RU2236412C2 - Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone - Google Patents
Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone Download PDFInfo
- Publication number
- RU2236412C2 RU2236412C2 RU2002119134/04A RU2002119134A RU2236412C2 RU 2236412 C2 RU2236412 C2 RU 2236412C2 RU 2002119134/04 A RU2002119134/04 A RU 2002119134/04A RU 2002119134 A RU2002119134 A RU 2002119134A RU 2236412 C2 RU2236412 C2 RU 2236412C2
- Authority
- RU
- Russia
- Prior art keywords
- derivatives
- oxidation
- benzyl
- morphinone
- take
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 28
- PFBSOANQDDTNGJ-YNHQPCIGSA-N morphinone Chemical class O([C@H]1C(C=C[C@H]23)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O PFBSOANQDDTNGJ-YNHQPCIGSA-N 0.000 title claims abstract description 26
- VFNHLTBRAFJQER-ISWURRPUSA-N (4r,4as,7ar,12bs)-4a,9-dihydroxy-3-methyl-2,4,7a,13-tetrahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-one Chemical class O=C([C@@H]1O2)C=C[C@@]3(O)[C@]4([H])N(C)CC[C@]13C1=C2C(O)=CC=C1C4 VFNHLTBRAFJQER-ISWURRPUSA-N 0.000 title claims abstract description 15
- UQCNKQCJZOAFTQ-ISWURRPUSA-N Oxymorphone Chemical class O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O UQCNKQCJZOAFTQ-ISWURRPUSA-N 0.000 title claims description 12
- 230000003647 oxidation Effects 0.000 claims abstract description 23
- 238000007254 oxidation reaction Methods 0.000 claims abstract description 23
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical class O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 claims abstract description 15
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims abstract description 14
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 11
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims abstract description 10
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 4
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 claims abstract 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 15
- 238000006243 chemical reaction Methods 0.000 claims description 12
- 150000002085 enols Chemical class 0.000 claims description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 8
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 8
- 150000003839 salts Chemical class 0.000 claims description 7
- 150000002148 esters Chemical class 0.000 claims description 5
- 239000001257 hydrogen Substances 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 229910052757 nitrogen Inorganic materials 0.000 claims description 4
- 238000005984 hydrogenation reaction Methods 0.000 claims description 3
- 239000003960 organic solvent Substances 0.000 claims description 3
- 230000020335 dealkylation Effects 0.000 claims description 2
- 238000006900 dealkylation reaction Methods 0.000 claims description 2
- 230000001590 oxidative effect Effects 0.000 claims description 2
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical class NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims 4
- 238000009795 derivation Methods 0.000 claims 2
- 229910052783 alkali metal Inorganic materials 0.000 claims 1
- -1 alkali metal salts Chemical class 0.000 claims 1
- 230000029936 alkylation Effects 0.000 claims 1
- 238000005804 alkylation reaction Methods 0.000 claims 1
- 150000001875 compounds Chemical class 0.000 abstract description 5
- CUILPNURFADTPE-UHFFFAOYSA-N hypobromous acid Chemical class BrO CUILPNURFADTPE-UHFFFAOYSA-N 0.000 abstract description 4
- 239000005557 antagonist Substances 0.000 abstract description 3
- 238000003745 diagnosis Methods 0.000 abstract description 3
- 239000003814 drug Substances 0.000 abstract description 3
- 229940127240 opiate Drugs 0.000 abstract description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 abstract description 2
- 125000004093 cyano group Chemical group *C#N 0.000 abstract description 2
- SATVIFGJTRRDQU-UHFFFAOYSA-N potassium hypochlorite Chemical class [K+].Cl[O-] SATVIFGJTRRDQU-UHFFFAOYSA-N 0.000 abstract description 2
- 239000011734 sodium Substances 0.000 abstract description 2
- 229910052708 sodium Inorganic materials 0.000 abstract description 2
- 239000000126 substance Substances 0.000 abstract 2
- 239000003795 chemical substances by application Substances 0.000 abstract 1
- 230000000694 effects Effects 0.000 abstract 1
- 230000007721 medicinal effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 14
- 238000004519 manufacturing process Methods 0.000 description 11
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 10
- 239000000047 product Substances 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 5
- 229960004126 codeine Drugs 0.000 description 5
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 5
- FQXXSQDCDRQNQE-UHFFFAOYSA-N markiertes Thebain Natural products COC1=CC=C2C(N(CC3)C)CC4=CC=C(OC)C5=C4C23C1O5 FQXXSQDCDRQNQE-UHFFFAOYSA-N 0.000 description 5
- 239000003401 opiate antagonist Substances 0.000 description 5
- 239000007800 oxidant agent Substances 0.000 description 5
- 229910052938 sodium sulfate Inorganic materials 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 229930003945 thebaine Natural products 0.000 description 5
- FQXXSQDCDRQNQE-VMDGZTHMSA-N thebaine Chemical compound C([C@@H](N(CC1)C)C2=CC=C3OC)C4=CC=C(OC)C5=C4[C@@]21[C@H]3O5 FQXXSQDCDRQNQE-VMDGZTHMSA-N 0.000 description 5
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- 235000011114 ammonium hydroxide Nutrition 0.000 description 4
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 4
- 229960005181 morphine Drugs 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- YYCRAERBSFHMPL-XFKAJCMBSA-N (4r,4as,7ar,12bs)-4a-hydroxy-9-methoxy-3-methyl-2,4,7a,13-tetrahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-one Chemical compound O=C([C@@H]1O2)C=C[C@@]3(O)[C@]4([H])N(C)CC[C@]13C1=C2C(OC)=CC=C1C4 YYCRAERBSFHMPL-XFKAJCMBSA-N 0.000 description 3
- 0 *C(C[C@@]1(C2)c(c(C3)ccc4O)c4O)C3C1C=CC2O Chemical compound *C(C[C@@]1(C2)c(c(C3)ccc4O)c4O)C3C1C=CC2O 0.000 description 3
- YYCRAERBSFHMPL-UHFFFAOYSA-N 14beta-Hydroxycodeinone Natural products O1C2C(=O)C=CC3(O)C4CC5=CC=C(OC)C1=C5C23CCN4C YYCRAERBSFHMPL-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229960002085 oxycodone Drugs 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- XYYVYLMBEZUESM-CMKMFDCUSA-N codeinone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=CC(=O)[C@@H]1OC1=C2C3=CC=C1OC XYYVYLMBEZUESM-CMKMFDCUSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- JGJLWPGRMCADHB-UHFFFAOYSA-N hypobromite Chemical compound Br[O-] JGJLWPGRMCADHB-UHFFFAOYSA-N 0.000 description 2
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Chemical compound Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 2
- 229960003086 naltrexone Drugs 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 206010012335 Dependence Diseases 0.000 description 1
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 1
- HLMSIZPQBSYUNL-IPOQPSJVSA-N Noroxymorphone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4 HLMSIZPQBSYUNL-IPOQPSJVSA-N 0.000 description 1
- 208000026251 Opioid-Related disease Diseases 0.000 description 1
- 239000008896 Opium Substances 0.000 description 1
- 238000006859 Swern oxidation reaction Methods 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- ZVTQWXCKQTUVPY-UHFFFAOYSA-N chloromethylcyclopropane Chemical compound ClCC1CC1 ZVTQWXCKQTUVPY-UHFFFAOYSA-N 0.000 description 1
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- NFDFQCUYFHCNBW-SCGPFSFSSA-N dienestrol Chemical compound C=1C=C(O)C=CC=1\C(=C/C)\C(=C\C)\C1=CC=C(O)C=C1 NFDFQCUYFHCNBW-SCGPFSFSSA-N 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 239000012433 hydrogen halide Substances 0.000 description 1
- 229910000039 hydrogen halide Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- UZHSEJADLWPNLE-GRGSLBFTSA-N naloxone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4CC=C UZHSEJADLWPNLE-GRGSLBFTSA-N 0.000 description 1
- 229960004127 naloxone Drugs 0.000 description 1
- 201000005040 opiate dependence Diseases 0.000 description 1
- 229960001027 opium Drugs 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 229960005118 oxymorphone Drugs 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 150000004965 peroxy acids Chemical class 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
Изобретение относится к способу получения производных морфинона, которые являются промежуточными соединениями для получения производных 14-гидроксиморфинона, которые, в свою очередь, используются для получения антагонистов опиатов-производных оксиморфона. Антагонисты опиатов являются важными лекарственными средствами для лечения и диагностики наркомании.The invention relates to a method for producing derivatives of morphinone, which are intermediate compounds for the preparation of derivatives of 14-hydroxymorphinone, which, in turn, are used to obtain antagonists of opiate derivatives of oxymorphone. Opiate antagonists are important drugs for the treatment and diagnosis of addiction.
Наиболее широко применяемым способом производства антагонистов опиатов - производных оксиморфона, таких как, например, налтрексон, и налоксон является способ, описанный в The Organic Chemistry of Drug Synthesis. Vol. 1 John Wiley&Sons, New York, 1990, p. 289-291, включающий окисление тебаина с получением 14-гидроксикодеинона, его гидрирование, последовательное деалкилирование метильных групп у фенильного кольца и азота с обработкой полученного нороксиморфона соответствующим алкилгалогенидом. Основным недостатком данного способа получения является необходимость использования в качестве исходного сырья дорогостоящего тебаина, т.к. количество производимого тебаина лимитировано вследствие очень низкого его содержания в опиумной смоле.The most widely used method for the production of opiate antagonists, oxymorphone derivatives, such as, for example, naltrexone, and naloxone is the method described in The Organic Chemistry of Drug Synthesis. Vol. 1 John Wiley & Sons, New York, 1990, p. 289-291, including the oxidation of thebaine to produce 14-hydroxycodeinone, its hydrogenation, sequential dealkylation of methyl groups on the phenyl ring and nitrogen with the treatment of the resulting noroxymorphone with the corresponding alkyl halide. The main disadvantage of this production method is the need to use expensive thebaine as a raw material, because the amount of thebaine produced is limited due to its very low content in opium resin.
В патентах США № 5922876 и № 6008355 раскрыты способы получения производных оксиморфона и оксикодона, включающие получение производных морфинона из морфина и кодеина прямым окислением по Сверну, а именно действием ДМСО с оксалилхлоридом в хлористом метилене при -78°С с получением технического продукта с последующей обработкой уксусным ангидридом и получением диенолацетатов, вторичным их окислением перекисью или надкислотой с получением производных 14-гидроксиморфинона, которые гидрируют с получением оксикодона и оксиморфона, которые далее деалкелируют по азоту с получением норпроизводных, которые алкилируют с получением антагонистов опиатов. Основным недостатком указанного способа является проведение окисления морфина и кодеина в производные морфинона в жестких условиях, что делает данный способ неудобным для использования его в промышленности.US Pat. Nos. 5922876 and 6008355 disclose methods for preparing derivatives of oxymorphone and oxycodone, including the preparation of morphinone derivatives from morphine and codeine by direct Swern oxidation, namely, the action of DMSO with oxalyl chloride in methylene chloride at -78 ° C to obtain a technical product with subsequent processing acetic anhydride and obtaining dienolacetates, their secondary oxidation with peroxide or peroxyacid to obtain derivatives of 14-hydroxymorphinone, which are hydrogenated to obtain oxycodone and oxymorphone, which then alkalize with nitrogen to produce nor derivatives that alkylate to produce opiate antagonists. The main disadvantage of this method is the oxidation of morphine and codeine to derivatives of morphinone under stringent conditions, which makes this method inconvenient for use in industry.
Помимо способа получения производных морфинона окислением кодеина и морфина, который описан выше как одна из стадий способа получения производных оксиморфона и оксикодона, наиболее широко применимым может считаться способ получения данных продуктов из тебаина реакцией галогеноводорода в присутствии катализатора с последующим гидролизом, описанный в патенте США № 4052402. Недостатком этого способа также является использование дорогостоящего тебаина.In addition to the method for producing morphinone derivatives by oxidation of codeine and morphine, which is described above as one of the steps of the method for producing derivatives of oxymorphone and oxycodone, the most widely applicable method for producing these products from thebaine by the reaction of hydrogen halide in the presence of a catalyst followed by hydrolysis, described in US patent No. 4052402 The disadvantage of this method is the use of expensive thebaine.
Производные морфинона также могут быть получены окислением морфина и кодеина различными сильными окислителями, однако селективность этого окисления низкая, причем в некоторых случаях окисление идет в сторону образования других продуктов (Beilstein, 27 II 136).Derivatives of morphinone can also be obtained by oxidation of morphine and codeine with various strong oxidizing agents, however, the selectivity of this oxidation is low, and in some cases the oxidation goes towards the formation of other products (Beilstein, 27 II 136).
Задачей настоящего изобретения является разработка более простого, технологичного, с использованием более доступного и дешевого исходного сырья способа получения производных морфинона, производных 14-гидроксиморфинона и способ получения антагонистов опиатов - производных оксиморфона, которые являются в настоящее время основными средствами терапии опиатной зависимости и диагностики наркомании.The objective of the present invention is to develop a simpler, more technologically advanced, using more affordable and cheaper feedstock method for producing morphinone derivatives, 14-hydroxymorphinone derivatives, and a method for producing opiate antagonists - oxymorphone derivatives, which are currently the main means of treatment of opiate addiction and diagnosis of drug addiction.
Авторами настоящего изобретения неожиданно было обнаружено, что использование таких сильных неспецифично окисляющих агентов, как соли хлорноватистой и бромноватистой кислот, в мягких условиях позволяет получить с высокими выходом и чистотой производные морфинона.The authors of the present invention unexpectedly found that the use of such strong nonspecific oxidizing agents as salts of hypochlorous and hydrobromic acids under mild conditions makes it possible to obtain morphinone derivatives with high yield and purity.
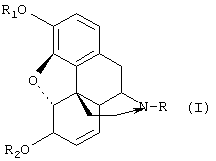
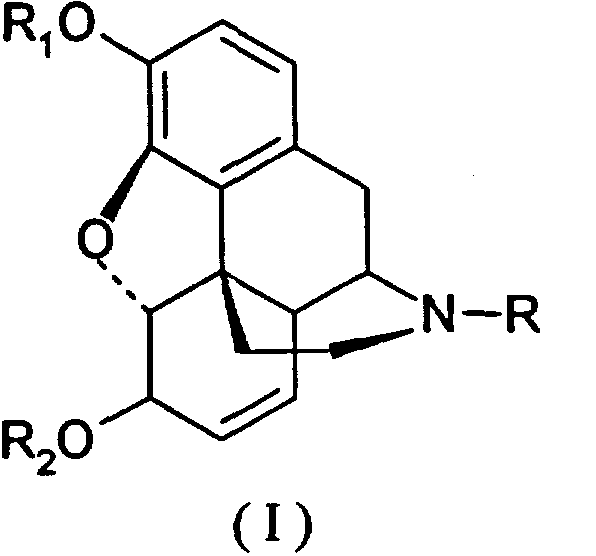
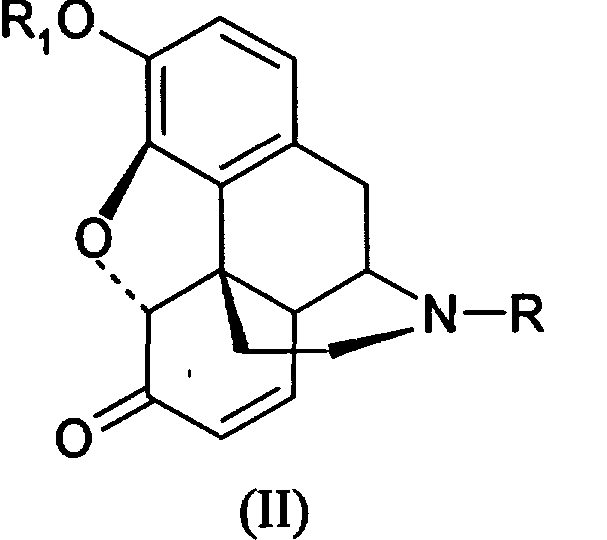
Таким образом, настоящее изобретение относится к способу получения производных морфинона, включающему окисление солями хлорноватистой и бромноватистой кислот производных морфина общей формулы I:Thus, the present invention relates to a method for producing derivatives of morphinone, comprising the oxidation by salts of hypochlorous and hydrobromic acids of morphine derivatives of the general formula I:
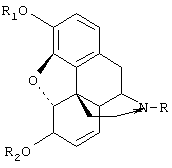
(I)(I)
где К представляет низший алкил, С3-С6циклоалкил, бензил или замещенный бензил, циан; R1 представляет алкил, бензил или алкилкарбонил; R2 представляет водород, алкил, бензил или алкилкарбонил.where K represents lower alkyl, C 3 -C 6 cycloalkyl, benzyl or substituted benzyl, cyan; R 1 represents alkyl, benzyl or alkylcarbonyl; R 2 represents hydrogen, alkyl, benzyl or alkylcarbonyl.
Окисление (по Схеме 1) предпочтительно ведут солями хлорноватистой и бромноватистой кислот, преимущественно полученными непосредственно перед окислением. Для окисления используются предпочтительно гипохлориты или гипобромиты натрия и калия.The oxidation (according to Scheme 1) is preferably carried out with salts of hypochlorous and hydrobromic acids, mainly obtained immediately before oxidation. For oxidation, preferably sodium or potassium hypochlorites or hypobromites are used.
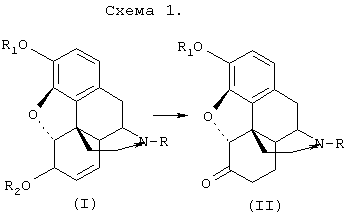
Окисление проводят простым смешиванием водного раствора окислителя и раствора исходного вещества либо его суспензии в органическом растворителе. Предпочтительно в качестве растворителя используют ацетонитрил. Процесс проводят при температуре в интервале от 0 до 20°С в течение от 30 мин до 2 часов. Далее целевой продукт выделяют, используя известные методики.The oxidation is carried out by simply mixing an aqueous solution of an oxidizing agent and a solution of the starting material or its suspension in an organic solvent. Preferably, acetonitrile is used as a solvent. The process is carried out at a temperature in the range from 0 to 20 ° C for from 30 minutes to 2 hours. Next, the target product is isolated using known methods.
В предпочтительном варианте осуществления изобретения соотношения органический растворитель - раствор окислителя выбирают таким образом, чтобы образовывалась двухфазная система, в которой целевой продукт находится в органической фазе, что облегчает его выделение. Предпочтительно указанное выше соотношение составляет 1:3.In a preferred embodiment of the invention, the organic solvent-oxidizer solution ratios are selected so that a two-phase system is formed in which the target product is in the organic phase, which facilitates its isolation. Preferably, the above ratio is 1: 3.
Другим объектом настоящего изобретения является способ получения производных 14-гидроксиморфинона. Общая схема способа представлена на Схеме 2.Another object of the present invention is a method for producing derivatives of 14-hydroxymorphinone. The general scheme of the method is presented in Scheme 2.
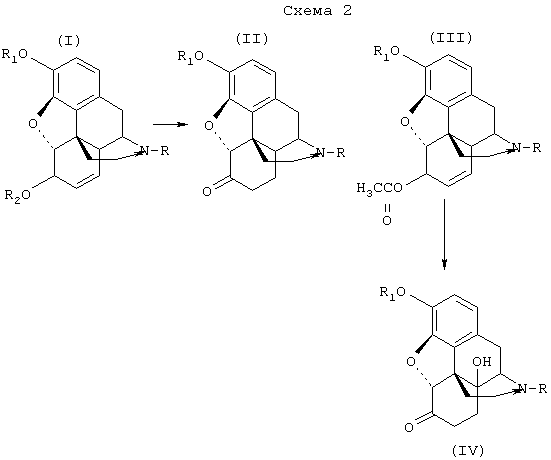
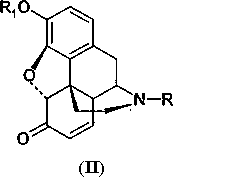
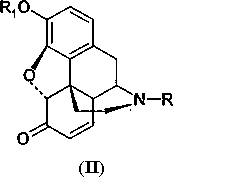
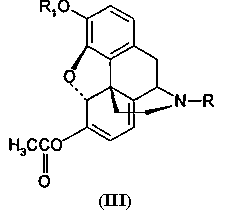
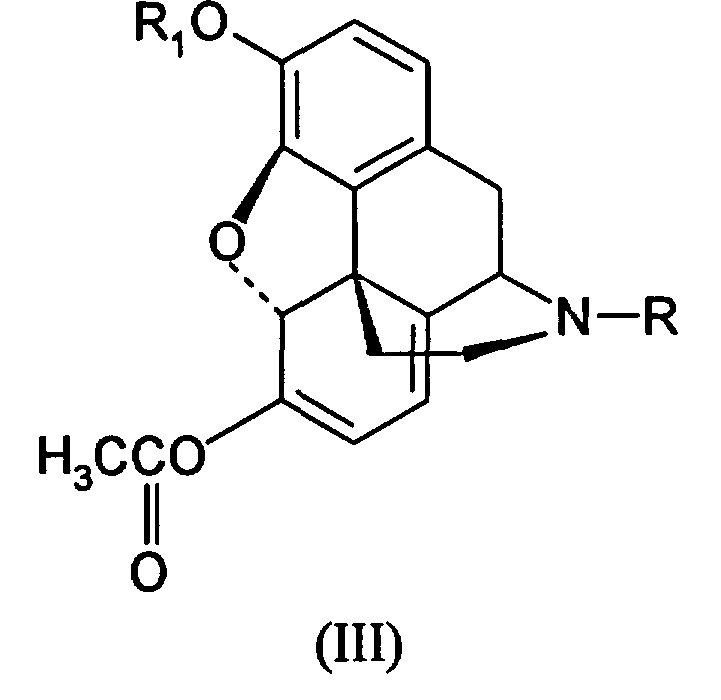
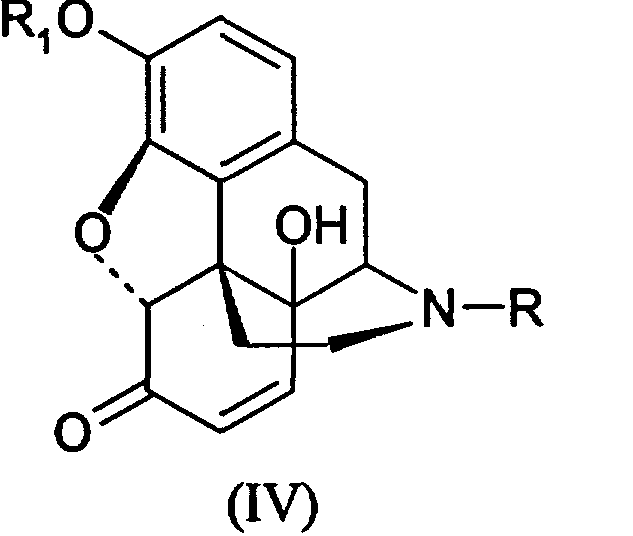
Производные морфина (I) окисляют гипохлоритом или гипобромитом с получением соединения (II) способом, представленным на Схеме 1. Далее производные морфинона (II) обрабатывают уксусным ангидридом с получением эфира енола (III), который окисляют далее перекисью водорода с получением производных 14-гидроксиморфинона. Вторую и третью стадии способа проводят по известным методикам, например, как описано в патенте США 6008355.Derivatives of morphine (I) are oxidized with hypochlorite or hypobromite to obtain compound (II) by the method presented in Scheme 1. Next, derivatives of morphinone (II) are treated with acetic anhydride to obtain enol (III) ester, which is further oxidized with hydrogen peroxide to obtain derivatives of 14-hydroxymorphinone . The second and third stages of the method are carried out by known methods, for example, as described in US patent 6008355.
Еще одним объектом настоящего изобретения является способ получения производных оксиморфона по общей схеме, представленной на Схеме 3:Another object of the present invention is a method for producing derivatives of oxymorphone according to the General scheme presented in Scheme 3:
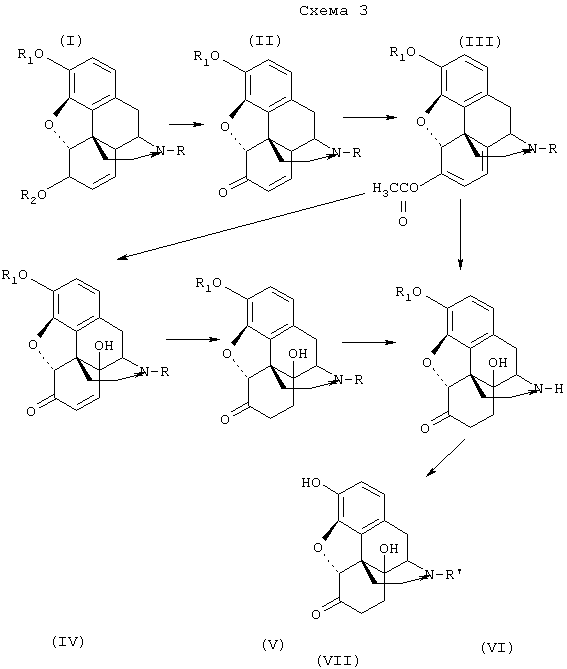
где R’ замещенный алкил или алкилкарбонил.where R ’substituted alkyl or alkylcarbonyl.
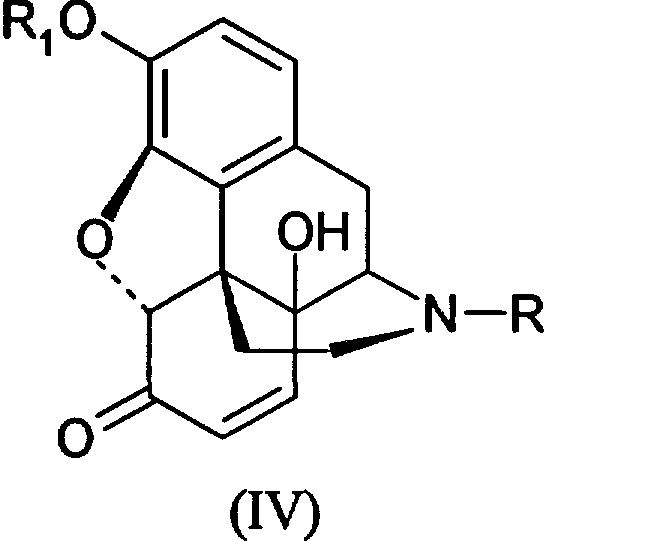
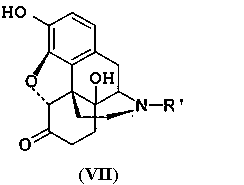
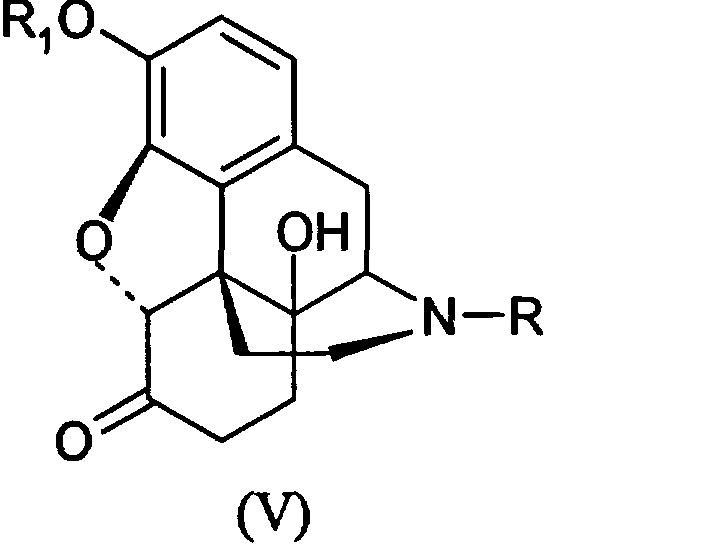
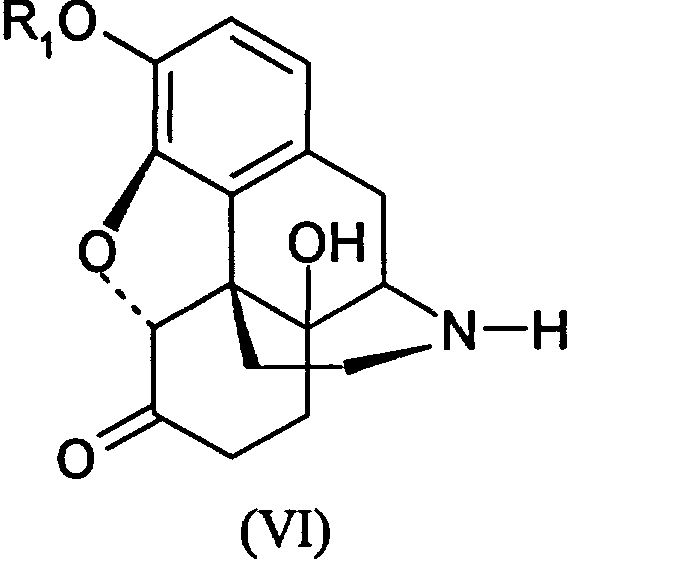
Первую стадию окисления производных морфина (I) в производные морфинона (II) проводят, как описано выше (Схема 1). Далее до производных 14-гидроксиморфинона (IV) процесс ведут, как показано на Схеме 2. Превращение производных 14-гидроксиморфинона (IV) в производные оксиморфона (V) производят гидрированием, далее (V) деалкелируют по азоту с получением норпроизводных (VI), которые алкилируют с получением антагонистов опиатов (VII), как показано ранее.The first stage of the oxidation of derivatives of morphine (I) to derivatives of morphinone (II) is carried out as described above (Scheme 1). Next, to the derivatives of 14-hydroxymorphinone (IV), the process is carried out as shown in Scheme 2. The conversion of the derivatives of 14-hydroxymorphinone (IV) to the derivatives of oxymorphone (V) is carried out by hydrogenation, then (V) is dealkelated with nitrogen to obtain the nor derivatives (VI), which alkylate to obtain opiate antagonists (VII), as shown previously.
Ниже приведен ряд конкретных примеров воплощения изобретения, которые даны для его иллюстрации.The following are a number of specific examples of embodiments of the invention, which are given to illustrate it.
ПримерыExamples
Пример 1. Получение кодеинона (II; R=СН3, R1=СН3).Example 1. Obtaining codeinon (II; R = CH 3 , R 1 = CH 3 ).
Готовят раствор окислителя, растворяя твердый гидроксид натрия (3,0 г) в 25 мл воды, и при охлаждении смесью лед/вода барботируют через этот раствор газообразный хлор, испаряя 2 мл жидкого хлора.An oxidizing solution is prepared by dissolving solid sodium hydroxide (3.0 g) in 25 ml of water, and when cooled with ice / water, chlorine gas is bubbled through this solution, evaporating 2 ml of liquid chlorine.
Отдельно готовят суспензию кодеина (5,0 г, 0,0168 моль) в ацетонитриле (8 мл). К суспензии добавляют раствор окислителя, поддерживая температуру реакционной массы 5-10°С. Реакционную массу выдерживают при этой температуре в течение часа и затем доводят температуру до комнатной. Отделяют органический слой. К органическому слою прибавляют хлористый метилен (25 мл) и промывают полученный раствор водой (2×25мл). Раствор продукта сушат сульфатом натрия и упаривают. После перекристаллизации из этилацетата получают 4,96 г целевого продукта (Тпл=184-185°С). Выход 94,3%.Separately, a suspension of codeine (5.0 g, 0.0168 mol) in acetonitrile (8 ml) was prepared. An oxidizer solution is added to the suspension, maintaining the temperature of the reaction mass 5-10 ° C. The reaction mass is kept at this temperature for one hour and then brought to room temperature. Separate the organic layer. Methylene chloride (25 ml) was added to the organic layer and the resulting solution was washed with water (2 × 25 ml). The product solution was dried with sodium sulfate and evaporated. After recrystallization from ethyl acetate, 4.96 g of the expected product are obtained ( mp = 184-185 ° C.). Yield 94.3%.
Пример 2. Получение 14-гидроксикодеинона(IV; R=СН3, R1=СН3).Example 2. Obtaining 14-hydroxycodeinone (IV; R = CH 3 , R 1 = CH 3 ).
К кодеинону (4,96 г; 0,0158 моль), полученному по методике, приведенной в Примере 1, прибавляют уксусный ангидрид (13,6 г; 0,158 моль) и толуол (5 мл) и кипятят с обратным холодильником в течение 3 часов. Полученный раствор охлаждают, разбавляют хлористым метиленом и добавляют водный раствор гидрокарбоната натрия (27,72 г; 0,33 моль), поддерживая температуру реакционной массы 5-10°С, доводят температуру до комнатной, отделяют органический слой, промывают его водой (2×50 мл) и сушат сульфатом натрия. Затем раствор упаривают досуха и остаток растирают с 50 мл низкокипящего петролейного эфира и фильтруют. Полученный технический диенолацетат кодеинона представляет собой осадок коричневого цвета, который без дальнейшей очистки подвергают окислению, смешивая с муравьиной кислотой (3,7 г; 0,1 моль), раствором перекиси водорода (3,87 г 30%) и водой (10 мл) и выдерживают реакционную массу при 35-40°С в течение 5 часов. Реакционную массу доводят до рН 9 аммиачной водой и экстрагируют хлористым метиленом (2×25 мл). После упаривания получают 3,55 г целевого продукта. Выход 72,0%.To codeinone (4.96 g; 0.0158 mol) obtained by the procedure described in Example 1, acetic anhydride (13.6 g; 0.158 mol) and toluene (5 ml) were added and refluxed for 3 hours . The resulting solution was cooled, diluted with methylene chloride and an aqueous solution of sodium bicarbonate (27.72 g; 0.33 mol) was added, maintaining the temperature of the reaction mixture 5-10 ° С, brought to room temperature, the organic layer was separated, washed with water (2 × 50 ml) and dried with sodium sulfate. Then the solution was evaporated to dryness and the residue was triturated with 50 ml of low boiling petroleum ether and filtered. The resulting technical codeinone dienol acetate is a brown precipitate, which is oxidized without further purification by mixing with formic acid (3.7 g; 0.1 mol), a solution of hydrogen peroxide (3.87 g 30%) and water (10 ml) and maintain the reaction mass at 35-40 ° C for 5 hours. The reaction mass was adjusted to pH 9 with ammonia water and extracted with methylene chloride (2 × 25 ml). After evaporation, 3.55 g of the expected product is obtained. Yield 72.0%.
Пример 3. Получение налтрексона (VII; R’=циклопропилметил, R1=H).Example 3. Obtaining naltrexone (VII; R '= cyclopropylmethyl, R 1 = H).
К 14-гидроксикодеинону (3,55 г; 0,0114 моль), полученному по методике, описанной в примере 2, помещенному в автоклав, добавляют метанол (15 мл) и палладий на угле (5%) (0,5 г) и гидрируют при 20 ати в течение 24 часов при комнатной температуре. Реакционную массу фильтруют и упаривают. Остаток смешивают с 20 мл гексаметилдисилазана и кипятят с обратным холодильником в течение 2 часов. Избыток гексаметилдисилазана отгоняют в вакууме. Остаток перекристаллизовывают из гексана (25 мл). Полученное 14-триметилсилилокси соединение смешивают с раствором бромциана в хлороформе (10 мл 1M) и кипятят полученную смесь в течение 20 часов. Реакционную массу промывают водой, сушат сульфатом натрия и упаривают. К полученному цианпроизводному добавляют раствор уксусной кислоты (60 мл, 80%) и реакционную массу кипятят в течение 2 часов. Смесь подщелачивают 25% раствором аммиака до рН 9. Выпавший осадок отфильтровывают, добавляют хлорметилциклопропан (7,0 г), йодид калия (2,0 г) и этанол (100 мл) и реакционную массу нагревают до кипения. Реакционную массу охлаждают, подщелачивают аммиачной водой до рН 9, экстрагируют хлористым метиленом (2×25 мл) и сушат сульфатом натрия. Раствор охлаждают до 0-5°С и добавляют раствор трибромида фосфора в хлористом метилене (50 мл 1М), реакционную массу перемешивают при комнатной температуре, подщелачивают аммиачной водой до рН 9 и отделяют органический слой, промывают его водой (2×30 мл), сушат сульфатом натрия и упаривают в вакууме досуха. Получают 2,17 г целевого продукта в виде основания. Выход 56,0%.To 14-hydroxycodeinone (3.55 g; 0.0114 mol) obtained by the procedure described in Example 2, placed in an autoclave, methanol (15 ml) and palladium on carbon (5%) (0.5 g) were added and hydrogenated at 20 ati for 24 hours at room temperature. The reaction mass is filtered and evaporated. The residue was mixed with 20 ml of hexamethyldisilazane and refluxed for 2 hours. Excess hexamethyldisilazane is distilled off in vacuo. The residue was recrystallized from hexane (25 ml). The resulting 14-trimethylsilyloxy compound was mixed with a solution of cyanogen bromide in chloroform (10 ml 1M) and the resulting mixture was boiled for 20 hours. The reaction mass is washed with water, dried with sodium sulfate and evaporated. A solution of acetic acid (60 ml, 80%) is added to the obtained cyano derivative, and the reaction mass is boiled for 2 hours. The mixture was made basic with a 25% ammonia solution to pH 9. The precipitate was filtered off, chloromethylcyclopropane (7.0 g), potassium iodide (2.0 g) and ethanol (100 ml) were added and the reaction mixture was heated to boiling. The reaction mass is cooled, alkalinized with ammonia water to pH 9, extracted with methylene chloride (2 × 25 ml) and dried with sodium sulfate. The solution is cooled to 0-5 ° C and a solution of phosphorus tribromide in methylene chloride (50 ml 1M) is added, the reaction mass is stirred at room temperature, alkalized with ammonia water to pH 9 and the organic layer is separated, washed with water (2 × 30 ml), dried with sodium sulfate and evaporated to dryness in vacuo. 2.17 g of the expected product is obtained in the form of a base. Yield 56.0%.
Claims (7)


Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2002119134/04A RU2236412C2 (en) | 2002-07-18 | 2002-07-18 | Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2002119134/04A RU2236412C2 (en) | 2002-07-18 | 2002-07-18 | Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2002119134A RU2002119134A (en) | 2004-02-27 |
| RU2236412C2 true RU2236412C2 (en) | 2004-09-20 |
Family
ID=33432893
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2002119134/04A RU2236412C2 (en) | 2002-07-18 | 2002-07-18 | Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2236412C2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2401270C2 (en) * | 2005-02-11 | 2010-10-10 | Цилаг Аг | Method of purifying noroxymorphone compounds |
| RU2422445C2 (en) * | 2006-12-14 | 2011-06-27 | Джонсон Мэттей Паблик Лимитед Компани | Method of preparing analgesics |
| RU2448709C1 (en) * | 2011-02-01 | 2012-04-27 | Учреждение Российской академии наук Институт проблем химико-энергетических технологий Сибирского отделения РАН (ИПХЭТ СО РАН) | Method for making naltrexone |
| RU2806817C2 (en) * | 2018-12-27 | 2023-11-07 | Алар Фармасьютикалс Инк. | Extended-release injection composition of naltrexone |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4052402A (en) * | 1976-03-15 | 1977-10-04 | Fabrica De Productos Quimicos Y Farmaceuticos Abello, S.A. | Process for synthesizing codeinone from thebaine |
| US5922876A (en) * | 1996-07-26 | 1999-07-13 | Penick Corporation | Preparation of oxymorphone from morphine |
-
2002
- 2002-07-18 RU RU2002119134/04A patent/RU2236412C2/en not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4052402A (en) * | 1976-03-15 | 1977-10-04 | Fabrica De Productos Quimicos Y Farmaceuticos Abello, S.A. | Process for synthesizing codeinone from thebaine |
| US5922876A (en) * | 1996-07-26 | 1999-07-13 | Penick Corporation | Preparation of oxymorphone from morphine |
| US6008355A (en) * | 1996-07-26 | 1999-12-28 | Penick Corporation | Preparation of oxycodone from codeine |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2401270C2 (en) * | 2005-02-11 | 2010-10-10 | Цилаг Аг | Method of purifying noroxymorphone compounds |
| RU2422445C2 (en) * | 2006-12-14 | 2011-06-27 | Джонсон Мэттей Паблик Лимитед Компани | Method of preparing analgesics |
| RU2448709C1 (en) * | 2011-02-01 | 2012-04-27 | Учреждение Российской академии наук Институт проблем химико-энергетических технологий Сибирского отделения РАН (ИПХЭТ СО РАН) | Method for making naltrexone |
| RU2806817C2 (en) * | 2018-12-27 | 2023-11-07 | Алар Фармасьютикалс Инк. | Extended-release injection composition of naltrexone |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2002119134A (en) | 2004-02-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5824448B2 (en) | Preparation of morphinan and morphinone compounds | |
| EP2222678B1 (en) | Processes for the production of (+)-'nal' morphinan compounds | |
| KR100941761B1 (en) | Reduction of α, β-unsaturated ketones in opioid compositions | |
| CN102325777B (en) | (+)-morphinan* N-oxide and its preparation method | |
| JP2001518444A5 (en) | ||
| JPS582956B2 (en) | 7- and 7-8-substituted 4,5α epoxymorphinan-6-one compounds | |
| US20100036128A1 (en) | PROCESS FOR MAKING MORPHINAN-6alpha-OLS | |
| JP2010531813A (en) | Conversion of thebaine to morphine derivatives. | |
| EP1421085B1 (en) | Process for the preparation of a 14-hydroxynormorphinone derivative | |
| JP2694156B2 (en) | Total synthesis of nortebaine, normorphine, and noroxymorphone enantiomers via N-nor intermediates | |
| AU2002331163A1 (en) | Process for the Preparation of a 14-hydroxynormorphinone Compound | |
| RU2236412C2 (en) | Method for preparing derivatives of morphinone, method for preparing derivatives of 14-hydroxymorphinone and method for preparing derivative of oxymorphone | |
| AU2009300384A1 (en) | Processes for increasing the yield of opiate alkaloid derivatives | |
| EP2342206B1 (en) | Processes for the hydrogenation of opiate alkaloid derivatives | |
| US20230357257A1 (en) | Novel process for the synthesis of noroxymorphone from morphine | |
| Kotick | Analgesic narcotic antagonists. 6. 7. beta., 8. beta.-Methano-and 7. beta., 8. beta.-epoxydihydrocodeinone | |
| EP2344508B1 (en) | Processes for the synthesis of opiate alkaloids with reduced impurity formation | |
| EP0009227B1 (en) | Fluorination process and fluoro analogs of hydrocodone and oxycodone | |
| JPH0234346B2 (en) | ||
| HK1063803B (en) | Process for the preparation of a 14-hydroxynormorphinone derivative | |
| HK1135107B (en) | Method of reducing alpha, beta- unsaturated ketones in opioid compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20080719 |