RU2160277C1 - Anssa-zirconocenes functioned with respect to cyclosilane bridges, and method of preparation thereof - Google Patents
Anssa-zirconocenes functioned with respect to cyclosilane bridges, and method of preparation thereof Download PDFInfo
- Publication number
- RU2160277C1 RU2160277C1 RU99113532A RU99113532A RU2160277C1 RU 2160277 C1 RU2160277 C1 RU 2160277C1 RU 99113532 A RU99113532 A RU 99113532A RU 99113532 A RU99113532 A RU 99113532A RU 2160277 C1 RU2160277 C1 RU 2160277C1
- Authority
- RU
- Russia
- Prior art keywords
- bridge
- alkyl
- silicon
- aryl
- solution
- Prior art date
Links
Images




Abstract
Description
Изобретение относится к новым мостичным цирконоценам (ЦЦ), а именно к анса-цирконоценам с циклосилановым мостиком, функционализированным непосредственно по мостику, которые могут быть использованы как катализаторы в химической промышленности при производстве полиолефинов (ПО). The invention relates to new bridge zirconocenes (CC), namely to ansa-zirconocenes with a cyclosilane bridge, functionalized directly along the bridge, which can be used as catalysts in the chemical industry in the production of polyolefins (PO).
Мостичные (анса-) ЦЦ, особенно ЦЦ с кремниевым мостиком, относятся к наиболее активным гомогенным металлоценовым катализаторам (МЦК) полимеризации олефинов, открытие которых произвело революцию в синтезе ПО благодаря их чрезвычайно высокой каталитической активности и стереоселективности. МЦК применяются совместно с полиметилалюмоксаном (МАО) в качестве сокатализатора (Sinclair K., Wilson R. Chemistry and Jndustry, 1994, N 7, p. 857). Bridged (ansa-) CCs, especially CCs with a silicon bridge, are among the most active homogeneous metallocene catalysts (MCCs) for the polymerization of olefins, the discovery of which revolutionized PO synthesis due to their extremely high catalytic activity and stereoselectivity. MCC are used in conjunction with polymethylaluminoxane (MAO) as a cocatalyst (Sinclair K., Wilson R. Chemistry and Jndustry, 1994, N 7, p. 857).
В настоящее время известно большое количество анса-металлоценов и цирконоценов. Наиболее близкими к предлагаемым анса-цирконоценам и способу их получения являются стерически затрудненные ЦЦ с кремниевым мостиком и способ их получения, описанные в работе Spaleck W. et al. Organometallics, 1994, 13, p. 954 - прототип, которые представляют собой бис-инденильные производные анса-цирконоценов C2 - симметрии с кремниевым мостиком формулы I, II или III.Currently, a large number of Ansa-metallocenes and zirconocenes are known. Closest to the proposed ansa-zirconocenes and the method for their preparation are sterically hindered CCs with a silicon bridge and the method for their preparation described in Spaleck W. et al. Organometallics, 1994, 13, p. 954 - a prototype, which are bis-indenyl derivatives of ansa-zirconocenes C 2 - symmetry with a silicon bridge of the formula I, II or III.
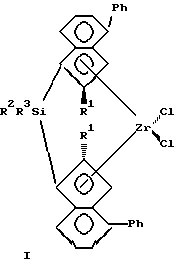
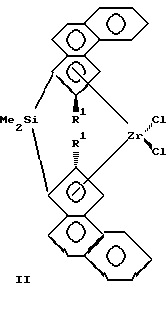

Ia R1=R2=R3=Me;
Ib R1=R2=Me, R3=Ph;
Ic R1=H, R2=R3=Me;
IIa R=Me;
IIb R=H.
Ia R 1 = R 2 = R 3 = Me;
Ib R 1 = R 2 = Me, R 3 = Ph;
Ic R 1 = H, R 2 = R 3 = Me;
IIa R = Me;
IIb R = H.
Получение известных соединений I-III представлено на примере 1 a-c схемой 1. The preparation of known compounds I-III is shown in Example 1 a-c of
Первой стадией синтеза является получение инденильных лигандов (4). Исходным соединением является 2-фенилбензилбромид (1), который конденсируют с малоновым или метилмалоновым эфиром в присутствии этилата натрия. Последующие реакции омыления эфира (в присутствии водной щелочи) и декарбоксилирования путем нагревания при 130oC дают соответствующую кислоту (2) с хорошим выходом (на неочищенную (2) 85% от теор.). Взаимодействием кислоты (2) с тионилхлоридом получают ее хлорангидрид, который подвергают циклизации в инданон (3) в присутствии AlCl3 в качестве катализатора Фриделя-Крафтса. Выход инданонов был удовлетворительный кроме нафтилинданона - промежуточного продукта при синтезе соединения III, так как более высокая реакционная способность нафтильного производного привела к побочным реакциям, и выход нафтилинданона был только 13%. Инданон (3) затем восстанавливают боргидридом натрия в смеси тетрагидрофурана (ТГФ) и метанола при 0oC, кипятят в присутствии p-толуолсульфокислоты и выделяют инден (4) методом препаративной ТСХ (тонкослойной хроматографии) на силикагеле с хорошим выходом.The first stage of synthesis is the preparation of indenyl ligands (4). The starting compound is 2-phenylbenzyl bromide (1), which is condensed with malonic or methyl malonic ether in the presence of sodium ethylate. Subsequent reactions of saponification of the ether (in the presence of aqueous alkali) and decarboxylation by heating at 130 ° C give the corresponding acid (2) in good yield (85% of theory per crude (2)). The reaction of acid (2) with thionyl chloride gives its acid chloride, which is cyclized to indanone (3) in the presence of AlCl 3 as a Friedel-Crafts catalyst. The yield of indanones was satisfactory except for naphthylindanone, an intermediate product in the synthesis of compound III, since the higher reactivity of the naphthyl derivative led to side reactions, and the yield of naphthylindanone was only 13%. Indanone (3) is then reduced with sodium borohydride in a mixture of tetrahydrofuran (THF) and methanol at 0 ° C, boiled in the presence of p-toluenesulfonic acid and inden (4) is isolated by preparative TLC (thin layer chromatography) on silica gel in good yield.
Следующей стадией является получение бисинденильных лигандов с кремниевым мостиком (5). Инден (4) растворяют в смеси сухого толуола и ТГФ в атмосфере аргона и прибавляют при комнатной температуре раствор н-бутиллития в гексане. Нагревают до 80oC, выдерживают при 80oC 1 час и охлаждают до 0oC, прибавляют дихлорид диметилсилана (а, с) или метилфенилсилана (b). Греют 1 час при 80oC и обрабатывают водой. Из органического слоя методом ТСХ выделяют соединение (5). Выход (5a) 70%, (5b) 44%, (5c) 62%.The next stage is the preparation of bisindenyl ligands with a silicon bridge (5). Indene (4) was dissolved in a mixture of dry toluene and THF in an argon atmosphere and a solution of n-butyllithium in hexane was added at room temperature. Heated to 80 o C, kept at 80 o C for 1 hour and cooled to 0 o C, dimethylsilane dichloride (a, c) or methylphenylsilane (b) was added. Heated for 1 hour at 80 o C and treated with water. Compound (5) is isolated from the organic layer by TLC. Yield (5a) 70%, (5b) 44%, (5c) 62%.
Заключительной стадией является получение цирконоценов 1 а-с. К раствору (5) в сухом толуоле в атмосфере аргона прибавляют при комнатной температуре раствор н-BuLi в гексане. Кипятят с обратным холодильником 3 часа, охлаждают до -25oC и прибавляют тетрахлорид циркония. Оставляют на 2 часа. Отфильтровывают циркониевый комплекс, представляющий собой смесь рацемической (активной) и мезо (неактивной) - форм (рац:мезо = 1:1). Перекристаллизовывают из метиленхлорида. Выход 1 а 33%, I b 10%, 1 с 15%
В работе, выбранной за прототип, проанализированы более ранние результаты по исследованию влияния структуры металлоценов на их поведение в реакции полимеризации олефинов, и с целью изучения влияния ароматических заместителей в инденильном ядре мостичных ЦЦ были синтезированы соединения I-III, которые были испытаны с МАО в качестве сокатализатора (Al : Zr = 15000 : 1) в полимеризации пропилена и этилена. Полимеризацию пропилена в работе-прототипе проводили в среде жидкого мономера при 70oC в термостатируемом стальном реакторе. МАО применяли в виде 10% -ного раствора в толуоле, ЦЦ растворяли в таком же растворе МАО. Полученные результаты показали (см. таблицу 1), что введение объемного ароматического заместителя в 4-положение инденильного ядра существенно улучшает все показатели процесса полимеризации по сравнению с незамещенным в этом положении (2a*) или 4-алкилзамещенным (4a*) ЦЦ, описанными ранее в других работах.The final stage is to obtain
The work selected for the prototype analyzed earlier results on the influence of the structure of metallocenes on their behavior in the polymerization of olefins, and in order to study the effect of aromatic substituents in the indenyl nucleus of bridge CC, compounds I-III were synthesized, which were tested with MAO as cocatalyst (Al: Zr = 15000: 1) in the polymerization of propylene and ethylene. The polymerization of propylene in the prototype work was carried out in a liquid monomer at 70 o C in a thermostatic steel reactor. MAO was used as a 10% solution in toluene, and CC was dissolved in the same MAO solution. The results showed (see table 1) that the introduction of a volume aromatic substituent in the 4-position of the indenyl nucleus significantly improves all indicators of the polymerization process compared with unsubstituted (2a * ) or 4-alkyl substituted (4a * ) CCs described previously in other works.
При проведении полимеризации этилена (в растворе) наивысшую каталитическую активность, как и в реакции с пропиленом, показали соединения Ia и III, они же показали и наивысший MW [2].When carrying out the polymerization of ethylene (in solution), the highest catalytic activity, as in the reaction with propylene, was shown by compounds Ia and III, and they also showed the highest M W [2].
Следует подчеркнуть, что беспрецедентно высокие показатели процесса полимеризации, полученные с использованием соединений Ia и III, по сравнению с другими мостичными цирконоценами позволяют считать соединения Ia и III наилучшими из известных на сегодня в качестве катализаторов полимеризации олефинов совместно с МАО, а сам факт открытия такого влияния ароматического заместителя в 4-ом положении инденильного кольца крупным вкладом в химию МЦК. It should be emphasized that the unprecedentedly high rates of the polymerization process obtained using compounds Ia and III, in comparison with other bridge zirconocenes, make it possible to consider compounds Ia and III as the best known today as catalysts for the polymerization of olefins together with MAO, and the fact of the discovery of such an effect aromatic substituent in the 4th position of the indenyl ring a major contribution to the chemistry of MCC.
Однако ЦЦ I-III, как и другие известные МЦК, являются синтетически труднодоступными. Так, соединение III, проявляющее наивысшую каталитическую активность, является наиболее труднодоступным из ЦЦ I-III из-за низких выходов промежуточных продуктов при синтезе. Кроме того, известные ЦЦ I-III отличаются высоким содержанием неактивной в реакции полимеризации мезо-формы (соотношение рац- и мезо-изомеров 1:1), от которой желательно освобождать металлокомплекс, что весьма затруднительно и часто невозможно. However, CC I-III, like other known MCC, are synthetically difficult to access. Thus, compound III, exhibiting the highest catalytic activity, is the most inaccessible of the CC I-III due to the low yields of intermediate products in the synthesis. In addition, the known CC I-III are characterized by a high content of the meso form inactive in the polymerization reaction (1: 1 ratio of the racem and meso isomers), from which it is desirable to release the metal complex, which is very difficult and often impossible.
Необходимо отметить еще один недостаток известных МЦК - все известные модификации структуры металлоценов осуществляются на самых ранних этапах их синтеза, что сильно тормозит исследования каталитической активности, так как нельзя заранее предвидеть ни выход на промежуточных стадиях, ни саму возможность получения как мостичного лиганда, так и конечного целевого продукта - металлокомплекса, а также будут ли полученные металлоцены растворимы в органических растворителях (толуоле), чтобы быть перспективными для получения гомогенных катализаторов. It is necessary to note one more drawback of the known MCCs - all known modifications of the structure of metallocenes are carried out at the very early stages of their synthesis, which greatly inhibits studies of catalytic activity, since it is impossible to foresee either the yield at intermediate stages or the very possibility of obtaining both a bridging ligand and the final the target product - the metal complex, and whether the metallocenes obtained will be soluble in organic solvents (toluene) in order to be promising for the preparation of homogeneous kat izatorov.
Задачей предлагаемого изобретения является создание новых анса-ЦЦ, обладающих высокой активностью и стереоселективностью в качестве катализатора полимеризации олефинов и имеющих принципиально новую структуру, а именно функционализированных непосредственно по кремниевому мостику, что позволит использовать их в качестве объекта исследований при изучении влияния функциональных заместителей в мостике, то есть в непосредственной близости от каталитического центра, а также разработка способа их получения с достаточно высоким выходом промежуточных и конечного продуктов с улучшенными методиками выделения промежуточных продуктов и с увеличенным содержанием активной рацемической формы в циркониевом комплексе. The objective of the invention is the creation of new ansa-CC, with high activity and stereoselectivity as a catalyst for the polymerization of olefins and having a fundamentally new structure, namely, functionalized directly on a silicon bridge, which will allow them to be used as an object of study when studying the influence of functional substituents in the bridge, that is, in the immediate vicinity of the catalytic center, as well as the development of a method for their production with a sufficiently high yield intermediate and final products with improved methods for the isolation of intermediate products and with an increased content of the active racemic form in the zirconium complex.
Решение поставленной задачи достигается:
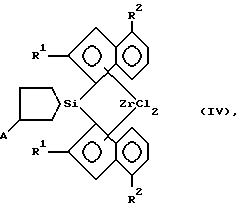
- предлагаемыми новыми анса-цирконоценами, функционализированными по циклосилановому мостику формулы IV:

R1 = H, CH3, С2H5;
R2 = H, алкил от С1 до С4, нормальный или разветвленный; арил;
А = BC8H14; MR3, где М - олово или кремний, R = алкил от C1 до C4, нормальный или разветвленный; арил.The solution to this problem is achieved:
- the proposed new ansa-zirconocenes functionalized on the cyclosilane bridge of formula IV:
R 1 = H, CH 3 , C 2 H 5 ;
R 2 = H, alkyl from C 1 to C 4 , straight or branched; aryl;
A = BC 8 H 14 ; MR 3 , where M is tin or silicon, R = C 1 to C 4 alkyl, straight or branched; aryl.
- и способом их получения, в котором литиевую соль индена подвергают взаимодействию с 1,1 - дихлор-2,5- дигидросилолом в диэтиловом эфире с последующим взаимодействием полученной дилитиевой соли соответствующего бисинденильного лиганда с кремниевым мостиком с тетрахлоридом циркония для получения соответствующего цирконоцена и нагреванием его в тетрагидрофуране с алкил(арил) производным моногидрида бора, олова или кремния. Цирконоцен нагревают в тетрагидрофуране с алкил(арил) производными (моно)гидрида бора, олова или кремния. - and a method for their preparation, in which the lithium salt of indene is reacted with 1,1 - dichloro-2,5-dihydrosilol in diethyl ether, followed by reaction of the obtained dilithium salt of the corresponding bisindenyl ligand with a silicon bridge with zirconium tetrachloride to obtain the corresponding zirconocene and heating it in tetrahydrofuran with an alkyl (aryl) derivative of boron, tin or silicon monohydride. Zirconocene is heated in tetrahydrofuran with alkyl (aryl) derivatives of (mono) boron, tin or silicon hydride.
Получение дилитиевой соли бис-инденильного продукта с кремниевым мостиком можно осуществлять в виде кристаллического аддукта с диэтиловым эфиром, который затем подвергают взаимодействию с тетрахлоридом циркония. The preparation of the dilithium salt of a bis-indenyl product with a silicon bridge can be carried out in the form of a crystalline adduct with diethyl ether, which is then reacted with zirconium tetrachloride.
При разработке предлагаемого изобретения первоначально были созданы новые ЦЦ с непредельным циклосилановым мостиком (описанные в отдельной заявке, подаваемой одновременно с данной), что и позволило получить анса-ЦЦ, функционализированные по мостику. When developing the proposed invention, new CCs with an unsaturated cyclosilane bridge (described in a separate application filed simultaneously with this one) were originally created, which made it possible to obtain ansa-CCs functionalized along the bridge.
Введение функциональных заместителей, особенно объемных, в мостик ЦЦ, то есть в непосредственной близости от каталитического центра, представляет большой интерес для химии МЦК, так как создает совершенно новые пространственные эффекты в металлокомплексе, которые неизбежно должны влиять на стереоселективные свойства катализатора. The introduction of functional substituents, especially bulky ones, into the CC bridge, i.e., in the immediate vicinity of the catalytic center, is of great interest for the chemistry of MCC, since it creates completely new spatial effects in the metal complex, which inevitably should affect the stereoselective properties of the catalyst.
Синтез предлагаемых ЦЦ IV описан в примерах и представлен на схеме 2. Полученные соединения IV (и промежуточные продукты) охарактеризованы данными элементного анализа и ЯМР - спектрами (1H, 13C).The synthesis of the proposed CC IV is described in the examples and is shown in
Соединения IV совместно с МАО в качестве сокатализатора были испытаны в реакции полимеризации пропилена. Процесс полимеризации проводили по методике, аналогичной описанной в работе-прототипе. Полученные результаты представлены в таблице 2. Compounds IV together with MAO as a cocatalyst were tested in the polymerization of propylene. The polymerization process was carried out according to the method similar to that described in the prototype work. The results are presented in table 2.
Примеры. Examples.
I. Синтез замещенного индена (42).I. Synthesis of substituted indene (4 2 ).
Синтез диэтилового эфира пропандикарбоновой кислоты CH3CH2CH(COOEt)2 (этилмалоновый эфир).Synthesis of propanedicarboxylic acid diethyl ester CH 3 CH 2 CH (COOEt) 2 (ethyl malonic ester).
К раствору этилата натрия, приготовленному из 16.2 г (0.705 г-атом) натрия и 350 мл абс. этанола добавили при перемешивании 53 мл (55.92 г, 0.349 моль) диэтилового эфира малоновой кислоты. К приготовленному таким образом раствору медленно при охлаждении на ледяной бане добавили 53 мл (77.38 г, 0.710 моль) этилбромида, дали смеси нагреться до комнатной температуры и перемешивали еще 30 минут. Этиловый спирт отогнали, добавили воду до полного растворения осадка, полученную смесь экстрагировали эфиром, объединенные эфирные вытяжки сушили сульфатом натрия, растворитель отогнали и оставшееся масло подвергли перегонке, что дало 52.33 г (80%) целевого продукта. To a sodium ethylate solution prepared from 16.2 g (0.705 g-atom) of sodium and 350 ml of abs. ethanol was added with stirring 53 ml (55.92 g, 0.349 mol) of malonic acid diethyl ether. To the solution thus prepared, while cooling in an ice bath, 53 ml (77.38 g, 0.710 mol) of ethyl bromide were slowly added, the mixture was allowed to warm to room temperature and was stirred for another 30 minutes. Ethyl alcohol was distilled off, water was added until the precipitate was completely dissolved, the resulting mixture was extracted with ether, the combined ether extracts were dried with sodium sulfate, the solvent was distilled off, and the remaining oil was distilled to give 52.33 g (80%) of the desired product.
Аналогично получали метилмалоновый эфир (вместо этилата натрия используют метилат натрия). Methyl malonic ester was prepared in a similar manner (sodium methylate was used instead of sodium ethylate).
Синтез 2-(гидроксиметил)дифенила 2-PhC6H4CH2OH (2-фенилбензиловый спирт).Synthesis of 2- (hydroxymethyl) diphenyl 2-PhC 6 H 4 CH 2 OH (2-phenylbenzyl alcohol).
К суспензии 5.7 г (150.0 ммоль) литийалюминийгидрида в 50 мл кипящего тетрагидрофурана в течение 4 часов добавили при перемешивании раствор 12.5 г (63,1 ммоль) 2-фенилбензойной кислоты в 100 мл абсолютного тетрагидрофурана, кипятили реакционную смесь еще 3 часа и оставили на ночь. Реакционную смесь осторожно при интенсивном охлаждении обработали ледяной водой до окончания выделения водорода и далее серной кислотой (1:1) до полного растворения осадка. Полученный таким образом раствор трижды экстрагировали эфиром, объединенные эфирные вытяжки сушили сульфатом натрия, растворитель отогнали и оставшееся масло охладили до начала кристаллизации. Хроматографически чистый продукт. Т.пл. 29oC. Выход 9.54 г (82%).To a suspension of 5.7 g (150.0 mmol) of lithium aluminum hydride in 50 ml of boiling tetrahydrofuran, a solution of 12.5 g (63.1 mmol) of 2-phenylbenzoic acid in 100 ml of absolute tetrahydrofuran was added with stirring over 4 hours, the reaction mixture was boiled for another 3 hours and left overnight . The reaction mixture was carefully treated with vigorous cooling with ice water until the evolution of hydrogen was complete and then with sulfuric acid (1: 1) until the precipitate was completely dissolved. The solution thus obtained was extracted three times with ether, the combined ether extracts were dried with sodium sulfate, the solvent was distilled off, and the remaining oil was cooled before crystallization began. Chromatographically pure product. Mp 29 o C. Yield 9.54 g (82%).
Аналогично получают 2-алкил и 2-нафтилбензиловый спирт (вместо-2-фенилбензойной кислоты используют 2-алкил или 2-нафтилбензойную кислоту). In a similar manner, 2-alkyl and 2-naphthylbenzyl alcohol are obtained (instead of-2-phenylbenzoic acid, 2-alkyl or 2-naphthylbenzoic acid is used).
Синтез 2-(бромометил)дифенила 2-PhC6H4CH2Br (2-фенилбензилбромид) (12).Synthesis of 2- (bromomethyl) diphenyl 2-PhC 6 H 4 CH 2 Br (2-phenylbenzyl bromide) (1 2 ).
Смесь 3.20 г (17.37 ммоль) 2-(гидроксиметил)дифенила, 5 мл конц. HBг и 1.2 мл конц. H2SO4 кипятили с обратным холодильником в течение 6 часов, охладили и вылили в 20 мл ледяной воды. Органическую фазу (нижний слой) отделили, промыли насыщенным раствором карбоната натрия; водную фазу нейтрализовали карбонатом натрия и трижды экстрагировали эфиром. Объединенную органическую фазу сушили сульфатом натрия, растворитель отогнали и оставшееся масло перегнали при уменьшенном давлении, собирая фракцию 160-165oC/7 мм рт. ст., что дало 3.64 г чистого продукта. Выход 85%. Осторожно! Сильный лакриматор!
Аналогично получали 2-алкил- и 2-нафтилбензилбромид.A mixture of 3.20 g (17.37 mmol) of 2- (hydroxymethyl) diphenyl, 5 ml conc. HBg and 1.2 ml conc. H 2 SO 4 was refluxed for 6 hours, cooled and poured into 20 ml of ice water. The organic phase (lower layer) was separated, washed with saturated sodium carbonate solution; the aqueous phase was neutralized with sodium carbonate and extracted three times with ether. The combined organic phase was dried with sodium sulfate, the solvent was distilled off, and the remaining oil was distilled under reduced pressure, collecting a fraction of 160-165 ° C / 7 mm Hg. Art., which gave 3.64 g of pure product. Yield 85%. Caution! Strong lacrimator!
Similarly, 2-alkyl and 2-naphthylbenzyl bromide were obtained.
Синтез 2-(2-дифенилметил)бутановой кислоты (Et)(2- PhC6H4CH2)CHCOOH (22).Synthesis of 2- (2-diphenylmethyl) butanoic acid (Et) (2-PhC 6 H 4 CH 2 ) CHCOOH (2 2 ).
К раствору этилата натрия, приготовленному из 1.40 г (60.9 мг-атом) натрия и 47 мл абс. этанола добавили при комнатной температуре и перемешивании раствор 11.70 г (62.2 ммоль) диэтилового эфира этилмалоновой кислоты в 15 мл абс. этанола. К приготовленному таким образом раствору при слабом кипении добавили по каплям раствор 15.60 г (63.1 ммоль) 2-(бромометил)дифенила в 20 мл абс. этанола, реакционную смесь кипятили еще 3 часа. Реакционную смесь охладили, обработали при охлаждении раствором 9 г КОН в 25 мл воды, снова кипятили в течение 4 часов и охладили. Растворители отогнали в вакууме водоструйного насоса и остаток подкислили концентрированной соляной кислотой до pH 1. Выпавший белый осадок отфильтровали и нагревали на бане из сплава Вуда в течение 1 часа при 130oC. Бледно-желтое масло использовали в дальнейшем без дополнительной очистки.To a sodium ethylate solution prepared from 1.40 g (60.9 mg atom) of sodium and 47 ml of abs. ethanol was added at room temperature with stirring a solution of 11.70 g (62.2 mmol) of ethyl malonic acid diethyl ether in 15 ml of abs. ethanol. To the solution thus prepared, at a slight boil, a solution of 15.60 g (63.1 mmol) of 2- (bromomethyl) diphenyl in 20 ml of abs. Was added dropwise. ethanol, the reaction mixture was boiled for another 3 hours. The reaction mixture was cooled, treated with cooling with a solution of 9 g of KOH in 25 ml of water, again boiled for 4 hours and cooled. The solvents were distilled off in a vacuum of a water-jet pump and the residue was acidified with concentrated hydrochloric acid to
Синтез хлорангидрида 2-(2-дифенилметил)бутановой кислоты (Et)(2-PhC6H4CH2)CHC(O)Cl.Synthesis of 2- (2-diphenylmethyl) butanoic acid chloride (Et) (2-PhC 6 H 4 CH 2 ) CHC (O) Cl.
К 7.20 г (28.2 ммоль) 2-(2- дифенилметил)бутановой кислоты добавили 20 мл свежеперегнанного тионилхлорида и полученный раствор кипятили с обратным холодильником в течение 1 часа. Избыток тионилхлорида отогнали; для полноты удаления тионилхлорида в смесь трижды вводили порции абс. толуола по 30 мл и отгоняли его в вакууме. Остаток растворили в 50 мл абс. толуола и использовали в дальнейшем без дополнительной обработки. To 7.20 g (28.2 mmol) of 2- (2-diphenylmethyl) butanoic acid was added 20 ml of freshly distilled thionyl chloride and the resulting solution was refluxed for 1 hour. Excess thionyl chloride was distilled off; to complete removal of thionyl chloride, abs. 30 ml of toluene and distilled off in vacuum. The residue was dissolved in 50 ml of abs. toluene and was used without further processing.
Синтез 4-фенил-2-этилинданона-1 2-Et-4-Ph-C9H6O(32).Synthesis of 4-phenyl-2-ethylindanone-1 2-Et-4-Ph-C 9 H 6 O (3 2 ).
Приготовленный как указано выше раствор хлорангидрида 2-(2- дифенилметил)бутановой кислоты в 50 мл абс. толуола в течение 1 часа добавили при перемешивании и охлаждении до 10oC к суспензии 5.70 г (42.7 ммоль) безводного хлорида алюминия в 50 мл абс.толуола. Смесь перемешивали при 80oC в течение 1 часа, вылили на 300 г колотого льда, подкислили конц. HCl до pH 1, органическую фазу отделили, а водную трижды экстрагировали эфиром. Объединенные органические фазы промыли водой, раствором соды и высушили над сульфатом натрия и удалили растворители в вакууме. Сырой продукт подвергли очистке методом препаративной колоночной хроматографии (силикагель, петролейный эфир - эфир 2:1). Светло-желтые кристаллы. Выход 4.65 г (70% в расчете на 2-(2- дифенилметил)бутановую кислоту). Аналогично получали 4-фенил-2- метилинданон, 4-фенилинданон, 4-алкил-2-алкилинданон и 4-нафтил-2- R1-инданон (R1 = H, CH3, C2H5).A solution of 2- (2-diphenylmethyl) butanoic acid chloride in 50 ml of abs. toluene was added over 1 hour with stirring and cooling to 10 ° C. to a suspension of 5.70 g (42.7 mmol) of anhydrous aluminum chloride in 50 ml of absolute toluene. The mixture was stirred at 80 o C for 1 hour, poured onto 300 g of crushed ice, acidified with conc. HCl to
Синтез 4-фенил-2-этилиндена 2-Et-4-PhlndH (42).Synthesis of 4-phenyl-2-ethylindene 2-Et-4-PhlndH (4 2 ).
К раствору 7.10 г (30.0 ммоль) 4-фенил-2-этилинданона-1 в смеси 30 мл абс. тетрагидрофурана и 15 мл абс. метанола медленно при перемешивании и охлаждении до 0oC добавляли небольшими порциями 1.70 г (44.9 ммоль) измельченного борогидрида натрия. Смесь перемешивали еще 16 часов, вылили на 100 г колотого льда, подкислили конц. HCl до pH 1, трижды экстрагировали эфиром. Органические вытяжки промыли насыщенным раствором поваренной соли и сушили сульфатом магния. Растворители отогнали в вакууме и оставшееся масло растворили в 100 мл сухого толуола, содержащего 0.3 г моногидрата n-толуолсульфокислоты, и кипятили в течение 2 часов. Охлажденный раствор промыли насыщенным водным раствором бикарбоната натрия, высушили сульфатом натрия, растворитель удалили в вакууме и остаток очистили препаративной колоночной хроматографией на силикагеле, используя в качестве элюента смесь петролейный эфир - дихлорометан 9:1. Бесцветное масло. 3.70 г (56% в расчете на 4-фенил-2-этилинданон-1).To a solution of 7.10 g (30.0 mmol) of 4-phenyl-2-ethylindanone-1 in a mixture of 30 ml of abs. tetrahydrofuran and 15 ml abs. Under stirring and cooling to 0 ° C, methanol was added slowly in small portions of 1.70 g (44.9 mmol) of ground sodium borohydride. The mixture was stirred for another 16 hours, poured onto 100 g of crushed ice, acidified with conc. HCl to
ЯМР 1H (400 МГц, CDCl3, 25oC) δ м.д.: 7.63-7.17 (м., 8H, ароматические протоны индена и фенильных групп); 6.59 (т., 4J = 1.6 Гц, 1H, олефиновый протон индена); 3.43 (с., 2H, аллильные протоны индена); 2.52 (уш.кв., 3J = 7.6 Гц, 2H, CH2 - протоны этильной группы); 1.24 (т., 3J = 7.6 Гц, 3H, CH3-протоны этильной группы);
13С{ 1H} (100 МГц, CDCl3, 25) δ м.д.: 152.72, 146.24, 141.37, 140.56, 137.46 (четвертичные ароматические и олефиновый углероды индена и фенильной группы); 128.44, 128.35, 126.98, 125.22, 125.19, 124.28, 119.09 (третичные ароматические и олефиновый углероды индена и фенильной группы); 41.00 (аллильный углерод индена); 24.26 (метиленовый углерод этильной группы); 13.28 (метильный углерод этильной группы). 1 H NMR (400 MHz, CDCl 3 , 25 ° C.) δ ppm: 7.63-7.17 (m, 8H, aromatic protons of indene and phenyl groups); 6.59 (i.e., 4 J = 1.6 Hz, 1H, indene olefin proton); 3.43 (s, 2H, allylic indene protons); 2.52 (br.s., 3 J = 7.6 Hz, 2H, CH2 - protons of the ethyl group); 1.24 (i.e., 3 J = 7.6 Hz, 3H, CH3-protons of the ethyl group);
13 C { 1 H} (100 MHz, CDCl 3 , 25) δ ppm: 152.72, 146.24, 141.37, 140.56, 137.46 (quaternary aromatic and olefinic carbons of indene and phenyl group); 128.44, 128.35, 126.98, 125.22, 125.19, 124.28, 119.09 (tertiary aromatic and olefin carbons of indene and phenyl group); 41.00 (allyl carbon of indene); 24.26 (methylene carbon ethyl group); 13.28 (methyl carbon of the ethyl group).
Аналогично получали другие замещенные индены (42) с R1 и R2, указанные на схеме 2.Other substituted indenes (4 2 ) with R 1 and R 2 indicated in
II. Синтез ЦЦ с непредельным циклосилановым мостиком. II. Synthesis of CC with unsaturated cyclosilane bridge.
Синтез 4-фенил-2-этилиндениллития 2-Et-4-PhlndLi. Synthesis of 4-phenyl-2-ethylindenyl lithium 2-Et-4-PhlndLi.
Синтез проводили в вакууммированной цельнопаянной аппаратуре типа сосудов Шленка. The synthesis was carried out in vacuum-soldered apparatus such as Schlenk vessels.
К раствору 11.00 г (49.93 ммоль) 4-фенил-2-этилиндена (42) в 70 мл диэтилового эфира добавили при охлаждении до -20oC и интенсивном перемешивании 23.8 мл (49.98 ммоль) 2.1 М раствора н-бутиллития в легком петролейном эфире, дали смеси нагреться до комнатной температуры и оставили на ночь. Примерно половину общего объема растворителей отогнали, желтый объемный осадок отделили фильтрованием от интенсивно-красного маточного раствора, трижды промыли на фильтре пентаном (порции по 70 мл), высушили в высоком вакууме и полученное снежно-белое твердое вещество растворили в 100 мл диэтилового эфира. Прозрачный желтый раствор отделили от тонкого осадка двукратной декантацией, оттитровали и использовали в виде 0.35 М раствора. Получено 105 мл 0.35 М раствора, выход 73.5% от теории. Аналогично получали литиевые соли с другими замещенными инденами.To a solution of 11.00 g (49.93 mmol) of 4-phenyl-2-ethylindene (4 2 ) in 70 ml of diethyl ether was added, while cooling to -20 ° C and vigorous stirring, 23.8 ml (49.98 mmol) of a 2.1 M solution of n-butyllithium in light petroleum ether, allowed the mixture to warm to room temperature and left overnight. About half of the total solvent volume was distilled off, the yellow volume precipitate was separated by filtration from the intense red mother liquor, washed three times with pentane on the filter (70 ml each), dried in high vacuum, and the resulting snow-white solid was dissolved in 100 ml of diethyl ether. The clear yellow solution was separated from the fine precipitate by double decantation, titrated and used as a 0.35 M solution. Received 105 ml of a 0.35 M solution, yield 73.5% of theory. Similarly, lithium salts with other substituted indenes were obtained.
Синтез 1,1'-(бутен-2-диил-1,4)силилиденбис(4-фенил-2- этилиндена) (1,4-CH2CH=CHCH2)Si(2-Et-4-PhlndH)2(52).Synthesis of 1,1 '- (butene-2-diyl-1,4) silylidenebis (4-phenyl-2-ethylindene) (1,4-CH 2 CH = CHCH 2 ) Si (2-Et-4-PhlndH) 2 (5 2 ).
К раствору 1.14 г (7.44 ммоль) 1,1-дихлоро-2,5-дигидросилола в 30 мл диэтилового эфира при охлаждении до -30 - -40oC и интенсивном перемешивании в течение 30 минут добавили 42.5 мл 0.35 М эфирного раствора (4-фенил-2-этилиндениллития) (14.86 ммоль), дали смеси постепенно нагреться до комнатной температуры и оставили на ночь. Раствор отфильтровали от выпавшего хлорида лития, растворитель отогнали, ввели 50 мл пентана и после повторного отфильтровывания и отгонки растворителя оставшееся слегка желтоватое стеклующееся масло высушили в высоком вакууме. Выход практически количественный. Полную очистку бидентатного лиганда (52) проводили в форме его дилитиевого производного.To a solution of 1.14 g (7.44 mmol) of 1,1-dichloro-2,5-dihydrosilol in 30 ml of diethyl ether, while cooling to -30 - -40 o C and vigorous stirring for 30 minutes was added 42.5 ml of a 0.35 M ether solution (4 -phenyl-2-ethylindenyl lithium) (14.86 mmol), allowed the mixture to gradually warm to room temperature and left overnight. The solution was filtered from the precipitated lithium chloride, the solvent was distilled off, 50 ml of pentane were added, and after repeated filtering and distillation of the solvent, the remaining slightly yellow glassy oil was dried under high vacuum. The output is almost quantitative. The bidentate ligand (5 2 ) was completely purified in the form of its dilithium derivative.
Аналогично осуществляли взаимодействие 1,1-дихлоро-2,5- дигидросилола с Li-солями других замещенных инденов. Similarly, 1,1-dichloro-2,5-dihydrosilol was reacted with the Li salts of other substituted indenes.
1,1'-(бутен-2-диил-1,4)силилиденбис(4-фенил-2- этилинденил)дилития аддукт с диэтиловым эфиром (1,4- CH2CH=CHCH2)Si(2-Et-4-Phlnd)2Li2•Et2O (add). К раствору полученного на предыдущей стадии лиганда (52) в смеси 10 мл диэтилового эфира и 40 мл пентана при охлаждении до -20oC и интенсивном перемешивании добавили 6.54 мл 2.16 М раствора н-бутиллития в гексане (14.12 ммоль) и дали смеси нагреться до комнатной температуры. Отделившееся первоначально желтое масло постепенно закристаллизовалось. Осадок отфильтровали, трижды промыли на фильтре той же смесью растворителей и полученное снежно-белое порошкообразное вещество высушили в высоком вакууме. Получено 3.19 г (5.26 ммоль) дилитиевой соли (add). Выход 70.7% в расчете на исходно взятый 4-фенил-2-этилиндениллитий.1,1 '- (butene-2-diyl-1,4) silylidenebis (4-phenyl-2-ethylindenyl) dilithium adduct with diethyl ether (1,4-CH 2 CH = CHCH 2 ) Si (2-Et-4 -Phlnd) 2 Li 2 • Et 2 O (add). To a solution of the ligand (5 2 ) obtained in the previous step in a mixture of 10 ml of diethyl ether and 40 ml of pentane was added 6.54 ml of a 2.16 M solution of n-butyllithium in hexane (14.12 mmol) while cooling to -20 ° C and the mixture was allowed to warm to room temperature. The initially separated yellow oil gradually crystallized. The precipitate was filtered off, washed three times on the filter with the same solvent mixture, and the resulting snow-white powdery substance was dried under high vacuum. Obtained 3.19 g (5.26 mmol) of dilithium salt (add). Yield 70.7% based on the initially taken 4-phenyl-2-ethylindenyl lithium.
NMR (THF-d8, 30oC) d м.д. 1H: 1.31 (т, CH2CH3); 2.18 (с, CH2Si); 3.04 (кв, CH2CH3); 6.03 (с, CH= CH); 6.32 (с, H3); 6.52 (м, H5, H6); 7.12 (т, пара-H); 7.30 (т, мета-H); 7.69 (м, H7), 7.79 (д, орто-H).NMR (THF-d 8 , 30 o C) d ppm 1 H: 1.31 (t, CH 2 CH 3 ); 2.18 (s, CH 2 Si); 3.04 (q, CH 2 CH 3 ); 6.03 (s, CH = CH); 6.32 (s, H 3 ); 6.52 (m, H 5 , H 6 ); 7.12 (t, para-H); 7.30 (t, meta-H); 7.69 (m, H 7 ); 7.79 (d, ortho-H).
13С: 17.58 (CH2CH3); 22.63 (CH2Si); 25.89 (CH2CH3); 95.97 (С3); 96.54 (С1); 113.82, 114.14, 119.86 (С5, C6, C7); 125.02 (пара-C); 128.22 (мета-C); 129.24 (орто-C); 132.60 (CH= CH); 130.34, 131.16, 137.77, 145.22, 147.36 (С3a, С7a, 2, С4, ипсо-C). 13 C: 17.58 (CH 2 CH 3 ); 22.63 (CH 2 Si); 25.89 (CH 2 CH 3 ); 95.97 (C 3 ); 96.54 (C 1 ); 113.82, 114.14, 119.86 (C 5 , C 6 , C 7 ); 125.02 (para-C); 128.22 (meta-C); 129.24 (ortho-C); 132.60 (CH = CH); 130.34, 131.16, 137.77, 145.22, 147.36 (C 3a , C 7a , 2 , C 4 , ipso-C).
Аналогично получали аддукты с диэтиловым эфиром дилитиевых солей других бидентатных лигандов с непредельным циклосилановым мостиком. Similarly, diethyl ether adducts of dilithium salts of other bidentate ligands with unsaturated cyclosilane bridge were obtained.
Синтез { η5,η5′-[1,1'-(Бутeн-2-диил-1,4)cилилиденбиc(4-фeнил-2- этилинденил)]}дихлороциркония (1,4-CH2CH=CHCH2)Si(2-Et-4-Phlnd)2ZrCl2, смесь рац- и мезо-форм (2:1).Synthesis of {η 5 , η 5 ′ - [1,1 '- (Butene-2-diyl-1,4) silylidenebis (4-phenyl-2-ethylindenyl)]} dichlorocirconium (1,4-CH 2 CH = CHCH 2 ) Si (2-Et-4-Phlnd) 2 ZrCl 2 , mixture of rac and meso forms (2: 1).
Суспензию 3.19 г (5.26 ммоль) эфирата дилитиевой соли (add) и 1.24 г (5.26 ммоль) ZrCl4 в 100 мл толуола перемешивали в течение суток при комнатной температуре, ввели еще 500 мл толуола, оранжевый маточный раствор отделили от очень мелкодисперсного осадка декантациями (3 недели), толуол отогнали и остаток перекристаллизовали из минимального объема хлористого метилена, что дало 2.40 г (3.53 ммоль) целевого комплекса, представляющего собой смесь рац- и мезо-форм в соотношении 2:1. Выход 67,1%.A suspension of 3.19 g (5.26 mmol) of dilithium salt ether (add) and 1.24 g (5.26 mmol) of ZrCl 4 in 100 ml of toluene was stirred overnight at room temperature, another 500 ml of toluene was added, the orange mother liquor was separated from the very finely divided precipitate by decantation ( 3 weeks), toluene was distilled off and the residue was recrystallized from the minimum volume of methylene chloride, which gave 2.40 g (3.53 mmol) of the target complex, which is a mixture of the rac and meso forms in a 2: 1 ratio. Yield 67.1%.
NMR 1H (CD2Cl2, 30oC) d м.д.: 1.11 (т, CH2CH3, рац); 1.21 (т, CH2CH3, мезо); 2.41 (м), 2.74 (м) (CH2CH3, рац); 2.76 (м, CH2CH3, мезо); 2.55 (ушир. с), 2.89 (ушир. с) (CH2Si, мезо); 2.61 (AB-система), 2.78 (AB-система, 2J(AB) = 18 Гц) (CH2Si, рац); 6.33 (с, HC=CH, рац); 6.33 (м), 6.36 (м) (HC= CH, мезо); 6.86 (с, H3, мезо); 6.97 (с, H3, рац); 6.93 (м), 7.10 - 7.65 (м) (H5, H6, H7, Ph).NMR 1 H (CD 2 Cl 2 , 30 ° C) d ppm: 1.11 (t, CH 2 CH 3 , rac); 1.21 (t, CH 2 CH 3 , meso); 2.41 (m), 2.74 (m) (CH 2 CH 3 , rac); 2.76 (m, CH 2 CH 3 , meso); 2.55 (broad s), 2.89 (broad s) (CH 2 Si, meso); 2.61 (AB system), 2.78 (AB system, 2 J (AB) = 18 Hz) (CH 2 Si, rac); 6.33 (s, HC = CH, rac); 6.33 (m), 6.36 (m) (HC = CH, meso); 6.86 (s, H 3 , meso); 6.97 (s, H 3 , rac); 6.93 (m), 7.10 - 7.65 (m) (H 5 , H 6 , H 7 , Ph).
C38H34Cl2Si1Zr1, М = 680,96. Найдено: Cl = 9,80%,. Вычислено: Cl = 10,43%.C 38 H 34 Cl 2 Si 1 Zr 1 , M = 680.96. Found: Cl = 9.80% ,. Calculated: Cl = 10.43%.
Аналогично получали ЦЦ с непредельным циклосилановым мостиком с другими замещенными бисинденильными лигандами. Similarly, CCs with unsaturated cyclosilane bridge with other substituted bisindenyl ligands were obtained.
Синтез { η5,η5′-[1,1'-(Бутен-2-диил-1,4)силилиденбисинденил]} дихлороциркония (1,4-CH2CH=CHCH2)Silnd2ZrCl2, смесь рац- и мезо-форм (1:1).Synthesis of {η 5 , η 5 ′ - [1,1 '- (Butene-2-diyl-1,4) silylidenebisindenyl]} dichloro zirconium (1,4-CH 2 CH = CHCH 2 ) Silnd 2 ZrCl 2 , a mixture of rac- and meso forms (1: 1).
Синтез индениллития IndLi. Synthesis of Inddenyllithium IndLi.
К раствору 11.62 г (100.00 ммоль) индена производства фирмы "Aldrich" в 50 мл диэтилового эфира добавили при охлаждении до -20oC и интенсивном перемешивании 53.2 мл (100.01 ммоль) 1.88 М раствора н-бутиллития в легком петролейном эфире, дали смеси нагреться до комнатной температуры и оставили на ночь. Примерно две трети общего объема растворителей отогнали, выпавший белый кристаллический осадок отделили фильтрованием от интенсивно-красного маточного раствора, трижды промыли на фильтре пентаном (порции по 70 мл), высушили в высоком вакууме. Снежно-белое кристаллическое вещество. Выход 10.90 г (89.27 ммоль, 89.3% от теории).To a solution of 11.62 g (100.00 mmol) of Aldrich manufactured indene in 50 ml of diethyl ether was added, while cooling to -20 ° C and vigorous stirring, 53.2 ml (100.01 mmol) of a 1.88 M solution of n-butyllithium in light petroleum ether, and the mixture was allowed to warm to room temperature and left overnight. About two thirds of the total solvent volume was distilled off, the white crystalline precipitate formed was separated by filtration from the intense red mother liquor, washed three times with pentane on the filter (70 ml each), and dried in high vacuum. Snow-white crystalline substance. Yield 10.90 g (89.27 mmol, 89.3% of theory).
Синтез 1,1'-(бутен-2-диил-1,4)силилиденбисиндена (1,4- CH2CH= CHCH2)Si(IndH)2 (52), R1=R2=H.Synthesis of 1,1 '- (butene-2-diyl-1,4) silylidenebisindene (1,4-CH 2 CH = CHCH 2 ) Si (IndH) 2 (5 2 ), R 1 = R 2 = H.
К раствору 2.86 г (37.51 ммоль) 1,1-дихлоро-2,5-дигидросилола в 30 мл диэтилового эфира при охлаждении до -30 - -40oC и интенсивном перемешивании в течение 30 минут добавили раствор 4.58 г (37.51 ммоль) индениллития в диэтиловом эфире, дали смеси постепенно нагреться до комнатной температуры и оставили на ночь. Раствор отфильтровали от выпавшего хлорида лития, растворитель отогнали, ввели 50 мл пентана и после повторного отфильтровывания и отгонки растворителя оставшееся слегка желтоватое масло высушили в высоком вакууме. Выход практически количественный. Полную очистку лиганда (52) (R1 = R2 = Н) проводили в форме его дилитиевого производного.A solution of 4.58 g (37.51 mmol) of indenyl lithium was added to a solution of 2.86 g (37.51 mmol) of 1,1-dichloro-2,5-dihydrosilol in 30 ml of diethyl ether while cooling to -30 - -40 ° C and vigorous stirring for 30 minutes. in diethyl ether, allowed the mixture to gradually warm to room temperature and left overnight. The solution was filtered from the precipitated lithium chloride, the solvent was distilled off, 50 ml of pentane were added, and after repeated filtering and distillation of the solvent, the remaining slightly yellowish oil was dried under high vacuum. The output is almost quantitative. Complete purification of the ligand (5 2 ) (R 1 = R 2 = N) was carried out in the form of its dilithium derivative.
Синтез [1,1'-(бутен-2-диил- 1,4)силилиденбисинденил] дилития (аддукт с диэтиловым эфиром) (1,4- CH2CH=CHCH2)Silnd2Li2 • Et2O (add, R1=R2=H).Synthesis of [1,1 '- (butene-2-diyl-1,4) silylidene bisindenyl] dilithium (adduct with diethyl ether) (1,4-CH 2 CH = CHCH 2 ) Silnd 2 Li 2 • Et 2 O (add, R 1 = R 2 = H).
К раствору 5.14 г (16.45 ммоль) полученного на предыдущей стадии лиганда (52) в 50 мл диэтилового эфира при охлаждении до -20oC и интенсивном перемешивании добавили 15.2 мл 2.16 М раствора н-бутиллития в гексане (16.45 ммоль), дали смеси нагреться до комнатной температуры и оставили на ночь. Выпавший белый осадок отфильтровали, трижды промыли на фильтре той же смесью растворителей и полученное снежно-белое порошкообразное вещество высушили в высоком вакууме. Получено 4.91 г (12.32 ммоль) дилитиевой соли (add). Выход 74.9% в расчете на исходно взятый индениллитий.To a solution of 5.14 g (16.45 mmol) of the ligand obtained in the previous step (5 2 ) in 50 ml of diethyl ether while cooling to -20 ° C and vigorous stirring was added 15.2 ml of a 2.16 M solution of n-butyllithium in hexane (16.45 mmol), warm to room temperature and left overnight. The precipitated white precipitate was filtered off, washed three times on the filter with the same solvent mixture, and the obtained snow-white powdery substance was dried in high vacuum. Received 4.91 g (12.32 mmol) of dilithium salt (add). Yield 74.9% calculated on the initial taken indenyl lithium.
NMR (THF-d8, 30oC) d м.д. 1H: 1.91 (с, CH2Si); 6.01 (с, CH=CH); 6.04 (д, 3J(H2-H3) = 3.2 Гц, H2); 6.47 (м, H5, H6); 6.88 (д, H3); 7.32 (м, H7); 7.64 (м, H4).NMR (THF-d 8 , 30 o C) d ppm 1 H: 1.91 (s, CH 2 Si); 6.01 (s, CH = CH); 6.04 (d, 3 J (H 2 -H 3 ) = 3.2 Hz, H 2 ); 6.47 (m, H 5 , H 6 ); 6.88 (d, H 3 ); 7.32 (m, H 7 ); 7.64 (m, H 4 ).
13С: 20.50 (CH2Si); 95.14 (С3); 98.26 (С1); 114.33, 114.63, 119.40, 121.67, 125.99 (C2, C4, С5, С6, C7); 132.78 (CH=CH); 133.89, 136.28 (C3a, C7a). 13 C: 20.50 (CH 2 Si); 95.14 (C 3 ); 98.26 (C 1 ); 114.33, 114.63, 119.40, 121.67, 125.99 (C 2 , C 4 , C 5 , C 6 , C 7 ); 132.78 (CH = CH); 133.89, 136.28 (C 3a , C 7a ).
Синтез { η5,η5′ -[1,1'-(Бутен-2-диил- 1,4)силилиденбисинденил]}дихлороциркония (1,4-CH2CH=CHCH2)SiInd2ZrCl2, смесь рац- и мезо-форм (1:1).Synthesis of {η 5 , η 5 ′ - [1,1 '- (Butene-2-diyl-1,4) silylidene bisindenyl]} dichloro-zirconium (1,4-CH 2 CH = CHCH 2 ) SiInd 2 ZrCl 2 , a mixture of rac- and meso forms (1: 1).
Суспензию 1.34 г (3.36 ммоль) эфирата дилитиевой соли (add) и 0.78 г (3.36 ммоль) ZrCl4 в 70 мл толуола перемешивали в течение суток, сухой остаток многократно экстрагировали толуолом, толуольный экстракт сконцентрировали, кристаллический желтый осадок отфильтровали от маточника, промыли толуолом, пентаном и высушили в высоком вакууме, что дало 1.02 г (2.17 ммоль) металлокомплекса, представляющего собой смесь рац- и мезо-форм в соотношении 1:1. Выход 64.5%.A suspension of 1.34 g (3.36 mmol) of dilithium salt ether (add) and 0.78 g (3.36 mmol) of ZrCl 4 in 70 ml of toluene was stirred during the day, the dry residue was repeatedly extracted with toluene, the toluene extract was concentrated, the crystalline yellow precipitate was filtered off from the mother liquor, washed with toluene , pentane and dried in high vacuum, which gave 1.02 g (2.17 mmol) of the metal complex, which is a mixture of the rac and meso forms in a 1: 1 ratio. Yield 64.5%.
NMR (CD2Cl2, 30oC) d м.д.:1H (CD2Cl2, 30oC) 2.29 (ушир. с), 2.64 (ушир. с) (CH2Si, мезо) 2.37 (AB-система), 2.56 (AB-система, 2J(AB) = 17 Гц) (CH2Si, рац); 6.11 (д, 3J(H2-H3) = 3.5 Гц, H2, рац); 6.17 (д, 3J(H2-H3) = 3.5 Гц, H2, мезо); 6.29 (с, CH=CH, рац); 6.29 (м), 6.30 (м) (CH=CH, мезо); 6.90 (д, H3, рац); 6.95 (д, H3, мезо); 6.93 (м), 7.22 (м) (H5, H6, мезо); 7.10 (м), 7.39 (м) (H5, H6 рац); 7.51 (м), 7.60 (м) (H4, H7, рац, мезо).NMR (CD 2 Cl 2 , 30 o C) d ppm: 1 H (CD 2 Cl 2 , 30 o C) 2.29 (broad s), 2.64 (broad s) (CH 2 Si, meso) 2.37 (AB system), 2.56 (AB system, 2 J (AB) = 17 Hz) (CH 2 Si, rac); 6.11 (d, 3 J (H 2 -H 3 ) = 3.5 Hz, H 2 , rac); 6.17 (d, 3 J (H 2 -H 3 ) = 3.5 Hz, H 2 , meso); 6.29 (s, CH = CH, rac); 6.29 (m), 6.30 (m) (CH = CH, meso); 6.90 (d, H 3 , rac); 6.95 (d, H 3 , meso); 6.93 (m), 7.22 (m) (H 5 , H 6 , meso); 7.10 (m), 7.39 (m) (H 5 , H 6 rats); 7.51 (m), 7.60 (m) (H 4 , H 7 , rac, meso).
13C: 16.45, 16.86 (CH2Si, мезо); 16.99 (CH2Si, рац); 87.94 (С1, рац); 89.08 (С1, мезо); 117.95, 118.14, 124.38, 126.34, 127.18, 128.00 (C2, C3, C4, C5, C6, С7, рац); 119.62, 119.85, 125.75, 126.06, 126.34, 127.54 (C2, C3, C4, C5, C6, С7, мезо); 130.65, 130.88 (CH=CH, мезо); 130.70 (CH=CH, рац); 125.89, 133.79 (С3a, С7a, рац); 128.13, 134.93 (С3a, С7a, мезо). C22H18Cl2Si1Zr1, М = 472,67. Найдено: Cl = 14,38%. Вычислено: Cl = 15,02%. 13 C: 16.45, 16.86 (CH 2 Si, meso); 16.99 (CH 2 Si, rac); 87.94 (C 1 , rac); 89.08 (C 1 , meso); 117.95, 118.14, 124.38, 126.34, 127.18, 128.00 (C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , rat); 119.62, 119.85, 125.75, 126.06, 126.34, 127.54 (C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , meso); 130.65, 130.88 (CH = CH, meso); 130.70 (CH = CH, rac); 125.89, 133.79 (C 3a , C 7a , rac); 128.13, 134.93 (C 3a , C 7a , meso). C 22 H 18 Cl 2 Si 1 Zr 1 , M = 472.67. Found: Cl = 14.38%. Calculated: Cl = 15.02%.
III. Синтез функционализированных по кремниевому мостику ЦЦ формулы IV. III. Synthesis of functionalized on a silicon bridge CC of the formula IV.
Синтез борированного анса-цирконоцена с незамещенными бисинденильными лигандами IVa ( η5:η5-1,1' (3-(9- борабициклононанил-9) силоландиил-1,1)бисинденил]дихлороциркония).Synthesis of boron ansa-zirconocene with unsubstituted bisindenyl ligands IVa (η 5 : η 5 -1.1 '(3- (9-borabicyclononanil-9) silolanediyl-1,1) bisindenyl] dichlorocirconium).
Раствор 125 мг (264,5 ммоль) анса-цирконоцена -1,1' [(Бут-2-диил-1,4)силилиден] -бисинденил-дихлороциркония (смесь рац- и мезо-формы) и 9-борабицикло [3, 3, 1] нонана (32 мг, 132,3 ммоль) в ТГФ смешали при комнатной температуре, нагрели при 70 - 80oC в запаянном сосуде, охладили, растворитель удалили, а остаток высушили в вакууме. Получено кристаллическое вещество желто-оранжевого цвета. Выход количественный. C30H33B1Cl2Si1Zr1. М = 594,69.A solution of 125 mg (264.5 mmol) of ansa-zirconocene -1,1 '[(But-2-diyl-1,4) silylidene] -bisindenyl-dichloro-zirconium (a mixture of the rac and meso forms) and 9-borabicyclo [3 , 3, 1] nonane (32 mg, 132.3 mmol) in THF was mixed at room temperature, heated at 70 - 80 o C in a sealed vessel, cooled, the solvent was removed, and the residue was dried in vacuo. Received a crystalline substance of yellow-orange color. The output is quantitative. C 30 H 33 B 1 Cl 2 Si 1 Zr 1 . M = 594.69.
Найдено: Zr = 15,88; Cl = 11,38. Вычислено: Zr = 15,34; Cl = 11,92. Found: Zr = 15.88; Cl = 11.38. Calculated: Zr = 15.34; Cl = 11.92.
Синтез станнилированного анса-цирконоцена с незамещенными бисинденильными лигандами IV b ([ η5:η5-1,1'(3-(триметилстанил) силоландиил-1,1) бисинденил] дихлороциркония).Synthesis of stannylated ansa-zirconocene with unsubstituted bisindenyl ligands IV b ([η 5 : η 5 -1.1 '(3- (trimethylstanyl) silolanediyl-1,1) bisindenyl] dichlorocirconium).
Смесь 125 мг (264,5 ммоль) анса-цирконоцена 7 (смесь рац- и мезо-форм) и 43 мг (264,5 ммоль) триметилстаннана в 30 мл ТГФ нагревали при 70 - 80oC в запаянном сосуде в течение 5 часов. Смесь охладили и растворитель удалили в вакууме. Полученное желто-оранжевое масло промыли пентаном (2 раза по 30 мл). Полученный сухой остаток растворили в эфире, профильтровали раствор и удалили эфир. Получено 155 мг (92% от теор.) комплекса IVb. C25H28Cl2Si1Sn1Zr1, M = 637,49.A mixture of 125 mg (264.5 mmol) of ansa-zirconocene 7 (a mixture of rac and meso forms) and 43 mg (264.5 mmol) of trimethylstannan in 30 ml of THF was heated at 70 - 80 ° C in a sealed vessel for 5 hours . The mixture was cooled and the solvent was removed in vacuo. The resulting yellow-orange oil was washed with pentane (2
Найдено: Zr = 14,90; Cl = 10,85. Вычислено: Zr = 14,31; Cl = 11,12. Found: Zr = 14.90; Cl = 10.85. Calculated: Zr = 14.31; Cl = 11.12.
Синтез борированного анса-цирконоцена с замещенными бисинденильными лигандами IVc ([ η5:η5-1,1' (3-(9-борабициклононанил-9) силоландиил-1,1) бис-4-фенил-2-этилинденил]дихлороциркония).Synthesis of boron ansa-zirconocene with substituted bisindenyl ligands IVc ([η 5 : η 5 -1.1 '(3- (9-borabicyclononanil-9) silolanediyl-1,1) bis-4-phenyl-2-ethylindenyl] dichlorocirconium) .
Синтез комплекса IVс проведен по методике синтеза цирконоцена IVа. Основное отличие и основная трудность заключаются в том, что продукт образуется в виде плохо кристаллизующегося масла. Твердый продукт получается в результате многократного промывания продуктов пентаном при -20 (-30)oC. Выход IVc 85% (от теоретического). C46H49B1Cl2Si1Zr1, M = 802,98.The synthesis of complex IVc was carried out by the method of synthesis of zirconocene IVa. The main difference and the main difficulty is that the product is formed in the form of poorly crystallizing oil. The solid product is obtained by repeatedly washing the products with pentane at -20 (-30) o C. IVc yield 85% (of theoretical). C 46 H 49 B 1 Cl 2 Si 1 Zr 1 , M = 802.98.
Найдено: Zr = 11,80; Cl = 8,25. Вычислено: Zr = 11,36; Cl = 8,83. Found: Zr = 11.80; Cl = 8.25. Calculated: Zr = 11.36; Cl = 8.83.
IV. Полимеризация пропилена. IV. Polymerization of propylene.
Методика. Methodology
Предварительно откачанный реактор объемом 0,25 л заполняют жидким пропиленом, добавляют 2-3 мл 10%-ого толуольного раствора МАО и предварительно растворенный в 10%-ом толуольном растворе МАО цирконоцен в количестве 0,1-110-6 моль. Нагревают до 50-70oC и ведут полимеризацию в течение 20-30 минут.A pre-evacuated 0.25 L reactor is filled with liquid propylene, 2-3 ml of a 10% toluene MAO solution and zirconocene pre-dissolved in a 10% MAO toluene solution in an amount of 0.1-110 -6 mol are added. Heated to 50-70 o C and conduct polymerization for 20-30 minutes.
Выход ИПП определяют взвешиванием. Молекулярно-массовые характеристики полипропилена получены методом гель-проникающей хроматографии на высокотемпературном гель-хроматографе "Waters 150-С". Параметры стереорегулярности определены из соотношения полос поглощения в ИК-спектрах D998/D973. Температуры плавления (Тпл) полученных полимеров определены методом дифференциальной сканирующей калориметрии на калориметре ДСК-910 ("DuPont Instrument"). Результаты опытов и свойства полученных полимеров приведены в таблице 2.The output of the IPP is determined by weighing. The molecular weight characteristics of polypropylene were obtained by gel permeation chromatography on a Waters 150-C high-temperature gel chromatograph. The stereoregularity parameters are determined from the ratio of the absorption bands in the IR spectra of D 998 / D 973 . The melting points ( Tm ) of the obtained polymers were determined by differential scanning calorimetry on a DSC-910 calorimeter (DuPont Instrument). The results of the experiments and the properties of the obtained polymers are shown in table 2.
Приведенные данные показывают, что предлагаемые ЦЦ с функционализированным циклосилановым мостиком обладают высокой каталитической активностью и стереоселективностью. The data presented show that the proposed CCs with a functionalized cyclosilane bridge have high catalytic activity and stereoselectivity.
Способ их получения отличается высокими выходами промежуточных и целевого продуктов, улучшенной методикой выделения дилитиевых производных мостичных лигандов и позволяет вдвое увеличить содержание активной рацемической формы в металлокомплексе. The method for their preparation is characterized by high yields of intermediate and target products, an improved procedure for the isolation of dilithium derivatives of bridge ligands and allows to double the content of the active racemic form in the metal complex.
Claims (3)

R1 = H, CH3, C2H5;
R2 = H, алкил от C1 до C4, нормальный или разветвленный, арил;
A = BC8H14, MR3, где M - олово или кремний, R - алкил от C1 до C4, нормальный или разветвленный, арил.1. Ansa-zirconocenes functionalized on a cyclosilane bridge, of general formula IV
R 1 = H, CH 3 , C 2 H 5 ;
R 2 = H, C 1 to C 4 alkyl, straight or branched, aryl;
A = BC 8 H 14 , MR 3 , where M is tin or silicon, R is C 1 to C 4 alkyl, straight or branched, aryl.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU99113532A RU2160277C1 (en) | 1999-06-21 | 1999-06-21 | Anssa-zirconocenes functioned with respect to cyclosilane bridges, and method of preparation thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU99113532A RU2160277C1 (en) | 1999-06-21 | 1999-06-21 | Anssa-zirconocenes functioned with respect to cyclosilane bridges, and method of preparation thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2160277C1 true RU2160277C1 (en) | 2000-12-10 |
Family
ID=20221698
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU99113532A RU2160277C1 (en) | 1999-06-21 | 1999-06-21 | Anssa-zirconocenes functioned with respect to cyclosilane bridges, and method of preparation thereof |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2160277C1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2337104C2 (en) * | 2002-10-25 | 2008-10-27 | Базелль Полиолефине Гмбх | METHOD FOR RACEMIC DIORGANOSILYL-BIS-(2-METHYLBENZO-[e]INDENYL)ZIRCONIUM COMPOUNDS DIASTEREOSELECTIVE SYNTHESIS, AND RACEMIC COMPOUND OF TRANSITION METAL |
| RU2425061C2 (en) * | 2006-02-02 | 2011-07-27 | Шеврон Филлипс Кемикал Компани Лп | Polymerisation catalysts for producing polymers with low level of long-chain branching |
| WO2015141675A1 (en) * | 2014-03-20 | 2015-09-24 | 日本ポリプロ株式会社 | Metallocene complex and method for manufacturing olefin polymer |
| CN105358588A (en) * | 2013-07-17 | 2016-02-24 | 埃克森美孚化学专利公司 | Process using substituted metallocene catalysts and products therefrom |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2095364C1 (en) * | 1991-09-06 | 1997-11-10 | Дсм Н.В. | Method of preparing bridge metallocene compound |
| RU2098423C1 (en) * | 1991-11-30 | 1997-12-10 | Хехст АГ | Metallocenes with benzofused indenyl derivatives, method of their synthesis, method of olefin polymer producing and catalyst for olefin polymerization |
| RU2118961C1 (en) * | 1992-06-27 | 1998-09-20 | Хехст АГ | Metallocenes with aryl-substituted indenyl derivatives, method of synthesis of olefin polymers and catalyst for olefins polymerization |
| RU2128183C1 (en) * | 1992-11-11 | 1999-03-27 | Дсм Н.В. | Indenyl compounds, method of polymerization of olefin, and polyolefin |
-
1999
- 1999-06-21 RU RU99113532A patent/RU2160277C1/en active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2095364C1 (en) * | 1991-09-06 | 1997-11-10 | Дсм Н.В. | Method of preparing bridge metallocene compound |
| RU2098423C1 (en) * | 1991-11-30 | 1997-12-10 | Хехст АГ | Metallocenes with benzofused indenyl derivatives, method of their synthesis, method of olefin polymer producing and catalyst for olefin polymerization |
| RU2118961C1 (en) * | 1992-06-27 | 1998-09-20 | Хехст АГ | Metallocenes with aryl-substituted indenyl derivatives, method of synthesis of olefin polymers and catalyst for olefins polymerization |
| RU2128183C1 (en) * | 1992-11-11 | 1999-03-27 | Дсм Н.В. | Indenyl compounds, method of polymerization of olefin, and polyolefin |
Non-Patent Citations (1)
| Title |
|---|
| Spaleck W. et.al., Organometallics, 1994. 13, с.954. * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2337104C2 (en) * | 2002-10-25 | 2008-10-27 | Базелль Полиолефине Гмбх | METHOD FOR RACEMIC DIORGANOSILYL-BIS-(2-METHYLBENZO-[e]INDENYL)ZIRCONIUM COMPOUNDS DIASTEREOSELECTIVE SYNTHESIS, AND RACEMIC COMPOUND OF TRANSITION METAL |
| RU2337104C9 (en) * | 2002-10-25 | 2009-08-27 | Базелль Полиолефине Гмбх | METHOD FOR RACEMIC DIORGANOSILYL-BIS-(2-METHYLBENZO-[e]INDENYL)ZIRCONIUM COMPOUNDS DIASTEREOSELECTIVE SYNTHESIS, AND RACEMIC COMPOUND OF TRANSITION METAL |
| RU2425061C2 (en) * | 2006-02-02 | 2011-07-27 | Шеврон Филлипс Кемикал Компани Лп | Polymerisation catalysts for producing polymers with low level of long-chain branching |
| CN105358588A (en) * | 2013-07-17 | 2016-02-24 | 埃克森美孚化学专利公司 | Process using substituted metallocene catalysts and products therefrom |
| CN105358588B (en) * | 2013-07-17 | 2018-05-18 | 埃克森美孚化学专利公司 | Method and product therefrom using substitution metallocene catalyst |
| WO2015141675A1 (en) * | 2014-03-20 | 2015-09-24 | 日本ポリプロ株式会社 | Metallocene complex and method for manufacturing olefin polymer |
| JP2015193605A (en) * | 2014-03-20 | 2015-11-05 | 日本ポリプロ株式会社 | Metallocene complex and method for producing olefin polymer |
| US9963522B2 (en) | 2014-03-20 | 2018-05-08 | Japan Polypropylene Corporation | Metallocene complex and method for producing olefin polymer |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3268903B2 (en) | Process for producing olefin polymers using metallocenes containing specifically substituted indenyl ligands | |
| JP3434288B2 (en) | Method for producing substituted indenes | |
| Piccolrovazzi et al. | Electronic effects in homogeneous indenylzirconium Ziegler-Natta catalysts | |
| RU2078771C1 (en) | Method for production of polyolefins | |
| RU2098423C1 (en) | Metallocenes with benzofused indenyl derivatives, method of their synthesis, method of olefin polymer producing and catalyst for olefin polymerization | |
| Stehling et al. | Ansa-Zirconocene polymerization catalysts with anelated ring ligands-effects on catalytic activity and polymer chain length | |
| US6268518B1 (en) | Metallocene compounds and their use in catalysts for the polymerization of olefins | |
| Mengele et al. | ansa-Metallocene derivatives. 27. Chiral zirconocene complexes with two dimethylsilylene bridges | |
| US6391991B1 (en) | Process for the preparation of amorphous polymers of propylene | |
| US5840947A (en) | Organometallic compound | |
| JP2006028191A (en) | Intermediates for the synthesis of metallocenes containing aryl-substituted indenyl derivatives as ligands and methods of use thereof | |
| JPH06340684A (en) | Metallocene having ligand of 2-substituted indenyl derivative, its production and method of using it | |
| KR20000076111A (en) | Process for the preparation of metallocene compounds | |
| Collins et al. | Synthesis of pure racemic isomers of ansa-titanocene dichlorides: conformational preferences are determined by substitution patterns on the cyclopentadienyl rings | |
| US5521265A (en) | Metallocenes and their use for olefin polymerization | |
| RU2161148C2 (en) | Method of synthesis of bis-cyclopentadienyl compounds, method of synthesis of cyclopentadienyl compounds, cyclopentadienyl compound | |
| RU2160276C1 (en) | Ansa-zirconocines with unsaturated cyclosilane bridge, and method of preparing thereof | |
| Halterman et al. | Application of the double Pauson-Khand cyclization to the synthesis of bis (cyclopentadienes): preparation of phenyl-bridged bis (tetrahydroindenyl) titanium and zirconium dichlorides | |
| RU2160277C1 (en) | Anssa-zirconocenes functioned with respect to cyclosilane bridges, and method of preparation thereof | |
| JP3998270B2 (en) | Method for producing cyclopentadienyl compound | |
| EP0557718B1 (en) | Catalyst for olefin polymerisation, process for preparing the same and its sue | |
| Schumann et al. | Synthesis and characterization of 1-and 2-(ω-alken-1-yl) indenes, their lithium salts and dichlorozirconium (IV) complexes | |
| Könemann et al. | Diastereoselective Synthesis of a Bis (tetrahydroindenyl) zirconocene Dichloride with “Ansa-Fused” Annulated Six-Membered Ring Systems: A Specific Solution of the Synthetic rac-/meso-Group 4 Bent Metallocene Problem | |
| JP2012012307A (en) | Method for producing crosslinked indene | |
| JP4127324B2 (en) | Method for producing 2-aryl substituted indenes |