 6H); LCMS[M+H] + m/z 403.3 (calculated for C 17 H 27 N 2 O 7 P, 402.16).
Пример 31Example 31
Получение соединения 31 Getting connection 31
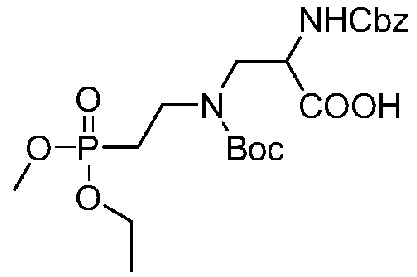
В 100-мл колбе с тремя горлышками соединение 30 (8,3 г, 20,85 ммоль) растворяли с использованием 50 мл растворителя дихлорметана и раствор помещали в низкотемпературный реактор, установленный на 0°C, в атмосфере газообразного азота, после чего капельно добавляли основание DIEA (6,7 г, 52,11 ммоль) при перемешивании и капельно добавляли Boc-ангидрид (5,0 г, 22,935 ммоль) в течение получаса; после завершения капельного добавления смесь помещали в условия комнатной температуры и перемешивали в течение 2 часов, и проводили мониторинг посредством TLC до завершения реакции (проявитель: DCM:MeOH=4:1). Последующая обработка: в реакционный раствор добавляли воду для промывания его три раза, органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, и полученный остаток очищали посредством ВЭЖХ. (Колонка: YMC-C18, 100 мм 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=214 нм)In a 100 ml three-necked flask, compound 30 (8.3 g, 20.85 mmol) was dissolved using 50 ml dichloromethane solvent and the solution was placed in a low temperature reactor set at 0° C. under nitrogen gas, after which DIEA base (6.7 g, 52.11 mmol) with stirring and Boc anhydride (5.0 g, 22.935 mmol) added dropwise over half an hour; after completion of the dropwise addition, the mixture was placed under room temperature conditions and stirred for 2 hours, and monitored by TLC until the reaction was completed (developer: DCM:MeOH=4:1). Post-treatment: Water was added to the reaction solution to wash it three times, the organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the resulting residue was purified by HPLC. (Column: YMC-C18, 100 mm 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0 мин, 40%B, 0-10 мин, 40%B, 10-20 мин, 40-45%B, 20-30 мин, 45%B, 30-45 мин, 45-80%B, 45-50 мин, 80-95%B. Фракцию с временем удержания 26-29,5 мин собирали и лиофилизировали с получением 4,02 г бесцветной маслянистой жидкости с выходом 38,3%. Данные 1H-ЯМР (CDCl3, 400 МГц):7,36-7,28 (м, 5H), 5,09 (с, 2H), 4,63-4,43 (м, 1H), 4..14-4,01 (м, 4H), 3,92-3,65 (м, 2H), 3,61-3,29 (м, 2H), 2,34-1,95 (м,2H), 1,45 (с,9H), 1,36-1,22 (м, 6H); LCMS[M+H]+m/z 503,4 (вычислено для C22H35N2O9P, 502,21).Gradient: 0 min, 40%B, 0-10 min, 40%B, 10-20 min, 40-45%B, 20-30 min, 45%B, 30-45 min, 45-80%B, 45 -50 min, 80-95%B. The fraction with a retention time of 26-29.5 minutes was collected and lyophilized to obtain 4.02 g of a colorless oily liquid with a yield of 38.3%. 1 H-NMR data (CDCl 3 , 400 MHz): 7.36-7.28 (m, 5H), 5.09 (s, 2H), 4.63-4.43 (m, 1H), 4. .14-4.01 (m, 4H), 3.92-3.65 (m, 2H), 3.61-3.29 (m, 2H), 2.34-1.95 (m, 2H) , 1.45 (s, 9H), 1.36-1.22 (m, 6H); LCMS[M+H] + m/z 503.4 (calculated for C 22 H 35 N 2 O 9 P, 502.21).
Пример 32Example 32
Получение соединения 32 Getting connection 32

В 150-мл круглодонной колбе 50 мл метанола использовали для растворения соединения 31 (4,0 г, 8,0 ммоль) после чего добавляли 800 мг Pd/C (содержащего 5% Pd) и заменяли газообразный водород, после чего реакционный раствор перемешивали при комнатной температуре в течение четырех часов; проводили мониторинг посредством TLC (проявитель: DCM:MeOH=4:1) до завершения реакции и реакцию останавливали. Фильтрацию через органическую мембрану использовали для удаления палладия на угле и проводили концентрирование при пониженном давлении с получением остатка, который прямо использовали на следующей стадии без необходимости в какой-либо очистке. LCMS[M+H]+m/z 369,4 (вычислено для C14H29N2O7P, 368,17).In a 150 ml round bottom flask, 50 ml of methanol was used to dissolve compound 31 (4.0 g, 8.0 mmol), after which 800 mg of Pd/C (containing 5% Pd) was added and hydrogen gas was replaced, after which the reaction solution was stirred at room temperature for four hours; monitored by TLC (developer: DCM:MeOH=4:1) until the completion of the reaction, and the reaction was stopped. Filtration through an organic membrane was used to remove the palladium-carbon and concentrated under reduced pressure to give a residue which was directly used in the next step without the need for any purification. LCMS[M+H] + m/z 369.4 (calculated for C 14 H 29 N 2 O 7 P, 368.17).
Пример 33Example 33
Получение соединения 33 Getting Compound 33

В 100-мл круглодонную колбу добавляли 30 мл ледяной уксусной кислоты для растворения неочищенного соединения 32, после чего добавляли малеиновый ангидрид (1,57 г, 16,0 ммоль) при перемешивании и реакции позволяли протекать при перемешивании при комнатной температуре в течение ночи. Реакцию останавливали на следующие сутки и ледяную уксусную кислоту удаляли посредством концентрирования при пониженном давлении с использованием масляного насоса при 45°C; затем полученный остаток очищали посредством ВЭЖХ. (Колонка: YMC-C18, 100 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=214 нм)To a 100 ml round bottom flask was added 30 ml of glacial acetic acid to dissolve the crude compound 32 , after which maleic anhydride (1.57 g, 16.0 mmol) was added with stirring and the reaction was allowed to proceed with stirring at room temperature overnight. The reaction was stopped the next day and glacial acetic acid was removed by concentration under reduced pressure using an oil pump at 45°C; then the resulting residue was purified by HPLC. (Column: YMC-C18, 100 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0 мин, 5%B, 0-10 мин, 5-15%B, 10-20 мин, 15-30%B, 20-35 мин, 30-55%B, 35-45 мин, 55%B, 45-55 мин, 55-80%B, 55-60 мин, 80-98%B. Фракцию с временем удержания 25-26,5 мин собирали и лиофилизировали с получением 3,35 г маслянистого вещества с выходом за две стадии 90%. Данные 1H-ЯМР (CDCl3, 400 МГц): 6,40 (д, J=12Hz, 1H), 6,35 (д, J=12Hz, 1H), 4,96 to 4,82 (м,1H), 4,19-3,99 (м, 4H), 3,98-3,61 (м, 2H), 3,60-3,36 (м, 2H), 2,22-2,00 (м, 2H), 1,45 (с, 9H), 1,33 (т, J=7,0Hz, 3H), 1,32 (т, J=7,0Hz, 3H); LCMS[M+H]+m/z 467,3 (вычислено для C18H31N2O10P, 466,17).Gradient: 0 min, 5%B, 0-10 min, 5-15%B, 10-20 min, 15-30%B, 20-35 min, 30-55%B, 35-45 min, 55%B , 45-55 min, 55-80%B, 55-60 min, 80-98%B. A fraction with a retention time of 25-26.5 minutes was collected and lyophilized to give 3.35 g of an oily substance with a 90% yield over two steps. 1 H-NMR data (CDCl 3 , 400 MHz): 6.40 (d, J=12Hz, 1H), 6.35 (d, J=12Hz, 1H), 4.96 to 4.82 (m, 1H ), 4.19-3.99 (m, 4H), 3.98-3.61 (m, 2H), 3.60-3.36 (m, 2H), 2.22-2.00 (m , 2H), 1.45 (s, 9H), 1.33 (t, J=7.0Hz, 3H), 1.32 (t, J=7.0Hz, 3H); LCMS[M+H] + m/z 467.3 (calculated for C 18 H 31 N 2 O 10 P, 466.17).
Пример 34Example 34
Получение соединения 34 Getting connection 34

В 100-мл круглодонной колбе соединение 33 (3,3 г, 7,07 ммоль) растворяли 40 мл толуола и 4 мл DMA при перемешивании, после чего добавляли триэтиламин (2,1 г, 21,2 ммоль) и устанавливали водосборник и обратный холодильник, и реакции позволяли протекать в условиях кипячения с обратным холодильником и перемешивания при 120°C в течение 3 часов до тех пор, пока мониторинг TLC не продемонстрировал, что реакция завершилась (проявитель: DCM:MeOH=4:1). Последующая обработка: реакционный раствор концентрировали при пониженном давлении при 45°C для удаления толуола и полученный остаток очищали посредством ВЭЖХ. (Колонка: YMC-C18, 100 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 200 мл/мин, детекция при λ=214 нм)In a 100 ml round bottom flask, compound 33 (3.3 g, 7.07 mmol) was dissolved with 40 ml of toluene and 4 ml of DMA with stirring, after which triethylamine (2.1 g, 21.2 mmol) was added and a trap and reflux were installed. refrigerator, and the reaction was allowed to proceed under reflux and stirring at 120°C for 3 hours until TLC monitoring showed that the reaction was complete (developer: DCM:MeOH=4:1). Post-treatment: The reaction solution was concentrated under reduced pressure at 45° C. to remove toluene, and the resulting residue was purified by HPLC. (Column: YMC-C18, 100 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0 мин, 5%B, 0-10 мин, 5-15%B, 10-20 мин, 15-30%B, 20-35 мин, 30-55%B, 35-45 мин, 55%B, 45-55 мин, 55-80%B, 55-60 мин, 80-98%B. Фракцию с временем удержания 27-29 мин собирали и лиофилизировали с получением 2,12 г белого порошкового твердого вещества (которое быстро впитывало влагу из воздуха и становилось вязким маслом), с выходом 67%. LCMS[M+H]+m/z 449,3 (вычислено для C18H29N2O9P, 448,16).Gradient: 0 min, 5%B, 0-10 min, 5-15%B, 10-20 min, 15-30%B, 20-35 min, 30-55%B, 35-45 min, 55%B , 45-55 min, 55-80%B, 55-60 min, 80-98%B. A fraction with a retention time of 27-29 minutes was collected and lyophilized to give 2.12 g of a white powdery solid (which rapidly absorbed moisture from the air and became a viscous oil) in 67% yield. LCMS[M+H] + m/z 449.3 (calculated for C 18 H 29 N 2 O 9 P, 448.16).
Пример 35Example 35
Получение соединения 35 Getting

100-мл круглодонную колбу, содержавшую соединение 34 (2,1 г, 4,69 ммоль), осушали с использованием масляного насоса, добавляли 20 мл сухого DMF и полученную перемешивали до полного растворения; далее добавляли DIEA (1,8 г, 14,07 ммоль), а затем EDCI (1,81 г, 9,38 ммоль) и HoAt (1,28 г, 9,38 ммоль), и активированное соединение перемешивали при комнатной температуре в течение пяти с половиной часов, после чего добавляли соединение 6 (2,66 г, 7,03 ммоль) и реакции позволяли протекать при комнатной температуре в течение ночи, а затем ее останавливали на следующие сутки. Последующая обработка: растворитель DMF удаляли посредством концентрирования при пониженном давлении при 45°C с использованием масляного насоса и полученный остаток очищали с использованием колоночной хроматографии (элюент: дихлорметан:метанол=от 20:1 до 10:1 до 5:1) с получением 1,01 г белого порошкового твердого вещества с выходом 26,6%. LCMS[M+H]+m/z 810,4 (вычислено для C36H56N7O12P, 809,37).A 100 ml round bottom flask containing compound 34 (2.1 g, 4.69 mmol) was dried using an oil pump, 20 ml of dry DMF was added and the resulting mixture was stirred until complete dissolution; further DIEA (1.8 g, 14.07 mmol) was added followed by EDCI (1.81 g, 9.38 mmol) and HoAt (1.28 g, 9.38 mmol) and the activated compound was stirred at room temperature for five and a half hours, after which compound 6 (2.66 g, 7.03 mmol) was added and the reaction was allowed to proceed at room temperature overnight, and then it was stopped the next day. Post-treatment: The DMF solvent was removed by concentration under reduced pressure at 45°C using an oil pump, and the resulting residue was purified using column chromatography (eluent: dichloromethane:methanol=20:1 to 10:1 to 5:1) to obtain 1 .01 g of a white powdery solid in 26.6% yield. LCMS[M+H] + m/z 810.4 (calculated for C 36 H 56 N 7 O 12 P, 809.37).
Пример 36Example 36
Получение соединения 36 Getting Compound 36

В 50-мл круглодонной колбе 15 мл сухого DMF использовали для растворения соединения 35 (1,0 г, 1,24 ммоль) при перемешивании, после чего добавляли DIEA (961 мг, 7,44 ммоль) и NPC (1,88 г, 6,2 ммоль) и реакционной смеси позволяли реагировать при комнатной температуре в течение ночи при перемешивании; на следующее утро мониторинг посредством TLC продемонстрировал, что реакция завершилась (проявитель: дихлорметан:метанол=5:1) и реакцию останавливали. Последующая обработка: реакционный раствор концентрировали при пониженном давлении с использованием масляного насоса при 45°C, и полученный остаток очищали посредством тонкослойной хроматографии (проявитель: дихлорметан:метанол=6:1) с получением 1,02 г белого порошкового твердого вещества с выходом 84,5%. LCMS[M+H]+m/z 975,3 (вычислено для C43H59N8O16P, 974,38).In a 50 ml round bottom flask, 15 ml dry DMF was used to dissolve compound 35 (1.0 g, 1.24 mmol) with stirring, after which DIEA (961 mg, 7.44 mmol) and NPC (1.88 g, 6.2 mmol) and the reaction mixture was allowed to react at room temperature overnight with stirring; the next morning monitoring by TLC showed that the reaction was complete (developer: dichloromethane:methanol=5:1) and the reaction was stopped. Post-treatment: The reaction solution was concentrated under reduced pressure using an oil pump at 45°C, and the resulting residue was purified by thin layer chromatography (developer: dichloromethane:methanol=6:1) to obtain 1.02 g of a white powdery solid with a yield of 84, 5%. LCMS[M+H] + m/z 975.3 (calculated for C 43 H 59 N 8 O 16 P, 974.38).
Пример 37Example 37
Получение соединения 37 Getting Compound 37

В 100-мл круглодонной колбе 10 мл сухого DMF использовали для растворения соединения 36 (1,0 г, 1,03 ммоль), после чего добавляли HoBt (28 мг, 0,21 ммоль) и DIEA (266 мг, 2,06 ммоль), и результирующей реакции позволяли протекать при перемешивании в течение получаса; далее добавляли MMAE (740 мг, 1,03 ммоль) и реакции позволяли протекать в течение ночи при комнатной температуре. Последующая обработка: реакционный раствор очищали посредством жидкофазной препаративной хроматографии. (Колонка: YMC-C18, 50 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 50 мл/мин, детекция при λ=214 нм)In a 100 ml round bottom flask, 10 ml dry DMF was used to dissolve compound 36 (1.0 g, 1.03 mmol) followed by the addition of HoBt (28 mg, 0.21 mmol) and DIEA (266 mg, 2.06 mmol ), and the resulting reaction was allowed to proceed with stirring for half an hour; further MMAE (740 mg, 1.03 mmol) was added and the reaction was allowed to proceed overnight at room temperature. Post-treatment: The reaction solution was purified by preparative liquid phase chromatography. (Column: YMC-C18, 50 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0 мин, 5%B, 0-10 мин, 5-15%B, 10-20 мин, 15-30%B, 20-35 мин, 30-55%B, 35-45 мин, 55-70%B, 45-55 мин, 70%B, 55-60 мин, 70-98%B.Gradient: 0 min, 5%B, 0-10 min, 5-15%B, 10-20 min, 15-30%B, 20-35 min, 30-55%B, 35-45 min, 55-70 %B, 45-55 min, 70%B, 55-60 min, 70-98%B.
Фракцию с временем удержания 38-40 мин собирали и лиофилизировали с получением 320 мг белого порошкового твердого вещества с выходом 20%. LCMS[M+H]+m/z 1553,8 (вычислено для C76H121N12O20P, 1552,85).The 38-40 min retention time fraction was collected and lyophilized to give 320 mg of a white powdery solid in 20% yield. LCMS[M+H] + m/z 1553.8 (calculated for C 76 H 121 N1 2 O 20 P, 1552.85).
Пример 38Example 38
Получение соединения 38 Getting connection 38

Соединение 37 (260 мг, 0,17 ммоль) растворяли в 25-мл круглодонной колбе с использованием 5 мл дистиллированного дихлорметана, после чего добавляли бромтриметилсилан (78 мг, 0,51 ммоль) на ледяной бане в газообразном азоте; реакции позволяли протекать в течение ночи при комнатной температуре при перемешивании и на следующее утро добавляли 1 мл метанола, после чего реакции позволяли протекать при перемешивании при комнатной температуре в течение одного часа и жидкофазный анализ использовали для подтверждения того, что соединение 37 полностью прореагировало. Последующая обработка: реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали посредством ВЭЖХ. (Колонка: YMC-C18, 30 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=214 нм)Compound 37 (260 mg, 0.17 mmol) was dissolved in a 25 ml round bottom flask using 5 ml of distilled dichloromethane, followed by the addition of bromotrimethylsilane (78 mg, 0.51 mmol) in an ice bath under nitrogen gas; the reaction was allowed to proceed overnight at room temperature with stirring and the
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0 мин, 5%B, 0-10 мин, 5-15%B, 10-20 мин, 15-30%B, 20-35 мин, 30-55%B, 35-45 мин, 55-70%B, 45-55 мин, 70%B, 55-60 мин, 70-98%B.Gradient: 0 min, 5%B, 0-10 min, 5-15%B, 10-20 min, 15-30%B, 20-35 min, 30-55%B, 35-45 min, 55-70 %B, 45-55 min, 70%B, 55-60 min, 70-98%B.
Фракцию с временем удержания 20-23 мин собирали и лиофилизировали с получением 140 мг белого порошкового твердого вещества с выходом 55%.The 20-23 min retention time fraction was collected and lyophilized to give 140 mg of a white powdery solid in 55% yield.
Пример 39Example 39
Получение соединения 39 Getting

В 25-мл круглодонной колбе 2 мл дихлорметана использовали для растворения соединения 38 (120 мг, 0,08 ммоль) при перемешивании, после чего капельно добавляли 800 микролитров трифторуксусной кислоты на ледяной бане; после капельного добавления реакционный раствор помещали в условия комнатной температуры и реакции позволяли протекать при перемешивании в течение одного часа до завершения реакции. Последующая обработка: реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали посредством ВЭЖХ. (Колонка: YMC-C18, 30 мм×450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=214 нм)In a 25 ml round bottom flask, 2 ml of dichloromethane was used to dissolve compound 38 (120 mg, 0.08 mmol) with stirring, after which 800 microliters of trifluoroacetic acid were added dropwise in an ice bath; after the dropwise addition, the reaction solution was placed under room temperature conditions, and the reaction was allowed to proceed with stirring for one hour to complete the reaction. Post-treatment: The reaction solution was concentrated under reduced pressure, and the resulting residue was purified by HPLC. (Column: YMC-C18, 30 mm×450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0 мин, 5%B, 0-10 мин, 5-15%B, 10-20 мин, 15-30%B, 20-35 мин, 30-55%B, 35-45 мин, 55-70%B, 45-55 мин, 70%B, 55-60 мин, 70-98%B.Gradient: 0 min, 5%B, 0-10 min, 5-15%B, 10-20 min, 15-30%B, 20-35 min, 30-55%B, 35-45 min, 55-70 %B, 45-55 min, 70%B, 55-60 min, 70-98%B.
Фракцию с временем удержания 18-19,5 мин собирали и лиофилизировали с получением 55 мг белого порошкового твердого вещества с выходом 49%. LC-MS m/z (ES+): 1397,8 (M+H)+.The 18-19.5 min retention time fraction was collected and lyophilized to give 55 mg of a white powdery solid in 49% yield. LC-MS m/z (ES + ): 1397.8 (M+H) + .
Пример 40Example 40
Получение соединения 40 Getting

Соединение 7 (332 мг, 0,836 ммоль) растворяли в сухом дихлорметане, добавляли пентафторфенол (184 мг, 1 ммоль) и дициклогексилкарбодиимид (2 ммоль) и проводили перемешивание при комнатной температуре в течение трех часов до устранения соединения 7. Добавляли MMAF (460 мг, 0,585 ммоль) и диизопропилэтиламин (DIEA, 0,27 мл, 1,67 ммоль) и растворяли в 15 мл сухого раствора дихлорметана и полученной реакции позволяли протекать при комнатной температуре в течение четырех часов в газообразном азоте. Раствор концентрировали при пониженном давлении и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=50:1) с получением 290 мг продукта. LC-MS m/z (ES+): 1168,8 (M+H)+.Compound 7 (332 mg, 0.836 mmol) was dissolved in dry dichloromethane, pentafluorophenol (184 mg, 1 mmol) and dicyclohexylcarbodiimide (2 mmol) were added and stirring was carried out at room temperature for three hours until compound 7 was eliminated. MMAF (460 mg, 0.585 mmol) and diisopropylethylamine (DIEA, 0.27 ml, 1.67 mmol) were added and dissolved in 15 ml dry dichloromethane solution and the resulting reaction was allowed to proceed at room temperature for four hours under nitrogen gas. The solution was concentrated under reduced pressure, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=50:1) to obtain 290 mg of a product. LC-MS m/z (ES + ): 1168.8 (M+H) + .
Пример 41Example 41
Получение соединения 41 Getting

100 мг соединения 39 растворяли в 10 мл сухого дихлорметана, добавляли 4 мл трифторуксусной кислоты и реакции позволяли протекать при комнатной температуре в течение двух, а затем проводили очистку посредством ВЭЖХ.100 mg of
(Колонка: YMC-C18, 30 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=215 нм)(Column: YMC-C18, 30 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 95%A, 10-25 мин, 95-70%A, 25-55 мин, 70-40%A.Gradient: 0-10 min, 95%A, 10-25 min, 95-70%A, 25-55 min, 70-40%A.
Фракцию с временем удержания 33 мин собирали и лиофилизировали с получением 50 мг продукта. LC-MS m/z (ES+): 956,6 (M+H)+.The 33 min retention time fraction was collected and lyophilized to give 50 mg of product. LC-MS m/z (ES + ): 956.6 (M+H) + .
Пример 42Example 42
Получение соединения 42 Getting Compound 42

Соединение 16 (400 мг, 0,76 ммоль) растворяли в сухом дихлорметане, добавляли пентафторфенол (184 мг, 1 ммоль) и дициклогексилкарбодиимид (2 ммоль) и проводили перемешивание при комнатной температуре в течение четырех часов до устранения соединения 16. Добавляли MMAF (389 мг, 0,49 ммоль) и диизопропилэтиламин (DIEA, 0,25 мл, 1,52 ммоль) и растворяли в 15 мл сухого дихлорметана и итоговой реакции позволяли протекать при комнатной температуре в течение четырех часов в газообразном азоте. Раствор концентрировали при пониженном давлении и полученный остаток подвергали разделению с использованием колоночной хроматографии на силикагеле (элюент: дихлорметан:метанол=50:1) с получением 330 мг продукта. LC-MS m/z (ES+): 1296,8 (M+H)+.Compound 16 (400 mg, 0.76 mmol) was dissolved in dry dichloromethane, pentafluorophenol (184 mg, 1 mmol) and dicyclohexylcarbodiimide (2 mmol) were added and stirring was carried out at room temperature for four hours until compound 16 was eliminated. MMAF (389 mg, 0.49 mmol) and diisopropylethylamine (DIEA, 0.25 ml, 1.52 mmol) were added and dissolved in 15 ml dry dichloromethane and the final reaction was allowed to proceed at room temperature for four hours under nitrogen gas. The solution was concentrated under reduced pressure, and the resulting residue was subjected to separation using silica gel column chromatography (eluent: dichloromethane:methanol=50:1) to obtain 330 mg of a product. LC-MS m/z (ES + ): 1296.8 (M+H) + .
Пример 43Example 43
Получение соединения 43 Getting Compound 43

100 мг соединения 41 растворяли в 10 мл сухого дихлорметана, добавляли 4 мл трифторуксусной кислоты добавляли и реакции позволяли протекать при комнатной температуре в течение двух часов с последующей очисткой посредством ВЭЖХ.100 mg of
(Колонка: YMC-C18, 30 мм × 450 мм, 10 мкм, ацетонитрил/вода (0,2% TFA), скорость потока 25 мл/мин, детекция при λ=215 нм)(Column: YMC-C18, 30 mm × 450 mm, 10 µm, acetonitrile/water (0.2% TFA),
Растворитель A: 0,2% TFA в водеSolvent A: 0.2% TFA in water
Растворитель B: АцетонитрилSolvent B: Acetonitrile
Градиент: 0-10 мин, 95%A, 10-25 мин, 95-70%A, 25-55 мин, 70-40%AGradient: 0-10 min, 95%A, 10-25 min, 95-70%A, 25-55 min, 70-40%A
Фракцию с временем удержания 30 мин собирали и лиофилизировали с получением 48 мг продукта. LC-MS m/z (ES+): 1028,5 (M+H)+.The 30 min retention time fraction was collected and lyophilized to give 48 mg of product. LC-MS m/z (ES + ): 1028.5 (M+H) + .
Пример 44Example 44
Конъюгат антитело-лекарственное средство H-11 получали согласно общему способу B, и обращено-фазовая ВЭЖХ продемонстрировала, что средняя величина соотношения лекарственное средство/антитело (DAR) составляла 7,2.The H-11 antibody-drug conjugate was prepared according to General Method B and reverse phase HPLC showed that the average drug/antibody ratio (DAR) was 7.2.
Пример 45Example 45
Конъюгат антитело-лекарственное средство H-20 получали согласно общему способу B, и обращено-фазовая ВЭЖХ продемонстрировала, что средняя величина соотношения лекарственное средство/антитело (DAR) составляла 7,2.The H-20 antibody-drug conjugate was prepared according to General Method B and reverse phase HPLC showed that the average drug/antibody ratio (DAR) was 7.2.
Пример 46Example 46
Конъюгат антитело-лекарственное средство H-29 получали согласно общему способу B, и обращено-фазовая ВЭЖХ продемонстрировала, что средняя величина соотношения лекарственное средство/антитело (DAR) составляла 6,8.The H-29 antibody-drug conjugate was prepared according to General Method B and reverse phase HPLC showed that the average drug/antibody ratio (DAR) was 6.8.
Пример 47Example 47
Конъюгат антитело-лекарственное средство H-39 получали согласно общему способу B, и обращено-фазовая ВЭЖХ продемонстрировала, что средняя величина соотношения лекарственное средство/антитело (DAR) составляла 7,0.The H-39 antibody-drug conjugate was prepared according to General Method B and reverse phase HPLC showed that the average drug/antibody ratio (DAR) was 7.0.
Пример 48Example 48
Конъюгат антитело-лекарственное средство H-41 получали согласно общему способу B, и обращено-фазовая ВЭЖХ продемонстрировала, что средняя величина соотношения лекарственное средство/антитело (DAR) составляла 7,5.The H-41 antibody-drug conjugate was prepared according to General Method B and reverse phase HPLC showed that the average drug/antibody ratio (DAR) was 7.5.
Пример 49Example 49
Конъюгат антитело-лекарственное средство H-43 получали согласно общему способу B, и обращено-фазовая ВЭЖХ продемонстрировала, что средняя величина соотношения лекарственное средство/антитело (DAR) составляла 7,7.The H-43 antibody-drug conjugate was prepared according to General Method B and reverse phase HPLC showed that the average drug/antibody ratio (DAR) was 7.7.
Пример 50Example 50
Синтез продолжали с получением следующих линкеров лекарственное средство-участок соединения:The synthesis was continued to give the following drug-site linkers:

где Ac обозначает неосновную аминокислоту или олигопептид, и аминогруппа в аминокислоте соединена с алкильной группой;where Ac denotes a non-basic amino acid or oligopeptide, and the amino group in the amino acid is connected to an alkyl group;
n=1,2, 3...;n=1,2, 3...;
В таблице 1 ниже обобщенно представлен синтез и характеристики участков присоединения лекарственного средства, содержащих различные аминокислоты и олигопептиды. В таблице в первой колонке слева представлен номер соединения, во второй колонке представлен класс аминокислоты, в третьей колонке представлена величина n, в четвертой колонке представлен общий способ синтеза и в пятой и шестой колонках показана вычисленная масса соединения лекарственное средство-участок присоединения и масса при определении при определении посредством масс-спектрометрии.Table 1 below summarizes the synthesis and characterization of drug attachment sites containing various amino acids and oligopeptides. In the table, the first column on the left is the compound number, the second column is the amino acid class, the third column is the value of n, the fourth column is the general synthesis method, and the fifth and sixth columns show the calculated mass of the compound drug-site of attachment and the mass at determination when determined by mass spectrometry.
Таблица 1Table 1
Сокращенные обозначения:Abbreviations:
Gly: L-глицин, Ala: L-аланин, Phe: L-фенилаланин, Trp: L-тирозин, Asp: L-аспарагиновая кислота, Ser: L-серин, Val: L- ВалинGly: L-glycine, Ala: L-alanine, Phe: L-phenylalanine, Trp: L-tyrosine, Asp: L-aspartic acid, Ser: L-serine, Val: L-Valine

Примеры 52-56Examples 52-56
Для оценки стабильности, гидрофильности и фармакологической активности ADC, полученных с использованием кислотного самостабилизирующегося участка присоединения, авторы настоящего изобретения получили соединения лекарственное средство-участок присоединения с кислотными стабилизирующими участками присоединения согласно примерам. Все эти соединения имели кислотный стабилизированный участок присоединения, который был соединен с цитотоксическим лекарственным средством MMAE или MMAF через расщепляющийся или не расщепляющийся линкер. В качестве сравнения типичное не самостабилизирующееся соединение лекарственное средство-участок присоединения (далее обозначаемое как MC-VC-PAB-MMAE) получали способами, приведенными в литературе, и получали соединение лекарственное средство-участок присоединения, имеющее основный самостабилизирующийся участок присоединения, как описано в данном патенте (далее обозначаемый как DPR-VC-PAB-MMAE).To evaluate the stability, hydrophilicity and pharmacological activity of ADCs prepared using an acidic self-stabilizing attachment site, the present inventors prepared drug-attachment compounds with acidic stabilizing attachment sites according to the examples. All of these compounds had an acid stabilized attachment site that was linked to the cytotoxic drug MMAE or MMAF via a cleavable or non-cleavable linker. As a comparison, a typical non-self-stabilizing compound drug-attachment site (hereinafter referred to as MC-VC-PAB-MMAE) was prepared by the methods described in the literature, and a compound drug-attachment site having a basic self-stabilizing attachment site was obtained, as described in this patent (hereinafter referred to as DPR-VC-PAB-MMAE).


Пример 52: Стабильность в плазме in vitro Example 52 In Vitro Plasma Stability
Результаты исследований стабильности в плазме, проведенные согласно способом, описанным общей методике D, были такими, как показано в таблице ниже: ADC с кислотными стабилизированными участками присоединения практически не утрачивают лекарственное средство в ходе инкубации в плазме, в то время как типичный ADC с участком присоединения MC продемонстрировал очень значительное снижение DAR после инкубации в течение 72 часов. Результаты эксперимента демонстрируют, что кислотный стабилизированный участок присоединения может значительно повышать стабильность ADC в плазме.The results of plasma stability studies conducted according to the method described in General Procedure D were as shown in the table below: ADCs with acid-stabilized attachment sites show little to no drug loss during plasma incubation, while a typical ADC with an attachment site MC showed a very significant decrease in DAR after incubation for 72 hours. The results of the experiment demonstrate that the acid stabilized attachment site can significantly increase the stability of ADC in plasma.
Пример 53: Детекция посредством SEC-ВЭЖХExample 53: Detection by SEC-HPLC
Образцы ADC, полученные посредством конъюгации, центрифугировали при 14000 об/мин в течение пяти минут, и проводили взятие супернатанта для анализа.ADC samples obtained by conjugation were centrifuged at 14,000 rpm for five minutes, and the supernatant was collected for analysis.
Устройство: Waters e2695 (2489UV/Vis)Device: Waters e2695 (2489UV/Vis)
Хроматография колонка: TSKgel G3000SWXL (7,8×300 мм, 5 мкм)Chromatography column: TSKgel G3000SWXL (7.8×300 mm, 5 µm)
Подвижная фаза: A: 50 мМ PB, 300 мМ NaCl, 200 мМ Arg, 5% IPA, pH 6,5Mobile phase: A: 50 mM PB, 300 mM NaCl, 200 mM Arg, 5% IPA, pH 6.5
Подвижную фазу A подвергали изократическому элюированию в течение 30 минут; скорость потока: 0,714 мл/мин, температура колонки: 25°C, длина волны детекции: 280 нм.Mobile phase A was subjected to isocratic elution for 30 minutes; flow rate: 0.714 ml/min, column temperature: 25°C, detection wavelength: 280 nm.
Степень агрегации каждого ADC определяли посредством SEC-ВЭЖХ и графики пиков SEC-ВЭЖХ представлены на фиг.2A-2H; соответствующие данные обобщенно представлены в таблице ниже, где уровень мономера значительно возрастал и степень агрегации значительно снижалась в ADC, которые включали кислотный стабилизированный участок присоединения.The degree of aggregation of each ADC was determined by SEC-HPLC and SEC-HPLC peak plots are shown in FIGS. 2A-2H; the corresponding data are summarized in the table below, where the level of monomer increased significantly and the degree of aggregation decreased significantly in ADCs that included an acid stabilized attachment site.
Пример 54Example 54
Эксперименты PK на мышах in vivo PK experiments in mice in vivo
Свойства PK для ADC (с нагрузкой 8) определяли с использованием модели на мышах. Лекарственное средство вводили посредством однократной внутривенной инъекции в хвост в дозе 2 мг/кг, и концентрацию тотальных антител (тотальные Ab) в крови оценивали в соответствии с общей методикой B; результаты представлены на фиг.3. Результаты эксперимента показывают, что в модели на мышах ADC с кислотным стабилизированным участком присоединения продемонстрировал значительно улучшенные свойства PK с концентрацией тотальных антител, поддерживаемой в течение более длительного периода времени.PK properties for ADC (load 8) were determined using a mouse model. The drug was administered by a single intravenous injection in the tail at a dose of 2 mg/kg, and the concentration of total antibodies (total Ab) in the blood was evaluated according to the General method B; the results are presented in Fig.3. The results of the experiment show that in a mouse model, ADC with an acid-stabilized attachment site showed significantly improved PK properties with total antibody concentration maintained for a longer period of time.
Пример 55Example 55
Анализ с использованием хроматографии гидрофобного взаимодействия (HIC)Analysis using hydrophobic interaction chromatography (HIC)
После полного восстановления IgG1-антитела до 8 тиолов на антитело согласно общей методике A, соответствующие конъюгаты антитело-лекарственное средство получали посредством неспецифического связывания. ADC с кислотными стабилизированными участками присоединения и ADC с общепринятыми участками присоединения (т.е. MC-VC-PAB-MMAE) с восемью элементами лекарственного средства на антитело далее разделяли и очищали посредством хроматографии гидрофобного взаимодействия и хроматографию (HIC) гидрофобного взаимодействия использовали для проведения анализа ADC согласно общей методике C. ADC с большей гидрофобностью и более высокими соотношениями лекарственное средство/молекула элюировались с более длительным временем удержания. Результаты представлены на фиг.4: ADC с кислотными участками присоединения продемонстрировали относительно короткое время удержания в HIC, причем конъюгат антитела MC-VC-PAB-MMAE демонстрировал наиболее длительное время удержания. Результаты эксперимента демонстрируют, что молекулы ADC, обладающие кислотным стабилизированным участком присоединения, демонстрируют лучшую гидрофильность.After complete reduction of the IgG1 antibody to 8 thiols per antibody according to General Method A, the corresponding antibody-drug conjugates were obtained by non-specific binding. ADCs with acid stabilized attachment sites and ADCs with conventional attachment sites (i.e. MC-VC-PAB-MMAE) with eight drug units per antibody were further separated and purified by hydrophobic interaction chromatography, and hydrophobic interaction chromatography (HIC) was used to perform analysis of ADCs according to General Method C. ADCs with greater hydrophobicity and higher drug/molecule ratios eluted with longer retention times. The results are shown in Figure 4: ADCs with acidic attachment sites showed a relatively short retention time in the HIC, with the MC-VC-PAB-MMAE antibody conjugate showing the longest retention time. The experimental results demonstrate that ADC molecules having an acidic stabilized attachment site show better hydrophilicity.
Пример 56Example 56
Эксперименты по определению эффективности лекарственного средства in vivo (анализ клеточной пролиферации) In vivo drug efficacy experiments (cell proliferation assay)
Клетки аденокарценомы поджелудочной железы человека (BxPc3) культивировали in vitro и инокулировали подкожно в бок мышей BALB/c с инокулируемым количеством клеток 5×106 и после вырастания опухолей до 100-200 мм3, животных распределяли на группы и им вводили однократную дозу ADC 3 мг/кг (инъецируемую через хвостовую вену) в то время как также получали контрольную группу носителя; мышей подвергали регулярному взвешиванию и измерению объема опухоли, и эффективность лекарственного средства в отношении модели BxPc3 оценивали путем исследования эффективности подавления опухоли посредством ADC, а также других индикаторов. Результаты эксперимента представлены на фиг.5 и 6. Результаты показывают, что ADC, обладающие кислотным стабилизированным участком присоединения, демонстрируют сходную эффективность с Dpr-MC-VC-PAB-MMAE при DAR=2, и значительно лучше, чем Mc-VC-PAB-MMAE, в этом отношении. При высокой нагрузке лекарственным средством DAR=8, те же ADC продемонстрировали высокую эффективность в модели с подкожным трансплантатом опухоли BxPc3 на мышах.Human pancreatic adenocarcinoma cells (BxPc3) were cultured in vitro and inoculated subcutaneously into the flank of BALB/c mice with an inoculated cell number of 5×10 6 and after tumor growth to 100-200 mm 3 , the animals were divided into groups and they were injected with a single dose of
Claims (36)



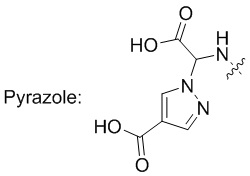


Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710462790.9 | 2017-06-19 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2020137091A RU2020137091A (en) | 2022-05-12 |
| RU2793125C2 true RU2793125C2 (en) | 2023-03-29 |
Family
ID=
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004010957A2 (en) * | 2002-07-31 | 2004-02-05 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| EA201490095A1 (en) * | 2011-06-21 | 2014-05-30 | Иммуноджен, Инк. | NEW DERIVATIVES OF MAYTANZINOIDS WITH PEPTIDE LINKER AND THEIR CONJUGATES |
| WO2016147031A2 (en) * | 2015-03-19 | 2016-09-22 | Hangzhou Dac Biotech Co., Ltd | Novel hydrophilic linkers and ligand-drug conjugates thereof |
| EA201691650A1 (en) * | 2014-02-17 | 2017-04-28 | Сиэтл Дженетикс, Инк. | HYDROPHILIC CONJUGATES ANTIBODY AND MEDICINE |
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004010957A2 (en) * | 2002-07-31 | 2004-02-05 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| EA201490095A1 (en) * | 2011-06-21 | 2014-05-30 | Иммуноджен, Инк. | NEW DERIVATIVES OF MAYTANZINOIDS WITH PEPTIDE LINKER AND THEIR CONJUGATES |
| EA201691650A1 (en) * | 2014-02-17 | 2017-04-28 | Сиэтл Дженетикс, Инк. | HYDROPHILIC CONJUGATES ANTIBODY AND MEDICINE |
| WO2016147031A2 (en) * | 2015-03-19 | 2016-09-22 | Hangzhou Dac Biotech Co., Ltd | Novel hydrophilic linkers and ligand-drug conjugates thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018288029B2 (en) | Antibody-drug conjugate having acidic self-stabilization junction | |
| CN108853514B (en) | Antibody drug conjugates with two different drugs | |
| JP2022105640A (en) | Complex of quaternized tubular lysine compounds | |
| CN111542344A (en) | Ligand-drug-conjugates comprising a single molecular weight polymyosine | |
| WO2019034176A1 (en) | Camptothecin-antibody conjugate | |
| JP2024503074A (en) | Camptothecin antibody-drug conjugate and method of use thereof | |
| TW201400131A (en) | Self-stabilizing linker conjugates | |
| CN107043406B (en) | Compounds, Linker-Drugs, and Ligand-Drug Conjugates | |
| CN111989339A (en) | Hydrophobic auristatin F compound and its conjugate | |
| CN111001012A (en) | Hydrophilic carbonate type antibody coupling drug | |
| WO2024012569A1 (en) | Linkers, conjugates and applications thereof | |
| US11319341B2 (en) | Immune-stimulating soluble doxorubicin-conjugated complex | |
| CN116847885A (en) | Double-cleaved ester linkers for antibody-drug conjugates | |
| RU2793125C2 (en) | Antibody-drug conjugate having an acid self-stabilizing binding site | |
| WO2024207177A9 (en) | Antibody, linkers, payload, conjugates and applications thereof | |
| CA3095067C (en) | An antibody-drug conjugate having an acidic self-stabilization junction | |
| HK40035178A (en) | Antibody-drug conjugate having acidic self-stabilization junction | |
| JP2025533577A (en) | Anti-CD33 antibodies and anti-CD33 antibody-drug conjugates and uses thereof | |
| WO2025113659A1 (en) | Trisaccharide linker, linker-payload comprising trisaccharide linker, and glycan chain-remodeled antibody-drug conjugate, and preparation methods therefor and uses thereof | |
| CN121422246A (en) | Anti-human ROR1 antibody-drug conjugates and their uses | |
| HK40076663B (en) | Selective drug release from internalized conjugates of biologically active compounds | |
| HK40043154A (en) | Hydrophobic auristatin f compounds and conjugates thereof |