KR20220169916A - 병렬적 다중 전위 증폭 방법 - Google Patents
병렬적 다중 전위 증폭 방법 Download PDFInfo
- Publication number
- KR20220169916A KR20220169916A KR1020220074560A KR20220074560A KR20220169916A KR 20220169916 A KR20220169916 A KR 20220169916A KR 1020220074560 A KR1020220074560 A KR 1020220074560A KR 20220074560 A KR20220074560 A KR 20220074560A KR 20220169916 A KR20220169916 A KR 20220169916A
- Authority
- KR
- South Korea
- Prior art keywords
- primer
- nucleic acid
- barcode
- nucleotide sequence
- dna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000003199 nucleic acid amplification method Methods 0.000 title claims abstract description 240
- 230000003321 amplification Effects 0.000 title claims abstract description 186
- 238000000034 method Methods 0.000 title claims abstract description 117
- 238000006073 displacement reaction Methods 0.000 title abstract description 11
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 175
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 173
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 173
- 238000002955 isolation Methods 0.000 claims abstract description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 claims description 202
- 238000012986 modification Methods 0.000 claims description 148
- 230000004048 modification Effects 0.000 claims description 148
- 125000003729 nucleotide group Chemical group 0.000 claims description 125
- 239000002773 nucleotide Substances 0.000 claims description 122
- 239000011616 biotin Substances 0.000 claims description 104
- 229960002685 biotin Drugs 0.000 claims description 104
- 235000020958 biotin Nutrition 0.000 claims description 94
- 238000007481 next generation sequencing Methods 0.000 claims description 91
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 claims description 52
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 claims description 52
- 238000005516 engineering process Methods 0.000 claims description 47
- 102000040430 polynucleotide Human genes 0.000 claims description 37
- 108091033319 polynucleotide Proteins 0.000 claims description 37
- 239000002157 polynucleotide Substances 0.000 claims description 37
- 239000000463 material Substances 0.000 claims description 34
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 26
- 238000003752 polymerase chain reaction Methods 0.000 claims description 24
- 238000011176 pooling Methods 0.000 claims description 19
- 239000011324 bead Substances 0.000 claims description 16
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 claims description 14
- 108010090804 Streptavidin Proteins 0.000 claims description 13
- 238000012163 sequencing technique Methods 0.000 claims description 13
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 12
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 claims description 10
- 238000000926 separation method Methods 0.000 claims description 10
- 108090000623 proteins and genes Proteins 0.000 claims description 8
- 235000000638 D-biotin Nutrition 0.000 claims description 7
- 239000011665 D-biotin Substances 0.000 claims description 7
- 229960000643 adenine Drugs 0.000 claims description 7
- 238000012216 screening Methods 0.000 claims description 7
- 229940104302 cytosine Drugs 0.000 claims description 6
- 229930024421 Adenine Natural products 0.000 claims description 5
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 claims description 5
- 150000001540 azides Chemical class 0.000 claims description 5
- 239000000126 substance Substances 0.000 claims description 5
- 229940113082 thymine Drugs 0.000 claims description 5
- 108010017826 DNA Polymerase I Proteins 0.000 claims description 4
- 102000004594 DNA Polymerase I Human genes 0.000 claims description 4
- 101710163270 Nuclease Proteins 0.000 claims description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 claims description 4
- 125000003277 amino group Chemical group 0.000 claims description 4
- 238000001943 fluorescence-activated cell sorting Methods 0.000 claims description 4
- 125000005980 hexynyl group Chemical group 0.000 claims description 4
- 238000000370 laser capture micro-dissection Methods 0.000 claims description 4
- 102000004169 proteins and genes Human genes 0.000 claims description 4
- 150000003573 thiols Chemical class 0.000 claims description 4
- 108090001008 Avidin Proteins 0.000 claims description 3
- 230000009977 dual effect Effects 0.000 claims description 3
- 108010087904 neutravidin Proteins 0.000 claims description 3
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 claims description 2
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 claims description 2
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 claims description 2
- 108010025076 Holoenzymes Proteins 0.000 claims description 2
- 101150054516 PRD1 gene Proteins 0.000 claims description 2
- 101100459905 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) NCP1 gene Proteins 0.000 claims description 2
- 101000865057 Thermococcus litoralis DNA polymerase Proteins 0.000 claims description 2
- 150000001345 alkine derivatives Chemical class 0.000 claims description 2
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 claims description 2
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 claims description 2
- 150000004662 dithiols Chemical class 0.000 claims description 2
- 150000002148 esters Chemical class 0.000 claims description 2
- 238000001489 optical tweezers Raman spectroscopy Methods 0.000 claims description 2
- 238000004458 analytical method Methods 0.000 abstract description 56
- 102000004190 Enzymes Human genes 0.000 abstract description 11
- 108090000790 Enzymes Proteins 0.000 abstract description 11
- 230000000379 polymerizing effect Effects 0.000 abstract description 5
- 108020004414 DNA Proteins 0.000 description 163
- 210000004027 cell Anatomy 0.000 description 159
- 239000000523 sample Substances 0.000 description 67
- 238000006243 chemical reaction Methods 0.000 description 65
- 239000000047 product Substances 0.000 description 61
- 230000008569 process Effects 0.000 description 29
- 230000027455 binding Effects 0.000 description 18
- 238000002474 experimental method Methods 0.000 description 17
- 238000004519 manufacturing process Methods 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- 102000053602 DNA Human genes 0.000 description 13
- 230000005764 inhibitory process Effects 0.000 description 13
- 206010028980 Neoplasm Diseases 0.000 description 12
- 230000007423 decrease Effects 0.000 description 12
- 239000012634 fragment Substances 0.000 description 12
- 210000001185 bone marrow Anatomy 0.000 description 11
- 238000003320 cell separation method Methods 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- 108091092584 GDNA Proteins 0.000 description 10
- 201000011510 cancer Diseases 0.000 description 10
- 210000002919 epithelial cell Anatomy 0.000 description 10
- 210000005259 peripheral blood Anatomy 0.000 description 10
- 239000011886 peripheral blood Substances 0.000 description 10
- 210000004556 brain Anatomy 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 108060002716 Exonuclease Proteins 0.000 description 8
- 238000010276 construction Methods 0.000 description 8
- 102000013165 exonuclease Human genes 0.000 description 8
- 238000013467 fragmentation Methods 0.000 description 8
- 238000006062 fragmentation reaction Methods 0.000 description 8
- 210000004072 lung Anatomy 0.000 description 8
- -1 rRNA Proteins 0.000 description 8
- 210000003719 b-lymphocyte Anatomy 0.000 description 7
- ZLAQATDNGLKIEV-UHFFFAOYSA-N 5-methyl-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CC1=CNC(=S)NC1=O ZLAQATDNGLKIEV-UHFFFAOYSA-N 0.000 description 6
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 6
- 210000002950 fibroblast Anatomy 0.000 description 6
- 238000013412 genome amplification Methods 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- 238000002156 mixing Methods 0.000 description 6
- 238000012545 processing Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 5
- 239000012530 fluid Substances 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 5
- RFLVMTUMFYRZCB-UHFFFAOYSA-N 1-methylguanine Chemical compound O=C1N(C)C(N)=NC2=C1N=CN2 RFLVMTUMFYRZCB-UHFFFAOYSA-N 0.000 description 4
- YSAJFXWTVFGPAX-UHFFFAOYSA-N 2-[(2,4-dioxo-1h-pyrimidin-5-yl)oxy]acetic acid Chemical compound OC(=O)COC1=CNC(=O)NC1=O YSAJFXWTVFGPAX-UHFFFAOYSA-N 0.000 description 4
- FZWGECJQACGGTI-UHFFFAOYSA-N 2-amino-7-methyl-1,7-dihydro-6H-purin-6-one Chemical compound NC1=NC(O)=C2N(C)C=NC2=N1 FZWGECJQACGGTI-UHFFFAOYSA-N 0.000 description 4
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 description 4
- OIVLITBTBDPEFK-UHFFFAOYSA-N 5,6-dihydrouracil Chemical compound O=C1CCNC(=O)N1 OIVLITBTBDPEFK-UHFFFAOYSA-N 0.000 description 4
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 4
- HYVABZIGRDEKCD-UHFFFAOYSA-N N(6)-dimethylallyladenine Chemical compound CC(C)=CCNC1=NC=NC2=C1N=CN2 HYVABZIGRDEKCD-UHFFFAOYSA-N 0.000 description 4
- 108091093037 Peptide nucleic acid Proteins 0.000 description 4
- 108010012306 Tn5 transposase Proteins 0.000 description 4
- 230000015556 catabolic process Effects 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000006731 degradation reaction Methods 0.000 description 4
- 210000002889 endothelial cell Anatomy 0.000 description 4
- 230000002068 genetic effect Effects 0.000 description 4
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 4
- DRAVOWXCEBXPTN-UHFFFAOYSA-N isoguanine Chemical compound NC1=NC(=O)NC2=C1NC=N2 DRAVOWXCEBXPTN-UHFFFAOYSA-N 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 244000005700 microbiome Species 0.000 description 4
- 238000005580 one pot reaction Methods 0.000 description 4
- 238000003908 quality control method Methods 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 210000000952 spleen Anatomy 0.000 description 4
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- 108700028369 Alleles Proteins 0.000 description 3
- 108091034117 Oligonucleotide Proteins 0.000 description 3
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000012790 confirmation Methods 0.000 description 3
- 238000004925 denaturation Methods 0.000 description 3
- 230000036425 denaturation Effects 0.000 description 3
- 210000004443 dendritic cell Anatomy 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 238000009396 hybridization Methods 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 3
- 239000004055 small Interfering RNA Substances 0.000 description 3
- 238000000527 sonication Methods 0.000 description 3
- 229940035893 uracil Drugs 0.000 description 3
- GUAHPAJOXVYFON-ZETCQYMHSA-N (8S)-8-amino-7-oxononanoic acid zwitterion Chemical compound C[C@H](N)C(=O)CCCCCC(O)=O GUAHPAJOXVYFON-ZETCQYMHSA-N 0.000 description 2
- WJNGQIYEQLPJMN-IOSLPCCCSA-N 1-methylinosine Chemical compound C1=NC=2C(=O)N(C)C=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O WJNGQIYEQLPJMN-IOSLPCCCSA-N 0.000 description 2
- HLYBTPMYFWWNJN-UHFFFAOYSA-N 2-(2,4-dioxo-1h-pyrimidin-5-yl)-2-hydroxyacetic acid Chemical compound OC(=O)C(O)C1=CNC(=O)NC1=O HLYBTPMYFWWNJN-UHFFFAOYSA-N 0.000 description 2
- SGAKLDIYNFXTCK-UHFFFAOYSA-N 2-[(2,4-dioxo-1h-pyrimidin-5-yl)methylamino]acetic acid Chemical compound OC(=O)CNCC1=CNC(=O)NC1=O SGAKLDIYNFXTCK-UHFFFAOYSA-N 0.000 description 2
- XQCZBXHVTFVIFE-UHFFFAOYSA-N 2-amino-4-hydroxypyrimidine Chemical compound NC1=NC=CC(O)=N1 XQCZBXHVTFVIFE-UHFFFAOYSA-N 0.000 description 2
- MWBWWFOAEOYUST-UHFFFAOYSA-N 2-aminopurine Chemical compound NC1=NC=C2N=CNC2=N1 MWBWWFOAEOYUST-UHFFFAOYSA-N 0.000 description 2
- XMSMHKMPBNTBOD-UHFFFAOYSA-N 2-dimethylamino-6-hydroxypurine Chemical compound N1C(N(C)C)=NC(=O)C2=C1N=CN2 XMSMHKMPBNTBOD-UHFFFAOYSA-N 0.000 description 2
- SMADWRYCYBUIKH-UHFFFAOYSA-N 2-methyl-7h-purin-6-amine Chemical compound CC1=NC(N)=C2NC=NC2=N1 SMADWRYCYBUIKH-UHFFFAOYSA-N 0.000 description 2
- HCGYMSSYSAKGPK-UHFFFAOYSA-N 2-nitro-1h-indole Chemical compound C1=CC=C2NC([N+](=O)[O-])=CC2=C1 HCGYMSSYSAKGPK-UHFFFAOYSA-N 0.000 description 2
- FTBBGQKRYUTLMP-UHFFFAOYSA-N 2-nitro-1h-pyrrole Chemical compound [O-][N+](=O)C1=CC=CN1 FTBBGQKRYUTLMP-UHFFFAOYSA-N 0.000 description 2
- KOLPWZCZXAMXKS-UHFFFAOYSA-N 3-methylcytosine Chemical compound CN1C(N)=CC=NC1=O KOLPWZCZXAMXKS-UHFFFAOYSA-N 0.000 description 2
- GJAKJCICANKRFD-UHFFFAOYSA-N 4-acetyl-4-amino-1,3-dihydropyrimidin-2-one Chemical compound CC(=O)C1(N)NC(=O)NC=C1 GJAKJCICANKRFD-UHFFFAOYSA-N 0.000 description 2
- XORHNJQEWQGXCN-UHFFFAOYSA-N 4-nitro-1h-pyrazole Chemical compound [O-][N+](=O)C=1C=NNC=1 XORHNJQEWQGXCN-UHFFFAOYSA-N 0.000 description 2
- MQJSSLBGAQJNER-UHFFFAOYSA-N 5-(methylaminomethyl)-1h-pyrimidine-2,4-dione Chemical compound CNCC1=CNC(=O)NC1=O MQJSSLBGAQJNER-UHFFFAOYSA-N 0.000 description 2
- WPYRHVXCOQLYLY-UHFFFAOYSA-N 5-[(methoxyamino)methyl]-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CONCC1=CNC(=S)NC1=O WPYRHVXCOQLYLY-UHFFFAOYSA-N 0.000 description 2
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical compound BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 2
- VKLFQTYNHLDMDP-PNHWDRBUSA-N 5-carboxymethylaminomethyl-2-thiouridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=S)NC(=O)C(CNCC(O)=O)=C1 VKLFQTYNHLDMDP-PNHWDRBUSA-N 0.000 description 2
- ZFTBZKVVGZNMJR-UHFFFAOYSA-N 5-chlorouracil Chemical compound ClC1=CNC(=O)NC1=O ZFTBZKVVGZNMJR-UHFFFAOYSA-N 0.000 description 2
- KSNXJLQDQOIRIP-UHFFFAOYSA-N 5-iodouracil Chemical compound IC1=CNC(=O)NC1=O KSNXJLQDQOIRIP-UHFFFAOYSA-N 0.000 description 2
- KELXHQACBIUYSE-UHFFFAOYSA-N 5-methoxy-1h-pyrimidine-2,4-dione Chemical compound COC1=CNC(=O)NC1=O KELXHQACBIUYSE-UHFFFAOYSA-N 0.000 description 2
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 2
- OZFPSOBLQZPIAV-UHFFFAOYSA-N 5-nitro-1h-indole Chemical compound [O-][N+](=O)C1=CC=C2NC=CC2=C1 OZFPSOBLQZPIAV-UHFFFAOYSA-N 0.000 description 2
- VYDWQPKRHOGLPA-UHFFFAOYSA-N 5-nitroimidazole Chemical compound [O-][N+](=O)C1=CN=CN1 VYDWQPKRHOGLPA-UHFFFAOYSA-N 0.000 description 2
- UJBCLAXPPIDQEE-UHFFFAOYSA-N 5-prop-1-ynyl-1h-pyrimidine-2,4-dione Chemical compound CC#CC1=CNC(=O)NC1=O UJBCLAXPPIDQEE-UHFFFAOYSA-N 0.000 description 2
- DCPSTSVLRXOYGS-UHFFFAOYSA-N 6-amino-1h-pyrimidine-2-thione Chemical compound NC1=CC=NC(S)=N1 DCPSTSVLRXOYGS-UHFFFAOYSA-N 0.000 description 2
- VKKXEIQIGGPMHT-UHFFFAOYSA-N 7h-purine-2,8-diamine Chemical compound NC1=NC=C2NC(N)=NC2=N1 VKKXEIQIGGPMHT-UHFFFAOYSA-N 0.000 description 2
- 229960005508 8-azaguanine Drugs 0.000 description 2
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 2
- 108091061744 Cell-free fetal DNA Proteins 0.000 description 2
- 208000005443 Circulating Neoplastic Cells Diseases 0.000 description 2
- 230000004544 DNA amplification Effects 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 2
- 229930010555 Inosine Natural products 0.000 description 2
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- SGSSKEDGVONRGC-UHFFFAOYSA-N N(2)-methylguanine Chemical compound O=C1NC(NC)=NC2=C1N=CN2 SGSSKEDGVONRGC-UHFFFAOYSA-N 0.000 description 2
- 108010004729 Phycoerythrin Proteins 0.000 description 2
- 108020004459 Small interfering RNA Proteins 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 210000002449 bone cell Anatomy 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000006037 cell lysis Effects 0.000 description 2
- 108091092259 cell-free RNA Proteins 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 230000000112 colonic effect Effects 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 238000010448 genetic screening Methods 0.000 description 2
- 229960003786 inosine Drugs 0.000 description 2
- 230000001678 irradiating effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000007403 mPCR Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- IZAGSTRIDUNNOY-UHFFFAOYSA-N methyl 2-[(2,4-dioxo-1h-pyrimidin-5-yl)oxy]acetate Chemical compound COC(=O)COC1=CNC(=O)NC1=O IZAGSTRIDUNNOY-UHFFFAOYSA-N 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- GJVFBWCTGUSGDD-UHFFFAOYSA-L pentamethonium bromide Chemical compound [Br-].[Br-].C[N+](C)(C)CCCCC[N+](C)(C)C GJVFBWCTGUSGDD-UHFFFAOYSA-L 0.000 description 2
- 150000002972 pentoses Chemical class 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 210000002105 tongue Anatomy 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- 210000003932 urinary bladder Anatomy 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 229940075420 xanthine Drugs 0.000 description 2
- QUTFFEUUGHUPQC-ILWYWAAHSA-N (2r,3r,4s,5r)-3,4,5,6-tetrahydroxy-2-[(4-nitro-2,1,3-benzoxadiazol-7-yl)amino]hexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC1=CC=C([N+]([O-])=O)C2=NON=C12 QUTFFEUUGHUPQC-ILWYWAAHSA-N 0.000 description 1
- VGIRNWJSIRVFRT-UHFFFAOYSA-N 2',7'-difluorofluorescein Chemical compound OC(=O)C1=CC=CC=C1C1=C2C=C(F)C(=O)C=C2OC2=CC(O)=C(F)C=C21 VGIRNWJSIRVFRT-UHFFFAOYSA-N 0.000 description 1
- RDVCCJOZVDOCSX-UHFFFAOYSA-N 2-(2-aminoethylamino)acetamide Chemical group NCCNCC(N)=O RDVCCJOZVDOCSX-UHFFFAOYSA-N 0.000 description 1
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 1
- GOLORTLGFDVFDW-UHFFFAOYSA-N 3-(1h-benzimidazol-2-yl)-7-(diethylamino)chromen-2-one Chemical compound C1=CC=C2NC(C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=NC2=C1 GOLORTLGFDVFDW-UHFFFAOYSA-N 0.000 description 1
- AUUIARVPJHGTSA-UHFFFAOYSA-N 3-(aminomethyl)chromen-2-one Chemical compound C1=CC=C2OC(=O)C(CN)=CC2=C1 AUUIARVPJHGTSA-UHFFFAOYSA-N 0.000 description 1
- LPXQRXLUHJKZIE-UHFFFAOYSA-N 8-azaguanine Chemical compound NC1=NC(O)=C2NN=NC2=N1 LPXQRXLUHJKZIE-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108091028075 Circular RNA Proteins 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- 229940123014 DNA polymerase inhibitor Drugs 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- XPDXVDYUQZHFPV-UHFFFAOYSA-N Dansyl Chloride Chemical compound C1=CC=C2C(N(C)C)=CC=CC2=C1S(Cl)(=O)=O XPDXVDYUQZHFPV-UHFFFAOYSA-N 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 208000005228 Pericardial Effusion Diseases 0.000 description 1
- 108091007412 Piwi-interacting RNA Proteins 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002594 Polyethylene Glycol 8000 Polymers 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 108010026552 Proteome Proteins 0.000 description 1
- 108020003224 Small Nucleolar RNA Proteins 0.000 description 1
- 102000042773 Small Nucleolar RNA Human genes 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 108020004417 Untranslated RNA Proteins 0.000 description 1
- 102000039634 Untranslated RNA Human genes 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000005336 allyloxy group Chemical group 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 210000004381 amniotic fluid Anatomy 0.000 description 1
- 238000012197 amplification kit Methods 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 108010058966 bacteriophage T7 induced DNA polymerase Proteins 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 238000010009 beating Methods 0.000 description 1
- 238000005842 biochemical reaction Methods 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000004958 brain cell Anatomy 0.000 description 1
- 210000000069 breast epithelial cell Anatomy 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 210000000621 bronchi Anatomy 0.000 description 1
- 210000000424 bronchial epithelial cell Anatomy 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 210000004922 colonic epithelial cell Anatomy 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 210000003618 cortical neuron Anatomy 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000009615 deamination Effects 0.000 description 1
- 238000006481 deamination reaction Methods 0.000 description 1
- 238000002242 deionisation method Methods 0.000 description 1
- 239000013578 denaturing buffer Substances 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- PGUYAANYCROBRT-UHFFFAOYSA-N dihydroxy-selanyl-selanylidene-lambda5-phosphane Chemical compound OP(O)([SeH])=[Se] PGUYAANYCROBRT-UHFFFAOYSA-N 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-K dioxido-sulfanylidene-sulfido-$l^{5}-phosphane Chemical compound [O-]P([O-])([S-])=S NAGJZTKCGNOGPW-UHFFFAOYSA-K 0.000 description 1
- 238000011978 dissolution method Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 210000001842 enterocyte Anatomy 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 210000000918 epididymis Anatomy 0.000 description 1
- 201000010063 epididymitis Diseases 0.000 description 1
- 230000001973 epigenetic effect Effects 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- IINNWAYUJNWZRM-UHFFFAOYSA-L erythrosin B Chemical compound [Na+].[Na+].[O-]C(=O)C1=CC=CC=C1C1=C2C=C(I)C(=O)C(I)=C2OC2=C(I)C([O-])=C(I)C=C21 IINNWAYUJNWZRM-UHFFFAOYSA-L 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 210000001508 eye Anatomy 0.000 description 1
- 210000000604 fetal stem cell Anatomy 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical group O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000013505 freshwater Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000010362 genome editing Methods 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 238000007490 hematoxylin and eosin (H&E) staining Methods 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- QDLAGTHXVHQKRE-UHFFFAOYSA-N lichenxanthone Natural products COC1=CC(O)=C2C(=O)C3=C(C)C=C(OC)C=C3OC2=C1 QDLAGTHXVHQKRE-UHFFFAOYSA-N 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000007257 malfunction Effects 0.000 description 1
- 210000005074 megakaryoblast Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- 239000002679 microRNA Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- 238000001000 micrograph Methods 0.000 description 1
- 230000009267 minimal disease activity Effects 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- STUHQDIOZQUPGP-UHFFFAOYSA-N morpholin-4-ium-4-carboxylate Chemical class OC(=O)N1CCOCC1 STUHQDIOZQUPGP-UHFFFAOYSA-N 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 210000001167 myeloblast Anatomy 0.000 description 1
- 210000004412 neuroendocrine cell Anatomy 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 238000001821 nucleic acid purification Methods 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 210000000963 osteoblast Anatomy 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 150000004893 oxazines Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 210000002571 pancreatic alpha cell Anatomy 0.000 description 1
- 210000002990 parathyroid gland Anatomy 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 210000004912 pericardial fluid Anatomy 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 210000004910 pleural fluid Anatomy 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 210000005132 reproductive cell Anatomy 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 210000004116 schwann cell Anatomy 0.000 description 1
- JRPHGDYSKGJTKZ-UHFFFAOYSA-K selenophosphate Chemical compound [O-]P([O-])([O-])=[Se] JRPHGDYSKGJTKZ-UHFFFAOYSA-K 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 210000004927 skin cell Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000002689 soil Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 239000012536 storage buffer Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- MPLHNVLQVRSVEE-UHFFFAOYSA-N texas red Chemical compound [O-]S(=O)(=O)C1=CC(S(Cl)(=O)=O)=CC=C1C(C1=CC=2CCCN3CCCC(C=23)=C1O1)=C2C1=C(CCC1)C3=[N+]1CCCC3=C2 MPLHNVLQVRSVEE-UHFFFAOYSA-N 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 210000000626 ureter Anatomy 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
Images



























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6848—Nucleic acid amplification reactions characterised by the means for preventing contamination or increasing the specificity or sensitivity of an amplification reaction
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1065—Preparation or screening of tagged libraries, e.g. tagged microorganisms by STM-mutagenesis, tagged polynucleotides, gene tags
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/06—Libraries containing nucleotides or polynucleotides, or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2521/00—Reaction characterised by the enzymatic activity
- C12Q2521/10—Nucleotidyl transfering
- C12Q2521/101—DNA polymerase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2535/00—Reactions characterised by the assay type for determining the identity of a nucleotide base or a sequence of oligonucleotides
- C12Q2535/122—Massive parallel sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2537/00—Reactions characterised by the reaction format or use of a specific feature
- C12Q2537/10—Reactions characterised by the reaction format or use of a specific feature the purpose or use of
- C12Q2537/143—Multiplexing, i.e. use of multiple primers or probes in a single reaction, usually for simultaneously analyse of multiple analysis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2563/00—Nucleic acid detection characterized by the use of physical, structural and functional properties
- C12Q2563/179—Nucleic acid detection characterized by the use of physical, structural and functional properties the label being a nucleic acid
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Plant Pathology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
일 양상은 제1 프라이머, 제2 프라이머 및 핵산 중합효소를 표적 핵산이 포함된 샘플에 첨가하여 핵산들을 증폭시키는 단계를 포함하는 핵산 증폭 방법에 관한 것이다. 일 양상에 따른 핵산 증폭 방법에 의하면, 바코드 서열을 포함하는 제1 프라이머를 이용하여 병렬적 다중 전위 증폭을 수행함으로써, MDA의 병렬성 및 처리량을 높여 NGS 라이브러리 제작 시간 및 비용을 혁신적으로 감소시키고, 제1 프라이머가 첨가되는 비율을 조절하여 바코드 편향 현상을 해결할 수 있으며, 레이저 기반 세포 분리 방식을 이용하여 증폭 품질도 향상시킬 수 있다. 따라서, 상기 핵산 증폭 방법은 다량의 단일 세포 전장 유전체 분석에 유용하게 이용될 수 있다.
Description
병렬적 다중 전위 증폭 방법에 관한 것이다.
미량(수 ng 이하의 양)의 DNA로부터 DNA를 증폭하는 기술 중에서 MDA(Multiple Displacement Amplification)는 널리 쓰이는 기술 중 하나이다. MDA의 응용 분야 중 하나는 단일 세포(single-cell) 유전체(genome) 분석이다. 단일 세포에는 극미량의 DNA만 존재하기 때문에, 이를 분석할 수 있는 수준의 양으로 증폭할 수 있는 전장 유전체 증폭(Whole genome amplification) 기술이 중요한데, MDA는 가장 많이 사용되어 온 전장 유전체 증폭 기술 중 하나이다. 단일 세포 유전체 분석이 중요한 이유는 동일해 보이는 세포들이라도 실은 서로 다른 유전체 염기서열을 갖고 있다는 이른바 세포 이질성(cell heterogeneity)이라는 현상 때문이다. 특히 변이가 많은 암이나, 뇌, 면역세포 등에서 세포 이질성이 더욱 두드러진다. 이 뿐만 아니라 인간과 함께 공존하고 있거나 환경에 존재하는 미생물(microorganism)이나 박테리아 혹은 병원균(pathogen)들은 포유동물에 비해서 생물학적 다양성(diversity)이 훨씬 크며, 그들의 유전체를 분석하는 마이크로바이옴(microbiome) 분야에서도 단일 세포 유전체 분석 기술이 크게 각광받고 있다. 특히 미생물들 중 다수가 실험실에서 배양할 수 없어(unculturable) 다량의 DNA를 확보하기 어렵기 때문에 MDA기술이 중요하다.
MDA의 또 다른 응용분야 중 하나는 단일 세포 뿐만 아니라 여러 개의 세포 또는 세포로부터 유래한 DNA(수 ng 이하)를 분석하는 분야이다. 상기 암, 뇌, 면역세포, 마이크로바이옴 등의 예에서 굳이 단일세포가 아니더라도 여러 개의 세포에 대해 한번의 MDA 증폭을 수행하여 세포 이질성을 파악할 수 있다. 그 외에도 샘플이 희귀(rare)하여 다량의 DNA를 얻을 수 없는 경우인 법의학(forensics), 정자(sperms), 배우자(gamete), 착상 전 유전 진단(preimplantation genetic diagnosis, PGD), 착상 전 유전자 스크리닝(preimplantation genetic screening, PGS), 착장 전 유전자 검사(preimplantation genetic testing), 순환 종양 세포(circulating tumor cells, CTC), 순환 종양 DNA(circulating tumor DNA) 및 무세포 DNA(cell free DNA)와 같은 응용분야에도 MDA는 유용하게 사용될 수 있다. 이하에서는 용어의 간결성을 위해 소량의 DNA 혹은 단일 세포 혹은 다수의 세포를 분석하는 경우를 모두 단일 세포라 통칭하여 표현한다.
MDA는 다른 전장 유전체 증폭 기술에 비해 유전체 커버리지(genome coverage)가 매우 높으며, 사용되는 phi29 DNA 중합효소는 높은 충실도(fidelity)를 갖고 있어 증폭 에러(amplification error)가 낮다는 장점이 있다. 이 두 가지 특장점으로 인해 MDA는 단일 염기서열 수준(single-nucleotide resolution)의 단일 세포 유전체 분석을 가능하게 하는 중요한 기술이었다. 여기서 단일 염기서열 수준의 유전체 분석이란, 분석하고자 하는 유전체 영역 중 80%가 넘는 유전체 영역에 대해 단일 염기서열 수준까지 염기서열을 파악해 낼 수 있는 기술을 지칭한다. 다른 경쟁 기술인 DOP-PCR은 증폭 균일도(amplification uniformity)는 높았지만, 유전체 커버리지가 낮아서 단일 염기서열 수준의 단일 세포 유전체 분석에는 적용하기 어려웠다. 또 다른 경쟁기술인 MALBAC은 증폭 균일도와, 유전체 커버리지는 높았지만, Bst 중합효소의 높은 증폭 에러로 인해서 단일염기 에러(false-positive SNVs)가 많아서 단일 염기서열 수준의 유전체 분석에는 적용하기 어려웠다.
하지만 현재 MDA기술과 같은 전장 유전체 증폭 기술의 한계점은 분석하고자 하는 세포수가 증가할수록 분석을 위한 비용 또한 선형적으로 증가한다는 것이다. 예를 들어 한 환자가 가지고 있는 1 cm3 의 암 조직에는 약 106 개의 서로 다른 암 세포들이 존재하는데, 이를 모두 각각의 세포로 분리하여 분석을 하기 위해서는 분석 비용이 기존 기술인 벌크 시퀀싱에 비해 106 배 많이 필요하고 이는 현실적으로 지불하기 어려운 비용이다.
이를 극복하기 위하여 다른 Tn5 유전자전위효소(transposase)를 이용하여 단일 세포의 gDNA에 바로 태깅(tagmentation)을 수행하며 세포를 구별할 수 있는 바코드(barcode) 또는 인덱스(index)를 삽입하여 병렬성(multiplexity) 또는 처리량(throughput)을 높이려는 노력이 있었다. 하지만 그 과정의 효율이 충분하지 않아, 충분한 유전체 커버리지를 얻지 못하였고, 단일 염기서열 수준의 단일 세포 유전체 분석이 어려웠다 (도 1 참조). 또 다른 경쟁기술로 Mission Bio사에서 Tapestri라고 불리는 제품이 출시된 바가 있다. Tapestri는 단일 세포에서 바로 병렬적(multiplex) PCR을 수행하여 단일 세포의 수백개의 영역에 대해 단일 염기변이(Single nucleotide variant)를 검출할 수 있게 하는 기술이다. 하지만 multiplex PCR 기반의 기술은 동시에 분석할 수 있는 유전체 영역의 넓이에 한계가 (100 kb 이내) 있어서 전장 유전체(Whole genome) 또는 전체 엑솜(Whole exome)과 같이 좀 더 넓은 유전체 영역을 분석하는 데에는 한계가 있었다.
따라서 단일 염기서열수준의 단일 세포 유전체 분석을 위해서는 MDA와 같은 전장 유전체 증폭 기술이 필수적이다. 하지만 MDA 증폭 산물(MDA product)을 차세대 염기서열 분석 기술(Next generation sequencing)로 시퀀싱하기 위해서는 개별 증폭 산물에 대해 각각 라이브러리 제작(Library preparation)을 거쳐야 하는데 이는 비용과 시간이 많이 드는 과정이다. 즉 여러 세포를 분석하고자 할수록 여러 MDA 증폭 산물에 대해 각각 라이브러리 제작 과정을 거쳐야 하며, 이는 높은 라이브러리 제작 비용 및 시간이라는 문제를 가져왔다. 만약 MDA증폭기술의 병렬성과 처리량을 높일 수 있는 기술이 개발된다면 단일 염기서열수준의 단일세포 유전체 분석을 훨씬 낮은 비용과 노력으로 수행할 수 있게 되어, 훨씬 더 많은 수의 이질적인 세포를 분석할 수 있게 될 것이다.
많은 수의 단일 세포를 분석하고자 할수록 라이브러리 제작 비용이 증가하는 문제는 모든 단일 세포 분석 분야에서 공통적으로 겪어온 어려움이다. 단일 세포 전사체(transcriptomics), 단일 세포 후성 유전체(epigenetics), 단일 세포 단백질체(proteomics) 및 단일 세포 다중체(multiomics) 등 모든 분야에서 비슷한 어려움을 겪어왔고, 이를 극복하기 위한 가장 널리 사용되어온 효과적인 수단은 바코딩(barcoding)을 이용한 병렬화 기술이다. 라이브러리 제작 과정은 다음과 같이 크게 두 가지의 목적으로 이루어져 있다: 1) 분석하고자 하는 DNA가 차세대 염기서열 분석기에서 시퀀싱될 수 있도록 어댑터를 삽입하는 과정, 2) 여러 샘플이 동시에 시퀀싱되었을 경우 샘플을 구별(identify)할 수 있도록 바코드 또는 인덱스를 삽입하는 과정. 단일 세포 분석 분야에서는 서로 다른 세포에 대해 서로 다른 바코드를 삽입하여 세포를 구분하여야 하기 때문에, 즉 2)의 목적을 달성하기 위해 개별 세포에 대해 각각 라이브러리 제작(Library preparation) 과정을 거쳐야 하며, 이로 인해 라이브러리 제작 과정의 비용이 증가하게 된다. 하지만 만약 2)의 목적을 라이브러리 제작 과정 이전에 달성할 수 있다면 여러 세포에서 유래한 DNA에 대해 한 번에 라이브러리 제작과정을 거쳐서 1)의 목적을 달성할 수 있을 것이다. 즉, 상기 언급한 바코딩을 이용한 병렬화 기술은 단일세포에서 유래한 DNA, RNA, 단백질, 휴성유전체와 같은 생물학적 분자들에 대해 미리 바코딩을 수행함으로써 서로 다른 세포에서 유래한 생물학적 분자들을 모두 합쳐서(pooling) 한 번에(one-pot) 라이브러리 제작 과정을 진행할 수 있도록 하여, 비용, 시간, 노동 등을 혁신적으로 줄일 수 있는 기술이다.
실제로 단일 세포 전사체(transcriptomics), 단일 세포 후성 유전체(epigenetics), 단일 세포 단백질체(proteomics) 및 단일 세포 다중체(multiomics) 등 많은 단일 세포 분석 분야에서 상기 언급한 바코딩을 이용한 병렬화 기술을 이용하여 분석 병렬성과 처리량을 혁신적으로 증가시킬 수 있었다. 단일 세포의 유전체를 sparse하게 살펴보는 것을 목표로 하는 경우에도 상기 언급한 Tn5 유전자전위효소를 이용한 방식으로 수백 내지 수만개의 세포를 병렬적으로 분석할 수 있었다. 여기서 유전체를 sparse하게 살펴본다는 것은 단일 세포의 전체 유전체를 살펴보는 것이 아니라 80%보다 낮은 일부의 유전체 영역에 대해 드문드문 살펴보는 것을 지칭한다.
하지만 단일 염기서열 수준의 단일 세포 유전체 분석을 위해서는 겨우 수 카피의 DNA(사람의 경우, 2 카피)에 대해서 손실 없이 바코드를 삽입해야 하는데 이는 달성하기 어려운 목적이다. 바코딩을 이용한 병렬화 기술이 효과를 발휘하려면, 바코드의 염기서열과 분석하고자 하는 세포 유전체에서 유래한 염기서열이 동시에 NGS 시퀀싱에서 읽혀야 시퀀싱된 세포 유래 염기서열의 정보를 바코드 정보와 매칭(match)시킬 수 있다. 하지만 현재 제일 널리 쓰이는 일루미나(Illumina)사의 NGS기술은 한 번에 시퀀싱할 수 있는 핵산의 길이 한계(read length 한계)가 최대 300 내지 600 bp정도이며, 단일 세포의 DNA를 이렇게 짧은 핵산 조각으로 단편화하여도 손실없이 바코드가 존재하도록 바코드를 삽입하는 것은 달성하기 어려운 과제이다. DNA와는 달리 RNA는 통상 전사물(transcript) 끝단에 A 테일(tail)이 존재하여 이를 PCR(polymerase chain reaction) 핸들로 삽입하여 전체 전사체에 대해 바코드를 삽입하는 것이 용이하지만, DNA는 매우 복잡한 염기서열의 긴 가닥으로 구성되어 있기 때문에, PCR 핸들로 사용할 수 있는 특정한 시퀀스가 존재하지 않으며, 전체 유전체에 대한 손실 없는 바코드 삽입이 어렵다.
즉, 수 카피밖에 존재하지 않는 단일 세포의 유전체를 단일 염기 수준으로 분석하는 동시에 바코드를 이용한 병렬화 기술을 적용하기 위해서는, 전체 유전체 영역에 대한 손실 없는 바코딩을 달성하기 위해 미량의 단일 세포 유전체를 미리 증폭(pre-amplification)하는 MDA와 같은 과정이 필수적이다. 하지만 MDA기술에 바코딩을 이용한 병렬화 기술을 도입하는 것 또한 쉽게 달성하기 어려운 일이다. 통상적인 PCR 반응에서는 바코딩을 이용한 병렬화 기술을 도입하기 위해 프라이머(primer)의 5'끝에 바코드를 추가하여도 PCR 반응에 큰 영향을 미치지 않았지만, MDA는 통상 N6(랜덤 헥사머)를 프라이머로 이용하는 기술로서 이 프라이머의 5’끝에 다른 시퀀스를 추가하면 MDA 반응 효율에 큰 악영향을 주게 된다. 이러한 현상의 원인은 아직 학계에 알려지지 않았으며, 양이온 농도를 증가시키거나 프라이머의 비특이적 혼성화(non-specific hybridization)를 줄이기 위해 헤어핀 구조를 삽입하는 것과 같은 간단한 해결전략으로는 문제가 해결되지 않는다. 따라서 MDA 과정에서 바코드를 삽입하기 어려워 그 후단의 과정인 라이브러리 제작 과정을 개별 세포에 대해 각각 수행해야 하였다.
최근 LIANTI라고 불리는 MDA와는 다른 전장 유전체 증폭 기술이 개발된 바 있고, LIANTI 결과물인 DNA 단편(DNA fragment)에 바코드를 바로 삽입할 수 있다고 주장된 바 있다. 하지만 LIANTI의 DNA 단편은 그 길이가 100 bp부터 2 kb까지 넓은 영역에 걸쳐 존재한다. 따라서 현재 short-read 시퀀싱 기술의 리드 길이 한계인 300 내지 600 bp의 시퀀싱으로 바코드가 달린 LIANTI 라이브러리를 단편화 없이 모두 읽기 어렵다는 한계가 있다. 하지만 단편화를 수행한다면 DNA 분자의 바코드 정보를 잃어버리기 때문에, LIANTI기술은 단일 염기서열 수준의 단일 세포 유전체 분석을 목적으로 할 때에, 병렬화가 어려운 기술이다.
한편, MDA 기술의 다른 어려움은, 단일 세포와 같이 미량의 DNA로부터 증폭을 수행할 경우 증폭 편향(amplification bias)이 심해서 증폭 품질이 우수한 결과물을 많이 얻기 어렵다는 점이다. 통상적으로 100개의 단일 세포들을 MDA로 증폭할 경우, 그중 겨우 5 내지 10개 정도의 세포만이 유전자 복제 수 변이(Copy number alteration, CNA)를 분석할 수 있는 수준의 결과물을 보이고, 나머지 세포들은 모두 QC(Quality control)를 통과하지 못하여 버려지게 된다. 이와 같은 문제는 MDA의 증폭 편향 문제, 혹은 증폭 퀄리티 문제라고 불리며, 이로 인해 특정한 수의 세포를 분석하기 위해서는 그보다 10 내지 20배 더 많은 세포에 대해 MDA 증폭을 해야 함으로, 비용 비효율성을 가져왔으며, MDA를 이용하여 다량의 단일 세포를 분석하는 것이 어려웠다.
따라서, MDA의 병렬성 및 처리량을 높임으로써, 다량의 단일 세포 전장 유전체 분석에 유용하게 이용될 수 있도록 바코드가 포함된 프라이머를 이용한 병렬적 다중 전위 증폭 방법을 개발하게 되었다.
제1 프라이머, 제2 프라이머 및 핵산 중합효소를 표적 핵산이 포함된 샘플에 첨가하여 핵산들을 증폭시키는 단계를 포함하는 핵산 증폭 방법으로서,
상기 제1 프라이머는 상기 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 상기 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 방법을 제공하는 것이다.
다른 양상은 제1 프라이머 및 제2 프라이머를 포함하는 핵산 증폭용 프라이머 세트로서,
상기 제1 프라이머는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 프라이머 세트를 제공하는 것이다.
또 다른 양상은 제1 프라이머 및 제2 프라이머를 포함하는 핵산 증폭용 키트로서,
상기 제1 프라이머는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 상기 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 키트를 제공하는 것이다.
일 양상은 제1 프라이머, 제2 프라이머 및 핵산 중합효소를 표적 핵산이 포함된 샘플에 첨가하여 핵산들을 증폭시키는 단계를 포함하는 핵산 증폭 방법으로서,
상기 제1 프라이머는 상기 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 상기 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 방법을 제공한다.
상기 "표적 핵산"은 상기 핵산 증폭 방법으로 증폭되어 NGS(Next generation sequencing) 라이브러리 제작 과정을 거쳐 시퀀싱될 수 있는 핵산을 의미하며, 전장 유전체(whole genome)를 포함할 수 있다. 일 양상에 따른 핵산 증폭 방법에 의하면, 상기 표적 핵산은 단일 염기 서열 수준으로 시퀀싱될 수 있다.
또한, 일 양상에 따른 핵산 증폭 방법에 의하면, 제1 프라이머 및 제2 프라이머를 이용하여 병렬적 다중 전위 증폭이 수행됨으로써, 다중 전위 증폭의 병렬성 및 처리량을 높여 NGS 라이브러리 제작 시간 및 비용을 혁신적으로 감소시킬 수 있다.
상기 표적 핵산은 DNA, RNA, 게놈 DNA, cDNA, 엑손, 인트론, 유전자의 일부분, 플라스미드(plasmid)의 일부분, 조절 서열, 원형 RNA, cfDNA(무세포 DNA), cfRNA(무세포 RNA), siRNA(작은 간섭 RNA), cffDNA(무세포 태아 DNA), mRNA, tRNA, rRNA, miRNA, snoRNA, piRNA, ncRNA, 잠금 핵산(LNA), 펩티드 핵산(PNA), 합성 폴리뉴클레오티드, 폴리뉴클레오티드 유사체 또는 이들의 조합으로부터 선택되는 것일 수 있다.
상기 표적 핵산은 단일 가닥, 이중 가닥 또는 삼중 가닥으로 이루어진 것일 수 있다. 또한, 상기 표적 핵산의 일부분만이 이중 가닥 또는 삼중 가닥으로 이루어진 것일 수 있다.
상기 표적 핵산은 전혀 알려지지 않은 염기 서열, 부분적으로 알려진 염기 서열 또는 완전히 알려진 염기 서열로 이루어진 것일 수 있다.
상기 표적 핵산은 자연적으로 발생하는 염기(우라실, 아데닌, 티민, 시토신, 구아닌)로 구성되는 폴리뉴클레오티드로 이루어진 것일 수 있고, 폴리뉴클레오티드 유사체로 이루어진 것일 수 있다.
상기 폴리뉴클레오티드 유사체는 i) 변형된 염기, ii) 변형된 펜토스, iii) 변형된 포스페이트 또는 iv) 변형된 염기 간 연결 부분을 포함할 수 있다.
상기 변형된 염기는 디아미노퓨린, 이소시토신, 이소구아닌, 슈도이소시토신, N9-(2-아미노-6-클로로퓨린), N9-(2,6-디아미노퓨린), N9-(7-데아자-구아닌), N9-(7-데아자-8-아자-구아닌) 및 N8-(7-데아자-8-아자-아데닌), 5-프로피닐-우라실, 2-티오-5-프로피닐-우라실, 5-플루오로우라실, 5-브로모우라실, 5-클로로우라실, 5-아이오도우라실, 히포잔틴, 잔틴, 4-아세틸시토신, 5-(카르복시히드록실메틸)우라실, 5-카르복시메틸아미노메틸-2-티오우리딘, 5-카르복시메틸아미노메틸우라실, 디히드로우라실, 베타-D-갈락토실퀘오신, 이노신, N6-이소펜테닐아데닌, 1-메틸구아닌, 1-메틸이노신, 2,2-디메틸구아닌, 2-메틸아데닌, 2-메틸구아닌, 2-티오티민, 2-아미노퓨린, 3-메틸 시토신, 5-메틸시토신, N6-아데닌, 7-메틸 구아닌, 5-메틸아미노메틸우라실, 5-메톡시아미노메틸-2-티오우라실, 베타-D-만노실퀘오신, 5'-메톡시카르복시메틸우라실, 5-메톡시우라실, 2-메틸티오-D46-이소펜테닐아데닌, 우라실-5-옥시아세트산 (v), 와이부톡소신, 슈도우라실, 퀘오신, 2-티오시토신, 5-메틸-2-티오우라실, 2-티오우라실, 4-티오우라실, 5-메틸우라실, 우라실-5-옥시아세트산 메틸에스테르, 우라실-5-옥시아세트산 (v), 5-메틸-2-티오우라실, 3-(3-아미노-3-N-2-카르복시프로필) 우라실, (acp3)w, 올리고뉴클레오티드 포스포르아미데이트, 나이트로피롤, 나이트로인돌, 아시클릭(acylic) 5-나이트로인돌, 4-나이트로피라졸, 4-나이트로이미다졸, C-5 프로피닐-C, C-5 프로피닐-U 또는 2-아미노 아데닌일 수 있다.
상기 변형된 펜토스는 잠금 핵산(LNA) 유사체(예를 들어, Bz-A-LNA, 5-Me-Bz-C-LNA, dmf-G-LNA 또는 T-LNA 등) 또는 2'- 또는 3'-변형체(여기서 2'- 또는 3'-위치는 수소, 하이드록시, 알콕시(예를 들어, 메톡시, 에톡시, 알릴옥시, 아이소프로폭시, 부톡시, 아이소부톡시 및 페녹시), 아지도, 아미도, 알킬아미노, 플루오로, 클로로 또는 브로모)일 수 있다.
상기 변형된 포스페이트는 인 원자가 +5 산화 상태로 존재하고 산소 원자 중 하나 이상이 비-산소 모이어티, 예를 들어, 황으로 대체된 포스페이트 유사체일 수 있다. 또 다른 변형된 포스페이트는 회합된 반대이온, 예를 들어 H+, NH4 +, Na+를 포함하는, 포스포로티오에이트, 포스포로다이티오에이트, 포스포로셀레노에이트, 포스포로다이셀레노에이트, 포스포로아닐로티오에이트, 포스포르아닐리데이트, 포스포르아미데이트 또는 보로노포스페이트일 수 있다.
상기 변형된 염기 간 연결 부분은 포스페이트 유사체, 아키랄(achiral)을 갖는 유사체, 하전되지 않은 소단위간 연결 또는 아키랄 소단위간 연결을 갖는 하전되지 않은 모르폴리노계 중합체일 수 있다. 일부 염기 간 연결 유사체는 모르폴리데이트, 아세탈 또는 폴리아마이드-연결된 헤테로사이클일 수 있다. 또한, 상기 염기가 펩티드 핵산(PNA)일 경우, 상기 변형된 염기 간 연결 부분은 2-아미노에틸글리신 아마이드 골격 중합체일 수 있다.
상기 표적 핵산은 단일 세포 또는 여러 세포에서 유래된 것일 수 있고, 생화학 처리(예를 들어, 중아황산염 또는 역전사)된 것일 수 있고, 환경에 남아있는 것일 수 있다.
상기 샘플은 상기 표적 핵산을 포함할 수 있고, 상기 샘플은 세포 샘플, 조직 샘플, 혈액 샘플, 소변 샘플, 타액 샘플, 림프액 샘플, 뇌척수액 샘플, 양수 샘플, 흉수 샘플, 심낭액 샘플, 복수 샘플, 안방수 샘플, 골수 샘플, 정액 샘플, 생검 샘플, 암 샘플, 종양 샘플, 범죄수사 샘플(forensic sample), 고고학 샘플, 고생물학 샘플, 감염 샘플, 생산 샘플, 식물 샘플, 미생물 샘플, 바이러스 샘플, 토양 샘플, 해양 샘플 및 담수 샘플로 이루어진 군에서 선택되는 하나 이상의 샘플일 수 있다.
상기 조직 샘플은 부고환, 눈, 근육, 피부, 힘줄, 정맥, 동맥, 혈액, 심장, 비장, 림프절, 골, 골수, 폐, 기관지, 기관, 장, 소장, 대장, 결장, 직장, 침샘, 혀, 방광, 맹장, 간, 췌장, 뇌, 위, 피부, 신장, 요관, 방광, 요도, 성샘, 고환, 난소, 자궁, 나팔관, 흉선, 피하수체, 갑상선, 부신 및 부갑상선으로 이루어진 군에서 선택되는 하나 이상의 조직에서 유래된 것일 수 있다. 또한, 상기 조직 샘플은 인간 또는 다른 유기체의 임의의 다양한 기관으로부터 유래된 것일 수 있다.
상기 세포 샘플은 동물 세포, 식물 세포, 진균 세포, 박테리아 세포 또는 원생동물 세포에서 유래된 것일 수 있고, 구체적으로 동물 세포에서 유래된 것일 수 있고, 보다 구체적으로 인간 세포에서 유래된 것일 수 있다.
상기 세포 샘플은 생식 세포(난자 세포, 정자 등), 난소 상피 세포, 난소 섬유아세포, 면역 세포, B 세포, T 세포, 자연 살해 세포, 수지상 세포, 암 세포, 진핵 세포, 줄기 세포, 혈액 세포, 근육 세포, 지방 세포, 피부 세포, 신경 세포, 골 세포, 췌장 세포, 내피 세포, 췌장 상피 세포, 췌장 알파 세포, 췌장 베타 세포, 췌장 내피 세포, 골수 림프아세포, 골수 B 림프아세포, 골수 대식세포, 골수 적혈모세포, 골수 수지상 세포, 골수 지방세포, 골수 골세포, 골수 연골세포, 전골아세포, 골수 거대핵모세포, 뇌 B 림프구, 뇌 신경교 세포, 뉴런, 뇌 성상세포, 신경외배엽 세포, 뇌 대식세포, 뇌 미세아교세포, 뇌 상피 세포, 피질 뉴런, 뇌 섬유아세포, 유방 상피 세포, 결장 상피 세포, 결장 B 림프구, 유방 근상피 세포, 유방 섬유아세포, 결장 장세포, 자궁경부 상피 세포, 유관 상피 세포, 혀 상피 세포, 편도 수지상 세포, 편도 B 림프구, 말초 혈액 림프아세포, 말초 혈액 T 림프아세포, 말초 혈액 피부 T 림프구, 말초 혈액 자연 살해 세포, 말초 혈액 B 림프아세포, 말초 혈액 단핵구, 말초 혈액 골수아세포, 말초 혈액 단핵모세포, 말초 혈액 전골아세포, 말초 혈액 대식세포, 말초 혈액 호염구, 간 내피 세포, 간 비만 세포, 간 상피 세포, 간 B 림프구, 비장 내피 세포, 비장 상피 세포, 비장 B 림프구, 간 세포, 간 섬유아세포, 폐 상피 세포, 기관지 상피 세포, 폐 섬유아세포, 폐 B 림프구, 폐 슈반 세포, 폐 편평 세포, 폐 대식세포, 폐 조골세포, 신경내분비 세포, 폐포, 위 상피 세포, 위 섬유아세포, 줄기 세포, 태아 세포, 종양 세포, 의심성 암 세포, 암 세포 및 유전자 편집 절차를 거친 세포로 이루어진 군에서 선택되는 하나 이상의 세포에서 유래된 것일 수 있다.
일 양상에 있어서, 상기 샘플은 공간분해능이 있는 분리(spatially resolved) 기술, 유전영동 디지털 분류(dielectrophoretic digital sorting), 형광 활성 세포 분류(fluorescence activated cell sorting, FACS), 유체역학 트랩(hydrodynamic traps), 액체 방울(droplet), 마이크로웰(microwell), 미세 유체역학(microfluidics), 미세조작(micromanipulation), Raman 핀셋(Raman tweezers) 및 CellRaft로 이루어진 군에서 선택되는 하나 이상의 기술로 분리된 것일 수 있다. 상기 기술들은 특정 기준(예컨대, 공간상에서의 위치, 표면 단백질들의 유무, H&E 염색 이미지 또는 in-situ 시퀀싱 이미지 등)에 따라 샘플들을 분류할 수 있고, 분류된 샘플들에 대해 각각 상기 핵산 증폭 방법을 수행하여, 각각의 샘플에 대한 유전체를 단일 염기 서열 수준으로 분석할 수 있다. 또한, 상기 기술은 인풋 gDNA의 양을 증가시킬 수 있어, 상기 핵산 증폭 방법의 증폭 품질을 높일 수 있다.
일 양상에 따르면, 상기 액체 방울, 마이크로웰 또는 미세 유체역학 기술은 생화학 반응을 위한 물리적으로 구별된(compartment) 반응물(reactor)의 수를 혁신적으로 증가시킬 수 있어, 상기 핵산 증폭 방법의 병렬성을 혁신적으로 증가시킬 수 있다. 구체적으로, 상기 액체 방울 또는 마이크로웰 내부에 단일 세포를 가둔 뒤, 각각의 구획화된 용액에서 상기 핵산 증폭 방법을 수행함으로써, 한 번에 다량의 단일 세포를 분석할 수 있다.
또한, 일 양상에 있어서, 상기 공간적 분리 기술은 LCM(Laser capture microdissectio), LMD(laser microdisection), LPC(Laser pressure catapulting) 및 PHLI-seq(phenotype-based high-throughput laser-aided isolation and sequencing)으로 이루어진 군에서 선택되는 하나 이상의 기술일 수 있다.
일 양상에 있어서, 상기 세포 샘플은 프로테니아제(proteinase) 프로테아제(protease), 열, 비드 비팅(bead beating), 세니테이션(sanitation), 50℃ 이상의 알칼라인(alkaline) 용해 또는 4℃ 이하 온도에서의 알칼라인 용해 방법을 이용하여 용해된 것일 수 있다. 또한, 상기 표적 핵산은 증폭시키기 위해 열 또는 알칼라인 변성(denaturation) 방법을 이용하여 단일 가닥으로 변성된 것일 수 있다.
상기 “증폭"은 핵산의 전부 또는 일부분이 추가적인 핵산 내에 복제되는 임의의 작용을 의미하며, “복제"와 동일한 의미로 사용될 수 있다. 상기 추가적인 핵산은 주형 핵산의 일부분과 실질적으로 동일하거나 실질적으로 상보적인 서열을 포함할 수 있다. 상기 추가적인 핵산 또는 주형 핵산은 각각 독립적으로 단일 가닥 또는 이중 가닥일 수 있다. 또한, 상기 증폭은 핵산의 선형 증폭 또는 기하급수적 증폭을 포함할 수 있다.
상기 "핵산 증폭 방법"은 다중 전위 증폭(multiple displacement amplification, MDA)일 수 있고, 구체적으로 병렬적 다중 전위 증폭일 수 있다. 상기 병렬적 다중 전위 증폭은 "bMDA(바코드 MDA)"와 호환성 있게 사용될 수 있다.
상기 용어 "다중 전위 증폭"은 가닥 전위(strand displacement) 기능을 가지는 핵산 중합효소를 이용하여 핵산을 기하급수적으로 증폭하는 방법을 의미한다.
일 양상에 있어서, 상기 핵산 증폭 방법을 통해 표적 핵산을 핵산 중합효소가 복제하여 복제된 핵산 가닥을 수득할 수 있고, 복제된 핵산 가닥이 표적 핵산으로부터 전위(displace)될 수 있다.
일 양상에 따르면, 상기 핵산 증폭 방법은 단일 세포 유전체를 단일 염기 서열 수준으로 분석할 수 있다. 또한, 상기 방법은 단일 세포 전사체, 후성전사체, 단백질체 등을 분석하는 기술과 함께 수행함으로써 단일 세포 다중체(multicomics) 분석 기술에 이용될 수 있다. 상기 단일 세포 다중체 분석 기술은 2차원 이상의 오믹스(omics) 정보를 분석함으로써, 개별 세포에 대한 다양한 정보를 동시에 분석하는 것에 이용될 수 있다. 따라서, 일 양상에 따른 핵산 증폭 방법은 단일 염기 서열 수준으로 단일 세포를 고처리량으로 분석할 수 있어, 단일 세포 다중체학 분석 기술의 처리량을 향상시킬 수 있다.
상기 "제1 프라이머"는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열 및 상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 폴리뉴클레오티드이다.
상기 "혼성화"는 2개의 단일 가닥 폴리뉴클레오티드가 비-공유 결합하여 안정한 이중 가닥 폴리뉴클레오티드를 형성하는 과정을 의미할 수 있다.
일 양상에 있어서, 상기 제1 염기 서열의 염기 개수는 4 내지 10 중 선택되는 하나의 정수일 수 있다. 상기 제1 염기 서열은 표적 핵산과 혼성화될 수 있도록 설계된 것일 수 있다.
또한, 일 양상에 있어서 상기 제1 염기 서열을 이루는 염기들은 A(아데닌), T(티민), G(구아닌), C(사이토신) 및 이들의 변형물로 이루어진 군에서 선택되는 염기일 수 있다.
상기 변형물은 디아미노퓨린, 이소시토신, 이소구아닌, 슈도이소시토신, N9-(2-아미노-6-클로로퓨린), N9-(2,6-디아미노퓨린), N9-(7-데아자-구아닌), N9-(7-데아자-8-아자-구아닌) 및 N8-(7-데아자-8-아자-아데닌), 5-프로피닐-우라실, 2-티오-5-프로피닐-우라실, 5-플루오로우라실, 5-브로모우라실, 5-클로로우라실, 5-아이오도우라실, 히포잔틴, 잔틴, 4-아세틸시토신, 5-(카르복시히드록실메틸)우라실, 5-카르복시메틸아미노메틸-2-티오우리딘, 5-카르복시메틸아미노메틸우라실, 디히드로우라실, 베타-D-갈락토실퀘오신, 이노신, N6-이소펜테닐아데닌, 1-메틸구아닌, 1-메틸이노신, 2,2-디메틸구아닌, 2-메틸아데닌, 2-메틸구아닌, 2-티오티민, 2-아미노퓨린, 3-메틸 시토신, 5-메틸시토신, N6-아데닌, 7-메틸 구아닌, 5-메틸아미노메틸우라실, 5-메톡시아미노메틸-2-티오우라실, 베타-D-만노실퀘오신, 5'-메톡시카르복시메틸우라실, 5-메톡시우라실, 2-메틸티오-D46-이소펜테닐아데닌, 우라실-5-옥시아세트산 (v), 와이부톡소신, 슈도우라실, 퀘오신, 2-티오시토신, 5-메틸-2-티오우라실, 2-티오우라실, 4-티오우라실, 5-메틸우라실, 우라실-5-옥시아세트산 메틸에스테르, 우라실-5-옥시아세트산 (v), 5-메틸-2-티오우라실, 3-(3-아미노-3-N-2-카르복시프로필) 우라실, (acp3)w, 올리고뉴클레오티드 포스포르아미데이트, 나이트로피롤, 나이트로인돌, 아시클릭(acylic) 5-나이트로인돌, 4-나이트로피라졸, 4-나이트로이미다졸, C-5 프로피닐-C, C-5 프로피닐-U 및 2-아미노 아데닌으로 이루어진 군에서 선택되는 하나 이상의 것일 수 있다.
일 양상에 따르면, 상기 제1 염기 서열은 표적 핵산에 따라 특정 염기 서열로 고정된 것일 수 있고, 다양한 염기 서열로 이루어진 것일 수 있으며, 랜덤(random) 염기 서열로 이루어진 것일 수 있다. 또한, 상기 제1 염기 서열은 표적 핵산과의 결합이 원활하도록, 랜덤 염기 서열에 특정한 고정된 염기 서열이 더 추가된 것일 수 있다.
또한, 일 양상에 있어서, 상기 제1 염기 서열은 핵산 분해효소(nuclease)에 저항성이 있는 염기를 포함하는 것일 수 있다. 상기 염기는 포스포로티올레이트 변형(phosphorothiolate modification)된 것일 수 있다. 상기 변형을 통해, 3'-5' 핵산 분해 기능을 가지는 핵산 중합효소로 표적 핵산을 증폭할 경우, 핵산 분해에 의한 프라이머 고갈을 방지할 수 있다.
일 양상에 있어서, 상기 제2 염기 서열의 염기 개수는 4 내지 35 중 선택되는 하나의 정수일 수 있다. 또한, 상기 제2 염기 서열의 종류는 10 내지 1,000,000개일 수 있다. 상기 제2 염기 서열은 한 번에 다량의 서로 다른 샘플 유래 표적 핵산을 구별할 수 있다.
또한, 일 양상에 있어서, 상기 제2 염기 서열은 바코드 서열을 포함할 수 있다. 상기 바코드 서열의 염기 개수는 4 내지 12 중 선택되는 하나의 정수일 수 있다. 상기 바코드 서열은 특정 샘플의 표적 핵산에 대하여 모두 같은 염기 서열을 갖고, 다른 샘플의 표적 핵산에 대하여 상이한 염기 서열을 갖도록 설계되어 표적 핵산이 유래된 샘플을 구별할 수 있다.
일 양상에 있어서, 상기 "제1 프라이머"는 상기 제2 염기 서열을 두 개 이상 포함할 수 있다.
일 양상에 있어서, 상기 “제2 프라이머"는 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 폴리뉴클레오티드이다.
일 양상에 있어서, 상기 제3 염기 서열의 염기 개수는 4 내지 10 중 선택되는 하나의 정수일 수 있다. 상기 제3 염기 서열은 표적 핵산과 혼성화될 수 있도록 설계된 것일 수 있다.
또한, 일 양상에 있어서 상기 제3 염기 서열을 이루는 염기들은 A(아데닌), T(티민), G(구아닌), C(사이토신) 및 이들의 변형물로 이루어진 군에서 선택되는 염기일 수 있다.
일 양상에 따르면, 상기 제3 염기 서열은 표적 핵산에 따라 특정 염기 서열로 고정된 것일 수 있고, 다양한 염기 서열로 이루어진 것일 수 있으며, 랜덤(random) 염기 서열로 이루어진 것일 수 있다. 또한, 상기 제3 염기 서열은 표적 핵산과의 결합이 원활하도록, 랜덤 염기 서열에 특정한 고정된 염기 서열이 더 추가된 것일 수 있다.
또한, 일 양상에 있어서, 상기 제3 염기 서열은 핵산 분해효소(nuclease)에 저항성이 있는 염기를 포함하는 것일 수 있다. 상기 염기는 포스포로티올레이트 변형(phosphorothiolate modification)된 것일 수 있다. 상기 변형을 통해, 3’-5’ 핵산 분해 기능을 가지는 핵산 중합효소로 표적 핵산을 증폭할 경우, 핵산 분해에 의한 프라이머 고갈을 방지할 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 상기 핵산 중합효소와 제1 프라이머의 친화도를 저해하는 물질이 결합된 것일 수 있다. 구체적으로, 상기 물질은 입체 장애(steric hindrance)를 나타내는 물리적 구조체일 수 있다. 상기 물질은 입체 장애를 통해 상기 핵산 중합효소와 제1 프라이머의 친화도를 저해하여 결합을 방지함으로써 핵산 증폭 효율을 높일 수 있다.
또한, 일 양상에 있어서, 바코드 서열 영역을 RNA로 제조하는 방법, 바코드 서열 영역과 바인딩 영역 사이에 링커(linker)를 추가하는 방법 또는 바코드 영역과 상보적인 서열의 ssDNA(single-strand DNA)를 혼성화시키는 방법을 통해서도, 상기 제1 프라이머와 상기 핵산 중합효소 간 친화도를 저해할 수 있고, 이를 통해 핵산 증폭 효율을 높일 수 있다.
일 양상에 있어서, 상기 핵산 증폭 방법은 상기 제1 프라이머로 증폭된 핵산을 선별하는 단계를 더 포함할 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 포획 물질과 결합된 것일 수 있다. 상기 포획 물질과 결합된 제1 프라이머로 핵산을 증폭한 뒤, 증폭된 핵산에서 포획 물질을 포획함으로써 제2 염기 서열을 포함하는 표적 핵산과 제2 염기 서열을 포함하지 않는 표적 핵산을 선별할 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 제1 염기 서열, 제2 염기 서열 및 포획 물질을 모두 포함하도록 개별적으로 합성될 수 있고, 제1 염기 서열 및 제2 염기 서열만을 합성한 뒤, 핵산 변형(modification)을 통해 포획 물질을 더 추가할 수도 있으며, 제1 염기 서열 및 제2 염기 서열을 각각 합성한 뒤 결찰(ligation) 또는 화학 반응을 통해 연결한 뒤, 핵산 변형을 통해 포획 물질을 더 추가할 수도 있다.
일 양상에 있어서, 상기 포획 물질은 비오틴(biotin), D-비오틴, 비오틴 dT, 비오틴-TEG, 듀얼 비오틴, PC-비오틴, 아크릴(acrylic), 티올(thiol), 디티올(dithiol), 아민기(amine group), 아크릴기(acrylic group), NHS 에스터, 아자이드(azide), 헥시닐(hexynyl), 옥타디닐(octadiynyl) dU, 히스 태그(his tag), 단백질 태그, polyA(폴리 아데닌), NGS 시퀀싱 어댑터를 이루는 염기 서열 중 10개 이상의 염기 서열 및 10개 이상의 염기 서열로 이루어진 폴리뉴클레오티드로 이루어진 군에서 선택되는 하나 이상일 수 있고, 구체적으로, 비오틴(biotin), D-비오틴, 비오틴 dT, 비오틴-TEG, 듀얼 비오틴 및 PC-비오틴으로 이루어진 군에서 선택되는 하나 이상일 수 있고, 보다 구체적으로, D-비오틴일 수 있다.
또한, 일 양상에 있어서, 상기 10개 이상의 염기 서열을 이루는 염기들은 A(아데닌), T(티민), G(구아닌), C(사이토신) 및 이들의 변형물로 이루어진 군에서 선택되는 염기일 수 있다.
일 양상에 따르면, 상기 포획 물질로 핵산 외의 분자를 이용할 경우, 긴 길이의 프라이머에 의한 가닥 전위 반응 저해 현상을 크게 줄일 수 있어, 가닥 전위 복제 효율을 현저하게 증가시킬 수 있다.
또한, 일 양상에 있어서, 상기 포획 물질로 비오틴을 이용할 경우, 바코드 서열을 포함하는 표적 핵산을 선별적으로 농축하기 위해 PCR을 사용하지 않기 때문에, PCR 편향 현상 및 증폭 오류를 최소화할 수 있다. 상기 PCR 편향 현상은 PCR 과정에서 특정 핵산이 다른 핵산에 비해 많이 증폭되거나 적게 증폭되는 현상을 의미하고, 증폭 오류는 PCR 과정에서 핵산 중합효소의 오류로 인해 핵산 염기 서열이 원본과 상이하게 복제되는 현상을 의미한다.
일 양상에 있어서, 상기 NGS 시퀀싱 어댑터는 NGS 시퀀싱을 위한 프라이머의 일부 염기 서열일 수 있다. 상기 어댑터를 제1 프라이머에 추가하고 PCR을 수행함으로써, 바코드 서열을 포함하는 증폭 산물만을 농축할 수 있다. 또한, 상기 NGS 시퀀싱 어댑터를 이루는 염기 서열 중 10개 이상의 염기 서열, 즉, NGS 시퀀싱 어댑터 중 일부가 상기 포획물질로 이용될 수 있다.
일 양상에 있어서, 상기 제1 프라이머에는 다른 고정된 염기 서열이 더 추가될 수 있다. 예를 들어, MALBAC(Multiple Annealing and Looping Based Amplification Cycles) 증폭 기술의 경우 표적 핵산을 증폭하기 위한 8 개의 염기로 이루어진 랜덤 염기 서열에 더하여 루프(loop)를 형성할 수 있는 특정 고정된 염기 서열이 더 추가되어 있다. MALBAC은 상기 프라이머를 이용하여 가닥 전위를 포함하는 DNA 증폭 반응을 수행할 수 있고, 이후, 특정 고정된 염기 서열을 가지는 프라이머를 이용하여 PCR을 수행하는데 이 PCR 과정 중에 특정 고정된 염기 서열을 양 끝에 가진 증폭 산물이 루프를 이루게 되어 증폭 편향을 줄일 수 있다. 일 양상에 따른 핵산 증폭 방법 또한, 마찬가지로 특정 목적의 고정된 염기 서열이 더 추가되어 있는 제1 프라이머를 이용하여 표적 핵산의 증폭이 가능하며, 간단한 예시로는 MALBAC 프라이머의 고정된 염기 서열과 랜덤 염기 서열 사이에 바코드 염기 서열을 더 포함하는 제1 프라이머를 이용하여 표적 핵산을 증폭할 수 있다. 상기 예시에서는 특정 고정된 염기 서열이 포획 물질의 역할을 대신할 수 있기 때문에 추가적인 포획 물질이 결합되어 있지 않은 제1 프라이머가 사용될 수도 있다. 상기 특정 고정된 염기 서열의 염기 개수는 목적에 따라 다양할 수 있지만, 통상적으로 5 내지 30 중 선택되는 하나의 정수일 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 플루오레세인이소티오시안산염(fluorescein isothiocyanate), 5,6-카복시메틸플루오레세인(5,6-carboxymethyl fluorescein), 텍사스 레드(Texas red), 2-NBDG(nitrobenz-2-oxa-1,3-diazol-4-yl), 쿠마린(coumarin), 댄실 클로라이드(dansyl chloride), 로다민(rhodamine), 아미노-메틸 쿠마린(amino-methyl coumarin), 에오신(Eosin), 에리트로신(Erythrosin), BODIPY, 캐스케이드 블루(Cascade Blue), 오레곤 그린(Oregon Green), 파이렌(pyrene), 리사민(lissamine), 잔텐(xanthene), 아크리딘(acridine), 옥사진(oxazines), 피코에리트린(phycoerythrin), Cy3, Cy3.5, Cy5, Cy5.5 및 Cy7으로 이루어진 군에서 선택되는 하나 이상의 형광 물질을 태깅하도록 변형된 것일 수 있고, 이를 통해 핵산 증폭 반응 산물 중 바코딩이 된 증폭 산물의 양을 정량할 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 UMI(Unique Molecular Identifier)를 포함하는 것일 수 있다. 상기 UMI는 핵산 증폭과정 중 표적 핵산 분자별로 고유한 염기서열을 갖고 있는 분자 바코드를 삽입하기 위한 매우 다양한 종류의 고유한 염기서열의 핵산을 갖고 있는 서열을 의미한다. 증폭과정에서 UMI를 삽입함으로써, 증폭산물에서 얻어진 염기서열이 증폭 편항(bias)으로부터 발생한 중복된 서열인지, 혹은 서로 다른 표적 분자로부터 유래한 서열인지 판단할 수 있으며, 이를 통해 UMI가 포함된 제1 프라이머는 증폭 편향(bias)을 감소시킬 수 있다.
일 양상에 따르면, 상기 선별은 아비딘(avidin), 뉴트라비딘(neutravidin), 스트렙트아비딘(streptavidin), 카복실기(carboxylic group), 티올(thiol), 알카인(alkyne), 디곡시제닌(digoxigenin), 아자이드(azide) 및 헥시닐(hexynyl)로 이루어진 군에서 선택되는 하나 이상의 물질을 이용한 방법 또는 PCR(Polymerase Chain reaction) 방법으로 수행되는 것일 수 있고, 구체적으로 아비딘, 뉴트라비딘 및 스트렙트아비딘으로 이루어진 군에서 선택되는 하나 이상의 물질을 이용한 방법으로 수행되는 것일 수 있고, 보다 구체적으로 스트렙트아비딘을 이용한 방법으로 수행되는 것일 수 있다.
일 양상에 따르면, 상기 스트렙트아비딘은 비오틴과 강력하게 결합할 수 있어 비오틴이 결합된 표적 핵산을 선별할 수 있다. 또한, 상기 스트렙트아비딘은 비오틴과 가역적으로 결합할 수 있어 선별 이후에 바코드 서열을 포함하는 증폭된 표적 핵산을 회수할 수 있다.
또한, 일 양상에 있어서 상기 포획 물질로 D-비오틴을 이용할 경우, 증폭된 표적 핵산을 변성(denaturation)시키지 않고 스트렙트아비딘으로 효과적으로 수득할 수 있다.
일 양상에 있어서, 상기 핵산 증폭 방법은 상기 샘플로부터 증폭된 핵산을 정제(purification)하는 단계;
정제된 핵산을 단편화(fragmentation)하는 단계; 및
단편화된 핵산들로 NGS(Next Generation Sequencing) 라이브러리를 제조하는 단계를 더 포함할 수 있다.
상기 "정제"는 SPRI(Solid phase Reversible immobilization) 기법을 이용하여 수행될 수 있다. 구체적으로, 0.8x 샘플 부피의 Ampure 자성 비드를 첨가하고 프로토콜에 따라 핵산을 정제할 수 있다.
상기 "단편화"는 음파처리(sonication) 또는 효소를 이용하여 수행될 수 있다. 또한, 상기 핵산은 50 bp 내지 10 kb의 길이로 단편화될 수 있다.
상기 "정제하는 단계"는 선별하는 단계 이전 또는 이후에 수행될 수 있으며, 상기 "정제하는 단계"가 선별하는 단계 이전에 수행될 경우, 상기"단편화하는 단계"는 선별하는 단계 이전 또는 이후에 수행될 수도 있다.
일 양상에 있어서, 상기 선별은 이중 가닥 또는 단일 가닥의 핵산을 선별하는 것일 수 있다. 선별된 핵산이 이중 가닥일 경우, 이중 가닥 핵산에서 비오틴을 제거한 뒤 NGS 라이브러리를 제조할 수 있고, 비오틴이 결합된 상태에서 NGS 라이브러리를 제조할 수도 있다. 5'-말단에 비오틴이 결합된 상태에서 NGS 라이브러리를 제조할 경우, 5'-비오틴이 존재하는 핵산 가닥에는 NGS 어댑터가 결찰(ligation)될 수 없고, 반대쪽 가닥에만 어댑터가 결찰됨으로써, NGS 시퀀싱 결과에서 바코드 서열이 나타나는 위치가 일정하게 고정될 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 10 몰%로 첨가되는 것일 수 있다. 구체적으로, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 1 몰%, 0.1 내지 3 몰%, 0.1 내지 5 몰%, 0.1 내지 7 몰%, 0.1 내지 10 몰%, 1 내지 3 몰%, 1 내지 5 몰%, 1 내지 7 몰%, 1 내지 10 몰%, 3 내지 5 몰%, 3 내지 7 몰%, 3 내지 10 몰%, 5 내지 7 몰%, 5 내지 10 몰% 또는 7 내지 10 몰%로 첨가되는 것일 수 있다.
일 양상에 따르면, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 상기 핵산 증폭 방법의 효율을 증가시킬 수 있다.
또한, 일 양상에 따르면, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 상기 핵산 증폭 방법의 증폭 편향(bias)을 감소시킬 수 있다. 구체적으로, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 선별 단계에서 샘플별 표적 핵산을 일정한 양으로 회수할 수 있으며, NGS 라이브러리 제조 단계에서 샘플별 시퀀싱 리드를 일정하게 조절할 수 있어 상기 핵산 증폭 방법의 증폭 편향을 감소시킬 수 있다. 또한, 상기 비율은 제2 염기 서열의 종류에 따라 다르게 조절될 수 있다.
일 양상에 있어서, 상기 핵산 중합효소는 DNA 중합효소 또는 RNA 중합효소일 수 있다. 또한, 상기 DNA 중합효소는 phi29 DNA 중합효소, Tts DNA 중합효소, 페이즈(phase) M2 DNA 중합효소, VENT DNA 중합효소, DNA 중합효소 I의 Klenow 단편(fragment), T5 DNA 중합효소, PRD1 DNA 중합효소, T4 DNA 중합효소 전효소(holoenzyme), T7 중합효소 유래 T7 Sequenase 및 Bst DNA 중합효소로 이루어진 군에서 선택되는 하나 이상의 DNA 중합효소일 수 있고, 구체적으로 phi29 DNA 중합효소 및 Bst DNA 중합효소로 이루어진 군에서 선택되는 하나 이상의 DNA 중합효소일 수 있고, 보다 구체적으로 phi29 DNA 중합효소일 수 있다.
일 양상에 있어서, 상기 DNA 중합효소는 10초 내지 10분당 0.1 내지 10 nmol이상의 dNTP를 중합할 수 있는 분량으로 첨가되는 것일 수 있고, 구체적으로는 1분당 1 nmol이상의 dNTP를 중합할 수 있는 분량으로 첨가되는 것일 수 있다.
일 양상에 있어서, 상기 샘플은 서로 다른 복수 개이고,
상기 표적 핵산은 서로 다른 복수 개이며,
상기 제1 프라이머는 서로 다른 복수 개이고,
상기 제2 프라이머는 서로 다른 복수 개이고,
상기 서로 다른 복수 개의 제1 프라이머들은 서로 다른 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드들인, 핵산 증폭 방법일 수 있다.
일 양상에 따르면, 서로 다른 고정된 제2 염기 서열을 포함하는 복수 개의 제1 프라이머로 상기 핵산 증폭 방법을 복수 회 수행함으로써, 서로 다른 샘플로부터 유래된 표적 핵산을 구별할 수 있다.
일 양상에 있어서, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 10 몰%로 첨가되는 것일 수 있다. 구체적으로, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 1 몰%, 0.1 내지 3 몰%, 0.1 내지 5 몰%, 0.1 내지 7 몰%, 0.1 내지 10 몰%, 1 내지 3 몰%, 1 내지 5 몰%, 1 내지 7 몰%, 1 내지 10 몰%, 3 내지 5 몰%, 3 내지 7 몰%, 3 내지 10 몰%, 5 내지 7 몰%, 5 내지 10 몰% 또는 7 내지 10 몰%로 첨가되는 것일 수 있다.
일 양상에 따르면, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 상기 핵산 증폭 방법의 효율을 증가시킬 수 있다.
또한, 일 양상에 따르면, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 상기 핵산 증폭 방법의 증폭 편향(bias)을 감소시킬 수 있다. 구체적으로, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 선별 단계에서 샘플별 표적 핵산을 일정한 양으로 회수할 수 있으며, NGS 라이브러리 제조 단계에서 샘플별 시퀀싱 리드를 일정하게 조절할 수 있어 상기 핵산 증폭 방법의 증폭 편향을 감소시킬 수 있다. 또한, 상기 비율은 제2 염기 서열의 종류에 따라 다르게 조절될 수 있다.
일 양상에 있어서, 상기 핵산 증폭 방법은 상기 복수 개의 제1 프라이머들로 증폭된 핵산들을 선별하는 단계를 더 포함할 수 있다.
또한, 일 양상에 있어서, 상기 복수 개의 제1 프라이머들은 포획 물질과 결합된 것일 수 있다. 상기 "포획 물질"은 전술한 범위 내일 수 있다.
일 양상에 있어서, 상기 핵산 증폭 방법은 상기 복수 개의 샘플로부터 증폭된 핵산을 정제(purification)하는 단계;
정제된 핵산을 단편화(fragmentation)하는 단계;
단편화된 핵산들을 풀링(pooling)하는 단계; 및
풀링된 핵산들로 NGS(Next Generation Sequencing) 라이브러리를 제조하는 단계를 더 포함할 수 있다.
상기 “정제하는 단계” 및 “단편화하는 단계"는 선별하는 단계 이전 또는 이후에 수행될 수 있으며, 상기 "정제하는 단계"가 선별하는 단계 이전에 수행될 경우, 상기 "단편화하는 단계"는 선별하는 단계 이전 또는 이후에 수행될 수 있다.
일 양상에 있어서, 상기 "풀링하는 단계"는 서로 다른 고정된 제2 염기 서열을 포함하는 복수 개의 제1 프라이머로부터 수득한 서로 다른 표적 핵산 증폭물을 한 번에 하나의 풀(pool)에 모으는 단계를 의미한다. 상기 풀링 단계는 선별 단계 이전 또는 이후, 정제 단계 이전 또는 이후, 단편화 단계 이전 또는 이후, NGS 라이브러리 제조 단계 이전 중 사용자가 원하는 시점에서 수행될 수 있다. 또한, 상기 풀링 단계에서 사용자가 더 많은 양의 시퀀싱 리드를 분석하고자 하는 경우, 샘플을 더 많이 풀링하고 적은 양을 분석하고자 하는 샘플은 적게 풀링함으로써 샘플별로 풀링하는 양을 조절할 수 있다.
일 양상에 있어서, 상기 풀링하는 단계 이후 핵산 정제 단계를 더 포함하여 핵산 증폭 방법을 수행할 수도 있다.
또한, 일 양상에 있어서, 상기 핵산 증폭의 처리량을 증가시키기 위하여, 동일한 제2 염기 서열을 가진 제1 프라이머를 각각의 액체 방울(droplet) 또는 마이크로웰(microwell)에 첨가하고 핵산 증폭 방법을 수행함으로써, 수천종류 이상의 샘플을 한 번에 증폭한 뒤 한 번에(One-pot) 풀링할 수도 있다.
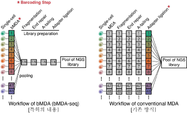
일 실시예에 따르면, 세포 용해 단계, bMDA 단계, bMDA 증폭 산물 처리 단계 및 bMDA-seq 단계로 이루어지는 핵산 증폭 방법을 수행함으로써, 여러 단일 세포에서 유래한 DNA에 대해 한 번에 NGS 라이브러리를 준비할 수 있음을 확인하였다. 상기 방법은 통상적으로 MDA 반응에 이용되는 랜덤 염기 서열 프라이머(제2 프라이머) 외에 바코드 서열을 포함하는 프라이머(제1 프라이머)를 첨가하여 수행되었고, 상기 바코드 서열로 인해 해당 NGS 리드가 어떤 샘플로부터 유래하였는지 판단할 수 있어 MDA 반응의 병렬성 및 처리량을 현저하게 높일 수 있다(실시예 1 참조).
다른 실시예에 따르면, 상기 핵산 증폭 방법에 있어서, 바코드 서열을 포함하는 프라이머(제1 프라이머)의 첨가 비율을 조절함으로써, 바코드 편향(bias) 현상을 감소시킬 수 있음을 확인하였다. 구체적으로, 바코드 편향 현상은 bMDA 증폭 편향, bMDA 증폭 산물에 포함된 바코딩 DNA 양의 편향 또는 NGS 라이브러리 변환 효율 편향을 의미할 수 있다. 구체적으로, 바코드가 포함된 프라이머의 비율이 모두 일정한 경우, 약 29.9%의 CV 값이 나타난 반면, 바코드가 포함된 프라이머의 비율을 각각 특정 비율로 다르게 설정할 경우, 최대 약 7.68%의 CV 값까지 줄일 수 있음을 확인하여, 상기 바코드 서열을 포함하는 프라이머의 첨가 비율을 각각 특정 비율로 다르게 설정할 경우, 바코드 편향 현상이 크게 감소함을 확인하였다(실시예 2 참조).
또 다른 실시예에 따르면, 세포 샘플의 분리 방식으로 레이저 기반 세포 분리 방식을 이용할 경우, bMDA의 증폭 품질이 향상됨을 확인하였다. 상기 레이저 기반 세포 분리 방식은, 물리적으로 인접한 주변 세포들을 함께 분리하여 분석함으로써, bMDA의 증폭 편향 현상을 최소화하고, 높은 품질의 증폭 산물을 제공할 수 있다(실시예 3 참조).
다른 양상은 제1 프라이머 및 제2 프라이머를 포함하는 핵산 증폭용 프라이머 세트로서,
상기 제1 프라이머는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 프라이머 세트를 제공한다.
상기 "제1 프라이머", "제2 프라이머", "표적 핵산", "샘플", "제1 염기 서열", "제2 염기 서열" 및 "제3 염기 서열" 등은 전술한 범위 내일 수 있다.
일 양상에 따르면, 상기 프라이머 세트로 핵산 증폭 방법을 수행할 경우, 서로 다른 샘플로부터 유래된 표적 핵산을 구별할 수 있다.
일 양상에 있어서,
상기 제1 프라이머는 서로 다른 복수 개이고,
상기 제2 프라이머는 서로 다른 복수 개이고,
상기 서로 다른 복수 개의 제1 프라이머들은 서로 다른 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 프라이머 세트일 수 있다.
일 양상에 따르면, 상기 프라이머 세트 내 제1 프라이머는 제2 프라이머 대비 0.1 내지 10 몰%로 첨가되는 것일 수 있다. 구체적으로, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 1 몰%, 0.1 내지 3 몰%, 0.1 내지 5 몰%, 0.1 내지 7 몰%, 0.1 내지 10 몰%, 1 내지 3 몰%, 1 내지 5 몰%, 1 내지 7 몰%, 1 내지 10 몰%, 3 내지 5 몰%, 3 내지 7 몰%, 3 내지 10 몰%, 5 내지 7 몰%, 5 내지 10 몰% 또는 7 내지 10 몰%로 첨가되는 것일 수 있다.
일 양상에 따르면, 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 핵산 증폭 방법의 효율을 증가시킬 수 있고, 선별 단계에서 샘플별 표적 핵산을 일정한 양으로 회수할 수 있으며, NGS 라이브러리 제조 단계에서 샘플별 시퀀싱 리드를 일정하게 조절할 수 있다. 또한, 상기 비율은 제2 염기 서열의 종류에 따라 다르게 조절될 수 있다.
또 다른 양상은 제1 프라이머 및 제2 프라이머를 포함하는 핵산 증폭용 키트로서,
상기 제1 프라이머는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 상기 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 키트를 제공한다.
상기 "제1 프라이머", "제2 프라이머", "표적 핵산", "샘플", "제1 염기 서열", "제2 염기 서열" 및 "제3 염기 서열" 등은 전술한 범위 내일 수 있다.
일 양상에 따르면, 상기 키트로 핵산 증폭 방법을 수행할 경우, 서로 다른 샘플로부터 유래된 표적 핵산을 구별할 수 있다.
일 양상에 있어서,
상기 제1 프라이머는 서로 다른 복수 개이고,
상기 제2 프라이머는 서로 다른 복수 개이고,
상기 서로 다른 복수 개의 제1 프라이머들은 서로 다른 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 키트일 수 있다.
일 양상에 따르면, 상기 키트 내 제1 프라이머는 제2 프라이머 대비 0.1 내지 10 몰%로 첨가되는 것일 수 있다. 구체적으로, 구체적으로, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 1 몰%, 0.1 내지 3 몰%, 0.1 내지 5 몰%, 0.1 내지 7 몰%, 0.1 내지 10 몰%, 1 내지 3 몰%, 1 내지 5 몰%, 1 내지 7 몰%, 1 내지 10 몰%, 3 내지 5 몰%, 3 내지 7 몰%, 3 내지 10 몰%, 5 내지 7 몰%, 5 내지 10 몰% 또는 7 내지 10 몰%로 첨가되는 것일 수 있다.
일 양상에 따르면, 상기 상기 제1 프라이머 및 제2 프라이머의 첨가되는 비율을 조절함으로써, 핵산 증폭 방법의 효율을 증가시킬 수 있고, 선별 단계에서 샘플별 표적 핵산을 일정한 양으로 회수할 수 있으며, NGS 라이브러리 제조 단계에서 샘플별 시퀀싱 리드를 일정하게 조절할 수 있다. 또한, 상기 비율은 제2 염기 서열의 종류에 따라 다르게 조절될 수 있다.
일 양상에 있어서, 상기 키트는 웰을 더 포함하고, 상기 제1 프라이머 및 제2 프라이머는 상기 웰에 분주(seeding)되어 있는 것일 수 있다. 구체적으로, 상기 웰은 8 스트립 튜브, 96 웰 플레이트 및 384 플레이트로 이루어진 군에서 선택되는 하나 이상의 것일 수 있다.
일 양상에 있어서, 상기 제1 프라이머 및 제2 프라이머는 동결 건조된 것일 수 있다. 상기 제1 프라이머 및 제2 프라이머가 동결 건조될 경우, 변질을 방지할 수 있다.
일 양상에 있어서, 상기 제1 프라이머 및 제2 프라이머가 1 ng 내지 1 μg 질량의 비드 또는 1 μm 내지 10 mm 직경의 비드에 결합된 상태로 제공될 수 있다. 상기 비드의 결합으로 인해, 파이펫팅(pipetting) 또는 분배(dispensing) 없이 서로 다른 바코드 서열을 가지는 프라이머를 웰 또는 액체 방울(droplet)으로 나누어서 핵산 증폭 방법을 수행할 수 있다. 상기 비드는 폴리스티렌(polystyrene)으로 만들어질 수 있고, 하이드로젤(hydrogel)로 만들어질 수도 있다. 또한, 상기 프라이머가 비드 표면에 공유결합으로 결합되어 제공될 수도 있고, 젤 속에 가둬져서 제공될 수도 있다.
일 양상에 있어서, 상기 키트를 이용하여 한 번 이상의 가닥 전위 반응이 일어나는 핵산 증폭 방법을 수행할 수 있다.
일 양상에 따른 제1 프라이머, 제2 프라이머 및 핵산 중합효소를 표적 핵산이 포함된 샘플에 첨가하여 핵산들을 증폭시키는 단계를 포함하는 핵산 증폭 방법에 의하면, 바코드 서열을 포함하는 제1 프라이머를 이용하여 병렬적 다중 전위 증폭을 수행함으로써, MDA의 병렬성 및 처리량을 높여 NGS 라이브러리 제작 시간 및 비용을 혁신적으로 감소시킬 수 있다. 또한, 상기 방법은 바코드 서열을 포함하는 프라이머와 바코드 서열을 포함하지 않는 프라이머의 비율을 조절함으로써, 바코드 편향 현상도 해결할 수 있으며, 레이저 기반 세포 분리 방식을 이용하여 증폭 품질도 향상시킬 수 있다. 따라서, 상기 핵산 증폭 방법은 다량의 단일 세포 전장 유전체 분석에 유용하게 이용될 수 있다.
도 1은 MDA와 Tn5 유전자전위효소(transposase)를 이용한 증폭 방법에서의 유전체 커버리지 값을 나타낸다.
도 2a는 bMDA(barcode multiple displacement amplification)를 위한 바코드가 포함된 프라이머의 구조를 나타낸다.
도 2b는 bMDA(barcode multiple displacement amplification)를 위한 바코드가 포함된 프라이머와 통상적인 N6 프라이머의 구성을 나타낸다
도 2c는 bMDA의 전체 과정을 개략적으로 도시한 흐름도를 나타낸다.
도 3은 bMDA 반응 이후에 서로 다른 표적 샘플(세포)에서 유래한 증폭 산물을 풀링하여 NGS 라이브러리 제작 과정을 한 번에(one-pot) 수행하는 공정을 도시한 흐름도를 나타낸다.
도 4a는 N6, 바코드 시퀀스 영역 및 Read 1(15-mer 뉴클레오티드)으로 구성된 R15B8N6 프라이머의 구조를 나타낸다.
도 4b는 N6 프라이머 외에 추가적인 시퀀스를 포함하는 프라이머(R15B8N6)를 사용할 경우, MDA 반응이 상당히 저해됨을 나타낸다.
도 4c는 R15B8N6 프라이머 내 Read 1이 PCR을 위한 핸들로 작용하여 바코딩된 DNA를 선별적으로 농축(enrichment)할 수 있음을 나타낸다.
도 5는 프라이머의 높은 농도가 MDA 반응 저해 현상의 원인 중 하나임을 나타낸다.
도 6은 프라이머의 긴 길이가 MDA 반응 저해 현상의 원인 중 하나임을 나타낸다.
도 7은 바코드가 포함된 프라이머의 농도 및 길이와 bMDA 반응 저해 현상과의 연관성을 정량적으로 분석한 결과를 나타낸다.
도 8a는 바코드가 포함된 프라이머가 DNA 중합효소와 결합하여 DNA 중합효소를 억제함으로써 bMDA 증폭 효율이 감소하는 모델을 나타낸다.
도 8b는 바코드가 포함된 프라이머가 다른 프라이머와 비특이적으로 혼성화(non-specific hybridization)되어 프라이머 다이머를 형성함으로써 MDA 증폭 효율이 떨어지는 모델을 나타낸다.
도 9는 phi29 DNA 중합효소의 양을 달리하여 측정한 bMDA의 증폭 효율 변화를 나타낸다.
도 10은 bMDA 반응 결과에 따른 라이브러리 복잡도(library complexity)의 계산 값 및 실험값을 나타낸다.
도 11은 bMDA-seq 후 측정되는 유전체 커버리지(genome coverage), 증폭 균일도(amplification uniformity), ADO (Allele Drop Out) 및 FPR (False positive rate)이 통상적인 MDA와 유사함을 나타낸다.
도 12는 MDA를 이용한 세포 처리 비용 및 유전체 분석 비용과 bMDA를 이용한 세포 처리 비용 및 유전체 분석 비용을 비교한 데이터를 나타낸다.
도 13a는 바코드가 포함된 프라이머 5’말단에 포획 물질로 비오틴이 결합되어 있는 구조 및 이로 인해 어댑터의 R1 시퀀스와 바코드 시퀀스간 라이게이션(ligation)이 방지될 수 있음을 나타낸다.
도 13b는 bMDA 바코드 시퀀스의 위치가 주로 Read 2에서 발견됨을 나타낸다.
도 14는 bMDA-seq 이후 NGS 결과에서 발견되는 바코드의 구성을 나타낸다.
도 15는 bMDA-seq 이후 바코드 스와핑(barcode swapping) 비율을 나타낸다.
도 16은 16개의 서로 다른 바코드 시퀀스에 따른 bMDA 증폭 산물에 포함된 바코딩 DNA 양(barcoded product mass) 및 NGS 라이브러리 변환 효율(library conversion yield)을 나타낸다.
도 17은 bMDA 반응 용액 중 바코드가 포함된 프라이머의 비율에 따른 NGS 리드(read) 수 변화를 나타낸다.
도 18은 바코드가 포함된 프라이머의 비율을 모두 일정하게 설정한 경우와, 각각 특정 비율로 다르게 설정한 경우에서의 바코드 편향 정도를 나타낸다.
도 19는 bMDA의 인풋으로 사용되는 템플릿(template)의 양의 변화에 따른 NGS 리드 수의 CV 값을 나타낸다.
도 20은 유체 기반의 단일세포 분리 기술과 레이저 기반의 단일세포 분리기술을 bMDA에 적용하였을 때의 차이를 비교한 도표를 나타낸다.
도 21은 MDA와 대비한 bMDA의 증폭 처리량 상승 및 유체 분리 방식과 대비한 레이저 기반 분리 방식의 증폭 품질 상승을 나타낸다.
도 22은 bMDA 및 레이저 기반 분리 방식을 이용할 경우 발생되는 비용 하락 효과를 나타낸다.
도 2a는 bMDA(barcode multiple displacement amplification)를 위한 바코드가 포함된 프라이머의 구조를 나타낸다.
도 2b는 bMDA(barcode multiple displacement amplification)를 위한 바코드가 포함된 프라이머와 통상적인 N6 프라이머의 구성을 나타낸다
도 2c는 bMDA의 전체 과정을 개략적으로 도시한 흐름도를 나타낸다.
도 3은 bMDA 반응 이후에 서로 다른 표적 샘플(세포)에서 유래한 증폭 산물을 풀링하여 NGS 라이브러리 제작 과정을 한 번에(one-pot) 수행하는 공정을 도시한 흐름도를 나타낸다.
도 4a는 N6, 바코드 시퀀스 영역 및 Read 1(15-mer 뉴클레오티드)으로 구성된 R15B8N6 프라이머의 구조를 나타낸다.
도 4b는 N6 프라이머 외에 추가적인 시퀀스를 포함하는 프라이머(R15B8N6)를 사용할 경우, MDA 반응이 상당히 저해됨을 나타낸다.
도 4c는 R15B8N6 프라이머 내 Read 1이 PCR을 위한 핸들로 작용하여 바코딩된 DNA를 선별적으로 농축(enrichment)할 수 있음을 나타낸다.
도 5는 프라이머의 높은 농도가 MDA 반응 저해 현상의 원인 중 하나임을 나타낸다.
도 6은 프라이머의 긴 길이가 MDA 반응 저해 현상의 원인 중 하나임을 나타낸다.
도 7은 바코드가 포함된 프라이머의 농도 및 길이와 bMDA 반응 저해 현상과의 연관성을 정량적으로 분석한 결과를 나타낸다.
도 8a는 바코드가 포함된 프라이머가 DNA 중합효소와 결합하여 DNA 중합효소를 억제함으로써 bMDA 증폭 효율이 감소하는 모델을 나타낸다.
도 8b는 바코드가 포함된 프라이머가 다른 프라이머와 비특이적으로 혼성화(non-specific hybridization)되어 프라이머 다이머를 형성함으로써 MDA 증폭 효율이 떨어지는 모델을 나타낸다.
도 9는 phi29 DNA 중합효소의 양을 달리하여 측정한 bMDA의 증폭 효율 변화를 나타낸다.
도 10은 bMDA 반응 결과에 따른 라이브러리 복잡도(library complexity)의 계산 값 및 실험값을 나타낸다.
도 11은 bMDA-seq 후 측정되는 유전체 커버리지(genome coverage), 증폭 균일도(amplification uniformity), ADO (Allele Drop Out) 및 FPR (False positive rate)이 통상적인 MDA와 유사함을 나타낸다.
도 12는 MDA를 이용한 세포 처리 비용 및 유전체 분석 비용과 bMDA를 이용한 세포 처리 비용 및 유전체 분석 비용을 비교한 데이터를 나타낸다.
도 13a는 바코드가 포함된 프라이머 5’말단에 포획 물질로 비오틴이 결합되어 있는 구조 및 이로 인해 어댑터의 R1 시퀀스와 바코드 시퀀스간 라이게이션(ligation)이 방지될 수 있음을 나타낸다.
도 13b는 bMDA 바코드 시퀀스의 위치가 주로 Read 2에서 발견됨을 나타낸다.
도 14는 bMDA-seq 이후 NGS 결과에서 발견되는 바코드의 구성을 나타낸다.
도 15는 bMDA-seq 이후 바코드 스와핑(barcode swapping) 비율을 나타낸다.
도 16은 16개의 서로 다른 바코드 시퀀스에 따른 bMDA 증폭 산물에 포함된 바코딩 DNA 양(barcoded product mass) 및 NGS 라이브러리 변환 효율(library conversion yield)을 나타낸다.
도 17은 bMDA 반응 용액 중 바코드가 포함된 프라이머의 비율에 따른 NGS 리드(read) 수 변화를 나타낸다.
도 18은 바코드가 포함된 프라이머의 비율을 모두 일정하게 설정한 경우와, 각각 특정 비율로 다르게 설정한 경우에서의 바코드 편향 정도를 나타낸다.
도 19는 bMDA의 인풋으로 사용되는 템플릿(template)의 양의 변화에 따른 NGS 리드 수의 CV 값을 나타낸다.
도 20은 유체 기반의 단일세포 분리 기술과 레이저 기반의 단일세포 분리기술을 bMDA에 적용하였을 때의 차이를 비교한 도표를 나타낸다.
도 21은 MDA와 대비한 bMDA의 증폭 처리량 상승 및 유체 분리 방식과 대비한 레이저 기반 분리 방식의 증폭 품질 상승을 나타낸다.
도 22은 bMDA 및 레이저 기반 분리 방식을 이용할 경우 발생되는 비용 하락 효과를 나타낸다.
이하 본 발명을 실시예를 통하여 보다 상세하게 설명한다. 그러나, 이들 실시예는 본 발명을 예시적으로 설명하기 위한 것으로 본 발명의 범위가 이들 실시예에 한정되는 것은 아니다.
실시예
실시예 1. bMDA-seq 수행 방법
(1) 세포 용해 단계
bMDA는 바코드가 포함된 프라이머를 이용한 MDA반응을, bMDA-seq은 bMDA를 포함한 단일세포로부터 NGS 라이브러리를 제작하는 전체 과정을 의미한다. 상기 bMDA-seq을 위한 첫 번째 단계는 세포를 분리한 뒤 용해하는 단계이다. 이를 위해, 7 μL의 용해 용액(변성 완충액(400 mM KOH, 10 mM EDTA 및 100 mM DTT) 3 μL, 2 μL PBS (REPLI-g Single Cell Kit, Qiagen), 1 μL 템플릿(template) 및 500 μM 랜덤 헥사머 1 μL)을 준비하고 10 내지 30분 간 4℃ 이하에서 반응시켰다. 상기 템플릿(template)은 세포, gDNA 등의 표적 샘플이 포함된 용액을 의미한다. 낮은 온도에서의 용해는 사이토신 디아민화(Cytosine deamination)와 같은 자발적인 DNA손상이 훨씬 적어 DNA 오류가 적은 장점이 있다. 이후 중화 완충액(400 mM HCl 및 600 mM Tris-HCl(pH 7.5)) 3 μL를 넣고 섞었다.
(2) bMDA 단계
1) 개요
여러 세포에서 유래한 DNA에 대해 한 번에 라이브러리 제작 과정을 거쳐 시퀀싱하기 위해, 바코드가 포함된 프라이머를 이용한 MDA 수행 방법을 고안하였다(bMDA).
상기 바코드가 포함된 프라이머는 바인딩 영역, 바코드 시퀀스 영역 및 포획 물질로 구성될 수 있다(도 2a 참조). 예를 들어, 상기 바코드가 포함된 프라이머는 하기와 같은 형태로 구성될 수 있다:
/5Biosg/JJJJJJNNNN*N*N.
(여기서, /5Biosg/는 5'-비오틴 변형으로 포획 물질이고, JJJJJJ는 6개의 염기로 이루어진 바코드 시퀀스 영역이고, NNNNNN은 6개의 랜덤 염기로 이루어진 바인딩 영역이며, *은 3'-5'핵산외부가수분해(exonuclease) 반응에 저항성이 있는 포스포로티올레이트(phosphorothiolate) 변형 뉴클레오티드이다.)
상기 바인딩 영역은 표적 샘플과 혼성화하여 증폭이 시작되도록 하는 시퀀스로, 6개의 무작위 뉴클레오티드로 이루어진 랜덤 헥사머(N6)를 사용하였다. DNA 중합 효소의 3'-5'핵산외부가수분해(exonuclease) 기능으로 인한 프라이머 고갈을 방지하기 위해, 상기 랜덤 헥사머 내 일부 뉴클레오티드간의 결합을 포스포로티올레이트(phosphorothiolate) 결합으로 변형하였다.
상기 바코드 시퀀스는 서로 다른 샘플로부터 유래된 DNA를 구별하기 위한 시퀀스로, 4 내지 12 뉴클레오티드로 이루어진 시퀀스를 사용하였다. 일례로, 6 뉴클레오티드로 이루어진 바코드 시퀀스를 사용할 경우, 46 = 1,024 종류의 서로 다른 바코드를 디자인함으로써, 1,024개의 세포를 병렬화하여 (multiplexing) 한 번에(one-pot) 분석할 수 있다.
상기 포획 물질은, 바코딩된 증폭 산물만을 선별적으로 포착하기 위한 물질로, 비오틴(biotin) 또는 10개 이상의 염기 서열로 이루어진 폴리뉴클레오티드를 사용하였다. 비오틴을 상기 포획물질로 사용할 경우, IDT사의 5'Biotin 변형된 올리고뉴클레오티드를 사용하였다. 10개 이상의 염기 서열로 이루어진 폴리뉴클레오티드를 포획물질로 사용할 경우, 일루미나(Illumina)사의 시퀀서(Sequencer)와 호환성을 위하여 Read 1서열 중 3'말단 영역의 일부 염기서열을 차용하여 ACGCTCTTCCGATCT(서열번호 1) 서열을 이용하였다.
비오틴은 스트렙트아비딘(streptavidin)과 강하게 결합하는 성질이 있어, 이를 이용해 바코딩된 DNA를 선별할 수 있으며, 10개 이상의 염기 서열로 이루어진 폴리뉴클레오티드는 PCR을 위한 핸들로 작용하여 바코딩된 DNA를 선별적으로 농축(enrichment)할 수 있다.
상기 바코드가 포함된 프라이머로 인한 MDA반응 저해 현상을 최소화하기 위해 통상적인 랜덤 헥사머(N6)와 변형된 프라이머를 특정 비율로 섞어서 실험하였으며, 2%의 변형된 프라이머를 포함한 프라이머 믹스가 가장 효과적임을 확인하였다(도 2b 참조).
상기 바코드가 포함된 프라이머를 사용하여 MDA를 수행할 경우(bMDA), 표적 샘플의 염기서열과 상보적인 랜덤 헥사머 시퀀스를 가진 프라이머가 표적 샘플에 혼성화되고, 프라이머 내의 바코드 시퀀스가 표적 샘플과 함께 증폭된다. 따라서, 서로 다른 표적 샘플을 서로 다른 바코드가 포함된 프라이머를 사용해 bMDA를 수행하고, 증폭 산물을 풀링(pooling)하여 한 번에 모은 뒤 짧은 길이로 단편화(fragmentation)시키면 바코드 시퀀스가 포함된 증폭 산물과 포함되지 않은 증폭 산물을 얻을 수 있다. 이 때, 스트렙트아비딘 자성 비드(streptavidin coated magenetic bead)를 사용하여 비오틴이 결합되어 있는, 즉 바코드가 포함된 DNA 조각(fragment) 만을 선별할 수 있다. 상기 스트렙트아비딘-비오틴 정제과정 이후 라이브러리 제작 과정을 거친 뒤 NGS(Next Generation Sequencing)를 수행하여 바코드 시퀀스를 확인함으로써, 해당 NGS 리드(read)가 어떤 샘플로부터 유래하였는지를 판단하는 역다중화(demultiplexing) 과정이 가능하다(도 2c 참조).
상기 bMDA는, 기존 MDA와 달리 MDA 증폭 과정에서 표적 샘플에 바로 DNA 바코딩을 수행함으로써, 기존 작업 흐름에 거의 변화를 가하지 않고 병렬화가 가능한 특징이 있다. bMDA 후 서로 다른 표적 샘플에서 유래한 증폭 산물을 풀링하여 한 번에 NGS 라이브러리 제작 과정을 수행함으로써, 라이브러리 제작 비용을 1/N(N=한 번에 분석하는 샘플 수 또는 바코드의 수)로 혁신적으로 절감할 수 있다(도 3 참조).
2) bMDA의 효율을 저해하는 원인 파악
그러나, 일반적으로 MDA 과정에 사용되는 N6 프라이머 외에 추가적인 시퀀스를 포함하는 프라이머를 사용할 경우, MDA 반응이 상당히 저해된다. 이를 확인하기 위해, N6 프라이머 50 μM과 R15B8N6 프라이머 50 μM을 각각 첨가하여 MDA를 수행한 뒤 증폭산물을 전기영동 하였다. 상기 R15B8N6 프라이머는 바인딩 영역으로 랜덤 헥사머(N6), 바코드 시퀀스 영역으로 8-mer (B8) 및 포획 물질로 15-mer의 뉴클레오티드(R15)로 구성된다(도 4a 참조). 상기 R15 서열로는 일루미나사의 Read 1 서열의 일부인 ACGCTCTTCCGATCT(서열번호 1) 서열을 이용하였다. 전기영동 결과, N6프라이머를 사용한 경우, 증폭산물이 통상적인 길이인 수 내지 수십 kb로 나타나 MDA증폭이 성공적으로 이루어졌음을 확인하였으나, 50 μM의 R15B8N6를 사용할 경우 증폭이 전혀 일어나지 않음을 확인하였다(도 4b 참조).
상기와 같은 bMDA 반응 저해 문제를 해결하기 위해 우선적으로, 이와 같은 현상의 원인을 파악하기 위한 두 가지 실험을 수행하였다.
① 프라이머 농도
프라이머의 농도가 bMDA 반응 효율을 저해하는지 여부를 확인하기 위하여, N6 프라이머 50 μM, N6 프라이머 5 μM 및 R15B8N6 프라이머 5 μM을 첨가하여 MDA를 수행하였다. 15개의 뉴클레오티드(R15)로 이루어진 고정된 DNA 시퀀스를 포획 물질로 하여 MDA가 수행될 경우, R15와 동일한 서열을 포함하는 프라이머(PCR primer)를 사용하여 PCR을 수행함으로써 바코딩된 DNA를 선별적으로 농축할 수 있다(도 4c 참조). 상기 PCR과정에서 바코딩이 되지 않은 DNA는 고정된 DNA시퀀스를 포함하지 않기 때문에 PCR 증폭이 이루어지지 않는다.
실험 결과, 5 μM의 R15B8N6 프라이머를 첨가할 경우 5 μM의 N6에 비해서는 수율은 낮지만 다소 성공적인 증폭산물이 관찰되었다(도 5 참조). 이는, R15B8N6 프라이머의 높은 농도가 상기 bMDA 반응 저해 현상의 원인 중 하나임을 의미한다. 그러나, 상기 실험 결과물에 대한 증폭 균일도(amplification uniformity)를 확인하기 위해 사람의 유전체 위치에 따른 유전자 복제 수 변이(CNA) 분석을 수행한 결과, 5 μM의 R15B8N6 프라이머를 이용하여 MDA를 수행할 경우, 5 μM의 N6를 이용하였을 때에 비해 증폭 편향(bias)이 매우 심하여 CNA 분석을 정상적으로 수행하기 어려움을 확인하였다(도 5 참조). 도 5의 예측 복제 수(Estimated Copy Number) 분석 결과에서 회색 점은 사람의 유전체 위치에 따라 얼마나 많은 NGS 시퀀스가 발견되는지를 나타낸 것으로, 증폭 편향이 낮아 유전체가 균일하게 증폭될수록 회색점들간의 변동(variation)이 낮은 결과를 보이는데, 5 μM의 R15B8N6 프라이머를 이용한 경우 회색점들간의 변동이 매우 심함을 확인할 수 있다. 이를 통해 R15B8N6 프라이머의 농도 이외에도 MDA증폭을 저해하는 다른 원인이 있음을 확인할 수 있다.
② 프라이머 길이
프라이머의 길이가 bMDA 반응 효율을 저해하는지 여부를 확인하기 위하여, N6 프라이머 50 μM, R15B8N6 프라이머 50 μM, N6 프라이머 50 μM + R15B8N6 프라이머 50 μM 및 N6 프라이머 500 μM을 첨가하여 MDA를 수행하였다.
그 결과, 일반적으로 MDA에 사용되는 프라이머 농도보다 10배 정도 높은 농도인 N6 프라이머 500 μM 처리군에서는 MDA 증폭 산물을 얻을 수 있었으나, 일반적으로 MDA에 사용되는 프라이머 농도인 N6 프라이머 50 μM 처리군에 R15B8N6 50 μM을 추가할 경우 MDA가 상당히 저해되었다(도 6 참조). R15B8N6 프라이머는 길이가 29-mer로서 N6프라이머에 비해 약 5배정도 길이가 길기 때문에, 프라이머의 길이가 반응에 영향을 주지 않는다면 R15B8N6 프라이머 50 μM을 추가하는 것은 N6 프라이머 250 μM을 추가하는 것과 비슷한 효과를 가져와야 한다. 하지만 N6 프라이머 50 μM에 R15B8N6 프라이머 50 μM을 추가할 경우 MDA증폭이 상당히 저해되나, R15B8N6 50 μM 보다도 더 많은 핵산을 포함하도록 N6 프라이머 500 μM로 증가시켰을 경우에도 MDA 증폭이 정상적으로 수행된다는 실험 결과는 R15B8N6 프라이머의 긴 길이가 bMDA 반응 저해 현상의 원인임을 의미한다.
③ 프라이머 농도 및 길이와 bMDA 반응 저해 현상 간 연관성의 정량적 분석
추가적으로, 바코드가 포함된 프라이머의 농도 및 길이와 MDA 반응 저해 현상과의 연관성을 정량적으로 분석하기 위하여, 바코드 시퀀스의 길이 및 농도를 달리하여 bMDA 증폭 효율(Amplification efficiency)을 분석하였다.
상기 bMDA 증폭 효율은 bMDA 반응 중 dsDNA(double-stranded DNA)와 반응하여 특이적으로 형광을 발하는 SYTOTM 13(Invitrogen)을 첨가하여 실시간으로 형광의 증가를 관찰하였으며(RT-MDA, Real time MDA), 형광 세기 증가의 최대 기울기를 증폭 효율로 정의하였다. 상기 증폭 효율값은 비교를 위해 랜덤 헥사머를 이용한 통상적인 MDA의 증폭 효율값으로 나누어 주어 최대값이 1이 되도록 정규화 하였다. 프라이머의 길이에 변화를 줄이기 위해 다양한 길이의 바코드가 포함된 프라이머를 사용하여 bMDA 반응을 수행하였으며, 구체적으로, 29-mer(ACGCTCTTCCGATCTGCCAAGACNNNN*N*N, 서열번호 2), 14-mer(GACTGTGTNNNN*N*N, 서열번호 3), 13-mer(ACTGTGTNNNN*N*N, 서열번호 4), 12-mer(CTGTGTNNNN*N*N, 서열번호 5), 11-mer(TGTGTNNNN*N*N, 서열번호 6) 및 10-mer(GTGTNNNN*N*N, 서열번호 7)로 이루어진 프라이머를 이용하였다. 이때, 상기 *는 포스포로티올레이트 변형을 의미한다. 바코드가 포함된 프라이머의 농도를 증가시키거나 감소시킬 ??에는 같은 양의 랜덤 헥사머를 감소시키거나 증가시켜 랜덤 헥사머를 포함하는 전체 프라이머의 양을 일정하게 유지시켰다.
분석 결과, 바코드가 포함된 프라이머의 비율(Proportion of barcoded primer)이 증가할수록, 또한 프라이머의 길이가 증가할수록 bMDA 증폭 효율이 극적으로 감소하는 것을 확인하였다(도 7 참조).
④ 프라이머 농도 및 길이에 따라 bMDA 반응이 저해되는 원인 분석
프라이머 농도 및 길이에 따라 bMDA 반응이 저해되는 원인으로, 하기 두 가지 모델을 고려해볼 수 있다.
첫 번째로, 바코드가 포함된 프라이머가 DNA 중합효소와 결합하여 DNA 중합효소의 억제제로 작용하는 경우이다(경쟁적 저해 모델, 도 8a 참조). 모든 DNA 중합효소는 기본적으로 DNA 결합 능력을 가지는데, 프라이머의 길이가 길어질수록 프라이머와 DNA 중합효소간 결합 친화도(affinity)가 증가한다. 따라서, 바코드 시퀀스가 길어질수록 DNA 중합효소가 바코드가 포함된 프라이머와 더 많이 결합하므로, DNA 중합효소가 더 많이 고갈(depletion)되어 bMDA 증폭 효율이 떨어질 수 있다.
두 번째로는, 바코드가 포함된 프라이머가 다른 프라이머와 비특이적으로 혼성화(non-specific hybridization)되는 경우이다(도 8b 참조). 이 경우, 프라이머 다이머(dimer)가 형성되어 bMDA 증폭 효율이 떨어질 수 있다.
상기 실험들을 통해, 바코드가 포함된 프라이머로 bMDA를 수행할 경우, 증폭 효율이 떨어지며, 프라이머의 농도 및 길이가 MDA 증폭 효율에 중요한 영향을 미침을 확인하였다. 바코드가 포함된 프라이머의 bMDA 증폭 효율 저해를 최소화하여 bMDA의 효율을 높일 수 있는 방안으로, 하기 5가지 방안을 고려해볼 수 있다.
3) bMDA의 효율을 상승시킬 수 있는 방법 파악
① DNA 중합효소 과량 첨가 방법
첫 번째로, DNA 중합효소를 과량으로 첨가하는 방법이 있다. 이는 상기에서 언급한 대로, 바코드 시퀀스가 길어질수록 바코드가 포함된 프라이머와 DNA 중합효소간 결합 친화도가 증가하여 DNA 중합효소가 더 많이 고갈되기 때문이다. 일례로, 통상 MDA를 위해 30℃에서 1분에 약 100 pmole의 dNTP를 중합할 수 있는 양의 효소를 첨가하는데, 30℃에서 1분에 약 1 nmole 이상의 dNTP를 중합할 수 있는 양의 효소를 첨가함으로써, 바코드가 포함된 프라이머에 의한 효소 고갈을 상쇄하여 bMDA의 증폭 효율 하락을 방지할 수 있다.
상기 아이디어를 검증하기 위하여, DNA 중합효소(phi29 DNA 중합효소)의 양을 달리하여 bMDA를 수행하여 효율값을 측정하였다. 상기 DNA 중합효소 양과 바코드가 포함된 프라이머의 비율에 따른 bMDA 반응 효율값은 상기 언급한 RT-MDA 기법을 이용하여 측정하였으며, 전체 데이터의 최대 반응 효율값이 1이 되도록 정규화(normalize)를 수행하였다. 바코드가 포함된 프라이머는 /5Biosg/ACTCTGNNNN*N*N(서열번호 8)을 이용하였다.
그 결과, DNA 중합효소의 양을 증가시켜도 증폭 효율 하락을 완전하게 방지하지 못함을 확인하였다(도 9 참조). 구체적으로, DNA 중합효소의 양(Enzyme quantity)이 1x이고, 바코드가 포함된 프라이머의 비율이 1%일 경우 bMDA 증폭 효율이 0.93의 값을 보이며, 중합효소의 양을 2x로 늘릴 경우 0.96, 4x로 늘릴 경우 0.78의 값을 보였다. 이는, DNA 중합효소의 양을 2x로 증가시키면 증폭 효율을 소폭 향상시킬 수 있으나, DNA 중합효소의 양을 4x로 증가시키면 증폭 효율이 오히려 다시 하락함을 의미한다. 상기와 같은 현상은 DNA 중합효소의 양을 늘리기 위해 bMDA 반응 용액에 함께 추가된 효소 보관 버퍼(enzyme storage buffer)로 인한 영향이 원인으로 추정된다.
② 바코드가 포함된 프라이머의 양을 줄이는 방법
두 번째 방법은, bMDA 반응 시 바코드가 포함된 프라이머의 양을 줄이는 방법이다. 다만, 이를 통해서 DNA 중합효소와 결합하는 프라이머의 양(또는 프라이머 다이머의 형성)을 줄일 수 있더라도, MDA 반응에서 프라이머의 농도가 떨어지게 되면 증폭 균일성이 낮아지는 문제가 발생한다(도 5 참조). 그 이유는 MDA의 반응 도중 가닥 치환(strand displacement)이 많이 일어날수록 MDA 증폭 속도와 증폭 균일도(amplficiation uniformity)가 높아지기 때문이며, 이 때문에 통상적인 MDA 반응에서는 프라이머 농도를 10 μM 이상의 높은 농도로 유지하게 된다. 따라서 이는 적절한 해결 방법이 아닐 가능성이 높다.
③ 바코드가 포함된 프라이머와 바코드가 포함되지 않은 프라이머를 특정 비율로 혼합하는 방법
세 번째 방법은, 바코드가 포함된 프라이머와 바코드가 포함되지 않고 바인딩 영역만이 존재하는 프라이머(예를 들어, 랜덤 핵사머 또는 공용 염기)를 특정 비율로 혼합하여 bMDA를 수행하는 방법이다. 상기 방법은 바코드가 포함된 프라이머의 양을 감소시켜 bMDA 증폭 효율 하락을 방지함과 동시에, MDA 반응에 필요한 N6를 포함하는 프라이머의 농도는 기존과 비슷하게 높은 상태로 유지함으로써, 증폭 균일성을 그대로 유지할 수 있다. 바코드가 포함된 프라이머가 N6를 포함하기 때문에, 상기 N6를 포함하는 프라이머의 농도란, 바코드가 포함된 프라이머의 농도와 통상적으로 MDA에 사용하는 N6 프라이머의 농도를 합한 것을 의미한다.
④ 바코드가 포함된 프라이머의 길이를 줄이는 방법
프라이머의 길이가 길어질수록 bMDA 증폭 효율이 낮아진다는 상기 실험 결과에 따르면(도 7 참조), 효율적인 bMDA를 위해서는 병렬화를 위한 바코드 시퀀스 및 bMDA 증폭 개시를 위한 N6 시퀀스 외에 프라이머 내 추가적인 핵산 시퀀스가 없는 것이 바람직함을 의미한다. 따라서, 바코드가 포함된 프라이머 내 포획 물질로 10개 이상의 염기 서열로 이루어진 폴리뉴클레오티드를 이용할 경우 bMDA 효율이 저해될 것이므로, 비오틴과 같은 생화학적 분자를 이용하여 바코딩된 DNA만을 선별적으로 농축하는 방식이 bMDA 효율 하락 문제를 해결하는데 매우 효과적인 방식이 될 수 있다. 또한 바코드 시퀀스의 길이도 줄어들수록 bMDA 증폭효율이 높아질 것이다.
⑤ 바코드가 포함된 프라이머의 길이와 비율을 적절히 조절하는 방법
i) 바코드가 포함된 프라이머의 적절한 길이 및 비율 결정
bMDA 증폭 효율을 높이고자 하는 측면에서는 바코드가 포함된 프라이머의 길이와 농도가 모두 최대한 줄어드는 것이 바람직한 반면, bMDA가 정상적으로 기능을 하는 측면에서는 프라이머의 길이와 농도가 모두 최대한 높아지는 것이 필요하다.
먼저, 바코드 길이가 충분히 길어야 바코딩으로 한 번에 병렬화할 수 있는 바코딩 용량이 증가한다. 여기서 바코드 길이(barcode length)란, 바코드가 포함된 프라이머 전체 길이에서 N6 길이에 해당하는 6을 뺀 값을 의미한다.
둘째로, 바코드가 포함된 프라이머의 비율이 충분히 높아야 라이브러리 복잡도(Library complexity)가 증가하게 된다. 라이브러리 복잡도란, bMDA 증폭 산물에서 바코딩된 증폭 산물만 모았을 경우, 인간 유전체를 얼마나 높은 커버리지 깊이(depth of coverage)로 분석할 수 있는지를 나타내는 값이다. 만약, 바코드가 포함된 프라이머의 비율이 지나치게 낮다면, 회수되는 바코드가 포함된 DNA 조각의 양이 적어 NGS 시퀀싱 결과 인간 유전체를 충분하게 커버하지 못하게 될 것이며, 따라서 단일 염기서열 수준의 단일 세포 유전체 분석이 불가능하게 될 것이다.
정리하면, bMDA 증폭 성능 및 증폭 균일성을 높이기 위해서는 바코드가 포함된 프라이머의 길이와 비율을 줄여야 하지만, 바코드의 길이를 줄일수록 바코드 용량이 떨어지고, 바코드가 포함된 프라이머의 비율을 줄일수록 bMDA 증폭 산물에서 바코딩된 증폭 산물의 양이 줄어들어 라이브러리 복잡도(Library complexity)가 감소하게 된다. 따라서, bMDA 증폭 효율을 높게 유지하면서 적절한 길이의 바코드 시퀀스를 가진 프라이머를 적절한 비율로 사용하는 기술자의 노력이 필수적이다.
바코드가 포함된 프라이머의 적절한 비율을 찾기 위해서 먼저 바코드가 포함된 프라이머가 전체 프라이머 내 2% 비율로 존재할 경우의 이론적인 라이브러리 복잡도를 계산하였다. 통상적으로 bMDA 증폭 산물은 10 kbp 정도의 길이 분포를 보이고, 평균 200bp 정도의 길이를 갖도록 DNA를 단편화 (fragmentation)할 경우, bMDA 증폭 산물 중 바코드가 포함된 DNA 조각은 전체의 0.04% 로 계산된다(바코드가 포함된 프라이머 비율 2% * 200 bp / 10 kbp = 0.04 %). 또한, 통상적으로 라이브러리 제작의 효율이 20 내지 40% 정도이므로, 최소값인 20%를 가정한다면 bMDA 증폭 산물 중 최종적으로 NGS 라이브러리로 변환되는 DNA의 비율은 0.008 % 로 계산된다(바코드가 포함된 DNA 조각 0.04% * 라이브러리 변환 효율 20% = 0.008 %).
상기 바코드가 포함된 DNA 조각의 비율(Biotinylated dsDNA fragment ratio)을 실험적으로 측정하기 위해서 bMDA 증폭 산물의 DNA 무게와 스트렙트아비딘-비오틴 정제 이후의 DNA 무게를 측정하여 나누어 주었다. 또한, 라이브러리 변환 효율(NGS library conversion rate)을 측정하기 위해서 라이브러리 제작 전후의 DNA 무게를 측정하였으며, 라이브러리 증폭 (Library amplification)을 위한 PCR이 DNA의 양을 사이클(PCR cycle)당 1.9배씩 증가시킨다고 가정하였다.
실제 실험을 통해서 측정한 결과, 바코드가 포함된 DNA 조각의 비율(Biotinylated dsDNA fragment ratio)이 평균 0.04%로 나타나며, 라이브러리 변환 효율(NGS library conversion rate)이 약 20%로 나타남을 확인하였다(도 10 참조).
통상적으로, MDA 반응을 통해 30 μg 이상의 다량의 증폭 산물을 획득할 수 있기 때문에, bMDA 반응 산물인 30 μg 중 약 2.4 ng의 DNA가 NGS 라이브러리로 변환된다(30 μg * bMDA의 NGS라이브러리 변환비율 0.008 % = 2.4 ng). 이는 인간 전체 유전체의 약 727x 에 해당하며(2.4 ng / 3.3 pg(인간 유전체 반수체의 무게) = 727), bMDA로부터 획득한 NGS 라이브러리를 이용하여 인간유전체를 727번까지 읽을 수 있음을 의미한다(라이브러리 복잡도 = 727x). 통상적으로, 단일 염기서열 수준의 단일세포 유전체 분석을 위해 10x 이상의 라이브러리 복잡도가 필요함을 생각하였을 때, 상기 결과는 바코드가 포함된 프라이머가 2 % 정도의 낮은 비율로 첨가되더라도 인간 유전체를 충분하게 커버하여 단일세포 수준의 유전체 분석에 문제가 없음을 의미한다.
바코드가 포함된 프라이머의 비율이 2%이면서 80% 이상의 bMDA 증폭 효율을 유지하는 바코드의 길이는 6-mer 이하로 나타난다(도 7 및 표 1 참조). 바코드의 길이를 6-mer로 하였을 때의 바코드 용량은 4,096 개로, 단일 염기서열 수준의 단일세포 유전체 분석을 목적으로 한 bMDA의 성능으로 충분히 높다고 판단하여, 바코드의 길이 6-mer, 바코드가 포함된 프라이머의 비율 2%를 bMDA 실험 조건으로 결정하였다.
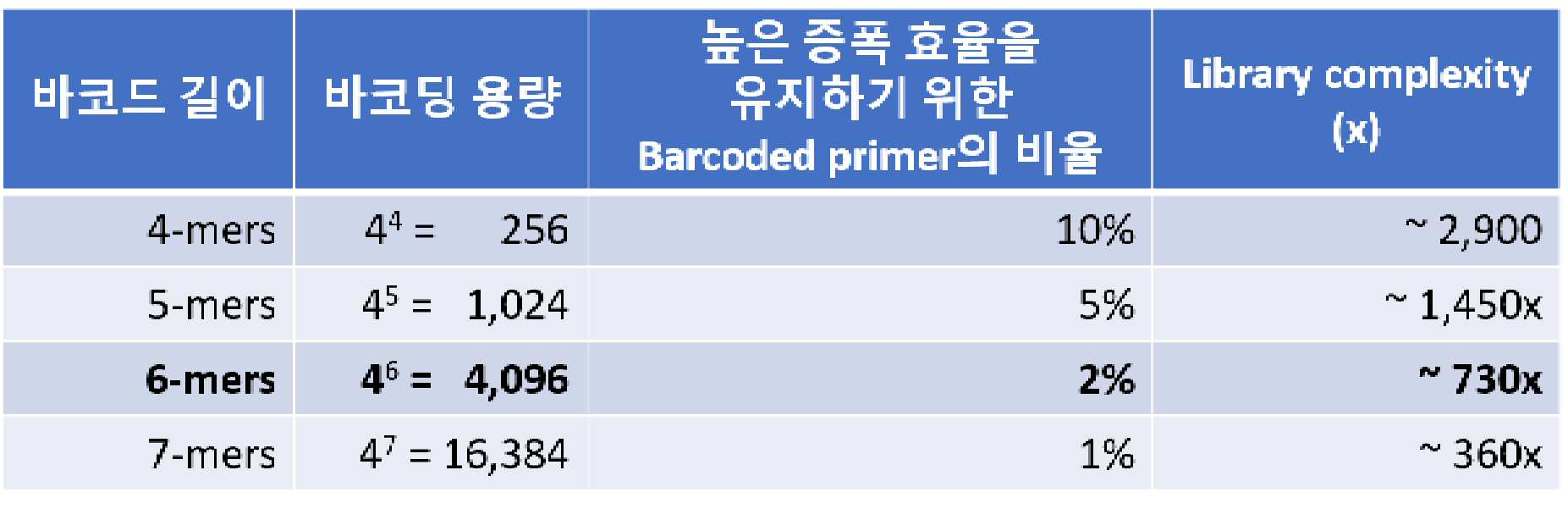
ii) 6-mer 길이의 바코드가 포함된 프라이머 2%를 첨가한 bMDA 결과 분석
6-mer 길이의 바코드가 포함된 프라이머를 전체 프라이머에 대비 2% 첨가한 조건에서 단일 세포에 대해 하기와 같은 조건으로 bMDA를 수행한 뒤, NGS를 이용하여 시퀀싱하였다. 이후, 유전체 커버리지(genome coverage), 커버리지 깊이 평균(mean depth of coverage), 증폭 균일도(coverage uniformity), 유전자 복제 수 변이(Copy number alteration, CNA) 분석 및 단일 염기변이(Single nucleotide variation, SNV) 분석 등 다양한 측면에서 통상적인 MDA와 bMDA의 차이를 측정하였다.
우선, 단일 세포를 분리하기 위해 HL-60 세포를 100 nm 두께의 ITO가 코팅된 슬라이드 글라스에 도말(smear)하였다. 상기 ITO는 적외선 레이저가 조사되었을 때 빛을 흡수하여 기화함으로써 레이저가 조사된 부분의 세포만을 선별적으로 분리하는 목적으로 사용된다. HL-60 세포가 도말된 슬라이드 글라스를 현미경 이미지로 관찰하면서 단일 세포가 존재하는 위치에 적외선 레이저를 조사하여 단일 세포를 분리하였다(도 11a 참조).
분리된 단일 세포를 상기 실시예 (1) 단계와 동일한 방법으로 용해시켜 세포 용해물(10 μL)을 얻은 후, 용해물에 40 μL의 마스터 믹스를 첨가하였다. 상기 마스터 믹스는 물 23 μL, 10X phi29 DNA 중합효소 완충액(500 mM Tris-HCl, 100 mM MgCl2, 100 mM (NH4)2SO4, 40 mM DTT, pH 7.5) 5 μL, 각각 25 mM의 dNTP 4 μL, 1 mM 랜덤 헥사머 2 μL, phi29 DNA 중합효소(Genomiphi V2 DNA amplification kit, Cytiva, cat. no. 25-6600-31) 2 μL, 40% (w/v) PEG 8000 3.2 μL, 1 M DTT 0.25 μL, 50 μM SYTO?? 13 Green Fluorescent Nucleic Acid Stain(Invitrogen) 0.5 μL 및 500 nM ROX 0.05 μL 로 구성하였다. 한편, bMDA 실험을 위해서 상기 랜덤 헥사머 대신 바코드가 포함된 프라이머를 동일한 농도로 이용하거나, 바코드가 포함된 프라이머와 랜덤 헥사머를 혼합하여 총 프라이머의 농도가 동일하도록 실험을 수행하였다.
상기와 같이 분리된 단일 세포 각각에 대해 서로 다른 바코드 서열을 가진 바코드가 포함된 프라이머를 이용하여 bMDA 증폭 산물을 얻었으며, 하기에 언급될 bMDA 증폭 산물 처리 단계에 따라 NGS 시퀀싱을 수행하였다.
또한, 비교를 위해서 통상적인 MDA 키트를 이용하는 방법(MDA kit), bMDA와 동일한 MDA 실험 조건에서 프라이머만 통상적인 랜덤 헥사머를 이용하는 방법(in-house MDA) 및 Tn5 유전자전위효소(transposase)를 이용하여 단일 세포의 gDNA에 바로 태깅(tagmentation)한 뒤, 단일 세포 유전체 분석을 수행하는 방법(Tn5 based)의 세 가지 방법으로 나누어 실험을 진행하였다. 상기 Tn5를 이용한 단일 세포 분석 방법은 Xi, L., Leong, P. & Mihajlovic, A. Preparing Single-cell DNA Library Using Nextera for Detection of CNV. Bio-protocol 9, (2019)의 논문의 프로토콜을 따라 수행하였다.
상기 실험에 따라 유전체 커버리지(genome coverage) 값을 측정한 결과, bMDA는 단일 염기서열수준의 유전체 분석이 가능할 수준으로 충분히 높은 유전체 커버리지인 86.7 ± 0.97%의 값을 보였으며, 이는 in-house MDA(84.7 ± 3.3% s.e.m.) 및 MDA kit(81.1%)와 비슷한 수준이다(도 11b 참조). 상기 유전체 커버리지(coverage breadth 혹은 genome coverage)란, NGS 분석결과를 인간 참조 유전체(reference human genome)에 정렬(alignment)하였을 때, 인간 유전체 전체를 몇 퍼센트나 커버할 수 있는지를 나타내는 값이다. bMDA가 보인 유전체 커버리지 값인 86.7%는 증폭 대상인 단일세포의 인간 유전체 정보의 86.7%를 획득할 수 있다는 의미이다. 이는, Tn5 기반의 단일세포 유전체 분석기술이 21.6 ± 3.7%의 유전체 커버리지를 보인 결과와는 대조되며, 즉 Tn5 기반의 유전체 분석 기술은 단일 염기서열 수준의 유전체 분석은 불가능하나 bMDA는 단일 염기서열 수준의 유전체 분석이 가능함을 의미한다.
또한, 커버된 인간 유전체 정보를 얼마나 깊이 있게 시퀀싱할 수 있는지 확인하기 위해 커버리지 깊이 평균(mean depth of coverage)을 측정한 결과, in-house MDA와 bMDA 모두 500x 이상의 커버리지 깊이 평균 값을 보이는 것을 확인하였다(도 11c 참조). 이는, bMDA 증폭 산물이 500x 이상의 라이브러리 복잡도를 가지고 있으며, 단일 염기서열 수준의 유전체 분석에 적합하다는 것을 의미한다. 여기서 커버리지 깊이 평균이란 NGS 시퀀싱 결과를 인간 참조 유전체에 정렬하였을 경우 인간 유전체 영역을 NGS 리드(read)가 얼마나 깊이 있게 커버하고 있는지를 나타내는 값이다. 예를 들어, 커버리지 깊이 평균이 500x 라는 것은, NGS 리드가 인간 유전체를 평균 500번 정도 커버하였다는 의미이다.
추가적으로, bMDA와 통상적인 MDA의 증폭 균일도(coverage uniformity)를 측정하기 위해 인간 유전체를 NGS 리드가 얼마나 균일하게 인간 유전체를 커버하고 있는지를 로렌츠 곡선(Lorenz curve)으로 도시하였다(도 11d 참조). 상기 로렌츠 곡선에서, 곡선 아래의 면적(AUC, Area Under Curve)값이 클수록 인간 유전체가 균일하게 커버된다는 의미이고, 이는 즉 bMDA 또는 MDA로 인한 증폭 편향(amplification bias)이 낮다는 의미이다. bMDA와 통상적인 MDA의 AUC값을 비교한 결과, 두 그룹간에는 통계적으로 중요한(statistically significant) 차이가 존재하지 않았다(도 11e 참조). 단일 세포를 분석한 경우 bMDA 그룹이 MDA 그룹에 비해 AUC가 다소 낮은 경향을 보였지만, 그 차이는 각 그룹 내에서의 차이(단일 세포와 단일 세포간의 차이)에 비해 작아서(one-way ANOVA, p=0.15) 유의미하지 않다고 판단하였다. 즉, bMDA는 바코드가 포함된 프라이머를 이용함으로 인해 증폭 균일도가 MDA에 비해 다소 낮을 수 있으나, 그 차이는 실질적으로 단일세포를 분석하는데 유의미하지 않으며, MDA와 bMDA가 모두 비슷한 기술적인 성능을 보인다고 결론내릴 수 있다.
bMDA는 또한 유전자 복제 수 변이(Copy number alteration, CNA)를 분석하는데 있어서도 통상적인 MDA와 비슷한 성능을 보였다(도 11f 참조). 66 pg의 gDNA에 대해서 bMDA를 수행한 결과, in-house MDA 또는 MDA kit와 마찬가지로 모두 정확한 CNA 결과를 제공하였다(Bulk 결과와 CNA 프로파일이 유사함). 하지만, 단일 세포를 표적으로 증폭을 수행하였을 경우에는, AUC가 높은 몇 샘플을 제외하고 대부분의 경우 정확한 CNA 분석을 제공하지 못하고 많은 위양성(false positive)과 위음성(false negative) 결과가 나타났다. 그러나, in-house MDA 또는 MDA 모두 유사한 결과를 보여주고 있다는 것에서 이는 bMDA의 변형된 프라이머로 인한 문제보다는 MDA 기반 기술이 가지고 있는 근본적인 한계로 해석된다. 실제로 단일 세포로부터 MDA분석을 수행할 경우, 약 5 내지 10%의 샘플만이 CNA 분석이 가능한 성공적인 증폭 결과를 보이고, 대부분의 샘플에서는 CNA 분석이 어려운 증폭결과를 보인다. 이는 반드시 극복되어야 할 MDA의 한계이며, 하기의 레이저 기반 세포 분리기술은 MDA가 가지고 있는 근원적 한계를 극복할 수 있는 한 방법이다.
마지막으로, bMDA 기술이 단일 염기변이(Single nucleotide variation, SNV) 분석에 적합한지 확인하기 위해 ADO(Allele Drop Out) 및 FPR(False positive rate) 값을 측정하였다. ADO 값은 단일 세포에 존재하는 두개의 대립 유전자(allele) 중 하나가 MDA 증폭 과정에서 소실되는 현상을 의미하며, FPR은 MDA 증폭 과정에서 원래는 존재하지 않았던 단일 염기변이가 DNA 중합효소의 오작동으로 인해 발생하는 현상의 비율을 의미한다.
측정 결과, 단일 염기변이 분석에 있어서 bMDA는 in-house MDA 또는 MDA kit와 비슷하거나 오히려 더 나은 성능을 보였다(도 11g 참조). 구체적으로, bMDA는 13.2 ± 1.6% (s.e.m.; n=3), MDA (in-house)는 19.0 ± 5.1% (s.e.m.; n=3), MDA kit는 27.7 ± 5.4% (s.e.m.; n=3)의 ADO 값을 보였다. 또한, bMDA는 1.1Х10-5 ± 0.35Х10-5 (s.e.m.; n=3) MDA (in-house)는 0.99Х10-5 ± 0.26Х10-5 (s.e.m.; n=3), MDA kit는 2.2Х10-5 ± 0.25Х10-5 (s.e.m.; n=3)의 FPR값을 보였다.
이와 같은 결과들을 토대로 상기 방법을 통해 수행된 bMDA가 통상적인 MDA 또는 상업화된 MDA 키트와 성능이 비슷하면서 더 나아가 병렬화가 가능하다는 것을 확인하였으며, 이로 인해 단일 염기서열 수준의 단일 세포 유전체 분석 비용 중 라이브러리 제작 과정의 비용을 상당히 낮춰 전체 비용이 3 내지 7배가량 감소할 수 있다(도 12 참조).
결론적으로, 병렬화가 가능한 bMDA를 위해서는, i) 10 μM 이상의 높은 프라이머 농도의 유지, ii) 바코드가 포함된 프라이머에 의한 증폭 효율 하락의 최소화, iii) 다량의 샘플을 바코딩할 수 있는 바코드 시퀀스의 충분한 길이 및 iv) 향후 시퀀싱하기에 충분한 양의 바코딩된 증폭 산물을 얻을 수 있도록 바코드가 포함된 프라이머의 양이 충분할 것의 4가지 요건이 만족되어야 하는데, 바코드가 포함된 프라이머와 랜덤 헥사머를 적절한 비율로 혼합하는 방법이 상기 4가지 요건이 모두 충족되는 최적의 해답임을 도출하였다.
⑥ 기타 bMDA 효율 상승 방법
DNA 중합효소와 바코드가 포함된 프라이머의 결합을 방지하기 위해 효소 또는 프라이머에 변형을 가함으로써 bMDA의 증폭 효율을 높일 수 있다. 예를 들어, 바코드 시퀀스 영역을 RNA로 제조하는 방법, 바코드 시퀀스 영역과 바인딩 영역 사이에 링커(linker)를 추가하는 방법, 프라이머에 입체 장애(steric hindrance)를 주기 위한 물리적 구조체를 결합하는 방법 또는 DNA 중합효소가 ssDNA에는 쉽게 결합하지 못하도록 조작하는 방법 등으로 bMDA의 증폭 효율을 높일 수 있다.
또한, bMDA 반응 시 바코드가 포함된 프라이머가 반응 용액에 점차 용해되도록 구성하여, 프라이머의 농도를 낮게 유지함으로써 bMDA의 증폭 효율을 높이는 방법도 가능하다.
(3) bMDA 증폭 산물 처리 단계
bMDA 증폭 산물의 NGS(Next generation sequencing)를 위해, 하기와 같은 처리 단계를 수행하였다.
1) 풀링(pooling) 단계
서로 다른 종류의 바코드가 포함된 프라이머 복수개를 이용하여 증폭된 bMDA 증폭 산물을 적절한 부피비로 혼합하였다(풀링). 구체적으로, 기술적 검증을 위한 실험으로 총 48개의 서로 다른 바코드가 포함된 프라이머를 디자인하였으며, MDA 증폭 산물을 동일한 부피비로 혼합하였다.
2) DNA 1차 정제 단계
상기 혼합물에서 DNA를 정제하기 위해 SPRI(Solid phase Reversible immobilization) 기법을 이용하였다. 구체적으로, 샘플 부피를 1x 라고 하였을 때, 0.8x 샘플 부피의 Ampure 자성 비드를 첨가하고 프로토콜에 따라 DNA를 정제하였고, 정제된 DNA를 0.4x 부피의 물에 용출(elute)하였다. 용출액은 1.5 시간 동안 20,000 g 로 원심분리하여 비드를 용출액으로부터 분리하였다.
3) 단편화(fragmentation) 단계
정제된 DNA를 음파처리(sonication) 또는 효소(DNA fragmentase)를 이용하여 50 bp 내지 10 kb의 적절한 길이로 단편화하였다. 상기 음파처리는 S220 Focused-ultrasonicator(Covaris) 장비를 이용하여 피크(peak) DNA 길이가 200 bp가 되도록 단편화하였으며, 그 이후에 SPRI 비드를 이용하여 다시 한번 더 정제를 수행하였다.
4) 비오틴 포획 및 비오틴 산물 정제 단계
bMDA 증폭 산물에서 바코딩된 DNA 단편을 선별적으로 포획하기 위해, Dynabeads MyOne Streptavidin T1(Invitrogen) 비드를 프로토콜에 따라 사용하였다.
그러나, 스트렙트아비딘과 비오틴의 결합은 매우 강력해서, 정제 후 DNA를 분리하여 회수하는 과정에서 어려움이 있다. 통상적으로 스트렙트아비딘과 비오틴의 결합을 끊기 위해서는 높은 온도, 탈이온화(deionization), 산성 조건 또는 단백질 환원 등의 방법들이 주로 사용되는데, 이와 같은 방법을 이용할 경우, dsDNA가 변성(denaturation)되거나 수득률(recovery rate)이 매우 감소하는 문제가 발생한다. 특히, dsDNA가 변성되면 그 뒤에 진행될 라이브러리 제작 과정이 불가능해지는 문제가 발생하며, 수득률이 감소하는 경우에는 라이브러리 복잡도를 낮춰 단일 염기서열 수준의 단일 세포 유전체 분석을 어렵게 만들게 되는 문제가 발생한다.
상기 문제점을 해결하기 위하여, SDS(Sodium Dodecyl Sulfate)와 과량의 D-비오틴을 포함하는 용출 완충액(SDS 1%, Tris-HCl(pH 8.0) 10mM, EDTA 1mM 및 D-Biotin 0.73 mM)을 첨가하여 54℃에서 1시간 동안 인큐베이션 하였다.
상기 조건 하에서 비드로부터 DNA를 분리한 결과, dsDNA의 변성 없이 스트렙트아비딘 비드로부터 DNA가 성공적으로 회수될 수 있음을 확인하였다(표 2 참조).
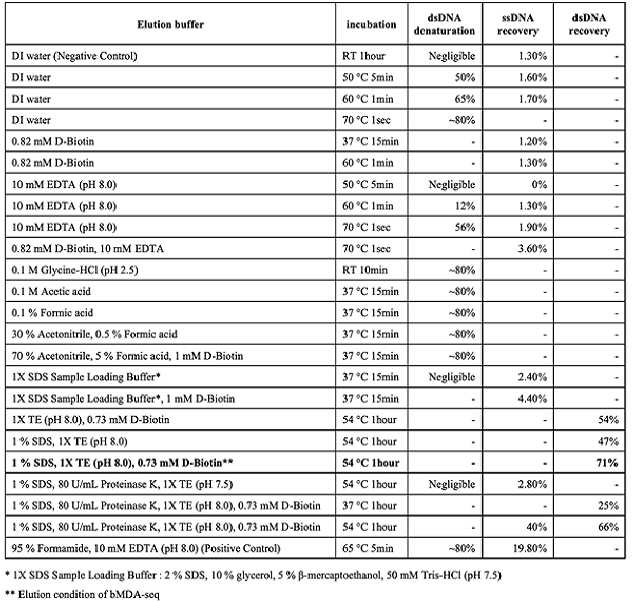
5) DNA 2차 정제 단계
상기 4)단계에서 수득한 DNA를 MinElute PCR Purification Kit (QIAGEN) 또는 Ampure SPRI bead를 이용하여 프로토콜에 따라 정제하였다.
6) 라이브러리 제작 단계
KAPA HyperPrep Kit(KK8500, KAPA Biosystems, Inc., USA)를 이용하여 라이브러리 제작을 수행하였다. 이때, 라이게이션(ligation)을 위한 인큐베이션을 8시간으로 연장한 것을 제외하고는, 제조사의 프로토콜에 따라 수행하였다.
상기 바코딩된 DNA의 5'말단에는 비오틴이 결합되어 있어, 라이브러리 제작 과정에서 어댑터 (Adapter)의 R1 가닥(strand)과 바코딩된 DNA의 비오틴이 포함된 DNA가닥간의 라이게이션(Ligation)이 방지된다(도 13a 참조). 이로 인해, 어댑터의 R2 가닥과 라이게이션된 DNA 분자만이 증폭되어 바코드가 Read 2에서 주로 발견되게 된다(도 13b 참조). 그러나, TA 또는 TC로 시작하는 바코드 시퀀스의 경우, Read 1에서 발견되는 비율이 다른 바코드 시퀀스에 비해 높게 나타났는데, 이는 바코드의 염기서열을 어떻게 디자인하는지가 bMDA 성능에 중요한 요소가 될 수 있음을 의미한다. 상기 실험에서 이용한 바코드가 포함된 프라이머 48개의 서열은 하기 표 3과 같다.
(표 3에서, /5Biosg/는 5’-비오틴 변형으로 포획 물질이고, NNNNNN은 6개의 랜덤 염기로 이루어진 바인딩 영역이며, *은 3’-5’핵산외부가수분해(exonuclease) 반응에 저항성이 있는 포스포로티올레이트(phosphorothiolate) 변형 뉴클레오티드이다.)
| 프라이머 서열 | |
| /5Biosg/ACTCTGNNNN*N*N(서열번호 8) | /5Biosg/CTTCCGNNNN*N*N(서열번호 9) |
| /5Biosg/CGAACTNNNN*N*N(서열번호 10) | /5Biosg/CCCGAANNNN*N*N(서열번호 11) |
| /5Biosg/CGGCTTNNNN*N*N(서열번호 12) | /5Biosg/CACGGCNNNN*N*N(서열번호 13) |
| /5Biosg/CTAGCCNNNN*N*N(서열번호 14) | /5Biosg/CGTGCANNNN*N*N(서열번호 15) |
| /5Biosg/CTATATNNNN*N*N(서열번호 16) | /5Biosg/CCCTTGNNNN*N*N(서열번호 17) |
| /5Biosg/CTTAGANNNN*N*N(서열번호 18) | /5Biosg/CTCATTNNNN*N*N(서열번호 19) |
| /5Biosg/AAACGANNNN*N*N(서열번호 20 | /5Biosg/ATCACCNNNN*N*N(서열번호 21) |
| /5Biosg/ATCCATNNNN*N*N(서열번호 22) | /5Biosg/CCAAAGNNNN*N*N(서열번호 23) |
| /5Biosg/ACATCGNNNN*N*N(서열번호 24) | /5Biosg/ACTAACNNNN*N*N(서열번호 25) |
| /5Biosg/CCTGTTNNNN*N*N(서열번호 26) | /5Biosg/AGCTCANNNN*N*N(서열번호 27) |
| /5Biosg/AATTGTNNNN*N*N(서열번호 28) | /5Biosg/ACCAGANNNN*N*N(서열번호 29) |
| /5Biosg/GTAGTTNNNN*N*N(서열번호 30) | /5Biosg/TGAAACNNNN*N*N(서열번호 31) |
| /5Biosg/TTATTGNNNN*N*N(서열번호 32) | /5Biosg/AAAGCTNNNN*N*N(서열번호 33) |
| /5Biosg/TATAAGNNNN*N*N(서열번호 34) | /5Biosg/TTACGTNNNN*N*N(서열번호 35) |
| /5Biosg/AATACANNNN*N*N(서열번호 36) | /5Biosg/ATTTAGNNNN*N*N(서열번호 37) |
| /5Biosg/TTCTACNNNN*N*N(서열번호 38) | /5Biosg/AAGTAANNNN*N*N(서열번호 39) |
| /5Biosg/TGACTANNNN*N*N(서열번호 40) | /5Biosg/TTAACANNNN*N*N(서열번호 41) |
| /5Biosg/TTGAATNNNN*N*N(서열번호 42) | /5Biosg/ACGGTANNNN*N*N(서열번호 43) |
| /5Biosg/TTTGCTNNNN*N*N(서열번호 44) | /5Biosg/TTTCAANNNN*N*N(서열번호 45) |
| /5Biosg/TACATANNNN*N*N(서열번호 46) | /5Biosg/TCTAGTNNNN*N*N(서열번호 47) |
| /5Biosg/TTGTGANNNN*N*N(서열번호 48) | /5Biosg/TCATGCNNNN*N*N(서열번호 49) |
| /5Biosg/TAAGTCNNNN*N*N(서열번호 50) | /5Biosg/TAACCGNNNN*N*N(서열번호 51) |
| /5Biosg/TCAGATNNNN*N*N(서열번호 52) | /5Biosg/TCCCAGNNNN*N*N(서열번호 53) |
| /5Biosg/TCGACGNNNN*N*N(서열번호 54) | /5Biosg/TAGAGCNNNN*N*N(서열번호 55) |
라이브러리 제작 이후, 일루미나(Illumina) 플랫폼을 사용하여 NGS를 수행한 결과, 예상한 바와 같이 대부분의 NGS Read 2에서(약 82%) 바코드 시퀀스가 발견되었다(도 14 참조). 즉, NGS 결과 중 Read 2의 첫 번째 6개의 염기는 바코드가 포함된 프라이머 내에 존재하는 바코드 시퀀스로 볼 수 있으므로, 이 정보를 이용하여 데이터를 역병렬화(demultiplex)하여 NGS 시퀀싱 결과가 어느 세포로부터 유래한 데이터인지를 분리할 수 있다(도 2c 참조).
한편, 여러 바코드 정보가 포함된 bMDA 증폭 산물을 풀링하여 한 번에 진행하는 일련의 실험 과정에서 바코드가 뒤바뀌는 바코드 스와핑(barcode swapping) 현상이 발생하는지 확인하기 위해, 총 48개의 바코드가 포함된 프라이머 중 1개의 프라이머는 쥐로부터 유래한 gDNA(NIH3T3 cell, cat. no. 21658, Korean Cell Line Bank)를 사용하였고, 나머지 47개의 프라이머는 사람으로부터 유래한 HL-60 세포의 gDNA를 사용하여 bMDA를 수행하였다.
그 결과, 쥐의 DNA를 템플릿(template)으로 사용한 경우에는 대부분의 NGS 리드가 쥐의 참조 유전체(mouse reference genome)에 매핑(mapping)되고 사람의 DNA를 템플릿으로 사용한 경우에는 대부분의 NGS 리드가 사람의 참조 유전체에 매핑됨을 확인하였다(도 15 참조). 이와 반대의 경우는 바코드 스와핑 현상이 발생한 것으로 생각할 수 있는데, 그 수치가 일루미나 NGS 플랫폼에 일반적으로 존재하는 인덱스 뒤바뀜(index hopping) 현상의 비율보다 작거나 같았다. 즉, bMDA-seq 과정을 거치더라도 바코드 스와핑 현상이 무시할 수 있을 정도로 낮게 측정되었다.
실시예 2. 바코드 편향(barcode bias) 현상의 확인 및 해결 방법 제시
(1) 바코드 편향 현상의 확인
바코드가 포함된 프라이머를 이용하여 bMDA를 수행할 경우, 크게 세 가지 종류의 바코드 편향 현상이 나타남을 확인하였다.
1) bMDA 증폭 편향
bMDA 증폭 편향은 바코드가 포함된 프라이머의 바코드 시퀀스가 표적 샘플의 시퀀스와 상보적(complement)일 경우, 해당 게놈 영역 주변에서 MDA 반응이 보다 활발하게 발생하는 현상을 의미한다.
2) bMDA 증폭 산물에 포함된 바코딩 DNA 양의 편향
이는, 바코드 시퀀스에 따라 바코드가 포함된 프라이머와 DNA 중합효소간 상호작용의 세기나, bMDA 반응 도중 표적 샘플에 프라이머가 혼성화될 확률이 달라질 수 있고, 이에 따라 bMDA 증폭 산물에서 바코딩된 DNA의 양이 바코드별로 서로 다른 현상을 의미한다.
서로 다른 16개의 바코드 시퀀스로 바코드가 포함된 프라이머를 디자인하여 bMDA를 수행한 뒤, bMDA 직후의 dsDNA의 농도와 비오틴 정제 이후의 dsDNA의 농도의 비를 측정해본 결과(Barcoded product Mass), 바코드마다 bMDA 증폭 산물에 포함된 바코딩 DNA의 양에 편향이 있음을 확인하였다(도 16 참조). 상기 바코드가 포함된 프라이머 16개의 서열은 하기 표 4와 같다.
(표 4에서, /5Biosg/는 5’-비오틴 변형으로 포획 물질이고, NNNNNN은 6개의 랜덤 염기로 이루어진 바인딩 영역이며, *은 3’-5’핵산외부가수분해(exonuclease) 반응에 저항성이 있는 포스포로티올레이트(phosphorothiolate) 변형 뉴클레오티드이다.)
| 프라이머 서열 | |
| /5Biosg/TAGTCNNNN*N*N(서열번호 56) | /5Biosg/AGAAGNNNN*N*N(서열번호 57) |
| /5Biosg/ACCGANNNN*N*N(서열번호 58) | /5Biosg/TGGCTNNNN*N*N(서열번호 59) |
| /5Biosg/AGCTANNNN*N*N(서열번호 60) | /5Biosg/GCTCTNNNN*N*N(서열번호 61) |
| /5Biosg/GTGACNNNN*N*N(서열번호 62) | /5Biosg/ACAGTNNNN*N*N(서열번호 63) |
| /5Biosg/ATGCCNNNN*N*N(서열번호 64) | /5Biosg/TACTGNNNN*N*N(서열번호 65) |
| /5Biosg/TCAAGNNNN*N*N(서열번호 66) | /5Biosg/CGTTANNNN*N*N(서열번호 67) |
| /5Biosg/CCTGGNNNN*N*N(서열번호 68) | /5Biosg/TTACCNNNN*N*N(서열번호 69) |
| /5Biosg/GATGTNNNN*N*N(서열번호 70) | /5Biosg/CTACANNNN*N*N(서열번호 71) |
3) NGS 라이브러리 변환 효율 편향
이는, 바코딩된 DNA만 선별적으로 포획한 뒤 NGS 라이브러리 제작 과정이 수행될 때, 바코드 시퀀스에 따라 엔드 리페어(end-repair), A-테일링(A-tailing) 또는 라이게이션의 효율이 달라 라이브러리 변환 수율 (Library Conversion Yield) 이 바코드별로 서로 다른 현상을 의미한다. (도 16 참조).
(2) 바코드 편향 현상의 해결 방법 제시
바코드 편향 현상을 고려하지 않고 샘플들을 풀링할 경우, 바코드 시퀀스에 따라 같은 부피에 존재하는 바코딩된 DNA의 양이 서로 달라지게 되며, 이를 시퀀싱하게 되면 리드(read) 수가 샘플마다 균일하지 않은 문제점이 발생한다. 이를 해결하기 위해 하기와 같은 방법들을 고려할 수 있다.
1) 바코드가 포함된 프라이머의 비율을 조절하는 방법
바코드가 포함된 프라이머와 랜덤 헥사머를 특정 비율로 혼합할 때, bMDA 증폭 산물에 포함된 바코딩된 DNA의 양 또는 NGS 라이브러리 변환 효율을 미리 고려하여, 바코드가 포함된 프라이머의 비율을 조절하여 bMDA를 수행함으로써 바코드 편향 현상을 해결할 수 있다.
bMDA 반응 용액 중 바코드가 포함된 프라이머의 비율(Relative proportion of barcoded primer)이 증가할수록 bMDA-seq 라이브러리 제작 후 NGS 시퀀싱 하였을 때의 NGS 리드 수(Normalized NGS read counts)가 상대적으로 증가함을 실험으로 확인하였는데(도 17 참조), 이를 통해 바코드가 포함된 프라이머의 비율이 증가할수록 bMDA 증폭 산물 내 바코딩 DNA 양을 증가시킬 수 있음을 검증하였다. 따라서, 바코드가 포함된 프라이머의 비율을 각각 적절하게 조절하여 얻은 증폭 산물을 모두 같은 부피로 혼합하면, 바코딩된 DNA의 양이 바코드가 포함된 프라이머의 종류별로 거의 일정하거나, NGS 리드 수가 바코드가 포함된 프라이머 종류별로 거의 일정하도록 만들 수 있어, 바코드 편향 현상을 해결할 수 있다.
상기와 같은 방법의 효과를 검증하기 위해, 바코드가 포함된 프라이머의 비율을 모두 일정하게 설정한 경우와, 각각 특정 비율로 다르게 설정한 경우에서의 바코드 편향 정도를 측정하였다. 구체적으로, 66 pg의 HL-60 gDNA를 48개의 서로 다른 바코드가 포함된 프라이머로 bMDA를 수행하였으며, 바코드가 포함된 프라이머의 비율이 일정한 실험군에서는 2%의 바코드가 포함된 프라이머를 이용하였고, 바코드가 포함된 프라이머의 비율을 적절하게 조절한 실험군에서는 하기 표 5와 같은 비율을 이용하였다.
(표 5에서, /5Biosg/는 5’-비오틴 변형으로 포획 물질이고, NNNNNN은 6개의 랜덤 염기로 이루어진 바인딩 영역이며, *은 3’-5’핵산외부가수분해(exonuclease) 반응에 저항성이 있는 포스포로티올레이트(phosphorothiolate) 변형 뉴클레오티드이다.)
| 프라이머 서열 | 비율 | 프라이머 서열 | 비율 |
| /5Biosg/ACTCTGNNNN*N*N(서열번호 8) | 1.30% | /5Biosg/CTTCCGNNNN*N*N(서열번호 9) | 1.76% |
| /5Biosg/CGAACTNNNN*N*N(서열번호 10) | 1.18% | /5Biosg/CCCGAANNNN*N*N(서열번호 11) | 1.32% |
| /5Biosg/CGGCTTNNNN*N*N(서열번호 12) | 0.98% | /5Biosg/CACGGCNNNN*N*N(서열번호 13) | 1.02% |
| /5Biosg/CTAGCCNNNN*N*N(서열번호 14) | 1.30% | /5Biosg/CGTGCANNNN*N*N(서열번호 15) | 0.92% |
| /5Biosg/CTATATNNNN*N*N(서열번호 16) | 1.44% | /5Biosg/CCCTTGNNNN*N*N(서열번호 17) | 1.62% |
| /5Biosg/CTTAGANNNN*N*N(서열번호 18) | 0.94% | /5Biosg/CTCATTNNNN*N*N(서열번호 19) | 1.54% |
| /5Biosg/AAACGANNNN*N*N(서열번호 20 | 1.06% | /5Biosg/ATCACCNNNN*N*N(서열번호 21) | 2.24% |
| /5Biosg/ATCCATNNNN*N*N(서열번호 22) | 1.92% | /5Biosg/CCAAAGNNNN*N*N(서열번호 23) | 0.82% |
| /5Biosg/ACATCGNNNN*N*N(서열번호 24) | 1.84% | /5Biosg/ACTAACNNNN*N*N(서열번호 25) | 1.68% |
| /5Biosg/CCTGTTNNNN*N*N(서열번호 26) | 1.02% | /5Biosg/AGCTCANNNN*N*N(서열번호 27) | 1.22% |
| /5Biosg/AATTGTNNNN*N*N(서열번호 28) | 0.92% | /5Biosg/ACCAGANNNN*N*N(서열번호 29) | 0.98% |
| /5Biosg/GTAGTTNNNN*N*N(서열번호 30) | 1.48% | /5Biosg/TGAAACNNNN*N*N(서열번호 31) | 1.02% |
| /5Biosg/TTATTGNNNN*N*N(서열번호 32) | 1.12% | /5Biosg/AAAGCTNNNN*N*N(서열번호 33) | 1.00% |
| /5Biosg/TATAAGNNNN*N*N(서열번호 34) | 0.88% | /5Biosg/TTACGTNNNN*N*N(서열번호 35) | 1.32% |
| /5Biosg/AATACANNNN*N*N(서열번호 36) | 1.26% | /5Biosg/ATTTAGNNNN*N*N(서열번호 37) | 1.60% |
| /5Biosg/TTCTACNNNN*N*N(서열번호 38) | 2.44% | /5Biosg/AAGTAANNNN*N*N(서열번호 39) | 0.90% |
| /5Biosg/TGACTANNNN*N*N(서열번호 40) | 1.34% | /5Biosg/TTAACANNNN*N*N(서열번호 41) | 1.46% |
| /5Biosg/TTGAATNNNN*N*N(서열번호 42) | 0.72% | /5Biosg/ACGGTANNNN*N*N(서열번호 43) | 1.32% |
| /5Biosg/TTTGCTNNNN*N*N(서열번호 44) | 1.04% | /5Biosg/TTTCAANNNN*N*N(서열번호 45) | 2.10% |
| /5Biosg/TACATANNNN*N*N(서열번호 46) | 1.06% | /5Biosg/TCTAGTNNNN*N*N(서열번호 47) | 1.62% |
| /5Biosg/TTGTGANNNN*N*N(서열번호 48) | 0.78% | /5Biosg/TCATGCNNNN*N*N(서열번호 49) | 1.18% |
| /5Biosg/TAAGTCNNNN*N*N(서열번호 50) | 0.86% | /5Biosg/TAACCGNNNN*N*N(서열번호 51) | 1.60% |
| /5Biosg/TCAGATNNNN*N*N(서열번호 52) | 1.00% | /5Biosg/TCCCAGNNNN*N*N(서열번호 53) | 1.48% |
| /5Biosg/TCGACGNNNN*N*N(서열번호 54) | 1.64% | /5Biosg/TAGAGCNNNN*N*N(서열번호 55) | 0.54% |
그 결과, 바코드가 포함된 프라이머의 비율이 모두 일정한 경우 (2 %), 약 29.9%(n=3)의 NGS 리드 수의 변동계수(CV, Coefficient of Variation) 값이 나타난 반면(Before correction), 바코드가 포함된 프라이머의 비율을 각각 특정 비율로 다르게 설정한 경우(After correction), 약 7.68%(n=3)의 CV 값이 나타났다(도 18 참조). 통상적인 NGS 어플리케이션에서 서로 다른 인덱스(index)로 제작된 라이브러리들의 농도와 길이를 기준으로, 서로 다른 라이브러리 간의 NGS 리드(read) 수를 균일하게 조정하는 일루미나 라이브러리 풀링 방식의 경우, 약 8.53%의 CV 값이 나타나는 것과 비교하였을 때, 바코드가 포함된 프라이머 비율을 조절하는 방식으로 바코드 편향 현상을 효과적으로 제거할 수 있었다.
또한, 이와 같은 방식으로 바코드가 포함된 프라이머의 비율이 조정 되어있는 시약을 미리 준비해두면, 서로 다른 바코드를 이용한 bMDA 증폭산물들을 모두 같은 부피로 풀링한 뒤에 NGS시퀀싱을 수행하더라도, NGS 시퀀싱 리드 수가 균일하도록 구성할 수 있어 사용자 편의성을 최대화할 수 있다. 즉, 사용자가 바코드 서열에 따른 차이를 고려하지 않고 일괄적인 실험 프로세스를 진행할 수 있다.
바코드가 포함된 프라이머의 비율을 효과적으로 조절하기 위해서는, bMDA 증폭 산물에 포함된 바코딩 DNA 양 또는 NGS 라이브러리 변환 효율을 실험을 통해 미리 데이터로 확보하거나, 이를 예측하는 알고리즘을 통해 추정한 값을 기준으로 삼을 수 있다. 또한, bMDA 증폭 산물에 포함된 바코딩 DNA 양이 샘플별로 거의 일정하도록 프라이머의 비율을 조절할 수도 있고, bMDA 증폭 산물에 포함된 바코딩 DNA 양 및 NGS 라이브러리 변환 효율을 모두 고려하여, NGS 리드 수가 샘플별로 거의 일정하도록 프라이머 비율을 조절할 수도 있다.
또한, 바코드가 포함된 프라이머의 비율을 조절하는 방법으로 바코드 편향을 교정(correction)할 경우, bMDA의 인풋(input)으로 사용되는 템플릿(template)의 양이 증가하거나 감소할 때, NGS 리드 수에 차이가 발생하는지를 확인하기 위한 실험을 수행하였다. 구체적으로, 상기 표 3의 48개의 서로 다른 바코드가 포함된 프라이머 중 5 내지 16개의 프라이머를 선택하고, 서로 다른 양의 HL-60 gDNA를 템플릿으로 하여 bMDA를 수행한 뒤 풀링하여 bMDA-seq을 수행하였으며, 풀링된 한 그룹의 bMDA-seq 산물 내에서의 바코드 간 NGS 리드 수의 변동계수(CV, Coefficient of Variation) 값을 측정하였다.
실험 결과, 바코드가 포함된 프라이머의 비율을 조절하여 바코드 표현을 교정할 때, 인풋 템플릿의 양이 증가하거나 감소하여도 NGS 리드 수에 큰 차이가 발생하지 않음을 확인하였다(도 19 참조). 사용자의 입장에서 샘플 별로 비슷한 수준의 NGS 리드를 확보하고자 하는 경우가 훨씬 흔하기 때문에, 증폭 산물을 같은 부피로 혼합하여 샘플 별로 비슷한 수의 NGS 리드를 확보할 수 있다는 것은 큰 장점이 될 수 있다.
상기와 같은 실험들을 통해, 바코드가 포함된 프라이머의 비율을 특정 기준에 따라 적절하게 조절하는 방법으로, 바코드 편향 현상을 해결하여 CV를 크게 낮출 수 있고, 증폭 산물을 같은 부피로 혼합하여 샘플 별로(또는 단일 세포 별로) 비슷한 NGS 리드 수를 확보할 수 있음을 확인하였다.
2) 기타 바코드 편향 현상 해결 방법
바코드 시퀀스에 따른 bMDA 편향을 줄이기 위한 방법으로, 샘플 내에 많이 존재하지 않는 바코드 시퀀스를 선정하거나, bMDA 편향에 크게 영향을 주지 않는 바코드 시퀀스를 선정할 수 있다. 또한, 바코드가 포함된 프라이머 내 바코드 시퀀스 영역만을 미리 dsDNA 형태로 제조함으로써 bMDA 편향을 최소화할 수 있다.
또 다른 방법으로는, 바코드 시퀀스에 따른 bMDA 증폭 산물에 포함된 바코딩 DNA 양 또는 NGS 라이브러리 변환 효율을 실험을 통해 미리 데이터로 확보하거나, 이를 예측하는 알고리즘을 통해 샘플 풀링 시 샘플별로 원하는 비율을 가지도록 서로 다른 부피로 풀링하는 방법이 있을 수 있다. 이때, bMDA 증폭 산물에 포함된 바코딩 DNA 양이 샘플별로 거의 일정하도록 조절하여 풀링할 수도 있고, bMDA 증폭 산물에 포함된 바코딩 DNA 양 및 NGS 라이브러리 변환 효율을 고려하여, NGS 리드 수가 샘플별로 거의 일정하도록 조절하여 풀링할 수도 있다.
실시예 3. 레이저 기반 세포 분리 방식의 bMDA 품질 향상 효과 확인
기존 단일 세포 분리 방식으로 유체를 이용한 방식(fluidic isolation)이 주로 사용되었는데, 상기 방식은 MDA 증폭 편향으로 인해 증폭 품질이 떨어지는 문제점이 존재한다(도 20 참조). 그 이유는, 앞서도 언급하였듯이, MDA증폭 기술의 한계로 인하여 단일 세포를 템플릿으로 하여 MDA를 수행할 경우, CNA 분석이 가능한 수준으로 증폭 균일도가 높은 세포가 전체의 5 내지 10%밖에 존재하지 않고, 대부분의 세포는 아주 심각한 증폭 편향을 보이기 때문이다(도 11f 참조).
상기와 같은 문제점을 해결하기 위해, 레이저를 이용하여 단일 세포가 아닌 유전체 정보가 비슷한 세포들을 여러 개 분리하여 한 번에 bMDA를 수행하는 방식을 사용할 수 있다. 레이저를 이용한 세포 분리는 상기 언급된 ITO가 코팅된 슬라이드 글라스를 현미경으로 관찰한 뒤, 분리하고자 하는 세포가 존재하는 영역에 적외선 레이저를 조사함으로써 이루어진다. 암조직을 일례로 들면, 공간적으로 인접한 암세포들은 공간적으로 멀리 떨어진 세포에 비해 상대적으로 비슷한(homogeneous) 유전체 정보를 갖고 있다. 따라서 단일 세포가 아니라 물리적으로 인접한 세포를 함께 분리하여 여러 개의 세포로부터 bMDA를 수행함으로써, bMDA 증폭 편향 문제를 혁신적으로 개선할 수 있다 (도 20 참조).
이와 같이 레이저를 이용하여 원하는 영역(Region of interest)을 분리하는 방식을 이용할 경우, 기존 단일 세포 분리 방식인 유체를 이용한 방식에 비해 성공률(yield, success rate)을 100%로 높일 수 있다(도 21 참조). 그 이유는, 유전적으로 비슷한 세포를 10개 정도만 보아도 CNA 분석이 가능한 수준으로 bMDA 증폭 편향을 낮출 수 있기 때문이다(도 11f 참조). 또한, bMDA가 MDA에 비해 처리량(throughput)이 약 5배가량 높기 때문에(도 21 참조), bMDA 및 레이저 기반 세포 분리 방식을 이용할 경우, MDA 및 유체를 이용한 세포 분리 방식에 비하여 총 15배 정도의 비용 하락 효과가 나타남을 확인하였다(도 22 참조).
상기 결과를 통해, 레이저 기반 세포 분리 방식이 물리적으로 인접한 세포들을 함께 분리하여 분석함으로써 bMDA의 증폭 편향 문제를 최소화하고, 높은 품질의 증폭 산물을 제공할 수 있음을 확인하였다. 이는, 물리적으로 인접한 세포일수록 물리적으로 거리가 먼 세포에 비해 유전적 유사성이 높으며, bMDA를 위해 사용되는 인풋 gDNA 양이 많아질수록 증폭 편향이 줄어들고 증폭 품질이 높아지기 때문이다. 즉, 상기 레이저 기반 세포 분리 방식은, 단일 세포를 분리하는 것은 아니나, 물리적으로 인접한 세포 간 유사성을 활용하여 여러 세포를 분리하면서 단일세포를 분석하는 것과 유사한 효과를 발휘할 수 있으며, 이를 통해 단일 세포 MDA에서 문제되었던 품질 관리(quality control, QC)를 통과하지 못하는 증폭 산물의 비율을 90 내지 95%에서 0%로 줄이고, 단일 세포 MDA의 비용 비효율성 문제를 경감할 수 있다.
또한, 상기 레이저 기반 세포 분리 방식은 물리적으로 인접한 세포를 분리하는 접근법 뿐 아니라 현미경 이미지 관찰을 통해 획득할 수 있는 크기, 모양, 색깔, 형광 신호 등의 여러 정보들을 기준으로 세포들을 분류 (classification)한 뒤, 그 중 분석하고자 하는 분류에 포함된 세포들을 한 번에 분리하는 접근법으로도 이용할 수 있다. 일례로, 혈액 도말(blood smear) 슬라이드를 현미경으로 관찰하여 분리하고자 하는 특정 종류의 백혈구를 분리하거나, FISH(fluorescence in situ hybridization) 염색 슬라이드를 형광 현미경으로 관찰하여 분리하고자 하는 신호를 보이는 세포를 분리하여 모은 뒤 여러 세포에 대해 한 번에 bMDA를 수행할 수 있다.
결론적으로, 상기 레이저 기반 세포 분리 방식의 증폭 품질 향상이 bMDA의 처리량 향상 및 가격 하락과 함께 시너지를 이루어 사용자에게 단일 세포 유전체 분석의 낮은 품질 및 낮은 병렬성의 문제를 해결하고, 증폭 편향 문제를 감소시키는 매우 효율적인 방식임을 검증하였다.
<110> Seoul National University R&DB Foundation
<120> Method for multiplex multiple displacement amplification
<130> SDP2022-1051
<150> KR 10-2021-0063272
<151> 2021-06-21
<160> 71
<170> KoPatentIn 3.0
<210> 1
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> R15 sequence
<400> 1
acgctcttcc gatct 15
<210> 2
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> 29-mer barcode primer
<220>
<221> misc_feature
<222> (27)..(29)
<223> Phosphorothiolate modification
<400> 2
acgctcttcc gatctgccaa gacnnnnnn 29
<210> 3
<211> 14
<212> DNA
<213> Artificial Sequence
<220>
<223> 14-mer barcode primer
<220>
<221> misc_feature
<222> (12)..(14)
<223> Phosphorothiolate modification
<400> 3
gactgtgtnn nnnn 14
<210> 4
<211> 13
<212> DNA
<213> Artificial Sequence
<220>
<223> 13-mer barcode primer
<220>
<221> misc_feature
<222> (11)..(13)
<223> Phosphorothiolate modification
<400> 4
actgtgtnnn nnn 13
<210> 5
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 5
ctgtgtnnnn nn 12
<210> 6
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 6
tgtgtnnnnn n 11
<210> 7
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> 10-mer barcode primer
<220>
<221> misc_feature
<222> (8)..(10)
<223> Phosphorothiolate modification
<400> 7
gtgtnnnnnn 10
<210> 8
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 8
actctgnnnn nn 12
<210> 9
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 9
cttccgnnnn nn 12
<210> 10
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 10
cgaactnnnn nn 12
<210> 11
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 11
cccgaannnn nn 12
<210> 12
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 12
cggcttnnnn nn 12
<210> 13
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 13
cacggcnnnn nn 12
<210> 14
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 14
ctagccnnnn nn 12
<210> 15
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 15
cgtgcannnn nn 12
<210> 16
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 16
ctatatnnnn nn 12
<210> 17
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 17
cccttgnnnn nn 12
<210> 18
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 18
cttagannnn nn 12
<210> 19
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 19
ctcattnnnn nn 12
<210> 20
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 20
aaacgannnn nn 12
<210> 21
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 21
atcaccnnnn nn 12
<210> 22
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 22
atccatnnnn nn 12
<210> 23
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 23
ccaaagnnnn nn 12
<210> 24
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 24
acatcgnnnn nn 12
<210> 25
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 25
actaacnnnn nn 12
<210> 26
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 26
cctgttnnnn nn 12
<210> 27
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 27
agctcannnn nn 12
<210> 28
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 28
aattgtnnnn nn 12
<210> 29
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 29
accagannnn nn 12
<210> 30
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 30
gtagttnnnn nn 12
<210> 31
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 31
tgaaacnnnn nn 12
<210> 32
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 32
ttattgnnnn nn 12
<210> 33
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 33
aaagctnnnn nn 12
<210> 34
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 34
tataagnnnn nn 12
<210> 35
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 35
ttacgtnnnn nn 12
<210> 36
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 36
aatacannnn nn 12
<210> 37
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 37
atttagnnnn nn 12
<210> 38
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 38
ttctacnnnn nn 12
<210> 39
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 39
aagtaannnn nn 12
<210> 40
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 40
tgactannnn nn 12
<210> 41
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 41
ttaacannnn nn 12
<210> 42
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 42
ttgaatnnnn nn 12
<210> 43
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 43
acggtannnn nn 12
<210> 44
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 44
tttgctnnnn nn 12
<210> 45
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 45
tttcaannnn nn 12
<210> 46
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 46
tacatannnn nn 12
<210> 47
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 47
tctagtnnnn nn 12
<210> 48
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 48
ttgtgannnn nn 12
<210> 49
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 49
tcatgcnnnn nn 12
<210> 50
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 50
taagtcnnnn nn 12
<210> 51
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 51
taaccgnnnn nn 12
<210> 52
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 52
tcagatnnnn nn 12
<210> 53
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 53
tcccagnnnn nn 12
<210> 54
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 54
tcgacgnnnn nn 12
<210> 55
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> 12-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (10)..(12)
<223> Phosphorothiolate modification
<400> 55
tagagcnnnn nn 12
<210> 56
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 56
tagtcnnnnn n 11
<210> 57
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 57
agaagnnnnn n 11
<210> 58
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 58
accgannnnn n 11
<210> 59
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 59
tggctnnnnn n 11
<210> 60
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 60
agctannnnn n 11
<210> 61
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 61
gctctnnnnn n 11
<210> 62
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 62
gtgacnnnnn n 11
<210> 63
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 63
acagtnnnnn n 11
<210> 64
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 64
atgccnnnnn n 11
<210> 65
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 65
tactgnnnnn n 11
<210> 66
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 66
tcaagnnnnn n 11
<210> 67
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 67
cgttannnnn n 11
<210> 68
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 68
cctggnnnnn n 11
<210> 69
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 69
ttaccnnnnn n 11
<210> 70
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 70
gatgtnnnnn n 11
<210> 71
<211> 11
<212> DNA
<213> Artificial Sequence
<220>
<223> 11-mer barcode primer
<220>
<221> misc_feature
<222> (1)
<223> 5' biotin modification
<220>
<221> misc_feature
<222> (9)..(11)
<223> Phosphorothiolate modification
<400> 71
ctacannnnn n 11
Claims (32)
- 제1 프라이머, 제2 프라이머 및 핵산 중합효소를 표적 핵산이 포함된 샘플에 첨가하여 핵산들을 증폭시키는 단계를 포함하는 핵산 증폭 방법으로서,
상기 제1 프라이머는 상기 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 상기 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 방법.
- 청구항 1에 있어서, 상기 제1 염기 서열 또는 제3 염기 서열의 염기 개수는 각각 독립적으로 4 내지 10 중 선택되는 하나의 정수인, 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 제1 염기 서열 또는 제3 염기 서열을 이루는 염기들은 A(아데닌), T(티민), G(구아닌), C(사이토신) 및 이들의 변형물로 이루어진 군에서 선택되는 염기인 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 제2 염기 서열의 염기 개수는 4 내지 35 중 선택되는 하나의 정수인, 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 10몰%로 첨가되는 것인 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 제1 프라이머는 상기 핵산 중합효소와 제1 프라이머의 친화도를 저해하는 물질이 결합된 것인, 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 제1 프라이머 또는 제2 프라이머는 핵산 분해효소(nuclease)에 저항성이 있는 염기가 결합된 것인 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 제1 프라이머로 증폭된 핵산을 선별하는 단계를 더 포함하는, 핵산 증폭 방법.
- 청구항 8에 있어서, 상기 제1 프라이머는 포획 물질과 결합된 것인, 핵산 증폭 방법.
- 청구항 9에 있어서, 상기 포획 물질은 비오틴(biotin), D-비오틴, 비오틴 dT, 비오틴-TEG, 듀얼 비오틴, PC-비오틴, 아크릴(acrylic), 티올(thiol), 디티올(dithiol), 아민기(amine group), 아크릴기(acrylic group), NHS 에스터, 아자이드(azide), 헥시닐(hexynyl), 옥타디닐(octadiynyl) dU, 히스 태그(his tag), 단백질 태그, polyA(폴리 아데닌), NGS 시퀀싱 어댑터를 이루는 염기 서열 중 10개 이상의 염기 서열 및 10개 이상의 염기 서열로 이루어진 폴리뉴클레오티드로 이루어진 군에서 선택되는 하나 이상인 것인, 핵산 증폭 방법.
- 청구항 8에 있어서, 상기 선별은 아비딘(avidin), 뉴트라비딘(neutravidin), 스트렙트아비딘(streptavidin), 카복실기(carboxylic group), 티올(thiol), 알카인(alkyne), 디곡시제닌(digoxigenin), 아자이드(azide) 및 헥시닐(hexynyl)로 이루어진 군에서 선택되는 하나 이상의 물질을 이용한 방법 또는 PCR(Polymerase Chain reaction) 방법으로 수행되는 것인, 핵산 증폭 방법.
- 청구항 8에 있어서, 상기 선별은 이중 가닥 또는 단일 가닥의 핵산을 선별하는 것인, 핵산 증폭 방법.
- 청구항 1에 있어서,
상기 샘플로부터 증폭된 핵산을 정제(purification)하는 단계;
정제된 핵산을 단편화(fragmentation)하는 단계; 및
단편화된 핵산으로 NGS(Next Generation Sequencing) 라이브러리를 제조하는 단계를 더 포함하는, 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 샘플은 공간분해능이 있는 분리(spatially resolved) 기술, 유전영동 디지털 분류(dielectrophoretic digital sorting), 형광 활성 세포 분류(fluorescence activated cell sorting, FACS), 유체역학 트랩(hydrodynamic traps), 액체 방울(droplet), 마이크로웰(microwell), 미세 유체역학(microfluidics), 미세조작(micromanipulation), Raman 핀셋(Raman tweezers) 및 CellRaft로 이루어진 군에서 선택되는 하나 이상의 기술로 분리된 것인, 핵산 증폭 방법.
- 청구항 14에 있어서, 상기 공간분해능이 있는 분리 기술은 LCM(Laser capture microdissectio), LMD(laser microdisection), LPC(Laser pressure catapulting) 및 PHLI-seq(phenotype-based high-throughput laser-aided isolation and sequencing)으로 이루어진 군에서 선택되는 하나 이상의 기술인 것인, 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 핵산 중합효소는 DNA 중합효소 또는 RNA 중합효소인, 핵산 증폭 방법.
- 청구항 16에 있어서, 상기 DNA 중합효소는 phi29 DNA 중합효소, Tts DNA 중합효소, 페이즈(phase) M2 DNA 중합효소, VENT DNA 중합효소, DNA 중합효소 I의 Klenow 단편(fragment), T5 DNA 중합효소, PRD1 DNA 중합효소, T4 DNA 중합효소 전효소(holoenzyme), T7 중합효소 유래 T7 Sequenase 및 Bst DNA 중합효소로 이루어진 군에서 선택되는 하나 이상의 DNA 중합효소인, 핵산 증폭 방법.
- 청구항 1에 있어서, 상기 샘플은 서로 다른 복수 개이고,
상기 표적 핵산은 서로 다른 복수 개이며,
상기 제1 프라이머는 서로 다른 복수 개이고,
상기 제2 프라이머는 서로 다른 복수 개이고,
상기 서로 다른 복수 개의 제1 프라이머들은 서로 다른 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드들인, 핵산 증폭 방법.
- 청구항 18에 있어서, 상기 제1 프라이머는 상기 제2 프라이머 대비 0.1 내지 10몰%로 첨가되는 것인 핵산 증폭 방법.
- 청구항 18에 있어서, 상기 복수 개의 제1 프라이머들로 증폭된 핵산들을 선별하는 단계를 더 포함하는, 핵산 증폭 방법.
- 청구항 20에 있어서, 상기 복수 개의 제1 프라이머들은 포획 물질과 결합된 것인, 핵산 증폭 방법.
- 청구항 18에 있어서,
상기 복수 개의 샘플로부터 증폭된 핵산들을 정제(purification)하는 단계;
정제된 핵산들을 단편화(fragmentation)하는 단계;
단편화된 핵산들을 풀링(pooling)하는 단계; 및
풀링된 핵산들로 NGS(Next Generation Sequencing) 라이브러리를 제조하는 단계를 더 포함하는, 핵산 증폭 방법.
- 제1 프라이머 및 제2 프라이머를 포함하는 핵산 증폭용 프라이머 세트로서,
상기 제1 프라이머는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 프라이머 세트.
- 청구항 23에 있어서,
상기 제1 프라이머는 서로 다른 복수 개이고,
상기 제2 프라이머는 서로 다른 복수 개이고,
상기 서로 다른 복수 개의 제1 프라이머들은 서로 다른 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 프라이머 세트.
- 청구항 24에 있어서, 상기 프라이머 세트 내 제1 프라이머는 제2 프라이머 대비 0.1 내지 10몰%로 포함되는 것인, 프라이머 세트.
- 제1 프라이머 및 제2 프라이머를 포함하는 핵산 증폭용 키트로서,
상기 제1 프라이머는 표적 핵산에 혼성화(hybridization)되는 제1 염기 서열; 및
상기 표적 핵산이 유래된 샘플을 구별하는 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드이고,
상기 제2 프라이머는 상기 표적 핵산에 혼성화되는 제3 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 키트.
- 청구항 26에 있어서,
상기 제1 프라이머는 서로 다른 복수 개이고,
상기 제2 프라이머는 서로 다른 복수 개이고,
상기 서로 다른 복수 개의 제1 프라이머들은 서로 다른 고정된 제2 염기 서열을 포함하는 염기 서열로 이루어진 폴리뉴클레오티드인, 키트.
- 청구항 27에 있어서, 상기 키트 내 제1 프라이머는 제2 프라이머 대비 0.1 내지 10몰%로 포함되는 것인, 키트.
- 청구항 26에 있어서, 상기 키트는 웰을 더 포함하고,
상기 제1 프라이머 및 제2 프라이머는 상기 웰에 분주되어 있는 것인, 키트.
- 청구항 29에 있어서, 상기 웰은 8 스트립 튜브, 96 웰 플레이트 및 384 플레이트로 이루어진 군에서 선택되는 하나 이상의 것인, 키트.
- 청구항 26에 있어서, 상기 제1 프라이머 및 제2 프라이머는 동결 건조된 것인, 키트.
- 청구항 26에 있어서, 상기 제1 프라이머 및 제2 프라이머가 1 μm 내지 10 mm 직경의 비드에 결합된 것인, 키트.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20210080035 | 2021-06-21 | ||
| KR1020210080035 | 2021-06-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20220169916A true KR20220169916A (ko) | 2022-12-28 |
Family
ID=84538431
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020220074560A Pending KR20220169916A (ko) | 2021-06-21 | 2022-06-20 | 병렬적 다중 전위 증폭 방법 |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP4321629A4 (ko) |
| KR (1) | KR20220169916A (ko) |
| WO (1) | WO2022270842A1 (ko) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150016036A (ko) | 2013-08-02 | 2015-02-11 | 삼성전자주식회사 | 핵산 증폭 방법 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9469874B2 (en) * | 2011-10-18 | 2016-10-18 | The Regents Of The University Of California | Long-range barcode labeling-sequencing |
| WO2015148219A1 (en) * | 2014-03-28 | 2015-10-01 | Clarient Diagnostic Services, Inc. | Accurate detection of rare genetic variants in next generation sequencing |
| WO2016118719A1 (en) * | 2015-01-23 | 2016-07-28 | Qiagen Sciences, Llc | High multiplex pcr with molecular barcoding |
| CA3016221C (en) * | 2016-04-07 | 2021-09-28 | Illumina, Inc. | Methods and systems for construction of normalized nucleic acid libraries |
| CN110770356B (zh) * | 2017-06-20 | 2024-06-07 | 生物辐射实验室股份有限公司 | 使用珠寡核苷酸的mda |
| EP3841202B1 (en) * | 2018-08-20 | 2023-10-04 | Bio-Rad Laboratories, Inc. | Nucleotide sequence generation by barcode bead-colocalization in partitions |
-
2022
- 2022-06-20 WO PCT/KR2022/008690 patent/WO2022270842A1/ko active Application Filing
- 2022-06-20 KR KR1020220074560A patent/KR20220169916A/ko active Pending
- 2022-06-20 EP EP22828689.4A patent/EP4321629A4/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150016036A (ko) | 2013-08-02 | 2015-02-11 | 삼성전자주식회사 | 핵산 증폭 방법 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4321629A4 (en) | 2025-05-07 |
| EP4321629A1 (en) | 2024-02-14 |
| WO2022270842A1 (ko) | 2022-12-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2770879C2 (ru) | Полногеномные библиотеки отдельных клеток для бисульфитного секвенирования | |
| US20220259638A1 (en) | Methods and compositions for high throughput sample preparation using double unique dual indexing | |
| US6897023B2 (en) | Method for determining relative abundance of nucleic acid sequences | |
| CN111849965B (zh) | 用于减少偏向性的多核苷酸衔接子设计 | |
| KR20190034164A (ko) | 단일 세포 전체 게놈 라이브러리 및 이의 제조를 위한 조합 인덱싱 방법 | |
| CA3183217A1 (en) | Compositions and methods for in situ single cell analysis using enzymatic nucleic acid extension | |
| EP2619329A1 (en) | Direct capture, amplification and sequencing of target dna using immobilized primers | |
| CN113528628A (zh) | 用于基因组应用和治疗应用的核酸分子的克隆复制和扩增的系统和方法 | |
| EP3262175A1 (en) | Methods and compositions for in silico long read sequencing | |
| US12286670B2 (en) | Full-length RNA sequencing | |
| WO2020193769A1 (en) | A high throughput sequencing method and kit | |
| KR20220169916A (ko) | 병렬적 다중 전위 증폭 방법 | |
| HK40072046A (en) | Single cell whole genome libraries for methylation sequencing | |
| HK40104820B (en) | Single cell whole genome libraries for methylation sequencing | |
| HK40104820A (en) | Single cell whole genome libraries for methylation sequencing | |
| HK40072046B (en) | Single cell whole genome libraries for methylation sequencing | |
| HK40076229A (en) | Methods and compositions for high throughput sample preparation using double unique dual indexing | |
| HK40027672A (en) | Single cell whole genome libraries for methylation sequencing | |
| HK40016494A (en) | Single cell whole genome libraries for methylation sequencing | |
| HK40016494B (en) | Single cell whole genome libraries for methylation sequencing | |
| WO2004013353A1 (en) | Analysis of biological samples |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0109 | Patent application |
Patent event code: PA01091R01D Comment text: Patent Application Patent event date: 20220620 |
|
| PA0201 | Request for examination | ||
| PG1501 | Laying open of application | ||
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection |
Comment text: Notification of reason for refusal Patent event date: 20250115 Patent event code: PE09021S01D |