JP2024178481A - Glycolipid inhibitors - Google Patents
Glycolipid inhibitors Download PDFInfo
- Publication number
- JP2024178481A JP2024178481A JP2021183784A JP2021183784A JP2024178481A JP 2024178481 A JP2024178481 A JP 2024178481A JP 2021183784 A JP2021183784 A JP 2021183784A JP 2021183784 A JP2021183784 A JP 2021183784A JP 2024178481 A JP2024178481 A JP 2024178481A
- Authority
- JP
- Japan
- Prior art keywords
- amino
- compound
- sulfonimidoyl
- butanoic acid
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000003112 inhibitor Substances 0.000 title claims description 7
- HVCOBJNICQPDBP-UHFFFAOYSA-N 3-[3-[3,5-dihydroxy-6-methyl-4-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyoxan-2-yl]oxydecanoyloxy]decanoic acid;hydrate Chemical compound O.OC1C(OC(CC(=O)OC(CCCCCCC)CC(O)=O)CCCCCCC)OC(C)C(O)C1OC1C(O)C(O)C(O)C(C)O1 HVCOBJNICQPDBP-UHFFFAOYSA-N 0.000 title 1
- 229930186217 Glycolipid Natural products 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 273
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 61
- 150000003839 salts Chemical class 0.000 claims abstract description 53
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 45
- 201000011510 cancer Diseases 0.000 claims abstract description 31
- 230000005764 inhibitory process Effects 0.000 claims abstract description 15
- 238000011282 treatment Methods 0.000 claims abstract description 10
- -1 (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoic acid Chemical compound 0.000 claims description 179
- 125000005843 halogen group Chemical group 0.000 claims description 36
- 238000000034 method Methods 0.000 claims description 28
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 28
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 27
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 26
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 7
- 238000004519 manufacturing process Methods 0.000 claims description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 3
- 239000003814 drug Substances 0.000 abstract description 24
- 229940079593 drug Drugs 0.000 abstract description 20
- 239000004480 active ingredient Substances 0.000 abstract description 11
- 201000010099 disease Diseases 0.000 abstract description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 9
- 239000000126 substance Substances 0.000 abstract description 5
- 208000014596 Berardinelli-Seip congenital lipodystrophy Diseases 0.000 abstract 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 237
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 135
- 239000000243 solution Substances 0.000 description 95
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 87
- 239000000203 mixture Substances 0.000 description 86
- 238000006243 chemical reaction Methods 0.000 description 62
- 230000000704 physical effect Effects 0.000 description 58
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 51
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 37
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 36
- 230000002829 reductive effect Effects 0.000 description 35
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- KJQFBVYMGADDTQ-CVSPRKDYSA-N L-buthionine-(S,R)-sulfoximine Chemical compound CCCCS(=N)(=O)CC[C@H](N)C(O)=O KJQFBVYMGADDTQ-CVSPRKDYSA-N 0.000 description 33
- 239000012044 organic layer Substances 0.000 description 28
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 27
- 239000007864 aqueous solution Substances 0.000 description 26
- 210000004027 cell Anatomy 0.000 description 26
- 238000010898 silica gel chromatography Methods 0.000 description 26
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 25
- 238000004128 high performance liquid chromatography Methods 0.000 description 24
- 238000004809 thin layer chromatography Methods 0.000 description 24
- 108010057281 Lipocalin 1 Proteins 0.000 description 23
- 230000014759 maintenance of location Effects 0.000 description 23
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 20
- 239000012071 phase Substances 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 229960003180 glutathione Drugs 0.000 description 18
- 229920006395 saturated elastomer Polymers 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 17
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 16
- 238000010511 deprotection reaction Methods 0.000 description 16
- 230000000144 pharmacologic effect Effects 0.000 description 15
- 230000000694 effects Effects 0.000 description 14
- 102000004190 Enzymes Human genes 0.000 description 13
- 108090000790 Enzymes Proteins 0.000 description 13
- 101001034527 Homo sapiens Glutamate-cysteine ligase catalytic subunit Proteins 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 238000005481 NMR spectroscopy Methods 0.000 description 12
- 238000006911 enzymatic reaction Methods 0.000 description 12
- 238000001816 cooling Methods 0.000 description 11
- 239000013078 crystal Substances 0.000 description 11
- 102100039696 Glutamate-cysteine ligase catalytic subunit Human genes 0.000 description 10
- 235000019270 ammonium chloride Nutrition 0.000 description 10
- 238000011156 evaluation Methods 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 230000000052 comparative effect Effects 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 239000003456 ion exchange resin Substances 0.000 description 8
- 229920003303 ion-exchange polymer Polymers 0.000 description 8
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 8
- 235000017557 sodium bicarbonate Nutrition 0.000 description 8
- 102100034580 AT-rich interactive domain-containing protein 1A Human genes 0.000 description 7
- 101000924266 Homo sapiens AT-rich interactive domain-containing protein 1A Proteins 0.000 description 7
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 7
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 7
- BVCZEBOGSOYJJT-UHFFFAOYSA-N ammonium carbamate Chemical compound [NH4+].NC([O-])=O BVCZEBOGSOYJJT-UHFFFAOYSA-N 0.000 description 7
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 7
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 7
- 239000003960 organic solvent Substances 0.000 description 7
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 6
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 6
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 5
- 101000830085 Homo sapiens Germ cell-less protein-like 1 Proteins 0.000 description 5
- 101000870644 Homo sapiens Glutamate-cysteine ligase regulatory subunit Proteins 0.000 description 5
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 5
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 5
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 150000002009 diols Chemical class 0.000 description 5
- MAVQFIBKMPVZMF-ZETCQYMHSA-N methyl (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-sulfanylbutanoate Chemical compound COC(=O)[C@H](CCS)NC(=O)OC(C)(C)C MAVQFIBKMPVZMF-ZETCQYMHSA-N 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 4
- JCMUOFQHZLPHQP-UHFFFAOYSA-N L-L-Ophthalmic acid Natural products OC(=O)CNC(=O)C(CC)NC(=O)CCC(N)C(O)=O JCMUOFQHZLPHQP-UHFFFAOYSA-N 0.000 description 4
- RITKHVBHSGLULN-WHFBIAKZSA-N L-gamma-glutamyl-L-cysteine Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(O)=O RITKHVBHSGLULN-WHFBIAKZSA-N 0.000 description 4
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 4
- 150000001204 N-oxides Chemical class 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 230000002950 deficient Effects 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 108010068906 gamma-glutamylcysteine Proteins 0.000 description 4
- 102000046788 human GCLC Human genes 0.000 description 4
- 102000005914 human glutamate-cysteine ligase modifier subunit Human genes 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 4
- IZDROVVXIHRYMH-UHFFFAOYSA-N methanesulfonic anhydride Chemical compound CS(=O)(=O)OS(C)(=O)=O IZDROVVXIHRYMH-UHFFFAOYSA-N 0.000 description 4
- JCMUOFQHZLPHQP-BQBZGAKWSA-N ophthalmic acid Chemical compound OC(=O)CNC(=O)[C@H](CC)NC(=O)CC[C@H](N)C(O)=O JCMUOFQHZLPHQP-BQBZGAKWSA-N 0.000 description 4
- 108010088490 ophthalmic acid Proteins 0.000 description 4
- HVVLQPOCRDLFGA-UHFFFAOYSA-N ophthalmic acid Natural products CCC(NC(=O)C(N)CCC(=O)O)C(=O)NCC(=O)O HVVLQPOCRDLFGA-UHFFFAOYSA-N 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- ASQMUMZEQLWJRC-UWTATZPHSA-N (3r)-4,4,4-trifluoro-3-hydroxybutanoic acid Chemical compound FC(F)(F)[C@H](O)CC(O)=O ASQMUMZEQLWJRC-UWTATZPHSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- IMWQVSSRLXGHOO-UHFFFAOYSA-N 4,4,4-trifluorobutyl methanesulfonate Chemical compound CS(=O)(=O)OCCCC(F)(F)F IMWQVSSRLXGHOO-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- 206010033128 Ovarian cancer Diseases 0.000 description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 239000004743 Polypropylene Substances 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 150000004696 coordination complex Chemical class 0.000 description 3
- 210000004748 cultured cell Anatomy 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 238000000105 evaporative light scattering detection Methods 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000000691 measurement method Methods 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 229920001155 polypropylene Polymers 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000001629 suppression Effects 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 238000002054 transplantation Methods 0.000 description 3
- ASQMUMZEQLWJRC-REOHCLBHSA-N (3s)-4,4,4-trifluoro-3-hydroxybutanoic acid Chemical compound FC(F)(F)[C@@H](O)CC(O)=O ASQMUMZEQLWJRC-REOHCLBHSA-N 0.000 description 2
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 description 2
- WSQXFIYFNWDJPH-UHFFFAOYSA-N 1-(2-bromoethyl)cyclobutan-1-ol Chemical compound OC1(CCBr)CCC1 WSQXFIYFNWDJPH-UHFFFAOYSA-N 0.000 description 2
- NDCSWRUDYCYSHC-UHFFFAOYSA-N 1-(2-hydroxyethyl)cyclobutan-1-ol Chemical compound OCCC1(O)CCC1 NDCSWRUDYCYSHC-UHFFFAOYSA-N 0.000 description 2
- HMUNWXXNJPVALC-UHFFFAOYSA-N 1-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C(CN1CC2=C(CC1)NN=N2)=O HMUNWXXNJPVALC-UHFFFAOYSA-N 0.000 description 2
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 2
- GDHPGAARGJPJSC-UHFFFAOYSA-N 2-(3,3-difluorocyclobutyl)ethanol Chemical compound OCCC1CC(F)(F)C1 GDHPGAARGJPJSC-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 2
- PYTQULGOMFBQNV-UHFFFAOYSA-N 4,4,4-trifluoro-3-(trifluoromethyl)butane-1,3-diol Chemical compound OCCC(O)(C(F)(F)F)C(F)(F)F PYTQULGOMFBQNV-UHFFFAOYSA-N 0.000 description 2
- JCJGOWYCFRASTM-UHFFFAOYSA-N 4,4,4-trifluoro-3-hydroxy-3-methylbutanoic acid Chemical compound FC(F)(F)C(O)(C)CC(O)=O JCJGOWYCFRASTM-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 208000003950 B-cell lymphoma Diseases 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 102000004263 Glutamate-Cysteine Ligase Human genes 0.000 description 2
- 108010081687 Glutamate-cysteine ligase Proteins 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 101100435489 Homo sapiens ARID1A gene Proteins 0.000 description 2
- 239000004201 L-cysteine Substances 0.000 description 2
- 235000013878 L-cysteine Nutrition 0.000 description 2
- 239000012448 Lithium borohydride Substances 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- PMZXXNPJQYDFJX-UHFFFAOYSA-N acetonitrile;2,2,2-trifluoroacetic acid Chemical compound CC#N.OC(=O)C(F)(F)F PMZXXNPJQYDFJX-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 2
- 238000005904 alkaline hydrolysis reaction Methods 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 238000005349 anion exchange Methods 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 229940041181 antineoplastic drug Drugs 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 2
- 229910001863 barium hydroxide Inorganic materials 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 2
- 239000000920 calcium hydroxide Substances 0.000 description 2
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000013375 chromatographic separation Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940000425 combination drug Drugs 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- ZWEDFBKLJILTMC-UHFFFAOYSA-N ethyl 4,4,4-trifluoro-3-hydroxybutanoate Chemical compound CCOC(=O)CC(O)C(F)(F)F ZWEDFBKLJILTMC-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229960002989 glutamic acid Drugs 0.000 description 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 238000007327 hydrogenolysis reaction Methods 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 238000010899 nucleation Methods 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000004749 rapidFire mass spectrometry Methods 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 125000005555 sulfoximide group Chemical group 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- OBETXYAYXDNJHR-SSDOTTSWSA-M (2r)-2-ethylhexanoate Chemical compound CCCC[C@@H](CC)C([O-])=O OBETXYAYXDNJHR-SSDOTTSWSA-M 0.000 description 1
- WQVSMPRULFEGHO-UHFFFAOYSA-N (4,4,4-trifluoro-3-hydroxy-3-methylbutyl) 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OCCC(C(F)(F)F)(C)O WQVSMPRULFEGHO-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- LJCZNYWLQZZIOS-UHFFFAOYSA-N 2,2,2-trichlorethoxycarbonyl chloride Chemical compound ClC(=O)OCC(Cl)(Cl)Cl LJCZNYWLQZZIOS-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 1
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 description 1
- ZRPAUEVGEGEPFQ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2 ZRPAUEVGEGEPFQ-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- YCPXWRQRBFJBPZ-UHFFFAOYSA-N 5-sulfosalicylic acid Chemical compound OC(=O)C1=CC(S(O)(=O)=O)=CC=C1O YCPXWRQRBFJBPZ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 238000009020 BCA Protein Assay Kit Methods 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 101710109147 Glutamate-cysteine ligase catalytic subunit Proteins 0.000 description 1
- 102100033398 Glutamate-cysteine ligase regulatory subunit Human genes 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical class NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- BHHGXPLMPWCGHP-UHFFFAOYSA-N Phenethylamine Chemical class NCCC1=CC=CC=C1 BHHGXPLMPWCGHP-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 208000008425 Protein deficiency Diseases 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 229920002536 Scavenger resin Polymers 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical class CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- XAKBSHICSHRJCL-UHFFFAOYSA-N [CH2]C(=O)C1=CC=CC=C1 Chemical group [CH2]C(=O)C1=CC=CC=C1 XAKBSHICSHRJCL-UHFFFAOYSA-N 0.000 description 1
- JLCHNBRGUPQWKF-UHFFFAOYSA-J [OH-].[C+4].[OH-].[OH-].[OH-] Chemical compound [OH-].[C+4].[OH-].[OH-].[OH-] JLCHNBRGUPQWKF-UHFFFAOYSA-J 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 229960003942 amphotericin b Drugs 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 125000000637 arginyl group Chemical class N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- DCFKHNIGBAHNSS-UHFFFAOYSA-N chloro(triethyl)silane Chemical compound CC[Si](Cl)(CC)CC DCFKHNIGBAHNSS-UHFFFAOYSA-N 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical class NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical class OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- BADXJIPKFRBFOT-UHFFFAOYSA-N dimedone Chemical compound CC1(C)CC(=O)CC(=O)C1 BADXJIPKFRBFOT-UHFFFAOYSA-N 0.000 description 1
- 150000004656 dimethylamines Chemical class 0.000 description 1
- 125000006222 dimethylaminomethyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002169 ethanolamines Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 210000004211 gastric acid Anatomy 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 230000005917 in vivo anti-tumor Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000009878 intermolecular interaction Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 108010082117 matrigel Proteins 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 150000003956 methylamines Chemical class 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229940037525 nasal preparations Drugs 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 125000005633 phthalidyl group Chemical group 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 150000003053 piperidines Chemical class 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- ZUFQCVZBBNZMKD-UHFFFAOYSA-M potassium 2-ethylhexanoate Chemical compound [K+].CCCCC(CC)C([O-])=O ZUFQCVZBBNZMKD-UHFFFAOYSA-M 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- QBERHIJABFXGRZ-UHFFFAOYSA-M rhodium;triphenylphosphane;chloride Chemical compound [Cl-].[Rh].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QBERHIJABFXGRZ-UHFFFAOYSA-M 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- VYPDUQYOLCLEGS-UHFFFAOYSA-M sodium;2-ethylhexanoate Chemical compound [Na+].CCCCC(CC)C([O-])=O VYPDUQYOLCLEGS-UHFFFAOYSA-M 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000003447 supported reagent Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DVFXLNFDWATPMW-IWOKLKJTSA-N tert-butyldiphenylsilyl Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO[Si](C=2C=CC=CC=2)(C=2C=CC=CC=2)C(C)(C)C)[C@@H](OP(O)(=O)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OC[C@@H]2[C@H](CC(O2)N2C3=NC=NC(N)=C3N=C2)OP(O)(=O)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)O)C1 DVFXLNFDWATPMW-IWOKLKJTSA-N 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- 125000006000 trichloroethyl group Chemical group 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical class OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 238000002525 ultrasonication Methods 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 239000006216 vaginal suppository Substances 0.000 description 1
- 239000000003 vaginal tablet Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
- A61K31/198—Alpha-amino acids, e.g. alanine or edetic acid [EDTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C381/00—Compounds containing carbon and sulfur and having functional groups not covered by groups C07C301/00 - C07C337/00
- C07C381/10—Compounds containing sulfur atoms doubly-bound to nitrogen atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
【課題】 がんなどのGCL関連疾患の進行抑制、再発抑制および/または治療において、GCL阻害活性を有する化合物を有効成分とする薬剤を提供すること。【解決手段】 一般式(I)【化1】TIFF2024178481000054.tif3993(式中、全ての記号は、明細書中に記載の記号と同じ意味を表す。)で示される化合物、またはその塩は、GCL阻害活性を有するため、がんなどのGCL関連疾患の進行抑制、再発抑制および/または治療において有用である。【選択図】 なし[Problem] To provide a drug containing as an active ingredient a compound having GCL inhibitory activity in the inhibition of progression, inhibition of recurrence and/or treatment of GCL-related diseases such as cancer. [Solution] A compound represented by general formula (I) [Chemical 1] TIFF2024178481000054.tif3993 (wherein all symbols have the same meaning as the symbols in the specification), or a salt thereof, has GCL inhibitory activity and is therefore useful in the inhibition of progression, inhibition of recurrence and/or treatment of GCL-related diseases such as cancer. [Selected Figure] None
Description
本開示は、グルタミン酸システインリガーゼ(以下、「GCL」と記載)阻害活性を有する化合物またはその塩およびそれらを有効成分として含有する医薬組成物等に関する。詳しくは、一般式(I): The present disclosure relates to a compound or its salt having glutamate cysteine ligase (hereinafter referred to as "GCL") inhibitory activity, and a pharmaceutical composition containing the compound or its salt as an active ingredient. In more detail, the general formula (I):

(式中、全ての記号は後記と同じ意味を表す。)で示される化合物またはその塩(以下、本開示化合物という。)およびそれらを有効成分として含有する医薬組成物等に関する。
(wherein all symbols have the same meaning as described below) or a salt thereof (hereinafter referred to as the disclosed compound), and pharmaceutical compositions containing them as an active ingredient, etc.
GCLは、グルタミン酸システインリガーゼ触媒サブユニット(以下、「GCLC」と記載)とグルタミン酸システインリガーゼ修飾サブユニット(以下、「GCLM」と記載)で構成される、グルタチオン(以下、「GSH」と記載)合成の律速酵素である。 GCL is the rate-limiting enzyme in the synthesis of glutathione (hereinafter referred to as "GSH") and is composed of a glutamate cysteine ligase catalytic subunit (hereinafter referred to as "GCLC") and a glutamate cysteine ligase modifying subunit (hereinafter referred to as "GCLM").
本酵素とがんとの関係が報告されている。例えば、GCLC阻害剤が、ARID1A欠損がんの治療に有用であること(特許文献1)やGCLCをノックアウトすることで、急性骨髄性白血病(AML)の腫瘍増殖が抑制されたことが報告されている(非特許文献1)。 The relationship between this enzyme and cancer has been reported. For example, it has been reported that GCLC inhibitors are useful for treating ARID1A-deficient cancers (Patent Document 1), and that knocking out GCLC suppresses tumor growth in acute myeloid leukemia (AML) (Non-Patent Document 1).
GCLC阻害剤としては、L-ブチオニンスルホキシイミン(以下、BSOと記載)が知られている。また、GCLC阻害剤として、BSO誘導体や低分子化合物に関する報告がある(特許文献2、非特許文献2~4)。
L-buthionine sulfoximine (hereinafter referred to as BSO) is known as a GCLC inhibitor. In addition, there have been reports of BSO derivatives and low molecular weight compounds as GCLC inhibitors (
本発明の課題は、GCLに対して阻害活性を有する化合物を提供することである。 The objective of the present invention is to provide a compound that has inhibitory activity against GCL.
本発明者らは、前記課題を解決すべく鋭意研究した結果、後述の一般式(I)で示される化合物が、GCLに対して阻害活性を有することを見出した。 As a result of intensive research aimed at solving the above problems, the present inventors have found that a compound represented by the general formula (I) described below has inhibitory activity against GCL.
すなわち、本開示は、一態様において、
[1]一般式(I):
That is, in one aspect, the present disclosure provides a method for producing a method for manufacturing a semiconductor device comprising:
[1] General formula (I):

(式中、R1は、水素原子、メチル基またはヒドロキシル基を表し、R2は、(1)-C(R3R4)-R5または(2)1~9個のR6で置換されていてもよいC3~C5シクロアルキル基を表し、R3またはR4は、それぞれ、(1)水素原子、(2)ヒドロキシル基、(3)ハロゲン原子または(4)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、R5は、(1)トリフルオロメチル基または(2)tert-ブチル基を表し、R6は、(1)ヒドロキシル基、(2)ハロゲン原子または(3)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、R6が複数の場合、複数のR6はそれぞれ同じでも異なっていてもよく、R7は、水素原子またはC1~C4アルキル基を表し、nは0または1を表す。)で示される化合物またはその塩、
[2]前記[1]に記載の一般式(I)で示される化合物またはその塩を含有する医薬組成物、
[3]GCL阻害剤である、前記[2]記載の医薬組成物等の実施態様を提供する。
(wherein R 1 represents a hydrogen atom, a methyl group or a hydroxyl group; R 2 represents (1) -C(R 3 R 4 )-R 5 or (2) a C3-C5 cycloalkyl group optionally substituted with 1 to 9 R 6 ; R 3 or R 4 each represents (1) a hydrogen atom, (2) a hydroxyl group, (3) a halogen atom or (4) a methyl group optionally substituted with 1 to 3 halogen atoms; R 5 represents (1) a trifluoromethyl group or (2) a tert-butyl group; R 6 represents (1) a hydroxyl group, (2) a halogen atom or (3) a methyl group optionally substituted with 1 to 3 halogen atoms; when there are multiple R 6s , the multiple R 6s may be the same or different; R 7 represents a hydrogen atom or a C1-C4 alkyl group; and n represents 0 or 1), or a salt thereof;
[2] A pharmaceutical composition comprising the compound represented by formula (I) or a salt thereof according to [1] above.
[3] The present invention provides an embodiment of the pharmaceutical composition etc. described in [2] above, which is a GCL inhibitor.
本開示化合物は、GCLに対して阻害活性を有することから、がんなどのGCLに関連する疾患(GCL関連疾患)の進行抑制、再発抑制および/または治療剤の有効成分として使用できる。 The disclosed compound has inhibitory activity against GCL and can therefore be used as an active ingredient in agents for inhibiting the progression or recurrence of and/or treating diseases associated with GCL, such as cancer (GCL-associated diseases).
以下、本開示を詳細に説明する。 This disclosure is explained in detail below.
本開示において、ハロゲン原子としては、フッ素、塩素、臭素およびヨウ素原子等が挙げられる。好ましくはフッ素または塩素原子であり、さらに好ましくはフッ素原子である。 In the present disclosure, examples of halogen atoms include fluorine, chlorine, bromine and iodine atoms. A fluorine or chlorine atom is preferred, and a fluorine atom is more preferred.
本開示において、1~3個のハロゲン原子で置換されていてもよいメチル基とは、例えば、1つ、2つまたは3つのハロゲン原子で置換されていてもよいメチル基であり、具体的にはフルオロメチル基、クロロメチル基、ブロモメチル基、ヨードメチル基、ジフルオロメチル基およびトリフルオロメチル基等が挙げられる。 In the present disclosure, a methyl group that may be substituted with 1 to 3 halogen atoms is, for example, a methyl group that may be substituted with 1, 2, or 3 halogen atoms, and specific examples thereof include a fluoromethyl group, a chloromethyl group, a bromomethyl group, an iodomethyl group, a difluoromethyl group, and a trifluoromethyl group.
本開示において、C1~C4アルキル基としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基およびtert-ブチル基が挙げられる。 In this disclosure, C1 to C4 alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, and tert-butyl groups.
本開示において、C3~C5シクロアルキル基としては、シクロプロピル基、シクロブチル基およびシクロペンチル基が挙げられる。 In this disclosure, C3-C5 cycloalkyl groups include cyclopropyl, cyclobutyl, and cyclopentyl groups.
本開示において、C3~C4シクロアルキル基としては、シクロプロピル基およびシクロブチル基が挙げられる。 In this disclosure, C3-C4 cycloalkyl groups include cyclopropyl and cyclobutyl groups.
本開示において、C4~C5シクロアルキル基としては、シクロブチル基およびシクロペンチル基が挙げられる。
などが挙げられる。
In the present disclosure, C4 to C5 cycloalkyl groups include cyclobutyl and cyclopentyl groups.
etc.
本開示において、R1として好ましくは水素原子である。 In the present disclosure, R 1 is preferably a hydrogen atom.
本開示において、nとして好ましくは1である。 In this disclosure, n is preferably 1.
本開示において、R5として好ましくは、トリフルオロメチル基である。 In the present disclosure, R 5 is preferably a trifluoromethyl group.
本開示において、R2として好ましくは(1)-C(R3R4)-CF3または(2)1~4個(好ましくは1~3個、より好ましくは1または2個)のR6で置換されていてもよいC3~C5シクロアルキル基(C3~C5シクロアルキル基として好ましくは、C3~C4シクロアルキル基またはC4~C5シクロアルキル基であり、より好ましくはシクロブチル基)である。 In the present disclosure, R 2 is preferably (1) -C(R 3 R 4 )-CF 3 or (2) a C3-C5 cycloalkyl group optionally substituted with 1 to 4 (preferably 1 to 3, more preferably 1 or 2) R 6 (the C3-C5 cycloalkyl group is preferably a C3-C4 cycloalkyl group or a C4-C5 cycloalkyl group, more preferably a cyclobutyl group).
本開示において、R3として好ましくは、水素原子、ハロゲン原子またはヒドロキシル基であり、より好ましくは、水素原子またはヒドロキシル基であり、さらに好ましくはヒドロキシル基である。 In the present disclosure, R3 is preferably a hydrogen atom, a halogen atom or a hydroxyl group, more preferably a hydrogen atom or a hydroxyl group, and even more preferably a hydroxyl group.
本開示において、R4として好ましくは、水素原子、ハロゲン原子、メチル基またはトリフルオロメチル基であり、より好ましくは、水素原子、メチル基またはトリフルオロメチル基である。 In the present disclosure, R 4 is preferably a hydrogen atom, a halogen atom, a methyl group, or a trifluoromethyl group, and more preferably a hydrogen atom, a methyl group, or a trifluoromethyl group.
本開示において、R6として好ましくは、ヒドロキシル基、ハロゲン原子、メチル基またはトリフルオロメチル基であり、より好ましくは、ヒドロキシル基またはハロゲン原子である。 In the present disclosure, R 6 is preferably a hydroxyl group, a halogen atom, a methyl group or a trifluoromethyl group, and more preferably a hydroxyl group or a halogen atom.
本開示において、R2として好ましい一実施形態は、 In the present disclosure, a preferred embodiment of R2 is

(式中、波線は、隣接する基との結合位置(CH2(CHR1)nとの結合位置)を示し、その他の記号は前記と同じ意味を表す。)である。 (In the formula, the wavy line indicates the bonding position to the adjacent group (the bonding position to CH 2 (CHR 1 ) n ), and the other symbols have the same meanings as above.)
本開示において、R2として好ましい一実施形態は、 In the present disclosure, a preferred embodiment of R2 is

(式中、mは1~3の整数を表し、pは0~3の整数を表し、R6aは、(1)ヒドロキシル基、(2)ハロゲン原子または(3)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、pが2または3のとき、複数のR6aはそれぞれ同じでも異なっていてもよい。また、その他の記号は前記と同じ意味を表す。)である。 (In the formula, m represents an integer of 1 to 3, p represents an integer of 0 to 3, R 6a represents (1) a hydroxyl group, (2) a halogen atom, or (3) a methyl group which may be substituted with 1 to 3 halogen atoms, and when p is 2 or 3, the multiple R 6a's may be the same or different, and the other symbols have the same meanings as defined above.)
本開示において、R2として好ましい一実施形態は、 In the present disclosure, a preferred embodiment of R2 is

(式中、m-1は1または2を表し、その他の記号は前記と同じ意味を表す。)、または (where m-1 represents 1 or 2, and the other symbols have the same meanings as above), or

(式中、m-2は1または2を表し、その他の記号は前記と同じ意味を表す。)である。 (In the formula, m-2 represents 1 or 2, and the other symbols have the same meanings as above.)
本開示において、R2として好ましい一実施形態は、 In the present disclosure, a preferred embodiment of R2 is

(式中、すべての記号は前記と同じ意味を表す。)である。 (In the formula, all symbols have the same meaning as above.)
本開示において、R6aとして好ましくは、ハロゲン原子、メチル基またはトリフルオロメチル基であり、より好ましくは、ハロゲン原子である。 In the present disclosure, R 6a is preferably a halogen atom, a methyl group or a trifluoromethyl group, and more preferably a halogen atom.
本開示において、mとして好ましくは、2である。 In this disclosure, m is preferably 2.
本開示において、m-1として好ましくは、2である。 In this disclosure, m-1 is preferably 2.
本開示において、m-2として好ましくは、1である。 In this disclosure, m-2 is preferably 1.
本開示において、pとして好ましくは、0~2であり、より好ましくは0または1であり、さらに好ましくは0である。
本開示において、R7として好ましくは、水素原子、エチル基またはイソプロピル基であり、より好ましくは水素原子である。
In the present disclosure, p is preferably 0 to 2, more preferably 0 or 1, and even more preferably 0.
In the present disclosure, R 7 is preferably a hydrogen atom, an ethyl group, or an isopropyl group, and more preferably a hydrogen atom.
本開示において、一般式(I)で示される化合物として好ましくは、一般式(I―1) In this disclosure, the compound represented by general formula (I) is preferably represented by general formula (I-1)

(式中、すべての記号は前記と同じ意味を表す。) で示される化合物である。 (wherein all symbols have the same meaning as above) is a compound represented by the formula:
本開示において、一般式(I)で示される化合物として好ましくは、一般式(I―2) In this disclosure, the compound represented by general formula (I) is preferably represented by general formula (I-2)

(式中、すべての記号は前記と同じ意味を表す。) で示される化合物である。 (wherein all symbols have the same meaning as above) is a compound represented by the formula:
本開示において、一般式(I)で示される化合物として好ましくは、一般式(II) In the present disclosure, the compound represented by general formula (I) is preferably represented by general formula (II)

(式中、すべての記号は前記と同じ意味を表す。) で示される化合物である。 (wherein all symbols have the same meaning as above) is a compound represented by the formula:
本開示において、一般式(I)で示される化合物として好ましくは、一般式(II-1) In the present disclosure, the compound represented by general formula (I) is preferably represented by general formula (II-1)

(式中、すべての記号は前記と同じ意味を表す。) で示される化合物である。 (wherein all symbols have the same meaning as above) is a compound represented by the formula:
本開示において、一般式(I)で示される化合物として好ましくは、一般式(II-2) In the present disclosure, the compound represented by general formula (I) is preferably represented by general formula (II-2)

(式中、記号は前記と同じ意味を表す。) で示される化合物である。 (wherein the symbols have the same meanings as above) is a compound represented by the formula:
本開示において、一般式(I)で示される化合物として好ましくは、一般式(II-3) In the present disclosure, the compound represented by general formula (I) is preferably represented by general formula (II-3)

(式中、R2aは (Wherein, R 2a is

(式中、記号は前記と同じ意味を表す。)、または (wherein the symbols have the same meanings as above), or

(式中、記号は前記と同じ意味を表す。)を表し、その他の記号は前記と同じ意味を表す。)で示される化合物である。 (wherein the symbols have the same meanings as above), and the other symbols have the same meanings as above. ) is a compound represented by the formula:
本開示において、一般式(I)で示される化合物として好ましくは、前記の一般式を含む各々の基の好ましい定義の組み合わせである。 In the present disclosure, the compound represented by general formula (I) is preferably a combination of the preferred definitions of each group including the general formula described above.
本開示において、一般式(I)で示される化合物の別の態様として最も好ましくは、後記の実施例に記載の実施例化合物またはその塩である。 In the present disclosure, another embodiment of the compound represented by general formula (I) is most preferably a compound of the examples described in the examples below, or a salt thereof.
本開示化合物は、GCL阻害活性を有する。本開示において、GCL阻害活性は、一実施形態において、GCLC阻害活性である。本開示化合物は、一実施形態において、BSOと同等以上のGCL阻害活性を有する。本開示化合物は、一実施形態において、BSOよりも細胞内でのGCL阻害作用が強い。本開示化合物は、一実施形態において、BSOよりも細胞内でのGCL阻害作用が約2倍以上、約3倍以上、約4倍以上、約5倍以上または約10倍以上強い。細胞内でのGCL阻害作用は、一般的な測定方法を用いて確認することができ、例えば、後述の薬理実施例2に記載の方法により測定することができる。 The disclosed compound has GCL inhibitory activity. In one embodiment, the GCL inhibitory activity in the present disclosure is GCLC inhibitory activity. In one embodiment, the disclosed compound has GCL inhibitory activity equal to or greater than that of BSO. In one embodiment, the disclosed compound has a stronger GCL inhibitory effect in cells than BSO. In one embodiment, the disclosed compound has a stronger GCL inhibitory effect in cells than BSO, about 2 times or more, about 3 times or more, about 4 times or more, about 5 times or more, or about 10 times or more. The GCL inhibitory effect in cells can be confirmed using a general measurement method, and can be measured, for example, by the method described in Pharmacological Example 2 below.
本開示化合物は、一実施形態において、薬物動態に優れる。動態に関する各パラメーター(AUC、CLtotal、T1/2および/またはBA等)は、一般的な測定方法を用いて確認することができる。 In one embodiment, the disclosed compound has excellent pharmacokinetics. Each pharmacokinetic parameter (AUC, CLtotal, T1/2 and/or BA, etc.) can be confirmed using a common measurement method.
本開示化合物は、一実施形態において、BSOよりも、強いインビボ作用を有し、BSOよりも低濃度から有効性を発揮する。インビボ作用(有効性等)は、例えば、一般的な測定方法を用いて確認することができ、例えば、後述の薬理実施例3に記載の方法により測定することができる。 In one embodiment, the disclosed compound has a stronger in vivo effect than BSO and is effective at a lower concentration than BSO. The in vivo effect (effectiveness, etc.) can be confirmed, for example, using a general measurement method, and can be measured, for example, by the method described in Pharmacological Example 3 below.
本開示においては、特に指示しない限り異性体はこれをすべて包含する。例えば、アルキル基、アルコキシ基およびアルキレン基などには直鎖のものおよび分岐鎖のものが含まれる。さらに、二重結合、環、縮合環における異性体(E、Z、シス、トランス体)、不斉炭素の存在などによる異性体(R、S体、α、β体、エナンチオマー、ジアステレオマー)、スルホキシイミン(Sulfoximine)の硫黄原子(不斉)の存在による異性体(R、S体、エナンチオマー、ジアステレオマー)、旋光性を有する光学活性体(D、L、d、l体)、クロマトグラフ分離による極性体(高極性体、低極性体)、平衡化合物、回転異性体、これらの任意の割合の混合物、ラセミ混合物は、すべて本開示に含まれる。また、本開示においては、互変異性による異性体をもすべて包含する。 In this disclosure, all isomers are included unless otherwise specified. For example, alkyl groups, alkoxy groups, and alkylene groups include straight-chain and branched-chain isomers. In addition, isomers in double bonds, rings, and condensed rings (E, Z, cis, and trans isomers), isomers due to the presence of asymmetric carbons (R, S, α, β, enantiomers, and diastereomers), isomers due to the presence of sulfur atoms (asymmetric) in sulfoximine (R, S, enantiomers, and diastereomers), optically active isomers with optical rotation (D, L, d, and l isomers), polar isomers (high polarity, low polarity) obtained by chromatographic separation, equilibrium compounds, rotational isomers, mixtures of these in any ratio, and racemic mixtures are all included in this disclosure. In addition, in this disclosure, all isomers due to tautomerism are also included.
本開示においては、特に断わらない限り、当業者にとって明らかなように記号 In this disclosure, unless otherwise specified, symbols that are clear to those skilled in the art
![]()
![]()
は紙面の向こう側(すなわちα-配置)に結合していることを表し、 indicates that it is bonded to the other side of the paper (i.e., α-configuration),
![]()
![]()
は紙面の手前側(すなわちβ-配置)に結合していることを表し、 indicates that it is bonded to the front side of the paper (i.e., β-configuration),
![]()
![]()
は、α-配置とβ-配置の任意の混合物であることを表す。
[塩]
一般式(I)等で示される化合物は、公知の方法で塩に変換される。
represents any mixture of α- and β-configurations.
[salt]
The compound represented by the formula (I) etc. can be converted into a salt by a known method.
塩として好ましくは薬学的に許容される塩である。 The salt is preferably a pharma- ceutically acceptable salt.
塩は、水溶性のものが好ましい。 The salt is preferably water-soluble.
薬学的に許容される塩としては、例えば、酸付加塩、アルカリ金属塩、アルカリ土類金属塩、アンモニウム塩またはアミン塩などが挙げられる。 Examples of pharma- ceutically acceptable salts include acid addition salts, alkali metal salts, alkaline earth metal salts, ammonium salts, or amine salts.
酸付加塩としては、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、リン酸塩および硝酸塩のような無機酸塩または酢酸塩、乳酸塩、酒石酸塩、安息香酸塩、クエン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、トリフルオロ酢酸塩、ベンゼンスルホン酸塩、トルエンスルホン酸塩、イセチオン酸塩、グルクロン酸塩、およびグルコン酸塩のような有機酸塩が挙げられる。 Acid addition salts include, for example, inorganic acid salts such as hydrochloride, hydrobromide, hydroiodide, sulfate, phosphate and nitrate, or organic acid salts such as acetate, lactate, tartrate, benzoate, citrate, methanesulfonate, ethanesulfonate, trifluoroacetate, benzenesulfonate, toluenesulfonate, isethionate, glucuronate and gluconate.
アルカリ金属塩としては、例えば、カリウム塩およびナトリウム塩などが挙げられる。 Examples of alkali metal salts include potassium salts and sodium salts.
アルカリ土類金属塩としては、例えば、カルシウム塩およびマグネシウム塩などが挙げられる。 Examples of alkaline earth metal salts include calcium salts and magnesium salts.
アンモニウム塩としては、例えば、テトラメチルアンモニウム塩などが挙げられる。 Examples of ammonium salts include tetramethylammonium salts.
アミン塩としては、例えば、トリエチルアミン塩、メチルアミン塩、ジメチルアミン塩、シクロペンチルアミン塩、ベンジルアミン塩、フェネチルアミン塩、ピペリジン塩、モノエタノールアミン塩、ジエタノールアミン塩、トリス(ヒドロキシメチル)アミノメタン塩、リジン塩、アルギニン塩およびN-メチル-D-グルカミン塩などが挙げられる。 Examples of amine salts include triethylamine salts, methylamine salts, dimethylamine salts, cyclopentylamine salts, benzylamine salts, phenethylamine salts, piperidine salts, monoethanolamine salts, diethanolamine salts, tris(hydroxymethyl)aminomethane salts, lysine salts, arginine salts, and N-methyl-D-glucamine salts.
また、本開示化合物は、任意の方法でN-オキシド体にすることができる。N-オキシド体とは、一般式(I)等で示される化合物の窒素原子が、酸化されたものを表す。 The disclosed compounds can be converted into N-oxides by any method. An N-oxide refers to a compound in which the nitrogen atom of a compound represented by general formula (I) or the like is oxidized.
一般式(I)等で示される化合物およびその塩は、溶媒和していない形態で存在してもよいし、水、エタノールなどの薬学的に許容できる溶媒と溶媒和した形態で存在してもよい。溶媒和物として好ましくは水和物である。一般式(I)等で示される化合物およびその塩は、溶媒和物に変換することができる。 The compounds represented by general formula (I) and their salts may exist in a non-solvated form or in a solvated form with a pharma- ceutically acceptable solvent such as water or ethanol. The solvates are preferably hydrates. The compounds represented by general formula (I) and their salts may be converted into solvates.
一般式(I)等で示される化合物は、適切な共結晶形成剤と共結晶を形成することができる。共結晶としては、薬学的に許容される共結晶形成剤と形成される、薬学的に許容されるものが好ましい。共結晶は、典型的に、2種以上の異なる分子がイオン結合とは異なる分子間相互作用で形成される結晶として定義される。また、共結晶は中性分子と塩の複合体であってもよい。共結晶は、公知の方法、例えば、融解結晶化により、溶媒からの再結晶により、または成分を一緒に物理的に粉砕することにより、調製することができる。適当な共結晶形成剤としては、WO2006/007448に記載のものを含む。 The compounds of formula (I) and the like can form co-crystals with a suitable co-crystal former. The co-crystal is preferably a pharma- ceutically acceptable one formed with a pharma- ceutically acceptable co-crystal former. A co-crystal is typically defined as a crystal formed by two or more different molecules with intermolecular interactions other than ionic bonds. A co-crystal may also be a complex of a neutral molecule and a salt. Co-crystals can be prepared by known methods, such as melt crystallization, recrystallization from a solvent, or by physically grinding the components together. Suitable co-crystal formers include those described in WO2006/007448.
本開示において、本開示化合物に関する言及はすべて、一般式(I)等で示される化合物、その塩、そのN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶、または一般式(I)等で示される化合物の塩のN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶を包含する。
[プロドラッグ]
一般式(I)等で示される化合物のプロドラッグとは、生体内において酵素や胃酸などによる反応により一般式(I)等で示される化合物に変換される化合物をいう。一般式(I)等で示される化合物のプロドラッグとしては、例えば、一般式(I)等で示される化合物がアミノ基を有する場合、該アミノ基がアシル化、アルキル化、リン酸化された化合物(例えば、一般式(I)等で示される化合物のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化、アセトキシメチル化、tert-ブチル化された化合物など);一般式(I)等で示される化合物が水酸基を有する場合、該水酸基がアシル化、アルキル化、リン酸化、ホウ酸化された化合物(例えば、一般式(I)等で示される化合物の水酸基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化、ジメチルアミノメチルカルボニル化された化合物など)が挙げられ;一般式(I)等で示される化合物がカルボキシ基を有する場合、該カルボキシ基がエステル化、アミド化された化合物(例えば、一般式(I)等で示される化合物のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、1-{(エトキシカルボニル)オキシ}エチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、1-{[(シクロヘキシルオキシ)カルボニル]オキシ}エチルエステル化、メチルアミド化された化合物など)などが挙げられる。これらの化合物はそれ自体公知の方法によって製造することができる。また、一般式(I等)で示される化合物のプロドラッグは水和物および非水和物のいずれであってもよい。また、一般式(I)等で示される化合物のプロドラッグは、廣川書店1990年刊「医薬品の開発」第7巻「分子設計」163~198頁に記載されているような、生理的条件において一般式(I)で示される化合物に変化するものであってもよい。
In this disclosure, all references to the disclosed compounds include a compound represented by general formula (I) or the like, a salt thereof, an N-oxide thereof, a solvate (e.g., hydrate) thereof, or a co-crystal thereof, or an N-oxide of a salt of a compound represented by general formula (I) or the like, a solvate (e.g., hydrate) thereof, or a co-crystal thereof.
[Prodrug]
The prodrug of the compound represented by the general formula (I) etc. refers to a compound which is converted into the compound represented by the general formula (I) etc. by a reaction with an enzyme, gastric acid or the like in a living body. Examples of the prodrug of the compound represented by the general formula (I) etc. include, when the compound represented by the general formula (I) etc. has an amino group, a compound in which the amino group has been acylated, alkylated or phosphorylated (for example, a compound in which the amino group of the compound represented by the general formula (I) etc. has been eicosanoylated, alanylated, pentylaminocarbonylated, (5-methyl-2-oxo-1,3-dioxolen-4-yl)methoxycarbonylated, tetrahydrofuranylated, pyrrolidylmethylated, pivaloyloxymethylated, acetoxymethylated or tert-butylated); when the compound represented by the general formula (I) etc. has a hydroxyl group, a compound in which the hydroxyl group has been acylated, alkylated, phosphorylated or borated (for example, a compound in which the hydroxyl group of the compound represented by the general formula (I) etc. has been acetylated, palmitoylated or propanoylated). yl, pivaloylated, succinylated, fumarylated, alanylated, dimethylaminomethylcarbonylated compounds, etc.); when the compound represented by the general formula (I) etc. has a carboxy group, the carboxy group is esterified or amidated (for example, the carboxy group of the compound represented by the general formula (I) etc. is ethyl esterified, phenyl esterified, carboxymethyl esterified, dimethylaminomethyl esterified, pivaloyloxymethyl esterified, 1-{(ethoxycarbonyl)oxy}ethyl esterified, phthalidyl esterified, (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl esterified, 1-{[(cyclohexyloxy)carbonyl]oxy}ethyl esterified, methylamidated compounds, etc.). These compounds can be produced by known methods. The prodrug of the compound represented by the general formula (I etc.) may be either a hydrate or a non-hydrate. In addition, the prodrug of the compound represented by the general formula (I) etc. may be one which is converted into the compound represented by the general formula (I) under physiological conditions, as described in "Drug Development", Vol. 7, "Molecular Design", pp. 163-198, published by Hirokawa Shoten in 1990.
さらに、一般式(I)等で示される化合物を構成する各原子は、その同位元素(例えば、2H、3H、11C、13C、14C、15N、16N、17O、18O、18F、35S、36Cl、77Br、125Iなど)などで置換されていてもよい。
[本開示化合物の製造方法]
本開示化合物は、公知の方法、例えば、以下に示す方法、これらに準ずる方法、Comprehensive Organic Transformations:A Guide to Functional Group Preparations、3rd Edition(Richard C.Larock、John Wiley & Sons Inc、2018)に記載された方法または実施例に示す方法等を適宜改良し、組み合わせて用いることで製造することができる。出発原料は、塩を用いてもよい。それぞれの反応を行う順序は、導入されている保護基や反応条件によって適宜入れ替えることができる。
Furthermore, each atom constituting the compound represented by general formula (I) etc. may be substituted with its isotope (e.g., 2H , 3H , 11C , 13C , 14C , 15N , 16N, 17O, 18O , 18F , 35S , 36Cl , 77Br , 125I , etc.).
[Method of producing the disclosed compound]
The disclosed compound can be produced by appropriately improving and combining known methods, such as the methods shown below, methods similar thereto, the methods described in Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 3rd Edition (Richard C. Larock, John Wiley & Sons Inc, 2018), or the methods shown in the Examples. The starting material may be a salt. The order of carrying out each reaction can be appropriately changed depending on the protective group introduced and the reaction conditions.
また、アミノ基、カルボキシル基または水酸基を有する化合物は、必要に応じて、これらの基に対して汎用される保護基、例えば、T.W.Greene、Protective Groups in Organic Synthesis、Wiley、New York、5th Edition、2014に記載の保護基で保護された化合物を用いて、適切な反応工程の後、公知の脱保護反応を行って製造することができる。 Compounds having an amino group, a carboxyl group, or a hydroxyl group can be produced, if necessary, by using a compound protected with a protecting group commonly used for these groups, for example, a protecting group described in T. W. Greene, Protective Groups in Organic Synthesis, Wiley, New York, 5th Edition, 2014, and carrying out a known deprotection reaction after an appropriate reaction step.
カルボキシル基の保護基としては、例えば、メチル、エチル、tert-ブチル、トリクロロエチル、ベンジル(Bn)、フェナシル、p-メトキシベンジル、トリチル、2-クロロトリチル等が挙げられる。 Examples of protecting groups for carboxyl groups include methyl, ethyl, tert-butyl, trichloroethyl, benzyl (Bn), phenacyl, p-methoxybenzyl, trityl, and 2-chlorotrityl.
アミノ基の保護基としては、例えば、ベンジルオキシカルボニル基、tert-ブトキシカルボニル基、アリルオキシカルボニル(Alloc)基、1-メチル-1-(4-ビフェニル)エトキシカルボニル(Bpoc)基、トリフルオロアセチル基、9-フルオレニルメトキシカルボニル基、ベンジル(Bn)基、p-メトキシベンジル基、ベンジルオキシメチル(BOM)基、2-(トリメチルシリル)エトキシメチル(SEM)基等が挙げられる。 Examples of protecting groups for amino groups include benzyloxycarbonyl, tert-butoxycarbonyl, allyloxycarbonyl (Alloc), 1-methyl-1-(4-biphenyl)ethoxycarbonyl (Bpoc), trifluoroacetyl, 9-fluorenylmethoxycarbonyl, benzyl (Bn), p-methoxybenzyl, benzyloxymethyl (BOM), and 2-(trimethylsilyl)ethoxymethyl (SEM) groups.
水酸基の保護基としては、例えば、メチル、トリチル、メトキシメチル(MOM)、1-エトキシエチル(EE)、メトキシエトキシメチル(MEM)、2-テトラヒドロピラニル(THP)、トリメチルシリル(TMS)、トリエチルシリル(TES)、tert-ブチルジメチルシリル(TBDMS)、tert-ブチルジフェニルシリル(TBDPS)、アセチル(Ac)、ピバロイル、ベンゾイル、ベンジル(Bn)、p-メトキシベンジル、アリルオキシカルボニル(Alloc)、2,2,2-トリクロロエトキシカルボニル(Troc)等が挙げられる。 Examples of hydroxyl protecting groups include methyl, trityl, methoxymethyl (MOM), 1-ethoxyethyl (EE), methoxyethoxymethyl (MEM), 2-tetrahydropyranyl (THP), trimethylsilyl (TMS), triethylsilyl (TES), tert-butyldimethylsilyl (TBDMS), tert-butyldiphenylsilyl (TBDPS), acetyl (Ac), pivaloyl, benzoyl, benzyl (Bn), p-methoxybenzyl, allyloxycarbonyl (Alloc), 2,2,2-trichloroethoxycarbonyl (Troc), etc.
脱保護反応は公知であり、以下の方法で行うことができる。例えば、
(1)アルカリ加水分解による脱保護反応、
(2)酸性条件下における脱保護反応、
(3)加水素分解による脱保護反応、
(4)シリル基の脱保護反応、
(5)金属を用いる脱保護反応、
(6)金属錯体を用いる脱保護反応、等が挙げられる。
The deprotection reaction is known and can be carried out by the following method. For example,
(1) Deprotection reaction by alkaline hydrolysis,
(2) Deprotection reaction under acidic conditions,
(3) deprotection reaction by hydrogenolysis;
(4) deprotection reaction of the silyl group,
(5) Deprotection reaction using a metal,
(6) Deprotection reaction using a metal complex, etc.
これらの方法を具体的に説明すると、
(1)アルカリ加水分解による脱保護反応は、例えば、有機溶媒(例えば、メタノール、テトラヒドロフラン(以下、THF)、ジオキサン等)中、アルカリ金属の水酸化物(例えば、水酸化ナトリウム、水酸化カリウム、水酸化リチウム等)、アルカリ土類金属の水酸化物(例えば、水酸化バリウム、水酸化カルシウム等)または炭酸塩(例えば、炭酸ナトリウム、炭酸カリウム等)あるいはその水溶液もしくはこれらの混合物を用いて、0~40℃で行われる。
To explain these methods in detail,
(1) The deprotection reaction by alkaline hydrolysis is carried out, for example, in an organic solvent (e.g., methanol, tetrahydrofuran (hereinafter, THF), dioxane, etc.) using an alkali metal hydroxide (e.g., sodium hydroxide, potassium hydroxide, lithium hydroxide, etc.), an alkaline earth metal hydroxide (e.g., barium hydroxide, calcium hydroxide, etc.) or a carbonate (e.g., sodium carbonate, potassium carbonate, etc.), or an aqueous solution thereof, or a mixture thereof, at 0 to 40° C.
(2)酸条件下での脱保護反応は、例えば、有機溶媒(例えば、ジクロロメタン、クロロホルム、ジオキサン、酢酸エチル、メタノール、イソプロピルアルコール、THF、アニソール等)中、有機酸(例えば、酢酸、トリフルオロ酢酸、メタンスルホン酸、p-トシル酸等)または無機酸(例えば、塩酸、硫酸等)もしくはこれらの混合物(例えば、臭化水素/酢酸等)中、2,2,2-トリフルオロエタノールの存在下または非存在下、0~100℃で行われる。 (2) The deprotection reaction under acidic conditions is carried out, for example, in an organic solvent (e.g., dichloromethane, chloroform, dioxane, ethyl acetate, methanol, isopropyl alcohol, THF, anisole, etc.), in an organic acid (e.g., acetic acid, trifluoroacetic acid, methanesulfonic acid, p-tosylic acid, etc.) or an inorganic acid (e.g., hydrochloric acid, sulfuric acid, etc.) or a mixture thereof (e.g., hydrogen bromide/acetic acid, etc.), in the presence or absence of 2,2,2-trifluoroethanol, at 0 to 100°C.
(3)加水素分解による脱保護反応は、例えば、溶媒(例えば、エーテル系(例えば、THF、ジオキサン、ジメトキシエタン、ジエチルエーテル等)、アルコール系(例えば、メタノール、エタノール等)、ベンゼン系(例えば、ベンゼン、トルエン等)、ケトン系(例えば、アセトン、メチルエチルケトン等)、ニトリル系(例えば、アセトニトリル等)、アミド系(例えば、N,N-ジメチルホルムアミド(以下、DMF)等)、水、酢酸エチル、酢酸またはそれらの2以上の混合溶媒等)中、触媒(例えば、パラジウム-炭素、パラジウム黒、水酸化パラジウム-炭素、酸化白金、ラネーニッケル等)の存在下、常圧または加圧下の水素雰囲気下またはギ酸アンモニウム存在下、0~200℃で行われる。 (3) The deprotection reaction by hydrogenolysis is carried out, for example, in a solvent (e.g., ethers (e.g., THF, dioxane, dimethoxyethane, diethyl ether, etc.), alcohols (e.g., methanol, ethanol, etc.), benzenes (e.g., benzene, toluene, etc.), ketones (e.g., acetone, methyl ethyl ketone, etc.), nitriles (e.g., acetonitrile, etc.), amides (e.g., N,N-dimethylformamide (DMF), etc.), water, ethyl acetate, acetic acid, or a mixture of two or more of these), in the presence of a catalyst (e.g., palladium-carbon, palladium black, palladium hydroxide-carbon, platinum oxide, Raney nickel, etc.), under normal or elevated pressure in a hydrogen atmosphere or in the presence of ammonium formate, at 0 to 200°C.
(4)シリル基の脱保護反応は、例えば、水と混和しうる有機溶媒(例えば、THF、アセトニトリル等)中、テトラブチルアンモニウムフルオライドを用いて、0~40℃で行われる。また、例えば、有機酸(例えば、酢酸、トリフルオロ酢酸、メタンスルホン酸、p-トシル酸等)または無機酸(例えば、塩酸、硫酸等)もしくはこれらの混合物(例えば、臭化水素/酢酸等)中、-10~100℃で行われる。 (4) The deprotection reaction of the silyl group is carried out, for example, in a water-miscible organic solvent (e.g., THF, acetonitrile, etc.) using tetrabutylammonium fluoride at 0 to 40°C. It is also carried out, for example, in an organic acid (e.g., acetic acid, trifluoroacetic acid, methanesulfonic acid, p-tosylic acid, etc.) or an inorganic acid (e.g., hydrochloric acid, sulfuric acid, etc.) or a mixture thereof (e.g., hydrogen bromide/acetic acid, etc.) at -10 to 100°C.
(5)金属を用いる脱保護反応は、例えば、酸性溶媒(例えば、酢酸、pH4.2~7.2の緩衝液またはそれらの溶液とTHF等の有機溶媒との混合液)中、粉末亜鉛の存在下、必要であれば超音波をかけながら、0~40℃で行われる。 (5) The deprotection reaction using a metal is carried out, for example, in an acidic solvent (e.g., acetic acid, a buffer solution having a pH of 4.2 to 7.2, or a mixture of such a solution with an organic solvent such as THF) in the presence of zinc powder at 0 to 40°C, with ultrasonication if necessary.
(6)金属錯体を用いる脱保護反応は、例えば、有機溶媒(例えば、ジクロロメタン、DMF、THF、酢酸エチル、アセトニトリル、ジオキサン、エタノール等)、水またはそれらの混合溶媒中、トラップ試薬(例えば、水素化トリブチルスズ、トリエチルシラン、ジメドン、モルホリン、ジエチルアミン、ピロリジン等)、有機酸(例えば、酢酸、ギ酸、2-エチルヘキサン酸等)および/または有機酸塩(例えば、2-エチルヘキサン酸ナトリウム、2-エチルヘキサン酸カリウム等)の存在下、ホスフィン系試薬(例えば、トリフェニルホスフィン等)の存在下または非存在下、金属錯体(例えば、テトラキストリフェニルホスフィンパラジウム(0)、二塩化ビス(トリフェニルホスフィン)パラジウム(II)、酢酸パラジウム(II)、塩化トリス(トリフェニルホスフィン)ロジウム(I)等)を用いて、0~40℃で行われる。 (6) The deprotection reaction using a metal complex is carried out, for example, in an organic solvent (e.g., dichloromethane, DMF, THF, ethyl acetate, acetonitrile, dioxane, ethanol, etc.), water or a mixture thereof, in the presence of a trapping reagent (e.g., tributyltin hydride, triethylsilane, dimedone, morpholine, diethylamine, pyrrolidine, etc.), an organic acid (e.g., acetic acid, formic acid, 2-ethylhexanoic acid, etc.) and/or an organic acid salt (e.g., sodium 2-ethylhexanoate, potassium 2-ethylhexanoate, etc.), in the presence or absence of a phosphine-based reagent (e.g., triphenylphosphine, etc.), using a metal complex (e.g., tetrakistriphenylphosphinepalladium(0), bis(triphenylphosphine)palladium(II) dichloride, palladium(II) acetate, tris(triphenylphosphine)rhodium(I) chloride, etc.), at 0 to 40°C.
一般式(I)で示される化合物は、反応工程式1で製造することができる。 The compound represented by general formula (I) can be produced by reaction scheme 1.

(反応工程式1中、PGはアミノ基の保護基を表し、Xは脱離基を表し、CO2Meはメトキシカルボニル基を表し、その他の記号は前記と同じ意味を表す。)
反応工程式1中、反応1-1はS-アルキル化反応である。S-アルキル化反応は公知であり、例えば、有機溶媒(DMF、ジメチルスルホキシド、クロロホルム、ジクロロメタン、ジエチルエーテル、THF、メチルtert-ブチルエーテル等)中、アルカリ金属の水酸化物(水酸化ナトリウム、水酸化カリウム、水酸化リチウム等)、アルカリ土類金属の水酸化物(水酸化バリウム、水酸化カルシウム等)もしくは炭酸塩(炭酸ナトリウム、炭酸カリウム等)またはその水溶液あるいはこれらの混合物の存在下、0~100℃で反応させることにより行なわれる。
(In reaction scheme 1, PG represents an amino-protecting group, X represents a leaving group, CO 2 Me represents a methoxycarbonyl group, and the other symbols have the same meanings as above.)
In reaction scheme 1, reaction 1-1 is an S-alkylation reaction. The S-alkylation reaction is known and is carried out, for example, in an organic solvent (DMF, dimethyl sulfoxide, chloroform, dichloromethane, diethyl ether, THF, methyl tert-butyl ether, etc.) in the presence of an alkali metal hydroxide (sodium hydroxide, potassium hydroxide, lithium hydroxide, etc.), an alkaline earth metal hydroxide (barium hydroxide, calcium hydroxide, etc.) or a carbonate (sodium carbonate, potassium carbonate, etc.) or an aqueous solution thereof, or a mixture thereof, at 0 to 100° C.
反応工程式1中、反応1-2はスルフィドからスルホキシイミンへの酸化反応である。例えば、有機溶媒(メタノール(MeOH))中、二酢酸ヨードベンゼンとカルバミン酸アンモニウムの存在下、0℃~室温で行われる。 In reaction scheme 1, reaction 1-2 is an oxidation reaction from sulfide to sulfoximine. For example, it is carried out in an organic solvent (methanol (MeOH)) in the presence of iodobenzene diacetate and ammonium carbamate at 0°C to room temperature.
反応工程式1中、反応1-3は脱保護反応であり、前記と同様の方法で実施することができる。 In reaction scheme 1, reactions 1-3 are deprotection reactions and can be carried out in the same manner as described above.
反応工程式1中、反応1-4は脱保護反応であり、前記と同様の方法で実施することができる。 In reaction scheme 1, reactions 1-4 are deprotection reactions and can be carried out in the same manner as described above.
本明細書中の各反応において、出発原料として用いた、一般式1a、一般式1bで示される化合物は公知であるか、あるいは公知の方法、例えば、Comprehensive Organic Transformations:A Guide to Functional Group Preparations、3rd Edition(Richard C.Larock、John Wiley & Sons Inc、2018)等に記載された方法、または公知の方法を一部改変した方法等を組み合わせて用いることで容易に製造することができる。 In each reaction in this specification, the compounds represented by general formula 1a and general formula 1b used as starting materials are publicly known or can be easily produced by combining publicly known methods, such as the methods described in Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 3rd Edition (Richard C. Larock, John Wiley & Sons Inc, 2018), or methods that are partially modified from publicly known methods.
本発明に用いられる化合物のうち、光学活性を有する化合物は、光学活性を有する出発原料または試薬を用いて製造するか、ラセミ体の製造中間体を光学分割し、次いで本開示化合物に導くか、あるいはラセミ体の本開示化合物を光学分割することで製造することができる。 Among the compounds used in the present invention, optically active compounds can be produced by using optically active starting materials or reagents, by optically resolving a racemic production intermediate and then leading to the compound disclosed herein, or by optically resolving a racemic compound disclosed herein.
この光学分割は公知であり、例えば、他の光学活性な化合物と塩・錯体などを形成させ、再結晶を行った後、目的とする化合物を単離するか、あるいは直接キラルカラムなどを用いて分離する方法などが挙げられる。 This optical resolution is well known, and examples of such methods include forming a salt or complex with another optically active compound, recrystallizing it, and then isolating the desired compound, or directly separating it using a chiral column.
本明細書中の各反応において、加熱を伴う反応は、当業者にとって明らかなように、水浴、油浴、砂浴またはマイクロウェーブを用いて行なうことができる。 In each reaction in this specification, reactions involving heating can be carried out using a water bath, oil bath, sand bath, or microwave, as will be apparent to those skilled in the art.
本明細書中の各反応において、適宜、高分子ポリマー(例えば、ポリスチレン、ポリアクリルアミド、ポリプロピレン、ポリエチレングリコール等)に担持させた固相担持試薬を用いてもよい。 In each reaction in this specification, a solid-phase supported reagent supported on a polymer (e.g., polystyrene, polyacrylamide, polypropylene, polyethylene glycol, etc.) may be used as appropriate.
本明細書中の各反応において、反応生成物は通常の精製手段、例えば、常圧下または減圧下における蒸留、シリカゲルまたはケイ酸マグネシウムを用いた高速液体クロマトグラフィー、薄層クロマトグラフィー、イオン交換樹脂、スカベンジャー樹脂あるいはカラムクロマトグラフィーまたは洗浄、再結晶などの方法により精製することができる。精製は反応ごとに行なってもよいし、いくつかの反応終了後に行なってもよい。
[毒性]
本開示化合物の毒性は低いものであるため、医薬品として安全に使用することができる。
[医薬品への適用]
本開示化合物は、GCLに対して阻害活性を有するため、GCLに関連する疾患(GCL関連疾患)、例えば、がんの有効な進行抑制、再発抑制または治療剤として処方することができる。
In each reaction in the present specification, the reaction product can be purified by a conventional purification method, for example, distillation under normal or reduced pressure, high performance liquid chromatography using silica gel or magnesium silicate, thin layer chromatography, ion exchange resin, scavenger resin or column chromatography, washing, recrystallization, etc. Purification may be carried out for each reaction or after completion of some reactions.
[toxicity]
The compounds of the present disclosure have low toxicity and can therefore be safely used as pharmaceuticals.
[Application to pharmaceuticals]
Since the compound of the present disclosure has an inhibitory activity against GCL, it can be prescribed as an effective agent for suppressing the progression or recurrence or for treating diseases associated with GCL (GCL-associated diseases), such as cancer.
本開示化合物が進行抑制、再発抑制および/または治療の対象とするがんには、何れの固形がんおよび血液がんも含まれるが、一例では、ARID1A欠損がんおよび/またはGCLに感受性を有するがん(GCL感受性がん)が挙げられる。 Cancers that the disclosed compounds are intended to inhibit progression, inhibit recurrence and/or treat include any solid cancer and blood cancer, but examples include ARID1A-deficient cancers and/or cancers sensitive to GCL (GCL-sensitive cancers).
本明細書で使用される場合、ARID1A欠損は、ARID1A遺伝子変異によって引き起こされるARID1A遺伝子欠損またはARID1Aタンパク質欠損を意味する。ARID1A欠損がんとは、ARID1A欠損を有するがんを意味する。ARID1A欠損がんの例としては、卵巣がん、子宮がん、胃がん、膀胱がん、胆管がん、肝がん、食道がん、肺がん、結腸がん、膵臓がん、乳がん、神経芽細胞腫、神経膠腫、皮膚がん、B細胞リンパ腫および腎がんなどがある。 As used herein, ARID1A deficiency refers to an ARID1A gene deficiency or ARID1A protein deficiency caused by an ARID1A gene mutation. ARID1A-deficient cancer refers to a cancer having an ARID1A deficiency. Examples of ARID1A-deficient cancers include ovarian cancer, uterine cancer, gastric cancer, bladder cancer, bile duct cancer, liver cancer, esophageal cancer, lung cancer, colon cancer, pancreatic cancer, breast cancer, neuroblastoma, glioma, skin cancer, B-cell lymphoma, and renal cancer.
GCL感受性がんとしては、例えば、急性骨髄性白血病、B細胞リンパ腫が挙げられる。 Examples of GCL-sensitive cancers include acute myeloid leukemia and B-cell lymphoma.
なお、本明細書において「がん治療」とは、例えば、(a)がん細胞の増殖を減少させるため、(b)がんに起因する症状を低減させるため、がん患者の生活の質を向上させるため、(c)既に投与されている他の抗がん剤またはがん治療補助薬の用量を低減させるため、および/または(d)がん患者の生存期間を延長させるために行われる治療を含む。また、「がんの進行抑制」とは、がんの進行を遅延、がんに関連する症状を安定化および症状の進行を後退させることを意味する。「再発抑制」とは、がん治療あるいは癌外科的切除術によってがん病変が完全にもしくは実質的に消滅または取り除かれた患者におけるがん再発を予防的に抑止することを意味する。 In this specification, "cancer treatment" includes, for example, treatments performed to (a) reduce the proliferation of cancer cells, (b) reduce symptoms caused by cancer and improve the quality of life of cancer patients, (c) reduce the dose of other anticancer drugs or cancer treatment adjuvant drugs already administered, and/or (d) extend the survival time of cancer patients. "Suppression of cancer progression" means delaying the progression of cancer, stabilizing symptoms related to cancer, and reversing the progression of symptoms. "Suppression of recurrence" means preventively suppressing cancer recurrence in patients in whom cancer lesions have completely or substantially disappeared or been removed by cancer treatment or surgical resection.
本開示化合物を上記の疾患の進行抑制、再発抑制および/または治療の目的に用いるには、有効成分である当該物質を、通常、各種の添加剤または溶媒などの薬学的に許容される担体とともに製剤化したうえで、全身的または局所的に、経口または非経口の形で投与される。ここで、薬学的に許容される担体とは、一般的に医薬品の製剤に用いられる、有効成分以外の物質を意味する。薬学的に許容される担体は、その製剤の投与量において薬理作用を示さず、無害で、有効成分の治療効果を妨げないものが好ましい。また、薬学的に許容される担体は、有効成分および製剤の有用性を高める、製剤化を容易にする、品質の安定化を図る、または使用性を向上させるなどの目的で用いることもできる。具体的には、薬事日報社2000年刊「医薬品添加物事典」(日本医薬品添加剤協会編集)などに記載されているような物質を、適宜目的に応じて選択すればよい。 To use the disclosed compound for the purpose of inhibiting the progression, inhibiting recurrence, and/or treating the above-mentioned disease, the substance, which is the active ingredient, is usually formulated with a pharma- ceutically acceptable carrier, such as various additives or solvents, and then administered systemically or locally, orally or parenterally. Here, the pharma- ceutically acceptable carrier means a substance other than the active ingredient that is generally used in the formulation of pharmaceuticals. A pharma- ceutically acceptable carrier is preferably one that does not exhibit pharmacological action at the dosage of the formulation, is harmless, and does not interfere with the therapeutic effect of the active ingredient. In addition, a pharma- ceutically acceptable carrier can also be used for the purpose of increasing the usefulness of the active ingredient and the formulation, facilitating formulation, stabilizing the quality, or improving usability. Specifically, a substance such as that described in "Dictionary of Pharmaceutical Additives" published by Yakuji Nipposha in 2000 (edited by the Japan Pharmaceutical Additives Association) may be appropriately selected according to the purpose.
投与に用いられる剤型としては、例えば、経口投与用製剤(例:錠剤、カプセル剤、顆粒剤、散剤、経口液剤、シロップ剤、経口ゼリー剤など)、口腔用製剤(例:口腔用錠剤、口腔用スプレー剤、口腔用半固形剤、含嗽剤など)、注射用製剤(例:注射剤など)、透析用製剤(例:透析用剤など)、吸入用製剤(例:吸入剤など)、眼科用製剤(例:点眼剤、眼軟膏剤など)、耳科用製剤(例:点耳剤など)、鼻科用製剤(例:点鼻剤など)、直腸用製剤(例:坐剤、直腸用半固形剤、腸注剤など)、腟用製剤(例:腟錠、腟用坐剤など)および皮膚用製剤(例:外用固形剤、外用液剤、スプレー剤、軟膏剤、クリーム剤、ゲル剤、貼付剤など)などが挙げられる。 Examples of dosage forms used for administration include oral preparations (e.g., tablets, capsules, granules, powders, oral liquids, syrups, oral jellies, etc.), oral preparations (e.g., oral tablets, oral sprays, oral semisolids, mouthwashes, etc.), injectable preparations (e.g., injectables, etc.), dialysis preparations (e.g., dialysis preparations, etc.), inhalation preparations (e.g., inhalants, etc.), ophthalmic preparations (e.g., eye drops, eye ointments, etc.), otic preparations (e.g., ear drops, etc.), nasal preparations (e.g., nasal drops, etc.), rectal preparations (e.g., suppositories, rectal semisolids, enteral injections, etc.), vaginal preparations (e.g., vaginal tablets, vaginal suppositories, etc.), and dermatological preparations (e.g., solids for external use, liquids for external use, sprays, ointments, creams, gels, patches, etc.).
[併用または配合剤]
本開示化合物または本開示化合物を有効成分として含む医薬組成物(以下、「本開示化合物等」と略記する。)は、(a)がんの進行抑制、再発抑制および/または治療効果の増強のために、(b)組み合わせて処方される他の薬剤の投与量の低減のために、(c)組み合わせて処方される他の薬剤の副作用の軽減のために、および/または(d)組み合わせて処方される他の薬剤の免疫増強作用を高めるために、すなわち、アジュバンドとして、一種以上の他の薬剤とともに組み合わせて処方してもよい。本開示において、他の薬剤(例えば、他の抗がん剤)とともに組み合わせて処方する場合の投与形態には、1つの製剤中に両成分を配合した配合剤の形態であっても、また別々の製剤としての投与形態であってもよい。その併用により、その他の薬剤の予防、症状進展抑制、再発抑制および/または治療効果を補完したり、投与量あるいは投与回数を維持ないし低減することができる。本開示化合物等と他の薬剤を別々に処方する場合には、一定期間同時投与し、その後、本開示化合物等のみあるいは他の薬剤のみを投与してもよい。また、本開示化合物等を先に投与し、その投与の後に他の薬剤を投与してもよいし、他の薬剤を先に投与し、本開示化合物等を後に投与してもよく、また、上記投与において、一定期間、両薬剤が同時に投与される期間があってもよい。また、各々の薬剤の投与方法は同じでも異なっていてもよい。薬剤の性質により、本開示化合物を含む製剤と他の薬剤を含む製剤のキットとして提供することもできる。ここで、他の薬剤の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、他の薬剤は任意の2種以上を適宜の割合で組み合わせて投与してもよい。また、前記他の薬剤には、現在までに見出されているものだけでなく今後見出されるものも含まれる。
[処方]
本開示化合物等または本開示化合物と他の薬剤の併用剤を上記の目的で用いるには、通常、全身的または局所的に、経口または非経口の形で投与される。投与量は、年齢、体重、症状、治療効果、投与方法、処理時間等により異なるが、通常、成人一人当たり、一回につき、1ngから2,000mgの範囲で一日一回から数回経口投与されるか、または成人一人当たり、一回につき、0.1ngから200mgの範囲で一日一回から数回非経口投与されるか、または一日30分から24時間の範囲で静脈内に持続投与される。もちろん前記したように、投与量は種々の条件により変動するため、上記投与量より少ない量で十分な場合もあるし、また範囲を越えて投与の必要な場合もある。
[Concomitant or combination drug]
The disclosed compound or a pharmaceutical composition containing the disclosed compound as an active ingredient (hereinafter abbreviated as "the disclosed compound, etc.") may be formulated in combination with one or more other drugs for the purposes of (a) suppressing the progression, suppressing recurrence, and/or enhancing the therapeutic effect of cancer, (b) reducing the dosage of other drugs prescribed in combination, (c) reducing the side effects of other drugs prescribed in combination, and/or (d) enhancing the immune enhancing effect of other drugs prescribed in combination, i.e., as an adjuvant. In the present disclosure, the dosage form when formulated in combination with other drugs (e.g., other anticancer drugs) may be a combination drug form in which both components are formulated in one formulation, or may be an administration form as separate formulations. By using the compound in combination, it is possible to complement the preventive, symptom progression suppressive, recurrence suppressive, and/or therapeutic effect of the other drug, or to maintain or reduce the dosage or number of administrations. When the disclosed compound, etc. and the other drug are formulated separately, they may be administered simultaneously for a certain period of time, and then only the disclosed compound, etc. or only the other drug may be administered. In addition, the compound disclosed herein may be administered first, and then the other drug may be administered, or the other drug may be administered first, and then the compound disclosed herein may be administered, or in the above administration, there may be a period during which both drugs are administered simultaneously for a certain period. In addition, the administration method of each drug may be the same or different. Depending on the nature of the drug, it may be provided as a kit of a formulation containing the compound disclosed herein and a formulation containing the other drug. Here, the dosage of the other drug may be appropriately selected based on the dose used in clinical practice. In addition, any two or more types of other drugs may be administered in combination at an appropriate ratio. In addition, the other drugs include not only those that have been discovered so far, but also those that will be discovered in the future.
[Prescription]
To use the disclosed compounds or the like or the combination of the disclosed compounds and other drugs for the above purpose, they are usually administered systemically or locally, orally or parenterally. The dosage varies depending on age, body weight, symptoms, therapeutic effect, administration method, treatment time, etc., but is usually orally administered once to several times a day in the range of 1ng to 2,000mg per dose per adult, or parenterally administered once to several times a day in the range of 0.1ng to 200mg per dose per adult, or continuously administered intravenously for 30 minutes to 24 hours a day. Of course, as mentioned above, the dosage varies depending on various conditions, so in some cases a dosage less than the above dosage is sufficient, and in other cases it is necessary to administer beyond the range.
本開示化合物は、薬学的有効量で哺乳動物(好ましくはヒト、より好ましくはヒト患者)へ投与される。 The disclosed compounds are administered to a mammal (preferably a human, more preferably a human patient) in a pharma- tically effective amount.
他に定義されない限り、本明細書中で使用される全ての技術的、科学的用語および略語は、本発明の分野に属する当業者によって普通に理解されるものと同様の意味を有する。 Unless otherwise defined, all technical and scientific terms and abbreviations used herein have the same meaning as commonly understood by one of ordinary skill in the art of the present invention.
また、本明細書において、明示的に引用される全ての特許文献および非特許文献もしくは参考文献の内容は、全て本明細書の一部としてここに引用し得る。 In addition, the contents of all patent and non-patent literature or references explicitly cited in this specification may be incorporated herein by reference.
本開示は、一態様において、下記の実施態様を提供する。
[1] 一般式(I)
In one aspect, the present disclosure provides the following embodiments.
[1] General formula (I)

(式中、
R1は、水素原子、メチル基またはヒドロキシル基を表し、
R2は、(1)-C(R3R4)-R5または(2)1~9個のR6で置換されていてもよいC3~C5シクロアルキル基を表し、
R3またはR4は、それぞれ、(1)水素原子、(2)ヒドロキシル基、(3)ハロゲン原子または(4)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、
R5は、(1)トリフルオロメチル基または(2)tert-ブチル基を表し、
R6は、(1)ヒドロキシル基、(2)ハロゲン原子または(3)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、R6が複数の場合、複数のR6はそれぞれ同じでも異なっていてもよく、
R7は、水素原子またはC1~C4アルキル基を表し、
nは0または1を表す。)で示される化合物またはその塩、
[2] R2が、(1)-C(R3R4)-CF3または(2)1~4個のR6で置換されていてもよいC3~C5シクロアルキル基(好ましくは、1~4個(好ましくは1~3個、より好ましくは1または2個)のR6で置換されていてもよいシクロブチル基)である、前記[1]記載の化合物またはその塩、
[3] R3が、水素原子、ハロゲン原子またはヒドロキシル基(より好ましくは、水素原子またはヒドロキシル基)である、前記[1]または[2]に記載の化合物またはその塩、
[4] R6が、ヒドロキシル基、ハロゲン原子、メチル基またはトリフルオロメチル基(より好ましくは、ヒドロキシル基またはハロゲン原子)である、前記[1]~[3]のいずれかに記載の化合物またはその塩、
[5] R2が、
(Wherein,
R 1 represents a hydrogen atom, a methyl group or a hydroxyl group;
R2 represents (1) -C( R3R4 ) -R5 or (2) a C3-C5 cycloalkyl group optionally substituted with 1 to 9 R6s ;
R3 and R4 each represent (1) a hydrogen atom, (2) a hydroxyl group, (3) a halogen atom, or (4) a methyl group optionally substituted with 1 to 3 halogen atoms;
R5 represents (1) a trifluoromethyl group or (2) a tert-butyl group;
R 6 represents (1) a hydroxyl group, (2) a halogen atom, or (3) a methyl group which may be substituted by 1 to 3 halogen atoms, and when there are a plurality of R 6s , the plurality of R 6s may be the same or different;
R7 represents a hydrogen atom or a C1-C4 alkyl group;
n represents 0 or 1; or a salt thereof,
[2] The compound according to the above [1], wherein R 2 is (1) -C(R 3 R 4 )-CF 3 or (2) a C3-C5 cycloalkyl group optionally substituted with 1 to 4 R 6 (preferably a cyclobutyl group optionally substituted with 1 to 4 (preferably 1 to 3, more preferably 1 or 2) R 6 ), or a salt thereof.
[3] The compound or salt thereof according to the above [1] or [2], wherein R3 is a hydrogen atom, a halogen atom or a hydroxyl group (more preferably a hydrogen atom or a hydroxyl group).
[4] The compound or salt thereof according to any one of the above [1] to [3], wherein R 6 is a hydroxyl group, a halogen atom, a methyl group or a trifluoromethyl group (more preferably a hydroxyl group or a halogen atom).
[5] R2 is,

(式中、記号は前記[1]と同じ意味を表す。)、または (wherein the symbols have the same meaning as in [1] above), or

(式中、mは1~3の整数を表し、pは0~3(好ましくは0~2)の整数を表し、R6aは、(1)ヒドロキシル基、(2)ハロゲン原子または(3)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、pが2または3のとき、複数のR6aはそれぞれ同じでも異なっていてもよい。)である、前記[1]または[2]に記載の化合物またはその塩、
[6] R2が、
(wherein m represents an integer of 1 to 3, p represents an integer of 0 to 3 (preferably 0 to 2), R 6a represents (1) a hydroxyl group, (2) a halogen atom, or (3) a methyl group which may be substituted by 1 to 3 halogen atoms, and when p is 2 or 3, a plurality of R 6a may be the same or different from each other), or a salt thereof according to the above [1] or [2].
[6] R2 is,

(式中、記号は前記[1]と同じ意味を表す。)である、前記[5]記載の化合物またはその塩、
[7] R4が、水素原子、ハロゲン原子、メチル基またはトリフルオロメチル基(より好ましくは、水素原子、メチル基またはトリフルオロメチル基)である、前記[1]~[6]のいずれかに記載の化合物またはその塩、
[8] R2が、
(wherein the symbols have the same meanings as in the above [1]), or a salt thereof,
[7] The compound or salt thereof according to any one of the above [1] to [6], wherein R 4 is a hydrogen atom, a halogen atom, a methyl group, or a trifluoromethyl group (more preferably a hydrogen atom, a methyl group, or a trifluoromethyl group).
[8] R2 is,

(式中、記号は前記[5]と同じ意味を表す。)である、前記[5]記載の化合物またはその塩、
[9] R6aが、ハロゲン原子、メチル基またはトリフルオロメチル基(より好ましくはハロゲン原子)である、前記[5]または[8]に記載の化合物またはその塩、
[10] mが2である、前記[5]、[8]または[9]に記載の化合物またはその塩、
[11] nが1である、前記[1]~[10]のいずれかに記載の化合物またはその塩、
[12] R1が水素原子である、前記[1]~[11]のいずれかに記載の化合物またはその塩、
[13] 一般式(I)で示される化合物が、一般式(II-3)
(wherein the symbols have the same meanings as in the above [5]), or a salt thereof,
[9] The compound or salt thereof according to the above [5] or [8], wherein R 6a is a halogen atom, a methyl group or a trifluoromethyl group (more preferably a halogen atom).
[10] The compound or salt thereof according to the above [5], [8] or [9], wherein m is 2.
[11] The compound or salt thereof according to any one of the above [1] to [10], wherein n is 1.
[12] The compound or salt thereof according to any one of the above [1] to [11], wherein R 1 is a hydrogen atom.
[13] The compound represented by general formula (I) is represented by general formula (II-3)

(式中、R2aは (Wherein, R 2a is

(式中、記号は前記[1]と同じ意味を表す。)、または (wherein the symbols have the same meanings as in [1] above), or

(式中、記号は前記[5]と同じ意味を表す。)を表し、その他の記号は前記[1]と同じ意味を表す。)で示される化合物である、前記[1]記載の化合物またはその塩、
[14] R4が、水素原子、ハロゲン原子、メチル基またはトリフルオロメチル基(より好ましくは、水素原子、メチル基またはトリフルオロメチル基)である、前記[13]に記載の化合物またはその塩、
[15] R6aが、ハロゲン原子、メチル基またはトリフルオロメチル基(より好ましくはハロゲン原子)であり、pが0または1である、前記[13]に記載の化合物またはその塩、
[16] R7が水素原子、エチル基またはイソプロピル基(より好ましくは水素原子)である、前記[1]~[15]のいずれかに記載の化合物またはその塩、
[17] 一般式(I)で示される化合物が、
(1)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタン酸、
(2)(2S)-2-アミノ-4-[S-(2-シクロペンチルエチル)スルホンイミドイル]ブタン酸、
(3)(2S)-2-アミノ-4-[S-(2-シクロブチルエチル)スルホンイミドイル]ブタン酸、
(4)(2S)-2-アミノ-4-[S-(2-シクロプロピルエチル)スルホンイミドイル]ブタン酸、
(5)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-メチルブチル)スルホンイミドイル]ブタン酸、
(6)(2S)-2-アミノ-4-{S-[2-(3,3-ジフルオロシクロブチル)エチル]スルホンイミドイル}ブタン酸、
(7)(2S)-2-アミノ-4-[S-(4,4-ジメチルペンチル)スルホンイミドイル]ブタン酸、
(8)(2S)-2-アミノ-4-{S-[2-(1-ヒドロキシシクロブチル)エチル]スルホンイミドイル}ブタン酸、
(9)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-ヒドロキシブチル)スルホンイミドイル]ブタン酸、
(10)(2S)-2-アミノ-4-{S-[2-(1-フルオロシクロブチル)エチル]スルホンイミドイル}ブタン酸、
(11)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-ヒドロキシ-3-メチルブチル)スルホンイミドイル]ブタン酸、
(12)(2S)-2-アミノ-4-{S-[4,4,4-トリフルオロ-3-ヒドロキシ-3-(トリフルオロメチル)ブチル]スルホンイミドイル}ブタン酸、
(13)(2S)-2-アミノ-4-[S-(3,3,4,4,4-ペンタフルオロブチル)スルホンイミドイル]ブタン酸、
(14)(S)-2-アミノ-4-((R,3R)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸、
(15)(S)-2-アミノ-4-((R,3S)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸、
(16)(S)-2-アミノ-4-((R)-2-(1-ヒドロキシシクロブチル)エチルスルホンイミドイル)ブタン酸、
(17)(S)-2-アミノ-4-((S)-4,4,4-トリフルオロブチルスルホンイミドイル)ブタン酸、
(18)エチル(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート、および
(19)イソプロピル(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート
からなる群から選択される化合物である、前記[1]記載の化合物またはその塩、
[18] 前記[1]記載の一般式(I)で示される化合物またはその塩を含有する医薬組成物、
[19] GCL阻害剤である、前記[18]記載の医薬組成物、
[20] GCL関連疾患(例えば、がん)の進行抑制、再発抑制および/または治療剤である、前記[18]または [19]記載の医薬組成物、
[21] 前記[1]記載の一般式(I)で示される化合物またはその塩の有効量を、がんの進行抑制、再発抑制および/または治療を必要とする患者に投与することを特徴とする、がんの進行抑制、再発抑制および/または治療方法、
[22] がんの進行抑制、再発抑制および/または治療に使用される、前記[1]記載の一般式(I)で示される化合物またはその塩、ならびに
[23] がんの進行抑制、再発抑制および/または治療剤を製造するための、前記[1]記載の一般式(I)で示される化合物またはその塩の使用。
(wherein the symbols have the same meanings as in the above [5]), and the other symbols have the same meanings as in the above [1]),
[14] The compound or salt thereof according to the above [13], wherein R 4 is a hydrogen atom, a halogen atom, a methyl group or a trifluoromethyl group (more preferably a hydrogen atom, a methyl group or a trifluoromethyl group).
[15] The compound or salt thereof according to the above [13], wherein R 6a is a halogen atom, a methyl group or a trifluoromethyl group (more preferably a halogen atom), and p is 0 or 1.
[16] The compound or salt thereof according to any one of the above [1] to [15], wherein R 7 is a hydrogen atom, an ethyl group or an isopropyl group (more preferably a hydrogen atom).
[17] The compound represented by formula (I),
(1) (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoic acid,
(2) (2S)-2-amino-4-[S-(2-cyclopentylethyl)sulfonimidoyl]butanoic acid,
(3) (2S)-2-amino-4-[S-(2-cyclobutylethyl)sulfonimidoyl]butanoic acid,
(4) (2S)-2-amino-4-[S-(2-cyclopropylethyl)sulfonimidoyl]butanoic acid,
(5) (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-methylbutyl)sulfonimidoyl]butanoic acid,
(6) (2S)-2-amino-4-{S-[2-(3,3-difluorocyclobutyl)ethyl]sulfonimidoyl}butanoic acid,
(7) (2S)-2-amino-4-[S-(4,4-dimethylpentyl)sulfonimidoyl]butanoic acid,
(8) (2S)-2-amino-4-{S-[2-(1-hydroxycyclobutyl)ethyl]sulfonimidoyl}butanoic acid,
(9) (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-hydroxybutyl)sulfonimidoyl]butanoic acid,
(10) (2S)-2-amino-4-{S-[2-(1-fluorocyclobutyl)ethyl]sulfonimidoyl}butanoic acid,
(11) (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-hydroxy-3-methylbutyl)sulfonimidoyl]butanoic acid,
(12) (2S)-2-amino-4-{S-[4,4,4-trifluoro-3-hydroxy-3-(trifluoromethyl)butyl]sulfonimidoyl}butanoic acid,
(13) (2S)-2-amino-4-[S-(3,3,4,4,4-pentafluorobutyl)sulfonimidoyl]butanoic acid,
(14) (S)-2-amino-4-((R,3R)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid,
(15) (S)-2-amino-4-((R,3S)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid,
(16) (S)-2-amino-4-((R)-2-(1-hydroxycyclobutyl)ethylsulfonimidoyl)butanoic acid,
(17) (S)-2-amino-4-((S)-4,4,4-trifluorobutylsulfonimidoyl)butanoic acid,
(18) the compound according to the above-mentioned [1], which is a compound selected from the group consisting of ethyl (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate, and (19) isopropyl (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate, or a salt thereof.
[18] A pharmaceutical composition comprising the compound represented by the general formula (I) or a salt thereof according to [1] above.
[19] The pharmaceutical composition according to the above [18], which is a GCL inhibitor.
[20] The pharmaceutical composition according to the above [18] or [19], which is an agent for inhibiting the progression, inhibiting the recurrence and/or treating a GCL-related disease (e.g., cancer).
[21] A method for inhibiting progression, suppressing recurrence and/or treating cancer, comprising administering an effective amount of the compound represented by the general formula (I) described in [1] above or a salt thereof to a patient in need of such inhibition, suppression of recurrence and/or treatment of cancer.
[22] A compound represented by the general formula (I) or a salt thereof according to the above [1], which is used for inhibiting the progression, inhibiting recurrence and/or treating cancer, and
[23] Use of the compound represented by the general formula (I) or a salt thereof described in the above [1] for the manufacture of an agent for inhibiting the progression, inhibiting the recurrence and/or treating cancer.
本開示は、一態様において、下記の実施態様を提供するが、本発明の範囲はこれに限定されない。本開示の記載に基づき種々の変更または修飾が当業者には可能であり、これらの変更または修飾も本発明に含まれる。
[合成実施例]
クロマトグラフィーによる分離の箇所およびTLCに示されるカッコ内の溶媒は、使用した溶出溶媒または展開溶媒を示し、割合は体積比を表す。
In one aspect, the present disclosure provides the following embodiments, but the scope of the present invention is not limited thereto. Based on the description of the present disclosure, various changes or modifications can be made by those skilled in the art, and these changes or modifications are also included in the present invention.
[Synthesis Example]
The solvents in parentheses shown in the chromatographic separations and TLCs indicate the eluting or developing solvents used, and the ratios are by volume.
NMRの箇所に示されているカッコ内の溶媒は、測定に使用した溶媒を示す。 The solvent in parentheses in the NMR section indicates the solvent used in the measurement.
本明細書中に用いた化合物名は、一般的にIUPACの規則に準じて命名を行なうコンピュータプログラム、ACD/Name(登録商標)を用いるか、ChemDraw Professional(バージョン18.0、PerkinElmer社製)を用いるか、またはIUPAC命名法に準じて命名したものである。 The compound names used in this specification were created using ACD/Name (registered trademark), a computer program that generally performs naming according to IUPAC rules, or ChemDraw Professional (Version 18.0, PerkinElmer), or in accordance with the IUPAC nomenclature system.
LC-MS/ELSDは、下記条件で行った。
条件A;カラム:YMC triart C18(粒子径:1.9 x 10-6 m, カラム長:30 x 2.0 mm I.D.);流速:1.0mL/min;カラム温度:30℃;移動相(A):0.1%トリフルオロ酢酸水溶液;移動相(B):0.1%TFA-アセトニトリル溶液;グラジエント(移動相(A):移動相(B)の比率を記載):[0分]95:5;[0.1分]95:5;[1.2分]5:95;[1.6分]5:95;検出器:UV(PDA)、ELSD、MS。
条件B;カラム:YMC triart C18(粒子径:1.9 x 10-6 m, カラム長:30 x 2.0 mm I.D.);流速:1.0mL/min;カラム温度:30℃;移動相(A):0.1%トリフルオロ酢酸水溶液;移動相(B):0.1%TFA-アセトニトリル溶液;グラジエント(移動相(A):移動相(B)の比率を記載):[0分]95:5;[0.1分]95:5;[1.2分]5:50;[1.21分]5:95;[1.6分]5:95;検出器:UV(PDA)、ELSD、MS。
LC-MS/ELSD was performed under the following conditions.
Condition A; Column: YMC triart C 18 (particle size: 1.9 x 10 -6 m, column length: 30 x 2.0 mm I.D.); Flow rate: 1.0 mL/min; Column temperature: 30°C; Mobile phase (A): 0.1% trifluoroacetic acid aqueous solution; Mobile phase (B): 0.1% TFA-acetonitrile solution; Gradient (ratio of mobile phase (A): mobile phase (B) is described): [0 min] 95:5; [0.1 min] 95:5; [1.2 min] 5:95; [1.6 min] 5:95; Detector: UV (PDA), ELSD, MS.
Condition B; Column: YMC triart C18 (particle size: 1.9 x 10-6 m, column length: 30 x 2.0 mm I.D.); Flow rate: 1.0 mL/min; Column temperature: 30°C; Mobile phase (A): 0.1% trifluoroacetic acid aqueous solution; Mobile phase (B): 0.1% TFA-acetonitrile solution; Gradient (ratio of mobile phase (A): mobile phase (B) is described): [0 min] 95:5; [0.1 min] 95:5; [1.2 min] 5:50; [1.21 min] 5:95; [1.6 min] 5:95; Detector: UV (PDA), ELSD, MS.
HPLC保持時間は、合成中間体については条件A、本開示化合物については条件Bでの保持時間を示す。 HPLC retention times are shown under condition A for synthetic intermediates and under condition B for the disclosed compounds.
SFC分取は、下記の何れかの条件で行った。
条件C;カラム:ダイセル CHIRALPAK IC(粒子径:5μm;カラム長:250x20 mm I.D.);流速:100mL/min;カラム温度:35℃;圧力:120bar;移動相(A):CO2;移動相(B):MeOH;アイソクラティック(移動相(A):移動相(B)=95:5);検出器:UV220nm。
条件D;カラム:ダイセル CHIRALPAK IC(粒子径:5μm;カラム長:250x20 mm I.D.);流速:100mL/min;カラム温度:35℃;圧力:120bar;移動相(A):CO2;移動相(B):MeOH;アイソクラティック(移動相(A):移動相(B)=90:10);検出器:UV210nm。
条件E;カラム:ダイセル CHIRALPAK IC(粒子径:5μm;カラム長:250x20 mm I.D.);流速:100mL/min;カラム温度:35℃;圧力:120bar;移動相(A):CO2;移動相(B):EtOH;アイソクラティック(移動相(A):移動相(B)=92:8);検出器:UV220nm。
参考例1:メチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[(4,4,4-トリフルオロブチル)チオ]ブタノアート
メチル(2S)-4-メルカプト-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート(CAS:690637-98-0)(750mg)のDMF(10mL)溶液に炭酸カリウム(415mg)および4,4,4-トリフルオロブチルメタンスルホナート(CAS:164523-19-7)(806mg)を加え、窒素雰囲気下、60℃で2時間撹拌した。反応液を水で希釈し、酢酸エチルおよびヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=8:2)によって精製することにより、以下の物性値を有する表題化合物(1.08g)を得た。
TLC:Rf 0.5(ヘキサン:酢酸エチル=3:1)
参考例2:メチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート
参考例1で製造した化合物(1.08g)のメタノール(10mL)溶液に二酢酸ヨードベンゼン(2.03g)およびカルバミン酸アンモニウム(351mg)を加え、室温で30分撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)によって精製することにより、以下の物性値を有する表題化合物(820mg)を得た。
TLC:Rf 0.6(酢酸エチル)
参考例3:(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタン酸
参考例2で製造した化合物(820mg)のTHF(6mL)溶液に水酸化リチウム一水和物(176mg)の水溶液(3mL)を0℃で加え、室温で1時間撹拌した。反応液をジオールシリカゲルカラムクロマトグラフィー(ジクロロメタン:メタノール=85:15)によって精製することにより、以下の物性値を有する表題化合物(764mg)を得た。
HPLC保持時間(分):0.845
実施例1:(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタン酸
SFC separation was performed under one of the following conditions:
Condition C; Column: Daicel CHIRALPAK IC (particle size: 5 μm; column length: 250 x 20 mm I.D.); flow rate: 100 mL/min; column temperature: 35° C.; pressure: 120 bar; mobile phase (A): CO 2 ; mobile phase (B): MeOH; isocratic (mobile phase (A):mobile phase (B) = 95:5); detector: UV 220 nm.
Condition D; Column: Daicel CHIRALPAK IC (particle size: 5 μm; column length: 250 x 20 mm I.D.); flow rate: 100 mL/min; column temperature: 35° C.; pressure: 120 bar; mobile phase (A): CO 2 ; mobile phase (B): MeOH; isocratic (mobile phase (A):mobile phase (B) = 90:10); detector: UV 210 nm.
Condition E; Column: Daicel CHIRALPAK IC (particle size: 5 μm; column length: 250 x 20 mm I.D.); flow rate: 100 mL/min; column temperature: 35° C.; pressure: 120 bar; mobile phase (A): CO 2 ; mobile phase (B): EtOH; isocratic (mobile phase (A):mobile phase (B) = 92:8); detector: UV 220 nm.
Reference Example 1: Methyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[(4,4,4-trifluorobutyl)thio]butanoate
Potassium carbonate (415 mg) and 4,4,4-trifluorobutyl methanesulfonate (CAS: 164523-19-7) (806 mg) were added to a solution of methyl (2S)-4-mercapto-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate (CAS: 690637-98-0) (750 mg) in DMF (10 mL), and the mixture was stirred at 60° C. for 2 hours under a nitrogen atmosphere. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to give the title compound (1.08 g) having the following physical properties.
TLC: Rf 0.5 (hexane:ethyl acetate=3:1)
Reference Example 2: Methyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate
To a solution of the compound (1.08 g) produced in Reference Example 1 in methanol (10 mL), iodobenzene diacetate (2.03 g) and ammonium carbamate (351 mg) were added and stirred at room temperature for 30 minutes. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (ethyl acetate) to give the title compound (820 mg) having the following physical properties.
TLC: Rf 0.6 (ethyl acetate)
Reference Example 3: (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoic acid
To a solution of the compound (820 mg) prepared in Reference Example 2 in THF (6 mL), an aqueous solution (3 mL) of lithium hydroxide monohydrate (176 mg) was added at 0° C., and the mixture was stirred at room temperature for 1 hour. The reaction solution was purified by diol silica gel column chromatography (dichloromethane:methanol=85:15) to obtain the title compound (764 mg) having the following physical properties.
HPLC retention time (min): 0.845
Example 1: (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoic acid

参考例3で製造した化合物(764mg)に5Nの塩酸水溶液(5mL)を加え、室温で30分撹拌した。反応液に水(5mL)を加え、イオン交換樹脂(商品名:DOWEX(H+form))に担持した。充分量の水で洗った後、8%のアンモニア水溶液により溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する本開示化合物(473mg)を得た。
MS(ESI,Pos.):293(M+H)+;
1H-NMR(D2O):δ 2.02, 2.20 - 2.38, 3.23 - 3.43, 3.77。
実施例1-1~1-4
4,4,4-トリフルオロブチルメタンスルホナートの代わりに対応するメタンスルホナートを用いて、参考例1→参考例2→参考例3→実施例1と同様の操作を行うことにより、以下の物性値を有する本開示化合物を得た。
実施例1-1:(2S)-2-アミノ-4-[S-(2-シクロペンチルエチル)スルホンイミドイル]ブタン酸
5N aqueous hydrochloric acid (5 mL) was added to the compound (764 mg) produced in Reference Example 3, and the mixture was stirred at room temperature for 30 minutes. Water (5 mL) was added to the reaction solution, and the mixture was supported on an ion exchange resin (trade name: DOWEX (H + form)). After washing with a sufficient amount of water, the resin was eluted with 8% aqueous ammonia. The fractions reacting with ninhydrin were collected and concentrated to obtain the compound of the present disclosure (473 mg) having the following physical properties.
MS (ESI, Pos.): 293 (M+H) + ;
1 H-NMR (D 2 O): δ 2.02, 2.20 - 2.38, 3.23 - 3.43, 3.77.
Examples 1-1 to 1-4
The same operations as in Reference Example 1 → Reference Example 2 → Reference Example 3 → Example 1 were carried out using the corresponding methanesulfonate instead of 4,4,4-trifluorobutyl methanesulfonate to obtain the presently disclosed compound having the following physical properties.
Example 1-1: (2S)-2-amino-4-[S-(2-cyclopentylethyl)sulfonimidoyl]butanoic acid

以下の物性値を有する本開示化合物(23mg)を得た。
HPLC保持時間(分):0.683;
MS(ESI,Pos.):263(M+H)+;
1H-NMR(D2O):δ 1.00 - 1.14, 1.37 - 1.60, 1.64 - 1.77, 1.77 - 1.86, 2.14 - 2.34, 3.16 - 3.39, 3.75。
実施例1-2:(2S)-2-アミノ-4-[S-(2-シクロブチルエチル)スルホンイミドイル]ブタン酸
The compound of the present disclosure (23 mg) having the following physical properties was obtained.
HPLC retention time (min): 0.683;
MS (ESI, Pos.): 263 (M+H) + ;
1H -NMR ( D2O ): δ 1.00 - 1.14, 1.37 - 1.60, 1.64 - 1.77, 1.77 - 1.86, 2.14 - 2.34, 3.16 - 3.39, 3.75.
Example 1-2: (2S)-2-amino-4-[S-(2-cyclobutylethyl)sulfonimidoyl]butanoic acid
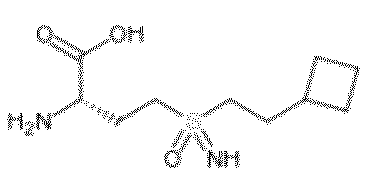
以下の物性値を有する本開示化合物(75mg)を得た。
HPLC保持時間(分):0.567;
MS(ESI,Pos.):249(M+H)+;
1H-NMR(D2O):δ 1.48 - 1.64, 1.68 - 1.85, 1.90 - 2.04, 2.14 - 2.37, 2.91 - 3.16, 3.17 - 3.44, 3.77。
実施例1-3:(2S)-2-アミノ-4-[S-(2-シクロプロピルエチル)スルホンイミドイル]ブタン酸
The compound of the present disclosure (75 mg) having the following physical properties was obtained.
HPLC retention time (min): 0.567;
MS (ESI, Pos.): 249 (M+H) + ;
1H -NMR ( D2O ): δ 1.48 - 1.64, 1.68 - 1.85, 1.90 - 2.04, 2.14 - 2.37, 2.91 - 3.16, 3.17 - 3.44, 3.77.
Example 1-3: (2S)-2-amino-4-[S-(2-cyclopropylethyl)sulfonimidoyl]butanoic acid

以下の物性値を有する本開示化合物(48mg)を得た。
HPLC保持時間(分):0.425;
MS(ESI,Pos.):235(M+H)+;
1H-NMR(D2O):δ -0.05 - 0.08, 0.30 - 0.44, 0.69, 1.47 - 1.65, 2.13 - 2.27, 3.14 - 3.35, 3.71。
実施例1-4:(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-メチルブチル)スルホンイミドイル]ブタン酸
The compound of the present disclosure (48 mg) having the following physical properties was obtained.
HPLC retention time (min): 0.425;
MS (ESI, Pos.): 235 (M+H) + ;
1 H-NMR (D 2 O): δ -0.05 - 0.08, 0.30 - 0.44, 0.69, 1.47 - 1.65, 2.13 - 2.27, 3.14 - 3.35, 3.71.
Example 1-4: (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-methylbutyl)sulfonimidoyl]butanoic acid

以下の物性値を有する本開示化合物(22mg)を得た。
HPLC保持時間(分):0.599;
MS(ESI,Pos.):291(M+H)+;
1H-NMR(D2O):δ 1.08, 1.73 - 1.87, 1.97 - 2.18, 2.20 - 2.34, 2.36 - 2.51, 3.21 - 3.44, 3.79。
参考例4:2-(3,3-ジフルオロシクロブチル)エチルメタンスルホナート
2-(3,3-ジフルオロシクロブチル)エタノール(CAS:1056467-54-9)(200mg)のジクロロメタン(2mL)溶液に氷冷下でジイソプロピルエチルアミン(0.38mL)およびメシル酸無水物(281mg)を加え、室温で1時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮することにより、以下の物性値を有する表題化合物(314mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=3:1)
実施例1-5:(2S)-2-アミノ-4-{S-[2-(3,3-ジフルオロシクロブチル)エチル]スルホンイミドイル}ブタン酸
The compound of the present disclosure (22 mg) having the following physical properties was obtained.
HPLC retention time (min): 0.599;
MS (ESI, Pos.): 291 (M+H) + ;
1H -NMR ( D2O ): δ 1.08, 1.73 - 1.87, 1.97 - 2.18, 2.20 - 2.34, 2.36 - 2.51, 3.21 - 3.44, 3.79.
Reference Example 4: 2-(3,3-difluorocyclobutyl)ethyl methanesulfonate
Diisopropylethylamine (0.38 mL) and mesylic anhydride (281 mg) were added to a solution of 2-(3,3-difluorocyclobutyl)ethanol (CAS: 1056467-54-9) (200 mg) in dichloromethane (2 mL) under ice cooling, and the mixture was stirred at room temperature for 1 hour. A saturated aqueous solution of ammonium chloride was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound (314 mg) having the following physical properties:
TLC: Rf 0.6 (hexane:ethyl acetate=3:1)
Example 1-5: (2S)-2-amino-4-{S-[2-(3,3-difluorocyclobutyl)ethyl]sulfonimidoyl}butanoic acid

4,4,4-トリフルオロブチルメタンスルホナートの代わりに参考例4で製造した化合物を用いて、参考例1→参考例2→参考例3→実施例1と同様の操作を行うことにより、以下の物性値を有する本開示化合物(61mg)を得た。
HPLC保持時間(分):0.523;
MS(ESI,Pos.):285(M+H)+;
1H-NMR(D2O): δ 1.86 - 2.00, 2.12 - 2.31, 2.57 - 2.73, 3.12 - 3.41, 3.76。
実施例1-6:(2S)-2-アミノ-4-[S-(4,4-ジメチルペンチル)スルホンイミドイル]ブタン酸
The compound of the present disclosure (61 mg) having the following physical properties was obtained by carrying out the same operations as in Reference Example 1 → Reference Example 2 → Reference Example 3 → Example 1 using the compound prepared in Reference Example 4 instead of 4,4,4-trifluorobutyl methanesulfonate.
HPLC retention time (min): 0.523;
MS (ESI, Pos.): 285 (M+H) + ;
1 H-NMR (D 2 O): δ 1.86 - 2.00, 2.12 - 2.31, 2.57 - 2.73, 3.12 - 3.41, 3.76.
Example 1-6: (2S)-2-amino-4-[S-(4,4-dimethylpentyl)sulfonimidoyl]butanoic acid

以下の物性値を有する本開示化合物(25mg)を得た。
HPLC保持時間(分):0.714;
MS(ESI,Pos.):265(M+H)+;
1H-NMR(D2O): δ 0.81, 1.17 - 1.29, 1.64 - 1.77, 2.15 - 2.30, 3.12 - 3.20, 3.20 - 3.39, 3.74。
参考例5:2-(1-ヒドロキシシクロブチル)エチルメタンスルホナート
1-(2-ヒドロキシエチル)シクロブタノール(CAS:83237-27-8)(200mg)のジクロロメタン(2mL)溶液に氷冷下でジイソプロピルエチルアミン(0.4mL)およびメシル酸無水物(329mg)を加え、室温で2時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮することにより、以下の物性値を有する表題化合物(335mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=2:1)
参考例6:メチル(2S)-4-{[2-(1-ヒドロキシシクロブチル)エチル]チオ}-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
メチル(2S)-4-メルカプト-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート(CAS:690637-98-0)(200mg)のDMF(4mL)溶液に炭酸カリウム(166mg)および参考例5で製造した化合物(202mg)を加え、窒素雰囲気下、60℃で2時間撹拌した。反応液を水で希釈し、酢酸エチルおよびヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=6:4)によって精製することにより、以下の物性値を有する表題化合物(236mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=1:1)
参考例7:メチル(2S)-4-{S-[2-(1-ヒドロキシシクロブチル)エチル]スルホンイミドイル}-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
参考例6で製造した化合物(235mg)のメタノール(5mL)溶液に二酢酸ヨードベンゼン(132mg)およびカルバミン酸アンモニウム(653mg)を加え、室温で30分撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:メタノール=95:5)によって精製することにより、以下の物性値を有する表題化合物(90mg)を得た。
TLC:Rf 0.2(酢酸エチル)
参考例8:(2S)-4-{S-[2-(1-ヒドロキシシクロブチル)エチル]スルホンイミドイル}-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタン酸
参考例7で製造した化合物(90mg)のTHF(1mL)溶液に水酸化リチウム一水和物(20mg)の水溶液(0.5mL)を0℃で加え、室温で1時間撹拌した。反応液をジオールシリカゲルカラムクロマトグラフィー(ジクロロメタン:メタノール=85:15)によって精製することにより、以下の物性値を有する表題化合物(62mg)を得た。
HPLC保持時間(分):0.773
実施例2:(2S)-2-アミノ-4-{S-[2-(1-ヒドロキシシクロブチル)エチル]スルホンイミドイル}ブタン酸
The compound of the present disclosure (25 mg) having the following physical properties was obtained.
HPLC retention time (min): 0.714;
MS (ESI, Pos.): 265 (M+H) + ;
1H -NMR ( D2O ): δ 0.81, 1.17 - 1.29, 1.64 - 1.77, 2.15 - 2.30, 3.12 - 3.20, 3.20 - 3.39, 3.74.
Reference Example 5: 2-(1-hydroxycyclobutyl)ethyl methanesulfonate
Diisopropylethylamine (0.4 mL) and mesylic anhydride (329 mg) were added to a solution of 1-(2-hydroxyethyl)cyclobutanol (CAS: 83237-27-8) (200 mg) in dichloromethane (2 mL) under ice cooling, and the mixture was stirred at room temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound (335 mg) having the following physical properties:
TLC: Rf 0.6 (hexane:ethyl acetate=2:1)
Reference Example 6: Methyl (2S)-4-{[2-(1-hydroxycyclobutyl)ethyl]thio}-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
Potassium carbonate (166 mg) and the compound prepared in Reference Example 5 (202 mg) were added to a solution of methyl (2S)-4-mercapto-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate (CAS: 690637-98-0) (200 mg) in DMF (4 mL), and the mixture was stirred at 60° C. for 2 hours under a nitrogen atmosphere. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=6:4) to give the title compound (236 mg) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=1:1)
Reference Example 7: Methyl (2S)-4-{S-[2-(1-hydroxycyclobutyl)ethyl]sulfonimidoyl}-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
To a solution of the compound (235 mg) produced in Reference Example 6 in methanol (5 mL), iodobenzene diacetate (132 mg) and ammonium carbamate (653 mg) were added and stirred at room temperature for 30 minutes. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (ethyl acetate:methanol=95:5) to give the title compound (90 mg) having the following physical properties.
TLC: Rf 0.2 (ethyl acetate)
Reference Example 8: (2S)-4-{S-[2-(1-hydroxycyclobutyl)ethyl]sulfonimidoyl}-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoic acid
To a solution of the compound (90 mg) prepared in Reference Example 7 in THF (1 mL), an aqueous solution (0.5 mL) of lithium hydroxide monohydrate (20 mg) was added at 0° C., and the mixture was stirred at room temperature for 1 hour. The reaction solution was purified by diol silica gel column chromatography (dichloromethane:methanol=85:15) to obtain the title compound (62 mg) having the following physical properties.
HPLC retention time (min): 0.773
Example 2: (2S)-2-amino-4-{S-[2-(1-hydroxycyclobutyl)ethyl]sulfonimidoyl}butanoic acid

参考例8で製造した化合物(62mg)に5Nの塩酸水溶液(1mL)を加え、室温で1時間撹拌した。反応液に水(1mL)を加え、イオン交換樹脂(商品名:DOWEX(H+form))に担持した。充分量の水で洗った後、8%のアンモニア水溶液により溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する本開示化合物(12mg)を得た。
MS(ESI,Pos.):265(M+H)+;
1H-NMR(D2O): δ 1.43 - 1.62, 1.63 - 1.77, 1.92 - 2.09, 2.20 - 2.36, 3.17 - 3.45, 3.79。
参考例9:エチル3-{[ジメチル(2-メチル-2-プロパニル)シリル]オキシ}-4,4,4-トリフルオロブタノアート
エチル4,4,4-トリフルオロ-3-ヒドロキシブタノアート(CAS:372-30-5)(600mg)のDMF(2mL)溶液にイミダゾール(439mg)およびtert-ブチルジメチルクロロシラン(632mg)を加え、室温で終夜撹拌した。反応液を水で希釈し、酢酸エチルおよびヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮し、以下の物性値を有する表題化合物を得た。
TLC:Rf 0.8(ヘキサン:酢酸エチル=5:1)
参考例10:3-{[ジメチル(2-メチル-2-プロパニル)シリル]オキシ}-4,4,4-トリフルオロ-1-ブタノール
参考例9で得られた残渣のTHF(10mL)溶液に氷冷下でリチウムボロヒドリド(140mg)およびメタノール(0.52mL)を加え、室温で終夜撹拌した。反応液に氷冷下、飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=8:2)によって精製することにより、以下の物性値を有する表題化合物(510mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=3:1)
参考例11:3-{[ジメチル(2-メチル-2-プロパニル)シリル]オキシ}-4,4,4-トリフルオロブチルメタンスルホナート
参考例10で製造した化合物(200mg)のジクロロメタン(2mL)溶液に氷冷下でジイソプロピルエチルアミン(0.2mL)およびメシル酸無水物(162mg)を加え、室温で2時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮することにより、以下の物性値を有する表題化合物(260mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=3:1)
参考例12:メチル(2S)-4-[(3-{[ジメチル(2-メチル-2-プロパニル)シリル]オキシ}-4,4,4-トリフルオロブチル)チオ]-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
メチル(2S)-4-メルカプト-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート(CAS:690637-98-0)(180mg)のDMF(8mL)溶液に炭酸カリウム(100mg)および参考例11で製造した化合物(267mg)を加え、窒素雰囲気下、60℃で2時間撹拌した。反応液を水で希釈し、酢酸エチルおよびヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=8:2)によって精製することにより、以下の物性値を有する表題化合物(350mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=3:1)
参考例13:メチル(2S)-4-[S-(3-{[ジメチル(2-メチル-2-プロパニル)シリル]オキシ}-4,4,4-トリフルオロブチル)スルホンイミドイル]-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
参考例12で製造した化合物(350mg)のメタノール(5mL)溶液に二酢酸ヨードベンゼン(690mg)およびカルバミン酸アンモニウム(140mg)を加え、室温で30分撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)によって精製することにより、以下の物性値を有する表題化合物(270mg)を得た。
TLC:Rf 0.5(ヘキサン:酢酸エチル=1:1)
参考例14:(2S)-4-[S-(3-{[ジメチル(2-メチル-2-プロパニル)シリル]オキシ}-4,4,4-トリフルオロブチル)スルホンイミドイル]-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタン酸
参考例13で製造した化合物(270mg)のTHF(2mL)溶液に水酸化リチウム一水和物(44mg)の水溶液(1mL)を0℃で加え、室温で1時間撹拌した。反応液をジオールシリカゲルカラムクロマトグラフィー(ジクロロメタン:メタノール=95:5)によって精製することにより、以下の物性値を有する表題化合物(200mg)を得た。
HPLC保持時間(分):1.114
実施例3:(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-ヒドロキシブチル)スルホンイミドイル]ブタン酸
A 5N aqueous hydrochloric acid solution (1 mL) was added to the compound (62 mg) produced in Reference Example 8, and the mixture was stirred at room temperature for 1 hour. Water (1 mL) was added to the reaction solution, and the mixture was supported on an ion exchange resin (trade name: DOWEX (H + form)). After washing with a sufficient amount of water, the resin was eluted with an 8% aqueous ammonia solution. The fractions reacting with ninhydrin were collected and concentrated to obtain the compound of the present disclosure (12 mg) having the following physical properties.
MS (ESI, Pos.): 265 (M+H) + ;
1H -NMR ( D2O ): δ 1.43 - 1.62, 1.63 - 1.77, 1.92 - 2.09, 2.20 - 2.36, 3.17 - 3.45, 3.79.
Reference Example 9: Ethyl 3-{[dimethyl(2-methyl-2-propanyl)silyl]oxy}-4,4,4-trifluorobutanoate
Imidazole (439 mg) and tert-butyldimethylchlorosilane (632 mg) were added to a solution of ethyl 4,4,4-trifluoro-3-hydroxybutanoate (CAS: 372-30-5) (600 mg) in DMF (2 mL), and the mixture was stirred at room temperature overnight. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound having the following physical properties.
TLC: Rf 0.8 (hexane:ethyl acetate=5:1)
Reference Example 10: 3-{[dimethyl(2-methyl-2-propanyl)silyl]oxy}-4,4,4-trifluoro-1-butanol
Lithium borohydride (140 mg) and methanol (0.52 mL) were added to a solution of the residue obtained in Reference Example 9 in THF (10 mL) under ice cooling, and the mixture was stirred at room temperature overnight. A saturated aqueous solution of ammonium chloride was added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to give the title compound (510 mg) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=3:1)
Reference Example 11: 3-{[dimethyl(2-methyl-2-propanyl)silyl]oxy}-4,4,4-trifluorobutyl methanesulfonate
To a solution of the compound (200 mg) prepared in Reference Example 10 in dichloromethane (2 mL), diisopropylethylamine (0.2 mL) and mesylic anhydride (162 mg) were added under ice cooling, and the mixture was stirred at room temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (260 mg) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=3:1)
Reference Example 12: Methyl (2S)-4-[(3-{[dimethyl(2-methyl-2-propanyl)silyl]oxy}-4,4,4-trifluorobutyl)thio]-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
Potassium carbonate (100 mg) and the compound prepared in Reference Example 11 (267 mg) were added to a solution of methyl (2S)-4-mercapto-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate (CAS: 690637-98-0) (180 mg) in DMF (8 mL), and the mixture was stirred at 60° C. for 2 hours under a nitrogen atmosphere. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to give the title compound (350 mg) having the following physical properties:
TLC: Rf 0.6 (hexane:ethyl acetate=3:1)
Reference Example 13: methyl (2S)-4-[S-(3-{[dimethyl(2-methyl-2-propanyl)silyl]oxy}-4,4,4-trifluorobutyl)sulfonimidoyl]-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
To a solution of the compound (350 mg) produced in Reference Example 12 in methanol (5 mL), iodobenzene diacetate (690 mg) and ammonium carbamate (140 mg) were added and stirred at room temperature for 30 minutes. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=1:1) to give the title compound (270 mg) having the following physical properties.
TLC: Rf 0.5 (hexane:ethyl acetate=1:1)
Reference Example 14: (2S)-4-[S-(3-{[dimethyl(2-methyl-2-propanyl)silyl]oxy}-4,4,4-trifluorobutyl)sulfonimidoyl]-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoic acid
To a solution of the compound (270 mg) prepared in Reference Example 13 in THF (2 mL), an aqueous solution (1 mL) of lithium hydroxide monohydrate (44 mg) was added at 0° C., and the mixture was stirred at room temperature for 1 hour. The reaction solution was purified by diol silica gel column chromatography (dichloromethane:methanol=95:5) to obtain the title compound (200 mg) having the following physical properties.
HPLC retention time (min): 1.114
Example 3: (2S)-2-Amino-4-[S-(4,4,4-trifluoro-3-hydroxybutyl)sulfonimidoyl]butanoic acid

参考例14で製造した化合物(200mg)に5Nの塩酸水溶液(2mL)を加え室温で1時間撹拌した。反応液に水(2mL)を加え、イオン交換樹脂(商品名:DOWEX(H+form))に担持した。充分量の水で洗った後、8%のアンモニア水溶液により溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する本開示題化合物(47mg)を得た。
MS(ESI,Pos.):293(M+H)+;
1H-NMR(D2O): δ 1.95 - 2.07, 2.15 - 2.34, 3.25 - 3.46, 3.77 - 3.82, 4.17。
実施例3-1:(2S)-2-アミノ-4-{S-[2-(1-フルオロシクロブチル)エチル]スルホンイミドイル}ブタン酸
5N aqueous hydrochloric acid (2 mL) was added to the compound (200 mg) produced in Reference Example 14, and the mixture was stirred at room temperature for 1 hour. Water (2 mL) was added to the reaction solution, and the mixture was supported on an ion exchange resin (trade name: DOWEX (H + form)). After washing with a sufficient amount of water, the resin was eluted with 8% aqueous ammonia. The fractions reacting with ninhydrin were collected and concentrated to obtain the subject compound of the present disclosure (47 mg) having the following physical properties.
MS (ESI, Pos.): 293 (M+H) + ;
1 H-NMR (D 2 O): δ 1.95 - 2.07, 2.15 - 2.34, 3.25 - 3.46, 3.77 - 3.82, 4.17.
Example 3-1: (2S)-2-amino-4-{S-[2-(1-fluorocyclobutyl)ethyl]sulfonimidoyl}butanoic acid

参考例11で製造した化合物の代わりに対応するメタンスルホナートを用いて、参考例12→参考例13→参考例14→実施例3と同様の操作を行うことにより、以下の物性値を有する本開示化合物(95mg)を得た。
HPLC保持時間(分):0.596;
MS(ESI,Pos.):267(M+H)+;
1H-NMR(D2O): δ 1.41 - 1.54, 1.67 - 1.81, 2.03 - 2.12, 2.14 - 2.34, 3.23 - 3.45, 3.75 - 3.85。
参考例15:4,4,4-トリフルオロ-3-ヒドロキシ-3-メチルブチル 4-メチルベンゼンスルホナート
4,4,4-トリフルオロ-3-ヒドロキシ-3-メチルブタン酸(CAS:338-03-4)(200mg)のTHF(2mL)溶液に氷冷下でボラン-THF錯体(0.92M、3.9mL)を加え、室温で終夜撹拌した。反応液に氷冷下、メタノールを加え、減圧濃縮した。得られた残渣のピリジン(2mL)溶液に塩化パラトルエンスルホニル(266mg)を加え、室温で3時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)によって精製することにより、以下の物性値を有する表題化合物(244mg)を得た。
TLC:Rf 0.5(ヘキサン:酢酸エチル=3:1)
実施例3-2:(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-ヒドロキシ-3-メチルブチル)スルホンイミドイル]ブタン酸
The compound of the present disclosure (95 mg) having the following physical properties was obtained by carrying out the same operations as in Reference Example 12 → Reference Example 13 → Reference Example 14 → Example 3 using the corresponding methanesulfonate instead of the compound prepared in Reference Example 11.
HPLC retention time (min): 0.596;
MS (ESI, Pos.): 267 (M+H) + ;
1H -NMR ( D2O ): δ 1.41 - 1.54, 1.67 - 1.81, 2.03 - 2.12, 2.14 - 2.34, 3.23 - 3.45, 3.75 - 3.85.
Reference Example 15: 4,4,4-trifluoro-3-hydroxy-3-methylbutyl 4-methylbenzenesulfonate
Borane-THF complex (0.92M, 3.9mL) was added to a solution of 4,4,4-trifluoro-3-hydroxy-3-methylbutanoic acid (CAS: 338-03-4) (200mg) in THF (2mL) under ice cooling, and the mixture was stirred overnight at room temperature. Methanol was added to the reaction solution under ice cooling, and the mixture was concentrated under reduced pressure. Paratoluenesulfonyl chloride (266mg) was added to a solution of the obtained residue in pyridine (2mL), and the mixture was stirred at room temperature for 3 hours. A saturated aqueous ammonium chloride solution was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane:ethyl acetate=3:1) to obtain the title compound (244mg) having the following physical properties.
TLC: Rf 0.5 (hexane:ethyl acetate=3:1)
Example 3-2: (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-hydroxy-3-methylbutyl)sulfonimidoyl]butanoic acid

参考例11で製造した化合物の代わりに参考例15で製造した化合物を用いて、参考例12→参考例13→参考例14→実施例3と同様の操作を行うことにより、以下の物性値を有する本開示化合物(62mg)を得た。
HPLC保持時間(分):0.329;
MS(ESI,Pos.):307(M+H)+;
1H-NMR(D2O): δ 1.32, 2.01 - 2.21, 2.24 - 2.34, 3.25 - 3.46, 3.76 - 3.84。
参考例16:4,4,4-トリフルオロ-3-ヒドロキシ-3-(トリフルオロメチル)ブチル 4-メチルベンゼンスルホナート
4,4,4-トリフルオロ-3-(トリフルオロメチル)-1,3-ブタンジオール(CAS:21379-33-9)(139mg)のピリジン(2mL)溶液に塩化パラトルエンスルホニル(149mg)を加え、室温で終夜撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)によって精製することにより、以下の物性値を有する表題化合物(244mg)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=3:1)
実施例3-3:(2S)-2-アミノ-4-{S-[4,4,4-トリフルオロ-3-ヒドロキシ-3-(トリフルオロメチル)ブチル]スルホンイミドイル}ブタン酸
The compound of the present disclosure (62 mg) having the following physical properties was obtained by carrying out the same operations as in Reference Example 12 → Reference Example 13 → Reference Example 14 → Example 3 using the compound prepared in Reference Example 15 instead of the compound prepared in Reference Example 11.
HPLC retention time (min): 0.329;
MS (ESI, Pos.): 307 (M+H) + ;
1H -NMR ( D2O ): δ 1.32, 2.01 - 2.21, 2.24 - 2.34, 3.25 - 3.46, 3.76 - 3.84.
Reference Example 16: 4,4,4-trifluoro-3-hydroxy-3-(trifluoromethyl)butyl 4-methylbenzenesulfonate
To a solution of 4,4,4-trifluoro-3-(trifluoromethyl)-1,3-butanediol (CAS: 21379-33-9) (139 mg) in pyridine (2 mL), paratoluenesulfonyl chloride (149 mg) was added and stirred at room temperature overnight. A saturated aqueous solution of ammonium chloride was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=3:1) to give the title compound (244 mg) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=3:1)
Example 3-3: (2S)-2-amino-4-{S-[4,4,4-trifluoro-3-hydroxy-3-(trifluoromethyl)butyl]sulfonimidoyl}butanoic acid

参考例11で製造した化合物の代わりに参考例16で製造した化合物を用いて、参考例12→参考例13→参考例14→実施例3と同様の操作を行うことにより、以下の物性値を有する本開示化合物(66mg)を得た。
HPLC保持時間(分):0.746;
MS(ESI,Pos.):361(M+H)+;
1H-NMR(D2O):δ 2.20 - 2.39, 2.39 - 2.49, 3.27 - 3.52, 3.80。
実施例3-4:(2S)-2-アミノ-4-[S-(3,3,4,4,4-ペンタフルオロブチル)スルホンイミドイル]ブタン酸
The compound of the present disclosure (66 mg) having the following physical properties was obtained by carrying out the same operations as in Reference Example 12 → Reference Example 13 → Reference Example 14 → Example 3 using the compound prepared in Reference Example 16 instead of the compound prepared in Reference Example 11.
HPLC retention time (min): 0.746;
MS (ESI, Pos.): 361 (M+H) + ;
1 H-NMR (D 2 O): δ 2.20 - 2.39, 2.39 - 2.49, 3.27 - 3.52, 3.80.
Example 3-4: (2S)-2-amino-4-[S-(3,3,4,4,4-pentafluorobutyl)sulfonimidoyl]butanoic acid

参考例11で製造した化合物の代わりに対応するアイオダイド(CAS:40723-80-6)を用いて、参考例12→参考例13→参考例14→実施例3と同様の操作を行うことにより、以下の物性値を有する本開示化合物(110mg)を得た。
HPLC保持時間(分):0.637;
MS(ESI,Pos.):313(M+H)+;
1H-NMR(D2O): δ 2.28, 2.58 - 2.80, 3.26 - 3.44, 3.44 - 3.62, 3.77。
参考例17:ベンジル(3R)-4,4,4-トリフルオロ-3-ヒドロキシブタノアート
(3R)-4,4,4-トリフルオロ-3-ヒドロキシブタン酸(CAS:108211-36-5)(1g)のDMF(10mL)溶液に炭酸カリウム(1.75g)および臭化ベンジル(826mL)を加え、室温で3時間撹拌した。反応液を水で希釈し、酢酸エチルとヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮することにより、以下の物性値を有する表題化合物(1.57g)を得た。
参考例18:ベンジル(3R)-4,4,4-トリフルオロ-3-[(トリエチルシリル)オキシ]ブタノアート
参考例17で得られた残渣(1.57g)のDMF(8mL)溶液にイミダゾール(861mg)およびTESCl(1.37mL)を加え、室温で終夜撹拌した。反応液を水で希釈し、酢酸エチルとヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=9:1)によって精製することにより、以下の物性値を有する表題化合物(2.3g)を得た。
TLC:Rf 0.8(ヘキサン:酢酸エチル=5:1)
参考例19:(3R)-4,4,4-トリフルオロ-3-[(トリエチルシリル)オキシ]-1-ブタノール
参考例18で得られた化合物(2.3g)のTHF(20mL)溶液に氷冷下でリチウムボロヒドリド(276mg)およびメタノール(1.0mL)を加え、室温で終夜撹拌した。反応液に氷冷下、飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=9:1)によって精製することにより、以下の物性値を有する表題化合物(1.32g)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=5:1)
参考例20:(3R)-4,4,4-トリフルオロ-3-[(トリエチルシリル)オキシ]ブチルメタンスルホナート
参考例19で製造した化合物(1.32g)のジクロロメタン(10mL)溶液に氷冷下でジイソプロピルエチルアミン(1.3mL)およびメシル酸無水物(1g)を加え、室温で2時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮することにより、以下の物性値を有する表題化合物(1.7g)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=5:1)
参考例21:メチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-({(3R)-4,4,4-トリフルオロ-3-[(トリエチルシリル)オキシ]ブチル}チオ)ブタノアート
メチル(2S)-4-メルカプト-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート(CAS:690637-98-0)(1g)のDMF(15mL)溶液に炭酸カリウム(554mg)および参考例20で製造した化合物(1.62g)を加え、窒素雰囲気下、60℃で2時間撹拌した。反応液を水で希釈し、酢酸エチルとヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=8:2)によって精製することにより、以下の物性値を有する表題化合物(1.9g)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=3:1)
参考例22:メチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-(S-{(3R)-4,4,4-トリフルオロ-3-[(トリエチルシリル)オキシ]ブチル}スルホンイミドイル)ブタノアート
参考例21で製造した化合物(1.9g)のメタノール(19mL)溶液に二酢酸ヨードベンゼン(2.6g)およびカルバミン酸アンモニウム(450mg)を加え、室温で30分撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)によって精製することにより、以下の物性値を有する表題化合物(1.65g)を得た。
TLC:Rf 0.5(ヘキサン:酢酸エチル=1:1)
参考例23:メチル(S)-4-((R,3R)-N-((ベンジルオキシ)カルボニル)-4,4,4-トリフルオロ-3-((トリエチルシリル)オキシ)ブチルスルホンイミドイル)-2-((tert-ブトキシカルボニル)アミノ)ブタノアート
参考例22で製造した化合物(1.65g)のジクロロメタン(10mL)溶液にピリジン(1.28mL)およびクロロギ酸ベンジル(0.67mL)を加え、室温で2時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=7:3)によって精製した。得られた化合物をSFC(条件E)で分取することにより、以下の物性値を有する表題化合物(680mg)を得た。
HPLC保持時間(分):1.418
参考例24:メチル(S)-2-((tert-ブトキシカルボニル)アミノ)-4-((R,3R)-4,4,4-トリフルオロ-3-((トリエチルシリル)オキシ)ブチルスルホンイミドイル)ブタノアート
参考例23で製造した化合物(680mg)のメタノール(8mL)溶液にパラジウム炭素(68mg)を加え、水素雰囲気下、室温で2時間撹拌した。反応液に酢酸エチル(8mL)を加え、セライト(商品名)でろ過することにより、以下の物性値を有する表題化合物(540mg)を得た。
HPLC保持時間(分):1.202
参考例25:(S)-2-((tert-ブトキシカルボニル)アミノ)-4-((R,3R)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸
参考例24で製造した化合物(540mg)のTHF(2mL)溶液に水酸化リチウム一水和物(78mg)の水溶液(1mL)を0℃で加え、室温で1時間撹拌した。反応液をジオールシリカゲルカラムクロマトグラフィー(ジクロロメタン:メタノール=85:15)によって精製することにより、以下の物性値を有する表題化合物(360mg)を得た。
HPLC保持時間(分):0.792
実施例4:(S)-2-アミノ-4-((R,3R)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸
The compound of the present disclosure (110 mg) having the following physical properties was obtained by carrying out the same operations as in Reference Example 12 → Reference Example 13 → Reference Example 14 → Example 3 using the corresponding iodide (CAS: 40723-80-6) instead of the compound produced in Reference Example 11.
HPLC retention time (min): 0.637;
MS (ESI, Pos.): 313 (M+H) + ;
1H -NMR ( D2O ): δ 2.28, 2.58 - 2.80, 3.26 - 3.44, 3.44 - 3.62, 3.77.
Reference Example 17: Benzyl (3R)-4,4,4-trifluoro-3-hydroxybutanoate
Potassium carbonate (1.75 g) and benzyl bromide (826 mL) were added to a solution of (3R)-4,4,4-trifluoro-3-hydroxybutanoic acid (CAS: 108211-36-5) (1 g) in DMF (10 mL), and the mixture was stirred at room temperature for 3 hours. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (1.57 g) having the following physical properties.
Reference Example 18: Benzyl (3R)-4,4,4-trifluoro-3-[(triethylsilyl)oxy]butanoate
Imidazole (861 mg) and TESCl (1.37 mL) were added to a solution of the residue (1.57 g) obtained in Reference Example 17 in DMF (8 mL), and the mixture was stirred at room temperature overnight. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=9:1) to give the title compound (2.3 g) having the following physical properties.
TLC: Rf 0.8 (hexane:ethyl acetate=5:1)
Reference Example 19: (3R)-4,4,4-trifluoro-3-[(triethylsilyl)oxy]-1-butanol
Lithium borohydride (276 mg) and methanol (1.0 mL) were added to a solution of the compound (2.3 g) obtained in Reference Example 18 in THF (20 mL) under ice cooling, and the mixture was stirred at room temperature overnight. A saturated aqueous solution of ammonium chloride was added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=9:1) to obtain the title compound (1.32 g) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=5:1)
Reference Example 20: (3R)-4,4,4-trifluoro-3-[(triethylsilyl)oxy]butyl methanesulfonate
To a solution of the compound (1.32 g) produced in Reference Example 19 in dichloromethane (10 mL), diisopropylethylamine (1.3 mL) and mesylic anhydride (1 g) were added under ice cooling, and the mixture was stirred at room temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (1.7 g) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=5:1)
Reference Example 21: Methyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-({(3R)-4,4,4-trifluoro-3-[(triethylsilyl)oxy]butyl}thio)butanoate
Potassium carbonate (554 mg) and the compound prepared in Reference Example 20 (1.62 g) were added to a solution of methyl (2S)-4-mercapto-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate (CAS: 690637-98-0) (1 g) in DMF (15 mL), and the mixture was stirred at 60° C. for 2 hours under a nitrogen atmosphere. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to give the title compound (1.9 g) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=3:1)
Reference Example 22: methyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-(S-{(3R)-4,4,4-trifluoro-3-[(triethylsilyl)oxy]butyl}sulfonimidoyl)butanoate
To a solution of the compound (1.9 g) produced in Reference Example 21 in methanol (19 mL), iodobenzene diacetate (2.6 g) and ammonium carbamate (450 mg) were added and stirred at room temperature for 30 minutes. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=1:1) to give the title compound (1.65 g) having the following physical properties.
TLC: Rf 0.5 (hexane:ethyl acetate=1:1)
Reference Example 23: methyl (S)-4-((R,3R)-N-((benzyloxy)carbonyl)-4,4,4-trifluoro-3-((triethylsilyl)oxy)butylsulfonimidoyl)-2-((tert-butoxycarbonyl)amino)butanoate
Pyridine (1.28 mL) and benzyl chloroformate (0.67 mL) were added to a solution of the compound (1.65 g) produced in Reference Example 22 in dichloromethane (10 mL), and the mixture was stirred at room temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=7:3). The resulting compound was separated by SFC (condition E) to obtain the title compound (680 mg) having the following physical properties.
HPLC retention time (min): 1.418
Reference Example 24: methyl (S)-2-((tert-butoxycarbonyl)amino)-4-((R,3R)-4,4,4-trifluoro-3-((triethylsilyl)oxy)butylsulfonimidoyl)butanoate
Palladium carbon (68 mg) was added to a solution of the compound (680 mg) prepared in Reference Example 23 in methanol (8 mL), and the mixture was stirred at room temperature under a hydrogen atmosphere for 2 hours. Ethyl acetate (8 mL) was added to the reaction mixture, and the mixture was filtered through Celite (trade name) to obtain the title compound (540 mg) having the following physical properties.
HPLC retention time (min): 1.202
Reference Example 25: (S)-2-((tert-butoxycarbonyl)amino)-4-((R,3R)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid
To a solution of the compound (540 mg) prepared in Reference Example 24 in THF (2 mL), an aqueous solution (1 mL) of lithium hydroxide monohydrate (78 mg) was added at 0° C., and the mixture was stirred at room temperature for 1 hour. The reaction solution was purified by diol silica gel column chromatography (dichloromethane:methanol=85:15) to give the title compound (360 mg) having the following physical properties.
HPLC retention time (min): 0.792
Example 4: (S)-2-amino-4-((R,3R)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid

参考例25で製造した化合物(360mg)に5Nの塩酸水溶液(3mL)を加え、室温で1時間撹拌した。反応液に水(3mL)を加え、イオン交換樹脂(商品名:DOWEX(H+form))に担持した。充分量の水で洗った後、8%のアンモニア水溶液により溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する本開示化合物(236mg)を得た。
MS(ESI,Pos.):293(M+H)+;
1H-NMR(D2O): δ 1.95 - 2.10, 2.16 - 2.36, 3.26 - 3.48, 3.75 - 3.82, 4.18。
参考例26:(3S)-4,4,4-トリフルオロ-3-[(トリエチルシリル)オキシ]ブチルメタンスルホナート
(3R)-4,4,4-トリフルオロ-3-ヒドロキシブタン酸の代わりに、(3S)-4,4,4-トリフルオロ-3-ヒドロキシブタン酸(CAS:128899-79-6)を用いて、参考例17→参考例18→参考例19→参考例20と同様の操作を行うことにより、以下の物性値を有する表題化合物(1.78g)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=5:1)
実施例5:(S)-2-アミノ-4-((R,3S)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸
5N aqueous hydrochloric acid (3 mL) was added to the compound (360 mg) produced in Reference Example 25, and the mixture was stirred at room temperature for 1 hour. Water (3 mL) was added to the reaction solution, and the mixture was supported on an ion exchange resin (trade name: DOWEX (H + form)). After washing with a sufficient amount of water, the mixture was eluted with 8% aqueous ammonia. The fractions reacting with ninhydrin were collected and concentrated to obtain the compound of the present disclosure (236 mg) having the following physical properties.
MS (ESI, Pos.): 293 (M+H) + ;
1 H-NMR (D 2 O): δ 1.95 - 2.10, 2.16 - 2.36, 3.26 - 3.48, 3.75 - 3.82, 4.18.
Reference Example 26: (3S)-4,4,4-trifluoro-3-[(triethylsilyl)oxy]butyl methanesulfonate
Instead of (3R)-4,4,4-trifluoro-3-hydroxybutanoic acid, (3S)-4,4,4-trifluoro-3-hydroxybutanoic acid (CAS: 128899-79-6) was used to carry out the same operations as in Reference Example 17 → Reference Example 18 → Reference Example 19 → Reference Example 20 to obtain the title compound (1.78 g) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=5:1)
Example 5: (S)-2-amino-4-((R,3S)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid

参考例20で製造した化合物の代わりに参考例26で製造した化合物を用いて、参考例21→参考例22→参考例23→参考例24→参考例25→実施例4と同様の操作を行うことにより、以下の物性値を有する表題化合物(185mg)を得た。
MS(ESI,Pos.):293(M+H)+;
1H-NMR(D2O): δ 1.95 - 2.07, 2.15 - 2.35, 3.25 - 3.46, 3.79, 4.17。
参考例27:メチル(2S)-4-{[2-(1-ヒドロキシシクロブチル)エチル]チオ}-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
メチル(2S)-4-メルカプト-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート(CAS:690637-98-0)(6.3g)のDMF(100mL)溶液に炭酸カリウム(3.5g)および1-(2-ブロモエチル)シクロブタノール(CAS:1909309-61-0)(5g)を加え、窒素雰囲気下、60℃で2時間撹拌した。反応液を水で希釈し、酢酸エチルおよびヘキサン(2:1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=6:4)によって精製することにより、以下の物性値を有する表題化合物(8.7g)を得た。
TLC:Rf 0.3(ヘキサン:酢酸エチル=2:1)
参考例28:メチル(2S)-4-{[2-(1-アセトキシシクロブチル)エチル]チオ}-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
参考例27で製造した化合物(8.7g)のジクロロメタン(150mL)溶液に、ジイソプロピルエチルアミン(10mL)、無水酢酸(4.3mL)および4-ジメチルアミノピリジン(CAS:1122-58-3)(610mg)を加え、室温で終夜撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=8:2)によって精製することにより、以下の物性値を有する表題化合物(9.35g)を得た。
TLC:Rf 0.6(ヘキサン:酢酸エチル=2:1)
参考例29:メチル(2S)-4-{S-[2-(1-アセトキシシクロブチル)エチル]スルホンイミドイル}-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)ブタノアート
参考例28で製造した化合物(9.35g)のメタノール(100mL)溶液に、二酢酸ヨードベンゼン(16.2g)およびカルバミン酸アンモニウム(2.8g)を加え、室温で1時間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)によって精製することにより、以下の物性値を有する表題化合物(8.35g)を得た。
TLC:Rf 0.5(酢酸エチル)
参考例30:メチル(S)-4-((R)-2-(1-アセトキシシクロブチル)-N-((ベンジルオキシ)カルボニル)エチルスルホンイミドイル)-2-((tert-ブトキシカルボニル)アミノ)ブタノアート
参考例29で製造した化合物(500mg)のジクロロメタン(4mL)溶液に、ピリジン(1mL)およびクロロギ酸ベンジル(0.25mL)を加え、室温で2時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=7:3)によって精製した。得られた化合物をSFC(条件D)で分取することにより、以下の物性値を有する標題化合物(250mg)を得た。
HPLC保持時間(分):1.208
参考例31:メチル(S)-4-((R)-2-(1-アセトキシシクロブチル)エチルスルホンイミドイル)-2-((tert-ブトキシカルボニル)アミノ)ブタノアート
参考例30で製造した化合物(250mg)のメタノール(4mL)溶液にパラジウム炭素(25mg)を加え水素雰囲気下、室温で2時間撹拌した。反応液に酢酸エチル(4mL)を加え、セライト(商品名)でろ過することにより、以下の物性値を有する表題化合物(180mg)を得た。
HPLC保持時間(分):0.967
参考例32:(S)-2-((tert-ブトキシカルボニル)アミノ)-4-((R)-2-(1-ヒドロキシシクロブチル)エチルスルホンイミドイル)ブタン酸
参考例31で製造した化合物(180mg)のTHF(2mL)溶液に水酸化リチウム一水和物(54mg)の水溶液(1mL)を0℃で加え、室温で2時間撹拌した。反応液をジオールシリカゲルカラムクロマトグラフィー(ジクロロメタン:メタノール=85:15)によって精製することにより、以下の物性値を有する表題化合物(156mg)を得た。
HPLC保持時間(分):0.837
実施例6:(S)-2-アミノ-4-((R)-2-(1-ヒドロキシシクロブチル)エチルスルホンイミドイル)ブタン酸
Using the compound prepared in Reference Example 26 instead of the compound prepared in Reference Example 20, the same operations as in Reference Example 21 → Reference Example 22 → Reference Example 23 → Reference Example 24 → Reference Example 25 → Example 4 were carried out to obtain the title compound (185 mg) having the following physical properties.
MS (ESI, Pos.): 293 (M+H) + ;
1H -NMR ( D2O ): δ 1.95 - 2.07, 2.15 - 2.35, 3.25 - 3.46, 3.79, 4.17.
Reference Example 27: methyl (2S)-4-{[2-(1-hydroxycyclobutyl)ethyl]thio}-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
Potassium carbonate (3.5 g) and 1-(2-bromoethyl)cyclobutanol (CAS: 1909309-61-0) (5 g) were added to a solution of methyl (2S)-4-mercapto-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate (CAS: 690637-98-0) (6.3 g) in DMF (100 mL), and the mixture was stirred at 60° C. for 2 hours under a nitrogen atmosphere. The reaction solution was diluted with water and extracted with ethyl acetate and hexane (2:1). The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=6:4) to give the title compound (8.7 g) having the following physical properties.
TLC: Rf 0.3 (hexane:ethyl acetate=2:1)
Reference Example 28: methyl (2S)-4-{[2-(1-acetoxycyclobutyl)ethyl]thio}-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
To a solution of the compound (8.7 g) produced in Reference Example 27 in dichloromethane (150 mL), diisopropylethylamine (10 mL), acetic anhydride (4.3 mL) and 4-dimethylaminopyridine (CAS: 1122-58-3) (610 mg) were added and stirred at room temperature overnight. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to give the title compound (9.35 g) having the following physical properties.
TLC: Rf 0.6 (hexane:ethyl acetate=2:1)
Reference Example 29: methyl (2S)-4-{S-[2-(1-acetoxycyclobutyl)ethyl]sulfonimidoyl}-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)butanoate
To a solution of the compound (9.35 g) produced in Reference Example 28 in methanol (100 mL), iodobenzene diacetate (16.2 g) and ammonium carbamate (2.8 g) were added and stirred at room temperature for 1 hour. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (ethyl acetate) to give the title compound (8.35 g) having the following physical properties.
TLC: Rf 0.5 (ethyl acetate)
Reference Example 30: methyl (S)-4-((R)-2-(1-acetoxycyclobutyl)-N-((benzyloxy)carbonyl)ethylsulfonimidoyl)-2-((tert-butoxycarbonyl)amino)butanoate
Pyridine (1 mL) and benzyl chloroformate (0.25 mL) were added to a solution of the compound (500 mg) produced in Reference Example 29 in dichloromethane (4 mL), and the mixture was stirred at room temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=7:3). The resulting compound was separated by SFC (condition D) to obtain the title compound (250 mg) having the following physical properties.
HPLC retention time (min): 1.208
Reference Example 31: methyl (S)-4-((R)-2-(1-acetoxycyclobutyl)ethylsulfonimidoyl)-2-((tert-butoxycarbonyl)amino)butanoate
Palladium carbon (25 mg) was added to a solution of the compound (250 mg) prepared in Reference Example 30 in methanol (4 mL), and the mixture was stirred at room temperature under a hydrogen atmosphere for 2 hours. Ethyl acetate (4 mL) was added to the reaction solution, and the mixture was filtered through Celite (trade name) to obtain the title compound (180 mg) having the following physical properties.
HPLC retention time (min): 0.967
Reference Example 32: (S)-2-((tert-butoxycarbonyl)amino)-4-((R)-2-(1-hydroxycyclobutyl)ethylsulfonimidoyl)butanoic acid
To a solution of the compound (180 mg) prepared in Reference Example 31 in THF (2 mL), an aqueous solution (1 mL) of lithium hydroxide monohydrate (54 mg) was added at 0° C., and the mixture was stirred at room temperature for 2 hours. The reaction solution was purified by diol silica gel column chromatography (dichloromethane:methanol=85:15) to obtain the title compound (156 mg) having the following physical properties.
HPLC retention time (min): 0.837
Example 6: (S)-2-Amino-4-((R)-2-(1-hydroxycyclobutyl)ethylsulfonimidoyl)butanoic acid

参考例32で製造した化合物(156mg)に5Nの塩酸水溶液(2mL)を加え、室温で2時間撹拌した。反応液に水(2mL)を加え、イオン交換樹脂(商品名:DOWEX(H+form))に担持した。充分量の水で洗った後、8%のアンモニア水溶液により溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する本開示化合物(60mg)を得た。
MS(ESI,Pos.):265(M+H)+;
1H-NMR(D2O):δ 1.44 - 1.59, 1.62 - 1.77, 1.93 - 2.08, 2.21 - 2.34, 3.15 - 3.31, 3.31 - 3.44 , 3.75 - 3.82。
参考例33:メチル(S)-4-((S)-N-((ベンジルオキシ)カルボニル)-4,4,4-トリフルオロブチルスルホンイミドイル)-2-((tert-ブトキシカルボニル)アミノ)ブタノアート
メチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート(215mg)のジクロロメタン(10mL)溶液に、ピリジン(1mL)およびクロロギ酸ベンジル(0.17mL)を0℃にて加えた後、室温で終夜撹拌した。反応液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)によって精製した。得られた化合物をSFC(条件C)で分取することにより、以下の物性値を有する表題化合物(87mg)を得た。
MS(ESI,Pos.):547(M+Na)+。
参考例34:メチル(S)-2-((tert-ブトキシカルボニル)アミノ)-4-((S)-4,4,4-トリフルオロブチルスルホンイミドイル)ブタノアート
参考例33で製造した化合物(79mg)のメタノール(4mL)溶液にパラジウム炭素(35mg)を加え水素雰囲気下、室温で終夜撹拌した。反応液に酢酸エチル(4mL)を加え、セライトでろ過した後、濃縮することにより、以下の物性値を有する表題化合物(60mg)を得た。
MS(ESI,Pos.):391(M+H)+。
参考例35:メチル(S)-2-アミノ-4-((S)-4,4,4-トリフルオロブチルスルホンイミドイル)ブタノアート
参考例34で製造した化合物(60mg)のメタノール(1mL)溶液に4N塩酸/酢酸エチル溶液(1mL)を加え、室温で3時間撹拌した。反応液を濃縮した後、イオン交換樹脂(商品名:DOWEX(H+fоrm))に担持させた。充分量のメタノールで洗浄した後、2Nアンモニア/メタノール溶液で溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する表題化合物(39mg)を得た。
MS(ESI,Pos.):291(M+H)+。
実施例7:(S)-2-アミノ-4-((S)-4,4,4-トリフルオロブチルスルホンイミドイル)ブタン酸
5N aqueous hydrochloric acid (2 mL) was added to the compound (156 mg) produced in Reference Example 32, and the mixture was stirred at room temperature for 2 hours. Water (2 mL) was added to the reaction solution, and the mixture was supported on an ion exchange resin (trade name: DOWEX (H + form)). After washing with a sufficient amount of water, the resin was eluted with 8% aqueous ammonia. The fractions reacting with ninhydrin were collected and concentrated to obtain the compound of the present disclosure (60 mg) having the following physical properties.
MS (ESI, Pos.): 265 (M+H) + ;
1 H-NMR (D 2 O): δ 1.44 - 1.59, 1.62 - 1.77, 1.93 - 2.08, 2.21 - 2.34, 3.15 - 3.31, 3.31 - 3.44, 3.75 - 3.82.
Reference Example 33: methyl (S)-4-((S)—N-((benzyloxy)carbonyl)-4,4,4-trifluorobutylsulfonimidoyl)-2-((tert-butoxycarbonyl)amino)butanoate
Pyridine (1 mL) and benzyl chloroformate (0.17 mL) were added to a solution of methyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate (215 mg) in dichloromethane (10 mL) at 0° C., and the mixture was stirred at room temperature overnight. Water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=5:1). The resulting compound was separated by SFC (condition C) to give the title compound (87 mg) having the following physical properties:
MS (ESI, Pos.): 547 (M+Na) + .
Reference Example 34: methyl (S)-2-((tert-butoxycarbonyl)amino)-4-((S)-4,4,4-trifluorobutylsulfonimidoyl)butanoate
Palladium carbon (35 mg) was added to a solution of the compound (79 mg) prepared in Reference Example 33 in methanol (4 mL), and the mixture was stirred overnight at room temperature under a hydrogen atmosphere. Ethyl acetate (4 mL) was added to the reaction mixture, which was then filtered through Celite and concentrated to give the title compound (60 mg) having the following physical properties:
MS (ESI, Pos.): 391 (M+H) + .
Reference Example 35: Methyl (S)-2-amino-4-((S)-4,4,4-trifluorobutylsulfonimidoyl)butanoate
A 4N hydrochloric acid/ethyl acetate solution (1 mL) was added to a methanol (1 mL) solution of the compound (60 mg) produced in Reference Example 34, and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated and then supported on an ion exchange resin (trade name: DOWEX (H+form)). After washing with a sufficient amount of methanol, the resin was eluted with a 2N ammonia/methanol solution. The fractions reacting with ninhydrin were collected and concentrated to give the title compound (39 mg) having the following physical properties.
MS (ESI, Pos.): 291 (M+H) + .
Example 7: (S)-2-amino-4-((S)-4,4,4-trifluorobutylsulfonimidoyl)butanoic acid

参考例35で製造した化合物(39mg)に水(0.5mL)を加え、0℃に冷却した。反応液に1N水酸化ナトリウム水溶液(0.14mL)を加え、30分撹拌した。反応液をイオン交換樹脂(商品名:DOWEX(NH4+form))に担持させ、充分量の水で溶出させた。ニンヒドリンに反応するフラクションを集め、濃縮することにより、以下の物性値を有する本開示化合物(33mg)を得た。
MS(ESI,Pos.):277(M+H)+;
1H-NMR(D2O):δ 1.98 - 2.06, 2.23 - 2.36, 3.23 - 3.41, 3.77。
参考例36:(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[(4,4,4-トリフルオロブチル)チオ]ブタン酸
参考例1で製造した化合物(70mg)のTHF(1mL)溶液に水酸化リチウム一水和物(16mg)の水溶液(0.5mL)を0℃で加え、室温で1時間撹拌した。反応液に1N塩酸水溶液を加えて酸性とし、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮することにより、以下の物性値を有する表題化合物(67mg)を得た。
HPLC保持時間(分):1.162
参考例37:エチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[(4,4,4-トリフルオロブチル)チオ]ブタノアート
参考例36で製造した化合物(67mg)のジクロロメタン(1mL)溶液に、エタノール(0.057mL)、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド(CAS:25952-53-8)(56mg)および4-ジメチルアミノピリジン(CAS:1122-58-3)(5mg)を加え、室温で3時間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=8:2)によって精製することにより、以下の物性値を有する表題化合物(57mg)を得た。
HPLC保持時間(分):1.288
参考例38:エチル(2S)-2-({[(2-メチル-2-プロパニル)オキシ]カルボニル}アミノ)-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート
参考例37で製造した化合物(57mg)のメタノール(3mL)溶液に二酢酸ヨードベンゼン(123mg)およびカルバミン酸アンモニウム(24mg)を加え、室温で30分撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:8)によって精製することにより、以下の物性値を有する表題化合物(61mg)を得た。
HPLC保持時間(分):1.05
実施例8:エチル(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート
Water (0.5 mL) was added to the compound (39 mg) produced in Reference Example 35, and the mixture was cooled to 0° C. 1N aqueous sodium hydroxide solution (0.14 mL) was added to the reaction solution, and the mixture was stirred for 30 minutes. The reaction solution was supported on an ion exchange resin (trade name: DOWEX (NH4 + form)) and eluted with a sufficient amount of water. The fractions reacting with ninhydrin were collected and concentrated to obtain the compound of the present disclosure (33 mg) having the following physical properties.
MS (ESI, Pos.): 277 (M+H) + ;
1 H-NMR (D 2 O): δ 1.98 - 2.06, 2.23 - 2.36, 3.23 - 3.41, 3.77.
Reference Example 36: (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[(4,4,4-trifluorobutyl)thio]butanoic acid
A solution (0.5 mL) of lithium hydroxide monohydrate (16 mg) was added to a solution (1 mL) of the compound (70 mg) prepared in Reference Example 1 in THF at 0° C., and the mixture was stirred at room temperature for 1 hour. The reaction mixture was acidified with a 1N aqueous hydrochloric acid solution, and extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (67 mg) having the following physical properties.
HPLC retention time (min): 1.162
Reference Example 37: Ethyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[(4,4,4-trifluorobutyl)thio]butanoate
To a solution of the compound (67 mg) produced in Reference Example 36 in dichloromethane (1 mL), ethanol (0.057 mL), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (CAS: 25952-53-8) (56 mg) and 4-dimethylaminopyridine (CAS: 1122-58-3) (5 mg) were added and stirred at room temperature for 3 hours. A saturated aqueous solution of sodium hydrogen carbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to give the title compound (57 mg) having the following physical properties.
HPLC retention time (min): 1.288
Reference Example 38: Ethyl (2S)-2-({[(2-methyl-2-propanyl)oxy]carbonyl}amino)-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate
To a solution of the compound (57 mg) produced in Reference Example 37 in methanol (3 mL), iodobenzene diacetate (123 mg) and ammonium carbamate (24 mg) were added and stirred at room temperature for 30 minutes. A saturated aqueous solution of sodium bicarbonate was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane:ethyl acetate=2:8) to give the title compound (61 mg) having the following physical properties.
HPLC retention time (min): 1.05
Example 8: Ethyl (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate

参考例38で製造した化合物(61mg)に4N塩酸-ジオキサン溶液(1mL)を加え、室温で1時間撹拌した。反応液を濃縮し、得られた残渣をアミノシリカゲルカラムクロマトグラフィー(酢酸エチル:メタノール=2:1)によって精製することにより、以下の物性値を有する本開示化合物(45mg)を得た。
MS(ESI,Pos.):305(M+H)+;
1H-NMR(CDCl3): δ 1.24 - 1.33, 1.90, 1.94 - 2.07, 2.09 - 2.22, 2.22 - 2.38, 3.07 - 3.16, 3.16 - 3.32, 3.58, 4.21。
実施例8-1:イソプロピル(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート
A 4N hydrochloric acid-dioxane solution (1 mL) was added to the compound (61 mg) prepared in Reference Example 38, and the mixture was stirred at room temperature for 1 hour. The reaction solution was concentrated, and the resulting residue was purified by aminosilica gel column chromatography (ethyl acetate:methanol=2:1) to give the disclosed compound (45 mg) having the following physical properties:
MS (ESI, Pos.): 305 (M+H) + ;
1H -NMR ( CDCl3 ): δ 1.24 - 1.33, 1.90, 1.94 - 2.07, 2.09 - 2.22, 2.22 - 2.38, 3.07 - 3.16, 3.16 - 3.32, 3.58, 4.21.
Example 8-1: Isopropyl (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate

エタノールの代わりに2-プロパノールを用いて、参考例37→参考例38→実施例8と同様の操作を行うことにより、以下の物性値を有する本開示化合物(43mg)を得た。
MS(ESI,Pos.):319(M+H)+;
1H-NMR(CDCl3):δ 1.27, 1.83 - 2.06, 2.09 - 2.22, 2.22 - 2.39, 3.11, 3.14 - 3.32, 3.53, 5.05。
[薬理実施例]
以下の薬理実施例において、比較例として、BSO(Toronto Research Chemicals)(CAS番号:83730-53-4)および非特許文献2に記載の化合物番号Ic((2S)-2-アミノ-4-[(2R,S)-2-カルボキシブチル-(R,S)-スルホンイミドイル]ブタン酸((2S)-2-Amino-4-[(2R,S)-2-carboxybutyl-(R,S)-sulfonimidoyl]butanoic acid))を使用し、それぞれ比較例1および比較例2とした。
[薬理実施例1]GCL酵素阻害活性評価
(GCL-化合物プレインキュベーション酵素阻害活性評価系(基質スタート))
200mmol/L Tris-HCl(pH 8.0)、20mmol/L MgCl2、150mmol/L KCl、0.1% BSAを含むアッセイバッファーを用いて、リコンビナントヒトGCLCおよびリコンビナントヒトGCLMを1:2のモル濃度比で混合し、37℃で60分間静置して複合体形成させたものをヒトGCL酵素溶液とした。次に、終濃度5nmol/L GCLCに相当するヒトGCL酵素複合体溶液にATPを終濃度0.2mmol/Lで、各化合物を終濃度0.001、0.003、0.01、0.03、0.1、0.3、1、3、10または30μmol/Lとなるよう添加した。このヒトGCL酵素複合体-ATP-化合物混合溶液を室温で60分間プレインキュベートした。さらに、基質であるL-グルタミン酸(L-glutamic acid、Nacalai tesque社)とL-システイン(L-cysteine、Nacalai tesque社)をそれぞれ終濃度1.2および0.2mmol/Lとなるように添加し、酵素反応を開始した。なお、リコンビナントヒトGCLCおよびリコンビナントヒトGCLMはそれぞれHisタグ付きであり、大腸菌に発現させた後にニッケルカラムおよび陰イオン交換カラムで精製したものを用いた。酵素反応は384ウェルポリプロピレン製のマイクロプレートを用いて室温で実施し、酵素非添加ウェルをBlank群とした。
The same operations as in Reference Example 37→Reference Example 38→Example 8 were carried out using 2-propanol instead of ethanol to give the presently disclosed compound (43 mg) having the following physical properties.
MS (ESI, Pos.): 319 (M+H) + ;
1H -NMR ( CDCl3 ): δ 1.27, 1.83 - 2.06, 2.09 - 2.22, 2.22 - 2.39, 3.11, 3.14 - 3.32, 3.53, 5.05.
[Pharmacological Examples]
In the following pharmacological examples, BSO (Toronto Research Chemicals) (CAS number: 83730-53-4) and compound number Ic ((2S)-2-Amino-4-[(2R,S)-2-carboxybutyl-(R,S)-sulfonimidoyl]butanoic acid) described in
[Pharmacological Example 1] Evaluation of GCL enzyme inhibitory activity
(GCL-compound preincubation enzyme inhibitory activity evaluation system (substrate start))
Using an assay buffer containing 200 mmol/L Tris-HCl (pH 8.0), 20 mmol/L MgCl 2 , 150 mmol/L KCl, and 0.1% BSA, recombinant human GCLC and recombinant human GCLM were mixed at a molar concentration ratio of 1:2, and the mixture was allowed to stand at 37°C for 60 minutes to form a complex, which was used as a human GCL enzyme solution. Next, ATP was added to a human GCL enzyme complex solution corresponding to a final concentration of 5 nmol/L GCLC at a final concentration of 0.2 mmol/L, and each compound was added to a final concentration of 0.001, 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 μmol/L. This human GCL enzyme complex-ATP-compound mixed solution was pre-incubated at room temperature for 60 minutes. Furthermore, the substrates L-glutamic acid (Nacalai Tesque) and L-cysteine (Nacalai Tesque) were added to final concentrations of 1.2 and 0.2 mmol/L, respectively, to initiate the enzyme reaction. The recombinant human GCLC and recombinant human GCLM were His-tagged and expressed in Escherichia coli and then purified using a nickel column and an anion exchange column. The enzyme reaction was carried out at room temperature using a 384-well polypropylene microplate, and wells to which no enzyme was added were used as the blank group.
酵素反応開始1時間後に、内部標準物質として20μmol/Lのオフタルミン酸(Ophthalmic acid)を含む1%ギ酸水溶液を添加して酵素反応を停止させた。酵素反応プレートの上部をアルミシールし、560gで5分間、室温にて遠心後、RapidFire-Mass Spectrometryシステムにて酵素反応生成物であるγ-グルタミルシステイン(γ-glutamylcysteine)および内部標準物質オフタルミン酸を定量した。それぞれの定量値の比をとり、Blank群の平均値を100%阻害、化合物非添加群の平均値を0%阻害として、各化合物濃度におけるγ-グルタミルシステイン産生阻害率を算出し、IC50値を求めた。 One hour after the start of the enzyme reaction, a 1% formic acid solution containing 20 μmol/L of ophthalmic acid as an internal standard was added to stop the enzyme reaction. The top of the enzyme reaction plate was sealed with aluminum, and the plate was centrifuged at 560 g for 5 minutes at room temperature, after which the enzyme reaction product γ-glutamylcysteine and the internal standard ophthalmic acid were quantified using a RapidFire-Mass Spectrometry system. The ratio of the respective quantitative values was calculated, and the average value of the blank group was set to 100% inhibition, and the average value of the compound-free group was set to 0% inhibition, to calculate the γ-glutamylcysteine production inhibition rate at each compound concentration, and the IC 50 value was obtained.
本開示化合物は優れたGCL酵素阻害活性を示した。表1に、本開示化合物の代表例として、各実施例において示される化合物のGCL酵素阻害活性(IC50値)を示す。 The compounds of the present disclosure exhibited excellent GCL enzyme inhibitory activity. Table 1 shows the GCL enzyme inhibitory activity ( IC50 value) of the compounds shown in each Example as representative examples of the compounds of the present disclosure.
非特許文献2において、比較例2として示される化合物は、BSOよりも、大腸菌GCLCへの結合親和性が約500倍(Ki値比)向上したことが報告されていたが、本アッセイ系においては、BSOと同等の阻害活性であった。
In

また、GCL酵素阻害活性は以下の方法においても評価できる。
(GCL酵素阻害活性評価系(酵素スタート))
200 mmol/L Tris-HCl(pH 8.0)、20 mmol/L MgCl2、150 mmol/L KCl,0.1% BSAを含むアッセイバッファーを用いて、リコンビナントヒトGCLCおよびリコンビナントヒトGCLMを1:2のモル濃度比で混合し、37℃で60分間静置して複合体形成させたものをヒトGCL酵素溶液とした。次に、ATP、基質であるL-グルタミン酸およびL-システインをそれぞれ終濃度0.2、1.2および0.2mmol/Lとなるよう調製し、各化合物を終濃度0.001、0.003、0.01、0.03、0.1、0.3、1、3、10または30μmol/Lとなるよう添加した。さらに続けて、上述のようにあらかじめ調製しておいたヒトGCL酵素複合体を終濃度5nmol/L GCLCに相当する濃度となるように添加し、酵素反応を開始した。なお、リコンビナントヒトGCLCおよびリコンビナントヒトGCLMはそれぞれHisタグ付きであり、大腸菌に発現させた後にニッケルカラムと陰イオン交換カラムで精製したものを用いた。酵素反応は384ウェルポリプロピレン製のマイクロプレートを用いて室温で実施し、酵素非添加ウェルをBlank群とした。
The GCL enzyme inhibitory activity can also be evaluated by the following method.
(GCL enzyme inhibitory activity evaluation system (enzyme start))
Using an assay buffer containing 200 mmol/L Tris-HCl (pH 8.0), 20 mmol/L MgCl2, 150 mmol/L KCl, and 0.1% BSA, recombinant human GCLC and recombinant human GCLM were mixed at a molar concentration ratio of 1:2, and allowed to stand at 37°C for 60 minutes to form a complex, which was used as a human GCL enzyme solution. Next, ATP, substrates L-glutamic acid, and L-cysteine were prepared to final concentrations of 0.2, 1.2, and 0.2 mmol/L, respectively, and each compound was added to a final concentration of 0.001, 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 μmol/L. Next, the human GCL enzyme complex prepared in advance as described above was added to a final concentration equivalent to 5 nmol/L GCLC to initiate the enzyme reaction. The recombinant human GCLC and recombinant human GCLM were His-tagged and expressed in E. coli and then purified using a nickel column and an anion exchange column. The enzyme reaction was carried out at room temperature using a 384-well polypropylene microplate, and the wells to which the enzyme was not added were used as the blank group.
酵素反応開始1時間後に、内部標準物質として20μmol/Lのオフタルミン酸を含む1%ギ酸水溶液を添加して酵素反応を停止させた。酵素反応プレートの上部をアルミシールし、560gで5分間、室温にて遠心後、RapidFire-Mass Spectrometryシステムにて酵素反応生成物であるγ-グルタミルシステインおよび内部標準物質オフタルミン酸を定量した。それぞれの定量値の比をとり、Blank群の平均値を100%阻害、化合物非添加群の平均値を0%阻害として、各化合物濃度におけるγ-グルタミルシステイン産生阻害率を算出し、IC50値を求めた。本系においても、本開示化合物は優れたGCL酵素阻害活性を示した。
[薬理実施例2]インビトロ試験
(細胞培養)
ヒト卵巣がん細胞株TOV21G(以下、細胞と記載)はATCC社より購入した。細胞は2mmol/L L-Glutamine、10%(vol%)FBS、1%(vol%)Penicillin-Streptomycin-Amphotericin B Suspensionを含むDMEM/Ham’s F-12(以下、10%FBS-DMEM/Ham’s F-12)を使用し、37℃、5%CO2および95%Air条件下で培養した。
[薬理実施例2-1]インビトロGSH濃度測定試験
(方法)
細胞を平底96ウェルプレートに2×103細胞/100μL/ウェルで播種した後、37℃、5%CO2および95%Air条件下で一晩インキュベーションした。細胞播種の翌日に各化合物を培養細胞に添加し、37℃、5%CO2および95%Air条件下で24時間培養後にGSH濃度をGSH-GloTM Glutathione Assay(Promega)にて、ATP濃度をCellTiter-Glo(登録商標) Luminescent Cell Viability Assay(Promega)にて測定した。細胞数補正の代替としてGSH濃度をATP濃度で除した数値を用いた。用量反応曲線を作成し、IC50値を算出した。
(結果)
BSO(比較例1)、比較例2、ならびに実施例1、実施例1-2、実施例2、実施例3、実施例8および実施例8-1でそれぞれ製造した各化合物のGCL阻害活性のIC50値はそれぞれ、1.5、1.3、0.2、0.2、0.3、0.1、0.007および0.01μmol/Lであった。比較例2がBSOと同等の阻害活性であったのに対し、本開示化合物はBSOよりも細胞系でのGCL阻害活性が向上していた。例えば、実施例1、実施例1-2、実施例1-5、実施例2、実施例3、実施例3-2、実施例3-3、実施例8および実施例8-1でそれぞれ製造した各化合物は、BSOの細胞系での活性に対して、GCL阻害活性として、各々7、7、3、6、14、12、10、206および106倍向上していることが確認された。
[薬理実施例2-2]インビトロ増殖試験
(方法)
細胞を平底96ウェルプレートに2×103細胞/100μL/ウェルで播種した後、37℃、5%CO2および95%Air条件下で一晩インキュベーションした。細胞播種の翌日に各化合物を培養細胞に添加し、37℃、5%CO2、95%Air条件下で48時間培養後にATP濃度をCellTiter-Glo(登録商標) Luminescent Cell Viability Assay(Promega)にて測定した。用量反応曲線を作成し、IC50値を算出した。
(結果)
BSO(比較例1)ならびに実施例1、実施例1-2、実施例2、実施例3、実施例8および実施例8-1でそれぞれ製造した各化合物の細胞増殖阻害のIC50値はそれぞれ、4.8、0.5、0.6、0.5、0.2、0.02および0.03μmol/Lであり、BSOの細胞系での活性に対して、増殖阻害活性として、各々11、9、11、27、292および166倍向上していることが確認された。
One hour after the start of the enzyme reaction, a 1% formic acid solution containing 20 μmol/L of ophthalmic acid as an internal standard was added to stop the enzyme reaction. The top of the enzyme reaction plate was sealed with aluminum, and the plate was centrifuged at 560 g for 5 minutes at room temperature, after which the enzyme reaction product γ-glutamylcysteine and the internal standard ophthalmic acid were quantified using a RapidFire-Mass Spectrometry system. The ratio of the respective quantified values was calculated, and the average value of the blank group was set to 100% inhibition, and the average value of the compound-free group was set to 0% inhibition, to calculate the γ-glutamylcysteine production inhibition rate at each compound concentration, and the IC 50 value was obtained. In this system, the compound of the present disclosure also showed excellent GCL enzyme inhibitory activity.
[Pharmacological Example 2] In vitro test
(Cell Culture)
Human ovarian cancer cell line TOV21G (hereinafter referred to as cells) was purchased from ATCC. The cells were cultured in DMEM/Ham's F-12 containing 2 mmol/L L-Glutamine, 10% (vol%) FBS, and 1% (vol%) Penicillin-Streptomycin-Amphotericin B Suspension (hereinafter referred to as 10% FBS-DMEM/Ham's F-12) under conditions of 37°C, 5% CO2 , and 95% air.
[Pharmacological Example 2-1] In vitro GSH concentration measurement test
(method)
The cells were seeded in a flat-bottom 96-well plate at 2 x 103 cells/100 μL/well and then incubated overnight at 37°C, 5% CO2 and 95% air. The day after cell seeding, each compound was added to the cultured cells, and after 24 hours of culture at 37°C, 5% CO2 and 95% air, the GSH concentration was measured using the GSH-Glo ™ Glutathione Assay (Promega) and the ATP concentration was measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). As an alternative to cell number correction, the value obtained by dividing the GSH concentration by the ATP concentration was used. A dose-response curve was created and the IC50 value was calculated.
(result)
The IC 50 values of the GCL inhibitory activity of each compound produced in BSO (Comparative Example 1), Comparative Example 2, and Examples 1, 1-2, 2, 3, 8, and 8-1 were 1.5, 1.3, 0.2, 0.2, 0.3, 0.1, 0.007, and 0.01 μmol/L, respectively. Comparative Example 2 had an inhibitory activity equivalent to that of BSO, whereas the compound of the present disclosure had an improved GCL inhibitory activity in a cell line compared to BSO. For example, it was confirmed that each compound produced in Examples 1, 1-2, 1-5, 2, 3, 3-2, 3-3, 8, and 8-1 had an improved GCL inhibitory activity of 7, 7, 3, 6, 14, 12, 10, 206, and 106 times, respectively, compared to the activity of BSO in a cell line.
[Pharmacological Example 2-2] In vitro proliferation test
(method)
The cells were seeded in a flat-bottom 96-well plate at 2 x 103 cells/100 μL/well and then incubated overnight at 37°C, 5% CO2 and 95% air. On the day after cell seeding, each compound was added to the cultured cells, and after 48 hours of culture at 37°C, 5% CO2 and 95% air, the ATP concentration was measured using CellTiter-Glo (registered trademark) Luminescent Cell Viability Assay (Promega). A dose-response curve was created and the IC50 value was calculated.
(result)
The IC50 values of cell proliferation inhibition of BSO (Comparative Example 1) and the compounds prepared in Examples 1, 1-2, 2, 3, 8 and 8-1 were 4.8, 0.5, 0.6, 0.5, 0.2, 0.02 and 0.03 μmol/L, respectively, and it was confirmed that the proliferation inhibitory activity was improved by 11, 9, 11, 27, 292 and 166 times, respectively, compared with the activity of BSO in the cell line.
薬理実施例2-1および薬理実施例2-2より、上記薬理実施例に記載の化合物に代表される本開示化合物は、BSOよりも細胞系での活性が向上した化合物であることが確認された。
[薬理実施例3]インビボ試験
(細胞培養)
ヒト卵巣がん細胞株TOV21G細胞はATCC社より購入した。同細胞は10%FBS-DMEM/Ham’s F-12を使用し、37℃、5%CO2および95%Air条件下で培養した。
(インビボゼノグラフト試験)
移植時7~8週齢の雌性CB17/Icr-Prkdcscid/CrlCrljマウスを実験に用いた。培養細胞をマトリゲルと等量ずつ混合した細胞懸濁液を目的濃度に調製し、マウス右側腹部皮下に2.5×106細胞/0.1mL/bodyの細胞を移植した。腫瘍が認められた個体について腫瘍の長径および短径を電子ノギスで測定し、以下の式に従って腫瘍体積を算出した。
腫瘍体積(mm3)=腫瘍の長径(mm)×(腫瘍の短径(mm))2×0.5
化合物は注射用水に溶解し、ゾンデを用いて1日1回経口投与を行った。
(インビボGSH濃度測定試験)
化合物投与前日または投与当日の平均腫瘍体積140~180mm3の個体を実験に用い、投与後24時間の腫瘍を濃度測定に供した。腫瘍重量を測定後、10倍量の5% 5-Sulfosalicylic acidを加え、氷冷下にてホモジナイザーで破砕した。遠心分離後の上清を使用し、GSH/GSSG-GloTM Assay(Promega)を用いて総GSH濃度を測定した。上清中のタンパク質量をPierceTM BCA Protein Assay Kitを用いて定量し、補正に用いた。
[薬理実施例3-1]TOV21G細胞皮下移植モデルにおける薬物動力学(PD)評価
単回投与後の腫瘍中GSH量を指標としたPD評価を実施した。TOV21G細胞を皮下移植後19日に群分けを行い、その翌日にBSO(比較例1)および実施例2で製造した化合物を各々単回経口投与した。群分け時の各群の腫瘍体積の平均値は、141~158mm3であった。BSOの用量を100、300および750mg/kg、実施例2で製造した化合物を30、100および300mg/kgとした。評価時点は、単回投与後24時間とした(図1)。BSOは用量依存的に腫瘍中GSHを低下させ、最も高用量である750mg/kgでは21%まで低下させた。実施例2で製造した化合物について、用量依存的に腫瘍中GSHを低下させ、最も高用量である300mg/kgではコントロールの20%まで低下させた。
From Pharmacological Example 2-1 and Pharmacological Example 2-2, it was confirmed that the compounds of the present disclosure typified by the compounds described in the above Pharmacological Examples are compounds having improved activity in cell lines compared to BSO.
[Pharmacological Example 3] In vivo test
(Cell Culture)
TOV21G cells, a human ovarian cancer cell line, were purchased from ATCC. The cells were cultured in 10% FBS-DMEM/Ham's F-12 under conditions of 37° C., 5% CO 2 and 95% air.
In vivo xenograft testing
Female CB17/Icr-Prkdc scid /CrlCrlj mice aged 7-8 weeks at the time of transplantation were used in the experiment. Cultured cells were mixed with equal amounts of Matrigel to prepare a cell suspension at the desired concentration, and 2.5 x 10 6 cells/0.1 mL/body of cells were transplanted subcutaneously into the right flank of the mouse. For individuals with tumors, the long and short diameters of the tumors were measured with electronic calipers, and the tumor volume was calculated according to the following formula.
Tumor volume (mm 3 )=longest axis of tumor (mm)×(shortest axis of tumor (mm)) 2 ×0.5
The compound was dissolved in water for injection and orally administered once a day using a sonde.
(In vivo GSH concentration measurement test)
Mice with an average tumor volume of 140-180 mm3 on the day before or on the day of compound administration were used in the experiment, and tumors 24 hours after administration were subjected to concentration measurement. After measuring the tumor weight, 10 times the amount of 5% 5-sulfosalicylic acid was added and the tumors were crushed with a homogenizer under ice cooling. The supernatant after centrifugation was used to measure the total GSH concentration using GSH/GSSG-Glo ™ Assay (Promega). The amount of protein in the supernatant was quantified using Pierce ™ BCA Protein Assay Kit and used for correction.
[Pharmacological Example 3-1] Pharmacokinetic (PD) evaluation in a TOV21G cell subcutaneous transplantation model
PD evaluation was performed using the amount of GSH in the tumor after a single administration as an index. Grouping was performed 19 days after subcutaneous implantation of TOV21G cells, and on the following day, BSO (Comparative Example 1) and the compound prepared in Example 2 were each orally administered in a single dose. The average tumor volume of each group at the time of grouping was 141 to 158 mm3 . The doses of BSO were 100, 300, and 750 mg/kg, and the compound prepared in Example 2 was 30, 100, and 300 mg/kg. The evaluation time point was 24 hours after the single administration (Figure 1). BSO dose-dependently reduced GSH in the tumor, and at the highest dose of 750 mg/kg, it reduced GSH to 21%. For the compound prepared in Example 2, GSH in the tumor was dose-dependently reduced, and at the highest dose of 300 mg/kg, it reduced GSH to 20% of the control.
次に、実施例3で製造した化合物について、単回投与後の腫瘍中GSH量を指標としたPD評価を実施した。TOV21Gを皮下移植後19日に群分けを行い、その3日後に同化合物を単回経口投与した。投与日の各群の腫瘍体積の平均値は、141~177mm3であった。実施例3で製造した化合物の用量を30、100および300mg/kgとし、評価時点を24時間とした。また、実施例2で製造した化合物の再現性確認および最大作用を確認するため、同化合物の300mg/kgおよび750mg/kg投与後24時間についても評価した。その結果、実施例3で製造した化合物が投薬後24時間において腫瘍中GSHを低下させることを確認した(図2)。実施例2で製造した化合物については、再現性を確認し、前回試験において100mg/kgおよび300mg/kgで用量反応性があったこと、ならびに今回試験の結果から300mg/kgで最大作用を示すと考えられた。 Next, the compound produced in Example 3 was subjected to PD evaluation using the amount of GSH in the tumor after a single administration as an index. Grouping was performed on the 19th day after subcutaneous implantation of TOV21G, and the compound was orally administered once 3 days later. The average tumor volume of each group on the day of administration was 141-177 mm3 . The doses of the compound produced in Example 3 were 30, 100, and 300 mg/kg, and the evaluation time point was 24 hours. In addition, in order to confirm the reproducibility and maximum effect of the compound produced in Example 2, the compound was also evaluated 24 hours after administration of 300 mg/kg and 750 mg/kg. As a result, it was confirmed that the compound produced in Example 3 reduced GSH in the tumor 24 hours after administration (Figure 2). The compound produced in Example 2 was confirmed to be reproducible, and it was found to have dose-responsiveness at 100 mg/kg and 300 mg/kg in the previous test, and it was considered to show maximum effect at 300 mg/kg based on the results of the current test.
以上から、最大作用を示す用量としてはBSOが750mg/kg、実施例2で製造した化合物が300mg/kg、実施例3で製造した化合物が100mg/kgであり、実施例2および実施例3でそれぞれ製造した各化合物は、それぞれ用量比でBSOより約3倍および約7.5倍、インビボ活性が向上していることが確認された。
[薬理実施例3-2]TOV21G細胞皮下移植モデルにおける抗腫瘍効果評価
実施例2および実施例3でそれぞれ製造した各化合物のインビボ抗腫瘍効果を評価した。各群の平均腫瘍体積が105~108mm3になった時点で群分けを行い、群分け日当日から最終観察日の前日まで各化合物を1日1回連日経口投与した。BSOの投与量を750mg/kg、実施例2で製造した化合物の投与量を300mg/kg、実施例3で製造した化合物の投与量を30および100mg/kgとした。本試験の最終評価日はDay23とした。
From the above, it was confirmed that the doses showing the maximum effect were 750 mg/kg for BSO, 300 mg/kg for the compound prepared in Example 2, and 100 mg/kg for the compound prepared in Example 3, and that the compounds prepared in Examples 2 and 3 had in vivo activities that were approximately 3-fold and approximately 7.5-fold higher than BSO in terms of dose ratio, respectively.
[Pharmacological Example 3-2] Evaluation of antitumor effect in a TOV21G cell subcutaneous transplantation model
The in vivo antitumor effects of each compound prepared in Example 2 and Example 3 were evaluated. When the average tumor volume of each group reached 105-108 mm3, the animals were divided into groups, and each compound was orally administered once a day from the day of grouping until the day before the final observation day. The dose of BSO was 750 mg/kg, the dose of the compound prepared in Example 2 was 300 mg/kg, and the doses of the compounds prepared in Example 3 were 30 and 100 mg/kg. The final evaluation day of this test was Day 23.
各群の腫瘍体積を図3に示した。Day23における各群の腫瘍体積の平均値は、媒体群で819mm3、BSO 750mg/kg群で463mm3、実施例2 300mg/kg群で396mm3、実施例3 30mg/kg群で453mm3、実施例3 100mg/kg群で414mm3であった。BSO 750mg/kg群の平均腫瘍増殖抑制率(TGImean)が44%であったのに対し、実施例2 300mg/kg群のTGImeanが52%、実施例3 30mg/kg群および100mg/kg群のTGImeanがそれぞれ45%および49%であった。以上から、最大作用を示す用量としてはBSOが750mg/kg、実施例2で製造した化合物が300mg/kg、実施例3で製造した化合物が30mg/kgであり、実施例2および実施例3でそれぞれ製造した各化合物は、それぞれ用量比でBSOより約3倍および約25倍インビボ活性が向上していることが確認された。
The tumor volume of each group is shown in Figure 3. The average tumor volume of each group on Day 23 was 819 mm 3 in the vehicle group, 463 mm 3 in the
以上から、実施例2および実施例3に代表される本開示化合物は、BSOよりもインビボ活性が向上した化合物であることが確認された。
[製剤実施例]
製剤例
以下の各成分を常法により混合した後打錠して、一錠中に10mgの活性成分を含有する錠剤約1万錠が得られる。
・(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタン酸 ……100g
・カルボキシメチルセルロースカルシウム(崩壊剤) …… 20g
・ステアリン酸マグネシウム(潤滑剤) …… 10g
・微結晶セルロース ……870g
From the above, it was confirmed that the compounds of the present disclosure represented by Examples 2 and 3 have improved in vivo activity compared to BSO.
[Formulation Examples]
Formulation examples
The following ingredients are mixed in a conventional manner and compressed into tablets to give approximately 10,000 tablets, each containing 10 mg of active ingredient.
(2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoic acid ... 100g
・Carboxymethylcellulose calcium (disintegrant) …… 20g
・Magnesium stearate (lubricant) …… 10g
・Microcrystalline cellulose...870g
本開示化合物は、GCL阻害活性を有するため、本開示化合物を有効成分として含む医薬品は、がんなどのGCL関連疾患の進行抑制、再発抑制および/または治療剤として有用である。 Since the disclosed compound has GCL inhibitory activity, pharmaceuticals containing the disclosed compound as an active ingredient are useful as agents for inhibiting the progression, inhibiting recurrence, and/or treating GCL-related diseases such as cancer.
Claims (13)

R1は、水素原子、メチル基またはヒドロキシル基を表し、
R2は、(1)-C(R3R4)-R5または(2)1~9個のR6で置換されていてもよいC3~C5シクロアルキル基を表し、
R3またはR4は、それぞれ、(1)水素原子、(2)ヒドロキシル基、(3)ハロゲン原子または(4)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、
R5は、(1)トリフルオロメチル基または(2)tert-ブチル基を表し、
R6は、(1)ヒドロキシル基、(2)ハロゲン原子または(3)1~3個のハロゲン原子で置換されていてもよいメチル基を表し、R6が複数の場合、複数のR6はそれぞれ同じでも異なっていてもよく、
R7は、水素原子またはC1~C4アルキル基を表し、
nは0または1を表す。)で示される化合物またはその塩。 General formula (I)
R 1 represents a hydrogen atom, a methyl group or a hydroxyl group;
R2 represents (1) -C( R3R4 ) -R5 or (2) a C3-C5 cycloalkyl group optionally substituted with 1 to 9 R6s ;
R3 and R4 each represent (1) a hydrogen atom, (2) a hydroxyl group, (3) a halogen atom, or (4) a methyl group optionally substituted with 1 to 3 halogen atoms;
R5 represents (1) a trifluoromethyl group or (2) a tert-butyl group;
R 6 represents (1) a hydroxyl group, (2) a halogen atom, or (3) a methyl group which may be substituted by 1 to 3 halogen atoms, and when there are a plurality of R 6s , the plurality of R 6s may be the same or different;
R7 represents a hydrogen atom or a C1-C4 alkyl group;
n represents 0 or 1, or a salt thereof.





(1)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタン酸、
(2)(2S)-2-アミノ-4-[S-(2-シクロペンチルエチル)スルホンイミドイル]ブタン酸、
(3)(2S)-2-アミノ-4-[S-(2-シクロブチルエチル)スルホンイミドイル]ブタン酸、
(4)(2S)-2-アミノ-4-[S-(2-シクロプロピルエチル)スルホンイミドイル]ブタン酸、
(5)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-メチルブチル)スルホンイミドイル]ブタン酸、
(6)(2S)-2-アミノ-4-{S-[2-(3,3-ジフルオロシクロブチル)エチル]スルホンイミドイル}ブタン酸、
(7)(2S)-2-アミノ-4-[S-(4,4-ジメチルペンチル)スルホンイミドイル]ブタン酸、
(8)(2S)-2-アミノ-4-{S-[2-(1-ヒドロキシシクロブチル)エチル]スルホンイミドイル}ブタン酸、
(9)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-ヒドロキシブチル)スルホンイミドイル]ブタン酸、
(10)(2S)-2-アミノ-4-{S-[2-(1-フルオロシクロブチル)エチル]スルホンイミドイル}ブタン酸、
(11)(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロ-3-ヒドロキシ-3-メチルブチル)スルホンイミドイル]ブタン酸、
(12)(2S)-2-アミノ-4-{S-[4,4,4-トリフルオロ-3-ヒドロキシ-3-(トリフルオロメチル)ブチル]スルホンイミドイル}ブタン酸、
(13)(2S)-2-アミノ-4-[S-(3,3,4,4,4-ペンタフルオロブチル)スルホンイミドイル]ブタン酸、
(14)(S)-2-アミノ-4-((R,3R)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸、
(15)(S)-2-アミノ-4-((R,3S)-4,4,4-トリフルオロ-3-ヒドロキシブチルスルホンイミドイル)ブタン酸、
(16)(S)-2-アミノ-4-((R)-2-(1-ヒドロキシシクロブチル)エチルスルホンイミドイル)ブタン酸、
(17)(S)-2-アミノ-4-((S)-4,4,4-トリフルオロブチルスルホンイミドイル)ブタン酸、
(18)エチル(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート、および
(19)イソプロピル(2S)-2-アミノ-4-[S-(4,4,4-トリフルオロブチル)スルホンイミドイル]ブタノアート
からなる群から選択される化合物である、請求項1記載の化合物またはその塩。 The compound represented by the general formula (I)
(1) (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoic acid,
(2) (2S)-2-amino-4-[S-(2-cyclopentylethyl)sulfonimidoyl]butanoic acid,
(3) (2S)-2-amino-4-[S-(2-cyclobutylethyl)sulfonimidoyl]butanoic acid,
(4) (2S)-2-amino-4-[S-(2-cyclopropylethyl)sulfonimidoyl]butanoic acid,
(5) (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-methylbutyl)sulfonimidoyl]butanoic acid,
(6) (2S)-2-amino-4-{S-[2-(3,3-difluorocyclobutyl)ethyl]sulfonimidoyl}butanoic acid,
(7) (2S)-2-amino-4-[S-(4,4-dimethylpentyl)sulfonimidoyl]butanoic acid,
(8) (2S)-2-amino-4-{S-[2-(1-hydroxycyclobutyl)ethyl]sulfonimidoyl}butanoic acid,
(9) (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-hydroxybutyl)sulfonimidoyl]butanoic acid,
(10) (2S)-2-amino-4-{S-[2-(1-fluorocyclobutyl)ethyl]sulfonimidoyl}butanoic acid,
(11) (2S)-2-amino-4-[S-(4,4,4-trifluoro-3-hydroxy-3-methylbutyl)sulfonimidoyl]butanoic acid,
(12) (2S)-2-amino-4-{S-[4,4,4-trifluoro-3-hydroxy-3-(trifluoromethyl)butyl]sulfonimidoyl}butanoic acid,
(13) (2S)-2-amino-4-[S-(3,3,4,4,4-pentafluorobutyl)sulfonimidoyl]butanoic acid,
(14) (S)-2-amino-4-((R,3R)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid,
(15) (S)-2-amino-4-((R,3S)-4,4,4-trifluoro-3-hydroxybutylsulfonimidoyl)butanoic acid,
(16) (S)-2-amino-4-((R)-2-(1-hydroxycyclobutyl)ethylsulfonimidoyl)butanoic acid,
(17) (S)-2-amino-4-((S)-4,4,4-trifluorobutylsulfonimidoyl)butanoic acid,
The compound according to claim 1, which is a compound selected from the group consisting of (18) ethyl (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate, and (19) isopropyl (2S)-2-amino-4-[S-(4,4,4-trifluorobutyl)sulfonimidoyl]butanoate, or a salt thereof.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021183784A JP2024178481A (en) | 2021-11-11 | 2021-11-11 | Glycolipid inhibitors |
| PCT/JP2022/041944 WO2023085367A1 (en) | 2021-11-11 | 2022-11-10 | Gcl inhibitor |
| TW111143041A TW202340142A (en) | 2021-11-11 | 2022-11-10 | Gcl inhibitor |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021183784A JP2024178481A (en) | 2021-11-11 | 2021-11-11 | Glycolipid inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2024178481A true JP2024178481A (en) | 2024-12-25 |
Family
ID=86335866
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021183784A Pending JP2024178481A (en) | 2021-11-11 | 2021-11-11 | Glycolipid inhibitors |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP2024178481A (en) |
| TW (1) | TW202340142A (en) |
| WO (1) | WO2023085367A1 (en) |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02503570A (en) * | 1988-03-21 | 1990-10-25 | コーネル・リサーチ・ファウンデーション、インコーポレイテッド | How to deplete glutathione |
-
2021
- 2021-11-11 JP JP2021183784A patent/JP2024178481A/en active Pending
-
2022
- 2022-11-10 TW TW111143041A patent/TW202340142A/en unknown
- 2022-11-10 WO PCT/JP2022/041944 patent/WO2023085367A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023085367A1 (en) | 2023-05-19 |
| TW202340142A (en) | 2023-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11672785B2 (en) | Tetrahydro-1H-pyrido [3,4-b]indole anti-estrogenic drugs | |
| US20230105745A1 (en) | Cycloalkyl and hetero-cycloalkyl inhibitors, preparation methods therefor, and use thereof | |
| JP7085566B2 (en) | Apoptosis inducer | |
| US4923986A (en) | Derivatives of physiologically active substance K-252 | |
| US7317040B2 (en) | Nitric oxide synthase inhibitor phosphate salt | |
| CA3115609C (en) | Processes of making and crystalline forms of a mdm2 inhibitor | |
| JP2022502385A (en) | Method for producing a compound for inhibiting the activity of SHP2 | |
| JP7173350B2 (en) | Urea compounds that antagonize the LPA1 receptor | |
| EP3686196B1 (en) | Polycyclic compound acting as ido inhibitor and/or ido-hdac dual inhibitor | |
| JP6907351B2 (en) | Carbonitrile derivatives as selective androgen receptor modulators | |
| JP2013514356A (en) | Pyrrolo [2,3-d] pyrimidine compound | |
| CZ20004450A3 (en) | Amidine derivatives | |
| KR20220052918A (en) | Peptide conjugates of cytotoxins as therapeutic agents | |
| AU2017339104B2 (en) | 2-amino-N-(arylsulfinyl)-acetamide compounds as inhibitors of bacterial aminoacyl-tRNA synthetase | |
| JP2002506056A (en) | Halogenated amidino amino acid derivatives useful as nitric oxide synthase inhibitors | |
| TWI843372B (en) | Compounds for mutant kras protein degradation and uses thereof | |
| CN114591201B (en) | Beta-elemene derivative with HDACi pharmacophore, and preparation method and application thereof | |
| KR101745225B1 (en) | Novel selective androgen receptor modulators | |
| AU2021219097A1 (en) | P2X3 modulators | |
| CA3214066A1 (en) | 1,3-substituted cyclobutyl derivatives and uses thereof | |
| US7125906B2 (en) | Indole derivatives having anti-angiogenetic activity | |
| JP6795525B2 (en) | Arylsulfonamide compounds as carbonic anhydrase inhibitors and their therapeutic use | |
| EP3040329B1 (en) | Aromatic compound and use thereof in the treatment of disorders associated with bone metabolism | |
| US10919911B2 (en) | Tricyclic ASK1 inhibitors | |
| CA3152836A1 (en) | Substituted urea dihydroorotate dehydrogenase inhibitors |