JP2016069652A - Cyclodextrin derivative and method for producing the same, and polymer of cyclodextrin derivative - Google Patents
Cyclodextrin derivative and method for producing the same, and polymer of cyclodextrin derivative Download PDFInfo
- Publication number
- JP2016069652A JP2016069652A JP2015191020A JP2015191020A JP2016069652A JP 2016069652 A JP2016069652 A JP 2016069652A JP 2015191020 A JP2015191020 A JP 2015191020A JP 2015191020 A JP2015191020 A JP 2015191020A JP 2016069652 A JP2016069652 A JP 2016069652A
- Authority
- JP
- Japan
- Prior art keywords
- cyclodextrin
- formula
- reaction
- polymer
- cyclodextrin derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
Description
本発明は、シクロデキストリン誘導体及びその製造方法、並びにシクロデキストリン誘導体の重合体に関する。 The present invention relates to a cyclodextrin derivative and a method for producing the same, and a polymer of the cyclodextrin derivative.
化学工場、石油精製工場、製鉄所など、多くの工場で環境汚染物質を含んだ排水が発生する。これらの汚染水を、海、河川ならびに大気中に放出することは規制されており、排出前に無害化する必要がある。 Many factories such as chemical factories, oil refineries, and steel mills generate wastewater containing environmental pollutants. The release of these polluted waters into the sea, rivers, and atmosphere is regulated and must be detoxified before discharge.
例えば、環境汚染物質の一つとしてフェノール類があるが、水道法や水質汚濁防止法などで厳しく規制されている。フェノール類は、有機合成化学工業における基礎物質であり、フェノール樹脂、ピクリン酸、染料、農薬等の原料として重要となっている。また、分析用試薬、防腐剤、医薬品等にも使用されており、汚染源としてはこれらを扱う事業場、研究施設、病院等からの排水がある。そのため、これらの排水からフェノール化合物を分離・除去することは、極めて重要な課題となっている。 For example, phenols are one of the environmental pollutants, but are strictly regulated by the Water Supply Law and Water Pollution Control Law. Phenols are basic substances in the organic synthetic chemical industry, and are important as raw materials for phenol resins, picric acid, dyes, agricultural chemicals and the like. It is also used in analytical reagents, preservatives, pharmaceuticals, etc., and the sources of pollution include wastewater from business sites, research facilities, hospitals, etc. that handle these. Therefore, separating and removing phenolic compounds from these wastewaters has become an extremely important issue.
このような問題点を解決する方法として、シクロデキストリンを利用する方法が提案されている。シクロデキストリンは、α−D−グルコース単位から構成される環状化合物である。シクロデキストリンは、直径数Åという分子サイズの小さな単分散環状オリゴマーであり、疎水性物質を分子内に取り込み包接化合物を形成することがよく知られている。 As a method for solving such problems, a method using cyclodextrin has been proposed. Cyclodextrin is a cyclic compound composed of α-D-glucose units. Cyclodextrin is a monodisperse cyclic oligomer having a small molecular size of several 直径 in diameter, and it is well known that a hydrophobic substance is incorporated into a molecule to form an inclusion compound.
シクロデキストリンを利用したフェノール化合物の除去方法としては、例えば、特許文献1,2に、シクロデキストリンをジイソシアネートで架橋して得られるポリマーによるフェノール化合物の除去方法が開示されている。また、特許文献3には、光架橋基を有するシクロデキストリンによるフェノール化合物の除去が開示されている。 As a method for removing a phenol compound using cyclodextrin, for example, Patent Documents 1 and 2 disclose a method for removing a phenol compound using a polymer obtained by crosslinking cyclodextrin with diisocyanate. Patent Document 3 discloses removal of a phenol compound by a cyclodextrin having a photocrosslinking group.
しかしながら、上述のようなシクロデキストリンによるフェノール化合物の除去方法は除去率が低く、フェノール化合物を十分に分離・除去できたとは言えない。 However, the removal method of the phenol compound by cyclodextrin as described above has a low removal rate, and it cannot be said that the phenol compound can be sufficiently separated and removed.
以上のような事情に鑑み、本発明の目的は、フェノール化合物を良好に除去可能な重合体及びその周辺技術を提供することにある。 In view of the circumstances as described above, an object of the present invention is to provide a polymer capable of removing a phenol compound satisfactorily and its peripheral technology.
本発明者らは、上記課題に鑑み鋭意検討した結果、シクロデキストリンに重合性官能基を付与して得られるモノマーをラジカル重合によりポリマー化することで、効率よくフェノール化合物を包接するとの知見を得て、本発明を完成するに至った。 As a result of intensive studies in view of the above problems, the present inventors have found that a monomer obtained by adding a polymerizable functional group to cyclodextrin is polymerized by radical polymerization, thereby efficiently including a phenol compound. As a result, the present invention has been completed.
上記目的を達成するため、本発明の一形態に係るシクロデキストリン誘導体は、式(1)に示す構造を有する。 In order to achieve the above object, a cyclodextrin derivative according to one embodiment of the present invention has a structure represented by Formula (1).
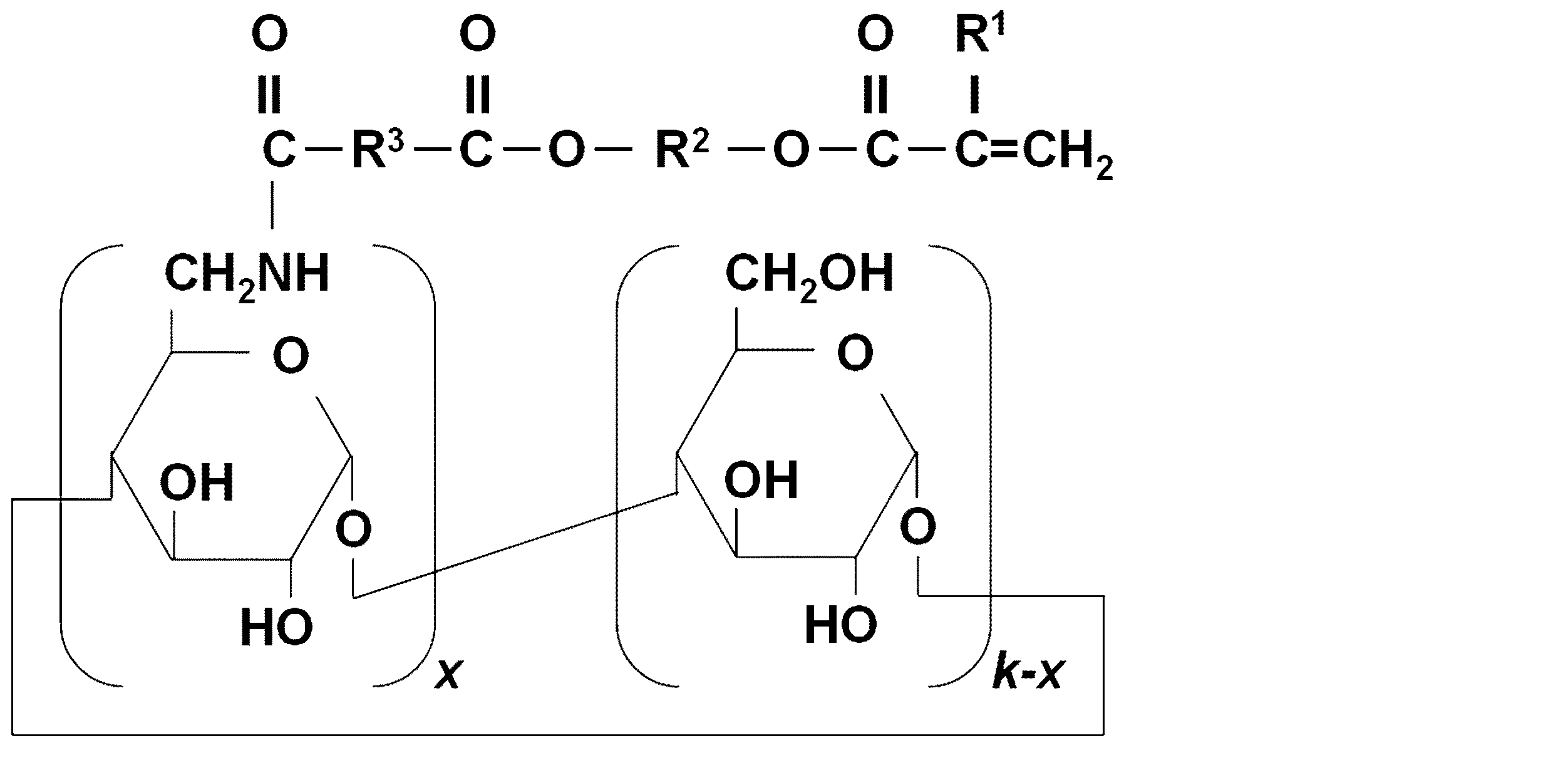
(式(1)中、xは1〜3の整数、kは6〜8の整数、R1は水素原子もしくはメチル基、R2は炭素原子数1〜4の炭化水素基、R3は炭素原子数1〜6の炭化水素基を示す。)
(In formula (1), x is an integer of 1 to 3, k is an integer of 6 to 8, R 1 is a hydrogen atom or a methyl group, R 2 is a hydrocarbon group having 1 to 4 carbon atoms, and R 3 is carbon. (A hydrocarbon group having 1 to 6 atoms is shown.)
また、上記シクロデキストリン誘導体は、式(2)に示す構造を有していてもよい。 The cyclodextrin derivative may have a structure represented by the formula (2).
(式(2)中、yは5〜7の整数、R1は水素原子もしくはメチル基、R2は炭素原子数1〜4の炭化水素基、R3は炭素原子数1〜6の炭化水素基を示す。)
(In the formula (2), y is an integer of 5 to 7, R 1 is a hydrogen atom or a methyl group, R 2 is a hydrocarbon group having 1 to 4 carbon atoms, and R 3 is a hydrocarbon having 1 to 6 carbon atoms. Group.)
本発明の一形態に係るシクロデキストリン誘導体の製造方法は、式(3)で表される化合物と式(4)で表される化合物とをアミド化することにより、上記シクロデキストリン誘導体を生成する工程を含む。 The method for producing a cyclodextrin derivative according to one aspect of the present invention includes the step of producing the cyclodextrin derivative by amidating the compound represented by the formula (3) and the compound represented by the formula (4). including.
(式(3)中、xは1〜3の整数、kは6〜8の整数を示す。)
(In formula (3), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
(式(4)中、R1は水素原子もしくはメチル基、R2は炭素原子数1〜4の炭化水素基、R3は炭素原子数1〜6の炭化水素基を示す。)
(In Formula (4), R 1 represents a hydrogen atom or a methyl group, R 2 represents a hydrocarbon group having 1 to 4 carbon atoms, and R 3 represents a hydrocarbon group having 1 to 6 carbon atoms.)
この構成によれば、上記シクロデキストリン誘導体を効率良く得ることができる。 According to this configuration, the cyclodextrin derivative can be obtained efficiently.
また、本発明の一形態に係るシクロデキストリン誘導体の製造方法は、(A)工程と(B)工程とを含む。
上記(A)工程では、式(5)で表される化合物にベンジルアミンを用いてベンジルアミノ化することにより式(6)で表される化合物を生成する。
上記(B)工程では、上記(A)工程で得られた上記式(6)で表される化合物を還元することにより上記式(3)で表される化合物を生成する。
Moreover, the manufacturing method of the cyclodextrin derivative which concerns on one form of this invention includes (A) process and (B) process.
In the step (A), a compound represented by the formula (6) is produced by benzylamination of the compound represented by the formula (5) using benzylamine.
In the step (B), the compound represented by the formula (3) is generated by reducing the compound represented by the formula (6) obtained in the step (A).
(式(5)中、xは1〜3の整数、kは6〜8の整数を示す。)
(In formula (5), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
(式(6)中、xは1〜3の整数、kは6〜8の整数を示す。)
(In formula (6), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
(式(3)中、xは1〜3の整数、kは6〜8の整数を示す。)
(In formula (3), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
この構成によれば、上記シクロデキストリン誘導体の中間体である式(3)で表される化合物を効率良く得ることができる。 According to this structure, the compound represented by Formula (3) which is an intermediate body of the said cyclodextrin derivative can be obtained efficiently.
本発明の一形態に係るシクロデキストリン誘導体の重合体は、式(1)又は式(2)で表されるシクロデキストリン誘導体を重合させることにより得られる。 The polymer of the cyclodextrin derivative according to one embodiment of the present invention can be obtained by polymerizing the cyclodextrin derivative represented by the formula (1) or the formula (2).
この構成によれば、得られた重合体により、低濃度のフェノール化合物含有水溶液からフェノールを効率良く除去することができる。 According to this configuration, the obtained polymer can efficiently remove phenol from a low concentration phenol compound-containing aqueous solution.
フェノール化合物を良好に除去可能な重合体及びその周辺技術を提供することができる。 The polymer which can remove a phenol compound favorably, and its peripheral technique can be provided.
[シクロデキストリン誘導体]
本実施形態に係るシクロデキストリン誘導体は、式(1)又は式(2)で表される化合物である。
[Cyclodextrin derivative]
The cyclodextrin derivative according to the present embodiment is a compound represented by formula (1) or formula (2).
式(1)中のxの値は、本実施形態に係るシクロデキストリン誘導体中の置換基を有するグルコース単位の数を表しており、すなわち当該置換基の数を表す。 The value of x in the formula (1) represents the number of glucose units having a substituent in the cyclodextrin derivative according to this embodiment, that is, the number of the substituent.
xの値は、1〜3の整数である。また、xの値は、1又は2であることが好ましく、1であることが更に好ましい。一方、xの値が3よりも多くなると、重合して得られたポリマー水溶液の粘度が高くなるため取扱いが困難となるか、あるいは水に不溶のポリマーとなるおそれがある。 The value of x is an integer from 1 to 3. Further, the value of x is preferably 1 or 2, and more preferably 1. On the other hand, when the value of x is more than 3, the viscosity of the polymer aqueous solution obtained by polymerization becomes high, so that the handling becomes difficult, or the polymer may be insoluble in water.
式(1)中のk−xの値は、本実施形態に係るシクロデキストリン誘導体中の置換基を持たないグルコース単位の数を表す。式(1)中のkの値は、6〜8の整数である。 The value of k−x in the formula (1) represents the number of glucose units having no substituent in the cyclodextrin derivative according to this embodiment. The value of k in Formula (1) is an integer of 6-8.
ここで、xの値が1であるときは、本実施形態に係るシクロデキストリン誘導体は式(2)に示す構造となる。式(2)中のyの値は、5〜7の整数である。 Here, when the value of x is 1, the cyclodextrin derivative according to this embodiment has a structure represented by Formula (2). The value of y in Formula (2) is an integer of 5-7.
式(1)、(2)中のR1は、水素原子もしくはメチル基を表す。また、式(1)、(2)中のR2は、炭素原子数1〜4の炭化水素基を表す。炭素原子数1〜4の炭化水素基としては、具体的に、メチレン基、エチレン基、プロピレン基、ブチレン基があげられる。 R 1 in the formulas (1) and (2) represents a hydrogen atom or a methyl group. Moreover, R < 2 > in Formula (1), (2) represents a C1-C4 hydrocarbon group. Specific examples of the hydrocarbon group having 1 to 4 carbon atoms include a methylene group, an ethylene group, a propylene group, and a butylene group.
式(1)、(2)中のR3は、炭素原子数1〜6の炭化水素基を表す。炭素原子数1〜6の炭化水素基としては、例えば、メチレン基、エチレン基、プロピレン基、ブチレン基、ペンチレン基、ヘキシレン基、シクロヘキシレン基、フェニレン基などがあげられる。 R 3 in the formulas (1) and (2) represents a hydrocarbon group having 1 to 6 carbon atoms. Examples of the hydrocarbon group having 1 to 6 carbon atoms include methylene group, ethylene group, propylene group, butylene group, pentylene group, hexylene group, cyclohexylene group, and phenylene group.
式(1)、(2)中のR3は、飽和炭化水素基であってもよく、不飽和炭化水素基であってもよい。さらに、式(1)、(2)中のR3は、炭素原子数が3〜6の場合には、環状炭化水素基であってもよい。 R 3 in the formulas (1) and (2) may be a saturated hydrocarbon group or an unsaturated hydrocarbon group. Further, R 3 in the formulas (1) and (2) may be a cyclic hydrocarbon group when the number of carbon atoms is 3 to 6.
式(1)で表される化合物としては、例えば、6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)シクロデキストリン、6−デオキシ−6−(2-メタクリロイルオキシエチルヘキサヒドロフタル酸アミド)シクロデキストリン、6−デオキシ−6−(2-メタクリロイルオキシエチルフタル酸アミド)シクロデキストリン、6−デオキシ−6−(2−アクリロイルオキシエチルコハク酸アミド)シクロデキストリン、6−デオキシ−6−(2−アクリロイルオキシエチルヘキサヒドロフタル酸アミド)シクロデキストリン、6−デオキシ−6−(2−アクリロイルオキシエチルフタル酸アミド)シクロデキストリンなどがあげられる。 Examples of the compound represented by the formula (1) include 6-deoxy-6- (2-methacryloyloxyethyl succinamide) cyclodextrin, 6-deoxy-6- (2-methacryloyloxyethyl hexahydrophthalic acid amide). ) Cyclodextrin, 6-deoxy-6- (2-methacryloyloxyethylphthalamide) cyclodextrin, 6-deoxy-6- (2-acryloyloxyethyl succinamide) cyclodextrin, 6-deoxy-6- (2 -Acryloyloxyethyl hexahydrophthalamide) cyclodextrin, 6-deoxy-6- (2-acryloyloxyethylphthalamide) cyclodextrin, and the like.
[シクロデキストリン]
本実施形態に係るシクロデキストリン誘導体の原料であるシクロデキストリンは、α−D−グルコース単位から構成される環状化合物であり、例えば、式(7)で表される。
[Cyclodextrin]
Cyclodextrin, which is a raw material of the cyclodextrin derivative according to this embodiment, is a cyclic compound composed of α-D-glucose units, and is represented by, for example, formula (7).
式(7)中のkの値は、α−D−グルコース単位の数を表す。式(7)において、k=6の場合、α−シクロデキストリン(以下、α−CDと記す場合もある。)が構成され、k=7の場合、β−シクロデキストリン(以下、β−CDと記す場合もある。)が構成され、k=8の場合、γ−シクロデキストリン(以下、γ−CDと記す場合もある。)が構成される。 The value of k in formula (7) represents the number of α-D-glucose units. In the formula (7), when k = 6, α-cyclodextrin (hereinafter sometimes referred to as α-CD) is constituted, and when k = 7, β-cyclodextrin (hereinafter referred to as β-CD). In the case where k = 8, γ-cyclodextrin (hereinafter sometimes referred to as γ-CD) is configured.
上記の各シクロデキストリン(α−CD、β−CD、γ−CD)は、各々単独で使用してもよく、混合して使用してもよい。また、入手性の観点からは、k=6であるα−CD又はk=7であるβ−CDの使用が好ましく、k=7であるβ−CDの使用が更に好ましい。 Each of the above cyclodextrins (α-CD, β-CD, γ-CD) may be used alone or as a mixture. From the viewpoint of availability, use of α-CD with k = 6 or β-CD with k = 7 is preferable, and use of β-CD with k = 7 is more preferable.
さらに、本実施形態に係るシクロデキストリンは、式(7)で表される構造に限られない。本実施形態に係るシクロデキストリンは、シクロデキストリン中の水酸基が他の官能基の付加により修飾されたものでもよい。そのようなシクロデキストリンとしては、例えば、アルキル基、ヒドロキシアルキル基、スルホ基、アリール基、アミノ基などによって置換されているシクロデキストリンがあげられる。 Furthermore, the cyclodextrin according to the present embodiment is not limited to the structure represented by the formula (7). The cyclodextrin according to the present embodiment may be one in which a hydroxyl group in the cyclodextrin is modified by adding another functional group. Examples of such cyclodextrins include cyclodextrins substituted with alkyl groups, hydroxyalkyl groups, sulfo groups, aryl groups, amino groups, and the like.
[シクロデキストリン誘導体の製造方法の概要]
本実施形態に係るシクロデキストリン誘導体は、式(3)で表される化合物(以下、アミノ化シクロデキストリンと呼称する。)と式(4)で表される化合物とを反応させることにより得られる。
[Outline of production method of cyclodextrin derivative]
The cyclodextrin derivative according to this embodiment can be obtained by reacting a compound represented by formula (3) (hereinafter referred to as aminated cyclodextrin) with a compound represented by formula (4).
式(3)中のx、kの値については、式(1)中のx、kと同等の値をとることができる。 About the value of x and k in Formula (3), the value equivalent to x and k in Formula (1) can be taken.
式(4)中のR1、R2、R3については、式(1)、(2)中のR1、R2、R3と同様のものをあげることができ、とりうる態様も、同様のものをあげることができる。 For R 1, R 2, R 3 in the formula (4), equation (1), (2) R 1, R 2, R 3 and may be mentioned the same ones as in, aspects that can be taken also The same can be mentioned.
式(4)で表される化合物としては、具体的に、2−メタクリロイルオキシエチルコハク酸、2−メタクリロイルオキシエチルヘキサヒドロフタル酸、2−メタクリロイルオキシエチルフタル酸、2−アクリロイルオキシエチルコハク酸、2−アクリロイルオキシエチルヘキサヒドロフタル酸、2−アクリロイルオキシエチルフタル酸があげられる。R1、R2、R3の炭素原子数が上記の各化合物より大きいものについては、入手が困難となる。 Specific examples of the compound represented by the formula (4) include 2-methacryloyloxyethyl succinic acid, 2-methacryloyloxyethyl hexahydrophthalic acid, 2-methacryloyloxyethyl phthalic acid, 2-acryloyloxyethyl succinic acid, Examples include 2-acryloyloxyethyl hexahydrophthalic acid and 2-acryloyloxyethyl phthalic acid. When the number of carbon atoms of R 1 , R 2 and R 3 is larger than the above compounds, it is difficult to obtain them.
本実施形態に係るシクロデキストリン誘導体は、具体的には、式(8)に示す反応経路2により、式(3)で表されるアミノ化シクロデキストリンと式(4)で表される化合物とをアミド化させることにより得られる。 Specifically, the cyclodextrin derivative according to the present embodiment comprises an aminated cyclodextrin represented by the formula (3) and a compound represented by the formula (4) by the reaction route 2 represented by the formula (8). Obtained by amidation.
式(8)中のCD−NH2は、式(3)で表されるアミノ化シクロデキストリンを表す。また、式(8)中のR1、R2、R3については、式(1)、(2)中のR1、R2、R3と同様のものをあげることができ、とりうる態様も、同様のものをあげることができる。 CD-NH 2 in formula (8) represents an aminated cyclodextrin represented by formula (3). Further, embodiments for R 1, R 2, R 3 in the formula (8), equation (1), may be mentioned the same as the R 1, R 2, R 3 in (2) may take The same can be mentioned.
式(8)中の(d)は、式(3)で表されるアミノ化シクロデキストリンと式(4)で表される化合物とをアミド化させて、本実施形態に係るシクロデキストリン誘導体を得る工程((C)工程)を表す。 (D) in the formula (8) is obtained by amidating the aminated cyclodextrin represented by the formula (3) and the compound represented by the formula (4) to obtain the cyclodextrin derivative according to this embodiment. Step (step (C)) is represented.
さらに、本実施形態に係るシクロデキストリン誘導体は、式(9)に示す反応経路1で得られる式(3)で表されるアミノ化シクロデキストリンを用いて製造することができる。 Furthermore, the cyclodextrin derivative according to this embodiment can be produced using an aminated cyclodextrin represented by the formula (3) obtained by the reaction route 1 shown in the formula (9).
式(9)中のCDは式(7)で表されるシクロデキストリンを表し、CD−Tsは式(5)で表される化合物(以下、トシル化シクロデキストリンと呼称する。)を表し、CD−NBnは式(6)で表される化合物(以下、ベンジルアミノ化シクロデキストリンと呼称する。)を表し、CD−NH2は式(3)で表されるアミノ化シクロデキストリンを表す。また、式(9)中のR4は、塩素原子もしくはイミダゾール基を表す。 CD in formula (9) represents a cyclodextrin represented by formula (7), CD-Ts represents a compound represented by formula (5) (hereinafter referred to as tosylated cyclodextrin), and CD. -NBn compounds represented by formula (6) (hereinafter, referred to as benzylamino cyclodextrin.) represent, CD-NH 2 represents an amino cyclodextrin represented by the formula (3). Further, R 4 in the formula (9) represents a chlorine atom or an imidazole group.
式(9)中の(a)は、式(7)で表されるシクロデキストリンにスルホン酸誘導体を用いてトシル化し、式(5)で表されるトシル化シクロデキストリンを得る工程((a−1)工程)を表す。 (A) in the formula (9) is a step of obtaining a tosylated cyclodextrin represented by the formula (5) by tosylating the cyclodextrin represented by the formula (7) with a sulfonic acid derivative ((a- 1) represents step).
式(9)中の(b)は、式(5)で表されるトシル化シクロデキストリンにベンジルアミンを用いてベンジルアミノ化し、式(6)で表されるベンジルアミノ化シクロデキストリンを得る工程((A)工程)を表す。 (B) in the formula (9) is a step of benzylamination of the tosylated cyclodextrin represented by the formula (5) using benzylamine to obtain a benzylaminated cyclodextrin represented by the formula (6) ( (A) Step).
式(9)中の(c)は、式(6)で表されるベンジルアミノ化シクロデキストリンを還元して、中間体である式(3)で表されるアミノ化シクロデキストリンを得る工程((B)工程)を表す。 (C) in the formula (9) is a step of reducing the benzyl aminated cyclodextrin represented by the formula (6) to obtain an aminated cyclodextrin represented by the formula (3) as an intermediate (( B) represents step).
式(5)中のx、kの値については、(a−1)工程により様々に変えることが可能である。 About the value of x and k in Formula (5), it is possible to change variously by the (a-1) process.
式(6)中のx、kの値については、式(5)のx、kと同等の値をとることができる。 As for the values of x and k in the equation (6), values equivalent to x and k in the equation (5) can be taken.
[シクロデキストリン誘導体の製造方法の詳細]
本実施形態に係るシクロデキストリン誘導体の製造方法は、上述のように、反応経路1〔(a−1)工程、(A)工程、(B)工程〕と、反応経路2〔(C)工程〕とを有する。以下、上記各反応経路及び各工程について詳細に説明する。
[Details of production method of cyclodextrin derivative]
As described above, the method for producing a cyclodextrin derivative according to this embodiment includes reaction path 1 [(a-1) step, (A) step, (B) step] and reaction route 2 [(C) step]. And have. Hereinafter, each reaction route and each step will be described in detail.
<反応経路1>
〔(a−1)工程(反応経路1の反応(a))〕
(a−1)工程は、式(7)で表されるシクロデキストリンを出発原料として、当該シクロデキストリンにスルホン酸誘導体を反応させ、式(5)で表されるトシル化シクロデキストリンを得るトシル化反応である。
<Reaction route 1>
[Step (a-1) (Reaction (a) in Reaction Path 1)]
In the step (a-1), tosylation is obtained by reacting the cyclodextrin represented by the formula (7) with a sulfonic acid derivative to obtain a tosylated cyclodextrin represented by the formula (5). It is a reaction.
上記トシル化反応で得られる式(5)で表されるトシル化シクロデキストリンについては、非特許文献1(Org. Synth., 77, 220 (2000))や非特許文献2(Org. Synth., 77, 225 (2000))で検討がなされており、式(7)で表されるシクロデキストリンにp−トルエンスルホニルクロライドやp−トルエンスルホニルイミダゾール等のスルホン酸誘導体を反応させることで得ることができる。 Regarding the tosylated cyclodextrin represented by the formula (5) obtained by the above tosylation reaction, Non-Patent Document 1 (Org. Synth., 77, 220 (2000)) and Non-Patent Document 2 (Org. Synth., 77, 225 (2000)), and can be obtained by reacting a cyclodextrin represented by the formula (7) with a sulfonic acid derivative such as p-toluenesulfonyl chloride or p-toluenesulfonylimidazole. .
本実施形態に係るシクロデキストリン誘導体は、式(7)で表されるシクロデキストリンとスルホン酸誘導体の仕込み比を変えることによって、式(1)中のxの値、すなわち置換基の導入率を様々に変えることが可能である。 The cyclodextrin derivative according to this embodiment varies the value of x in formula (1), that is, the introduction rate of substituents, by changing the charge ratio of cyclodextrin and sulfonic acid derivative represented by formula (7). It is possible to change to
上記トシル化反応において、スルホン酸誘導体の使用量は、式(7)で表されるシクロデキストリンに対しモル比で1倍量〜30倍量とすることができ、3倍量〜20倍量が好ましい。スルホン酸誘導体の使用量を1倍量以上とすることにより、反応を十分に進行させることができる。また、スルホン酸誘導体の使用量を30倍量以下とすることにより、目的とする置換基導入率を効率良く得ることができる。 In the tosylation reaction, the sulfonic acid derivative can be used in a molar ratio of 1 to 30 times the cyclodextrin represented by formula (7), and 3 to 20 times the amount. preferable. By making the use amount of the sulfonic acid derivative 1 time or more, the reaction can sufficiently proceed. Moreover, the target substituent introduction | transduction rate can be obtained efficiently by the usage-amount of a sulfonic acid derivative being 30 times or less.
上記トシル化反応は溶媒の存在下で行うことができる。当該溶媒としては、式(7)で表されるシクロデキストリンを溶解し、スルホン酸誘導体と反応しない、又は反応性の低いものを用いることができる。例えば、水酸化ナトリウム水溶液、ピリジン又はピコリンなどがあげられる。これらの溶媒は、各々単独で用いてもよく、混合して用いてもよい。 The tosylation reaction can be performed in the presence of a solvent. As the solvent, a solvent that dissolves the cyclodextrin represented by the formula (7) and does not react with the sulfonic acid derivative or has low reactivity can be used. For example, sodium hydroxide aqueous solution, pyridine, picoline, etc. are mentioned. These solvents may be used alone or in combination.
上記トシル化反応の反応温度は、通常−20℃〜60℃であり、−5℃〜25℃が好ましい。反応温度を−20℃以上とすることにより、反応を十分に進行させることができる。また、反応温度を60℃以下とすることにより、副反応(分解反応)を防ぐことができ、これにより式(5)で表されるトシル化シクロデキストリンを効率良く得ることができる。 The reaction temperature of the tosylation reaction is usually −20 ° C. to 60 ° C., preferably −5 ° C. to 25 ° C. By setting the reaction temperature to −20 ° C. or higher, the reaction can sufficiently proceed. Moreover, by making reaction temperature 60 degrees C or less, a side reaction (decomposition reaction) can be prevented and the tosylated cyclodextrin represented by Formula (5) can be obtained efficiently.
上記トシル化反応において、式(7)で表されるシクロデキストリンの濃度は、通常1%〜20%であり、2%〜10%が好ましい。当該濃度を1%以上とすることにより、使用する溶媒の量を減少させることができ、これにより生産効率を向上させることができる。また、当該濃度を20%以下とすることにより、トシル基の導入率を抑えることができ、これにより式(5)中のxの値を3以下に制御することができる。 In the tosylation reaction, the concentration of cyclodextrin represented by formula (7) is usually 1% to 20%, preferably 2% to 10%. By setting the concentration to 1% or more, the amount of the solvent to be used can be reduced, thereby improving the production efficiency. Moreover, the introduction | transduction rate of a tosyl group can be suppressed by making the said density | concentration 20% or less, and, thereby, the value of x in Formula (5) can be controlled to 3 or less.
上記トシル化反応の反応時間は、通常30分〜12時間であり、1時間〜6時間が好ましい。 The reaction time of the tosylation reaction is usually 30 minutes to 12 hours, preferably 1 hour to 6 hours.
上記トシル化反応により得られた生成物は、塩酸等の酸の添加や、アセトン等の溶媒に再沈殿させることで析出させることができる。当該生成物の精製には、シリカゲルクロマトグラフィーや再結晶などの一般的な精製方法を用いることができる。 The product obtained by the tosylation reaction can be precipitated by adding an acid such as hydrochloric acid or by reprecipitation in a solvent such as acetone. For purification of the product, general purification methods such as silica gel chromatography and recrystallization can be used.
〔(A)工程(反応経路1の反応(b))〕
(A)工程は、(a−1)工程で得た式(5)で表されるトシル化シクロデキストリンとベンジルアミンとを反応させ、式(6)で表されるベンジルアミノ化シクロデキストリンを得るベンジルアミノ化反応である。
[Step (A) (Reaction (b) in Reaction Path 1)]
In the step (A), the tosylated cyclodextrin represented by the formula (5) obtained in the step (a-1) and benzylamine are reacted to obtain the benzyl aminated cyclodextrin represented by the formula (6). Benzyl amination reaction.
上記ベンジルアミノ化反応は、溶媒の存在下で行うことができる。当該溶媒としては、式(5)で表されるトシル化シクロデキストリンを溶解させることができるものを用いることができる。そのような溶媒としては、例えば、N,N−ジメチルホルムアミド、N,N−ジメチルスルホキシド、N,N−ジメチルアセトアミドなどがあげられる。これらの溶媒は、各々単独で用いてもよく、混合して用いてもよい。 The benzyl amination reaction can be performed in the presence of a solvent. As the solvent, a solvent capable of dissolving the tosylated cyclodextrin represented by the formula (5) can be used. Examples of such a solvent include N, N-dimethylformamide, N, N-dimethylsulfoxide, N, N-dimethylacetamide and the like. These solvents may be used alone or in combination.
上記ベンジルアミノ化反応のベンジルアミンの使用量は、式(5)で表されるトシル化シクロデキストリンに対し、モル比で1倍量〜30倍量とすることができ、2倍量〜20倍量とすることが好ましい。ベンジルアミンの使用量を1倍量以上とすることにより、反応を十分に進行させることができる。また、ベンジルアミンの使用量が30倍量以下であると経済的である。 The amount of benzylamine used in the benzylamination reaction can be 1 to 30 times the molar amount of the tosylated cyclodextrin represented by the formula (5), and 2 to 20 times. It is preferable to use an amount. By making the amount of benzylamine used more than 1 time, the reaction can be sufficiently advanced. Moreover, it is economical that the usage-amount of benzylamine is 30 times or less.
上記ベンジルアミノ化反応の反応温度は、通常25℃〜120℃であり、40℃〜100℃が好ましい。反応温度を25℃以上とすることにより、反応を十分に進行させることができる。また、反応温度を120℃以下とすることにより、生成物の分解を防ぐことができる。 The reaction temperature of the benzyl amination reaction is usually 25 ° C to 120 ° C, preferably 40 ° C to 100 ° C. By setting the reaction temperature to 25 ° C. or higher, the reaction can sufficiently proceed. Moreover, decomposition | disassembly of a product can be prevented by making reaction temperature into 120 degrees C or less.
上記ベンジルアミノ化反応において、式(5)で表されるトシル化シクロデキストリンの反応時の濃度は、通常5%〜60%であり、10%〜40%が好ましい。当該濃度を5%以上とすることにより、反応を十分に進行させることができ、あるいは使用する溶媒の量を減少させることができる。これにより、生産効率を向上させることができる。また、当該濃度を60%以下とすることにより、反応液の粘度上昇を抑えることができ、これにより反応液の取り扱いを容易にすることができる。 In the benzylamination reaction, the concentration of the tosylated cyclodextrin represented by the formula (5) during the reaction is usually 5% to 60%, preferably 10% to 40%. By setting the concentration to 5% or more, the reaction can proceed sufficiently, or the amount of solvent used can be reduced. Thereby, production efficiency can be improved. In addition, by setting the concentration to 60% or less, an increase in the viscosity of the reaction solution can be suppressed, whereby the handling of the reaction solution can be facilitated.
上記ベンジルアミノ化反応の反応時間は、通常30分〜24時間であり、1時間〜12時間が好ましい。 The reaction time for the benzylamination reaction is usually 30 minutes to 24 hours, preferably 1 to 12 hours.
上記ベンジルアミノ化反応により得られた生成物の精製には、再沈殿や再結晶などの一般的な精製方法を用いることができる。 For purification of the product obtained by the benzylamination reaction, a general purification method such as reprecipitation or recrystallization can be used.
〔(B)工程(反応経路1の反応(c))〕
(B)工程は、(A)工程で得た式(6)で表されるベンジルアミノ化シクロデキストリンを還元し、式(3)で表されるアミノ化シクロデキストリンを得る還元反応である。
[Step (B) (Reaction (c) in Reaction Path 1)]
Step (B) is a reduction reaction in which the benzyl aminated cyclodextrin represented by formula (6) obtained in step (A) is reduced to obtain an aminated cyclodextrin represented by formula (3).
上記還元反応は、金属触媒の存在下で行うことができる。当該金属触媒としては、例えば、パラジウム触媒を用いることができる。パラジウム触媒を用いる場合は、パラジウムを活性炭に担持させたパラジウム活性炭素を用いることが好ましい。 The reduction reaction can be performed in the presence of a metal catalyst. As the metal catalyst, for example, a palladium catalyst can be used. When using a palladium catalyst, it is preferable to use palladium activated carbon in which palladium is supported on activated carbon.
上記パラジウム活性炭素の使用量は、式(6)で表されるベンジルアミノ化シクロデキストリンに対し、通常0.1質量%〜50質量%とすることができ、1質量%〜30質量%が好ましい。パラジウム活性炭素の使用量を0.1質量%以上とすることにより、反応を十分に進行させることができる。また、パラジウム活性炭素の使用量が50質量%以下であると経済的である。 The amount of the palladium activated carbon used can be usually 0.1% by mass to 50% by mass with respect to the benzylaminated cyclodextrin represented by the formula (6), and preferably 1% by mass to 30% by mass. . By setting the amount of palladium activated carbon used to be 0.1% by mass or more, the reaction can sufficiently proceed. Moreover, it is economical that the usage-amount of palladium activated carbon is 50 mass% or less.
上記還元反応は、溶媒の存在下で行うことができる。当該溶媒としては、式(6)で表されるベンジルアミノ化シクロデキストリン及び式(3)で表されるアミノ化シクロデキストリンを溶解させることができるものを用いることができる。そのような溶媒としては、例えば、N,N−ジメチルホルムアミド、N,N−ジメチルスルホキシド、N,N−ジメチルアセトアミド、水などがあげられる。これらの溶媒は、各々単独で用いてもよく、混合して用いてもよい。 The reduction reaction can be performed in the presence of a solvent. As the said solvent, what can dissolve the benzyl aminated cyclodextrin represented by Formula (6) and the aminated cyclodextrin represented by Formula (3) can be used. Examples of such a solvent include N, N-dimethylformamide, N, N-dimethylsulfoxide, N, N-dimethylacetamide, water and the like. These solvents may be used alone or in combination.
上記還元反応の反応温度は、通常25℃〜120℃であり、40℃〜100℃が好ましい。反応温度を25℃以上とすることにより、反応を十分に進行させることができる。また、反応温度を120℃以下とすることにより、生成物の分解を防ぐことができる。 The reaction temperature of the reduction reaction is usually 25 ° C to 120 ° C, preferably 40 ° C to 100 ° C. By setting the reaction temperature to 25 ° C. or higher, the reaction can sufficiently proceed. Moreover, decomposition | disassembly of a product can be prevented by making reaction temperature into 120 degrees C or less.
上記還元反応において、式(6)で表されるベンジルアミノ化シクロデキストリンの反応時の濃度は、通常1%〜30%であり、2%〜20%が好ましい。当該濃度を1%以上とすることにより、反応を十分に進行させることができ、あるいは使用する溶媒の量を減少させることができる。これにより、生産効率を向上させることができる。また、当該濃度を30%以下とすることにより、反応液の粘度上昇を抑えることができ、これにより反応液の取り扱いを容易にすることができる。 In the above reduction reaction, the concentration of the benzylaminated cyclodextrin represented by formula (6) during the reaction is usually 1% to 30%, preferably 2% to 20%. By setting the concentration to 1% or more, the reaction can sufficiently proceed, or the amount of solvent used can be reduced. Thereby, production efficiency can be improved. In addition, by setting the concentration to 30% or less, an increase in the viscosity of the reaction solution can be suppressed, whereby the handling of the reaction solution can be facilitated.
上記還元反応の反応時間は、通常1時間〜48時間であり、2時間〜24時間が好ましい。 The reaction time of the reduction reaction is usually 1 hour to 48 hours, and preferably 2 hours to 24 hours.
上記還元反応により得られた生成物の精製には、再沈殿や再結晶などの一般的な精製方法を用いることができる。 For purification of the product obtained by the reduction reaction, a general purification method such as reprecipitation or recrystallization can be used.
<反応経路2>
〔(C)工程(反応経路2の反応(d))〕
(C)工程は、(B)工程で得た中間体である式(3)で表されるアミノ化シクロデキストリンに式(4)で表される化合物を縮合させて、式(1)又は式(2)で表されるシクロデキストリン誘導体を得るアミド化反応である。
<Reaction route 2>
[Step (C) (Reaction (d) in Reaction Path 2)]
In the step (C), the compound represented by the formula (4) is condensed with the aminated cyclodextrin represented by the formula (3) which is the intermediate obtained in the step (B), and then the formula (1) or the formula This is an amidation reaction to obtain a cyclodextrin derivative represented by (2).
上記アミド化反応は、溶媒の存在下で行うことができる。当該溶媒としては、式(3)で表されるアミノ化シクロデキストリンと式(4)で表される化合物を溶解し、反応しないものを用いることができる。そのような溶媒としては、例えば、N,N−ジメチルホルムアミド、N,N−ジメチルスルホキシド、N,N−ジメチルアセトアミドなどがあげられる。 The amidation reaction can be performed in the presence of a solvent. As the solvent, a solvent in which the aminated cyclodextrin represented by the formula (3) and the compound represented by the formula (4) are dissolved and does not react can be used. Examples of such a solvent include N, N-dimethylformamide, N, N-dimethylsulfoxide, N, N-dimethylacetamide and the like.
上記アミド化反応は、副反応を避けるために縮合剤の存在下で反応を行うことが好ましい。当該縮合剤としては、例えば、N,N'−ジシクロヘキシルカルボジイミド等のカルボジイミド系縮合剤、ベンゾトリアゾール−1−イルオキシ−トリスジメチルアミノホスホニウム塩等のホスホニウム系縮合剤、1−ヒドロキシベンゾトリアゾール等のトリアゾール系縮合剤、4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド等のトリアジン系縮合剤などがあげられ、特にアミド結合を選択的に形成させる4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリドが好ましい。 The amidation reaction is preferably performed in the presence of a condensing agent in order to avoid side reactions. Examples of the condensing agent include carbodiimide condensing agents such as N, N′-dicyclohexylcarbodiimide, phosphonium condensing agents such as benzotriazol-1-yloxy-trisdimethylaminophosphonium salt, and triazole such as 1-hydroxybenzotriazole. Condensing agents, triazine condensing agents such as 4- (4,6-dimethoxy-1,3,5-triazin-2-yl) -4-methylmorpholinium chloride, and the like. 4- (4,6-dimethoxy-1,3,5-triazin-2-yl) -4-methylmorpholinium chloride to be formed is preferred.
上記縮合剤の使用量は、式(3)で示されるアミノ化シクロデキストリンに対し、モル比で1倍量〜5倍量用いることができる。上記縮合剤の使用量を当該モル比で1倍量以上とすることにより、反応を十分に進行させることができる。また、上記縮合剤の使用量が当該モル比で5倍量以下であると経済的である。 The amount of the condensing agent used can be 1 to 5 times the molar ratio of the aminated cyclodextrin represented by formula (3). By making the use amount of the condensing agent 1 time or more by the molar ratio, the reaction can be sufficiently advanced. Moreover, it is economical when the amount of the condensing agent used is 5 times or less in terms of the molar ratio.
上記アミド化反応の反応温度は、通常−20℃〜100℃であり、0℃〜80℃が好ましい。反応温度を−20℃以上とすることにより、反応を十分に進行させることができる。また、反応温度を100℃以下とすることにより、重合等の副反応を防ぐことができる。 The reaction temperature of the amidation reaction is usually −20 ° C. to 100 ° C., preferably 0 ° C. to 80 ° C. By setting the reaction temperature to −20 ° C. or higher, the reaction can sufficiently proceed. Moreover, side reactions, such as superposition | polymerization, can be prevented by making reaction temperature into 100 degrees C or less.
上記アミド化反応において、式(3)で示されるアミノ化シクロデキストリンの反応時の濃度は、通常1%〜30%であり、2%〜20%が好ましい。当該濃度を1%以上とすることにより、反応を十分に進行させることができ、あるいは使用する溶媒の量を減少させることができる。これにより、生産効率を向上させることができる。また、当該濃度を30%以下とすることにより、反応液の粘度上昇を抑えることができ、これにより反応液の取り扱いを容易にすることができる。 In the amidation reaction, the concentration of the aminated cyclodextrin represented by the formula (3) during the reaction is usually 1% to 30%, preferably 2% to 20%. By setting the concentration to 1% or more, the reaction can sufficiently proceed, or the amount of solvent used can be reduced. Thereby, production efficiency can be improved. In addition, by setting the concentration to 30% or less, an increase in the viscosity of the reaction solution can be suppressed, whereby the handling of the reaction solution can be facilitated.
上記アミド化反応の反応時間は、通常30分〜24時間であり、1時間〜12時間が好ましい。 The reaction time of the amidation reaction is usually 30 minutes to 24 hours, preferably 1 hour to 12 hours.
上記アミド化反応により得られた生成物の精製には、再沈殿や再結晶などの一般的な精製方法を用いることができる。 For purification of the product obtained by the amidation reaction, a general purification method such as reprecipitation or recrystallization can be used.
以上の製造方法により、式(1)又は式(2)で表されるシクロデキストリン誘導体を得ることができる。また、式(2)で表されるシクロデキストリン誘導体を得るためには、(C)工程のアミド化反応を行う前に精製を行い、xの値が2以上の式(3)で表されるアミノ化シクロデキストリンを除去しておくことが好ましい。特に、(a−1)工程で得られる式(5)で表されるトシル化シクロデキストリンを精製し、xの値が2以上のものを除去しておくことが更に好ましい。 The cyclodextrin derivative represented by the formula (1) or the formula (2) can be obtained by the above production method. Moreover, in order to obtain the cyclodextrin derivative represented by the formula (2), purification is performed before the amidation reaction in the step (C), and the value of x is represented by the formula (3) of 2 or more. It is preferable to remove the aminated cyclodextrin. In particular, it is more preferable to purify the tosylated cyclodextrin represented by the formula (5) obtained in the step (a-1) and remove those having a value of x of 2 or more.
式(3)で表されるアミノ化シクロデキストリンは市販品を用いても良いが、式(1)又は式(2)で表されるシクロデキストリン誘導体の純度に影響するので高純度品を用いることが好ましい。 A commercially available product may be used as the aminated cyclodextrin represented by the formula (3), but since it affects the purity of the cyclodextrin derivative represented by the formula (1) or the formula (2), a high-purity product should be used. Is preferred.
本発明のシクロデキストリン誘導体を得る製造方法は、上述の製造方法に制限されない。反応経路1で得られる式(3)で表される中間体のアミノ化シクロデキストリンは、一般的には、例えば Langmuir 1999、 15、 5489−5495等に記載の製造方法によっても合成される。当該製造方法において、アミノ化シクロデキストリンは、式(10)に示す反応経路3により得られる。 The production method for obtaining the cyclodextrin derivative of the present invention is not limited to the production method described above. The intermediate aminated cyclodextrin represented by the formula (3) obtained in the reaction route 1 is generally also synthesized by a production method described in, for example, Langmuir 1999, 15, 5589-5495. In the production method, the aminated cyclodextrin is obtained by the reaction route 3 shown in the formula (10).
式(10)中のCD、CD−Ts及びCD−NH2は、式(9)中のCD、CD−Ts及びCD−NH2と同様の化合物を表し、CD−N3はアジ化シクロデキストリンを表す。 CD in Equation (10), CD-Ts and CD-NH 2 is, CD in Equation (9) represents the same compound CD-Ts and CD-NH 2, CD-N 3 is azide cyclodextrin Represents.
式(10)に示すように、反応経路3では、反応(e)と反応(f)の経路と、反応(g)のみの経路がある。反応(e)のアジ化反応を行う製造方法は、原料のアジ化ナトリウムが爆発性を有することから製造には不向きである。一方、アンモニアを反応させる反応(g)は、副反応として加水分解が生じやすく、収率が低くなる。そのため、アミノ化シクロデキストリンの製造方法としては、本発明の反応経路1が好ましい。 As shown in the equation (10), the reaction path 3 includes a path for the reaction (e) and the reaction (f) and a path for only the reaction (g). The production method for carrying out the azination reaction (e) is unsuitable for production since the raw material sodium azide has explosive properties. On the other hand, in the reaction (g) in which ammonia is reacted, hydrolysis tends to occur as a side reaction, resulting in a low yield. Therefore, the reaction route 1 of the present invention is preferable as a method for producing an aminated cyclodextrin.
[シクロデキストリン誘導体の重合体]
本実施形態に係るシクロデキストリン誘導体の重合体は、上述の式(1)又は式(2)で表されるシクロデキストリン誘導体を重合することにより得られる重合体である。
[Polymer of cyclodextrin derivative]
The polymer of the cyclodextrin derivative according to this embodiment is a polymer obtained by polymerizing the cyclodextrin derivative represented by the above formula (1) or formula (2).
上記重合体の分子量は、通常、数平均分子量5000〜500000程度であるが特に限定されず、各用途に要求される性能が発揮し得るように重合条件等を調整して適宜決定することができる。 The molecular weight of the polymer is usually a number average molecular weight of about 5,000 to 500,000, but is not particularly limited, and can be appropriately determined by adjusting the polymerization conditions and the like so that the performance required for each application can be exhibited. .
上記重合体を生成するための重合反応は、窒素、二酸化炭素、アルゴン、ヘリウム等の不活性雰囲気下、ラジカル重合開始剤の存在下で、例えば、塊状重合、懸濁重合、乳化重合、溶液重合等の公知の方法により行うことができる。精製等の観点からは、溶液重合が好ましい。この重合反応により、上記重合体が得られる。 The polymerization reaction for producing the polymer is performed in the presence of a radical polymerization initiator in an inert atmosphere such as nitrogen, carbon dioxide, argon, helium, etc., for example, bulk polymerization, suspension polymerization, emulsion polymerization, solution polymerization. It can carry out by well-known methods, such as. From the viewpoint of purification and the like, solution polymerization is preferable. The polymer is obtained by this polymerization reaction.
上記重合反応は、式(1)又は式(2)で表されるシクロデキストリン誘導体単独で行っても良く、その他の一般的なモノマーとの共重合体でも良い。その他の一般的なモノマーとしては、例えば、2−ヒドロキシエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、グリセロール(メタ)アクリレート、グリシジル(メタ)アクリレート、(メタ)アクリロイルオキシエチルホスホリルコリン、メチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリレート等の各種(メタ)アクリル酸エステル;メチルビニルエーテル等の各種ビニルエーテル;アクリルアミド、N,N'−ジメチルアクリルアミド、(メタ)アクリル酸、アリルアルコール、アクリロニトリル、酢酸ビニル、N−ビニルピロリドン、スチレン、塩化ビニル、イタコン酸、イタコン酸エステル、フマル酸、フマル酸エステル、マレイン酸、マレイン酸エステル等の各種ラジカル重合性モノマーがあげられる。以下、上記重合体を構成するモノマーあるいはそれらの混合物を、単量体組成物と呼称する。 The polymerization reaction may be performed with the cyclodextrin derivative represented by formula (1) or formula (2) alone, or may be a copolymer with other general monomers. Other common monomers include, for example, 2-hydroxyethyl (meth) acrylate, diethylaminoethyl (meth) acrylate, polyethylene glycol mono (meth) acrylate, glycerol (meth) acrylate, glycidyl (meth) acrylate, (meth) Various (meth) acrylic esters such as acryloyloxyethyl phosphorylcholine, methyl (meth) acrylate, dimethylaminoethyl (meth) acrylate; various vinyl ethers such as methyl vinyl ether; acrylamide, N, N′-dimethylacrylamide, (meth) acrylic acid , Allyl alcohol, acrylonitrile, vinyl acetate, N-vinyl pyrrolidone, styrene, vinyl chloride, itaconic acid, itaconic acid ester, fumaric acid, fumaric acid ester, malein Various radically polymerizable monomers such as acid and maleic acid ester are listed. Hereinafter, the monomer constituting the polymer or a mixture thereof is referred to as a monomer composition.
上記重合反応に用いることができるラジカル重合開始剤としては、例えば、2,2−アゾビス(2−アミジノプロピル)二塩酸塩、2,2−アゾビス(2−(5−メチル−2−イミダゾリン−2−イル)プロパン)二塩酸塩、4,4−アゾビス(4−シアノ吉草酸)、2,2−アゾビスイソブチルアミド二水和物、2,2−アゾビス(2,4−ジメチルバレロニトリル)、2,2−アゾビスイソブチロニトリル(AIBN)、ジメチル−2,2'−アゾビスイソブチレート、1−((1−シアノ−1−メチルエチル)アゾ)ホルムアミド、2,2'−アゾビス(2−メチル−N−フェニルプロピオンアミヂン)ジハイドロクロライド、2,2'−アゾビス(2−メチル−N−(2−ヒドロキシエチル)−プロピオンアミド)、2,2'−アゾビス(2−メチルプロピオンアミド)ジハイドレート、4,4'−アゾビス(4−シアノペンタン酸)、2,2'−アゾビス(2−(ヒドロキシメチル)プロピオニトリル)等のアゾ系ラジカル重合開始剤があげられる。また、例えば、過酸化ベンゾイル、ジイソプロピルペルオキシジカーボネート、t−ブチルペルオキシ−2−エチルヘキサノエート、t−ブチルペルオキシピバレート、t−ブチルペルオキシジイソブチレート、過酸化ラウロイル、t−ブチルペルオキシネオデカノエート、コハク酸ペルオキシド(=サクシニルペルオキシド)、グルタルペルオキシド、サクシニルペルオキシグルタレート、t−ブチルペルオキシマレート、t−ブチルペルオキシピバレート、ジ−2−エトキシエチルペルオキシカーボネート、3−ヒドロキシ−1,1−ジメチルブチルペルオキシピバレート等の有機過酸化物があげられる。さらに、過硫酸アンモニウム、過硫酸カリウム、過硫酸ナトリウム等の過硫酸化物が挙げられる。これらのラジカル重合開始剤は、各々単独で用いてもよく、混合して用いてもよい。 Examples of the radical polymerization initiator that can be used in the polymerization reaction include 2,2-azobis (2-amidinopropyl) dihydrochloride, 2,2-azobis (2- (5-methyl-2-imidazoline-2). -Yl) propane) dihydrochloride, 4,4-azobis (4-cyanovaleric acid), 2,2-azobisisobutyramide dihydrate, 2,2-azobis (2,4-dimethylvaleronitrile), 2,2-azobisisobutyronitrile (AIBN), dimethyl-2,2′-azobisisobutyrate, 1-((1-cyano-1-methylethyl) azo) formamide, 2,2′-azobis (2-methyl-N-phenylpropionamidin) dihydrochloride, 2,2′-azobis (2-methyl-N- (2-hydroxyethyl) -propionamide), 2,2′-azobis ( Azo radical polymerization initiators such as 2-methylpropionamido) dihydrate, 4,4′-azobis (4-cyanopentanoic acid), 2,2′-azobis (2- (hydroxymethyl) propionitrile) . Further, for example, benzoyl peroxide, diisopropyl peroxydicarbonate, t-butylperoxy-2-ethylhexanoate, t-butylperoxypivalate, t-butylperoxydiisobutyrate, lauroyl peroxide, t-butylperoxyneodeca Noate, succinic peroxide (= succinyl peroxide), glutar peroxide, succinyl peroxyglutarate, t-butyl peroxymalate, t-butyl peroxypivalate, di-2-ethoxyethyl peroxycarbonate, 3-hydroxy-1,1 -Organic peroxides such as dimethylbutyl peroxypivalate. Furthermore, persulfate oxides, such as ammonium persulfate, potassium persulfate, and sodium persulfate, are mentioned. These radical polymerization initiators may be used alone or in combination.
ラジカル重合開始剤の使用量は、単量体組成物100質量部に対して通常0.001質量部〜10質量部であり、0.01質量部〜5.0質量部が好ましい。 The usage-amount of a radical polymerization initiator is 0.001 mass part-10 mass parts normally with respect to 100 mass parts of monomer compositions, and 0.01 mass part-5.0 mass parts are preferable.
上記重合反応は、溶媒の存在下で行うことができる。当該溶媒としては、単量体組成物を溶解し、反応しないものが使用できる。そのような溶媒としては、例えば、水、メタノール、エタノール、n−プロパノール、イソプロパノール等のアルコール系溶媒、N,N−ジメチルスルホキシド、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ピリジン、などがあげられる。特に、N,N−ジメチルスルホキシド、N,N−ジメチルホルムアミド又はこれらの溶媒と水との混合溶媒が好ましい。 The polymerization reaction can be performed in the presence of a solvent. As the solvent, a solvent that dissolves the monomer composition and does not react can be used. Examples of such solvents include alcohol solvents such as water, methanol, ethanol, n-propanol, and isopropanol, N, N-dimethyl sulfoxide, N, N-dimethylformamide, N, N-dimethylacetamide, pyridine, and the like. Can be given. In particular, N, N-dimethylsulfoxide, N, N-dimethylformamide or a mixed solvent of these solvents and water is preferable.
上記重合反応において、反応時の溶液濃度は、1質量%〜50質量%とすることができる。当該溶液濃度は、反応物である単量体組成物の濃度と、生成物である重合体の濃度との両方を含む。当該溶液濃度を1質量%以上とすることにより、製造効率を向上させることができる。また、当該溶液濃度を50質量%以下とすることにより、反応液の高粘度化を防ぎ、反応を十分に進行させることができる。 In the above polymerization reaction, the solution concentration during the reaction can be 1% by mass to 50% by mass. The concentration of the solution includes both the concentration of the monomer composition that is a reactant and the concentration of the polymer that is a product. Manufacturing efficiency can be improved by making the said solution concentration into 1 mass% or more. In addition, by setting the solution concentration to 50% by mass or less, it is possible to prevent the reaction solution from becoming highly viscous and allow the reaction to proceed sufficiently.
上記重合反応により得られる重合体の精製には、再沈殿法、透析法、限外濾過法等の一般的な精製方法を用いることができる。 For purification of the polymer obtained by the polymerization reaction, a general purification method such as a reprecipitation method, a dialysis method, or an ultrafiltration method can be used.
上記各精製方法に用いることができる溶媒としては、シクロデキストリン誘導体と反応しないものが使用でき、例えば、水、メタノール、エタノール、n−プロパノール、イソプロパノール等が挙げられる。特に、水、メタノール、エタノール又はこれらの混合溶媒が好ましい。 As a solvent that can be used in each of the above purification methods, a solvent that does not react with a cyclodextrin derivative can be used, and examples thereof include water, methanol, ethanol, n-propanol, and isopropanol. In particular, water, methanol, ethanol or a mixed solvent thereof is preferable.
以下、実施例及び比較例を挙げて本発明を更に具体的に説明するが、本発明はこれら実施例の範囲に限定されるものではない。なお、例中の各種測定は以下の通り行った。 EXAMPLES Hereinafter, although an Example and a comparative example are given and this invention is demonstrated further more concretely, this invention is not limited to the range of these Examples. Various measurements in the examples were performed as follows.
[1H−NMR分析]
日本電子社製、商品名「JNM−AL400」を用い、溶媒:D2O、試料濃度:10mg/g、積算回数:128回の条件で測定を行った。
[1 H-NMR analysis]
Measurement was carried out under the conditions of solvent: D 2 O, sample concentration: 10 mg / g, integration number: 128 times, using a product name “JNM-AL400” manufactured by JEOL Ltd.
[質量分析(ESI−MS)]
Waters社製、商品名「Q−micro2695」を用い、試料濃度:100ppm、溶媒:10mM酢酸アンモニウム含有10%アセトニトリル水溶液、検出モード:ESI+、キャピラリー電圧:3.54V、コーン電圧:30V、イオン源ヒーター:120℃、脱溶媒ガス:350℃の条件で測定を行った。
[Mass Spectrometry (ESI-MS)]
Product name “Q-micro2695” manufactured by Waters Inc., sample concentration: 100 ppm, solvent: 10 mM aqueous solution containing 10 mM ammonium acetate, detection mode: ESI +, capillary voltage: 3.54 V, cone voltage: 30 V, ion source heater : Measurement was performed under the conditions of 120 ° C. and solvent removal gas: 350 ° C.
[数平均分子量の測定(GPC)]
移動相の調製:臭化リチウム2.61gとリン酸2.94gとをDMF1Lに溶解させた。測定試料の調製:サンプル2mgを移動相1gに溶解させた。その他の測定条件は以下の通りである。カラム:KD−806M(昭和電工社製)、標準物質:PEO(東ソー社製)、検出:視差屈折率計RI−8020(東ソー社製)、数平均分子量(Mn)の算出:分子量計算プログラム(SC−8020用GPCプログラム)、流速:1.0mL/分、カラム温度:40℃、試料溶液注入量:100μL,測定時間:30分の条件で測定を行った。
[Measurement of number average molecular weight (GPC)]
Preparation of mobile phase: 2.61 g of lithium bromide and 2.94 g of phosphoric acid were dissolved in 1 L of DMF. Preparation of measurement sample: 2 mg of sample was dissolved in 1 g of mobile phase. Other measurement conditions are as follows. Column: KD-806M (manufactured by Showa Denko KK), standard material: PEO (manufactured by Tosoh Corporation), detection: parallax refractometer RI-8020 (manufactured by Tosoh Corporation), calculation of number average molecular weight (Mn): molecular weight calculation program ( Measurement was performed under the conditions of GPC program for SC-8020), flow rate: 1.0 mL / min, column temperature: 40 ° C., sample solution injection amount: 100 μL, measurement time: 30 minutes.
[フェノール化合物の測定(HPLC)]
移動相の調製:イオン交換水700mLとアセトニトリル(和光純薬工業社製)300mLとを混合させて調製した。測定試料の調製:サンプル0.1gを移動相0.9gに溶解させた。その他の測定条件は以下の通りである。カラム:Inertsil ODS−3V(GL Science社製)、検出:紫外可視検出器UV−8020(東ソー社製)、流速:1.0mL/分、カラム温度:40℃、試料溶液注入量:20μL、測定時間:30分の条件で測定を行った。
[Measurement of phenolic compounds (HPLC)]
Preparation of mobile phase: Prepared by mixing 700 mL of ion-exchanged water and 300 mL of acetonitrile (manufactured by Wako Pure Chemical Industries, Ltd.). Preparation of measurement sample: 0.1 g of sample was dissolved in 0.9 g of mobile phase. Other measurement conditions are as follows. Column: Inertsil ODS-3V (manufactured by GL Science), detection: UV-visible detector UV-8020 (manufactured by Tosoh Corporation), flow rate: 1.0 mL / min, column temperature: 40 ° C., sample solution injection amount: 20 μL, measurement Time: Measurement was performed under the condition of 30 minutes.
[実施例1−1]
<6−O−トシル−β−シクロデキストリンの合成>
β−シクロデキストリン(β−CD、東京化成工業社製)250.0g、水酸化ナトリウム(和光純薬工業社製)125.0gをイオン交換水7500gに溶解させ、0℃以下に冷却した。その後、p−トルエンスルホン酸クロライド(p−TsCl、東京化成工業社製)150.0gを加え、3時間トシル化反応を行った。反応後、p−TsCl100.0gを追加し、さらに2時間トシル化反応を行った。トシル化反応終了後、未反応のp−TsClをろ別し、ろ液を回収した。ろ液を4℃に冷却し、濃塩酸(和光純薬工業社製)の10%水溶液3165.2gを加え、目的物を析出させた。析出物をろ過により回収し、ろ紙上の沈殿物をイオン交換水1000.0gとアセトン(中国精油社製)500.0gで洗浄した後、50℃で減圧乾燥した。減圧乾燥により得られた白色粉末に、イオン交換水894.6gを加え、85℃で2時間攪拌した。攪拌後、室温まで冷却し、2時間静置した。静置後、ろ過により沈殿物を回収し、ろ紙上の沈殿物をイオン交換水894.6gとアセトン894.6gで洗浄した後、50℃で減圧乾燥することで式(5)のk=7に相当する6−O−トシル−β−シクロデキストリンの白色粉末を得た。
[Example 1-1]
<Synthesis of 6-O-tosyl-β-cyclodextrin>
250.0 g of β-cyclodextrin (β-CD, manufactured by Tokyo Chemical Industry Co., Ltd.) and 125.0 g of sodium hydroxide (manufactured by Wako Pure Chemical Industries, Ltd.) were dissolved in 7500 g of ion-exchanged water and cooled to 0 ° C. or lower. Thereafter, 150.0 g of p-toluenesulfonic acid chloride (p-TsCl, manufactured by Tokyo Chemical Industry Co., Ltd.) was added, and a tosylation reaction was performed for 3 hours. After the reaction, 100.0 g of p-TsCl was added, and a tosylation reaction was further performed for 2 hours. After completion of the tosylation reaction, unreacted p-TsCl was filtered off, and the filtrate was recovered. The filtrate was cooled to 4 ° C., and 3165.2 g of 10% aqueous solution of concentrated hydrochloric acid (manufactured by Wako Pure Chemical Industries, Ltd.) was added to precipitate the target product. The precipitate was collected by filtration, and the precipitate on the filter paper was washed with 1000.0 g of ion-exchanged water and 500.0 g of acetone (manufactured by China Seiyaku Co., Ltd.), and then dried at 50 ° C. under reduced pressure. To the white powder obtained by drying under reduced pressure, 894.6 g of ion-exchanged water was added and stirred at 85 ° C. for 2 hours. After stirring, the mixture was cooled to room temperature and allowed to stand for 2 hours. After standing, the precipitate is recovered by filtration, and the precipitate on the filter paper is washed with 894.6 g of ion-exchanged water and 894.6 g of acetone, and then dried under reduced pressure at 50 ° C. to obtain k = 7 in formula (5). A white powder of 6-O-tosyl-β-cyclodextrin corresponding to
<6−デオキシ−6−ベンジルアミノ−β−シクロデキストリンの合成>
上記により得られた6−O−トシル−β−シクロデキストリン100.0gとN,N−ジメチルホルムアミド(DMF、和光純薬工業社製)200.0gとを混合し、40℃に昇温して溶解させた。その後、ベンジルアミン(東京化成工業社製)99.8gを加えて75℃に昇温し、7時間ベンジルアミノ化反応を行った。ベンジルアミノ化反応終了後、40℃以下まで冷却し、15wt%アセトニトリル水溶液2000.0gを加えることで白色の沈殿物を得た。沈殿物をろ過で回収し、ろ過物をアセトン1000.0gで洗浄した後、50℃で減圧乾燥した。減圧乾燥により得られた白色粉末にイオン交換水1000.0gを加えて激しく攪拌し、スラリー状の白色液体を得た。白色液体をろ過することにより水を除去し、ろ紙上の結晶をイオン交換水500.0gとアセトン1000.0gで洗浄した後、50℃で減圧乾燥することで式(6)のk=7に相当する6−デオキシ−6−ベンジルアミノ−β−シクロデキストリンの白色粉末を得た。
<Synthesis of 6-deoxy-6-benzylamino-β-cyclodextrin>
100.0 g of 6-O-tosyl-β-cyclodextrin obtained as described above and 200.0 g of N, N-dimethylformamide (DMF, manufactured by Wako Pure Chemical Industries, Ltd.) are mixed and heated to 40 ° C. Dissolved. Thereafter, 99.8 g of benzylamine (manufactured by Tokyo Chemical Industry Co., Ltd.) was added, the temperature was raised to 75 ° C., and benzylamination reaction was performed for 7 hours. After completion of the benzylamination reaction, the mixture was cooled to 40 ° C. or lower, and 2000.0 g of a 15 wt% aqueous acetonitrile solution was added to obtain a white precipitate. The precipitate was collected by filtration, and the filtrate was washed with 1000.0 g of acetone and then dried under reduced pressure at 50 ° C. To the white powder obtained by drying under reduced pressure, 1000.0 g of ion-exchanged water was added and stirred vigorously to obtain a slurry-like white liquid. Water is removed by filtering the white liquid, and the crystals on the filter paper are washed with 500.0 g of ion-exchanged water and 1000.0 g of acetone, and then dried under reduced pressure at 50 ° C. to obtain k = 7 in formula (6). The corresponding 6-deoxy-6-benzylamino-β-cyclodextrin white powder was obtained.
<6−デオキシ−6−アミノ−β−シクロデキストリンの合成>
上記により得られた6−デオキシ−6−ベンジルアミノ−β−シクロデキストリン78.0gをN,N−ジメチルアセトアミド(DMAc、和光純薬工業社製)780.0gに溶解させ、65℃に昇温した。昇温後、イオン交換水780.0gに分散させたパラジウム活性炭素(10%)(和光純薬工業社製)7.8gを加え、反応容器内を水素雰囲気下にし、70℃で7時間水素添加分解反応を行った。水素添加分解反応終了後、イオン交換水780.0gを加えて40℃以下に冷却し、セライトを敷いたろ紙でろ過してろ液を回収した。さらに、ろ紙上の活性炭をイオン交換水1950.0gで洗浄し、ろ液を回収した。回収したろ液の減圧蒸留を行い、約300gまで濃縮した後、濃縮液をアセトン1950.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物を酢酸エチル(和光純薬工業社製)780.0gで2回洗浄した後、50℃で減圧乾燥した。減圧乾燥により得られた粉末に、イオン交換水361.3gを加え、50℃で1時間攪拌した。攪拌後、冷却せずにろ過してろ液を回収した。さらに、ろ過物をイオン交換水180.6gで洗浄し、ろ液を回収した。回収したろ液をアセトン2709.6gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物を酢酸エチル780.0gで洗浄した後、ろ過物を50℃で減圧乾燥することで式(3)のk=7に相当する6−デオキシ−6−アミノ−β−シクロデキストリンの粉末を得た。
<Synthesis of 6-deoxy-6-amino-β-cyclodextrin>
78.0 g of 6-deoxy-6-benzylamino-β-cyclodextrin obtained as described above was dissolved in 780.0 g of N, N-dimethylacetamide (DMAc, manufactured by Wako Pure Chemical Industries, Ltd.), and the temperature was raised to 65 ° C. did. After heating, 7.8 g of palladium activated carbon (10%) (manufactured by Wako Pure Chemical Industries, Ltd.) dispersed in 780.0 g of ion-exchanged water was added, the inside of the reaction vessel was placed in a hydrogen atmosphere, and hydrogen was maintained at 70 ° C for 7 hours. Addition decomposition reaction was performed. After completion of the hydrogenolysis reaction, 780.0 g of ion-exchanged water was added and the mixture was cooled to 40 ° C. or lower, and filtered through a filter paper covered with celite to collect the filtrate. Furthermore, the activated carbon on the filter paper was washed with 1950.0 g of ion exchange water, and the filtrate was recovered. The collected filtrate was distilled under reduced pressure and concentrated to about 300 g, and then the concentrated solution was added to 1950.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed twice with 780.0 g of ethyl acetate (manufactured by Wako Pure Chemical Industries, Ltd.) and then dried under reduced pressure at 50 ° C. To the powder obtained by drying under reduced pressure, 361.3 g of ion-exchanged water was added and stirred at 50 ° C. for 1 hour. After stirring, the filtrate was collected by filtration without cooling. Further, the filtrate was washed with 180.6 g of ion exchange water, and the filtrate was recovered. The collected filtrate was added to 2709.6 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 780.0 g of ethyl acetate, and then the filtrate was dried under reduced pressure at 50 ° C., thereby corresponding to 6-deoxy-6-amino corresponding to k = 7 in formula (3). A powder of -β-cyclodextrin was obtained.
<6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−β−シクロデキストリンの合成>
上記により得られた6−デオキシ−6−アミノ−β−シクロデキストリン60.0gに、式(4)のR1=メチル基、R2=エチレン基、R3=エチレン基に相当する2−メタクリロイロキシエチルコハク酸(HO−MS、共栄社化学社製)16.04gを溶解させたDMF300.0gを加え、均一に分散させた。その後、4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド(DMT−MM、和光純薬工業社製)20.5gを加え、室温で8時間アミド化反応を行った。アミド化反応終了後、反応液をアセトン1500.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトニトリル900.0gで洗浄した。得られたろ過物に75wt%アセトニトリル水溶液1600.0gを加え、十分に攪拌して分散させた。分散液をろ過し、ろ過物をアセトン300.0gで2回洗浄し、40℃で減圧乾燥させることで式(1)のk=7、x=1、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリンを99%、式(1)のk=7、x=2、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するジ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリンを1%含有した6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリンの白色粉末を得た。
<Synthesis of 6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin>
To 60.0 g of 6-deoxy-6-amino-β-cyclodextrin obtained above, 2 -methacrylic acid corresponding to R 1 = methyl group, R 2 = ethylene group, R 3 = ethylene group in formula (4) 300.0 g of DMF in which 16.04 g of leuoxyethyl succinic acid (HO-MS, manufactured by Kyoeisha Chemical Co., Ltd.) was dissolved was added and dispersed uniformly. Thereafter, 20.5 g of 4- (4,6-dimethoxy-1,3,5-triazin-2-yl) -4-methylmorpholinium chloride (DMT-MM, manufactured by Wako Pure Chemical Industries, Ltd.) was added at room temperature. The amidation reaction was carried out for 8 hours. After completion of the amidation reaction, the reaction solution was added to 1500.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 900.0 g of acetonitrile. To the obtained filtrate, 1600.0 g of a 75 wt% aqueous acetonitrile solution was added, and the mixture was sufficiently stirred and dispersed. The dispersion was filtered, and the filtrate was washed twice with 300.0 g of acetone and dried under reduced pressure at 40 ° C., whereby k = 7, x = 1, R 1 = methyl group, R 2 = ethylene of formula (1). Group, R 3 = 99% mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin corresponding to ethylene group, k = 7 in formula (1), x = 2, 6 containing 1% of di-6-deoxy-6- (2-methacryloyloxyethylsuccinamide) -β-cyclodextrin corresponding to R 1 = methyl group, R 2 = ethylene group, R 3 = ethylene group A white powder of deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin was obtained.
得られた生成物についての1H NMR測定及びESI−MS測定の結果は以下の通りである。
1H NMR(溶媒:D2O、標準ピーク:HOD):2.0ppm(3H,CH 3 )、2.4−2.8ppm(4H,CO−(CH 2 ) 2 −CO)、3.5−4.1ppm(42H,−CH(OH)−,−CH 2 OH)、4.3−4.5ppm(4H,−O−(CH 2 ) 2 −O−)、4.6−5.0ppm(20H,OH)、5.1ppm(7H,−CH(OH)−)、5.7−6.3ppm(2H,CH 2 )
ESI−MS(M+H+):1347(モノ−6−デオキシ(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)、1558(ジ−6−デオキシ(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)
The results of 1 H NMR measurement and ESI-MS measurement of the obtained product are as follows.
1 H NMR (solvent: D 2 O, standard peak: HOD): 2.0 ppm (3H, CH 3 ), 2.4-2.8 ppm (4H, CO— (CH 2 ) 2 —CO), 3.5 -4.1ppm (42H, - CH (OH ) -, - CH 2 OH), 4.3-4.5ppm (4H, -O- (CH 2) 2 -O -), 4.6-5.0ppm (20H, OH), 5.1ppm ( 7H, - CH (OH) -), 5.7-6.3ppm (2H, CH 2)
ESI-MS (M + H + ): 1347 (mono-6-deoxy (2-methacryloyloxyethyl succinamide) -β-cyclodextrin), 1558 (di-6-deoxy (2-methacryloyloxyethyl succinamide)- β-cyclodextrin)
[実施例2−1]
<モノ−6−O−トシル−β−シクロデキストリンの合成>
実施例1−1と同様の手順で得られた上記6−O−トシル−β−シクロデキストリンの白色粉末102.0gに、イオン交換水644.1gとアセトニトリル787.2gとを加え、撹拌しながら2時間還流した。還流後、室温まで冷却し、2時間静置した。静置後、ろ過により沈殿物を回収し、55wt%アセトニトリル水溶液894.6gで洗浄した後、50℃で減圧乾燥することで式(5)のk=7、x=1に相当するモノ−6−O−トシル−β−シクロデキストリンの白色粉末を得た。
[Example 2-1]
<Synthesis of Mono-6-O-tosyl-β-cyclodextrin>
To 102.0 g of the white powder of 6-O-tosyl-β-cyclodextrin obtained in the same procedure as in Example 1-1, 644.1 g of ion-exchanged water and 787.2 g of acetonitrile were added and stirred. Refluxed for 2 hours. After refluxing, it was cooled to room temperature and allowed to stand for 2 hours. After standing, the precipitate was collected by filtration, washed with 894.6 g of 55 wt% acetonitrile aqueous solution, and then dried under reduced pressure at 50 ° C., so that mono-6 corresponding to k = 7 and x = 1 in formula (5) A white powder of -O-tosyl-β-cyclodextrin was obtained.
<モノ−6−デオキシ−6−ベンジルアミノ−β−シクロデキストリンの合成>
上記により得られたモノ−6−O−トシル−β−シクロデキストリン80.0gとDMF160.0gとを混合し、40℃に昇温して溶解させた。その後、ベンジルアミン79.8gを加えて75℃に昇温し、7時間ベンジルアミノ化反応を行った。ベンジルアミノ化反応終了後、40℃以下まで冷却し、15wt%アセトニトリル水溶液1600.0gを加えることで、白色の沈殿物を得た。沈殿物をろ過で回収し、ろ過物をアセトン800.0gで洗浄した後、50℃で減圧乾燥した。減圧乾燥により得られた白色粉末に、イオン交換水800gを加えて激しく攪拌し、スラリー状の白色液体を得た。白色液体をろ過することにより水を除去し、ろ紙上の結晶をイオン交換水400gとアセトン800gで洗浄した後、50℃で減圧乾燥することで式(6)のk=7、x=1に相当するモノ−6−デオキシ−6−ベンジルアミノ−β−シクロデキストリンの白色粉末を得た。
<Synthesis of mono-6-deoxy-6-benzylamino-β-cyclodextrin>
Mono-6-O-tosyl-β-cyclodextrin 80.0 g obtained above and DMF 160.0 g were mixed and heated to 40 ° C. to dissolve. Thereafter, 79.8 g of benzylamine was added, the temperature was raised to 75 ° C., and a benzylamination reaction was performed for 7 hours. After completion of the benzylamination reaction, the mixture was cooled to 40 ° C. or lower, and 1600.0 g of a 15 wt% aqueous acetonitrile solution was added to obtain a white precipitate. The precipitate was collected by filtration, and the filtrate was washed with 800.0 g of acetone and dried under reduced pressure at 50 ° C. To the white powder obtained by drying under reduced pressure, 800 g of ion-exchanged water was added and stirred vigorously to obtain a slurry-like white liquid. Water is removed by filtering the white liquid, and the crystals on the filter paper are washed with 400 g of ion-exchanged water and 800 g of acetone, and then dried under reduced pressure at 50 ° C., so that k = 7 and x = 1 in formula (6). The corresponding mono-6-deoxy-6-benzylamino-β-cyclodextrin white powder was obtained.
<モノ−6−デオキシ−6−アミノ−β−シクロデキストリンの合成>
上記により得られたモノ−6−デオキシ−6−ベンジルアミノ−β−シクロデキストリン64.0gをDMAc640.0gに溶解させ、65℃に昇温した。昇温後、イオン交換水640.0gに分散させたパラジウム活性炭素(10%)6.4gを加え、反応容器内を水素雰囲気下にし、70℃で7時間水素添加分解反応させた。水素添加分解反応終了後、イオン交換水640.0gを加えて40℃以下に冷却し、セライトを敷いたろ紙でろ過してろ液を回収した。さらに、ろ紙上の活性炭をイオン交換水1280.0gで洗浄し、ろ液を回収した。回収したろ液の減圧蒸留を行い、約250gまで濃縮した後、濃縮液をアセトン1600.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物を酢酸エチル780.0gで2回洗浄した後、50℃で減圧乾燥した。減圧乾燥により得られた粉末にイオン交換水296.4gを加えて50℃で1時間攪拌した。1時間攪拌後、冷却せずにろ過してろ液を回収した。さらに、ろ過物をイオン交換水148.2gで洗浄し、ろ液を回収した。回収したろ液をアセトン2223.3gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物を酢酸エチル640.0gで洗浄した後、ろ過物を50℃で減圧乾燥することで式(3)のk=7、x=1に相当するモノ−6−デオキシ−6−アミノ−β−シクロデキストリンの粉末を得た。
<Synthesis of mono-6-deoxy-6-amino-β-cyclodextrin>
Mono-6-deoxy-6-benzylamino-β-cyclodextrin 64.0 g obtained above was dissolved in 640.0 g of DMAc, and the temperature was raised to 65 ° C. After the temperature elevation, 6.4 g of palladium activated carbon (10%) dispersed in 640.0 g of ion-exchanged water was added, the inside of the reaction vessel was placed in a hydrogen atmosphere, and a hydrogenolysis reaction was performed at 70 ° C. for 7 hours. After completion of the hydrogenolysis reaction, 640.0 g of ion-exchanged water was added, the mixture was cooled to 40 ° C. or lower, and filtered through a filter paper covered with celite to collect the filtrate. Furthermore, the activated carbon on the filter paper was washed with 1280.0 g of ion exchange water, and the filtrate was recovered. The collected filtrate was distilled under reduced pressure and concentrated to about 250 g, and then the concentrated solution was added to 1600.0 g of acetone to precipitate a precipitate. The precipitate was recovered by filtration, and the filtrate was washed twice with 780.0 g of ethyl acetate and then dried under reduced pressure at 50 ° C. To the powder obtained by drying under reduced pressure, 296.4 g of ion-exchanged water was added and stirred at 50 ° C. for 1 hour. After stirring for 1 hour, the filtrate was recovered by filtration without cooling. Further, the filtrate was washed with 148.2 g of ion exchange water, and the filtrate was recovered. The collected filtrate was added to 2223.3 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 640.0 g of ethyl acetate, and then the filtrate was dried at 50 ° C. under reduced pressure to obtain mono-6 corresponding to k = 7 and x = 1 in formula (3). A powder of -deoxy-6-amino-β-cyclodextrin was obtained.
<モノ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−β−シクロデキストリンの合成>
上記により得られたモノ−6−デオキシ−6−アミノ−β−シクロデキストリン50.0gに、HO−MS12.54gを溶解させたDMF250.0gを加え、均一に分散させた。その後、DMT−MM17.08gを加え、室温で8時間アミド化反応を行った。アミド化反応終了後、反応液をアセトン1250.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトニトリル750.0gで洗浄した。得られたろ過物に75wt%アセトニトリル水溶液1333.3gを加え、十分に攪拌して分散させた。分散液をろ過し、ろ過物をアセトン250.0gで2回洗浄し、40℃で減圧乾燥させることで式(2)のy=6、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するモノ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−β−シクロデキストリンの白色粉末を得た。
<Synthesis of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin>
250.0 g of DMF in which 12.54 g of HO-MS was dissolved was added to 50.0 g of mono-6-deoxy-6-amino-β-cyclodextrin obtained as described above and dispersed uniformly. Thereafter, 17.08 g of DMT-MM was added, and an amidation reaction was performed at room temperature for 8 hours. After completion of the amidation reaction, the reaction solution was added to 1250.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 750.0 g of acetonitrile. To the obtained filtrate was added 1333.3 g of a 75 wt% acetonitrile aqueous solution, and the mixture was sufficiently stirred and dispersed. The dispersion was filtered, and the filtrate was washed twice with 250.0 g of acetone and dried under reduced pressure at 40 ° C., whereby y = 6, R 1 = methyl group, R 2 = ethylene group, R 3 in formula (2). = White powder of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin corresponding to ethylene group was obtained.
得られた生成物についての1H NMR測定及びESI−MS測定の結果は以下の通りである。
1H NMR(溶媒:D2O、標準ピーク:HOD):2.0ppm(3H,CH 3 )、2.4−2.8ppm(4H,CO−(CH 2 ) 2 −CO)、3.5−4.1ppm(42H,−CH(OH)−,−CH 2 OH)、4.3−4.5ppm(4H,−O−(CH 2 ) 2 −O−)、4.6−5.0ppm(20H,OH)、5.1ppm(7H,−CH(OH)−)、5.7−6.3ppm(2H,CH 2 )
ESI−MS(M+H+):1347
The results of 1 H NMR measurement and ESI-MS measurement of the obtained product are as follows.
1 H NMR (solvent: D 2 O, standard peak: HOD): 2.0 ppm (3H, CH 3 ), 2.4-2.8 ppm (4H, CO— (CH 2 ) 2 —CO), 3.5 -4.1ppm (42H, - CH (OH ) -, - CH 2 OH), 4.3-4.5ppm (4H, -O- (CH 2) 2 -O -), 4.6-5.0ppm (20H, OH), 5.1ppm ( 7H, - CH (OH) -), 5.7-6.3ppm (2H, CH 2)
ESI-MS (M + H < + > ): 1347
[実施例3−1]
<モノ−6−デオキシ−6−(2-メタクリロイルオキシエチルヘキサヒドロフタル酸アミド)−β−シクロデキストリンの合成>
実施例2−1と同様の手順で得られたモノ−6−デオキシ−6−アミノ−β−シクロデキストリン2.0gに、式(4)のR1=メチル基、R2=エチレン基、R3=シクロへキシレン基に相当する2−メタクリロイロキシエチルヘキサヒドロフタル酸(HO−HH、共栄社化学社製)HO−HH1.0gを溶解させたDMF10.0gを加え、均一に溶解させた。その後、DMT−MM0.68gを加え、室温で8時間アミド化反応を行った。アミド化反応終了後、反応液をアセトン50.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトニトリル30.0gで洗浄した。得られたろ過物に75wt%アセトニトリル水溶液53.3gを加え、十分に攪拌して分散させた。分散液をろ過し、ろ過物をアセトン10.0gで2回洗浄し、40℃で減圧乾燥させることで式(2)のy=6、R1=メチル基、R2=エチレン基、R3=シクロへキシレン基に相当するモノ−6−デオキシ−6−(2-メタクリロイルオキシエチルヘキサヒドロフタル酸アミド)−β−シクロデキストリンの白色粉末を得た。
[Example 3-1]
<Synthesis of mono-6-deoxy-6- (2-methacryloyloxyethylhexahydrophthalamide) -β-cyclodextrin>
To a mono-6-deoxy-6-amino-β-cyclodextrin (2.0 g) obtained by the same procedure as in Example 2-1, R 1 = methyl group, R 2 = ethylene group, R of formula (4) 3 = 2-methacryloyloxyethyl hexahydrophthalic acid (HO-HH, manufactured by Kyoeisha Chemical Co., Ltd.) HO-HH corresponding to cyclohexylene group was added to 10.0 g of DMF and dissolved uniformly. Thereafter, 0.68 g of DMT-MM was added, and an amidation reaction was performed at room temperature for 8 hours. After completion of the amidation reaction, the reaction solution was added to 50.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 30.0 g of acetonitrile. To the obtained filtrate was added 53.3 g of a 75 wt% acetonitrile aqueous solution, and the mixture was sufficiently stirred and dispersed. The dispersion was filtered, and the filtrate was washed twice with 10.0 g of acetone and dried under reduced pressure at 40 ° C., whereby y = 6, R 1 = methyl group, R 2 = ethylene group, R 3 in formula (2). = White powder of mono-6-deoxy-6- (2-methacryloyloxyethylhexahydrophthalamide) -β-cyclodextrin corresponding to cyclohexylene group was obtained.
得られた生成物についての1H NMR測定及びESI−MS測定の結果は以下の通りである。
1H NMR(溶媒:D2O、標準ピーク:HOD):1.0−2.0ppm(8H,−CH 2 −)2.0ppm(3H,CH 3 )、2.6−3.1ppm(2H,CO−CH−CH−CO)、3.3−4.2ppm(42H,−CH(OH)−,−CH 2 OH)、4.2−4.5ppm(4H,−O−(CH 2 ) 2 −O−)、4.6−5.0ppm(20H,OH)、5.1ppm(7H,−CH(OH)−)、5.8−6.2ppm(2H,CH 2 )
ESI−MS(M+Na+):1423
The results of 1 H NMR measurement and ESI-MS measurement of the obtained product are as follows.
1 H NMR (solvent: D 2 O, standard peak: HOD): 1.0-2.0 ppm (8H, —CH 2 — ) 2.0 ppm (3H, CH 3 ), 2.6-3.1 ppm (2H , CO- CH-CH -CO), 3.3-4.2ppm (42H, - CH (OH) -, - CH 2 OH), 4.2-4.5ppm (4H, -O- (CH 2) 2 -O -), 4.6-5.0ppm (20H , OH), 5.1ppm (7H, - CH (OH) -), 5.8-6.2ppm (2H, CH 2)
ESI-MS (M + Na < + > ): 1423
[実施例4−1]
<6−O−トシル−α−シクロデキストリンの合成>
α−シクロデキストリン(α−CD、シクロケム社製)10.0gをピコリン(和光純薬工業社製)80.0gに溶解後、窒素置換を行い、0℃に冷却した。別容器でp−TsCl 10.0gをピリジン(和光純薬工業社製)15.0gに溶解させた。α−CDを溶解させたピコリン溶液を5℃以下に保ちながら、p−TsClを溶解させたピリジン溶液を滴下してトシル化反応を行った。滴下終了後、室温で2時間攪拌を継続した。トシル化反応終了後、アセトン115.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン600gで洗浄した後、減圧乾燥して白色〜黄色粉末を得た。得られた粉末を100.0gの水に分散溶解させ、ダイヤイオンHP−20(三菱化学社製)を充填したカラムに通液し、十分な水で洗浄した。次いで十分な量の25%エタノール水溶液を通液させて目的物を溶出させた。回収した25%エタノール水溶液を減圧留去することで、式(5)のk=6に相当する6−O−トシル−α−シクロデキストリンの白色粉末を得た。
[Example 4-1]
<Synthesis of 6-O-tosyl-α-cyclodextrin>
After 10.0 g of α-cyclodextrin (α-CD, manufactured by Cyclochem) was dissolved in 80.0 g of picoline (manufactured by Wako Pure Chemical Industries, Ltd.), nitrogen substitution was performed and the mixture was cooled to 0 ° C. In a separate container, 10.0 g of p-TsCl was dissolved in 15.0 g of pyridine (Wako Pure Chemical Industries, Ltd.). While maintaining the picoline solution in which α-CD was dissolved at 5 ° C. or lower, a pyridine solution in which p-TsCl was dissolved was dropped to perform a tosylation reaction. After completion of the dropwise addition, stirring was continued at room temperature for 2 hours. After completion of the tosylation reaction, the mixture was added to 115.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 600 g of acetone and then dried under reduced pressure to obtain a white to yellow powder. The obtained powder was dispersed and dissolved in 100.0 g of water, passed through a column filled with Diaion HP-20 (manufactured by Mitsubishi Chemical Corporation), and washed with sufficient water. Next, a sufficient amount of 25% aqueous ethanol solution was passed through to elute the target product. The collected 25% ethanol aqueous solution was distilled off under reduced pressure to obtain a white powder of 6-O-tosyl-α-cyclodextrin corresponding to k = 6 in formula (5).
<6−デオキシ−6−ベンジルアミノ−α−シクロデキストリンの合成>
上記により得られた6−O−トシル−α−シクロデキストリン7.0gとDMF14.0g、ベンジルアミン12.0gを加えて75℃に昇温し、7時間ベンジルアミノ化反応を行った。ベンジルアミノ化反応終了後、40℃以下まで冷却し、アセトン165.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン165.0gで洗浄した後、50℃で減圧乾燥することで式(6)のk=6に相当する6−デオキシ−6−ベンジルアミノ−α−シクロデキストリンの白色粉末を得た。
<Synthesis of 6-deoxy-6-benzylamino-α-cyclodextrin>
7.0 g of 6-O-tosyl-α-cyclodextrin obtained above, 14.0 g of DMF, and 12.0 g of benzylamine were added, the temperature was raised to 75 ° C., and a benzylamination reaction was performed for 7 hours. After completion of the benzylamination reaction, the reaction mixture was cooled to 40 ° C. or lower and poured into 165.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 165.0 g of acetone, and then dried under reduced pressure at 50 ° C., whereby 6-deoxy-6-benzylamino-α-corresponding to k = 6 in formula (6). A white powder of cyclodextrin was obtained.
<6−デオキシ−6−アミノ−α−シクロデキストリンの合成>
上記により得られた6−デオキシ−6−ベンジルアミノ−α−シクロデキストリン5.0gをDMAc50.0gに溶解させた。イオン交換水50.0gに分散させたパラジウム活性炭素(10%)0.5gを加え、反応容器内を水素雰囲気下にし、70℃で7時間水素添加分解反応を行った。水素添加分解反応終了後、イオン交換水25.0gを加えて40℃以下に冷却し、セライトを敷いたろ紙でろ過してろ液を回収した。さらに、ろ紙上の活性炭をイオン交換水100.0gで洗浄し、ろ液を回収した。回収したろ液の減圧蒸留を行い、約20gまで濃縮した後、濃縮液をアセトン125.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン100.0gで洗浄した後、50℃で減圧乾燥することで式(3)のk=6に相当する6−デオキシ−6−アミノ−α−シクロデキストリンの粉末を得た。
<Synthesis of 6-deoxy-6-amino-α-cyclodextrin>
5.0 g of 6-deoxy-6-benzylamino-α-cyclodextrin obtained as described above was dissolved in 50.0 g of DMAc. 0.5 g of palladium activated carbon (10%) dispersed in 50.0 g of ion-exchanged water was added, the inside of the reaction vessel was placed in a hydrogen atmosphere, and a hydrogenolysis reaction was performed at 70 ° C. for 7 hours. After completion of the hydrogenolysis reaction, 25.0 g of ion-exchanged water was added, the mixture was cooled to 40 ° C. or lower, and filtered through a filter paper covered with celite, and the filtrate was recovered. Furthermore, the activated carbon on the filter paper was washed with 100.0 g of ion exchange water, and the filtrate was recovered. The collected filtrate was distilled under reduced pressure and concentrated to about 20 g, and then the concentrated solution was poured into 125.0 g of acetone to precipitate a precipitate. The precipitate was recovered by filtration, and the filtrate was washed with 100.0 g of acetone and then dried under reduced pressure at 50 ° C. to thereby obtain 6-deoxy-6-amino-α-cyclo which corresponds to k = 6 in formula (3). A dextrin powder was obtained.
<6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリンの合成>
上記により得られた6−デオキシ−6−アミノ−α−シクロデキストリン3.5gに、式(4)のHO−MS3.32g、DMF17.5gを加え、均一に溶解させた。その後、DMT−MM2.99gを加え、室温で8時間アミド化反応を行った。アミド化反応終了後、反応液をアセトン87.5gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトニトリル35.0gとアセトン35.0gで洗浄し、40℃で減圧乾燥させることで式(1)のk=6、x=1、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリンを75%、式(1)のk=6、x=2、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するジ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリンを24%、式(1)のk=6、x=3、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリンを1%含有した6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリンの白色粉末を得た。
<Synthesis of 6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin>
To 3.5 g of 6-deoxy-6-amino-α-cyclodextrin obtained as described above, 3.32 g of HO-MS of formula (4) and 17.5 g of DMF were added and dissolved uniformly. Thereafter, 2.99 g of DMT-MM was added, and an amidation reaction was performed at room temperature for 8 hours. After completion of the amidation reaction, the reaction solution was added to 87.5 g of acetone to precipitate a precipitate. The precipitate is recovered by filtration, and the filtrate is washed with 35.0 g of acetonitrile and 35.0 g of acetone and dried under reduced pressure at 40 ° C., so that k = 6, x = 1, R 1 = methyl group in formula (1). , R 2 = ethylene group, R 3 = 75% mono-6-deoxy-6- (2-methacryloyloxyethylsuccinamide) -α-cyclodextrin corresponding to ethylene group, k = 6 in formula (1) X = 2, R 1 = methyl group, R 2 = ethylene group, R 3 = 24 corresponding to di-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin %, Mono-6-deoxy-6- (2-methacryloyloxyethyl succinate corresponding to k = 6, x = 3, R 1 = methyl group, R 2 = ethylene group, R 3 = ethylene group in formula (1) Acid amide) -α To give a white powder of a cyclodextrin containing 1% 6-deoxy-6- (2-methacryloyloxyethyl succinic acid amide)-.alpha.-cyclodextrin.
得られた生成物についての1H NMR測定及びESI−MS測定の結果は以下の通りである。
1H NMR(溶媒:D2O、標準ピーク:HOD):2.0ppm(4H,CH 3 )、2.4−2.8ppm(5H,CO−(CH 2 ) 2 −CO)、3.4−4.2ppm(36H,−CH(OH)−,−CH 2 OH)、4.2−4.6ppm(5H,−O−(CH 2 ) 2 −O−)、4.7−4.9ppm(20H,OH)、5.1ppm(6H,−CH(OH)−)、5.7−6.3ppm(3H,CH 2 )
ESI−MS(M+H+):1185(モノ−6−デオキシ(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリン)、1397(ジ−6−デオキシ(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリン)、1609(トリ−6−デオキシ(2−メタクリロイルオキシエチルコハク酸アミド)−α−シクロデキストリン)
The results of 1 H NMR measurement and ESI-MS measurement of the obtained product are as follows.
1 H NMR (solvent: D 2 O, standard peak: HOD): 2.0 ppm (4H, CH 3 ), 2.4-2.8 ppm (5H, CO— (CH 2 ) 2 —CO), 3.4 -4.2ppm (36H, - CH (OH ) -, - CH 2 OH), 4.2-4.6ppm (5H, -O- (CH 2) 2 -O -), 4.7-4.9ppm (20H, OH), 5.1ppm ( 6H, - CH (OH) -), 5.7-6.3ppm (3H, CH 2)
ESI-MS (M + H + ): 1185 (mono-6-deoxy (2-methacryloyloxyethyl succinamide) -α-cyclodextrin), 1397 (di-6-deoxy (2-methacryloyloxyethyl succinamide)- α-cyclodextrin), 1609 (tri-6-deoxy (2-methacryloyloxyethyl succinamide) -α-cyclodextrin)
[実施例5−1]
<モノ−6−O−トシル−α−シクロデキストリンの合成>
α−CD10.0gをピコリン80.0gに溶解後、窒素置換を行い、0℃に冷却した。別容器でp−TsCl 10.0gをピリジン15.0gに溶解させた。α−CDを溶解させたピコリン溶液を5℃以下に保ちながら、p−TsClを溶解させたピリジン溶液を滴下してトシル化反応を行った。滴下終了後、室温で2時間攪拌を継続した。トシル化反応終了後、アセトン115.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン600gで洗浄した後、減圧乾燥して白色〜黄色粉末を得た。得られた粉末を100.0gの水に分散溶解させ、ダイヤイオンHP−20を充填したカラムに通液し、十分な水で洗浄した。次いで20%エタノール水溶液を通液させて目的物を溶出させた。回収した20%エタノール水溶液を減圧留去することで、式(5)のk=6、x=1に相当するモノ−6−O−トシル−α−シクロデキストリンの白色粉末を得た。
[Example 5-1]
<Synthesis of mono-6-O-tosyl-α-cyclodextrin>
After 10.0 g of α-CD was dissolved in 80.0 g of picoline, nitrogen substitution was performed and the mixture was cooled to 0 ° C. In a separate container, 10.0 g of p-TsCl was dissolved in 15.0 g of pyridine. While maintaining the picoline solution in which α-CD was dissolved at 5 ° C. or lower, a pyridine solution in which p-TsCl was dissolved was dropped to perform a tosylation reaction. After completion of the dropwise addition, stirring was continued at room temperature for 2 hours. After completion of the tosylation reaction, the mixture was added to 115.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 600 g of acetone and then dried under reduced pressure to obtain a white to yellow powder. The obtained powder was dispersed and dissolved in 100.0 g of water, passed through a column filled with Diaion HP-20, and washed with sufficient water. Next, a 20% aqueous ethanol solution was passed through to elute the target product. The collected 20% aqueous ethanol solution was distilled off under reduced pressure to obtain a white powder of mono-6-O-tosyl-α-cyclodextrin corresponding to k = 6 and x = 1 in formula (5).
<モノ−6−デオキシ−6−ベンジルアミノ−α−シクロデキストリンの合成>
上記により得られたモノ−6−O−トシル−α−シクロデキストリン3.2gとDMF6.4g、ベンジルアミン3.65gを加えて75℃に昇温し、7時間ベンジルアミノ化反応を行った。ベンジルアミノ化反応終了後、40℃以下まで冷却し、アセトン66.3gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン66.3gで洗浄した後、50℃で減圧乾燥することで式(6)のk=6、x=1に相当するモノ−6−デオキシ−6−ベンジルアミノ−α−シクロデキストリンの白色粉末を得た。
<Synthesis of mono-6-deoxy-6-benzylamino-α-cyclodextrin>
Mono-6-O-tosyl-α-cyclodextrin (3.2 g) obtained above, 6.4 g of DMF and 3.65 g of benzylamine were added, the temperature was raised to 75 ° C., and a benzylamination reaction was performed for 7 hours. After completion of the benzylamination reaction, the reaction mixture was cooled to 40 ° C. or lower and charged into 66.3 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 66.3 g of acetone, and then dried under reduced pressure at 50 ° C., whereby mono-6-deoxy-6 corresponding to k = 6 and x = 1 in formula (6). -A white powder of benzylamino-α-cyclodextrin was obtained.
<モノ−6−デオキシ−6−アミノ−α−シクロデキストリンの合成>
上記により得られたモノ−6−デオキシ−6−ベンジルアミノ−α−シクロデキストリン2.8gをDMAc28.0gに溶解させた。イオン交換水28.0gに分散させたパラジウム活性炭素(10%)0.28gを加え、反応容器内を水素雰囲気下にし、70℃で7時間水素添加分解反応を行った。水素添加分解反応終了後、イオン交換水14.0gを加えて40℃以下に冷却し、セライトを敷いたろ紙でろ過してろ液を回収した。さらに、ろ紙上の活性炭をイオン交換水56.0gで洗浄し、ろ液を回収した。回収したろ液の減圧蒸留を行い、約10gまで濃縮した後、濃縮液をアセトン70.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン70.0gで洗浄した後、50℃で減圧乾燥することで式(3)のk=7に相当するモノ−6−デオキシ−6−アミノ−α−シクロデキストリンの粉末を得た。
<Synthesis of mono-6-deoxy-6-amino-α-cyclodextrin>
2.8 g of mono-6-deoxy-6-benzylamino-α-cyclodextrin obtained as described above was dissolved in 28.0 g of DMAc. 0.28 g of palladium activated carbon (10%) dispersed in 28.0 g of ion-exchanged water was added, the inside of the reaction vessel was placed in a hydrogen atmosphere, and a hydrogenolysis reaction was performed at 70 ° C. for 7 hours. After completion of the hydrogenolysis reaction, 14.0 g of ion-exchanged water was added, the mixture was cooled to 40 ° C. or lower, and filtered through a filter paper covered with celite to collect the filtrate. Further, the activated carbon on the filter paper was washed with 56.0 g of ion exchange water, and the filtrate was recovered. The collected filtrate was distilled under reduced pressure and concentrated to about 10 g, and then the concentrated solution was put into 70.0 g of acetone to precipitate a precipitate. The precipitate was collected by filtration, and the filtrate was washed with 70.0 g of acetone, and then dried under reduced pressure at 50 ° C. to thereby obtain mono-6-deoxy-6-amino-α corresponding to k = 7 in formula (3). -A powder of cyclodextrin was obtained.
<モノ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリンの合成>
上記により得られたモノ−6−デオキシ−6−アミノ−β−シクロデキストリン2.4gに、式(4)のHO−MS1.14g、DMF12.0gを加え、均一に溶解させた。その後、DMT−MM1.03gを加え、室温で8時間アミド化反応を行った。アミド化反応終了後、反応液をアセトン60.0gに投入し、沈殿を析出させた。沈殿物をろ過で回収し、ろ過物をアセトン60.0gで洗浄し、40℃で減圧乾燥させることで式(2)のy=5、R1=メチル基、R2=エチレン基、R3=エチレン基に相当するモノ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリンの白色粉末を得た。
<Synthesis of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin>
To 2.4 g of mono-6-deoxy-6-amino-β-cyclodextrin obtained as described above, 1.14 g of HO-MS of formula (4) and 12.0 g of DMF were added and dissolved uniformly. Thereafter, 1.03 g of DMT-MM was added, and an amidation reaction was performed at room temperature for 8 hours. After completion of the amidation reaction, the reaction solution was added to 60.0 g of acetone to precipitate a precipitate. The precipitate is recovered by filtration, the filtrate is washed with 60.0 g of acetone, and dried under reduced pressure at 40 ° C., whereby y = 5, R 1 = methyl group, R 2 = ethylene group, R 3 in formula (2). = White powder of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin corresponding to ethylene group was obtained.
得られた生成物についての1H NMR測定及びESI−MS測定の結果は以下の通りである。
1H NMR(溶媒:D2O、標準ピーク:HOD):2.0ppm(3H,CH 3 )、2.4−2.8ppm(4H,CO−(CH 2 ) 2 −CO)、3.4−4.2ppm(36H,−CH(OH)−,−CH 2 OH)、4.2−4.6ppm(4H,−O−(CH 2 ) 2 −O−)、4.7−4.9ppm(20H,OH)、5.1ppm(6H,−CH(OH)−)、5.7−6.3ppm(2H,CH 2 )
ESI−MS(M+H+):1185
The results of 1 H NMR measurement and ESI-MS measurement of the obtained product are as follows.
1 H NMR (solvent: D 2 O, standard peak: HOD): 2.0 ppm (3H, CH 3 ), 2.4-2.8 ppm (4H, CO— (CH 2 ) 2 —CO), 3.4 -4.2ppm (36H, - CH (OH ) -, - CH 2 OH), 4.2-4.6ppm (4H, -O- (CH 2) 2 -O -), 4.7-4.9ppm (20H, OH), 5.1ppm ( 6H, - CH (OH) -), 5.7-6.3ppm (2H, CH 2)
ESI-MS (M + H < + > ): 1185
[実施例6]
<シクロデキストリン誘導体の重合体の合成>
実施例1−1、2−1、3−1、4−1、5−1で得られたシクロデキストリン誘導体である白色粉末を、DMFかあるいはDMFと水の混合溶媒に溶解し、4つ口フラスコに入れて30分間窒素を吹込んだ。その後、50℃に加温し、DMFに溶解させておいたt−ブチルパーオキシネオデカノエイト(PB−ND、日油株式会社製)を加えることで、ラジカル重合反応を開始した。PB−NDはDMFで共洗いした。50℃で4時間ラジカル重合反応させた後、70℃に昇温してさらに3時間ラジカル重合反応させた。ラジカル重合反応終了後、室温まで冷却し、イオン交換水:エタノール(和光純薬工業社製)=25:75(v/v)の混合溶媒135.0gに再沈殿した。得られた沈殿物をろ過により回収し、減圧乾燥することでシクロデキストリン誘導体の重合体の白色粉末を得た。得られた重合体のGPC測定を行い、分子量を測定した。
[Example 6]
<Synthesis of cyclodextrin derivative polymer>
The white powder as the cyclodextrin derivative obtained in Examples 1-1, 2-1, 3-1, 4-1, 5-1 was dissolved in DMF or a mixed solvent of DMF and water, Nitrogen was bubbled through the flask for 30 minutes. Then, the radical polymerization reaction was started by heating to 50 ° C. and adding t-butyl peroxyneodecanoate (PB-ND, manufactured by NOF Corporation) dissolved in DMF. PB-ND was washed with DMF. After the radical polymerization reaction at 50 ° C. for 4 hours, the temperature was raised to 70 ° C. and the radical polymerization reaction was further performed for 3 hours. After completion of the radical polymerization reaction, the reaction solution was cooled to room temperature and reprecipitated in 135.0 g of a mixed solvent of ion-exchanged water: ethanol (Wako Pure Chemical Industries, Ltd.) = 25:75 (v / v). The obtained precipitate was collected by filtration and dried under reduced pressure to obtain a white powder of a polymer of cyclodextrin derivative. GPC measurement of the obtained polymer was performed, and molecular weight was measured.
[実施例6−1]
シクロデキストリン誘導体は、実施例1−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)とジ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)との混合物3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF11.29gである。PB−NDの量は、0.018gである。PB−ND溶解用のDMFは0.35gである。PB−NDの共洗い用のDMFは0.37gである。
[Example 6-1]
The cyclodextrin derivatives were mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 1-1 and di-6-deoxy-6- (2- 3.0 g of a mixture with methacryloyloxyethyl succinamide) -β-cyclodextrin). The solvent for dissolving the cyclodextrin derivative is 11.29 g of DMF. The amount of PB-ND is 0.018 g. The DMF for dissolving PB-ND is 0.35 g. The DMF for washing with PB-ND is 0.37 g.
[実施例6−2]
シクロデキストリン誘導体は、実施例1−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)とジ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)との混合物3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF25.57gである。PB−NDの量は、0.037gである。PB−ND溶解用のDMFは0.70gである。PB−NDの共洗い用のDMFは0.73gである。
[Example 6-2]
The cyclodextrin derivatives were mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 1-1 and di-6-deoxy-6- (2- 3.0 g of a mixture with methacryloyloxyethyl succinamide) -β-cyclodextrin). The solvent for dissolving the cyclodextrin derivative is 25.57 g of DMF. The amount of PB-ND is 0.037 g. The DMF for dissolving PB-ND is 0.70 g. The DMF for co-washing with PB-ND is 0.73 g.
[実施例6−3]
シクロデキストリン誘導体は、実施例1−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)とジ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)との混合物3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF8.29gと水3.0gとの混合溶媒である。PB−NDの量は、0.018gである。PB−ND溶解用のDMFは0.35gである。PB−NDの共洗い用のDMFは0.37gである。
[Example 6-3]
The cyclodextrin derivatives were mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 1-1 and di-6-deoxy-6- (2- 3.0 g of a mixture with methacryloyloxyethyl succinamide) -β-cyclodextrin). The solvent for dissolving the cyclodextrin derivative is a mixed solvent of 8.29 g of DMF and 3.0 g of water. The amount of PB-ND is 0.018 g. The DMF for dissolving PB-ND is 0.35 g. The DMF for washing with PB-ND is 0.37 g.
[実施例6−4]
シクロデキストリン誘導体は、実施例1−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)とジ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)との混合物3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF18.82gと水6.75gとの混合溶媒である。PB−NDの量は、0.037gである。PB−ND溶解用のDMFは0.70gである。PB−NDの共洗い用のDMFは0.73gである。
[Example 6-4]
The cyclodextrin derivatives were mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 1-1 and di-6-deoxy-6- (2- 3.0 g of a mixture with methacryloyloxyethyl succinamide) -β-cyclodextrin). The solvent for dissolving the cyclodextrin derivative is a mixed solvent of 18.82 g of DMF and 6.75 g of water. The amount of PB-ND is 0.037 g. The DMF for dissolving PB-ND is 0.70 g. The DMF for co-washing with PB-ND is 0.73 g.
[実施例6−5]
シクロデキストリン誘導体は、実施例2−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF11.29gである。PB−NDの量は、0.018gである。PB−ND溶解用のDMFは0.35gである。PB−NDの共洗い用のDMFは0.37gである。
[Example 6-5]
The cyclodextrin derivative is 3.0 g of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 2-1. The solvent for dissolving the cyclodextrin derivative is 11.29 g of DMF. The amount of PB-ND is 0.018 g. The DMF for dissolving PB-ND is 0.35 g. The DMF for washing with PB-ND is 0.37 g.
[実施例6−6]
シクロデキストリン誘導体は、実施例2−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF25.57gである。PB−NDの量は、0.037gである。PB−ND溶解用のDMFは0.70gである。PB−NDの共洗い用のDMFは0.73gである。
[Example 6-6]
The cyclodextrin derivative is 3.0 g of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 2-1. The solvent for dissolving the cyclodextrin derivative is 25.57 g of DMF. The amount of PB-ND is 0.037 g. The DMF for dissolving PB-ND is 0.70 g. The DMF for co-washing with PB-ND is 0.73 g.
[実施例6−7]
シクロデキストリン誘導体は、実施例2−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF8.29gと水3.0gとの混合溶媒である。PB−NDの量は、0.018gである。PB−ND溶解用のDMFは0.35gである。PB−NDの共洗い用のDMFは0.37gである。
[Example 6-7]
The cyclodextrin derivative is 3.0 g of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 2-1. The solvent for dissolving the cyclodextrin derivative is a mixed solvent of 8.29 g of DMF and 3.0 g of water. The amount of PB-ND is 0.018 g. The DMF for dissolving PB-ND is 0.35 g. The DMF for washing with PB-ND is 0.37 g.
[実施例6−8]
シクロデキストリン誘導体は、実施例2−1で得られたモノ−6−デオキシ−6−(2−メタクリロイルオキシエチルコハク酸アミド)−β−シクロデキストリン)3.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF18.82gと水6.75gとの混合溶媒である。PB−NDの量は、0.037gである。PB−ND溶解用のDMFは0.70gである。PB−NDの共洗い用のDMFは0.73gである。
[Example 6-8]
The cyclodextrin derivative is 3.0 g of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -β-cyclodextrin) obtained in Example 2-1. The solvent for dissolving the cyclodextrin derivative is a mixed solvent of 18.82 g of DMF and 6.75 g of water. The amount of PB-ND is 0.037 g. The DMF for dissolving PB-ND is 0.70 g. The DMF for co-washing with PB-ND is 0.73 g.
[実施例6−9]
シクロデキストリン誘導体は、実施例3−1で得られたモノ−6−デオキシ(2−メタクリロイルオキシエチルヘキサヒドロフタル酸アミド)−β−シクロデキストリン1.6gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF6.02gである。PB−NDの量は、0.010gである。PB−ND溶解用のDMFは0.19gである。PB−NDの共洗い用のDMFは0.20gである。
[Example 6-9]
The cyclodextrin derivative is 1.6 g of mono-6-deoxy (2-methacryloyloxyethylhexahydrophthalamide) -β-cyclodextrin obtained in Example 3-1. The solvent for dissolving the cyclodextrin derivative is 6.02 g DMF. The amount of PB-ND is 0.010 g. The DMF for dissolving PB-ND is 0.19 g. The DMF for washing with PB-ND is 0.20 g.
[実施例6−10]
シクロデキストリン誘導体は、実施例4−1で得られたモノ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリンと、ジ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリンと、トリ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリンとの混合物2.0gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF7.52gである。PB−NDの量は、0.012gである。PB−ND溶解用のDMFは0.23gである。PB−NDの共洗い用のDMFは0.24gである。
[Example 6-10]
The cyclodextrin derivatives were mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin obtained in Example 4-1, di-6-deoxy-6- (2 -Methacryloyloxyethyl succinamide) -α-cyclodextrin and tri-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin 2.0 g. The solvent for dissolving the cyclodextrin derivative is 7.52 g of DMF. The amount of PB-ND is 0.012 g. The DMF for dissolving PB-ND is 0.23 g. The DMF for washing with PB-ND is 0.24 g.
[実施例6−11]
シクロデキストリン誘導体は、実施例5−1で得られたモノ−6−デオキシ−6−(2−メタクリロイロキシエチルコハク酸アミド)−α−シクロデキストリン1.5gである。シクロデキストリン誘導体を溶解させる溶媒は、DMF5.64gである。PB−NDの量は、0.009gである。PB−ND溶解用のDMFは0.17gである。PB−NDの共洗い用のDMFは0.18gである。
[Example 6-11]
The cyclodextrin derivative is 1.5 g of mono-6-deoxy-6- (2-methacryloyloxyethyl succinamide) -α-cyclodextrin obtained in Example 5-1. The solvent for dissolving the cyclodextrin derivative is 5.64 g of DMF. The amount of PB-ND is 0.009 g. The DMF for dissolving PB-ND is 0.17 g. The DMF for co-washing with PB-ND is 0.18 g.
[比較例6−1]
<シクロデキストリン架橋ポリマーの合成>
β−CD2.0gをDMF13.5gに溶解させ、ジラウリン酸ジ−n−ブチルすずを1滴加えた。窒素置換を行い、4.5gのDMFに溶解させた2,4トリレンジイソシアネート(TDI)1.2gをゆっくり滴下し、70℃で24時間反応させた。反応終了後、反応液を100mLのクロロホルム中に投入して1晩攪拌させた。ろ過にて析出物を回収し、60℃で減圧することでシクロデキストリン架橋ポリマーを得た。
[Comparative Example 6-1]
<Synthesis of cyclodextrin cross-linked polymer>
2.0 g of β-CD was dissolved in 13.5 g of DMF, and 1 drop of di-n-butyltin dilaurate was added. After nitrogen substitution, 1.2 g of 2,4tolylene diisocyanate (TDI) dissolved in 4.5 g of DMF was slowly added dropwise and reacted at 70 ° C. for 24 hours. After completion of the reaction, the reaction solution was put into 100 mL of chloroform and stirred overnight. The precipitate was collected by filtration, and the cyclodextrin crosslinked polymer was obtained by reducing the pressure at 60 ° C.
[合成結果]
表1は、実施例6−1〜6−11に係るシクロデキストリン誘導体の重合体と比較例6−1のシクロデキストリン架橋ポリマーについて、使用したシクロデキストリン誘導体と使用した溶媒、シクロデキストリン誘導体の重合体とシクロデキストリン架橋ポリマーの数平均分子量Mnとを示している。
[Composition result]
Table 1 shows the cyclodextrin derivative polymer according to Examples 6-1 to 6-11 and the cyclodextrin crosslinked polymer of Comparative Example 6-1. And the number average molecular weight Mn of the cyclodextrin crosslinked polymer.
[実施例7]
<シクロデキストリン誘導体の重合体によるフェノールの除去実験>
シクロデキストリン誘導体の重合体の白色粉末と、フェノール(和光純薬工業社製)5.9mgと、イオン交換水とをサンプル管に秤量し、均一に混合溶解させた後、ウェイブローターで一晩攪拌した。一晩攪拌することで、フェノールを包接したシクロデキストリン誘導体の重合体の白色沈殿を得た。得られた白色沈殿を0.2μmセルロースアセテートフィルター(ADVANTEC社製)で除去し、得られたろ液のHPLC測定を行い、ろ液中のフェノールを算出した。
[Example 7]
<Phenol removal experiment using polymer of cyclodextrin derivative>
A white powder of a polymer of cyclodextrin derivative, 5.9 mg of phenol (manufactured by Wako Pure Chemical Industries, Ltd.), and ion-exchanged water are weighed in a sample tube, uniformly mixed and dissolved, and then stirred overnight with a waveblower. did. By stirring overnight, a white precipitate of a polymer of a cyclodextrin derivative encapsulating phenol was obtained. The obtained white precipitate was removed with a 0.2 μm cellulose acetate filter (manufactured by ADVANTEC), and the obtained filtrate was subjected to HPLC measurement to calculate phenol in the filtrate.
[実施例7−1]
シクロデキストリン誘導体の重合体は、実施例6−1で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-1]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-1. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−2]
シクロデキストリン誘導体の重合体は、実施例6−2で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-2]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-2. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−3]
シクロデキストリン誘導体の重合体は、実施例6−3で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-3]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-3. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−4]
シクロデキストリン誘導体の重合体は、実施例6−4で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-4]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-4. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−5]
シクロデキストリン誘導体の重合体は、実施例6−5で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-5]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-5. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−6]
シクロデキストリン誘導体の重合体は、実施例6−6で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-6]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-6. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−7]
シクロデキストリン誘導体の重合体は、実施例6−7で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-7]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-7. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−8]
シクロデキストリン誘導体の重合体は、実施例6−8で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 7-8]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-8. The amount of ion-exchanged water charged is 1.83 g.
[実施例7−9]
シクロデキストリン誘導体の重合体は、実施例6−9で得られた重合体0.18gである。イオン交換水の仕込み量は1.82gである。
[Example 7-9]
The polymer of the cyclodextrin derivative is 0.18 g of the polymer obtained in Example 6-9. The amount of ion-exchanged water charged is 1.82 g.
[実施例7−10]
シクロデキストリン誘導体の重合体は、実施例6−10で得られた重合体0.15gである。イオン交換水の仕込み量は1.85gである。
[Example 7-10]
The polymer of the cyclodextrin derivative is 0.15 g of the polymer obtained in Example 6-10. The amount of ion-exchanged water charged is 1.85 g.
[実施例7−11]
シクロデキストリン誘導体の重合体は、実施例6−11で得られた重合体0.15gである。イオン交換水の仕込み量は1.85gである。
[Example 7-11]
The polymer of the cyclodextrin derivative is 0.15 g of the polymer obtained in Example 6-11. The amount of ion-exchanged water charged is 1.85 g.
[比較例7−1]
シクロデキストリン誘導体の重合体の代わりに、比較例6−1で得られたシクロデキストリン架橋ポリマー0.23gを用いた。イオン交換水の仕込み量は1.77gである。
[Comparative Example 7-1]
Instead of the polymer of the cyclodextrin derivative, 0.23 g of the cyclodextrin crosslinked polymer obtained in Comparative Example 6-1 was used. The amount of ion-exchanged water charged is 1.77 g.
[比較例7−2]
シクロデキストリン誘導体の重合体の代わりに、式(7)で表されるβ−CD0.14gを用いた。イオン交換水の仕込み量は1.86gである。
[Comparative Example 7-2]
Instead of the cyclodextrin derivative polymer, β-CD 0.14 g represented by the formula (7) was used. The amount of ion-exchanged water charged is 1.86 g.
[実験結果]
表2は、実施例7−1〜7−11、比較例7−1、7−2に係るフェノール除去実験について、使用したシクロデキストリン誘導体の重合体と、フェノールおよび水の仕込み量と、攪拌後の溶液の様子、フェノールの除去率を示している。
[Experimental result]
Table 2 shows the polymer of the cyclodextrin derivative used for the phenol removal experiments according to Examples 7-1 to 7-11 and Comparative Examples 7-1 and 7-2, the amounts of phenol and water charged, and after stirring. This shows the state of the solution and the phenol removal rate.
[実施例8]
<シクロデキストリン誘導体の重合体によるクレゾールの除去実験>
シクロデキストリン誘導体の重合体の白色粉末と、クレゾール(和光純薬工業社製)6.8mgと、イオン交換水とをサンプル管に秤量し、均一に混合溶解させた後、ウェイブローターで一晩攪拌した。一晩攪拌することで、クレゾールを包接したシクロデキストリン誘導体の重合体の白色沈殿を得た。得られた白色沈殿を0.2μmセルロースアセテートフィルターで除去し、得られたろ液のHPLC測定を行い、ろ液中のクレゾールを算出した。
[Example 8]
<Removal experiment of cresol by polymer of cyclodextrin derivative>
A white powder of a polymer of a cyclodextrin derivative, 6.8 mg of cresol (manufactured by Wako Pure Chemical Industries, Ltd.) and ion-exchanged water are weighed into a sample tube, uniformly mixed and dissolved, and then stirred overnight with a waveblower. did. By stirring overnight, a white precipitate of a polymer of cyclodextrin derivatives enclosing cresol was obtained. The obtained white precipitate was removed with a 0.2 μm cellulose acetate filter, and the obtained filtrate was subjected to HPLC measurement to calculate cresol in the filtrate.
[実施例8−1]
シクロデキストリン誘導体の重合体は、実施例6−1で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-1]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-1. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−2]
シクロデキストリン誘導体の重合体は、実施例6−2で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-2]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-2. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−3]
シクロデキストリン誘導体の重合体は、実施例6−3で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-3]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-3. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−4]
シクロデキストリン誘導体の重合体は、実施例6−4で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-4]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-4. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−5]
シクロデキストリン誘導体の重合体は、実施例6−5で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-5]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-5. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−6]
シクロデキストリン誘導体の重合体は、実施例6−6で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-6]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-6. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−7]
シクロデキストリン誘導体の重合体は、実施例6−7で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-7]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-7. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−8]
シクロデキストリン誘導体の重合体は、実施例6−8で得られた重合体0.17gである。イオン交換水の仕込み量は1.83gである。
[Example 8-8]
The polymer of the cyclodextrin derivative is 0.17 g of the polymer obtained in Example 6-8. The amount of ion-exchanged water charged is 1.83 g.
[実施例8−9]
シクロデキストリン誘導体の重合体は、実施例6−9で得られた重合体0.18gである。イオン交換水の仕込み量は1.82gである。
[Example 8-9]
The polymer of the cyclodextrin derivative is 0.18 g of the polymer obtained in Example 6-9. The amount of ion-exchanged water charged is 1.82 g.
[実施例8−10]
シクロデキストリン誘導体の重合体は、実施例6−10で得られた重合体0.15gである。イオン交換水の仕込み量は1.85gである。
[Example 8-10]
The polymer of the cyclodextrin derivative is 0.15 g of the polymer obtained in Example 6-10. The amount of ion-exchanged water charged is 1.85 g.
[実施例8−11]
シクロデキストリン誘導体の重合体は、実施例6−11で得られた重合体0.15gである。イオン交換水の仕込み量は1.85gである。
[Example 8-11]
The polymer of the cyclodextrin derivative is 0.15 g of the polymer obtained in Example 6-11. The amount of ion-exchanged water charged is 1.85 g.
[比較例8−1]
シクロデキストリン誘導体の重合体の代わりに、比較例6−1で得られたシクロデキストリン架橋ポリマー0.23gを用いた。イオン交換水の仕込み量は1.77gである。
[Comparative Example 8-1]
Instead of the polymer of the cyclodextrin derivative, 0.23 g of the cyclodextrin crosslinked polymer obtained in Comparative Example 6-1 was used. The amount of ion-exchanged water charged is 1.77 g.
[比較例8−2]
シクロデキストリン誘導体の重合体の代わりに、式(7)で表されるβ−CD0.14gを用いた。イオン交換水の仕込み量は1.86gである。
[Comparative Example 8-2]
Instead of the cyclodextrin derivative polymer, β-CD 0.14 g represented by the formula (7) was used. The amount of ion-exchanged water charged is 1.86 g.
[実験結果]
表3は、実施例8−1〜8−11、比較例8−1、8−2に係るクレゾール除去実験について、使用したシクロデキストリン誘導体の重合体と、クレゾールおよび水の仕込み量と、攪拌後の溶液の様子、クレゾールの除去率を示している。
[Experimental result]
Table 3 shows the polymer of the cyclodextrin derivative used for the experiments for removing cresol according to Examples 8-1 to 8-11 and Comparative Examples 8-1 and 8-2, the charged amounts of cresol and water, and after stirring. The state of the solution and the removal rate of cresol are shown.
表2および表3より、実施例7−1〜7−11、8−1〜8〜11において、実施例6−1〜6−11に係るシクロデキストリン誘導体の重合体を用いることにより、水溶液中のフェノールあるいはクレゾール(以下、フェノール化合物と呼称する。)を分子内あるいは分子間に取り込んで包接化合物が形成されており、攪拌後に沈殿物による白濁が認められた。また、HPLC測定から、フェノールの除去率は67%〜98%、クレゾールの除去率は61%〜97%となり、水溶液中のフェノール化合物を効率良く除去できていることがわかった。さらに、シクロデキストリン誘導体の重合体の数平均分子量Mnが大きいほうが、フェノール化合物の包接能が高く、除去率も特に高いことがわかった。一方、比較例である7−1、8−1はTDIにより高密度に架橋されたポリマーであり、フェノール化合物を包接しにくく、除去率は54%〜56%であり、比較例7−2、8−2では、重合体ではないβ−CDを用いているため包接化合物を形成しても沈殿物を生じず、フェノール化合物の除去率は0%であった。 From Tables 2 and 3, in Examples 7-1 to 7-11 and 8-1 to 8-11, by using the polymer of cyclodextrin derivatives according to Examples 6-1 to 6-11, An inclusion compound was formed by incorporating phenol or cresol (hereinafter referred to as a phenol compound) in the molecule or between the molecules, and white turbidity due to precipitates was observed after stirring. From the HPLC measurement, the phenol removal rate was 67% to 98%, and the cresol removal rate was 61% to 97%, indicating that the phenolic compound in the aqueous solution was efficiently removed. Furthermore, it was found that the higher the number average molecular weight Mn of the polymer of cyclodextrin derivatives, the higher the inclusion ability of the phenol compound and the higher the removal rate. On the other hand, Comparative Examples 7-1 and 8-1 are polymers cross-linked with high density by TDI, are difficult to include phenol compounds, and the removal rate is 54% to 56%. Comparative Example 7-2, In 8-2, since β-CD, which is not a polymer, was used, no precipitate was formed even when the inclusion compound was formed, and the phenol compound removal rate was 0%.
以上、本発明の実施形態について説明したが、本発明は上述の実施形態にのみ限定されるものではなく、本発明の要旨を逸脱しない範囲内において種々変更を加え得ることは勿論である。 The embodiment of the present invention has been described above, but the present invention is not limited to the above-described embodiment, and it is needless to say that various modifications can be made without departing from the gist of the present invention.
Claims (5)
(式(1)中、xは1〜3の整数、kは6〜8の整数、R1は水素原子もしくはメチル基、R2は炭素原子数1〜4の炭化水素基、R3は炭素原子数1〜6の炭化水素基を示す。) A cyclodextrin derivative represented by the formula (1).
(In formula (1), x is an integer of 1 to 3, k is an integer of 6 to 8, R 1 is a hydrogen atom or a methyl group, R 2 is a hydrocarbon group having 1 to 4 carbon atoms, and R 3 is carbon. (A hydrocarbon group having 1 to 6 atoms is shown.)
式(2)で表されるシクロデキストリン誘導体。
(式(2)中、yは5〜7の整数、R1は水素原子もしくはメチル基、R2は炭素原子数1〜4の炭化水素基、R3は炭素原子数1〜6の炭化水素基を示す。) The cyclodextrin derivative according to claim 1,
A cyclodextrin derivative represented by the formula (2).
(In the formula (2), y is an integer of 5 to 7, R 1 is a hydrogen atom or a methyl group, R 2 is a hydrocarbon group having 1 to 4 carbon atoms, and R 3 is a hydrocarbon having 1 to 6 carbon atoms. Group.)
シクロデキストリン誘導体の製造方法。
(式(3)中、xは1〜3の整数、kは6〜8の整数を示す。)
(式(4)中、R1は水素原子もしくはメチル基、R2は炭素原子数1〜4の炭化水素基、R3は炭素原子数1〜6の炭化水素基を示す。) The manufacturing method of a cyclodextrin derivative which produces | generates the cyclodextrin derivative of Claim 1 or 2 by amidating the compound represented by Formula (3) and the compound represented by Formula (4).
(In formula (3), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
(In Formula (4), R 1 represents a hydrogen atom or a methyl group, R 2 represents a hydrocarbon group having 1 to 4 carbon atoms, and R 3 represents a hydrocarbon group having 1 to 6 carbon atoms.)
式(5)で表される化合物にベンジルアミンを用いてベンジルアミノ化することにより式(6)で表される化合物を生成する(A)工程と、
前記(A)工程で得られた前記式(6)で表される化合物を還元することにより前記式(3)で表される化合物を生成する(B)工程と
を含むシクロデキストリン誘導体の製造方法。
(式(5)中、xは1〜3の整数、kは6〜8の整数を示す。)
(式(6)中、xは1〜3の整数、kは6〜8の整数を示す。) A method for producing a cyclodextrin derivative according to claim 3,
(A) a step of producing a compound represented by the formula (6) by benzylamination of the compound represented by the formula (5) using benzylamine;
And (B) step of producing a compound represented by the formula (3) by reducing the compound represented by the formula (6) obtained in the step (A). .
(In formula (5), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
(In formula (6), x represents an integer of 1 to 3, and k represents an integer of 6 to 8.)
シクロデキストリン誘導体の重合体。 A polymer of a cyclodextrin derivative obtained by polymerizing the cyclodextrin derivative according to claim 1 or 2.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014199300 | 2014-09-29 | ||
| JP2014199300 | 2014-09-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016069652A true JP2016069652A (en) | 2016-05-09 |
| JP6561728B2 JP6561728B2 (en) | 2019-08-21 |
Family
ID=55866181
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015191020A Active JP6561728B2 (en) | 2014-09-29 | 2015-09-29 | Cyclodextrin derivative and process for producing the same, and polymer of cyclodextrin derivative |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6561728B2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020037673A (en) * | 2018-08-30 | 2020-03-12 | 日本食品化工株式会社 | Cyclodextrin derivative and method for producing the same |
| WO2020080194A1 (en) | 2018-10-15 | 2020-04-23 | 国立大学法人大阪大学 | Cyclic oligosaccharide and method for producing same |
| JP2021181573A (en) * | 2017-03-02 | 2021-11-25 | 国立大学法人大阪大学 | Host-group-containing polymerizable monomer, polymer material, method for producing the same, and clathrate compound and method for producing the same |
| CN116178590A (en) * | 2022-12-09 | 2023-05-30 | 杭州楠大环保科技有限公司 | Microbial nutrition synergist suitable for wastewater treatment |
| CN116284504A (en) * | 2023-01-04 | 2023-06-23 | 昆明理工大学 | A series of cross-linked cyclodextrin polymers and their preparation methods and applications |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007046041A (en) * | 2005-07-13 | 2007-02-22 | Meiwa Kasei Kk | Cyclodextrin compound containing photocrosslinking group, method for producing the same and adsorbent containing the same |
| JP2012229359A (en) * | 2011-04-27 | 2012-11-22 | Neos Co Ltd | Method for producing cyclodextrin polymer and method for selectively adsorbing and removing halogenated aromatic compound contained in medium by using the same |
-
2015
- 2015-09-29 JP JP2015191020A patent/JP6561728B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007046041A (en) * | 2005-07-13 | 2007-02-22 | Meiwa Kasei Kk | Cyclodextrin compound containing photocrosslinking group, method for producing the same and adsorbent containing the same |
| JP2012229359A (en) * | 2011-04-27 | 2012-11-22 | Neos Co Ltd | Method for producing cyclodextrin polymer and method for selectively adsorbing and removing halogenated aromatic compound contained in medium by using the same |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL COMMUNICATION, vol. 48, JPN6019023155, 2012, pages 892 - 894, ISSN: 0004058889 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021181573A (en) * | 2017-03-02 | 2021-11-25 | 国立大学法人大阪大学 | Host-group-containing polymerizable monomer, polymer material, method for producing the same, and clathrate compound and method for producing the same |
| US12054571B2 (en) | 2017-03-02 | 2024-08-06 | Osaka University | Host-group-containing polymerizable monomer, polymer material, method for producing same, and clathrate compound and method for producing same |
| JP2020037673A (en) * | 2018-08-30 | 2020-03-12 | 日本食品化工株式会社 | Cyclodextrin derivative and method for producing the same |
| JP7461717B2 (en) | 2018-08-30 | 2024-04-04 | 日本食品化工株式会社 | Cyclodextrin derivatives and their production method |
| WO2020080194A1 (en) | 2018-10-15 | 2020-04-23 | 国立大学法人大阪大学 | Cyclic oligosaccharide and method for producing same |
| KR20210075975A (en) | 2018-10-15 | 2021-06-23 | 고꾸리쯔 다이가꾸 호우징 오사까 다이가꾸 | Cyclic oligosaccharide and manufacturing method thereof |
| US11492418B2 (en) | 2018-10-15 | 2022-11-08 | Osaka University | Cyclic oligosaccharide and method for producing same |
| CN116178590A (en) * | 2022-12-09 | 2023-05-30 | 杭州楠大环保科技有限公司 | Microbial nutrition synergist suitable for wastewater treatment |
| CN116178590B (en) * | 2022-12-09 | 2024-03-29 | 杭州楠大环保科技有限公司 | A microbial nutrient synergist suitable for wastewater treatment |
| CN116284504A (en) * | 2023-01-04 | 2023-06-23 | 昆明理工大学 | A series of cross-linked cyclodextrin polymers and their preparation methods and applications |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6561728B2 (en) | 2019-08-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6561728B2 (en) | Cyclodextrin derivative and process for producing the same, and polymer of cyclodextrin derivative | |
| JP6624660B1 (en) | Polymer material and method for producing the same | |
| US11535691B2 (en) | Macromolecular material, method for producing same, and polymerizable monomer composition | |
| KR20220115564A (en) | Novel sulfobetaine monomers, methods for their preparation, and uses thereof | |
| Thakur | Cellulose-based graft copolymers: structure and chemistry | |
| KR102378719B1 (en) | Powdery water-soluble cationic polymer composition | |
| Gebru et al. | Humic acid removal using cellulose acetate membranes grafted with poly (methyl methacrylate) and aminated using tetraethylenepentamine | |
| WO2015079788A1 (en) | Method for preparing metal nanoparticles and metal nanoparticle | |
| JP2018034111A (en) | Metal adsorbent | |
| WO2001096424A1 (en) | Process for producing high-purity vinylpyrrolidone polymer | |
| JPWO2018003821A1 (en) | Copolymer and its use | |
| Tizzotti et al. | Synthesis of thermosensitive guar‐based hydrogels with tunable physico‐chemical properties by click chemistry | |
| JPH03203916A (en) | Copolymer of 3-methacryloyloxy-2-hydroxypropyltrimethyl- ammonium chloride monomer and vinyl monomer | |
| JP6894747B2 (en) | Polymer | |
| KR20240161641A (en) | Method for producing polyethylene glycol derivatives | |
| Kumar et al. | The Graft Copolymerization of N-(Hydroxymethyl) Acrylamide onto Xanthan Gum and Its Anti-Oxidative Property | |
| EP4206243B1 (en) | Packing material and column for size exclusion chromatography | |
| JP7523768B2 (en) | Polymer production method and polymerization catalyst | |
| JP4743476B2 (en) | Acryloylmorpholine-sulfur dioxide copolymer and process for producing the same | |
| Birnbaum et al. | Synthesis of α-biotinyl poly (ethylene glycol-bN-isopropylacrylamide) block copolymers with different fluorescent dyes at the ω-side | |
| JP4534012B2 (en) | Polyethylene glycol-aminoalkylcarbamoylcyclodextrin copolymer, process for producing the same, and inclusion complex compound using the same | |
| JP2024042233A (en) | Copolymers and deodorizing compositions | |
| Marambio et al. | Swelling behavior and metal ion retention from aqueous solution of hydrogels based on N‐1‐vinyl‐2‐pyrrolidone and N‐hydroxymethylacrylamide | |
| JP2012245500A (en) | Water treatment agent | |
| Jayachandran et al. | Synthesis and Characterization of Polymer Brushes of Poly (N, N-dimethylacrylamide) from Polystyrene Latex by Aqueous Atom Transfer Radical Polymerization. Volume 35, Number 11, May, 21, 2002, pp 4247− 4257. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180723 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190625 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190708 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6561728 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |