JPWO2009125858A1 - 2-Lipid dispersion preparation containing indolinone derivative - Google Patents
2-Lipid dispersion preparation containing indolinone derivative Download PDFInfo
- Publication number
- JPWO2009125858A1 JPWO2009125858A1 JP2010507295A JP2010507295A JPWO2009125858A1 JP WO2009125858 A1 JPWO2009125858 A1 JP WO2009125858A1 JP 2010507295 A JP2010507295 A JP 2010507295A JP 2010507295 A JP2010507295 A JP 2010507295A JP WO2009125858 A1 JPWO2009125858 A1 JP WO2009125858A1
- Authority
- JP
- Japan
- Prior art keywords
- lipid
- lipid dispersion
- dispersion preparation
- preparation according
- liposome
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Dispersion Chemistry (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Z−3−[2,4−(ジメチルピロール−5−イル)メチルイデニル]−2−インドリノンを含有する脂質分散体製剤により、非経口投与時に安全性、利便性、効果に優れた抗悪性腫瘍剤を提供する。Anti-malignant tumor agent excellent in safety, convenience, and effect during parenteral administration by lipid dispersion preparation containing Z-3- [2,4- (dimethylpyrrol-5-yl) methylidenyl] -2-indolinone I will provide a.
Description
本発明は、2−インドリノン誘導体を含有する脂質分散体製剤及び当該脂質分散体製剤を含有する抗腫瘍剤に関する。 The present invention relates to a lipid dispersion preparation containing a 2-indolinone derivative and an antitumor agent containing the lipid dispersion preparation.
現在までにがん治療のために多くの薬物が開発され、医療に用いられている。しかし、現在に至るまで腫瘍組織に対してのみ選択的に殺細胞効果を示す抗がん剤は開発されていない。抗がん剤をそのまま血管内に投与した場合、血中から速く消失する、標的以外の臓器にも分布することがあり、必ずしも効果的にがん組織に集積するとは限らない。このため抗がん剤の多くは、がん組織に対する抗腫瘍作用(主作用)を十分に発揮できず、またしばしば望ましくない正常組織への作用(副作用)を伴い、重篤な毒性を引き起こしている。抗がん剤の主作用の増強、副作用の軽減は、現在のがん化学療法の重要な課題であり、薬物をがん組織、がん細胞に効率的に集積する薬物送達システム(DDS,Drug Delivery System)の開発が強く求められている。
リポソームやO/Wエマルションに代表される脂質分散体は生体成分由来のリン脂質を膜脂質とする閉鎖小胞であり、生体に投与したときの毒性、抗原性が低い特徴を持つ。また脂質分散体内に薬物を封入することにより、その薬物の血中安定性及び生体内分布を変化させて標的組織への到達性を改善できることが示されている(非特許文献1、特許文献1)。またがん組織に多く存在する新生血管の血管壁は既存の血管のそれと比較して透過性が高く、平均粒子径50〜200nm程度の脂質分散体の小胞はがん組織に集積しやすいことが知られている(非特許文献2)。従って抗腫瘍活性薬物を脂質分散体内に封入した製剤は主作用を増強し、副作用を軽減することが大いに期待されるDDSのひとつである。
本発明で使用するZ−3−[2,4−(ジメチルピロール−5−イル)メチルイデニル]−2−インドリノン(ファイザー社,大鵬薬品工業株式会社 開発コード:SU5416)は血管内皮細胞増殖因子(VEGF)のリン酸化を阻害することにより腫瘍組織における血管新生阻害作用を示し、幅広いスペクトルのヒト腫瘍に対して強い増殖抑制効果を示すことが知られており、下記一般式(1)で表される化合物である(特許文献2、非特許文献3)。
Lipid dispersions typified by liposomes and O / W emulsions are closed vesicles that use phospholipids derived from biological components as membrane lipids, and are characterized by low toxicity and low antigenicity when administered to the living body. It has also been shown that encapsulating a drug in a lipid dispersion can improve the reach to the target tissue by changing the blood stability and biodistribution of the drug (Non-patent
Z-3- [2,4- (dimethylpyrrol-5-yl) methylidenyl] -2-indolinone (Pfizer, Taiho Pharmaceutical Co., Ltd., development code: SU5416) used in the present invention is vascular endothelial growth factor (VEGF). It is known that it exhibits an angiogenesis-inhibiting action in tumor tissues by inhibiting phosphorylation of), and has a strong growth-inhibiting effect on a broad spectrum of human tumors. It is a compound (
本発明の課題は抗悪性腫瘍作用を示すSU5416の非経口投与時に適した安全性、利便性に優れた脂質分散体製剤を提供することにある。
また本発明の課題はSU5416を含有する抗腫瘍剤を提供することにある。The subject of this invention is providing the lipid dispersion formulation excellent in the safety | security and convenience suitable at the time of parenteral administration of SU5416 which shows an antineoplastic action.
Another object of the present invention is to provide an antitumor agent containing SU5416.
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、SU5416が脂溶性であり、脂質膜に親和性が高いことを着想し、SU5416をリポソーム又はO/Wエマルションといった脂質分散体に封入した結果、クレモホールや有機溶媒を含有しない投与形態を確立し、本発明を完成するに至った。さらに、脂質分散体のサイズなどを適切に調節することによって、SU5416をマウスの血管内に投与したときの抗腫瘍活性が有意に増大し、かつ毒性の増加は認められないことを見出した。
即ち、以下の発明に係るものである。
1.Z−3−[2,4−(ジメチルピロール−5−イル)メチルイデニル]−2−インドリノンを含有する非経口投与用の脂質分散体製剤。
2.上記脂質膜を構成する脂質成分の少なくとも1種がリン脂質である脂質分散体製剤。
3.リン脂質が、精製卵黄ホスファチジルコリン、精製大豆ホスファチジルコリン、水素添加精製卵黄ホスファチジルコリン、水素添加精製大豆ホスファチジルコリン、ジパルミトイルホスファチジルコリン、ジステアロイルホスファチジルコリン及び1−パルミトイル−2−オレオイルホスファチジルコリンからなる群より選択される少なくとも1種である上記脂質分散体製剤。
4.脂質分散粒子の平均粒子径が1μm以下である上記脂質分散体製剤。
5.製剤中のリン脂質濃度が1〜100mMである上記脂質分散体製剤。
6.製剤中のZ−3−[2,4−(ジメチルピロール−5−イル)メチルイデニル]−2−インドリノンの濃度が0.2〜20mMである上記脂質分散体製剤。
7.脂質膜の表面がポリエチレングリコール誘導体又はPRP配列を含むペプチドで修飾されている上記脂質分散体製剤。
8.脂質膜の表面がPRP配列を含むペプチドで修飾されている上記脂質分散体製剤。
9.PRP配列を含むペプチドがAPRPGである上記脂質分散体製剤。
10.上記のいずれかに記載の脂質分散体製剤を含有する非経口投与用の抗腫瘍剤。As a result of intensive research to solve the above problems, the present inventors have conceived that SU5416 is fat-soluble and has a high affinity for lipid membranes, and SU5416 is a lipid dispersion such as a liposome or an O / W emulsion. As a result of encapsulating in the body, a dosage form not containing cremophor or an organic solvent was established, and the present invention was completed. Furthermore, it was found that by appropriately adjusting the size of the lipid dispersion and the like, the antitumor activity was significantly increased when SU5416 was administered into the blood vessels of mice, and no increase in toxicity was observed.
That is, the present invention relates to the following invention.
1. A lipid dispersion preparation for parenteral administration containing Z-3- [2,4- (dimethylpyrrol-5-yl) methylidenyl] -2-indolinone.
2. A lipid dispersion preparation wherein at least one lipid component constituting the lipid membrane is a phospholipid.
3. The phospholipid is at least one selected from the group consisting of purified egg yolk phosphatidylcholine, purified soybean phosphatidylcholine, hydrogenated purified egg yolk phosphatidylcholine, hydrogenated purified soy phosphatidylcholine, dipalmitoylphosphatidylcholine, distearoylphosphatidylcholine and 1-palmitoyl-2-oleoylphosphatidylcholine. Said lipid dispersion formulation which is a seed.
4). The above lipid dispersion preparation, wherein the average particle size of the lipid dispersed particles is 1 μm or less.
5. The said lipid dispersion formulation whose phospholipid density | concentration in a formulation is 1-100 mM.
6). The above lipid dispersion preparation, wherein the concentration of Z-3- [2,4- (dimethylpyrrol-5-yl) methylidenyl] -2-indolinone in the preparation is 0.2 to 20 mM.
7). The lipid dispersion preparation, wherein the lipid membrane surface is modified with a polyethylene glycol derivative or a peptide containing a PRP sequence.
8). The lipid dispersion preparation described above, wherein the surface of the lipid membrane is modified with a peptide containing a PRP sequence.
9. The above lipid dispersion preparation, wherein the peptide containing the PRP sequence is APRPG.
10. An antitumor agent for parenteral administration containing the lipid dispersion preparation according to any one of the above.
本発明は、脂質分散体製剤は利便性を高めるとともに、毒性を上昇させることなく、抗腫瘍効果を高めることができる。従って、本発明の脂質分散体製剤は抗腫瘍剤として格別顕著な効果を示し、癌患者の生存期間をより延長させる。 The lipid dispersion preparation of the present invention can improve the antitumor effect without increasing toxicity while enhancing convenience. Therefore, the lipid dispersion preparation of the present invention exhibits a particularly remarkable effect as an antitumor agent, and further extends the survival period of cancer patients.
本発明の脂質分散体の具体的形態として、リポソーム及びO/Wエマルションが挙げられる。
本発明に用いられるリポソームとは、前記したように細胞膜を構成しているリン脂質を水中に分散させて形成される脂質二分子膜により囲まれた内水相部分を有する閉鎖小胞であり、そのサイズや脂質二分子の数によって多重相リポソーム(Multilamellar Vesicle:MLV)、大きな一枚膜リポソーム(Large Unilamellar Vesicle:LUV)及び小さな一枚膜リポソーム(Small Unilamellar Vesicle:SUV)の3種類に分類される。本発明ではいずれの種類のリポソームも使用可能である。本発明のリポソームは生体内投与前に安定なリポソーム構造を形成しなければならない。
リポソームを構成するリン脂質としては、例えば精製卵黄フォスファチジルコリン(以下EPCとする)、精製大豆フォスファチジルコリン(以下SPCとする)、水素添加精製卵黄ホスファチジルコリン(相転移温度50〜60℃、以下HEPC)、水素添加精製大豆ホスファチジルコリン(相転移温度約55℃、以下HSPC)、ジパルミトイルホスファチジルコリン(相転移温度約41℃、以下DPPC)、ジステアロイルホスファチジルコリン(相転移温度約58℃、以下DSPC)及び1−パルミトイル−2−オレオイルホスファチジルコリン(相転移温度−3℃、以下POPC)から選ばれ、より好ましくはDPPC及びDSPCがあげられる。これらは1種又は2種以上混合して用いることができ、相転移温度の低いリン脂質、例えばPOPCを用いる場合は2種以上混合して調製する。
本発明に用いられるリポソームは、これらリン脂質に加えて、好ましくはリポソームの安定性を改善することが報告されているコレステロール誘導体などの安定化剤と混合して使用される。コレステロール誘導体とリン脂質のモル比は、1:1〜10が好ましく、1:2.5〜5がさらに好ましい。
また、等張化剤として、例えば、グリセリン、ブドウ糖、塩化ナトリウムなどの添加も可能である。さらに、パラベン類、クロロブタノール、ベンジルアルコール、プロピレングリコールなどの防腐剤を加えてもよい。
本発明で用いられるリン脂質とSU5416とのモル比は、1:0.01〜0.1が好ましく、1:0.025〜0.075がさらに好ましく、1:0.04〜0.06が特に好ましい。
本発明のリポソーム製剤は公知の方法により調製される。公知のリポソーム製剤の調製方法としては、例えば、逆相蒸発法[Proc.Natl.Acad.Sci.USA,75,4194(1978),WO97/48398]、凍結融解法[Arch.Biochem.Biophys,212,186(1981)]、pH勾配法[Biochem.Biophys.Acta,816,294(1985)、特開平7−165560]などがあげられる。
これらの中で、逆相蒸発法は、水に難溶な薬物をリポソーム内に高い包含率で封入する際に有用であり、SU5416においても最適な方法のひとつである。逆相蒸発法による本発明のリポソーム製剤の調製方法としては、例えば、SU5416と脂質成分をクロロホルム、エーテル、エタノールなどの溶媒に溶解後、ナス型フラスコに入れ、減圧下溶媒留去し脂質薄膜を形成する。次いで、緩衝液を薄膜に加え、凍結融解を行い、内水相が緩衝液である大きな多重相リポソーム(MLV)を調製する。さらに、エクストルージョン法などにより、小さな一枚膜リポソーム(SUV)とする。以上の操作により、薬物をリポソームの脂質膜に高率かつ定量的に封入することができる。
本発明に用いられるO/Wエマルションとは、膜相脂質であるリン脂質が、内相脂質を取り巻いた脂質粒子を含有するものであり、内相脂質中及び膜相脂質中に薬物が溶解した状態で存在する。
O/Wエマルションを構成する膜相脂質としては、界面活性を有する必要があり、種々のリン脂質を使用することができる。例えばEPC、SPC、HEPC、HSPC、DPPC、DSPC及びPOPCから選ばれる。これらは1種又は2種以上混合して用いることができる。
一方、O/Wエマルションを構成する内相脂質としては薬物を溶解できる脂質が望ましく、精製ダイズ油、コレステロール、コレステロールエステル、トリオレイン、トリカプリリン、トリ(カプリル・カプリン酸)グリセリン、オレイン酸、オレイン酸エチル、トコフェロールなどを挙げることができる。
O/Wエマルションは粒子間で凝集しやすい性質があるため、ポリビニルピロリドン、血清アルブミンなどの高分子を外水相に溶解して、凝集を抑制する方法をとることができる。また、等張化剤として、例えば、グリセリン、ブドウ糖、塩化ナトリウムなどの添加も可能である。さらに、パラベン類、クロロブタノール、ベンジルアルコール、プロピレングリコールなどの防腐剤を加えてもよい。
本発明のO/Wエマルションで用いられる脂質とSU5416とのモル比は、1:0.01〜0.5が好ましく、1:0.02〜0.2がさらに好ましく、1:0.04〜0.1が特に好ましい。
リポソーム及びO/Wエマルションの粒子サイズは内包されている薬物の生体内分布、腫瘍組織への送達性に大きな影響を与えることが報告されている[Biological and Pharmaceutical Bulletin,17,935(1994)]。従って、本発明においては、薬物を内包した脂質分散体の粒子径を適切かつ均一にするために、サイジング処理を行うのが好ましく、例えば、エクストルーダー(リペックスバイオメンブラン社製など)を使用し、適切た孔径のメンブランフィルターに数回通過させることにより、或いは超音波ホモジナイザーを使用することにより調製する。
リポソームの平均粒子径は、好ましくは1μm(1000nm)以下、より好ましくは50〜500nm、更に好ましくは100〜300nm、特に好ましくは75〜150nm、最も好ましくは75〜125nmである。O/Wエマルションの平均粒子径は、好ましくは1μm(1000nm)以下、より好ましくは50〜500nm、更に好ましくは100〜300nm、特に好ましくは150〜300nmである。
製剤中のリン脂質およびSU5416の濃度は、SU5416の脂質分散体への封入率を高め、SU5416の析出を防止し、溶液の粘度を非経口投与に適した範囲とするために調整する必要があり、リン脂質濃度は1〜100mM、好ましくは5〜50mMとし、SU5416濃度は0.2〜20mM、好ましくは0.5〜10mMとすることが望ましい。
またリポソーム及びO/Wエマルションの膜表面を、必要に応じ、ポリエチレングリコール(PEG)誘導体で修飾することによって、リポソームの血中での安定性を改善することができる。多くの場合、PEG誘導体としては、分子量500〜20000のPEGとリン脂質の共有結合体を用い、分子量2000〜5000のPEGとジステアロイルホスファチジルエタノールアミン(以下DSPE)の結合体(以下DSPE−PEG)が最も広汎に使われている。
また、ペプチド、レクチン、抗体、糖質、糖タンパク、糖脂質などのリガンドで修飾することによって、リポソーム及びO/Wエマルションの血中での安定性、組織分布、腫瘍組織への移行性等をさらに向上させることができる。特に、PRP配列を含むペプチドはがん組織に多く存在する新生血管に特異的に集積することが確認されており、VEGF受容体の一つであるfms−like tyrosine kinase−1(flt−1)へ親和性を持つことが示唆されている[Oncogene,21,2662(2002)、WO00/23476]。ポリエチレングリコール誘導体及びPRP配列を含むペプチドで膜表面を修飾することにより、血中で安定であり、かつ腫瘍組織への移行性が高いリポソーム及びO/Wエマルションを得ることができる。
ここで、用いられるPRP配列を含むペプチドは、プロリン−アルギニン−プロリンのアミノ酸配列を含むペプチドであればよく、ペプチド合成分野において公知の方法により作成することができる。例えばAPRPG(アラニン−プロリン−アルギニン−プロリン−グリシン)、GPRPL(グリシン−プロリン−アルギニン−プロリン−ロイシン)、GPRPR(グリシン−プロリン−アルギニン−プロリン−アルギニン)を例示することができる。
以上の操作によって、本発明の脂質分散体製剤が調製され、必要に応じ、超遠心処理、ゲル濾過処理、限外濾過処理及び透析処理を単独又は適宜組み合わせて行うことによって、リポソーム及びO/Wエマルションに内包されなかった薬物を除去することができる。
上記の方法により得られる本発明の脂質分散体製剤はそのままでも使用できるが、保存期間、保存条件などを考慮して、マンニトール、トレハロース、ラクトース、グリシンなどの賦形剤を加えて凍結乾燥することもできる。またグリセリンなどの凍結保存剤を加え、凍結保存してもよい。
本発明の脂質分散体製剤は、一般的には、使用時に生理的に許容される水溶液で懸濁又は希釈して注射剤(静脈内、筋肉内、皮下、腹腔内投与製剤)として用いられるが、点鼻剤、吸入剤、点眼剤、坐剤、経皮吸収剤、経粘膜吸収剤などとして使用することもできる。最終製剤は、悪性腫瘍〔ヒトなどの哺乳動物の肺がん、消化器がん(食道がん、胃がん、直腸がん、結腸がん)、乳がん、頭頸部がん、肝臓がん、胆嚢がん、膵臓がん、婦人科がん(子宮がん、子宮頸がん、卵巣がんなど)、泌尿器がん(腎がん、膀胱がん、前立腺がん)、脳腫瘍、白血病、メラノーマ、悪性リンパ腫など〕の治療、緩解のための有効量が投与されるように設計されていればよい。例えば、静脈内投与リポソーム製剤としてSU5416が0.01〜10000mg含有されていればよい。本発明のリポソーム製剤に含まれるSU5416の投与量は、0.01〜1000mg/dayとするのが好ましい。Specific examples of the lipid dispersion of the present invention include liposomes and O / W emulsions.
The liposome used in the present invention is a closed vesicle having an inner aqueous phase portion surrounded by a lipid bilayer formed by dispersing phospholipids constituting a cell membrane in water as described above, Depending on its size and the number of lipid bimolecules, it is classified into three types: multi-phase liposome (MLV), large unilamellar liposome (LUV), and small unilamellar liposome (SUV). The Any kind of liposome can be used in the present invention. The liposome of the present invention must form a stable liposome structure before in vivo administration.
Examples of the phospholipid constituting the liposome include purified egg yolk phosphatidylcholine (hereinafter referred to as EPC), purified soybean phosphatidylcholine (hereinafter referred to as SPC), hydrogenated purified egg yolk phosphatidylcholine (
In addition to these phospholipids, the liposomes used in the present invention are preferably used in admixture with stabilizers such as cholesterol derivatives that have been reported to improve liposome stability. The molar ratio of cholesterol derivative and phospholipid is preferably 1: 1 to 10, and more preferably 1: 2.5 to 5.
Further, as an isotonic agent, for example, glycerin, glucose, sodium chloride and the like can be added. Furthermore, preservatives such as parabens, chlorobutanol, benzyl alcohol, propylene glycol and the like may be added.
The molar ratio of the phospholipid used in the present invention to SU5416 is preferably 1: 0.01 to 0.1, more preferably 1: 0.025 to 0.075, and 1: 0.04 to 0.06. Particularly preferred.
The liposome preparation of the present invention is prepared by a known method. As a method for preparing a known liposome preparation, for example, the reverse phase evaporation method [Proc. Natl. Acad. Sci. USA, 75, 4194 (1978), WO 97/48398], freeze-thaw method [Arch. Biochem. Biophys, 212, 186 (1981)], pH gradient method [Biochem. Biophys. Acta, 816, 294 (1985), JP-A-7-165560] and the like.
Among these, the reverse phase evaporation method is useful when encapsulating a poorly water-soluble drug in liposomes at a high inclusion rate, and is also one of the most suitable methods for SU5416. As a method for preparing the liposome preparation of the present invention by the reverse phase evaporation method, for example, SU5416 and a lipid component are dissolved in a solvent such as chloroform, ether, ethanol, etc., and then placed in an eggplant type flask. Form. Next, a buffer solution is added to the thin film, freeze-thawed, and large multiphase liposomes (MLV) whose inner aqueous phase is a buffer solution are prepared. Furthermore, small unilamellar liposomes (SUV) are obtained by an extrusion method or the like. By the above operation, the drug can be encapsulated in the lipid membrane of the liposome at a high rate and quantitatively.
The O / W emulsion used in the present invention is one in which a phospholipid that is a membrane phase lipid contains lipid particles surrounding the inner phase lipid, and the drug is dissolved in the inner phase lipid and in the membrane phase lipid. Exists in a state.
The membrane phase lipid constituting the O / W emulsion needs to have surface activity, and various phospholipids can be used. For example, it is selected from EPC, SPC, HEPC, HSPC, DPPC, DSPC and POPC. These can be used alone or in combination.
On the other hand, as the internal phase lipid constituting the O / W emulsion, a lipid capable of dissolving a drug is desirable. Refined soybean oil, cholesterol, cholesterol ester, triolein, tricaprylin, tri (caprylic / capric acid) glycerin, oleic acid, olein Examples thereof include ethyl acid and tocopherol.
Since the O / W emulsion tends to aggregate between particles, a method of suppressing aggregation by dissolving a polymer such as polyvinylpyrrolidone or serum albumin in the outer aqueous phase can be used. Further, as an isotonic agent, for example, glycerin, glucose, sodium chloride and the like can be added. Furthermore, preservatives such as parabens, chlorobutanol, benzyl alcohol, propylene glycol and the like may be added.
The molar ratio of the lipid used in the O / W emulsion of the present invention to SU5416 is preferably 1: 0.01 to 0.5, more preferably 1: 0.02 to 0.2, and 1: 0.04 to 0.1 is particularly preferred.
It has been reported that the particle size of liposomes and O / W emulsions has a great influence on the biodistribution of encapsulated drugs and the delivery to tumor tissues [Biological and Pharmaceutical Bulletin, 17, 935 (1994)]. . Therefore, in the present invention, it is preferable to perform sizing treatment in order to make the particle size of the lipid dispersion encapsulating the drug appropriate and uniform, for example, using an extruder (manufactured by Lipex Biomembrane, etc.). Prepare by passing several times through a membrane filter of appropriate pore size or by using an ultrasonic homogenizer.
The average particle diameter of the liposome is preferably 1 μm (1000 nm) or less, more preferably 50 to 500 nm, still more preferably 100 to 300 nm, particularly preferably 75 to 150 nm, and most preferably 75 to 125 nm. The average particle size of the O / W emulsion is preferably 1 μm (1000 nm) or less, more preferably 50 to 500 nm, still more preferably 100 to 300 nm, and particularly preferably 150 to 300 nm.
The concentration of phospholipid and SU5416 in the formulation should be adjusted to increase the encapsulation rate of SU5416 in the lipid dispersion, prevent the precipitation of SU5416, and bring the solution viscosity to a range suitable for parenteral administration. The phospholipid concentration is 1 to 100 mM, preferably 5 to 50 mM, and the SU5416 concentration is 0.2 to 20 mM, preferably 0.5 to 10 mM.
Moreover, the stability in the blood of a liposome can be improved by modifying the membrane surface of a liposome and O / W emulsion with a polyethylene glycol (PEG) derivative as needed. In many cases, a PEG derivative having a molecular weight of 500 to 20000 and a phospholipid covalent conjugate is used, and a molecular weight of 2000 to 5000 PEG and distearoylphosphatidylethanolamine (hereinafter DSPE) conjugate (hereinafter DSPE-PEG). Is the most widely used.
In addition, by modifying with ligands such as peptides, lectins, antibodies, carbohydrates, glycoproteins and glycolipids, the stability of liposomes and O / W emulsions in blood, tissue distribution, transferability to tumor tissues, etc. Further improvement can be achieved. In particular, it has been confirmed that a peptide containing a PRP sequence is specifically accumulated in a new blood vessel present in a large amount in cancer tissue, and is one of VEGF receptors, fms-like tyrosine kinase-1 (flt-1). Has been suggested [Oncogene, 21, 2662 (2002), WO 00/23476]. By modifying the membrane surface with a polyethylene glycol derivative and a peptide containing a PRP sequence, it is possible to obtain liposomes and O / W emulsions that are stable in blood and highly transferable to tumor tissues.
Here, the peptide containing the PRP sequence used may be a peptide containing the amino acid sequence of proline-arginine-proline, and can be prepared by a known method in the field of peptide synthesis. For example, APRPG (alanine-proline-arginine-proline-glycine), GPRPL (glycine-proline-arginine-proline-leucine), GPRPR (glycine-proline-arginine-proline-arginine) can be exemplified.
Through the above operations, the lipid dispersion preparation of the present invention is prepared, and if necessary, by performing ultracentrifugation treatment, gel filtration treatment, ultrafiltration treatment and dialysis treatment alone or in combination, liposomes and O / W Drugs not encapsulated in the emulsion can be removed.
The lipid dispersion preparation of the present invention obtained by the above method can be used as it is, but in consideration of the storage period, storage conditions, etc., an excipient such as mannitol, trehalose, lactose, glycine is added and lyophilized. You can also. Further, a cryopreservation agent such as glycerin may be added and stored frozen.
The lipid dispersion preparation of the present invention is generally used as an injection (intravenous, intramuscular, subcutaneous, intraperitoneal administration preparation) after being suspended or diluted with a physiologically acceptable aqueous solution at the time of use. They can also be used as nasal drops, inhalants, eye drops, suppositories, transdermal absorbents, transmucosal absorbents, and the like. The final formulation consists of malignant tumors [lung cancer in mammals such as humans, digestive organ cancer (esophageal cancer, stomach cancer, rectal cancer, colon cancer), breast cancer, head and neck cancer, liver cancer, gallbladder cancer, Pancreatic cancer, gynecological cancer (uterine cancer, cervical cancer, ovarian cancer, etc.), urological cancer (renal cancer, bladder cancer, prostate cancer), brain tumor, leukemia, melanoma, malignant lymphoma, etc. It suffices if it is designed so that an effective amount for treatment and remission is administered. For example, SU5416 should just contain 0.01-10000 mg as an intravenous administration liposome formulation. The dosage of SU5416 contained in the liposome preparation of the present invention is preferably 0.01 to 1000 mg / day.
以下に実施例及び試験例を挙げて本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
[実施例1]
(ポリエチレングリコール誘導体で修飾したSU5416リポソーム製剤、以下PEG−Lip−SU5416とする)
DPPC30μmol、POPC30μmol、コレステロール15μmol、DSPE−PEG6μmol、SU5416(Chloroform溶液)3μmolをナス型フラスコに入れ(脂質構成: DPPC/POPC/コレステロール/DSPE−PEG/SU5416=10/10/5/2/1)、5mLのクロロホルムを加えた。エバポレーターで減圧下、クロロホルムを留去し、ナス型フラスコの内壁面に脂質薄膜を形成させた。さらにデシケーター内で1時間真空乾燥した。次に20mM HEPES〔4−(2−hydroxyethyl)−1−piperazineethanesulfonic acid〕緩衝液(pH7.4)1.5mLで水和後、凍結融解を3回行った。バス型ソニケーターを用いて10分間超音波処理した後、エクストルーダー(リペックスバイオメンブラン社製)を用い、孔径100nmのポリカーボネートメンブランフィルター(ヌクレポア)に5回通過させてリポソーム製剤を調製した。リポソームの平均粒子径は約120nmであった。
[実施例2]
(ポリエチレングリコール誘導体及びAPRPGペプチドで修飾したSU5416リポソーム製剤、以下APRPG−Lip−SU5416とする)
DPPC30μmol、POPC30μmol、コレステロール15μmol、DSPE−PEG化したAPRPG配列を含むペプチド(DSPE−PEG−APRPG)6μmol、SU5416(Chloroform溶液)3μmolをナス型フラスコに入れ(脂質構成:DPPC/POPC/コレステロール/DSPE−PEG−APRPG/SU5416=10/10/5/2/1)、5mLのクロロホルムを加えた。以下、実施例1の方法に従い、リポソーム製剤を調製した。リポソームの平均粒子径は約130nmであった。
[実施例3]
(EPCとトリオレインから成るSU5416エマルション製剤、以下O/W−EPC−SU5416とする)
EPCのクロロホルム溶液(100mM)とトリオレイン(油状)を、EPC:トリオレインのモル比が2:8となるようにマイクロシリンジを用いて50mLナス型フラスコに分取した。次に、SU5416のクロロホルム溶液(50mM)を、EPC:トリオレイン:SU5416のモル比が2:8:1になるように必要量をナス型フラスコに添加した。さらに少量のクロロホルムを加え、脂質溶液が均一になるように軽く混和した。ロータリーエバポレーターで減圧下、クロロホルムを留去し、ナス型フラスコの底に脂質の薄膜を形成した。80℃に保ちながら、0.3Mグルコース/1% ポリビニルピロリドン溶液を加え、ボルテックスミキサーで激しく攪拌することにより水和させた。粒子径を均質にするために、超音波ホモジナイザー(SONIFER Model250,BRANSON)で超音波処理を90分間行った。最終的な脂質濃度は、EPCとして20mMである。エマルションの平均粒子径は約240nmであった。
[実施例4]
(PEG誘導体で修飾した、DPPC、コレステロール、コレステロールオレエートから成るSU5416O/Wエマルション製剤、以下O/W−DPPC−PEG−SU5416とする)
DPPC、コレステロール、コレステロールオレエートのクロロホルム溶液(いずれも100mM)とDSPE−PEGのクロロホルム/メタノール(1/3)溶液(100mM)を、DPPC:コレステロール:コレステロールオレエート:DSPE−PEGのモル比が10:10:20:2となるようにマイクロシリンジを用いて50mLナス型フラスコに分取した。次に、SU5416のクロロホルム溶液(50mMを、DPPC:コレステロール:コレステロールオレエート:DSPE−PEG:SU5416のモル比が10:10:20:2:1となるように必要量をナス型フラスコに添加した。さらに少量のクロロホルムを加え、脂質溶液が均一になるように軽く混和した。ロータリーエバポレーターで減圧下、クロロホルムを留去し、ナス型フラスコの底に脂質の薄膜を形成した。80℃に保ちながら、0.3M HEPES/1% ポリビニルピロリドン溶液を加え、ボルテックスミキサーで激しく攪拌することにより水和させた。粒子径を均質にするために、超音波ホモジナイザー(SONIFER Model250,BRANSON)で超音波処理を90分間行った。最終的な脂質濃度は、DPPCとして10mMである。エマルションの平均粒子径は約160nmであった。
[比較例1]
(脂質分散体としていないSU5416溶液、以下Free SU5416製剤とする)
ポリエチレングリコール400、クレモホールEL(Polyoxyl 35castor oil)、ベンジルアルコール、無水エタノールを重量比45:31.5:2:21.5で混合して、Free SU5416用の溶媒とした。これにSU5416を1.2mg/mLとなるように溶解した。動物に投与する直前に0.45%塩化ナトリウム溶液で4倍希釈した。
[試験例1]
(SU5416の脂質分散体封入率と経時安定性)
実施例1及び実施例2で調製したリポソーム溶液を、それぞれセファデックスG−25を充てんしたPD−10カラムを用いたゲル濾過法により、6画分に分画した。各画分に10% reduced Triton X−100溶液50μL及び20mM HEPES bufferを加え混合し、さらに超音波処理を行うことによりリポソーム膜を可溶化した。HPLCを用いて吸収波長440nmの条件下における溶液の吸光度を測定し、検量線から各画分のSU5416濃度を求めた。一方、各画分について濁度(吸収波長750nm)を測定し、リポソーム濃度を決定した(図1及び図2、fraction1−3)。
実施例3で調製したO/Wエマルション溶液を、上記と同様の方法でSU5416濃度、O/Wエマルション濃度を求めた。
実施例1、実施例2及び実施例3の各画分のSU5416濃度及び脂質分散体濃度から脂質分散体へのSU5416封入率を求めた結果、脂質分散体へのSU5416の封入率は、実施例1で得たPEG−Lip−SU5416は86.0%、実施例2で得たAPRPG−Lip−SU5416は85.8%、実施例3で得たO/W−EPC−SU5416は86.9%であり、高い効率で封入されていることが確認された。また、いずれの脂質分散体製剤についても、平均粒子径は調製後14日間変化しなかった(図3及び図4)。また、いずれの脂質分散体製剤についても、SU5416封入率が調製直後から14日間変化せず製剤として安定であることが明らかとなった(図5及び図6)。
HPLC条件
検出器:紫外可視吸光光度計(測定波長:440nm)
カラム:TSKGEL ODS−80TS 4.6mm×25cm (TOSOH)
カラム温度:40℃
移動相:Methanol/35mMKH2PO4 (pH3.0) 混液(75:25)
流速 :1.0mL/min (Pressure 約16kgf/cm2)
注入量:10又は20μL
測定時間 :25min(ピークは約15分のところで検出)
[試験例2]
(脂質分散体SU5416製剤とFree SU5416製剤の溶血性)
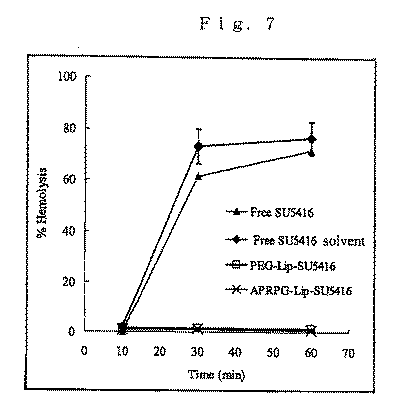
マウスから採取した血液を遠心分離(2000×g,5min)して上清を採取した。残渣にリン酸緩衝生理食塩水(PBS,pH7.4)を加え混合した後、遠心分離(2000×g,5min)して上清を採取し、この操作を5回繰り返した。20mM HEPES緩衝生理食塩水で5%(v/v)に希釈し、赤血球溶液とした。赤血球溶液180μLに実施例1及び実施例2で調製した脂質分散体SU5416リポソーム溶液、比較例1で調製したFree SU5416溶液ならびにFree SU5416溶解用の溶媒のそれぞれ20μLを加え、ボルテックスミキサーで混合した。その後37℃で10,30,60minインキュベートし、遠心分離(10,000×g,5min)を行った。上清100μLを取り、マイクロプレートリーダーで570nmの波長を測定し、溶血率を算出した。
その結果、図7に示すように、Free SU5416溶液及びFree SU5416の溶媒では顕著な溶血が見られた一方、PEG−Lip−SU5416及びAPRPG−Lip−SU5416溶液ではほとんど溶血は認められなかった。この理由として、リポソーム製剤中にクレモホールEL及び有機溶媒が含まれていないため、赤血球膜を破壊する作用がないことが考えられた。
[試験例3]
(脂質分散体SU5416製剤とFree SU5416製剤の抗腫瘍効果と毒性評価)
抗腫瘍効果の評価に用いるマウス大腸がん細胞株Colon26NL17を5%CO2存在下、37℃10%FBS(Fetal Bovine Serum、ウシ胎児血清)−DMEM(Dulbecco’s Modified Eagle’s Medium、ダルベッコ改変イーグル培地)/Ham F12=1/1で培養し、継代した。5×106cells/mLに調製したColon26NL17を4週齢雄性Balb/Cマウスの左腹側部に0.2mL皮下投与(1×106cells/mouse)して固形がん担がんマウスを作成した。Colon26NL17移植後5,7,9,11及び13日目の計5回、コントロールとしてHEPES buffered saline及び実施例2で調製されたAPRPG−Lip−SU5416溶液或いは比較例1で調製されたFree SU5416溶液の0.2mL/body/dayをマウスの尾静脈内に投与した(投与量はいずれもSU5416として3mg/kg)。がん移植4日後から腫瘍容積を測定することにより、SU5416封入リポソーム溶液の抗腫瘍効果を調べた。腫瘍容積は以下の式を用いて算出した。
腫瘍容積(Tumor volume)=0.4×a×b2
(a:腫瘍部位の長径、b:腫瘍部位の短径)
その結果、図8に示すように、APRPG−Lip−SU5416処置群は、Control処置群、リポソーム化していないSU5416処置群(Free SU5416)に比べ有意な腫瘍増殖抑制効果を示した。
また副作用の指標として体重変化を調べた結果、図9に示すように、すべての処置群において顕著な体重減少はみられなかった。
これらの結果より、SU5416のリポソーム製剤はリポソーム化していない溶液と比較して有意に抗腫瘍活性を増強することが示された。抗がん活性の増強の原因は、SU5416をリポソーム製剤とすることでSU5416の体内動態を改善し、問題点であった血中半減期の延長、さらに標的化による新生血管への集積性の向上などが考えられる。Hereinafter, the present invention will be described in detail with reference to Examples and Test Examples, but the present invention is not limited thereto.
[Example 1]
(SU5416 liposome preparation modified with polyethylene glycol derivative, hereinafter referred to as PEG-Lip-SU5416)
30 μmol of DPPC, 30 μmol of POPC, 15 μmol of cholesterol, 6 μmol of DSPE-PEG, 3 μmol of SU5416 (Chloroform solution) are placed in an eggplant type flask (lipid composition: DPPC / POPC / cholesterol / DSPE-PEG / SU5416 = 10/10/5/2/1), 5 mL of chloroform was added. Chloroform was distilled off under reduced pressure with an evaporator to form a lipid thin film on the inner wall surface of the eggplant type flask. Furthermore, it was vacuum-dried in a desiccator for 1 hour. Next, it was hydrated with 1.5 mL of 20 mM HEPES [4- (2-hydroxyethyl) -1-piperazine etheric acid] buffer (pH 7.4), and freeze-thawed three times. After ultrasonic treatment for 10 minutes using a bath sonicator, a liposome preparation was prepared by passing 5 times through a polycarbonate membrane filter (Nuclepore) with a pore size of 100 nm using an extruder (manufactured by Lipex Biomembrane). The average particle size of the liposome was about 120 nm.
[Example 2]
(SU5416 liposome preparation modified with polyethylene glycol derivative and APRPG peptide, hereinafter referred to as APRPG-Lip-SU5416)
[Example 3]
(SU5416 emulsion formulation consisting of EPC and triolein, hereinafter referred to as O / W-EPC-SU5416)
A chloroform solution (100 mM) of EPC and triolein (oil) were fractionated into a 50 mL eggplant type flask using a microsyringe so that the molar ratio of EPC: triolein was 2: 8. Next, the required amount of SU5416 in chloroform (50 mM) was added to the eggplant type flask so that the molar ratio of EPC: triolein: SU5416 was 2: 8: 1. Further, a small amount of chloroform was added and lightly mixed so that the lipid solution was uniform. Chloroform was distilled off under reduced pressure using a rotary evaporator to form a lipid thin film on the bottom of the eggplant-shaped flask. While maintaining at 80 ° C., 0.3 M glucose / 1% polyvinylpyrrolidone solution was added and hydrated by vigorous stirring with a vortex mixer. In order to make the particle diameter uniform, ultrasonic treatment was performed for 90 minutes with an ultrasonic homogenizer (
[Example 4]
(SU5416 O / W emulsion preparation modified with PEG derivative and consisting of DPPC, cholesterol, cholesterol oleate, hereinafter referred to as O / W-DPPC-PEG-SU5416)
DPPC, cholesterol, cholesterol oleate in chloroform (100 mM each) and DSPE-PEG in chloroform / methanol (1/3) (100 mM) in a molar ratio of DPPC: cholesterol: cholesterol oleate: DSPE-PEG of 10 : 10: 20: 2 was dispensed into a 50 mL eggplant-shaped flask using a microsyringe. Next, the required amount of SU5416 in chloroform (50 mM, DPPC: cholesterol: cholesterol oleate: DSPE-PEG: SU5416 molar ratio was added to the eggplant type flask was 10: 10: 20: 2: 1). A small amount of chloroform was added, and the mixture was lightly mixed so that the lipid solution was uniform, and the chloroform was distilled off under reduced pressure using a rotary evaporator to form a thin lipid film on the bottom of the eggplant-shaped flask. Then, 0.3M HEPES / 1% polyvinylpyrrolidone solution was added and hydrated by vigorous stirring with a vortex mixer.To homogenize the particle size, sonication was performed with an ultrasonic homogenizer (
[Comparative Example 1]
(SU5416 solution not lipid dispersion, hereinafter referred to as Free SU5416 formulation)
Polyethylene glycol 400, Cremophor EL (Polyoxyl 35 castor oil), benzyl alcohol, and absolute ethanol were mixed at a weight ratio of 45: 31.5: 2: 21.5 to obtain a solvent for Free SU5416. SU5416 was dissolved in this so that it might become 1.2 mg / mL. Immediately before administration to animals, it was diluted 4-fold with 0.45% sodium chloride solution.
[Test Example 1]
(SU5416 lipid dispersion encapsulation rate and stability over time)
The liposome solution prepared in Example 1 and Example 2 was fractionated into 6 fractions by gel filtration using a PD-10 column filled with Sephadex G-25. To each fraction, 50 μL of 10% reduced Triton X-100 solution and 20 mM HEPES buffer were added and mixed, and sonication was performed to solubilize the liposome membrane. The absorbance of the solution was measured using HPLC at an absorption wavelength of 440 nm, and the SU5416 concentration of each fraction was determined from the calibration curve. On the other hand, turbidity (absorption wavelength 750 nm) was measured for each fraction, and the liposome concentration was determined (FIGS. 1 and 2, fractions 1-3).
The SU5416 concentration and O / W emulsion concentration of the O / W emulsion solution prepared in Example 3 were determined in the same manner as described above.
As a result of obtaining the SU 5416 encapsulation rate in the lipid dispersion from the SU 5416 concentration and the lipid dispersion concentration of each fraction of Example 1, Example 2 and Example 3, the encapsulation rate of SU 5416 in the lipid dispersion is PEG-Lip-SU5416 obtained in 1 was 86.0%, APRPG-Lip-SU5416 obtained in Example 2 was 85.8%, and O / W-EPC-SU5416 obtained in Example 3 was 86.9%. It was confirmed that it was sealed with high efficiency. Moreover, about any lipid dispersion formulation, the average particle diameter did not change for 14 days after preparation (FIGS. 3 and 4). In addition, for any lipid dispersion preparation, it was revealed that the SU5416 encapsulation rate did not change for 14 days from immediately after preparation and was stable as a preparation (FIGS. 5 and 6).
HPLC condition detector: UV-visible spectrophotometer (measurement wavelength: 440 nm)
Column: TSKGEL ODS-80TS 4.6 mm x 25 cm (TOSOH)
Column temperature: 40 ° C
Mobile phase: Methanol / 35 mM KH 2 PO 4 (pH 3.0) mixed solution (75:25)
Flow rate: 1.0 mL / min (Pressure about 16 kgf / cm 2 )
Injection volume: 10 or 20 μL
Measurement time: 25 min (a peak is detected at about 15 minutes)
[Test Example 2]
(Hemolytic properties of lipid dispersion SU5416 preparation and Free SU5416 preparation)
The blood collected from the mouse was centrifuged (2000 × g, 5 min) and the supernatant was collected. Phosphate buffered saline (PBS, pH 7.4) was added to the residue and mixed, then centrifuged (2000 × g, 5 min), the supernatant was collected, and this operation was repeated 5 times. It was diluted to 5% (v / v) with 20 mM HEPES buffered saline to obtain an erythrocyte solution. 20 μL each of the lipid dispersion SU5416 liposome solution prepared in Example 1 and Example 2, the Free SU5416 solution prepared in Comparative Example 1 and the solvent for dissolving Free SU5416 was added to 180 μL of the erythrocyte solution, and mixed with a vortex mixer. Thereafter, the cells were incubated at 37 ° C. for 10, 30, 60 min, and centrifuged (10,000 × g, 5 min). 100 μL of the supernatant was taken, the wavelength of 570 nm was measured with a microplate reader, and the hemolysis rate was calculated.
As a result, as shown in FIG. 7, remarkable hemolysis was observed in the Free SU5416 solution and the Free SU5416 solvent, while almost no hemolysis was observed in the PEG-Lip-SU5416 and APRPG-Lip-SU5416 solutions. The reason for this was considered that since the liposome preparation does not contain Cremophor EL and an organic solvent, it does not have an action of destroying the erythrocyte membrane.
[Test Example 3]
(Anti-tumor effect and toxicity evaluation of lipid dispersion SU5416 preparation and Free SU5416 preparation)
Mouse colon cancer cell line Colon26NL17 used for evaluation of antitumor effect was modified in the presence of 5% CO 2 at 37 ° C. and 10% FBS (Fetal Bovine Serum, fetal bovine serum) -DMEM (Dulbecco's Modified Eagle's Medium, Dulbecco modification) Eagle medium) / Ham F12 = 1/1 and subcultured. Colon 26NL17 prepared to 5 × 10 6 cells / mL was subcutaneously administered (1 × 10 6 cells / mouse) to the left ventral part of 4-week-old male Balb / C mice to give solid cancer-bearing mice. Created. Five times on
Tumor volume = 0.4 × a × b2
(A: major axis of the tumor site, b: minor axis of the tumor site)
As a result, as shown in FIG. 8, the APRPG-Lip-SU5416 treatment group showed a significant tumor growth inhibitory effect compared to the Control treatment group and the SU5416 treatment group (Free SU5416) that was not made into liposomes.
As a result of examining the change in body weight as an index of side effects, as shown in FIG. 9, no significant weight loss was observed in all treatment groups.
These results indicate that the SU5416 liposome preparation significantly enhances the antitumor activity as compared to the non-liposomal solution. The cause of the enhancement of the anticancer activity is to improve the pharmacokinetics of SU5416 by using SU5416 as a liposome preparation, prolong the blood half-life, which has been a problem, and improve the accumulation in new blood vessels by targeting And so on.
本発明の脂質分散体製剤は利便性を高めるとともに、毒性を上昇させることなく、抗腫瘍効果を高めることができる。従って、本発明の脂質分散体製剤は抗腫瘍剤として格別顕著な効果を示すことが期待され、癌患者の生存期間をより延長させるような治療効果の高い治療法への応用が期待される。 The lipid dispersion preparation of the present invention can enhance the convenience and enhance the antitumor effect without increasing toxicity. Therefore, the lipid dispersion preparation of the present invention is expected to exhibit a particularly remarkable effect as an antitumor agent, and is expected to be applied to a therapeutic method having a high therapeutic effect that further extends the survival period of cancer patients.
Claims (10)
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008099662 | 2008-04-07 | ||
| JP2008099662 | 2008-04-07 | ||
| PCT/JP2009/057409 WO2009125858A1 (en) | 2008-04-07 | 2009-04-06 | Lipid dispersion preparation comprising 2-indolinone derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JPWO2009125858A1 true JPWO2009125858A1 (en) | 2011-08-04 |
Family
ID=41161989
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010507295A Pending JPWO2009125858A1 (en) | 2008-04-07 | 2009-04-06 | 2-Lipid dispersion preparation containing indolinone derivative |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JPWO2009125858A1 (en) |
| TW (1) | TW200946112A (en) |
| WO (1) | WO2009125858A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5914418B2 (en) * | 2013-06-26 | 2016-05-11 | 富士フイルム株式会社 | Lipid particle, nucleic acid delivery carrier, composition for producing nucleic acid delivery carrier, lipid particle production method and gene introduction method |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60208910A (en) * | 1984-03-31 | 1985-10-21 | Green Cross Corp:The | Preparation of composite of hardly water-soluble drug and phospholipid |
| JPS63500456A (en) * | 1985-08-13 | 1988-02-18 | カリフオルニア バイオテクノロジ− インコ−ポレイテツド | Microemulsion used in medicine |
| JPH0789874A (en) * | 1993-07-27 | 1995-04-04 | Terumo Corp | Carrier recognizing injured part of intravascular wall |
| JPH11279082A (en) * | 1998-03-30 | 1999-10-12 | Taisho Pharmaceut Co Ltd | Fat emulsion for injection |
| JP2004331630A (en) * | 2003-05-12 | 2004-11-25 | Nippon Fine Chem Co Ltd | PEG-binding peptide |
| JP2005225818A (en) * | 2004-02-13 | 2005-08-25 | Otsuka Pharmaceut Factory Inc | Pharmaceutical composition of paclitaxel or docetaxel |
| JP2006063008A (en) * | 2004-08-26 | 2006-03-09 | Konica Minolta Medical & Graphic Inc | Liposome preparation for cancer treatment and production method thereof |
| JP2006517917A (en) * | 2002-12-19 | 2006-08-03 | アルザ・コーポレーシヨン | Method of treating angiogenic tissue growth |
| WO2007027941A2 (en) * | 2005-08-31 | 2007-03-08 | Abraxis Bioscience, Llc. | Compositions and methods for preparation of poorly water soluble drugs with increased stability |
| WO2007088952A1 (en) * | 2006-01-31 | 2007-08-09 | Taiho Pharmaceutical Co., Ltd. | Liposome preparation comprising substance having anti-tumor activity |
-
2009
- 2009-04-06 JP JP2010507295A patent/JPWO2009125858A1/en active Pending
- 2009-04-06 WO PCT/JP2009/057409 patent/WO2009125858A1/en not_active Ceased
- 2009-04-07 TW TW098111447A patent/TW200946112A/en unknown
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60208910A (en) * | 1984-03-31 | 1985-10-21 | Green Cross Corp:The | Preparation of composite of hardly water-soluble drug and phospholipid |
| JPS63500456A (en) * | 1985-08-13 | 1988-02-18 | カリフオルニア バイオテクノロジ− インコ−ポレイテツド | Microemulsion used in medicine |
| JPH0789874A (en) * | 1993-07-27 | 1995-04-04 | Terumo Corp | Carrier recognizing injured part of intravascular wall |
| JPH11279082A (en) * | 1998-03-30 | 1999-10-12 | Taisho Pharmaceut Co Ltd | Fat emulsion for injection |
| JP2006517917A (en) * | 2002-12-19 | 2006-08-03 | アルザ・コーポレーシヨン | Method of treating angiogenic tissue growth |
| JP2004331630A (en) * | 2003-05-12 | 2004-11-25 | Nippon Fine Chem Co Ltd | PEG-binding peptide |
| JP2005225818A (en) * | 2004-02-13 | 2005-08-25 | Otsuka Pharmaceut Factory Inc | Pharmaceutical composition of paclitaxel or docetaxel |
| JP2006063008A (en) * | 2004-08-26 | 2006-03-09 | Konica Minolta Medical & Graphic Inc | Liposome preparation for cancer treatment and production method thereof |
| WO2007027941A2 (en) * | 2005-08-31 | 2007-03-08 | Abraxis Bioscience, Llc. | Compositions and methods for preparation of poorly water soluble drugs with increased stability |
| WO2007088952A1 (en) * | 2006-01-31 | 2007-08-09 | Taiho Pharmaceutical Co., Ltd. | Liposome preparation comprising substance having anti-tumor activity |
Non-Patent Citations (1)
| Title |
|---|
| JPN6012065487; Yasufumi Katanasaka et al.: Cancer Letters vol.270, No.2, 2008, p.260-268 * |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200946112A (en) | 2009-11-16 |
| WO2009125858A1 (en) | 2009-10-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Gu et al. | Nanotechnology-mediated immunochemotherapy combined with docetaxel and PD-L1 antibody increase therapeutic effects and decrease systemic toxicity | |
| Castañeda-Reyes et al. | Development, characterization and use of liposomes as amphipathic transporters of bioactive compounds for melanoma treatment and reduction of skin inflammation: A review | |
| CN1960729B (en) | Irinotecan preparations | |
| KR101309440B1 (en) | Agent for enhancing anti-tumor effect comprising oxaliplatin liposome preparation, and anti-tumor agent comprising the liposome preparation | |
| Dong et al. | Dual‐loaded liposomes tagged with hyaluronic acid have synergistic effects in triple‐negative breast cancer | |
| CN108348538B (en) | Tumor therapeutic drug containing gemcitabine liposome composition and kit | |
| CN111956614A (en) | Paclitaxel liposome and preparation method thereof | |
| US12370214B2 (en) | Combined pharmaceutical formulation comprising drug-containing liposome composition and platinum preparation | |
| Wang et al. | A novel α-enolase-targeted drug delivery system for high efficacy prostate cancer therapy | |
| JP2003530362A (en) | Lipid-based systems for targeting diagnostic agents | |
| Xie et al. | A novel estrogen-targeted PEGylated liposome co-delivery oxaliplatin and paclitaxel for the treatment of ovarian cancer | |
| ES2984148T3 (en) | Combination drug containing a liposomal composition encapsulating a drug and an immune checkpoint inhibitor | |
| Tan et al. | Bioactive fatty acid analog-derived hybrid nanoparticles confer antibody-independent chemo-immunotherapy against carcinoma | |
| CN102579337A (en) | Long circulation lipid nano-suspension containing docetaxel and preparation method thereof | |
| WO2005021012A1 (en) | Drug carrier having gemcitabine enclosed therein | |
| CN1980671B (en) | Liposome preparation containing slightly water-soluble camptothecin | |
| JPWO2009125858A1 (en) | 2-Lipid dispersion preparation containing indolinone derivative | |
| Xiao et al. | Liposomal co-delivery system encapsulating celastrol and paclitaxel displays highly enhanced efficiency and low toxicity against pancreatic cancer | |
| EP3395370B1 (en) | Liposome and liposome composition | |
| US20250268827A1 (en) | Combined medicinal drug including liposome composition encompassing topotecan or salt thereof and dna damage repair inhibitor | |
| WO2011037252A1 (en) | Liposome preparation containing spicamycin derivative | |
| CN113116822A (en) | Paclitaxel loaded anti-tumor transmembrane liposome composition and preparation method thereof | |
| BR102017018582A2 (en) | LIPOSOMES, OBTAINING PROCESS, ANTI-TUMOR COMPOSITIONS AND USES | |
| BR102017018582B1 (en) | LIPOSOMES, PROCESS OF OBTAINING, ANTI-TUMORAL COMPOSITIONS AND USES | |
| HK40014043A (en) | Liposomal formulation for use in the treatment of cancer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120703 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120830 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20121218 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130218 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130402 |