JPWO2022153295A5 - - Google Patents
Download PDFInfo
- Publication number
- JPWO2022153295A5 JPWO2022153295A5 JP2023541633A JP2023541633A JPWO2022153295A5 JP WO2022153295 A5 JPWO2022153295 A5 JP WO2022153295A5 JP 2023541633 A JP2023541633 A JP 2023541633A JP 2023541633 A JP2023541633 A JP 2023541633A JP WO2022153295 A5 JPWO2022153295 A5 JP WO2022153295A5
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- tumor
- sak
- cell composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 210000004027 cell Anatomy 0.000 claims description 924
- 239000000203 mixture Substances 0.000 claims description 289
- 206010028980 Neoplasm Diseases 0.000 claims description 206
- 230000014509 gene expression Effects 0.000 claims description 136
- 210000004881 tumor cell Anatomy 0.000 claims description 129
- 108010002350 Interleukin-2 Proteins 0.000 claims description 106
- 238000000034 method Methods 0.000 claims description 96
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 claims description 96
- 230000001472 cytotoxic effect Effects 0.000 claims description 90
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims description 78
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 claims description 66
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 claims description 66
- 238000011282 treatment Methods 0.000 claims description 58
- 102000004127 Cytokines Human genes 0.000 claims description 56
- 108090000695 Cytokines Proteins 0.000 claims description 56
- 238000000338 in vitro Methods 0.000 claims description 51
- 239000012190 activator Substances 0.000 claims description 50
- 102000001398 Granzyme Human genes 0.000 claims description 48
- 108060005986 Granzyme Proteins 0.000 claims description 48
- 229940126546 immune checkpoint molecule Drugs 0.000 claims description 46
- 239000003112 inhibitor Substances 0.000 claims description 43
- 108010021064 CTLA-4 Antigen Proteins 0.000 claims description 41
- 229940045513 CTLA4 antagonist Drugs 0.000 claims description 41
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 claims description 38
- 229930192851 perforin Natural products 0.000 claims description 38
- 102000004503 Perforin Human genes 0.000 claims description 37
- 108010056995 Perforin Proteins 0.000 claims description 37
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 claims description 32
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 claims description 32
- 238000001727 in vivo Methods 0.000 claims description 29
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 claims description 28
- 102100028990 C-X-C chemokine receptor type 3 Human genes 0.000 claims description 27
- 101000916050 Homo sapiens C-X-C chemokine receptor type 3 Proteins 0.000 claims description 27
- 230000001404 mediated effect Effects 0.000 claims description 23
- 102100038077 CD226 antigen Human genes 0.000 claims description 21
- 101000884298 Homo sapiens CD226 antigen Proteins 0.000 claims description 21
- 108010018951 Interleukin-8B Receptors Proteins 0.000 claims description 21
- 208000032839 leukemia Diseases 0.000 claims description 21
- 102100028989 C-X-C chemokine receptor type 2 Human genes 0.000 claims description 20
- 238000004519 manufacturing process Methods 0.000 claims description 20
- 102000009410 Chemokine receptor Human genes 0.000 claims description 19
- 108050000299 Chemokine receptor Proteins 0.000 claims description 19
- 102000019034 Chemokines Human genes 0.000 claims description 18
- 108010012236 Chemokines Proteins 0.000 claims description 18
- 230000000903 blocking effect Effects 0.000 claims description 15
- 230000004936 stimulating effect Effects 0.000 claims description 15
- 230000035755 proliferation Effects 0.000 claims description 14
- 238000012546 transfer Methods 0.000 claims description 14
- 230000001965 increasing effect Effects 0.000 claims description 13
- 206010027406 Mesothelioma Diseases 0.000 claims description 11
- 239000003550 marker Substances 0.000 claims description 11
- 208000034578 Multiple myelomas Diseases 0.000 claims description 9
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 9
- 208000023958 prostate neoplasm Diseases 0.000 claims description 5
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 claims description 4
- 102000015212 Fas Ligand Protein Human genes 0.000 claims description 4
- 108010039471 Fas Ligand Protein Proteins 0.000 claims description 4
- 102100025618 C-X-C chemokine receptor type 6 Human genes 0.000 claims description 3
- 238000011285 therapeutic regimen Methods 0.000 claims description 3
- 101000856683 Homo sapiens C-X-C chemokine receptor type 6 Proteins 0.000 claims description 2
- 102000003675 cytokine receptors Human genes 0.000 claims description 2
- 108010057085 cytokine receptors Proteins 0.000 claims description 2
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 claims 1
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 claims 1
- 230000001902 propagating effect Effects 0.000 claims 1
- 102000000588 Interleukin-2 Human genes 0.000 description 103
- 239000003446 ligand Substances 0.000 description 92
- 210000001744 T-lymphocyte Anatomy 0.000 description 71
- 238000011467 adoptive cell therapy Methods 0.000 description 61
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 49
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 49
- 150000007523 nucleic acids Chemical class 0.000 description 45
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 41
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 41
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 39
- 230000000694 effects Effects 0.000 description 39
- 239000012636 effector Substances 0.000 description 37
- 239000013598 vector Substances 0.000 description 36
- -1 ULBP256 Proteins 0.000 description 34
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 32
- 239000000427 antigen Substances 0.000 description 32
- 210000000822 natural killer cell Anatomy 0.000 description 32
- 108090000623 proteins and genes Proteins 0.000 description 31
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 30
- 210000004405 cytokine-induced killer cell Anatomy 0.000 description 30
- 102000005962 receptors Human genes 0.000 description 30
- 108020003175 receptors Proteins 0.000 description 30
- 108091007433 antigens Proteins 0.000 description 29
- 102000036639 antigens Human genes 0.000 description 29
- 208000009329 Graft vs Host Disease Diseases 0.000 description 28
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 description 28
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 description 28
- 208000024908 graft versus host disease Diseases 0.000 description 28
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 27
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 27
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 26
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 26
- 230000035772 mutation Effects 0.000 description 26
- 101100510617 Caenorhabditis elegans sel-8 gene Proteins 0.000 description 24
- 201000011510 cancer Diseases 0.000 description 24
- 102000003812 Interleukin-15 Human genes 0.000 description 23
- 108090000172 Interleukin-15 Proteins 0.000 description 23
- 108020004707 nucleic acids Proteins 0.000 description 23
- 102000039446 nucleic acids Human genes 0.000 description 23
- 241000699670 Mus sp. Species 0.000 description 22
- 108091028043 Nucleic acid sequence Proteins 0.000 description 22
- 108091008874 T cell receptors Proteins 0.000 description 22
- 108010061299 CXCR4 Receptors Proteins 0.000 description 21
- 102000012000 CXCR4 Receptors Human genes 0.000 description 21
- 102100030704 Interleukin-21 Human genes 0.000 description 21
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 21
- 230000000259 anti-tumor effect Effects 0.000 description 21
- 210000004899 c-terminal region Anatomy 0.000 description 21
- 230000001976 improved effect Effects 0.000 description 21
- 230000002401 inhibitory effect Effects 0.000 description 21
- 108010074108 interleukin-21 Proteins 0.000 description 21
- 210000000265 leukocyte Anatomy 0.000 description 20
- 101000991061 Homo sapiens MHC class I polypeptide-related sequence B Proteins 0.000 description 18
- 102100030301 MHC class I polypeptide-related sequence A Human genes 0.000 description 18
- 102100030300 MHC class I polypeptide-related sequence B Human genes 0.000 description 18
- 230000001419 dependent effect Effects 0.000 description 18
- 102100028970 HLA class I histocompatibility antigen, alpha chain E Human genes 0.000 description 17
- 101100395313 Homo sapiens HLA-E gene Proteins 0.000 description 17
- 101000607306 Homo sapiens UL16-binding protein 1 Proteins 0.000 description 17
- 101000607318 Homo sapiens UL16-binding protein 3 Proteins 0.000 description 17
- 102100040012 UL16-binding protein 1 Human genes 0.000 description 17
- 102100040011 UL16-binding protein 3 Human genes 0.000 description 17
- 230000004913 activation Effects 0.000 description 17
- 238000000684 flow cytometry Methods 0.000 description 17
- 229910052618 mica group Inorganic materials 0.000 description 17
- 108020003285 Isocitrate lyase Proteins 0.000 description 16
- 230000000735 allogeneic effect Effects 0.000 description 16
- 108090000765 processed proteins & peptides Proteins 0.000 description 16
- 238000005138 cryopreservation Methods 0.000 description 15
- 210000004698 lymphocyte Anatomy 0.000 description 15
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 14
- 210000001185 bone marrow Anatomy 0.000 description 14
- 239000010445 mica Substances 0.000 description 14
- 102000004169 proteins and genes Human genes 0.000 description 14
- 230000011664 signaling Effects 0.000 description 14
- 210000003719 b-lymphocyte Anatomy 0.000 description 13
- JGPOSNWWINVNFV-UHFFFAOYSA-N carboxyfluorescein diacetate succinimidyl ester Chemical compound C=1C(OC(=O)C)=CC=C2C=1OC1=CC(OC(C)=O)=CC=C1C2(C1=C2)OC(=O)C1=CC=C2C(=O)ON1C(=O)CCC1=O JGPOSNWWINVNFV-UHFFFAOYSA-N 0.000 description 13
- 238000010257 thawing Methods 0.000 description 13
- 102000043129 MHC class I family Human genes 0.000 description 12
- 108091054437 MHC class I family Proteins 0.000 description 12
- 238000003556 assay Methods 0.000 description 12
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 11
- 230000003013 cytotoxicity Effects 0.000 description 11
- 231100000135 cytotoxicity Toxicity 0.000 description 11
- 230000001177 retroviral effect Effects 0.000 description 11
- 241000700605 Viruses Species 0.000 description 10
- 210000004369 blood Anatomy 0.000 description 10
- 239000008280 blood Substances 0.000 description 10
- 230000004083 survival effect Effects 0.000 description 10
- 230000001225 therapeutic effect Effects 0.000 description 10
- 102100025248 C-X-C motif chemokine 10 Human genes 0.000 description 9
- 101001002657 Homo sapiens Interleukin-2 Proteins 0.000 description 9
- 102100029740 Poliovirus receptor Human genes 0.000 description 9
- 230000003213 activating effect Effects 0.000 description 9
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 9
- 238000011161 development Methods 0.000 description 9
- 230000018109 developmental process Effects 0.000 description 9
- 238000011534 incubation Methods 0.000 description 9
- 239000002609 medium Substances 0.000 description 9
- 239000013612 plasmid Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 102000004196 processed proteins & peptides Human genes 0.000 description 9
- 210000000952 spleen Anatomy 0.000 description 9
- 108010074708 B7-H1 Antigen Proteins 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 8
- 102000017578 LAG3 Human genes 0.000 description 8
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 8
- 102100021669 Stromal cell-derived factor 1 Human genes 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 231100000433 cytotoxic Toxicity 0.000 description 8
- 230000012010 growth Effects 0.000 description 8
- 210000002865 immune cell Anatomy 0.000 description 8
- 230000001939 inductive effect Effects 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 230000003993 interaction Effects 0.000 description 8
- 108010048507 poliovirus receptor Proteins 0.000 description 8
- 229920001184 polypeptide Polymers 0.000 description 8
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 230000000638 stimulation Effects 0.000 description 8
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 8
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 7
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 7
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 7
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 7
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 239000002458 cell surface marker Substances 0.000 description 7
- 238000002784 cytotoxicity assay Methods 0.000 description 7
- 231100000263 cytotoxicity test Toxicity 0.000 description 7
- 210000004443 dendritic cell Anatomy 0.000 description 7
- 239000013604 expression vector Substances 0.000 description 7
- 239000012894 fetal calf serum Substances 0.000 description 7
- 230000003394 haemopoietic effect Effects 0.000 description 7
- 230000028993 immune response Effects 0.000 description 7
- 238000009169 immunotherapy Methods 0.000 description 7
- 230000014759 maintenance of location Effects 0.000 description 7
- 210000000581 natural killer T-cell Anatomy 0.000 description 7
- 239000002245 particle Substances 0.000 description 7
- 210000005259 peripheral blood Anatomy 0.000 description 7
- 239000011886 peripheral blood Substances 0.000 description 7
- 230000003389 potentiating effect Effects 0.000 description 7
- 210000003289 regulatory T cell Anatomy 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- 101710098275 C-X-C motif chemokine 10 Proteins 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- 102000037978 Immune checkpoint receptors Human genes 0.000 description 6
- 108091008028 Immune checkpoint receptors Proteins 0.000 description 6
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 6
- 230000006907 apoptotic process Effects 0.000 description 6
- 230000001413 cellular effect Effects 0.000 description 6
- 230000009089 cytolysis Effects 0.000 description 6
- 230000001086 cytosolic effect Effects 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 201000005787 hematologic cancer Diseases 0.000 description 6
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 6
- 230000002147 killing effect Effects 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 238000010186 staining Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 230000005748 tumor development Effects 0.000 description 6
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 6
- 102100025279 C-X-C motif chemokine 11 Human genes 0.000 description 5
- 102100039396 C-X-C motif chemokine 16 Human genes 0.000 description 5
- 102100036170 C-X-C motif chemokine 9 Human genes 0.000 description 5
- 102100038078 CD276 antigen Human genes 0.000 description 5
- 101000889133 Homo sapiens C-X-C motif chemokine 16 Proteins 0.000 description 5
- 101000617130 Homo sapiens Stromal cell-derived factor 1 Proteins 0.000 description 5
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 5
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 description 5
- 108060003951 Immunoglobulin Proteins 0.000 description 5
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 5
- 102000004890 Interleukin-8 Human genes 0.000 description 5
- 108090001007 Interleukin-8 Proteins 0.000 description 5
- 206010060862 Prostate cancer Diseases 0.000 description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 5
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 5
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 5
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 description 5
- 210000000612 antigen-presenting cell Anatomy 0.000 description 5
- 238000001574 biopsy Methods 0.000 description 5
- 238000012512 characterization method Methods 0.000 description 5
- DJZCTUVALDDONK-HQMSUKCRSA-N concanamycin A Chemical compound O1C(=O)\C(OC)=C\C(\C)=C\[C@@H](C)[C@@H](O)[C@@H](CC)[C@@H](O)[C@H](C)C\C(C)=C\C=C\[C@H](OC)[C@H]1[C@@H](C)[C@@H](O)[C@H](C)[C@]1(O)O[C@H](\C=C\C)[C@@H](C)[C@H](O[C@@H]2O[C@H](C)[C@@H](OC(N)=O)[C@H](O)C2)C1 DJZCTUVALDDONK-HQMSUKCRSA-N 0.000 description 5
- DJZCTUVALDDONK-UHFFFAOYSA-N concanamycin A Natural products O1C(=O)C(OC)=CC(C)=CC(C)C(O)C(CC)C(O)C(C)CC(C)=CC=CC(OC)C1C(C)C(O)C(C)C1(O)OC(C=CC)C(C)C(OC2OC(C)C(OC(N)=O)C(O)C2)C1 DJZCTUVALDDONK-UHFFFAOYSA-N 0.000 description 5
- 230000000139 costimulatory effect Effects 0.000 description 5
- 230000001461 cytolytic effect Effects 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 5
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 5
- 239000012737 fresh medium Substances 0.000 description 5
- 102000018358 immunoglobulin Human genes 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 230000010534 mechanism of action Effects 0.000 description 5
- 238000013508 migration Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 210000005087 mononuclear cell Anatomy 0.000 description 5
- 229960003301 nivolumab Drugs 0.000 description 5
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 5
- 108010056030 retronectin Proteins 0.000 description 5
- 230000028327 secretion Effects 0.000 description 5
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 5
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 description 4
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 description 4
- 208000026310 Breast neoplasm Diseases 0.000 description 4
- 102100032912 CD44 antigen Human genes 0.000 description 4
- 102100025278 Coxsackievirus and adenovirus receptor Human genes 0.000 description 4
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 4
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 4
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 4
- 102100037907 High mobility group protein B1 Human genes 0.000 description 4
- 101000858060 Homo sapiens C-X-C motif chemokine 11 Proteins 0.000 description 4
- 101000947172 Homo sapiens C-X-C motif chemokine 9 Proteins 0.000 description 4
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 description 4
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 4
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 4
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 4
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 4
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 4
- 101001136592 Homo sapiens Prostate stem cell antigen Proteins 0.000 description 4
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 4
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 4
- 206010025323 Lymphomas Diseases 0.000 description 4
- 102000003735 Mesothelin Human genes 0.000 description 4
- 108090000015 Mesothelin Proteins 0.000 description 4
- 102100023123 Mucin-16 Human genes 0.000 description 4
- 102100036735 Prostate stem cell antigen Human genes 0.000 description 4
- 239000012979 RPMI medium Substances 0.000 description 4
- 101710088580 Stromal cell-derived factor 1 Proteins 0.000 description 4
- 230000006044 T cell activation Effects 0.000 description 4
- 230000001154 acute effect Effects 0.000 description 4
- 238000002617 apheresis Methods 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 229950002916 avelumab Drugs 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 238000004113 cell culture Methods 0.000 description 4
- 238000002659 cell therapy Methods 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 230000003828 downregulation Effects 0.000 description 4
- 229950009791 durvalumab Drugs 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 230000002349 favourable effect Effects 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- 238000007710 freezing Methods 0.000 description 4
- 230000008014 freezing Effects 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 210000000987 immune system Anatomy 0.000 description 4
- 229960005386 ipilimumab Drugs 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 102000006240 membrane receptors Human genes 0.000 description 4
- 230000001394 metastastic effect Effects 0.000 description 4
- 206010061289 metastatic neoplasm Diseases 0.000 description 4
- 230000005012 migration Effects 0.000 description 4
- 229960002621 pembrolizumab Drugs 0.000 description 4
- 238000003752 polymerase chain reaction Methods 0.000 description 4
- 108091033319 polynucleotide Proteins 0.000 description 4
- 102000040430 polynucleotide Human genes 0.000 description 4
- 239000002157 polynucleotide Substances 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 238000010361 transduction Methods 0.000 description 4
- 230000026683 transduction Effects 0.000 description 4
- 238000011269 treatment regimen Methods 0.000 description 4
- 230000003612 virological effect Effects 0.000 description 4
- SUGXUUGGLDCZKB-UHFFFAOYSA-N 3,4-dichloroisocoumarin Chemical compound C1=CC=C2C(Cl)=C(Cl)OC(=O)C2=C1 SUGXUUGGLDCZKB-UHFFFAOYSA-N 0.000 description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 3
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 3
- 108010001857 Cell Surface Receptors Proteins 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- 101150089023 FASLG gene Proteins 0.000 description 3
- 102000003886 Glycoproteins Human genes 0.000 description 3
- 108090000288 Glycoproteins Proteins 0.000 description 3
- 101000858088 Homo sapiens C-X-C motif chemokine 10 Proteins 0.000 description 3
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 3
- 101000599940 Homo sapiens Interferon gamma Proteins 0.000 description 3
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 3
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 3
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 3
- 108010074328 Interferon-gamma Proteins 0.000 description 3
- 102000003814 Interleukin-10 Human genes 0.000 description 3
- 108090000174 Interleukin-10 Proteins 0.000 description 3
- 241000713666 Lentivirus Species 0.000 description 3
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 3
- 102100022430 Melanocyte protein PMEL Human genes 0.000 description 3
- 208000002030 Merkel cell carcinoma Diseases 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 102000000812 NK Cell Lectin-Like Receptor Subfamily K Human genes 0.000 description 3
- 108010001657 NK Cell Lectin-Like Receptor Subfamily K Proteins 0.000 description 3
- 102000002356 Nectin Human genes 0.000 description 3
- 108060005251 Nectin Proteins 0.000 description 3
- 206010029266 Neuroendocrine carcinoma of the skin Diseases 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 108020004511 Recombinant DNA Proteins 0.000 description 3
- 208000006265 Renal cell carcinoma Diseases 0.000 description 3
- 206010041067 Small cell lung cancer Diseases 0.000 description 3
- 108010083312 T-Cell Antigen Receptor-CD3 Complex Proteins 0.000 description 3
- 238000002619 cancer immunotherapy Methods 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 210000004748 cultured cell Anatomy 0.000 description 3
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 description 3
- 230000004069 differentiation Effects 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 230000017188 evasion or tolerance of host immune response Effects 0.000 description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 239000012642 immune effector Substances 0.000 description 3
- 229940121354 immunomodulator Drugs 0.000 description 3
- 108091008042 inhibitory receptors Proteins 0.000 description 3
- 230000004807 localization Effects 0.000 description 3
- 210000002540 macrophage Anatomy 0.000 description 3
- 230000003211 malignant effect Effects 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 210000001616 monocyte Anatomy 0.000 description 3
- 210000000066 myeloid cell Anatomy 0.000 description 3
- 210000000440 neutrophil Anatomy 0.000 description 3
- 238000004806 packaging method and process Methods 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 239000013603 viral vector Substances 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 102100026887 Beta-defensin 103 Human genes 0.000 description 2
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 2
- 108050006947 CXC Chemokine Proteins 0.000 description 2
- 102000019388 CXC chemokine Human genes 0.000 description 2
- 102100024533 Carcinoembryonic antigen-related cell adhesion molecule 1 Human genes 0.000 description 2
- 101710190843 Carcinoembryonic antigen-related cell adhesion molecule 1 Proteins 0.000 description 2
- 206010057248 Cell death Diseases 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 108091007741 Chimeric antigen receptor T cells Proteins 0.000 description 2
- 238000001712 DNA sequencing Methods 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 108010001517 Galectin 3 Proteins 0.000 description 2
- 102100039558 Galectin-3 Human genes 0.000 description 2
- 206010061968 Gastric neoplasm Diseases 0.000 description 2
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 2
- 102100030385 Granzyme B Human genes 0.000 description 2
- 229940121832 Granzyme B inhibitor Drugs 0.000 description 2
- 101001009603 Homo sapiens Granzyme B Proteins 0.000 description 2
- 101001055157 Homo sapiens Interleukin-15 Proteins 0.000 description 2
- 101000946860 Homo sapiens T-cell surface glycoprotein CD3 epsilon chain Proteins 0.000 description 2
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 2
- 102100037850 Interferon gamma Human genes 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 208000032818 Microsatellite Instability Diseases 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- 206010028851 Necrosis Diseases 0.000 description 2
- 102100035488 Nectin-2 Human genes 0.000 description 2
- 108010069196 Neural Cell Adhesion Molecules Proteins 0.000 description 2
- 102100023616 Neural cell adhesion molecule L1-like protein Human genes 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 102100035794 T-cell surface glycoprotein CD3 epsilon chain Human genes 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- 101710165471 Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 230000001464 adherent effect Effects 0.000 description 2
- 108700025316 aldesleukin Proteins 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 229960003852 atezolizumab Drugs 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000022534 cell killing Effects 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 208000013056 classic Hodgkin lymphoma Diseases 0.000 description 2
- 210000001228 classical NK T cell Anatomy 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 238000012136 culture method Methods 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 239000002619 cytotoxin Substances 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 230000000040 effect on leukemia Effects 0.000 description 2
- 229940056913 eftilagimod alfa Drugs 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 230000008029 eradication Effects 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000012239 gene modification Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 230000005017 genetic modification Effects 0.000 description 2
- 235000013617 genetically modified food Nutrition 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 238000003306 harvesting Methods 0.000 description 2
- 210000002443 helper t lymphocyte Anatomy 0.000 description 2
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 2
- 102000056003 human IL15 Human genes 0.000 description 2
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 2
- 210000004964 innate lymphoid cell Anatomy 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000003810 lymphokine-activated killer cell Anatomy 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 208000021039 metastatic melanoma Diseases 0.000 description 2
- 230000033607 mismatch repair Effects 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 230000017074 necrotic cell death Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 210000004882 non-tumor cell Anatomy 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- 230000008488 polyadenylation Effects 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- 229940066453 tecentriq Drugs 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 230000009261 transgenic effect Effects 0.000 description 2
- 206010044412 transitional cell carcinoma Diseases 0.000 description 2
- 102000035160 transmembrane proteins Human genes 0.000 description 2
- 108091005703 transmembrane proteins Proteins 0.000 description 2
- 229950007217 tremelimumab Drugs 0.000 description 2
- 208000023747 urothelial carcinoma Diseases 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 1
- JPSHPWJJSVEEAX-OWPBQMJCSA-N (2s)-2-amino-4-fluoranylpentanedioic acid Chemical compound OC(=O)[C@@H](N)CC([18F])C(O)=O JPSHPWJJSVEEAX-OWPBQMJCSA-N 0.000 description 1
- XOQABDOICLHPIS-UHFFFAOYSA-N 1-hydroxy-2,1-benzoxaborole Chemical compound C1=CC=C2B(O)OCC2=C1 XOQABDOICLHPIS-UHFFFAOYSA-N 0.000 description 1
- QRBLKGHRWFGINE-UGWAGOLRSA-N 2-[2-[2-[[2-[[4-[[2-[[6-amino-2-[3-amino-1-[(2,3-diamino-3-oxopropyl)amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2s,3r,4r,5s)-4-carbamoyl-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)- Chemical compound N=1C(C=2SC=C(N=2)C(N)=O)CSC=1CCNC(=O)C(C(C)=O)NC(=O)C(C)C(O)C(C)NC(=O)C(C(O[C@H]1[C@@]([C@@H](O)[C@H](O)[C@H](CO)O1)(C)O[C@H]1[C@@H]([C@](O)([C@@H](O)C(CO)O1)C(N)=O)O)C=1NC=NC=1)NC(=O)C1=NC(C(CC(N)=O)NCC(N)C(N)=O)=NC(N)=C1C QRBLKGHRWFGINE-UGWAGOLRSA-N 0.000 description 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 229940125559 AB154 Drugs 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 229940125554 ASP-8374 Drugs 0.000 description 1
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 102100026882 Alpha-synuclein Human genes 0.000 description 1
- 101710145634 Antigen 1 Proteins 0.000 description 1
- 101001005269 Arabidopsis thaliana Ceramide synthase 1 LOH3 Proteins 0.000 description 1
- 101001005312 Arabidopsis thaliana Ceramide synthase LOH1 Proteins 0.000 description 1
- 108091008875 B cell receptors Proteins 0.000 description 1
- 102100027205 B-cell antigen receptor complex-associated protein alpha chain Human genes 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 1
- 101710117995 B-lymphocyte antigen CD19 Proteins 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 229940125556 BGB-A1217 Drugs 0.000 description 1
- 229940125565 BMS-986016 Drugs 0.000 description 1
- 229940125557 BMS-986207 Drugs 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 101710125296 Beta-defensin 3 Proteins 0.000 description 1
- 208000031648 Body Weight Changes Diseases 0.000 description 1
- 241000701822 Bovine papillomavirus Species 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- 108090000342 C-Type Lectins Proteins 0.000 description 1
- 102000003930 C-Type Lectins Human genes 0.000 description 1
- 101710098272 C-X-C motif chemokine 11 Proteins 0.000 description 1
- 102100039398 C-X-C motif chemokine 2 Human genes 0.000 description 1
- 102100036189 C-X-C motif chemokine 3 Human genes 0.000 description 1
- 102100036150 C-X-C motif chemokine 5 Human genes 0.000 description 1
- 101710085500 C-X-C motif chemokine 9 Proteins 0.000 description 1
- 102000002086 C-type lectin-like Human genes 0.000 description 1
- 108050009406 C-type lectin-like Proteins 0.000 description 1
- 101710185679 CD276 antigen Proteins 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 1
- 102100035793 CD83 antigen Human genes 0.000 description 1
- 102000024905 CD99 Human genes 0.000 description 1
- 108060001253 CD99 Proteins 0.000 description 1
- 229940125558 COM-902 Drugs 0.000 description 1
- 108091008928 CXC chemokine receptors Proteins 0.000 description 1
- 102000054900 CXCR Receptors Human genes 0.000 description 1
- 101150004010 CXCR3 gene Proteins 0.000 description 1
- 101150066398 CXCR4 gene Proteins 0.000 description 1
- 108010061304 CXCR6 Receptors Proteins 0.000 description 1
- 101100297347 Caenorhabditis elegans pgl-3 gene Proteins 0.000 description 1
- 101100408682 Caenorhabditis elegans pmt-2 gene Proteins 0.000 description 1
- 102100028801 Calsyntenin-1 Human genes 0.000 description 1
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 1
- 102000011727 Caspases Human genes 0.000 description 1
- 108010076667 Caspases Proteins 0.000 description 1
- 231100000023 Cell-mediated cytotoxicity Toxicity 0.000 description 1
- 206010057250 Cell-mediated cytotoxicity Diseases 0.000 description 1
- 102100030099 Chloride anion exchanger Human genes 0.000 description 1
- 102100028757 Chondroitin sulfate proteoglycan 4 Human genes 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 102100032768 Complement receptor type 2 Human genes 0.000 description 1
- 208000037845 Cutaneous squamous cell carcinoma Diseases 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 108010049207 Death Domain Receptors Proteins 0.000 description 1
- 102000009058 Death Domain Receptors Human genes 0.000 description 1
- 102100036466 Delta-like protein 3 Human genes 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 101150064015 FAS gene Proteins 0.000 description 1
- 229940125570 FS118 Drugs 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 229920001917 Ficoll Polymers 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 102100021260 Galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferase 1 Human genes 0.000 description 1
- 102100031351 Galectin-9 Human genes 0.000 description 1
- 101710121810 Galectin-9 Proteins 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 102100032530 Glypican-3 Human genes 0.000 description 1
- 102100021186 Granulysin Human genes 0.000 description 1
- 101710168479 Granulysin Proteins 0.000 description 1
- 108010007712 Hepatitis A Virus Cellular Receptor 1 Proteins 0.000 description 1
- 108010007707 Hepatitis A Virus Cellular Receptor 2 Proteins 0.000 description 1
- 102100034459 Hepatitis A virus cellular receptor 1 Human genes 0.000 description 1
- 101710083479 Hepatitis A virus cellular receptor 2 homolog Proteins 0.000 description 1
- 101710168537 High mobility group protein B1 Proteins 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101000914489 Homo sapiens B-cell antigen receptor complex-associated protein alpha chain Proteins 0.000 description 1
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 1
- 101000912247 Homo sapiens Beta-defensin 103 Proteins 0.000 description 1
- 101000889128 Homo sapiens C-X-C motif chemokine 2 Proteins 0.000 description 1
- 101000947193 Homo sapiens C-X-C motif chemokine 3 Proteins 0.000 description 1
- 101000947186 Homo sapiens C-X-C motif chemokine 5 Proteins 0.000 description 1
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 1
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 1
- 101000916489 Homo sapiens Chondroitin sulfate proteoglycan 4 Proteins 0.000 description 1
- 101000941929 Homo sapiens Complement receptor type 2 Proteins 0.000 description 1
- 101000928513 Homo sapiens Delta-like protein 3 Proteins 0.000 description 1
- 101000894906 Homo sapiens Galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferase 1 Proteins 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101001014668 Homo sapiens Glypican-3 Proteins 0.000 description 1
- 101001025337 Homo sapiens High mobility group protein B1 Proteins 0.000 description 1
- 101100072411 Homo sapiens IL21 gene Proteins 0.000 description 1
- 101001015037 Homo sapiens Integrin beta-7 Proteins 0.000 description 1
- 101000960952 Homo sapiens Interleukin-1 receptor accessory protein Proteins 0.000 description 1
- 101001055222 Homo sapiens Interleukin-8 Proteins 0.000 description 1
- 101001008874 Homo sapiens Mast/stem cell growth factor receptor Kit Proteins 0.000 description 1
- 101000620359 Homo sapiens Melanocyte protein PMEL Proteins 0.000 description 1
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 1
- 101001109508 Homo sapiens NKG2-A/NKG2-B type II integral membrane protein Proteins 0.000 description 1
- 101001098352 Homo sapiens OX-2 membrane glycoprotein Proteins 0.000 description 1
- 101000582950 Homo sapiens Platelet factor 4 Proteins 0.000 description 1
- 101000586618 Homo sapiens Poliovirus receptor Proteins 0.000 description 1
- 101000610551 Homo sapiens Prominin-1 Proteins 0.000 description 1
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 description 1
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 1
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 1
- 101000610604 Homo sapiens Tumor necrosis factor receptor superfamily member 10B Proteins 0.000 description 1
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 1
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- 229940125561 IBI-939 Drugs 0.000 description 1
- 102000039990 IL-2 family Human genes 0.000 description 1
- 108091069192 IL-2 family Proteins 0.000 description 1
- 101150039708 IL15 gene Proteins 0.000 description 1
- 102000037977 Immune checkpoint ligands Human genes 0.000 description 1
- 108091008029 Immune checkpoint ligands Proteins 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 1
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 1
- 102100033016 Integrin beta-7 Human genes 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 102000018682 Interleukin Receptor Common gamma Subunit Human genes 0.000 description 1
- 108010066719 Interleukin Receptor Common gamma Subunit Proteins 0.000 description 1
- 102100039880 Interleukin-1 receptor accessory protein Human genes 0.000 description 1
- 102000004556 Interleukin-15 Receptors Human genes 0.000 description 1
- 108010017535 Interleukin-15 Receptors Proteins 0.000 description 1
- 102100030692 Interleukin-20 Human genes 0.000 description 1
- 102100039064 Interleukin-3 Human genes 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108010038452 Interleukin-3 Receptors Proteins 0.000 description 1
- 102000010790 Interleukin-3 Receptors Human genes 0.000 description 1
- 102100026236 Interleukin-8 Human genes 0.000 description 1
- 102000002698 KIR Receptors Human genes 0.000 description 1
- 108010043610 KIR Receptors Proteins 0.000 description 1
- ZQISRDCJNBUVMM-UHFFFAOYSA-N L-Histidinol Natural products OCC(N)CC1=CN=CN1 ZQISRDCJNBUVMM-UHFFFAOYSA-N 0.000 description 1
- ZQISRDCJNBUVMM-YFKPBYRVSA-N L-histidinol Chemical compound OC[C@@H](N)CC1=CNC=N1 ZQISRDCJNBUVMM-YFKPBYRVSA-N 0.000 description 1
- 229940125563 LAG3 inhibitor Drugs 0.000 description 1
- 101150030213 Lag3 gene Proteins 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 102100020862 Lymphocyte activation gene 3 protein Human genes 0.000 description 1
- 229940125568 MGD013 Drugs 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108010048043 Macrophage Migration-Inhibitory Factors Proteins 0.000 description 1
- 102100037791 Macrophage migration inhibitory factor Human genes 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 1
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102000003792 Metallothionein Human genes 0.000 description 1
- 108090000157 Metallothionein Proteins 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 1
- 102100022682 NKG2-A/NKG2-B type II integral membrane protein Human genes 0.000 description 1
- 102100035487 Nectin-3 Human genes 0.000 description 1
- 108090000028 Neprilysin Proteins 0.000 description 1
- 102000003729 Neprilysin Human genes 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 108020003217 Nuclear RNA Proteins 0.000 description 1
- 102000043141 Nuclear RNA Human genes 0.000 description 1
- 102100037589 OX-2 membrane glycoprotein Human genes 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 208000037273 Pathologic Processes Diseases 0.000 description 1
- 208000037581 Persistent Infection Diseases 0.000 description 1
- LTQCLFMNABRKSH-UHFFFAOYSA-N Phleomycin Natural products N=1C(C=2SC=C(N=2)C(N)=O)CSC=1CCNC(=O)C(C(O)C)NC(=O)C(C)C(O)C(C)NC(=O)C(C(OC1C(C(O)C(O)C(CO)O1)OC1C(C(OC(N)=O)C(O)C(CO)O1)O)C=1NC=NC=1)NC(=O)C1=NC(C(CC(N)=O)NCC(N)C(N)=O)=NC(N)=C1C LTQCLFMNABRKSH-UHFFFAOYSA-N 0.000 description 1
- 108010035235 Phleomycins Proteins 0.000 description 1
- 102100030304 Platelet factor 4 Human genes 0.000 description 1
- 101710182846 Polyhedrin Proteins 0.000 description 1
- 206010036711 Primary mediastinal large B-cell lymphomas Diseases 0.000 description 1
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 1
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 1
- 102100040120 Prominin-1 Human genes 0.000 description 1
- 229940125566 REGN3767 Drugs 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 1
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 1
- 241000714474 Rous sarcoma virus Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- 101000668858 Spinacia oleracea 30S ribosomal protein S1, chloroplastic Proteins 0.000 description 1
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 101000898746 Streptomyces clavuligerus Clavaminate synthase 1 Proteins 0.000 description 1
- 229940125564 Sym022 Drugs 0.000 description 1
- 102100035721 Syndecan-1 Human genes 0.000 description 1
- 230000006043 T cell recruitment Effects 0.000 description 1
- 229940126547 T-cell immunoglobulin mucin-3 Drugs 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 1
- 102000043124 TIM family Human genes 0.000 description 1
- 108091054435 TIM family Proteins 0.000 description 1
- 229940125567 TSR-033 Drugs 0.000 description 1
- 210000000447 Th1 cell Anatomy 0.000 description 1
- 108020004566 Transfer RNA Proteins 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 1
- 102100040112 Tumor necrosis factor receptor superfamily member 10B Human genes 0.000 description 1
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 1
- 102000003425 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 229940127174 UCHT1 Drugs 0.000 description 1
- 108090000848 Ubiquitin Proteins 0.000 description 1
- 102000044159 Ubiquitin Human genes 0.000 description 1
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 1
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 1
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 208000010115 WHIM syndrome Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 210000001766 X chromosome Anatomy 0.000 description 1
- 210000002593 Y chromosome Anatomy 0.000 description 1
- 230000006786 activation induced cell death Effects 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 108091005764 adaptor proteins Proteins 0.000 description 1
- 102000035181 adaptor proteins Human genes 0.000 description 1
- 101150063416 add gene Proteins 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 210000005057 airway smooth muscle cell Anatomy 0.000 description 1
- 229960005310 aldesleukin Drugs 0.000 description 1
- 108090000185 alpha-Synuclein Proteins 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 102000023732 binding proteins Human genes 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 230000004579 body weight change Effects 0.000 description 1
- 230000004856 capillary permeability Effects 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 190000008236 carboplatin Chemical compound 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000005890 cell-mediated cytotoxicity Effects 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 230000003399 chemotactic effect Effects 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 108091008034 costimulatory receptors Proteins 0.000 description 1
- 239000002577 cryoprotective agent Substances 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 231100000050 cytotoxic potential Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 229940127276 delta-like ligand 3 Drugs 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 229940125436 dual inhibitor Drugs 0.000 description 1
- 238000001493 electron microscopy Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 229940115924 etigilimab Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 238000011124 ex vivo culture Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 108010052621 fas Receptor Proteins 0.000 description 1
- 102000018823 fas Receptor Human genes 0.000 description 1
- 229940124981 favezelimab Drugs 0.000 description 1
- 210000004700 fetal blood Anatomy 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 238000000799 fluorescence microscopy Methods 0.000 description 1
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012595 freezing medium Substances 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 1
- 230000017323 hematopoietic stem cell migration to bone marrow Effects 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 102000043321 human CTLA4 Human genes 0.000 description 1
- 102000043557 human IFNG Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229940121569 ieramilimab Drugs 0.000 description 1
- 102000027596 immune receptors Human genes 0.000 description 1
- 108091008915 immune receptors Proteins 0.000 description 1
- 230000037189 immune system physiology Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 238000003312 immunocapture Methods 0.000 description 1
- 239000000677 immunologic agent Substances 0.000 description 1
- 229940124541 immunological agent Drugs 0.000 description 1
- 238000013394 immunophenotyping Methods 0.000 description 1
- 238000012744 immunostaining Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000012606 in vitro cell culture Methods 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 108090000681 interleukin 20 Proteins 0.000 description 1
- 229940076264 interleukin-3 Drugs 0.000 description 1
- 102000010681 interleukin-8 receptors Human genes 0.000 description 1
- 108010038415 interleukin-8 receptors Proteins 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 210000001821 langerhans cell Anatomy 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000004777 loss-of-function mutation Effects 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 201000008443 lung non-squamous non-small cell carcinoma Diseases 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 208000019420 lymphoid neoplasm Diseases 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 210000001806 memory b lymphocyte Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000010232 migration assay Methods 0.000 description 1
- 230000001617 migratory effect Effects 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 230000009054 pathological process Effects 0.000 description 1
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012660 pharmacological inhibitor Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 238000011518 platinum-based chemotherapy Methods 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 229940087463 proleukin Drugs 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000012342 propidium iodide staining Methods 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 230000009145 protein modification Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000028617 response to DNA damage stimulus Effects 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 201000010106 skin squamous cell carcinoma Diseases 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 230000009211 stress pathway Effects 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 230000001502 supplementing effect Effects 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940061918 tebotelimab Drugs 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229950007133 tiragolumab Drugs 0.000 description 1
- 230000009258 tissue cross reactivity Effects 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 102000042286 type I cytokine receptor family Human genes 0.000 description 1
- 108091052247 type I cytokine receptor family Proteins 0.000 description 1
- 210000000037 type II NK T lymphocyte Anatomy 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229940020434 vibostolimab Drugs 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 229940055760 yervoy Drugs 0.000 description 1
- 108010065816 zeta chain antigen T cell receptor Proteins 0.000 description 1
Description
本発明は免疫療法の分野を対象とする。具体的には、本発明は癌の治療に有用な、養子細胞療法のための改良された細胞組成物と方法を提供する。 The present invention is directed to the field of immunotherapy. Specifically, the present invention provides improved cell compositions and methods for adoptive cell therapy useful in the treatment of cancer.
養子細胞移植(ACT、養子細胞療法とも称される)は、癌免疫療法で最も有望な戦略の1種であり、黒色腫、子宮頸癌、リンパ腫、白血病、胆管癌及び神経芽細胞腫等の様々な種類の癌に対する有効性が証明されている。 Adoptive cell transfer (ACT, also known as adoptive cell therapy) is one of the most promising strategies in cancer immunotherapy and has proven effective against various types of cancer, including melanoma, cervical cancer, lymphoma, leukemia, cholangiocarcinoma, and neuroblastoma.
ACTは免疫細胞の抗腫瘍活性の増強を試みる細胞療法である。これは対象から抽出された免疫細胞に基づくが、通常はエクスビボで処理し、大規模に増殖させた後、患者に戻す(自家療法)又は別の個人に移植する(同種異系療法)。ACTでは、Tリンパ球(T細胞)、ナチュラルキラー(NK)細胞、樹状細胞及び幹細胞等の様々な免疫細胞を使用することができる。 ACT is a cellular therapy that attempts to enhance the anti-tumor activity of immune cells. It is based on immune cells extracted from the subject, usually treated ex vivo, expanded on a large scale, and then either returned to the patient (autologous therapy) or transplanted into another individual (allogeneic therapy). A variety of immune cells can be used in ACT, including T lymphocytes (T cells), natural killer (NK) cells, dendritic cells, and stem cells.
腫瘍浸潤リンパ球(TIL)は、患者の腫瘍生検から単離され、エクスビボで増殖されてACT組成物を産生する腫瘍特異的T細胞である。このために、高濃度のIL-2、抗CD3抗体及び照射アロ反応性フィーダー細胞を使用してTILをエクスビボで増殖することが奨励されており、5日目にIL-2(抗CD3ではない)を補充する。8~15日間の増殖で得られた細胞をその後、支持的なIL-2投与と共に患者に戻す。しかし、ACT用のTIL組成物の産生は技術的に困難であり、IL-2とフィーダー細胞の使用によって望ましくない変異が生じる。更に、このアプローチは、腫瘍試料の入手可能性や、各試料から通常得られる初期TILの数が少ないことによって制限される。 Tumor infiltrating lymphocytes (TILs) are tumor-specific T cells isolated from patient tumor biopsies and expanded ex vivo to produce ACT compositions. For this purpose, it has been suggested to expand TILs ex vivo using high concentrations of IL-2, anti-CD3 antibodies and irradiated alloreactive feeder cells, supplemented with IL-2 (but not anti-CD3) on day 5. The cells obtained after 8-15 days of expansion are then returned to the patient with supportive IL-2 administration. However, the production of TIL compositions for ACT is technically challenging and the use of IL-2 and feeder cells introduces undesirable mutations. Furthermore, this approach is limited by the availability of tumor samples and the small number of initial TILs that are usually obtained from each sample.
ACT組成物の産生に使用することができる他の原料は末梢血単核細胞(PBMC)であり、そこからリンパ球集団を単離、増殖、及び/又は様々な手順によって操作し、その有効性を高めることができる。T細胞受容体(TCR)媒介の主要組織適合性複合体(MHC)-II依存的細胞傷害性を発揮するために、治療対象に対して自家性又は組織適合性の必要があるT細胞とは異なり、NK細胞(CD56又はCD16の発現とTCR-CD3複合体の欠如を特徴とする)やNK-T細胞等の他のリンパ球集団は、非MHCII制限的に作用する。しかし、このような集団は通常、有効性が限られているという特徴があり、臨床効果を示すためには大量の投与を必要とする。 Another source that can be used for the production of ACT compositions is peripheral blood mononuclear cells (PBMCs), from which lymphocyte populations can be isolated, expanded, and/or manipulated by various procedures to enhance their efficacy. Unlike T cells, which need to be autologous or histocompatible with the subject to be treated in order to exert T cell receptor (TCR)-mediated major histocompatibility complex (MHC)-II-dependent cytotoxicity, other lymphocyte populations, such as NK cells (characterized by expression of CD56 or CD16 and lack of TCR-CD3 complex) and NK-T cells, act in a non-MHCII-restricted manner. However, such populations are usually characterized by limited efficacy and require large doses to demonstrate clinical efficacy.
例えば、ナチュラルキラー(NK)細胞前駆体やTリンパ球等の様々なリンパ球種で構成されているリンホカイン活性化キラー(LAK)細胞は、サイトカイン(通常はIL-2)の存在下でPBMCを培養することによって産生される。このような不均一な細胞組成物は、IL-2とのインキュベーション後にインビトロで腫瘍細胞のHLA非依存的溶解を示す。LAK細胞は特定の個人において腫瘍退縮を媒介するのに非常に効果的であるが、癌退縮を媒介するには非常に多くの細胞を必要とするため、この治療アプローチを実行するのが難しい場合がある。更に、必要な高用量のIL-2は有毒な副作用を媒介し、その最も一般的なものは、重大な体液貯留を引き起こす毛細血管透過性漏出症候群である。 For example, lymphokine-activated killer (LAK) cells, which are composed of various lymphocyte types, including natural killer (NK) cell precursors and T lymphocytes, are produced by culturing PBMCs in the presence of cytokines, usually IL-2. Such heterogeneous cell compositions exhibit HLA-independent lysis of tumor cells in vitro after incubation with IL-2. Although LAK cells are highly effective in mediating tumor regression in certain individuals, this therapeutic approach can be difficult to implement due to the large number of cells required to mediate cancer regression. Furthermore, the high doses of IL-2 required mediate toxic side effects, the most common of which is capillary permeability leak syndrome, which causes significant fluid retention.
サイトカイン誘導キラー(CIK)細胞は、循環前駆体からインビトロで増殖させた非MHC制限的な細胞傷害性抗腫瘍細胞である。CIK細胞はT細胞とNK細胞の両方の特徴を共有する。PBMC、リンパ球除去又は臍帯血から開始するCIK細胞産生用の増殖プロトコルでは、0日目に1000U/mLのヒト組換えヒトインターフェロン-ガンマ(rIFN-γ)を連続的に添加した後、+1日目に、CD3(OKT3)に対するモノクローナル抗体を50ng/mLと500IU/mLのIL-2を添加する必要がある。IL-2と新鮮な完全X-VIVO培地(抗CD3抗体ではない)を5日毎に14~21日間添加する。この方法の最後に、2種の主要な細胞集団の産生が報告されている。1種の集団はCD3、CD8、NKG2D及びCD56陽性細胞を含み(通常、CIKと称される)、もう1種の集団はCD3、CD8及びNKG2D陽性細胞とCD56陰性細胞を含み、細胞傷害活性が欠如していることが報告されている(Introna et al, Int. J. Mol. Sci. 2018, 19, 358)。 Cytokine-induced killer (CIK) cells are non-MHC-restricted cytotoxic antitumor cells expanded in vitro from circulating precursors. CIK cells share characteristics of both T and NK cells. The expansion protocol for CIK cell production starting from PBMC, lymphodepleted or umbilical cord blood requires the sequential addition of 1000 U/mL human recombinant human interferon-gamma (rIFN-γ) on day 0, followed by the addition of 50 ng/mL of a monoclonal antibody against CD3 (OKT3) and 500 IU/mL of IL-2 on day +1. IL-2 and fresh complete X-VIVO medium (no anti-CD3 antibody) are added every 5 days for 14-21 days. At the end of this method, the production of two major cell populations has been reported. One population contains CD3, CD8, NKG2D, and CD56 positive cells (usually referred to as CIK), and the other population contains CD3, CD8, and NKG2D positive cells and CD56 negative cells, and has been reported to lack cytotoxic activity (Introna et al, Int. J. Mol. Sci. 2018, 19, 358).
CIKの相対的な安全性プロファイルにも関わらず、臨床診療で使用すると有効性に関して極端な不均一性がもたらされた。治療を受けた患者426名に対するCIK細胞の抗腫瘍効果を調査した11種の臨床試験の結果から、奏効が報告された384名(90.1%)の内、24名(5.6%)のみが完全奏効、27名(6.3%)が部分奏効(PR)、40名(9.4%)がやや有効であることが分かった。一方、161名(37.8%)の患者は安定、129名(30.3%)は進行であったが、これはCIK細胞療法の全体的な有効性は中程度であることを示している(Introna et al,同書)。 Despite the relative safety profile of CIK, its use in clinical practice has led to extreme heterogeneity regarding efficacy. Results from 11 clinical trials investigating the antitumor effect of CIK cells in 426 treated patients showed that of 384 (90.1%) who reported responses, only 24 (5.6%) had a complete response, 27 (6.3%) had a partial response (PR), and 40 (9.4%) had a minor response. Meanwhile, 161 (37.8%) patients had stable disease and 129 (30.3%) had progressive disease, indicating that the overall efficacy of CIK cell therapy is moderate (Introna et al., ibid.).
ACT用の様々な細胞組成物の単離及び/又はエクスビボ増殖のためのプロトコルが開示されている。通常、このようなプロトコルには、サイトカインの存在下での初期刺激及び/又はTCR/CD3アクチベーター等の他の刺激、及びそのようなサイトカインとアクチベーターの異なる組み合わせの存在下での増殖又は伝播が含まれることが多い。更に、そのようなプロトコルには、例えば、増殖の前後のいずれかに、アフェレーシス又はフローサイトメトリー又は対応する表面マーカーを標的とするビーズベースの方法によって所望の細胞集団を単離又は富化する追加の工程が含まれることもある。 Protocols for the isolation and/or ex vivo expansion of various cell compositions for ACT are disclosed. Typically, such protocols often include an initial stimulation in the presence of cytokines and/or other stimuli such as TCR/CD3 activators, and expansion or propagation in the presence of different combinations of such cytokines and activators. Furthermore, such protocols may include additional steps of isolating or enriching the desired cell population, for example, by apheresis or flow cytometry or bead-based methods targeting the corresponding surface markers, either before or after expansion.
例えば、US2020138861号明細書では、ドナーの末梢血からCD3陰性及びCD56陽性NK細胞を単離することを含む、炎症性腸疾患を治療するための養子移植手順が開示されている。US10149863号明細書では、単離された単核細胞をインビトロで培養し、T細胞の増殖と活性化を刺激する培養サイトカインを添加し、NKT様細胞表面マーカーを使用する細胞選別技法を用いてNKT様細胞を選別することを含む、単離哺乳動物NKT様細胞の調製方法が開示されている。 For example, US2020138861 discloses an adoptive transfer procedure for treating inflammatory bowel disease, which involves isolating CD3-negative and CD56-positive NK cells from the peripheral blood of a donor. US10149863 discloses a method for preparing isolated mammalian NKT-like cells, which involves culturing isolated mononuclear cells in vitro, adding culture cytokines that stimulate T cell proliferation and activation, and selecting the NKT-like cells using a cell sorting technique that uses NKT-like cell surface markers.
US2012258085号明細書は、腫瘍を有する対象由来の末梢血の細胞試料を用意することと、増殖した細胞集団の少なくとも35%が活性化NK細胞とNK様T細胞を含むまで細胞をインキュベート及び増殖させることと、増殖した細胞を回収することを含む、表現型CD3-CD56+を有する増殖及び活性化されたNK細胞と表現型CD3+CD56+を有するNK様T細胞を取得する方法に関する。特に、US2012258085号明細書では、PBMCをモノクローナル抗CD3抗体(OKT-3、10ng/mL)と500U/mLのIL-2の存在下で最初に培養し、5日後に、5%ヒト血清とIL-2を含むがOKT-3を含まない新鮮な培地を培養物に補充し、これを培養終了まで2~3日毎に行う方法が開示されている。 US2012258085 relates to a method for obtaining expanded and activated NK cells with the phenotype CD3 − CD56 + and NK-like T cells with the phenotype CD3 + CD56 + , comprising providing a cell sample of peripheral blood from a tumor-bearing subject, incubating and expanding the cells until at least 35% of the expanded cell population comprises activated NK cells and NK- like T cells , and harvesting the expanded cells. In particular, US2012258085 discloses a method in which PBMCs are first cultured in the presence of a monoclonal anti-CD3 antibody (OKT-3, 10 ng/mL) and 500 U/mL IL-2, and after 5 days, the culture is replenished with fresh medium containing 5% human serum and IL-2 but without OKT-3, and this is repeated every 2-3 days until the end of the culture.
多くのACT組成物の有効性が限られているため、遺伝子改変細胞の使用が提案されている。このような例としては、共有腫瘍関連抗原(TAA)由来のエピトープを認識するクローン化組換えTCRαβ鎖を発現するか、又はTCRζ鎖のシグナル伝達ドメインと共刺激分子(CD28やCD137/4-1BB等)に融合した腫瘍抗原を認識する免疫グロブリン可変領域で構成されるキメラ抗原受容体(CAR)を発現するPBMC(又はNK細胞等の他のリンパ球集団)から増殖したT細胞が挙げられる。例えば、US2020345779号とUS2020223920号では、αβT細胞、γδT細胞、NK細胞、NKT細胞、B細胞、自然リンパ球系細胞(ILC)、CIK細胞、細胞傷害性Tリンパ球(CTL)、LAK細胞及び制御性T細胞等の様々な細胞集団で発現することができるCARポリペプチドが開示されている。 Due to the limited efficacy of many ACT compositions, the use of genetically modified cells has been proposed. Examples of such include T cells expanded from PBMCs (or other lymphocyte populations such as NK cells) that express a cloned recombinant TCRαβ chain that recognizes an epitope derived from a shared tumor-associated antigen (TAA) or express a chimeric antigen receptor (CAR) composed of a signaling domain of the TCRζ chain and an immunoglobulin variable region that recognizes a tumor antigen fused to a costimulatory molecule (such as CD28 or CD137/4-1BB). For example, US2020345779 and US2020223920 disclose CAR polypeptides that can be expressed in various cell populations such as αβT cells, γδT cells, NK cells, NKT cells, B cells, innate lymphoid cells (ILCs), CIK cells, cytotoxic T lymphocytes (CTLs), LAK cells, and regulatory T cells.
近年の癌管理における開発にも関わらず、癌免疫療法で用いることができる更なる手段、改良方法及び細胞組成物が依然として必要とされている。特に、一次的又は二次的な治療に対する非反応性は癌患者の間で非常に蔓延しており、多くの既存の治療はかなりの有害作用を特徴としている。従って、有効性及び/又は安全性を高める治療手法の開発は非常に有益である。更に、生存率の向上及び/又は腫瘍の結合又は保持の強化を示すACT用の細胞組成物を得ることが有益となるであろう。 Despite recent developments in cancer management, there remains a need for further tools, improved methods and cell compositions that can be used in cancer immunotherapy. In particular, non-responsiveness to primary or secondary treatments is highly prevalent among cancer patients and many existing treatments are characterized by significant adverse effects. Therefore, the development of therapeutic approaches that increase efficacy and/or safety would be highly beneficial. Furthermore, it would be beneficial to have cell compositions for ACT that exhibit improved survival and/or enhanced tumor binding or retention.
本発明は免疫療法の分野を対象とする。具体的には、本発明は癌の治療に有用な、養子細胞療法(ACT)のための改良された細胞組成物と方法を提供する。より具体的には、本発明の実施形態では、高割合の活性化CD8+細胞、特にCD8+NKG2D+グランザイムB+細胞を含み、細胞傷害性の増強を特徴とする細胞組成物(本明細書では超活性化キラー細胞(SAK)と称する)の使用を採用する。本発明は更に、末梢血単核細胞(PBMC)からSAK組成物を産生するための方法、及び癌管理におけるその利用に関する。有利な実施形態によれば、本発明は更に、生存率の向上及び/又は腫瘍の結合及び保持の強化を示す遺伝子改変SAK組成物に関する。 The present invention is directed to the field of immunotherapy. In particular, the present invention provides improved cell compositions and methods for adoptive cell therapy (ACT), useful in the treatment of cancer. More particularly, embodiments of the present invention employ the use of cell compositions (referred to herein as superactivated killer cells (SAK)) that contain a high percentage of activated CD8 + cells, particularly CD8 + NKG2D + Granzyme B + cells, and that are characterized by enhanced cytotoxicity. The present invention further relates to methods for producing SAK compositions from peripheral blood mononuclear cells (PBMCs), and their use in cancer management. According to advantageous embodiments, the present invention further relates to genetically modified SAK compositions that exhibit improved survival and/or enhanced tumor binding and retention.
本発明は、部分的には、様々な腫瘍細胞に対して非常に強力な抗腫瘍細胞傷害活性を発揮することができるSAK組成物の開発に基づく。驚くべきことに、本明細書に開示の独特の培養方法によってPBMCから産生されたSAK細胞は、他のPBMC由来のエフェクター細胞組成物(これまで知られているACTプロトコルに従って産生された、サイトカイン誘導キラー細胞(CIK)及びインターロイキン-2(IL-2)増殖T細胞組成物が含まれる)よりも腫瘍細胞の根絶において有意に有効であった。更に、産生されたSAK細胞は、ヒトリンパ腫(Raji)生着マウスモデルにおいて、インビボでの腫瘍発生の阻害に非常に有効であることが分かった。 The present invention is based, in part, on the development of a SAK composition capable of exerting highly potent antitumor cytotoxic activity against a variety of tumor cells. Surprisingly, SAK cells produced from PBMCs by the unique culture method disclosed herein were significantly more effective at eradicating tumor cells than other PBMC-derived effector cell compositions, including cytokine-induced killer cells (CIK) and interleukin-2 (IL-2)-expanded T cell compositions produced according to previously known ACT protocols. Furthermore, the produced SAK cells were found to be highly effective in inhibiting tumor development in vivo in a human lymphoma (Raji) engrafted mouse model.
本明細書に開示のように、産生されたSAK集団は、CD8+細胞画分全体の表面で検出される高いNKG2Dレベルを特徴とし、NKG2Dリガンドを発現する腫瘍に対して特に有効であることが分かった。本明細書で更に開示のように、SAK集団はFasL、グランザイムB及びパーフォリンの高発現レベルを特徴とするが、その抗腫瘍細胞傷害活性は主にグランザイムBとパーフォリンによって媒介され、細胞間接触が必要であるにも関わらず、抗FasL中和抗体の使用に対して殆ど非感受性であることが分かった。これは、細胞傷害活性が実質的にFAS媒介であると報告された、これまで知られているACTの細胞集団とは対照的である。更に、この細胞は、エフェクター免疫細胞の生存、増殖及び/又は活性にプラスの影響を与えることが知られている受容体の大量の発現や、疲労及び/又は活性化誘発細胞死と関連していることが知られている抑制性チェックポイント分子の発現の最小化等、細胞表面受容体の有利なプロファイルを示すことが実証された。本明細書で更に実証するように、SAK細胞は、様々な腫瘍細胞に対してMHC依存的細胞傷害活性と非MHC依存的細胞傷害活性の両方を保有することが分かった。更に、SAK細胞の養子移植により安全性が向上し、移植片対宿主病(GVHD)を引き起こすリスクが顕著に減少することが示され、凍結保存後も高レベルの活性が維持された。また、本発明は、部分的には、抗腫瘍活性が増強された遺伝子改変SAK組成物の産生にも基づく。 As disclosed herein, the generated SAK population is characterized by high NKG2D levels detected on the surface of the entire CD8 + cell fraction and has been found to be particularly effective against tumors expressing NKG2D ligands. As further disclosed herein, the SAK population is characterized by high expression levels of FasL, granzyme B and perforin, but its antitumor cytotoxic activity is found to be largely insensitive to the use of anti-FasL neutralizing antibodies, despite being mediated primarily by granzyme B and perforin and requiring cell-cell contact. This is in contrast to previously known ACT cell populations, whose cytotoxic activity was reported to be substantially FAS-mediated. Furthermore, the cells have been demonstrated to display a favorable cell surface receptor profile, including abundant expression of receptors known to positively affect survival, proliferation and/or activity of effector immune cells, and minimal expression of inhibitory checkpoint molecules known to be associated with exhaustion and/or activation-induced cell death. As further demonstrated herein, SAK cells have been found to possess both MHC-dependent and non-MHC-dependent cytotoxic activity against a variety of tumor cells. Moreover, adoptive transfer of SAK cells has been shown to improve safety, significantly reducing the risk of causing graft-versus-host disease (GVHD), and maintains high levels of activity after cryopreservation. The present invention is also based, in part, on the production of genetically modified SAK compositions with enhanced antitumor activity.
従って、本発明の実施形態では、改善された治療特性を特徴とするACT用細胞組成物が提供される。他の実施形態では、本発明は、ACT用細胞組成物を産生するための改良方法に関する。幾つかの実施形態によれば、本発明の細胞組成物は、アフェレーシスの先行工程又は他の精製工程を行わず、腫瘍生検由来の腫瘍特異的細胞を単離する必要もなく、本明細書に開示のエクスビボ増殖プロトコルによってPBMC試料から直接有利に産生することができる。幾つかの実施形態では、本発明の細胞組成物は、IL-2とCD3アクチベーター(例えば、CD3特異的刺激抗体)の存在下でのインキュベーション又は培養を少なくとも9日間、典型的には10~16日間(例えば、10~12日間)行って試料の細胞を増殖させる方法によって産生する。本明細書に開示の有利な実施形態によれば、有効量のIL-2とCD3アクチベーターの常時存在下で細胞を増殖して、有効量のIL-2とCD3アクチベーターを増殖期間を通じて定期的(通常は2~4日毎)に細胞に補充するようにする。 Thus, in embodiments of the present invention, a cell composition for ACT is provided that is characterized by improved therapeutic properties. In other embodiments, the present invention relates to an improved method for producing a cell composition for ACT. According to some embodiments, the cell composition of the present invention can be advantageously produced directly from a PBMC sample by the ex vivo expansion protocol disclosed herein, without a preceding step of apheresis or other purification steps, and without the need to isolate tumor-specific cells from a tumor biopsy. In some embodiments, the cell composition of the present invention is produced by a method in which the cells of the sample are expanded by incubation or culture in the presence of IL-2 and a CD3 activator (e.g., a CD3-specific stimulating antibody) for at least 9 days, typically 10-16 days (e.g., 10-12 days). According to advantageous embodiments disclosed herein, the cells are expanded in the constant presence of an effective amount of IL-2 and a CD3 activator, such that the cells are periodically replenished with an effective amount of IL-2 and a CD3 activator throughout the expansion period (usually every 2-4 days).
一様相では、インビトロ増殖PBMC(例えば、少なくとも5×106~10×108個の生細胞)を含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であって、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。 In one aspect, an ACT cell composition is provided that comprises in vitro expanded PBMCs (e.g., at least 5 x 10 6 to 10 x 10 8 viable cells), 70-85% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% are CD56 - cells, and up to 5% are CD3 - cells, and that exhibits significant cytotoxic activity against tumor cells in vitro and in vivo.
一実施形態では、細胞の7~12%がCD3+CD4+細胞であり、20~28%がCD3+CD56+細胞であり、10~14%がCD3+CD8+CD56+細胞である。他の実施形態では、組成物中のCD3+CD8+細胞の量は、組成物中のCD3+CD4+細胞の量の6~8倍である。例示的な実施形態では、細胞組成物は、CD3+CD4+細胞に対するCD3+CD8+細胞の比率が7~8であることを特徴とする。他の実施形態では、組成物は3%未満のCD3-CD56+細胞を含む。他の実施形態では、CD3+CD8+細胞の少なくとも40%は、パーフォリン、Fasリガンド(FasL)、CXCR4及びCXCR2から成る群から選択される少なくとも1種のマーカーの表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞は、DNAM-1+、CTLA-4-細胞として更に特徴付けられる。他の実施形態では、CD3+CD8+細胞の少なくとも90%がCXCR3の表面発現を特徴とし、CD3+CD8+細胞の少なくとも30%がCXCR6の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の50%以下が、CTLA-4、PD-1及びTIGITから成る群から選択される複数の免疫チェックポイント分子の表面発現を特徴とする。 In one embodiment, 7-12% of the cells are CD3 + CD4 + cells, 20-28% are CD3 + CD56 + cells, and 10-14% are CD3 + CD8 + CD56 + cells. In another embodiment, the amount of CD3 + CD8 + cells in the composition is 6-8 times the amount of CD3 + CD4 + cells in the composition. In an exemplary embodiment, the cell composition is characterized by a ratio of CD3 + CD8 + cells to CD3 + CD4 + cells of 7-8. In another embodiment, the composition comprises less than 3% CD3 - CD56 + cells. In another embodiment, at least 40% of the CD3 + CD8 + cells are characterized by surface expression of at least one marker selected from the group consisting of perforin, Fas ligand (FasL), CXCR4, and CXCR2 . In other embodiments, the CD3 + CD8 + cells are further characterized as DNAM-1 + , CTLA-4 − cells. In other embodiments, at least 90% of the CD3 + CD8 + cells are characterized by surface expression of CXCR3 and at least 30% of the CD3 + CD8 + cells are characterized by surface expression of CXCR6. In other embodiments, no more than 50% of the CD3 + CD8 + cells are characterized by surface expression of multiple immune checkpoint molecules selected from the group consisting of CTLA-4, PD-1, and TIGIT.
他の実施形態では、細胞組成物は、腫瘍細胞に対してグランザイムB及び/又はパーフォリンによって媒介される非MHC制限細胞傷害活性を示す。他の実施形態では、細胞組成物は、造血器腫瘍細胞と固形腫瘍細胞に対してグランザイムB及び/又はパーフォリンによって媒介される非MHC制限細胞傷害活性を示す。他の実施形態では、細胞傷害活性は、FAS-FasL相互作用によって実質的に媒介されない。他の実施形態では、細胞組成物は、GVHDを実質的に誘発することなく、インビボで非組織適合性対象における腫瘍の発生を阻害又は防止することができる。 In other embodiments, the cell composition exhibits non-MHC restricted cytotoxic activity mediated by granzyme B and/or perforin against tumor cells. In other embodiments, the cell composition exhibits non-MHC restricted cytotoxic activity mediated by granzyme B and/or perforin against hematopoietic tumor cells and solid tumor cells. In other embodiments, the cytotoxic activity is not substantially mediated by FAS-FasL interactions. In other embodiments, the cell composition is capable of inhibiting or preventing the development of tumors in non-histocompatible subjects in vivo without substantially inducing GVHD.
他の実施形態では、細胞組成物は、インビトロで0.25:1の比率で24時間以内に造血器腫瘍細胞を特異的に根絶することができる。他の実施形態では、細胞組成物は、インビトロで造血器腫瘍細胞と0.25:1の比率で24時間インキュベートした場合に、有意に造血器腫瘍細胞を特異的に根絶することができる、及び/又は、組成物細胞と腫瘍細胞の比率を1:1としてインビトロで24時間インキュベートすると腫瘍細胞の80~90%が根絶されるように造血器腫瘍細胞を特異的に根絶することができる。他の実施形態では、腫瘍細胞は少なくとも1種のNKG2Dリガンドを発現する。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。他の実施形態では、細胞組成物は5×106~10×109個の生細胞を含む。 In another embodiment, the cell composition is capable of specifically eradicating hematopoietic tumor cells in vitro within 24 hours at a ratio of 0.25:1. In another embodiment, the cell composition is capable of significantly specifically eradicating hematopoietic tumor cells when incubated in vitro with hematopoietic tumor cells at a ratio of 0.25:1 for 24 hours and/or is capable of specifically eradicating hematopoietic tumor cells such that 80-90% of tumor cells are eradicated when incubated in vitro for 24 hours at a ratio of 1:1 composition cells to tumor cells. In another embodiment, the tumor cells express at least one NKG2D ligand. In another embodiment, the tumor cells express multiple NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In another embodiment, the cell composition comprises between 5x106 and 10x109 viable cells.
他の実施形態では、細胞組成物は、有効量のIL-2とCD3アクチベーターの存在下で少なくとも9日間PBMCを増殖させることを含む方法によって調製し、有効量のIL-2とCD3アクチベーターを48~96時間毎に補充する。他の実施形態では、インキュベーションは他のサイトカイン又は抗体の非存在下で行う。 In another embodiment, the cell composition is prepared by a method comprising expanding PBMCs in the presence of an effective amount of IL-2 and a CD3 activator for at least 9 days, and replenishing the effective amount of IL-2 and a CD3 activator every 48-96 hours. In another embodiment, the incubation is performed in the absence of other cytokines or antibodies.
他の実施形態では、細胞組成物の細胞を遺伝子改変する。他の実施形態では、キメラ抗原受容体(CAR)、サイトカイン、ケモカイン、サイトカイン又はケモカイン受容体、又はそれらの任意の組み合わせの内の少なくとも1種を発現するように細胞を遺伝子改変する。他の実施形態では、BCMA、CD47、PDL-1、メソテリン、EpCAM、CD34、CD44、PSCA、MUC16、CD276、CD123、CD19、CD20及びEFFRvIIIから成る群から選択される抗原に対するCARを発現するように細胞組成物を遺伝子改変する。他の実施形態では、細胞組成物は、CD19特異的CAR及びCD123特異的CARのいずれか1種を発現する。他の実施形態では、C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体を発現するように細胞を遺伝子改変する。他の実施形態では、細胞組成物は、外因性IL-2、外因性IL-15、外因性IL-21のいずれか1種、又はそれらの任意の組み合わせを発現する。他の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように細胞組成物を改変する。 In another embodiment, the cells of the cell composition are genetically modified. In another embodiment, the cells are genetically modified to express at least one of a chimeric antigen receptor (CAR), a cytokine, a chemokine, a cytokine or chemokine receptor, or any combination thereof. In another embodiment, the cell composition is genetically modified to express a CAR for an antigen selected from the group consisting of BCMA, CD47, PDL-1, mesothelin, EpCAM, CD34, CD44, PSCA, MUC16, CD276, CD123, CD19, CD20, and EFFRvIII. In another embodiment, the cell composition expresses one of a CD19-specific CAR and a CD123-specific CAR. In another embodiment, the cells are genetically modified to express a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain. In another embodiment, the cell composition expresses one of exogenous IL-2, exogenous IL-15, exogenous IL-21, or any combination thereof. In other embodiments, the cell composition is modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21.
他の様相では、ACT用細胞組成物を産生するための方法であって、
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを48~96時間毎に補充することと
を含み、培養物中の生細胞の数を少なくとも20倍増加させ、少なくとも5×106~10×108個の生細胞を含む細胞組成物を得る方法が提供される。
In another aspect, a method for producing a cell composition for ACT, comprising:
a. providing a PBMC sample;
b. A method is provided which comprises expanding PBMCs in the constant presence of effective amounts of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, for at least 9 days, with replenishing effective amounts of IL-2 and a CD3 activator every 48-96 hours, thereby increasing the number of viable cells in the culture by at least 20 fold, and obtaining a cell composition comprising at least 5 x 10 to 10 x 10 viable cells.
他の実施形態では、唯一の外因的に添加された細胞刺激因子としてのIL-2とCD3アクチベーターの常時存在下で増殖を行う。他の実施形態では、工程bは、500~1500IU/mLのIL-2と10~60ng/mLの抗CD3抗体の存在下で細胞を増殖させることを含み、これらは48~96時間毎に10~16日間再補充する。他の実施形態では、更なる富化、刺激又は増殖工程に細胞を供しない。他の実施形態では、生細胞の数を少なくとも30倍増加させるように増殖を行う。他の実施形態では、5×106~10×109個の生存可能な単核細胞(その70~85%はNKG2DとグランザイムBを発現するCD3+CD8+細胞である)を含む細胞組成物を産生するように増殖を行う。他の実施形態では、インビトロで造血器腫瘍細胞と0.25:1の比率で24時間インキュベートした場合に、有意に造血器腫瘍細胞を特異的に根絶することができる細胞組成物を産生するように増殖を行う。他の実施形態では、得られた細胞組成物は、インビトロで0.25:1の比率で24時間以内に造血器腫瘍細胞を特異的に根絶することができる。他の実施形態では、工程(b)は細胞を遺伝子改変することを更に含む。他の実施形態では、この方法は、CAR、サイトカイン、ケモカイン、サイトカイン又はケモカインの受容体、又はそれらの任意の組み合わせの内の少なくとも1種を発現するように細胞を遺伝子改変することを含む。他の実施形態では、この方法は、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように細胞を改変することを含む。他の実施形態では、この方法は、得られた細胞組成物を凍結保存することを更に含む。 In another embodiment, the expansion is carried out in the constant presence of IL-2 and a CD3 activator as the only exogenously added cell stimulatory factors. In another embodiment, step b comprises expanding the cells in the presence of 500-1500 IU/mL IL-2 and 10-60 ng/mL anti-CD3 antibody, which are replenished every 48-96 hours for 10-16 days. In another embodiment, the cells are not subjected to further enrichment, stimulation or expansion steps. In another embodiment, the expansion is carried out to increase the number of viable cells by at least 30-fold. In another embodiment, the expansion is carried out to produce a cell composition comprising 5×10 6 to 10×10 9 viable mononuclear cells, of which 70-85% are CD3 + CD8 + cells expressing NKG2D and granzyme B. In another embodiment, the expansion is carried out to produce a cell composition capable of specifically eradicating hematopoietic tumor cells when incubated in vitro with hematopoietic tumor cells at a ratio of 0.25:1 for 24 hours. In other embodiments, the resulting cell composition is capable of specifically eradicating hematopoietic malignant cells in vitro at a ratio of 0.25:1 within 24 hours. In other embodiments, step (b) further comprises genetically modifying the cells. In other embodiments, the method comprises genetically modifying the cells to express at least one of a CAR, a cytokine, a chemokine, a receptor for a cytokine or chemokine, or any combination thereof. In other embodiments, the method comprises modifying the cells to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In other embodiments, the method further comprises cryopreserving the resulting cell composition.
他の実施形態では、この方法によって調製された細胞組成物が提供される。他の実施形態では、
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを2~4日間毎に補充することとで、培養物中の生細胞の数を少なくとも20倍増加させ、それによって少なくとも5×106~10×108個の生細胞を含む細胞組成物を得ることと、
c.得られた細胞組成物を回収することと
を含む方法によって調製される、ACT用細胞組成物が提供される。
In another embodiment, a cell composition prepared by this method is provided.
a. providing a PBMC sample;
b. expanding the PBMCs for at least 9 days in the constant presence of an effective amount of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, whereby the number of viable cells in the culture is increased by at least 20 fold by replenishing the effective amount of IL-2 and the CD3 activator every 2-4 days, thereby obtaining a cell composition comprising at least 5 x 106 to 10 x 108 viable cells;
and c. recovering the resulting cell composition.
他の様相では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む細胞組成物が提供される。 In another aspect, a cell composition is provided that includes leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor that includes a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21.
他の実施形態では、細胞の70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞である。他の実施形態では、有効量のIL-2とCD3アクチベーターの常時存在下で少なくとも9日間PBMCを増殖させることを含み、有効量のIL-2とCD3アクチベーターを48~96時間補充する方法によって細胞組成物を調製する。 In another embodiment, 70-85% of the cells are CD3 + CD8 + cells that express NKG2D and granzyme B, at least 70% are CD56 − cells, and up to 5% are CD3 − cells. In another embodiment, the cell composition is prepared by a method comprising expanding PBMCs in the constant presence of an effective amount of IL-2 and a CD3 activator for at least 9 days, supplemented with an effective amount of IL-2 and a CD3 activator for 48-96 hours.
他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療用である本発明の細胞組成物が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療用するためのACT細胞組成物であって、少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であり、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。他の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む、腫瘍の治療を必要とする対象において腫瘍の治療に使用するための細胞組成物が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療に使用するための、本明細書に開示の方法によって調製される細胞組成物が提供される。 In another embodiment, a cell composition of the present invention is provided for treating a tumor in a subject in need of such treatment. In another embodiment, an ACT cell composition for treating a tumor in a subject in need of such treatment is provided, the ACT cell composition comprising at least 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, 70-85% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% are CD56 − cells, and up to 5% are CD3 − cells, and exhibits significant cytotoxic activity against tumor cells in vitro and in vivo. In another embodiment, a cell composition for use in treating a tumor in a subject in need of such treatment is provided, the cell composition comprising leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In other embodiments, there is provided a cell composition prepared by the methods disclosed herein for use in treating a tumor in a subject in need thereof.
様々な実施形態では、腫瘍は造血器腫瘍又は固形腫瘍である。他の実施形態では、腫瘍は白血病、多発性骨髄腫、前立腺腫瘍又は中皮腫から成る群から選択される。他の実施形態では、腫瘍は少なくとも1種のNKG2Dリガンドの発現を特徴とする。他の実施形態では、腫瘍は、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。他の実施形態では、腫瘍は、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3を発現する。他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド、CXCR2リガンド及びCXCR3リガンドから成る群から選択される少なくとも1種のケモカインの発現を特徴とする。 In various embodiments, the tumor is a hematopoietic tumor or a solid tumor. In other embodiments, the tumor is selected from the group consisting of leukemia, multiple myeloma, prostate tumor, or mesothelioma. In other embodiments, the tumor is characterized by expression of at least one NKG2D ligand. In other embodiments, the tumor expresses multiple NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, the tumor expresses MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, the tumor is characterized by expression of at least one chemokine selected from the group consisting of CXCR6 ligand, CXCR4 ligand, CXCR2 ligand, and CXCR3 ligand.
他の実施形態では、腫瘍は、CTLA-4、PD-1及びTIGITから成る群から選択される抑制性免疫チェックポイント分子の少なくとも1種のリガンドの発現を特徴とする。他の実施形態では、腫瘍は、少なくとも1種のチェックポイント分子阻害剤による治療に対して抵抗性である。他の実施形態では、腫瘍は、少なくとも1種のCTLA-4特異的遮断抗体による治療に対して抵抗性である。他の実施形態では、対象は、チェックポイント分子阻害剤を用いた治療レジメンを受けていない。他の実施形態では、使用は、Lag-3、Tim-3又はそれらの組み合わせに対する少なくとも1種のチェックポイント分子阻害剤を対象に投与することを更に含む。 In another embodiment, the tumor is characterized by expression of at least one ligand of an inhibitory immune checkpoint molecule selected from the group consisting of CTLA-4, PD-1, and TIGIT. In another embodiment, the tumor is resistant to treatment with at least one checkpoint molecule inhibitor. In another embodiment, the tumor is resistant to treatment with at least one CTLA-4 specific blocking antibody. In another embodiment, the subject has not received a treatment regimen with a checkpoint molecule inhibitor. In another embodiment, the use further comprises administering to the subject at least one checkpoint molecule inhibitor against Lag-3, Tim-3, or a combination thereof.
他の実施形態では、細胞組成物は凍結保存及び解凍を経ている。様々な実施形態では、細胞組成物は、対象に対して自家性、組織適合性同種異系、又は非組織適合性同種異系である。特定の実施形態では、細胞組成物は、対象に対して非組織適合性同種異系である。他の実施形態では、腫瘍はMHCI発現及び/又は活性のダウンレギュレーションを特徴とする。 In other embodiments, the cell composition has undergone cryopreservation and thawing. In various embodiments, the cell composition is autologous, histocompatible allogeneic, or non-histocompatible allogeneic to the subject. In certain embodiments, the cell composition is non-histocompatible allogeneic to the subject. In other embodiments, the tumor is characterized by downregulation of MHC I expression and/or activity.
他の様相では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量の本発明の細胞組成物と接触させ、それによって対象の腫瘍を治療することを含む方法が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量のACT細胞組成物と接触させることを含む方法が提供されるが、ACT細胞組成物は、少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%はNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%はCD56-細胞であり、最大5%はCD3-細胞であって、組成物はインビトロとインビボで腫瘍細胞に対して有意な細胞傷害活性を示し、それによって対象の腫瘍を治療する。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量の細胞組成物と接触させることを含む方法が提供されるが、該細胞組成物は、腫瘍の治療を必要とする対象において腫瘍を治療するために使用される、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含み、それによって対象の腫瘍を治療する。他の様相では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を本明細書に開示する方法により調製される細胞組成物の治療有効量と接触させ、それによって対象の腫瘍を治療することを含む方法が提供される。 In another aspect, a method of treating a tumor in a subject in need of such treatment is provided comprising contacting tumor cells with a therapeutically effective amount of a cell composition of the invention, thereby treating the tumor in the subject. In another embodiment, a method of treating a tumor in a subject in need of such treatment is provided comprising contacting tumor cells with a therapeutically effective amount of an ACT cell composition comprising at least 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, 70-85% of which are CD3 + CD8 + cells that express NKG2D and granzyme B, at least 70% of which are CD56 − cells, and up to 5% of which are CD3 − cells, and the composition exhibits significant cytotoxic activity against tumor cells in vitro and in vivo, thereby treating the tumor in the subject. In another embodiment, a method of treating a tumor in a subject in need of tumor treatment is provided comprising contacting tumor cells with a therapeutically effective amount of a cell composition comprising: (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) a leukocyte genetically modified to express at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21, for use in treating a tumor in a subject in need of tumor treatment, thereby treating the tumor in the subject. In another aspect, a method of treating a tumor in a subject in need of tumor treatment is provided comprising contacting tumor cells with a therapeutically effective amount of a cell composition prepared by the methods disclosed herein, thereby treating the tumor in the subject.
様々な実施形態では、細胞組成物は、対象に対して自家性、組織適合性同種異系、又は非組織適合性同種異系である。特定の実施形態では、細胞組成物は、対象に対して非組織適合性同種異系である。他の実施形態では、接触はインビボで行う。他の実施形態では、接触はエクスビボで行う。他の実施形態では、細胞組成物は、腫瘍細胞と接触させる前に凍結保存及び解凍を経ている。他の実施形態では、腫瘍は造血器腫瘍である。他の実施形態では、腫瘍は固形腫瘍である。他の実施形態では、腫瘍は白血病、多発性骨髄腫、前立腺腫瘍又は中皮腫から成る群から選択される。他の実施形態では、腫瘍は少なくとも1種のNKG2Dリガンドの発現を特徴とする。他の実施形態では、腫瘍は、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。他の実施形態では、腫瘍は、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3を発現する。 In various embodiments, the cell composition is autologous, histocompatible allogeneic, or non-histocompatible allogeneic to the subject. In certain embodiments, the cell composition is non-histocompatible allogeneic to the subject. In other embodiments, the contacting occurs in vivo. In other embodiments, the contacting occurs ex vivo. In other embodiments, the cell composition has undergone cryopreservation and thawing prior to contacting with the tumor cells. In other embodiments, the tumor is a hematopoietic tumor. In other embodiments, the tumor is a solid tumor. In other embodiments, the tumor is selected from the group consisting of leukemia, multiple myeloma, prostate tumor, or mesothelioma. In other embodiments, the tumor is characterized by expression of at least one NKG2D ligand. In other embodiments, the tumor expresses a plurality of NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, the tumor expresses MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3.
他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド、CXCR2リガンド及びCXCR3リガンドから成る群から選択される少なくとも1種のケモカインの発現を特徴とする。他の実施形態では、腫瘍は、CTLA-4、PD-1及びTIGITから成る群から選択される抑制性免疫チェックポイント分子の少なくとも1種のリガンドの発現を特徴とする。他の実施形態では、腫瘍は、少なくとも1種のチェックポイント分子阻害剤による治療に対して抵抗性である。他の実施形態では、腫瘍は、少なくとも1種のCTLA-4特異的遮断抗体による治療に対して抵抗性である。他の実施形態では、対象は、チェックポイント分子阻害剤を用いた治療レジメンを受けていない。他の実施形態では、使用は、Lag-3、Tim-3又はそれらの組み合わせに対する少なくとも1種のチェックポイント分子阻害剤を対象に投与することを更に含む。他の実施形態では、腫瘍はMHCI発現及び/又は活性のダウンレギュレーションを特徴とする。 In another embodiment, the tumor is characterized by expression of at least one chemokine selected from the group consisting of CXCR6 ligand, CXCR4 ligand, CXCR2 ligand, and CXCR3 ligand. In another embodiment, the tumor is characterized by expression of at least one ligand of an inhibitory immune checkpoint molecule selected from the group consisting of CTLA-4, PD-1, and TIGIT. In another embodiment, the tumor is resistant to treatment with at least one checkpoint molecule inhibitor. In another embodiment, the tumor is resistant to treatment with at least one CTLA-4 specific blocking antibody. In another embodiment, the subject has not received a treatment regimen with a checkpoint molecule inhibitor. In another embodiment, the use further comprises administering to the subject at least one checkpoint molecule inhibitor against Lag-3, Tim-3, or a combination thereof. In another embodiment, the tumor is characterized by downregulation of MHCI expression and/or activity.
本発明の他の目的、特徴及び利点は、以下の説明と図面から明らかになるであろう。 Other objects, features and advantages of the present invention will become apparent from the following description and drawings.
本発明は癌の治療に有用な、養子細胞療法(ACT)のための改良された細胞組成物と方法に関する。より具体的には、本発明の実施形態では、細胞傷害性の増強(例えば、非組織適合性腫瘍細胞に対する)を特徴とする細胞組成物(本明細書では超活性化キラー細胞(SAK)と称する)の使用を採用する。本発明は更に、SAK組成物を産生するための方法、及び癌管理におけるその利用に関する。有利な実施形態によれば、本発明は更に、生存率の向上及び/又は腫瘍の結合及び保持の強化を示す遺伝子改変SAK組成物に関する。 The present invention relates to improved cell compositions and methods for adoptive cell therapy (ACT), useful in the treatment of cancer. More specifically, embodiments of the invention employ the use of cell compositions, referred to herein as superactivated killer cells (SAK), characterized by enhanced cytotoxicity (e.g., against non-histocompatible tumor cells). The present invention further relates to methods for producing SAK compositions and their use in cancer management. According to advantageous embodiments, the present invention further relates to genetically modified SAK compositions that exhibit improved survival and/or enhanced tumor binding and retention.
本発明は、部分的には、MHC認識を必要とせずに、様々な腫瘍細胞に対して非常に強力に抗腫瘍細胞傷害活性を発揮することができるSAK組成物の開発に基づく。驚くべきことに、本明細書に開示の独特の培養方法によって末梢血単核細胞(PBMC)から産生されたSAK細胞は、他のPBMC由来のエフェクター細胞組成物(これまで知られているACTプロトコルに従って産生された、サイトカイン誘導キラー細胞(CIK)及びインターロイキン-2(IL-2)増殖T細胞組成物が含まれる)よりも腫瘍細胞の根絶において有意に有効であった。更に、産生されたSAK細胞は、インビボでの腫瘍発生の阻害に非常に有効であり、安全性が向上し、移植片対宿主病(GVHD)を引き起こすリスクが顕著に減少し、凍結保存に適合することも分かった。更に、この細胞は、細胞表面受容体(免疫チェックポイント分子やケモカイン受容体を含む)の有利なプロファイルを示すことが実証された。特に、SAK細胞は、DNAM-1を含む活性化受容体の高発現と、抑制性チェックポイント分子(例えば、CTLA-4)の発現の最小化を特徴とすることが分かった。また、本発明は、部分的には、腫瘍特異的活性の増強及び/又は延長を伴う改良治療手法に適した遺伝子改変SAK細胞組成物の開発にも基づく。 The present invention is based, in part, on the development of a SAK composition capable of exerting highly potent antitumor cytotoxic activity against a variety of tumor cells without the need for MHC recognition. Surprisingly, SAK cells generated from peripheral blood mononuclear cells (PBMCs) by the unique culture method disclosed herein were significantly more effective in eradicating tumor cells than other PBMC-derived effector cell compositions, including cytokine-induced killer cells (CIK) and interleukin-2 (IL-2) expanded T cell compositions generated according to previously known ACT protocols. Furthermore, the generated SAK cells were found to be highly effective in inhibiting tumor development in vivo, with improved safety, a significantly reduced risk of causing graft-versus-host disease (GVHD), and compatible with cryopreservation. Furthermore, the cells were demonstrated to exhibit a favorable profile of cell surface receptors, including immune checkpoint molecules and chemokine receptors. In particular, SAK cells have been found to be characterized by high expression of activating receptors, including DNAM-1, and minimal expression of inhibitory checkpoint molecules (e.g., CTLA-4). The present invention is also based, in part, on the development of genetically modified SAK cell compositions suitable for improved therapeutic approaches with enhanced and/or prolonged tumor-specific activity.
従って、本発明の実施形態では、改善された治療特性を特徴とするACT用細胞組成物が提供される。他の実施形態では、本発明は、ACT用細胞組成物を産生するための改良方法に関する。幾つかの実施形態によれば、本発明の細胞組成物は、アフェレーシスの先行工程又は他の精製工程を行わず、腫瘍生検由来の腫瘍特異的細胞を単離する必要もなく、本明細書に開示のエクスビボ増殖プロトコルによってPBMC試料から直接有利に産生することができる。幾つかの実施形態では、IL-2とCD3アクチベーター(例えば、CD3特異的刺激抗体)の存在下でのインキュベーション又は培養を少なくとも9日間、典型的には9~16日間(より典型的には10~12日間)行って試料の細胞(例えば、0.25×106~5×106個のPBMC)を増殖させる。抗CD3抗体は遊離懸濁液の形態で好都合に使用することができ、プレートに結合させる必要も、同種異系フィーダー細胞によって提示する必要もないことを理解されたい。 Thus, in embodiments of the invention, a cell composition for ACT is provided that is characterized by improved therapeutic properties. In other embodiments, the invention relates to an improved method for producing a cell composition for ACT. According to some embodiments, the cell composition of the invention can be advantageously produced directly from a PBMC sample by the ex vivo expansion protocol disclosed herein, without a preceding step of apheresis or other purification steps, and without the need to isolate tumor-specific cells from a tumor biopsy. In some embodiments, incubation or culture in the presence of IL-2 and a CD3 activator (e.g., a CD3-specific stimulating antibody) is carried out for at least 9 days, typically 9-16 days (more typically 10-12 days), to expand the cells of the sample (e.g., 0.25x106 to 5x106 PBMCs). It is to be understood that the anti-CD3 antibody can be conveniently used in the form of a free suspension and does not need to be plate-bound or presented by allogeneic feeder cells.
特に、有効量のIL-2とCD3アクチベーターの常時存在下で有利に細胞を増殖し、全増殖期間を通じて十分な量で維持されるようにする(例えば、少なくとも9日間、500~1500IU/mLのIL-2と20~40ng/mLの抗CD3抗体)。従って、本発明によれば、刺激因子の常時存在は、培地にIL-2とCD3アクチベーターを1~5日毎(通常は2~4日毎)に定期的に補充することによって維持する。例えば、初期濃度約1000IU/mLの組換えヒトIL-2(rhIL-2)と約30ng/mLの抗ヒトCD3抗体(例えば、OKT-3)(これらは、培養中2~4日毎(例えば、0日目、4日目、6日目及び8日目)に再補充する)の存在下、約1×106個(細胞)/mLの濃度でPBMCを培養することができる。 In particular, the cells are advantageously expanded in the constant presence of effective amounts of IL-2 and CD3 activator, so as to be maintained in sufficient amounts throughout the entire expansion period (e.g., 500-1500 IU/mL IL-2 and 20-40 ng/mL anti-CD3 antibody for at least 9 days). Thus, according to the present invention, the constant presence of stimulatory factors is maintained by periodically supplementing the culture medium with IL-2 and CD3 activator every 1-5 days (usually every 2-4 days). For example, PBMCs can be cultured at a concentration of about 1 x 106 cells/mL in the presence of an initial concentration of about 1000 IU/mL recombinant human IL-2 (rhIL-2) and about 30 ng/mL anti-human CD3 antibody (e.g., OKT-3), which are replenished every 2-4 days during culture (e.g., days 0, 4, 6 and 8 ).
更なる有利な実施形態によれば、唯一の細胞刺激因子としてのIL-2とCD3アクチベーターの存在下で増殖を行う。従って、幾つかの実施形態では、サイトカイン(例えば、IFN-γ)等の外因的に添加された更なる細胞刺激因子及び/又はフィーダー細胞の非存在下で増殖を行う。他の実施形態では、この方法は、培養物中の生細胞の数を少なくとも20倍、典型的には平均で少なくとも30倍(例えば、30~55倍)増加させるように行う。他の実施形態では、この方法によって、培養後11日以内に生細胞数を25~55倍増加させる。他の実施形態では、この方法は、少なくとも1.5×108個、典型的には2×108~4×108個の生細胞、最も典型的には5×106~10×108個の生細胞、最大約10×109個の生細胞を産生するように行う。 According to further advantageous embodiments, the expansion is carried out in the presence of IL-2 and a CD3 activator as the only cell stimulatory factor. Thus, in some embodiments, the expansion is carried out in the absence of exogenously added additional cell stimulatory factors, such as cytokines (e.g., IFN-γ) and/or feeder cells. In other embodiments, the method is carried out to increase the number of viable cells in the culture by at least 20-fold, typically by an average of at least 30-fold (e.g., 30-55-fold). In other embodiments, the method increases the number of viable cells by 25-55-fold within 11 days of culture. In other embodiments, the method is carried out to produce at least 1.5×10 8 , typically 2×10 8 to 4×10 8 viable cells, most typically 5×10 6 to 10×10 8 viable cells, up to about 10×10 9 viable cells.
一様相では、インビトロ増殖PBMCを含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であって、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。 In one aspect, an ACT cell composition is provided comprising in vitro expanded PBMCs, 70-85% of which are CD3 + CD8 + cells that express NKG2D and granzyme B, at least 70% of which are CD56 - cells, and up to 5% of which are CD3 - cells, and which exhibit significant cytotoxic activity against tumor cells in vitro and in vivo.
他の様相では、ACT用細胞組成物を製造するための方法であって、
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを48~96時間毎に補充することと
を含み、培養物中の生細胞の数を少なくとも20倍増加させ、少なくとも5×106~10×108個の生細胞を含む細胞組成物を得る方法が提供される。
In another aspect, a method for producing a cell composition for ACT, comprising:
a. providing a PBMC sample;
b. A method is provided which comprises expanding PBMCs in the constant presence of effective amounts of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, for at least 9 days, with replenishing effective amounts of IL-2 and a CD3 activator every 48-96 hours, thereby increasing the number of viable cells in the culture by at least 20 fold, and obtaining a cell composition comprising at least 5 x 10 to 10 x 10 viable cells.
他の実施形態では、この方法によって調製された細胞組成物が提供される。他の実施形態では、
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを2~4日間毎に補充することとで、培養物中の生細胞の数を少なくとも20倍増加させ、それによって少なくとも5×106~10×108個の生細胞を含む細胞組成物を得ることと、
c.得られた細胞組成物を回収することと
を含む方法によって調製される、ACT用細胞組成物が提供される。
In another embodiment, a cell composition prepared by this method is provided.
a. providing a PBMC sample;
b. expanding the PBMCs for at least 9 days in the constant presence of an effective amount of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, whereby the number of viable cells in the culture is increased by at least 20 fold by replenishing the effective amount of IL-2 and the CD3 activator every 2-4 days, thereby obtaining a cell composition comprising at least 5 x 106 to 10 x 108 viable cells;
and c. recovering the resulting cell composition.
他の様相では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む細胞組成物が提供される。 In another aspect, a cell composition is provided that includes leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor that includes a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21.
他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療に使用するための本発明の細胞組成物が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療に使用するためのACT細胞組成物であって、少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であり、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。他の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む、腫瘍の治療を必要とする対象において腫瘍の治療に使用するための細胞組成物が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療に使用するための、本明細書に開示の方法によって調製される細胞組成物が提供される。 In another embodiment, a cell composition of the present invention is provided for use in treating a tumor in a subject in need of such treatment. In another embodiment, an ACT cell composition is provided for use in treating a tumor in a subject in need of such treatment, comprising at least 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, 70-85% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% are CD56 − cells, and up to 5% are CD3 − cells, and which exhibit significant cytotoxic activity against tumor cells in vitro and in vivo. In another embodiment, a cell composition is provided for use in treating a tumor in a subject in need of such treatment, comprising leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In other embodiments, there is provided a cell composition prepared by the methods disclosed herein for use in treating a tumor in a subject in need thereof.
他の様相では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量の本発明の細胞組成物と接触させ、それによって対象の腫瘍を治療することを含む方法が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量のACT細胞組成物と接触させることを含む方法が提供されるが、ACT細胞組成物は、少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%はNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%はCD56-細胞であり、最大5%はCD3-細胞であって、組成物はインビトロとインビボで腫瘍細胞に対して有意な細胞傷害活性を示し、それによって対象の腫瘍を治療する。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量の細胞組成物と接触させることを含む方法が提供されるが、該細胞組成物は、腫瘍の治療を必要とする対象において腫瘍を治療するために使用される、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含み、それによって対象の腫瘍を治療する。 In another aspect, a method of treating a tumor in a subject in need of such treatment is provided comprising contacting tumor cells with a therapeutically effective amount of a cell composition of the invention, thereby treating the tumor in the subject. In another embodiment, a method of treating a tumor in a subject in need of such treatment is provided comprising contacting tumor cells with a therapeutically effective amount of an ACT cell composition comprising at least 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, 70-85% of which are CD3 + CD8 + cells that express NKG2D and granzyme B, at least 70% of which are CD56 − cells, and up to 5% of which are CD3 − cells, and the composition exhibits significant cytotoxic activity against tumor cells in vitro and in vivo, thereby treating the tumor in the subject. In another embodiment, a method of treating a tumor in a subject in need of such treatment is provided, comprising contacting tumor cells with a therapeutically effective amount of a cell composition comprising leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21, for use in treating a tumor in a subject in need of such treatment, thereby treating the tumor in the subject.
上述の及び他の様相と実施形態を以下で更に詳細に説明し、例示する。 These and other aspects and embodiments are described in further detail and illustrated below.
調製方法
幾つかの実施形態では、本発明は、本発明の細胞組成物を産生するための方法に関する。幾つかの実施形態では、ACT用細胞組成物を産生するための方法であって
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを2~4日間毎に補充することとで、培養物中の生細胞の数を少なくとも20倍増加させ、それによって少なくとも5×106~10×108個の生細胞を含む細胞組成物を得ることと、
c.得られた細胞組成物を回収することと
を含む方法が提供される。
Preparation Methods In some embodiments, the present invention relates to methods for producing the cell compositions of the present invention. In some embodiments, a method for producing a cell composition for ACT comprises the steps of: a. providing a PBMC sample;
b. expanding the PBMCs for at least 9 days in the constant presence of an effective amount of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, whereby the number of viable cells in the culture is increased by at least 20 fold by replenishing the effective amount of IL-2 and the CD3 activator every 2-4 days, thereby obtaining a cell composition comprising at least 5 x 106 to 10 x 108 viable cells;
and c. recovering the resulting cell composition.
他の実施形態では、この方法は、
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを48~96時間毎に補充することと
を含み、培養物中の生細胞の数を少なくとも20倍増加させ、少なくとも5×106~10×108個の生細胞を含む細胞組成物を得る。
In another embodiment, the method comprises:
a. providing a PBMC sample;
b. Expanding the PBMCs in the constant presence of an effective amount of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, for at least 9 days, wherein the effective amount of IL-2 and the CD3 activator are replenished every 48-96 hours, thereby increasing the number of viable cells in the culture by at least 20 fold and obtaining a cell composition comprising at least 5 x 10 to 10 x 10 viable cells.
本明細書で使用される「PBMC試料」又は「末梢血単核細胞試料」又は「未分画PBMC」という用語は、全PBMC、即ち、所定の亜集団において実質的に富化されていない、丸い核を有する生存可能な白血球の集団を意味する。通常、本発明に係るPBMC試料は末梢血から得られており、選択工程には供されておらず、接着PBMC(本質的に90%超の単球で構成されている)又は非接着PBMC(T細胞、B細胞、ナチュラルキラー(NK)細胞、NKT細胞及びDC前駆体を含む)のみを含む。 As used herein, the term "PBMC sample" or "peripheral blood mononuclear cell sample" or "unfractionated PBMC" refers to total PBMCs, i.e., a population of viable white blood cells with round nuclei that are not substantially enriched in any given subpopulation. Typically, the PBMC sample according to the invention is obtained from peripheral blood, has not been subjected to a selection process, and contains only adherent PBMCs (consisting essentially of more than 90% monocytes) or non-adherent PBMCs (including T cells, B cells, natural killer (NK) cells, NKT cells, and DC precursors).
通常、PBMCは、遠心分離(例えば、バフィーコート由来)、密度勾配遠心分離(例えば、フィコール・ハイパック経由)又は当技術分野で既知の他の方法(例えば、末梢血からの抽出又は部分精製)によって得ることができる。例えば、PBMCは、血液層を分離する親水性多糖であるフィコールを使用して全血から抽出することができ、PBMCは血漿層の下に細胞環を形成する。更に、PBMCは、赤血球を優先的に溶解する低張溶解によって全血から抽出することができる。このような手順は当業者には知られている。 Typically, PBMCs can be obtained by centrifugation (e.g., from buffy coats), density gradient centrifugation (e.g., via Ficoll-Hypaque), or other methods known in the art (e.g., extraction or partial purification from peripheral blood). For example, PBMCs can be extracted from whole blood using Ficoll, a hydrophilic polysaccharide that separates blood layers, with the PBMCs forming a ring of cells below the plasma layer. Additionally, PBMCs can be extracted from whole blood by hypotonic lysis, which preferentially lyses red blood cells. Such procedures are known to those skilled in the art.
本明細書で使用される「増殖」という用語は、細胞複製によって細胞集団の細胞数が増加することを意味する。特に、本明細書に開示のPBMC増殖は、本発明に係るACT細胞組成物を産生するための、PBMC内の細胞集団のポリクローナル活性化と増殖を含むインビトロ又はエクスビボ培養方法を意味する。通常、例えば、ヒト対象の治療用の臨床グレードのACT組成物を産生するために、特定のcGMPグレードの環境及びcGMPグレードの培地中で増殖を行う。幾つかの実施形態では、9~16日間又は11~16日間、例えば、9日間、10日間、11日間、12日間、13日間、14日間、15日間、又は16日間増殖を行う。 As used herein, the term "expansion" refers to an increase in cell number of a cell population by cell replication. In particular, PBMC expansion as disclosed herein refers to an in vitro or ex vivo culture method that includes polyclonal activation and expansion of a cell population within a PBMC to produce an ACT cell composition according to the present invention. Typically, expansion is performed in a specific cGMP-grade environment and cGMP-grade medium to produce, for example, a clinical grade ACT composition for the treatment of a human subject. In some embodiments, expansion is performed for 9-16 days or 11-16 days, e.g., 9 days, 10 days, 11 days, 12 days, 13 days, 14 days, 15 days, or 16 days.
より具体的には、増殖方法中にインターロイキン-2(IL-2)とCD3アクチベーターを培地に補充する。幾つかの実施形態では、細胞組成物を上述の方法によって調製し、有効量のIL-2とCD3アクチベーターを48~96時間毎、例えば、48時間毎、49時間毎、50時間毎、51時間毎、52時間毎、53時間毎、54時間毎、55時間毎、56時間毎、57時間毎、58時間毎、59時間毎、60時間毎、61時間毎、62時間毎、63時間毎、64時間毎、65時間毎、66時間毎、67時間毎、68時間毎、69時間毎、70時間毎、71時間毎、72時間毎、73時間毎、74時間毎、75時間毎、76時間毎、77時間毎、78時間毎、79時間毎、80時間毎、81時間毎、82時間毎、83時間毎、84時間毎、85時間毎、86時間毎、87時間毎、88時間毎、89時間毎、90時間毎、91時間毎、92時間毎、93時間毎、94時間毎、95時間毎,又は96時間毎に補充する。 More specifically, the medium is supplemented with interleukin-2 (IL-2) and a CD3 activator during the expansion method. In some embodiments, the cell composition is prepared by the method described above, and an effective amount of IL-2 and a CD3 activator is administered every 48 to 96 hours, e.g., every 48 hours, 49 hours, 50 hours, 51 hours, 52 hours, 53 hours, 54 hours, 55 hours, 56 hours, 57 hours, 58 hours, 59 hours, 60 hours, 61 hours, 62 hours, 63 hours, 64 hours, 65 hours, 66 hours, 67 hours, etc. , every 68 hours, 69 hours, 70 hours, 71 hours, 72 hours, 73 hours, 74 hours, 75 hours, 76 hours, 77 hours, 78 hours, 79 hours, 80 hours, 81 hours, 82 hours, 83 hours, 84 hours, 85 hours, 86 hours, 87 hours, 88 hours, 89 hours, 90 hours, 91 hours, 92 hours, 93 hours, 94 hours, 95 hours, or 96 hours.
サイトカインIL-2は培養中のリンパ球の成長因子として作用し、例えば、活性化T細胞の細胞死を防ぐために必要である。IL-2の活性は、アルファ(CD25)、ベータ(CD122)及びガンマ(CD132)と称される3本の鎖で構成される複合体であるIL-2受容体(IL-2R)への結合及びそれを介したシグナル伝達によって媒介される。二量体IL-2RはメモリーCD8+T細胞とNK細胞によって発現されるが、制御性T細胞と活性化T細胞は高レベルの三量体IL-2Rを発現する。本明細書で使用される「インターロイキン2」又は「IL-2」とは、哺乳動物のIL-2、好ましくはヒトIL-2(例えば、組換え産生されたヒトIL-2)を意味する。幾つかの実施形態では、ヒトIL-2の生物学的等価物として当技術分野で認識されており、IL-2受容体に選択的に結合し、ヒトリンパ球の増大や増殖をサポートする際に同等の活性を発揮することができるポリペプチド(例えば、IL-2融合タンパク質、コンジュゲート及びバリアント)を使用することもできる。IL-2は組換えによって(例えば、本明細書に開示の方法によって)産生することができ、様々な供給元から市販されている。例えば、組換えヒトIL-2(rhIL-2)はR&D Systems社から購入することができる。Proleukin(登録商標)(アルデスロイキン)は高度に精製されたrhIL-2タンパク質であり、癌免疫療法での臨床使用が承認されている。 The cytokine IL-2 acts as a growth factor for lymphocytes in culture and is necessary, for example, to prevent cell death of activated T cells. The activity of IL-2 is mediated by binding to and signaling through the IL-2 receptor (IL-2R), a complex composed of three chains designated alpha (CD25), beta (CD122), and gamma (CD132). Dimeric IL-2R is expressed by memory CD8 + T cells and NK cells, whereas regulatory and activated T cells express high levels of trimeric IL-2R. As used herein, "interleukin 2" or "IL-2" refers to mammalian IL-2, preferably human IL-2 (e.g., recombinantly produced human IL-2). In some embodiments, polypeptides (e.g., IL-2 fusion proteins, conjugates, and variants) recognized in the art as biological equivalents of human IL-2 and capable of selectively binding to the IL-2 receptor and exerting equivalent activity in supporting the expansion and proliferation of human lymphocytes can also be used. IL-2 can be produced recombinantly (e.g., by the methods disclosed herein) and is commercially available from a variety of sources. For example, recombinant human IL-2 (rhIL-2) can be purchased from R&D Systems. Proleukin® (aldesleukin) is a highly purified rhIL-2 protein that has been approved for clinical use in cancer immunotherapy.
CD3複合体はT細胞受容体(TCR)鎖と結合し、T細胞の表面にTCR-CD3複合体を形成する。従って、CD3はT細胞マーカーと考えられているが、NK-T細胞等の他の特定の細胞集団もCD3表面発現(CD3+細胞)を特徴とする一方、NK細胞、B細胞及び骨髄系細胞等の他の細胞型は通常、CD3表面発現の欠如(CD3-細胞)を特徴とするものではない。哺乳動物では、TCR-CD3複合体は、TCR鎖及びCD3-ゼータ(ζ鎖)と複合体を形成するCD3γ鎖、CD3δ鎖、及び2個のCD3ε鎖で構成される。本明細書で使用される「CD3アクチベーター」とは、リンパ球表面のCD3分子(典型的にはヒトCD3分子)に結合することによって、ポリクローナル刺激及び増殖を誘導又は媒介することができる剤である。CD3アクチベーターの例としては、刺激性CD3特異的抗体、その抗原結合部分、及びそのコンジュゲートが挙げられる。通常、CD3アクチベーターは刺激性抗CD3モノクローナル抗体(mAb)であり、通常はCD3ε等のCD3シグナル伝達ドメインを指向する。特定の実施形態では、本発明の実施形態で使用される有利なCD3アクチベーターは、ヒトCD3ε鎖に対するマウスモノクローナル抗体であるOKT3である。T細胞受容体/CD3複合体を架橋する他の抗体の例としては、HIT3a及びUCHT1抗CD3mAbが挙げられる。CD3アクチベーターの更なる例は、CD3リガンド及びCD3結合マイトジェンである。このような抗体や剤は様々な供給元から市販されている。例えば、OKT3及び他の抗CD3mAbは、Biolegend社やeBioscience社から入手可能である。 The CD3 complex binds to the T cell receptor (TCR) chain to form a TCR-CD3 complex on the surface of T cells. Thus, although CD3 is considered a T cell marker, certain other cell populations, such as NK-T cells, are also characterized by CD3 surface expression (CD3 + cells), whereas other cell types, such as NK cells, B cells, and myeloid cells, are not typically characterized by a lack of CD3 surface expression (CD3 − cells). In mammals, the TCR-CD3 complex is composed of the CD3 γ chain, the CD3 δ chain, and two CD3 ε chains, which form a complex with the TCR chain and the CD3-zeta (ζ chain). As used herein, a “CD3 activator” is an agent capable of inducing or mediating polyclonal stimulation and proliferation by binding to the CD3 molecule (typically a human CD3 molecule) on the surface of lymphocytes. Examples of CD3 activators include stimulatory CD3-specific antibodies, antigen-binding portions thereof, and conjugates thereof. Typically, the CD3 activator is a stimulatory anti-CD3 monoclonal antibody (mAb), typically directed against a CD3 signaling domain, such as CD3ε. In a particular embodiment, a preferred CD3 activator for use in embodiments of the invention is OKT3, a murine monoclonal antibody against the human CD3ε chain. Other examples of antibodies that cross-link the T cell receptor/CD3 complex include the HIT3a and UCHT1 anti-CD3 mAbs. Further examples of CD3 activators are CD3 ligands and CD3-binding mitogens. Such antibodies and agents are commercially available from a variety of sources. For example, OKT3 and other anti-CD3 mAbs are available from Biolegend and eBioscience.
本発明の実施形態によれば、他のサイトカイン又は抗体の非存在下でPBMCを増殖する。換言すれば、IL-2とCD3アクチベーター以外のサイトカイン及び抗体は培養物に補充(外部から添加)しない。従って、本発明に係る増殖方法は、他種の細胞組成物を産生するのに使用される他の増殖プロトコルとは区別することができる(例えば、CIK細胞は特にIFN-γの存在下で(例えば、PBMC)の増殖によって産生する)。しかし、増殖方法中に、サイトカインを含む様々な因子が培養細胞によって産生され、培地に分泌される場合があることを理解されたい。 According to an embodiment of the invention, PBMCs are expanded in the absence of other cytokines or antibodies. In other words, no cytokines or antibodies other than IL-2 and CD3 activators are supplemented (exogenously added) to the culture. Thus, the expansion method of the present invention can be distinguished from other expansion protocols used to produce other types of cell compositions (e.g., CIK cells, particularly by expansion of (e.g., PBMCs) in the presence of IFN-γ). However, it should be understood that various factors, including cytokines, may be produced by the cultured cells and secreted into the medium during the expansion method.
更に、本発明に係る増殖方法は、IL-2及びCD3アクチベーターの常時存在下で行い、IL-2及びCD3アクチベーターが増殖方法全体を通じて有効量で維持されるようにこれらを連続的又は断続的に補充する。従って、本発明に係る増殖方法は、他種の細胞組成物を産生するのに使用される他の増殖プロトコルとは区別することができる(例えば、CIK細胞の産生では、増殖の2日目にIL-2とOKT3を添加するが、その後OKT3を培養物に補充することはない)。 Furthermore, the expansion method of the present invention is carried out in the constant presence of IL-2 and CD3 activator, which are continuously or intermittently replenished so that IL-2 and CD3 activator are maintained in effective amounts throughout the expansion method. Thus, the expansion method of the present invention is distinguishable from other expansion protocols used to produce other types of cell compositions (e.g., in the production of CIK cells, IL-2 and OKT3 are added on the second day of expansion, but OKT3 is not subsequently supplemented to the culture).
他の実施形態では、唯一の外因的に添加された細胞刺激因子としてのIL-2とCD3アクチベーターの常時存在下で増殖を行う。従って、共刺激分子(例えば、CD28)、抗原(例えば、MHC-抗原複合体)等(これらは培養細胞への外因性源である)に対する抗体等、インビトロ又はエクスビボ増殖方法中にリンパ球の活性化及び増殖を誘導するために使用される他の細胞刺激因子は人為的に導入(補充)されない。特定の細胞刺激因子は、増殖方法中に培養細胞によって内因的に産生される場合があることを理解されたい。標準的な組織培養培地(例えば、RPMI等の完全培地)に含まれる様々な因子は、これらの実施形態によれば、外因的に添加された細胞刺激因子とは見なされないことが更に理解されよう。例えば、組織培養血清、例えば、最終濃度が5~15%、典型的には7~12%又は8~13%、より典型的には約10%のウシ胎仔血清(FCS)存在下で増殖を好都合に行うことができる。 In other embodiments, the expansion is carried out in the constant presence of IL-2 and CD3 activators as the only exogenously added cell stimulatory factors. Thus, other cell stimulatory factors used to induce lymphocyte activation and proliferation during the in vitro or ex vivo expansion method, such as antibodies against costimulatory molecules (e.g., CD28), antigens (e.g., MHC-antigen complexes), etc. (which are of exogenous source to the cultured cells), are not artificially introduced (supplemented). It should be understood that certain cell stimulatory factors may be endogenously produced by the cultured cells during the expansion method. It should be further understood that various factors contained in standard tissue culture media (e.g., complete media such as RPMI) are not considered exogenously added cell stimulatory factors according to these embodiments. For example, expansion can be conveniently carried out in the presence of tissue culture serum, e.g., fetal calf serum (FCS) at a final concentration of 5-15%, typically 7-12% or 8-13%, more typically about 10%.
本明細書に開示のように、培養物中の生細胞の数を少なくとも20倍(又は幾つかの実施形態では30~60倍)増加させるように増殖を有利に行い、それによって、5×106~10×108個の生細胞(又は他の実施形態では、最大10×109個の生細胞)を含む細胞組成物を得る。本明細書で使用される「生細胞」という用語は、壊死を起こしていない細胞、又は初期又は後期のアポトーシス状態にない細胞を意味する。細胞生存率を求めるためのアッセイは、フローサイトメトリーによって検出可能なヨウ化プロピジウム(PI)染色を使用する等、当技術分野では既知である。従って、一実施形態では、生細胞はヨウ化プロピジウム摂取を示さず、ホスファチジルセリンを発現しない細胞である。光、蛍光又は電子顕微鏡技法を用いるか、又は色素トリパンブルーの取り込みによって壊死を更に特定することができる。 As disclosed herein, expansion is advantageously performed to increase the number of viable cells in culture by at least 20-fold (or in some embodiments, 30-60-fold), thereby obtaining a cell composition comprising 5×10 6 to 10×10 8 viable cells (or in other embodiments, up to 10×10 9 viable cells). As used herein, the term "viable cells" refers to cells that are not undergoing necrosis or are not in an early or late apoptotic state. Assays for determining cell viability are known in the art, such as using propidium iodide (PI) staining detectable by flow cytometry. Thus, in one embodiment, viable cells are cells that do not exhibit propidium iodide uptake and do not express phosphatidylserine. Necrosis can be further identified using light, fluorescence or electron microscopy techniques, or by uptake of the dye trypan blue.
更に、本発明の産生方法は、増殖工程によって得られる細胞組成物を回収する工程を含む。例えば、細胞を回収し、遠心分離又は他の洗浄工程に供して、残留抗体とサイトカインを除去することができる。幾つかの実施形態では、腫瘍細胞と接触させる前、又は対象へ投与する前に、細胞を適切な希釈剤又は媒体(例えば、PBS)に再懸濁することができる。 Additionally, the production methods of the present invention include a step of harvesting the cell composition obtained by the expansion step. For example, the cells can be harvested and subjected to centrifugation or other washing steps to remove residual antibodies and cytokines. In some embodiments, the cells can be resuspended in an appropriate diluent or medium (e.g., PBS) prior to contacting with the tumor cells or administering to the subject.
幾つかの実施形態では、細胞は、富化(例えば、増殖工程前のアフェレーシス、リンパ球アフェレーシス、又は白血球の免疫捕捉)、刺激(例えば、抗原特異的)及び/又は増殖(例えば、フィーダー細胞の存在下での急速増殖プロトコル、又は他の/追加のサイトカイン存在下での増殖)の更なる工程には供されていない。 In some embodiments, the cells have not been subjected to further steps of enrichment (e.g., apheresis prior to the expansion step, lymphoapheresis, or immunocapture of leukocytes), stimulation (e.g., antigen-specific) and/or expansion (e.g., rapid expansion protocols in the presence of feeder cells, or expansion in the presence of other/additional cytokines).
他の様相では、本発明の細胞組成物を産生する方法であって、
a)PBMC試料を提供することと、
b)有効量のIL-2とCD3アクチベーターの常時存在下、少なくとも9日間PBMCを増殖させ、そのとき有効量のIL-2とCD3アクチベーターを2~4日毎に補充することと、
c)得られた細胞組成物を回収することと
を含む方法が提供される。
In another aspect, there is provided a method for producing a cell composition of the invention, comprising the steps of:
a) providing a PBMC sample;
b) expanding the PBMCs in the constant presence of effective amounts of IL-2 and a CD3 activator for at least 9 days, whereby effective amounts of IL-2 and a CD3 activator are replenished every 2-4 days;
and c) recovering the resulting cell composition.
幾つかの実施形態では、本明細書に開示の本発明の細胞組成物を得るように増殖を行う。他の実施形態では、唯一の外因的に添加された細胞刺激因子としてのIL-2とCD3アクチベーターの常時存在下で(他のサイトカイン及び抗体の非存在下で)増殖を行う。 In some embodiments, expansion is performed to obtain the cell compositions of the invention disclosed herein. In other embodiments, expansion is performed in the constant presence of IL-2 and CD3 activator as the only exogenously added cell stimulators (in the absence of other cytokines and antibodies).
他の実施形態では、この方法は、細胞を遺伝子改変して本明細書に開示の細胞組成物を得ることを更に含む。幾つかの実施形態では、得られる細胞組成物は凍結保存に適していることが有利である。従って、ACT用の特定の他の細胞組成物とは対照的に、本発明の細胞組成物は効力を実質的に失うことなく、増殖後に凍結し、対象への投与前に解凍することができる。従って、他の実施形態では、この方法は得られた細胞組成物を凍結させることを更に含む。例えば、液体窒素を使用して凍結保存プロトコルを行うことができ、適切な培地(例えば、90%FCS+10%ジメチルスルホキシド(DMSO)を含む)中に細胞を懸濁させるが、これに限定されない。必要に応じて、市販のシステム又は賦形剤(例えば、CryoSure(登録商標)USPグレードの凍結保護剤)を用いる。 In other embodiments, the method further comprises genetically modifying the cells to obtain the cell composition disclosed herein. In some embodiments, the resulting cell composition is advantageously suitable for cryopreservation. Thus, in contrast to certain other cell compositions for ACT, the cell compositions of the present invention can be frozen after expansion and thawed prior to administration to a subject without substantial loss of potency. Thus, in other embodiments, the method further comprises freezing the resulting cell composition. For example, the cryopreservation protocol can be performed using liquid nitrogen and suspending the cells in an appropriate medium (e.g., including, but not limited to, 90% FCS + 10% dimethyl sulfoxide (DMSO)). If necessary, commercially available systems or excipients (e.g., CryoSure® USP grade cryoprotectant) can be used.
他の実施形態では、工程bは、500~1500IU/mLのIL-2と10~60ng/mLの抗CD3抗体の存在下で細胞を増殖させることを含み、これらを2~4日毎に10~16日間再補充する。他の実施形態では、細胞は更なる富化、刺激又は増殖工程(例えば、工程bの前又は後)に供されていない。他の実施形態では、生細胞の数を少なくとも30倍増加させるように増殖を行う。他の実施形態では、少なくとも5×106~10×108個(例えば、5×106~10×109個)の生存可能な単核細胞を含む細胞組成物を産生するように増殖を行い、該細胞の70~80%(又は他の実施形態では70~85%)はNKG2DとグランザイムBを発現するCD3+CD8+細胞である。他の実施形態では、インビトロで腫瘍細胞と0.25:1の比率で24時間インキュベートした場合に、有意に造血器腫瘍細胞を特異的に根絶することができる細胞組成物を産生するように増殖を行う。他の実施形態では、工程bは、細胞を遺伝子改変して、例えば、本明細書に開示の遺伝子改変細胞組成物を産生することを更に含む。細胞をCD3刺激因子とIL-2の補充に供する前に、細胞を改変(例えば、形質導入又はトランスフェクト)することが有利である。 In other embodiments, step b comprises expanding the cells in the presence of 500-1500 IU/mL IL-2 and 10-60 ng/mL anti-CD3 antibody, which are replenished every 2-4 days for 10-16 days. In other embodiments, the cells are not subjected to further enrichment, stimulation or expansion steps (e.g., before or after step b). In other embodiments, expansion is performed to increase the number of viable cells by at least 30-fold. In other embodiments, expansion is performed to produce a cell composition comprising at least 5×10 6 to 10×10 8 (e.g., 5×10 6 to 10×10 9 ) viable mononuclear cells, of which 70-80% (or 70-85% in other embodiments) are CD3 + CD8 + cells expressing NKG2D and granzyme B. In other embodiments, expansion is performed to produce a cell composition capable of specifically eradicating hematopoietic tumor cells when incubated in vitro with tumor cells at a ratio of 0.25:1 for 24 hours. In other embodiments, step b further comprises genetically modifying the cells, e.g., to produce a genetically modified cell composition as disclosed herein. It may be advantageous to modify (e.g., transduce or transfect) the cells prior to subjecting them to CD3 stimulatory factors and IL-2 supplementation.
細胞組成物
他の実施形態では、本明細書に開示の方法によって調製される細胞組成物が提供される。他の実施形態では、本明細書に開示のSAK細胞を含む細胞組成物が提供される。他の実施形態では、本発明は、本明細書に開示の養子細胞移植(ACT)組成物に関する。他の実施形態では、ACT細胞組成物であって、5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~80%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であって、インビトロ及びインビボで非組織適合性腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。他の実施形態では、ACT細胞組成物であって、少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であって、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。
Cell Compositions In another embodiment, a cell composition prepared by the methods disclosed herein is provided. In another embodiment, a cell composition comprising SAK cells as disclosed herein is provided. In another embodiment, the invention relates to an adoptive cell transfer (ACT) composition as disclosed herein. In another embodiment, an ACT cell composition is provided, the ACT cell composition comprising 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, 70-80% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% of which are CD56 − cells, and up to 5% of which are CD3 − cells, and which exhibit significant cytotoxic activity against non-histocompatible tumor cells in vitro and in vivo. In another embodiment, an ACT cell composition is provided comprising at least 5 x 10 to 10 x 10 viable in vitro expanded PBMCs, 70 to 85% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% of which are CD56 - cells, and up to 5% of which are CD3 - cells, and which exhibits significant cytotoxic activity against tumor cells in vitro and in vivo.
特に明記しない限り、本明細書で使用される「養子移植」という用語は、予め感作された免疫学的剤(例えば、細胞又は血清)をレシピエントに移植する受動免疫療法の形態を意味する。「養子移植免疫療法」、「養子細胞療法」及び「養子細胞免疫療法」という語句は本明細書では互換的に使用され、本発明の細胞組成物等のエフェクター免疫担当細胞を、それを必要とする対象に投与(養子移植)して癌の発症又は症状を緩和又は改善する治療的又は予防的レジメン又はモダリティを意味する。従って、本発明に係るACT組成物は、無菌且つ適切な(例えば、cGMPグレードの)条件下で産生し、腫瘍性障害を患っているヒト対象に抗腫瘍レジメンの一環として投与する有効量(例えば、少なくとも5×106個の細胞、最大約10×109個の細胞)を含む。 Unless otherwise specified, the term "adoptive transfer" as used herein refers to a form of passive immunotherapy in which a pre-sensitized immunological agent (e.g., cells or serum) is transferred to a recipient. The terms "adoptive transfer immunotherapy", "adoptive cell therapy" and "adoptive cellular immunotherapy" are used interchangeably herein to refer to a therapeutic or prophylactic regimen or modality in which effector immunocompetent cells, such as the cell composition of the present invention, are administered (adoptive transfer) to a subject in need thereof to alleviate or ameliorate the onset or symptoms of cancer. Thus, the ACT composition of the present invention is produced under sterile and appropriate (e.g., cGMP grade) conditions and comprises an effective amount (e.g., at least 5×10 6 cells, up to about 10×10 9 cells) to be administered to a human subject suffering from a neoplastic disorder as part of an anti-tumor regimen.
本明細書で使用される「インビトロ増殖PBMC」という用語は、インビトロ又はエクスビボでの増殖工程を経て、本明細書に開示の細胞組成物をもたらす、末梢血由来の単核細胞を意味する。従って、このような細胞及び組成物は、腫瘍由来リンパ球(TIL、末梢血からではなく腫瘍生検から得られるもの)、白血球細胞株、及び多能性幹細胞からインビトロで分化した白血球等の他の供給源から取得又は由来する白血球組成物とは区別することができる。 As used herein, the term "in vitro expanded PBMCs" refers to mononuclear cells derived from peripheral blood that have undergone an in vitro or ex vivo expansion process to result in the cell compositions disclosed herein. Such cells and compositions are therefore distinct from leukocyte compositions obtained or derived from other sources, such as tumor-derived lymphocytes (TILs, obtained from tumor biopsies rather than from peripheral blood), leukocyte cell lines, and leukocytes differentiated in vitro from pluripotent stem cells.
Tリンパ球(T細胞)は、免疫応答に関与する様々な異なる細胞型の1種であり、CD3の表面発現(CD3+細胞)を特徴とする。T細胞の活性は、主要組織適合性複合体(MHC)分子に関連してT細胞に提示される抗原によって制御される。次に、T細胞受容体(TCR)がMHC抗原複合体に結合する。抗原がMHCと複合体を形成すると、MHC抗原複合体はT細胞上の特定のTCRに結合し、それによって該T細胞の活性が変化する。抗原提示細胞によるTリンパ球の適切な活性化には、TCRの刺激だけでなく、その共受容体の複合的且つ協調的な関与が必要である。 T lymphocytes (T cells) are one of several different cell types involved in the immune response and are characterized by surface expression of CD3 (CD3 + cells). T cell activity is controlled by antigens that are presented to the T cell in association with major histocompatibility complex (MHC) molecules. The T cell receptor (TCR) then binds to the MHC-antigen complex. Once the antigen is complexed with MHC, the MHC-antigen complex binds to a specific TCR on the T cell, thereby altering the activity of the T cell. Proper activation of T lymphocytes by antigen-presenting cells requires not only stimulation of the TCR but also the combined and coordinated engagement of its co-receptors.
ヘルパーT細胞(TH細胞)は、B細胞の形質細胞やメモリーB細胞への成熟、細胞傷害性T細胞やマクロファージの活性化等の免疫学的方法において他の白血球を支援する。このような細胞は、その表面にCD4糖タンパク質を発現するため、CD4+T細胞(又はCD3+CD4+細胞)としても知られている。ヘルパーT細胞は、抗原提示細胞(APC)の表面に発現するMHCクラスII分子によってペプチド抗原が提示されると活性化される。一旦活性化されると、急速に分裂し、活発な免疫応答を調節又は支援するサイトカインと称される小型タンパク質を分泌する。 Helper T cells (TH cells) assist other white blood cells in immunological processes such as maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. These cells are also known as CD4 + T cells (or CD3 + CD4 + cells) because they express the CD4 glycoprotein on their surface. Helper T cells become activated when peptide antigens are presented to them by MHC class II molecules expressed on the surface of antigen-presenting cells (APCs). Once activated, they divide rapidly and secrete small proteins called cytokines that regulate or support a vigorous immune response.
細胞傷害性T細胞(TC細胞、又はCTL)は、ウイルス感染細胞や腫瘍細胞を破壊し、移植拒絶反応にも関与する。このような細胞は、表面にCD8糖タンパク質を発現するため、CD8+T細胞(又はCD3+CD8+細胞)としても知られている。このような細胞は、全ての有核細胞の表面に存在するMHCクラスI分子に関連する抗原に結合することによって、その標的を認識する。 Cytotoxic T cells (TC cells, or CTLs) destroy virus-infected and tumor cells and also play a role in transplant rejection. These cells are also known as CD8 + T cells (or CD3 + CD8 + cells) because they express the CD8 glycoprotein on their surface. These cells recognize their targets by binding to antigens associated with MHC class I molecules, which are present on the surface of all nucleated cells.
ナチュラルキラー細胞(NK細胞)は、CD56、CD16及び/又はCD57の発現と、T細胞受容体-CD3複合体の欠如によって定められる末梢血リンパ球のサブセットである。NK細胞は、特定の細胞溶解酵素の活性化によって「自己」MHC/HLA抗原を発現できない細胞に結合して死滅させる能力、NK活性化受容体のリガンドを発現する腫瘍細胞又は他の疾患細胞を死滅させる能力、及び免疫応答を刺激又は阻害するサイトカインを放出する能力を特徴とする。 Natural killer cells (NK cells) are a subset of peripheral blood lymphocytes defined by the expression of CD56, CD16 and/or CD57 and the absence of T cell receptor-CD3 complexes. NK cells are characterized by their ability to bind and kill cells that fail to express "self" MHC/HLA antigens by activation of specific cytolytic enzymes, to kill tumor cells or other diseased cells that express ligands for NK activating receptors, and to release cytokines that stimulate or inhibit the immune response.
ナチュラルキラーT細胞(NKT細胞又はNK-T細胞)は、TCRを発現するCD1d拘束性T細胞である。従来のMHC分子によって提示されるペプチド抗原を検出する従来のT細胞とは異なり、NK-T細胞は、非古典的MHC分子であるCD1dによって提示される脂質抗原を認識する。インバリアント又はI型NK-T細胞は非常に限られたTCRレパートリーを発現するが、非古典的又は非インバリアントII型NKT細胞はより不均一なTCRαβの利用を示す。適応性又はインバリアント(I型)NKT細胞は、TCR Va24-Ja18、Vb11、CD1d、CD3、CD4、CD8、aGalCer、CD161及びCD56のマーカーの少なくとも1種以上の発現によって特定することができる。 Natural killer T cells (NKT cells or NK-T cells) are CD1d-restricted T cells that express the TCR. Unlike conventional T cells, which detect peptide antigens presented by conventional MHC molecules, NK-T cells recognize lipid antigens presented by the non-classical MHC molecule, CD1d. Invariant or type I NK-T cells express a very limited TCR repertoire, whereas non-classical or non-invariant type II NKT cells show more heterogeneous TCRαβ utilization. Adaptive or invariant (type I) NKT cells can be identified by expression of at least one or more of the following markers: TCR Va24-Ja18, Vb11, CD1d, CD3, CD4, CD8, aGalCer, CD161, and CD56.
CD56としても知られる神経細胞接着分子(NCAM)は、所謂同種親和性相互作用とヘテロ親和性相互作用の両方に関与する免疫グロブリンスーパーファミリーのメンバーである。CD56には3種の主要なアイソフォーム(NCAM-120、NCAM-140及びNCAM-180)が存在し、全て1種の単一遺伝子からの選択的スプライシングによって産生され、細胞内ドメインの長さが異なる。CD56の発現は、末梢血から単離されたCD56+細胞の大部分を構成するNK細胞と最も厳密に関連している。しかし、CD56は、例えば、他のリンパ系細胞(ガンマデルタ(γδ)T細胞及び樹状細胞(DC)が挙げられるが、これらに限定されない)でも検出されている。 Neural cell adhesion molecule (NCAM), also known as CD56, is a member of the immunoglobulin superfamily involved in both so-called homophilic and heterophilic interactions. There are three major isoforms of CD56 (NCAM-120, NCAM-140, and NCAM-180), all produced by alternative splicing from a single gene, which differ in the length of the intracellular domain. CD56 expression is most closely associated with NK cells, which constitute the majority of CD56 + cells isolated from peripheral blood. However, CD56 has also been detected on other lymphoid cells, including, but not limited to, gamma delta (γδ) T cells and dendritic cells (DCs).
本明細書において、当業者に既知の方法(例えば、細胞を細胞表面マーカーに特異的に結合する抗体と接触させ、その後、そのような接触した細胞のフローサイトメトリー分析を行って、抗体が細胞に特異的に結合するかどうかを決定する)を用いて検出するに十分な量で細胞表面上に細胞表面マーカーを発現する場合、この細胞を細胞表面マーカーに対して「陽性」であると見なす。細胞は細胞表面マーカーのメッセンジャーRNAを発現する場合があるが、本明細書に記載の組成物及び方法に対して陽性であると見なされるためには、細胞はその表面に目的のマーカーを発現しなければならないことは理解されたい。同様に、当業者に既知の方法(例えば、細胞を細胞表面マーカーに特異的に結合する抗体と接触させ、その後、そのような接触した細胞のフローサイトメトリー分析を行って、抗体が細胞に結合するかどうかを決定する)を用いて検出するに十分な量で細胞表面上に細胞表面マーカーを発現しない場合、この細胞を細胞表面マーカーに対して「陰性」であると見なす。 As used herein, a cell is considered "positive" for a cell surface marker if it expresses the cell surface marker on the cell surface in an amount sufficient to be detected using methods known to one of skill in the art (e.g., contacting the cell with an antibody that specifically binds to the cell surface marker, followed by flow cytometric analysis of the contacted cells to determine whether the antibody specifically binds to the cell). It should be understood that while a cell may express messenger RNA for the cell surface marker, a cell must express the marker of interest on its surface to be considered positive for the compositions and methods described herein. Similarly, a cell is considered "negative" for a cell surface marker if it does not express the cell surface marker on the cell surface in an amount sufficient to be detected using methods known to one of skill in the art (e.g., contacting the cell with an antibody that specifically binds to the cell surface marker, followed by flow cytometric analysis of the contacted cells to determine whether the antibody specifically binds to the cell).
様々な実施形態では、本発明の細胞組成物は、高い相対CD8+細胞レベルと低い相対CD4+細胞レベルを特徴とする。幾つかの実施形態では、組成物はCD3+CD8+細胞のCD3+CD4+細胞に対する比率が高い(少なくとも6:1、例えば、6.5~8.5:1、典型的には7~8:1)ことを特徴とする。換言すれば、組成物中のCD3+CD8+細胞の量は、組成物中のCD3+CD4+細胞の量の6~8倍、典型的には7~8倍である。他の実施形態では、細胞組成物は高いNK-T細胞レベルを特徴とする。他の実施形態では、細胞はT細胞に対するNK-T細胞の比率が高いことを特徴とする。幾つかの実施形態では、細胞組成物はCD3+CD8+細胞に対するCD3+CD8+CD56+細胞の発生率が15~30%、典型的には19~25%又は20~22%であることを特徴とする。更に他の実施形態では、細胞組成物はCD56+細胞に対するCD8+細胞の比率が高い(例えば、2~4)ことを特徴とする。様々な実施形態では、このような高いレベル及び比率は、本明細書に示すように、元のPBMC試料に対する増強、又はCIK細胞調製物等の他のACT組成物に対する増強と関連することがある。各々の可能性は本発明の別個の実施形態を表す。 In various embodiments, the cell compositions of the invention are characterized by high relative CD8 + cell levels and low relative CD4 + cell levels. In some embodiments, the compositions are characterized by a high ratio of CD3 + CD8 + cells to CD3 + CD4 + cells (at least 6:1, e.g., 6.5-8.5:1, typically 7-8:1). In other words, the amount of CD3 + CD8 + cells in the composition is 6-8 times, typically 7-8 times, the amount of CD3 + CD4 + cells in the composition. In other embodiments, the cell compositions are characterized by high NK-T cell levels. In other embodiments, the cells are characterized by a high ratio of NK-T cells to T cells. In some embodiments, the cell compositions are characterized by an incidence of CD3 + CD8 + CD56 + cells relative to CD3 + CD8 + cells of 15-30%, typically 19-25% or 20-22%. In yet other embodiments, the cell compositions are characterized by a high ratio of CD8 + cells to CD56 + cells (e.g., 2-4). In various embodiments, such high levels and ratios may be associated with an enhancement relative to the original PBMC sample, or an enhancement relative to other ACT compositions, such as CIK cell preparations, as described herein. Each possibility represents a separate embodiment of the present invention.
例えば、幾つかの実施形態では、本発明の細胞組成物は、細胞傷害性CD3+CD8+細胞が平均して少なくとも60%、典型的には75%(例えば、70~80%、70~85%、73~76%又は73~78%)存在することを特徴とする。他の実施形態では、細胞組成物は、CD3+CD8+細胞が70~85%存在することを特徴とする。他の実施形態では、細胞組成物は、CD3+CD4+細胞が最大15%(例えば、7~12%、11~13%、9~12%又は8~11%)、典型的にはCD3+CD4+細胞が平均して最大10%存在することを特徴とする。他の実施形態では、平均してCD3+CD8+細胞の少なくとも15%がCD8+CD56+細胞である(例えば、15~30%又は17~20%)。他の実施形態では、細胞組成物は、CD3+CD8+CD56+細胞が少なくとも9%、典型的には10~14%であることを特徴とする。他の実施形態では、細胞組成物はCD3+CD8+CD56+細胞が7~17%存在することを特徴とする。更に他の実施形態では、細胞組成物はCD3+CD56+細胞を約8~35%、典型的には20~28%又は22~26%含む。他の実施形態では、細胞組成物はCD3+CD56+細胞を15~30%含む。 For example, in some embodiments, the cell compositions of the invention are characterized by the presence of an average of at least 60%, typically 75% (e.g., 70-80%, 70-85%, 73-76% or 73-78%) cytotoxic CD3 + CD8 + cells. In other embodiments, the cell compositions are characterized by the presence of 70-85% CD3 + CD8 + cells. In other embodiments, the cell compositions are characterized by the presence of up to 15% CD3 + CD4 + cells (e.g., 7-12%, 11-13%, 9-12% or 8-11%), typically up to 10% CD3 + CD4 + cells. In other embodiments, on average at least 15% of the CD3 + CD8 + cells are CD8 + CD56 + cells (e.g., 15-30% or 17-20%). In another embodiment, the cell composition is characterized by at least 9%, typically 10-14%, CD3 + CD8 + CD56 + cells. In another embodiment, the cell composition is characterized by the presence of 7-17% CD3 + CD8 + CD56 + cells. In yet another embodiment, the cell composition comprises about 8-35%, typically 20-28% or 22-26%, CD3 + CD56 + cells. In another embodiment, the cell composition comprises 15-30% CD3 + CD56 + cells .
幾つかの実施形態では、細胞組成物は活性化マーカーとケモカイン受容体の高発現を特徴とする。幾つかの実施形態では、細胞はNKG2D、グランザイムB、パーフォリン、FASL、CXCR4及びCXCR2から成る群から選択される少なくとも1種、典型的には複数種の受容体を発現し、各々の可能性は本発明の別個の実施形態を表す。例えば、本明細書に示すように、増殖細胞組成物の実質的に全てのCD8+細胞が活性化マーカーNKG2DとグランザイムBを発現することが分かった(以下の実施例4及び5を参照)。従って、本発明は、その実施形態において、CD8+細胞が典型的にはNKG2DとグランザイムBの発現によっても特徴付けられる改良細胞組成物を提供する。 In some embodiments, the cell composition is characterized by high expression of activation markers and chemokine receptors. In some embodiments, the cells express at least one, and typically more than one, receptor selected from the group consisting of NKG2D, Granzyme B, Perforin, FASL, CXCR4, and CXCR2, each possibility representing a separate embodiment of the invention. For example, as shown herein, it has been found that substantially all CD8 + cells of an expanded cell composition express the activation markers NKG2D and Granzyme B (see Examples 4 and 5 below). Thus, in its embodiments, the invention provides improved cell compositions in which the CD8 + cells are also typically characterized by expression of NKG2D and Granzyme B.
ナチュラルキラーグループ2メンバーD(NKG2D)は、C型レクチン様受容体のNKG2ファミリーに属する膜貫通タンパク質である。ヒトにおいては、NKG2DはNK細胞、γδT細胞及びCD8+αβT細胞によって発現され、NKG2Dリガンドを発現する標的細胞に対するエフェクター細胞の機能を促進する活性化受容体として作用する。このようなリガンドは、MHCクラスI鎖関連タンパク質(MIC)又はUL-16結合タンパク質(ULBP)ファミリー(例えば、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3)に属する誘導自己タンパク質であり、これらは完全に存在しないか又は正常細胞の表面に低レベルでのみ存在し、感染細胞、悪性細胞、形質転換細胞、老化細胞及びストレスを受けた細胞で過剰発現する。その発現は様々な工程で(様々なストレス経路によって、最も顕著にはDNA損傷応答によって)調節される。NKG2Dは、その表面膜発現を促進及び安定化するDNAX活性化タンパク質10(DAP10)と結合している。NKG2Dにはその細胞質ドメインにシグナル伝達モチーフがなく、シグナル伝達はDAP10のリン酸化を介したライゲーションの際に生じ、これによって下流のシグナル伝達エフェクター分子が動員され、最終的には細胞障害性が引き起こされる。 Natural killer group 2 member D (NKG2D) is a transmembrane protein that belongs to the NKG2 family of C-type lectin-like receptors. In humans, NKG2D is expressed by NK cells, γδ T cells, and CD8 + αβ T cells and acts as an activating receptor that promotes effector cell function against target cells expressing NKG2D ligands. Such ligands are inducible self-proteins that belong to the MHC class I chain-related protein (MIC) or UL-16 binding protein (ULBP) family (e.g., MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3), which are completely absent or present only at low levels on the surface of normal cells and overexpressed in infected, malignant, transformed, senescent, and stressed cells. Its expression is regulated in various steps (by various stress pathways, most notably by the DNA damage response). NKG2D is associated with DNAX-activating protein 10 (DAP10), which promotes and stabilizes its surface membrane expression. NKG2D lacks signaling motifs in its cytoplasmic domain, and signaling occurs upon phosphorylation-mediated ligation of DAP10, which recruits downstream signaling effector molecules and ultimately leads to cytotoxicity.
更なる実施形態によれば、CD8+細胞はパーフォリン及び/又はFASLを更に発現することができる。他の実施形態では、CD8+細胞はCXCR4及び/又はCXCR2を更に発現することができる。他の実施形態では、CD8+細胞はCXCR3及び/又はCXCR6を更に発現することができる。各々の可能性は本発明の別個の実施形態を表す。他の実施形態では、CD3+CD8+細胞の少なくとも40%は、パーフォリン、FasL、CXCR4及びCXCR2から成る群から選択される少なくとも1種のマーカーの表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の少なくとも90%がCXCR3の表面発現を特徴とし、CD3+CD8+細胞の少なくとも30%がCXCR6の表面発現を特徴とする。 According to further embodiments, the CD8 + cells can further express perforin and/or FASL. In other embodiments, the CD8 + cells can further express CXCR4 and/or CXCR2. In other embodiments, the CD8 + cells can further express CXCR3 and/or CXCR6. Each possibility represents a separate embodiment of the invention. In other embodiments, at least 40% of the CD3 + CD8 + cells are characterized by surface expression of at least one marker selected from the group consisting of perforin, FasL, CXCR4, and CXCR2. In other embodiments, at least 90% of the CD3 + CD8 + cells are characterized by surface expression of CXCR3 and at least 30% of the CD3 + CD8 + cells are characterized by surface expression of CXCR6.
CXCケモカイン受容体は、CXCケモカインファミリーのサイトカインに特異的に結合して応答する内在性膜タンパク質である。構造的には、このような受容体は、細胞膜を7回貫通するため、7回膜貫通(7-TM)タンパク質として知られるGタンパク質関連受容体として特徴付けられる。 CXC chemokine receptors are integral membrane proteins that specifically bind and respond to cytokines of the CXC chemokine family. Structurally, such receptors are characterized as G protein-linked receptors, known as seven-transmembrane (7-TM) proteins, because they span the cell membrane seven times.
CXCR4(フーシンとしても知られる)は、その標準リガンドとしてのCXCL12(又はSDF-1)として知られるケモカインの受容体であり、リンパ球に対する強力な走化性活性を備えた分子である。CXCR4は幅広い細胞分布を有し、様々な造血細胞型(例えば、好中球、単球、T細胞及びB細胞、樹状細胞、ランゲルハンス細胞及びマクロファージ)で発現する。CXCR4活性化は細胞遊走と細胞増殖の両方を促進することができ、CXCL12は、造血幹細胞の骨髄へのホーミングと造血幹細胞の静止において重要であることも知られている。CXCR4の更なるリガンドが幾つか開示されており、例えば、損傷関連分子パターン分子である高移動度グループボックス1タンパク質(HMGB1)、マクロファージ遊走阻害因子(MIF)、自然免疫の調節に関与するサイトカイン、細胞外ユビキチン(eUb)及びベータディフェンシン-3(HBD3)が挙げられる。 CXCR4 (also known as fusin) is the receptor for the chemokine known as CXCL12 (or SDF-1) as its canonical ligand, a molecule with strong chemotactic activity for lymphocytes. CXCR4 has a wide cellular distribution and is expressed on various hematopoietic cell types (e.g., neutrophils, monocytes, T and B cells, dendritic cells, Langerhans cells and macrophages). CXCR4 activation can promote both cell migration and cell proliferation, and CXCL12 is also known to be important in hematopoietic stem cell homing to the bone marrow and in hematopoietic stem cell quiescence. Several additional ligands for CXCR4 have been disclosed, including the damage-associated molecular pattern molecule high mobility group box 1 protein (HMGB1), macrophage migration inhibitory factor (MIF), a cytokine involved in the regulation of innate immunity, extracellular ubiquitin (eUb) and beta defensin-3 (HBD3).
インターロイキン8受容体ベータ(IL8RB)としても知られるCXCR2は、好中球及び他の特定の細胞型の表面に発現する。CXCR2は、損傷又は感染部位への好中球の動員に関与する主要なサイトカインであり、マクロファージや上皮細胞、気道平滑筋細胞、内皮細胞等の他の細胞型によって産生されるケモカインであるIL-8(CXCL8としても知られる)に高い親和性で結合する。CXCR2の他のリガンドとしては、例えば、CXCL2、CXCL3及びCXCL5が挙げられる。 CXCR2, also known as interleukin 8 receptor beta (IL8RB), is expressed on the surface of neutrophils and certain other cell types. CXCR2 binds with high affinity to IL-8 (also known as CXCL8), a chemokine produced by macrophages and other cell types, including epithelial cells, airway smooth muscle cells, and endothelial cells, which is the primary cytokine involved in the recruitment of neutrophils to sites of injury or infection. Other ligands for CXCR2 include, for example, CXCL2, CXCL3, and CXCL5.
CXCR3とそのリガンド、CXCL9、CXCL10及びCXCL11は、インターフェロン誘発炎症反応時の重要な免疫化学誘引物質である。CXCR3(GPR9/CD183)は、様々な細胞型で発現するインターフェロン誘導性ケモカイン受容体であるが、単球、Th1T細胞、CD8+T細胞、NKT細胞、NK細胞、樹状細胞、及び一部の癌細胞で優先的に発現する。ヒトにおけるCXCR3には、CXCR3-A、CXCR3-B、及びケモカイン受容体3代替物(CXCR3-alt)の3種のアイソフォームが存在する。CXCR3-AはCXCケモカインCXCL9(MIG)、CXCL10(IP-10)、及びCXCL11(I-TAC)に結合し、CXCR3-BはCXCL9、CXCL10、及びCXCL11に加えてCXCL4にも結合することができる。 CXCR3 and its ligands, CXCL9, CXCL10, and CXCL11, are important immunochemoattractants during interferon-induced inflammatory responses. CXCR3 (GPR9/CD183) is an interferon-inducible chemokine receptor expressed in a variety of cell types, but is preferentially expressed in monocytes, Th1 T cells, CD8 + T cells, NKT cells, NK cells, dendritic cells, and some cancer cells. There are three isoforms of CXCR3 in humans: CXCR3-A, CXCR3-B, and chemokine receptor 3 alternative (CXCR3-alt). CXCR3-A binds to the CXC chemokines CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (I-TAC), whereas CXCR3-B can bind to CXCL9, CXCL10, and CXCL11 as well as CXCL4.
CXCR6(STRL33及びTYMSTRとしても知られる)は、例えば、ナイーブCD8+細胞、CD3-CD16-/lowCD56+及びCD3-CD16lowCD56-NK細胞、NKT細胞、活性化CD4+及びCD8+T細胞、及びγδT細胞の30~40%で発現するケモカイン受容体である。CXCR6はCXCL16にのみ結合し、移植片対宿主病におけるT細胞動員、感染後の肝臓常在CD8T細胞の維持、肺疾患重症度の調節等、様々な免疫方法で役割を果たす。そのリガンドは、腫瘍細胞の増殖、遊走、浸潤及び転移において重要な役割を果たすα-ケモカインサブファミリーに属する走化性サイトカインである。特に、CXCR6の活性化は腫瘍微小環境での細胞傷害性T細胞の生存と増殖に重要であり、それによってT細胞の消耗を軽減することが示唆された。 CXCR6 (also known as STRL33 and TYMSTR) is a chemokine receptor expressed, for example, on naive CD8 + cells, CD3 − CD16 −/low CD56 + and CD3 − CD16 low CD56 − NK cells, NKT cells, activated CD4 + and CD8 + T cells, and 30-40% of γδ T cells. CXCR6 binds only to CXCL16 and plays a role in various immune methods, such as T cell recruitment in graft-versus-host disease, maintenance of liver-resident CD8 T cells after infection, and modulating lung disease severity. Its ligands are chemotactic cytokines belonging to the α-chemokine subfamily that play important roles in tumor cell proliferation, migration, invasion, and metastasis. In particular, it has been suggested that activation of CXCR6 is important for the survival and proliferation of cytotoxic T cells in the tumor microenvironment, thereby attenuating T cell exhaustion.
更に、本発明に係る細胞組成物はDNAM-1の表面発現を更に特徴とする。他の実施形態では、CD3+CD8+細胞の少なくとも90%、93%、95%、97%、典型的には実質的に全て(最大100%)がDNAM-1の表面発現を特徴とする。 Additionally, the cell compositions of the present invention are further characterized by surface expression of DNAM-1, hi other embodiments, at least 90%, 93%, 95%, 97%, and typically substantially all (up to 100%) of the CD3 + CD8 + cells are characterized by surface expression of DNAM-1.
DNAXアクセサリー分子-1(DNAM-1、CD226)は、免疫グロブリンスーパーファミリーの膜貫通ネクチン様糖タンパク質であり、NK細胞とT細胞で活性化受容体として作用する。そのリガンドであるCD112(ポリオウイルス受容体(PVR)としても知られる)とCD155(ネクチン-2としても知られる)も同様にネクチンファミリーのメンバーであり、DNAM-1-CD112/CD155軸は、主にNK細胞媒介の腫瘍細胞の死滅における重要な役割で知られている。DNAM-1/PVR軸は同種異系活性化T細胞のNK細胞媒介溶解に関与する一方、自家状況ではNKG2Dが主要な受容体として現れることが提案されている。 DNAX accessory molecule-1 (DNAM-1, CD226) is a transmembrane nectin-like glycoprotein of the immunoglobulin superfamily that acts as an activating receptor in NK and T cells. Its ligands, CD112 (also known as poliovirus receptor (PVR)) and CD155 (also known as nectin-2), are also members of the nectin family, and the DNAM-1-CD112/CD155 axis is primarily known for its key role in NK cell-mediated tumor cell killing. It has been proposed that the DNAM-1/PVR axis is involved in NK cell-mediated lysis of allogeneically activated T cells, whereas NKG2D appears as the primary receptor in the autologous setting.
他の実施形態では、本発明の細胞組成物は、抑制性免疫チェックポイント分子の発現の低下を特徴とする。 In other embodiments, the cell compositions of the present invention are characterized by reduced expression of inhibitory immune checkpoint molecules.
免疫チェックポイント分子は、自己寛容を誘導することによって、免疫系が自分自身の体を攻撃するのを防ぐ免疫回避機序を表す。通常、免疫チェックポイント受容体はT細胞上に存在し、抗原提示細胞上に発現する免疫チェックポイントリガンドと相互作用する。しかし、一部の癌は免疫チェックポイント標的を刺激して攻撃から自身を保護することができる。T細胞はMHC分子上に提示された抗原を認識し、活性化されて免疫反応を引き起こすが、これと並行して起こる免疫チェックポイント受容体とリガンドとの相互作用がT細胞の活性化を制御する。 Immune checkpoint molecules represent an immune evasion mechanism that prevents the immune system from attacking one's own body by inducing self-tolerance. Normally, immune checkpoint receptors are present on T cells and interact with immune checkpoint ligands expressed on antigen-presenting cells. However, some cancers can stimulate immune checkpoint targets to protect themselves from attack. T cells recognize antigens presented on MHC molecules and become activated to trigger an immune response, while the interaction of immune checkpoint receptors with ligands in parallel controls T cell activation.
免疫チェックポイント受容体には共刺激受容体と抑制性受容体があり、T細胞の活性化と免疫反応は両受容体間のバランスによって制御される。本明細書で使用される「抑制性免疫チェックポイント分子」という用語は、特に、T細胞等の免疫エフェクター細胞に発現し、それぞれのリガンドによる結合で抗腫瘍免疫応答の抑制的調節又は阻害を媒介することができる、抑制性免疫チェックポイント受容体を包含する。その例としては、CTLA-4、PD-1、VISTA、B7-H3、B7-H4、HVEM、KIRファミリー受容体、TIM-1、TIM-3、LAG-3、及びTIGITが挙げられるが、これらに限定されない。 Immune checkpoint receptors include costimulatory receptors and inhibitory receptors, and T cell activation and immune responses are controlled by the balance between these two receptors. As used herein, the term "inhibitory immune checkpoint molecule" specifically encompasses inhibitory immune checkpoint receptors that are expressed on immune effector cells such as T cells and that can mediate downregulation or inhibition of antitumor immune responses upon binding with their respective ligands. Examples include, but are not limited to, CTLA-4, PD-1, VISTA, B7-H3, B7-H4, HVEM, KIR family receptors, TIM-1, TIM-3, LAG-3, and TIGIT.
特定の実施形態によれば、本発明のACT組成物のCD3+CD8+細胞は、CTLA-4、PD-1、TIGIT、Lag-3及びTim-3から成る群から選択される1種以上の免疫チェックポイント分子の表面発現の低下を特徴とする。様々な実施形態では、このような表面レベルの低下は、本明細書に示すように、元のPBMC試料に対する低下、又はCIK細胞調製物等の他のACT組成物に対する低下と関連することがある。各々の可能性は本発明の別個の実施形態を表す。 According to certain embodiments, the CD3 + CD8 + cells of the ACT compositions of the invention are characterized by reduced surface expression of one or more immune checkpoint molecules selected from the group consisting of CTLA-4, PD-1, TIGIT, Lag-3, and Tim-3. In various embodiments, such reduced surface levels may be associated with a reduction relative to the original PBMC sample, or a reduction relative to other ACT compositions, such as CIK cell preparations, as shown herein. Each possibility represents a separate embodiment of the present invention.
他の実施形態では、CD3+CD8+細胞はCTLA-4の実質的な表面発現の欠如を特徴とする。他の実施形態では、CD3+CD8+細胞の少なくとも95%、97%及び最大約100%がCTLA-4-細胞である。細胞傷害性Tリンパ球抗原4(CTLA-4/CD152)は、制御性T細胞で構成的に発現する共受容体であり、活性化(癌において特に顕著な現象)の後に従来のCD4+及びCD8+T細胞でアップレギュレートされる。CTLA-4は、活性化受容体CD28よりも高い親和性と結合力でAPC上のCD80とCD86に結合するため、そのリガンドに対してCD28を打ち負かしてT細胞阻害を促進することができる。 In other embodiments, the CD3 + CD8 + cells are characterized by a substantial lack of surface expression of CTLA-4. In other embodiments, at least 95%, 97%, and up to about 100% of the CD3 + CD8 + cells are CTLA-4 − cells. Cytotoxic T-lymphocyte antigen 4 (CTLA-4/CD152) is a co-receptor constitutively expressed on regulatory T cells and is upregulated on conventional CD4 + and CD8 + T cells following activation, an event particularly prominent in cancer. CTLA-4 binds to CD80 and CD86 on APCs with higher affinity and avidity than the activating receptor CD28, and is therefore able to outcompete CD28 for its ligand and promote T-cell inhibition.
他の実施形態では、CD3+CD8+細胞の25%、20%、15%又は10%以下がPD-1の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約15%がPD-1の表面発現を特徴とする。プログラム細胞死タンパク質1(PD1)は、制御性T細胞、B細胞、ナチュラルキラー細胞及び一部の骨髄細胞集団だけでなく、活性化中に全てのT細胞によって発現される。PD1は持続的な抗原遭遇時に高い持続的な発現レベルを示すことが多く、これは慢性感染症や癌の状況で発生することがある。このような状況では、PD1が防御免疫を制限する場合がある。PD1はPD-L1とPD-L2の2種のリガンドを有し、腫瘍微小環境でのPD-L1の発現は一般に腫瘍の免疫回避に関連している。 In other embodiments, no more than 25%, 20%, 15%, or 10% of the CD3 + CD8 + cells are characterized by surface expression of PD-1. In other embodiments, about 15% of the CD3 + CD8 + cells are characterized by surface expression of PD-1. Programmed cell death protein 1 (PD1) is expressed by all T cells during activation, as well as regulatory T cells, B cells, natural killer cells, and some myeloid cell populations. PD1 often shows high and sustained expression levels upon persistent antigen encounter, which may occur in the setting of chronic infection or cancer. In such settings, PD1 may limit protective immunity. PD1 has two ligands, PD-L1 and PD-L2, and expression of PD-L1 in the tumor microenvironment is commonly associated with tumor immune evasion.
他の実施形態では、CD3+CD8+細胞の30%、25%、22%、20%又は18%以下がTIGITの表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約20~22%がTIGITの表面発現を特徴とする。T細胞免疫グロブリンとITIMドメイン(TIGIT)は、CD4+T細胞、CD8+T細胞及びTregs等のNK細胞及びT細胞に発現する抑制性受容体である。TIGIT発現は通常、ナイーブ細胞では低いが、活性化の際にはT細胞とNK細胞の両方がTIGITをアップレギュレートすることが示されている。TIGITはCD155(ポリオウイルス受容体:PVR、NECL-5)、CD112及びCD113の3種のリガンドを有し、これらは全てネクチン及びNECL分子のファミリーに属する。 In other embodiments, no more than 30%, 25%, 22%, 20%, or 18% of the CD3 + CD8 + cells are characterized by surface expression of TIGIT. In other embodiments, about 20-22% of the CD3 + CD8 + cells are characterized by surface expression of TIGIT. T cell immunoglobulin with ITIM domain (TIGIT) is an inhibitory receptor expressed on NK cells and T cells, including CD4 + T cells, CD8 + T cells, and Tregs . TIGIT expression is normally low on naive cells, but both T cells and NK cells have been shown to upregulate TIGIT upon activation. TIGIT has three ligands, CD155 (poliovirus receptor: PVR, NECL-5), CD112, and CD113, all of which belong to the nectin and NECL families of molecules.
他の実施形態では、CD3+CD8+細胞の55%、53%、51%、50%又は48%以下がLag-3の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約50~51%がLag-3の表面発現を特徴とする。リンパ球活性化遺伝子3(LAG3、CD223としても知られる)は、活性化T細胞、TIL、Tregs、NK細胞、B細胞及び形質細胞様DCで発現する免疫グロブリンスーパーファミリーのメンバーである。LAG3はMHC-IIと相互作用して、同じMHC分子のTCRとCD4への結合を禁止して、免疫応答におけるTCRシグナル伝達を直接妨げる。更なるLAG3リガンドとしては、FGL-1、α-シヌクレイン原線維(α-syn)、レクチンのガレクチン-3(Gal-3)及びリンパ節類洞内皮細胞C型レクチン(LSECtin)が挙げられる。LAG3は、他の免疫チェックポイント分子と同様に、T細胞の細胞増殖、活性化及び恒常性を負に制御し、Treg抑制機能に役割を果たすことが報告されている。例えば、T細胞では、LAG3はサイトカインとグランザイムの産生と増殖を抑制し、同時に制御性T細胞への分化を促進する。 In other embodiments, up to 55%, 53%, 51%, 50%, or 48% of the CD3 + CD8 + cells are characterized by surface expression of Lag-3. In other embodiments, about 50-51% of the CD3 + CD8 + cells are characterized by surface expression of Lag-3. Lymphocyte activation gene 3 (LAG3, also known as CD223) is a member of the immunoglobulin superfamily expressed on activated T cells, TILs, Tregs , NK cells, B cells, and plasmacytoid DCs. LAG3 interacts with MHC-II to inhibit binding of the same MHC molecule to TCR and CD4, directly interfering with TCR signaling in immune responses. Additional LAG3 ligands include FGL-1, α-synuclein fibrils (α-syn), the lectin galectin-3 (Gal-3), and lymph node sinusoidal endothelial cell C-type lectin (LSECtin). LAG3, like other immune checkpoint molecules, has been reported to negatively regulate T cell proliferation, activation, and homeostasis, and play a role in Treg suppressive function. For example, in T cells, LAG3 suppresses the production and proliferation of cytokines and granzymes, while promoting differentiation into regulatory T cells.
他の実施形態では、CD3+CD8+細胞の55%、54%、52%、50%又は48%以下がTim-3の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約50~54%がTim-3の表面発現を特徴とする。TIMファミリーのメンバーであるT細胞免疫グロブリンとムチンドメイン含有タンパク質3(TIM3)は、インターフェロンγ(IFNγ)産生CD4+及びCD8+T細胞によって発現されると共に、制御性T細胞、骨髄細胞、NK細胞及びマスト細胞等の他の多くの細胞型によって発現される。TIM3は、複数の異なるリガンド(例えば、ガレクチン9、ホスファチジルセリン、癌胎児性抗原関連細胞接着分子1(CEACAM1)及び高移動度タンパク質B1(HMGB1))を有することが報告されている。TIM3は複数の共抑制受容体(チェックポイント受容体)を含むモジュールの一部であり、これらの受容体は、慢性ウイルス感染症や癌における機能不全又は「疲弊した」T細胞で共発現及び共制御される。 In other embodiments, up to 55%, 54%, 52%, 50%, or 48% of the CD3 + CD8 + cells are characterized by surface expression of Tim-3. In other embodiments, about 50-54% of the CD3 + CD8 + cells are characterized by surface expression of Tim-3. T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3), a member of the TIM family, is expressed by interferon-γ (IFNγ)-producing CD4 + and CD8 + T cells, as well as by many other cell types, including regulatory T cells, myeloid cells, NK cells, and mast cells. TIM3 has been reported to have several different ligands, such as galectin 9, phosphatidylserine, carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), and high mobility group protein B1 (HMGB1). TIM3 is part of a module that contains multiple co-inhibitory receptors (checkpoint receptors), which are co-expressed and co-regulated in dysfunctional or "exhausted" T cells in chronic viral infections and cancer.
他の実施形態では、細胞組成物は腫瘍細胞に対して細胞傷害活性を発揮する。幾つかの実施形態では、腫瘍細胞は造血起源である。「造血起源の腫瘍細胞」という語句は、血液細胞又はその前駆体に由来する悪性細胞を意味する。他の実施形態では、腫瘍細胞はリンパ系起源である。他の実施形態では、腫瘍細胞は骨髄起源である。特定の実施形態では、腫瘍細胞は白血病細胞、例えば、急性骨髄性白血病(AML)細胞である。他の特定の実施形態では、腫瘍細胞は多発性骨髄腫細胞である。 In other embodiments, the cell composition exerts cytotoxic activity against tumor cells. In some embodiments, the tumor cells are of hematopoietic origin. The phrase "tumor cells of hematopoietic origin" refers to malignant cells derived from blood cells or their precursors. In other embodiments, the tumor cells are of lymphoid origin. In other embodiments, the tumor cells are of myeloid origin. In certain embodiments, the tumor cells are leukemia cells, e.g., acute myeloid leukemia (AML) cells. In other specific embodiments, the tumor cells are multiple myeloma cells.
例示的な実施形態によれば、細胞組成物は、造血器腫瘍細胞とインビトロで(それぞれ)少なくとも0.25:1の比率で24時間インキュベートした場合、造血器腫瘍細胞を特異的に根絶(死滅)することができる。更なる例示的な実施形態によれば、細胞組成物は、造血器腫瘍細胞とインビトロで1:1の比率で24時間インキュベートした場合に、造血器腫瘍細胞の少なくとも50%、典型的には60~99%、より典型的には約80~90%を特異的に根絶することができる。他の実施形態では、腫瘍細胞は少なくとも1種、典型的には複数のNKG2Dリガンド(例えば、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3が挙げられるが、これらに限定されない)を発現する。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256及びULBP3を発現する。他の実施形態では、本明細書に示すように、細胞組成物は、同じ条件下でCIK組成物よりも高い程度まで造血器腫瘍細胞を特異的に根絶することができる(例えば、エフェクター細胞と腫瘍細胞の比率が1:1の場合は6~10倍高くなり、エフェクター細胞と腫瘍細胞の比率が0.5:1の場合は最大で約15倍高くなる)。他の実施形態では、腫瘍細胞は固形腫瘍細胞である。特定の実施形態では、細胞は前立腺腫瘍細胞又は中皮腫細胞である。各々の可能性は本発明の別個の実施形態を表す。 According to exemplary embodiments, the cell composition is capable of specifically eradicating (killing) hematopoietic tumor cells when incubated in vitro with the hematopoietic tumor cells at a ratio of at least 0.25:1 (respectively) for 24 hours. According to further exemplary embodiments, the cell composition is capable of specifically eradicating at least 50%, typically 60-99%, more typically about 80-90% of the hematopoietic tumor cells when incubated in vitro with the hematopoietic tumor cells at a ratio of 1:1 for 24 hours. In other embodiments, the tumor cells express at least one, typically multiple NKG2D ligands (e.g., but not limited to, MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3). In other embodiments, the tumor cells express MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, as shown herein, the cell composition can specifically eradicate hematopoietic tumor cells to a greater extent than the CIK composition under the same conditions (e.g., 6-10 fold greater at a 1:1 effector to tumor cell ratio, and up to about 15 fold greater at a 0.5:1 effector to tumor cell ratio). In other embodiments, the tumor cells are solid tumor cells. In certain embodiments, the cells are prostate tumor cells or mesothelioma cells. Each possibility represents a separate embodiment of the present invention.
本明細書で使用される、腫瘍細胞に対する「特異的根絶」という用語は、標的腫瘍細胞に対して選択的(又は優先的)である、標的化された腫瘍細胞死を意味する。通常、根絶は腫瘍細胞によって誘導される細胞傷害活性によって発揮される(例えば、本明細書に開示の条件下での細胞組成物と腫瘍細胞との接触時)。 As used herein, the term "specific eradication" of tumor cells refers to targeted tumor cell death that is selective (or preferential) for the target tumor cells. Typically, eradication is exerted by cytotoxic activity induced by the tumor cells (e.g., upon contact of the cell composition with the tumor cells under conditions disclosed herein).
本明細書で使用される「細胞障害性」及び「細胞障害活性」という用語は、特に細胞媒介細胞障害性又は細胞溶解、即ち、免疫細胞、特にエフェクター免疫細胞の細胞死滅活性を意味する。免疫系の顕著な細胞傷害性エフェクター細胞には、NK細胞、細胞傷害性T細胞(例えば、CD8+T細胞)及びNK-T細胞がある。細胞傷害性細胞(例えば、CTL又はNK細胞)は、1種以上の異なる機序(例えば、細胞溶解性顆粒に蓄えられた1種以上の細胞毒素の放出、又はFasリガンド(FasL)の発現が挙げられるが、これに限定されない)を用いた標的細胞のアポトーシス又は溶解によって標的細胞を死滅させることができる。細胞毒素の例としては、例えば、パーフォリン、グランザイム及びグラニュライシンが挙げられる。現在知られているグランザイムは、グランザイムA、B、H、K及びMである。このような細胞障害性又は細胞溶解活性は、標準的な技法を用いて、例えば、生細胞の選択的標識を使用するアッセイによって測定及び定量化することができる。例えば、二酢酸サクシニミジルエステル(CFSE)及び/又はヨウ化プロピジウムベースのアッセイ、又は当技術分野で既知の他の適切なアッセイを用いることができるが、これに限定されない。 The terms "cytotoxicity" and "cytotoxic activity" as used herein refer in particular to cell-mediated cytotoxicity or cytolysis, i.e., the cell-killing activity of immune cells, particularly effector immune cells. Prominent cytotoxic effector cells of the immune system include NK cells, cytotoxic T cells (e.g., CD8 + T cells) and NK-T cells. Cytotoxic cells (e.g., CTL or NK cells) can kill target cells by apoptosis or lysis of the target cell using one or more different mechanisms, including, but not limited to, release of one or more cytotoxins stored in cytolytic granules or expression of Fas ligand (FasL). Examples of cytotoxins include, for example, perforin, granzymes and granulysin. Currently known granzymes are granzymes A, B, H, K and M. Such cytotoxic or cytolytic activity can be measured and quantified using standard techniques, for example, by assays using selective labeling of live cells. For example, but without limitation, succinimidyl ester diacetate (CFSE) and/or propidium iodide based assays, or other suitable assays known in the art, may be used.
他の実施形態では、細胞傷害活性はグランザイムB及び/又はパーフォリンによって媒介される。他の実施形態では、細胞傷害活性はFAS-FasL相互作用によっては実質的に媒介されない。 In other embodiments, the cytotoxic activity is mediated by granzyme B and/or perforin. In other embodiments, the cytotoxic activity is not substantially mediated by FAS-FasL interactions.
パーフォリンは、Ca+2依存的に標的細胞に移動する孔形成細胞溶解性タンパク質である。パーフォリンは、死滅させる予定の細胞のごく近傍で細胞溶解性顆粒を放出すると、標的細胞の細胞膜に細孔を形成し、そこからグランザイムと関連分子が侵入し、アポトーシスを誘導する。グランザイムB(グランザイム2及び細胞傷害性Tリンパ球関連セリンエステラーゼ1としても知られる)は、カスパーゼの切断と活性化により、細胞媒介免疫応答において標的細胞のアポトーシスを迅速に誘導する特に重要なセリンプロテアーゼである。FAS受容体は、Fas、FasR、アポトーシス抗原1(APO-1又はAPT)、分化クラスター95(CD95)又は腫瘍壊死因子受容体スーパーファミリーメンバー6(TNFRSF6)としても知られ、そのリガンドであるFasリガンド(FasL)と結合すると、プログラム細胞死(アポトーシス)を引き起こす細胞表面の死受容体である。免疫エフェクター細胞によるFasLの発現、及びそれに続くFAS発現標的腫瘍細胞との接触時のFAS-FasL相互作用は、パーフォリン非依存的に腫瘍細胞のアポトーシスを誘導することができる。このようなパーフォリン非依存的な細胞傷害活性は、パーフォリンモノマーの重合はできないが、細胞媒介細胞溶解が依然として起こるCa2+非依存的な系で検討することができる。 Perforin is a pore-forming cytolytic protein that translocates to target cells in a Ca +2- dependent manner. Upon releasing cytolytic granules in close proximity to the cell to be killed, perforin creates pores in the plasma membrane of the target cell through which granzymes and related molecules can enter and induce apoptosis. Granzyme B (also known as granzyme 2 and cytotoxic T lymphocyte-associated serine esterase 1) is a particularly important serine protease that rapidly induces apoptosis of target cells in cell-mediated immune responses by cleaving and activating caspases. The FAS receptor, also known as Fas, FasR, apoptosis antigen 1 (APO-1 or APT), cluster of differentiation 95 (CD95) or tumor necrosis factor receptor superfamily member 6 (TNFRSF6), is a cell surface death receptor that triggers programmed cell death (apoptosis) upon binding to its ligand, Fas ligand (FasL). Expression of FasL by immune effector cells, and subsequent FAS-FasL interaction upon contact with FAS-expressing target tumor cells, can induce tumor cell apoptosis in a perforin-independent manner. Such perforin-independent cytotoxic activity can be examined in a Ca2+ -independent system in which polymerization of perforin monomers is not possible, yet cell-mediated cytolysis still occurs.
本明細書において、特定の剤又は経路(例えば、パーフォリン、グランザイムB又はFAS-FasL)によって媒介される細胞傷害活性とは、剤の関与又は活性によって誘発される、及び/又は実質的に剤の関与又は活性に依存する活性を意味する。このような活性には、剤が対応するエフェクター細胞内で発現された後に活性化されることが必要である。従って、剤又は経路に対する特定の薬理学的阻害剤又は抗体の存在下で細胞傷害活性は、阻害剤又は抗体の非存在下での同じ条件下での活性と比較して有意に低下する(典型的には少なくとも50%、より典型的には少なくとも60%、70%、80%、90%及び最大100%の阻害)が、これは、活性が剤(例えば、パーフォリン又はグランザイムB)によって媒介されていることを示す。同様に、有意な低下(統計的に有意、又は幾つかの実施形態では5%、10%、20%又は30%以下の低下)がないことは、活性が剤(例えば、FasL)によって媒介されていないことを示す。例えば、本明細書に開示の細胞障害性アッセイは、3,4-ジクロロイソクマリン(DCI)又は他の市販のグランザイムB阻害剤、コンカナマイシンA(CMA)又は他の市販のパーフォリン阻害剤、又はSigma社又は他の供給業者から市販されている抗FasL(又は抗FAS)ブロック抗体の存在下で行うことができる。このようなアッセイの非限定的な例は以下の実施例にて記載する。 As used herein, cytotoxic activity mediated by a particular agent or pathway (e.g., perforin, granzyme B, or FAS-FasL) refers to activity that is triggered by and/or substantially dependent on the involvement or activity of the agent. Such activity requires that the agent is activated after expression in the corresponding effector cell. Thus, in the presence of a particular pharmacological inhibitor or antibody to the agent or pathway, the cytotoxic activity is significantly reduced (typically at least 50%, more typically at least 60%, 70%, 80%, 90%, and up to 100% inhibition) compared to the activity under the same conditions in the absence of the inhibitor or antibody, indicating that the activity is mediated by the agent (e.g., perforin or granzyme B). Similarly, the absence of a significant reduction (statistically significant, or in some embodiments, a reduction of 5%, 10%, 20%, or 30% or less) indicates that the activity is not mediated by the agent (e.g., FasL). For example, the cytotoxicity assays disclosed herein can be performed in the presence of 3,4-dichloroisocoumarin (DCI) or other commercially available granzyme B inhibitors, concanamycin A (CMA) or other commercially available perforin inhibitors, or anti-FasL (or anti-FAS) blocking antibodies available from Sigma or other suppliers. Non-limiting examples of such assays are described in the Examples below.
本明細書に示すように、本発明に係る細胞組成物は、様々な腫瘍細胞に対してMHC制限細胞傷害活性と非MHC制限細胞傷害活性の両方を有することが見出された。従って、一実施形態では、細胞傷害活性はMHC依存的である(即ち、細胞上のMHC分子の存在又は活性が必要である)。他の実施形態では、細胞傷害活性は非MHC依存的である。他の実施形態では、細胞傷害活性は実質的にMHCI非依存的である。他の実施形態では、細胞傷害活性は実質的にMHCII非依存的である。 As shown herein, the cell compositions of the present invention have been found to have both MHC-restricted and non-MHC-restricted cytotoxic activity against a variety of tumor cells. Thus, in one embodiment, the cytotoxic activity is MHC-dependent (i.e., requires the presence or activity of an MHC molecule on the cell). In another embodiment, the cytotoxic activity is non-MHC-dependent. In another embodiment, the cytotoxic activity is substantially MHC I-independent. In another embodiment, the cytotoxic activity is substantially MHC II-independent.
他の実施形態では、本発明の細胞組成物は、非腫瘍細胞に対して実質的な細胞傷害活性を示さずに、腫瘍特異的細胞傷害活性を発揮することができる。他の実施形態では、本発明の細胞組成物は、移植片対宿主(GVH)反応又は移植片対宿主病(GVHD)を実質的に誘導しない。従って、幾つかの実施形態では、本発明の細胞組成物は治療する対象と組織適合性である必要はない。従って、自家性、組織適合性同種異系、及び非組織適合性同種異系PBMC試料を本発明の組成物の調製に使用することができる。 In other embodiments, the cell compositions of the present invention can exert tumor-specific cytotoxic activity without substantial cytotoxic activity against non-tumor cells. In other embodiments, the cell compositions of the present invention do not substantially induce graft-versus-host (GVH) reactions or graft-versus-host disease (GVHD). Thus, in some embodiments, the cell compositions of the present invention need not be histocompatible with the subject being treated. Thus, autologous, histocompatible allogeneic, and non-histocompatible allogeneic PBMC samples can be used to prepare the compositions of the present invention.
本明細書で使用される「移植片対宿主病」又は「GVHD」という用語は、移植レシピエントの組織に対する移植白血球による攻撃に起因する炎症性疾患を意味する。GVHDは通常、免疫低下宿主において移植片の免疫担当細胞によって非自己であると認識された場合に発生し、急性及び/又は慢性GVHDを包含する。ACTの状況では、GVHDという用語は、特に、養子移植された免疫細胞によって誘発される、レシピエント対象の非腫瘍細胞、組織及び/又は器官への損傷に起因する病理過程及び臨床症状を意味する。GVHDは、上皮組織、特に皮膚、肝臓及び消化管粘膜に対する炎症を介した損傷として現れることが多い。 As used herein, the term "graft-versus-host disease" or "GVHD" refers to an inflammatory disease resulting from an attack by transplanted leukocytes against the tissues of the transplant recipient. GVHD typically occurs in an immunocompromised host when the graft is recognized as non-self by immunocompetent cells of the graft and encompasses acute and/or chronic GVHD. In the context of ACT, the term GVHD specifically refers to the pathological process and clinical symptoms resulting from damage induced by adoptively transferred immune cells to non-tumor cells, tissues and/or organs of the recipient subject. GVHD often manifests as inflammation-mediated damage to epithelial tissues, particularly the skin, liver and gastrointestinal mucosa.
「組織適合性」という用語は、異なる個体間の組織の類似性を意味する。組織適合性のレベルは、患者とドナーがどの程度適合しているかを表す。主要な組織適合性決定因子はヒト白血球抗原(HLA)である。HLAタイピングは、潜在的なドナーと潜在的なレシピエントの間で行い、両者のHLAがどの程度一致するかを確認する。本明細書で使用される「組織適合性」という用語は、6種のHLA抗原(2種のA抗原、2種のB抗原、及び2種のDR抗原)の全てがドナーとレシピエントとの間で同一である実施形態を意味する。 The term "histocompatibility" refers to the similarity of tissues between different individuals. The level of histocompatibility represents how well the patient and donor are matched. The major histocompatibility determinants are the human leukocyte antigens (HLA). HLA typing is performed between a potential donor and a potential recipient to determine how well their HLAs match. As used herein, the term "histocompatibility" refers to an embodiment in which all six HLA antigens (two A antigens, two B antigens, and two DR antigens) are identical between the donor and the recipient.
本明細書で更に示すように、本発明の細胞組成物は、インビボで腫瘍細胞(非組織適合性腫瘍細胞を含む)に対して有意な細胞傷害活性を示す。例えば、本明細書の実施例8に示すように、本発明に係る組成物はリンパ系腫瘍細胞に対して顕著な細胞傷害活性を示し、腫瘍の発生を少なくとも78%~97%阻害した。従って、本発明は、幾つかの実施形態において、脾臓における造血器腫瘍の発生を少なくとも50%、少なくとも60%、少なくとも70%又は少なくとも80%、90%、95%又は97%阻害することができる細胞組成物に関し、各々の可能性は本発明の別個の実施形態を表す。更に、本発明に係る有利な組成物(本発明の組成物)は、インビボでの非組織適合性腫瘍細胞に対して、PBMC(一般に、インビボで腫瘍細胞を根絶するのに実質的に有効ではないと考えられている)又はCIKやLAK等の種々のPBMC由来組成物によって発揮されるよりも有意に高い細胞傷害活性を示す。 As further shown herein, the cell compositions of the present invention exhibit significant cytotoxic activity against tumor cells (including non-histocompatible tumor cells) in vivo. For example, as shown in Example 8 herein, the compositions of the present invention exhibited significant cytotoxic activity against lymphoid tumor cells, inhibiting tumor development by at least 78% to 97%. Thus, in some embodiments, the present invention relates to cell compositions that can inhibit the development of hematopoietic tumors in the spleen by at least 50%, at least 60%, at least 70%, or at least 80%, 90%, 95%, or 97%, each possibility representing a separate embodiment of the present invention. Furthermore, advantageous compositions of the present invention (compositions of the present invention) exhibit significantly higher cytotoxic activity against non-histocompatible tumor cells in vivo than that exerted by PBMCs (generally considered to be substantially ineffective in eradicating tumor cells in vivo) or various PBMC-derived compositions such as CIK and LAK.
他の実施形態では、ACT細胞組成物であって、5×106~10×109個の生存可能なインビトロ増殖PBMCを含み、その70~85%がNKG2D、DNAM-1+及びグランザイムBを発現するCD3+CD8+細胞であり、少なくとも65%がCD56-細胞であり、最大8%がCD3-細胞であって、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。他の実施形態では、生細胞の5~15%がCD3+CD4+細胞であり、15~30%がCD3+CD56+細胞であり、7~17%がCD3+CD8+CD56+細胞である。他の実施形態では、細胞組成物は、CD3+CD4+細胞に対するCD3+CD8+細胞の比率が7~8であることを特徴とし、及び/又は5%未満のCD3-CD56+細胞を含む。 In another embodiment, an ACT cell composition is provided that comprises 5x106-10x109 viable in vitro expanded PBMCs, of which 70-85% are CD3 + CD8 + cells expressing NKG2D, DNAM-1 + and granzyme B, at least 65% are CD56- cells, and up to 8% are CD3- cells, and that exhibit significant cytotoxic activity against tumor cells in vitro and in vivo. In another embodiment, 5-15% of the viable cells are CD3 + CD4 + cells, 15-30% are CD3 + CD56 + cells, and 7-17% are CD3 + CD8 + CD56 + cells. In another embodiment, the cell composition is characterized by a ratio of CD3 + CD8+ cells to CD3 + CD4 + cells of 7-8, and/or comprises less than 5 % CD3 - CD56 + cells.
遺伝子改変細胞
他の実施形態では、本発明の細胞組成物は遺伝子改変されている。一実施形態では、腫瘍特異性をもたらす少なくとも1種の受容体、例えば、組換えT細胞受容体(TCR)又はキメラ抗原受容体(CAR)を発現するように細胞が改変されている。他の実施形態では、本発明の組換え構築物、又は以下に開示の組換え構築物の独特な組み合わせを含む細胞組成物が提供される。
Genetically Modified Cells In other embodiments, the cell compositions of the invention are genetically modified. In one embodiment, the cells are modified to express at least one receptor that confers tumor specificity, such as a recombinant T cell receptor (TCR) or a chimeric antigen receptor (CAR). In other embodiments, cell compositions are provided that include a recombinant construct of the invention, or a unique combination of recombinant constructs disclosed below.
CARは、標的となる抗原に強く結合する分子の結合部位(即ち「結合部分」)と、リンパ球の活性化につながるシグナル伝達の開始を担う従来の免疫受容体の細胞質ドメイン(「シグナル伝達部分」)を結合する。最も一般的には、使用される結合部分は、標的となる抗原に対して高い親和性を有するモノクローナル抗体(mAb)のFab(抗原結合)断片の構造に由来する。Fabは2種の遺伝子の産物であるため、対応する配列は通常、短いリンカー断片を介して結合され、これにより重鎖が軽鎖由来のペプチドの上に折り畳まれて天然構造になり、単鎖断片可変(scFv)領域が形成される。本来のCAR系として知られている多くの系は抗体断片をT細胞に結合するため、「Tボディ」とも称されていた。他の考えられる抗原結合部分としては、ホルモン又はサイトカイン分子のシグナル伝達部分、膜受容体の細胞外ドメイン、及びライブラリーのスクリーニング(例えば、ファージディスプレイ)に由来するペプチドが挙げられる。本発明の組成物に使用されるCARに適した抗原標的は、本明細書に開示の疾患特異的抗原である。 CARs combine a binding site (i.e., "binding moiety") of a molecule that binds strongly to the target antigen with the cytoplasmic domain ("signaling moiety") of a conventional immune receptor that is responsible for initiating signal transduction leading to lymphocyte activation. Most commonly, the binding moiety used is derived from the structure of a Fab (antigen-binding) fragment of a monoclonal antibody (mAb) that has high affinity for the target antigen. Since Fab is the product of two genes, the corresponding sequences are usually linked via a short linker fragment, which allows the heavy chain to fold onto a peptide derived from the light chain in its native conformation, forming a single-chain fragment variable (scFv) region. Many of the original known CAR systems were also called "T-bodies" because they bind antibody fragments to T cells. Other possible antigen-binding moieties include signaling moieties of hormone or cytokine molecules, extracellular domains of membrane receptors, and peptides derived from library screening (e.g., phage display). Antigen targets suitable for the CARs used in the compositions of the invention are the disease-specific antigens disclosed herein.
「抗体」という用語は、インタクトな分子と抗原結合部位を含むその断片の両方を包含することを意味する。本発明によって開示される抗体は完全に合成であってもよく、その場合、抗体のポリペプチド鎖は合成され、場合によっては受容体として本明細書に開示のポリペプチドに結合するように最適化される。このような抗体はキメラ抗体又はヒト化抗体であってもよく、構造が完全に四量体であってもよく、又は二量体であって単一の重鎖と単一の軽鎖のみを含むものであってもよい。 The term "antibody" is meant to encompass both intact molecules and fragments thereof that contain an antigen-binding site. The antibodies disclosed by the present invention may be fully synthetic, in which case the polypeptide chains of the antibody are synthesized and, optionally, optimized to bind to the polypeptides disclosed herein as receptors. Such antibodies may be chimeric or humanized, and may be fully tetrameric in structure, or dimeric, containing only a single heavy chain and a single light chain.
モノクローナル抗体とポリクローナル抗体を産生する方法は、当技術分野では周知である。幾つかの既知の方法のいずれか1種によって抗体を産生することができ、抗体分子のインビボ産生の誘導、免疫グロブリンライブラリーのスクリーニング、又は培養中の連続細胞株によるモノクローナル抗体分子の産生を利用することができる。このような方法としては、ハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術、及びエプスタイン・バーウイルス(EBV)ハイブリドーマ技術が挙げられるが、これらに限定されない。インビボで抗体を産生する従来の方法に加えて、当技術分野で周知の方法により、ファージディスプレイ技術を用いてインビトロで抗体を産生することができる(例えば、Current Protocols in Immunology, Colligan et al (Eds.), John Wiley & Sons, Inc. (1992-2000), Chapter 17, Section 17.1)。ペプチドリンカーをコードするオリゴヌクレオチドによって連結された重鎖可変ドメインと軽鎖可変ドメインをコードするDNA配列を含む構造遺伝子を構築することによって単鎖Fvを調製する。構造遺伝子を発現ベクターに挿入した後、大腸菌等の宿主細胞に導入する。組換え宿主細胞は、2種の可変ドメインを架橋するリンカーペプチドを有する単一ポリペプチド鎖を合成する。単鎖Fvを産生するための十分なガイダンスが当技術分野の文献に提供されている。 Methods for producing monoclonal and polyclonal antibodies are well known in the art. Antibodies can be produced by any one of several known methods, including inducing in vivo production of antibody molecules, screening immunoglobulin libraries, or producing monoclonal antibody molecules by continuous cell lines in culture. Such methods include, but are not limited to, hybridoma technology, human B-cell hybridoma technology, and Epstein-Barr Virus (EBV) hybridoma technology. In addition to traditional methods for producing antibodies in vivo, antibodies can be produced in vitro using phage display technology by methods well known in the art (e.g., Current Protocols in Immunology, Colligan et al (Eds.), John Wiley & Sons, Inc. (1992-2000), Chapter 17, Section 17.1). Single-chain Fvs are prepared by constructing a structural gene that includes DNA sequences encoding the heavy and light chain variable domains linked by an oligonucleotide encoding a peptide linker. The structural gene is inserted into an expression vector, which is then introduced into a host cell such as E. coli. The recombinant host cell synthesizes a single polypeptide chain with a linker peptide bridging the two variable domains. Ample guidance for producing single-chain Fvs is provided in the literature in the art.
細胞質ドメインに関して、CARの設計は、共刺激分子のシグナル伝達ドメイン、例えば、CD80、CD86、CD40、CD83、4-1BB(CD137)、CD28及び/又はCD3ζシグナル伝達ドメインを単独で、又はCARの状況において有用な他の所望の細胞質ドメインと組み合わせて含むように行うことができる。他の実施形態では、CARは、4-1BB、CD28、CD3ζ、又はそれらの組み合わせから成る群から選択される共刺激分子のシグナル伝達ドメインを含む。例示的な一実施形態では、本発明のCAR改変細胞は、所望のCAR、例えば、抗CD19結合ドメイン、膜貫通ドメイン、及び細胞質シグナル伝達ドメインを含むCARを含むレンチウイルスベクター等のウイルスベクターを細胞に導入することによって産生することができる。或いは、ゲノムに組み込まれずに細胞内で安定に維持されるベクターを使用する。他の実施形態では、本発明のCAR改変細胞は、そのようなCAR分子をコードする遺伝子を細胞内に形質導入又はトランスフェクションすることによって産生することができる。 With respect to the cytoplasmic domain, the CAR can be designed to include a signaling domain of a costimulatory molecule, e.g., CD80, CD86, CD40, CD83, 4-1BB (CD137), CD28, and/or CD3ζ signaling domain, alone or in combination with other desired cytoplasmic domains useful in the context of the CAR. In other embodiments, the CAR includes a signaling domain of a costimulatory molecule selected from the group consisting of 4-1BB, CD28, CD3ζ, or a combination thereof. In an exemplary embodiment, the CAR-modified cells of the invention can be produced by introducing a viral vector, such as a lentiviral vector, into a cell that includes a desired CAR, e.g., a CAR that includes an anti-CD19 binding domain, a transmembrane domain, and a cytoplasmic signaling domain. Alternatively, a vector that is stably maintained in the cell without being integrated into the genome is used. In other embodiments, the CAR-modified cells of the invention can be produced by transducing or transfecting a gene encoding such a CAR molecule into the cell.
特定の実施形態では、CAR、例えば、CD19特異的CAR又はCD123特異的CARを発現するように細胞を遺伝子改変する。CD19分子(分化クラスター19)としても知られるBリンパ球抗原CD19、Bリンパ球表面抗原B4、T細胞表面抗原Leu-12及びCVID3は、ヒトにおいては遺伝子CD19によってコードされる膜貫通タンパク質である。ヒトにおいては、CD19は全てのB系列細胞で発現する。CD19はヒトB細胞において2種の主要な役割を果たす。一方では、細胞質シグナル伝達タンパク質を膜に動員するアダプタータンパク質として作用し、他方では、CD19/CD21複合体内で作用して、B細胞受容体シグナル伝達経路の閾値を低下させる。全てのB細胞に存在するため、Bリンパ球の発生、リンパ腫診断のバイオマーカーであり、白血病免疫療法の標的として利用することができる。インターロイキン-3受容体(CD123)は、免疫系で重要な可溶性サイトカインであるインターロイキン-3のシグナル伝達を助ける細胞上に存在する分子である。受容体をコードする遺伝子は、X染色体とY染色体の偽常染色体領域に位置する。この受容体はI型サイトカイン受容体ファミリーに属し、特有なα鎖と一般のβ(βc又はCD131)サブユニットが対になったヘテロ二量体である。αサブユニットの遺伝子は40キロベースの長さで、12個のエクソンを有する。 In certain embodiments, the cells are genetically modified to express a CAR, e.g., a CD19-specific CAR or a CD123-specific CAR. The B-lymphocyte antigen CD19, also known as the CD19 molecule (cluster of differentiation 19), B-lymphocyte surface antigen B4, T-cell surface antigen Leu-12 and CVID3, is a transmembrane protein that in humans is encoded by the gene CD19. In humans, CD19 is expressed on all B-lineage cells. CD19 plays two major roles in human B cells. On the one hand, it acts as an adaptor protein that recruits cytoplasmic signaling proteins to the membrane, and on the other hand, it acts within the CD19/CD21 complex to lower the threshold of the B-cell receptor signaling pathway. As it is present on all B cells, it is a biomarker for B-lymphocyte development, lymphoma diagnosis, and can be used as a target for leukemia immunotherapy. The interleukin-3 receptor (CD123) is a molecule present on cells that helps signal interleukin-3, a soluble cytokine important in the immune system. The gene encoding the receptor is located in the pseudoautosomal regions of the X and Y chromosomes. This receptor belongs to the type I cytokine receptor family and is a heterodimer consisting of a unique α chain paired with a common β (βc or CD131) subunit. The gene for the α subunit is 40 kilobases long and has 12 exons.
ある例示的な実施形態では、CARは、4-1BBとCD3ζシグナル伝達ドメインに連結された抗CD19抗体のscFvを含む。他の例示的な実施形態では、CARは更なるドメイン(例えば、CD8由来のヒンジ領域及び/又は膜貫通領域が挙げられるが、これに限定されない)を更に含むことができる。他の例示的な実施形態では、CARは、CD28とCD3ζシグナル伝達ドメインに連結された抗CD19抗体のscFvを含むことができる。本発明の実施形態によって使用することができる特定のCAR分子とそれをコードする構築物の非限定的な例を、以下の実施例8及び9に記載する。 In one exemplary embodiment, the CAR comprises an scFv of an anti-CD19 antibody linked to 4-1BB and CD3ζ signaling domains. In other exemplary embodiments, the CAR can further comprise additional domains, such as, but not limited to, a hinge region and/or a transmembrane region from CD8. In other exemplary embodiments, the CAR can comprise an scFv of an anti-CD19 antibody linked to CD28 and CD3ζ signaling domains. Non-limiting examples of specific CAR molecules and constructs encoding same that can be used in accordance with embodiments of the present invention are described in Examples 8 and 9 below.
他の実施形態では、他の標的、例えば、BCMA、CD47、PDL-1、メソテリン、EpCAM、CD34、CD44、PSCA、MUC16、CD276、CD123、CD19、CD20及びEFFRvIIIに対するCARを使用することができる。更に他の実施形態では、他の様々なマーカーをCAR標的として使用することができ、例えば、AFP、Axl、B7-H6、BCMA、ビオチン、CD10、CD117、CD123、CD133、CD138、CD19、CD20、CD200、CD22、CD276、CD30、CD33、CD34、CD38、CD44、CD5、CD70、CD79A、CD80、CD86、CD99、CEA、CLDN18.2、c-Met、CS1、DLL3、DR5、EGFR、EGFRvIII、EpCAM、EphA2、ErbB2、ERBB3、FAP、線維芽細胞活性化タンパク質(FAP)、FRα、GD2、GPC3、IL1RAP、ITGB7、LeY、メソテリン、MUC1、MUC16、PD-1、PD-L1、PMEL、PSCA、PSMA、及びVEGFR2が挙げられるが、これらに限定されない。様々な受容体の配列が入手可能であり、遺伝子改変用の核酸構築物の産生に使用することができる。例えば、T細胞組成物を改変するのに使用される、CD19又はCD123に対するCAR構築物は、それぞれUS20140271635号及びUS20140322212号に開示されているが、これに限定されない。 In other embodiments, CARs can be used against other targets, such as BCMA, CD47, PDL-1, mesothelin, EpCAM, CD34, CD44, PSCA, MUC16, CD276, CD123, CD19, CD20, and EFFRvIII. In still other embodiments, a variety of other markers can be used as CAR targets, such as AFP, Axl, B7-H6, BCMA, biotin, CD10, CD117, CD123, CD133, CD138, CD19, CD20, CD200, CD22, CD276, CD30, CD33, CD34, CD38, CD44, CD5, CD70, CD79A, CD80, CD86, CD99, CEA, CLD Examples of receptors include, but are not limited to, N18.2, c-Met, CS1, DLL3, DR5, EGFR, EGFRvIII, EpCAM, EphA2, ErbB2, ERBB3, FAP, fibroblast activation protein (FAP), FRα, GD2, GPC3, IL1RAP, ITGB7, LeY, mesothelin, MUC1, MUC16, PD-1, PD-L1, PMEL, PSCA, PSMA, and VEGFR2. Sequences for a variety of receptors are available and can be used to generate nucleic acid constructs for genetic modification. For example, CAR constructs against CD19 or CD123 for use in modifying T cell compositions are disclosed, but are not limited to, in US20140271635 and US20140322212, respectively.
他の実施形態では、例えば、癌の治療に有用な腫瘍関連抗原(TAA)又は他の標的に対する、組換えTCRをコードする構築物を使用することができる。よく知られたTAAの非限定的な例は、MART-1、gp100209-217、gp100154-163、CSPG4、NY-ESO、MAGE-A1、及びチロシナーゼである。このようなTCR構築物は当技術分野で知られており、組換え法によって取得又は産生することができる。 In other embodiments, constructs encoding recombinant TCRs against tumor associated antigens (TAA) or other targets useful, for example, in the treatment of cancer, can be used. Non-limiting examples of well-known TAAs are MART-1, gp100 209-217 , gp100 154-163 , CSPG4, NY-ESO, MAGE-A1, and tyrosinase. Such TCR constructs are known in the art and can be obtained or produced by recombinant methods.
他の実施形態では、少なくとも1種のサイトカイン、ケモカイン、又はその受容体を発現するように細胞が改変されている。一実施形態では、改変ケモカイン受容体を発現するように細胞を遺伝子改変する。一実施形態では、腫瘍の局在化又は保持を増強するようにケモカイン受容体は改変されている。本明細書において、腫瘍局在化又は保持の増強は、養子移植されたエフェクター細胞が腫瘍微小環境で遊走、浸透及び/又は増殖する能力に関連し、そのようなトランスジェニック受容体がない場合のレベルと比較して、養子移植後の腫瘍において特定可能なエフェクター細胞の相対数の増大として現れる。例えば(例えば、治療対象の腫瘍が骨髄に由来するか、又は骨髄内に存在する場合)、改変ケモカイン受容体は、トランスジェニック細胞の骨髄保持を有利に増強する。特定の実施形態では、改変ケモカイン受容体は、C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体である(例えば、本明細書に記載の腫瘍局在化及び/又は保持の増強をもたらす)。例えば、CXCR4遺伝子でのWHIM変異(WHIM症候群の患者で特定)は、コードされた受容体のC末端トランケーション(例えば、15~20個のC末端アミノ酸(aa)の除去)と関連している。特定の実施形態では、改変CXCR4は、例えば、R334X変異(本明細書ではWHIM R334x変異とも称され、残基333の後にトランケーション変異を含むCXCR4であり、後述の配列番号8によって例示)に起因する19個のC末端aaトランケーションを特徴とする。非限定的な例によると、このような改変細胞は、骨髄に局在する造血器腫瘍や転移の治療に特に有用である。 In other embodiments, the cells are modified to express at least one cytokine, chemokine, or receptor thereof. In one embodiment, the cells are genetically modified to express a modified chemokine receptor. In one embodiment, the chemokine receptor is modified to enhance tumor localization or retention. As used herein, enhanced tumor localization or retention refers to the ability of adoptively transferred effector cells to migrate, penetrate, and/or proliferate in the tumor microenvironment, and is manifested as an increase in the relative number of identifiable effector cells in the tumor after adoptive transfer, compared to levels in the absence of such transgenic receptor. For example, (e.g., when the tumor to be treated originates from or resides in bone marrow), the modified chemokine receptor advantageously enhances bone marrow retention of the transgenic cells. In certain embodiments, the modified chemokine receptor is a modified CXCR4 receptor that includes a mutation or truncation of the C-terminal tail domain (e.g., resulting in enhanced tumor localization and/or retention as described herein). For example, WHIM mutations in the CXCR4 gene (identified in patients with WHIM syndrome) are associated with C-terminal truncations of the encoded receptor (e.g., removal of 15-20 C-terminal amino acids (aa)). In certain embodiments, modified CXCR4 is characterized by a 19 C-terminal aa truncation, for example, due to an R334X mutation (also referred to herein as a WHIM R334x mutation, which is a CXCR4 containing a truncation mutation after residue 333, exemplified by SEQ ID NO: 8 below). By way of non-limiting example, such modified cells are particularly useful for the treatment of hematopoietic tumors and metastases localized to the bone marrow.
他の実施形態では、IL-2ファミリーの少なくとも1種のサイトカインを発現するように細胞が遺伝子改変されている。例えば、IL-2、IL-15及び/又はIL-21を外因的に(例えば、構成的又はアップレギュレートするように)発現するように細胞を改変することができる。各々の可能性は本発明の別個の実施形態を表す。他の実施形態では、細胞組成物は、外因性IL-2、外因性IL-15、外因性IL-21のいずれか1種、又はそれらの任意の組み合わせを発現する。「外因性」という用語は、細胞又は生物体内のタンパク質、遺伝子、核酸、又はポリヌクレオチドに関して使用される場合、人為的又は天然手段によって細胞又は生物に導入されたタンパク質、遺伝子、核酸、又はポリヌクレオチドを意味する。外因性核酸は、異なる生物体又は細胞に由来するものであってもよく、生物体又は細胞内に天然に存在する核酸の1個以上の追加のコピーであってもよい。非限定的な例として、外因性核酸は、天然細胞とは異なる染色体位置にある核酸、又は天然に見出されるものとは異なる核酸配列に隣接する核酸である。外因性核酸はエピソームベクター等の染色体外核酸とすることもできる。 In other embodiments, the cells are genetically modified to express at least one cytokine of the IL-2 family. For example, the cells can be modified to exogenously express (e.g., constitutively or upregulated) IL-2, IL-15, and/or IL-21. Each possibility represents a separate embodiment of the invention. In other embodiments, the cell composition expresses any one of exogenous IL-2, exogenous IL-15, exogenous IL-21, or any combination thereof. The term "exogenous" when used with reference to a protein, gene, nucleic acid, or polynucleotide in a cell or organism means a protein, gene, nucleic acid, or polynucleotide that has been introduced into the cell or organism by artificial or natural means. An exogenous nucleic acid may be from a different organism or cell, or may be one or more additional copies of a nucleic acid that is naturally present in the organism or cell. As a non-limiting example, an exogenous nucleic acid is a nucleic acid that is at a different chromosomal location than in a native cell, or that is adjacent to a different nucleic acid sequence than that found in nature. The exogenous nucleic acid can also be an extrachromosomal nucleic acid, such as an episomal vector.
IL-15は、上述のIL-2と構造的及び機能的に類似したサイトカインである。IL-2と同様に、IL-15は、IL-2/IL-15受容体ベータ鎖(CD122)と共通ガンマ鎖(ガンマ-C、CD132)で構成される複合体に結合し、それを通じてシグナル伝達する。IL-15はIL15遺伝子によってコードされており、NK細胞の増殖を誘導する。IL-21はヒトのIL21遺伝子によってコードされており、NK細胞や細胞傷害性T細胞等の免疫系の増殖を誘導する。 IL-15 is a cytokine structurally and functionally similar to IL-2 mentioned above. Like IL-2, IL-15 binds to and signals through a complex composed of the IL-2/IL-15 receptor beta chain (CD122) and the common gamma chain (gamma-C, CD132). IL-15 is encoded by the IL15 gene and induces the proliferation of NK cells. IL-21 is encoded by the human IL21 gene and induces the proliferation of immune system members such as NK cells and cytotoxic T cells.
幾つかの実施形態では、細胞は複数の改変を特徴とする。非限定的で有利な実施形態によれば、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように改変された細胞組成物を本明細書で開示する。本明細書に開示のように、幾つかの実施形態では、そのような改変SAK細胞組成物は、造血器腫瘍及び白血病等の骨髄に局在する腫瘍の治療に特に有用である。幾つかの実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインをコードする核酸構築物によるトランスフェクション、形質導入又は感染によって細胞が修飾されている。他の実施形態では、構築物はウイルスベクターである。他の実施形態では、構築物は、少なくとも1種の翻訳調節配列、例えば、配列内リボソーム進入部位(IRES)配列、又は2Aペプチド配列等の自己切断配列を更に含む。例示的な実施形態では、翻訳調節配列は、要素(i)と(ii)をコードする核酸配列の間、及び/又は要素(ii)と(iii)の間に位置する。他の特定の実施形態では、要素は構成的に発現される。 In some embodiments, the cells are characterized by multiple modifications. According to non-limiting, advantageous embodiments, disclosed herein are cell compositions modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation in the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. As disclosed herein, in some embodiments, such modified SAK cell compositions are particularly useful for treating tumors localized in the bone marrow, such as hematopoietic tumors and leukemias. In some embodiments, the cells are modified by transfection, transduction, or infection with a nucleic acid construct encoding (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation in the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In other embodiments, the construct is a viral vector. In other embodiments, the construct further comprises at least one translational regulatory sequence, e.g., an internal ribosome entry site (IRES) sequence, or a self-cleaving sequence, such as a 2A peptide sequence. In exemplary embodiments, the translational regulatory sequence is located between the nucleic acid sequences encoding elements (i) and (ii) and/or between elements (ii) and (iii). In other specific embodiments, the elements are constitutively expressed.
他の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む細胞組成物が提供される。 In another embodiment, a cell composition is provided that includes leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21.
他の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインをコードする核酸構築物が提供される。更に他の実施形態では、本明細書に開示の本発明の核酸構築物が提供される。 In another embodiment, a nucleic acid construct is provided that encodes (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In yet another embodiment, a nucleic acid construct of the invention disclosed herein is provided.
特に、所望の遺伝子産物を送達及び発現するために使用される発現構築物又はベクターを含む核酸構築物の調製は、当技術分野で知られている。単離された核酸配列は、その天然源から遺伝子全体(即ち、完全遺伝子)又はその一部として得ることができる。核酸分子は、組換えDNA技術(例えば、ポリメラーゼ連鎖反応(PCR)増幅、クローニング)又は化学合成によって産生することもできる(例えば、Sambrook et al., 2001, Molecular Cloning: A Laboratory Manual, Cold Springs Harbor Laboratory, New York; Ausubel, et al., 1989, Chapters 2 and 4を参照)。核酸配列としては、天然の核酸配列及びその相同体が挙げられ、例えば、天然の対立遺伝子変異体や、機能的ペプチドをコードする核酸分子の能力を実質的に妨げないような方法で、ヌクレオチドが挿入、欠失、置換、及び/又は反転された改変核酸配列が挙げられるが、これらに限定されない。ポリヌクレオチド又はオリゴヌクレオチド配列はタンパク質の遺伝コードから推定可能であるが、コードの縮重を考慮する必要があると共に、所謂「Wobble rule」による、コドンの3番目の位置における古典的な塩基対に対する例外を考慮する必要がある。おおよそヌクレオチドを含むポリヌクレオチドは、同一又は同等のタンパク質をもたらすことができる。組換え産生法を用いて、例えば、大腸菌又は酵母等の微生物の選択宿主細胞は、ポリペプチド又はポリペプチド類似体をコードする特定のDNA配列を含むハイブリッドウイルス又はプラスミドDNAベクターで形質転換し、DNA配列の転写及び翻訳の際に宿主内でポリペプチドを合成する。 In particular, the preparation of nucleic acid constructs, including expression constructs or vectors used to deliver and express a desired gene product, is known in the art. An isolated nucleic acid sequence can be obtained from its natural source as an entire gene (i.e., a complete gene) or a portion thereof. Nucleic acid molecules can also be produced by recombinant DNA techniques (e.g., polymerase chain reaction (PCR) amplification, cloning) or chemical synthesis (see, e.g., Sambrook et al., 2001, Molecular Cloning: A Laboratory Manual, Cold Springs Harbor Laboratory, New York; Ausubel, et al., 1989, Chapters 2 and 4). Nucleic acid sequences include naturally occurring nucleic acid sequences and their homologs, including, but not limited to, naturally occurring allelic variants and modified nucleic acid sequences in which nucleotides have been inserted, deleted, substituted, and/or inverted in a manner that does not substantially interfere with the ability of the nucleic acid molecule to encode a functional peptide. A polynucleotide or oligonucleotide sequence can be deduced from the genetic code of a protein, taking into account the degeneracy of the code and the exception to the classical base pairing at the third position of a codon due to the so-called "Wobble rule". Polynucleotides containing approximately nucleotides can result in the same or equivalent protein. Using recombinant production methods, a selected host cell, for example a microorganism such as E. coli or yeast, is transformed with a hybrid virus or plasmid DNA vector containing a specific DNA sequence encoding a polypeptide or polypeptide analog, and the polypeptide is synthesized within the host upon transcription and translation of the DNA sequence.
本発明の実施形態に従って構築物にクローン化可能なサイトカイン、ケモカイン受容体、CAR及び他の遺伝要素の例示的な配列は、以下の実施例8及び9に記載する。例えば、ヒトIL-2の例示的な核酸配列は配列番号3で示され、ヒトIL-15の例示的な核酸配列は配列番号4で示され、野生型CXCR4の例示的な核酸配列は配列番号7で示され、改変C’切断型CXCR4の例示的な核酸配列は配列番号8で示され、CD19特異的CARの例示的な核酸配列は配列番号9で示され、CD123特異的CARの例示的な核酸配列は配列番号1で示される。他の実施形態では、少なくとも90%の配列同一性、例えば、93%、95%、97%、又は99%の相同性を保持する、これらの配列の機能的に同等な変異体及び相同体を使用することができる。 Exemplary sequences of cytokines, chemokine receptors, CARs and other genetic elements that can be cloned into constructs according to embodiments of the invention are described in Examples 8 and 9 below. For example, an exemplary nucleic acid sequence for human IL-2 is set forth in SEQ ID NO:3, an exemplary nucleic acid sequence for human IL-15 is set forth in SEQ ID NO:4, an exemplary nucleic acid sequence for wild-type CXCR4 is set forth in SEQ ID NO:7, an exemplary nucleic acid sequence for modified C'-truncated CXCR4 is set forth in SEQ ID NO:8, an exemplary nucleic acid sequence for a CD19-specific CAR is set forth in SEQ ID NO:9, and an exemplary nucleic acid sequence for a CD123-specific CAR is set forth in SEQ ID NO:1. In other embodiments, functionally equivalent variants and homologues of these sequences that retain at least 90% sequence identity, e.g., 93%, 95%, 97%, or 99% homology, can be used.
当技術分野で知られているように、構築物は他の制御配列又は選択可能マーカーを含むこともできる。核酸構築物(本明細書では「発現ベクター」とも称される)は、このベクターを原核生物、真核生物又は場合によっては両方(例えば、シャトルベクター)における複製及び統合に適したものにする追加の配列を含むことができる。更に、典型的なクローニングベクターには、転写及び翻訳開始配列、転写及び翻訳ターミネーター、及びポリアデニル化シグナルが含まれる場合もある。 As known in the art, the construct may also include other control sequences or selectable markers. The nucleic acid construct (also referred to herein as an "expression vector") may include additional sequences that render the vector suitable for replication and integration in prokaryotes, eukaryotes, or possibly both (e.g., shuttle vectors). Additionally, a typical cloning vector may also include transcription and translation initiation sequences, transcription and translation terminators, and a polyadenylation signal.
既に記載した要素に加え、本発明の発現ベクターは通常、クローン化核酸の発現レベルを高めること、又は組換えDNAを保有する細胞の特定を容易にすることを目的とした他の特殊な要素を含むことができる。例えば、多くの動物ウイルスには、許容細胞型におけるウイルスゲノムの染色体外複製を促進するDNA配列が含まれている。このようなウイルスレプリコンを保持するプラスミドは、プラスミド上又は宿主細胞のゲノムに保持される遺伝子によって適切な因子が提供される限り、エピソーム的に複製される。 In addition to the elements already described, the expression vectors of the invention can typically contain other specialized elements intended to enhance the level of expression of the cloned nucleic acid or to facilitate identification of cells harboring the recombinant DNA. For example, many animal viruses contain DNA sequences that promote extrachromosomal replication of the viral genome in permissive cell types. Plasmids carrying such viral replicons will replicate episomally as long as the appropriate factors are provided by genes carried on the plasmid or in the genome of the host cell.
ベクターは真核生物レプリコンを含む場合も含まない場合もある。真核生物レプリコンが存在する場合、ベクターは適切な選択可能マーカーを用いて真核細胞内で増幅可能である。ベクターが真核生物レプリコンを含まない場合、エピソーム増幅は不可能である。その代わりに、組換えDNAは操作された細胞のゲノムに組み込まれ、プロモーターが所望の核酸の発現を指示する。 A vector may or may not contain a eukaryotic replicon. If a eukaryotic replicon is present, the vector is amplifiable in a eukaryotic cell using an appropriate selectable marker. If the vector does not contain a eukaryotic replicon, episomal amplification is not possible. Instead, the recombinant DNA is integrated into the genome of the engineered cell, and the promoter directs expression of the desired nucleic acid.
哺乳類発現ベクターの例としては、pcDNA3、pcDNA3.1(+/-)、pGL3、pZeoSV2(+/-)、pSecTag2、pDisplay、pEF/myc/cyto、pCMV/myc/cyto、pCR3.1、pSinRep5、DH26S、DHBB、pNMT1、pNMT41、及びpNMT81(これらはInvitrogen社から入手可能)、pCI(Promega社から入手可能)、pMbac、pPbac、pBK-RSV及びpBK-CMV(これらはStrategene社から入手可能)、pTRES(Clontech社から入手可能)及びその誘導体が挙げられるが、これらに限定されない。これらは、本明細書に記載の実施形態において有用な構築物のベクター骨格として機能することができる。 Examples of mammalian expression vectors include, but are not limited to, pcDNA3, pcDNA3.1(+/-), pGL3, pZeoSV2(+/-), pSecTag2, pDisplay, pEF/myc/cyto, pCMV/myc/cyto, pCR3.1, pSinRep5, DH26S, DHBB, pNMT1, pNMT41, and pNMT81 (available from Invitrogen), pCI (available from Promega), pMbac, pPbac, pBK-RSV and pBK-CMV (available from Strategene), pTRES (available from Clontech) and derivatives thereof, which can serve as vector backbones for constructs useful in the embodiments described herein.
レトロウイルス等の真核生物ウイルスの調節エレメントを含む発現ベクターも使用することができる。SV40ベクターとしては、例えば、pSVT7やpMT2が挙げられる。ウシパピローマウイルス由来のベクターとしては、pBV-1MTHAが挙げられ、エプスタイン・バーウイルス由来のベクターとしては、pHEBO及びp2O5が挙げられる。他のベクターの例としては、pMSG、pAV009/A+、pMTO10/A+、pMAMneo-5、バキュロウイルスpDSVE、及びSV40初期プロモーター、SV40後期プロモーター、メタロチオネインプロモーター、マウス乳癌ウイルスプロモーター、ラウス肉腫ウイルスプロモーター、ポリヘドリンプロモーター、又は真核細胞での発現に有効であることが示されている他のプロモーターの指示下でタンパク質の発現を可能にする他の任意のベクターが挙げられる。これらは、本発明の構築物のベクター骨格として機能することができる。 Expression vectors containing regulatory elements of eukaryotic viruses such as retroviruses can also be used. SV40 vectors include, for example, pSVT7 and pMT2. Vectors derived from bovine papilloma virus include pBV-1MTHA, and Epstein-Barr virus include pHEBO and p2O5. Other examples of vectors include pMSG, pAV009/A + , pMTO10/A + , pMAMneo-5, baculovirus pDSVE, and any other vector that allows expression of a protein under the direction of the SV40 early promoter, SV40 late promoter, metallothionein promoter, mouse mammary tumor virus promoter, Rous sarcoma virus promoter, polyhedrin promoter, or other promoters shown to be effective for expression in eukaryotic cells. These can serve as vector backbones for the constructs of the present invention.
上述のように、ウイルスは非常に特殊な感染病原体であり、多くの場合、宿主の防御機構を回避するように進化してきた。通常、ウイルスは特定の種類の細胞に感染し、増殖する。ウイルスベクターのターゲティング特異性は、その天然の特異性を利用して、所定の細胞型を特異的に標的にし、それによって感染細胞に組換え遺伝子を導入する。従って、本発明によって使用されるベクターの種類は、形質転換される細胞の種類に依存する。形質転換される細胞型に応じて適切なベクターを選択する能力は、十分に当業者の能力の範囲内であるため、選択の考慮事項の一般的な説明は本明細書では行わない。 As mentioned above, viruses are very specialized infectious agents that have evolved to often evade the host's defense mechanisms. Typically, viruses infect and grow in specific types of cells. The targeting specificity of viral vectors exploits their natural specificity to specifically target a given cell type, thereby introducing a recombinant gene into the infected cell. Thus, the type of vector used by the present invention depends on the type of cell to be transformed. The ability to select an appropriate vector depending on the cell type to be transformed is well within the capabilities of one of skill in the art, and therefore a general discussion of selection considerations is not provided herein.
レトロウイルス由来のベクターとしては、例えば、レンチウイルスベクターが挙げられる。「レンチウイルスベクター」及び「組換えレンチウイルスベクター」は、レンチウイルスとして知られるレトロウイルスベクターのサブセットに由来する。レンチウイルスベクターとは、目的の核酸分子を担持し、特定の実施形態においては、目的の核酸分子の発現を指示することができる核酸構築物を意味する。レンチウイルスベクターには、少なくとも1つの転写プロモーター/エンハンサー又は遺伝子座定義要素、又は選択的スプライシング、核RNA輸送、メッセンジャーの翻訳後修飾、又はタンパク質の転写後修飾等の他の手段によって遺伝子発現を制御する他の要素が含まれる。このようなベクター構築物には、パッケージングシグナル、末端反復配列(LTRS)又はその一部、及び使用するレンチウイルスベクターに適切なプラス鎖及びマイナス鎖プライマー結合部位(これらがレトロウイルスベクターにまだ存在しない場合)も含む必要がある。必要に応じて、組換えレンチウイルスベクターは、ポリアデニル化を指示するシグナル、Neo、TK、ハイグロマイシン、フレオマイシン、ヒスチジノール、又はDHFR等の選択可能マーカー、及び1種以上の制限部位と翻訳終結配列を含むこともできる。例として、そのようなベクターは通常、5’LTR、tRNA結合部位、パッケージングシグナル、第2鎖DNA合成の起点、及び3’LTR又はその一部を含む。 Vectors derived from retroviruses include, for example, lentiviral vectors. "Lentiviral vector" and "recombinant lentiviral vector" are derived from a subset of retroviral vectors known as lentiviruses. By lentiviral vector is meant a nucleic acid construct that carries and, in certain embodiments, directs the expression of a nucleic acid molecule of interest. Lentiviral vectors include at least one transcriptional promoter/enhancer or locus-defining element or other elements that control gene expression by other means such as alternative splicing, nuclear RNA transport, post-translational modification of messengers, or post-transcriptional modification of proteins. Such vector constructs should also include packaging signals, long terminal repeats (LTRS) or portions thereof, and plus-strand and minus-strand primer binding sites appropriate for the lentiviral vector used (if these are not already present in the retroviral vector). Optionally, the recombinant lentiviral vector can also include a signal directing polyadenylation, a selectable marker such as Neo, TK, hygromycin, phleomycin, histidinol, or DHFR, and one or more restriction sites and a translation termination sequence. By way of example, such vectors typically include a 5'LTR, a tRNA binding site, a packaging signal, an origin of second strand DNA synthesis, and a 3'LTR or a portion thereof.
「レンチウイルスベクター粒子」は本発明内で利用することができ、少なくとも1種の目的の遺伝子を保有するレンチウイルスを意味する。レトロウイルスは選択可能マーカーを含むこともある。組換えレンチウイルスは、感染時にその遺伝物質(RNA)をDNAに逆転写し、この遺伝物質を宿主細胞のDNAに組み込むことができる。レンチウイルスベクター粒子は、レンチウイルスエンベロープ、非レンチウイルスエンベロープ(例えば、両種指向性又はVSV-Gエンベロープ)、又はキメラエンベロープを有することができる。 "Lentiviral vector particle" may be utilized within the present invention and refers to a lentivirus carrying at least one gene of interest. Retroviruses may also include a selectable marker. Recombinant lentiviruses can reverse transcribe their genetic material (RNA) into DNA upon infection and integrate this genetic material into the DNA of a host cell. Lentiviral vector particles may have a lentiviral envelope, a non-lentiviral envelope (e.g., an amphotropic or VSV-G envelope), or a chimeric envelope.
幾つかの実施形態では、改変細胞組成物は、本明細書に開示の本発明のSAK細胞組成物又はACT細胞組成物である。例えば、ACT細胞組成物は少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%はCD3-細胞であり、組成物はインビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示し、本明細書に開示の遺伝子改変に供することができる。 In some embodiments, the modified cell composition is an inventive SAK cell composition or an ACT cell composition as disclosed herein, for example, the ACT cell composition comprises at least 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, of which 70-85% are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% are CD56 − cells, and up to 5% are CD3 − cells, the composition exhibits significant cytotoxic activity against tumor cells in vitro and in vivo, and is amenable to genetic modification as disclosed herein.
他の実施形態では、他の白血球調製物又は集団(例えば、T細胞、NK細胞、NK-T細胞及びDC、及び当技術分野で既知の様々なACT組成物(例えば、TIL、CIK及びLAK)が挙げられるが、これらに限定されない)を改変に使用することができる。しかし、本明細書に開示の本発明の改良構築物によって改変されていない白血球の使用は、本発明の教示及び原理とは区別されるべきであることを理解されたい。特定の実施形態によれば、そのような代替白血球組成物の使用は明確に除外される。 In other embodiments, other leukocyte preparations or populations (e.g., but not limited to, T cells, NK cells, NK-T cells, and DCs, and various ACT compositions known in the art (e.g., TIL, CIK, and LAK)) can be used for modification. However, it should be understood that the use of leukocytes that have not been modified by the improved constructs of the present invention disclosed herein should be distinguished from the teachings and principles of the present invention. According to certain embodiments, the use of such alternative leukocyte compositions is expressly excluded.
治療用途
他の様相では、本発明の細胞組成物は癌の治療に使用することができる。他の様相では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、腫瘍細胞を治療有効量の本発明の細胞組成物と接触させ、それによって対象の腫瘍を治療することを含む方法が提供される。他の実施形態では、腫瘍の治療を必要とする対象において腫瘍の治療に使用するためのACT組成物であって、少なくとも5×106~10×108個の生存可能なインビトロ増殖末梢血単核細胞(PBMC)を含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であり、インビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示すACT組成物が提供される。他の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む、腫瘍の治療を必要とする対象において腫瘍の治療に使用するための細胞組成物が提供される。
Therapeutic Uses In another aspect, the cell compositions of the present invention can be used to treat cancer. In another aspect, a method of treating a tumor in a subject in need of such treatment is provided, comprising contacting tumor cells with a therapeutically effective amount of the cell composition of the present invention, thereby treating the tumor in the subject. In another embodiment, an ACT composition for use in treating a tumor in a subject in need of such treatment is provided, the ACT composition comprising at least 5×10 6 to 10×10 8 viable in vitro expanded peripheral blood mononuclear cells (PBMCs), 70-85% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% of which are CD56 − cells, and up to 5% of which are CD3 − cells, and which exhibit significant cytotoxic activity against tumor cells in vitro and in vivo. In another embodiment, there is provided a cell composition for use in treating a tumor in a subject in need thereof comprising: (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) a leukocyte genetically modified to express at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21.
本明細書で使用される「腫瘍」及び「癌」という用語は同義的に使用され、固形腫瘍と非固形悪性腫瘍の両方を意味する場合がある。他の実施形態では、腫瘍は造血器腫瘍である。リンパ系の造血器腫瘍の非限定的な例としては、例えば、白血病、急性リンパ性白血病、急性リンパ芽球性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、ヘアリー細胞リンパ腫及びバーキットリンパ腫が挙げられる。骨髄系の造血器腫瘍の非限定的な例としては、例えば、急性及び慢性骨髄性白血病、骨髄異形成症候群及び前骨髄球性白血病、線維肉腫や横紋筋肉腫等の間葉起源の腫瘍が挙げられる。他の実施形態では、腫瘍は固形腫瘍である。固形腫瘍の非限定的な例としては、例えば、肺腫瘍、胃腫瘍、卵巣腫瘍、乳房腫瘍、結腸直腸腫瘍、乳房腫瘍、膵臓腫瘍、及び腎腫瘍が挙げられる。様々な実施形態では、腫瘍は白血病、多発性骨髄腫、前立腺癌、中皮腫、肺癌及び膵臓癌から成る群から選択され、各々の可能性が本発明の別個の実施形態を表す。特定の実施形態では、腫瘍は白血病、多発性骨髄腫、前立腺癌及び中皮腫から成る群から選択される。特定の実施形態では、腫瘍はAMLである。 As used herein, the terms "tumor" and "cancer" are used interchangeably and may refer to both solid and non-solid malignant tumors. In another embodiment, the tumor is a hematopoietic tumor. Non-limiting examples of lymphoid hematopoietic tumors include, for example, leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T-cell lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma, and Burkitt's lymphoma. Non-limiting examples of myeloid hematopoietic tumors include, for example, acute and chronic myeloid leukemia, myelodysplastic syndromes and promyelocytic leukemia, tumors of mesenchymal origin such as fibrosarcoma and rhabdomyosarcoma. In another embodiment, the tumor is a solid tumor. Non-limiting examples of solid tumors include, for example, lung tumors, gastric tumors, ovarian tumors, breast tumors, colorectal tumors, breast tumors, pancreatic tumors, and renal tumors. In various embodiments, the tumor is selected from the group consisting of leukemia, multiple myeloma, prostate cancer, mesothelioma, lung cancer, and pancreatic cancer, with each possibility representing a separate embodiment of the present invention. In certain embodiments, the tumor is selected from the group consisting of leukemia, multiple myeloma, prostate cancer, and mesothelioma. In certain embodiments, the tumor is AML.
他の実施形態では、腫瘍は少なくとも1種のNKG2Dリガンドの発現を特徴とする。幾つかの実施形態では、NKG2Dリガンドは、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3から成る群から選択される。他の実施形態では、腫瘍は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。他の実施形態では、腫瘍は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3を発現する。各々の可能性は本発明の別個の実施形態を表す。 In other embodiments, the tumor is characterized by expression of at least one NKG2D ligand. In some embodiments, the NKG2D ligand is selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, the tumor expresses multiple NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, the tumor expresses MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. Each possibility represents a separate embodiment of the present invention.
好都合には、対象の腫瘍によるNKG2Dリガンドやチェックポイント分子リガンド等のマーカーの発現は、腫瘍細胞を(例えば、固形腫瘍生検から、又は血液や尿等の体液から)得て、腫瘍細胞の表面上のマーカーの発現を適切なアッセイ(例えば、免疫染色及びフローサイトメトリーが挙げられるが、これらに限定されない)によって検査して確認することができる。このようなアッセイの非限定的な例を本明細書に示す。 Advantageously, expression of markers such as NKG2D ligands or checkpoint molecule ligands by a tumor of interest can be confirmed by obtaining tumor cells (e.g., from a solid tumor biopsy or from a bodily fluid such as blood or urine) and examining the expression of the markers on the surface of the tumor cells by a suitable assay (e.g., including but not limited to, immunostaining and flow cytometry). Non-limiting examples of such assays are provided herein.
他の実施形態では、腫瘍は、少なくとも1種の抑制性免疫チェックポイント分子及び/又はそのリガンドの発現を特徴とする。幾つかの実施形態では、腫瘍は、CTLA-4、PD-1、TIGIT、Lag-3、Tim-3及びそれらの組み合わせの内の少なくとも1種のリガンドを発現することができ、各々の可能性は本発明の別個の実施形態を表す。他の実施形態では、腫瘍は、CTLA-4、PD-1、及びTIGITから成る群から選択される抑制性免疫チェックポイント分子の少なくとも1種のリガンドの発現を特徴とする。他の実施形態では、腫瘍は、CTLA-4、PD-1、及びTIGITから成る群から選択される複数の抑制性免疫チェックポイント分子のリガンドの発現を特徴とする。他の実施形態では、腫瘍は、少なくとも1種のCTLA-4リガンド(例えば、CD80及び/又はCD86)の発現を特徴とする。 In other embodiments, the tumor is characterized by expression of at least one inhibitory immune checkpoint molecule and/or its ligand. In some embodiments, the tumor can express at least one ligand of CTLA-4, PD-1, TIGIT, Lag-3, Tim-3, and combinations thereof, each possibility representing a separate embodiment of the invention. In other embodiments, the tumor is characterized by expression of at least one ligand of an inhibitory immune checkpoint molecule selected from the group consisting of CTLA-4, PD-1, and TIGIT. In other embodiments, the tumor is characterized by expression of ligands of multiple inhibitory immune checkpoint molecules selected from the group consisting of CTLA-4, PD-1, and TIGIT. In other embodiments, the tumor is characterized by expression of at least one CTLA-4 ligand (e.g., CD80 and/or CD86).
他の実施形態では、腫瘍は、少なくとも1種のチェックポイント分子阻害剤による治療に対して抵抗性である。他の実施形態では、腫瘍は、CTLA-4、PD-1、TIGIT、Lag-3、Tim-3及びそれらのリガンド及び組み合わせから成る群から選択される少なくとも1種のチェックポイント分子に対する少なくとも1種の阻害剤(例えば、遮断抗体)による治療に対して抵抗性であり、各々の可能性は本発明の別個の実施形態を表す。 In other embodiments, the tumor is resistant to treatment with at least one checkpoint molecule inhibitor. In other embodiments, the tumor is resistant to treatment with at least one inhibitor (e.g., a blocking antibody) of at least one checkpoint molecule selected from the group consisting of CTLA-4, PD-1, TIGIT, Lag-3, Tim-3, and ligands and combinations thereof, each possibility representing a separate embodiment of the invention.
例示的な実施形態によれば、腫瘍は、イピリムマブ(YERVOY(登録商標))及びトレメリムマブから成る群から選択される少なくとも1種のCTLA4特異的阻害剤(例えば、遮断抗体)による治療に対して抵抗性である。他の実施形態では、腫瘍は、アテゾリズマブ(TECENTRIQ(登録商標))、デュルバルマブ(IMFINZI(登録商標))、アベルマブ(BAVENCIO(登録商標))、ニボルマブ(OPDIVO(登録商標))及びペムブロリズマブ(KEYTRUDA(登録商標))から成る群から選択される少なくとも1種のPD1特異的阻害剤(例えば、PD-1又はPD-L1に対する遮断抗体)による治療に対して抵抗性である。他の実施形態では、腫瘍は、チラゴルマブ(MTIG7192A)、オシペルリマブ(BGB-A1217)、ビボストリマブ(MK-7684)、ドムバナリマブ(AB-154)、BMS-986207、EOS-448、ASP-8374、COM-902、エチギリマブ(MPH-313)、IBI-939、AGEN-1307、CASC-674、抗PVR抗体(NB-6253)、及びPH-804から成る群から選択される少なくとも1種のTIGIT特異的阻害剤による治療に対して抵抗性である。他の実施形態では、腫瘍は、PRS-332、P13B02-3、LBL-007、エフティラギモドα(IMP321)、LAG525(IMP701)、MK-4280、REGN3767、レラトリマブ(BMS-986016)、BI 754111、FS118、テボテリマブ(MGD013)、TSR-033、INCAGN2385、Sym022及びXmAb22841から成る群から選択される少なくとも1種のLAG3特異的阻害剤による治療に対して抵抗性である。他の実施形態では、腫瘍は、少なくとも1種のTIM3特異的阻害剤、例えば、ICAGN02390、Sym023、LY3321367、MGB453、TSR022、BGBA425、及びBMS986258による治療に対して抵抗性である。 According to an exemplary embodiment, the tumor is resistant to treatment with at least one CTLA4-specific inhibitor (e.g., a blocking antibody) selected from the group consisting of ipilimumab (YERVOY®) and tremelimumab. In other embodiments, the tumor is resistant to treatment with at least one PD1-specific inhibitor (e.g., a blocking antibody against PD-1 or PD-L1) selected from the group consisting of atezolizumab (TECENTRIQ®), durvalumab (IMFINZI®), avelumab (BAVENCIO®), nivolumab (OPDIVO®), and pembrolizumab (KEYTRUDA®). In other embodiments, the tumor is resistant to treatment with at least one TIGIT-specific inhibitor selected from the group consisting of tiragolumab (MTIG7192A), osipellimab (BGB-A1217), vibostolimab (MK-7684), domvanalimab (AB-154), BMS-986207, EOS-448, ASP-8374, COM-902, etigilimab (MPH-313), IBI-939, AGEN-1307, CASC-674, anti-PVR antibody (NB-6253), and PH-804. In other embodiments, the tumor is resistant to treatment with at least one LAG3-specific inhibitor selected from the group consisting of PRS-332, P13B02-3, LBL-007, eftilagimod alfa (IMP321), LAG525 (IMP701), MK-4280, REGN3767, leratolimab (BMS-986016), BI 754111, FS118, tebotelimab (MGD013), TSR-033, INCAGN2385, Sym022, and XmAb22841. In other embodiments, the tumor is resistant to treatment with at least one TIM3-specific inhibitor, e.g., ICAGN02390, Sym023, LY3321367, MGB453, TSR022, BGBA425, and BMS986258.
他の実施形態では、腫瘍は、複数のチェックポイント阻害剤による治療、又は複数のチェックポイント分子の阻害剤による治療に対して抵抗性である。例えば、腫瘍は、R07121661等のTIM3とPD1の両方を標的とする二重阻害剤による治療、又はニボルマブとイピリムマブとの併用療法に対して抵抗性となり得るが、これに限定されない。 In other embodiments, the tumor is resistant to treatment with multiple checkpoint inhibitors or treatment with inhibitors of multiple checkpoint molecules. For example, but not limited to, the tumor may be resistant to treatment with a dual inhibitor that targets both TIM3 and PD1, such as R07121661, or combination therapy with nivolumab and ipilimumab.
他の実施形態では、対象は1種以上のチェックポイント分子阻害剤による治療レジメンを受けている。他の実施形態では、対象はチェックポイント分子阻害剤による治療レジメンを受けていない。例えば、YERVOY(商標)(イピリムマブ)は静脈内注入用のヒトCTLA-4遮断抗体であり、切除不能又は転移性黒色腫の治療が対象である。OPDIVO(登録商標)(ニボルマブ)は、切除不能又は転移性黒色腫の治療に適応されるPD-1遮断抗体である。KEYTRUDA(登録商標)(ペムブロリズマブ)は、黒色腫、NSCLC、頭頸部扁平上皮癌、古典的ホジキンリンパ腫(cHL)、縦隔原発B細胞性大細胞型リンパ腫、尿路上皮癌、マイクロサテライト不安定性が高い又はミスマッチ修復欠損のある癌、マイクロサテライト不安定性が高い又はミスマッチ修復欠損のある結腸直腸癌、胃癌、食道癌、子宮頸癌、肝細胞癌(HCC)、メルケル細胞癌(MCC)、腎細胞癌(RCC)、子宮内膜癌、腫瘍変異負荷の高い癌、皮膚扁平上皮癌、及び三種陰性乳癌の治療に適応されるPD-1遮断抗体である。TECENTRIQ(登録商標)(アテゾリズマブ)は、局所進行性又は転移性尿路上皮癌、転移性NSCLC、EGFR又はALKゲノム腫瘍異常のない転移性非扁平上皮NSCLC、小細胞肺癌(SCLC)、進展期小細胞肺癌(ES-SCLC)、切除不能又は転移性HCC、及び黒色腫の治療に適応されるプログラム死リガンド1(PD-L1)遮断抗体である。IMFINZI(登録商標)(デュルバルマブ)は、プラチナベースの化学療法と放射線療法を同時に実施した後に疾患が進行しない切除不能なステージIIIのNSCLCの成人患者の治療に適応されるPD-L1遮断抗体であり、ES-SCLC成人患者の第一選択治療として、エトポシドとカルボプラチン又はシスプラチンのいずれかとの併用を行う。BAVENCIO(登録商標)(アベルマブ)は、MCC、UC、及びRCCの治療に適応されるPD-L1遮断抗体である。 In other embodiments, the subject is receiving a therapeutic regimen with one or more checkpoint molecule inhibitors. In other embodiments, the subject is not receiving a therapeutic regimen with a checkpoint molecule inhibitor. For example, YERVOY™ (ipilimumab) is a human CTLA-4 blocking antibody for intravenous infusion indicated for the treatment of unresectable or metastatic melanoma. OPDIVO® (nivolumab) is a PD-1 blocking antibody indicated for the treatment of unresectable or metastatic melanoma. KEYTRUDA® (pembrolizumab) is a PD-1 blocking antibody indicated for the treatment of melanoma, NSCLC, squamous cell carcinoma of the head and neck, classical Hodgkin lymphoma (cHL), primary mediastinal large B-cell lymphoma, urothelial carcinoma, microsatellite instability high or mismatch repair deficient cancers, microsatellite instability high or mismatch repair deficient colorectal cancer, gastric cancer, esophageal cancer, cervical cancer, hepatocellular carcinoma (HCC), Merkel cell carcinoma (MCC), renal cell carcinoma (RCC), endometrial cancer, cancers with high tumor mutation burden, cutaneous squamous cell carcinoma, and triple-negative breast cancer. TECENTRIQ® (atezolizumab) is a programmed death-ligand 1 (PD-L1) blocking antibody indicated for the treatment of locally advanced or metastatic urothelial carcinoma, metastatic NSCLC, metastatic non-squamous NSCLC without EGFR or ALK genomic tumor aberrations, small cell lung cancer (SCLC), extensive stage small cell lung cancer (ES-SCLC), unresectable or metastatic HCC, and melanoma. IMFINZI® (durvalumab) is a PD-L1 blocking antibody indicated for the treatment of adult patients with unresectable stage III NSCLC whose disease has not progressed after concurrent platinum-based chemotherapy and radiation therapy, in combination with etoposide and either carboplatin or cisplatin, as first-line treatment of adult patients with ES-SCLC. BAVENCIO® (avelumab) is a PD-L1 blocking antibody indicated for the treatment of MCC, UC, and RCC.
他の実施形態では、上述の方法又は使用は、本明細書に開示の少なくとも1種のチェックポイント分子阻害剤の投与(本発明の細胞組成物と同時又は逐次組み合わせ)を更に含む。例えば(例えば、腫瘍がLag-3、Tim-3又はTIGITの少なくとも1種のリガンドの表面発現を特徴とする場合)、上述の方法又は使用は、Lag-3、Tim-3又はTIGITそれぞれに対する少なくとも1種のチェックポイント分子阻害剤の投与を更に含む。他の実施形態では、上述の方法又は使用は、Lag-3、Tim-3、又はそれらの組み合わせに対する少なくとも1種のチェックポイント分子阻害剤の投与を更に含む。更に他の実施形態では、本発明の細胞組成物を唯一の治療剤として使用することができる。例えば、本発明の組成物は、CTLA-4又はPD-1の少なくとも1種のリガンドの表面発現を特徴とする腫瘍の治療における単独療法として使用することができるが、これに限定されない。各々の可能性は本発明の別個の実施形態を表す。 In other embodiments, the above-mentioned method or use further comprises administration (concurrently or sequentially in combination with the cell composition of the present invention) of at least one checkpoint molecule inhibitor disclosed herein. For example (e.g., where the tumor is characterized by surface expression of at least one ligand of Lag-3, Tim-3, or TIGIT), the above-mentioned method or use further comprises administration of at least one checkpoint molecule inhibitor against each of Lag-3, Tim-3, or TIGIT. In other embodiments, the above-mentioned method or use further comprises administration of at least one checkpoint molecule inhibitor against Lag-3, Tim-3, or a combination thereof. In yet other embodiments, the cell composition of the present invention can be used as the only therapeutic agent. For example, but not limited to, the composition of the present invention can be used as a monotherapy in the treatment of tumors characterized by surface expression of at least one ligand of CTLA-4 or PD-1. Each possibility represents a separate embodiment of the present invention.
他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド、CXCR2リガンド及びCXCR3リガンドから成る群から選択される少なくとも1種のケモカインの発現を特徴とする。他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド及びCXCR3リガンドから成る群から選択される少なくとも1種のケモカインの発現を特徴とする。他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド、CXCR2リガンド及びCXCR3リガンドから成る群から選択される複数のケモカインの発現を特徴とする。他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド及びCXCR3リガンドから成る群から選択される複数のケモカインの発現を特徴とする。特定の実施形態では、腫瘍は、CXCR4リガンド(例えば、CXCL12)の発現を特徴とする。他の特定の実施形態では、腫瘍は、CXCR6リガンド(例えば、CXCL16)の発現を特徴とする。例えば、CXCL16発現腫瘍としては、非小細胞肺癌(NSCLC)腫瘍、胃腫瘍、卵巣腫瘍、乳房腫瘍、結腸直腸腫瘍、乳房腫瘍、膵臓腫瘍、及び腎臓腫瘍を挙げることができるが、これらに限定されず、各々の可能性は本発明の別個の実施形態を表す。 In other embodiments, the tumor is characterized by expression of at least one chemokine selected from the group consisting of CXCR6 ligand, CXCR4 ligand, CXCR2 ligand, and CXCR3 ligand. In other embodiments, the tumor is characterized by expression of at least one chemokine selected from the group consisting of CXCR6 ligand, CXCR4 ligand, and CXCR3 ligand. In other embodiments, the tumor is characterized by expression of multiple chemokines selected from the group consisting of CXCR6 ligand, CXCR4 ligand, CXCR2 ligand, and CXCR3 ligand. In other embodiments, the tumor is characterized by expression of multiple chemokines selected from the group consisting of CXCR6 ligand, CXCR4 ligand, and CXCR3 ligand. In certain embodiments, the tumor is characterized by expression of a CXCR4 ligand (e.g., CXCL12). In other certain embodiments, the tumor is characterized by expression of a CXCR6 ligand (e.g., CXCL16). For example, CXCL16-expressing tumors can include, but are not limited to, non-small cell lung cancer (NSCLC) tumors, gastric tumors, ovarian tumors, breast tumors, colorectal tumors, breast tumors, pancreatic tumors, and renal tumors, with each possibility representing a separate embodiment of the present invention.
他の実施形態では、接触をインビボで行う。従って、本発明の方法は、対象に治療有効量の本発明の細胞組成物(例えば、ACT細胞組成物)を投与することを含むことができ、組成物は、少なくとも5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~85%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であって、組成物はインビトロ及びインビボで腫瘍細胞に対して有意な細胞傷害活性を示す。他の実施形態では、接触をエクスビボで行う。他の実施形態では、IL-2の投与と同時に細胞を対象に投与する。更に他の実施形態では(例えば、改良遺伝子改変細胞を使用する場合)、IL-2の同時投与を行わずに細胞を有利に使用することができ、それによって安全性が高まり、副作用が低減する。 In other embodiments, the contacting is performed in vivo. Thus, the methods of the invention can include administering to a subject a therapeutically effective amount of a cell composition of the invention (e.g., an ACT cell composition), the composition comprising at least 5×10 6 to 10×10 8 viable in vitro expanded PBMCs, 70-85% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% of which are CD56 − cells, and up to 5% of which are CD3 − cells, and the composition exhibits significant cytotoxic activity against tumor cells in vitro and in vivo. In other embodiments, the contacting is performed ex vivo. In other embodiments, the cells are administered to the subject simultaneously with administration of IL-2. In still other embodiments (e.g., when improved genetically modified cells are used), the cells can be advantageously used without co-administration of IL-2, thereby increasing safety and reducing side effects.
「有効量」又は「治療有効量」とは、本発明の方法において有益な結果を発揮するのに十分な量を意味する。より具体的には、治療有効量は、障害(例えば、癌)の症状を予防、軽減又は改善するのに有効な、又は治療を受ける対象の生存を延長するのに有効な活性成分(例えば、エフェクター細胞)の量を意味する。例えば、対象に投与するための本発明のACT組成物は、本明細書に開示のPBMCから増殖した生細胞を有効量である少なくとも5×106~10×108個、典型的には最大約10×109個含む。腫瘍細胞とエクスビボで接触させるための有効量の細胞組成物は、例えば、培養中の各腫瘍細胞に対して少なくとも約0.25~1個のエフェクター細胞を含むことができる。 By "effective amount" or "therapeutically effective amount" is meant an amount sufficient to exert a beneficial result in the methods of the invention. More specifically, a therapeutically effective amount means an amount of active ingredient (e.g., effector cells) effective to prevent, reduce or ameliorate symptoms of a disorder (e.g., cancer) or to prolong the survival of the subject being treated. For example, an ACT composition of the invention for administration to a subject comprises an effective amount of at least 5x106 to 10x108 , typically up to about 10x109 , viable cells expanded from PBMCs as disclosed herein. An effective amount of a cell composition for contacting tumor cells ex vivo can comprise, for example, at least about 0.25 to 1 effector cell for each tumor cell in culture.
インビトロ細胞培養方法の状況において、IL-2及びCD3アクチベーター等の細胞刺激因子の有効量は、本明細書に開示の有利な表現型調節(例えば、細胞増殖、活性化マーカー(例えば、DNAM-1、NKG2D)のアップレギュレーション、及び細胞傷害能力の増強が挙げられるが、これらに限定されない)を発揮するのに十分な量である。例えば、IL-2の有効量としては組換えヒトIL-2の場合、例えば、500~1500、600~1300、750~1500又は500~1200IU/mLとすることができ、抗CD3抗体の有効量としてはOKT3の場合、例えば、10~60、20~40、10~40又は20~60ng/mLとすることができるが、これに限定されない。 In the context of an in vitro cell culture method, an effective amount of a cell stimulator, such as IL-2 and a CD3 activator, is an amount sufficient to exert the beneficial phenotypic modulation disclosed herein, including, but not limited to, cell proliferation, upregulation of activation markers (e.g., DNAM-1, NKG2D), and enhanced cytotoxicity. For example, an effective amount of IL-2 can be, for example, 500-1500, 600-1300, 750-1500, or 500-1200 IU/mL for recombinant human IL-2, and an effective amount of an anti-CD3 antibody can be, for example, but not limited to, 10-60, 20-40, 10-40, or 20-60 ng/mL for OKT3.
治療有効量の決定は、特に本明細書に記載の詳細な開示を考慮すると、十分に当業者の能力の範囲内である。本発明の方法で使用される任意の調製物に関して、用量又は治療有効量は、最初にインビトロアッセイ及び細胞培養アッセイから推定することができる。例えば、動物モデルにおいて用量を処方して、所望の濃度又は力価を得ることができる。このような情報を用いて、ヒトにおける有用な用量をより正確に決定することができる。 Determination of a therapeutically effective amount is well within the capabilities of those skilled in the art, especially in light of the detailed disclosure provided herein. For any preparation used in the methods of the invention, the dose or therapeutically effective amount can be estimated initially from in vitro and cell culture assays. For example, a dose can be formulated in animal models to achieve a desired concentration or potency. Such information can be used to more accurately determine useful doses in humans.
本明細書に記載の活性成分の毒性及び治療有効性は、インビトロ、細胞培養物又は実験動物における標準的な薬学的手順によって確認することができる。このようなインビトロアッセイ、細胞培養アッセイ及び動物試験から得たデータは、ヒトにおいて用いる用量範囲を処方する上で使用することができる。投与量は使用する剤形や利用する投与経路に応じて変化し得る。正確な処方、投与経路及び投与量は、患者の状態を考慮して個々の医師が選択することができる(例えば、Fingl, E. et al. (1975), "The Pharmacological Basis of Therapeutics," Ch. 1, p.1.を参照)。 Toxicity and therapeutic efficacy of the active ingredients described herein can be determined by standard pharmaceutical procedures in vitro, in cell cultures, or in experimental animals. The data obtained from such in vitro assays, cell culture assays, and animal studies can be used in formulating a range of dosages for use in humans. Dosages may vary depending on the dosage form employed and the route of administration utilized. The exact formulation, route of administration, and dosage can be chosen by the individual physician in view of the patient's condition (see, e.g., Fingl, E. et al. (1975), "The Pharmacological Basis of Therapeutics," Ch. 1, p.1.).
他の実施形態では、腫瘍は、MHCI発現及び/又は活性のダウンレギュレーションを特徴とする。本明細書に示すように、本発明に係る細胞組成物は、様々な腫瘍細胞に対してMHC依存的細胞傷害活性と非MHC依存的細胞傷害活性の両方を有することが見出された。従って、このような細胞組成物は、治療抵抗性の腫瘍(例えば、免疫療法又はチェックポイント分子阻害剤に対して抵抗性)の治療、又は、例えば、MHCI分子の発現の低下、機能喪失突然変異、又は腫瘍に特徴的なMHCI経路の他の障害に起因する、MHCI抗原提示障害に関連する免疫回避表現型を示す腫瘍の治療にも有利に使用することができる。 In other embodiments, the tumor is characterized by downregulation of MHCI expression and/or activity. As shown herein, the cell compositions of the present invention have been found to have both MHC-dependent and non-MHC-dependent cytotoxic activity against a variety of tumor cells. Thus, such cell compositions can also be advantageously used to treat therapy-resistant tumors (e.g., resistant to immunotherapy or checkpoint molecule inhibitors) or tumors that exhibit an immune evasion phenotype associated with impaired MHCI antigen presentation, e.g., due to reduced expression of MHCI molecules, loss-of-function mutations, or other defects in the MHCI pathway characteristic of tumors.
他の実施形態では、細胞組成物は、腫瘍細胞と接触する前に、凍結保存とその後の解凍プロトコルを経ている。本明細書に示すように、本発明の原理に従った細胞組成物は、凍結保存及び解凍後に非常に強力な細胞障害性を示した。また、この活性は、更なる増殖又は操作工程を必要とせずに、解凍時間付近であっても現れた(解凍24時間以内に、対応する非凍結保存細胞の細胞傷害活性の70~80%を保持)。従って、本発明の方法の他の実施形態では、解凍プロトコルは細胞組成物を腫瘍細胞と接触させてから1~3日以内、例えば、36時間、24時間、12時間又は6時間以内に行う。各々の可能性は本発明の別個の実施形態を表す。他の実施形態では、細胞組成物は、対象に対して自家性、組織適合性同種異系、又は非組織適合性同種異系とすることができ、各々の可能性は本発明の別個の実施形態を表す。 In other embodiments, the cell composition has undergone a cryopreservation and subsequent thawing protocol prior to contact with the tumor cells. As shown herein, cell compositions according to the principles of the invention have demonstrated highly potent cytotoxicity after cryopreservation and thawing, and this activity was manifested even near the time of thawing (70-80% of the cytotoxic activity of the corresponding non-cryopreserved cells was retained within 24 hours of thawing) without the need for further expansion or manipulation steps. Thus, in other embodiments of the methods of the invention, the thawing protocol is performed within 1-3 days, e.g., within 36 hours, 24 hours, 12 hours, or 6 hours, of contacting the cell composition with the tumor cells. Each possibility represents a separate embodiment of the invention. In other embodiments, the cell composition can be autologous, histocompatible allogeneic, or non-histocompatible allogeneic to the subject, each possibility representing a separate embodiment of the invention.
更なる実施形態
一様相では、本発明は、ACT用細胞組成物を産生するための方法であって、
a.PBMC試料を提供することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間PBMCを増殖させ、このとき、有効量のIL-2とCD3アクチベーターを2~4日間毎に補充することとで、培養物中の生細胞の数を少なくとも20倍増加させ、それによって少なくとも5×106~10×108個の生細胞を含む細胞組成物を得ることと、
c.得られた細胞組成物を回収することと
を含む方法を提供する。
In a further aspect, the invention provides a method for producing a cell composition for ACT, comprising the steps of:
a. providing a PBMC sample;
b. expanding the PBMCs for at least 9 days in the constant presence of an effective amount of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, whereby the number of viable cells in the culture is increased by at least 20 fold by replenishing the effective amount of IL-2 and the CD3 activator every 2-4 days, thereby obtaining a cell composition comprising at least 5 x 106 to 10 x 108 viable cells;
and c. recovering the resulting cell composition.
他の実施形態では、唯一の外因的に添加された細胞刺激因子としてのIL-2とCD3アクチベーターの常時存在下で増殖を行う。特定の実施形態では、工程bは、500~1500IU/mLのIL-2と10~60ng/mLの抗CD3抗体の存在下で細胞を増殖させることを含み、これらは2~4日間毎に10~16日間再補充する。他の実施形態では、細胞は更なる富化、刺激又は増殖工程に供されていない。他の実施形態では、生細胞の数を少なくとも30倍増加させるように増殖を行う。他の実施形態では、5×106~10×108個の生存可能な単核細胞(その70~80%はNKG2DとグランザイムBを発現するCD3+CD8+細胞である)を含む細胞組成物を産生するように増殖を行う。他の実施形態では、インビトロで造血器腫瘍細胞と0.25:1の比率で24時間インキュベートした場合に、有意に造血器腫瘍細胞を特異的に根絶することができる細胞組成物を産生するように増殖を行う。他の実施形態では、本明細書に開示のように、本発明の細胞組成物を産生するように増殖を行う。 In other embodiments, the expansion is carried out in the constant presence of IL-2 and a CD3 activator as the only exogenously added cell stimulatory factors. In a particular embodiment, step b comprises expanding the cells in the presence of 500-1500 IU/mL IL-2 and 10-60 ng/mL anti-CD3 antibody, which are replenished every 2-4 days for 10-16 days. In other embodiments, the cells are not subjected to further enrichment, stimulation or expansion steps. In other embodiments, the expansion is carried out to increase the number of viable cells by at least 30-fold. In other embodiments, the expansion is carried out to produce a cell composition comprising 5×10 6 to 10×10 8 viable mononuclear cells, of which 70-80% are CD3 + CD8 + cells expressing NKG2D and granzyme B. In other embodiments, the expansion is carried out to produce a cell composition capable of specifically eradicating hematopoietic tumor cells when incubated in vitro with hematopoietic tumor cells at a ratio of 0.25:1 for 24 hours. In other embodiments, expansion is performed to produce the cell compositions of the invention as disclosed herein.
他の実施形態では、工程bは細胞を遺伝子改変することを更に含む。他の実施形態では、この方法は、CAR及び/又は少なくとも1種のサイトカイン、ケモカイン、又はその受容体を発現するように細胞を遺伝子改変することを含む。例示的実施形態では、この方法は、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように細胞を改変することを含む。他の実施形態では、この方法は、工程cで回収した細胞組成物を凍結保存する工程を更に含む。 In other embodiments, step b further comprises genetically modifying the cells. In other embodiments, the method comprises genetically modifying the cells to express a CAR and/or at least one cytokine, chemokine, or receptor thereof. In an exemplary embodiment, the method comprises modifying the cells to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In other embodiments, the method further comprises cryopreserving the cell composition collected in step c.
他の実施形態では、本明細書に開示の方法によって調製される細胞組成物が提供される。 In other embodiments, a cell composition prepared by the methods disclosed herein is provided.
他の様相では、ACT細胞組成物であって、5×106~10×108個の生存可能なインビトロ増殖PBMCを含み、その70~80%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であり、インビトロ及びインビボで非組織適合性腫瘍細胞に対して有意な細胞傷害活性を示すACT細胞組成物が提供される。 In another aspect, an ACT cell composition is provided comprising 5x106 to 10x108 viable in vitro expanded PBMCs, 70-80% of which are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% of which are CD56- cells, and up to 5% of which are CD3- cells, and which exhibit significant cytotoxic activity against non-histocompatible tumor cells in vitro and in vivo.
一実施形態では、生細胞の7~12%がCD3+CD4+細胞であり、20~28%がCD3+CD56+細胞であり、10~14%がCD3+CD8+CD56+細胞である。他の実施形態では、組成物は、CD3+CD4+細胞に対するCD3+CD8+細胞の比率が7~8であることを特徴とする。他の(追加の又は代替の)実施形態では、組成物はCD3-CD56+細胞を3%未満含む。 In one embodiment, 7-12% of the live cells are CD3 + CD4 + cells, 20-28% are CD3 + CD56 + cells, and 10-14% are CD3 + CD8 + CD56 + cells. In another embodiment, the composition is characterized by a ratio of CD3 + CD8 + cells to CD3 + CD4 + cells of 7 to 8. In another (additional or alternative) embodiment, the composition comprises less than 3% CD3 - CD56 + cells.
ある実施形態によれば、CD3+CD8+細胞の少なくとも40%は、パーフォリン、FasL、CXCR4及びCXCR2から成る群から選択される少なくとも1種のマーカーの表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の少なくとも40%は、パーフォリン、FasL、CXCR4及びCXCR2から成る群から選択される複数のマーカーの表面発現を特徴とする。幾つかかの実施形態では、CD3+CD8+細胞の約40%がFasLを発現し、CD3+CD8+細胞の約50%がパーフォリンを発現し、CD3+CD8+細胞の約90%がCXCR2を発現し、CD3+CD8+細胞の約100%がCXCR4、グランザイムB及びNKG2Dを発現する。 According to certain embodiments, at least 40% of the CD3 + CD8 + cells are characterized by surface expression of at least one marker selected from the group consisting of perforin, FasL, CXCR4 and CXCR2. In other embodiments, at least 40% of the CD3 + CD8 + cells are characterized by surface expression of multiple markers selected from the group consisting of perforin, FasL, CXCR4 and CXCR2. In some embodiments, about 40% of the CD3 + CD8 + cells express FasL, about 50% of the CD3 + CD8 + cells express perforin, about 90% of the CD3 + CD8 + cells express CXCR2, and about 100% of the CD3 + CD8 + cells express CXCR4, granzyme B and NKG2D.
他の実施形態では、CD3+CD8+細胞の少なくとも30%、典型的には少なくとも約40%が、CXCR3、CXCR6及びDNAM-1から成る群から選択される少なくとも1種のマーカーの表面発現を特徴とする。一実施形態では、CD3+CD8+細胞の少なくとも90%がCXCR3の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の少なくとも90%、典型的には実質的に全てがDNAM-1の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の少なくとも30%がCXCR6の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約90%がCXCR3の表面発現を特徴とし、CD3+CD8+細胞の実質的に全てがDNAM-1の表面発現を特徴とし、CD3+CD8+細胞の約30%がCXCR6の表面発現を特徴とする。従って、本発明の実施形態に係るACT細胞組成物は、NKG2D+、グランザイムB+、パーフォリン+、FasL+、CXCR4+、CXCR2+、CXCR3+、CXCR6+及びDNAM-1+で定義されるCD3+CD8+細胞集団を含む。 In other embodiments, at least 30%, typically at least about 40%, of the CD3 + CD8 + cells are characterized by surface expression of at least one marker selected from the group consisting of CXCR3, CXCR6, and DNAM-1. In one embodiment, at least 90% of the CD3 + CD8 + cells are characterized by surface expression of CXCR3. In another embodiment, at least 90%, typically substantially all of the CD3 + CD8 + cells are characterized by surface expression of DNAM-1. In another embodiment, at least 30% of the CD3 + CD8 + cells are characterized by surface expression of CXCR6. In another embodiment, about 90% of the CD3 + CD8 + cells are characterized by surface expression of CXCR3, substantially all of the CD3 + CD8 + cells are characterized by surface expression of DNAM-1, and about 30% of the CD3 + CD8 + cells are characterized by surface expression of CXCR6. Thus, an ACT cell composition according to an embodiment of the invention comprises a CD3 +CD8+ cell population defined as NKG2D + , Granzyme B + , Perforin + , FasL + , CXCR4 + , CXCR2 + , CXCR3 + , CXCR6 + and DNAM-1 + .
他の実施形態では、CD3+CD8+細胞の55%以下、典型的には最大約50%が、CTLA-4、PD-1、TIGIT、Lag-3及びTim-3から成る群から選択される1種以上の免疫チェックポイント分子の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約50%以下が、CTLA-4、PD-1、TIGIT、Lag-3及びTim-3から成る群から選択される複数の免疫チェックポイント分子の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞は、CTLA-4の実質的な表面発現の欠如を特徴とする。他の実施形態では、CD3+CD8+細胞の約50~51%がLag-3の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約50~54%がTim-3の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約15%がPD-1の表面発現を特徴とする。他の実施形態では、CD3+CD8+細胞の約20~22%がTIGITの表面発現を特徴とする。従って、本発明の実施形態に係るACT細胞組成物は、CTLA-4-、PD-1-、TIGIT-、Lag-3+及びTim-3+で定義されるCD3+CD8+細胞集団を含む。他の実施形態では、本発明の実施形態に係るACT細胞組成物は、CTLA-4-、PD-1-、及びTIGIT-で定義されるCD3+CD8+細胞集団を含む。各々の可能性は本発明の別個の実施形態を表す。 In other embodiments, up to 55% of the CD3 + CD8 + cells, typically up to about 50%, are characterized by surface expression of one or more immune checkpoint molecules selected from the group consisting of CTLA-4, PD-1, TIGIT, Lag-3, and Tim-3. In other embodiments, up to about 50% of the CD3 + CD8 + cells are characterized by surface expression of multiple immune checkpoint molecules selected from the group consisting of CTLA-4, PD-1, TIGIT, Lag-3, and Tim-3. In other embodiments, the CD3 + CD8 + cells are characterized by a substantial lack of surface expression of CTLA-4. In other embodiments, about 50-51% of the CD3 + CD8 + cells are characterized by surface expression of Lag-3. In other embodiments, about 50-54% of the CD3 + CD8 + cells are characterized by surface expression of Tim-3. In another embodiment, about 15% of the CD3 + CD8 + cells are characterized by surface expression of PD-1. In another embodiment, about 20-22% of the CD3 + CD8 + cells are characterized by surface expression of TIGIT. Thus, ACT cell compositions according to embodiments of the invention comprise a CD3 + CD8 + cell population defined as CTLA-4 − , PD-1 − , TIGIT − , Lag-3 + and Tim-3 + . In another embodiment, ACT cell compositions according to embodiments of the invention comprise a CD3 + CD8 + cell population defined as CTLA-4 − , PD-1 − , and TIGIT − . Each possibility represents a separate embodiment of the present invention.
幾つかの実施形態では、細胞組成物は、造血器腫瘍細胞に対してMHC依存的(又はMHC制限)細胞傷害活性を示す。他の実施形態では、細胞組成物は、造血器腫瘍細胞及び固形腫瘍細胞に対して非MHC依存的(又は非MHC制限)細胞傷害活性を示す。他の実施形態では、細胞傷害活性はグランザイムB及び/又はパーフォリンによって媒介される。他の実施形態では、細胞傷害活性はFAS-FasL相互作用によって実質的に媒介されない。他の実施形態では、細胞組成物は、MHC依存的細胞傷害活性と非MHC依存的細胞傷害活性の両方を有する。 In some embodiments, the cell composition exhibits MHC-dependent (or MHC-restricted) cytotoxic activity against hematopoietic tumor cells. In other embodiments, the cell composition exhibits non-MHC-dependent (or non-MHC-restricted) cytotoxic activity against hematopoietic tumor cells and solid tumor cells. In other embodiments, the cytotoxic activity is mediated by granzyme B and/or perforin. In other embodiments, the cytotoxic activity is not substantially mediated by FAS-FasL interactions. In other embodiments, the cell composition has both MHC-dependent and non-MHC-dependent cytotoxic activity.
他の実施形態では、組成物は、インビトロで造血器腫瘍細胞と0.25:1の比率で24時間インキュベートした場合に、有意に造血器腫瘍細胞を特異的に根絶することができる。他の実施形態では、組成物は造血器腫瘍細胞を特異的に根絶することができ、組成物細胞と腫瘍細胞の比率を1:1としてインビトロで24時間インキュベートすると腫瘍細胞の80~90%が根絶する。他の実施形態では、腫瘍細胞は少なくとも1種のNKG2Dリガンドを発現する。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。 In another embodiment, the composition is capable of specifically eradicating hematopoietic tumor cells when incubated in vitro for 24 hours at a ratio of 0.25:1. In another embodiment, the composition is capable of specifically eradicating hematopoietic tumor cells, eradicating 80-90% of the tumor cells when incubated in vitro for 24 hours at a ratio of 1:1 composition cells to tumor cells. In another embodiment, the tumor cells express at least one NKG2D ligand. In another embodiment, the tumor cells express multiple NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3.
他の実施形態では、組成物は、GVHDを実質的に誘発せずに、インビボで非組織適合性腫瘍細胞を特異的に根絶することができる。他の実施形態では、組成物は、GVHDを実質的に誘発せずに、インビボで非組織適合性対象における腫瘍発生を阻害又は防止することができる。 In other embodiments, the composition is capable of specifically eradicating non-histocompatible tumor cells in vivo without substantially inducing GVHD. In other embodiments, the composition is capable of inhibiting or preventing tumor development in a non-histocompatible subject in vivo without substantially inducing GVHD.
他の実施形態では、組成物は、凍結保存及びその後の解凍プロトコルの後、少なくとも70%、典型的には少なくとも80%の抗腫瘍細胞傷害活性を保持する。 In other embodiments, the composition retains at least 70%, typically at least 80%, of its antitumor cytotoxic activity after cryopreservation and subsequent thawing protocol.
他の実施形態では、本明細書に開示の方法によって組成物を調製する。幾つかの実施形態によれば、細胞組成物は、有効量のIL-2とCD3アクチベーターの常時存在下で少なくとも9日間PBMCを増殖させることを含む方法によって調製し、有効量のIL-2とCD3アクチベーターを2~4日毎に補充する。他の実施形態では、細胞組成物のインキュベーションは、他のサイトカイン又は抗体の非存在下で行う。 In other embodiments, the composition is prepared by the methods disclosed herein. According to some embodiments, the cell composition is prepared by a method comprising expanding PBMCs in the constant presence of an effective amount of IL-2 and a CD3 activator for at least 9 days, and replenishing the effective amount of IL-2 and a CD3 activator every 2-4 days. In other embodiments, the cell composition is incubated in the absence of other cytokines or antibodies.
他の実施形態では、細胞組成物を遺伝子改変する。他の実施形態では、CARを発現するように細胞組成物を遺伝子改変する。他の実施形態では、CARは、BCMA、CD47、PDL-1、メソテリン、EpCAM、CD34、CD44、PSCA、MUC16、CD276、CD123、CD19、CD20及びEFFRvIIIから成る群から選択される抗原を指向する。特定の実施形態では、組成物の細胞は、CD19特異的CAR又はCD123特異的CARを発現する。他の実施形態では、少なくとも1種のサイトカイン、ケモカイン、又はその受容体を発現するように細胞組成物を遺伝子改変する。他の実施形態では、改変ケモカイン受容体を発現するように細胞組成物を遺伝子改変する。他の実施形態では、改変ケモカイン受容体は、C末端テールドメインの変異又はトランケーションを含むCXCR4受容体である。他の実施形態では、細胞組成物はIL-2、IL-15及び/又はIL-21を外因的に発現する。特定の実施形態では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように細胞組成物は改変されている。 In other embodiments, the cell composition is genetically modified. In other embodiments, the cell composition is genetically modified to express a CAR. In other embodiments, the CAR is directed to an antigen selected from the group consisting of BCMA, CD47, PDL-1, mesothelin, EpCAM, CD34, CD44, PSCA, MUC16, CD276, CD123, CD19, CD20, and EFFRvIII. In certain embodiments, the cells of the composition express a CD19-specific CAR or a CD123-specific CAR. In other embodiments, the cell composition is genetically modified to express at least one cytokine, chemokine, or receptor thereof. In other embodiments, the cell composition is genetically modified to express a modified chemokine receptor. In other embodiments, the modified chemokine receptor is a CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain. In other embodiments, the cell composition exogenously expresses IL-2, IL-15, and/or IL-21. In certain embodiments, the cell composition is modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21.
他の実施形態では、この方法は、工程cで得られる細胞組成物の凍結保存を更に含む。他の実施形態では、方法は、凍結保存した細胞組成物を解凍プロトコルに供する(例えば、それを必要とする対象へ投与する前)ことを更に含む。他の実施形態では、この方法は、工程cの後に、凍結保存とその後の解凍プロトコルを更に含み、細胞組成物は、凍結保存前の細胞組成物の抗腫瘍細胞傷害活性特性の少なくとも70%、典型的には少なくとも80%を保持する。 In other embodiments, the method further comprises cryopreserving the cellular composition obtained in step c. In other embodiments, the method further comprises subjecting the cryopreserved cellular composition to a thawing protocol (e.g., prior to administration to a subject in need thereof). In other embodiments, the method further comprises cryopreservation and subsequent thawing protocols after step c, wherein the cellular composition retains at least 70%, typically at least 80%, of the antitumor cytotoxic activity properties of the cellular composition prior to cryopreservation.
他の様相では、(i)CD19特異的CAR又はCD123特異的CAR、(ii)C末端テールドメインの変異又はトランケーションを含む改変CXCR4受容体、並びに(iii)IL-2、IL-15及びIL-21から成る群から選択される少なくとも1種のサイトカインを発現するように遺伝子改変された白血球を含む細胞組成物が提供される。他の実施形態では、細胞組成物は、細胞の70~80%がNKG2DとグランザイムBを発現するCD3+CD8+細胞であり、少なくとも70%がCD56-細胞であり、最大5%がCD3-細胞であることを特徴とする。他の実施形態によれば、細胞組成物は、有効量のIL-2とCD3アクチベーターの常時存在下で少なくとも9日間PBMCを増殖させることを含む方法によって調製し、有効量のIL-2とCD3アクチベーターを2~4日毎に補充する。 In another aspect, a cell composition is provided comprising leukocytes genetically modified to express (i) a CD19-specific CAR or a CD123-specific CAR, (ii) a modified CXCR4 receptor comprising a mutation or truncation of the C-terminal tail domain, and (iii) at least one cytokine selected from the group consisting of IL-2, IL-15, and IL-21. In another embodiment, the cell composition is characterized in that 70-80% of the cells are CD3 + CD8 + cells expressing NKG2D and granzyme B, at least 70% are CD56- cells, and up to 5% are CD3- cells. According to another embodiment, the cell composition is prepared by a method comprising expanding PBMCs in the constant presence of an effective amount of IL-2 and a CD3 activator for at least 9 days, and replenishing the effective amount of IL-2 and the CD3 activator every 2-4 days.
幾つかの実施形態では、本明細書に開示の本発明の細胞組成物は、腫瘍の治療を必要とする対象において腫瘍の治療に使用するためのものである。一実施形態では、腫瘍は造血器腫瘍である。他の実施形態では、腫瘍は固形腫瘍である。幾つかの実施形態では、腫瘍は、白血病、多発性骨髄腫、前立腺腫瘍又は中皮腫から成る群から選択される。他の実施形態では、腫瘍は少なくとも1種のNKG2Dリガンドの発現を特徴とする。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3を発現する。 In some embodiments, the cell compositions of the present invention disclosed herein are for use in treating a tumor in a subject in need of such treatment. In one embodiment, the tumor is a hematopoietic tumor. In another embodiment, the tumor is a solid tumor. In some embodiments, the tumor is selected from the group consisting of leukemia, multiple myeloma, prostate tumor, or mesothelioma. In another embodiment, the tumor is characterized by expression of at least one NKG2D ligand. In another embodiment, the tumor cells express multiple NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In another embodiment, the tumor cells express MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3.
他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド、CXCR3リガンド及びCXCR2リガンドから成る群から選択される少なくとも1種のケモカインの発現を特徴とする。他の実施形態では、腫瘍は、CXCR6リガンド、CXCR4リガンド、及びCXCR3リガンドから成る群から選択される少なくとも1種のケモカインの発現を特徴とする。他の実施形態では、腫瘍はCXCR6リガンドの発現を特徴とする。特定の実施形態では、腫瘍はCXCR6リガンドであるCXCL16の発現を特徴とする。 In other embodiments, the tumor is characterized by expression of at least one chemokine selected from the group consisting of a CXCR6 ligand, a CXCR4 ligand, a CXCR3 ligand, and a CXCR2 ligand. In other embodiments, the tumor is characterized by expression of at least one chemokine selected from the group consisting of a CXCR6 ligand, a CXCR4 ligand, and a CXCR3 ligand. In other embodiments, the tumor is characterized by expression of a CXCR6 ligand. In certain embodiments, the tumor is characterized by expression of the CXCR6 ligand, CXCL16.
他の実施形態では、腫瘍は、少なくとも1種の抑制性免疫チェックポイント分子及び/又はそのリガンドの発現を特徴とする。幾つかの実施形態では、腫瘍は、CTLA-4、PD-1、TIGIT、Lag-3、Tim-3及びそれらの組み合わせの内の少なくとも1種のリガンドを発現することがあり、各々の可能性は本発明の別個の実施形態を表す。他の実施形態では、腫瘍は、CTLA-4、PD-1、及びTIGITから成る群から選択される抑制性免疫チェックポイント分子の少なくとも1種のリガンドの発現を特徴とする。他の実施形態では、腫瘍は、CTLA-4、PD-1、及びTIGITから成る群から選択される複数の抑制性免疫チェックポイント分子のリガンドの発現を特徴とする。他の実施形態では、腫瘍は、少なくとも1種のCTLA-4リガンド(例えば、CD80及び/又はCD86)の発現を特徴とする。 In other embodiments, the tumor is characterized by expression of at least one inhibitory immune checkpoint molecule and/or its ligand. In some embodiments, the tumor may express at least one ligand of CTLA-4, PD-1, TIGIT, Lag-3, Tim-3, and combinations thereof, each possibility representing a separate embodiment of the invention. In other embodiments, the tumor is characterized by expression of at least one ligand of an inhibitory immune checkpoint molecule selected from the group consisting of CTLA-4, PD-1, and TIGIT. In other embodiments, the tumor is characterized by expression of ligands of multiple inhibitory immune checkpoint molecules selected from the group consisting of CTLA-4, PD-1, and TIGIT. In other embodiments, the tumor is characterized by expression of at least one CTLA-4 ligand (e.g., CD80 and/or CD86).
他の実施形態では、腫瘍は、少なくとも1種のチェックポイント分子阻害剤による治療に対して抵抗性である。幾つかの実施形態では、腫瘍は、CTLA-4、PD-1、TIGIT、Lag-3、Tim-3及びそれらの組み合わせから成る群から選択される少なくとも1種のチェックポイント分子の阻害剤による治療に対して抵抗性となり得るが、各々の可能性は本発明の別の実施形態を表す。他の実施形態では、腫瘍は、CTLA-4、PD-1、及びTIGITから成る群から選択される少なくとも1種のチェックポイント分子の阻害剤による治療に対して抵抗性である。他の実施形態では、腫瘍は、CTLA-4、PD-1、及びTIGITの阻害剤による治療に対して抵抗性である。 In other embodiments, the tumor is resistant to treatment with at least one checkpoint molecule inhibitor. In some embodiments, the tumor may be resistant to treatment with an inhibitor of at least one checkpoint molecule selected from the group consisting of CTLA-4, PD-1, TIGIT, Lag-3, Tim-3, and combinations thereof, each possibility representing a separate embodiment of the invention. In other embodiments, the tumor is resistant to treatment with an inhibitor of at least one checkpoint molecule selected from the group consisting of CTLA-4, PD-1, and TIGIT. In other embodiments, the tumor is resistant to treatment with an inhibitor of CTLA-4, PD-1, and TIGIT.
例えば、本発明の細胞組成物は、CTLA-4の実質的な発現を欠いていることが本明細書で示されており、従って、唯一の治療剤としてのCTLA-4遮断抗体(例えば、イピリムマブ及び/又はトレメリムマブ)による治療に対して抵抗性の腫瘍の治療に特に有利となり得る。従って、他の実施形態では、対象はチェックポイント分子阻害剤による治療レジメンを受けていない。 For example, the cell compositions of the invention are shown herein to lack substantial expression of CTLA-4 and may therefore be particularly advantageous for treating tumors that are resistant to treatment with a CTLA-4 blocking antibody (e.g., ipilimumab and/or tremelimumab) as the sole therapeutic agent. Thus, in other embodiments, the subject has not received a treatment regimen with a checkpoint molecule inhibitor.
他の例では、本発明の細胞組成物は、Tim-3及び/又はLag-3を発現する腫瘍の治療に使用することができ、例えば、それぞれTim-3阻害剤及び/又はLag-3阻害剤を用いた併用療法に使用することができる。従って、他の実施形態では、対象は、例えば、PD-1、TIGIT、Lag-3、Tim-3又はそれらの組み合わせに対する少なくとも1種のチェックポイント分子阻害剤による治療レジメンを受けているが、各々の可能性が本発明の別個の実施形態を表す。他の実施形態では、上述の使用は、例えば、PD-1、TIGIT、Lag-3、Tim-3又はそれらの組み合わせに対する少なくとも1種のチェックポイント分子阻害剤の投与を更に含み、各々の可能性が本発明の別個の実施形態を表す。他の実施形態では、この使用は、Lag-3、Tim-3又はそれらの組み合わせに対する少なくとも1種のチェックポイント分子阻害剤の投与を更に含む。幾つかの実施形態では、本発明に係る細胞組成物の使用によって、既存の抗癌療法、例えば、免疫チェックポイント阻害剤の用量を低下又は最小限にすることができる。 In other examples, the cell compositions of the present invention can be used to treat tumors expressing Tim-3 and/or Lag-3, e.g., in combination therapy with a Tim-3 inhibitor and/or a Lag-3 inhibitor, respectively. Thus, in other embodiments, the subject is undergoing a treatment regimen with at least one checkpoint molecule inhibitor, e.g., against PD-1, TIGIT, Lag-3, Tim-3, or a combination thereof, each possibility representing a separate embodiment of the present invention. In other embodiments, the above-described use further comprises administration of at least one checkpoint molecule inhibitor, e.g., against PD-1, TIGIT, Lag-3, Tim-3, or a combination thereof, each possibility representing a separate embodiment of the present invention. In other embodiments, the use further comprises administration of at least one checkpoint molecule inhibitor, e.g., against Lag-3, Tim-3, or a combination thereof. In some embodiments, the use of the cell compositions of the present invention can reduce or minimize the dose of existing anti-cancer therapies, e.g., immune checkpoint inhibitors.
他の様相では、腫瘍の治療を必要とする対象において腫瘍を治療する方法であって、本明細書に開示の本発明の治療有効量の細胞組成物に腫瘍細胞を接触させることを含む方法が提供される。 In another aspect, there is provided a method of treating a tumor in a subject in need thereof, the method comprising contacting tumor cells with a therapeutically effective amount of a cell composition of the invention disclosed herein.
一実施形態では、細胞組成物は、対象に対して自家性、組織適合性同種異系、又は非組織適合性同種異系である。特定の実施形態では、細胞組成物は、対象に対して非組織適合性同種異系である。他の実施形態では、接触をインビボで行う。他の実施形態では、接触をエクスビボで行う。他の実施形態では、腫瘍は造血器腫瘍である。他の実施形態では、腫瘍は固形腫瘍である。他の実施形態では、腫瘍は少なくとも1種のNKG2Dリガンドの発現を特徴とする。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3から成る群から選択される複数のNKG2Dリガンドを発現する。他の実施形態では、腫瘍細胞は、MICA、MICB、HLAE、ULBP1、ULBP256、及びULBP3を発現する。 In one embodiment, the cell composition is autologous, histocompatible allogeneic, or non-histocompatible allogeneic to the subject. In certain embodiments, the cell composition is non-histocompatible allogeneic to the subject. In other embodiments, the contacting occurs in vivo. In other embodiments, the contacting occurs ex vivo. In other embodiments, the tumor is a hematopoietic tumor. In other embodiments, the tumor is a solid tumor. In other embodiments, the tumor is characterized by expression of at least one NKG2D ligand. In other embodiments, the tumor cells express a plurality of NKG2D ligands selected from the group consisting of MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3. In other embodiments, the tumor cells express MICA, MICB, HLAE, ULBP1, ULBP256, and ULBP3.
本発明の幾つかの実施形態をより十分に説明するために以下の実施例を提示する。しかし、これらの実施例は本発明の広い範囲を限定するものとして解釈すべきではない。 The following examples are presented to more fully illustrate certain embodiments of the present invention. However, these examples should not be construed as limiting the broad scope of the invention.
超活性化キラー(SAK)細胞の産生と特徴付け
フィコール・ハイパック密度遠心分離(Sigma社製)によって末梢血単核細胞(PBMC)をヒト血液から単離した。PBMCをRPMI培地(完全培地+MEM NEAA×1及び0.1mMの2-メルカプトエタノール)に1×106個(細胞)/mLの濃度で再懸濁させ、0日目に1000IU/mLの組換えヒトIL-2(rhIL-2、R&DSystems社製)と30ng/mLの抗ヒトCD3抗体(OKT-3、eBioscience社製)で刺激した。4日目、6日目及び8日目に新鮮な培地+rhIL-2と抗CD3を添加して培養を維持した。細胞を少なくとも11日間増殖させ、様々な時点で回収して更なる分析に備えた。
Generation and Characterization of Superactivated Killer (SAK) Cells Peripheral blood mononuclear cells (PBMCs) were isolated from human blood by Ficoll-Hypaque density centrifugation (Sigma). PBMCs were resuspended at a concentration of 1x106 cells/mL in RPMI medium (complete medium + 1x MEM NEAA and 0.1 mM 2-mercaptoethanol) and stimulated with 1000 IU/mL recombinant human IL-2 (rhIL-2, R&D Systems) and 30 ng/mL anti-human CD3 antibody (OKT-3, eBioscience) on day 0. Cultures were maintained by adding fresh medium + rhIL-2 and anti-CD3 on days 4, 6, and 8. Cells were expanded for at least 11 days and harvested at various time points for further analysis.
増殖率を評価するために、回収した細胞をトリパンブルーで染色し、0日目、4日目、6日目、8日目及び11日目に生細胞の総数をカウントした。結果を図1Aに示すが、9回の別々の実験から得た平均±標準偏差を示す。図1Aに示すように、細胞は絶えず増殖し、11日間の培養後に30.55倍の増殖率に達した。 To assess the proliferation rate, the harvested cells were stained with trypan blue and the total number of viable cells was counted on days 0, 4, 6, 8 and 11. The results are shown in Figure 1A and represent the mean ± standard deviation from nine separate experiments. As shown in Figure 1A, the cells proliferated continuously, reaching a proliferation rate of 30.55-fold after 11 days of culture.
次に、フローサイトメトリーによる免疫表現型分析を以下のように行った。様々な時点で細胞を回収し、洗浄し、様々なモノクローナル抗体で4℃にて30分間染色し、フローサイトメトリーで分析した。次の抗体、即ち、FITC抗CD4、APC抗CD8、Percp抗CD3、及びPE抗CD56(全てBiolegend社製)を使用した。結果を図1B~1Dに示すが、各図には9回の別々の実験から得た平均値±標準偏差を示す。 Immunophenotyping by flow cytometry was then performed as follows: Cells were harvested at various time points, washed, stained with various monoclonal antibodies for 30 min at 4°C, and analyzed by flow cytometry. The following antibodies were used: FITC anti-CD4, APC anti-CD8, Percp anti-CD3, and PE anti-CD56 (all from Biolegend). The results are shown in Figures 1B-1D, where each figure shows the mean ± standard deviation from nine separate experiments.
図1B~1Dに示すように、増殖細胞集団全体におけるCD3+CD8+細胞とCD3+CD56+細胞の割合(%)は経時的に徐々に上昇したが、CD3+CD4+細胞の割合(%)は経時的に低下した。11日目の様々な亜集団の平均値と割合を表1にまとめる(9回の独立した実験の平均値±SD)。11日目の亜集団を表2で更に特徴付ける(全組成物からの各亜集団の%、5回の独立した実験の平均値±SD)。CD8+CD56+/CD8+は、総CD8+集団の内のCD8+CD56+のパーセンタイルを表す。 As shown in Figures 1B-1D, the percentages of CD3 + CD8 + and CD3 + CD56 + cells in the total expanded cell population gradually increased over time, whereas the percentages of CD3 + CD4 + cells decreased over time. The means and percentages of the various subpopulations at day 11 are summarized in Table 1 (mean ± SD of 9 independent experiments). The subpopulations at day 11 are further characterized in Table 2 (% of each subpopulation from total composition, mean ± SD of 5 independent experiments). CD8 + CD56 + /CD8 + represents the percentile of CD8 + CD56 + within the total CD8 + population.


25名の個々のドナーから得た試料の包括的分析から、組成物は平均74.6±1.6%のCD3+CD8+細胞と12±1%のCD3+CD4+細胞によって特徴付けられた。更に、CD3+CD8+細胞とCD3+CD4+細胞の比率は少なくとも6:1、典型的には約7~8:1であった。得られた細胞(本明細書では超活性化キラー(SAK)細胞と称する)を、以下の実施例で詳述するように更なる分析に供した。 Comprehensive analysis of samples from 25 individual donors revealed that the composition was characterized by an average of 74.6±1.6% CD3 + CD8 + cells and 12±1% CD3 + CD4 + cells. Furthermore, the ratio of CD3 + CD8 + cells to CD3 + CD4 + cells was at least 6:1, and typically about 7-8:1. The resulting cells, referred to herein as superactivated killer (SAK) cells, were subjected to further analysis as detailed in the Examples below.
比較として、ACT用のPBMC由来エフェクター細胞(即ち、サイトカイン誘導キラー細胞(CIK))を調製するための他の既知のプロトコルも試験した。CIKの調製では、PBMCをRPMI培地(完全培地+MEM NEAA×1及び0.1mMの2-メルカプトエタノール)に1×106個(細胞)/mLの濃度で再懸濁させ、0日目に1000IU/mLの組換えヒトIFN-γ(Peprotec社製)で刺激した。1日目に、50ng/mLの抗ヒトCD3抗体(OKT-3、eBioscience社製)と500IU/mLのrhIL-2(R and D system社製)を添加した。4日目、6日目及び8日目に新鮮な培地+500IU/mLのrhIL-2を添加して培養を維持した。細胞を少なくとも11日間増殖させた。11日目にCIK細胞を回収し、更なるアッセイに備えた。上で詳述したように、SAK細胞も同時に増殖させた。11日目のCIK集団の特徴付けを表3に示す。 As a comparison, other known protocols for preparing PBMC-derived effector cells for ACT (i.e., cytokine-induced killer cells (CIK)) were also tested. For preparation of CIK, PBMC were resuspended in RPMI medium (complete medium + 1x MEM NEAA and 0.1 mM 2-mercaptoethanol) at a concentration of 1x106 cells/mL and stimulated with 1000 IU/mL recombinant human IFN-γ (Peprotec) on day 0. On day 1, 50 ng/mL anti-human CD3 antibody (OKT-3, eBioscience) and 500 IU/mL rhIL-2 (R and D system) were added. Cultures were maintained with fresh medium + 500 IU/mL rhIL-2 on days 4, 6, and 8. Cells were allowed to grow for at least 11 days. CIK cells were harvested on day 11 for further assays. SAK cells were also grown in parallel as detailed above. Characterization of the CIK population on day 11 is shown in Table 3.
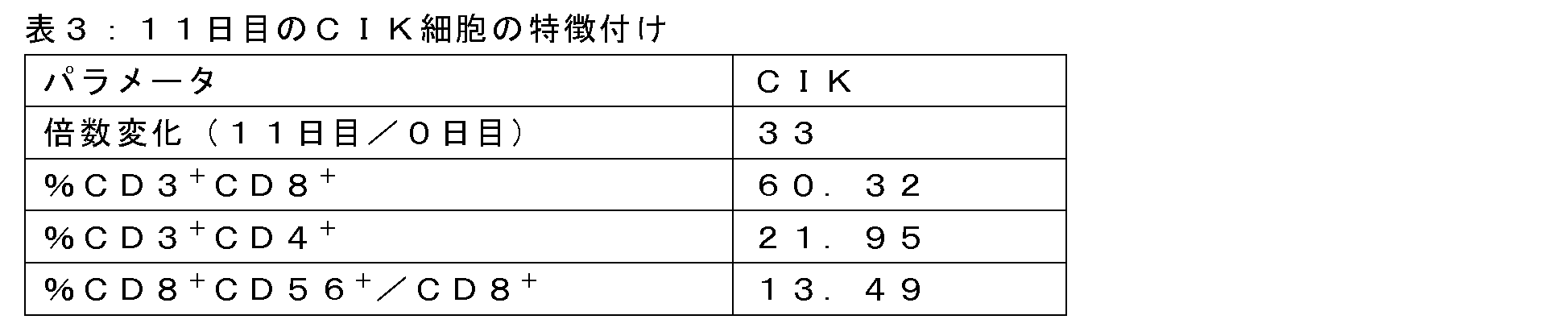
表1~3から分かるように、SAK細胞は、増殖11日目では表現型がCIK細胞とは異なっており、SAK細胞組成物では、CIK細胞組成物に比べて全CD8+細胞の内のCD8+CD56+細胞の割合が有意に高く、CIK細胞組成物に比べてCD3+CD4+細胞の割合が有意に低い。 As can be seen from Tables 1-3, SAK cells were phenotypically distinct from CIK cells on day 11 of expansion, with the SAK cell composition having a significantly higher percentage of CD8 + CD56 + cells among total CD8 + cells than the CIK cell composition and a significantly lower percentage of CD3 + CD4 + cells than the CIK cell composition.
更に、ACTを目的とするCIK細胞は、更なる亜集団の相対的割合(例えば、CD3-細胞とCD56+細胞の発生率(これはCIK細胞においてより高い)が挙げられるが、これに限定されない)によって本明細書に記載のSAK細胞と更に区別される。CIK細胞組成物は通常、21日間の増殖後に、上述のCIKプロトコルに本質的に相当する方法によって癌患者に投与する。このような組成物は、典型的には平均含量が少なくとも30%のCD56+細胞、より典型的には最大約50%~80%のCD56+細胞を特徴とし、平均含量が少なくとも5%、より典型的には最大約10%~70%のCD3-細胞を特徴とする。例えば、これまでに報告された様々な例示的CIK組成物は、21日目において、40~80%のCD3+CD56+細胞、20~60%のCD3+CD56-細胞、及び1~10%のCD3-CD56+細胞;35%のCD3+CD56+細胞、65%のCD3+CD56-細胞、及び2.23%のCD3-CD56+細胞(平均値、90%の総CD3+細胞);又は31.6%のCD3+CD56+細胞、41.5%のCD3+CD56-細胞、及び11.7%のCD3-CD56+細胞(中央値)を含むことを特徴としていた。 Moreover, CIK cells intended for ACT are further distinguished from SAK cells described herein by the relative proportions of additional subpopulations, including but not limited to, the incidence of CD3 − cells and CD56 + cells, which is higher in CIK cells. CIK cell compositions are typically administered to cancer patients after 21 days of expansion in a manner essentially corresponding to the CIK protocol described above. Such compositions are typically characterized by an average content of at least 30% CD56 + cells, more typically up to about 50%-80% CD56 + cells, and an average content of at least 5%, more typically up to about 10%-70% CD3 − cells. For example, various exemplary CIK compositions reported thus far have been characterized as comprising, at day 21, 40-80% CD3 + CD56 + cells, 20-60% CD3 + CD56- cells, and 1-10% CD3 - CD56 + cells; 35% CD3 + CD56 + cells, 65% CD3 + CD56 - cells, and 2.23% CD3 - CD56 + cells (mean values, 90% of total CD3 + cells); or 31.6% CD3 + CD56 + cells, 41.5% CD3 + CD56 - cells, and 11.7% CD3 - CD56 + cells (median values).
SAK細胞の抗腫瘍細胞傷害活性
様々な腫瘍細胞(本明細書では標的細胞とも称される)に対するエフェクター細胞(SAK)の影響を評価するために、カルボキシフルオレセイン二酢酸スクシニミジルエステル(CFSE)ベースのアッセイを用いて細胞障害性アッセイを行った。SAK細胞を培養11日目に回収した。標的細胞を製造業者の指示に従ってCFSE(eBioscience社製)で標識し、96ウェルプレートに2×104個(細胞)/ウェルの濃度で播種した。エフェクター対標的(E:T)比率を0.25:1、0.5:1、1:1、2:1、5:1、10:1及び30:1としてエフェクター細胞を標的細胞に添加した。24時間後、CFSE+PI-で標識した標的細胞をフローサイトメトリーによって分析した。生細胞のパーセンタイルは、(エフェクター細胞を含むCFSE+PI-細胞の数/エフェクター細胞を含まないCFSE+PI-細胞の数)×100で計算した。
Antitumor cytotoxic activity of SAK cells To evaluate the effect of effector cells (SAK) on various tumor cells (also referred to herein as target cells), cytotoxicity assays were performed using a carboxyfluorescein diacetate succinimidyl ester (CFSE)-based assay. SAK cells were harvested on day 11 of culture. Target cells were labeled with CFSE (eBioscience) according to the manufacturer's instructions and seeded at a concentration of 2 × 104 cells/well in 96-well plates. Effector cells were added to target cells at effector-to-target (E:T) ratios of 0.25:1, 0.5:1, 1:1, 2:1, 5:1, 10:1, and 30:1. After 24 h, CFSE + PI - labeled target cells were analyzed by flow cytometry. The percentile of viable cells was calculated as (number of CFSE + PI − cells with effector cells/number of CFSE + PI − cells without effector cells)×100.
THP-1白血病細胞とRPMI8266多発性骨髄腫細胞を用いて得られた結果をそれぞれ図2A~2Bに示すが、各比率における3回の平均値±標準偏差を示す。図2A~2Bに示すように、SAK細胞は、0.25:1という低いE:T比率であっても、両方の型の造血器腫瘍細胞株、THP-1及びRPMI8266に対して強力な細胞傷害活性を示した。E:T比率を2:1として使用した場合、SAK細胞は約90%の顕著な死滅効率を示し、抗腫瘍細胞療法としてのSAK細胞の効力が示された。 The results obtained using THP-1 leukemia cells and RPMI8266 multiple myeloma cells are shown in Figures 2A-2B, respectively, and show the mean ± standard deviation of triplicates at each ratio. As shown in Figures 2A-2B, SAK cells exhibited potent cytotoxic activity against both types of hematopoietic tumor cell lines, THP-1 and RPMI8266, even at an E:T ratio as low as 0.25:1. When an E:T ratio of 2:1 was used, SAK cells showed a remarkable killing efficiency of about 90%, demonstrating the efficacy of SAK cells as an antitumor cell therapy.
PC3前立腺癌細胞やH28中皮腫癌細胞等の様々な固形腫瘍細胞株について実験を繰り返し、造血MV4-11白血病細胞と比較した。結果を図3A~3Cに示すが、各比率における3回の平均値±標準偏差を示す。図3A~3Cに示すように、SAK細胞は、造血MV4-11細胞と両方の固形腫瘍細胞株に対して顕著な効力を示した。E:T比を上げると、全ての癌細胞株、特にMV4-11白血病細胞に対するSAK細胞の有効性が高まった。例えば、MV4-11細胞に対して90%の効率を得るには2:1のE:T比率が必要である一方、PC3前立腺癌細胞とH28中皮腫癌細胞に対して90%の効率を得るには30:1のE:T比率が必要であった。 The experiment was repeated with various solid tumor cell lines, including PC3 prostate cancer cells and H28 mesothelioma cancer cells, and compared with hematopoietic MV4-11 leukemia cells. The results are shown in Figures 3A-3C, which show the mean ± standard deviation of triplicates at each ratio. As shown in Figures 3A-3C, SAK cells showed significant efficacy against hematopoietic MV4-11 cells and both solid tumor cell lines. Increasing the E:T ratio increased the efficacy of SAK cells against all cancer cell lines, especially MV4-11 leukemia cells. For example, an E:T ratio of 2:1 was required to achieve 90% efficiency against MV4-11 cells, while an E:T ratio of 30:1 was required to achieve 90% efficiency against PC3 prostate cancer cells and H28 mesothelioma cancer cells.
次に、フローサイトメトリーを用いてSAK細胞組成物をCD3+CD56+細胞とCD3+CD56-細胞に分離し、各集団の細胞傷害活性を元のSAK細胞組成物の細胞傷害活性と比較した。この目的のため、上で詳述したように、各集団を様々なE:T比率(0.25:1、0.5:1、1:1及び2:1)でMV4-11細胞とインキュベートした。注目すべきことに、試験した集団の細胞障害能力と完全なSAK組成物の細胞障害能力との間には、試験したE:T比率のいずれにおいても有意差は見出されなかった。更に、増殖10日目と12日目に回収したSAK細胞組成物を用いて同様の結果が得られた。 The SAK cell compositions were then separated into CD3 + CD56 + and CD3 + CD56 - cells using flow cytometry and the cytotoxic activity of each population was compared to that of the original SAK cell composition. For this purpose, each population was incubated with MV4-11 cells at various E:T ratios (0.25:1, 0.5:1, 1:1 and 2:1) as detailed above. Of note, no significant differences were found between the cytotoxic potential of the tested populations and that of the intact SAK composition at any of the E:T ratios tested. Moreover, similar results were obtained with SAK cell compositions harvested on days 10 and 12 of expansion.
従って、SAK細胞は、MHCII非依存的に様々な腫瘍細胞に対して強力な抗腫瘍細胞傷害活性を示した。更に、細胞傷害活性はCD56非依存的のように思われる。 Thus, SAK cells exhibited potent antitumor cytotoxic activity against various tumor cells in an MHCII-independent manner. Furthermore, the cytotoxic activity appears to be CD56-independent.
SAK細胞は、他のPBMC由来エフェクター細胞と比較して、有意に向上した抗腫瘍細胞傷害活性を示す
SAK細胞が優れた抗腫瘍活性を発揮するという発見の後、SAK細胞の細胞傷害活性を他の2種のPBMC由来エフェクター細胞、即ち、サイトカイン誘導キラー細胞(CIK)及び活性化リンパ球組成物(IL-2増殖T細胞)と比較した。
SAK cells exhibit significantly improved antitumor cytotoxic activity compared to other PBMC-derived effector cells Following the discovery that SAK cells exert superior antitumor activity, the cytotoxic activity of SAK cells was compared with two other PBMC-derived effector cells, namely, cytokine-induced killer cells (CIK) and an activated lymphocyte composition (IL-2-proliferated T cells).
CIKを調製するため、PBMCをRPMI培地(完全培地+MEM NEAA×1及び0.1mMの2-メルカプトエタノール)に1×106個(細胞)/mLの濃度で再懸濁させ、0日目に1000IU/mLの組換えヒトIFN-γ(Peprotec社製)で刺激した。50ng/mLの抗ヒトCD3抗体(OKT-3、eBioscience社製)と500IU/mLのrhIL-2(R and D system社製)を1日目に添加した。4日目、6日目及び8日目に新鮮な培地+500IU/mLのrhIL-2を添加して培養を維持した。細胞を少なくとも11日間増殖させた。11日目にCIK細胞を回収して更なるアッセイに備えた。0日目にPBMCを50ng/mLの抗ヒトCD3及び300IU/mLのrhIL-2とインキュベートし、300IU/mLのrhIL-2を含む新鮮な培地を更に添加した後、IL-2増殖T細胞培養物を得た。 To prepare CIK, PBMCs were resuspended in RPMI medium (complete medium + 1x MEM NEAA and 0.1 mM 2-mercaptoethanol) at a concentration of 1x106 cells/mL and stimulated with 1000 IU/mL recombinant human IFN-γ (Peprotec) on day 0. 50 ng/mL anti-human CD3 antibody (OKT-3, eBioscience) and 500 IU/mL rhIL-2 (R and D system) were added on day 1. Cultures were maintained by adding fresh medium + 500 IU/mL rhIL-2 on days 4, 6 and 8. Cells were expanded for at least 11 days. CIK cells were harvested on day 11 for further assays. On day 0, PBMCs were incubated with 50 ng/mL anti-human CD3 and 300 IU/mL rhIL-2, and IL-2-expanded T cell cultures were obtained after further addition of fresh medium containing 300 IU/mL rhIL-2.
未処理のMV4-11細胞を対照として設定した場合、エフェクター細胞としてSAK、CIK又はIL-2増殖T細胞を使用し、標的細胞としてMV4-11白血病細胞を使用して、CFSEベースの細胞障害性アッセイを基本的には実施例2に記載のように行った。結果を図4A~4Bに示すが、各比率における3回の平均値±標準偏差を示す。 CFSE-based cytotoxicity assays were performed essentially as described in Example 2 using SAK, CIK or IL-2 expanded T cells as effector cells and MV4-11 leukemia cells as target cells, with untreated MV4-11 cells set as control. The results are shown in Figures 4A-4B, showing the mean ± standard deviation of triplicates at each ratio.
予想外にも、図4A~4Bに示すように、SAK細胞は、対照エフェクター細胞よりも腫瘍細胞の根絶において有意に有効であることが分かった。CIK細胞と比較した場合(図4A)、SAK細胞が有意な細胞傷害活性を示すのに必要なE:T比率は低く、SAK細胞は、試験した各E:T比率で優れた細胞傷害活性を示した。例えば、E:T比率が5:1の場合、SAK細胞は90%の効率を示したが、同じ比率でCIK細胞の効率は僅かに40%であった(図4A)。SAK細胞は、IL-2増殖T細胞と比較して更に有効であることが分かった。例えば、E:T比率が0.25:1であっても、SAK細胞は60%の効率を示したが、同じ比率においてIL-2増殖T細胞は全く有効ではなかった。また、E:T比率が2:1であっても、SAK細胞は90%以上の死滅活性を示す一方、IL-2増殖T細胞の活性は25%に過ぎなかった(図4B)。 Unexpectedly, as shown in Figures 4A-4B, SAK cells were found to be significantly more effective at eradicating tumor cells than control effector cells. When compared to CIK cells (Figure 4A), a lower E:T ratio was required for SAK cells to exhibit significant cytotoxic activity, and SAK cells exhibited superior cytotoxic activity at each E:T ratio tested. For example, at an E:T ratio of 5:1, SAK cells were 90% efficient, whereas CIK cells were only 40% efficient at the same ratio (Figure 4A). SAK cells were found to be even more effective compared to IL-2-expanded T cells. For example, even at an E:T ratio of 0.25:1, SAK cells were 60% efficient, whereas IL-2-expanded T cells were completely ineffective at the same ratio. Also, even at an E:T ratio of 2:1, SAK cells exhibited over 90% killing activity, whereas IL-2-expanded T cells were only 25% active (Figure 4B).
従って、SAK細胞は、従来既知のPBMC由来のエフェクター細胞と比較して、非常に効率的な抗腫瘍細胞傷害活性を発揮することが予想外に見出された。 Therefore, it was unexpectedly found that SAK cells exert highly efficient antitumor cytotoxic activity compared to previously known PBMC-derived effector cells.
SAK細胞と腫瘍細胞の免疫表現型の特徴付け
CD8+SAK細胞でのNKG2Dの発現を次のように評価した。実施例1に記載のように調製したSAK細胞を増殖11日目に回収し、洗浄し、FITC標識抗NKG2DとAPC標識抗CD8モノクローナル抗体(Biolegend社製)で4℃にて30分間染色し、フローサイトメトリーによって分析した。CD8+細胞上の抗体による染色を検出し、アイソタイプ抗体対照染色と比較した。
Immunophenotypic characterization of SAK and tumor cells NKG2D expression on CD8 + SAK cells was assessed as follows: SAK cells prepared as described in Example 1 were harvested on day 11 of growth, washed, stained with FITC-labeled anti-NKG2D and APC-labeled anti-CD8 monoclonal antibodies (Biolegend) for 30 minutes at 4°C, and analyzed by flow cytometry. Antibody staining on CD8 + cells was detected and compared to isotype antibody control staining.
結果を図5に示す。図5に示すように、CD8+SAK細胞はNKG2Dを高度に発現する。注目すべきことに、CD8+細胞の100%がNKG2Dを発現することが判明し、アイソタイプ対照と比較して平均蛍光強度が8.4倍増加した。 The results are shown in Figure 5. As shown in Figure 5, CD8 + SAK cells highly express NKG2D. Remarkably, 100% of CD8 + cells were found to express NKG2D, with an 8.4-fold increase in mean fluorescence intensity compared to the isotype control.
SAK細胞はNKG2D受容体の発現を示したので、フローサイトメトリーを用いて様々な種類の腫瘍細胞で各種NKG2Dリガンドの発現を試験した。結果を以下の表4にまとめる。 Because SAK cells showed expression of the NKG2D receptor, we tested the expression of various NKG2D ligands in various tumor cell types using flow cytometry. The results are summarized in Table 4 below.

表4に示すように、MV411白血病細胞は、ULBP3、HLAE、ULBP1、ULBP256、及びMICA及び/又はMICB等の様々なNKG2Dリガンドをその表面に高度に発現する。これに対し、H28中皮腫細胞は、これらのマーカーを有意に低く、即ち、非有意なレベルで発現する(例えば、ULBP256)か、又は検出不可能なレベルで発現する(例えば、MICA及びMICB)。 As shown in Table 4, MV411 leukemia cells highly express various NKG2D ligands on their surface, such as ULBP3, HLAE, ULBP1, ULBP256, and MICA and/or MICB. In contrast, H28 mesothelioma cells express these markers at significantly lower, i.e., non-significant levels (e.g., ULBP256) or at undetectable levels (e.g., MICA and MICB).
従って、SAK細胞はNKG2Dの高発現を特徴とし、MV4-11白血病細胞等のNKG2Dリガンドの高発現を特徴とする標的腫瘍細胞に対して特に有効であることが分かった。 Therefore, SAK cells are characterized by high expression of NKG2D and have been found to be particularly effective against target tumor cells characterized by high expression of NKG2D ligands, such as MV4-11 leukemia cells.
SAKの細胞傷害活性は、FasL-Fas相互作用ではなく、主にグランザイムBとパーフォリンの分泌によって媒介される
SAK細胞の死滅機序を調べるため、基本的には実施例4に記載のように、CD8+SAK細胞でのFASL、グランザイムB及びパーフォリンの発現レベルを調べた。即ち、SAK細胞を増殖11日目に回収し、洗浄し、PE抗FasL APC抗パーフォリン抗体又はPE抗グランザイムモノクローナル抗体(全てBiolegend社製)で4℃にて30分間染色し、フローサイトメトリーによって分析した。CD8+細胞上の抗体による染色を検出し、アイソタイプ抗体対照染色と比較した。
Cytotoxic activity of SAK is mediated primarily by secretion of granzyme B and perforin, not FasL-Fas interactions To examine the mechanism of killing of SAK cells, expression levels of FASL, granzyme B, and perforin in CD8 + SAK cells were examined essentially as described in Example 4. Briefly, SAK cells were harvested on day 11 of growth, washed, stained with PE anti-FasL APC anti-perforin antibody or PE anti-granzyme monoclonal antibody (all from Biolegend) for 30 min at 4°C, and analyzed by flow cytometry. Antibody staining on CD8 + cells was detected and compared to isotype antibody control staining.
図6A~6Cに示す結果は、CD8+細胞傷害性細胞集団によるFasL、グランザイムB及びパーフォリンの高発現を実証する。平均蛍光強度(MFI)で表される、マーカー特異的抗体で染色したCD8+細胞での発現レベルをアイソタイプ対照抗体の発現レベルと比較したものを以下の表5にまとめる。表5に示すように、CD8+細胞の40%がFasL陽性であり、50%がパーフォリン陽性であり、100%がグランザイムBを発現する。 The results shown in Figures 6A-6C demonstrate high expression of FasL, Granzyme B and Perforin by the CD8 + cytotoxic cell population. Expression levels, expressed as mean fluorescence intensity (MFI), in CD8 + cells stained with marker-specific antibodies compared to isotype control antibody expression levels are summarized below in Table 5. As shown in Table 5, 40% of the CD8 + cells are FasL positive, 50% are Perforin positive and 100% express Granzyme B.

次に、標的誘導パーフォリン分泌を次のように調べた。5×103個のSAK細胞(11日目)をMV4-11細胞に0.25:1(E:T)の比率で24時間添加した。24時間後、酵素結合免疫吸着測定法(ELISA)キット(Abcam社製)を使用して培地上清中のパーフォリンの濃度を調べた。図7に示すように、標的MV4-11腫瘍細胞とのインキュベーション後にパーフォリン分泌レベルが上昇した。 Next, target-induced perforin secretion was examined as follows: 5 × 10 3 SAK cells (day 11) were added to MV4-11 cells at a ratio of 0.25:1 (E:T) for 24 h. After 24 h, the concentration of perforin in the culture supernatant was examined using an enzyme-linked immunosorbent assay (ELISA) kit (Abcam). As shown in Figure 7, the level of perforin secretion was increased after incubation with target MV4-11 tumor cells.
死滅機序を更に特徴付けるため、様々な阻害剤の存在下、基本的には実施例2に記載のようにCFSEベースの細胞障害性アッセイを行った。この目的のため、試験した阻害剤の有無に関わらず、白血病MV4-11細胞をSAK細胞と共に培養し、未処理のMV4-11細胞を対照として設定した。次の阻害剤、即ち、3,4-ジクロロイソクマリン(DCI:グランザイムB阻害剤、Sigma社製)、コンカナマイシンA(CMA:パーフォリン阻害剤、Sigma社製)及び抗FasL遮断抗体(Biolegend社製)を使用した。標的細胞とのインキュベーションの2時間前に、50μMのDCI、1000mMのCMA又は5μg/mLの抗FasLをSAK細胞に添加した。白血病MV4-11標的細胞に対するSAKの細胞傷害活性を、(エフェクター細胞の存在下でのCFSE+PI-細胞の数/エフェクター細胞の非存在下でのCFSE+PI-細胞の数)×100によって計算される生細胞の割合で表した。 To further characterize the killing mechanism, CFSE-based cytotoxicity assays were performed in the presence of various inhibitors, essentially as described in Example 2. For this purpose, leukemic MV4-11 cells were cultured with SAK cells with or without the tested inhibitors, and untreated MV4-11 cells were set as controls. The following inhibitors were used: 3,4-dichloroisocoumarin (DCI: granzyme B inhibitor, Sigma), concanamycin A (CMA: perforin inhibitor, Sigma) and anti-FasL blocking antibody (Biolegend). 50 μM DCI, 1000 mM CMA or 5 μg/mL anti-FasL were added to SAK cells 2 hours prior to incubation with target cells. The cytotoxic activity of SAK against leukemic MV4-11 target cells was expressed as the percentage of viable cells calculated by (number of CFSE + PI − cells in the presence of effector cells/number of CFSE + PI − cells in the absence of effector cells) × 100.
結果を図8A~8Cに示すが、各比率における3回の平均±標準偏差を示す。図に示すように、グランザイムB阻害剤とパーフォリン阻害剤はSAK細胞の細胞傷害活性を有意に低下させたが(それぞれ図8Aと8C)、抗FasL抗体とのインキュベーションは生細胞の割合の減少に殆ど又は全く影響を及ぼさなかった(図8B)。 The results are shown in Figures 8A-8C, which show the mean ± standard deviation of triplicates for each ratio. As shown in the figures, granzyme B inhibitors and perforin inhibitors significantly reduced the cytotoxic activity of SAK cells (Figures 8A and 8C, respectively), whereas incubation with anti-FasL antibody had little or no effect on the reduction in the percentage of viable cells (Figure 8B).
従って、特定の理論又は作用機序に束縛されたくはないが、SAK細胞は、FasL-Fas相互作用よりもグランザイムB及びパーフォリン分泌に主に依存する抗腫瘍細胞傷害活性を示すと思われる。 Thus, without wishing to be bound by any particular theory or mechanism of action, SAK cells appear to exhibit antitumor cytotoxic activity that is primarily dependent on granzyme B and perforin secretion rather than FasL-Fas interactions.
SAK遊走能力の特徴付け
SAK細胞集団を更に特徴付けるために、次のようにフローサイトメトリーによってケモカイン受容体発現の分析を行った。細胞を様々な時点で回収し、洗浄し、APC抗CXCR4(12G5クローン)、APC抗CXCR3、PE抗CXCR6又はFITC抗CXCR2モノクローナル抗体、更にはFITC又はAPC(全てBiolegend社製)でそれぞれ標識した抗CD8抗体で4℃にて30分間染色し、フローサイトメトリーによって分析した。
Characterization of SAK migratory capacity To further characterize the SAK cell population, analysis of chemokine receptor expression was performed by flow cytometry as follows: Cells were harvested at various time points, washed, stained with APC anti-CXCR4 (12G5 clone), APC anti-CXCR3, PE anti-CXCR6 or FITC anti-CXCR2 monoclonal antibodies, as well as anti-CD8 antibodies labeled with FITC or APC (all from Biolegend), respectively, for 30 min at 4° C., and analyzed by flow cytometry.
図9Aに示す結果から、増殖11日目のSAK細胞でのケモカイン受容体CXCR2、CXCR3(上部パネル、それぞれ左と右)、CXCR4及びCXCR6(下部パネル、それぞれ左と右)の発現が分かる。特に、実質的に全てのCD3+CD8+細胞はCXCR4の表面発現を特徴とすることが分かり、残りのケモカイン受容体は平均してCD3+CD8+細胞の約90%(CXCR2とCXCR3)又は約30%(CXCR6)で見出された。 The results shown in Figure 9A show the expression of the chemokine receptors CXCR2, CXCR3 (upper panels, left and right, respectively), CXCR4 and CXCR6 (lower panels, left and right, respectively) on SAK cells at day 11 of expansion. Notably, it was found that virtually all CD3 + CD8 + cells were characterized by surface expression of CXCR4, while the remaining chemokine receptors were found on average in approximately 90% (CXCR2 and CXCR3) or approximately 30% (CXCR6) of CD3 + CD8 + cells.
SAK細胞上のケモカイン受容体の機能を試験するために遊走アッセイを行った。この目的のため、ケモカインCXCL12、IP-10又はrhIL-8(PeproTech EC社製)に応答したSAK細胞の遊走を、孔径5μmのTranswell(Costar社製)を用いて評価した。1%ウシ胎仔血清(FCS)を含むRPMI培地にSAK細胞を再懸濁させた。細胞(2×105個/ウェル)を総量100μLで上部チャンバに添加し、100ng/mLのCXCL12又はIL-8、又は500ng/mLのIP-10を補充した600μLのRPMIを下部チャンバに添加した。4時間以内に下部コンパートメントに遊走する細胞の量をFACSによって求め、上部チャンバに流入した細胞の割合で表した。 Migration assays were performed to test the function of chemokine receptors on SAK cells. For this purpose, migration of SAK cells in response to the chemokines CXCL12, IP-10 or rhIL-8 (PeproTech EC) was assessed using 5 μm pore size Transwells (Costar). SAK cells were resuspended in RPMI medium containing 1% fetal calf serum (FCS). Cells (2× 105 cells/well) were added to the upper chamber in a total volume of 100 μL, and 600 μL of RPMI supplemented with 100 ng/mL CXCL12 or IL-8, or 500 ng/mL IP-10, was added to the lower chamber. The amount of cells migrating to the lower compartment within 4 h was determined by FACS and expressed as a percentage of cells entering the upper chamber.
図9Bに示す結果から、SAK細胞がケモカインIL-8、SDF-1及びIP-10に向かって遊走することが分かる。特に、試験した濃度では細胞の3%がIL-8に向かって遊走したが、細胞の約40%はSDF-1に向かって遊走し、約30%はIP-10に向かって遊走した。従って、SAK集団では、CXCR4及びCXCR3依存的な遊走がCXCR2依存的な遊走よりも顕著であると思われる。SAK細胞でのCXCR3とCXCR6の発現は、SAK細胞がCXCR3リガンドCXCL9及びCXCL11と、CXCR6リガンドCXCL16に向かって遊走する可能性があることも示す。 The results shown in Figure 9B indicate that SAK cells migrate toward the chemokines IL-8, SDF-1, and IP-10. Notably, at the concentrations tested, 3% of the cells migrated toward IL-8, whereas approximately 40% of the cells migrated toward SDF-1 and approximately 30% migrated toward IP-10. Thus, CXCR4- and CXCR3-dependent migration appears to be more prominent than CXCR2-dependent migration in the SAK population. Expression of CXCR3 and CXCR6 in SAK cells also indicates that SAK cells may migrate toward the CXCR3 ligands CXCL9 and CXCL11, and the CXCR6 ligand CXCL16.
要約すると、この結果は、SAK細胞が腫瘍組織、特にCXCR4、CXCR6及びCXCR6リガンドを発現する腫瘍に動員される能力を示す。更に興味深いのは、CXCR6等の特定のケモカイン受容体が腫瘍微小環境での細胞傷害性T細胞の生存と増殖に特に重要であることが示唆され、それによってT細胞の消耗が抑制されるという所見である。従って、特定の理論又は作用機序に束縛されたくはないが、SAK細胞でのCXCR6の発現は、表現型の活性化、及び腫瘍微小環境での生存及び増殖の可能性の向上を示すことがある。 In summary, the results demonstrate the ability of SAK cells to be recruited to tumor tissues, particularly tumors expressing CXCR4, CXCR6 and CXCR6 ligands. Of further interest is the finding that certain chemokine receptors, such as CXCR6, are suggested to be particularly important for the survival and proliferation of cytotoxic T cells in the tumor microenvironment, thereby inhibiting T cell exhaustion. Thus, without wishing to be bound by any particular theory or mechanism of action, expression of CXCR6 on SAK cells may indicate phenotypic activation and improved likelihood of survival and proliferation in the tumor microenvironment.
インビボでの養子移植
異種移植モデルを産生するために、NOD SCIDガンマ(NSG)マウス(成熟T細胞、B細胞及びNK細胞を欠く免疫不全マウス)を300cGyで照射した。24時間後、RAJI細胞(ヒトBリンパ芽球様株)を最終体積200μLで背側尾静脈を介して静脈内(iv)注射した(1×105個(細胞)/マウス)。
To generate the xenograft model, NOD SCID gamma (NSG) mice (immunodeficient mice lacking mature T, B and NK cells) were irradiated with 300 cGy. 24 hours later, RAJI cells (a human B lymphoblastoid line) were injected intravenously (iv) via the dorsal tail vein in a final volume of 200 μL (1 × 10 cells/mouse).
次に、マウスに20×106個のSAK細胞を静脈内(iv)注射し、5日目、6日目及び7日目に10μgのIL-2を腹腔内(ip)注射した。対照マウスにはPBSを静脈内(iv)注射し、10μgのIL-2を腹腔内(ip)注射した。マウスを12日目に屠殺し、分析用に骨髄(BM)と脾臓を採取した。これらの器官から細胞を単離し、抗ヒトCD20抗体(生着した腫瘍細胞を特異的に認識する)で染色し、BMと脾臓における生着細胞の割合をFACSによって評価した。結果を下の表6に示す。阻害%は100-(SAKマウスのCD20/対照マウスのCD20×100)で計算した。 Mice were then injected intravenously (iv) with 20x106 SAK cells and intraperitoneally (ip) with 10μg IL-2 on days 5, 6 and 7. Control mice were injected intravenously (iv) with PBS and intraperitoneally (ip) with 10μg IL-2. Mice were sacrificed on day 12 and bone marrow (BM) and spleen were harvested for analysis. Cells were isolated from these organs and stained with anti-human CD20 antibody (which specifically recognizes engrafted tumor cells) and the percentage of engrafted cells in BM and spleen was assessed by FACS. The results are shown in Table 6 below. % inhibition was calculated as 100-(CD20 in SAK mice/CD20 in control mice x 100).

表6に示すように、SAK細胞の養子移植は、骨髄と脾臓におけるRaji細胞の生着を有意に阻害し、特に顕著な有効性によって脾臓における腫瘍発生の約78%阻害をもたらした。 As shown in Table 6, adoptive transfer of SAK cells significantly inhibited engraftment of Raji cells in the bone marrow and spleen, with particularly remarkable efficacy resulting in approximately 78% inhibition of tumor development in the spleen.
上で詳述したように実験を繰り返したが、BM細胞及び脾臓細胞と共に、屠殺したNSGマウスから採取した血液を抗ヒトCD19抗体で染色した。結果を以下の表7に示すが、阻害%は100-(SAKマウスのCD19/対照マウスのCD19×100)で計算した。図10A~10Cで可視化したが、SAK細胞を脾臓(図10A)、BM(図10B)及び血液(図10C)に移植した後のNSGマウスに対する対照NSGマウスにおけるCD19+Raji細胞の割合を示す。 The experiment was repeated as detailed above, but blood drawn from sacrificed NSG mice was stained with anti-human CD19 antibody along with BM and spleen cells. The results are shown in Table 7 below, where % inhibition was calculated as 100-(CD19 in SAK mice/CD19 in control mice x 100). Visualized in Figures 10A-10C, the percentage of CD19 + Raji cells in NSG mice versus control NSG mice following transfer of SAK cells into the spleen (Figure 10A), BM (Figure 10B), and blood (Figure 10C) are shown.

表7に示すように、SAK細胞の養子移植によって、骨髄、血液及び脾臓におけるRaji細胞の生着が有意に阻害され、脾臓における腫瘍発生の約97%阻害をもたらす顕著な有効性が得られた。これらの知見から、SAK細胞のみが腫瘍の増殖と生着を有効に阻害できることが分かる。 As shown in Table 7, adoptive transfer of SAK cells significantly inhibited engraftment of Raji cells in bone marrow, blood, and spleen, and was highly effective in inhibiting tumor development in the spleen by approximately 97%. These findings indicate that only SAK cells can effectively inhibit tumor growth and engraftment.
遺伝子改変SAK
改良された遺伝子改変SAK細胞を以下に記載のように調製した。
Genetically modified SAK
The improved genetically modified SAK cells were prepared as described below.
核酸構築物の産生には、レトロウイルスMSGV-1D3-28Z All ITAMインタクト発現ベクター(#107226、Addgene社製)を以下の改変を加えて使用した。レトロウイルスベクター骨格であるpMSGV1は、マウス幹細胞ウイルス(MSCV)の末端反復配列を使用するMSCVベースのスプライスGAGベクター(pMSGV)の誘導体である。制限酵素NcoI及びSalIを使用して、プラスミドから「1D3-28Z All ITAMインタクト」インサートを除去した。以下に詳述のような様々なcDNAインサートをPCRによって増幅し、NcoI及びSalI制限部位を有するプラスミドに連結した。得られた構築物A~D(以下の表8に詳述)をDNA配列決定によって確認した。 For the production of nucleic acid constructs, the retroviral MSGV-1D3-28Z All ITAM intact expression vector (#107226, Addgene) was used with the following modifications. The retroviral vector backbone, pMSGV1, is a derivative of the murine stem cell virus (MSCV)-based splice GAG vector (pMSGV) that uses the MSCV long terminal repeat sequences. The "1D3-28Z All ITAM intact" insert was removed from the plasmid using restriction enzymes NcoI and SalI. The various cDNA inserts, as detailed below, were amplified by PCR and ligated into plasmids with NcoI and SalI restriction sites. The resulting constructs A-D (detailed in Table 8 below) were confirmed by DNA sequencing.

抗ヒトCD123CAR(構築物Aに含まれる)をコードする核酸配列は次の通りである。
ATGGCCCTGCCTGTGACAGCCCTGCTGCTGCCTCTGGCTCTGCTGCTGCATGCCGCTAGACCCGATATTGTCCTCACTCAATCGCCGGACTCACTGGCGGTGTCCCTCGGAGAGAGGGCGACGATCAATTGCCGGGCTTCCGAATCCGTCGATAACTACGGAAACACCTTTATGCACTGGTACCAACAGAAGCCAGGACAGCCACCAAAGCTGTTGATCTACCGCGCTTCAAACCTTGAGTCGGGTGTGCCGGACCGCTTCAGCGGCAGCGGTTCCAGAACCGACTTTACCCTCACCATCAGCTCGCTGCAGGCCGAAGATGTCGCCGTCTATTACTGCCAACAGAGCAACGAAGATCCGCCTACTTTCGGACAGGGGACTAAACTGGAAATCAAGGGCGGAGGAGGCTCGGGTGGAGGAGGATCGGGAGGAGGCGGGTCCGGTGGTGGCGGATCGCAAATCCAGCTGGTGCAGTCCGGCGCAGAAGTGAAGAAGCCGGGAGCGTCCGTGAAAGTGAGCTGCAAGGCCTCAGGGTACATCTTCACCAATTACGGCATGAATTGGGTGCGGCAGGCACCCGGACAGCGCCTGGAGTGGATGGGCTGGATCAACACTTACACCGGGGAAAGCACGTACTCGGCCGACTTCAAAGGACGGGTGACCATTACCCTGGATACCTCGGCCTCAACCGCTTACATGGAGCTCTCATCACTTAGATCCGAGGACACTGCCGTCTACTACTGTGCAAGGAGCGGAGGCTACGACCCTATGGACTATTGGGGACAAGGCACTACTGTGACTGTGTCGTCCACCACGACGCCAGCGCCGCGACCACCAACACCGGCGCCCACCATCGCGTCGCAGCCCCTGTCCCTGCGCCCAGAGGCGTGCCGGCCAGCGGCGGGGGGCGCAGTGCACACGAGGGGGCTGGACTTCGCCTGTGATATCTACATCTGGGCGCCCTTGGCCGGGACTTGTGGGGTCCTTCTCCTGTCACTGGTTATCACCCTTTACTGCTAA(配列番号1)。
The nucleic acid sequence encoding the anti-human CD123CAR (contained in construct A) is as follows:
ATGGCCCTGCCTGTGACAGCCCTGCTGCTGCCTCTGGCTCTGCTGCTGCTGCTGCATGCCGCTAGACCCGATATTGTCCTCACTCAATCGCCGGACTCACTGGCGGTGTCCCTCGGAGAGAGGGCGA CGATCAATTGCCGGGCTTCCGAATCCGTCGATAACTACGGAAACACCTTTATGCACTGGTACCAACAGAAGCCAGGACAGCCACCAAAGCTGTTGATCTACCGCGCTTCAAACCTTGAGTC GGGTGTGCCGGACCG CTTCAGCGGCAGCGGTTCCAGAACCGACTTTACCTCACCATCAGCTCGCTGCAGGCCGAAGATGTCGCCGTCTATTACTGCCAACAGAGCAACGAAGATCCGCCTACTTTCGGACAGGGG ACTAAAACTGGAAATCAAAGGGCGGAGGAGGCTCGGGTGGAGGAGGATCGGGAGGAGGCGGGTCCGGTGGTGGCGGATCGCAATCCAGCTGGTGCAGTCCGGCGCAGAAGTGAAGAAGCCG GAGCGTCCGTGAAAG TGAGCTGCAAGGCCTCAGGGTACATCTTCACCAATTACGGCATGAATTGGGTGCGGCAGGCACCCGGACAGCGCCTGGAGTGGATGGGCTGGATCAACACTTACACCGGGGAAAGCACGTA CTCGGCCGACTTCAAAGGACGGGTGACCATTACCCTGGATAACCTCGGCCTCAACCGCTTACATGGAGCTCTCACTCACTTAGATCCGAGGACACTGCCGTCTACTGTGCAAGGAGCGGA GGCTACGACCCTATG GACTATTGGGGACAAGGCACTACTGTGACTGTGTCGTCCCACCACGACGCCAGCGCCGCGACCACCAACACCGGCGCCCACCATCGCGTCGCAGCCCCTGTCCCTGCGCCCAGAGGCGTGCC GGCCAGCGGCGGGGGCGCAGTGCACACGAGGGGGCTGGACTTCGCCTGTGATATCTACATCTGGGCGCCCTTGGCCGGGACTTGTGGGTCCTTCTCCTGTCACTGGTTATCACCCTTTTA CTGCTAA (SEQ ID NO: 1).
IRES核酸配列(構築物A及びBに含まれる)は次の通りである。
GAATTCCCGGGTCGACCTGCAGAAGCTTAAAACAGCTCTGGGGTTGTACCCACCCCAGAGGCCCACGTGGCGGCTAGTACTCCGGTATTGCGGTACCCTTGTACGCCTGTTTTATACTCCCTTCCCGTAACTTAGACGCACAAAACCAAGTTCAATAGAAGGGGGTACAAACCAGTACCACCACGAACAAGCACTTCTGTTTCCCCGGTGATGTCGTATAGACTGCTTGCGTGGTTGAAAGCGACGGATCCGTTATCCGCTTATGTACTTCGAGAAGCCCAGTACCACCTCGGAATCTTCGATGCGTTGCGCTCAGCACTCAACCCCAGAGTGTAGCTTAGGCTGATGAGTCTGGACATCCCTCACCGGTGACGGTGGTCCAGGCTGCGTTGGCGGCCTACCTATGGCTAACGCCATGGGACGCTAGTTGTGAACAAGGTGTGAAGAGCCTATTGAGCTACATAAGAATCCTCCGGCCCCTGAATGCGGCTAATCCCAACCTCGGAGCAGGTGGTCACAAACCAGTGATTGGCCTGTCGTAACGCGCAAGTCCGTGGCGGAACCGACTACTTTGGGTGTCCGTGTTTCCTTTTATTTTATTGTGGCTGCTTATGGTGACAATCACAGATTGTTATCATAAAGCGAATTGGATTGCGGCCGC(配列番号2)。
The IRES nucleic acid sequence (contained in constructs A and B) is as follows:
GAATTCCCGGGTCGACCTGCAGAAGCTTAAAACAGCTCTGGGGTTGTACCCCACCAGAGGCCCACGTGGCGGCTAGTACTCCGGTATTGCGGTACCCTTGTACGCCTGTTTTATATACTCCC TTCCCGTAACTTAGACGCACAAAAACCCAAGTTCAATAGAAGGGGGTA CAAACCAGTACCACCACGAACAAGCACTTCTGTTTCCCCGGTGATGTCGTATAGACTGCTTGCGTGGTTGAAAGCGACGGATCCGTTATCCGCTTATGTACTTCGAGAAGCCCAGTACCAC CTCGGAATCTTCGATGCGTTGCGCTCAGCACTCAACCCCAGAGGTGT AGCTTAGGCTGATGAGTCTGGACATCCCTCACCGGTGACGGTGGTCCAGGCTGCGTTGGCGGCCTACCTATGGCTAACGCCATGGGACGCTAGTTGTGAACAAGGTGTGAAGAGCCCTATTG AGCTACATAAGAATCCTCCGGCCCCTGAATGCGGCTAATCCCAAACC TCGGAGCAGGTGGTCACAAACCAGTGATTGGCCTGTCGTAACGCGCAAGTCCGTGGCGGAACCGACTACTTTGGGTGTCCGTGTTTCCTTTTATTTTATTGTGGCTGCTTATGGTGACAAT CACAGATTGTTATCATAAAGCGAATTGGATTGCGGCCGC (SEQ ID NO: 2).
ヒトIL-2(受入番号BC066257、構築物A及びBに含まれる)の核酸配列は次の通りである。
ATGTACAGGATGCAACTCCTGTCTTGCATTGCACTAAGTCTTGCACTTGTCACAAACAGTGCACCTACTTCAAGTTCTACAAAGAAAACACAGCTACAACTGGAGCATTTACTGCTGGATTTACAGATGATTTTGAATGGAATTAATAATTACAAGAATCCCAAACTCACCAGGATGCTCACATTTAAGTTTTACATGCCCAAGAAGGCCACAGAACTGAAACATCTTCAGTGTCTAGAAGAAGAACTCAAACCTCTGGAGGAAGTGCTAAATTTAGCTCAAAGCAAAAACTTTCACTTAAGACCCAGGGACTTAATCAGCAATATCAACGTAATAGTTCTGGAACTAAAGGGATCTGAAACAACATTCATGTGTGAATATGCTGATGAGACAGCAACCATTGTAGAATTTCTGAACAGATGGATTACCTTTTGTCAAAGCATCATCTCAACACTGACTTGA(配列番号3)。
The nucleic acid sequence of human IL-2 (accession number BC066257, contained in constructs A and B) is as follows:
ATGTACAGGATGCAAACTCCTGTCTTGCATTGCACTAAGTCTTGCACTTGTCACAAACAGTGCACCTACTTCAAGTTCTACAAAGAAAACACAGCTACAACTGGAGCATTTACTGCTGGATT TACAGATGATTTTGAATGGAATTAATAATTACAAGAATCCCAAACTCACCAGGATGCTCACATTTAAGTTTTACATGCCCAAGAAGGCCACAGAACTGAAACATCTTCAGTGTC TAGAAGAAGAACTCAAACCTCTGGAGGAAGTGCTAAATTTAGCTCAAAGCAAAAACTTTTCACTTAAGACCCAGGGACTTAATCAGCAATATCGTAATAGTTCTGGAACTAAAGGGATC TGAAACAACATTCATGTGTGAATATGCTGATGAGACAGCAACCATTGTAGAATTTCTGAACAGATGGATTACCTTTTGTCAAGCATCATCTCAACACTGACTTGA (SEQ ID NO: 3).
ヒトIL-15(受入番号BC100961.1、構築物Cに含まれる)の核酸配列は以下の通りである。
ATGAGAATTTCGAAACCACATTTGAGAAGTATTTCCATCCAGTGCTACTTGTGTTTACTTCTAAACAGTCATTTTCTAACTGAAGCTGGCATTCATGTCTTCATTTTGGGCTGTTTCAGTGCAGGGCTTCCTAAAACAGAAGCCAACTGGGTGAATGTAATAAGTGATTTGAAAAAAATTGAAGATCTTATTCAATCTATGCATATTGATGCTACTTTATATACGGAAAGTGATGTTCACCCCAGTTGCAAAGTAACAGCAATGAAGTGCTTTCTCTTGGAGTTACAAGTTATTTCACTTGAGTCCGGAGATGCAAGTATTCATGATACAGTAGAAAATCTGATCATCCTAGCAAACAACAGTTTGTCTTCTAATGGGAATGTAACAGAATCTGGATGCAAAGAATGTGAGGAACTGGAGGAAAAAAATATTAAAGAATTTTTGCAGAGTTTTGTACATATTGTCCAAATGTTCATCAACACTTCT(配列番号4)。
The nucleic acid sequence of human IL-15 (accession number BC100961.1, contained in construct C) is as follows:
ATGAGAATTTCGAAACCACATTTGAGAAGTATTTCCATCCAGTGCTACTTGTGTTTACTTCTAAACAGTCATTTTCTAACTGAAGCTGGCATTCATGTCTTCATTTTGGGCTGTTTCAGTG CAGGGCTTCCTAAAACAGAAGCCAACTGGGTGAATGTAATAAGTGATTTGAAAAAAAATTGAAGATCTTATTCAATCTATTGCATATTGATGCTACTTTATACGGAAAGTGATGTTCACCC CAGTT GCAAAGTAACAGCAAGTGCTTTCTCTTGGAGTTACAAGTTATTTCACTTGAGTCCGGAGATGCAAGTATTCATGATACAGTAGAAATCTGATCATCCTAGCAAACAAGTTTGTC TTCTAATGGGAATGTAACAGAATCTGGATGCAAAGAATGTGAGGAACTGGAGGGAAAAATATTAAAGAATTTTTGCAGAGTTTGTACATATTGTCCAAATGTTCATCAACACTTCT (SEQ ID NO. 4).
2A要素(構築物Cに含まれる)の核酸配列は次の通りである。
GAGGGCAGGGGAAGTCTACTAACATGCGGGGACGTGGAGGAAAATCCCGGCCCA(配列番号5)。
The nucleic acid sequence of the 2A element (contained in construct C) is as follows:
GAGGGCAGGGGAAGTCTAACTAACATGCGGGGACGTGGAGGAAAATCCCGGCCCA (SEQ ID NO:5).
GFP(構築物B及びCに含まれる)の核酸配列は次の通りである。
ATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAA(配列番号6)。
The nucleic acid sequence of GFP (contained in constructs B and C) is as follows:
ATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACG GCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCCACCCT CGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTT AAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGA ACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCAT CAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCAC TACCAGCAGAACACCCCCATCGGCGACGGCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCCAACGAGAAGCGCGATCCACATGGTCCTGCTGG AGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAA (SEQ ID NO: 6).
野生型CXCR4ケモカイン受容体は次のアミノ酸配列を有する。
MSIPLPLLQIYTSDNYTEEMGSGDYDSMKEPCFREENANFNKIFLPTIYSIIFLTGIVGNGLVILVMGYQKKLRSMTDKYRLHLSVADLLFVITLPFWAVDAVANWYFGNFLCKAVHVIYTVNLYSSVLILAFISLDRYLAIVHATNSQRPRKLLAEKVVYVGVWIPALLLTIPDFIFANVSEADDRYICDRFYPNDLWVVVFQFQHIMVGLILPGIVILSCYCIIISKLSHSKGHQKRKALKTTVILILAFFACWLPYYIGISIDSFILLEIIKQGCEFENTVHKWISITEALAFFHCCLNPILYAFLGAKFKTSAQHALTSVSRGSSLKILSKGKRGGHSSVSTESESSSFHSS(配列番号7)。改変C’切断CXCR4(配列番号7の19個のC末端アミノ酸が除去されているWHIM R334x変異、NM_001008540.2、構築物Dに含まれる)をコードする核酸配列は次の核酸配列を有する。
ATGTCCATTCCTTTGCCTCTTTTGCAGATATACACTTCAGATAACTACACCGAGGAAATGGGCTCAGGGGACTATGACTCCATGAAGGAACCCTGTTTCCGTGAAGAAAATGCTAATTTCAATAAAATCTTCCTGCCCACCATCTACTCCATCATCTTCTTAACTGGCATTGTGGGCAATGGATTGGTCATCCTGGTCATGGGTTACCAGAAGAAACTGAGAAGCATGACGGACAAGTACAGGCTGCACCTGTCAGTGGCCGACCTCCTCTTTGTCATCACGCTTCCCTTCTGGGCAGTTGATGCCGTGGCAAACTGGTACTTTGGGAACTTCCTATGCAAGGCAGTCCATGTCATCTACACAGTCAACCTCTACAGCAGTGTCCTCATCCTGGCCTTCATCAGTCTGGACCGCTACCTGGCCATCGTCCACGCCACCAACAGTCAGAGGCCAAGGAAGCTGTTGGCTGAAAAGGTGGTCTATGTTGGCGTCTGGATCCCTGCCCTCCTGCTGACTATTCCCGACTTCATCTTTGCCAACGTCAGTGAGGCAGATGACAGATATATCTGTGACCGCTTCTACCCCAATGACTTGTGGGTGGTTGTGTTCCAGTTTCAGCACATCATGGTTGGCCTTATCCTGCCTGGTATTGTCATCCTGTCCTGCTATTGCATTATCATCTCCAAGCTGTCACACTCCAAGGGCCACCAGAAGCGCAAGGCCCTCAAGACCACAGTCATCCTCATCCTGGCTTTCTTCGCCTGTTGGCTGCCTTACTACATTGGGATCAGCATCGACTCCTTCATCCTCCTGGAAATCATCAAGCAAGGGTGTGAGTTTGAGAACACTGTGCACAAGTGATTTCCATCACCGAGGCCCTAGCTTTCTTCCACTGTTGTCTGAACCCCATCCTCTATGCTTTCCTTGGAGCCAAATTTAAAACCTCTGCCCAGCACGCACTCACCTCTGTGAGCAGAGGGTCCAGCCTCAAGATCCTCTCCAAAGGAAAGTAA(配列番号8)。
The wild-type CXCR4 chemokine receptor has the following amino acid sequence:
MSIPLPLLQIYTSDNYTEEMGSGDYDSMKEPCFREENANFNKIFLPTIYSIIFLTGIVGNGLVILVMGYQKKLRSMTDKYRLHLSVADLLFVITLPFWAVDAVANWYFGNFLCKAVHVHVIYT VNLYSSVLILAFISLDRYLAIVHATNSQRPRKLLAEKVVYVGVWIPALLLTIPDFIFANVS The nucleic acid sequence encoding the modified C' truncated CXCR4 (WHIM R334x mutation in which the 19 C-terminal amino acids of SEQ ID NO:7 have been removed, contained in NM_001008540.2, construct D) has the following nucleic acid sequence:
ATGTCCATTCCTTTGCCTCTTTTGCAGATATACACTTCAGATAACTACACCGAGGAAATGGGCTCAGGGGACTATGACTCCATGAAGGAACCCTGTTTCCGTGAAGAAAATGCTAATTTCA ATAAAATCTTCCTGCCCACCATCTACTCCATCATCTTCTTAACTGGCATTGTGGGCAATGGATTGGTCATCCTGGTCATGGGTTACCAGAAGAAACTGAGAAGCATGACGGACAAGTACAG GCTGCACCTGTCA GTGGCCGACCTCCTCTTTGTCATCACGCTTCCCTTCTGGGCAGTTGATGCCGTGGCAAACTGGTACTTTGGGAACTTCCTATGCAAGGCAGTCCATGTCATCTACACAGTCAACCTCTACA GCAGTGTCCTCATCCTGGCCTTCATCAGTCTGGACCGCTACCTGGCCATCGTCCACGCCACCAACAGTCAGAGGCCAAGGAAGCTGTTGGCTGAAAAGGTGGTCTATGTTGGCGTCTGGAT CCCTGCCCTCCTG CTGACTATTCCCGACTTCATCTTTGCCAACGTCAGTGAGGCAGATGACAGATATATCTGTGACCGCTTCTACCCCAATGACTTGTGGGTGGTTGTGTTCCAGTTTCAGCACATCATGGTTG GCCTTATCCTGCCTGGTATTGTCATCCTGTCCTGCTATTGCATTATCCATCCAAGCTGTCACACTCCAAGGGCCACCAGAAGCGCAAGGCCCTCAAGACCACAGTCATCCTCCATCCTGGC TTTCTTCGCCTGT TGGCTGCCTTACTACATTGGGATCAGCATCGACTCCTTCATCCTCCTGGAAATCATCAAGCAAGGGTGTGAGTTTGAGAACACTGTGCACAAGTGATTTCCATCACCGAGGCCCTAGCTTT CTTCCACTGTTGTCTGAACCCCATCCTCTATGCTTTCCTTGGAGCCAAATTTAAAACCTCTGCCCAGCACGCACTCACCTCTGTGAGCAGAGGGTCCAGCCTCAAAGATCCTCTCCAAAGGA AAGTAA (SEQ ID NO: 8).
形質導入用のレトロウイルス粒子を調製するために、eco-phoenix細胞を5×105個(細胞)の密度で6ウェルプレートに接種し、感染当日に60~70%のコンフルエンスまで到達させた。製造業者のプロトコルに従って、リポフェクタミントランスフェクション試薬を使用し、37℃、5%CO2で5時間、レトロウイルスプラスミドMSGV(試験した構築物を含む)2μg/ウェルをEco-phoenix細胞にトランスフェクトした。48時間後、レトロウイルス粒子を含む上清を回収し、0.45μmフィルター(米国マサチューセッツ州、ビルリカ、EMD Millipore社製)で濾過して細胞片を除去した。上清を使用してPG13細胞を形質導入し、PG13ベースのパッケージング細胞株を産生し、改変MSGVウイルスを産生した。 To prepare retroviral particles for transduction, eco-phoenix cells were seeded at a density of 5x105 cells in 6-well plates and allowed to reach 60-70% confluence on the day of infection. Eco-phoenix cells were transfected with 2μg/well of retroviral plasmid MSGV (containing the tested construct) for 5 hours at 37°C, 5% CO2 using Lipofectamine transfection reagent according to the manufacturer's protocol. After 48 hours, the supernatant containing retroviral particles was harvested and filtered through a 0.45μm filter (EMD Millipore, Billerica, MA, USA) to remove cell debris. The supernatant was used to transduce PG13 cells to produce a PG13-based packaging cell line and produce the modified MSGV virus.
PG13形質導入は、RetroNectin(登録商標)(Takara Shuzo社製)によって媒介させた。非組織培養24ウェルプレートをRetroNectin(登録商標)(PBSで1:40に希釈)でコーティングした。プレートを4℃で一晩コーティングした後、2%BSAで室温(RT)にて30分間ブロックし、使用前にPBSで1回洗浄した。トランスフェクトしたeco-phoenix細胞由来のウイルス含有上清を、RetroNectin(登録商標)コーティングプレートに添加し、2時間遠心分離した。次の工程では、PG13細胞をウェルに添加し、ウイルスと共に24時間インキュベートした。3日後、陽性PG13細胞(FACSによって検出)を選択し、増殖させた。 PG13 transduction was mediated by RetroNectin® (Takara Shuzo). Non-tissue culture 24-well plates were coated with RetroNectin® (diluted 1:40 in PBS). Plates were coated overnight at 4°C, then blocked with 2% BSA for 30 min at room temperature (RT) and washed once with PBS before use. Virus-containing supernatants from transfected eco-phoenix cells were added to RetroNectin®-coated plates and centrifuged for 2 h. In the next step, PG13 cells were added to the wells and incubated with the virus for 24 h. After 3 days, positive PG13 cells (detected by FACS) were selected and expanded.
得られたウイルス粒子を使用して、基本的には実施例1に記載のように調製したSAK細胞を増殖方法中に形質導入した。具体的には、新たに単離したPBMCを、30ng/mLのOKT3と1000IU/mLのIL-2の存在下で48時間刺激した。刺激した後、上述のようにレトロウイルスベクターでプレコートした培養皿に移すことによって、レトロウイルスベクター(上述のようにPG13細胞によって産生する)を細胞に形質導入した。この目的のために、非組織培養24ウェルプレートをRetroNectin(登録商標)でコーティングし、ウイルス含有上清(PG13由来)をRetroNectin(登録商標)コーティングプレートに添加し、2時間遠心分離した。次の工程では、刺激した細胞をウェルに添加し、ウイルス粒子と共に24時間インキュベートした。その後、実施例1に記載のように、30ngの抗CD3と1000IU/mLのIL-2を用いて細胞を更に増殖させた。APC抗CXCR4抗体、CD123-Fc及びPE抗Fc抗体を使用したFACS、又はGFP分析によって陽性(構築物含有)細胞を検出及び定量し、対応する外因性遺伝子を発現することを確認した。 The obtained viral particles were used to transduce SAK cells during the expansion method, which were prepared essentially as described in Example 1. Specifically, freshly isolated PBMCs were stimulated for 48 hours in the presence of 30 ng/mL OKT3 and 1000 IU/mL IL-2. After stimulation, the cells were transduced with the retroviral vector (produced by PG13 cells as described above) by transferring them to culture dishes precoated with the retroviral vector as described above. For this purpose, non-tissue culture 24-well plates were coated with RetroNectin®, and the virus-containing supernatant (from PG13) was added to the RetroNectin®-coated plates and centrifuged for 2 hours. In the next step, the stimulated cells were added to the wells and incubated with the viral particles for 24 hours. The cells were then further expanded with 30 ng anti-CD3 and 1000 IU/mL IL-2 as described in Example 1. Positive (construct-containing) cells were detected and quantified by FACS using APC anti-CXCR4 antibody, CD123-Fc and PE anti-Fc antibodies, or GFP analysis, confirming expression of the corresponding exogenous gene.
同様に、構築物A及びDを2A配列と共にプラスミド骨格にクローン化し、CD123特異的CAR、改変CXCR4受容体、及びIL-2(抗CD123-IRES-IL-2-2A-CXCR4whim)を構成的に発現することができる核酸構築物を産生する。結合した構築物を上述のようにSAK細胞に形質導入する。同様に、構築物CXCR4whim-IRES-IL-2を産生し、新たに調製したSAK細胞に形質導入する。 Similarly, constructs A and D are cloned into a plasmid backbone along with the 2A sequence to produce a nucleic acid construct capable of constitutively expressing a CD123-specific CAR, a modified CXCR4 receptor, and IL-2 (anti-CD123-IRES-IL-2-2A-CXCR4whim). The combined construct is transduced into SAK cells as described above. Similarly, construct CXCR4whim-IRES-IL-2 is produced and transduced into freshly prepared SAK cells.
基本的にはそれぞれ実施例2及び7に記載のように、様々な構築物を形質導入した細胞をインビトロ及びインビボでの抗腫瘍活性についてアッセイする。 Cells transduced with the various constructs will be assayed for antitumor activity in vitro and in vivo essentially as described in Examples 2 and 7, respectively.
CD19特異的CARを発現する遺伝子改変SAK細胞の細胞傷害活性
CD19CARを発現する遺伝子改変SAK細胞(CD19発現B細胞悪性腫瘍を認識するように改変)を基本的には実施例8に記載のように調製し、次のように変更した。
Cytotoxic Activity of Genetically Modified SAK Cells Expressing CD19-Specific CAR Genetically modified SAK cells expressing CD19 CAR (modified to recognize CD19-expressing B-cell malignancies) were prepared essentially as described in Example 8 with the following modifications.
scFv標的ドメイン(CD19に特異的)、CD8由来ヒンジ領域、CD8由来膜貫通領域、4-1BB/CD137共刺激ドメイン、及びCD3ζ鎖細胞内シグナル伝達ドメインを発現するようにベクターを操作した。核酸構築物の産生には、CD19CAR配列を有するレトロウイルスMSGV-1D3-28Z All ITAMインタクト発現ベクターを使用した。制限酵素NcoI及びSalIを使用して、プラスミドから「1D3-28Z All ITAMインタクト」インサートを除去した。CD19CARcDNAをPCRによって増幅し、NcoI及びSalI制限部位を有するプラスミドに連結した。得られた構築物をDNA配列決定によって確認した。 The vector was engineered to express the scFv targeting domain (specific for CD19), the CD8-derived hinge region, the CD8-derived transmembrane region, the 4-1BB/CD137 costimulatory domain, and the CD3 ζ chain intracellular signaling domain. The retroviral MSGV-1D3-28Z All ITAM intact expression vector carrying the CD19CAR sequence was used to generate the nucleic acid construct. The "1D3-28Z All ITAM intact" insert was removed from the plasmid using restriction enzymes NcoI and SalI. The CD19CAR cDNA was amplified by PCR and ligated into a plasmid carrying NcoI and SalI restriction sites. The resulting construct was verified by DNA sequencing.
CD19CAR(センス鎖)をコードする核酸配列は次の通りである。
gccaccatggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgctcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgagactctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagctggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtagcgaaaccacttactataactcttccctgaagtcacgggtcaccatttcaaaggataactcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgccgtgtattactgtgccaagcattactactatggagggtcctacgccatggactactggggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcgggagcggtggaggtggctccggaggtggcggaagcgaaatcgtgatgacccagagccctgcaaccctgtccctttctcccggggaacgggctaccctttcttgtcgggcatcacaagatatctcaaaatacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatctaccacacctctcgcctgcatagcgggattcccgcacgctttagcgggtctggaagcgggaccgactacactctgaccatctcatctctccagcccgaggacttcgccgtctacttctgccagcagggtaacaccctgccgtacaccttcggccagggcaccaagcttgagatcaaaaccactactcccgctccaaggccacccacccctgccccgaccatcgcctctcagccgctttccctgcgtccggaggcatgtagacccgcagctggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttgggcccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcctgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggaggaaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagtacgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctcgg(配列番号9)。
The nucleic acid sequence encoding CD19CAR (sense strand) is as follows:
gccaccatggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgctcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgagactc tgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagctggattagaca gcctcccggaaagggactggagtggatcggagtgatttggggtagcgaaaccacttactataactctcttccctgaagtcacgggtcaccatttcaaaggataactcaaagaatcaagtgagc ctcaagctctcatcagtcaccgccgctgacaccgccgtgtattactgtgccaagcattactacta tggaggtcctacgccatggactactgggggccaggaactctggtcactgtgtcatctggtggaggaggtagcggaggagcgggagcggtggaggtggctccggaggtggcggaagcgaa atcgtgatgacccagagccctgcaacctgtccctttctcccgggaacggggctaccctttcttg tcggcatcacaagatatctcaaaatacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatccacacctctcgcctgcatagcgggattcccgcacgctttagc gggtctggaagcgggaccgactacactctgaccatctcatctctccagcccgaggacttcgccgt ctacttctgccagcaggtaacaccctgccgtacaccttcggccaggcaccaagcttgagatcaaaaccactactcccgctccaaggccacccacccctgccccgaccatcgcctctcag ccgctttccctgcgtccggaggcatgtagacccgcagctggtgggggccgtgcatacccggggtct tgacttcgcctgcgatatctacttgggcccctctggctggtacttgcgggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcggaagaagctgctgtacatcttt aagcaacccttcatgaggcctgtgcagactactcaagaggaggacggctgttcatgccggttccc agaggaggaggaaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggag tacgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaa tccccaagagggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccaggactc agcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctcgg (SEQ ID NO: 9).
形質導入用のレトロウイルス粒子の調製は、実施例8に記載のように行った。陽性(構築物含有)細胞の検出及び定量化は、CD19-FcとPE抗Fc抗体を使用したFACS分析によって行い、CD19CAR遺伝子を発現することを確認した。 Preparation of retroviral particles for transduction was performed as described in Example 8. Detection and quantification of positive (construct-containing) cells was performed by FACS analysis using CD19-Fc and PE anti-Fc antibodies, confirming expression of the CD19CAR gene.
CD19陽性細胞(Toledo)とCD19陰性細胞(U937)に対するCD19CAR SAK細胞の細胞傷害活性を対照SAK細胞と比較して測定した。CFSEベースの細胞傷害性アッセイは、エフェクター細胞としてSAK及びCD19CAR SAK細胞を使用し、標的細胞としてToledo及びU937細胞を使用して、基本的には実施例2に記載のように行った。CD19+(Toledo)細胞とCD19-(U937)細胞についての結果をそれぞれ図11Aと11Bに示すが、各比率における3回の平均±標準偏差を示す。 The cytotoxic activity of CD19CAR SAK cells against CD19 positive cells (Toledo) and CD19 negative cells (U937) was measured in comparison with control SAK cells. CFSE-based cytotoxicity assays were performed essentially as described in Example 2 using SAK and CD19CAR SAK cells as effector cells and Toledo and U937 cells as target cells. The results for CD19 + (Toledo) and CD19 - (U937) cells are shown in Figures 11A and 11B, respectively, and represent the mean ± standard deviation of triplicates at each ratio.
図11Aに示すように、CD19CAR SAK細胞は、対照SAK細胞よりもToledo細胞(CD19陽性細胞)を根絶する上で有意に有効であることが分かった。 As shown in FIG. 11A, CD19CAR SAK cells were found to be significantly more effective at eradicating Toledo cells (CD19 positive cells) than control SAK cells.
CD19CAR SAK細胞は、試験した各々のE:T比率において優れた細胞傷害活性を示した。例えば、E:T比率が1:1の場合、CD19CAR SAK細胞は93%の効率を示したが、同じ比率における対照SAK細胞の効率は54%に過ぎなかった。これに対し、U937細胞に対するCD19CAR SAKと対照SAKの細胞傷害活性は同一であり、これは、CD19CAR細胞の活性の増強がCD19発現腫瘍細胞に特異的であることを意味する。 CD19CAR SAK cells showed excellent cytotoxic activity at each E:T ratio tested. For example, at an E:T ratio of 1:1, CD19CAR SAK cells showed 93% efficiency, whereas control SAK cells at the same ratio showed only 54% efficiency. In contrast, the cytotoxic activity of CD19CAR SAK and control SAK against U937 cells was identical, meaning that the enhanced activity of CD19CAR cells is specific to CD19-expressing tumor cells.
これらの結果から、CAR SAKが従来のCAR T細胞の有効な代替物となり得ること、特に従来のCAR T細胞では治療効果が乏しい固形腫瘍に対して有効となり得ることが分かる。 These results indicate that CAR SAK can be an effective alternative to conventional CAR T cells, particularly for solid tumors for which conventional CAR T cells have poor therapeutic effects.
インビボのアロ反応性
SAK細胞が移植片対宿主(GVH)反応と移植片対宿主病(GVHD)を誘発する可能性を特徴付けるため、NSGマウスにSAK細胞又はPBMCを注射し、GVHDの発症を1ヶ月間モニターした。この目的のため、先ずNSGマウスを300cGyで照射した。24時間後、SAK細胞又はPBMC(ヒト血液から精製)を最終体積200μLで背側尾静脈を介して静脈内(iv)注射した(マウス1匹当たり5×106個の細胞)。急性GVHD臨床スコアを週に3回計算した。次の4種のパラメータ(姿勢、活動性、毛皮の質感、及び皮膚の完全性)の各々を0(存在しない場合)又は2(存在する場合)でスコア化した。マウス体重も週に3回測定した。結果を図12A~12Cに示す。
In vivo alloreactivity To characterize the potential of SAK cells to induce graft-versus-host (GVH) responses and graft-versus-host disease (GVHD), NSG mice were injected with SAK cells or PBMCs and monitored for the development of GVHD for one month. For this purpose, NSG mice were first irradiated with 300 cGy. 24 hours later, SAK cells or PBMCs (purified from human blood) were injected intravenously (iv) via the dorsal tail vein in a final volume of 200 μL (5× 106 cells per mouse). Acute GVHD clinical scores were calculated three times a week. Each of the following four parameters (posture, activity, fur texture, and skin integrity) was scored as 0 (if absent) or 2 (if present). Mouse weights were also measured three times a week. The results are shown in Figures 12A-12C.
注射後約15日で、PBMCを投与したマウスはGVHDの臨床徴候を示し始めた(図12C)。マウスは毛づくろいがされていないため、汚らしく見えた。体重も減り始めた。PBMCで処理した全てのマウスは重度の急性GVHDを発症し、17日以内に死亡した(図12A)。対照的に、SAK細胞で処理したマウスではGVHDの徴候が最小限に抑えられ、全てのマウスが生存した。これらのマウスは、PBMC群と比較して体重減少が軽度であった(図12B)。 Approximately 15 days after injection, mice receiving PBMCs began to show clinical signs of GVHD (Figure 12C). The mice were not groomed and looked dirty. They also began to lose weight. All mice treated with PBMCs developed severe acute GVHD and died within 17 days (Figure 12A). In contrast, mice treated with SAK cells showed minimal signs of GVHD and all survived. These mice experienced milder weight loss compared to the PBMC group (Figure 12B).
これらの結果は、組織適合性のない対象の治療を含め、臨床における同種異系使用へのSAK細胞の適合性を示すと共に、SAK細胞が既存の養子細胞移植治療プロトコルに対する有効な改善となり得ることを示す。 These results demonstrate the suitability of SAK cells for allogeneic clinical use, including treatment of histoincompatible subjects, and suggest that SAK cells may be a useful improvement to existing adoptive cell transfer treatment protocols.
凍結保存したSAK細胞の細胞障害性の評価
SAK細胞を市販の治療製品として使用するには、その産生及び凍結方法を臨床使用前に有利に評価する必要がある。解凍したSAK細胞の細胞傷害活性を評価するため、凍結24時間後に該細胞を回収し、MV411及びTHP-1白血病標的細胞と様々な比率で共培養した。
Evaluation of Cytotoxicity of Cryopreserved SAK Cells For SAK cells to be used as a commercial therapeutic product, their production and freezing methods must be advantageously evaluated prior to clinical use. To evaluate the cytotoxic activity of thawed SAK cells, the cells were harvested 24 hours after freezing and co-cultured with MV411 and THP-1 leukemia target cells at various ratios.
この目的のため、増殖11日目の新鮮なSAK細胞(実施例1に記載)を冷凍培地(90%ウシ胎仔血清(FCS)+10%ジメチルスルホキシド(DMSO)を含む)中で凍結させ、液体窒素に移して凍結保存させた。得られた凍結保存SAK細胞を解凍24時間後に細胞障害性アッセイに使用し、11日目の新鮮なSAK細胞(凍結とその後の解凍を経ていない)の細胞障害活性と比較した。結果を図13A~13Bに示す。 For this purpose, fresh SAK cells (described in Example 1) at day 11 of growth were frozen in freezing medium (containing 90% fetal calf serum (FCS) + 10% dimethyl sulfoxide (DMSO)) and transferred to liquid nitrogen for cryopreservation. The resulting cryopreserved SAK cells were used in cytotoxicity assays 24 hours after thawing and compared to the cytotoxic activity of fresh SAK cells at day 11 (which had not been subjected to freezing and subsequent thawing). The results are shown in Figures 13A-13B.
図13A及び13Bに示すように、解凍したSAKは新鮮なSAKと比較して細胞傷害活性が低下したが、これは、凍結融解サイクルがSAK細胞の生存率及び/又は活性にある程度影響を及ぼし得ることを示す。しかし、凍結保存されたSAK細胞は、E:T比率が低くてもMV411(図13B)及びTHP-1(図13A)細胞に対して有意且つ強力な細胞傷害活性を示し、対応する新鮮なSAK細胞の細胞傷害活性の少なくとも約70~80%を保持した。 As shown in Figures 13A and 13B, thawed SAK had reduced cytotoxic activity compared to fresh SAK, indicating that freeze-thaw cycles may affect the viability and/or activity of SAK cells to some extent. However, cryopreserved SAK cells showed significant and potent cytotoxic activity against MV411 (Figure 13B) and THP-1 (Figure 13A) cells even at low E:T ratios, retaining at least about 70-80% of the cytotoxic activity of the corresponding fresh SAK cells.
従って、この結果から、SAK細胞が調製直後だけでなく、凍結保存後も使用できる可能性があることが分かる。更に、細胞組成物は、追加の増殖又は操作の工程を必要とせずに、解凍直後に非常に強力な細胞障害性を示すことが本明細書で実証されている。従って、SAK細胞を含む細胞組成物は、貯蔵寿命の長い安定した製品として製造及び流通することができ、これは凍結保存によって容易になる。これは、凍結保存に対して感受性が高いことが報告されているNK細胞等の他の免疫細胞集団に基づく特定の細胞療法とは対照的であり、固形腫瘍の臨床治療における多数の失敗に関連していることが示唆される特性である(Mark et al., Nat Commun 11, 5224 (2020))。 The results therefore indicate that SAK cells may be used not only immediately after preparation, but also after cryopreservation. Furthermore, it is demonstrated herein that the cell composition exhibits very potent cytotoxicity immediately after thawing, without the need for additional growth or manipulation steps. Thus, cell compositions containing SAK cells can be manufactured and distributed as stable products with long shelf lives, which is facilitated by cryopreservation. This is in contrast to certain cell therapies based on other immune cell populations, such as NK cells, which have been reported to be highly sensitive to cryopreservation, a property suggested to be associated with numerous failures in the clinical treatment of solid tumors (Mark et al., Nat Commun 11, 5224 (2020)).
SAK細胞での免疫チェックポイント分子の発現
CD8+SAK細胞での免疫チェックポイント分子の存在を次のように評価した。実施例1に記載のように調製したSAK細胞を増殖11日目に回収し、洗浄し、PE標識抗Tim-3抗体(図14上部パネル、左)、APC標識抗CTLA-4抗体(図14上部パネル、右)、APC標識抗PD-1抗体(図14中央パネル、左)、APC標識抗TIGIT抗体(図14中央パネル、右)又はPE標識抗Lag-3抗体(図14下部パネル、左)のいずれかで4℃にて30分間染色し、フローサイトメトリーによって分析した。APC又はPE標識抗CD8モノクローナル抗体(Biolegend社製)とアイソタイプ対照抗体も使用した。抑制性チェックポイント分子Tim-3、CTLA-4、PD-1、TIGIT及びLag-3に加えて、活性化受容体DNAM-1(図14下部パネル、右)の存在もSAK細胞上で評価した。
Expression of immune checkpoint molecules in SAK cells The presence of immune checkpoint molecules in CD8 + SAK cells was evaluated as follows. SAK cells prepared as described in Example 1 were harvested on day 11 of growth, washed, and stained with either PE-labeled anti-Tim-3 antibody ( FIG. 14 , upper panel, left), APC-labeled anti-CTLA-4 antibody ( FIG. 14 , upper panel, right), APC-labeled anti-PD-1 antibody ( FIG. 14 , center panel, left), APC-labeled anti-TIGIT antibody ( FIG. 14 , center panel, right), or PE-labeled anti-Lag-3 antibody ( FIG. 14 , lower panel, left) for 30 minutes at 4° C., and analyzed by flow cytometry. APC- or PE-labeled anti-CD8 monoclonal antibody (Biolegend) and isotype control antibody were also used. In addition to the inhibitory checkpoint molecules Tim-3, CTLA-4, PD-1, TIGIT and Lag-3, the presence of the activating receptor DNAM-1 (FIG. 14 lower panel, right) was also assessed on SAK cells.
図14に示すように、注目すべきは、実質的に全てのCD8+細胞が活性化受容体DNAM-1を発現することが分かった。これに対し、抑制性免疫チェックポイント分子は検出不可能であるか、又は検出されても低いレベルであった。特に、SAK細胞はCTLA-4を全く発現せず、発現したPD-1とTIGITのレベルが低かったが、これは、対応するリガンドを発現する腫瘍による阻害の可能性が低いことを示す。 Notably, as shown in Figure 14, virtually all CD8 + cells were found to express the activating receptor DNAM-1. In contrast, inhibitory immune checkpoint molecules were either undetectable or detected at low levels. Notably, SAK cells did not express any CTLA-4 and expressed low levels of PD-1 and TIGIT, indicating a low likelihood of inhibition by tumors expressing the corresponding ligands.
異なるドナーの試料中の様々な表面マーカーを発現する細胞の割合を定量すると、CD8+細胞の99%~100%がDNAM-1を発現し、細胞の約0%がCTLA-4を発現し、細胞の12%~15%がPD-1を発現、細胞の20%~22%がTIGITを発現することが分かった。Lag-3とTim-3の表面発現は、それぞれCD8+細胞の約51%~63%及び54%~97%で見られた。 Quantification of the percentage of cells expressing various surface markers in samples from different donors revealed that 99%-100% of CD8 + cells expressed DNAM-1, approximately 0% of cells expressed CTLA-4, 12%-15% of cells expressed PD-1, and 20%-22% of cells expressed TIGIT. Surface expression of Lag-3 and Tim-3 was found in approximately 51%-63% and 54%-97% of CD8 + cells, respectively.
次に、SAK細胞上のチェックポイント分子の発現をCIK細胞上のチェックポイント分子の発現と比較した。基本的には実施例1に記載したように、同じドナーから組成物を調製し、細胞を11日目に回収し、上述したようにフローサイトメトリーによる表現型分析に供した。結果を以下の表9に示すが、各細胞型における各表示マーカーの染色時に測定された平均蛍光強度(MFI)を示すと共に、CIK細胞と比較したSAK細胞における相対発現(倍数増加、SAK細胞のMFI/CIK細胞のMFI、「SAK/CIK」と表示)を示す。 Next, expression of checkpoint molecules on SAK cells was compared to expression of checkpoint molecules on CIK cells. Compositions were prepared from the same donors essentially as described in Example 1, and cells were harvested on day 11 and subjected to phenotypic analysis by flow cytometry as described above. The results are shown in Table 9 below, which shows the mean fluorescence intensity (MFI) measured upon staining for each indicated marker in each cell type, as well as the relative expression in SAK cells compared to CIK cells (fold increase, MFI of SAK cells/MFI of CIK cells, denoted as "SAK/CIK").
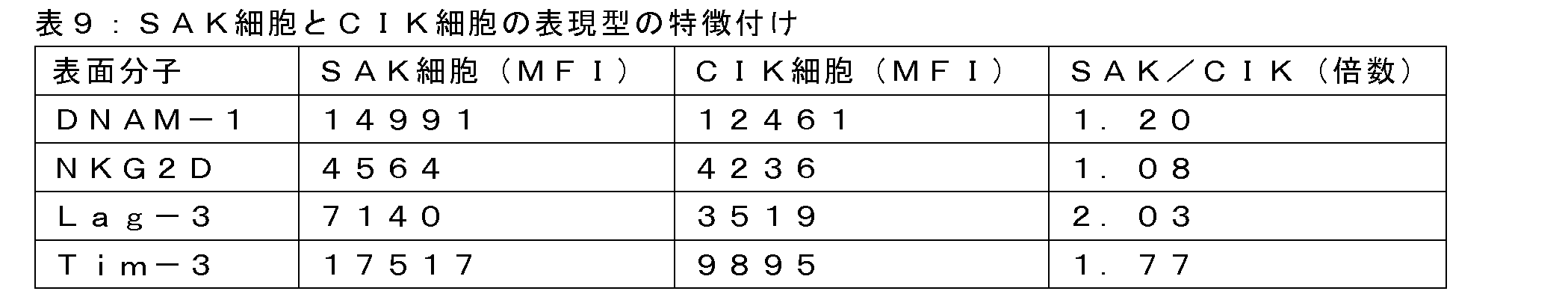
表9に示すように、種々の増殖プロトコルによって調製した細胞は、表面マーカーの独特の発現プロファイルに関する様々な表現型を示した。特に、SAK細胞は、11日目に回収した対応するCIK細胞よりも顕著に高いレベルの試験マーカーを示す。従って、この結果から、容易に利用可能な方法によってSAK細胞を容易に特定し、他の細胞組成物と区別できることが実証される。また、11日目のCIK細胞よりもSAK細胞において高いレベルでリンパ球活性化中に上昇することが知られている表面マーカーの存在は、改良増殖方法中にSAK細胞が「超活性化」表現型に適応することを更に示唆する。 As shown in Table 9, cells prepared by the various growth protocols exhibited various phenotypes with respect to unique expression profiles of surface markers. In particular, SAK cells exhibit significantly higher levels of the tested markers than the corresponding CIK cells harvested on day 11. Thus, the results demonstrate that SAK cells can be readily identified and distinguished from other cell compositions by readily available methods. Additionally, the presence of surface markers known to be elevated during lymphocyte activation at higher levels in SAK cells than in CIK cells on day 11 further suggests that SAK cells adopt a "hyperactivated" phenotype during the improved growth method.
NK細胞とT細胞の機能は、抑制性受容体(例えば、PD-1、TIGIT、Lag-3、Tim-3及び/又はCTLA-4)と活性化受容体(例えば、DNAM-1)のバランスによって調節且つ維持されることが知られている。更に、抑制性免疫チェックポイント受容体の遮断は、T細胞の消耗を効果的に逆転させ、T細胞の抗腫瘍能力を回復させる上で重要となり得る。従って、抑制性免疫チェックポイント分子の遮断がSAK細胞活性にも影響を及ぼし得ることについて関心が高まっている。 It is known that NK and T cell functions are regulated and maintained by the balance between inhibitory receptors (e.g., PD-1, TIGIT, Lag-3, Tim-3 and/or CTLA-4) and activating receptors (e.g., DNAM-1). Furthermore, blockade of inhibitory immune checkpoint receptors may be important in effectively reversing T cell exhaustion and restoring the antitumor capabilities of T cells. Thus, there is growing interest that blockade of inhibitory immune checkpoint molecules may also affect SAK cell activity.
注目すべきは、SAK細胞は11日間の増殖後に、CIK細胞と比較して腫瘍細胞に対する細胞傷害能力が顕著に向上することが本明細書で実証されたことである(例えば、実施例3)。特定の理論又は作用機序に束縛されたくはないが、本明細書に開示の様々な受容体とエフェクター分子との独特のバランスを特徴とするSAK細胞の「超活性化」表現型は、腫瘍細胞に対する好ましい機能的能力と関連している場合がある。 Of note, it has been demonstrated herein that SAK cells have significantly enhanced cytotoxicity against tumor cells compared to CIK cells after 11 days of growth (e.g., Example 3). Without wishing to be bound by a particular theory or mechanism of action, the "hyperactivated" phenotype of SAK cells, characterized by a unique balance of various receptors and effector molecules disclosed herein, may be associated with favorable functional capabilities against tumor cells.
要約すると、本明細書に開示の有利なプロトコルによって産生したSAK細胞は、免疫チェックポイント分子やケモカイン受容体等の細胞表面受容体の有利なプロファイルを示すことが分かった。特に、SAK細胞は、免疫エフェクターの生存、増殖及び/又は活性にプラスの影響を与えることが知られているDNAM-1とCXCR6の発現を特徴とすることが分かった。特定の理論又は作用機序に束縛されたくはないが、これらの結果は、腫瘍回避を最小限に抑えることができる、活性化表現型を特徴とするACT組成物を産生する能力と一致する。 In summary, SAK cells produced by the advantageous protocols disclosed herein were found to exhibit a favorable profile of cell surface receptors, such as immune checkpoint molecules and chemokine receptors. In particular, SAK cells were found to be characterized by expression of DNAM-1 and CXCR6, which are known to positively affect immune effector survival, proliferation and/or activity. Without wishing to be bound by a particular theory or mechanism of action, these results are consistent with the ability to produce ACT compositions characterized by an activated phenotype that can minimize tumor evasion.
主要組織適合性複合体クラスI(MHCI)の関与
MV4-11白血病細胞(混合型B骨髄単球性白血病のヒト細胞株、本明細書ではMV411とも称する)に対するSAK細胞の死滅機序をMHCクラスIの阻害剤を使用して調べた。細胞傷害性アッセイは基本的には実施例2に記載のように行い、SAK細胞を添加する1時間前に、MV411細胞を1μg/mLの抗MHCI抗体(W6/32クローン、Biolegend社製)と共にインキュベートした。
Involvement of Major Histocompatibility Complex Class I (MHCI) The killing mechanism of SAK cells against MV4-11 leukemia cells (a human cell line of mixed B-myelomonocytic leukemia, also referred to herein as MV411) was examined using an inhibitor of MHC class I. Cytotoxicity assays were performed essentially as described in Example 2, except that MV411 cells were incubated with 1 μg/mL anti-MHCI antibody (W6/32 clone, Biolegend) 1 hour before the addition of SAK cells .
従って、この結果から、SAK細胞が白血病細胞に対してMHCI依存的細胞傷害活性と非MHCI依存的細胞傷害活性の両方を示すことが分かる。注目すべきことに、エフェクターSAK細胞は組織適合性ではないため、両方の活性はHLA対立遺伝子の適合性を必要としなかった。従って、本発明に係るSAK細胞を含む組成物は、ダウンレギュレートされたMHC-I発現及び/又は活性を特徴とする免疫回避性腫瘍又は治療抵抗性腫瘍の治療にも有利に使用することができる。 Thus, the results show that SAK cells exhibit both MHCI-dependent and non-MHCI-dependent cytotoxic activity against leukemia cells. Notably, both activities did not require compatibility of HLA alleles, as the effector SAK cells were not histocompatible. Thus, compositions comprising SAK cells according to the present invention can also be advantageously used to treat immune-evasive or therapy-resistant tumors characterized by downregulated MHC-I expression and/or activity.
要約すると、本明細書に記載の結果から、組織適合性対象と非組織適合性対象の両方における様々な起源の腫瘍の治療に対するSAK細胞の適合性が実証される。本明細書で実証したように、SAK細胞は、造血器腫瘍と固形腫瘍に対して顕著な抗腫瘍活性を示し、HLA適合性がない場合でも抗腫瘍効力を保持し、更には、移植片対宿主病(GVHD)を引き起こすリスクを顕著に抑制することで明らかなように、安全性の向上を示した。特定の理論又は作用機序に束縛されたくはないが、様々な腫瘍細胞に対するSAK細胞の細胞傷害活性は、本明細書で示すように、NKG2D及び/又はT細胞受容体(TCR)媒介活性を伴うことがある。 In summary, the results described herein demonstrate the suitability of SAK cells for the treatment of tumors of various origins in both histocompatible and non-histocompatible subjects. As demonstrated herein, SAK cells exhibited significant antitumor activity against hematopoietic and solid tumors, retained antitumor efficacy even in the absence of HLA compatibility, and further demonstrated improved safety as evidenced by a significant reduction in the risk of inducing graft-versus-host disease (GVHD). Without wishing to be bound by a particular theory or mechanism of action, the cytotoxic activity of SAK cells against various tumor cells may involve NKG2D and/or T cell receptor (TCR)-mediated activity, as shown herein.
特定の実施形態の上述の説明は、本発明の一般的な性質を完全に明らかにするものであるため、他者は現在の知見を応用することによって、過度の実験を行わずに一般概念から逸脱することなく、そのような特定の実施形態を様々な用途に容易に改変及び/又は適応させることができる。従って、そのような適応や改変は、開示された実施形態の均等物の意味及び範囲内で理解されるべきであり、また、そのように意図されている。本明細書で使用される表現又は用語は説明を目的としたものであり、限定を目的とするものではないことを理解されたい。開示された様々な機能を実施するための手段、材料及び工程は、本発明から逸脱することなく、様々な代替形態をとることができる。 The above description of the specific embodiments fully reveals the general nature of the present invention, so that others, applying current knowledge, can easily modify and/or adapt such specific embodiments to various applications without undue experimentation and without departing from the general concept. Such adaptations and modifications should therefore be understood and are intended to be within the meaning and scope of equivalents of the disclosed embodiments. It should be understood that the expressions or terms used herein are for the purpose of description and not for the purpose of limitation. The means, materials and steps for carrying out the various disclosed functions may take various alternative forms without departing from the invention.
配列番号1: 抗ヒトCD123 CAR
配列番号2: IRES核酸配列
配列番号5: 2Aエレメント
配列番号6: GFP
配列番号9: CD19 CAR
SEQ ID NO: 1: Anti-human CD123 CAR
SEQ ID NO:2: IRES nucleic acid sequence SEQ ID NO:5: 2A element SEQ ID NO:6: GFP
SEQ ID NO: 9: CD19 CAR
Claims (15)
a.末梢血単核細胞(PBMC)試料を用意することと、
b.有効量のIL-2とCD3アクチベーターの常時存在下、且つ他のサイトカイン又は抗体の非存在下で少なくとも9日間にわたりPBMCを増殖させることと
を含み、培養物中の生細胞数を少なくとも20倍に増加させ、少なくとも5×106~10×108個の生細胞を含む細胞組成物を得る、方法。 1. A method for producing a cell composition for adoptive cell transfer (ACT), comprising:
a. providing a peripheral blood mononuclear cell (PBMC) sample;
b. Propagating the PBMCs in the constant presence of an effective amount of IL-2 and a CD3 activator, and in the absence of other cytokines or antibodies, for at least 9 days, thereby increasing the number of viable cells in the culture by at least 20 fold and obtaining a cell composition comprising at least 5 x 10 to 10 x 10 viable cells.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163136217P | 2021-01-12 | 2021-01-12 | |
| US63/136,217 | 2021-01-12 | ||
| PCT/IL2022/050038 WO2022153295A1 (en) | 2021-01-12 | 2022-01-11 | Improved adoptive cell transfer therapy for cancer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2024502170A JP2024502170A (en) | 2024-01-17 |
| JPWO2022153295A5 true JPWO2022153295A5 (en) | 2024-11-06 |
Family
ID=82447153
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023541633A Pending JP2024502170A (en) | 2021-01-12 | 2022-01-11 | Improved adoptive cell transfer therapy for cancer |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20240307536A1 (en) |
| EP (1) | EP4277638A4 (en) |
| JP (1) | JP2024502170A (en) |
| IL (1) | IL304373A (en) |
| WO (1) | WO2022153295A1 (en) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009097119A2 (en) * | 2008-01-29 | 2009-08-06 | Fred Hutchinson Cancer Research Center | Identification of cd8+ t cells that are cd161hi and/or il18r(alpha)hi and have rapid drug efflux capacity |
| CN102428173B (en) * | 2009-03-26 | 2014-07-30 | 细胞维护北欧制药公司 | Expansion of NK cells |
| KR20230024984A (en) * | 2020-06-12 | 2023-02-21 | 엔카르타, 인크. | Genetically Modified Natural Killer Cells for CD70-Directed Cancer Immunotherapy |
-
2022
- 2022-01-11 JP JP2023541633A patent/JP2024502170A/en active Pending
- 2022-01-11 US US18/271,883 patent/US20240307536A1/en active Pending
- 2022-01-11 WO PCT/IL2022/050038 patent/WO2022153295A1/en not_active Ceased
- 2022-01-11 EP EP22739254.5A patent/EP4277638A4/en active Pending
-
2023
- 2023-07-10 IL IL304373A patent/IL304373A/en unknown
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7527049B2 (en) | CAR expression vector and CAR-expressing T cells | |
| US12365906B2 (en) | Immunomodulatory fusion proteins and uses thereof | |
| JP7026161B2 (en) | Immunomodulatory fusion proteins and their use | |
| KR102483822B1 (en) | Tagged chimeric effector molecules and receptors thereof | |
| EP3025719B1 (en) | Combination immunotherapy of antigen-recognizing receptors and hematopoietic cells for the treatment of diseases | |
| CN108137670A (en) | NY-ESO-1 specific TCRs and methods of use thereof | |
| KR20140023931A (en) | Method and compositions for cellular immunotherapy | |
| JP2016528899A (en) | Anti-CD30 chimeric antigen receptor and use thereof | |
| US20210379149A1 (en) | Increasing or Maintaining T-Cell Subpopulations in Adoptive T-Cell Therapy | |
| JP2024509917A (en) | Method for selective stimulation of T cells in solid tumors using orthogonal IL-2 delivery by oncolytic viruses | |
| WO2019140278A1 (en) | Immunotherapy targeting core binding factor antigens | |
| US20240307536A1 (en) | Improved adoptive cell transfer therapy for cancer | |
| JP2024541976A (en) | Chimeric adaptor polypeptides | |
| JPWO2022153295A5 (en) | ||
| Biondi | Enhancing AML CAR CIK therapeutic potency increasing the localization of engineered cells in the malignant niche and its selectivity by LSCs specific targeting | |
| Özkazanç Ünsal | Development of a novel in vitro screening method using genetically modified NK-92 cells against various tumor cells | |
| HK1235426B (en) | Car expression vector and car-expressing t cells |