JP2008516996A - Antiviral oligonucleotide - Google Patents
Antiviral oligonucleotide Download PDFInfo
- Publication number
- JP2008516996A JP2008516996A JP2007537092A JP2007537092A JP2008516996A JP 2008516996 A JP2008516996 A JP 2008516996A JP 2007537092 A JP2007537092 A JP 2007537092A JP 2007537092 A JP2007537092 A JP 2007537092A JP 2008516996 A JP2008516996 A JP 2008516996A
- Authority
- JP
- Japan
- Prior art keywords
- oligonucleotide
- virus
- antiviral
- rep
- oligonucleotides
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 108091034117 Oligonucleotide Proteins 0.000 title claims abstract description 736
- 230000000840 anti-viral effect Effects 0.000 title claims abstract description 222
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims abstract description 345
- 125000003729 nucleotide group Chemical group 0.000 claims abstract description 206
- 239000002773 nucleotide Substances 0.000 claims abstract description 197
- 241000282414 Homo sapiens Species 0.000 claims abstract description 78
- 238000011282 treatment Methods 0.000 claims abstract description 58
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 claims abstract description 38
- 230000009385 viral infection Effects 0.000 claims abstract description 33
- 208000036142 Viral infection Diseases 0.000 claims abstract description 32
- 230000002265 prevention Effects 0.000 claims abstract description 30
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 20
- 241000282412 Homo Species 0.000 claims abstract description 17
- 239000000203 mixture Substances 0.000 claims description 218
- 241000700605 Viruses Species 0.000 claims description 185
- 230000000694 effects Effects 0.000 claims description 113
- 230000003993 interaction Effects 0.000 claims description 70
- 239000008194 pharmaceutical composition Substances 0.000 claims description 66
- 239000003814 drug Substances 0.000 claims description 61
- 241000700588 Human alphaherpesvirus 1 Species 0.000 claims description 51
- 230000000295 complement effect Effects 0.000 claims description 45
- 108010067390 Viral Proteins Proteins 0.000 claims description 34
- 208000015181 infectious disease Diseases 0.000 claims description 32
- 230000009471 action Effects 0.000 claims description 29
- 239000003795 chemical substances by application Substances 0.000 claims description 27
- 239000003937 drug carrier Substances 0.000 claims description 27
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical group NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 24
- 210000002966 serum Anatomy 0.000 claims description 20
- 241000701074 Human alphaherpesvirus 2 Species 0.000 claims description 19
- 230000002378 acidificating effect Effects 0.000 claims description 18
- 241000713772 Human immunodeficiency virus 1 Species 0.000 claims description 17
- 238000001727 in vivo Methods 0.000 claims description 16
- 238000006467 substitution reaction Methods 0.000 claims description 15
- 241000701044 Human gammaherpesvirus 4 Species 0.000 claims description 14
- 238000004519 manufacturing process Methods 0.000 claims description 14
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 14
- 238000009826 distribution Methods 0.000 claims description 13
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 13
- 201000011510 cancer Diseases 0.000 claims description 11
- 231100000419 toxicity Toxicity 0.000 claims description 10
- 230000001988 toxicity Effects 0.000 claims description 10
- 241000700586 Herpesviridae Species 0.000 claims description 9
- 241001493065 dsRNA viruses Species 0.000 claims description 9
- 230000008685 targeting Effects 0.000 claims description 9
- 239000000843 powder Substances 0.000 claims description 8
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical group CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 claims description 8
- 241000711573 Coronaviridae Species 0.000 claims description 7
- 241000711549 Hepacivirus C Species 0.000 claims description 7
- 241000701085 Human alphaherpesvirus 3 Species 0.000 claims description 7
- 208000002606 Paramyxoviridae Infections Diseases 0.000 claims description 7
- 230000004700 cellular uptake Effects 0.000 claims description 7
- 238000011200 topical administration Methods 0.000 claims description 7
- 241000712461 unidentified influenza virus Species 0.000 claims description 7
- 208000031886 HIV Infections Diseases 0.000 claims description 6
- 241001631646 Papillomaviridae Species 0.000 claims description 6
- 238000011321 prophylaxis Methods 0.000 claims description 6
- 241000701242 Adenoviridae Species 0.000 claims description 5
- 241000712892 Arenaviridae Species 0.000 claims description 5
- 241000709687 Coxsackievirus Species 0.000 claims description 5
- 241000711950 Filoviridae Species 0.000 claims description 5
- 241000700739 Hepadnaviridae Species 0.000 claims description 5
- 241000713340 Human immunodeficiency virus 2 Species 0.000 claims description 5
- 241000150452 Orthohantavirus Species 0.000 claims description 5
- 241000712464 Orthomyxoviridae Species 0.000 claims description 5
- 241000711504 Paramyxoviridae Species 0.000 claims description 5
- 241000701945 Parvoviridae Species 0.000 claims description 5
- 241000150350 Peribunyaviridae Species 0.000 claims description 5
- 241000709664 Picornaviridae Species 0.000 claims description 5
- 241000700625 Poxviridae Species 0.000 claims description 5
- 241000702247 Reoviridae Species 0.000 claims description 5
- 241000712907 Retroviridae Species 0.000 claims description 5
- 241000710924 Togaviridae Species 0.000 claims description 5
- 108700010877 adenoviridae proteins Proteins 0.000 claims description 5
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 201000003075 Crimean-Congo hemorrhagic fever Diseases 0.000 claims description 4
- 241000725619 Dengue virus Species 0.000 claims description 4
- 241001115402 Ebolavirus Species 0.000 claims description 4
- 241000709661 Enterovirus Species 0.000 claims description 4
- 241000710781 Flaviviridae Species 0.000 claims description 4
- 206010023927 Lassa fever Diseases 0.000 claims description 4
- 241001115401 Marburgvirus Species 0.000 claims description 4
- 241000711931 Rhabdoviridae Species 0.000 claims description 4
- 241000710886 West Nile virus Species 0.000 claims description 4
- 241000710951 Western equine encephalitis virus Species 0.000 claims description 4
- 241001455657 Human betaherpesvirus 6A Species 0.000 claims description 3
- 241001455656 Human betaherpesvirus 6B Species 0.000 claims description 3
- 241000342334 Human metapneumovirus Species 0.000 claims description 3
- 241000713124 Rift Valley fever virus Species 0.000 claims description 3
- 241000710772 Yellow fever virus Species 0.000 claims description 3
- 229920001519 homopolymer Polymers 0.000 claims description 3
- 229940051021 yellow-fever virus Drugs 0.000 claims description 3
- 229910052770 Uranium Inorganic materials 0.000 claims description 2
- 241000724205 Rice stripe tenuivirus Species 0.000 claims 2
- 230000003449 preventive effect Effects 0.000 claims 1
- 238000012986 modification Methods 0.000 abstract description 97
- 230000004048 modification Effects 0.000 abstract description 74
- 241001465754 Metazoa Species 0.000 abstract description 35
- 239000003443 antiviral agent Substances 0.000 abstract description 23
- 238000007385 chemical modification Methods 0.000 abstract description 2
- 238000009472 formulation Methods 0.000 description 80
- 238000000034 method Methods 0.000 description 63
- 230000003612 virological effect Effects 0.000 description 62
- 239000002502 liposome Substances 0.000 description 61
- 238000012360 testing method Methods 0.000 description 55
- -1 i.e. Polymers 0.000 description 54
- 150000001875 compounds Chemical class 0.000 description 49
- 230000027455 binding Effects 0.000 description 46
- 229940079593 drug Drugs 0.000 description 44
- 230000001965 increasing effect Effects 0.000 description 44
- 229920000642 polymer Polymers 0.000 description 44
- 239000004094 surface-active agent Substances 0.000 description 44
- 210000004027 cell Anatomy 0.000 description 42
- 230000000692 anti-sense effect Effects 0.000 description 41
- 150000007523 nucleic acids Chemical class 0.000 description 39
- 108090000623 proteins and genes Proteins 0.000 description 39
- 239000000839 emulsion Substances 0.000 description 38
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 38
- 102000039446 nucleic acids Human genes 0.000 description 35
- 108020004707 nucleic acids Proteins 0.000 description 35
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 33
- 150000003839 salts Chemical class 0.000 description 32
- 241000725303 Human immunodeficiency virus Species 0.000 description 31
- 239000006166 lysate Substances 0.000 description 31
- 102000004169 proteins and genes Human genes 0.000 description 30
- 108020004414 DNA Proteins 0.000 description 29
- 102000053602 DNA Human genes 0.000 description 29
- 235000018102 proteins Nutrition 0.000 description 29
- 229920001223 polyethylene glycol Polymers 0.000 description 27
- 238000002360 preparation method Methods 0.000 description 27
- 150000002632 lipids Chemical class 0.000 description 26
- 210000000605 viral structure Anatomy 0.000 description 25
- 239000002202 Polyethylene glycol Substances 0.000 description 24
- 239000002552 dosage form Substances 0.000 description 23
- 235000000346 sugar Nutrition 0.000 description 23
- 230000005764 inhibitory process Effects 0.000 description 22
- 229920002477 rna polymer Polymers 0.000 description 22
- 230000015572 biosynthetic process Effects 0.000 description 21
- 201000010099 disease Diseases 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 21
- 239000004530 micro-emulsion Substances 0.000 description 21
- 239000000126 substance Substances 0.000 description 21
- 102100034343 Integrase Human genes 0.000 description 20
- 239000002253 acid Substances 0.000 description 20
- 239000000074 antisense oligonucleotide Substances 0.000 description 20
- 238000012230 antisense oligonucleotides Methods 0.000 description 20
- 230000007246 mechanism Effects 0.000 description 20
- 239000000243 solution Substances 0.000 description 20
- 238000003556 assay Methods 0.000 description 19
- 239000007924 injection Substances 0.000 description 18
- 238000002347 injection Methods 0.000 description 18
- 230000035515 penetration Effects 0.000 description 18
- 238000013459 approach Methods 0.000 description 17
- 238000012552 review Methods 0.000 description 17
- 108091028664 Ribonucleotide Proteins 0.000 description 16
- 241000700584 Simplexvirus Species 0.000 description 16
- 230000001419 dependent effect Effects 0.000 description 16
- 235000014113 dietary fatty acids Nutrition 0.000 description 16
- 239000003623 enhancer Substances 0.000 description 16
- 229930195729 fatty acid Natural products 0.000 description 16
- 239000000194 fatty acid Substances 0.000 description 16
- 239000002336 ribonucleotide Substances 0.000 description 16
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 15
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 15
- NAGJZTKCGNOGPW-UHFFFAOYSA-K dioxido-sulfanylidene-sulfido-$l^{5}-phosphane Chemical compound [O-]P([O-])([S-])=S NAGJZTKCGNOGPW-UHFFFAOYSA-K 0.000 description 15
- 230000014509 gene expression Effects 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- 239000003921 oil Substances 0.000 description 14
- 239000012071 phase Substances 0.000 description 14
- 238000003786 synthesis reaction Methods 0.000 description 14
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 13
- 238000010521 absorption reaction Methods 0.000 description 13
- 239000002609 medium Substances 0.000 description 13
- 108020004999 messenger RNA Proteins 0.000 description 13
- 230000010412 perfusion Effects 0.000 description 13
- 238000002962 plaque-reduction assay Methods 0.000 description 13
- 210000003491 skin Anatomy 0.000 description 13
- 210000002845 virion Anatomy 0.000 description 13
- 241000701022 Cytomegalovirus Species 0.000 description 12
- 101710203526 Integrase Proteins 0.000 description 12
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 12
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 12
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 12
- 150000004665 fatty acids Chemical class 0.000 description 12
- 230000002401 inhibitory effect Effects 0.000 description 12
- 150000004713 phosphodiesters Chemical class 0.000 description 12
- 230000010076 replication Effects 0.000 description 12
- 125000002652 ribonucleotide group Chemical group 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 239000003833 bile salt Substances 0.000 description 11
- 238000002875 fluorescence polarization Methods 0.000 description 11
- 235000019198 oils Nutrition 0.000 description 11
- 229940126585 therapeutic drug Drugs 0.000 description 11
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 10
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 10
- 229960004150 aciclovir Drugs 0.000 description 10
- 125000000217 alkyl group Chemical group 0.000 description 10
- 230000008901 benefit Effects 0.000 description 10
- 210000000234 capsid Anatomy 0.000 description 10
- 239000002738 chelating agent Substances 0.000 description 10
- 229940104302 cytosine Drugs 0.000 description 10
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 10
- 239000003995 emulsifying agent Substances 0.000 description 10
- 150000002148 esters Chemical class 0.000 description 10
- 239000000499 gel Substances 0.000 description 10
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 10
- 230000002458 infectious effect Effects 0.000 description 10
- 238000001802 infusion Methods 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- RMLUKZWYIKEASN-UHFFFAOYSA-M sodium;2-amino-9-(2-hydroxyethoxymethyl)purin-6-olate Chemical compound [Na+].O=C1[N-]C(N)=NC2=C1N=CN2COCCO RMLUKZWYIKEASN-UHFFFAOYSA-M 0.000 description 10
- 210000001519 tissue Anatomy 0.000 description 10
- 102000004506 Blood Proteins Human genes 0.000 description 9
- 108010017384 Blood Proteins Proteins 0.000 description 9
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical class OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 9
- 239000004793 Polystyrene Substances 0.000 description 9
- 230000036436 anti-hiv Effects 0.000 description 9
- 230000015556 catabolic process Effects 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 238000006731 degradation reaction Methods 0.000 description 9
- 206010022000 influenza Diseases 0.000 description 9
- 230000003834 intracellular effect Effects 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- 238000004806 packaging method and process Methods 0.000 description 9
- 239000002245 particle Substances 0.000 description 9
- 241001529453 unidentified herpesvirus Species 0.000 description 9
- KWVJHCQQUFDPLU-YEUCEMRASA-N 2,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC KWVJHCQQUFDPLU-YEUCEMRASA-N 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 241000700721 Hepatitis B virus Species 0.000 description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 8
- 101710163270 Nuclease Proteins 0.000 description 8
- 208000024777 Prion disease Diseases 0.000 description 8
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 8
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 8
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 8
- 230000001413 cellular effect Effects 0.000 description 8
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 8
- 239000004615 ingredient Substances 0.000 description 8
- 125000005647 linker group Chemical group 0.000 description 8
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 8
- 239000002777 nucleoside Substances 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 229910052708 sodium Inorganic materials 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- RUDATBOHQWOJDD-UHFFFAOYSA-N (3beta,5beta,7alpha)-3,7-Dihydroxycholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)CC2 RUDATBOHQWOJDD-UHFFFAOYSA-N 0.000 description 7
- 208000030507 AIDS Diseases 0.000 description 7
- 108091023037 Aptamer Proteins 0.000 description 7
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 7
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 7
- 108020004459 Small interfering RNA Proteins 0.000 description 7
- 230000003321 amplification Effects 0.000 description 7
- 239000002246 antineoplastic agent Substances 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 125000002091 cationic group Chemical group 0.000 description 7
- 235000012000 cholesterol Nutrition 0.000 description 7
- 239000012091 fetal bovine serum Substances 0.000 description 7
- 230000002209 hydrophobic effect Effects 0.000 description 7
- 239000002736 nonionic surfactant Substances 0.000 description 7
- 238000003199 nucleic acid amplification method Methods 0.000 description 7
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- 150000003230 pyrimidines Chemical class 0.000 description 7
- 229960000329 ribavirin Drugs 0.000 description 7
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 6
- 241000283690 Bos taurus Species 0.000 description 6
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 6
- 108010050904 Interferons Proteins 0.000 description 6
- 102000014150 Interferons Human genes 0.000 description 6
- 239000005639 Lauric acid Substances 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- 241001494479 Pecora Species 0.000 description 6
- 206010061603 Respiratory syncytial virus infection Diseases 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 229920002472 Starch Polymers 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 6
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 6
- 239000003963 antioxidant agent Substances 0.000 description 6
- 235000006708 antioxidants Nutrition 0.000 description 6
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 6
- 235000010980 cellulose Nutrition 0.000 description 6
- 229920002678 cellulose Polymers 0.000 description 6
- 239000001913 cellulose Substances 0.000 description 6
- RUDATBOHQWOJDD-BSWAIDMHSA-N chenodeoxycholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)CC1 RUDATBOHQWOJDD-BSWAIDMHSA-N 0.000 description 6
- 229960001091 chenodeoxycholic acid Drugs 0.000 description 6
- 238000000576 coating method Methods 0.000 description 6
- 239000006071 cream Substances 0.000 description 6
- 229940127089 cytotoxic agent Drugs 0.000 description 6
- 239000012634 fragment Substances 0.000 description 6
- 229960002963 ganciclovir Drugs 0.000 description 6
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 6
- 238000009396 hybridization Methods 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 238000012417 linear regression Methods 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 6
- 150000003904 phospholipids Chemical class 0.000 description 6
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- JJICLMJFIKGAAU-UHFFFAOYSA-M sodium;2-amino-9-(1,3-dihydroxypropan-2-yloxymethyl)purin-6-olate Chemical compound [Na+].NC1=NC([O-])=C2N=CN(COC(CO)CO)C2=N1 JJICLMJFIKGAAU-UHFFFAOYSA-M 0.000 description 6
- 239000008107 starch Substances 0.000 description 6
- 229940032147 starch Drugs 0.000 description 6
- 235000019698 starch Nutrition 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 229940104230 thymidine Drugs 0.000 description 6
- 210000003501 vero cell Anatomy 0.000 description 6
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 5
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 5
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 5
- 241000282472 Canis lupus familiaris Species 0.000 description 5
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 5
- 239000004380 Cholic acid Substances 0.000 description 5
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 5
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 5
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 5
- 241000450599 DNA viruses Species 0.000 description 5
- 241000196324 Embryophyta Species 0.000 description 5
- 241000283073 Equus caballus Species 0.000 description 5
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- 235000021314 Palmitic acid Nutrition 0.000 description 5
- 229920002125 Sokalan® Polymers 0.000 description 5
- 235000021355 Stearic acid Nutrition 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 230000009286 beneficial effect Effects 0.000 description 5
- 239000003613 bile acid Substances 0.000 description 5
- 210000000170 cell membrane Anatomy 0.000 description 5
- 235000019416 cholic acid Nutrition 0.000 description 5
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 5
- 229960002471 cholic acid Drugs 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 229960003724 dimyristoylphosphatidylcholine Drugs 0.000 description 5
- 238000012377 drug delivery Methods 0.000 description 5
- 238000005538 encapsulation Methods 0.000 description 5
- 229960002949 fluorouracil Drugs 0.000 description 5
- 230000004927 fusion Effects 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 229940029575 guanosine Drugs 0.000 description 5
- 229940079322 interferon Drugs 0.000 description 5
- 210000004877 mucosa Anatomy 0.000 description 5
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 5
- 239000002105 nanoparticle Substances 0.000 description 5
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 5
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 5
- 229940098695 palmitic acid Drugs 0.000 description 5
- 239000003961 penetration enhancing agent Substances 0.000 description 5
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 5
- 239000002719 pyrimidine nucleotide Substances 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 239000003381 stabilizer Substances 0.000 description 5
- 239000008117 stearic acid Substances 0.000 description 5
- 238000007920 subcutaneous administration Methods 0.000 description 5
- 239000000829 suppository Substances 0.000 description 5
- 230000000699 topical effect Effects 0.000 description 5
- 230000009447 viral pathogenesis Effects 0.000 description 5
- 239000011782 vitamin Substances 0.000 description 5
- 235000013343 vitamin Nutrition 0.000 description 5
- 229940088594 vitamin Drugs 0.000 description 5
- 229930003231 vitamin Natural products 0.000 description 5
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 description 4
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 4
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 4
- CITHEXJVPOWHKC-UUWRZZSWSA-N 1,2-di-O-myristoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCC CITHEXJVPOWHKC-UUWRZZSWSA-N 0.000 description 4
- AXTGDCSMTYGJND-UHFFFAOYSA-N 1-dodecylazepan-2-one Chemical compound CCCCCCCCCCCCN1CCCCCC1=O AXTGDCSMTYGJND-UHFFFAOYSA-N 0.000 description 4
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 4
- PEHVGBZKEYRQSX-UHFFFAOYSA-N 7-deaza-adenine Chemical compound NC1=NC=NC2=C1C=CN2 PEHVGBZKEYRQSX-UHFFFAOYSA-N 0.000 description 4
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 4
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 4
- 229930024421 Adenine Natural products 0.000 description 4
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical class OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 229920002307 Dextran Polymers 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 230000005526 G1 to G0 transition Effects 0.000 description 4
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 4
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 4
- 239000005642 Oleic acid Substances 0.000 description 4
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 4
- 108091030071 RNAI Proteins 0.000 description 4
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 4
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 239000012190 activator Substances 0.000 description 4
- 229960000643 adenine Drugs 0.000 description 4
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 4
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 4
- 125000000129 anionic group Chemical group 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 229940114079 arachidonic acid Drugs 0.000 description 4
- 235000021342 arachidonic acid Nutrition 0.000 description 4
- 239000011324 bead Substances 0.000 description 4
- 229940093761 bile salts Drugs 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 230000035587 bioadhesion Effects 0.000 description 4
- 239000000227 bioadhesive Substances 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 4
- 229960003964 deoxycholic acid Drugs 0.000 description 4
- 239000005547 deoxyribonucleotide Substances 0.000 description 4
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 4
- 229960002086 dextran Drugs 0.000 description 4
- 230000029087 digestion Effects 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 4
- 230000009368 gene silencing by RNA Effects 0.000 description 4
- RZRNAYUHWVFMIP-HXUWFJFHSA-N glycerol monolinoleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-HXUWFJFHSA-N 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 208000006454 hepatitis Diseases 0.000 description 4
- 231100000283 hepatitis Toxicity 0.000 description 4
- KUQWZSZYIQGTHT-UHFFFAOYSA-N hexa-1,5-diene-3,4-diol Chemical compound C=CC(O)C(O)C=C KUQWZSZYIQGTHT-UHFFFAOYSA-N 0.000 description 4
- 210000000987 immune system Anatomy 0.000 description 4
- 238000010348 incorporation Methods 0.000 description 4
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 4
- 230000002452 interceptive effect Effects 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 4
- 229960004488 linolenic acid Drugs 0.000 description 4
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 230000010534 mechanism of action Effects 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- 229960000485 methotrexate Drugs 0.000 description 4
- 239000011859 microparticle Substances 0.000 description 4
- 230000003278 mimic effect Effects 0.000 description 4
- 229940074096 monoolein Drugs 0.000 description 4
- 150000003833 nucleoside derivatives Chemical class 0.000 description 4
- 125000003835 nucleoside group Chemical group 0.000 description 4
- 238000007911 parenteral administration Methods 0.000 description 4
- 244000052769 pathogen Species 0.000 description 4
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 229920000771 poly (alkylcyanoacrylate) Polymers 0.000 description 4
- 238000003752 polymerase chain reaction Methods 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 230000000241 respiratory effect Effects 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 238000012163 sequencing technique Methods 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 229960004274 stearic acid Drugs 0.000 description 4
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 4
- 239000002562 thickening agent Substances 0.000 description 4
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 4
- 210000001215 vagina Anatomy 0.000 description 4
- 229960003636 vidarabine Drugs 0.000 description 4
- 230000029812 viral genome replication Effects 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical compound OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 description 3
- RFVNOJDQRGSOEL-UHFFFAOYSA-N 2-hydroxyethyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCO RFVNOJDQRGSOEL-UHFFFAOYSA-N 0.000 description 3
- WOKDXPHSIQRTJF-UHFFFAOYSA-N 3-[3-[3-[3-[3-[3-[3-[3-[3-(2,3-dihydroxypropoxy)-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]propane-1,2-diol Chemical compound OCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)CO WOKDXPHSIQRTJF-UHFFFAOYSA-N 0.000 description 3
- 208000035657 Abasia Diseases 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 241000271566 Aves Species 0.000 description 3
- 208000011691 Burkitt lymphomas Diseases 0.000 description 3
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 3
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 3
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 3
- 108090000565 Capsid Proteins Proteins 0.000 description 3
- 101710205625 Capsid protein p24 Proteins 0.000 description 3
- 239000004971 Cross linker Substances 0.000 description 3
- 230000004543 DNA replication Effects 0.000 description 3
- 208000034423 Delivery Diseases 0.000 description 3
- 241000725618 Duck hepatitis B virus Species 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 206010016654 Fibrosis Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108010035713 Glycodeoxycholic Acid Proteins 0.000 description 3
- WVULKSPCQVQLCU-UHFFFAOYSA-N Glycodeoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(=O)NCC(O)=O)C)C1(C)C(O)C2 WVULKSPCQVQLCU-UHFFFAOYSA-N 0.000 description 3
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 3
- 206010061598 Immunodeficiency Diseases 0.000 description 3
- 208000032420 Latent Infection Diseases 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 description 3
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 208000037581 Persistent Infection Diseases 0.000 description 3
- 101710177166 Phosphoprotein Proteins 0.000 description 3
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 3
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- 101710149279 Small delta antigen Proteins 0.000 description 3
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 3
- 108020005202 Viral DNA Proteins 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000005600 alkyl phosphonate group Chemical group 0.000 description 3
- 150000008051 alkyl sulfates Chemical class 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 230000003622 anti-hsv Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 3
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 230000007882 cirrhosis Effects 0.000 description 3
- 208000019425 cirrhosis of liver Diseases 0.000 description 3
- 229960004106 citric acid Drugs 0.000 description 3
- 235000015165 citric acid Nutrition 0.000 description 3
- 230000009918 complex formation Effects 0.000 description 3
- 239000008139 complexing agent Substances 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 229960000633 dextran sulfate Drugs 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 206010014599 encephalitis Diseases 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 229940071106 ethylenediaminetetraacetate Drugs 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 239000003925 fat Substances 0.000 description 3
- 235000019197 fats Nutrition 0.000 description 3
- 150000002191 fatty alcohols Chemical class 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 239000013022 formulation composition Substances 0.000 description 3
- 229960005102 foscarnet Drugs 0.000 description 3
- 238000001502 gel electrophoresis Methods 0.000 description 3
- WVULKSPCQVQLCU-BUXLTGKBSA-N glycodeoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 WVULKSPCQVQLCU-BUXLTGKBSA-N 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 125000001475 halogen functional group Chemical group 0.000 description 3
- 208000002672 hepatitis B Diseases 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- 238000010255 intramuscular injection Methods 0.000 description 3
- 239000007927 intramuscular injection Substances 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- 239000007928 intraperitoneal injection Substances 0.000 description 3
- 238000007913 intrathecal administration Methods 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000007758 minimum essential medium Substances 0.000 description 3
- 201000011216 nasopharynx carcinoma Diseases 0.000 description 3
- 229960002446 octanoic acid Drugs 0.000 description 3
- 230000003285 pharmacodynamic effect Effects 0.000 description 3
- 150000008300 phosphoramidites Chemical class 0.000 description 3
- 125000004437 phosphorous atom Chemical group 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 230000010287 polarization Effects 0.000 description 3
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 3
- 229920000058 polyacrylate Polymers 0.000 description 3
- 229920000768 polyamine Polymers 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 239000005017 polysaccharide Substances 0.000 description 3
- 230000001737 promoting effect Effects 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 238000000159 protein binding assay Methods 0.000 description 3
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 230000003252 repetitive effect Effects 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 239000002356 single layer Substances 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 159000000000 sodium salts Chemical class 0.000 description 3
- 230000007928 solubilization Effects 0.000 description 3
- 238000005063 solubilization Methods 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 210000002784 stomach Anatomy 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- AWDRATDZQPNJFN-VAYUFCLWSA-N taurodeoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS(O)(=O)=O)C)[C@@]2(C)[C@@H](O)C1 AWDRATDZQPNJFN-VAYUFCLWSA-N 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 231100001274 therapeutic index Toxicity 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 229940035893 uracil Drugs 0.000 description 3
- 230000009677 vaginal delivery Effects 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- OQQOAWVKVDAJOI-UHFFFAOYSA-N (2-dodecanoyloxy-3-hydroxypropyl) dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCCCCCC OQQOAWVKVDAJOI-UHFFFAOYSA-N 0.000 description 2
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 2
- STGXGJRRAJKJRG-JDJSBBGDSA-N (3r,4r,5r)-5-(hydroxymethyl)-3-methoxyoxolane-2,4-diol Chemical compound CO[C@H]1C(O)O[C@H](CO)[C@H]1O STGXGJRRAJKJRG-JDJSBBGDSA-N 0.000 description 2
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 2
- LVNGJLRDBYCPGB-UHFFFAOYSA-N 1,2-distearoylphosphatidylethanolamine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(COP([O-])(=O)OCC[NH3+])OC(=O)CCCCCCCCCCCCCCCCC LVNGJLRDBYCPGB-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- FZWGECJQACGGTI-UHFFFAOYSA-N 2-amino-7-methyl-1,7-dihydro-6H-purin-6-one Chemical compound NC1=NC(O)=C2N(C)C=NC2=N1 FZWGECJQACGGTI-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- OHXPGWPVLFPUSM-KLRNGDHRSA-N 3,7,12-trioxo-5beta-cholanic acid Chemical compound C1CC(=O)C[C@H]2CC(=O)[C@H]3[C@@H]4CC[C@H]([C@@H](CCC(O)=O)C)[C@@]4(C)C(=O)C[C@@H]3[C@]21C OHXPGWPVLFPUSM-KLRNGDHRSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- HFQQYIUTYJVYFZ-UHFFFAOYSA-N 4-methylpentyl 2-cyanoprop-2-enoate Chemical compound CC(C)CCCOC(=O)C(=C)C#N HFQQYIUTYJVYFZ-UHFFFAOYSA-N 0.000 description 2
- RYVNIFSIEDRLSJ-UHFFFAOYSA-N 5-(hydroxymethyl)cytosine Chemical compound NC=1NC(=O)N=CC=1CO RYVNIFSIEDRLSJ-UHFFFAOYSA-N 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- HCGHYQLFMPXSDU-UHFFFAOYSA-N 7-methyladenine Chemical compound C1=NC(N)=C2N(C)C=NC2=N1 HCGHYQLFMPXSDU-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102100023321 Ceruloplasmin Human genes 0.000 description 2
- 229920001661 Chitosan Polymers 0.000 description 2
- 241000282552 Chlorocebus aethiops Species 0.000 description 2
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 241000792859 Enema Species 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 208000018522 Gastrointestinal disease Diseases 0.000 description 2
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 2
- 229930182566 Gentamicin Natural products 0.000 description 2
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 2
- 108091093094 Glycol nucleic acid Proteins 0.000 description 2
- 229930186217 Glycolipid Natural products 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 208000005176 Hepatitis C Diseases 0.000 description 2
- 206010019973 Herpes virus infection Diseases 0.000 description 2
- 208000029433 Herpesviridae infectious disease Diseases 0.000 description 2
- 101000666657 Homo sapiens Rho-related GTP-binding protein RhoQ Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 206010027906 Monocytosis Diseases 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 206010034133 Pathogen resistance Diseases 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920001244 Poly(D,L-lactide) Polymers 0.000 description 2
- 229920002730 Poly(butyl cyanoacrylate) Polymers 0.000 description 2
- 229920002724 Poly(ethyl cyanoacrylate) Polymers 0.000 description 2
- 229920002319 Poly(methyl acrylate) Polymers 0.000 description 2
- 229920002723 Poly(methyl cyanoacrylate) Polymers 0.000 description 2
- 229920000148 Polycarbophil calcium Polymers 0.000 description 2
- 102000029797 Prion Human genes 0.000 description 2
- 108091000054 Prion Proteins 0.000 description 2
- 102000007327 Protamines Human genes 0.000 description 2
- 108010007568 Protamines Proteins 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 101500027983 Rattus norvegicus Octadecaneuropeptide Proteins 0.000 description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 2
- 102100038339 Rho-related GTP-binding protein RhoQ Human genes 0.000 description 2
- MEFKEPWMEQBLKI-AIRLBKTGSA-O S-adenosyl-L-methionine Chemical compound O[C@@H]1[C@H](O)[C@@H](C[S+](CC[C@H]([NH3+])C([O-])=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MEFKEPWMEQBLKI-AIRLBKTGSA-O 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 101710172711 Structural protein Proteins 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- WBWWGRHZICKQGZ-UHFFFAOYSA-N Taurocholic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(=O)NCCS(O)(=O)=O)C)C1(C)C(O)C2 WBWWGRHZICKQGZ-UHFFFAOYSA-N 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 108010059993 Vancomycin Proteins 0.000 description 2
- 241000700647 Variola virus Species 0.000 description 2
- ZHAFUINZIZIXFC-UHFFFAOYSA-N [9-(dimethylamino)-10-methylbenzo[a]phenoxazin-5-ylidene]azanium;chloride Chemical compound [Cl-].O1C2=CC(=[NH2+])C3=CC=CC=C3C2=NC2=C1C=C(N(C)C)C(C)=C2 ZHAFUINZIZIXFC-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 230000002745 absorbent Effects 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 230000001070 adhesive effect Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 2
- 229960003942 amphotericin b Drugs 0.000 description 2
- 239000003945 anionic surfactant Substances 0.000 description 2
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 2
- 229940124411 anti-hiv antiviral agent Drugs 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000011668 ascorbic acid Chemical class 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 210000000941 bile Anatomy 0.000 description 2
- 230000002051 biphasic effect Effects 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000003560 cancer drug Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 229960000724 cidofovir Drugs 0.000 description 2
- 239000000084 colloidal system Substances 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 239000002537 cosmetic Substances 0.000 description 2
- 210000004748 cultured cell Anatomy 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 230000000593 degrading effect Effects 0.000 description 2
- 229960002997 dehydrocholic acid Drugs 0.000 description 2
- 229940124447 delivery agent Drugs 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 229960005160 dimyristoylphosphatidylglycerol Drugs 0.000 description 2
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 2
- 208000037771 disease arising from reactivation of latent virus Diseases 0.000 description 2
- 230000006806 disease prevention Effects 0.000 description 2
- BPHQZTVXXXJVHI-AJQTZOPKSA-N ditetradecanoyl phosphatidylglycerol Chemical compound CCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@@H](O)CO)OC(=O)CCCCCCCCCCCCC BPHQZTVXXXJVHI-AJQTZOPKSA-N 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 239000013583 drug formulation Substances 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 210000001163 endosome Anatomy 0.000 description 2
- 239000007920 enema Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000010408 film Substances 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- 229960000961 floxuridine Drugs 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 229960004675 fusidic acid Drugs 0.000 description 2
- 150000002270 gangliosides Chemical class 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 229960002518 gentamicin Drugs 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- VKOBVWXKNCXXDE-UHFFFAOYSA-N icosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCC(O)=O VKOBVWXKNCXXDE-UHFFFAOYSA-N 0.000 description 2
- 229960004716 idoxuridine Drugs 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 238000000099 in vitro assay Methods 0.000 description 2
- 229960000905 indomethacin Drugs 0.000 description 2
- 208000037797 influenza A Diseases 0.000 description 2
- 239000008011 inorganic excipient Substances 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 238000007917 intracranial administration Methods 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 238000007914 intraventricular administration Methods 0.000 description 2
- 230000002262 irrigation Effects 0.000 description 2
- 238000003973 irrigation Methods 0.000 description 2
- QRWOVIRDHQJFDB-UHFFFAOYSA-N isobutyl cyanoacrylate Chemical compound CC(C)COC(=O)C(=C)C#N QRWOVIRDHQJFDB-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 229940070765 laurate Drugs 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229910021645 metal ion Inorganic materials 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000000693 micelle Substances 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 239000012457 nonaqueous media Substances 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 230000001293 nucleolytic effect Effects 0.000 description 2
- 239000007764 o/w emulsion Substances 0.000 description 2
- 238000002515 oligonucleotide synthesis Methods 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 239000008012 organic excipient Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 239000008251 pharmaceutical emulsion Substances 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical compound C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 2
- 229960002895 phenylbutazone Drugs 0.000 description 2
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920001308 poly(aminoacid) Polymers 0.000 description 2
- 229950005134 polycarbophil Drugs 0.000 description 2
- 229920000223 polyglycerol Polymers 0.000 description 2
- 229920002720 polyhexylacrylate Polymers 0.000 description 2
- 229920002704 polyhistidine Polymers 0.000 description 2
- 150000004804 polysaccharides Chemical class 0.000 description 2
- 229920002717 polyvinylpyridine Polymers 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 229940048914 protamine Drugs 0.000 description 2
- 235000004252 protein component Nutrition 0.000 description 2
- 230000006916 protein interaction Effects 0.000 description 2
- 230000004850 protein–protein interaction Effects 0.000 description 2
- 210000002345 respiratory system Anatomy 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000000344 soap Substances 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 229940023144 sodium glycolate Drugs 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 239000013589 supplement Substances 0.000 description 2
- 229920001059 synthetic polymer Polymers 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- WBWWGRHZICKQGZ-GIHLXUJPSA-N taurocholic acid Chemical compound C([C@@H]1C[C@H]2O)[C@@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@@H]([C@@H](CCC(=O)NCCS(O)(=O)=O)C)[C@@]2(C)[C@H](O)C1 WBWWGRHZICKQGZ-GIHLXUJPSA-N 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- JEJAMASKDTUEBZ-UHFFFAOYSA-N tris(1,1,3-tribromo-2,2-dimethylpropyl) phosphate Chemical compound BrCC(C)(C)C(Br)(Br)OP(=O)(OC(Br)(Br)C(C)(C)CBr)OC(Br)(Br)C(C)(C)CBr JEJAMASKDTUEBZ-UHFFFAOYSA-N 0.000 description 2
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- RUDATBOHQWOJDD-UZVSRGJWSA-N ursodeoxycholic acid Chemical compound C([C@H]1C[C@@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)CC1 RUDATBOHQWOJDD-UZVSRGJWSA-N 0.000 description 2
- MYPYJXKWCTUITO-LYRMYLQWSA-N vancomycin Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C(O)=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-N 0.000 description 2
- 229960003165 vancomycin Drugs 0.000 description 2
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 description 2
- 244000000009 viral human pathogen Species 0.000 description 2
- 239000007762 w/o emulsion Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 2
- NFLGAXVYCFJBMK-RKDXNWHRSA-N (+)-isomenthone Natural products CC(C)[C@H]1CC[C@@H](C)CC1=O NFLGAXVYCFJBMK-RKDXNWHRSA-N 0.000 description 1
- YIMATHOGWXZHFX-WCTZXXKLSA-N (2r,3r,4r,5r)-5-(hydroxymethyl)-3-(2-methoxyethoxy)oxolane-2,4-diol Chemical compound COCCO[C@H]1[C@H](O)O[C@H](CO)[C@H]1O YIMATHOGWXZHFX-WCTZXXKLSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- MDKGKXOCJGEUJW-VIFPVBQESA-N (2s)-2-[4-(thiophene-2-carbonyl)phenyl]propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C(=O)C1=CC=CS1 MDKGKXOCJGEUJW-VIFPVBQESA-N 0.000 description 1
- QGVQZRDQPDLHHV-DPAQBDIFSA-N (3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthrene-3-thiol Chemical compound C1C=C2C[C@@H](S)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 QGVQZRDQPDLHHV-DPAQBDIFSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- PHIQHXFUZVPYII-ZCFIWIBFSA-N (R)-carnitine Chemical compound C[N+](C)(C)C[C@H](O)CC([O-])=O PHIQHXFUZVPYII-ZCFIWIBFSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- VDVMOGXIBBDZNI-DLEQIPTRSA-N (Z)-octadec-9-enoic acid propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O.CCCCCCCC\C=C/CCCCCCCC(O)=O.CCCCCCCC\C=C/CCCCCCCC(O)=O.CCCCCCCC\C=C/CCCCCCCC(O)=O.CCCCCCCC\C=C/CCCCCCCC(O)=O VDVMOGXIBBDZNI-DLEQIPTRSA-N 0.000 description 1
- AVZIYOYFVVSTGQ-RBWRNIRVSA-N (z)-octadec-9-enoic acid;propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O AVZIYOYFVVSTGQ-RBWRNIRVSA-N 0.000 description 1
- FJXSLZRUXGTLPF-HKIWRJGFSA-N (z)-octadec-9-enoic acid;propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O FJXSLZRUXGTLPF-HKIWRJGFSA-N 0.000 description 1
- UFSCXDAOCAIFOG-UHFFFAOYSA-N 1,10-dihydropyrimido[5,4-b][1,4]benzothiazin-2-one Chemical compound S1C2=CC=CC=C2N=C2C1=CNC(=O)N2 UFSCXDAOCAIFOG-UHFFFAOYSA-N 0.000 description 1
- PTFYZDMJTFMPQW-UHFFFAOYSA-N 1,10-dihydropyrimido[5,4-b][1,4]benzoxazin-2-one Chemical compound O1C2=CC=CC=C2N=C2C1=CNC(=O)N2 PTFYZDMJTFMPQW-UHFFFAOYSA-N 0.000 description 1
- LRANPJDWHYRCER-UHFFFAOYSA-N 1,2-diazepine Chemical compound N1C=CC=CC=N1 LRANPJDWHYRCER-UHFFFAOYSA-N 0.000 description 1
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Chemical class OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- KSQPABRRLYOJAM-UHFFFAOYSA-N 1-cyclohexyl-3-methyl-1-nitrosourea Chemical compound CNC(=O)N(N=O)C1CCCCC1 KSQPABRRLYOJAM-UHFFFAOYSA-N 0.000 description 1
- CBXRMKZFYQISIV-UHFFFAOYSA-N 1-n,1-n,1-n',1-n',2-n,2-n,2-n',2-n'-octamethylethene-1,1,2,2-tetramine Chemical compound CN(C)C(N(C)C)=C(N(C)C)N(C)C CBXRMKZFYQISIV-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- DBPWSSGDRRHUNT-CEGNMAFCSA-N 17α-hydroxyprogesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 DBPWSSGDRRHUNT-CEGNMAFCSA-N 0.000 description 1
- UHUHBFMZVCOEOV-UHFFFAOYSA-N 1h-imidazo[4,5-c]pyridin-4-amine Chemical compound NC1=NC=CC2=C1N=CN2 UHUHBFMZVCOEOV-UHFFFAOYSA-N 0.000 description 1
- LDGWQMRUWMSZIU-LQDDAWAPSA-M 2,3-bis[(z)-octadec-9-enoxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)C)OCCCCCCCC\C=C/CCCCCCCC LDGWQMRUWMSZIU-LQDDAWAPSA-M 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- DSAYAFZWRDYBQY-UHFFFAOYSA-N 2,5-dimethylhexa-1,5-diene Chemical compound CC(=C)CCC(C)=C DSAYAFZWRDYBQY-UHFFFAOYSA-N 0.000 description 1
- 102100027962 2-5A-dependent ribonuclease Human genes 0.000 description 1
- 108010000834 2-5A-dependent ribonuclease Proteins 0.000 description 1
- PBUUPFTVAPUWDE-UGZDLDLSSA-N 2-[[(2S,4S)-2-[bis(2-chloroethyl)amino]-2-oxo-1,3,2lambda5-oxazaphosphinan-4-yl]sulfanyl]ethanesulfonic acid Chemical compound OS(=O)(=O)CCS[C@H]1CCO[P@](=O)(N(CCCl)CCCl)N1 PBUUPFTVAPUWDE-UGZDLDLSSA-N 0.000 description 1
- BRLJKBOXIVONAG-UHFFFAOYSA-N 2-[[5-(dimethylamino)naphthalen-1-yl]sulfonyl-methylamino]acetic acid Chemical compound C1=CC=C2C(N(C)C)=CC=CC2=C1S(=O)(=O)N(C)CC(O)=O BRLJKBOXIVONAG-UHFFFAOYSA-N 0.000 description 1
- JRYMOPZHXMVHTA-DAGMQNCNSA-N 2-amino-7-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrrolo[2,3-d]pyrimidin-4-one Chemical compound C1=CC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O JRYMOPZHXMVHTA-DAGMQNCNSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- WKMPTBDYDNUJLF-UHFFFAOYSA-N 2-fluoroadenine Chemical compound NC1=NC(F)=NC2=C1N=CN2 WKMPTBDYDNUJLF-UHFFFAOYSA-N 0.000 description 1
- XBBVURRQGJPTHH-UHFFFAOYSA-N 2-hydroxyacetic acid;2-hydroxypropanoic acid Chemical compound OCC(O)=O.CC(O)C(O)=O XBBVURRQGJPTHH-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- OALHHIHQOFIMEF-UHFFFAOYSA-N 3',6'-dihydroxy-2',4',5',7'-tetraiodo-3h-spiro[2-benzofuran-1,9'-xanthene]-3-one Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(I)=C(O)C(I)=C1OC1=C(I)C(O)=C(I)C=C21 OALHHIHQOFIMEF-UHFFFAOYSA-N 0.000 description 1
- PDBUTMYDZLUVCP-UHFFFAOYSA-N 3,4-dihydro-1,4-benzoxazin-2-one Chemical compound C1=CC=C2OC(=O)CNC2=C1 PDBUTMYDZLUVCP-UHFFFAOYSA-N 0.000 description 1
- PLIKISMHPLKIRY-UHFFFAOYSA-N 3-acetamido-6-[2-(4-isothiocyanatophenyl)ethenyl]cyclohexa-2,4-diene-1,1-disulfonic acid Chemical compound C1=CC(NC(=O)C)=CC(S(O)(=O)=O)(S(O)(=O)=O)C1C=CC1=CC=C(N=C=S)C=C1 PLIKISMHPLKIRY-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ASFAFOSQXBRFMV-LJQANCHMSA-N 3-n-(2-benzyl-1,3-dihydroxypropan-2-yl)-1-n-[(1r)-1-(4-fluorophenyl)ethyl]-5-[methyl(methylsulfonyl)amino]benzene-1,3-dicarboxamide Chemical compound N([C@H](C)C=1C=CC(F)=CC=1)C(=O)C(C=1)=CC(N(C)S(C)(=O)=O)=CC=1C(=O)NC(CO)(CO)CC1=CC=CC=C1 ASFAFOSQXBRFMV-LJQANCHMSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 description 1
- ZMGMDXCADSRNCX-UHFFFAOYSA-N 5,6-dihydroxy-1,3-diazepan-2-one Chemical class OC1CNC(=O)NCC1O ZMGMDXCADSRNCX-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- IZZIWIAOVZOBLF-UHFFFAOYSA-N 5-methoxysalicylic acid Chemical compound COC1=CC=C(O)C(C(O)=O)=C1 IZZIWIAOVZOBLF-UHFFFAOYSA-N 0.000 description 1
- ZLAQATDNGLKIEV-UHFFFAOYSA-N 5-methyl-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CC1=CNC(=S)NC1=O ZLAQATDNGLKIEV-UHFFFAOYSA-N 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- 108091027075 5S-rRNA precursor Proteins 0.000 description 1
- KXBCLNRMQPRVTP-UHFFFAOYSA-N 6-amino-1,5-dihydroimidazo[4,5-c]pyridin-4-one Chemical compound O=C1NC(N)=CC2=C1N=CN2 KXBCLNRMQPRVTP-UHFFFAOYSA-N 0.000 description 1
- DCPSTSVLRXOYGS-UHFFFAOYSA-N 6-amino-1h-pyrimidine-2-thione Chemical compound NC1=CC=NC(S)=N1 DCPSTSVLRXOYGS-UHFFFAOYSA-N 0.000 description 1
- NJBMMMJOXRZENQ-UHFFFAOYSA-N 6H-pyrrolo[2,3-f]quinoline Chemical compound c1cc2ccc3[nH]cccc3c2n1 NJBMMMJOXRZENQ-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- LOSIULRWFAEMFL-UHFFFAOYSA-N 7-deazaguanine Chemical compound O=C1NC(N)=NC2=C1CC=N2 LOSIULRWFAEMFL-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HRYKDUPGBWLLHO-UHFFFAOYSA-N 8-azaadenine Chemical compound NC1=NC=NC2=NNN=C12 HRYKDUPGBWLLHO-UHFFFAOYSA-N 0.000 description 1
- LPXQRXLUHJKZIE-UHFFFAOYSA-N 8-azaguanine Chemical compound NC1=NC(O)=C2NN=NC2=N1 LPXQRXLUHJKZIE-UHFFFAOYSA-N 0.000 description 1
- 229960005508 8-azaguanine Drugs 0.000 description 1
- OYHQOLUKZRVURQ-HZJYTTRNSA-M 9-cis,12-cis-Octadecadienoate Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC([O-])=O OYHQOLUKZRVURQ-HZJYTTRNSA-M 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 102100034082 Alkaline ceramidase 3 Human genes 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- BMFMQGXDDJALKQ-BYPYZUCNSA-N Argininic acid Chemical class NC(N)=NCCC[C@H](O)C(O)=O BMFMQGXDDJALKQ-BYPYZUCNSA-N 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Chemical class 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- 108010087504 Beta-Globulins Proteins 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 206010006448 Bronchiolitis Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 description 1
- QYOVMAREBTZLBT-KTKRTIGZSA-N CCCCCCCC\C=C/CCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO Chemical compound CCCCCCCC\C=C/CCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO QYOVMAREBTZLBT-KTKRTIGZSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 230000010736 Chelating Activity Effects 0.000 description 1
- 201000006082 Chickenpox Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- 208000002691 Choroiditis Diseases 0.000 description 1
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 1
- 208000006154 Chronic hepatitis C Diseases 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 108091028732 Concatemer Proteins 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 241000700626 Cowpox virus Species 0.000 description 1
- 241001635223 Coxsackievirus B2 Species 0.000 description 1
- 241000709675 Coxsackievirus B3 Species 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 206010011831 Cytomegalovirus infection Diseases 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 239000003109 Disodium ethylene diamine tetraacetate Substances 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 1
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 1
- 201000011001 Ebola Hemorrhagic Fever Diseases 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 206010015866 Extravasation Diseases 0.000 description 1
- 208000001860 Eye Infections Diseases 0.000 description 1
- 239000001263 FEMA 3042 Chemical class 0.000 description 1
- 102000008946 Fibrinogen Human genes 0.000 description 1
- 108010049003 Fibrinogen Proteins 0.000 description 1
- MPJKWIXIYCLVCU-UHFFFAOYSA-N Folinic acid Natural products NC1=NC2=C(N(C=O)C(CNc3ccc(cc3)C(=O)NC(CCC(=O)O)CC(=O)O)CN2)C(=O)N1 MPJKWIXIYCLVCU-UHFFFAOYSA-N 0.000 description 1
- 229940123457 Free radical scavenger Drugs 0.000 description 1
- 101150050733 Gnas gene Proteins 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 229920000569 Gum karaya Polymers 0.000 description 1
- 108010078851 HIV Reverse Transcriptase Proteins 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 108700010908 HIV-1 proteins Proteins 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 206010019663 Hepatic failure Diseases 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 208000007514 Herpes zoster Diseases 0.000 description 1
- 101000798828 Homo sapiens Alkaline ceramidase 3 Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 241000701041 Human betaherpesvirus 7 Species 0.000 description 1
- 101100427508 Human cytomegalovirus (strain AD169) UL39 gene Proteins 0.000 description 1
- 241001502974 Human gammaherpesvirus 8 Species 0.000 description 1
- 241000701027 Human herpesvirus 6 Species 0.000 description 1
- 241000702617 Human parvovirus B19 Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 241000712431 Influenza A virus Species 0.000 description 1
- 206010022004 Influenza like illness Diseases 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 208000000913 Kidney Calculi Diseases 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 208000008771 Lymphadenopathy Diseases 0.000 description 1
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 201000005505 Measles Diseases 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- NFLGAXVYCFJBMK-UHFFFAOYSA-N Menthone Chemical compound CC(C)C1CCC(C)CC1=O NFLGAXVYCFJBMK-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 208000005647 Mumps Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 206010029148 Nephrolithiasis Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 229940122313 Nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- WTAYIFXKJBMZLY-XZABIIKCSA-N OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O WTAYIFXKJBMZLY-XZABIIKCSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000027868 Paget disease Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 229920002230 Pectic acid Chemical class 0.000 description 1
- JNTOCHDNEULJHD-UHFFFAOYSA-N Penciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(CCC(CO)CO)C=N2 JNTOCHDNEULJHD-UHFFFAOYSA-N 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Chemical class OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 201000007100 Pharyngitis Diseases 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 108010020346 Polyglutamic Acid Chemical class 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 208000003971 Posterior uveitis Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 101150093191 RIR1 gene Proteins 0.000 description 1
- 101150030723 RIR2 gene Proteins 0.000 description 1
- 230000026279 RNA modification Effects 0.000 description 1
- 230000007022 RNA scission Effects 0.000 description 1
- 206010037742 Rabies Diseases 0.000 description 1
- 230000010799 Receptor Interactions Effects 0.000 description 1
- 108020005091 Replication Origin Proteins 0.000 description 1
- 206010062106 Respiratory tract infection viral Diseases 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 241000282849 Ruminantia Species 0.000 description 1
- 241000315672 SARS coronavirus Species 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- ABBQHOQBGMUPJH-UHFFFAOYSA-M Sodium salicylate Chemical compound [Na+].OC1=CC=CC=C1C([O-])=O ABBQHOQBGMUPJH-UHFFFAOYSA-M 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 241000934878 Sterculia Species 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- ULUAUXLGCMPNKK-UHFFFAOYSA-N Sulfobutanedioic acid Chemical compound OC(=O)CC(C(O)=O)S(O)(=O)=O ULUAUXLGCMPNKK-UHFFFAOYSA-N 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 206010043275 Teratogenicity Diseases 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 206010070863 Toxicity to various agents Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 101150093137 UL13 gene Proteins 0.000 description 1
- 101150068034 UL30 gene Proteins 0.000 description 1
- 101150100826 UL40 gene Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- HDOVUKNUBWVHOX-QMMMGPOBSA-N Valacyclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCOC(=O)[C@@H](N)C(C)C)C=N2 HDOVUKNUBWVHOX-QMMMGPOBSA-N 0.000 description 1
- 206010046980 Varicella Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 108010059722 Viral Fusion Proteins Proteins 0.000 description 1
- 208000028227 Viral hemorrhagic fever Diseases 0.000 description 1
- 235000018936 Vitellaria paradoxa Nutrition 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 208000003152 Yellow Fever Diseases 0.000 description 1
- LEBBDRXHHNYZIA-LDUWYPJVSA-N [(2s,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] n-[(z)-1,3-dihydroxyoctadec-4-en-2-yl]carbamate Chemical compound CCCCCCCCCCCCC\C=C/C(O)C(CO)NC(=O)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O LEBBDRXHHNYZIA-LDUWYPJVSA-N 0.000 description 1
- RLXCFCYWFYXTON-JTTSDREOSA-N [(3S,8S,9S,10R,13S,14S,17R)-3-hydroxy-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-16-yl] N-hexylcarbamate Chemical group C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC(OC(=O)NCCCCCC)[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 RLXCFCYWFYXTON-JTTSDREOSA-N 0.000 description 1
- HMNZFMSWFCAGGW-XPWSMXQVSA-N [3-[hydroxy(2-hydroxyethoxy)phosphoryl]oxy-2-[(e)-octadec-9-enoyl]oxypropyl] (e)-octadec-9-enoate Chemical compound CCCCCCCC\C=C\CCCCCCCC(=O)OCC(COP(O)(=O)OCCO)OC(=O)CCCCCCC\C=C\CCCCCCCC HMNZFMSWFCAGGW-XPWSMXQVSA-N 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- MCGSCOLBFJQGHM-SCZZXKLOSA-N abacavir Chemical compound C=12N=CN([C@H]3C=C[C@@H](CO)C3)C2=NC(N)=NC=1NC1CC1 MCGSCOLBFJQGHM-SCZZXKLOSA-N 0.000 description 1
- 229960004748 abacavir Drugs 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- XVIYCJDWYLJQBG-UHFFFAOYSA-N acetic acid;adamantane Chemical compound CC(O)=O.C1C(C2)CC3CC1CC2C3 XVIYCJDWYLJQBG-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 229940023476 agar Drugs 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000005083 alkoxyalkoxy group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 150000004996 alkyl benzenes Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Chemical class OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 150000001408 amides Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 125000005122 aminoalkylamino group Chemical group 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000002280 amphoteric surfactant Substances 0.000 description 1
- 229960001830 amprenavir Drugs 0.000 description 1
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- 230000003941 amyloidogenesis Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 229940031955 anhydrous lanolin Drugs 0.000 description 1
- 229920006318 anionic polymer Polymers 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 229940037157 anticorticosteroids Drugs 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 238000011225 antiretroviral therapy Methods 0.000 description 1
- 229940124522 antiretrovirals Drugs 0.000 description 1
- 239000003903 antiretrovirus agent Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000002820 assay format Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 239000003212 astringent agent Substances 0.000 description 1
- 229960000892 attapulgite Drugs 0.000 description 1
- 206010064097 avian influenza Diseases 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- 230000037429 base substitution Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N benzo-alpha-pyrone Natural products C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000007698 birth defect Effects 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 125000001369 canonical nucleoside group Chemical group 0.000 description 1
- LDVVMCZRFWMZSG-UHFFFAOYSA-N captan Chemical compound C1C=CCC2C(=O)N(SC(Cl)(Cl)Cl)C(=O)C21 LDVVMCZRFWMZSG-UHFFFAOYSA-N 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 238000003763 carbonization Methods 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 229960004203 carnitine Drugs 0.000 description 1
- IVUMCTKHWDRRMH-UHFFFAOYSA-N carprofen Chemical compound C1=CC(Cl)=C[C]2C3=CC=C(C(C(O)=O)C)C=C3N=C21 IVUMCTKHWDRRMH-UHFFFAOYSA-N 0.000 description 1
- 229960003184 carprofen Drugs 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000034303 cell budding Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 239000013553 cell monolayer Substances 0.000 description 1
- 229920003086 cellulose ether Polymers 0.000 description 1
- 229940106189 ceramide Drugs 0.000 description 1
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 230000003196 chaotropic effect Effects 0.000 description 1
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000035606 childbirth Effects 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229960002155 chlorothiazide Drugs 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 150000004775 coumarins Chemical class 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 108700007153 dansylsarcosine Proteins 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- HABLENUWIZGESP-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O.CCCCCCCCCC(O)=O HABLENUWIZGESP-UHFFFAOYSA-N 0.000 description 1
- STORWMDPIHOSMF-UHFFFAOYSA-N decanoic acid;octanoic acid;propane-1,2,3-triol Chemical compound OCC(O)CO.CCCCCCCC(O)=O.CCCCCCCCCC(O)=O STORWMDPIHOSMF-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 210000004207 dermis Anatomy 0.000 description 1
- 238000011033 desalting Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- GUJOJGAPFQRJSV-UHFFFAOYSA-N dialuminum;dioxosilane;oxygen(2-);hydrate Chemical compound O.[O-2].[O-2].[O-2].[Al+3].[Al+3].O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O GUJOJGAPFQRJSV-UHFFFAOYSA-N 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 229960001193 diclofenac sodium Drugs 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- 125000005594 diketone group Chemical group 0.000 description 1
- OGQYPPBGSLZBEG-UHFFFAOYSA-N dimethyl(dioctadecyl)azanium Chemical compound CCCCCCCCCCCCCCCCCC[N+](C)(C)CCCCCCCCCCCCCCCCCC OGQYPPBGSLZBEG-UHFFFAOYSA-N 0.000 description 1
- BPHQZTVXXXJVHI-UHFFFAOYSA-N dimyristoyl phosphatidylglycerol Chemical compound CCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCCCCCCCC BPHQZTVXXXJVHI-UHFFFAOYSA-N 0.000 description 1
- MWRBNPKJOOWZPW-CLFAGFIQSA-N dioleoyl phosphatidylethanolamine Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(COP(O)(=O)OCCN)OC(=O)CCCCCCC\C=C/CCCCCCCC MWRBNPKJOOWZPW-CLFAGFIQSA-N 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 235000019301 disodium ethylene diamine tetraacetate Nutrition 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 229940095399 enema Drugs 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 229940125532 enzyme inhibitor Drugs 0.000 description 1
- CHNUOJQWGUIOLD-NFZZJPOKSA-N epalrestat Chemical compound C=1C=CC=CC=1\C=C(/C)\C=C1/SC(=S)N(CC(O)=O)C1=O CHNUOJQWGUIOLD-NFZZJPOKSA-N 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- ITSGNOIFAJAQHJ-BMFNZSJVSA-N esorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)C[C@H](C)O1 ITSGNOIFAJAQHJ-BMFNZSJVSA-N 0.000 description 1
- 229950002017 esorubicin Drugs 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000036251 extravasation Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 208000011323 eye infectious disease Diseases 0.000 description 1
- 229960004396 famciclovir Drugs 0.000 description 1
- GGXKWVWZWMLJEH-UHFFFAOYSA-N famcyclovir Chemical compound N1=C(N)N=C2N(CCC(COC(=O)C)COC(C)=O)C=NC2=C1 GGXKWVWZWMLJEH-UHFFFAOYSA-N 0.000 description 1
- 150000002194 fatty esters Chemical class 0.000 description 1
- ZPAKPRAICRBAOD-UHFFFAOYSA-N fenbufen Chemical compound C1=CC(C(=O)CCC(=O)O)=CC=C1C1=CC=CC=C1 ZPAKPRAICRBAOD-UHFFFAOYSA-N 0.000 description 1
- 229960001395 fenbufen Drugs 0.000 description 1
- 229940012952 fibrinogen Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- LPEPZBJOKDYZAD-UHFFFAOYSA-N flufenamic acid Chemical compound OC(=O)C1=CC=CC=C1NC1=CC=CC(C(F)(F)F)=C1 LPEPZBJOKDYZAD-UHFFFAOYSA-N 0.000 description 1
- 229960004369 flufenamic acid Drugs 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229920001002 functional polymer Polymers 0.000 description 1
- 230000000799 fusogenic effect Effects 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical class OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 230000035784 germination Effects 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- UPWGQKDVAURUGE-UHFFFAOYSA-N glycerine monooleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC(CO)CO UPWGQKDVAURUGE-UHFFFAOYSA-N 0.000 description 1
- 229940074049 glyceryl dilaurate Drugs 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 125000005908 glyceryl ester group Chemical group 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- KWLMIXQRALPRBC-UHFFFAOYSA-L hectorite Chemical compound [Li+].[OH-].[OH-].[Na+].[Mg+2].O1[Si]2([O-])O[Si]1([O-])O[Si]([O-])(O1)O[Si]1([O-])O2 KWLMIXQRALPRBC-UHFFFAOYSA-L 0.000 description 1
- 229910000271 hectorite Inorganic materials 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 208000010710 hepatitis C virus infection Diseases 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- XDZLHTBOHLGGCJ-UHFFFAOYSA-N hexyl 2-cyanoprop-2-enoate Chemical compound CCCCCCOC(=O)C(=C)C#N XDZLHTBOHLGGCJ-UHFFFAOYSA-N 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 239000002835 hiv fusion inhibitor Substances 0.000 description 1
- QRMZSPFSDQBLIX-UHFFFAOYSA-N homovanillic acid Chemical compound COC1=CC(CC(O)=O)=CC=C1O QRMZSPFSDQBLIX-UHFFFAOYSA-N 0.000 description 1
- 235000012907 honey Nutrition 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 239000000416 hydrocolloid Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 229960002899 hydroxyprogesterone Drugs 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 239000003547 immunosorbent Substances 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 229910003480 inorganic solid Inorganic materials 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 150000002496 iodine Chemical class 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 239000002563 ionic surfactant Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000000111 isothermal titration calorimetry Methods 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 235000010494 karaya gum Nutrition 0.000 description 1
- 239000000231 karaya gum Substances 0.000 description 1
- 229940039371 karaya gum Drugs 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 201000010666 keratoconjunctivitis Diseases 0.000 description 1
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 1
- 229960001627 lamivudine Drugs 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 239000008141 laxative Substances 0.000 description 1
- 229940125722 laxative agent Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 235000001510 limonene Nutrition 0.000 description 1
- 229940087305 limonene Drugs 0.000 description 1
- 229940049918 linoleate Drugs 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 231100000835 liver failure Toxicity 0.000 description 1
- 208000007903 liver failure Diseases 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 208000030500 lower respiratory tract disease Diseases 0.000 description 1
- 208000018555 lymphatic system disease Diseases 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 229950000547 mafosfamide Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Chemical class 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 208000027202 mammary Paget disease Diseases 0.000 description 1
- 238000005399 mechanical ventilation Methods 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000034217 membrane fusion Effects 0.000 description 1
- 229930007503 menthone Natural products 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 238000006198 methoxylation reaction Methods 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 125000006362 methylene amino carbonyl group Chemical group [H]N(C([*:2])=O)C([H])([H])[*:1] 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 231100000324 minimal toxicity Toxicity 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- RZRNAYUHWVFMIP-UHFFFAOYSA-N monoelaidin Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-UHFFFAOYSA-N 0.000 description 1
- 229910052901 montmorillonite Inorganic materials 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 208000010805 mumps infectious disease Diseases 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 1
- 229940105132 myristate Drugs 0.000 description 1
- BLYOHBPLFYXHQA-UHFFFAOYSA-N n,n-bis(prop-2-enyl)prop-2-enamide Chemical compound C=CCN(CC=C)C(=O)C=C BLYOHBPLFYXHQA-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-M naphthalene-1-sulfonate Chemical compound C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-M 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 229940042880 natural phospholipid Drugs 0.000 description 1
- 238000002663 nebulization Methods 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 210000003757 neuroblast Anatomy 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000007171 neuropathology Effects 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 229940042402 non-nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 239000002726 nonnucleoside reverse transcriptase inhibitor Substances 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- PIDFXJFGSDJONG-UHFFFAOYSA-N octadecanoic acid;propan-2-yl tetradecanoate Chemical compound CCCCCCCCCCCCCC(=O)OC(C)C.CCCCCCCCCCCCCCCCCC(O)=O PIDFXJFGSDJONG-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 229940124276 oligodeoxyribonucleotide Drugs 0.000 description 1
- 239000003605 opacifier Substances 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 229960003752 oseltamivir Drugs 0.000 description 1
- VSZGPKBBMSAYNT-RRFJBIMHSA-N oseltamivir Chemical compound CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 VSZGPKBBMSAYNT-RRFJBIMHSA-N 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 238000002640 oxygen therapy Methods 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 229910052625 palygorskite Inorganic materials 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000007918 pathogenicity Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 229940002988 pegasys Drugs 0.000 description 1
- 108010092853 peginterferon alfa-2a Proteins 0.000 description 1
- 229960001179 penciclovir Drugs 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- ONTNXMBMXUNDBF-UHFFFAOYSA-N pentatriacontane-17,18,19-triol Chemical compound CCCCCCCCCCCCCCCCC(O)C(O)C(O)CCCCCCCCCCCCCCCC ONTNXMBMXUNDBF-UHFFFAOYSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000008255 pharmaceutical foam Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000010587 phase diagram Methods 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- 150000002991 phenoxazines Chemical class 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 125000001095 phosphatidyl group Chemical group 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003905 phosphatidylinositols Chemical class 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical compound [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- KQOXLKOJHVFTRN-UHFFFAOYSA-N pleconaril Chemical compound O1N=C(C)C=C1CCCOC1=C(C)C=C(C=2N=C(ON=2)C(F)(F)F)C=C1C KQOXLKOJHVFTRN-UHFFFAOYSA-N 0.000 description 1
- 229960000471 pleconaril Drugs 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229920001553 poly(ethylene glycol)-block-polylactide methyl ether Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920000447 polyanionic polymer Polymers 0.000 description 1
- 229920005646 polycarboxylate Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 239000010318 polygalacturonic acid Chemical class 0.000 description 1
- 229920002643 polyglutamic acid Chemical class 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920002714 polyornithine Polymers 0.000 description 1
- 108010055896 polyornithine Proteins 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000002213 purine nucleotide Substances 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- RXTQGIIIYVEHBN-UHFFFAOYSA-N pyrimido[4,5-b]indol-2-one Chemical compound C1=CC=CC2=NC3=NC(=O)N=CC3=C21 RXTQGIIIYVEHBN-UHFFFAOYSA-N 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- LKUNXBRZDFMZOK-UHFFFAOYSA-N rac-1-monodecanoylglycerol Chemical compound CCCCCCCCCC(=O)OCC(O)CO LKUNXBRZDFMZOK-UHFFFAOYSA-N 0.000 description 1
- GHBFNMLVSPCDGN-UHFFFAOYSA-N rac-1-monooctanoylglycerol Chemical compound CCCCCCCC(=O)OCC(O)CO GHBFNMLVSPCDGN-UHFFFAOYSA-N 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 238000002108 rapid electrokinetic patterning Methods 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 229940100618 rectal suppository Drugs 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000015227 regulation of liquid surface tension Effects 0.000 description 1
- 230000017610 release of virus from host Effects 0.000 description 1
- 125000006853 reporter group Chemical group 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000003307 reticuloendothelial effect Effects 0.000 description 1
- 229940064914 retrovir Drugs 0.000 description 1
- 150000003291 riboses Chemical class 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 239000005060 rubber Substances 0.000 description 1
- 201000005404 rubella Diseases 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 208000008864 scrapie Diseases 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 206010040872 skin infection Diseases 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- NRHMKIHPTBHXPF-TUJRSCDTSA-M sodium cholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 NRHMKIHPTBHXPF-TUJRSCDTSA-M 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- VMSNAUAEKXEYGP-YEUHZSMFSA-M sodium glycodeoxycholate Chemical compound [Na+].C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 VMSNAUAEKXEYGP-YEUHZSMFSA-M 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 229960004025 sodium salicylate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- JAJWGJBVLPIOOH-IZYKLYLVSA-M sodium taurocholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 JAJWGJBVLPIOOH-IZYKLYLVSA-M 0.000 description 1
- 229940045946 sodium taurodeoxycholate Drugs 0.000 description 1
- JGMJQSFLQWGYMQ-UHFFFAOYSA-M sodium;2,6-dichloro-n-phenylaniline;acetate Chemical compound [Na+].CC([O-])=O.ClC1=CC=CC(Cl)=C1NC1=CC=CC=C1 JGMJQSFLQWGYMQ-UHFFFAOYSA-M 0.000 description 1
- IWQPOPSAISBUAH-VOVMJQHHSA-M sodium;2-[[(2z)-2-[(3r,4s,5s,8s,9s,10s,11r,13r,14s,16s)-16-acetyl-3,11-dihydroxy-4,8,10,14-tetramethyl-2,3,4,5,6,7,9,11,12,13,15,16-dodecahydro-1h-cyclopenta[a]phenanthren-17-ylidene]-6-methylheptanoyl]amino]ethanesulfonate Chemical compound [Na+].C1C[C@@H](O)[C@@H](C)[C@@H]2CC[C@]3(C)[C@@]4(C)C[C@H](C(C)=O)/C(=C(C(=O)NCCS([O-])(=O)=O)/CCCC(C)C)[C@@H]4C[C@@H](O)[C@H]3[C@]21C IWQPOPSAISBUAH-VOVMJQHHSA-M 0.000 description 1
- YXHRQQJFKOHLAP-FVCKGWAHSA-M sodium;2-[[(4r)-4-[(3r,5r,8r,9s,10s,12s,13r,14s,17r)-3,12-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonate Chemical compound [Na+].C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 YXHRQQJFKOHLAP-FVCKGWAHSA-M 0.000 description 1
- FIWQZURFGYXCEO-UHFFFAOYSA-M sodium;decanoate Chemical compound [Na+].CCCCCCCCCC([O-])=O FIWQZURFGYXCEO-UHFFFAOYSA-M 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000003445 sucroses Chemical class 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical group NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003457 sulfones Chemical group 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004354 sulfur functional group Chemical group 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 229960004492 suprofen Drugs 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 229920002258 tannic acid Chemical class 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 231100000211 teratogenicity Toxicity 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 235000007586 terpenes Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- TUNFSRHWOTWDNC-UHFFFAOYSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 1
- JYKSTGLAIMQDRA-UHFFFAOYSA-N tetraglycerol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO JYKSTGLAIMQDRA-UHFFFAOYSA-N 0.000 description 1
- 229950000329 thiouracil Drugs 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 125000002640 tocopherol group Chemical class 0.000 description 1
- 235000019149 tocopherols Nutrition 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- ZMANZCXQSJIPKH-UHFFFAOYSA-O triethylammonium ion Chemical compound CC[NH+](CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-O 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940096973 urethral suppository Drugs 0.000 description 1
- 239000006217 urethral suppository Substances 0.000 description 1
- 229940054870 urso Drugs 0.000 description 1
- 229960001661 ursodiol Drugs 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 229940093257 valacyclovir Drugs 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000007332 vesicle formation Effects 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000033041 viral attachment to host cell Effects 0.000 description 1
- 230000007419 viral reactivation Effects 0.000 description 1
- 230000010464 virion assembly Effects 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 239000008307 w/o/w-emulsion Substances 0.000 description 1
- 229960005080 warfarin Drugs 0.000 description 1
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
- 229960001028 zanamivir Drugs 0.000 description 1
- ARAIBEBZBOPLMB-UFGQHTETSA-N zanamivir Chemical compound CC(=O)N[C@@H]1[C@@H](N=C(N)N)C=C(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO ARAIBEBZBOPLMB-UFGQHTETSA-N 0.000 description 1
- 229960002555 zidovudine Drugs 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2121/00—Preparations for use in therapy
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16611—Simplexvirus, e.g. human herpesvirus 1, 2
- C12N2710/16622—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/16011—Orthomyxoviridae
- C12N2760/16111—Influenzavirus A, i.e. influenza A virus
- C12N2760/16122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
抗ウイルス活性を有するランダム配列オリゴヌクレオチドおよびその抗ウイルス薬としての使用について記載する。多くの場合、オリゴヌクレオチドは、40より大きいヌクレオチド長を有し、かつ例えば修飾されたヌクレオチド間結合およびリボース部分の2'位の修飾などの化学修飾を含む。ヒトおよび動物のウイルス感染の予防または処置のための使用、ならびにヒトおよび動物のオンコウイルスを原因とする癌の予防または処置のための使用も記載する。これら使用は典型的に、そのような処置を必要とするヒトまたは動物に対して、配列非依存的な作用様式により作用する、薬理学的に許容される治療有効量の少なくとも一種のオリゴヌクレオチドを投与することを含む。Described are random sequence oligonucleotides with antiviral activity and their use as antiviral agents. In many cases, the oligonucleotide has a nucleotide length greater than 40 and includes chemical modifications such as, for example, a modified internucleotide linkage and a modification at the 2 ′ position of the ribose moiety. Also described are uses for the prevention or treatment of viral infections in humans and animals, and use for the prevention or treatment of cancers caused by human and animal oncoviruses. These uses typically provide a pharmacologically acceptable therapeutically effective amount of at least one oligonucleotide that acts in a sequence-independent manner on a human or animal in need of such treatment. Administration.
Description
本発明の分野
本発明は、抗ウイルス活性を有するオリゴヌクレオチド、並びにヒト及び動物のウイルスにより起きるウイルス感染、癌遺伝子ウイルスにより起きる癌及び病因がウイルスに基づくそのほかの疾患における治療剤としてのその使用に関する。
FIELD OF THE INVENTION The present invention relates to oligonucleotides having antiviral activity and their use as therapeutics in viral infections caused by human and animal viruses, cancers caused by oncogene viruses and other diseases whose etiology is virus-based. .
発明の背景
以下の記述は、単に読者の理解を助けるために提供されるのであって、記述された情報又は引用された文献のいかなるものも本発明の先行技術を構成することを容認するものではない。
BACKGROUND OF THE INVENTION The following description is provided solely to aid the reader's understanding and is not intended to allow any of the described information or cited documents to constitute prior art to the present invention. Absent.
人類を苦しめる多数の重大な感染症はウイルスによって引き起こされる。狂犬病、天然痘、灰白髄炎、ウイルス性出血熱、肝炎、黄熱病、免疫不全症、及び種々の脳炎を含む、これら多数の疾患は、致命的であることが多い。そのほかの疾患も、呼吸障害及び消化器障害と同様にインフルエンザ、麻疹、おたふく風邪、水痘のように伝染性であり、急性の不快感を生じさせるという点で重要である。風疹やサイトメガロウイルスのようなものは、先天異常を生じさせる。最後に、ヒトや動物で癌を生じさせうる腫瘍ウイルスとして知られるウイルスがある。 Many serious infections that afflict humanity are caused by viruses. Many of these diseases are often fatal, including rabies, smallpox, leukomyelitis, viral hemorrhagic fever, hepatitis, yellow fever, immunodeficiency, and various encephalitis. Other diseases are important in that they are contagious, like influenza, measles, mumps and chickenpox, as well as respiratory and gastrointestinal disorders, and cause acute discomfort. Things like rubella and cytomegalovirus can cause birth defects. Finally, there are viruses known as tumor viruses that can cause cancer in humans and animals.
ウイルスの中でも、ヘルペスウイルス科は大変興味深い。ヘルペスウイルス科は遍在する部類の十二面体の二本鎖DNAのウイルスである。性状分析された100を超えるヘルペスウイルス(HHV)科のうち8種類だけがヒトに感染する。これらの中で最もよく知られているのは、単純疱疹ウイルス1(HSV-1)、単純疱疹ウイルス2(HSV-2)、水痘帯状疱疹ウイルス(水痘又は帯状疱疹)、サイトメガロウイルス(CMV)及びエプステイン・バーウイルス(EBV)である。ヘルペスウイルスのヒトにおける蔓延は高く、全世界人口の少なくとも1/3が冒され、米国では人口の70〜80%が何らかのヘルペス感染を有する。ヘルペス感染の病態は、通常は口の周りや顔にすぐに直る病変を生じさせるだけであるHSV-1の場合のように、通常危険ではない一方で、これらのウイルスが、さらに危険な症状の原因になりうることも知られており、それは、性器潰瘍及び分泌物から、脳炎(致死率15%)や播種性の感染(致死率40%)を招く可能性がある致命的な感染まで異なる。 Among viruses, Herpesviridae is very interesting. The herpesviridae is a ubiquitous class of dodecahedron double-stranded DNA viruses. Only 8 of the over 100 herpesviridae (HHV) families that have been characterized infect humans. The best known of these are herpes simplex virus 1 (HSV-1), herpes simplex virus 2 (HSV-2), varicella zoster virus (varicella or shingles), cytomegalovirus (CMV) And Epstein-Barr virus (EBV). Herpes viruses are highly prevalent in humans, affecting at least one third of the world's population, and 70-80% of the population in the United States has some herpes infection. While the pathology of herpes infections is usually not dangerous, as is the case with HSV-1, which usually only causes lesions that immediately heal around the mouth and face, these viruses are more dangerous It is also known to be causative and varies from genital ulcers and secretions to fatal infections that can lead to encephalitis (15% mortality) and disseminated infection (40% mortality) .
ヘルペスウイルスは自然界では播種性が高く、ヒトに対して病原性が高い。たとえば、エプステイン・バーウイルス(EBV)は、小児期後期もしくは青年期又は成人初期で感染性単球増多症を生じさせることが知られている。急性感染性単球増多症の顕著な特徴は、喉の痛み、発熱、頭痛、リンパ節症、扁桃腺肥大及び末梢血における異型のリンパ球分裂である。そのほかの徴候には軽い肝炎、脾臓肥大及び脳炎が挙げられることが多い。EBVはまた、癌:バーキットリンパ腫(BL)及び鼻咽腔癌(NPC)に関係する。赤道アフリカの流行域では、BLは最も一般的な小児悪性腫瘍であり、小児における癌のおよそ80%を占める。NPCは北アメリカの白人ではほどほどに認められるが、中国南部では25〜55歳の年齢で生じる最も一般的な癌の1つである。サイトメガロウイルスと同様に、EBVは、移植後のリンパ増殖性疾患に関係しており、それは、固形臓器又は骨髄の移植後の慢性免疫抑制の致命的な合併症になりうる。 Herpesviruses are highly disseminated in nature and highly pathogenic to humans. For example, Epstein-Barr virus (EBV) is known to cause infectious monocytosis in late childhood or adolescence or early adulthood. Prominent features of acute infectious monocytosis are sore throat, fever, headache, lymphadenopathy, tonsil hypertrophy and atypical lymphocyte division in peripheral blood. Other signs often include mild hepatitis, spleen enlargement and encephalitis. EBV is also implicated in cancer: Burkitt lymphoma (BL) and nasopharyngeal carcinoma (NPC). In the epidemic region of Equatorial Africa, BL is the most common childhood malignancy, accounting for approximately 80% of cancers in children. NPC is modestly observed in North American whites, but is one of the most common cancers that occurs in the age of 25-55 years in southern China. Like cytomegalovirus, EBV is associated with lymphoproliferative disease after transplantation, which can be a fatal complication of chronic immunosuppression after solid organ or bone marrow transplantation.
たとえば、中心性網脈絡膜炎又は角結膜炎のような皮膚や眼の感染を含むそのほかの疾患もHSVに関係する。米国では1年におよそ300,000例の眼のHSV感染が診断される。 For example, other diseases involving skin and eye infections such as central choroiditis or keratoconjunctivitis are also associated with HSV. In the United States, approximately 300,000 ocular HSV infections are diagnosed per year.
AIDS(後天性免疫不全症候群)はヒト免疫不全ウイルス(HIV)により引き起こされる。身体の免疫系の細胞を殺す又は損傷することによって、HIVは感染及び特定の癌と闘う身体の能力を漸進的に破壊する。現在、世界中でおよそ4200万人の人々がHIV/AIDSと共に暮らしている。2002年にはHIV/AIDSに関係した原因で総計310万人が死亡した。抗HIV薬物療法の最終的な目標はウイルスが再生して免疫系を損傷するのを妨げることである。HIVに対する過去15年間にわたる闘いで実質的な進歩は遂げられているが、治療は未だに医学をかわしている。今日、内科医は、この疾患を管理するのに3種の異なる薬剤クラスの多数の抗ウイルス剤を有している。通常2又は3種のクラスの薬剤が、HAART(高活性抗レトロウイルス治療)として知られる多様な組み合わせで調合される。HAART療法は通常2つのヌクレオシド逆転写酵素阻害剤を、プロテアーゼ阻害剤又は非ヌクレオシド逆転写酵素阻害剤のいずれかの第3の薬剤と共に含む。臨床試験によって、HAARTは、ウイルス負荷を軽減し、薬剤耐性の可能性をできるだけ抑える最も有効な手段であることが判っている。 AIDS (Acquired Immunodeficiency Syndrome) is caused by the human immunodeficiency virus (HIV). By killing or damaging cells of the body's immune system, HIV progressively destroys the body's ability to fight infection and certain cancers. Currently, around 42 million people around the world live with HIV / AIDS. In 2002, a total of 3.1 million people died from HIV / AIDS-related causes. The ultimate goal of anti-HIV drug therapy is to prevent the virus from regenerating and damaging the immune system. Substantial progress has been made in the fight over HIV over the past 15 years, but treatment still evades medicine. Today, physicians have a number of antiviral agents in three different drug classes to manage this disease. Usually two or three classes of drugs are formulated in various combinations known as HAART (Highly Active Antiretroviral Therapy). HAART therapy usually includes two nucleoside reverse transcriptase inhibitors with a third agent, either a protease inhibitor or a non-nucleoside reverse transcriptase inhibitor. Clinical trials have shown that HAART is the most effective means of reducing viral load and minimizing the potential for drug resistance.
HAARTは一般にウイルス負荷として知られる体内のHIVの量を減らすことが示されているが、何万という患者がこの療法で重大な問題に遭遇している。副作用には重篤なものもあり、脂肪代謝の異常、腎臓結石及び心疾患が挙げられる。たとえば、悪心、吐き気及び不眠のようなそのほかの副作用はさほど重篤ではないが、一生、慢性的な薬剤療法を必要とするHIV患者にとっては依然として問題である。 Although HAART has been shown to reduce the amount of HIV in the body, commonly known as viral load, tens of thousands of patients have encountered serious problems with this therapy. Some side effects are serious and include abnormal fat metabolism, kidney stones, and heart disease. Other side effects such as nausea, nausea and insomnia, for example, are less severe, but remain a problem for HIV patients who need chronic medication for life.
現在認可されている抗HIV剤は、HIVが感染したCD4+T細胞に入り、逆転写酵素又はプロテアーゼいずれかのウイルス酵素の機能を阻止することによって作用する。HIVは再生するためにこれらの酵素を必要としている。しかしながら、HIVは度々突然変異して、逆転写酵素又はプロテアーゼの阻害剤がこれらの耐性株に対して効かないようにする。いったん耐性が生じると、ウイルス負荷が上昇し、効かない薬剤を別の抗レトロウイルス剤に交換する必要を強いられる。残念ながら、ウイルスがあるクラスの1つの薬剤に耐性になると、そのクラスの別の薬剤の有効性も低くなる。多数の抗HIV剤が類似の様式で作用するので、交差耐性として知られるこの現象が生じる。薬剤の交差耐性の発生は、患者にとって利用できる治療選択肢が減るので極めて望ましくない。 Currently approved anti-HIV agents enter CD4 + T cells infected with HIV and act by blocking the function of viral enzymes, either reverse transcriptase or protease. HIV needs these enzymes to regenerate. However, HIV is often mutated to prevent reverse transcriptase or protease inhibitors from working against these resistant strains. Once resistance develops, the viral load increases and forces the ineffective drug to be replaced with another antiretroviral agent. Unfortunately, when a virus becomes resistant to one drug in a class, the effectiveness of another drug in that class is also reduced. This phenomenon, known as cross-resistance, occurs because many anti-HIV agents act in a similar manner. The occurrence of drug cross-resistance is highly undesirable because it reduces the treatment options available to the patient.
従って、ウイルスが耐性を発現していないほかの作用機序を介して作用する、HIVに対して有効なそのほかの抗ウイルス剤の開発に対して大きなニーズがある。ヨーロッパで新たにHIVと診断された患者の10人に1人が市場にある認可された薬剤の少なくとも1種にすでに耐性であるHIV株に感染しているという最近のデータを考えると、このことは、特に重要になりつつある。 Therefore, there is a great need for the development of other antiviral agents effective against HIV that act through other mechanisms of action where the virus does not develop resistance. Considering recent data that 1 in 10 newly diagnosed HIV patients in Europe are already infected with HIV strains that are already resistant to at least one of the approved drugs on the market. Is becoming particularly important.
呼吸器多核体ウイルス(RSV)は、上下気道に感染を起こす。それは、マイナス鎖のエンベロープ型RNAウイルスであり、感染性が高い。それは幼年の小児を一般に冒し、幼児の下部気道疾病の最も一般的な原因である。RSVの感染は通常、中程度から重篤な風邪のような症状と関係する。しかしながら、いかなる年齢でも、特に高齢の又は免疫障害のある患者では重篤な下部気道疾患が生じる可能性がある。重篤な感染のある小児は酸素療法、及び特定の症例では機械的人工呼吸を必要とする可能性がある。全米医師会によれば、RSV感染が原因であることが多い、細気管支炎のために入院する小児の数が増えている。RSV感染はまた、骨髄移植センタ-の患者における地域関連型の呼吸器ウイルス感染のおよそ1/3の原因である。高齢集団では、RSV感染は最近では、重篤度においてインフルエンザウイルス感染に非常に類似すると認識されている。 Respiratory polynuclear virus (RSV) causes infection of the upper and lower respiratory tract. It is a negative-strand envelope RNA virus and is highly infectious. It affects young children in general and is the most common cause of infant lower airway disease. RSV infection is usually associated with moderate to severe cold-like symptoms. However, severe lower respiratory tract disease can occur at any age, especially in elderly or immunocompromised patients. Children with severe infection may need oxygen therapy and, in certain cases, mechanical ventilation. According to the National Medical Association, an increasing number of children are hospitalized for bronchiolitis, often due to RSV infection. RSV infection is also responsible for approximately one third of community-related respiratory viral infections in patients at bone marrow transplant centers. In the elderly population, RSV infection has recently been recognized as very similar to influenza virus infection in severity.
流感としても知られているインフルエンザ(INF)はインフルエンザウイルスにより生じる伝染病である。それはヒトの気道(鼻、喉及び肺)を攻撃する。米国では年に平均約36,000人がインフルエンザで死亡し、年に114,000人がインフルエンザのために入院している。インフルエンザは近年、以前に動物にのみ見つかっているより高い病原性を有する株(例えば鳥インフルエンザ)の出現が、さらに深刻な懸念となっている。 Influenza (INF), also known as flu, is a contagious disease caused by influenza viruses. It attacks the human respiratory tract (nose, throat and lungs). In the United States, an average of about 36,000 people die each year from influenza, and 114,000 people are hospitalized for influenza each year. In recent years, the emergence of strains with higher pathogenicity (eg, bird flu) that have previously been found only in animals has become a more serious concern.
あらゆる感染性疾患において、所定の療法の有効性は、宿主の免疫応答に依存することが多い。潜在性感染を確立するすべてのヘルペスウイルスの能力により、免疫障害のある患者において再活性化した感染が極めて高い発生率で生じるので、これはヘルペスウイルスに特にあてはまる。腎移植患者では、40〜70%がHSVの潜在感染を再活性化させ、80〜100%がCMV感染を再活性化させる。そのようなウイルスの再活性化は、AIDS患者でも認められる。 In any infectious disease, the effectiveness of a given therapy often depends on the host's immune response. This is especially true for herpes viruses because the ability of all herpesviruses to establish a latent infection results in a very high incidence of reactivated infections in immunocompromised patients. In kidney transplant patients, 40-70% reactivate latent infection with HSV and 80-100% reactivate CMV infection. Such virus reactivation is also observed in AIDS patients.
B型肝炎ウイルス(HBV)は、ウイルスの肝炎ウイルス科に属するDNAウイルスである。HBVはヒトにおいてB型肝炎を発症させる。世界で20億人が感染していると推定される(3人に1人)。約3億5000万人は慢性的に感染したままであり、毎年、推定100万人がB型肝炎及びその合併症で死亡している。HBVは、生涯にわたる感染、肝硬変、肝癌、肝不全及び死を引き起こす。ウイルスは血液及び体液を介して伝染する。これは、血液と血液の直接的接触、無防備な性交、不潔な針の使用を介して、又、出産過程で感染した女性から新生児へと起こりうる。感染する健常な成人のほとんど(90%)は回復し、将来のB型肝炎感染への防御抗体を作る。少数(5〜10%)がウイルスを始末することができず、慢性の感染を煩うが、幼児の90%及び50%までの幼年小児はウイルスに感染した際、慢性の感染を煩う。α-インターフェロンは、使用する治療剤の最も頻度の多いタイプである。インフルエンザ様の症状、鬱、発疹、そのほかの反応及び血球数の異常を含む重大な副作用がこの治療に関係する。もう1つの治療選択肢には、その使用に関連した多数の副作用を有する3TCが挙げられる。ここ数年 、3TCで治療した患者がHBVの耐性株を発現していることを示す報告の数が増えている。このことは、HBVとHIVを同時感染した患者集団で特に問題である。このウイルスに対する新たな抗ウイルス剤を開発する緊急の必要性が明らかにある。 Hepatitis B virus (HBV) is a DNA virus belonging to the hepatitis virus family of viruses. HBV causes hepatitis B in humans. It is estimated that 2 billion people are infected worldwide (1 in 3). About 350 million people remain chronically infected and an estimated 1 million people die each year from hepatitis B and its complications. HBV causes lifelong infection, cirrhosis, liver cancer, liver failure and death. Viruses are transmitted through blood and body fluids. This can occur through direct contact between blood, unprotected sexual intercourse, the use of filthy needles, and from a woman who is infected during childbirth to a newborn. Most healthy adults (90%) who are infected recover and make protective antibodies against future hepatitis B infections. A small number (5-10%) are unable to clean up the virus and suffer from chronic infections, but up to 90% and 50% of young children suffer from chronic infection when infected by the virus. α-interferon is the most frequent type of therapeutic agent used. Serious side effects including flu-like symptoms, depression, rash, other reactions and abnormal blood counts are associated with this treatment. Another treatment option includes 3TC, which has a number of side effects associated with its use. In recent years, an increasing number of reports have shown that patients treated with 3TC express resistant strains of HBV. This is particularly problematic in patient populations co-infected with HBV and HIV. There is clearly an urgent need to develop new antiviral agents against this virus.
C型肝炎ウイルス(HCV)の感染は、感染患者数が400万人を超えると思われる米国で最も一般的な血液由来の慢性感染である。この一般的なウイルスの感染は、米国における肝硬変及び肝癌の主な原因であり、今や肝臓移植の主な理由である。感染からの回復は一般的ではなく、感染患者の約85パーセントは慢性のウイルス保有者になり10〜20パーセントは肝硬変を発症する。現在、世界で1億7000万人が慢性保有者であると推定される。疾病対策センタ-によれば、米国だけで毎年、8,000〜10,000人が慢性C型肝炎で死亡し、約1,000人が肝臓移植を受けている。C型肝炎に利用できるワクチンはない。インターフェロンα、又はインターフェロンとリバビリンの併用による長い治療法は約40パーセントの患者にしか有効ではなく、有意な副作用を生じさせる。 Hepatitis C virus (HCV) infection is the most common blood-borne chronic infection in the United States, with more than 4 million infected cases. This common viral infection is the leading cause of cirrhosis and liver cancer in the United States and is now the main reason for liver transplantation. Recovery from infection is uncommon, with approximately 85 percent of infected patients becoming chronic virus carriers and 10 to 20 percent developing cirrhosis. Currently, it is estimated that 170 million people worldwide are chronic carriers. According to the Centers for Disease Control, every year in the United States alone, 8,000-10,000 people die from chronic hepatitis C and about 1,000 people undergo liver transplants. There is no vaccine available for hepatitis C. Long treatments with interferon alpha or a combination of interferon and ribavirin are effective only in about 40 percent of patients and produce significant side effects.
今日、ウイルス感染の治療の見通しは一般によくない。一般に、ウイルスに対する治療は平凡な有効性しか有さず、有効投与量の投与の妨げになるか又は長期の治療を妨げる強い副作用と関係している。これらの問題を例示する3つの臨床状況は、ヘルペスウイルス、HIV及びRSVの感染である。 Today, the prospects for treating viral infections are generally not good. In general, treatment for viruses has only mediocre efficacy and is associated with strong side effects that prevent administration of effective doses or prevent long-term treatment. Three clinical situations that illustrate these problems are herpes virus, HIV and RSV infection.
ヘルペスウイルスの場合、診療所での使用が現在認可されている5つの主な治療薬がある:イドクスウリジン、ビダラビン、アシクロビル、フォスカーネット及びガンシクロビルである。限られた有効性を有するが、これらの治療も副作用を伴う。イドクスウリジンで治療を受けた患者の35%でアレルギー反応が報告されており、ビダラビンは患者の15%で胃腸障害を生じさせ、ヌクレオシド類縁体であるアシクロビル、フォスカーネット及びガンシクロビルは宿主細胞でのDNA複製に影響を及ぼす。ガンシクロビルの場合、この薬剤で治療を受けたAIDS患者の40%で好中球減少症及び血小板減少症が報告されている。 In the case of herpesviruses, there are five main therapies currently approved for use in the clinic: idoxuridine, vidarabine, acyclovir, foscarnet and ganciclovir. Although having limited effectiveness, these treatments also have side effects. Allergic reactions have been reported in 35% of patients treated with idoxuridine, vidarabine causes gastrointestinal disorders in 15% of patients, and the nucleoside analogs acyclovir, foscarnet and ganciclovir are host cells. Affects DNA replication. In the case of ganciclovir, neutropenia and thrombocytopenia have been reported in 40% of AIDS patients treated with this drug.
HIV感染を治療するために現在利用可能な多数の異なる薬剤があるが、これらはすべて、患者が妥当な生活の質を得るには、多大な補足的投薬を必要とするに十分なほど強力な副作用と関係している。HIVの薬剤耐性株の追加的問題(ヘルペスウイルス科感染にも見られる問題)は通常、治療カクテルの周期的変更を要し、場合によっては感染を極めて治療しにくくする。 There are a number of different drugs currently available to treat HIV infection, all of which are powerful enough to require a large amount of supplemental medication in order for the patient to obtain a reasonable quality of life. It is related to side effects. Additional problems with drug-resistant strains of HIV (problems also found in herpesviridae infections) usually require periodic changes in treatment cocktails, and in some cases make infections extremely difficult to treat.
乳児でのRSV感染の治療は、新薬開発の緊急の必要性のもう1つの例である。この場合、通常の治療法は、隔離テントの中で微粒子のエアゾールを用いた吸入によるリバビリンの送達である。リバビリンは、穏やかな効果しないだけでなく、その使用は重大な副作用に関係している。加えて、リバビリンには催奇形性が知られているので、薬剤の流出の可能性が病院職員に大きな懸念を抱かせてきた。 Treatment of RSV infection in infants is another example of the urgent need for new drug development. In this case, the usual treatment is delivery of ribavirin by inhalation using a particulate aerosol in an isolation tent. Not only does ribavirin have a mild effect, but its use is associated with serious side effects. In addition, since ribavirin is known for its teratogenicity, the potential for drug spills has caused great concern to hospital staff.
開発中の新しく出現する抗ウイルス性薬剤については、以下の3つの特徴、1.改善された有効性、2.軽減された副作用のリスク及び3.突然変異によってウイルスが克服しにくい作用メカニズムを取り入れることが極めて望ましいことは明らかである。 The newly developed antiviral drugs under development have the following three characteristics: Improved efficacy, 2. Clearly, it is highly desirable to incorporate a reduced risk of side effects and a mechanism of action that is difficult for viruses to overcome by mutation.
種々のアンチセンス手法によって特定のウイルスを阻害する試みが行われている。 Attempts have been made to inhibit specific viruses by various antisense techniques.
Zamecnikらは、逆転写酵素プライマ-部位及びスプライスドナー/アクセプタ部位を特異的に標的としたONを用いた(Zamecnik et al., (1986) Proc. Natl. Acad. Sci. USA 83:4143)(Goodchild & Zamecnik, 1989, 米国特許第4,806,463号) Zamecnik et al. Used ON specifically targeting reverse transcriptase primer sites and splice donor / acceptor sites (Zamecnik et al., (1986) Proc. Natl. Acad. Sci. USA 83: 4143) ( Goodchild & Zamecnik, 1989, US Pat. No. 4,806,463)
Crooke及び共同研究者らは(Crooke et al., (1992) Antimicrob. Agents Chemother., 36:527-532)HSV-1に対するアンチセンスを記載した。 Crooke and coworkers (Crooke et al., (1992) Antimicrob. Agents Chemother., 36: 527-532) described antisense to HSV-1.
Draperらは(1993, 米国特許第5,248,670号)Cat配列を有し、HSV-1遺伝子、UL13、UL39、及びUL40にハイブリッド形成する抗HSV活性を有するアンチセンスオリゴヌクレオチドを報告した。 (1993, US Pat. No. 5,248,670) reported an antisense oligonucleotide having a Cat sequence and having anti-HSV activity that hybridizes to the HSV-1 gene, UL13, UL39, and UL40.
Keanらは(Biochemistry, 34:14617-14620, 1995)、抗HSV作用剤としての、アンチセンスメチルホスホネートリゴマーの調査を報告した。 Kean et al. (Biochemistry, 34: 14617-14620, 1995) reported a survey of antisense methylphosphonate ligomers as anti-HSV agonists.
Peymanらは(Biol. Chem. Hoppe. Seyler, Mar, 376:195-198, 1995)、その抗ウイルス特性について、HSV-1のIE110及びUL30のmRNAに関する特異的アンチセンスオリゴヌクレオチドを報告した。 Peyman et al. (Biol. Chem. Hoppe. Seyler, Mar, 376: 195-198, 1995) have reported specific antisense oligonucleotides for HSV-1 IE110 and UL30 mRNA for their antiviral properties.
IE1、IE2又はDNAポリメラーゼをコードするCMVのmRNAを標的とするオリゴヌクレオチド又はオリゴヌクレオチド類縁体が、Andersonら(1997, 米国特許第5,591,720号)により報告された。 Oligonucleotides or oligonucleotide analogs targeting the mRNA of CMV encoding IE1, IE2 or DNA polymerase were reported by Anderson et al. (1997, US Pat. No. 5,591,720).
Hanecakらは(1999, 米国特許第5,952,490号)、保存されたGカルテット配列及び十分な数の隣接ヌクレオチドを有し、HSV-1のようなウイルスの活性を有意に阻害する修飾オリゴヌクレオチドを記載した。 (1999, US Pat. No. 5,952,490) described a modified oligonucleotide that has a conserved G quartet sequence and a sufficient number of flanking nucleotides and significantly inhibits the activity of a virus such as HSV-1. .
Jairathらは(Antiviral Res., 33:201-213, 1997)RSVに対するアンチセンスオリゴヌクレオチドを報告した。 Jairath et al. (Antiviral Res., 33: 201-213, 1997) reported an antisense oligonucleotide against RSV.
Torrenceらは(1999, 米国特許第5,998,602号)、RSVのアンチゲノム鎖(mRNA鎖)の一本鎖部分に相補的なアンチセンスを含む化合物、リンカー及びRNA分解酵素Lのオリゴヌクレオチド活性化剤を報告した。 Torrence et al. (1999, US Pat. No. 5,998,602) describe a compound containing an antisense complementary to a single-stranded portion of an RSV antigenome strand (mRNA strand), a linker, and an oligonucleotide activator for RNase L. reported.
Qiらは(Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi, 14:253-256, 2000)、コクサッキーウイルスB3におけるアンチセンスPS-オリゴヌクレオチド(PS-ON)を調べたことを報告した。 Qi et al. (Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi, 14: 253-256, 2000) reported that they investigated an antisense PS-oligonucleotide (PS-ON) in Coxsackievirus B3.
国際公開公報第9203051号(Roizman and Maxwell)は、HSVウイルスゲノムの肝要な領域又はそのmRNA転写物に相補的であり、抗ウイルス活性を示すメチルホスホネートアンチセンスオリゴマーを記載している。 WO9203051 (Roizman and Maxwell) describes a methylphosphonate antisense oligomer that is complementary to an essential region of the HSV viral genome or its mRNA transcript and exhibits antiviral activity.
グアノシン/チミジン又はグアノシン-リッチのホスホロチオエートのオリゴデオキシヌクレオチド(GT-PS-ON)は抗ウイルス活性を有することが報告されている。論文には、「幾つかの異なるPS含有のGTリッチON(B106〜140、I100〜12及びG106〜57)すべて長さは26又は27ntは、36(B106〜96、B106〜97)又は45ntから成るGTリッチONと同様にHIV-2の力価を下げるのに有効であった(表4)」。(Fennewald et al., Antiviral Res., 26:37-54, 1995) Guanosine / thymidine or guanosine-rich phosphorothioate oligodeoxynucleotides (GT-PS-ON) have been reported to have antiviral activity. The article states that "Several different PS-containing GT-rich ONs (B106-140, I100-12 and G106-57) all 26 or 27 nt in length from 36 (B106-96, B106-97) or 45 nt It was effective in lowering the titer of HIV-2 as well as GT Rich ON (Table 4). (Fennewald et al., Antiviral Res., 26: 37-54, 1995)
米国特許第6,184,369号には、高い比率でグアノシン塩基を含有する抗HIV、抗HSV及び抗CMVのオリゴヌクレオチドが記載されている。好ましい態様では、オリゴヌクレオチドは三次構造を有し、この構造は、グアノシン四分子によって安定化される。さらなる態様では、この発明のオリゴヌクレオチド組成物は2つの隣接したデオキシグアノシンの2以上の種類を有する。この特許は、少なくとも2つの位置で少なくとも2つのG残基を包含するGリッチのオリゴデオキシヌクレオチド(ODN)を請求している。 US Pat. No. 6,184,369 describes anti-HIV, anti-HSV and anti-CMV oligonucleotides containing a high proportion of guanosine bases. In a preferred embodiment, the oligonucleotide has a tertiary structure, which is stabilized by a guanosine tetramolecule. In a further embodiment, the oligonucleotide composition of this invention has two or more types of two adjacent deoxyguanosines. This patent claims a G-rich oligodeoxynucleotide (ODN) that includes at least two G residues at at least two positions.
Cohenらは(米国特許第5,264,423号及び同第5,276,019号)HIVの複製の阻害を記載し、さらに詳しくは、正常な生きている細胞の存在下で外来の核酸の複製を妨げるのに使用することができるPS-ODN類縁体を記載している。Cohenらは、ウイルス配列に特異的なアンチセンスPS-ODNの抗ウイルス活性を記載している。彼らはまた14、18、21及び28量体のポリA、ポリT及びポリCのPS-ODNを調べたことを記載し、これらPS-ODNの抗ウイルス効果を示している。 Cohen et al. (US Pat. Nos. 5,264,423 and 5,276,019) describe inhibition of HIV replication, and more particularly, used to prevent replication of foreign nucleic acids in the presence of normal living cells. PS-ODN analogs that can be used are described. Cohen et al. Describe the antiviral activity of antisense PS-ODN specific for viral sequences. They also described examining the PS-ODNs of 14, 18, 21, and 28-mer poly A, poly T and poly C, indicating the antiviral effect of these PS-ODNs.
Matsukuraらは(Matsukura et al., Proc. Natl. Acad. Sci. USA, 84:7706-7710, 1987)、上記Cohenらの米国特許に記載された結果を後になって公開した。 Matsukura et al. (Matsukura et al., Proc. Natl. Acad. Sci. USA, 84: 7706-7710, 1987) later published the results described in the Cohen et al. US patent.
Gaoらは(Gao et al., J. Biol. Chem, 264:11521-11526, 1989)、7、15、21及び28ヌクレオチドのサイズにおけるポリA、ポリT及びポリCのPS-ODNを調べることによるPS-ODNによってHSV-2の複製が阻害されることを記載している。 Gao et al. (Gao et al., J. Biol. Chem, 264: 11521-11526, 1989) examine PS-ODN of poly A, poly T and poly C at sizes of 7, 15, 21 and 28 nucleotides. Describes that PS-ODN inhibits HSV-2 replication.
Archambault, Stein及び Cohenは(Archambault et al., Arch. Virol., 139:97109, 1994)28ヌクレオチドのPS-ODNポリCがHSV-1に対して有効ではないことを報告している。 Archambault, Stein and Cohen (Archambault et al., Arch. Virol., 139: 97109, 1994) report that 28 nucleotide PS-ODN poly C is not effective against HSV-1.
Steinらは(Stein et al., AIDS Res. Hum. Retrovir., 5:639-646, 1989)、一般に21〜28ヌクレオチドの長さの抗HIV ODNに関する追加データに関する結果を公表した。 Stein et al. (Stein et al., AIDS Res. Hum. Retrovir., 5: 639-646, 1989) published results on additional data on anti-HIV ODN, generally 21-28 nucleotides in length.
Marshalらは(Marshall et al., Proc. Natl. Acad. Sci. USA, 89:6265-6269, 1992)、長さ4〜28ヌクレオチドのホスホロチオエート及びホスホロチオエートポリCオリゴの抗HIV-1効果を記載している。 Marshal et al. (Marshall et al., Proc. Natl. Acad. Sci. USA, 89: 6265-6269, 1992) describe the anti-HIV-1 effect of phosphorothioate and phosphorothioate poly C oligos 4 to 28 nucleotides in length. ing.
Stein及びChengは(Stein et al., Science, 261:1004-1012, 1993)、概説論文の中で28ヌクレオチドの非特異的ODNの抗ウイルス活性について言及し、「PSオリゴの抗HIV特性は、非配列特異的な効果によって有意に影響され、すなわち、阻害効果は塩基配列に依存しない」と述べている。 Stein and Cheng (Stein et al., Science, 261: 1004-1012, 1993) mentioned in a review article the antiviral activity of a non-specific ODN of 28 nucleotides, “The anti-HIV properties of PS oligos are It is significantly affected by non-sequence-specific effects, ie the inhibitory effect is independent of the base sequence ”.
概説論文の中で、Lebedeva及びSteinは(Lebedeva et al., Annul. Rev. Pharmacol., 41:403-419, 2001)、ウイルスタンパク質も含めて、PS-ODNの活性を結合する種々の非特異的タンパク質を報告している。彼らは、「これらの分子は生物学的に極めて活性が高く、相対的に人為物をアンチセンスと間違えやすいことが多い」と述べている。 Among the review papers, Lebedeva and Stein (Lebedeva et al., Annul. Rev. Pharmacol., 41: 403-419, 2001), various nonspecifics that bind the activity of PS-ODN, including viral proteins Have reported a specific protein. They state that "these molecules are extremely biologically active and relatively easy to mistake artifacts for antisense."
Reinらは(米国特許第6,316,190号)、融合相手に連結され、抗ウイルス性化合物として使用することができるHIVのヌクレオカプシドに結合するGTリッチのONデコイを報告した。同様に、Campbellらは(Campbell et al., J. Virol., 73:2270-2279, 1999)、HIVのヌクレオカプシドに特異的に結合するが、抗ウイルス活性には関係しないTGTGTモチーフを持つPO-ODNを報告した。 Rein et al. (US Pat. No. 6,316,190) reported a GT-rich ON decoy that binds to a nucleocapsid of HIV that can be linked to a fusion partner and used as an antiviral compound. Similarly, Campbell et al. (Campbell et al., J. Virol., 73: 2270-2279, 1999), PO- with a TTGGT motif that binds specifically to the nucleocapsid of HIV but is not related to antiviral activity. Reported ODN.
Fengらは(Feng et al., J. Virol., 76:11757-11762, 2002)、組換えHIVのヌクレオカプシドに結合するが、抗HIV活性に関するデータの参照もないA(n)及びTG(n)のPO-ODNを記載した。 Feng et al. (Feng et al., J. Virol., 76: 11757-11762, 2002) bind to the nucleocapsid of recombinant HIV but have no reference to data on anti-HIV activity A (n) and TG (n ) PO-ODN.
抗癌剤、抗ウイルス剤として又はそのほかの疾患を治療するために開発されたアンチセンスODNは通常、長さ、およそ20ヌクレオチドである。概説論文(Stein, CA, J. Clin. Invest., 108:641-644, 2001)では、「アンチセンスオリゴヌクレオチドの長さは最適化されなければならない:アンチセンスオリゴヌクレオチドが長すぎるか又は短かすぎれば、特異性の要素は失われる。現在、アンチセンスオリゴヌクレオチドの最適な長さは大雑把に16〜20ヌクレオチドであると思われる」と確認されている。同様に、もう1つの概説論文(Crooke, ST., Methods Enzymol., 313:3-45, 2000)では、「RNA及びRNA二本鎖形成に比べて、ホスホロチオエートオリゴヌクレオチドは、単位当たりおよそ-2.2°低いTmを有する。このことは、インビトロで有効であるには、ホスホロチオエートオリゴヌクレオチドは通常、長さ17〜20量体であらねばならない」と述べられている。 Antisense ODNs developed as anticancer agents, antiviral agents, or to treat other diseases are usually approximately 20 nucleotides in length. In the review paper (Stein, CA, J. Clin. Invest., 108: 641-644, 2001) “The length of the antisense oligonucleotide must be optimized: the antisense oligonucleotide is either too long or too short If it is too much, the element of specificity is lost, and it is now confirmed that the optimal length of an antisense oligonucleotide is roughly 16-20 nucleotides. " Similarly, in another review paper (Crooke, ST., Methods Enzymol., 313: 3-45, 2000), “phosphorothioate oligonucleotides are approximately −2.2 per unit compared to RNA and RNA duplex formation. It has a low T m, which means that in order to be effective in vitro, phosphorothioate oligonucleotides usually must be 17-20 mer in length.
Caruthers及び共同研究者らは(Marshall et al., Proc. Natl. Acad. Sci. USA, 89:6265-6269, 1992)、12量体のポリシチジン-PS2-ODN及び14量体のPS2-ODNに関するホスホロジチオエートODNの抗HIV活性を報告した。抗HIV活性について別のサイズは調べなかった。彼らはまた、12、14、20及び28量体のポリシチジンPS2-ODNについてHIV逆転写酵素(RT)の阻害剤を報告した。後になって(Marshal et al., Science,259:1564-1570, 1993)、HIV RTの配列特異的阻害を示す結果を報告した。同じグループは幾つかの特許の中でPS2-ODNに関するデータを公表した。米国特許第5,218,103号及び同第5,684,148号では、PS2-ODNの構造及び合成を記載している。米国特許第5,452,496号、同第5,278,302号、及び同第5,695,979号では15塩基以下のPS2-ODNに関してHIV RTの阻害が記載されている。米国特許第5,750,666号及び同第5,602,244号では、PS2-ODNのアンチセンス活性が記載されている。 Caruthers and co-workers (Marshall et al., Proc. Natl. Acad. Sci. USA, 89: 6265-6269, 1992) are concerned with 12-mer polycytidine-PS2-ODN and 14-mer PS2-ODN. The anti-HIV activity of phosphorodithioate ODN was reported. Another size was not examined for anti-HIV activity. They also reported inhibitors of HIV reverse transcriptase (RT) for 12, 14, 20, and 28-mer polycytidine PS2-ODN. Later (Marshal et al., Science, 259: 1564-1570, 1993) reported results showing sequence specific inhibition of HIV RT. The same group published data on PS2-ODN in several patents. US Pat. Nos. 5,218,103 and 5,684,148 describe the structure and synthesis of PS2-ODN. US Pat. Nos. 5,452,496, 5,278,302, and 5,695,979 describe inhibition of HIV RT for PS2-ODN of 15 bases or less. US Pat. Nos. 5,750,666 and 5,602,244 describe the antisense activity of PS2-ODN.
リボースの2'位が修飾されたオリゴヌクレオチドおよびそれらをアンチセンス戦略において使用することは、例えば、以下に引用する参照に記載されているように評価されている。 Oligonucleotides modified at the 2 ′ position of ribose and their use in antisense strategies have been evaluated, for example, as described in the references cited below.
lnoueおよび共同研究者(Inoue et al. (1985) Nucleic Acid Res. 16:165168)は、オリゴ(2'-O-メチルリボヌクレオチド)の合成および性質について記載している。同一グループ(Inoue et al. (1987) FEBS Letter 215:327-330)は、オリゴヌクレオチドが全て2'-O-メチルリボヌクレオチドを含む場合、RNase Hが仲介しないmRNA切断が起こることを報告した。混合オリゴヌクレオチド、即ち非修飾および2'-O-メチルリボヌクレオチドを有するオリゴヌクレオチドを用いて、その研究者等は、RNAおよび2'-O-メチルリボヌクレオチドにより形成された核酸複合体の、配列特異的なRNAse H加水分解を報告している。 lnoue and coworkers (Inoue et al. (1985) Nucleic Acid Res. 16: 165168) describe the synthesis and properties of oligos (2′-O-methyl ribonucleotides). The same group (Inoue et al. (1987) FEBS Letter 215: 327-330) reported that RNase H-mediated mRNA cleavage occurs when all oligonucleotides contain 2'-O-methyl ribonucleotides. Using mixed oligonucleotides, i.e., oligonucleotides with unmodified and 2'-O-methyl ribonucleotides, the investigators have sequenced the nucleic acid complex formed by RNA and 2'-O-methyl ribonucleotides. Specific RNAse H hydrolysis has been reported.
RNase H仲介による標的mRNAの切断を支持しない、完全2'-O-メチル化およびホスホロチオエート化オリゴヌクレオチドを使用して、活性アンチセンスオリゴヌクレオチドが、RNase H-依存性機序によるICAM-1発現を阻害するか否かを決定した(Chiang et al., (1991) J. Biol. Chem. 266:18162-18171)。彼等は、これらのアンチセンスオリゴヌクレオチドが、治療薬として有用であり得ることを述べた。 Using fully 2'-O-methylated and phosphorothioated oligonucleotides that do not support RNase H-mediated cleavage of the target mRNA, active antisense oligonucleotides promote ICAM-1 expression through an RNase H-dependent mechanism. It was determined whether to inhibit (Chiang et al., (1991) J. Biol. Chem. 266: 18162-18171). They stated that these antisense oligonucleotides can be useful as therapeutic agents.
2'-O-メチル、2'-O-プロピル、2'-O-ペンチルおよび2'-フルオロを含む2'-糖修飾を伴うオリゴヌクレオチドのアンチセンス活性を分析した。均一に2'-修飾したオリゴヌクレオチドのアンチセンス活性の評価により、これらの化合物は遺伝子発現の阻害に全く効果的ではないことが明らかとなった。活性は、化合物が少なくとも5個の2'-デオキシリボヌクレオチド残基の伸展を含む場合に回復した。この最小限度のデオキシリボヌクレオチド長は、インビトロでの有効なRNase H活性に必要な最小の長さと完全に相関した(Monia et al., 1993, J. Biol. Chem. 268:14514.)。 The antisense activity of oligonucleotides with 2′-sugar modifications including 2′-O-methyl, 2′-O-propyl, 2′-O-pentyl and 2′-fluoro was analyzed. Evaluation of the antisense activity of uniformly 2′-modified oligonucleotides revealed that these compounds were not at all effective in inhibiting gene expression. Activity was restored when the compound contained an extension of at least 5 2'-deoxyribonucleotide residues. This minimal deoxyribonucleotide length correlated perfectly with the minimum length required for effective RNase H activity in vitro (Monia et al., 1993, J. Biol. Chem. 268: 14514.).
Yuら((1996) Bioorganic. Med. Chem. 4:1685-1692)は、ホスホロチオエート、ホスホジエステルまたは2'-O-メチル修飾糖部分を含む混合バックボーンを有するハイブリッドアンチセンスオリゴヌクレオチドは、p24ELISA定量化により測定した特異的抗-HIV活性を有することを報告した。 Yu et al. ((1996) Bioorganic. Med. Chem. 4: 1685-1692) described hybrid antisense oligonucleotides with mixed backbones containing phosphorothioate, phosphodiester or 2'-O-methyl modified sugar moieties, p24 ELISA quantification. Reported to have specific anti-HIV activity measured by.
ホスホロチオエート化2'-O-メチル-オリゴリボヌクレオチドが、その変異型ヒトβ-グロブリン遺伝子によって安定的に形質転換された哺乳動物細胞内で発現したIVS2-654プレmRNAの異常なスプライス部位を標的とした場合に、正確なスプライシングが効果的に回復したことが報告されている(Sierakowska, et al (1996) Proc. Natl. Acad. Sci. USA 93:12840-12844.)。 Phosphorothioated 2'-O-methyl-oligoribonucleotide targets an aberrant splice site of IVS2-654 pre-mRNA expressed in mammalian cells stably transformed with its mutant human β-globulin gene In this case, it has been reported that accurate splicing is effectively restored (Sierakowska, et al (1996) Proc. Natl. Acad. Sci. USA 93: 12840-12844.).
総説、Agrawal ((1999) Biochim. Biophys. Acta 1489:53-68)は、最適な活性のためには、アンチセンスオリゴヌクレオチドは、ヌクレアーゼに関する増大した安定性または標的RNAに対する高い親和性のみでなく、様々な性質を組み合わせる必要があることを示唆している。そのような性質は、RNase H活性を含む。後の再考において、AgrawalおよびKandimalla ((2000) Mol. Med. Today-6:72-81)は、2'-O-メチル修飾を含む混合バックボーンオリゴヌクレオチドは、それらのRNAse H活性を含む改善された性質のために、第二世代アンチセンスオリゴヌクレオチドの選択肢となっていると述べている。アンチセンスオリゴは、標的RNAに結合してRNAse Hを活性化する能力など、所定の重要な性質を有すると想定される(Agrawal and Kandimalla, 2001, Current Cancer Drug Target 1:197-209.)。殆どのアンチセンスアプローチでは、アンチセンス効能を増大させるために、RNase Hによる標的RNA切断が所望される(Kurreck, 2003, Eur. J. Biochem. 270:1628-1644.)。 A review, Agrawal ((1999) Biochim. Biophys. Acta 1489: 53-68), shows that for optimal activity, antisense oligonucleotides not only have increased stability for nucleases or high affinity for target RNA. Suggests that various properties need to be combined. Such properties include RNase H activity. In a later review, Agrawal and Kandimalla ((2000) Mol. Med. Today-6: 72-81) found that mixed backbone oligonucleotides containing 2'-O-methyl modifications were improved including their RNAse H activity. Because of its unique properties, it has become an option for second generation antisense oligonucleotides. Antisense oligos are assumed to have certain important properties, such as the ability to bind to target RNA and activate RNAse H (Agrawal and Kandimalla, 2001, Current Cancer Drug Target 1: 197-209.). In most antisense approaches, target RNA cleavage by RNase H is desired to increase antisense efficacy (Kurreck, 2003, Eur. J. Biochem. 270: 1628-1644.).
多数の研究が、アンチセンスオリゴヌクレオチドにおける2'-O-メトキシエチル修飾の使用について記載している。一例は、Zellweger et al. ((2001) J. Pharmacol. Experimental Therapeutics 298:934-940)に記載されているギャップ2'修飾オリゴヌクレオチドアンチセンスを使用した研究である。別の一例は、RNase H非依存的な2'-O-メトキシエチルアンチセンスを使用した翻訳開始複合体の形成の阻害を示している(Baker et al. 1997) J. Biol. Chem. 272:1994-12000.)。 Numerous studies have described the use of 2′-O-methoxyethyl modifications in antisense oligonucleotides. An example is a study using gap 2 ′ modified oligonucleotide antisense described in Zellweger et al. ((2001) J. Pharmacol. Experimental Therapeutics 298: 934-940). Another example shows inhibition of translation initiation complex formation using RNase H-independent 2'-O-methoxyethyl antisense (Baker et al. 1997) J. Biol. Chem. 272: 1994-12000.).
Kuwasaki et al. (2003) J. Antimicrob. Chemother. 51:813-819は、高度なヌクレアーゼ耐性の、ヌクレオシド上に2'-O-メチル基を含みかつヌクレオチド間結合上に硫黄基を含む二量体ヘアピングアノシン−四重鎖の構造、およびその培養細胞内の抗-HIV-1活性を記載している。 Kuwasaki et al. (2003) J. Antimicrob. Chemother. 51: 813-819 is a highly nuclease-resistant dimer containing a 2'-O-methyl group on the nucleoside and a sulfur group on the internucleotide linkage. Describes the structure of the body hairpin anosine-quadruplex and its anti-HIV-1 activity in cultured cells.
Mou および Gray (2002) (Nucleic Acid Res. 30:749-758)は、典型的なホスホロチオエート-DNAオリゴマーと比較して、2'-O-メチル修飾の追加は、非特異的なタンパク質結合特性を低下させることを示している。36mersオリゴヌクレオチドに関するg5pのタンパク質結合親和性は、dA36<rA36<2'-O-MeA36<S-rA36<<S-2'-O-MeA36<S-dA36 (ここで、d=デオキシ、r=リボ、2'-O-Me=2'-O-メチル、S=ホスホロチオエート)の順序で増大した。この順序は、これらのオリゴマー修飾に関して、血漿タンパク質、例えばヒト血清アルブミン、γ-グロブリンおよびフィブリノーゲンの非特異的結合に関する、Kandimallaら((1998) Bioorganic Med Chem Lett. 8:2103-2108)に報告されたS-RNA<<S-2'-O-MeRNA<S-DNAの順序と一致していた。 Mou and Gray (2002) (Nucleic Acid Res. 30: 749-758) show that the addition of 2'-O-methyl modification improves nonspecific protein binding properties compared to typical phosphorothioate-DNA oligomers. It shows that it decreases. The protein binding affinity of g5p for the 36mers oligonucleotide is: dA 36 <rA 36 <2'-O-MeA 36 <S-rA 36 <<S-2'-O-MeA 36 <S-dA 36 (where d = deoxy, r = ribo, 2′-O-Me = 2′-O-methyl, S = phosphorothioate). This order is reported to Kandimalla et al. ((1998) Bioorganic Med Chem Lett. 8: 2103-2108) for non-specific binding of plasma proteins such as human serum albumin, γ-globulin and fibrinogen with respect to these oligomer modifications. The order of S-RNA <<S-2'-O-MeRNA<S-DNA was consistent.
米国特許第5,591,623号および米国特許第5,514,788号は、細胞内付着分子の合成または代謝の調節を介した治療の可能性がある疾病の処置および診断のための組成物および方法を記載している。好ましい態様によれば、細胞内付着遺伝子をコードする核酸との特異的ハイブリダイズが可能なオリゴヌクレオチドについて記載する。本発明は、2'-O-メチルホスホロチオエートオリゴヌクレオチドの合成およびそのアンチセンスとしての使用について記載する。 US Pat. No. 5,591,623 and US Pat. No. 5,514,788 describe compositions and methods for the treatment and diagnosis of potentially therapeutic diseases through modulation of intracellular adhesion molecule synthesis or metabolism. According to a preferred embodiment, oligonucleotides capable of specific hybridization with nucleic acids encoding intracellular adhesion genes are described. The present invention describes the synthesis of 2′-O-methyl phosphorothioate oligonucleotides and their use as antisense.
米国特許第5,652,355号、米国特許第6,143,881号および米国特許第6,346,614号は、核酸分解に抵抗し、RNAまたはDNAと安定な二重鎖を形成しかつRNAとハイブリダイズした際にRNase Hを活性化する、ハイブリッドオリゴヌクレオチド(デオキシ-およびリボヌクレオチドのセグメントを含む)を記載している。ホスホロチオエート2'-O-メチル-オリゴヌクレオチドの特性の一つは、RNase Hを活性化しないことであることが示されている。一局面において、本発明は、ウイルス、病原有機体または細胞遺伝子の発現の阻害に有効なハイブリッドオリゴヌクレオチドを提供する。本発明のこの局面に従ったオリゴヌクレオチドの特徴は、デオキシリボヌクレオチドの存在である。本発明に従ったオリゴヌクレオチドは、少なくとも一つのデオキシリボヌクレオチドを含む。本発明のこの局面に従ったオリゴヌクレオチドのヌクレオチド配列は、ウイルス、病原有機体または細胞遺伝子に由来する核酸配列に相補的である。 U.S. Pat.Nos. 5,652,355, 6,143,881 and 6,346,614 resist nucleolytic degradation, form stable duplexes with RNA or DNA and activate RNase H when hybridized with RNA Hybrid oligonucleotides (including deoxy- and ribonucleotide segments) are described. One of the properties of phosphorothioate 2′-O-methyl-oligonucleotide has been shown to be that it does not activate RNase H. In one aspect, the present invention provides hybrid oligonucleotides that are effective in inhibiting the expression of viruses, pathogenic organisms or cellular genes. A feature of the oligonucleotide according to this aspect of the invention is the presence of deoxyribonucleotides. The oligonucleotide according to the invention comprises at least one deoxyribonucleotide. The nucleotide sequence of the oligonucleotide according to this aspect of the invention is complementary to a nucleic acid sequence derived from a virus, pathogenic organism or cellular gene.
米国特許第5,591,721号および米国特許第6,608,035号は、ヌクレオチド配列が標的遺伝子と相補的なオリゴヌクレオチドの経口投与による、動物内の遺伝子発現を下方調節する方法を記載している。従って、該特許に記載されている性質により、このようなオリゴヌクレオチドは、動物内の特定の遺伝子の発現を制御または下方調節する能力により、治療的に有用であると言われている。本発明の方法において有用なハイブリッドDNA/RNAオリゴヌクレオチドは、核酸分解に抵抗し、RNAまたはDNAと安定な二重鎖を形成しおよび好まくはRNAとハイブリダイズするとRNase Hを活性化する。本発明に従ったオリゴヌクレオチドは、ウイルス、病原有機体内の様々な遺伝子の発現の阻害に、または細胞遺伝子の発現の阻害に有効であることが報告されている。従って、本発明の方法に従ったオリゴヌクレオチドは、ウイルス、病原有機体または細胞遺伝子由来の核酸配列に相補的なヌクレオチド配列を有する。 US Pat. No. 5,591,721 and US Pat. No. 6,608,035 describe methods for down-regulating gene expression in animals by oral administration of oligonucleotides whose nucleotide sequences are complementary to the target gene. Thus, due to the properties described in the patent, such oligonucleotides are said to be therapeutically useful due to their ability to control or down-regulate the expression of certain genes in animals. Hybrid DNA / RNA oligonucleotides useful in the methods of the invention resist nucleolytic degradation, form stable duplexes with RNA or DNA, and preferably activate RNase H when hybridized with RNA. Oligonucleotides according to the present invention have been reported to be effective in inhibiting the expression of various genes in viruses, pathogenic organisms or in inhibiting the expression of cellular genes. Thus, an oligonucleotide according to the method of the invention has a nucleotide sequence that is complementary to a nucleic acid sequence derived from a virus, pathogenic organism or cellular gene.
米国特許第6,608,035号には、ホスホロチオエートオリゴヌクレオチドは胃内で6時間後には安定ではないが、3'末端および5'末端に2'-O-メチルリボヌクレオチドを含みかつ内部にデオキシリボヌクレオチドを含むハイブリッドホスホロチオエートオリゴヌクレオチドは、胃内でより安定であり、但し一部は分解されることを示すデータを示している。 In US Pat. No. 6,608,035, a phosphorothioate oligonucleotide is not stable in the stomach after 6 hours, but contains a 2′-O-methyl ribonucleotide at the 3 ′ and 5 ′ ends and a deoxyribonucleotide internally. The data show that phosphorothioate oligonucleotides are more stable in the stomach but are partially degraded.
発明の概要
本発明は、オリゴヌクレオチド(ON)、例えば高度に修飾されたオリゴヌクレオチドを含むオリゴデオキシヌクレオチド(ODN)が、幅広く適合可能な、配列非依存的な抗ウイルス活性を有し得るという発見に関する。有利な修飾は、修飾されたヌクレオチド間結合および2'-修飾を含む。抗ウイルス活性を有することを目的として、オリゴヌクレオチドが任意のウイルス配列に相補的であるかまたは特定のヌクレオチド分布を有する必要はない。このようなオリゴヌクレオチドはランダマーとして調製してもよく、従って調製物中、例えば32またはそれ以上のヌクレオチド長の15マイクロモルのランダマーの調製物中に、任意の特定の配列が多くて数コピー存在すると考えられる。
SUMMARY OF THE INVENTION The present invention finds that oligonucleotides (ON), such as oligodeoxynucleotides (ODN), including highly modified oligonucleotides, can have broadly adaptable, sequence-independent antiviral activity. About. Advantageous modifications include modified internucleotide linkages and 2′-modifications. In order to have antiviral activity, the oligonucleotide need not be complementary to any viral sequence or have a specific nucleotide distribution. Such oligonucleotides may be prepared as randomers, so there are at most several copies of any particular sequence in the preparation, for example in the preparation of a 15 micromolar randomer with a length of 32 or more nucleotides. I think that.
加えて、本発明者らは、異なる長さのオリゴヌクレオチドが、異なる抗ウイルス効果を有することを発見している。例えば、ホスホロチオエートヌクレオチド間結合で修飾された際、最大の抗ウイルス効果を産み出す抗ウイルスオリゴヌクレオチドの長さは、典型的に40〜120ヌクレオチドの範囲内にある。オリゴヌクレオチドの抗ウイルス特性に関する本発見の観点から、本発明は、多数の異なるウイルスに対する活性を有し得、かつ抗ウイルス薬として幅広い範囲で選択することも可能な、オリゴヌクレオチド抗ウイルス薬を提供する。そのような抗ウイルス薬は、現在入手可能な抗ウイルス治療の限られた選択肢の観点から、特に有利である。 In addition, the inventors have discovered that oligonucleotides of different lengths have different antiviral effects. For example, the length of an antiviral oligonucleotide that produces the maximum antiviral effect when modified with phosphorothioate internucleotide linkages is typically in the range of 40-120 nucleotides. In view of this discovery regarding the antiviral properties of oligonucleotides, the present invention provides oligonucleotide antiviral agents that can have activity against a number of different viruses and that can also be selected as a broad range of antiviral agents. To do. Such antiviral drugs are particularly advantageous in view of the limited options of currently available antiviral treatments.
従って、本発明のON、例えばODNはウイルス感染の処置もしくは予防、またはウイルス、例えばオンコウイルス(例、レトロウイルス、パピローマウイルスおよびヘルペスウイルス)により誘導される腫瘍もしくは癌の処置もしくは予防に関する治療法、ならびにその病因がウイルスを基礎とする他の疾病の処置または予防に有用である。そのような処置は、例えばヒトおよび動物の免疫抑制患者のウイルス感染の予防または処置を含む、多くの種類の患者および処置に適用される。 Accordingly, ONs of the invention, such as ODNs, are therapeutic or therapeutic for the treatment or prevention of viral infections, or the treatment or prevention of tumors or cancers induced by viruses such as oncoviruses (e.g. retroviruses, papillomaviruses and herpesviruses) As well as the treatment or prevention of other diseases whose etiology is based on viruses. Such treatment applies to many types of patients and treatments including, for example, prevention or treatment of viral infections in human and animal immunosuppressed patients.
本発明の第一の局面は、抗ウイルス性が基本的には配列非依存的に、例えば配列相補的ではない作用様式で発生する抗ウイルスオリゴヌクレオチド、例えば精製オリゴヌクレオチド、およびそのようなオリゴヌクレオチドを含有する調合物に関する。 A first aspect of the present invention is that antiviral oligonucleotides in which antiviral properties occur essentially in a sequence independent manner, eg, in a mode of action that is not sequence complementary, such as purified oligonucleotides, and such oligonucleotides. Relates to a formulation containing
本発明において有用なオリゴヌクレオチドは、様々な長さ、例えば少なくとも6、10、14、15、20、21、22、23、24、25、26、27、28、29、30、35、38、40、45、50、60、70、80、90、100、110、120、140、160またはそれ以上のヌクレオチド長を有し得る。同様に、オリゴヌクレオチドは、例えば前記した値の任意の二つを、その範囲の終点を含むものとして定義する範囲内、例えば10〜20、20〜30、20〜40、30〜40、30〜50、40〜50、40〜60、40〜80、50〜60、50〜70、60〜70、70〜80、60〜120および80〜120範囲内のヌクレオチド長を有し得る。特定の態様において、最小の長さまたは長さの範囲は、本抗ウイルスオリゴヌクレオチドに関して本明細書に列挙した、他の任意のオリゴヌクレオチドの詳細と組み合わされる。 Oligonucleotides useful in the invention can be of various lengths, e.g. at least 6, 10, 14, 15, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 38, It may have a nucleotide length of 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 140, 160 or more. Similarly, oligonucleotides may be used in a range that defines any two of the above values as including the end of the range, such as 10-20, 20-30, 20-40, 30-40, 30- It may have a nucleotide length in the range of 50, 40-50, 40-60, 40-80, 50-60, 50-70, 60-70, 70-80, 60-120 and 80-120. In certain embodiments, the minimum length or length range is combined with any other oligonucleotide details listed herein for the antiviral oligonucleotide.
抗ウイルスヌクレオチドは、様々な修飾、例えば安定化修飾を含んでもよく、従って少なくとも一つの修飾をホスホジエステル結合内に、ならびに/または糖、および/もしくは塩基上に含んでもよい。例えば、オリゴヌクレオチドは、1個または複数のホスホロチオエート結合、ホスホロジチオエート結合および/またはメチルホスホネート結合を含んでもよい。例えば、合成条件が化学的に適合可能である修飾など、化学的に適合可能な異なる修飾結合を組み合わせることができる。修飾結合が有用である一方、オリゴヌクレオチドは、ホスホジエステル結合、例えば少なくとも一つのホスホジエステル結合、または少なくとも5、10、20、30%もしくはそれ以上のホスホジエステル結合を含み得る。更なる有用な修飾は、糖の2'位における修飾、2'-O-メチル修飾のような2'-O-アルキル修飾、2'-アミノ修飾、2'-フルオロのような2'-ハロ修飾;非環式ヌクレオチド類似体を含むが、それらに限定されない。他の修飾も当該技術分野にて公知であり、使用することができる。特定の態様において、オリゴヌクレオチドは、全体に修飾結合、例えばホスホロチオエートを有する;3'-および/または5'-キャップを有する;末端3'-5'結合を含む;オリゴヌクレオチドは、リンカーにより連結された2個またはそれ以上のオリゴヌクレオチド配列からなる鎖状体であるか、または該鎖状体を含む。 Antiviral nucleotides may include various modifications, such as stabilizing modifications, and thus may include at least one modification within the phosphodiester linkage and / or on the sugar and / or base. For example, the oligonucleotide may comprise one or more phosphorothioate linkages, phosphorodithioate linkages and / or methylphosphonate linkages. Different modified bonds that are chemically compatible can be combined, for example, modifications in which the synthesis conditions are chemically compatible. While modified linkages are useful, oligonucleotides can include phosphodiester linkages, such as at least one phosphodiester linkage, or at least 5, 10, 20, 30% or more phosphodiester linkages. Further useful modifications are modifications at the 2 ′ position of the sugar, 2′-O-alkyl modifications such as 2′-O-methyl modifications, 2′-amino modifications, 2′-halo such as 2′-fluoro. Modifications; including but not limited to acyclic nucleotide analogs. Other modifications are known in the art and can be used. In certain embodiments, the oligonucleotide has a modified bond, eg, phosphorothioate, has a 3′- and / or 5′-cap; contains a terminal 3′-5 ′ bond; the oligonucleotides are linked by a linker Or a chain consisting of two or more oligonucleotide sequences.
本発明は更に、1個または複数のヌクレオチド残基において、オリゴヌクレオチドの性質を変更して、より高い安定性、血清とのより低い相互作用、より高い細胞取り込み、ウイルスタンパク質とのより高い相互作用、送達用に調合するための改善された能力、検出可能なシグナル、より高い抗ウイルス活性、より良好な薬物動態学的性質、特定の組織分布、より低い毒性からなる群より選択される1個または複数の性質を獲得する分子に対して結合または複合化されているオリゴヌクレオチドを提供する。 The invention further modifies the nature of the oligonucleotide at one or more nucleotide residues to provide higher stability, lower interaction with serum, higher cellular uptake, higher interaction with viral proteins. 1 selected from the group consisting of improved ability to formulate for delivery, detectable signal, higher antiviral activity, better pharmacokinetic properties, specific tissue distribution, lower toxicity Alternatively, an oligonucleotide is provided that is bound or conjugated to a molecule that acquires multiple properties.
所定の態様において、オリゴヌクレオチドは、少なくとも10、20、30、40、50、60、70、80、90、95または100%の修飾結合、例えばホスホロチオエート、ホスホロジチオエートおよび/またはメチルホスホネートを含む。 In certain embodiments, the oligonucleotide comprises at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95 or 100% modified linkages such as phosphorothioate, phosphorodithioate and / or methylphosphonate. .
所定の態様において、少なくとも10、20、30、40、50、60、70、80、90もしくは95%、または全部のヌクレオチドが、リボースの2'位において、例えば2'-OMe、2'-F、2'-アミノで修飾されている。 In certain embodiments, at least 10, 20, 30, 40, 50, 60, 70, 80, 90 or 95%, or all nucleotides, are at the 2 ′ position of ribose, for example 2′-OMe, 2′-F. , Modified with 2'-amino.
所定の態様において、修飾結合はオリゴヌクレオチドの2'-修飾と組み合わされ、例えば、少なくとも30%の修飾結合と少なくとも30%の2'-修飾;または各々少なくとも40%と40%、少なくとも50%と50%、少なくとも60%と60%、少なくとも70%と70%、少なくとも80%と80%、少なくとも90%と90%、100%と100%のように組み合わされる。所定の態様において、オリゴヌクレオチドは、少なくとも30、40、50、60、70、80、90または100%の修飾結合と、少なくとも30、40、50、60、70、80、90または100%の2'-修飾とを含み、ここで、該態様は、列挙した各修飾結合の割合と2'-修飾の割合との組み合わせを含む(例、少なくとも50%の修飾結合と少なくとも80%の2'-修飾、および少なくとも80%の修飾結合と100%の2'-修飾)。記載した組み合わせの割合の各々の特定の態様において、修飾結合はホスホロチオエート結合である;修飾結合がホスホロジチオエート結合である;2'-修飾が2'-OMeである;2'-修飾が2'-フルオロである;2'-修飾が2'-OMeと2'-フルオロの組み合わせである;修飾結合がホスホロチオエート結合でありかつ2'-修飾が2'-OMeである;修飾結合がホスホロチオエート結合でありかつ2'-修飾が2'-フルオロである;修飾結合がホスホロジチオエート結合でありかつ2'-修飾が2'-OMeである;修飾結合がホスホロジチオエート結合でありかつ2'-修飾が2'-フルオロである;修飾結合がホスホロジチオエート結合でありかつ2'-修飾が2'-OMeと2'-フルオロの組み合わせである。特定パーセントの修飾結合と特定の割合の2'-修飾を組み合わせる本明細書に記載されたオリゴヌクレオチドの所定の態様において、オリゴヌクレオチドは少なくとも15、20、25、30、35、40、50、60、70、80、90、100、110もしくは120ヌクレオチド長であるか、または特定の長さの任意の二つを、その範囲の終点を含むものとして定義する範囲内の長さである。 In certain embodiments, the modified linkage is combined with a 2′-modification of the oligonucleotide, eg, at least 30% modified linkage and at least 30% 2′-modification; or at least 40% and 40%, at least 50%, respectively. Combined as 50%, at least 60% and 60%, at least 70% and 70%, at least 80% and 80%, at least 90% and 90%, 100% and 100%. In certain embodiments, the oligonucleotide has at least 30, 40, 50, 60, 70, 80, 90 or 100% modified linkage and at least 30, 40, 50, 60, 70, 80, 90 or 100% 2 Wherein the embodiment includes a combination of the percentage of each modified bond listed and the percentage of 2'-modification (e.g., at least 50% modified bonds and at least 80% 2'- Modification, and at least 80% modified bonds and 100% 2'-modification). In each particular embodiment of the described combination proportions, the modified bond is a phosphorothioate bond; the modified bond is a phosphorodithioate bond; 2′-modification is 2′-OMe; 2′-modification is 2 '-Fluoro; 2'-modification is a combination of 2'-OMe and 2'-fluoro; the modified bond is a phosphorothioate bond and the 2'-modification is 2'-OMe; the modified bond is a phosphorothioate bond And the 2'-modification is 2'-fluoro; the modified bond is a phosphorodithioate bond and the 2'-modification is 2'-OMe; the modified bond is a phosphorodithioate bond and 2 The '-modification is 2'-fluoro; the modified bond is a phosphorodithioate bond and the 2'-modification is a combination of 2'-OMe and 2'-fluoro. In certain embodiments of the oligonucleotides described herein that combine a certain percentage of modified linkages and a certain percentage of 2'-modifications, the oligonucleotide is at least 15, 20, 25, 30, 35, 40, 50, 60 , 70, 80, 90, 100, 110 or 120 nucleotides in length, or a length within a range defining any two of the specified lengths as including the end of the range.
所定の態様において、1、2、3、4、5、6、7、8、9または10個のヌクレオチド間結合を除いた全部のヌクレオチド間結合、および/またはリボース部分の2'位が、例えばホスホロチオエート、ホスホロジチオエートまたはメチルホスホネート結合により、ならびに/またはリボース部分の2'-OMe、2'-F、および/もしくは2'-アミノ修飾により修飾される。 In certain embodiments, all internucleotide linkages except 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 internucleotide linkages, and / or the 2 ′ position of the ribose moiety are, for example, It is modified by phosphorothioate, phosphorodithioate or methylphosphonate linkages and / or by 2'-OMe, 2'-F, and / or 2'-amino modifications of the ribose moiety.
数個の態様において、オリゴヌクレオチドは、少なくとも1、2、3もしくは4個のリボヌクレオチドを含むか、または少なくとも10、20、30、40、50、60、70、80、90%もしくは100%ものリボヌクレオチドを含む。 In some embodiments, the oligonucleotide comprises at least 1, 2, 3 or 4 ribonucleotides, or at least 10, 20, 30, 40, 50, 60, 70, 80, 90% or 100% Contains ribonucleotides.
特定の態様において、オリゴヌクレオチドは、鎖内に(即ち、鎖バックボーンの一部を形成する)および/または側鎖部分として、例えば1、2、3、4、5、6、7、8、9、10個もしくはそれ以上もの、または5、10、20%もしくはそれ以上までの鎖部分および/または側鎖部分に非ヌクレオチド基を含む。 In certain embodiments, the oligonucleotide is present in the strand (ie, forming part of the strand backbone) and / or as a side chain portion, eg, 1, 2, 3, 4, 5, 6, 7, 8, 9 , 10 or more, or up to 5, 10, 20% or more of the chain and / or side chain moieties containing non-nucleotide groups.
所定の態様において、オリゴヌクレオチドは、5、8、10、15、20、25、30ヌクレオチドよりも長い自己相補的な配列を有さない;オリゴヌクレオチドは、触媒活性、例えばRNAに対する切断活性を有さない;オリゴヌクレオチドは、RNAi機序を誘導しない。 In certain embodiments, the oligonucleotide has no self-complementary sequence longer than 5, 8, 10, 15, 20, 25, 30 nucleotides; the oligonucleotide has catalytic activity, eg, cleavage activity on RNA. No; oligonucleotides do not induce RNAi mechanisms.
特定の態様において、オリゴヌクレオチドは、1個または複数のウイルスタンパク質に結合する;オリゴヌクレオチドの配列(またはその一部、例えば少なくとも20、30、40、50、60、70%もしくはそれ以上の配列)は、ウイルスゲノムに由来する;ウイルスゲノム由来の配列を伴うオリゴヌクレオチドの活性は、同一長のランダマーオリゴヌクレオチドもしくはランダムオリゴヌクレオチドに優位ではない;オリゴヌクレオチドは、ウイルス配列に相補的な部分と、ウイルス配列に相補的ではない部分とを含む;オリゴヌクレオチドの配列は、ウイルスパッケージング配列、またはアプタマー相互作用に関与する他のウイルス配列に由来する;別に示さない限り、オリゴヌクレオチド配列は、A(x)、C(x)、G(x)、T(x)、U(x)、l(x)、AC(x)、AG(x)、AT(x)、AU(x)、CG(x)、CT(x)、CU(x)、GT(x)、GU(x)、TU(x)、Al(x)、IC(x)、IG(X)、IT(x)IU(x)を含み、ここで、xは、2、3、4、5、6、...60...120である(特定の態様において、オリゴヌクレオチドは、少なくとも15、20、25、29、30、32、34、35、36、38、40、45、46、50、60、70、80、90、100、110、120、140もしくは160ヌクレオチド長、もしくは列挙した値の任意の二つを、その範囲の終点を含むものとして定義する範囲内の長さであるか、または特定した反復配列の長さが、少なくとも、直前に特定した長さもしくは長さの範囲内である);オリゴヌクレオチドは、例えば、上記の単量体および/または二量体の各組み合わせが、一度に2、3または4回反復される各組み合わせを含む、反復配列の組み合わせ(例、上記に特定した反復配列)を含む;オリゴヌクレオチドが一本鎖(RNAまたはDNA)である;オリゴヌクレオチドが二本鎖(RNAまたはDNA)である;オリゴヌクレオチドが少なくとも一つのGカルテット(Gquartet)またはCpG部分を含む;オリゴヌクレオチドがウイルスmRNAに相補的な部分を含みかつ少なくとも29、37もしくは38ヌクレオチド長(または上記に特定した他の長さ)である;オリゴヌクレオチドが、少なくとも一つの非ワトソン-クリックオリゴヌクレオチド、および/または他の一つのヌクレオチドとの非ワトソン-クリック結合に参加する少なくとも一つヌクレオチド、および/または他のヌクレオチドと塩基対を形成できない少なくとも一つのヌクレオチドとを含む;オリゴヌクレオチドがランダムオリゴヌクレオチドである、オリゴヌクレオチドがランダマーであるか、またはランダマー部分を含み、例えば少なくとも5、10、15、20、25、30、35、40もしくはそれ以上の連続したオリゴヌクレオチドの長さを有するか、もしくは上記にオリゴヌクレオチド長に関して特定した長さを有するランダマー部分を含むか、または少なくとも10、20、30、40、50、60、70、80、90%もしくは全部のヌクレオチドがランダマーである;オリゴヌクレオチドが、1個または複数のヌクレオチド残基において、オリゴヌクレオチドの性質を変更する、例えばより高い安定性(血清中での安定性または特定の溶液中での安定性など)、血清とのより低い相互作用、より高い細胞取り込み、ウイルスタンパク質とのより高い相互作用、送達用に調合するための改善された能力、検出可能なシグナル、改善された薬物動態学的性質、特定の組織分布、および/またはより低い毒性を提供する分子に対して結合または複合化している。 In certain embodiments, the oligonucleotide binds to one or more viral proteins; the sequence of the oligonucleotide (or a portion thereof, eg, at least 20, 30, 40, 50, 60, 70% or more sequences) Is derived from the viral genome; the activity of oligonucleotides with sequences derived from the viral genome is not superior to random-length or random oligonucleotides of the same length; the oligonucleotide comprises a portion complementary to the viral sequence and the virus The sequence of the oligonucleotide is derived from a viral packaging sequence or other viral sequence involved in aptamer interaction; unless otherwise indicated, the oligonucleotide sequence is A (x ), C (x), G (x), T (x), U (x), l (x), AC (x), AG (x), AT (x), AU (x), CG (x ), CT (x), CU (x), GT (x), GU (x), TU (x), Al (x), IC (x), IG (X), IT (x) IU (x) Where x is 2, 3, 4, 5, 6, ... 60 ... 120 (in certain embodiments, the oligonucleotide is at least 15, 20, 25, 29, 30, 32, 34, 35, 36, 38, 40, 45, 46, 50, 60, 70, 80, 90, 100, 110, 120, 140 or 160 nucleotides in length, or any two of the listed values, The length within the range defined as including the end of the range, or the length of the identified repetitive sequence is at least within the length or length range specified immediately above); For example, each combination of the above monomers and / or dimers comprises a combination of repetitive sequences (eg, the repetitive sequences specified above), including each combination repeated 2, 3, or 4 times at a time. The oligonucleotide is single stranded (RNA or DNA); The oligonucleotide is double stranded (RNA or DNA); the oligonucleotide comprises at least one G quartet or CpG moiety; the oligonucleotide comprises a part complementary to the viral mRNA and at least 29, 37 or 38 Nucleotide length (or other length as specified above); at least one non-Watson-Crick oligonucleotide and / or at least one participating in non-Watson-Crick binding with one other nucleotide One nucleotide, and / or at least one nucleotide that cannot base pair with another nucleotide; the oligonucleotide is a random oligonucleotide, the oligonucleotide is a randomer, or includes a randomer moiety, such as at least 5, 10, 15, 20, 25 30, 35, 40 or more continuous oligonucleotides, or comprising a randomer moiety having a length specified above with respect to the oligonucleotide length, or at least 10, 20, 30, 40, 50, 60, 70, 80, 90% or all nucleotides are randomers; the oligonucleotide alters the properties of the oligonucleotide at one or more nucleotide residues, eg, higher stability (in serum Stability or in a particular solution), lower interaction with serum, higher cellular uptake, higher interaction with viral proteins, improved ability to be formulated for delivery, detection Bind to molecules that provide possible signals, improved pharmacokinetic properties, specific tissue distribution, and / or lower toxicity It is compounded.
シトシンおよび/またはチミジンおよび/または他のピリミジンを含む、ピリミジンヌクレオチドのみ(または、少なくとも主として)を含有するホスホロチオエート化ONは、低pHに耐性を有し、またポリシトシンオリゴヌクレオチドは多数のヌクレアーゼに対して増大した耐性を示し、それにより抗ウイルスONの経口投与のための二つの重要な性質を提供することが発見された。従って、所定の態様において、オリゴヌクレオチドは、少なくとも80、90、もしくは95または100%の修飾されたヌクレオチド間結合(例、ホスホロチオエートまたはホスホロジチオエート)を有し、ピリミジン含有量が50%より多い、60%より多い、70%より多い、80%より多い、90%より多いもしくは100%であり、即ちピリミジンオリゴヌクレオチドであるか、またはシトシン含有量が50%より多い、60%より多い、70%より多い、80%より多い、90%より多いもしくは100%であり;即ちポリシトシンオリゴヌクレオチドである。所定の態様において、長さは、少なくとも29、30、32、34、36、38、40、45、50、60、70もしくは80ヌクレオチド、または20〜28、25〜35、29〜40、30〜40、35〜45、40〜50、45〜55、50〜60、55〜65、60〜70、65〜75もしくは70〜80の範囲内、または列挙した値の任意の二つを、その範囲の終点を含むものとして定義する範囲内の長さである。特定の態様において、オリゴヌクレオチドは、少なくとも50、60、70、80または90%シトシンである;少なくとも50、60、70、80または90%チミジン(および、上記に列挙した総ピリミジン含有量を有し得る)である。特定の態様において、オリゴヌクレオチドは、列挙した割合のシトシンおよびチミジンのいずれかを含み、残りのピリミジンヌクレオチドは、シトシンおよびチミジンのもう一方である。また所定の態様において、オリゴヌクレオチドは、少なくとも10、12、14、16、18、20、25、30、35、40またはそれ以上の連続するピリミジンヌクレオチド、例えばCヌクレオチド、Tヌクレオチド、またはCTジヌクレオチド対を含む。所定の態様において、ピリミジンオリゴヌクレオチドは、ピリミジンヌクレオチドのみからなる;少なくとも1、2、3、4、5、6、7、8、9または10個の非ピリミジン部分を含む;1〜5、6〜10、11〜15または少なくとも16個の非ピリミジンバックボーン部分を含む;少なくとも一つの、1〜20、1〜5、6〜10、11〜15または16〜20個の非ヌクレオチド部分を含む;少なくとも一つの、1〜20、1〜5、6〜10、11〜15または16〜20個のプリンヌクレオチドを含む。好ましくは、非ヌクレオチド部分が存在する態様において、そのような部分間の結合、またはそのような部分とヌクレオチドとの間の結合は、オリゴヌクレオチドのサイズおよび化学的性質に適切な条件下でのゲル電気泳動により評価して、40-mer ポリCオリゴヌクレオチドのPS結合と同様に、酸性条件に対して少なくとも25、35、50、70、90または100%耐性である。 Phosphorothioated ON containing only (or at least primarily) pyrimidine nucleotides, including cytosine and / or thymidine and / or other pyrimidines, is resistant to low pH, and polycytosine oligonucleotides are resistant to numerous nucleases. Have been found to exhibit increased resistance and thereby provide two important properties for oral administration of antiviral ON. Thus, in certain embodiments, the oligonucleotide has at least 80, 90, or 95 or 100% modified internucleotide linkages (eg, phosphorothioate or phosphorodithioate) and has a pyrimidine content greater than 50%. More than 60%, more than 70%, more than 80%, more than 90% or 100%, i.e. it is a pyrimidine oligonucleotide or has a cytosine content of more than 50%, more than 60%, 70 Greater than%, greater than 80%, greater than 90% or 100%; ie a polycytosine oligonucleotide. In certain embodiments, the length is at least 29, 30, 32, 34, 36, 38, 40, 45, 50, 60, 70 or 80 nucleotides, or 20-28, 25-35, 29-40, 30- 40, 35-45, 40-50, 45-55, 50-60, 55-65, 60-70, 65-75 or 70-80, or any two of the listed values within the range The length within the range defined as including the end point of. In certain embodiments, the oligonucleotide is at least 50, 60, 70, 80 or 90% cytosine; at least 50, 60, 70, 80 or 90% thymidine (and has a total pyrimidine content as listed above). Get). In certain embodiments, the oligonucleotide comprises any of the listed proportions of cytosine and thymidine and the remaining pyrimidine nucleotides are the other of cytosine and thymidine. Also, in certain embodiments, the oligonucleotide is at least 10, 12, 14, 16, 18, 20, 25, 30, 35, 40 or more consecutive pyrimidine nucleotides, such as C nucleotides, T nucleotides, or CT dinucleotides Includes pairs. In certain embodiments, the pyrimidine oligonucleotide consists solely of pyrimidine nucleotides; comprises at least 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 non-pyrimidine moieties; 1-5, 6- 10, 11-15 or at least 16 non-pyrimidine backbone moieties; at least one 1-20, 1-5, 6-10, 11-15 or 16-20 non-nucleotide moieties; at least one Contains 1-20, 1-5, 6-10, 11-15 or 16-20 purine nucleotides. Preferably, in embodiments where non-nucleotide moieties are present, the linkage between such moieties, or between such moieties and the nucleotide, is a gel under conditions appropriate to the size and chemistry of the oligonucleotide. As assessed by electrophoresis, it is at least 25, 35, 50, 70, 90 or 100% resistant to acidic conditions, similar to PS binding of 40-mer polyC oligonucleotides.
オリゴヌクレオチドは、組み合わせて例えば混合物として使用してもよい。そのような組み合わせまたは混合物は、例えば、少なくとも2、3、4、5、10、20、50、100、1000、10,000、100,000、1,000,000またはそれ以上の異なるオリゴヌクレオチドを含み得、例えばオリゴヌクレオチドの任意の組み合わせは、本明細書において記載されている。そのような組み合わせまたは混合物は、例えば、異なる配列、および/または異なる長さ、および/または異なる修飾、および/または異なるリンカーまたは複合分子を有し得る。そのような組み合わせまたは混合物の複数の特定の態様において、複数のオリゴヌクレオチドは最小の長さを有し、またはオリゴヌクレオチドに関して上述したような範囲内の長さを有する。このような組み合わせまたは混合物の特定の態様において、少なくとも一つの、複数の、または各オリゴヌクレオチドは、個々の抗ウイルスオリゴヌクレオチドに関して本明細書において詳細する任意の他の性質を有し得る(両立する任意の組み合わせを有してもよい)。 Oligonucleotides may be used in combination, for example as a mixture. Such combinations or mixtures can include, for example, at least 2, 3, 4, 5, 10, 20, 50, 100, 1000, 10,000, 100,000, 1,000,000 or more different oligonucleotides, such as any of the oligonucleotides Combinations of are described herein. Such combinations or mixtures can have, for example, different sequences, and / or different lengths, and / or different modifications, and / or different linkers or complex molecules. In certain embodiments of such combinations or mixtures, the plurality of oligonucleotides have a minimum length or have a length within the range as described above for oligonucleotides. In certain embodiments of such combinations or mixtures, at least one, multiple, or each oligonucleotide may have any other properties detailed herein for individual antiviral oligonucleotides (compatible) Any combination).
所定の態様において、オリゴヌクレオチドの配列は、標的ウイルスのゲノム配列の任意の等しい長さ部分に対して完全には相補的でなく、または標的ウイルスのゲノム配列の任意の等しい長さ部分に対して95、90、80、70、60もしくは50%未満の相補性を有する。オリゴヌクレオチド配列は、本質的にポリA、ポリC、ポリG、ポリT、GカルテットまたはTGリッチ配列からなるわけではない。 In certain embodiments, the sequence of the oligonucleotide is not completely complementary to any equal length portion of the target virus genomic sequence, or to any equal length portion of the target virus genomic sequence. Have less than 95, 90, 80, 70, 60 or 50% complementarity. The oligonucleotide sequence does not consist essentially of a poly A, poly C, poly G, poly T, G quartet or TG rich sequence.
本発明のオリゴに関連して使用されるように、用語「TGリッチ」は、抗ウイルスオリゴヌクレオチドの配列が、少なくとも50パーセントのTおよびGヌクレオチドからなり、またはそのように特定された場合、少なくとも60、70、80、90もしくは95%、または100%ものTおよびGからなることを示す。 As used in connection with the oligos of the present invention, the term “TG rich” refers to an antiviral oligonucleotide sequence comprising at least 50 percent T and G nucleotides, or at least as specified. 60, 70, 80, 90 or 95%, or 100% T and G.
関連する局面において、本発明は、本明細書に記載する少なくとも二種の、例えば少なくとも2、3、4、5、7、10、50、100、1000、10,000、100,000、1,000,000またはそれ以上の異なる抗ウイルスオリゴヌクレオチドを含有する抗ウイルスオリゴヌクレオチドの混合物を提供する。 In related aspects, the present invention provides at least two, eg, at least 2, 3, 4, 5, 7, 10, 50, 100, 1000, 10,000, 100,000, 1,000,000 or more different as described herein. A mixture of antiviral oligonucleotides containing antiviral oligonucleotides is provided.
本明細書においてオリゴヌクレオチドまたは他の物質に関連して使用されるように、用語「抗ウイルス」は系において、さもなければ少なくとも一種のウイルスの感染性ウイルス粒子の形成に適切な系において、ウイルス粒子の産生を阻害する、即ち形成される感染性ウイルス粒子数を低下させる、オリゴヌクレオチドまたは他の物質の存在の効果を指す。本発明の所定の態様において、抗ウイルスオリゴヌクレオチドは、多数の異なるウイルスに対して抗ウイルス活性を有すると想定される。 As used herein in connection with oligonucleotides or other substances, the term “antiviral” refers to viral in a system, otherwise in a system suitable for the formation of infectious viral particles of at least one virus. Refers to the effect of the presence of oligonucleotides or other substances that inhibit the production of particles, ie reduce the number of infectious virus particles formed. In certain embodiments of the invention, the antiviral oligonucleotide is assumed to have antiviral activity against a number of different viruses.
用語「抗ウイルスオリゴヌクレオチド製剤」は、抗ウイルス薬としての使用に適合された少なくとも一種の抗ウイルスオリゴヌクレオチドを含有する調製物を指す。製剤は、一種または複数種のオリゴヌクレオチドを含有し、インビボで抗ウイルス薬としての製剤の用途を妨害しない他の物質を含有し得る。そのような他の物質は、希釈剤、賦形剤、担体物質、および/または他の抗ウイルス物質を含むが、それらに限定されるわけではない。 The term “antiviral oligonucleotide formulation” refers to a preparation containing at least one antiviral oligonucleotide adapted for use as an antiviral agent. The formulation contains one or more oligonucleotides and may contain other substances that do not interfere with the use of the formulation as an antiviral agent in vivo. Such other materials include, but are not limited to, diluents, excipients, carrier materials, and / or other antiviral materials.
本明細書において使用される場合、用語「薬学的組成物」は、生理学的または薬学的に許容される担体または賦形剤を含有する抗ウイルスオリゴヌクレオチド製剤を指す。そのような組成物は、組成物を所望の対象、例えばヒトへの投与に対して不適切なものとしない他の成分も含み得る。 As used herein, the term “pharmaceutical composition” refers to an antiviral oligonucleotide formulation containing a physiologically or pharmaceutically acceptable carrier or excipient. Such compositions can also include other ingredients that do not render the composition unsuitable for administration to a desired subject, eg, a human.
本発明の文脈において、特に限定しなければ、用語「オリゴヌクレオチド(ON)」は、オリゴデオキシヌクレオチド(ODN)もしくはオリゴデオキシリボヌクレオチドまたはオリゴリボヌクレオチドを意味する。従って、「オリゴヌクレオチド」は、リボ核酸(RNA)および/またはデオキシリボ核酸(DNA)、および/またはそれらの類似体のオリゴマーまたはポリマーを指す。この用語は、天然に存在する核酸塩基、糖およびヌクレオシド間の共有結合(バックボーン)からなるオリゴヌクレオチド、ならびに同様に機能する、天然に存在しない部分を有するオリゴヌクレオチドを含む。このような修飾または置換オリゴヌクレオチドは、所望の性質、例えば、向上した細胞取り込み、核酸標的に対する向上した親和性およびヌクレアーゼ存在下での安定性の増大などにより、天然の形態よりも好ましい場合がある。使用し得る修飾の例は、本明細書に記載されている。バックボーンおよび/または他の修飾を含むオリゴヌクレオチドも、オリゴヌクレオシドと称され得る。 In the context of the present invention, unless otherwise limited, the term “oligonucleotide (ON)” means an oligodeoxynucleotide (ODN) or oligodeoxyribonucleotide or oligoribonucleotide. Thus, “oligonucleotide” refers to an oligomer or polymer of ribonucleic acid (RNA) and / or deoxyribonucleic acid (DNA), and / or analogs thereof. The term includes naturally occurring nucleobases, oligonucleotides consisting of covalent bonds (backbones) between sugars and nucleosides, as well as oligonucleotides having non-naturally occurring moieties that function similarly. Such modified or substituted oligonucleotides may be preferred over the natural form due to the desired properties, such as improved cellular uptake, improved affinity for nucleic acid targets and increased stability in the presence of nucleases. . Examples of modifications that can be used are described herein. Oligonucleotides containing backbone and / or other modifications can also be referred to as oligonucleosides.
本発明の文脈において、「修飾されたヌクレオチド間結合」という表現は、天然に存在するポリヌクレオチドに一般に見られるホスホジエステル結合とは異なる、オリゴヌクレオチドのヌクレオチドまたはヌクレオチド類似体間の結合を指す。例としては、ホスホロチオエート結合、ホスホロジチオエート結合およびメチルホスホネート結合がある。 In the context of the present invention, the expression “modified internucleotide linkage” refers to a linkage between nucleotides or nucleotide analogues of an oligonucleotide that is different from the phosphodiester linkages commonly found in naturally occurring polynucleotides. Examples are phosphorothioate linkages, phosphorodithioate linkages and methylphosphonate linkages.
オリゴヌクレオチドの特定の長さの詳細、例えば少なくとも20ヌクレオチド長は、オリゴヌクレオチドが少なくとも20個の結合したヌクレオチドを含むことを意味する。明確に否定しない場合、オリゴヌクレオチドは、オリゴヌクレオチド鎖のバックボーンの一部を形成する、付加的な非ヌクレオチド部分も含んでもよい。別に示さない限り、バックボーンに非ヌクレオチド部分が存在する場合、少なくとも10個の結合したヌクレオチドは連続している。 A specific length detail of an oligonucleotide, eg, at least 20 nucleotides long, means that the oligonucleotide comprises at least 20 linked nucleotides. If not explicitly denied, the oligonucleotide may also contain additional non-nucleotide moieties that form part of the backbone of the oligonucleotide chain. Unless otherwise indicated, when there is a non-nucleotide moiety in the backbone, at least 10 linked nucleotides are contiguous.
抗ウイルス製剤、薬学的組成物、または他の材料に関連して使用される、「抗ウイルス薬としての使用に適合された」という表現は、物質が抗ウイルス効果を示し、例えばヒト対象のような対象に投与するための、インビボ系でのウイルス産生阻害における使用に対して該物質を不適切なものとする、いずれの成分または物質も含有しないことを示す。 As used in connection with antiviral formulations, pharmaceutical compositions, or other materials, the expression “adapted for use as an antiviral agent” means that the substance exhibits an antiviral effect, such as in a human subject. It does not contain any component or substance that renders the substance inappropriate for use in inhibiting virus production in an in vivo system for administration to a subject.
抗ウイルスオリゴヌクレオチドの抗ウイルス作用に関連して本明細書において使用される場合、「配列非依存的な作用様式」とは、特定の生物学的活性(例、抗ウイルス活性)が、オリゴヌクレオチド内の特定のオリゴヌクレオチド配列に依存しないこと示す。例えば、活性は、アンチセンス活性または配列依存的なアプタマー相互作用を起こす特定の配列のような、配列依存的なハイブリダイゼーションに依存しない。同様に、「配列相補的ではない作用様式」という表現は、それにより物質が抗ウイルス効果を示す機序が、相補的な核酸配列のハイブリダイゼーション、例えばアンチセンス効果を原因としないことを示す。逆に、「配列相補的な作用様式」は、物質の抗ウイルス効果が、相補的な核酸配列のハイブリダイゼーションまたは配列特異的なアプタマー相互作用に関与することを意味する。従って、物質の抗ウイルス活性が「配列非依存的な作用様式を原因とする」、または活性が「主として配列相補的な作用様式を原因としない」と示される場合、オリゴヌクレオチドの活性は、本明細書において提供する4つの試験のうち少なくとも一つを満たすことを意味する(実施例10参照)。特定の態様において、オリゴヌクレオチドは、試験1、試験2、試験3、試験4または試験5を満たす;オリゴヌクレオチドは、二つの試験の組み合わせ、即ち試験1および2;試験1および3;試験1および4、試験1および5、試験2および3、試験2および4、試験2および5、試験3および4、試験3および5、または試験4および5を満たす;オリゴヌクレオチドは、3つの試験の組み合わせ、即ち試験1、2および3、試験1、2および4、試験1、2および5、試験1、3および4、試験1、3および5、試験2、3および4、試験2、3および5、試験3、4および5を満たす;オリゴヌクレオチドは、試験1、2、3および4の全てを満たす。 As used herein in connection with the antiviral activity of an antiviral oligonucleotide, a “sequence-independent mode of action” is a specific biological activity (eg, antiviral activity) where the oligonucleotide Independence from specific oligonucleotide sequences. For example, activity does not depend on sequence-dependent hybridization, such as antisense activity or specific sequences that cause sequence-dependent aptamer interactions. Similarly, the expression “mode of action that is not sequence complementary” indicates that the mechanism by which the substance exhibits an antiviral effect is not due to hybridization of complementary nucleic acid sequences, eg, an antisense effect. Conversely, “sequence-complementary mode of action” means that the antiviral effect of the substance is involved in the hybridization of complementary nucleic acid sequences or sequence-specific aptamer interactions. Thus, if the antiviral activity of a substance is “due to a sequence-independent mode of action” or the activity is indicated as “not primarily due to a sequence-complementary mode of action”, the activity of the oligonucleotide is Means meeting at least one of the four tests provided in the specification (see Example 10). In certain embodiments, the oligonucleotide meets Test 1, Test 2, Test 3, Test 4 or Test 5; the oligonucleotide is a combination of two tests, namely Test 1 and 2; Test 1 and 3; Test 1 and 4, test 1 and 5, test 2 and 3, test 2 and 4, test 2 and 5, test 3 and 4, test 3 and 5, or test 4 and 5; oligonucleotide is a combination of three tests, Test 1, 2 and 3, test 1, 2 and 4, test 1, 2 and 5, test 1, 3 and 4, test 1, 3 and 5, test 2, 3 and 4, test 2, 3 and 5, Satisfy tests 3, 4 and 5; oligonucleotide meets all of tests 1, 2, 3 and 4.
抗ウイルス物質の投与に関連して本明細書において使用される場合、用語「対象」は、例えば哺乳動物のような動物、例、ヒト、ヒトではない霊長類、牛、豚、羊、馬、犬および猫を含む高等生物;鳥(鳥綱);ならびに植物、例えば果樹を指す。 As used herein in connection with administration of antiviral agents, the term “subject” refers to an animal such as a mammal, eg, a human, a non-human primate, a cow, a pig, a sheep, a horse, Refers to higher organisms, including dogs and cats; birds (Birds); and plants, such as fruit trees.
関連する局面は、抗ウイルスオリゴヌクレオチドランダマーまたは少なくとも一種のランダマーを含有するランダマー調合物に関し、ここでランダマーの抗ウイルス活性は、主として配列非依存的に、例えば配列相補的ではない作用様式により生じる。このようなランダマー調合物は、例えば、異なる長さ、例えば少なくとも2、3、5、10もしくはそれ以上の異なる長さのランダマーの混合物を含むか、または本明細書に記載する他の混合物を含み得る。 A related aspect relates to an antiviral oligonucleotide randomer or a randomer formulation containing at least one randomer, wherein the antiviral activity of the randomer occurs primarily in a sequence independent manner, for example by a mode of action that is not sequence complementary. Such randomer formulations include, for example, a mixture of randomers of different lengths, such as at least 2, 3, 5, 10 or more different lengths, or include other mixtures described herein. obtain.
オリゴヌクレオチド配列に関連して本明細書において使用される場合、用語「ランダム」は、ウイルスmRNAに相補的ではない配列またはONを特徴付け、ヘアピンを形成しないように選択され、またその内部にパリンドローム配列を含まない。用語「ランダム」が、特定のウイルスに向けたオリゴヌクレオチドの抗ウイルス活性の文脈で使用される場合、用語は、特定のウイルスのウイルスmRNAに対する相補性の不在を暗示する。相補性の不在は、例えば、複数のウイルス、特定のウイルスファミリー由来のウイルスまたは感染性ヒトウイルスのように、より広範囲に亘り得る。 As used herein in connection with oligonucleotide sequences, the term `` random '' characterizes a sequence or ON that is not complementary to viral mRNA, is selected so as not to form a hairpin, and has a Paris therein. Does not contain any nucleotide sequence. When the term “random” is used in the context of the antiviral activity of an oligonucleotide directed to a particular virus, the term implies the absence of complementarity to the viral mRNA of the particular virus. The absence of complementarity can be more widespread, for example, multiple viruses, viruses from specific virus families or infectious human viruses.
本出願において、用語「ランダマー」は、各位置に、例えばNNNNNNNNNNのようなゆらぎ(N)を有する一本鎖DNAを意味するものとする。各塩基は、このONが実質的に同一サイズの、異なるランダムに生成した配列の集団として実際に存在するように、ゆらぎとして合成される。このようなランダマーの調製は、通常、特定の長さ(主により短い長さ)に前後するサイズの分布を生成することが認識されている;明確に否定しない場合、本文脈において、このような調製物は特定の長さのランダマーとして見なされる。 In the present application, the term “lander” shall mean single-stranded DNA having fluctuations (N), such as NNNNNNNNNN, at each position. Each base is synthesized as a fluctuation so that this ON actually exists as a collection of different randomly generated sequences of substantially the same size. It is recognized that the preparation of such randomers usually produces a distribution of sizes around a certain length (mainly shorter); unless explicitly denied, in this context The preparation is considered as a randomer of a certain length.
「ウイルスゲノム由来の」という表現は、特定の配列が、ウイルスゲノムヌクレオチド配列またはその相補体と少なくとも70%の同一性を有するヌクレオチド塩基配列を有する(例、そのようなウイルスゲノム配列と同一または相補的)か、または対応するRNA配列であることを示す。本発明の特定の態様において、用語は、配列がオリゴヌクレオチドが指向する特定のウイルスのウイルスゲノム配列またはその相補的な配列と少なくとも70%の同一性であることを示す。特定の態様において、同一性は、少なくとも80、90、95、98、99または100%である。 The expression “derived from a viral genome” has a nucleotide sequence whose specific sequence has at least 70% identity to the viral genomic nucleotide sequence or its complement (eg, identical or complementary to such viral genomic sequence). Or a corresponding RNA sequence. In certain embodiments of the invention, the term indicates that the sequence is at least 70% identical to the viral genome sequence of the particular virus to which the oligonucleotide is directed or its complementary sequence. In certain embodiments, identity is at least 80, 90, 95, 98, 99 or 100%.
本発明はまた、治療有効量の薬理学的に許容される抗ウイルスオリゴヌクレオチド、または本明細書に記載する、例えば少なくとも6ヌクレオチド長の、または本明細書に列挙する他の長さのオリゴヌクレオチドの混合物、ここで、オリゴヌクレオチドの抗ウイルス活性は、主として配列非依存的に、例えば、配列相補的ではない作用様式により生じる、および薬学的に許容される担体を含有する抗ウイルス薬学的組成物を提供する。特定の態様において、オリゴヌクレオチドまたはオリゴヌクレオチドの組み合わせもしくは混合物は、個々のオリゴヌクレオチドまたはオリゴヌクレオチドの組み合わせもしくは混合物に関して上記に特定した通りである。特定の態様において、薬学的組成物は、ヒト、またはヒトではない動物、例えばヒトではない霊長類への投与が認可されている。 The invention also provides a therapeutically effective amount of a pharmacologically acceptable antiviral oligonucleotide, or an oligonucleotide as described herein, eg, of at least 6 nucleotides in length, or other lengths listed herein. , Wherein the antiviral activity of the oligonucleotide occurs primarily in a sequence independent manner, eg, by a mode of action that is not sequence complementary, and contains a pharmaceutically acceptable carrier I will provide a. In certain embodiments, the oligonucleotides or combinations or mixtures of oligonucleotides are as specified above for individual oligonucleotides or combinations or mixtures of oligonucleotides. In certain embodiments, the pharmaceutical compositions are approved for administration to humans or non-human animals, such as non-human primates.
特定の態様において、薬学的組成物は、ウイルス病因を伴う疾病の処置、制御または予防に適合している;プリオン疾病の処置、制御、または予防に適合している;眼内投与、経口消化、経腸投与、吸入、皮内、皮下、筋内、腹腔内、くも膜下腔内、気管内、もしくは静脈内注入、または局所投与による送達に適合している。 In certain embodiments, the pharmaceutical composition is adapted for the treatment, control or prevention of diseases associated with viral pathogenesis; adapted for the treatment, control or prevention of prion diseases; intraocular administration, oral digestion, Suitable for enteral administration, inhalation, intradermal, subcutaneous, intramuscular, intraperitoneal, intrathecal, intratracheal, or intravenous infusion, or local delivery.
特定の態様において、薬学的組成物は、経口消化、経口粘膜送達、鼻腔内滴下または噴霧、眼内注入、結膜下注入、点眼、点耳、吸入により、気管内注入もしくは噴霧、気管支内注入もしくは噴霧、胸膜内注入、腹腔内注入灌流もしくは洗浄、くも膜下腔内注入もしくは灌流、頭蓋内注入もしくは灌流、筋内注入、静脈内注入もしくは灌流、動脈内注入もしくは灌流、リンパ管内注入もしくは灌流、皮下注入もしくは灌流、皮内注入、局所皮膚適用、臓器灌流により、外科手術中の局所適用により、腫瘍内注入、局所適用、胃注入灌流もしくは洗浄、腸内注入もしくは灌流、大腸内注入灌流もしくは洗浄、直腸注入灌流もしくは洗浄、直腸坐薬もしくは浣腸により、尿道坐薬もしくは注入により、膀胱内注入灌流もしくは洗浄または関節内注入からなる群より選択されるが、これらに限定されない様式による送達用に調合され得る。 In certain embodiments, the pharmaceutical composition is administered by oral digestion, oral mucosal delivery, intranasal instillation or spray, intraocular injection, subconjunctival injection, eye drop, ear drop, inhalation, intratracheal injection or spray, intrabronchial injection or Nebulization, intrapleural injection, intraperitoneal injection, perfusion or lavage, intrathecal injection or perfusion, intracranial injection or perfusion, intramuscular injection, intravenous injection or perfusion, intraarterial injection or perfusion, intralymphatic injection or perfusion, subcutaneous Infusion or perfusion, intradermal injection, topical skin application, organ perfusion, local application during surgery, intratumoral injection, topical application, gastric infusion perfusion or irrigation, intestinal injection or perfusion, intestinal infusion perfusion or irrigation, Rectal infusion perfusion or lavage, rectal suppository or enema, urethral suppository or infusion, intravesical infusion perfusion or lavage, or intraarticular injection Is selected from the group consisting of, it may be formulated for delivery by a manner which is not limited thereto.
特定の態様において、組成物は、例えば特定の細胞または組織を標的とした送達系;リポソーム調合物、別の一抗ウイルス薬、例えば非ヌクレオチド抗ウイルスポリマー、アンチセンス分子、siRNAまたは小分子薬を含有する。 In certain embodiments, the composition comprises, for example, a delivery system targeted to a specific cell or tissue; a liposomal formulation, another antiviral drug, such as a non-nucleotide antiviral polymer, an antisense molecule, siRNA or a small molecule drug. contains.
特定の態様において、抗ウイルスオリゴヌクレオチド、オリゴヌクレオチド調製物、オリゴヌクレオチド製剤または抗ウイルス薬学的組成物は、インビトロで、標的ウイルス(例、任意の特定のウイルス、または本明細書に示すようなウイルス群のウイルス)に関して、10、5、2、1、0.50、0.20、0.10、0.09.0.08、0.07、0.75、0.06、0.05、0.045、0.04、0.035、0.03、0.025、0.02、0.015もしくは0.01μMであるかまたはそれ未満のIC50を有する。 In certain embodiments, the antiviral oligonucleotide, oligonucleotide preparation, oligonucleotide formulation or antiviral pharmaceutical composition is a target virus (e.g., any particular virus, or virus as indicated herein) in vitro. Group virus) is 10, 5, 2, 1, 0.50, 0.20, 0.10, 0.09.0.08, 0.07, 0.75, 0.06, 0.05, 0.045, 0.04, 0.035, 0.03, 0.025, 0.02, 0.015 or 0.01 μM. Or an IC 50 of less than that.
製剤、薬学的組成物、予防または処置のための使用、および予防または処置のための方法の特定の態様において、組成物または製剤は、ウイルス病因を伴う疾病の処置、制御、または予防に適合している;プリオン疾病の処置、制御、または予防に適合している;眼内、経口消化、経腸、吸入、または皮内、皮下、筋内もしくは静脈内注入からなる群より選択される送達様式に適合しており;更に、特定の細胞との親和性を増大させる分子を含むか、該分子に関連し得る送達系を含む;更に、少なくとも一種の他の抗ウイルス薬を組み合わせて含む;および/または更に抗ウイルスポリマーを組み合わせて含む。 In certain embodiments of the formulation, pharmaceutical composition, use for prevention or treatment, and method for prevention or treatment, the composition or formulation is adapted for treatment, control, or prevention of a disease associated with viral pathogenesis. Suitable for treatment, control or prevention of prion diseases; delivery mode selected from the group consisting of intraocular, oral digestion, enteral, inhalation, or intradermal, subcutaneous, intramuscular or intravenous infusion Further comprising a molecule that increases or can be associated with a specific cell; further comprising at least one other antiviral agent in combination; and In combination with / or further antiviral polymers.
特定の態様において、薬学的組成物は、本明細書に記載したような少なくとも一種のポリピリミジンオリゴヌクレオチドを含有する。ポリピリミジンオリゴヌクレオチドについて発見された低pHに対する耐性を考慮すると;所定の態様において、この組成物は、インビボの酸性部位への送達、例えば経口送達または経膣送達に適合している。 In certain embodiments, the pharmaceutical composition contains at least one polypyrimidine oligonucleotide as described herein. Given the resistance to low pH found for polypyrimidine oligonucleotides; in certain embodiments, the composition is adapted for delivery to acidic sites in vivo, such as oral or vaginal delivery.
このようなポリピリミジンオリゴヌクレオチドを含有する経口投与用の組成物および調合物の特定の態様において、組成物または製剤は、粉末、顆粒、微粒子、ナノ粒子、水または非水性媒体中の縣濁液または溶液、エマルション(例、ミクロエマルジョン)、カプセル、ジェルカプセル、サシェ、錠剤もしくは小錠剤の形態で調製される。所定の態様において、増粘剤、矯味矯臭剤、希釈剤、乳化剤、分散補助剤または結合剤が含まれてもよい。いくつかの態様において、経口製剤は、本発明のオリゴヌクレオチドが、1個または複数の浸透促進剤、界面活性剤および/またはキレート剤、例えばそれらに限定されるわけではないが、脂肪酸および/またはそのエステルもしくは塩(例えば、アラキドン酸、ウンデカン酸、オレイン酸、ラウリン酸、カプリル酸、カプリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、リノール酸、リノレン酸、ジカプレート、トリカプレート、モノオレイン、ジラウリン、グリセリル1-モノカプレート、1-ドデシルアザシクロヘプタン-2-オン、アシルカルニチン、アシルコリン、またはモノグリセリド、ジグリセリドもしくはその薬学的に許容される塩(例、ナトリウム)、胆汁酸および/またはその塩(例えば、ケノデオキシコール酸(CDCA)およびウルソデオキシコール酸(ursodeoxychenedeoxycholic acid)(UDCA)、コール酸、デヒドロコール酸、デオキシコール酸、グルコール酸、グリコール酸(glycholic acid)、グリコデオキシコール酸、タウロコール酸、タウロデオキシコール酸、タウロ-24、25-ジヒドロ-フシジン酸ナトリウム、グリコジヒドロフシジン酸ナトリウム)と共に投与される製剤である。いくつかの態様は、浸透促進剤の組み合わせ、例えばラウリン酸、カプリン酸およびUDCAのナトリウム塩のような脂肪酸/塩と胆汁酸/塩との組み合わせを含む。更なる例示的な浸透促進剤は、ポリオキシエチレン-9-ラウリルエーテル、ポリオキシエチレン-20-セチルエーテルを含む。 In certain embodiments of compositions and formulations for oral administration containing such polypyrimidine oligonucleotides, the composition or formulation is a suspension in powder, granules, microparticles, nanoparticles, water or a non-aqueous medium. Alternatively, it is prepared in the form of a solution, emulsion (eg, microemulsion), capsule, gel capsule, sachet, tablet or small tablet. In certain embodiments, thickeners, flavoring agents, diluents, emulsifiers, dispersion aids or binders may be included. In some embodiments, the oral formulation comprises the oligonucleotide of the invention as one or more penetration enhancers, surfactants and / or chelating agents, such as, but not limited to, fatty acids and / or Its esters or salts (e.g. arachidonic acid, undecanoic acid, oleic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin Glyceryl 1-monocaprate, 1-dodecylazacycloheptan-2-one, acylcarnitine, acylcholine, or monoglyceride, diglyceride or a pharmaceutically acceptable salt thereof (eg, sodium), bile acid and / or salt thereof ( For example, chenodeoxycholic acid (CDCA) and urso Ursodeoxychenedeoxycholic acid (UDCA), cholic acid, dehydrocholic acid, deoxycholic acid, glycholic acid, glycolic acid, glycodeoxycholic acid, taurocholic acid, taurodeoxycholic acid, tauro-24, 25 -Dihydro-sodium fusidate, sodium glycodihydrofusidate) Some embodiments are permeation enhancer combinations, eg fatty acids / salts such as the sodium salt of lauric acid, capric acid and UDCA And a bile acid / salt combination, further exemplary penetration enhancers include polyoxyethylene-9-lauryl ether, polyoxyethylene-20-cetyl ether.
本発明のオリゴヌクレオチドが顆粒形態(噴霧乾燥粒子を含む)で調製されるかまたは錯化されて微粒子またはナノ粒子を形成する特定の態様において、ポリアミノ酸;ポリイミン;ポリアクリレート;ポリアルキルアクリレート、ポリオキセタン、ポリアルキルシアノアクリレート;カチオン化ゼラチン、アルブミン、デンプン、アクリレート、ポリエチレングリコール(PEG)およびデンプン;ポリアルキルシアノアクリレート;DEAE-誘導体化ポリイミン、ポルラン、セルロース、およびデンプンから選択されるか、またはより詳細には、キトサン、N-トリメチルキトサン(N-trimethytchitosan)、ポリ-L-リシン、ポリヒスチジン、ポリオリチン(polyorithine)、ポリスペルミン、プロタミン、ポリビニルピリジン、ポリチオジエチルアミノ-メチルエチレンP(TDAE)、ポリアミノスチレン(例、p-アミノ)、ポリ(メチルシアノアクリレート)、ポリ(エチルシアノアクリレート)、ポリ(ブチルシアノアクリレート)、ポリ(イソブチルシアノアクリレート)、ポリ(イソヘキシルシアノアクリレート)、DEAE-メタクリレート、DEAE-ヘキシルアクリレート、DEAE-アクリルアミド、DEAE-アルブミンおよびDEAE-デキストラン、ポリメチルアクリレート、ポリヘキシルアクリレート、ポリ(D、L-乳酸)、ポリ(DL-乳酸-グリコール酸(PLGA)、アルギネートおよびポリエチレングリコール(PEG)から選択される錯化剤が使用されるが、それらに限定されるわけではない。 In certain embodiments where the oligonucleotides of the invention are prepared in granular form (including spray-dried particles) or complexed to form microparticles or nanoparticles, polyamino acids; polyimines; polyacrylates; polyalkylacrylates, poly Oxetane, polyalkyl cyanoacrylate; selected from cationized gelatin, albumin, starch, acrylate, polyethylene glycol (PEG) and starch; polyalkyl cyanoacrylate; DEAE-derivatized polyimine, porlan, cellulose, and starch, or more Specifically, chitosan, N-trimethytchitosan, poly-L-lysine, polyhistidine, polyorithine, polyspermine, protamine, polyvinylpyridine, polythiodiethylamino-methylethylene P (TDA E), polyaminostyrene (e.g., p-amino), poly (methyl cyanoacrylate), poly (ethyl cyanoacrylate), poly (butyl cyanoacrylate), poly (isobutyl cyanoacrylate), poly (isohexyl cyanoacrylate), DEAE -Methacrylate, DEAE-hexyl acrylate, DEAE-acrylamide, DEAE-albumin and DEAE-dextran, polymethyl acrylate, polyhexyl acrylate, poly (D, L-lactic acid), poly (DL-lactic acid-glycolic acid (PLGA), alginate And complexing agents selected from polyethylene glycol (PEG) are used, but are not limited thereto.
特定の態様において、組成物は、経膣投与に適合している。このような態様において、組成物は、錠剤、溶液、クリーム、ジェル、坐薬の形態で調製され得るが、それらに限定されるわけではない。 In certain embodiments, the composition is adapted for vaginal administration. In such embodiments, the composition may be prepared in the form of tablets, solutions, creams, gels, suppositories, but is not limited thereto.
特定の態様において、組成物は、局所投与に適合している。 In certain embodiments, the composition is adapted for topical administration.
本明細書において使用される場合、用語「ポリピリミジンオリゴヌクレオチド」または「ピリミジンオリゴヌクレオチド」は、50%より多いピリミジンヌクレオチドを含むオリゴヌクレオチドを意味する。 As used herein, the term “polypyrimidine oligonucleotide” or “pyrimidine oligonucleotide” means an oligonucleotide comprising more than 50% pyrimidine nucleotides.
本発明のオリゴヌクレオチドおよび組成物のインビボでの投与に関して使用されるように、用語「酸性部位」は、pH7未満の部位を意味する。例としては、胃(一般にpH1〜2)、膣(一般にpH4〜5であるが、より低い場合もある)、およびより低い程度では、皮膚(一般にpH4〜6)が含まれる。 As used with respect to in vivo administration of the oligonucleotides and compositions of the invention, the term “acidic site” means a site below pH 7. Examples include stomach (generally pH 1-2), vagina (generally pH 4-5, but may be lower), and to a lesser extent skin (generally pH 4-6).
本明細書において使用される場合、「経口送達に適合している」および同様の表現は、組成物が酸性のpHに十分耐性を有して、臨床的に過剰な活性の損失、例えば胃の酸性を原因とする、初回通過による50%未満(または40%、30%、20%、10%または5%未満と示されている)の過剰な損失を起こすことなく、経口投与できることを示す。 As used herein, “compatible with oral delivery” and similar expressions means that the composition is well tolerated to acidic pH and has a clinically excessive loss of activity, such as gastric Indicates that it can be administered orally without excessive loss of less than 50% (or indicated as less than 40%, 30%, 20%, 10% or 5%) due to acidity due to acidity.
本明細書において使用される場合、「経膣投与に適合している」および同様の表現は、組成物が適切に投与された場合に、本組成物が臨床的に許容できない程度(例、特定の保持時間中、50%、40%、30%、20%、10%未満または5%)に分解せず、(吸収された物質を除いて)膣内に少なくとも1時間(または、指定された場合、少なくとも2時間、4時間、8時間、12時間、1日または2日)実質的に留まるように調製されていることを示す。このような組成物の保持は、例えば、物理的形態または接着特性等の、多数の異なる因子または因子の組み合わせによるものであり得る。 As used herein, “compatible with vaginal administration” and similar expressions refer to the extent to which the composition is clinically unacceptable when the composition is properly administered (eg, specific 50%, 40%, 30%, 20%, less than 10% or 5%) during the retention time of at least 1 hour (or specified) in the vagina (excluding absorbed substances) If at least 2 hours, 4 hours, 8 hours, 12 hours, 1 day or 2 days). Such composition retention may be due to a number of different factors or combinations of factors such as, for example, physical form or adhesive properties.
特定のウイルスまたはウイルス群に関連して、本明細書において抗ウイルスオリゴヌクレオチドおよび調合物等に関連して使用されるように、用語「標的とする」は、ウイルスまたはウイルス群を阻止するためにオリゴヌクレオチドを選択することを示す。特定の組織または細胞タイプに関連して使用される場合、本用語は、オリゴヌクレオチド、調合物、または送達系が、オリゴヌクレオチドが特定の組織または細胞タイプ内またはその近位に優先的に存在し、および/または優先的に抗ウイルス効果を示すように選択されることを示す。 As used herein in connection with antiviral oligonucleotides and formulations, etc. in connection with a particular virus or group of viruses, the term “target” is used to block a virus or group of viruses. Indicates that an oligonucleotide is selected. When used in connection with a particular tissue or cell type, the term means that the oligonucleotide, formulation, or delivery system is preferentially present in or near a particular tissue or cell type. And / or preferentially selected to exhibit an antiviral effect.
本明細書において使用される場合、用語「送達系」は、オリゴヌクレオチド(例えば、アンチセンスオリゴ、siRNAまたは本明細書に記載するオリゴヌクレオチド)と組み合わせると、インビボで意図する位置と接触するオリゴヌクレオチドの量を増大させる、ならびに/または標的におけるその存在の持続時間を、例えば送達系の不在下での量および/もしくは持続時間と比較して、少なくとも20、50もしくは100%、またはそれ以上延長させる、ならびに/または副作用の原因となる相互作用を防止または低減する、成分を指す。 As used herein, the term “delivery system” refers to an oligonucleotide that contacts the intended location in vivo when combined with an oligonucleotide (eg, an antisense oligo, siRNA, or oligonucleotide described herein). And / or extend the duration of its presence in the target by at least 20, 50 or 100%, or more, for example, compared to the amount and / or duration in the absence of the delivery system And / or an ingredient that prevents or reduces interactions that cause side effects.
抗ウイルス薬および他の薬物または試験化合物に関連して本明細書において使用される場合、用語「小分子」は、分子量が1500ダルトンまたはそれ未満である分子を意味する。いくつかの場合において、分子量は、1000、800、600、500もしくは400ダルトンまたはそれ未満である。 As used herein in connection with antiviral drugs and other drugs or test compounds, the term “small molecule” means a molecule having a molecular weight of 1500 daltons or less. In some cases, the molecular weight is 1000, 800, 600, 500 or 400 daltons or less.
別の局面において、本発明は、少なくとも一種の抗ウイルスオリゴヌクレオチド、抗ウイルスオリゴヌクレオチド混合物、抗ウイルスオリゴヌクレオチド製剤または抗ウイルス薬学的組成物を含むキットを提供し、該キットは、そのようなオリゴヌクレオチド、オリゴヌクレオチド混合物またはオリゴヌクレオチド製剤を、ラベルを貼ったパッケージ内に含み、ここで、オリゴヌクレオチドの抗ウイルス活性は、主として配列非依存的に、例えば配列相補的ではない作用様式により生じ、パッケージ上のラベルは、抗ウイルスオリゴヌクレオチドが、少なくとも一種のウイルスに対して使用し得ることを示す。 In another aspect, the present invention provides a kit comprising at least one antiviral oligonucleotide, antiviral oligonucleotide mixture, antiviral oligonucleotide formulation or antiviral pharmaceutical composition, the kit comprising such an oligonucleotide. Nucleotides, oligonucleotide mixtures or oligonucleotide preparations are contained in a labeled package, where the antiviral activity of the oligonucleotide occurs mainly in a sequence-independent manner, for example by a mode of action that is not sequence complementary, The upper label indicates that the antiviral oligonucleotide can be used against at least one virus.
特定の態様において、キットは、本明細書に記載する少なくとも一種の抗ウイルスオリゴヌクレオチドを含有する薬学的組成物を含む。一態様において、キットは、少なくとも二種の異なる抗ウイルスオリゴヌクレオチドの混合物を含む。一態様において、抗ウイルスオリゴヌクレオチドは、動物のインビボでの使用に適合し、および/またはラベルは、オリゴヌクレオチドまたは組成物が動物での使用に許容および/または認可されていることを示す;動物はヒトのような哺乳動物、または牛、豚、反すう動物、羊、もしくは馬のようなヒトではない哺乳動物である;動物は、ヒトではない動物である;動物は、鳥である、キットは、例えば米国食品医薬品局(U.S. Food and Drug Administration)または同等の機関のような規制機関により、動物、例えばヒトでの使用が認可されている。 In certain embodiments, the kit comprises a pharmaceutical composition containing at least one antiviral oligonucleotide described herein. In one embodiment, the kit comprises a mixture of at least two different antiviral oligonucleotides. In one aspect, the antiviral oligonucleotide is compatible with the in vivo use of the animal, and / or the label indicates that the oligonucleotide or composition is acceptable and / or approved for use in an animal; Is a mammal such as a human or a non-human mammal such as a cow, pig, ruminant, sheep, or horse; the animal is a non-human animal; the animal is a bird, the kit is Approved for use in animals, eg, humans, by regulatory agencies such as the US Food and Drug Administration or equivalent.
別の局面において、本発明は、抗ウイルス薬として使用するための、抗ウイルスオリゴヌクレオチド、例えば配列相補的ではない抗ウイルスオリゴヌクレオチドを選択するための方法を提供する。本方法は、複数の異なるランダムオリゴヌクレオチドを合成し、オリゴヌクレオチドのウイルスの感染性ビリオンを産生する能力を阻害する活性を試験し、抗ウイルス薬として使用するための、薬学的に許容されるレベルの活性を有するオリゴヌクレオチドを選択することを含む。 In another aspect, the invention provides a method for selecting an antiviral oligonucleotide, eg, an antiviral oligonucleotide that is not sequence complementary, for use as an antiviral agent. The method synthesizes a plurality of different random oligonucleotides, tests the activity of the oligonucleotide to inhibit the ability of the virus to produce infectious virions, and is used at an pharmaceutically acceptable level. Selecting an oligonucleotide having the following activity:
特定の態様において、異なるランダムオリゴヌクレオチドは、異なる長さのランダマーを含む;ランダムオリゴヌクレオチドは、異なる配列を有するかまたは共通する配列、複数の最短のオリゴの配列を有し得る;および/または異なるランダムオリゴヌクレオチドは、少なくとも5ヌクレオチド長のランダマーセグメントを含む複数のオリゴヌクレオチドを含むか、もしくは異なるランダムオリゴヌクレオチドは、異なる長さの複数のランダマーを含む。例えば抗ウイルスオリゴヌクレオチドに関して本明細書に記載する他のオリゴヌクレオチドは、特定の系で試験し得る。 In certain embodiments, different random oligonucleotides comprise different lengths of randomers; random oligonucleotides may have different sequences or have a common sequence, sequences of multiple shortest oligos; and / or different Random oligonucleotides comprise a plurality of oligonucleotides comprising randomer segments that are at least 5 nucleotides in length, or different random oligonucleotides comprise a plurality of randomers of different lengths. Other oligonucleotides described herein, for example with respect to antiviral oligonucleotides, can be tested in specific systems.
更に別の局面において、本発明は、対象においてウイルス感染を予防または処置する方法を提供し、該方法は、そのような処置を必要とする対象に、本明細書に記載する治療有効量の少なくとも一種の薬理学的に許容されるオリゴヌクレオチド、例えば少なくとも6ヌクレオチド長の配列相補的ではないオリゴヌクレオチドを投与するか、またはそのようなオリゴヌクレオチドを含有する抗ウイルス薬学的組成物もしくは製剤または混合物を投与することによる。更なる態様において、本発明は、ウイルス感染の予防または処置のための使用を提供し、該使用は、そのような処置を必要とする対象に、本明細書に記載する治療有効量の少なくとも一種の薬理学的に許容されるオリゴヌクレオチド、例えば少なくとも6ヌクレオチド長の配列相補的ではないオリゴヌクレオチドを投与するか、またはそのようなオリゴヌクレオチドを含有する抗ウイルス薬学的組成物もしくは製剤または混合物を投与することによる。特定の態様において、ウイルスは、本発明を用いた抑制に適した、本明細書に列挙されている任意のウイルスであり得る;感染は、ウイルス感染に関連するものとして本明細書に示される疾病または病状に関係する;対象は、本明細書に示される種類の対象、例えばヒト、ヒトではない動物、ヒトではない哺乳動物、鳥、植物等である;処置は、ウイルス疾病、またはウイルス病因を伴う疾病、例えば本明細書の「発明の背景」に示される疾病に関する。 In yet another aspect, the invention provides a method of preventing or treating a viral infection in a subject, said method comprising at least a therapeutically effective amount described herein in a subject in need of such treatment. Administering a kind of pharmacologically acceptable oligonucleotide, for example an oligonucleotide that is not sequence complementary at least 6 nucleotides in length, or an antiviral pharmaceutical composition or formulation or mixture containing such an oligonucleotide By administering. In a further aspect, the present invention provides a use for the prevention or treatment of a viral infection, said use comprising at least one therapeutically effective amount as described herein in a subject in need of such treatment. Administering a pharmacologically acceptable oligonucleotide, e.g. an oligonucleotide that is not sequence complementary at least 6 nucleotides in length, or an antiviral pharmaceutical composition or formulation or mixture containing such an oligonucleotide By doing. In certain embodiments, the virus can be any virus listed herein suitable for suppression using the present invention; the infection is a disease indicated herein as being associated with a viral infection. A subject is a subject of the type indicated herein, eg, a human, a non-human animal, a non-human mammal, a bird, a plant, etc .; a treatment is associated with a viral disease, or a viral etiology Related diseases, such as those indicated in the “Background of the Invention” herein.
また別の局面において、本発明は、対象において酸性環境内のウイルス感染を予防または処置する方法を提供し、本方法は、そのような処置を必要とする対象に、治療有効量の少なくとも一種の薬理学的に許容される本発明の抗ウイルス薬学的組成物を投与することを含み、該組成物は、インビボの酸性部位への投与に適合している。 In yet another aspect, the present invention provides a method for preventing or treating a viral infection in an acidic environment in a subject, the method comprising a therapeutically effective amount of at least one of a subject in need of such treatment. Administration of a pharmacologically acceptable antiviral pharmaceutical composition of the invention, said composition being adapted for administration to an acidic site in vivo.
また別の局面において、本発明は、対象における酸性環境内のウイルス感染の予防または処置における使用を提供し、本使用は、そのような処置を必要とする対象に、治療有効量の少なくとも一種の薬理学的に許容される本発明の抗ウイルス薬学的組成物を投与することを含み、組成物は、インビボの酸性部位への投与に適合している。 In yet another aspect, the present invention provides use in the prevention or treatment of a viral infection in an acidic environment in a subject, the use comprising at least one therapeutically effective amount in a subject in need of such treatment. Administering a pharmacologically acceptable antiviral pharmaceutical composition of the invention, the composition being adapted for administration to an acidic site in vivo.
特定の態様において、本明細書に記載されている抗ウイルスオリゴヌクレオチド(またはオリゴヌクレオチド製剤もしくは薬学的組成物)が投与される;投与は、本明細書に記載されているような方法で行われる;本明細書に記載するような送達系または方法が使用される;ウイルス感染は、DNAウイルスまたはRNAウイルスの感染である;ウイルスは、パルボウイルス科、パポバウイルス科、アデノウイルス科、ヘルペスウイルス科、ポックスウイルス科、ヘパドナウイルス科またはパピローマウイルス科である;ウイルスは、アレナウイルス科、ブニヤウイルス科、カルシウイルス科、コロナウイルス科、フィロウイルス科、フラウイルス科、オルトミクソウイルス科、パラミクソウイルス科、ピコルナウイルス科、レオウイルス科、ラブドウイルス科、レトロウイルス科またはトガウイルス科である;ヘルペスウイルス科ウイルスは、EBV、HSV-1、HSV-2、CMV、VZV、HHV-6、HHV-7またはHHV-8である;ウイルスは、HIV-1またはHIV-2である;ウイルスは、呼吸器多核体ウイルス(RSV)である;ウイルスは、パラインフルエンザ-3ウイルスである;ウイルスは、インフルエンザウイルス、例えばインフルエンザAである;ウイルスは、HBVである;ウイルスは、天然痘ウイルスまたは牛痘ウイルスである;ウイルスは、コロナウイルスである;ウイルスは、SARSウイルスである;ウイルスは、ウエストナイルウイルスである;ウイルスは、ハンタウイルスである;ウイルスは、パラインフルエンザウイルスである;ウイルスは、コクサッキーウイルスである;ウイルスは、ライノウイルスである;ウイルスは、黄熱病ウイルスである;ウイルスは、デングウイルスである;ウイルスは、C型肝炎ウイルスである;ウイルスは、エボラウイルスである;ウイルスは、マールブルグウイルスである;ウイルスは、ラッサ熱ウイルスである;ウイルスは、水痘帯状疱疹ウイルスである;ウイルスは、エプスタイン・バーウイルスである;ウイルスは、ヒトヘルペスウイルス6Aまたは6Bである;ウイルスは、HBVである;ウイルスは、パラインフルエンザウイルスである;ウイルスは、ヒトメタニューモウイルスである;ウイルスは、リフトバレー熱ウイルスである;ウイルスは、クリミア・コンゴ出血熱ウイルスである;ウイルスは、西部馬脳炎ウイルスである。 In certain embodiments, an antiviral oligonucleotide (or oligonucleotide formulation or pharmaceutical composition) described herein is administered; administration is performed in a manner as described herein. A delivery system or method as described herein is used; the viral infection is an infection of a DNA virus or RNA virus; the virus is a parvoviridae, papovaviridae, adenoviridae, herpesviridae, Poxviridae, Hepadnaviridae or Papillomaviridae; viruses are Arenaviridae, Bunyaviridae, Calciviridae, Coronaviridae, Filoviridae, Flaviridae, Orthomyxoviridae, Paramyxoviridae , Picornaviridae, Reoviridae, La Grape Rusaceae, Retroviridae or Togaviridae; Herpesviridae virus is EBV, HSV-1, HSV-2, CMV, VZV, HHV-6, HHV-7 or HHV-8; The virus is a respiratory multinuclear virus (RSV); the virus is a parainfluenza-3 virus; the virus is an influenza virus, such as influenza A; the virus is The virus is a smallpox virus or cowpox virus; the virus is a coronavirus; the virus is a SARS virus; the virus is a West Nile virus; the virus is a hantavirus; Is a parainfluenza virus; the virus is a Coxsackie virus; the virus is a rhinovirus; The virus is a dengue virus; the virus is a hepatitis C virus; the virus is an Ebola virus; the virus is a Marburg virus; the virus is a Lassa fever virus; The varicella-zoster virus; the virus is Epstein-Barr virus; the virus is human herpesvirus 6A or 6B; the virus is HBV; the virus is a parainfluenza virus; the virus is human meta The virus is Rift Valley fever virus; the virus is Crimea-Congo hemorrhagic fever virus; the virus is Western equine encephalitis virus.
特定の態様において、本発明のオリゴヌクレオチドは、インフルエンザウイルスを標的とする。 In certain embodiments, the oligonucleotides of the invention target influenza viruses.
特定の態様において、オリゴヌクレオチドは、ポリピリミジンオリゴヌクレオチド(またはそのようなポリピリミジンオリゴヌクレオチドを含有する製剤もしくは薬学的組成物)であり、これは例えば本明細書に記載する経口または経膣投与に適合し得る。 In certain embodiments, the oligonucleotide is a polypyrimidine oligonucleotide (or a formulation or pharmaceutical composition containing such a polypyrimidine oligonucleotide), for example for oral or vaginal administration as described herein. Can fit.
同様に、関連する局面において、本発明は、ヒトまたは動物におけるオンコウイルスに起因する癌の予防的処置方法を提供し、本方法は、そのような治療を必要とするヒトまたは動物に、少なくとも6ヌクレオチド長(または本明細書に記載する他の長さ)の治療有効量の薬理学的に許容される少なくとも一種のランダムオリゴヌクレオチド、またはそのようなオリゴヌクレオチドを含有する製剤もしくは薬学的組成物を投与することによる。一つの態様において、本発明のオリゴヌクレオチドの混合物。 Similarly, in a related aspect, the invention provides a method for the prophylactic treatment of cancer caused by oncoviruses in humans or animals, the method comprising at least 6 humans or animals in need of such therapy. A therapeutically effective amount of at least one pharmacologically acceptable random oligonucleotide of nucleotide length (or other length as described herein), or a formulation or pharmaceutical composition containing such an oligonucleotide. By administering. In one embodiment, a mixture of oligonucleotides of the invention.
同様に、関連する局面において、本発明は、ヒトまたは動物におけるオンコウイルスに起因する癌の予防的処置のための使用を提供し、本使用は、そのような処置を必要とするヒトまたは動物に、少なくとも6ヌクレオチド長(または本明細書に記載する他の長さ)の治療有効量の薬理学的に許容される少なくとも一種のランダムオリゴヌクレオチド、またはそのようなオリゴヌクレオチドを含有する製剤もしくは薬学的組成物を投与することによる。一つの態様において、本発明のオリゴヌクレオチドの混合物を投与する。 Similarly, in a related aspect, the present invention provides use for the prophylactic treatment of cancer caused by oncovirus in a human or animal, the use being directed to a human or animal in need of such treatment. A therapeutically effective amount of at least one pharmacologically acceptable random oligonucleotide at least 6 nucleotides in length (or other length described herein), or a formulation or pharmaceutical containing such an oligonucleotide By administering the composition. In one embodiment, a mixture of oligonucleotides of the invention is administered.
特定の態様において、オリゴヌクレオチドは、本発明のために本明細書に記載するようなものであり、例えば本明細書に記載する長さを有する;本明細書に記載するような投与方法が用いられる;本明細書に記載するような使用が用いられる;本明細書に記載するような送達系が用いられる。 In certain embodiments, the oligonucleotide is as described herein for the present invention, eg, has a length as described herein; the administration method as described herein is used. Uses as described herein are used; delivery systems as described herein are used.
用語「治療有効量」は、意図されるタイプの典型的な対象に投与した場合に、感染性ウイルス粒子の産生の治療的または予防的に有意な低下をもたらすに十分な量を指す。対象に対する抗ウイルスオリゴヌクレオチド投与に関与する局面において、典型的には、オリゴヌクレオチド、製剤または組成物は、治療有効量で投与されるべきである。 The term “therapeutically effective amount” refers to an amount sufficient to provide a therapeutically or prophylactically significant reduction in the production of infectious viral particles when administered to a typical subject of the intended type. In aspects involving antiviral oligonucleotide administration to a subject, typically the oligonucleotide, formulation or composition should be administered in a therapeutically effective amount.
本明細書に記載するオリゴヌクレオチド製剤、薬学的組成物、処置および予防方法、ならびに/または処置および予防のための使用を含む特定の態様において、配列非依存的な作用様式を有するオリゴヌクレオチドは、トランスフェクション剤と関連していない;配列非依存的な作用様式を有するオリゴヌクレオチドは、リポソームおよび/または非リポソーム脂質粒子内に封入されていない。所定の態様において、配列非依存的な作用様式を有するオリゴヌクレオチドは、薬学的組成物中に存在するか、または主として配列依存的な作用様式で作用する抗ウイルスオリゴヌクレオチド、例えばアンチセンスオリゴヌクレオチドもしくはsiRNAと共に(同時にまたは連続的に)投与される。ここで、配列依存的な作用様式を有するオリゴヌクレオチドは、トランスフェクション剤と関連されるか、および/またはリポソームおよび/または非リポソーム脂質粒子内に封入され得る。 In certain embodiments, including oligonucleotide formulations, pharmaceutical compositions, methods of treatment and prevention, and / or uses for treatment and prevention described herein, an oligonucleotide having a sequence-independent mode of action comprises Not associated with transfection agents; oligonucleotides having a sequence-independent mode of action are not encapsulated within liposomes and / or non-liposomal lipid particles. In certain embodiments, an oligonucleotide having a sequence-independent mode of action is present in a pharmaceutical composition, or an antiviral oligonucleotide that acts primarily in a sequence-dependent mode of action, such as an antisense oligonucleotide or It is administered with siRNA (simultaneously or sequentially). Here, oligonucleotides having a sequence-dependent mode of action can be associated with transfection agents and / or encapsulated in liposomes and / or non-liposomal lipid particles.
別の局面において、配列非依存性の、例えば配列相補的ではない相互作用が、有効な抗ウイルス活性をもたらすという発見は、ウイルス成分、1個または複数のウイルスタンパク質(例、感染した宿主有機体のウイルス培養物から抽出もしくは精製した、または組換え法により生成した)に対するオリゴヌクレオチドの結合を変更する化合物を同定するスクリーニング方法を提供する。例えば、本方法は、試験化合物が1個または複数のウイルス成分に対するオリゴヌクレオチドの結合を低下させるか否かの測定に関与し得る。 In another aspect, the discovery that sequence-independent, e.g., non-sequence complementary, interactions results in effective antiviral activity is the detection of viral components, one or more viral proteins (e.g., infected host organisms). Screening methods for identifying compounds that alter the binding of oligonucleotides to (extracted or purified from, or produced by, recombinant methods). For example, the method can involve determining whether a test compound reduces binding of the oligonucleotide to one or more viral components.
本明細書において使用される用語「スクリーニング」は、複数の化合物をアッセイして、所望の性質を所有するか否かを決定することを指す。複数の化合物は、例えば少なくとも10、100、1000、10,000またはそれ以上の試験化合物であり得る。 The term “screening” as used herein refers to assaying a plurality of compounds to determine whether they possess the desired property. The plurality of compounds can be, for example, at least 10, 100, 1000, 10,000 or more test compounds.
特定の態様において、多様なアッセイフォーマットおよび検出方法のいずれかを使用して、そのような結合の変化を同定することができる。例えばオリゴヌクレオチドを、スクリーニングするべき化合物の存在下および不在下で、ウイルス成分と接触させて(例、別個の反応において)、化合物の不在下と比較して、化合物の存在下でオリゴのウイルス成分に対する結合における差異が生じるか否かを測定する。そのような差異の存在は、化合物がランダムオリゴヌクレオチドのウイルス成分に対する結合を変更することを示す。または、オリゴヌクレオチドをウイルス成分に結合させ、加えた試験化合物による置換を測定するか、または逆に、試験化合物を結合させ、加えたオリゴヌクレオチドによる置換を測定するといった競合的置換を用いることができる。 In certain embodiments, any of a variety of assay formats and detection methods can be used to identify such binding changes. For example, the oligonucleotide is contacted with a viral component in the presence and absence of the compound to be screened (e.g., in a separate reaction) and compared to the absence of the compound in the presence of the compound in the viral component of the oligo. Measure whether a difference in binding to occurs. The presence of such differences indicates that the compound alters the binding of the random oligonucleotide to the viral component. Alternatively, competitive displacement can be used, such as binding the oligonucleotide to the viral component and measuring displacement by the added test compound, or conversely, binding the test compound and measuring displacement by the added oligonucleotide. .
特定の態様において、オリゴヌクレオチドは、抗ウイルスオリゴヌクレオチドに関して本明細書に記載した通りである;オリゴヌクレオチドは、少なくとも6、8、10、15、20、25、29、30、32、34、36、38、40、46、50、60、70、80、90、100、110もしくは120ヌクレオチド長を有するか、または少なくとも抗ウイルスオリゴヌクレオチドに関して本明細書に特定した他の長さを有するか、または前記の値の任意の二つをその範囲の終点を含むものとして定義する範囲内の長さを有する;試験化合物は、小分子である。試験化合物は、400、500、600、800、1000、1500、2000、2500または3000ダルトン未満の分子量を有するか、または前記の値の任意の二つを、その範囲の終点を含むものとして定義する範囲内の分子量を有する;ウイルス抽出物またはウイルス成分は、本明細書に列挙するウイルスを由来とする;少なくとも100、1000、10,000、20,000、50,000または100,000の化合物がスクリーニングされる;オリゴヌクレオチドは、インビトロで、10、5、2、1、0.500、0.200、0.100、0.075、0.05、0.045、0.04、0.035、0.03、0.025、0.02、0.015または0.01μMと等しいかまたはそれ未満のIC50を有する。 In certain embodiments, the oligonucleotide is as described herein for the antiviral oligonucleotide; the oligonucleotide is at least 6, 8, 10, 15, 20, 25, 29, 30, 32, 34, 36 , 38, 40, 46, 50, 60, 70, 80, 90, 100, 110 or 120 nucleotides in length, or at least other lengths specified herein for antiviral oligonucleotides, or Having a length within the range defining any two of the values as including the end of the range; the test compound is a small molecule. A test compound has a molecular weight of less than 400, 500, 600, 800, 1000, 1500, 2000, 2500 or 3000 Daltons, or any two of the above values are defined as including endpoints of that range A viral extract or viral component is derived from the viruses listed herein; at least 100, 1000, 10,000, 20,000, 50,000 or 100,000 compounds are screened; In vitro, it has an IC 50 equal to or less than 10, 5, 2, 1, 0.500, 0.200, 0.100, 0.075, 0.05, 0.045, 0.04, 0.035, 0.03, 0.025, 0.02, 0.015 or 0.01 μM.
本発明は更に、表21に示すオリゴヌクレオチドを提供する。 The present invention further provides the oligonucleotides shown in Table 21.
本発明は更に、REP 1001、REP 2001、REP 3007、REP 2004、REP 2005、REP 2006、REP 2007、REP 2008、REP 2017、REP 2018、REP 2020、REP 2021、REP 2024、REP 2036、A20、G20、C20、REP 2029、REP 2031、REP 2030、REP 2033、REP 2055、REP 2056、REP 2057、REP 2060およびREP 2107のいずれか一つに示されている抗ウイルスオリゴヌクレオチドを提供する。 The present invention further includes REP 1001, REP 2001, REP 3007, REP 2004, REP 2005, REP 2006, REP 2007, REP 2008, REP 2017, REP 2018, REP 2020, REP 2021, REP 2024, REP 2036, A20, G20 , C20, REP 2029, REP 2031, REP 2030, REP 2033, REP 2055, REP 2056, REP 2057, REP 2060, and REP 2107.
本明細書で使用される用語「ウイルス構成成分」は、ウイルスにコードされた生成物又はウイルス感染の結果感染した宿主細胞により産生される生成物を言う。そのような構成成分には、タンパク質及びそのほかの生体分子を挙げることができる。そのようなウイルス構成成分は、たとえば、ウイルスの培養、感染した宿主生物、たとえば、動物又は植物から得ることができ、又は組換え系(原核生物又は真核生物)においてウイルスの配列、同様に、ウイルスがコードするタンパク質に相当するアミノ酸配列を有する合成タンパク質から産生することができる。用語「ウイルス培養抽出物」は、ウイルス特異的生成物を包含するウイルスにより感染させられた細胞からの抽出物を言う。同様に、「ウイルスタンパク質」は、通常ウイルスにコードされたウイルス特異的タンパク質を言うが、ウイルス感染の結果として宿主の配列により部分的にコードされうるウイルス特異的タンパク質も言う。 The term “viral component” as used herein refers to a product encoded by a virus or a product produced by a host cell infected as a result of a viral infection. Such components can include proteins and other biomolecules. Such viral components can be obtained, for example, from viral cultures, infected host organisms such as animals or plants, or viral sequences in recombinant systems (prokaryotes or eukaryotes), as well It can be produced from a synthetic protein having an amino acid sequence corresponding to the protein encoded by the virus. The term “virus culture extract” refers to an extract from cells infected by a virus, including virus-specific products. Similarly, "viral protein" refers to a virus-specific protein that is normally encoded by a virus, but also refers to a virus-specific protein that can be partially encoded by a host sequence as a result of viral infection.
関連する局面では、本発明は、前出の方法により同定される抗ウイルス性化合物、たとえば、新規の抗ウイルス性化合物を提供する。 In a related aspect, the present invention provides antiviral compounds identified by the above methods, for example, novel antiviral compounds.
さらなる局面では、本発明は、オリゴヌクレオチドのプールを、たとえば、固定相媒体に結合した少なくとも1つのウイルス構成成分に接触させ、ウイルス構成成分に結合したオリゴヌクレオチドを回収することによってオリゴヌクレオチドのプールから少なくとも1つのウイルス構成成分に結合するオリゴヌクレオチドを精製する方法を提供する。一般に、回収には、ウイルス構成成分からのオリゴヌクレオチドの置き換えが含まれる。本方法は、回収したオリゴヌクレオチド(すなわち、ウイルスタンパク質に結合したオリゴヌクレオチド)の配列決定及び/又は抗ウイルス活性試験を含むこともできる。 In a further aspect, the present invention relates to a pool of oligonucleotides from a pool of oligonucleotides, for example by contacting the pool of oligonucleotides with at least one viral component bound to a stationary phase medium and recovering the oligonucleotide bound to the viral component. Methods are provided for purifying oligonucleotides that bind to at least one viral component. In general, recovery involves the replacement of oligonucleotides from viral components. The method can also include sequencing and / or antiviral activity testing of recovered oligonucleotides (ie, oligonucleotides bound to viral proteins).
特定の態様では、プールの結合したオリゴヌクレオチドは、たとえば、イオン性の置き換え体を用いて、任意の適当な方法によって固定相媒体から置き換えられ、置き換えられたオリゴヌクレオチドを回収する。通常、種々の置換方法について、オリゴヌクレオチドが高い結合親和性の順に一様に置き換わるように厳密性を高くすること(たとえば、段階的な又は連続的な勾配があるように塩のような置換剤の濃度を高めることによって)によって置換を実施することができる。多くの場合、低い厳密性の洗浄を行って弱く結合したオリゴヌクレオチドを取り除き、置き換わった、さらに強く結合したオリゴヌクレオチドを含有する1以上の分画を回収する。場合によっては、さらに使用するために、非常に強く結合するオリゴヌクレオチド(たとえば、さらに厳密な置換条件による置換で生じた分画におけるオリゴヌクレオチド)を含有する分画を選択することが望ましい。 In certain embodiments, the pool-bound oligonucleotide is displaced from the stationary phase medium by any suitable method, eg, using an ionic displacement body, and the displaced oligonucleotide is recovered. Typically, for various substitution methods, increasing the stringency so that the oligonucleotides are uniformly replaced in order of high binding affinity (eg, salt-like substitution agents so that there is a stepped or continuous gradient) Can be performed by increasing the concentration of In many cases, low stringency washing is performed to remove weakly bound oligonucleotides and one or more fractions containing displaced, more strongly bound oligonucleotides are recovered. In some cases, it may be desirable to select a fraction containing oligonucleotides that bind very strongly (eg, oligonucleotides in fractions resulting from substitution under more stringent substitution conditions) for further use.
同様に、本発明は、オリゴヌクレオチドのプールを1以上のウイルスタンパク質に接触させ、ウイルスタンパク質に結合したオリゴヌクレオチドを増幅させて濃縮したオリゴヌクレオチドプールを提供することによる、少なくとも1つのウイルス構成成分に結合するオリゴヌクレオチドのプールからオリゴヌクレオチドを濃縮する方法を提供する。接触及び増幅は、次の回のオリゴヌクレオチドのプールとして前の回の濃縮したオリゴヌクレオチドプールを用いて、複数回、たとえば1、2、3、4、5、10以上の追加回数行うことができる。方法はまた、接触及び増幅の1以上の回数に続いて、濃縮されたオリゴヌクレオチドプールにおけるオリゴヌクレオチドの配列決定及び抗ウイルス活性試験をも含むことができる。 Similarly, the invention relates to at least one viral component by contacting a pool of oligonucleotides with one or more viral proteins and amplifying and concentrating the oligonucleotides bound to the viral proteins to enrich the oligonucleotide pool. Methods are provided for concentrating oligonucleotides from a pool of binding oligonucleotides. Contacting and amplification can be performed multiple times, for example, 1, 2, 3, 4, 5, 10 or more additional times, using the previous round of concentrated oligonucleotide pool as the next pool of oligonucleotides. . The method can also include sequencing of the oligonucleotides in the enriched oligonucleotide pool and antiviral activity testing following one or more times of contacting and amplification.
本方法は、たとえば、置換剤を用いて、上述のように、種々の技法によりウイルス構成成分(たとえば、固相媒体に結合したウイルスタンパク質)からオリゴヌクレオチドを置換することを含む。上記で示したように、更なる使用、たとえば、結合及び増幅のさらなる回のために、さらに強く結合するオリゴヌクレオチドを選択することは有利でありうる。方法は1以上の濃縮されたオリゴヌクレオチド、たとえば、高親和性オリゴヌクレオチドを選択することをさらに含み、さらに使用することができる。特定の態様では、選択には、関心のある特定のウイルスについて、宿主のゲノム配列(たとえば、ヒト)に相補的な配列を有するオリゴヌクレオチドの排除が包含されうる。そのような排除は、たとえば、配列配置プログラム(たとえば、BLAST検索)を用いて配列データベースにおける特定の宿主に由来する配列とオリゴヌクレオチド配列を比較すること、及び宿主配列と同一の、又は特定のレベルで同一性を持つ配列を有するオリゴヌクレオチドを排除することを含むことができる。そのような宿主相補性配列の排除及び/又は宿主配列に相補的でない1以上のオリゴヌクレオチドの選択は、本発明のそのほかの局面についても実施することができる。 The method includes, for example, substituting an oligonucleotide from a viral component (eg, a viral protein bound to a solid phase medium) by various techniques, as described above, using, for example, a displacing agent. As indicated above, it may be advantageous to select oligonucleotides that bind more strongly for further use, eg, further rounds of binding and amplification. The method can further include selecting and further using one or more concentrated oligonucleotides, eg, high affinity oligonucleotides. In certain embodiments, selection can include the elimination of oligonucleotides having sequences complementary to the host genomic sequence (eg, human) for the particular virus of interest. Such exclusion includes, for example, using a sequence placement program (eg, a BLAST search) to compare the oligonucleotide sequence with a sequence from a particular host in a sequence database, and at the same or a specific level as the host sequence. And excluding oligonucleotides having sequences with identity. The elimination of such host complementarity sequences and / or selection of one or more oligonucleotides that are not complementary to the host sequence can also be performed for other aspects of the invention.
オリゴヌクレオチドを同定する、精製する又は濃縮する、前出の方法では、オリゴヌクレオチドは本明細書に記載されるような種類である。上記の方法は、たとえば、オリゴヌクレオチドのランダマー調製物から、高親和性オリゴヌクレオチドを同定する、精製する又は濃縮するために有利である。 In the previous method of identifying, purifying or concentrating oligonucleotides, the oligonucleotides are of the type as described herein. The above methods are advantageous, for example, for identifying, purifying or enriching high affinity oligonucleotides from oligonucleotide randomer preparations.
関連する局面では、本発明は、当初のオリゴヌクレオチドプールから抗ウイルス性オリゴヌクレオチドを同定する、精製する又は濃縮する前出の方法のいずれかの方法を用いて同定される1以上のオリゴヌクレオチドを包含する抗ウイルス性オリゴヌクレオチド調製物に関するものであり、その際、オリゴヌクレオチド調製物中のオリゴヌクレオチドは、当初のオリゴヌクレオチドプールにおけるオリゴヌクレオチドの平均結合親和性よりも、高い、1以上のウイルスタンパク質に対する平均結合親和性を呈する。 In a related aspect, the invention relates to one or more oligonucleotides identified using any of the previous methods for identifying, purifying or enriching antiviral oligonucleotides from an initial oligonucleotide pool. One or more viral proteins, wherein the oligonucleotide in the oligonucleotide preparation is higher than the average binding affinity of the oligonucleotide in the original oligonucleotide pool Exhibits an average binding affinity for.
特定の態様では、オリゴヌクレオチドの平均結合親和性は、当初のオリゴヌクレオチドプールにおけるオリゴヌクレオチドの平均結合親和性よりも少なくとも2倍、3倍、5倍、10倍、20倍、50倍又は100倍高く、又はさらに高く;結合親和性の中央値は、当初のオリゴプールの結合親和性の中央値に比べて、少なくとも2倍、3倍、5倍、10倍、20倍、50倍又は100倍高く、その際、中央値は真ん中の値を言う。 In certain embodiments, the average binding affinity of the oligonucleotide is at least 2-fold, 3-fold, 5-fold, 10-fold, 20-fold, 50-fold or 100-fold greater than the average binding affinity of the oligonucleotides in the original oligonucleotide pool. Higher or even higher; median binding affinity is at least 2-fold, 3-fold, 5-fold, 10-fold, 20-fold, 50-fold or 100-fold compared to the median binding affinity of the original oligopool In that case, the median is the middle value.
さらにもう1つの局面では、本発明は、少なくとも1つの抗ウイルス性オリゴヌクレオチド及び少なくとも1つの非ヌクレオチド抗ウイルス性ポリマーを包含する抗ウイルス性ポリマーミックスを提供する。特定の態様では、オリゴヌクレオチドは抗ウイルス性オリゴヌクレオチドについて本明細書で記載されるようなオリゴヌクレオチドであり及び/又は抗ウイルス性ポリマーは本明細書で記載されるような抗ウイルス性ポリマーはであり、又はさもなければ、当該技術で既知である抗ウイルス性ポリマーは、又は以下に特定する抗ウイルス性ポリマーはである。 In yet another aspect, the present invention provides an antiviral polymer mix comprising at least one antiviral oligonucleotide and at least one non-nucleotide antiviral polymer. In certain embodiments, the oligonucleotide is an oligonucleotide as described herein for an antiviral oligonucleotide and / or the antiviral polymer is an antiviral polymer as described herein. Yes, or else is an antiviral polymer known in the art, or an antiviral polymer specified below.
さらにもう1つの局面では、本発明は、ランダマーが少なくとも長さ6ヌクレオチドであるオリゴヌクレオチドランダマーを提供する。特定の態様では、ランダマーは抗ウイルス性オリゴヌクレオチドについて上記で特定されたような長さを有し;ランダマーは、少なくとも1つのホスホロチオエート結合を包含し、ランダマーは、少なくとも1つのホスホロジチオエート結合又は本明細書で列記されるようなそのほかの修飾を包含し;ランダマーオリゴヌクレオチドは少なくとも1つの非ランダマー断片(たとえば、選択されたウイルスの核酸配列に相補的な断片)を包含し、それは、オリゴヌクレオチドについて上記で特定されたような長さを有することができ;ランダマーは、少なくとも5、10、15、20、50、100、200、500又は700μM、1、5、7、10、20、50、100、200、500又は700mM若しくは1Mのランダマーを含有する調製物中若しくは調製物のプール中にあり、又は包含的な端点として前出から2つの異なる値をとることにより画定される範囲内にあり、又は列記された尺度若しくは尺度の範囲の1つで合成される。 In yet another aspect, the invention provides an oligonucleotide randomer, wherein the randomer is at least 6 nucleotides in length. In certain embodiments, the randomer has a length as specified above for the antiviral oligonucleotide; the randomer includes at least one phosphorothioate linkage, and the randomer includes at least one phosphorodithioate linkage or Including other modifications as listed herein; a randomer oligonucleotide includes at least one non-randomer fragment (eg, a fragment complementary to a selected viral nucleic acid sequence), which is an oligonucleotide Can have a length as specified above; the randomer is at least 5, 10, 15, 20, 50, 100, 200, 500 or 700 μM, 1, 5, 7, 10, 20, 50, In preparations or pools of preparations containing 100, 200, 500 or 700 mM or 1 M randomer, or as an inclusive endpoint Are in a range defined by taking two different values from or synthesized with one of the listed scales or scale ranges.
同様に、本発明は、少なくとも1つのランダマー、たとえば、上述のようなランダマーを合成することによって抗ウイルス性ランダマーを調製する方法を提供する。 Similarly, the present invention provides a method of preparing an antiviral randomer by synthesizing at least one randomer, eg, a randomer as described above.
上記で示したように、ウイルス感染又はウイルス感染のリスク又は特定のウイルスへの標的化に関与するいかなる局面についても、特定の態様では、ウイルスは上記で列記されたようなものである。 As indicated above, for any aspect involved in viral infection or risk of viral infection or targeting to a particular virus, in certain embodiments, the virus is as listed above.
表現「ヒト及び動物のウイルス」は、限定することなく、一般にDNAウイルス及びRNAウイルスを包含することが意図される。DNAウイルスには、たとえば、パルボウイルス科、パポバウイルス科、アデノウイルス科、ヘルペスウイルス科、ポックスウイルス科、肝炎ウイルス科、及びパピローマウイルス科が挙げられる。RNAウイルスには、たとえば、アレナウイルス科、ブンヤウイルス科、カルシウイルス科、コロナウイルス科、フィロウイルス科、フラビウイルス科、オルソミクソウイルス科、パラミクソウイルス科、ピコルナウイルス科、レオウイルス科、ラブドウイルス科、レトロウイルス科又はトガウイルス科が挙げられる。 The expression “human and animal viruses” is generally intended to encompass, without limitation, DNA viruses and RNA viruses. DNA viruses include, for example, Parvoviridae, Papovaviridae, Adenoviridae, Herpesviridae, Poxviridae, Hepatitis Viridae, and Papillomaviridae. RNA viruses include, for example, Arenaviridae, Bunyaviridae, Calciviridae, Coronaviridae, Filoviridae, Flaviviridae, Orthomyxoviridae, Paramyxoviridae, Picornaviridae, Reoviridae, Rhabdoviridae, Retroviridae, or Togaviridae.
もう1つの分子又は部分と結合又は複合化することによるオリゴヌクレオチドの特性の改変に関連して、特性の改変は、結合又は複合化した分子又は部分のない同一オリゴヌクレオチドに比べて、評価される。 In connection with altering the properties of an oligonucleotide by binding or conjugating with another molecule or moiety, the property alteration is assessed relative to the same oligonucleotide without the bound or complexed molecule or portion. .
追加の態様は、詳細な説明及び請求の範囲から明らかになるであろう。 Additional aspects will be apparent from the detailed description and from the claims.
好ましい態様の詳細な説明
本発明は、配列非依存的な機序により作用する抗ウイルスオリゴヌクレオチドの同定および使用に関し、また多数のウイルスに対する使用の発見を含み、抗ウイルス活性は、より大きいオリゴヌクレオチドにおいてより大きく、典型的に40ヌクレオチドまたはそれ以上の長さのオリゴヌクレオチドにおいて最適である。
Detailed Description of Preferred Embodiments The present invention relates to the identification and use of antiviral oligonucleotides that act by a sequence-independent mechanism and includes the discovery of use against a number of viruses, where antiviral activity is greater In oligonucleotides, typically 40 nucleotides or longer in length.
本発明によれば、少なくとも一種の修飾されたヌクレオチド間結合を含むオリゴヌクレオチドを提供し、該オリゴヌクレオチドは、標的ウイルスに対する抗ウイルス活性を有し、該活性は、大部分は配列非依存的な作用様式で作用する。 According to the present invention there is provided an oligonucleotide comprising at least one modified internucleotide linkage, said oligonucleotide having antiviral activity against the target virus, said activity being largely sequence independent. Acts in a mode of action.
本発明によれば、オリゴヌクレオチド内の少なくとも50%のヌクレオチドが、リボース部分の2'位において修飾され、かつ少なくとも50%のヌクレオチド間結合が修飾されているオリゴヌクレオチドを提供し、該オリゴヌクレオチドは、標的ウイルスに対する抗ウイルス活性を有し、該活性は、大部分は配列非依存的な作用様式で作用する。一態様においては、各々50%、80%である。一態様においては、各々80%、80%である。一態様においては、各々90%、90%である。一態様においては、各々100%、100%である。 According to the present invention, there is provided an oligonucleotide wherein at least 50% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety and at least 50% of the internucleotide linkage is modified, the oligonucleotide comprising: Have antiviral activity against the target virus, which acts largely in a sequence-independent manner of action. In one embodiment, they are 50% and 80%, respectively. In one embodiment, they are 80% and 80%, respectively. In one embodiment, they are 90% and 90%, respectively. In one embodiment, they are 100% and 100%, respectively.
長さおよび非自己相補性
本発明は更に、少なくとも15ヌクレオチド長のオリゴヌクレオチドを提供する。一態様において、少なくとも20ヌクレオチド長。一態様において、少なくとも25ヌクレオチド長。一態様において、少なくとも30ヌクレオチド長。一態様において、少なくとも35ヌクレオチド長。一態様において、少なくとも40ヌクレオチド長。一態様において、少なくとも45ヌクレオチド長。一態様において、少なくとも50ヌクレオチド長。一態様において、少なくとも60ヌクレオチド長。一態様において、少なくとも80ヌクレオチド長。
Length and Non-Self Complementarity The present invention further provides oligonucleotides that are at least 15 nucleotides in length. In one embodiment, at least 20 nucleotides in length. In one embodiment, at least 25 nucleotides in length. In one embodiment, at least 30 nucleotides in length. In one embodiment, at least 35 nucleotides in length. In one embodiment, at least 40 nucleotides in length. In one embodiment, at least 45 nucleotides in length. In one embodiment, at least 50 nucleotides in length. In one embodiment, at least 60 nucleotides in length. In one embodiment, at least 80 nucleotides in length.
本発明は更に、20〜30ヌクレオチド長のオリゴヌクレオチドを提供する。一態様において、30〜40ヌクレオチド長。一態様において、40〜50ヌクレオチド長。一態様において、50〜60ヌクレオチド長。一態様において、60〜70ヌクレオチド長のオリゴヌクレオチド。一態様において、70〜80ヌクレオチド長。 The present invention further provides oligonucleotides 20-30 nucleotides in length. In one embodiment, 30-40 nucleotides in length. In one embodiment, 40-50 nucleotides in length. In one embodiment, 50-60 nucleotides in length. In one embodiment, an oligonucleotide 60-70 nucleotides in length. In one embodiment, 70-80 nucleotides in length.
本発明は更に、5より大きい連続するヌクレオチドからなる自己相補的な配列を有さないオリゴヌクレオチドを提供する。一態様において、10より大きい連続するヌクレオチド。一態様において、20より大きい連続するヌクレオチド。 The invention further provides oligonucleotides that do not have a self-complementary sequence of contiguous nucleotides greater than 5. In one embodiment, more than 10 contiguous nucleotides. In one embodiment, greater than 20 contiguous nucleotides.
本発明は更に、触媒活性を有さないオリゴヌクレオチドを提供する。 The present invention further provides oligonucleotides that do not have catalytic activity.
ランダム
本発明は更に、標的ウイルスに対する抗ウイルス活性を有するオリゴヌクレオチドを提供し、該オリゴヌクレオチドの配列は、該標的ウイルスのゲノム配列の等しい長さのいずれの部分とも相補的ではない。
Random The invention further provides an oligonucleotide having antiviral activity against a target virus, the sequence of the oligonucleotide not being complementary to any part of equal length of the target virus genomic sequence.
本発明は更に、ヒト病原ウイルスのゲノム配列の等しい長さのいずれの部分とも相補的ではないオリゴヌクレオチドを提供する。 The present invention further provides oligonucleotides that are not complementary to any part of equal length of the genomic sequence of the human pathogenic virus.
本発明は更に、2005年1月1日に配列決定されたようなヒト病原ウイルスのゲノム配列の等しい長さのいずれの部分とも相補的ではないオリゴヌクレオチドを提供する。 The present invention further provides oligonucleotides that are not complementary to any part of equal length of the genomic sequence of a human pathogenic virus as sequenced on January 1, 2005.
本発明は更に、ヒトのゲノム配列の等しい長さのいずれの部分とも相補的ではないオリゴヌクレオチドを提供する。 The present invention further provides oligonucleotides that are not complementary to any portion of equal length in the human genomic sequence.
本発明は更に、牛、馬、豚、羊、鳥、犬、猫および魚からなる群より選択される1種または複数種の動物のゲノム配列の等しい長さのいずれの部分とも相補的ではないオリゴヌクレオチドを提供する。 The present invention is further not complementary to any part of equal length of the genomic sequence of one or more animals selected from the group consisting of cattle, horses, pigs, sheep, birds, dogs, cats and fish. Oligonucleotides are provided.
RNAおよび他の鎖部分の制限
本発明は更に、少なくとも30%のヌクレオチドがリボヌクレオチドであるオリゴヌクレオチドを提供する。一態様において、少なくとも50%のヌクレオチドがリボヌクレオチドである。一態様において、少なくとも70%のヌクレオチドがリボヌクレオチドである。一態様において、少なくとも80%のヌクレオチドがリボヌクレオチドである。一態様において、少なくとも90%のヌクレオチドがリボヌクレオチドである。一態様において、全部のヌクレオチドがリボヌクレオチドである。
Restriction of RNA and other strand moieties The present invention further provides oligonucleotides in which at least 30% of the nucleotides are ribonucleotides. In one embodiment, at least 50% of the nucleotides are ribonucleotides. In one embodiment, at least 70% of the nucleotides are ribonucleotides. In one embodiment, at least 80% of the nucleotides are ribonucleotides. In one embodiment, at least 90% of the nucleotides are ribonucleotides. In one embodiment, all nucleotides are ribonucleotides.
本発明は更に、1〜4個の非ヌクレオチド鎖部分を含むオリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide comprising 1 to 4 non-nucleotide strand moieties.
ランダマー
本発明は更に、ランダマー配列の少なくとも10個の連続するヌクレオチドを提供する。一態様において、ランダマー配列の少なくとも20個のヌクレオチド。一態様において、ランダマー配列の少なくとも30個のヌクレオチド。一態様において、ランダマー配列の少なくとも40個のヌクレオチド。
Randomers The present invention further provides at least 10 consecutive nucleotides of a randomer sequence. In one embodiment, at least 20 nucleotides of the randomer sequence. In one embodiment, at least 30 nucleotides of the randomer sequence. In one embodiment, at least 40 nucleotides of the randomer sequence.
本発明は更に、ランダマーオリゴヌクレオチドであるオリゴヌクレオチドを提供する。 The present invention further provides oligonucleotides that are randomer oligonucleotides.
ホモポリマー
本発明は更に、少なくとも10個の連続するAヌクレオチドからなるホモポリマー配列を含むオリゴヌクレオチドを提供する。一態様において、少なくとも10個の連続するTヌクレオチド。一態様において、少なくとも10個の連続するUヌクレオチド。一態様において、少なくとも10個の連続するCヌクレオチド。一態様において、少なくとも10個の連続するGヌクレオチド。一態様において、少なくとも10個の連続するIヌクレオチド類似体。
Homopolymer The present invention further provides an oligonucleotide comprising a homopolymer sequence consisting of at least 10 consecutive A nucleotides. In one embodiment, at least 10 consecutive T nucleotides. In one embodiment, at least 10 consecutive U nucleotides. In one embodiment, at least 10 consecutive C nucleotides. In one embodiment, at least 10 consecutive G nucleotides. In one embodiment, at least 10 consecutive I nucleotide analogues.
ヘテロ二量体
本発明は更に、少なくとも10ヌクレオチド長のポリAT配列を含むオリゴヌクレオチドを提供する。一態様において、少なくとも10ヌクレオチド長のポリAC配列。一態様において、少なくとも10ヌクレオチド長のポリAG配列。一態様において、少なくとも10ヌクレオチド長のポリAU配列。一態様において、少なくとも10ヌクレオチド長のポリAI配列。一態様において、少なくとも10ヌクレオチド長のポリGC配列。一態様において、少なくとも10ヌクレオチド長のポリGT配列。一態様において、少なくとも10ヌクレオチド長のポリGU配列。一態様において、少なくとも10ヌクレオチド長のポリGI配列。一態様において、少なくとも10ヌクレオチド長のポリCT配列。一態様において、少なくとも10ヌクレオチド長のポリCU配列。一態様において、少なくとも10ヌクレオチド長のポリCI配列。一態様において、少なくとも10ヌクレオチド長のポリTI配列。
Heterodimers The present invention further provides oligonucleotides comprising a polyAT sequence at least 10 nucleotides in length. In one embodiment, a poly AC sequence that is at least 10 nucleotides in length. In one embodiment, a poly AG sequence that is at least 10 nucleotides in length. In one embodiment, a poly AU sequence that is at least 10 nucleotides in length. In one embodiment, a polyAI sequence that is at least 10 nucleotides in length. In one embodiment, a poly GC sequence that is at least 10 nucleotides in length. In one embodiment, a poly GT sequence that is at least 10 nucleotides in length. In one embodiment, a polyGU sequence that is at least 10 nucleotides in length. In one embodiment, a polyGI sequence that is at least 10 nucleotides in length. In one embodiment, a poly CT sequence that is at least 10 nucleotides in length. In one embodiment, a polyCU sequence that is at least 10 nucleotides in length. In one embodiment, a polyCI sequence that is at least 10 nucleotides in length. In one embodiment, a polyTI sequence that is at least 10 nucleotides in length.
psおよびps2を含む、修飾結合
本発明は更に、修飾された結合が、ホスホロチオエート結合、ホスホロジチオエート結合およびボラノホスフェート結合からなる群から選択されるオリゴヌクレオチドを提供する。
Modified linkage comprising ps and ps2 The present invention further provides an oligonucleotide wherein the modified linkage is selected from the group consisting of a phosphorothioate linkage, a phosphorodithioate linkage and a boranophosphate linkage.
本発明は更に、少なくとも50%のヌクレオチド間結合が、修飾された結合であるオリゴヌクレオチドを提供する。一態様において、少なくとも80%のヌクレオチド間結合が修飾された結合である。一態様において、少なくとも90%のヌクレオチド間結合が修飾された結合である。一態様において、全部のヌクレオチド間結合が修飾された結合である。 The invention further provides oligonucleotides in which at least 50% of the internucleotide linkages are modified linkages. In one embodiment, at least 80% of the internucleotide linkages are modified linkages. In one embodiment, at least 90% of the internucleotide linkages are modified linkages. In one embodiment, all internucleotide linkages are modified linkages.
本発明は更に、少なくとも50%のヌクレオチド間結合がホスホロチオエート結合であるオリゴヌクレオチドを提供する。一態様において、少なくとも80%のヌクレオチド間結合がホスホロチオエート結合である。一態様において、少なくとも90%のヌクレオチド間結合がホスホロチオエート結合である。一態様において、全てのヌクレオチド間結合がホスホロチオエート結合である。 The invention further provides oligonucleotides wherein at least 50% of the internucleotide linkages are phosphorothioate linkages. In one embodiment, at least 80% of the internucleotide linkages are phosphorothioate linkages. In one embodiment, at least 90% of the internucleotide linkages are phosphorothioate linkages. In one embodiment, all internucleotide linkages are phosphorothioate linkages.
本発明は更に、少なくとも50%のヌクレオチド間結合がホスホロジチオエート結合であるオリゴヌクレオチドを提供する。一態様において、少なくとも80%のヌクレオチド間結合がホスホロジチオエート結合である。一態様において、全てのヌクレオチド間結合がホスホロジチオエート結合である。 The invention further provides oligonucleotides in which at least 50% of the internucleotide linkages are phosphorodithioate linkages. In one embodiment, at least 80% of the internucleotide linkages are phosphorodithioate linkages. In one embodiment, all internucleotide linkages are phosphorodithioate linkages.
2'-修飾、修飾結合との組み合わせ
本発明は更に、少なくとも1個のホスホジエステル結合を含むオリゴヌクレオチドを提供する。一態様において、該オリゴヌクレオチドは、少なくとも10%のホスホジエステル結合を含む。一態様において、該オリゴヌクレオチドは、少なくとも20%ホスホジエステル結合を含む。
2′-Modification, Combination with Modified Bond The present invention further provides an oligonucleotide comprising at least one phosphodiester bond. In one embodiment, the oligonucleotide comprises at least 10% phosphodiester linkage. In one embodiment, the oligonucleotide comprises at least 20% phosphodiester linkage.
一態様において、該オリゴヌクレオチド内の少なくとも50%のヌクレオチドが、リボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の少なくとも60%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の少なくとも70%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の少なくとも80%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の少なくとも90%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の100%のヌクレオチドがリボース部分の2'位において修飾されている。 In one embodiment, at least 50% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 60% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 70% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 80% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 90% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety. In one embodiment, 100% of the nucleotides in the oligonucleotide are modified at the 2 ′ position of the ribose moiety.
本発明は更に、オリゴヌクレオチド内の、少なくとも50%のヌクレオチド間結合が修飾されかつ少なくとも50%のヌクレオチドがリボース部分の2'位において修飾されているオリゴヌクレオチドを提供する。一態様において、該オリゴヌクレオチド内の少なくとも60%のヌクレオチド間結合が修飾され、かつ少なくとも60%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の少なくとも70%のヌクレオチド間結合が修飾され、かつ少なくとも70%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の少なくとも80%のヌクレオチド間結合が修飾され、かつ少なくとも80%のヌクレオチドがリボース部分の2'位において修飾されている。一態様において、該オリゴヌクレオチド内の全てのヌクレオチド間結合が修飾され、かつ全てのヌクレオチドがリボース部分の2'位において修飾されている。 The present invention further provides oligonucleotides in which at least 50% of the internucleotide linkages in the oligonucleotide are modified and at least 50% of the nucleotides are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 60% of the internucleotide linkages within the oligonucleotide are modified and at least 60% of the nucleotides are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 70% of the internucleotide linkages within the oligonucleotide are modified and at least 70% of the nucleotides are modified at the 2 ′ position of the ribose moiety. In one embodiment, at least 80% of the internucleotide linkages within the oligonucleotide are modified and at least 80% of the nucleotides are modified at the 2 ′ position of the ribose moiety. In one embodiment, all internucleotide linkages within the oligonucleotide are modified and all nucleotides are modified at the 2 ′ position of the ribose moiety.
本発明は更に、オリゴヌクレオチド内の少なくとも15%のヌクレオチドが、リボース部分の2'位において2'-0Me部分を含むオリゴヌクレオチドを提供する。一態様において、該オリゴヌクレオチド内の少なくとも20%のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の少なくとも30%のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の少なくとも50%のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の少なくとも60%のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の少なくとも70%のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の少なくとも80%のヌクレオチドが、リボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の少なくとも90%のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。一態様において、該オリゴヌクレオチド内の全部のヌクレオチドがリボース部分の2'位において2'-0Me部分を含む。 The invention further provides an oligonucleotide wherein at least 15% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 20% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 30% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 50% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 60% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 70% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 80% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, at least 90% of the nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety. In one embodiment, all nucleotides in the oligonucleotide comprise a 2′-0Me moiety at the 2 ′ position of the ribose moiety.
多様な特徴
本発明は更に、リンカーで連結された2個またはそれ以上のオリゴヌクレオチド配列からなる鎖状体であるオリゴヌクレオチドを提供する。
Various Features The present invention further provides an oligonucleotide that is a chain consisting of two or more oligonucleotide sequences linked by a linker.
本発明は更に、1個または複数のヌクレオチド残基において、オリゴヌクレオチドの性質を変更して、より高い安定性、血清とのより低い相互作用、より高い細胞取り込み、ウイルスタンパク質とのより高い相互作用、送達用に調製するための改善された能力、検出可能なシグナル、より高い抗ウイルス活性、より良好な薬物動態学的性質、特定の組織分布、より低い毒性からなる群より選択される1個または複数の性質を獲得する分子に対して結合または複合しているオリゴヌクレオチドを提供する。 The invention further modifies the nature of the oligonucleotide at one or more nucleotide residues to provide higher stability, lower interaction with serum, higher cellular uptake, higher interaction with viral proteins. 1 selected from the group consisting of improved ability to prepare for delivery, detectable signal, higher antiviral activity, better pharmacokinetic properties, specific tissue distribution, lower toxicity Alternatively, an oligonucleotide is provided that is bound or conjugated to a molecule that acquires multiple properties.
本発明は更に、二本鎖であるオリゴヌクレオチドを提供する。 The invention further provides oligonucleotides that are double stranded.
本発明は更に、二本鎖または一本鎖であり、かつ非ワトソン-クリック型の相互作用を介してハイブリダイズすることが可能な、少なくとも1個の塩基を含むオリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide comprising at least one base that is double-stranded or single-stranded and is capable of hybridizing via non-Watson-Crick interactions.
本発明は更に、ウイルスmRNAに対して相補的な部分を含むオリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide comprising a portion that is complementary to a viral mRNA.
本発明は更に、1個または複数のウイルス成分に結合するオリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide that binds to one or more viral components.
本発明は更に、1個または複数の宿主成分と相互作用し、該相互作用がウイルスの活性または産生の阻害をもたらす、オリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide that interacts with one or more host components, which interaction results in inhibition of viral activity or production.
本発明は更に、その配列の少なくとも一部が、ウイルスゲノムに由来するオリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide wherein at least part of the sequence is derived from the viral genome.
本発明は更に、その配列の少なくとも一部がウイルスゲノムに由来し、かつ配列相補的ではない作用様式が大部分である抗ウイルス活性を有するオリゴヌクレオチドを提供する。 The present invention further provides an oligonucleotide having antiviral activity, wherein at least part of the sequence is derived from the viral genome and the mode of action is largely non-sequence complementary.
本発明は更に、その配列の少なくとも一部が、ウイルスパッケージング配列またはアプタマー相互作用に関与する他のウイルス配列に由来する、オリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide wherein at least part of the sequence is derived from a viral packaging sequence or other viral sequences involved in aptamer interactions.
本発明は更に、その配列の少なくとも一部が、ウイルス成分もしくは宿主成分またはその両方とのアプタマー相互作用に関与する、オリゴヌクレオチドを提供する。 The invention further provides an oligonucleotide wherein at least part of the sequence is involved in aptamer interaction with a viral component or a host component or both.
活性レベル
本発明は更に、標的ウイルスに関して0.10μmまたはそれ未満のIC50を有するオリゴヌクレオチドを提供する。一態様において、該オリゴヌクレオチドは、標的ウイルスに関して0.05μmまたはそれ未満のIC50を有する。一態様において、該オリゴヌクレオチドは、標的ウイルスに関して0.025μmまたはそれ未満のIC50を有する。一態様において、該オリゴヌクレオチドは、標的ウイルスに関して0.015μmまたはそれ未満のIC50を有する。
Activity Level The present invention further provides oligonucleotides having an IC 50 of 0.10 μm or less for the target virus. In one embodiment, the oligonucleotide has an IC 50 of 0.05 μm or less for the target virus. In one embodiment, the oligonucleotide has an IC 50 of 0.025 μm or less for the target virus. In one embodiment, the oligonucleotide has an IC 50 of 0.015 μm or less for the target virus.
標的ウイルス
本発明は更に、DNAウイルスを標的とするオリゴヌクレオチドを提供する。一態様において、RNAウイルス。一態様において、ヘルペスウイルス科の一メンバー。一態様において、HSV-1。一態様において、HSV-2。一態様において、CMV。一態様において、ヘパドナウイルス科の一メンバー。一態様において、HBV。一態様において、パルボウイルス科の一メンバー。一態様において、ポックスウイルス科の一メンバー。一態様において、パピローマウイルス科の一メンバー。一態様において、アデノウイルス科の一メンバー。一態様において、レトロウイルス科の一メンバー。一態様において、HIV-1。一態様において、HIV-2。一態様において、パラミクソウイルス科の一メンバー。一態様において、RSV。一態様において、パラインフルエンザウイルス。一態様において、ブニヤウイルス科の一メンバー。一態様において、ハンタウイルス。一態様において、ピコルナウイルス科の一メンバー。一態様において、コクサッキーウイルス。一態様において、ライノウイルス。一態様において、フラビウイルス科の一メンバー。一態様において、黄熱病ウイルス。一態様において、デングウイルス。一態様において、ウエストナイルウイルス。一態様において、C型肝炎ウイルス。一態様において、フィロウイルス科の一メンバー。一態様において、エボラウイルス。一態様において、マールブルグウイルス。一態様において、オルトミクソウイルス科の一メンバー。一態様において、インフルエンザウイルス。一態様において、トガウイルス科の一メンバー。一態様において、コロナウイルス科の一メンバー。一態様において、レオウイルス科の一メンバー。一態様において、ラブドウイルス科の一メンバー。一態様において、アレナウイルス科の一メンバー。一態様において、カルシウイルス科の一メンバー。一態様において、水痘帯状疱疹ウイルス。一態様において、エプスタイン・バーウイルス。一態様において、ヘルペスウイルス6Aまたは6B。一態様において、ヘパドナウイルス科の一メンバー。一態様において、ヒトメタニューモウイルス。一態様において、リフトバレーウイルス。一態様において、クリミア・コンゴ出血熱ウイルス。一態様において、西部馬脳炎ウイルス。一態様において、ラッサ熱ウイルス。
Target Virus The present invention further provides oligonucleotides that target DNA viruses. In one embodiment, an RNA virus. In one embodiment, a member of the Herpesviridae family. In one embodiment, HSV-1. In one embodiment, HSV-2. In one embodiment, CMV. In one embodiment, a member of the Hepadnaviridae family. In one embodiment, HBV. In one embodiment, a member of the Parvoviridae family. In one embodiment, a member of the Poxviridae family. In one embodiment, a member of the Papillomaviridae family. In one embodiment, a member of the adenoviridae family. In one embodiment, a member of the Retroviridae family. In one embodiment, HIV-1. In one embodiment, HIV-2. In one embodiment, a member of the Paramyxoviridae family. In one embodiment, RSV. In one embodiment, parainfluenza virus. In one embodiment, a member of the Bunyaviridae family. In one embodiment, a hantavirus. In one embodiment, a member of the Picornaviridae family. In one embodiment, Coxsackie virus. In one embodiment, rhinovirus. In one embodiment, a member of the Flaviviridae family. In one embodiment, yellow fever virus. In one embodiment, dengue virus. In one embodiment, West Nile virus. In one embodiment, hepatitis C virus. In one embodiment, a member of the Filoviridae family. In one embodiment, Ebola virus. In one embodiment, Marburg virus. In one embodiment, a member of the Orthomyxoviridae family. In one aspect, an influenza virus. In one embodiment, a member of the Togaviridae family. In one embodiment, a member of the Coronaviridae family. In one embodiment, a member of the Reoviridae family. In one embodiment, a member of the Rhabdoviridae family. In one embodiment, a member of the Arenaviridae family. In one embodiment, a member of the Calciviridae family. In one embodiment, varicella-zoster virus. In one embodiment, Epstein-Barr virus. In one embodiment, herpes virus 6A or 6B. In one embodiment, a member of the Hepadnaviridae family. In one embodiment, a human metapneumovirus. In one aspect, Rift Valley virus. In one embodiment, Crimea Congo hemorrhagic fever virus. In one embodiment, western equine encephalitis virus. In one embodiment, Lassa fever virus.
オリゴヌクレオチド
本発明は更に、少なくとも20個の結合したヌクレオチドを含み、少なくとも80%の結合が修飾されており;かつ少なくとも80%のヌクレオチドが、リボース糖部分の2'-修飾を含む、オリゴヌクレオチドを提供する。一態様において、このオリゴヌクレオチドは抗ウイルス活性を有している。
Oligonucleotides The present invention further comprises an oligonucleotide comprising at least 20 linked nucleotides, wherein at least 80% of the linkages are modified; and at least 80% of the nucleotides comprise a 2′-modification of the ribose sugar moiety. provide. In one embodiment, the oligonucleotide has antiviral activity.
一態様において、少なくとも90%のヌクレオチド間結合が修飾されている。一態様において、全部のヌクレオチド間結合が修飾されている。一態様において、少なくとも90%のヌクレオチドがリボース糖の2'-修飾を含む。一態様において、全部のヌクレオチドがリボース糖の2'-修飾を含む。 In one embodiment, at least 90% of the internucleotide linkages are modified. In one embodiment, all internucleotide linkages are modified. In one embodiment, at least 90% of the nucleotides contain a 2′-modification of the ribose sugar. In one embodiment, every nucleotide comprises a 2′-modification of the ribose sugar.
本発明は更に、2'-修飾が2'-OMe置換であるオリゴヌクレオチドを提供する。一態様において、少なくとも90%のヌクレオチドが2'-OMe置換を含む。一態様において、全部のヌクレオチドが2'-OMe置換を含む。 The invention further provides an oligonucleotide wherein the 2′-modification is a 2′-OMe substitution. In one embodiment, at least 90% of the nucleotides contain 2′-OMe substitutions. In one embodiment, all nucleotides contain 2′-OMe substitutions.
本発明は更に、2'-修飾が2'-メトキシエトキシ置換であるオリゴヌクレオチドを提供する。一態様において、少なくとも15%のヌクレオチドが2'-メトキシエトキシ置換を含む。一態様において、少なくとも50%のヌクレオチドが2'-メトキシエトキシ置換を含む。一態様において、少なくとも90%のヌクレオチドが2'-メトキシエトキシ置換を含む。一態様において、全部のヌクレオチドが2'-メトキシエトキシ置換を含む。 The invention further provides an oligonucleotide wherein the 2'-modification is a 2'-methoxyethoxy substitution. In one embodiment, at least 15% of the nucleotides contain 2'-methoxyethoxy substitution. In one embodiment, at least 50% of the nucleotides contain 2′-methoxyethoxy substitution. In one embodiment, at least 90% of the nucleotides contain 2'-methoxyethoxy substitution. In one embodiment, all nucleotides contain 2′-methoxyethoxy substitutions.
本発明は更に、少なくとも40ヌクレオチド長のオリゴヌクレオチドを提供する。一態様において、少なくとも50ヌクレオチド長。一態様において、少なくとも60ヌクレオチド長。一態様において、少なくとも80ヌクレオチド長。 The present invention further provides oligonucleotides that are at least 40 nucleotides in length. In one embodiment, at least 50 nucleotides in length. In one embodiment, at least 60 nucleotides in length. In one embodiment, at least 80 nucleotides in length.
本発明は更に、30〜40ヌクレオチド長のオリゴヌクレオチドを提供する。一態様において、40〜50ヌクレオチド長。一態様において、50〜60ヌクレオチド長。一態様において、60〜70ヌクレオチド長。一態様において、70〜80ヌクレオチド長。 The present invention further provides oligonucleotides of 30-40 nucleotides in length. In one embodiment, 40-50 nucleotides in length. In one embodiment, 50-60 nucleotides in length. In one embodiment, 60-70 nucleotides in length. In one embodiment, 70-80 nucleotides in length.
本発明は更に、5個より多い連続するヌクレオチドからなる自己相補的な配列を有さないヌクレオチドを提供する。一態様において、10個より多い連続するヌクレオチド。一態様において、20個より多い連続するヌクレオチド。 The invention further provides nucleotides that do not have a self-complementary sequence of more than 5 consecutive nucleotides. In one embodiment, more than 10 contiguous nucleotides. In one embodiment, more than 20 contiguous nucleotides.
本発明は更に、触媒活性を有さないオリゴヌクレオチドを提供する。 The present invention further provides oligonucleotides that do not have catalytic activity.
鎖部分の制限
本発明は更に、1〜4個の非ヌクレオチド鎖部分を更に含むオリゴヌクレオチドを提供する。
Chain Part Restriction The present invention further provides an oligonucleotide further comprising 1 to 4 non-nucleotide chain moieties.
混合物
本発明は更に、少なくとも2種の異なる本発明の抗ウイルスオリゴヌクレオチドの混合物を含む、オリゴヌクレオチド混合物を提供する。一態様において、少なくとも10種の異なる抗ウイルスオリゴヌクレオチド。一態様において、少なくとも100種の異なる抗ウイルスオリゴヌクレオチド。一態様において、少なくとも1000種の異なる抗ウイルスオリゴヌクレオチド。一態様において、少なくとも106種の異なる抗ウイルスオリゴヌクレオチド。
Mixtures The invention further provides an oligonucleotide mixture comprising a mixture of at least two different antiviral oligonucleotides of the invention. In one embodiment, at least 10 different antiviral oligonucleotides. In one embodiment, at least 100 different antiviral oligonucleotides. In one embodiment, at least 1000 different antiviral oligonucleotides. In one embodiment, at least 106 different antiviral oligonucleotides.
本発明は更に、複数の該異なるオリゴヌクレオチドが、少なくとも10ヌクレオチド長である混合物を提供する。一態様において、少なくとも20ヌクレオチド長である。一態様において、少なくとも30ヌクレオチド長である。一態様において、少なくとも40ヌクレオチド長である。一態様において、少なくとも50ヌクレオチド長である。一態様において、少なくとも60ヌクレオチド長である。一態様において、少なくとも70ヌクレオチド長である。一態様において、少なくとも80ヌクレオチド長である。一態様において、少なくとも120ヌクレオチド長である。 The invention further provides a mixture wherein the plurality of different oligonucleotides are at least 10 nucleotides in length. In one embodiment, it is at least 20 nucleotides in length. In one embodiment, it is at least 30 nucleotides in length. In one embodiment, it is at least 40 nucleotides in length. In one embodiment, it is at least 50 nucleotides in length. In one embodiment, it is at least 60 nucleotides in length. In one embodiment, it is at least 70 nucleotides in length. In one embodiment, it is at least 80 nucleotides in length. In one embodiment, it is at least 120 nucleotides in length.
薬学的組成物
本発明は更に、治療有効量の少なくとも一種の薬理学的に許容される抗ウイルスオリゴヌクレオチド、ポリピリミジンまたはオリゴヌクレオチド混合物、および薬学的に許容される担体とを含有する抗ウイルス薬学的組成物を提供し、該オリゴヌクレオチドまたは該混合物中のオリゴヌクレオチドの抗ウイルス活性は、主として配列非依存的な作用様式により生じる。
Pharmaceutical Composition The present invention further comprises an antiviral pharmaceutical comprising a therapeutically effective amount of at least one pharmacologically acceptable antiviral oligonucleotide, polypyrimidine or oligonucleotide mixture, and a pharmaceutically acceptable carrier. The antiviral activity of the oligonucleotides or oligonucleotides in the mixture arises primarily through a sequence-independent mode of action.
本発明は更に、ウイルス病因を伴う疾病の処置、制御または予防に適合された抗ウイルス薬学的組成物を提供する。 The present invention further provides antiviral pharmaceutical compositions adapted for the treatment, control or prevention of diseases associated with viral pathogenesis.
本発明は更に、プリオン疾病の処置、制御または予防に適合された抗ウイルス薬学的組成物を提供する。 The invention further provides an antiviral pharmaceutical composition adapted for the treatment, control or prevention of prion diseases.
本発明は更に、眼内、経口消化、経腸、吸入、皮内注入、皮下注入、筋内注入、腹腔内注入、くも膜下注入、気管内注入および静脈内注入からなる群より選択される様式による送達に適合された抗ウイルス薬学的組成物を提供する。 The invention is further of a mode selected from the group consisting of intraocular, oral digestion, enteral, inhalation, intradermal injection, subcutaneous injection, intramuscular injection, intraperitoneal injection, subarachnoid injection, intratracheal injection and intravenous injection. An antiviral pharmaceutical composition adapted for delivery by is provided.
本発明は更に、送達系を更に含む抗ウイルス薬学的組成物を提供する。一態様において、該送達系は、特定の細胞または特定の組織を標的とする。一態様において、該組成物は、更に少なくとも一種の他の抗ウイルス薬を、組み合わせて含有する。一態様において、該組成物は、更に非ヌクレオチド抗ウイルスポリマーを、組み合わせて含有する。一態様において、該組成物は、抗ウイルスアンチセンスオリゴヌクレオチドを、組み合わせて含有する。一態様において、該組成物は、更に抗ウイルスRNAi-誘導オリゴヌクレオチドを含有する。一態様において、該抗ウイルスRNAi-誘導オリゴヌクレオチドは、siRNAである。 The present invention further provides an antiviral pharmaceutical composition further comprising a delivery system. In one embodiment, the delivery system targets a specific cell or a specific tissue. In one embodiment, the composition further contains at least one other antiviral agent in combination. In one embodiment, the composition further comprises a combination of non-nucleotide antiviral polymers. In one embodiment, the composition contains a combination of antiviral antisense oligonucleotides. In one embodiment, the composition further comprises an antiviral RNAi-derived oligonucleotide. In one embodiment, the antiviral RNAi-derived oligonucleotide is an siRNA.
本発明は更に、標的ウイルスに関して0.10μMまたはそれ未満のIC50を有する抗ウイルス薬学的組成物を提供する。一態様において、標的ウイルスに関して0.05μMまたはそれ未満のIC50を有する抗ウイルス薬学的組成物を提供する。一態様において、標的ウイルスに関して0.025μMまたはそれ未満のIC50を有する抗ウイルス薬学的組成物を提供する。一態様において、標的ウイルスに関して0.015μMまたはそれ未満のIC50を有する抗ウイルス薬学的組成物を提供する。 The present invention further provides antiviral pharmaceutical compositions having an IC 50 of 0.10 μM or less for the target virus. In one embodiment, an antiviral pharmaceutical composition is provided that has an IC 50 of 0.05 μM or less for the target virus. In one embodiment, an antiviral pharmaceutical composition is provided that has an IC 50 of 0.025 μM or less for the target virus. In one embodiment, an antiviral pharmaceutical composition is provided that has an IC 50 of 0.015 μM or less for the target virus.
キット
本発明は更に、少なくとも一種の抗ウイルスオリゴヌクレオチド、混合物または抗ウイルス薬学的組成物を、ラベルを貼ったパッケージ内に含み、該オリゴヌクレオチドの抗ウイルス活性が主として配列相補的ではない作用様式により生じ、かつ該パッケージ上のラベルが該抗ウイルスオリゴヌクレオチドを少なくとも一種のウイルスに対して使用できることを示す、キットを提供する。
Kits The present invention further includes at least one antiviral oligonucleotide, mixture or antiviral pharmaceutical composition in a labeled package, wherein the antiviral activity of the oligonucleotide is primarily non-sequence complementary. A kit is provided that occurs and the label on the package indicates that the antiviral oligonucleotide can be used against at least one virus.
本発明は更に、少なくとも二種の異なる抗ウイルスオリゴヌクレオチドの混合物を含むキットを提供する。 The present invention further provides a kit comprising a mixture of at least two different antiviral oligonucleotides.
本発明は更に、規制機関によりヒトでの使用が認可されたキットを提供する。 The present invention further provides kits approved for human use by regulatory agencies.
本発明は更に、規制機関により、少なくとも一種のヒトではない動物での使用が許可されたキット提供する。一態様において、該ヒトではない動物は、霊長類である。一態様において、該ヒトではない動物は、猫である。一態様において、該ヒトではない動物は、牛である。一態様において、該ヒトではない動物は、羊である。一態様において、該ヒトではない動物は、犬である。一態様において、該ヒトではない動物は、豚である。一態様において、該ヒトではない動物は、馬である。 The present invention further provides kits that have been approved for use in regulatory animals by at least one non-human animal. In one embodiment, the non-human animal is a primate. In one embodiment, the non-human animal is a cat. In one embodiment, the non-human animal is a cow. In one embodiment, the non-human animal is a sheep. In one embodiment, the non-human animal is a dog. In one embodiment, the non-human animal is a pig. In one embodiment, the non-human animal is a horse.
処置の方法
本発明は更に、本発明による少なくとも一種のオリゴヌクレオチド、または本発明による薬学的組成物の、対象におけるウイルス感染の予防または処置のための医薬の製造における使用を提供する。
Method of treatment The invention further provides the use of at least one oligonucleotide according to the invention, or a pharmaceutical composition according to the invention, in the manufacture of a medicament for the prevention or treatment of a viral infection in a subject.
一態様において、該対象はヒトである。一態様において、該対象はヒトではない動物である。一態様において、該ヒトではない動物は霊長類である。一態様において、該ヒトではない動物は猫である。一態様において、該ヒトではない動物は牛である。一態様において、該ヒトではない動物は羊である。一態様において、該ヒトではない動物は犬である。一態様において、該ヒトではない動物は豚である。一態様において、該ヒトではない動物は馬である。一態様において、該対象は植物である。 In one embodiment, the subject is a human. In one embodiment, the subject is a non-human animal. In one embodiment, the non-human animal is a primate. In one embodiment, the non-human animal is a cat. In one embodiment, the non-human animal is a cow. In one embodiment, the non-human animal is a sheep. In one embodiment, the non-human animal is a dog. In one embodiment, the non-human animal is a pig. In one embodiment, the non-human animal is a horse. In one embodiment, the subject is a plant.
本発明は更に、本発明による少なくとも一種のオリゴヌクレオチド、または本発明による薬学的組成物の、ヒトまたはヒトではない動物内でオンコウイルスに起因する癌の予防的処置のための医薬の製造における使用を提供する。 The invention further relates to the use of at least one oligonucleotide according to the invention, or a pharmaceutical composition according to the invention in the manufacture of a medicament for the prophylactic treatment of cancer caused by oncoviruses in humans or non-human animals. I will provide a.
一態様において、該オリゴヌクレオチドはヒトに投与される。一態様において、該オリゴヌクレオチドは、ヒトではない動物に投与される。一態様において、該ヒトではない動物は霊長類である。一態様において、該ヒトではない動物は猫である。一態様において、該ヒトではない動物は牛である。一態様において、該ヒトではない動物は羊である。一態様において、該ヒトではない動物は犬である。一態様において、該ヒトではない動物は豚である。一態様において、該ヒトではない動物は馬である。 In one embodiment, the oligonucleotide is administered to a human. In one embodiment, the oligonucleotide is administered to an animal that is not human. In one embodiment, the non-human animal is a primate. In one embodiment, the non-human animal is a cat. In one embodiment, the non-human animal is a cow. In one embodiment, the non-human animal is a sheep. In one embodiment, the non-human animal is a dog. In one embodiment, the non-human animal is a pig. In one embodiment, the non-human animal is a horse.
ポリピリミジンオリゴ関連 Polypyrimidine oligo-related
本発明は更に、少なくとも50%のピリミジン残基を含むオリゴヌクレオチドを提供する。一態様においては、少なくとも80%。一態様においては、少なくとも90%。一態様においては、ピリミジン残基のみである。 The invention further provides an oligonucleotide comprising at least 50% pyrimidine residues. In one aspect, at least 80%. In one aspect, at least 90%. In one embodiment, there are only pyrimidine residues.
本発明は更に、ピリミジン残基がシトシン残基であるオリゴヌクレオチドを提供する。一態様においては、チミン残基。一態様においては、シトシン残基またはチミン残基である。 The present invention further provides an oligonucleotide wherein the pyrimidine residue is a cytosine residue. In one aspect, a thymine residue. In one embodiment, it is a cytosine residue or a thymine residue.
本発明は更に、治療有効量の少なくとも一種の薬理学的に許容されるポリピリミジンオリゴヌクレオチドまたはポリピリミジンオリゴヌクレオチド混合物、および薬学的に許容される担体とを含有し、該オリゴヌクレオチドまたは該混合物中のオリゴヌクレオチドの抗ウイルス活性が主として配列非依存的な作用様式により生じる、抗ウイルス薬学的組成物を提供する。一態様において、該オリゴヌクレオチドは、少なくとも一つの修飾されたヌクレオチド間結合を含む。 The present invention further comprises a therapeutically effective amount of at least one pharmacologically acceptable polypyrimidine oligonucleotide or polypyrimidine oligonucleotide mixture, and a pharmaceutically acceptable carrier, wherein the oligonucleotide or the mixture An antiviral pharmaceutical composition is provided in which the antiviral activity of the oligonucleotides of the present invention occurs primarily through a sequence independent mode of action. In one embodiment, the oligonucleotide comprises at least one modified internucleotide linkage.
一態様において、該組成物は、インビボの酸性部位への投与に適合している。
一態様において、該組成物は、更に浸透促進剤を含有する。
一態様において、該組成物は、更に界面活性剤を含有する。
一態様において、該組成物は、粉末形態である。
一態様において、該組成物は、顆粒形態である。
一態様において、該組成物は、微粒子形態である。
一態様において、該組成物は、ナノ粒子形態である。
一態様において、該組成物は、縣濁液または溶液形態である。
一態様において、該組成物は、エマルション形態である。
一態様において、該組成物は、経口投与に適合している。
一態様において、該組成物は、経膣投与に適合している。
一態様において、該組成物は、少なくとも一種のポリCオリゴヌクレオチドを含有する。
一態様において、該組成物は、少なくとも一種のポリTオリゴヌクレオチドを含有する。
一態様において、該組成物は、少なくとも一種のポリCTオリゴヌクレオチドを含有する。
一態様において、該組成物は、ヒトへの投与が認可されている。
一態様において、該組成物は、哺乳動物への投与が認可されている。
一態様において、該組成物は、非哺乳動物への投与が認可されている。
In one aspect, the composition is adapted for administration to an acidic site in vivo.
In one embodiment, the composition further contains a penetration enhancer.
In one embodiment, the composition further contains a surfactant.
In one embodiment, the composition is in powder form.
In one embodiment, the composition is in granular form.
In one embodiment, the composition is in particulate form.
In one embodiment, the composition is in nanoparticulate form.
In one embodiment, the composition is in the form of a suspension or solution.
In one embodiment, the composition is in the form of an emulsion.
In one embodiment, the composition is adapted for oral administration.
In one embodiment, the composition is adapted for vaginal administration.
In one embodiment, the composition contains at least one poly C oligonucleotide.
In one embodiment, the composition contains at least one poly T oligonucleotide.
In one embodiment, the composition contains at least one poly CT oligonucleotide.
In one embodiment, the composition is approved for human administration.
In one embodiment, the composition is approved for administration to a mammal.
In one aspect, the composition is approved for administration to a non-mammal.
本発明は更に、インビボの酸性部位への投与に適合された、少なくとも一種の薬理学的に許容されるポリピリミジンオリゴヌクレオチドを含有する薬学的組成物の、対象においてウイルス感染を予防または処置するための医薬の製造における使用を提供する。 The present invention further provides a pharmaceutical composition comprising at least one pharmacologically acceptable polypyrimidine oligonucleotide adapted for administration to an acidic site in vivo for preventing or treating viral infection in a subject. For use in the manufacture of a medicament.
一態様において、該対象はヒトである。一態様において、該対象は哺乳動物である。一態様において、該対象は非哺乳動物である。 In one embodiment, the subject is a human. In one embodiment, the subject is a mammal. In one embodiment, the subject is a non-mammal.
「発明の背景」に記載したように、多数のアンチセンスオリゴヌクレオチド(ON)の抗ウイルス活性が試験されている。しかしながら、これらのアンチセンスONは配列特異的であり、典型的に約16〜20のヌクレオチド長である。 As described in “Background of the Invention”, the antiviral activity of a number of antisense oligonucleotides (ON) has been tested. However, these antisense ONs are sequence specific and are typically about 16-20 nucleotides in length.
実施例1および実施例2の結果に示されるように、ランダムPS-ONの抗ウイルス効果は、配列特異的ではない。これらの試験に使用したPS-ONの容積および濃度を考慮すると、混合物中に特定のランダム配列が1コピーより多く存在することは、理論的にほぼ不可能である。このことは、これらPS-ONランダマーにアンチセンス効果は存在し得ないことを意味する。後者の例では、抗ウイルス効果がPS-ONの配列特異性により生じたと仮定すると、そのような効果は、1個のみの分子により生じる必要があり、それは可能であるとは思われない。例えば、40塩基長のONランダマーに関して、集団中の特定の配列はいずれも、理論的には総断片の1/440または4.1×10-41のみ存在する。1モル=6.022×1023分子を前提とし、本発明者らの最大規模の合成が現在、15マイクロモルの規模で行われている事実により、可能な配列の全ては存在しないと思われ、また、各配列は、1個のコピーのみとして存在する可能性が最も高い。勿論、本発明の教示を適用する当業者は、そのような配列特異的なONの配列を有するONも使用し得るが、本発明で発見された配列非依存的活性を利用する。従って、本発明は、非配列相補的なONに限定されないが、ウイルス感染を処置するための、配列特異的なアンチセンスおよびRNAi(例、siRNA)ONに関する先行技術に開示されているものの特許請求権を放棄する。 As shown in the results of Example 1 and Example 2, the antiviral effect of random PS-ON is not sequence specific. Given the volume and concentration of PS-ON used in these studies, it is theoretically almost impossible to have more than one copy of a particular random sequence in the mixture. This means that these PS-ON randomers cannot have an antisense effect. In the latter case, assuming that the antiviral effect was caused by the sequence specificity of PS-ON, such an effect needs to be caused by only one molecule, and it does not seem possible. For example, 40 with respect to the base length of ON randomers, any particular sequence in the population, theoretically exists only 1/4 40 or 4.1 × 10 -41 Total fragments. Based on the assumption that 1 mole = 6.022 × 10 23 molecules, our largest scale synthesis is currently carried out on a 15 micromole scale, and it is believed that not all possible sequences exist, and Each sequence is most likely to exist as only one copy. Of course, those skilled in the art applying the teachings of the present invention may use ONs having such sequence-specific ON sequences, but take advantage of the sequence-independent activities discovered in the present invention. Thus, the present invention is not limited to non-sequence complementary ONs, but claims of those disclosed in the prior art for sequence-specific antisense and RNAi (eg, siRNA) ON to treat viral infections. Give up the right.
適用可能なウイルス(例えば、本明細書にデータが記載されているものを含む)に関しては、ランダマーのサイズが増大するにつれて、オリゴヌクレオチド長さが40ヌクレオチド迄、および更に40ヌクレオチドを越える迄、その抗ウイルス能が増加する。現在のホスホロアミダイトベースのオリゴヌクレオチド合成の限界を原因として、比較的大きいPS-ON(例、80-および120-mers)は、所望のサイズよりも小さい断片が相当混入することを指摘せねばならない。比較的大きいオリゴ(80および120bp)に見られる比較的弱い効果(塩基毎ベースで)は、これら集団中の完全長ランダマーの濃度が比較的低いことを反映し得、また適切な部位における利用可能性の低下も反映し得る。適切な純度(例、少なくとも完全長75%)の比較的大きいランダマー(>40塩基)が合成または精製され、および/またはこれらの任意のONの送達が、例えば標的放出または持続放出を提供する送達系によって促進されるならば、はるかに増大した抗ウイルス活性を達成することが可能であり得るだろう。 For applicable viruses (including, for example, those whose data are described herein), as the size of the randomer increases, the oligonucleotide length increases to 40 nucleotides and even more than 40 nucleotides. Increases antiviral ability. Due to the limitations of current phosphoramidite-based oligonucleotide synthesis, it must be pointed out that relatively large PS-ONs (eg 80- and 120-mers) are considerably contaminated with fragments smaller than the desired size. Don't be. The relatively weak effects (on a base-by-base basis) seen with relatively large oligos (80 and 120 bp) can reflect the relatively low concentration of full-length randomers in these populations and are available at the appropriate site It can also reflect a decline in sex. A relatively large randomer (> 40 bases) of appropriate purity (eg, at least 75% full length) is synthesized or purified, and / or delivery of any of these ONs provides, for example, targeted release or sustained release It would be possible to achieve much increased antiviral activity if facilitated by the system.
本発明において、ランダマー(または本明細書に記載するような他の抗ウイルスオリゴヌクレオチド)は、以下を含むが、それらに限定されるわけではない数個の機序によりウイルス複製を遮断し得る:1. ビリオンの吸収または受容体相互作用を阻止して、感染を防止する、2. ウイルスアセンブリーをドーピングするかまたはウイルスゲノムをカプシドにパッケージングして(ウイルスDNAまたはRNAを競合させてパッケージングする)、不完全ビリオンとする、3. パッケージングまたはカプシドタンパク質が他の構造タンパク質と相互作用する間、カプシドの形成を混乱させまたは防止して、その結果ウイルス放出を阻害するか、または不完全ビリオンを放出させる、4. キーとなるウイルス成分に結合し、それらの活性を阻害または低下させる、5. ウイルス増殖に必要なキーとなる宿主成分に結合する。 In the present invention, a randomer (or other antiviral oligonucleotide as described herein) can block viral replication by several mechanisms including, but not limited to: 1. Prevent virion absorption or receptor interaction to prevent infection 2. Dope viral assembly or package viral genome in capsid (competitive packaging with viral DNA or RNA 3. Incomplete virions 3. While the packaging or capsid protein interacts with other structural proteins, it disrupts or prevents the formation of the capsid, thereby inhibiting viral release or incomplete Release virions, 4. bind to key viral components and inhibit or reduce their activity, 5. Binds to key host components required for virus growth.
上記に示したような、ウイルス阻害を達成する機序に限定されることなく、ONのウイルス複製に対する阻害特性を説明および/または予想し得る数個の可能な機序が存在する。これらのうちの第一の機序は、ONの一般的なアプタマー効果が、それらを細胞表面上のタンパク質またはウイルスタンパク質のいずれかに結合させて、ウイルス吸収および融合を防止することである。効果に関するサイズ閾値は、相互作用に必要な所定の累積電荷の結果であると考えられる。 Without being limited to the mechanisms for achieving viral inhibition, as indicated above, there are several possible mechanisms that can explain and / or predict the inhibitory properties of ON against viral replication. The first of these is that the general aptamer effect of ON binds them to either cell surface or viral proteins to prevent viral absorption and fusion. The size threshold for the effect is considered to be the result of a predetermined cumulative charge required for interaction.
第二の可能な機序は、ウイルスのパッケージングおよび/またはアセンブリーを阻害することにより、ONが細胞内で機能し得ることである。所定のサイズ閾値を越えるONは、正常なカプシド/核酸相互作用と競合するかまたは妨害して、新しいウイルス内への機能的ウイルスゲノムのパッケージングを防止し得る。または、ONは正常なカプシドの形成を阻止して正常なウイルス発芽を阻止し、ウイルス安定性を変更し得るか、または内部移行により適切なビリオン分解を阻止し得る。 A second possible mechanism is that ON can function intracellularly by inhibiting viral packaging and / or assembly. An ON that exceeds a predetermined size threshold can compete with or interfere with normal capsid / nucleic acid interactions to prevent packaging of the functional viral genome into a new virus. Alternatively, ON can prevent normal capsid formation to prevent normal virus germination, alter virus stability, or prevent proper virion degradation by internalization.
作用機序は未だ完全に証明されていないが、アッセイの結果により、本ONが、臨床的関連物質、アシクロビル、ガンシクロビル、リバビリンおよびプロテアーゼ阻害剤と比較して、ウイルス阻害に多大な抗力を有し得ることが示される。従って、本発明によるONは、ウイルス感染の処置または予防に使用し得る。処置するウイルス感染は、ヒト、動物および植物ウイルスを原因とし得る。本抗ウイルスオリゴヌクレオチドの安定性および/または活性を増大させるために、オリゴヌクレオチドの化学修飾を有利に使用することができる。RNA上のリボースの2'位におけるメトキシ化および他の修飾は、RNAをヌクレアーゼに対して安定化し、ホスホロチオエート化核酸に見られるタンパク質結合を最小にし、および標的配列を伴うこれらオリゴの融点を上昇させることが証明されている。2'-Oメチル化および他の2'-修飾は、現在、アンチセンスオリゴヌクレオチドの性質を改善させるために使用されているが、これらの修飾が全リボース上に存在する場合、オリゴヌクレオチドはRNase H活性を引き出さず、完全2'-修飾オリゴヌクレオチドをアンチセンス活性に関する比較的弱い候補とする。このことにより、オリゴヌクレオチドの両端のみに2'修飾を含み、オリゴのRNase Hを活性化させる能力を維持する、2'-Oメチルおよび他の2'修飾「ギャップマー(gapmers)」を使用する結果となっている。本発明者らの知識によれば、ホスホロチオエート結合およびオリゴヌクレオチドの各リボース糖上が2'-O-メチルまたは他の2'修飾されたリボヌクレオチドを伴う配列非特異的な抗ウイルスオリゴヌクレオチドは報告されていない。 Although the mechanism of action is not yet fully proven, the results of the assay indicate that the ON has a greater resistance to virus inhibition compared to clinically relevant substances, acyclovir, ganciclovir, ribavirin and protease inhibitors. Shown to get. Thus, the ON according to the present invention can be used for the treatment or prevention of viral infection. Viral infections to be treated can be caused by human, animal and plant viruses. In order to increase the stability and / or activity of the antiviral oligonucleotides, chemical modification of the oligonucleotides can be advantageously used. Methoxylation and other modifications at the 2 'position of ribose on RNA stabilizes RNA against nucleases, minimizes protein binding found in phosphorothioated nucleic acids, and increases the melting point of these oligos with target sequences It has been proven. 2'-O methylation and other 2'-modifications are currently used to improve the properties of antisense oligonucleotides, but if these modifications are present on all riboses, the oligonucleotide It does not elicit H activity and makes fully 2′-modified oligonucleotides relatively weak candidates for antisense activity. This uses 2'-O methyl and other 2 'modified "gapmers" that contain 2' modifications only at both ends of the oligonucleotide and maintain the ability of the oligo to activate RNase H It is the result. According to our knowledge, non-sequence anti-specific oligonucleotides with phosphorothioate linkages and 2'-O-methyl or other 2 'modified ribonucleotides on each ribose sugar of the oligonucleotide are reported. It has not been.
本明細書において記載するように、本発明者らは、40塩基PS-ONランダマーが、数種の異なるウイルスの強力な阻害剤であることを見出した。本発明者らは、チオエート化バックボーンは、ONランダマーに増大した疎水性の性質を付与し、それによりONランダマーのウイルス融合タンパク質の疎水性ドメインとの相互作用が可能になり得るという非限定的な仮説を提案する。これら疎水性ドメインは、HSV、HIV、インフルエンザ、RSVおよびエボラを含む多数の異なるウイルスの膜融合活性に不可欠であると考えられている。HIVの場合、このような疎水性ドメインは、融合阻害剤の開発のための標的として使用されている。 As described herein, the inventors have found that the 40 base PS-ON randomer is a potent inhibitor of several different viruses. The inventors have provided a non-limiting indication that a thioated backbone may confer increased hydrophobic properties to the ON randomer, thereby allowing interaction with the hydrophobic domain of the ON randomer's viral fusion protein. Propose a hypothesis. These hydrophobic domains are believed to be essential for the membrane fusion activity of a number of different viruses including HSV, HIV, influenza, RSV and Ebola. In the case of HIV, such hydrophobic domains have been used as targets for the development of fusion inhibitors.
従って、ホスホロチオエート結合ならびに2'-O-メチルおよび他の2'糖修飾を含むリボヌクレオチド修飾を本発明のオリゴヌクレオチドに組み込むことは、配列非特異的な抗ウイルスオリゴヌクレオチドの性質の改善に有用である。結果は、PS-ONの各リボースの2'位の修飾はその抗ウイルス活性を有意に変更しないが、PS-ONの血清タンパク質との一般的な相互作用を低下させ、低いpHに対する耐性を有意に向上させることを示す。これらの性質は、薬物動態学的性能を改善し、および通常PS-ONに見られる毒性の副作用を低減することが期待される。例えば、これらのpH耐性は、PS-ONを経口送達により適したものとする。また、これらの血清タンパク質との相互作用の低下は、抗ウイルス活性に影響を与えずに、その薬物動態学的挙動を改善することが期待される。このように、各結合のホスホロチオエート化、および各リボヌクレオチドのリボースの2'位における修飾、例えば2'-O-メチル修飾を有するオリゴヌクレオチドは、抗ウイルス活性を有し、従来のアンチセンスオリゴヌクレオチドに所望される性質であるRNase H活性を引き起こさないが、該性質は本発明に記載する活性には完全に不必要である。結果はまた、PS-ONの各リボースの'2位における修飾は、同一の修飾を含むが両端のみ(ギャップマー)が修飾されたPS-ONと比較して、ONのヌクレアーゼ耐性をより高めることを示している。ギャップマーは、アンチセンス技術において優先的に使用されている。各リボースの'2-位に修飾を含むPS-ONのヌクレアーゼ耐性は、薬物動態学的および/または経口利用性の改善のような有益な性質を示す筈である。 Thus, incorporation of phosphorothioate linkages and ribonucleotide modifications, including 2'-O-methyl and other 2 'sugar modifications, into the oligonucleotides of the invention is useful for improving the properties of sequence-nonspecific antiviral oligonucleotides. is there. The results show that modification of the 2 'position of each ribose in PS-ON does not significantly alter its antiviral activity, but reduces the general interaction of PS-ON with serum proteins and significantly reduces resistance to low pH To improve. These properties are expected to improve pharmacokinetic performance and reduce the toxic side effects normally seen with PS-ON. For example, these pH tolerances make PS-ON more suitable for oral delivery. Also, the reduction in interaction with these serum proteins is expected to improve its pharmacokinetic behavior without affecting antiviral activity. Thus, an oligonucleotide having phosphorothioation of each bond and modification at the 2 ′ position of the ribose of each ribonucleotide, for example 2′-O-methyl modification, has antiviral activity and is a conventional antisense oligonucleotide Does not cause RNase H activity, which is a desired property, but is completely unnecessary for the activity described in the present invention. The results also show that the modification at position 2 of each ribose of PS-ON increases the nuclease resistance of ON compared to PS-ON that contains the same modification but is modified at both ends only (gapmer). Is shown. Gapmers are preferentially used in antisense technology. The nuclease resistance of PS-ON containing a modification at the '2-position of each ribose should exhibit beneficial properties such as improved pharmacokinetics and / or oral availability.
加えて、各リボースの2'位に修飾を含むPS-ONが所望の性質を示すと共に、リボースの2'位の多数が修飾されたPS-ON、例えばリボースの少なくとも50%の修飾、およびより好ましくは80%もしくはそれ以上の修飾を含むPS-ONも、所望の性質を示すと思われる。 In addition, PS-ONs containing modifications at the 2 ′ position of each ribose exhibit the desired properties, and PS-ONs that are modified at a number of the 2 ′ positions of ribose, such as at least 50% modification of ribose, and more PS-ON, preferably containing 80% or more modifications, would also exhibit the desired properties.
上述したように、本オリゴヌクレオチドの活性は、ランダマーが例えばアンチセンス活性を有さないため、ハイブリダイゼーションにより核酸を標的としない。従って、本発明者らは、オリゴヌクレオチドはタンパク質を標的とするものと確信する。ホスホロチオエートオリゴヌクレオチドに2'-O-メチルリボース修飾を加えると、タンパク質結合活性が低下するため(Kandimalla et al., 1998, Bioorganic Med Chem Lett. 8:2103-2108; Mouet al., 2002, Nucleic Acid Res. 30:749-758)、これらの修飾が抗ウイルス活性を低下させることが予想された。意外にも、本発明者らは、ホスホロチオエートオリゴヌクレオチドに対して2'-O-メチルリボース修飾を加えても、抗ウイルス活性に影響を及ぼさないことを見出した。 As described above, the activity of the oligonucleotide does not target the nucleic acid by hybridization because the randomer does not have, for example, antisense activity. The inventors are therefore convinced that oligonucleotides target proteins. Adding 2'-O-methylribose modification to phosphorothioate oligonucleotides reduces protein binding activity (Kandimalla et al., 1998, Bioorganic Med Chem Lett. 8: 2103-2108; Mouet al., 2002, Nucleic Acid Res. 30: 749-758), these modifications were expected to reduce antiviral activity. Surprisingly, the inventors have found that adding 2'-O-methyl ribose modifications to phosphorothioate oligonucleotides does not affect antiviral activity.
本明細書において、多数の異なるオリゴヌクレオチドに関するアッセイの結果を記載する。別に示さない限り、試験したオリゴヌクレオチドは、2'-H部分(2'-デオキシ)を有し、従ってODNである。しかしながら、本発明の配列非依存的な活性は、そのような2'-H部分を含むオリゴヌクレオチドに限定されず、2'-OH部分ならびに他の2'-修飾、例えば2'-O-メチルおよび2'-フルオロを有するヌクレオチド含有のオリゴにも存在する。 Described herein are the results of the assay for a number of different oligonucleotides. Unless otherwise indicated, the tested oligonucleotides have a 2'-H moiety (2'-deoxy) and are therefore ODN. However, the sequence-independent activity of the present invention is not limited to oligonucleotides containing such 2′-H moieties, but 2′-OH moieties as well as other 2′-modifications such as 2′-O-methyl. And also in oligonucleotides containing nucleotides with 2'-fluoro.
本明細書の記載において、以下を含む多数の略語を使用する:
選択された略語
ON:オリゴヌクレオチド
ODN:オリゴデオキシヌクレオチド
PS:ホスホロチオエート
PS2:ホスホロジチオエート
PRA:プラーク減少アッセイ
PFU:プラーク形成単位
INF A:インフルエンザAウイルス
HIV:ヒト免疫不全ウイルス(特定しない場合、HIV-1およびHIV-2の双方を含む)
HSV:単純ヘルペスウイルス(特定しない場合、HSV-2およびHSV-3の双方を含む)
RSV:呼吸器多核体ウイルス
COX:コクサッキーウイルス
DHBV:アヒルB型肝炎ウイルス
In the description herein, a number of abbreviations are used including:
Selected abbreviation
ON: Oligonucleotide
ODN: oligodeoxynucleotide
PS: phosphorothioate
PS2: phosphorodithioate
PRA: plaque reduction assay
PFU: Plaque formation unit
INF A: Influenza A virus
HIV: Human immunodeficiency virus (including both HIV-1 and HIV-2 if not specified)
HSV: Herpes simplex virus (including both HSV-2 and HSV-3 if not specified)
RSV: Respiratory polynuclear virus
COX: Coxsackie virus
DHBV: duck hepatitis B virus
広いスペクトルの抗ウイルス活性
上記で議論された結論及び本明細書で報告されるデータに従って、ランダムON及びONランダマーは、ウイルスゲノムの集合及び/又はパッケージング及び/又はカプシド形成が複製に必要な工程であるウイルスとの広いスペクトルの抗ウイルス活性を有すると思われる。従って、この仮説を調べるために、サイズの異なる幾つかのPS-ONを選択して、種々のウイルス感染の細胞モデルで試験した。多数のそのような試験を実施例において本明細書で記載するが、それには、CMV、HIV-1、RSV、コクサッキーウイルスB2、DHBV、ハンタウイルス、パラインフルエンザウイルス、及びワクシニアウイルスによる試験が含まれ、並びに実施例1及び2に記載されるHSV-1及びHSV-2における試験も含まれる。
Broad spectrum antiviral activity In accordance with the conclusions discussed above and the data reported herein, random ON and ON randomers can be used to replicate viral genome assembly and / or packaging and / or capsid formation. It appears to have a broad spectrum of antiviral activity with the virus. Therefore, to test this hypothesis, several PS-ONs of different sizes were selected and tested in various cell models of viral infection. A number of such tests are described herein in the Examples, including tests with CMV, HIV-1, RSV, Coxsackievirus B2, DHBV, Hantavirus, parainfluenza virus, and vaccinia virus; Also included are tests in HSV-1 and HSV-2 described in Examples 1 and 2.
広いスペクトルの抗ウイルス活性における結論
様々なウイルスによる有効性試験は、ランダムON及びランダマーが種々の異なるウイルスに対して阻害特性を示すことを明らかにしている。さらに、これらの試験は、小さいランダマーよりも大きいランダマーの方がウイルス阻害についてさらに大きな有効性を示すという結論を指示している。このことは、カプシド形成するウイルスすべてにおけるランダムON又はONランダマーについて共通するサイズ依存性及び/又は電荷依存性のメカニズムを示唆している。
Conclusions on broad spectrum antiviral activity Efficacy studies with various viruses reveal that random ONs and randomers exhibit inhibitory properties against a variety of different viruses. In addition, these studies indicate the conclusion that larger randomers show greater efficacy for viral inhibition than smaller randomers. This suggests a common size-dependent and / or charge-dependent mechanism for random ON or ON randomers in all encapsidating viruses.
HSV及びCMVは双方ともヘルペスウイルスファミリーの二本鎖DNAウイルスであるが、HIVは、レトロウイルス由来のRNAウイルスであり、又、RSVはパラミクソウイルス由来のRNAウイルスである。ONランダマーが種々の異なるウイルスのウイルス機能を阻害できるという事実を考えると、上述のように列記したメカニズムに限定されることなく、以下のメカニズムが合理的である:A)ON/ONランダマーはアプタマー効果を介してウイルス機能を阻害しており、原形質膜とのウイルスの融合を妨げる;及び/又は、B)ON/ONランダマーはビリオンの集合又はウイルスDNAのカプシド内へのパッケージングを妨げる又はドープして結果として欠陥のあるビリオンを生じる;及び/又は、C)ON/ONランダマーは、ウイルスの集合及び/又はパッケージング及び/又は遺伝子発現に必要とされる宿主のタンパク質又は構成成分を妨害している。 HSV and CMV are both double-stranded DNA viruses of the herpesvirus family, while HIV is a retrovirus-derived RNA virus, and RSV is a paramyxovirus-derived RNA virus. Given the fact that ON randomers can inhibit the viral function of a variety of different viruses, the following mechanisms are reasonable without being limited to the mechanisms listed above: A) ON / ON randomers are aptamers Inhibits viral function through effects and prevents viral fusion with the plasma membrane; and / or B) ON / ON randomers prevent virion assembly or packaging of viral DNA into the capsid or Doping results in defective virions; and / or C) ON / ON randomers interfere with host proteins or components required for viral assembly and / or packaging and / or gene expression is doing.
抗ウイルス活性の必要条件
無作為化されたDNA配列がウイルス阻害にとって十分であると思われるので、ホスホロチオエート非存在下で抗ウイルス活性を維持できるかどうか、又、有効性は、ランダム配列又はウイルスゲノムで見い出される特異的配列のいずれの選択によって増強されるかを見るのは興味深い。
Prerequisites for antiviral activity Since randomized DNA sequences appear to be sufficient for viral inhibition, whether antiviral activity can be maintained in the absence of phosphorothioate, and efficacy depends on random sequence or viral genome It is interesting to see which of the specific sequences found in can be enhanced by selection.
従って、ランダマーの抗ウイルス有効性に対するその効果に関してDNA及びRNAの修飾を調べた。ランダマーは配列非依存的な、例えば非配列相補的なメカニズムを介して作用するので、これらの実験は、核酸構成におけるわずかな変化及び抗ウイルス有効性における電荷分布を調べるように設計した。 Therefore, DNA and RNA modifications were examined for their effects on the antiviral efficacy of randomers. Since randomers act through a sequence-independent, eg non-sequence complementary mechanism, these experiments were designed to examine minor changes in nucleic acid composition and charge distribution in antiviral efficacy.
異なるヌクレオチド/ヌクレオシド修飾を含むONがHSV-1を阻止できるか否かを試験するために、実施例に記載したようにHSV-1PRA中でREP 2024、2026、2059および2060を試験した。REP 2024 (ONの両端の4塩基上のリボースに2-O-メチル修飾を含むPS-ON)、REP 2026 (ONの両端の4塩基間の結合にメチルホスホネート修飾を含むPO-ON)、REP 2059 (20塩基長のRNA PS-ONランダマー)およびREP 2060(30塩基長のRNA PS-ONランダマー)は、抗-HSV-1活性を示した。アッセイをVERO細胞内で、HSV-1 (KOS株)を用いてプラーク減少アッセイとして実施した。濃度を上昇させて、PS-ONを試験した。直線回帰から計算したIC50値は、各々0.14、3.41、1.36および0.80であった。 To test whether ONs containing different nucleotide / nucleoside modifications can block HSV-1, REPs 2024, 2026, 2059 and 2060 were tested in HSV-1PRA as described in the Examples. REP 2024 (PS-ON with 2-O-methyl modification on ribose on 4 bases at both ends of ON), REP 2026 (PO-ON with methyl phosphonate modification at the bond between 4 bases at both ends of ON), REP 2059 (20 base RNA PS-ON randomer) and REP 2060 (30 base RNA PS-ON randomer) showed anti-HSV-1 activity. The assay was performed in VERO cells as a plaque reduction assay using HSV-1 (KOS strain). PS-ON was tested at increasing concentrations. IC 50 values calculated from linear regression were 0.14, 3.41, 1.36 and 0.80, respectively.
後者の例では、抗ウイルス効果がDNAホスホロチオエート主鎖から成るONによってのみ生じるのであれば、そのような効果はたった1つの分子で生じることになる。しかし、そのほかの主鎖及び修飾も陽性の抗ウイルス活性を提供した。当然、本発明の教示を適用する当業者は、異なる化合物ONも使用できる。ホスホロチオエートのような、しかし、それに限定されないONの修飾は、抗ウイルス活性に有益であると思われる。このことは、媒体及び細胞内での双方でのONの必要とされる電荷及び/又はDNAの安定化に対する必要条件によるのが最も可能性が高く、又、PS-ONのキラル性によってもよい。 In the latter example, if the antiviral effect occurs only with ON consisting of a DNA phosphorothioate backbone, such an effect will occur with only one molecule. However, other backbones and modifications also provided positive antiviral activity. Of course, one of ordinary skill in the art applying the teachings of the present invention can also use different compounds ON. Modifications of ON, such as but not limited to phosphorothioate, may be beneficial for antiviral activity. This is most likely due to the required charge and / or DNA stabilization requirements of the ON both in the medium and in the cell, and may also depend on the chirality of the PS-ON. .
化合物REP 2026は、分解から保護する両端で未修飾のPOヌクレオチド及び4つのメチルホスホネート結合を含む中心部分を有する一方で、抗ウイルス活性を示した。このことは、分解から保護する一方で抗ウイルス剤としてPO-ONを使用できることを示している。末端でヌクレオチドを修飾し、及び/又は後に記載するような好適な送達システムを使用することによって、この保護を達成することができる。 Compound REP 2026 exhibited antiviral activity while having a central portion containing unmodified PO nucleotides and four methylphosphonate linkages at both ends protecting from degradation. This indicates that PO-ON can be used as an antiviral agent while protecting against degradation. This protection can be achieved by modifying the nucleotides at the termini and / or using a suitable delivery system as described below.
一般に、使用されるDNAの配列組成は、ランダマーであれ、ランダム配列であれ、又は特異的なHSV-1配列であれ、全体としての有効性にはほとんど影響しない。しかしながら、中間の長さで、HSV-1配列はランダム配列よりもほぼ3倍強力だった。このデータは、特異的なアンチセンスが特異的なHSV配列に対して機能的に存在する一方で、本明細書で解明された配列非依存的なメカニズム(非アンチセンスメカニズム)がこの活性の優勢な部分を表してもよいことを示唆している。実際、ONが40塩基に増殖するにつれて、本質的に抗ウイルス活性はすべて配列非依存的な(例えば非アンチセンス)効果に起因しうる。 In general, the sequence composition of the DNA used, whether randomer, random or specific HSV-1 sequences, has little impact on overall effectiveness. However, at medium length, the HSV-1 sequence was almost three times stronger than the random sequence. This data shows that while specific antisense exists functionally for specific HSV sequences, the sequence-independent mechanism elucidated herein (non-antisense mechanism) predominates this activity. This suggests that this part may be expressed. Indeed, as ON grows to 40 bases, essentially all antiviral activity can be attributed to sequence-independent (eg, non-antisense) effects.
低毒性ランダマー
ONランダマーを用いた目標の1つは、毒性を下げることである。動物では、異なる配列が、たとえば、一般毒性、血清タンパク質との相互作用及び免疫系との相互作用のような異なる応答を誘発することが知られている(Monteith et al., Toxicol. Sci., 46:365-375,1998)。従って、ONの混合物は、任意の特定の配列のレベルが極めて低いので、毒性効果を下げてもよく、そのため、配列又はヌクレオチド組成による有意な相互作用の可能性は低い。
Low toxicity randomer
One goal with ON randomers is to reduce toxicity. In animals, different sequences are known to elicit different responses such as general toxicity, interactions with serum proteins and interactions with the immune system (Monteith et al., Toxicol. Sci., 46: 365-375,1998). Thus, a mixture of ONs may reduce the toxic effect because the level of any particular sequence is very low, so the possibility of significant interaction by sequence or nucleotide composition is low.
薬学的組成物
本発明のONは、ウイルス性疾患を処置するのに(又はその予防に)有用な治療組成物又は製剤の形態であってもよく、ヒト又は非ヒト動物において、及び/又は特定のウイルス又はウイルス群に対して使用するために規制当局により認可されることができる。これらONは、生理学的に及び/又は薬学的に許容される担体とともに組み合わせた場合、薬学的組成物の一部として使用してもよい。担体の特性は投与経路に依存してもよい。本発明の薬学的組成物は、そのほかの活性因子及び/又は活性を向上させる作用剤も含有してもよい。
Pharmaceutical Compositions ONs of the present invention may be in the form of therapeutic compositions or formulations useful for treating (or preventing) viral diseases, in human or non-human animals and / or specific Can be approved by regulatory authorities for use against other viruses or virus groups. These ONs may be used as part of a pharmaceutical composition when combined with a physiologically and / or pharmaceutically acceptable carrier. The characteristics of the carrier may depend on the route of administration. The pharmaceutical composition of the present invention may also contain other active factors and / or agents that improve the activity.
薬学的組成物又は製剤において使用される、又は動物を処置する方法を実践するために使用される本発明のONの投与は、種々の従来の方法、たとえば、眼内、経口摂取、腸内、吸入、又は皮膚、皮下、筋肉内、腹腔内、くも膜下、気管内、又は静脈内への注入において行うことができる。 Administration of ONs of the present invention used in pharmaceutical compositions or formulations or used to practice methods of treating animals can be performed in a variety of conventional ways, such as intraocular, oral ingestion, enteral, It can be done by inhalation or infusion into the skin, subcutaneous, intramuscular, intraperitoneal, subarachnoid, intratracheal, or intravenous.
本発明の薬学的組成物又はオリゴヌクレオチド製剤はさらに、たとえば、限定なしで、リファンピン、リバビリン、プレコナリル、シドフォビル、アシクロビル、ペンシクロビル、ガンシクロビル、バラシクロビル、ファムシクロビル、フォスカーネット、ビダラビン、アマンタジン、ザナミビル、オセルタミビル、レスキモド、抗プロテアーゼ、ペグ化したインターフェロン(Pegasys(商標))抗HIVプロテアーゼ(例えばロピニビル、サキニビル、アンプレナビル)、HIV融合阻害剤、ヌクレオチドHIV RT阻害剤(たとえば、AZT、ラミブジン、アバカビル)、非ヌクレオチドHIV RT阻害剤、ドコノソル、インターフェロン類、ブチル化ヒドロキシトルエン(BHT)及びハイパーリシンのようなウイルス疾患の治療用化学療法剤を含有してもよい。たとえば、本発明のONとの相乗効果を生じるために、そのような追加因子及び/又は作用剤が薬学的組成物に包含されてもよい。 The pharmaceutical composition or oligonucleotide formulation of the present invention further includes, for example, without limitation, rifampin, ribavirin, pleconaril, cidofovir, acyclovir, penciclovir, ganciclovir, valacyclovir, famciclovir, foscarnet, vidarabine, amantadine, zanamivir, Oseltamivir, resquimod, anti-protease, pegylated interferon (Pegasys ™) anti-HIV protease (eg, lopinivil, saquinivir, amprenavir), HIV fusion inhibitor, nucleotide HIV RT inhibitor (eg, AZT, lamivudine, abacavir) , Non-nucleotide HIV RT inhibitors, doconosol, interferons, butylated hydroxytoluene (BHT) and chemotherapeutic agents for the treatment of viral diseases such as hyperlysine. For example, such additional factors and / or agents may be included in the pharmaceutical composition to produce a synergistic effect with the ON of the present invention.
本発明の薬学的組成物又はオリゴヌクレオチド製剤はさらに、たとえば、限定なしで、ポリ陰イオン作用剤、硫酸化多糖類、ヘパリン、硫酸デキストラン、ポリ硫酸ペントサン、硫酸ポリビニルアルコール、アセマナン、ポリヒドロキシカルボキシレート類、硫酸セルロース、スルホン化ベンゼン環又はナフタレン環を含有するポリマー類及びナフタレンスルホネートポリマー、アセチルフタロイルセルロース、ポリ-L-リジン、カプリン酸ナトリウム、陽イオン系両親媒系物質、コール酸のようなポリマーを含有してもよい。ポリマーは場合によっては、ビリオン自体に結合する又は吸着することによって細胞におけるビリオンの侵入に影響を及ぼすことが知られている。抗ウイルス性ポリマーのこの特性は、ビリオンへの結合又は吸着についてONと競合するのに有用であることができ、その結果、細胞外活性に比べて、ONの細胞内活性が高められる。 The pharmaceutical composition or oligonucleotide formulation of the present invention may further include, for example, without limitation, polyanionic agents, sulfated polysaccharides, heparin, dextran sulfate, polypentosane sulfate, polyvinyl alcohol sulfate, acemanane, polyhydroxycarboxylate. , Cellulose sulfate, polymers containing sulfonated benzene rings or naphthalene rings and naphthalene sulfonate polymers, acetylphthaloyl cellulose, poly-L-lysine, sodium caprate, cationic amphiphiles, cholic acid, etc. A polymer may be contained. It is known that polymers sometimes affect virion entry in cells by binding or adsorbing to virions themselves. This property of the antiviral polymer can be useful in competing with ON for binding or adsorption to virions, resulting in increased intracellular activity of ON compared to extracellular activity.
例示的な脂質封入および送達
PS-ON(ならびに他の修飾結合を含むオリゴヌクレオチド)は、天然のホスホジエステルと比較して内因性ヌクレアーゼに対する耐性が高いが、完全に安定ではなく、血液および組織内でゆっくりと分解する。PSオリゴヌクレオチド薬物の臨床的適用における限界は、そのi.v.投与による補体の活性化の性向である。一般に、リポソームおよび他の送達系は、薬物の毒性を低下させ、血漿中での滞留時間を延長し、および浸透亢進(hyperpermeable)血管系を介した担体の管外遊出により疾病組織へより多量の活性薬物を送達することによって、ONを含む薬物の治療係数を向上させる。更に、PS-ONの場合、脂質封入により、循環中、タンパク質結合部位との可能な相互作用が防止される(Klimuk et al.(2000) J Pharmacol Exp Ther 292:480-488)。
Exemplary lipid encapsulation and delivery
PS-ON (and oligonucleotides containing other modified linkages) are more resistant to endogenous nucleases than natural phosphodiesters, but are not completely stable and degrade slowly in blood and tissues. A limitation in the clinical application of PS oligonucleotide drugs is their propensity to activate complement by iv administration. In general, liposomes and other delivery systems reduce drug toxicity, prolong plasma residence time, and allow greater outflow to diseased tissue by extravasation of the carrier through the hyperpermeable vasculature. By delivering active drugs, the therapeutic index of drugs including ON is improved. In addition, in the case of PS-ON, lipid encapsulation prevents possible interactions with protein binding sites during circulation (Klimuk et al. (2000) J Pharmacol Exp Ther 292: 480-488).
本明細書において記載する本発明者らが得た結果によれば、アプローチとしては、それらに限定されるわけではないが、親油性分子、極性脂質、リポソーム、単分子膜、二分子膜、小胞、プログラム可能な融合小胞、ミセル、シクロデキストリン、PEG、イオン注入、粉末注入およびナノ粒子(例えばPIBCA、PIHCA、PHCA、ゼラチン、PEG-PLAのような)のような送達系を使用して、本明細書において記載したON、ならびに/またはアンチセンスおよびsiRNAオリゴヌクレオチドを送達する。このような送達系の使用は、それらに限定されるわけではない1個または複数の以下の利点を提供する:活性化合物の動物およびヒト内での毒性の低下、IC50の低下、薬物送達の観点からの作用持続時間の延長ならびにオリゴヌクレオチドの血清タンパク質との非特異的結合からの保護。 According to the results obtained by the inventors described herein, approaches include, but are not limited to, lipophilic molecules, polar lipids, liposomes, monolayers, bilayers, small molecules Using delivery systems like vesicles, programmable fusion vesicles, micelles, cyclodextrins, PEG, ion injection, powder injection and nanoparticles (such as PIBCA, PIHCA, PHCA, gelatin, PEG-PLA) Delivers ON, and / or antisense and siRNA oligonucleotides as described herein. The use of such delivery systems provides one or more of the following advantages, including but not limited to: reduced toxicity of active compounds in animals and humans, reduced IC 50 , drug delivery Prolonged duration of action from a point of view and protection from non-specific binding of oligonucleotides to serum proteins.
従って、本発明者らは、PS-ONランダマーの抗ウイルス活性が、サイズの増大と共に増加することを示した。更に、この活性は、ウイルスタンパク質(ウイルス溶解物中の)に対する親和性の増大と相関する。ホスホロチオエート修飾がタンパク質-DNA相互作用における親和性を増大させることが当技術分野にて周知であるため、本発明者らは、ウイルス溶解物との相互作用の測定と同じFP-ベースのアッセイを用いて、牛胎児血清(FBS)に対する益々増大するPS-ONランダマーの結合能力を試験した。このアッセイでは、250ugの熱不活性化されていないFBSを、結合(mP値)が飽和した条件下で、蛍光標識した20塩基PS-ONランダマーと複合させた。増大するサイズ(REP 2003、REP 2004、REP 2006およびREP 2007)の非標識PS-ONランダマーを使用して、FBSの標識したベイトとの相互作用と競合させた。この試験の結果は、PS-ONランダマーのサイズが増大するにつれ、FBSに対する親和性が増大することを明白に示す。この結果は、最も高い活性の抗ウイルスPS-ONはまた、最も高い親和性でタンパク質に結合するであろうことを示唆している。 Thus, the inventors have shown that the antiviral activity of PS-ON randomers increases with increasing size. Furthermore, this activity correlates with increased affinity for viral proteins (in viral lysates). Since it is well known in the art that phosphorothioate modifications increase affinity in protein-DNA interactions, we use the same FP-based assay as measuring interaction with viral lysates. Thus, the increasing ability of PS-ON randomer to bind fetal bovine serum (FBS) was tested. In this assay, 250 ug of non-heat inactivated FBS was conjugated with a fluorescently labeled 20 base PS-ON randomer under conditions of saturated binding (mP value). Increasing sizes (REP 2003, REP 2004, REP 2006 and REP 2007) of unlabeled PS-ON randomers were used to compete with FBS labeled bait interactions. The results of this study clearly show that the affinity for FBS increases as the size of the PS-ON randomer increases. This result suggests that the most active antiviral PS-ON will also bind to proteins with the highest affinity.
しかしながら、ホスホロチオエートアンチセンスオリゴヌクレオチドの主な治療的問題点は、Kandimallaおよび共同研究者(Kandimalla et al.(1998) Bioorg. Med. Chem. Lett. 8:2103-2108)により記載されるように、主として、タンパク質(特に、血清タンパク質)との増大した相互作用を原因とする副作用であることが、当技術分野にて公知である。従って、ある場合には、抗ウイルスONを作用部位へ送達する一方で、その血清タンパク質との相互作用を防止することができる適切な送達系の使用が有益であろう。加えて、本発明の配列非依存的ONと、アンチセンスオリゴヌクレオチドおよびsiRNAsのような他の種類の抗ウイルスONとの組み合わせ使用に適切な送達系を用いることが有益であり得る。 However, the main therapeutic problems of phosphorothioate antisense oligonucleotides are as described by Kandimalla and co-workers (Kandimalla et al. (1998) Bioorg. Med. Chem. Lett. 8: 2103-2108) It is known in the art to be a side effect mainly due to increased interaction with proteins (especially serum proteins). Thus, in some cases it may be beneficial to use an appropriate delivery system that can deliver antiviral ON to the site of action while preventing its interaction with serum proteins. In addition, it may be beneficial to use a delivery system suitable for combined use of the sequence-independent ON of the present invention with other types of antiviral ONs such as antisense oligonucleotides and siRNAs.
送達系の所定の効果を示すために、本発明者らは、リポソームベースの二つの異なる送達技術を試験した:それらはサイトフェンクチン(Cytofectin)およびDOTAPである。本発明者らは本発明者らのインビトロでのFP-ベースの相互作用アッセイにおいて、DOTAPおよびサイトフェンクチンによるREP 2006の血清タンパク質との相互作用からの保護を測定した。封入されていないREP 2006は、結合した蛍光オリゴを血清から競合することができたが、REP 2006がDOTAPおよびサイトフェンクチンのいずれかで封入されると、血清の結合においてもはや競合が不可能であった。これらのデータにより、封入は、オリゴを血清との相互作用から保護し、副作用が低減された、より良好な薬物動態学的挙動をもたらすことが示唆される。 To show the predetermined effect of the delivery system, we tested two different delivery technologies based on liposomes: Cytofectin and DOTAP. We measured the protection of REP 2006 from interaction with serum proteins by DOTAP and cytofenctin in our in vitro FP-based interaction assay. Unencapsulated REP 2006 was able to compete with serum for bound fluorescent oligos, but once REP 2006 was encapsulated with either DOTAP or cytofenctin, it could no longer compete for serum binding. there were. These data suggest that encapsulation results in better pharmacokinetic behavior with protection of the oligo from serum interaction and reduced side effects.
本発明者らはまた、PS-ONランダマーREP 2006(サイトフェンクチンおよびDOTAPのいずれかで封入された)の、高濃度の血清(50%)の存在下での293A細胞への送達を、蛍光定量法を用いて、標識したREP 2006の細胞内濃度を測定することにより測定した。これらの結果は、そのような送達薬がREP 2006の細胞内濃度を上昇させ、かつDOTAPの場合、細胞内REP 2006のレベルが24時間後顕著に上昇したことを示す。最後に、本発明者らは、本発明者らのインビトロでのFPベースの相互作用アッセイにおいて、DOTAPおよびサイトフェンクチンによる、REP 2006の血清タンパク質相互作用からの保護を測定した。封入されていないREP 2006は、結合した蛍光オリゴを血清から競合することができたが、REP 2006がDOTAPおよびサイトフェンクチンのいずれかで封入されると、血清の結合においてもはや競合が不可能であった。これらのデータにより、封入は、オリゴを血清との相互作用から保護し、および副作用が低減されたより良好な薬物動態学的挙動をもたらすであろうことが示唆される。 We have also demonstrated the delivery of PS-ON randomer REP 2006 (encapsulated with either cytofenctin and DOTAP) to 293A cells in the presence of high concentrations of serum (50%). It was measured by measuring the intracellular concentration of labeled REP 2006 using a quantitative method. These results indicate that such delivery agents increased the intracellular concentration of REP 2006, and in the case of DOTAP, the level of intracellular REP 2006 was significantly increased after 24 hours. Finally, we measured the protection of REP 2006 from serum protein interactions by DOTAP and cytofenctin in our in vitro FP-based interaction assay. Unencapsulated REP 2006 was able to compete with serum for bound fluorescent oligos, but once REP 2006 was encapsulated with either DOTAP or cytofenctin, it could no longer compete for serum binding. there were. These data suggest that encapsulation will result in better pharmacokinetic behavior with protection of the oligo from serum interaction and reduced side effects.
同様に、オリゴヌクレオチドの脂質封入の効果を示すために、本発明者らは、培養細胞を蛍光標識したランダマーに暴露し、次に溶解細胞中の蛍光強度を2ラウンドの洗浄の後、試験することにより、追加のPS-ONランダマーの取り込みを監視した。250nM REP 2004-FLに暴露した細胞の細胞内取り込みを、送達系なしでおよび以下の脂質ベースの送達系の一つの内部に封入した後、試験した:リポフェクタミン(商標) (Invitrogen)、ポリフェクト(商標) (Qiagen)およびオリゴフェクタミン(商標)(Invitrogen)。4時間後、細胞をPBSで2回洗浄し、MPER溶解試薬(PROMEGA)を用いて溶解した。脂質系を伴うおよび伴わない同数の暴露細胞からの相対的な蛍光収率を検出した。本発明者らは、試験した3種の全薬剤の存在下、送達系を用いない場合と比較して、細胞内PS-ON濃度の有意な上昇があったことを観察した。 Similarly, to demonstrate the effect of oligonucleotide lipid encapsulation, we expose cultured cells to fluorescently labeled randomers and then test the fluorescence intensity in lysed cells after two rounds of washing. Thus, the uptake of additional PS-ON randomers was monitored. Intracellular uptake of cells exposed to 250 nM REP 2004-FL was tested without a delivery system and encapsulated within one of the following lipid-based delivery systems: Lipofectamine ™ (Invitrogen), Polyfect ™ ) (Qiagen) and Oligofectamine ™ (Invitrogen). After 4 hours, the cells were washed twice with PBS and lysed using MPER lysis reagent (PROMEGA). The relative fluorescence yield from the same number of exposed cells with and without lipid system was detected. We observed that in the presence of all three drugs tested, there was a significant increase in intracellular PS-ON concentration compared to the case without the delivery system.
試験結果に従えば、送達系の使用は、オリゴヌクレオチドを血清相互作用から保護する役割をし、副作用を低減し、組織分布を増大させ、および/またはONの細胞内送達を有意に増大することができる。 According to test results, the use of a delivery system serves to protect the oligonucleotide from serum interactions, reduces side effects, increases tissue distribution, and / or significantly increases intracellular delivery of ON. Can do.
送達システムを使用することにおけるもう1つの可能性のある利益は、ビリオンの細胞侵入の増加のような異なるメカニズムを介してウイルス感染の増幅を妨げるために、吸着のような、感染性ビリオンとの相互作用からONを保護することである。 Another potential benefit in using the delivery system is that with infectious virions, such as adsorption, to prevent amplification of viral infections through different mechanisms such as increased cell invasion of virions. Protect ON from interaction.
もう1つのアプローチは、特異的な細胞との親和性を高める分子に送達システムを会合させることによって細胞特異的な送達を達成することであり、そのような分子は、制約なしで、抗体、受容体、リガンド、ビタミン、ホルモン及びペプチドである。 Another approach is to achieve cell-specific delivery by associating the delivery system with a molecule that enhances affinity with specific cells, such molecules without limitation, antibody, receptor Body, ligand, vitamins, hormones and peptides.
送達システムに関する追加の選択肢は以下に提供される。 Additional options for delivery systems are provided below.
結合されたON
特定の態様では、本発明のONは、ウイルス複製を阻害するというそれらの能力を危うくすることなく、多数の方法で修飾される。たとえば、ONは1以上のそのヌクレオチド残基にてもう1つの部分と結合されるか、又は複合化される。従って、本発明のオリゴヌクレオチドの修飾は、活性を高める、細胞の分布を高める、特異的に又は非特異的に細胞膜を透過する輸送を増やす、又は分解若しくは排泄に対して保護する、又は有利な特性を提供する1以上の部分又は抱合体にオリゴヌクレオチドを化学的に連結することを含むことができる。そのような有利な特性には、たとえば、さらに低い血清相互作用、さらに高いウイルス-タンパク質相互作用、送達用に配合される能力、検出可能なシグナル、改善された薬物動態特性、及びさらに低い毒性が挙げられる。そのような共役基は第1級又は第2級のヒドロキシル基のような官能基と共有結合することができる。たとえば、共役部分には、ステロイド分子、非芳香族親油性分子、ペプチド、コレステロール、ビスコレステロール、抗体、PEG、タンパク質、水溶性ビタミン、脂溶性ビタミン、もう1つのON、又はONの活性及び/又は生物利用性を改善するそのほかの任意の分子を挙げることができる。
Combined ON
In certain embodiments, ONs of the invention are modified in a number of ways without compromising their ability to inhibit viral replication. For example, ON is bound to or complexed with another moiety at one or more of its nucleotide residues. Thus, the modification of the oligonucleotides of the invention increases activity, increases cell distribution, increases transport across the cell membrane specifically or non-specifically, or protects against degradation or excretion, or is advantageous. It may include chemically linking the oligonucleotide to one or more moieties or conjugates that provide the property. Such advantageous properties include, for example, lower serum interactions, higher virus-protein interactions, the ability to be formulated for delivery, detectable signal, improved pharmacokinetic properties, and lower toxicity. Can be mentioned. Such conjugated groups can be covalently linked to functional groups such as primary or secondary hydroxyl groups. For example, the conjugated moiety may include steroid molecules, non-aromatic lipophilic molecules, peptides, cholesterol, bis-cholesterol, antibodies, PEG, proteins, water-soluble vitamins, fat-soluble vitamins, another ON, or ON activity and / or Mention may be made of any other molecule that improves bioavailability.
さらに詳細には、本発明の典型的な共役基には、干渉物質、リポータ分子、ポリアミン、ポリアミド、ポリエチレングリコール、ポリエーテル、SATE、t-ブチル-SATE、オリゴマーの薬力学的特性を高める基、及びオリゴマーの薬物動態特性を高める基を挙げることができる。典型的な共役基には、コレステロール類、脂質、リン脂質、ビオチン、フェナジン、葉酸塩、フェナントリジン、アントラキノン、アクリジン、フルオレセイン類、ローダミン類、クマリン類、蛍光ヌクレオ塩基及び染料を挙げることができる。 More specifically, exemplary conjugated groups of the present invention include interfering substances, reporter molecules, polyamines, polyamides, polyethylene glycols, polyethers, SATE, t-butyl-SATE, groups that enhance the pharmacodynamic properties of oligomers, And groups that enhance the pharmacokinetic properties of the oligomer. Typical conjugated groups can include cholesterols, lipids, phospholipids, biotin, phenazine, folate, phenanthridine, anthraquinone, acridine, fluoresceins, rhodamines, coumarins, fluorescent nucleobases and dyes. .
本発明に関連して、薬力学的特性を高める基には、オリゴマーの分解耐性を高める基及び/又は血清の相互作用に対して保護する基が挙げられる。本発明に関連して、薬物動態特性を高める基には、オリゴマーの取り込み、分布、代謝又は排泄を改善する基が挙げられる。典型的な共役基は1992年10月23日に出願され、参考として本明細書に組み入れられる国際特許出願PCT/US92/09196号に記載されている。 In the context of the present invention, groups that enhance pharmacodynamic properties include groups that enhance the degradation resistance of oligomers and / or groups that protect against serum interactions. In the context of the present invention, groups that enhance pharmacokinetic properties include groups that improve oligomer uptake, distribution, metabolism or excretion. Exemplary conjugated groups are described in International Patent Application No. PCT / US92 / 09196, filed Oct. 23, 1992, which is incorporated herein by reference.
共役部分には、たとえば、コレステロール部分(Letsinger et al., Proc. Natl. Acad. Sci. USA, 86:6553-6556, 1989)、コール酸(Manoharan et al., Bioorg. Med. Chem. Let., 4:1053-1060, 1994)、チオエーテル、たとえば、ヘキシル-S-トリチルチオール(Manoharan et al., Ann. N.Y. Acad. Sci., 660:306-309, 1992、Manoharan et al., Bioorg. Med. Chem. Let., 3, 2765-2770, 1993)、チオコレステロール(Oberhauser et al., Nucl. Acids Res., 20:533-538, 1992)、脂肪族鎖、たとえば、ドデカジオール若しくはウンデシル残基(Saison-Behmoaras et al., EMBO J., 10:1111-1118, 1991、Kabanov et al., FEBS Lett., 259:327-330, 1990、Svinarchuk et al., Biochimie, 75, 49-54, 1993)、リン脂質、たとえば、ジ-ヘキサデシル-rac-グリセロール又はトリエチル-アンモニウム1,2-ジ-o-ヘキサデシル-rac-グリセロ-3-H-ホスホネート(Manoharan et al., Tetrahedron Lett., 36:3651-3654,1995; Shea et al., Nucl. Acids Res.,18:3777-3783,1990)、ポリアミン若しくはポリエチレングリコール鎖(Manoharan et al., Nucleosides & Nucleotides,14:969-973, 1995)、又はアダマンタン酢酸(Manoharan et al., Tetrahedron Lett., 36:3651-3654, 1995)、パールミチル部分(Mishra et al., Biochim. Biophys. Acta, 1264, 229-237, 1995)、又はオクタデシルアミン若しくはヘキシルアミノカルボニル-オキシコレステロール部分(Crooke et al., J. Pharmacol Exp. Ther., 277:923-937, 1996)のような脂質部分が挙げられるが、これらに限定されない。 Conjugate moieties include, for example, cholesterol moieties (Letsinger et al., Proc. Natl. Acad. Sci. USA, 86: 6553-6556, 1989), cholic acid (Manoharan et al., Bioorg. Med. Chem. Let. , 4: 1053-1060, 1994), thioethers such as hexyl-S-tritylthiol (Manoharan et al., Ann. NY Acad. Sci., 660: 306-309, 1992, Manoharan et al., Bioorg. Med) Chem. Let., 3, 2765-2770, 1993), thiocholesterol (Oberhauser et al., Nucl. Acids Res., 20: 533-538, 1992), aliphatic chains such as dodecadiol or undecyl residues (Saison-Behmoaras et al., EMBO J., 10: 1111-1118, 1991, Kabanov et al., FEBS Lett., 259: 327-330, 1990, Svinarchuk et al., Biochimie, 75, 49-54, 1993), phospholipids such as di-hexadecyl-rac-glycerol or triethyl-ammonium 1,2-di-o-hexadecyl-rac-glycero-3-H-phosphonate (Manoharan et al., Tetrahedron Lett., 36: 3651-3654,1995; Shea e t al., Nucl. Acids Res., 18: 3777-3783, 1990), polyamine or polyethylene glycol chains (Manoharan et al., Nucleosides & Nucleotides, 14: 969-973, 1995), or adamantane acetic acid (Manoharan et al ., Tetrahedron Lett., 36: 3651-3654, 1995), pearl mityl moiety (Mishra et al., Biochim. Biophys. Acta, 1264, 229-237, 1995), or octadecylamine or hexylaminocarbonyl-oxycholesterol moiety (Crooke et al., J. Pharmacol Exp. Ther., 277: 923-937, 1996), but not limited thereto.
本オリゴヌクレオチドは、たとえば、限定することなく、アスピリン、ワルファリン、フェニルブタゾン、イブプロフェン、スプロフェン、フェンブフェン、ケトプロフェン、(S)-(+)-プラノプロフェン、カルプロフェン、ダンシルサルコシン、2,3,5-トリオド安息香酸、フルフェナム酸、フォリン酸、ベンゾチアジアジド、クロロチアジド、ジアゼピン、インドメタシン、バルビツール酸塩、セファロプロリン、サルファ剤、抗糖尿病剤、抗菌剤又は抗生剤のような活性のある薬剤物質に共役してもよい。 The oligonucleotide may be, for example, without limitation, aspirin, warfarin, phenylbutazone, ibuprofen, suprofen, fenbufen, ketoprofen, (S)-(+)-planoprofen, carprofen, dansylsarcosine, 2,3,5 -Active drug substances such as triodobenzoic acid, flufenamic acid, folinic acid, benzothiadiazide, chlorothiazide, diazepine, indomethacin, barbiturates, cephaloproline, sulfa drugs, antidiabetics, antibacterials or antibiotics May be conjugated.
典型的なオリゴヌクレオチド抱合体の調製を記載する典型的な米国特許には、たとえば、米国特許第4,828,979; 4,948,882; 5,218,105; 5,525,465; 5,541 ,313; 5,545,730; 5,552,538; 5,578,717, 5,580,731; 5,580,731; 5,591,584; 5,109,124; 5,118,802; 5,138,045; 5,414,077; 5,486,603; 5,512,439;5,578,718; 5,608,046; 4,587,044; 4,605,735; 4,667,025; 4,762,779; 4,789,737;4,824,941 ; 4,835,263; 4,876,335; 4,904,582; 4,958,013; 5,082,830; 5,112,963;5,214,136; 5,082,830; 5,112,963; 5,214,136; 5,245,022; 5,254,469; 5,258,506; 5,262,536; 5,272,250; 5,292,873; 5,317,098; 5,371 ,241 , 5,391 ,723; 5,416,203,5,451 ,463; 5,510,475; 5,512,667; 5,514,785; 5,565,552; 5,567,810; 5,574,142; 5,585,481; 5,587,371; 5,595,726; 5,597,696; 5,599,923; 5,599,928; および5,688,941号が挙げられ、それぞれ参考としてその全体を本明細書に組み入れる。 Typical U.S. patents describing the preparation of typical oligonucleotide conjugates include, for example, U.S. Pat.Nos. 4,828,979; 4,948,882; 5,218,105; 5,525,465; 5,541 313; ; 5,118,802; 5,138,045; 5,414,077; 5,486,603; 5,512,439; 5,578,718; 5,608,046; 4,587,044; 4,605,735; 4,667,025; 4,762,779; 4,789,737; 4,824,941; 4,835,263; 4,876,335; 4,904,582,4 5,258,506; 5,262,536; 5,272,250; 5,292,873; 5,317,098; 5,371, 241, 5,391 723; 5,416,203; 5,451, 463; 5,510,475; 5,512,667; 5,514,785; 5,565,552; 5,567,810; No. 5,688,941, each of which is incorporated herein by reference in its entirety.
もう1つのアプローチは、たとえば、s-アセチルチオ-エチル又はs-ピバシロイルチオ-エチルのような酵素的に切断可能な、電荷を中和する付加体による修飾によって親油性プロオリゴヌクレオチドのような抗ウイルス性ONを調製することである(Vives et al., Nucl. Acids Res., 27:4071-4076, 1999)。そのような修飾は、ONの細胞への取り込みを高め、従って細胞内で活性であるONにとって有益であることが示されている。 Another approach is to use anti-viral properties such as lipophilic pro-oligonucleotides by modification with charge-neutralizing adducts such as, for example, s-acetylthio-ethyl or s-pivacylylthio-ethyl. ON is to be prepared (Vives et al., Nucl. Acids Res., 27: 4071-4076, 1999). Such modifications have been shown to be beneficial for ONs that increase the uptake of ONs into cells and are therefore active in cells.
非特異的ONの設計
もう1つのアプローチにおいて、ヒト(又はそのほかの対象生物)のゲノムに対して低い、好ましくはできれば最低の相同性を示す抗ウイルス性ONを設計する。目標は、ヒト又は動物のゲノム配列及びmRNAとの相互作用による毒性が最低を示すONを得ることである。第1工程は、たとえば、手動で、又はさらに通例ではコンピュータプログラムを用いて、無作為方式にてヌクレオチド、A、C、G、Tを並べることにより、ONの所望の長さの配列を製造する。第2の工程は、GenBank及び/又はアンサンブルヒューマンゲノムデータベースのようなヒト配列のライブラリとONの配列を比較する。所望であれば、配列の生成及び比較を繰り返して行い、対象ゲノムとの所望の低い相同性を有する配列を同定することができる。望ましくは、ONの配列は、ゲノム全体とのできるだけ最低の相同性であり、一方、好ましくは自己相互反応をできるだけ抑える。
Designing non-specific ONs In another approach, an antiviral ON is designed that exhibits a low, preferably the lowest possible homology to the human (or other target organism) genome. The goal is to obtain an ON with minimal toxicity due to interaction with human or animal genomic sequences and mRNA. The first step produces a sequence of the desired length of ON, for example, manually or more commonly using a computer program, by aligning nucleotides, A, C, G, T in a random fashion . The second step compares the sequence of ON with a library of human sequences, such as GenBank and / or Ensemble Human Genome Database. If desired, sequence generation and comparison can be repeated to identify sequences having the desired low homology with the target genome. Desirably, the sequence of ON has the lowest possible homology with the entire genome, while preferably minimizing self-interaction.
アンチセンス活性を持つ非特異的ON
もう1つのアプローチでは、抗ウイルス性の非特異的配列部分をアンチセンス配列部分に結合して最終的なONの活性を高める。ONの非特異的部分は本発明に記載されている。アンチセンス部分はウイルスmRNAに相補的である。
Nonspecific ON with antisense activity
In another approach, an antiviral non-specific sequence portion is attached to the antisense sequence portion to enhance the final ON activity. Non-specific parts of ON are described in the present invention. The antisense portion is complementary to the viral mRNA.
Gリッチモチーフ活性を持つ非特異的ON
もう1つのアプローチでは、抗ウイルス性の非特異的配列部分を、最終的なONの活性を改善するモチーフ部分に結合させる。ONの非特異的部分は本発明に記載されている。モチーフ部分は、非限定例として、免疫系の刺激剤として文献に記載されているCpG、Gカルテット、及び/又はCGを挙げることができる。Agrawal et al., Curr. Cancer Drug Targets, 3: 197-209, 2001。
Non-specific ON with G-rich motif activity
In another approach, an antiviral non-specific sequence moiety is attached to a motif moiety that improves the activity of the final ON. Non-specific parts of ON are described in the present invention. Non-limiting examples of the motif portion include CpG, G quartet, and / or CG described in the literature as stimulators of the immune system. Agrawal et al., Curr. Cancer Drug Targets, 3: 197-209, 2001.
非ワトソン・クリックON
もう1つのアプローチは、1以上の種類の非ワトソン・クリック型ヌクレオチド/ヌクレオシドから構成されるONを使用することである。そのようなONは、PS-ONに類似する以下の特性の一部を持つPS-ONを模倣することができる:(a)全体の変化;(b)単位間の空間;(c)鎖の長さ;(d)一方の側での負の電荷の蓄積による正味の双極子;(e)タンパク質に結合する能力;(f)ウイルスタンパク質に結合する能力;(g)細胞に侵入する能力;(h)許容される治療指数、(i)抗ウイルス活性。ONは好ましいホスホロチオエートの主鎖を有するが、それに限定されない。非ワトソン・クリック型ヌクレオチド/ヌクレオシドの例は、Kool, Acc. Chem. Res., 35:936-943, 2002及び合成の脱塩基部位を含有するONを記載しているTakeshita et al., J. Biol. Chem., 262:10171-10179, 1987に記載されている。
Non Watson Crick ON
Another approach is to use an ON composed of one or more types of non-Watson-Crick nucleotides / nucleosides. Such ONs can mimic PS-ONs with some of the following properties similar to PS-ONs: (a) global changes; (b) inter-unit space; (c) chain Length; (d) net dipole due to accumulation of negative charge on one side; (e) ability to bind protein; (f) ability to bind viral protein; (g) ability to enter cells; (H) Acceptable therapeutic index, (i) Antiviral activity. ON has a preferred phosphorothioate backbone, but is not limited thereto. Examples of non-Watson-Crick nucleotide / nucleosides are Kool, Acc. Chem. Res., 35: 936-943, 2002 and Takeshita et al., J., describing ON containing a synthetic abasic site. Biol. Chem., 262: 10171-10179, 1987.
抗ウイルス性ポリマー
もう1つのアプローチは、ホスホロチオエートONの活性を模倣するポリマーを使用することである。文献に記載されているように、幾つかの陰イオン性ポリマーは抗ウイルス性の阻害活性を有することが示された。これらのポリマーは幾つかのクラスに属する:(1)多糖類の硫酸エステル(デキストリン及び硫酸デキストラン;硫酸セルロース);(2)スルホン化されたベンゼン環又はナフタレン環を含有するポリマー及びスルホン酸ナフタレンポリマー;(3)ポリカルボキシレート(アクリル酸ポリマー);及びアセチルフタロイルセルロース(Neurath et al., BMC Infect. Dis., 2:27, 2002)及び(4)脱塩基オリゴヌクレオチド(Takeshita et al., 1987, J. Biol. Chem., 262:10171-10179)。非ヌクレオチドの抗ウイルス性ポリマーのそのほかの例は文献に記載されている。本明細書に記載されるポリマーは、本明細書に記載されるPS-ONを模倣し、PS-ONに類似する以下の特性を有する:(a)鎖の長さ;(b)一方の側での負の電荷の蓄積による正味の双極子;(c)タンパク質に結合する能力;(d)ウイルスタンパク質に結合する能力;(e)許容される治療指数、(f)抗ウイルス活性。PS-ONの効果を模倣するために、抗ウイルス性ポリマーは好ましくは、PS-ONに比較されるように、その単位間で類似の空間を示すポリ陰イオンであってもよい。それは、単独で又は送達システムと組み合わせて、細胞に侵入する能力を有してもよい。
Antiviral polymers Another approach is to use polymers that mimic the activity of phosphorothioate ON. As described in the literature, some anionic polymers have been shown to have antiviral inhibitory activity. These polymers belong to several classes: (1) polysaccharide sulfates (dextrin and dextran sulfate; cellulose sulfate); (2) polymers containing sulfonated benzene or naphthalene rings and sulfonated naphthalene polymers. (3) polycarboxylate (acrylic acid polymer); and acetylphthaloylcellulose (Neurath et al., BMC Infect. Dis., 2:27, 2002) and (4) abasic oligonucleotide (Takeshita et al., 1987, J. Biol. Chem., 262: 10171-10179). Other examples of non-nucleotide antiviral polymers are described in the literature. The polymers described herein mimic the PS-ON described herein and have the following properties similar to PS-ON: (a) chain length; (b) one side A net dipole due to the accumulation of negative charge at (c) the ability to bind to a protein; (d) the ability to bind to a viral protein; (e) an acceptable therapeutic index; (f) antiviral activity. In order to mimic the effect of PS-ON, the antiviral polymer may preferably be a polyanion that exhibits a similar space between its units, as compared to PS-ON. It may have the ability to enter cells alone or in combination with a delivery system.
二本鎖PS-ONの抗ウイルス活性
ランダム配列及びその相補体(PSが修飾されている又はされていないのいずれか)をどこかに記載したように蛍光で標識し、本発明に記載しているように蛍光分極により精製したHSV-1及びHIV-1に結合する能力を調べる。アクリルアミドゲル電気泳動により、ハイブリッド形成を検証した。修飾していないREP2017(2017U)は一本鎖(ss)又は二本鎖(ds)のいずれもHSV-1又はHIV-1の溶解物における結合活性を有さなかった。しかしながら、PS修飾したREP2017は、一本鎖又は二本鎖のいずれかでHSV-1及びHIV-1との相互作用が可能であった。
Antiviral activity of double-stranded PS-ON Random sequences and their complements (either PS modified or not) are fluorescently labeled as described elsewhere and described in the present invention. The ability to bind to HSV-1 and HIV-1 purified by fluorescence polarization is examined. Hybridization was verified by acrylamide gel electrophoresis. Unmodified REP2017 (2017U) had no binding activity in HSV-1 or HIV-1 lysates, either single stranded (ss) or double stranded (ds). However, PS-modified REP2017 was able to interact with HSV-1 and HIV-1 either single-stranded or double-stranded.
本明細書で記載される我々の結果に従って、アプローチは有効な抗ウイルス剤として二本鎖ONを使用することである。好ましくは、そのようなONは、ホスホロチオエート主鎖を有するが、本明細書で一本鎖ONについて記載されるような抗ウイルス活性及び/又は安定性及び/又は送達特性を高める他の及び/又は追加的な修飾も有してもよい。 According to our results described herein, the approach is to use double stranded ON as an effective antiviral agent. Preferably, such ONs have a phosphorothioate backbone, but other and / or enhance antiviral activity and / or stability and / or delivery properties as described herein for single chain ONs. There may also be additional modifications.
創薬のためのインビトロアッセイ
蛍光分極に基づいてインビトロアッセイを開発し、PS-ONのウイルス構成成分、たとえば、ウイルスタンパク質に結合する能力を測定する。タンパク質(又はもう1つの相互作用因子)が蛍光的に標識されたベイトに結合すると、溶液中でベイトの三次元のタンブリングが遅れる。標識されたベイトに由来する励起された光の分極における固有の上昇によってこのタンブリングの遅延を測定する。従って、上昇した分極(大きさのない測定値[mP]として報告される)は増加した結合に相関する。
In vitro assays for drug discovery An in vitro assay is developed based on fluorescence polarization to measure the ability of PS-ON to bind to viral components, such as viral proteins. When protein (or another interactor) binds to a fluorescently labeled bait, the three-dimensional tumbling of the bait is delayed in solution. This tumbling delay is measured by the intrinsic rise in the polarization of the excited light derived from the labeled bait. Thus, increased polarization (reported as an unmeasured measure [mP]) correlates with increased binding.
一つの方法体系は、非可撓性リンカー(3'-(6-フルオレセイン)CPG)を使用して3'末端をFITCで標識したPS-ONランダマーを、ベイトとして使用することである。このPS-ONランダマーを、アッセイ緩衝液(10mM Tris、pH7.2、80mM NaCl、10mM EDTA、100mM b-メルカプトエタノールおよび1%トゥイーン20)中で2nMに希釈する。次いで、このオリゴを、適切な相互作用物(interactor)と共に混合する。この場合、本発明者らは、0.5M KClおよび0.5% Triton X-100 (HSV-1およびHIV-1)または10mM Tris、pH7.5、150mM NaCl、1mM EDTAおよび0.1% Triton X-100 (RSV)中に懸濁させた、ショ糖傾斜精製したHSV-1 (Maclntyre株)、HIV-1(Mn株)またはRSV(A2株)の溶解物を使用する。ベイトとの相互作用の後、複合体を様々な非標識PS-ONでチャレンジして、ベイトをその複合体から置換する能力を評価する。 One methodology is to use PS-ON randomers labeled as FITC at the 3 'end with FITC using an inflexible linker (3'-(6-fluorescein) CPG) as the bait. The PS-ON randomer is diluted to 2 nM in assay buffer (10 mM Tris, pH 7.2, 80 mM NaCl, 10 mM EDTA, 100 mM b-mercaptoethanol and 1% Tween 20). The oligo is then mixed with the appropriate interactor. In this case, we have 0.5M KCl and 0.5% Triton X-100 (HSV-1 and HIV-1) or 10 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA and 0.1% Triton X-100 (RSV Sucrose gradient purified HSV-1 (Maclntyre strain), HIV-1 (Mn strain) or RSV (A2 strain) lysate suspended in Following interaction with the bait, the complex is challenged with various unlabeled PS-ONs to assess the ability to displace the bait from the complex.
予備試験において、異なるサイズの3種のベイト;6 (REP 2032-FL)、10 (REP 2003-FL)および20塩基(REP 2004-FL)を使用し、ベイトのHSV-1、HIV-1およびRSV溶解物と相互作用する能力を試験した。異なるサイズのベイトに対するウイルス溶解物の結合を、蛍光偏光により測定した。任意のウイルス溶解物の存在下、結合の程度は、使用するベイトのサイズに依存し、ウイルス溶解物の存在下、2004-FLはmP(結合)において最大のシフトを示した。本発明者らは、サイズ依存的なPS-ONランダマーの抗ウイルス効力と類似することに気付いた。次いで、このベイトを使用して、異なるサイズのPS-ONの、ベイトと溶解物との相互作用に競合する能力を評価した。 In the preliminary test, three baits of different sizes were used; 6 (REP 2032-FL), 10 (REP 2003-FL) and 20 bases (REP 2004-FL), and bait HSV-1, HIV-1 and The ability to interact with RSV lysate was tested. Viral lysate binding to different sized baits was measured by fluorescence polarization. In the presence of any virus lysate, the extent of binding was dependent on the size of the bait used, and in the presence of virus lysate, 2004-FL showed the greatest shift in mP (binding). The inventors have found that it is similar to the antiviral efficacy of size-dependent PS-ON randomers. This bait was then used to evaluate the ability of different sized PS-ONs to compete for bait and lysate interactions.
REP 2004-FLのHSV-1、HIV-1、およびRSV溶解物との相互作用を、増大するサイズのPS-ONを用いてチャレンジした。ウイルス溶解物に対するPS-ONランダマーの親和性の測定は、蛍光偏光により検出した。ベイトとしてREP 2004-FLを使用して、HSV-1溶解物、HIV-1溶解物またはRSV溶解物との複合体形成を、増大する濃度のREP 2003、REP 2004、REP 2006またはREP 2007でチャレンジした。試験した各ウイルス溶解物に関して、本発明者らは、REP 2003はベイトを溶解物から競合除去できないことに気が付いた。REP 2004(非標識ベイト)競合物により引き出された比較的弱い競合から明らかとなったように、ベイト相互作用は非常に強力であった。しかしながら、競合物PS-ONのサイズが20塩基を越えて増大すると、そのベイト置換能力もより増強することが観察された。このことは、PS-ONランダマーのサイズが増大するにつれ、ウイルス溶解物中のタンパク質成分に対する親和性が増大することを示す。この現象は、より大きいPS-ONランダマーの、HSV-1、HSV-2、CMV、HIV-1およびRSVに対する抗ウイルス活性の増大を反映している。 The interaction of REP 2004-FL with HSV-1, HIV-1 and RSV lysates was challenged with increasing sizes of PS-ON. Measurement of the affinity of PS-ON randomer for virus lysates was detected by fluorescence polarization. Challenge complex formation with HSV-1 lysate, HIV-1 lysate or RSV lysate with increasing concentrations of REP 2003, REP 2004, REP 2006 or REP 2007 using REP 2004-FL as bait did. For each virus lysate tested, we found that REP 2003 could not competitively remove the bait from the lysate. The bait interaction was very strong, as evidenced by the relatively weak competition drawn by the REP 2004 (unlabeled bait) competitor. However, it was observed that as the size of competitor PS-ON increased beyond 20 bases, its bait replacement capacity was also enhanced. This indicates that the affinity for protein components in the virus lysate increases as the size of the PS-ON randomer increases. This phenomenon reflects the increased antiviral activity of larger PS-ON randomers against HSV-1, HSV-2, CMV, HIV-1 and RSV.
ベイト競合における効力と、PS-ONランダマーの抗ウイルス活性との間の類似性は、このアッセイの典型が抗ウイルス活性の優れた予測物であることを示す。このアッセイは強固であり、実行が容易で、非常に安定であるため、特定の標的の同定に基づくのではなく、1個または複数の成分、例えば、ウイルスタンパク質との相互作用能力に基づいて、新規な抗ウイルス分子を同定するハイスループットスクリーニングの非常に良好な候補とする。 The similarity between the potency in bait competition and the antiviral activity of PS-ON randomers indicates that the typical of this assay is a good predictor of antiviral activity. Because this assay is robust, easy to perform, and very stable, it is based on the ability to interact with one or more components, for example, viral proteins, rather than based on the identification of a specific target. It is a very good candidate for high throughput screening to identify new antiviral molecules.
本明細書において記載した例示的な方法は、ウイルス溶解物との相互作用の測定に蛍光偏光を使用するが、タンパク質相互作用を監視するための多数の技術が当該技術分野にて公知であり、本方法に使用することができる。それらは表面プラズモン共鳴、蛍光共鳴エネルギー移動(FRET)、酵素免疫吸着測定法(ELISA)、ゲル電気泳動(移動度シフト測定のため)、等熱滴定および差分走査熱量測定ならびにカラムクロマトグラフィーを含むが、それらに限定されるわけではない。これらの他の異なる技術をONのウイルス溶解物または成分との相互作用の測定に適用することができ、従って抗ウイルス活性を有する化合物のスクリーニングに有用であり得る。 The exemplary methods described herein use fluorescence polarization to measure interaction with virus lysates, but numerous techniques for monitoring protein interactions are known in the art, It can be used in this method. They include surface plasmon resonance, fluorescence resonance energy transfer (FRET), enzyme immunosorbent assay (ELISA), gel electrophoresis (for mobility shift measurements), isothermal titration and differential scanning calorimetry, and column chromatography. However, it is not limited to them. These other different techniques can be applied to measure the interaction of ON with viral lysates or components and may thus be useful in screening for compounds with antiviral activity.
所望の供給源、たとえば、コンビナトリアルケミストリにより合成された又は天然物質の精製により単離されたライブラリから新規の化合物をスクリーニングするのに、本明細書に記載された方法を使用する。それを、a)新規のONの適当なサイズ、修飾及び主鎖を決定するのに;b)新規のポリマーを含む新規の分子を調べるのに;c)新規のON又は新規の化合物に対する特定のウイルスの感受性を予測するのに;又はd)特定のウイルスを最大限阻害するための一揃いの化合物を決定するのに用いられる。 The methods described herein are used to screen new compounds from libraries of interest, eg, synthesized by combinatorial chemistry or isolated by purification of natural materials. A) to determine the appropriate size, modification and backbone of the new ON; b) to investigate new molecules containing the new polymer; c) to identify new ONs or specific compounds for new compounds Used to predict virus susceptibility; or d) to determine a set of compounds to maximally inhibit a particular virus.
大きなサイズのPS-ONランダマーとの溶解物の大きな親和性は、PS-ONランダマーの作用の抗ウイルスメカニズムが、ビリオンの感染又は複製の修正のいずれかを妨げる1以上のウイルスタンパク質構成成分との相互作用に基づくことを示唆している。それはまた、この相互作用が、電荷(サイズ)依存性であり、配列に依存していないことを示唆している。これらPS-ONランダマーは、幾つかの異なるファミリーにまたがって複数のウイルスにわたってサイズ依存性の活性を有するので、我々は、PS-ONは、共通する、電荷依存性のタンパク質-タンパク質相互作用、タンパク質-DNA/RNA相互作用及び/又は分子-分子相互作用を妨害することを示唆している。これらの相互作用には以下を挙げることができる(これらに限定されない)。
カプシド形成中の個々のカプシドサブユニット間の相互作用。
カプシド/ヌクレオカプシドタンパク質とウイルスゲノムとの間の相互作用。
出芽中のカプシドと糖タンパク質との間の相互作用。
感染中の糖タンパク質と受容体との間の相互作用。
ウイルス複製に関与するそのほかの鍵となるウイルス構成成分間の相互作用。
The large affinity of the lysate with the large size PS-ON randomer is due to the fact that the antiviral mechanism of action of the PS-ON randomer is associated with one or more viral protein components that prevent either virion infection or replication correction. It is based on the interaction. It also suggests that this interaction is charge (size) dependent and sequence independent. Because these PS-ON randomers have size-dependent activity across multiple viruses across several different families, we believe that PS-ON is a common, charge-dependent protein-protein interaction, protein -Suggests interfering with DNA / RNA interactions and / or molecule-molecule interactions. These interactions can include (but are not limited to):
Interaction between individual capsid subunits during capsid formation.
Interaction between capsid / nucleocapsid protein and viral genome.
Interaction between budding capsid and glycoprotein.
Interaction between glycoprotein and receptor during infection.
Interaction between other key viral components involved in virus replication.
タンパク質-タンパク質相互作用のこれら複数の同時阻害剤は、抗ウイルス性の阻害の新規のメカニズムを代表する。 These multiple simultaneous inhibitors of protein-protein interactions represent a novel mechanism of antiviral inhibition.
ウイルス溶解物相互作用に対するPS-ONの配列組成の効果
本発明者らは、異なる配列のPS-ONの幾つかのウイルス溶解物と相互作用する能力をモニターした。各場合において、本明細書において前に記載したように20merのPS-ONを3’末端にてFITCで標識する。調べたPS-ONは、A20、G20、T20、C20、AC10、AG10、TC10、TG10、REP 2004及びREP 2017から成った。これら配列のそれぞれをアッセイ緩衝液で4nMに希釈し1ugのHSV-1、HIV-1又はRSVの溶解物の存在下インキュベートした。蛍光分極によって相互作用を測定した。
Effect of sequence composition of PS-ON on viral lysate interactions We monitored the ability of different sequences of PS-ON to interact with several viral lysates. In each case, a 20mer PS-ON is labeled with FITC at the 3 'end as previously described herein. The PS-ON examined consisted of A20, G20, T20, C20, AC10, AG10, TC10, TG10, REP 2004 and REP 2017. Each of these sequences was diluted to 4 nM with assay buffer and incubated in the presence of 1 ug of HSV-1, HIV-1 or RSV lysate. The interaction was measured by fluorescence polarization.
試験する全配列との相互作用のプロファイルは、全ウイルス溶解物で同様であり、結合相互作用の性質は、非常に類似することが示される。異なる配列組成を有する20mer PS-ON (A20、C20、G20、T20、AC10、AG10、TC10、TG10、REP 2004、REP 2017)の、ウイルス溶解物に結合する能力を、蛍光偏光法により測定した。FITCで標識したPS-ON3'を、HSV-1、HIV-1またはRSV溶解物1ugの存在下でインキュベートした。各溶解物のうち、均一組成(A20、G20、T20、C20)のPS-ONは最も弱い相互作用物であり、これらのうちA20は有意な差をもって最も弱い相互作用物であった。試験した他のPS-ONについては、TG10を除いたそれら全部が、類似する、強力な相互作用を示し、TG10は各溶解物中で一貫して最も強い相互作用を示した。これらPS-ONの結合プロファイルは、3個のウイルス溶解物の全部について同様であった。 The profile of interaction with all sequences tested is similar for all virus lysates, indicating that the nature of the binding interaction is very similar. The ability of 20mer PS-ONs with different sequence compositions (A20, C20, G20, T20, AC10, AG10, TC10, TG10, REP 2004, REP 2017) to bind to viral lysates was measured by fluorescence polarization. PS-ON3 ′ labeled with FITC was incubated in the presence of 1 ug of HSV-1, HIV-1 or RSV lysate. Among each lysate, PS-ON of uniform composition (A20, G20, T20, C20) was the weakest interactant, and among these, A20 was the weakest interactant with a significant difference. For the other PS-ONs tested, all except TG10 showed similar strong interactions, which were consistently the strongest interaction in each lysate. The binding profiles of these PS-ONs were similar for all three virus lysates.
HIV-IにおけるPS-ONランダマーの標的同定
実施例9に記載した蛍光偏光法により、PS-ONランダマーの、精製HIV-1タンパク質に結合する能力を試験した。精製HIV-1p24または精製HIV-1gp41の量を増大させて、REP 2004-FLと反応させた。本発明等は、これら両方のタンパク質について、これら両方のタンパク質との相互作用を示す蛍光偏光におけるタンパク質濃度依存的なシフトが存在することに注目する。
Target identification of PS-ON randomers in HIV-I The ability of PS-ON randomers to bind to purified HIV-1 protein was tested by fluorescence polarization as described in Example 9. The amount of purified HIV-1p24 or purified HIV-1 gp41 was increased and reacted with REP 2004-FL. The present invention and others note that there is a protein concentration dependent shift in fluorescence polarization that shows an interaction with both of these proteins.
ある範囲のサイズのPS-ONランダマーの、これらのタンパク質に結合する能力も、REP 2032、REP 2003、REP 2004、REP 2006およびREP 2007の蛍光バージョンを用いて試験した。本発明者らは、p24については、p24とのサイズ依存的な相互作用が存在しないことを観察したが、しかし、本発明者らは、20塩基未満のランダマーに対して、20塩基を越えるPS-ONランダマーのgp41に対する結合が増加したことを観察した。このことは、PS-ONランダマーの長さが20塩基を越えると、gp41の多数のコピーが個々のランダマーに結合し、それらの偏光を増大させ得ることを示唆している。 A range of sizes of PS-ON randomers were also tested for their ability to bind to these proteins using fluorescent versions of REP 2032, REP 2003, REP 2004, REP 2006 and REP 2007. We have observed that for p24 there is no size-dependent interaction with p24, but we have a PS of greater than 20 bases for randomers of less than 20 bases. We observed increased binding of the -ON randomer to gp41. This suggests that when the length of PS-ON randomers exceeds 20 bases, multiple copies of gp41 can bind to individual randomers and increase their polarization.
このことは、それが、ウイルス合成の間構造タンパク質を隔離するさらに大きなONの能力を明らかにし、新しいビリオン形成への利用性を限定しているので、有意な所見である。 This is a significant finding as it reveals the greater ability of ON to sequester structural proteins during viral synthesis and limits its utility for new virion formation.
高親和性のオリゴヌクレオチド
もう1つのアプローチは、ウイルスタンパク質のようなウイルス構成成分に対する、ONの標準プールにおける平均親和性よりも高い親和性を有するONを濃縮する又は精製する方法である。従って、方法は1以上のウイルス構成成分に対する高い親和性を示し、たとえば、そのように高い結合親和性に寄与する三次元形状を有する1以上の非配列相補的ONを提供する。理論的根拠は、ONがウイルス構成成分との結合で直鎖分子として作用する一方でそのようなウイルス構成成分との相互作用を高める三次元形状にも折り畳むことができるということである。特定の技術に限定することなく、以下の方法によって高い親和性のONを精製又は濃縮することができる。
High Affinity Oligonucleotides Another approach is to concentrate or purify ONs with a higher affinity for viral components, such as viral proteins, than the average affinity in the standard pool of ONs. Thus, the method exhibits a high affinity for one or more viral components, for example providing one or more non-sequence complementary ONs having a three-dimensional shape that contributes to such a high binding affinity. The rationale is that ON can be folded into a three-dimensional shape that acts as a linear molecule in association with virus components while enhancing interaction with such virus components. Without being limited to a specific technique, high affinity ON can be purified or concentrated by the following method.
高い親和性のON又は複数の高い親和性のONを精製する方法の1つは、ONを結合する親和性マトリクスとしてウイルスタンパク質を結合させた固定相媒体を用い、次いで、漸増する厳密性条件下(たとえば、漸増する濃度の塩又はそのほかのカオトロピック剤、及び/又は漸増する温度及び/又はpHの変化)でそれを溶出することができる。そのような方法はたとえば、以下によって行うことができる。
(a)固定相に結合したウイルスタンパク質又は数種のウイルスタンパク質又はウイルス溶解物を有する交換カラムにONのプールを装填すること;
(b)たとえば、漸増する塩溶液のような置換剤溶液を使用することによってカラムから結合したONを置換すること(溶出すること);
(c)異なる塩濃度で溶出されたONの分画を回収すること;
(d)高い塩濃度で溶出されたONがウイルスタンパク質との高い親和性を有するように、異なる分画から、さらに好ましくは高い塩濃度の分画から溶出されたONをクローニングし、配列決定すること;及び
(e)結合アッセイ及び/又はウイルス阻害アッセイ、たとえば本明細書で記載されたような蛍光分極結合アッセイ及び/又は細胞ウイルス阻害アッセイ及び/又は動物ウイルス阻害アッセイにおいて配列決定されたONの活性を調べること。
One method of purifying high affinity ONs or multiple high affinity ONs is to use a stationary phase medium with viral proteins attached as the affinity matrix to bind the ONs, then increasing stringent conditions. It can be eluted with (for example, increasing concentrations of salt or other chaotropic agent, and / or increasing temperature and / or pH changes). Such a method can be performed, for example, by:
(A) loading a pool of ONs into an exchange column having viral proteins or several viral proteins or virus lysates bound to a stationary phase;
(B) displace (elute) bound ON from the column, eg, by using a displacer solution such as increasing salt solution;
(C) collecting ON fractions eluted at different salt concentrations;
(D) Clone and sequence ON eluted from different fractions, more preferably from higher salt concentration fractions, so that the ON eluted at high salt concentrations has a high affinity with viral proteins. And (e) a binding assay and / or a virus inhibition assay, eg, a ON sequence sequenced in a fluorescence polarization binding assay and / or a cellular virus inhibition assay and / or an animal virus inhibition assay as described herein. Check for activity.
第2の例では、SELEX方法論(Morris et al., Biochemistry, 95:2902-2907, 1998)に由来する及びそれから改変された方法を用いて、高い親和性のONを精製することができる。そのような方法の実施の1つは以下のように行うことができる。
(a)たとえば1014〜1016の異なる配列のような膨大な数の配列を含有する合成ランダムONの集団であるONの出発プールを提供すること。各ON分子は、各末端でのプライマ-結合配列に隣接してランダム配列の断片を含有してポリメラーゼ鎖反応(PCR)を促進する。本質的にすべての分子のヌクレオチド配列が固有なので、集団では膨大な数の構造が試料採取される。これらの構造が、たとえば、所定のウイルス標的分子に結合する能力のような各分子の生化学的特性を決定する;
(b)ウイルスタンパク質又は数種のウイルスタンパク質又はウイルス溶解物にONを接触させること;
(c)たとえば、元々のゲルシフト及びニトロセルロースろ過のような、結合したONと結合しないONを区分化することができる区分化技術を用いて、ウイルスタンパク質に結合したONを選択すること。これらの方法のいずれかが結合していない種から結合した種を物理的に分離し、最も結合した配列を優先的に回収することができる。また、小さなタンパク質に結合するONを選択するには、標的を固相支持体に結合させ、親和性精製マトリクスとしてその支持体を用いることが望ましい。結合しないそれらの分子は洗い流され、結合したものは遊離した標的とともに溶出され、再び、結合した種と結合しない種を物理的に分離する。
(d)ONの隣接配列とハイブリッド形成するプライマ-を用いたPCRを用いて、溶出された結合ONを増幅すること;
(e)ウイルスタンパク質への最高の結合親和性を示すONを優先的に回収するために、工程(b)、(c)及び(d)を複数回(すなわち、複数サイクル又は複数回の濃縮及び増幅)繰り返す。数サイクルの濃縮及び増幅の後、集団では所望の生化学的特性を示す配列が優勢である:
(f)濃縮サイクル、たとえば、最後のサイクルから選択した1以上のONをクローニングし、配列決定すること;及び
(g)アッセイ、たとえば本明細書で記載されたような蛍光分極結合アッセイ及び/又は細胞ウイルス阻害アッセイ及び/又は動物ウイルス阻害アッセイにおいて配列決定されたONの結合及び/又は活性を調べること。
In a second example, high affinity ONs can be purified using methods derived from and modified from the SELEX methodology (Morris et al., Biochemistry, 95: 2902-2907, 1998). One implementation of such a method can be performed as follows.
(A) To provide a starting pool of ONs that are a population of synthetic random ONs containing a vast number of sequences, eg, 10 14 to 10 16 different sequences. Each ON molecule contains a fragment of random sequence adjacent to the primer-binding sequence at each end to facilitate the polymerase chain reaction (PCR). Since the nucleotide sequence of essentially every molecule is unique, a huge number of structures are sampled in a population. These structures determine the biochemical properties of each molecule, such as the ability to bind to a given viral target molecule;
(B) contacting ON with a viral protein or several viral proteins or lysates;
(C) Select ONs that are bound to viral proteins using a partitioning technique that can partition unbound and unbound ONs, such as the original gel shift and nitrocellulose filtration. The bound species can be physically separated from the unbound species by any of these methods, and the most bound sequences can be preferentially recovered. Also, to select ONs that bind to small proteins, it is desirable to bind the target to a solid support and use that support as an affinity purification matrix. Those molecules that do not bind are washed away, those that bind are eluted with the free target, and again again physically separate the bound and unbound species.
(D) amplifying the eluted bound ON using PCR with a primer that hybridizes to the flanking sequence of the ON;
(E) Steps (b), (c) and (d) are repeated multiple times (ie multiple cycles or multiple enrichments and Amplification) Repeat. After several cycles of enrichment and amplification, the population is dominated by sequences that exhibit the desired biochemical properties:
(F) cloning and sequencing one or more ONs selected from the enrichment cycle, eg, the last cycle; and (g) an assay, eg, a fluorescence polarization binding assay as described herein, and / or To examine the binding and / or activity of ON sequenced in cell virus inhibition assays and / or animal virus inhibition assays.
もう1つのアプローチは、スプリット合成方法論の修飾を適用して、Yang et al., Nucl. Acids Res., 30(e132): 1-8, 2002に記載されているように1-ビーズ1-PS-ON及び1-ビーズ1-PS2-ONを創製する。ウイルスタンパク質への特異的なビーズの結合及び選択を行うことができる。選択されたビーズのPCRに基づいた同定タグを用いて、双方の核酸塩基の配列決定及びチオエート/ジチオエート結合の位置決めを行うことができる。このアプローチによって、ウイルスタンパク質に結合する強力な抗ウイルス特性を呈するPS-ON又はPS2-ONの同定を迅速に且つ都合よく行うことができる。 Another approach is to apply a modification of the split synthesis methodology to 1-bead 1-PS as described in Yang et al., Nucl. Acids Res., 30 (e132): 1-8, 2002. -ON and 1-Bead 1-PS2-ON are created. Binding and selection of specific beads to viral proteins can be performed. PCR-based identification tags of selected beads can be used to sequence both nucleobases and locate thioate / dithioate bonds. This approach allows rapid and convenient identification of PS-ON or PS2-ON that exhibits potent antiviral properties that bind to viral proteins.
ウイルスタンパク質に結合する高親和性の具体的配列がいったん決定されると(たとえば、個々の増幅及び配列決定によって)1つ以上のそのような高親和性の配列を選択し、合成して(たとえば、化学合成又は酵素的合成のいずれか)、薬物動態特性を改善することを含む活性を改善するために修飾されうる高親和性ONの調製を提供する。そのような高親和性ONを本発明では使用することができる。 Once the specific sequence of high affinity that binds to the viral protein is determined (eg, by individual amplification and sequencing), one or more such high affinity sequences are selected and synthesized (eg, , Either chemical synthesis or enzymatic synthesis), provides for the preparation of high affinity ONs that can be modified to improve activity, including improving pharmacokinetic properties. Such a high affinity ON can be used in the present invention.
プリオン病
もう1つのアプローチは、プリオン病の治療、進行の制御又は予防のための本発明の別の態様で使用される。この致死的神経変性疾患は感染性であり、ヒト及び動物の双方を冒す。細胞性プリオンタンパク質、PrPCのスクレイピーアイソフォーム、PrPSCへの構造変化がプリオン病の発症及び進行の必須段階であるとみなされている。プリオン病の神経病理学にはアミロイドポリマーが関係している。
Prion disease Another approach is used in another aspect of the invention for the treatment, control or prevention of prion disease. This fatal neurodegenerative disease is infectious and affects both humans and animals. Structural changes to cellular prion protein, PrPC scrapie isoform, and PrPSC are considered essential steps in the onset and progression of prion disease. Amyloid polymers are involved in the neuropathology of prion diseases.
プリオンタンパク質断片と二本鎖核酸とのインキュベートの結果、アミロイド線維形成物を生じる(Nandi et al., J. Mol. Biol., 322:153-161, 2002)。ホスホロチオエート類のようにタンパク質に親和性を有するONは、二本鎖核酸とPrPCとの相互作用と競合させる又はそれを阻害するのに使用され、その結果、アミロイドポリマー形成物を止める。異なるサイズ及び異なる組成のそのようなONを適当な送達形態で用いて、プリオン病に罹った患者を治療することができ、又はリスクの高い状況を予防することができる。そのような干渉するONは、試験ONの存在下で、Nandi et al.,(前記)により記載されたように、アミロイドポリマーの折り畳み変化を測定することによって同定することができる。 Incubation of prion protein fragments with double stranded nucleic acids results in amyloid fibril formation (Nandi et al., J. Mol. Biol., 322: 153-161, 2002). ONs that have an affinity for proteins, such as phosphorothioates, are used to compete with or inhibit the interaction of double-stranded nucleic acids with PrPC, thus stopping amyloid polymer formation. Such ONs of different sizes and different compositions can be used in appropriate delivery forms to treat patients with prion disease or to prevent high-risk situations. Such interfering ONs can be identified by measuring the amyloid polymer folding change, as described by Nandi et al., (Supra) in the presence of a test ON.
想定されるウイルスの病因
もう1つのアプローチは、以下の例で限定することなく記載されるように、想定されるウイルス病因のある疾患又は症状の治療又は予防のための本発明のもう1つの態様で使用される。ウイルスは、ウイルスの一次感染に関係のない疾患及び症状において推定上の原因物質である。たとえば、関節炎はHCV(Olivieri et al., Rheum. Dis. Clin. North Am., 29:111-122, 2003)、パルボウイルスB19、HIV、HSV、CMV、EBV及びVZV(Stahl et al., Clin Rheumatol., 19:281-286, 2000)に関係している。そのほかのウイルスも様々な疾患において役目を担っているとして同定されている。たとえば、パーキンソン病におけるインフルエンザA(Takahashi et al., Jpn. J. Infect. Dis., 52:89-98, 1999)、多発性硬化症におけるコロナウイルス、EBV及びそのほかのウイルス(Talbot et al., Curr Top Microbiol. Immunol., 253, 247-71, 2001)、慢性疲労症候群におけるEBV、CMV及びHSV-6(Lerner et al., Drugs Today, 38:549-561, 2002)、喘息(Walter et al., J. Clin. Invest., 110:165-175, 2002)及びパージェット病におけるパラミクソウイルス、及びGuillain-Barre症候群におけるHBV、HSV及びインフルエンザ。
Possible viral pathogenesis Another approach is another aspect of the invention for the treatment or prevention of a disease or condition with a possible viral pathogenesis, as described without limitation in the examples below. Used in. Viruses are putative causative agents in diseases and conditions that are not related to the primary infection of the virus. For example, arthritis can occur in HCV (Olivieri et al., Rheum. Dis. Clin. North Am., 29: 111-122, 2003), parvovirus B19, HIV, HSV, CMV, EBV and VZV (Stahl et al., Clin Rheumatol., 19: 281-286, 2000). Other viruses have also been identified as playing a role in various diseases. For example, influenza A in Parkinson's disease (Takahashi et al., Jpn. J. Infect. Dis., 52: 89-98, 1999), coronavirus, EBV and other viruses in multiple sclerosis (Talbot et al., Curr Top Microbiol. Immunol., 253, 247-71, 2001), EBV, CMV and HSV-6 in chronic fatigue syndrome (Lerner et al., Drugs Today, 38: 549-561, 2002), asthma (Walter et al , J. Clin. Invest., 110: 165-175, 2002) and Paramyxovirus in Paget's disease, and HBV, HSV and influenza in Guillain-Barre syndrome.
これらの病因のために、本発明を用いた関連するウイルスの阻害は、相当する疾患若しくは症状、又は疾患の少なくとも症状の一部の進展を遅延させる、減速させる又は防ぐことができる。 Because of these etiologies, inhibition of related viruses using the present invention can delay, slow down or prevent the corresponding disease or condition, or at least a portion of the disease from developing.
オリゴヌクレオチドの修飾及び合成
上記の要約で示したように、修飾したオリゴヌクレオチドは本発明で有用である。そのような修飾されたオリゴヌクレオチドには、たとえば、修飾した主鎖又は非天然のヌクレオシド間結合を含有するオリゴヌクレオチドが挙げられる。修飾した主鎖を有するオリゴヌクレオチドには、主鎖にリン原子を保持するもの及び主鎖にリン原子を有さないものが挙げられる。
Oligonucleotide Modifications and Synthesis As indicated in the summary above, modified oligonucleotides are useful in the present invention. Such modified oligonucleotides include, for example, oligonucleotides containing modified backbones or non-natural internucleoside linkages. Oligonucleotides having a modified main chain include those that retain a phosphorus atom in the main chain and those that do not have a phosphorus atom in the main chain.
そのような修飾されたオリゴヌクレオチドの主鎖には、たとえば、ホスホロチオエート類、キラルホスホロチオエート類、ホスホロジチオエート類、ホスホトリエステル類、アミノアルキルホスホトリエステル類、3'-アルキレンホスホネート類、5'-アルキレンホスホネート類及びキラルホスホネート類を含むメチル又はそのほかのアルキルホスホネート類、ホスフィネート類、3'-アミノホスホロアミダイト及びアミノアルキルホスホロアミダイトを含むホスホロアミダイト類、チオノホスホロアミダイト類、チオノアルキルホスホネート類、チオノアルキルホスホトリエステル類、セレノホスフェート類、カルバラニルホスフェート通常の3',5'結合を有するボラノ-ホスフェート類、2',5'結合したこれらの類縁体、並びに1以上のヌクレオチド間結合が、3'〜3'、5'〜5'又は2'〜2'の結合である反転した極性を有するものが挙げられる。反転した極性を有するオリゴヌクレオチドには通常、3'のほとんどのヌクレオチド間結合で単一の3'〜3'結合、すなわち、脱塩基(ヌクレオ塩基を失っている又はそれに代わってヒドロキシル基を有する)である単一の反転ヌクレオシド残基が挙げられる。種々の塩、混合塩及び遊離の酸の形態も挙げられる。 Such modified oligonucleotide backbones include, for example, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkyl phosphotriesters, 3′-alkylene phosphonates, 5 ′ -Methyl or other alkyl phosphonates including alkylene phosphonates and chiral phosphonates, phosphinates, phosphoramidites including 3'-amino phosphoramidites and aminoalkyl phosphoramidites, thionophosphoramidites, thiono Alkyl phosphonates, thionoalkyl phosphotriesters, selenophosphates, carvalanyl phosphate borano-phosphates with conventional 3 ', 5' linkages, 2 ', 5' linked analogs, and one or more Between nucleotides If found 3'~3 ', 5'~5' include those having a polarity obtained by inverting a bond or 2'~2 '. Oligonucleotides with reversed polarity typically have a single 3 'to 3' linkage with most 3 'internucleotide linkages, ie abasic (losing nucleobase or having a hydroxyl group instead) And a single inverted nucleoside residue. Various salts, mixed salts and free acid forms are also mentioned.
上記で示されるようなリンを含有する結合を持つオリゴヌクレオチドの調製は、たとえば、米国特許第3,687,808; 4,469,863; 4,476,301;5,023,243; 5,177,196; 5,188,897; 5,264,423; 5,276,019; 5,278,302; 5,286,717;5,321 ,131 ; 5,399,676; 5,405,939; 5,453,496; 5,455,233; 5,466,677; 5,476,925;5,519,126; 5,536,821 ; 5,541 ,306; 5,550,111; 5,563,253; 5,571 ,799; 5,587,361; 5,194,599; 5,565,555; 5,527,899; 5,721,218; 5,672,697; および5,625,050号に記載されており、参考としてその全体を本明細書に組み入れる。 Preparation of oligonucleotides having linkages containing phosphorus as shown above is described, for example, in U.S. Pat.Nos. 3,687,808; 4,469,863; 4,476,301; 5,023,243; 5,177,196; 5,188,897; 5,405,939; 5,453,496; 5,455,233; 5,466,677; 5,476,925; 5,519,126; 5,536,821; 5,541,306; 5,550,111; 5,563,253; 5,571,799; 5,587,361; 5,194,599; 5,565,555; 5,527,899; 5,721,218; 5,672,697; and 5,625,050 The entirety is incorporated herein.
ホスホジエステルを包含しない一部の典型的なオリゴヌクレオチドの主鎖は、短鎖アルキル又はシクロアルキルヌクレオチド間結合、混合へテロ原子及びアルキル又はシクロアルキルヌクレオチド間結合、又は1以上の短鎖へテロ原子の又は複素環のヌクレオチド間結合により形成される主鎖を有する。これらには、モルホリノ結合(ヌクレオシドの糖部分から一部が形成される)、シロキサン主鎖、硫化物、スルホキシド及びスルホンの主鎖;ホルムアセチル及びチオホルムアセチルの主鎖;メチレンホルムアセチル及びチオホルムアセチルの主鎖;リボセチル主鎖;アルケン含有主鎖;スルファメート主鎖;メチレンイミノ及びメチレンヒドラジノ主鎖;スルホネート及びスルホンアミド主鎖;アミド主鎖;及び混合されたN、O、S及びCH2の成分部分を有するそのほかのものを有するものが挙げられる。特に有利なのは、1以上の荷電された部分を包含する主鎖結合である。先行するオリゴヌクレオチドの調製を記載する米国特許の例には、米国特許第5,034,506; 5,166,315; 5,185,444; 5,214,134; 5,216,141 ; 5,235,033; 5,264,562; 5,264,564; 5,405,938; 5,434,257; 5,466,677; 5,470,967; 5,489, 677; 5,541 ,307; 5,561 ,225; 5,596,086; 5,602,240; 5,610,289; 5,602,240; 5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312; 5,633,360; 5,677,437; 5,792,608; 5,646,269; 5,677,439号が挙げられ、それぞれ参考としてその全体を本明細書に組み入れる。 Some typical oligonucleotide backbones that do not include phosphodiesters include short alkyl or cycloalkyl internucleotide bonds, mixed heteroatoms and alkyl or cycloalkyl internucleotide bonds, or one or more short heteroatoms. Or a main chain formed by a heterocycle internucleotide linkage. These include morpholino linkages (partially formed from the nucleoside sugar moiety), siloxane backbones, sulfides, sulfoxides and sulfone backbones; formacetyl and thioformacetyl backbones; methyleneformacetyl and thioform Acetyl backbone; ribocetyl backbone; alkene-containing backbone; sulfamate backbone; methyleneimino and methylenehydrazino backbone; sulfonate and sulfonamide backbone; amide backbone; and mixed N, O, S and CH 2 The thing which has the other thing which has these component parts is mentioned. Particularly advantageous are backbone bonds that include one or more charged moieties. Examples of U.S. patents describing the preparation of prior oligonucleotides include U.S. Pat.Nos. 5,034,506; 5,166,315; 5,185,444; 5,214,134; 5,216,141; 5,561,225; 5,596,086; 5,602,240; 5,610,289; 5,602,240; 5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312; 5,633,360; 5,677,437; 5,792,608; 5,646,269; 5,677,439;
修飾されたオリゴヌクレオチドは1以上の置換された糖部分も含有してもよい。たとえば、そのようなオリゴヌクレオチドは、以下の2'修飾:OH;F;O-、S-、又はN-アルキル;O-、S-、又はN-アルケニル;O-、S-、又はN-アルキニル;又はO-アルキル-O-アルキルの1つを包含することができ、その際、アルキル、アルケニル及びアルキニルは、置換された又は置換されないC1〜C10のアルキル又はC2〜C10のアルケニル及びアルキニル、あるいは2'-o-(o-カルボラン-1-イル)メチルであってもよい。特定の例は、O[(CH2)nO]mCH3、O(CH2)-OCH3、O(CH2)nNH2、O(CH2)nCH3、O(CH2)nONH2及びO(CH2)nON[CH2)nCH3]2であり、その際、n及びmは1〜10である。そのほかの典型的なオリゴヌクレオチドは、以下の2'修飾:C1〜C10の低級アルキル、置換低級アルキル、アルケニル、アルキニル、アルカリル、アラルキル、O-アルカリル又はO-アラルキル、SH、SCH3、OCN、Cl、Br、CN、CF3、OCF3、SOCH3、SO2CH3、ONO2、NO2、N3、NH2、ヘテロシクロアルキル、ヘテロシクロアルカリル、アミノアルキルアミノ、ポリアルキルアミノ、置換シリル、リポータ基、干渉物質、オリゴヌクレオチドの薬物動態特性を改善するための基、又はオリゴヌクレオチドの薬力学的特性を改善するための基の1つを包含する。例には、2'-メトキシエトキシ(2'-O-(2-メトキシエチル) 又は2'-MOEとしても知られる2'-O-CH2CH2OCH3)(Martin et al., Helv. Chim. Acta., 78:486-504, 1995)、すなわち、アルコキシアルコキシ基;2'-ジメチル-アミノキシエトキシ、すなわち、2'-DMAOEとしても知られるO(CH2)2ON(CH3)2基;及び2'-ジメチルアミノエトキシエトキシ(2'-O-ジメチルアミノエトキシエチル又は2'-DMAEOEとしても知られる)すなわち、2'-O-CH2-O-CH2-N(CH2)2が含まれる。 Modified oligonucleotides may also contain one or more substituted sugar moieties. For example, such oligonucleotides may have the following 2 ′ modifications: OH; F; O-, S-, or N-alkyl; O-, S-, or N-alkenyl; O-, S-, or N- alkynyl; or O- alkyl -O- 1 one to be able to encompass alkyl, in which alkyl, alkenyl and alkynyl, alkyl or C 2 -C 10 of C 1 -C 10 not substituted or substituted It may be alkenyl and alkynyl, or 2'-o- (o-carboran-1-yl) methyl. Specific examples are O [(CH 2 ) n O] m CH 3 , O (CH 2 ) -OCH 3 , O (CH 2 ) n NH 2 , O (CH 2 ) n CH 3 , O (CH 2 ) n ONH 2 and O (CH 2 ) n ON [CH 2 ) n CH 3 ] 2 , where n and m are 1 to 10. Other exemplary oligonucleotides include the following 2 ′ modifications: C 1 -C 10 lower alkyl, substituted lower alkyl, alkenyl, alkynyl, alkaryl, aralkyl, O-alkaryl or O-aralkyl, SH, SCH 3 , OCN , Cl, Br, CN, CF 3 , OCF 3 , SOCH 3 , SO 2 CH 3 , ONO 2 , NO 2 , N 3 , NH 2 , heterocycloalkyl, heterocycloalkaryl, aminoalkylamino, polyalkylamino, It includes one of a substituted silyl, a reporter group, an interfering substance, a group for improving the pharmacokinetic properties of the oligonucleotide, or a group for improving the pharmacodynamic properties of the oligonucleotide. Examples include 2′-methoxyethoxy (2′-O—CH 2 CH 2 OCH 3 ), also known as 2′-O- (2-methoxyethyl) or 2′-MOE (Martin et al., Helv. Chim. Acta., 78: 486-504, 1995), ie, alkoxyalkoxy groups; 2'-dimethyl-aminoxyethoxy, ie, O (CH 2 ) 2 ON (CH 3 ), also known as 2'-DMAOE 2 groups; and 2′-dimethylaminoethoxyethoxy (also known as 2′-O-dimethylaminoethoxyethyl or 2′-DMAEOE), ie 2′-O—CH 2 —O—CH 2 —N (CH 2 ) 2 is included.
そのほかの修飾には、2'-ヒドロキシ基が糖環の3'又は4'の炭素に結合し、それによって二環式糖部分を形成するロックされた核酸(LNA)が挙げられる。結合は、2'の酸素原子と4'の炭素原子を架橋するメチレン(-CH2-) 基であることができ、その際、nは1又は2である。LNA及びその調製は、国際公開公報第98/39352号及び同第99/14226号に記載されており、参考としてその全体を本明細書に組み入れる。 Other modifications include locked nucleic acids (LNA) in which the 2′-hydroxy group is attached to the 3 ′ or 4 ′ carbon of the sugar ring, thereby forming a bicyclic sugar moiety. The bond can be a methylene (—CH 2 —) group bridging the 2 ′ oxygen atom and the 4 ′ carbon atom, where n is 1 or 2. LNA and its preparation are described in WO 98/39352 and 99/14226, which are hereby incorporated by reference in their entirety.
そのほかの修飾には、Orum et al., (2001) Curr. Opin. Mol. Ther., 3:239-243に記載されているようなロックされた核酸のようなイオウ-窒素架橋修飾が挙げられる。 Other modifications include sulfur-nitrogen bridge modifications such as locked nucleic acids as described in Orum et al., (2001) Curr. Opin. Mol. Ther., 3: 239-243. .
そのほかの修飾には、2'-メトキシ(2'-O-CH3)、2'-メトキシエチル(2'O-CH2-CH3)、2'-エチル、2'-エトキシ、2'-アミノプロポキシ(2'-OCH2CH2CH2NH2)、2'-アリル(2'-CH2-CH=CH2)、2'-O-アリル(2'-O-CH2-CH=CH2)及び2'フルオロ(2'-F) が挙げられる。 Other modifications include 2'-methoxy (2'-O-CH 3 ), 2'-methoxyethyl (2'O-CH 2 -CH 3 ), 2'-ethyl, 2'-ethoxy, 2'- Aminopropoxy (2'-OCH 2 CH 2 CH 2 NH 2 ), 2'-allyl (2'-CH 2 -CH = CH 2 ), 2'-O-allyl (2'-O-CH 2 -CH = CH 2 ) and 2 ′ fluoro (2′-F).
2'修飾は、アラビノ位(上)であっても、リボ(下)位であってもよい。類似の修飾をオリゴヌクレオチドの他の位置で行ってもよく、特に3'末端ヌクレオチド上の糖の3'位又は2'-5'結合オリゴヌクレオチドで、及び5'末端ヌクレオチドの5'位で行ってもよい。オリゴヌクレオチドは、ペントフラノシル糖の代わりにシクロブチル部分として糖模倣体を有してもよい。そのような修飾された糖構造の調製を記載する典型的な米国特許には、たとえば、米国特許第4,981 ,957; 5,118,800; 5,319,080; 5,359,044; 5,393, 878; 5,446,137; 5,466,786; 5,514,785; 5,519,134; 5,567, 811 ; 5,576,427; 5,591 ,722; 5,597,909; 5,610,300; 5,627, 053; 5,639,873; 5,646,265; 5,658,873; 5,670,633; 5,792, 747; および5,700,920号が挙げられる。 The 2 ′ modification may be in the arabino position (up) or in the ribo (down) position. Similar modifications may be made at other positions on the oligonucleotide, particularly at the 3 ′ position of the sugar on the 3 ′ terminal nucleotide or at the 2′-5 ′ linked oligonucleotide and at the 5 ′ position of the 5 ′ terminal nucleotide. May be. Oligonucleotides may have sugar mimetics as cyclobutyl moieties instead of pentofuranosyl sugars. Typical U.S. patents describing the preparation of such modified sugar structures include, for example, U.S. Pat.Nos. 4,981, 957; 5,118,800; 5,319,080; 5,359,044; 5,393, 878; 5,446,137; 5,466,786; 5,514,785; 5,519,134; 5,567, No. 811; 5,576,427; 5,591, 722; 5,597,909; 5,610,300; 5,627, 053; 5,639,873; 5,646,265; 5,658,873; 5,670,633; 5,792, 747; and 5,700,920.
さらにそのほかの修飾には、リンカーで連結された複数のオリゴヌクレオチドから成るONコンカテマーが挙げられる。リンカーはたとえば、修飾されたヌクレオチド及び非ヌクレオチド単位から成ってもよい。場合によっては、リンカーはONコンカテマーに自由度を提供する。そのようなONコンカテマーの使用は、さらに小さなオリゴヌクレオチド基礎単位を連結して所望の長さを得ることにより最終分子を合成する容易な方法を提供することができる。たとえば12個の炭素のリンカー(C12ホスホロアミダイト)を用いて2以上のONコンカテマーを連結し、長さ、安定性及び自由度を提供する。 Still other modifications include ON concatamers consisting of multiple oligonucleotides linked by a linker. A linker may consist of, for example, modified nucleotide and non-nucleotide units. In some cases, the linker provides a degree of freedom for the ON concatemer. The use of such ON concatamers can provide an easy way to synthesize the final molecule by linking smaller oligonucleotide building blocks to obtain the desired length. For example, a 12 carbon linker (C12 phosphoramidite) is used to link two or more ON concatamers to provide length, stability and freedom.
本明細書で使用されるとき、「未修飾の」又は「天然の」塩基(ヌクレオ塩基)には、プリン塩基のアデニン(A)及びグアニン(G)並びにピリミジン塩基のチミン(T)、シトシン(C)及びウラシル(U)が包含される。オリゴヌクレオチドは、塩基修飾又は塩基置換も包含してもよい。修飾された塩基は、5-メチルシトシン(5-me-C)、5-ヒドロキシメチルシトシン、キサンチン、ヒポキサンチン、2-アミノアデニン、アデニン及びグアニンの6-メチル及びそのほかのアルキル誘導体、アデニン及びグアニンの2-プロピル及びそのほかのアルキル誘導体、2-チオウラシル、2-チオチミン及び2-チオシトシン、5-ハロウラシル及びシトシン、5-プロピニル(-C≡C-CH3)ウラシル及びシトシン及びピリミジン塩基のそのほかのアルキル誘導体、6-アゾウラシル、シトシン及びチミン、5-ウラシル(偽ウラシル)、4-チオウラシル、8-ハロ、8-アミノ、8-チオール、8-チオアルキル、8-ヒドロキシル及びそのほかの8-置換されたアデニン及びグアニン、5-ハロ、特に、5-ブロモ、5-トリフルオロメチル及びそのほかの5-置換されたウラシル及びシトシン、7-メチルグアニン及び7-メチルアデニン、2-F-アデニン、2-アミノ-アデニン、8-アザグアニン及び8-アザアデニン、7-デアザグアニン及び7-デアザアデニン及び3-デアザグアニン及び3-デアザアデニンのような合成塩基及び天然塩基を包含してもよい。追加の修飾された塩基には、フェノキサジンシチジン(1H-ピリミド[5,4-b][1,4]ベンゾキサジン-2(3H)-オン)、フェノチアジンシチジン(1H-ピリミド[5,4-b][1,4]ベンゾチアジン-2(3H)-オン)、置換されたフェノキサジンシチジン(たとえば、9-(2-アミノエトキシ)-H-ピリミド[5,4-b][1,4]ベンゾキサジン-2(3H)-オン)、カルバゾールシチジン(2H-ピリミド[4,5-b]インドール-2-オン)、ピリドインドールシチジン(H-ピリド[3',2':4,5]ピロロ[2,3-d]ピリミジン-2-オン)のようなG-クランプのような三環式ピリミジン類が挙げられる。修飾された塩基にはプリン又はピリミジンが、たとえば、7-デアザアデニン、7-デアザグアノシン、2-アミノピリジン及び2-ピリドンのようなそのほかの複素環で置き換えたものを挙げてもよい。さらなるヌクレオチドには、米国特許第3,687,808号に記載されたもの、ポリマー科学および技術のコンサイス百科事典(The Concise Encyclopedia Of Polymer Science And Engineering) p858-859, Kroschwitz, J. I., ed. John Wiley & Sons, 1990 に記載されているもの、Englisch et al., Angewandte Chemie, International Edition,30:613, 1991 に記載されるもの、及びアンチセンス研究と応用(Antisense Research and Applications)15章 Sanghvi Y.S., p289-302, Crooke, S. T. and Lebleu, B., ed., CRC Press, 1993 に記載されているものが挙げられる。 As used herein, “unmodified” or “natural” bases (nucleobases) include the purine bases adenine (A) and guanine (G) and the pyrimidine bases thymine (T), cytosine ( C) and uracil (U) are included. Oligonucleotides may also include base modifications or base substitutions. Modified bases include 5-methylcytosine (5-me-C), 5-hydroxymethylcytosine, xanthine, hypoxanthine, 2-aminoadenine, adenine and 6-methyl and other alkyl derivatives of guanine, adenine and guanine 2-propyl and other alkyl derivatives of 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5-halouracil and cytosine, 5-propynyl (-C≡C-CH 3 ) uracil and other alkyls of cytosine and pyrimidine bases Derivatives, 6-azouracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 8-halo, 8-amino, 8-thiol, 8-thioalkyl, 8-hydroxyl and other 8-substituted adenines And guanine, 5-halo, especially 5-bromo, 5-trifluoromethyl and other 5-substituted uracils and cytosines , 7-methylguanine and 7-methyladenine, 2-F-adenine, 2-amino-adenine, 8-azaguanine and 8-azaadenine, 7-deazaguanine and 7-deazaadenine and 3-deazaguanine and 3-deazaadenine Bases and natural bases may be included. Additional modified bases include phenoxazine cytidine (1H-pyrimido [5,4-b] [1,4] benzoxazin-2 (3H) -one), phenothiazine cytidine (1H-pyrimido [5,4-b ] [1,4] benzothiazin-2 (3H) -one), substituted phenoxazine cytidine (eg 9- (2-aminoethoxy) -H-pyrimido [5,4-b] [1,4] benzoxazine -2 (3H) -one), carbazole cytidine (2H-pyrimido [4,5-b] indol-2-one), pyridoindole cytidine (H-pyrido [3 ′, 2 ′: 4,5] pyrrolo [ And tricyclic pyrimidines such as G-clamp such as 2,3-d] pyrimidin-2-one). Modified bases may include those in which purine or pyrimidine is replaced by other heterocycles such as, for example, 7-deazaadenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone. Additional nucleotides include those described in US Pat. No. 3,687,808, The Concise Encyclopedia Of Polymer Science And Engineering, p858-859, Kroschwitz, JI, ed. John Wiley & Sons, 1990. , Englisch et al., Angewandte Chemie, International Edition, 30: 613, 1991, and Antisense Research and Applications, Chapter 15 Sanghvi YS, p289-302, Crooke, ST and Lebleu, B., ed., CRC Press, 1993.
もう1つの修飾には、ホスホロジチオエート結合が挙げられる。ホスホロジチオエートON(PS2-ON)及びPS-ONがタンパク質に対して類似の結合親和性を有すること(Tonkinson et al., Antisense Res. Dev., 4:269-278, 1994)(Cheng et al., J. Mol. Recogn., 10:101-107, 1997)は知っているが、及びONの作用の可能性のあるメカニズムがウイルスタンパク質に結合することであるのは知っているが、本発明で記載される抗ウイルス性ONにホスホロジチオエートを包含することが望ましい。 Another modification includes a phosphorodithioate linkage. Phosphorodithioate ON (PS2-ON) and PS-ON have similar binding affinity for proteins (Tonkinson et al., Antisense Res. Dev., 4: 269-278, 1994) (Cheng et al., J. Mol. Recogn., 10: 101-107, 1997) and know that the potential mechanism of action of ON is binding to viral proteins, It is desirable to include phosphorodithioate in the antiviral ON described in the present invention.
ONを修飾するもう1つのアプローチは、Yu et al., Bioorg. Med. Chem., 8:275-284, 2000及びInagawa et al., FEBS Lett., 25:48-52, 2002に記載されているような立体定義された又は立体濃縮されたONを製造することである。従来の方法で調製されたONは、ヌクレオチド間結合に関与するリン原子のまわりの不斉性によるジアステレオマーの混合物から成る。これは、ONとウイルスタンパク質のようなウイルス構成成分との間の結合の安定性に影響を及ぼす可能性がある。以前のデータは、タンパク質の結合が有意に立体依存性であることを示していた(Yu et al.)。従って、立体定義された又は立体濃縮されたONを使用することによって、タンパク質の結合特性を改善し、抗ウイルス有効性を改善することができた。 Another approach to modify ON is described in Yu et al., Bioorg. Med. Chem., 8: 275-284, 2000 and Inagawa et al., FEBS Lett., 25: 48-52, 2002. Is to produce a stereo-defined or stereo-enriched ON. The ON prepared by conventional methods consists of a mixture of diastereomers due to asymmetry around the phosphorus atom involved in internucleotide linkage. This can affect the stability of binding between ON and viral components such as viral proteins. Previous data has shown that protein binding is significantly sterically dependent (Yu et al.). Thus, by using stereo-defined or stereo-enriched ON, it was possible to improve the binding properties of the protein and improve the antiviral efficacy.
上述のような修飾の組込みを多数の様々な組込みパターン及びレベルで利用することができる。すなわち、オリゴヌクレオチドでの各ヌクレオチド又は結合で特定の修飾が包含される必要はなく、単一のオリゴヌクレオチド又は単一のヌクレオチドにおいてさえ、様々な修飾を組み合わせて利用することができる。 Modification incorporation as described above can be utilized in a number of different incorporation patterns and levels. That is, specific modifications need not be included at each nucleotide or bond in the oligonucleotide, and various modifications can be utilized in combination, even on a single oligonucleotide or single nucleotide.
例として、また上記の説明によれば、ホスホロチオエートまたはジチオエート結合を含む修飾オリゴヌクレオチドは、1個または複数の置換された糖部分、特に糖部分における2'-エチル、2'-エトキシ、2'-メトキシ、2'-アミノプロポキシ、2'-アリル、2'-フルオロ、2'-ペンチル、2'-プロピル、2'-ジメチルアミノオキシエトキシおよび2'-ジメチルアミノエトキシエトキシを含むが、これらに限定されない修飾も含んでもよい。2'-修飾は、アラビノ(上方)位またはリボ(下方)位に存在してもよい。好ましい2'-アラビノ修飾は、2'-フルオロである。同様の修飾を、オリゴヌクレオチド上の他の位置に、特に3'末端ヌクレオチド上にまたは2'-5'結合オリゴヌクレオチド中の糖の3'位、および5'末端ヌクレオチドの5'位にしてもよい。オリゴヌクレオチドは、ペントフラノシル糖に代わって、シクロブチル部分のような糖擬似も有してもよい。更にONは、非環式プロピレングリコールホスホジエステルバックボーンを伴うグリコール核酸(GNA)からなる構造またはその一部を含む構造を有してもよい(Zhang L, et al (2005) J. Am. Chem. Soc. 127(12):4174-5)。このようなGNAは、ホスホロチオエート結合を含んでもよく、かつピリミジン塩基のみを含んでもよい。 By way of example and according to the above description, modified oligonucleotides containing phosphorothioate or dithioate linkages can include one or more substituted sugar moieties, particularly 2'-ethyl, 2'-ethoxy, 2'- in the sugar moiety. Including, but not limited to, methoxy, 2'-aminopropoxy, 2'-allyl, 2'-fluoro, 2'-pentyl, 2'-propyl, 2'-dimethylaminooxyethoxy and 2'-dimethylaminoethoxyethoxy Modifications that are not made may also be included. The 2'-modification may be in the arabino (up) position or ribo (down) position. A preferred 2'-arabino modification is 2'-fluoro. Similar modifications can be made at other positions on the oligonucleotide, especially on the 3 'terminal nucleotide or in the 2'-5' linked oligonucleotide at the 3 'position of the sugar and the 5' position of the 5 'terminal nucleotide. Good. Oligonucleotides may also have sugar mimetics such as cyclobutyl moieties in place of the pentofuranosyl sugar. Furthermore, the ON may have a structure consisting of a glycol nucleic acid (GNA) with an acyclic propylene glycol phosphodiester backbone or a part thereof (Zhang L, et al (2005) J. Am. Chem. Soc. 127 (12): 4174-5). Such GNAs may contain phosphorothioate linkages and may contain only pyrimidine bases.
オリゴヌクレオチドの合成
当該技術で既知の方法を用いて、本オリゴヌクレオチドを合成することができる。たとえば、標準のホスホロアミダイト試薬をヨウ素による酸化と共に用いて、自動DNAシンセサイザー(たとえば、Applied Biosystems モデル380B)にて、非置換又は置換のホスホジエステル(P=O)オリゴヌクレオチドを合成することができる。ホスホジエステルオリゴヌクレオチドについて、ホスフィト結合の段階的チオエート化のために0.2Mの311-1,2-ベンゾジチオール-3-オン1,1-ジオキシドのアセトニトリル溶液により標準的な酸化瓶を置き換えることを除いて、ホスホロチオエート(P=S)を合成することができる。チオエート化待機工程を68秒に増やすことができ、その後、キャッピング工程が続く。CPGカラムからの切断及び55℃(18時間)における濃縮水酸化アンモニウム中での脱ブロックの後、2.5容積のエタノールにより、0.5MのNaCl溶液から2回沈殿させることによりオリゴヌクレオチドを精製することができる。
Oligonucleotide Synthesis The oligonucleotides can be synthesized using methods known in the art. For example, an unsubstituted or substituted phosphodiester (P = O) oligonucleotide can be synthesized on an automated DNA synthesizer (eg, Applied Biosystems model 380B) using standard phosphoramidite reagents with oxidation by iodine. . For phosphodiester oligonucleotides, except replacing the standard oxidation bottle with acetonitrile solution of 0.2M 311-1,2-benzodithiol-3-one 1,1-dioxide for stepwise thioation of phosphite linkages Thus, phosphorothioate (P = S) can be synthesized. The thioate waiting step can be increased to 68 seconds, followed by a capping step. After cleavage from a CPG column and deblocking in concentrated ammonium hydroxide at 55 ° C (18 hours), the oligonucleotide can be purified by precipitating twice from 0.5 M NaCl solution with 2.5 volumes of ethanol. it can.
米国特許第5,508,270号に記載されるように、ホスフィネートオリゴヌクレオチドを調製することができ;米国特許第4,469,863号に記載されるように、アルキルホスホネートオリゴヌクレオチドを調製することができ;米国特許第5,610,289号及び同第5,625,050号に記載されるように、3'-デオキシ-3'-メチレンホスホネートオリゴヌクレオチドを調製することができ;米国特許第5,256,775号及び同第5,366,878号に記載されるように、ホスホロアミダイトオリゴヌクレオチドを調製することができ;公表されたPCT出願、PCT/US94/00902号及び同PCT/US93/06976(それぞれ国際公開公報第94/17093号及び同第94/02499号として公表)に記載されるように、アルキルホスホノチオエートオリゴヌクレオチドを調製することができ;米国特許第5,476,925号に記載されるように、3'-デオキシ-3'-アミノホスホロアミダイトオリゴヌクレオチドを調製することができ;米国特許第5,023,243号に記載されるように、ホスホトリエステルオリゴヌクレオチドを調製することができ;米国特許第5,130,302号及び同第5,177,198号に記載されるように、ボラノホスフェートオリゴヌクレオチドを調製することができ;米国特許第5,378,825号、同第5,386,023号、同第5,489,677号、同第5,602,240号及び同第5,610,289号に記載されるように、MMI結合オリゴヌクレオチドとしても同定されるメチレンメチルイミノ結合オリゴヌクレオチド、MDII結合オリゴヌクレオチドとしても同定されるメチレンジメチルヒドラゾ結合オリゴヌクレオチド、及びアミド-3結合オリゴヌクレオチドとしても同定されるメチレンカルボニルアミノ結合オリゴヌクレオチド、及びアミド-4結合オリゴヌクレオ-シドとしても同定されるメチレンアミノカルボニル結合オリゴヌクレオチド、並びにたとえば別のMMI及びP=O又はP=Sの結合を有する混合主鎖化合物を調製することができ;米国特許第5,264,562号及び同第5,264,564号に記載されるように、ホルムアセタール及びチオホルムアセタール結合オリゴヌクレオチドを調製することができ;並びに米国特許第5,223,618号に記載されるように、エチレンオキシド結合オリゴヌクレオチドを調製することができる。引用した特許及び特許出願はそれぞれ、その全体を参考として本明細書に組み入れられる。 Phosphinate oligonucleotides can be prepared as described in US Pat. No. 5,508,270; alkyl phosphonate oligonucleotides can be prepared as described in US Pat. No. 4,469,863; US Pat. No. 5,610,289 3′-deoxy-3′-methylene phosphonate oligonucleotides can be prepared as described in US Pat. Nos. 5,625,050; phosphonates as described in US Pat. Nos. 5,256,775 and 5,366,878. Loamidite oligonucleotides can be prepared; published PCT applications, PCT / US94 / 00902 and PCT / US93 / 06976 (published as WO94 / 17093 and 94/02499, respectively) Alkylphosphonothioate oligonucleotides can be prepared as described in US Pat. No. 5,476,925, as described in US Pat. No. 5,476,925. 3′-amino phosphoramidite oligonucleotides can be prepared; phosphotriester oligonucleotides can be prepared as described in US Pat. No. 5,023,243; US Pat. Nos. 5,130,302 and 5,177,198 Boranophosphate oligonucleotides can be prepared as described in US Pat. Nos. 5,378,825, 5,386,023, 5,489,677, 5,602,240, and 5,610,289. , Methylenemethylimino-linked oligonucleotides that are also identified as MMI-linked oligonucleotides, methylenedimethylhydrazo-linked oligonucleotides that are also identified as MDII-linked oligonucleotides, and methylenecarbonylamino bonds that are also identified as amide-3 linked oligonucleotides Oligonucleotide and amide-4-linked oligo Methyleneaminocarbonyl-linked oligonucleotides that are also identified as gonnucleosides can be prepared, as well as mixed backbone compounds having, for example, another MMI and P═O or P═S linkage; US Pat. No. 5,264,562 and the like Form acetal and thioform acetal linked oligonucleotides can be prepared as described in US Pat. No. 5,264,564; and ethylene oxide linked oligonucleotides can be prepared as described in US Pat. No. 5,223,618. Each cited patent and patent application is hereby incorporated by reference in its entirety.
オリゴヌクレオチド製剤及び薬学的組成物
本オリゴヌクレオチドをオリゴヌクレオチド製剤又は薬学的組成物に調製することができる。従って、本オリゴヌクレオチドは、そのほかの分子、分子構造又は、たとえば、リポソーム、受容体標的分子、取り込み、分布及び/又は吸収を助けるための経口、経腸、局所又はそのほかの製剤のような化合物の混合物と混合されてもよく、内包されてもよく、共役されてもよく、さもなければ会合されてもよい。そのような取り込み、分布及び/又は吸収を助ける製剤の調製を記載する典型的な米国特許には、たとえば、米国特許第5,108,921 ; 5,354,844; 5,416,016; 5,459,127; 5,521 ,291 ; 5,543,158; 5,547,932; 5,583,020; 5,591 ,721; 4,426,330; 4,534,899; 5,013,556; 5,108,921; 5,213,804; 5,227,170; 5,264,221 ; 5,356,633; 5,395,619; 5,416,016; 5,417,978; 5,462,854; 5,469,854; , 5,512,295; 5,527,528; 5,534,259; 5,543,152; 5,556,948; 5,580,575; および5,595,756号が挙げられ、それぞれその全体を参考として本明細書に組み入れられる。
Oligonucleotide preparations and pharmaceutical compositions The oligonucleotides can be prepared into oligonucleotide preparations or pharmaceutical compositions. Thus, the present oligonucleotides can be used for other molecules, molecular structures or compounds such as liposomes, receptor targeting molecules, oral, enteral, topical or other formulations to aid uptake, distribution and / or absorption. It may be mixed with a mixture, encapsulated, conjugated, or otherwise associated. Typical U.S. patents describing the preparation of formulations that assist in such uptake, distribution and / or absorption include, for example, U.S. Pat.Nos. 5,108,921; 5,354,844; 5,416,016; 5,459,127; , 721; 4,426,330; 4,534,899; 5,013,556; 5,108,921; 5,213,804; 5,227,170; 5,264,221; 5,356,633; 5,395,619; 5,416,016; 5,417,978; 5,462,854; 5,469,854;, 5,512,295; 5,527,528; 5,534,259; 5,543,152; 5,556,948; 5,580,575; and No. 5,595,756 can be mentioned, respectively Which is incorporated herein by reference in its entirety.
本発明のオリゴヌクレオチド、製剤及び組成物は、ヒトを含む動物に投与する際、生物学的に活性のある代謝産物又はその残余を提供する(直接的に又は間接的に)することが可能である、薬学的に許容される塩、エステル又はそのようなエステルの塩を包含する。従って、たとえば、開示もまた、本発明の化合物のプロドラッグ及び薬学的に許容される塩、そのようなプロドラッグの薬学的に許容される塩、及びそのほかの生物等価物に引き付けられる。 The oligonucleotides, formulations and compositions of the present invention are capable of providing (directly or indirectly) biologically active metabolites or residues thereof when administered to animals, including humans. It includes certain pharmaceutically acceptable salts, esters or salts of such esters. Thus, for example, the disclosure is also drawn to prodrugs and pharmaceutically acceptable salts of the compounds of the invention, pharmaceutically acceptable salts of such prodrugs, and other bioequivalents.
用語「プロドラッグ」は、内因性の酵素の作用又はそのほかの化学物質及び/又は条件によって体内又は細胞内で活性形態(すなわち、薬剤)に変換される不活性形態で調製される治療剤を指す。特定の態様では、本オリゴヌクレオチドのプロドラッグ版は、その全体を参考として本明細書に組み入れられる、Gosselinらの国際公開公報第93/24510号及びImbachらの国際公開公報第94/26764号及び米国特許第5,770,713号に開示されている方法に従って、SATE[(S-アセチル-2-チオエチル)ホスフェート]誘導体として調製される。 The term “prodrug” refers to a therapeutic agent prepared in an inactive form that is converted into the active form (ie, drug) in the body or cells by the action of endogenous enzymes or other chemicals and / or conditions. . In certain embodiments, prodrug versions of the oligonucleotides are disclosed in Gosselin et al. WO 93/24510 and Imbach et al. WO 94/26764 and incorporated herein by reference in their entirety. Prepared as a SATE [(S-acetyl-2-thioethyl) phosphate] derivative according to the method disclosed in US Pat. No. 5,770,713.
用語「薬学的に許容される塩」は、本化合物の生理学的に、且つ薬学的に許容される塩、すなわち、母型化合物の所望の生物活性を保持し、且つ、望ましくない毒性効果をそれに付与しない塩を言う。そのような薬学的に許容される塩の多数は既知であり、本発明で使用することができる。 The term “pharmaceutically acceptable salt” refers to a physiologically and pharmaceutically acceptable salt of the compound, ie, retains the desired biological activity of the parent compound and imparts undesirable toxic effects thereto. Say salt that does not give. Many of such pharmaceutically acceptable salts are known and can be used in the present invention.
オリゴヌクレオチドについて、薬学的に許容される塩の有用な例には、たとえば、ナトリウム、カリウム、アンモニウム、マグネシウム、カルシウムのような陽イオンと共に形成される塩、スペルミン及びスペルミジンなどのようなポリアミン類;たとえば、塩酸、臭化水素酸、硫酸、リン酸、硝酸などのような無機酸と共に形成される酸付加塩;たとえば、酢酸、シュウ酸、酒石酸、コハク酸、マレイン酸、フマル酸、グルコン酸、クエン酸、リンゴ酸、アスコルビン酸、安息香酸、タンニン酸、パルミチン酸、アルギニン酸、ポリグルタミン酸、ナフタレンスルホン酸、メタンスルホン酸、p-トルエンスルホン酸、ナフタレンスルホン酸、ポリガラクツロン酸などのような有機酸と共に形成される塩;並びに塩素、臭素及びヨウ素のような元素陰イオンから形成される塩が挙げられるが、これらに限定されない。 For oligonucleotides, useful examples of pharmaceutically acceptable salts include, for example, salts formed with cations such as sodium, potassium, ammonium, magnesium, calcium, polyamines such as spermine and spermidine; For example, acid addition salts formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid; for example, acetic acid, oxalic acid, tartaric acid, succinic acid, maleic acid, fumaric acid, gluconic acid, Organics such as citric acid, malic acid, ascorbic acid, benzoic acid, tannic acid, palmitic acid, arginic acid, polyglutamic acid, naphthalenesulfonic acid, methanesulfonic acid, p-toluenesulfonic acid, naphthalenesulfonic acid, polygalacturonic acid Salts formed with acids; and like chlorine, bromine and iodine Salts formed from elemental anions but not limited thereto.
本発明はまた、本発明の抗ウイルス性オリゴヌクレオチドを含有する薬学的に許容される組成物及び製剤を包含する。局所治療又は全身性の治療が所望なのかどうか、及び治療すべき場所によって、多数の方法でそのような薬学的に許容される組成物を投与してもよい。たとえば、投与は、局所(眼並びに膣及び直腸への送達を含む粘膜)であってもよく;噴霧器による粉末又はエアゾールの吸入又は吸い込みによる肺へのものであってもよく;気管支内;鼻内;上皮及び経皮;経口;又は非経口であってもよい。非経口投与には、静脈内、動脈内、皮下、腹腔内、又は筋肉内の注射又は点滴;あるいはたとえば、くも膜下又は脳室内のような頭蓋内投与が挙げられる。 The invention also encompasses pharmaceutically acceptable compositions and formulations containing the antiviral oligonucleotides of the invention. Such pharmaceutically acceptable compositions may be administered in a number of ways depending upon whether local or systemic treatment is desired and upon the place to be treated. For example, administration may be local (mucosal including ocular and vaginal and rectal delivery); may be into the lungs by inhalation or inhalation of powder or aerosol by nebulizer; intrabronchial; intranasal Epithelial and transdermal; oral; or parenteral. Parenteral administration includes intravenous or intraarterial, subcutaneous, intraperitoneal, or intramuscular injection or infusion; or, for example, intracranial administration such as subarachnoid or intraventricular.
局所投与用の薬学的に許容される組成物及び製剤には、経皮貼付剤、軟膏、ローション、クリーム、ジェル、ドロップ、座薬、スプレー、液体及び粉末が挙げられる。従来の薬学的担体、水溶液、粉末又は油性の基剤、濃厚剤などが必要であってもよいし、望ましくてもよい。被覆したコンドーム、手袋なども有用である。好ましい局所用製剤には、本発明のオリゴヌクレオチドが、たとえば、脂質、リポソーム、脂肪酸、脂肪酸エステル、ステロイド、キレート剤、及び界面活性剤のような局所送達剤との混合剤であるようなものが挙げられる。好ましい脂質及びリポソームには、中性の(たとえば、ジオレオイルホスファチジルDOPEエタノールアミン、ジミリストイルホスファチジルコリンDMPC、ジステアロイルホスファチジルコリン)、陰性の(たとえば、ジミリストイルホスファチジルグリセロールDMPG)及び陽イオンの(たとえば、ジオレオイルテトラメチルアミノプロピルDOTAP及びジオレオイルホスファチジルエタノールアミンDOTMA)が挙げられる。オリゴヌクレオチドは、リポソーム内に内包されてもよいし、それとの、特に陽イオンのリポソームとの複合体を形成してもよい。又は、オリゴヌクレオチドは、脂質、特に陽イオン脂質と複合体を形成してもよい。好ましい脂肪酸及びエステル類には、アラキドン酸、オレイン酸、エイコサン酸、ラウリン酸、カプリル酸、カプリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、リノール酸、リノレン酸、ジカプレート、トリカプレート、モノオレイン、ジラウリン、グリセリル 1-モノカプレート、1-ドデシルアザシクロヘプタン-2-オン、アシルカルニチン、アシルコリン、若しくはC1〜10のアルキルエステル(たとえば、イソプロピルミリステ-トIPM)、モノグリセリド、ジグリセリド又はこれらの薬学的に許容される塩が挙げられるが、これらに限定されない。 Pharmaceutically acceptable compositions and formulations for topical administration include transdermal patches, ointments, lotions, creams, gels, drops, suppositories, sprays, liquids and powders. Conventional pharmaceutical carriers, aqueous solutions, powders or oily bases, thickeners and the like may be necessary or desirable. Coated condoms and gloves are also useful. Preferred topical formulations are those in which the oligonucleotides of the invention are admixtures with topical delivery agents such as lipids, liposomes, fatty acids, fatty acid esters, steroids, chelating agents, and surfactants. Can be mentioned. Preferred lipids and liposomes include neutral (eg dioleoyl phosphatidyl DOPE ethanolamine, dimyristoyl phosphatidylcholine DMPC, distearoyl phosphatidylcholine), negative (eg dimyristoyl phosphatidylglycerol DMPG) and cationic (eg di Oleoyltetramethylaminopropyl DOTAP and dioleoylphosphatidylethanolamine DOTMA). The oligonucleotide may be encapsulated within the liposome or may form a complex with it, particularly with a cationic liposome. Alternatively, the oligonucleotide may form a complex with a lipid, particularly a cationic lipid. Preferred fatty acids and esters include arachidonic acid, oleic acid, eicosanoic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurate, glyceryl 1-monocaprate, 1-dodecylazacycloheptan-2-one, acylcarnitines, acylcholines, or alkyl esters of C 1 to 10 (e.g., isopropyl myristate stearate - DOO IPM), monoglyceride, diglyceride, or a pharmaceutically Include, but are not limited to, chemically acceptable salts.
経口投与用の組成物及び製剤には、粉末又は顆粒、微粒子、ナノ粒子、水若しくは非水性媒体における懸濁液若しくは溶液、カプセル、ジェルカプセル、サック、錠剤又は微小錠剤が挙げられる。濃厚剤、香味剤、希釈剤、乳化剤、分散助剤又は結合剤が望ましくてもよい。好ましい経口製剤は、本発明のオリゴヌクレオチドが1以上の浸透増強剤界面活性剤及びキレート剤と合わせて混合されるものである。典型的な界面活性剤には、脂肪酸及び/又はそのエステル類又は塩、胆汁酸及び/又はその塩が挙げられる。典型的な胆汁酸/塩には、ケノデオキシコール酸(CDCA)、及びウルソデオキシケネデオキシコール酸(UDCA)、コール酸、デヒドロコール酸、デオキシコール酸、グルコール酸、グリコール酸、グリコデオキシコール酸、タウロコール酸、タウロデオキシコール酸、タウロ-24,25-ジヒドロ-フシジン酸ナトリウム、グリコジヒドロフシジン酸ナトリウムが挙げられる。典型的な脂肪酸には、アラキドン酸、ウンデカン酸、オレイン酸、ラウリル酸、カプリル酸、カプリン酸、ミリスチル酸、パルミチン酸、ステアリン酸、リノール酸、リノレン酸、ジカプレート、トリカプレート、モノオレイン、ジラウリン、グリセリル1-モノカプレート、1-ドデシルアザシクロヘプタン-2-オン、アクリルカルニチン、アクリルコリン、モノグリセリド、ジグリセリド又はこれらの薬学的に許容される塩(たとえば、ナトリウム)が挙げられる。また、好ましいのは、浸透増強剤の併用、たとえば、胆汁酸/塩と併用する脂肪酸/塩である。特に好ましい併用は、ラウリル酸、カプリン酸及びUDCAのナトリウム塩である。さらに典型的な浸透増強剤には、ポリオキシエチレン-9-ラウリルエーテル、ポリオキシエチレン-20-セチルエーテルが挙げられる。本発明のオリゴヌクレオチドは、スプレー乾燥した粒子を含む顆粒形態で経口的に送達してもよいし、又は複合化して微粒子又はナノ粒子を形成してもよい。オリゴヌクレオチド錯化剤には、ポリアミノ酸;ポリイミン類;ポリアクリレート類;ポリアルキルアクリレート類、ポリオキシエタン類、ポリアルキルシアノアクリレート類;カチオン化ゼラチン、アルブミン、デンプン、アクリレート類、ポリエチレングリコール(PEG)及びデンプン;ポリアルキルシアノアクリレート類;DEAE-誘導体化ポリイミン類、ポルラン類、セルロース及びデンプンが挙げられる。特に有利な錯化剤には、キトサン、N-トリメチルキトサン、ポリ-L-リジン、ポリヒスチジン、ポリオルニチン、ポリスペルミン、プロタミン、ポリビニルピリジン、ポリチオジエチルアミノメチルエチレンP(TDAE)、ポリアミノスチレン(たとえば、p-アミノ)、ポリ(メチルシアノアクリレート)、ポリ(エチルシアノアクリレート)、ポリ(ブチルシアノアクリレート)、ポリ(イソブチルシアノアクリレート)、ポリ(イソヘキシルシアノアクリレート)、DEAE-メタクリレート、DEAE-ヘキシルアクリレート、DEAE-アクリルアミド、DEAE-アルブミン及びDEAE-デキストラン、ポリメチルアクリレート、ポリヘキシルアクリレート、ポリ(D,L-乳酸)、ポリ(DL-乳酸-コ-グリコール酸(PLGA)、アルギネート並びにポリエチレングリコール(PEG)が挙げられる。 Compositions and formulations for oral administration include powders or granules, microparticles, nanoparticles, suspensions or solutions in water or non-aqueous media, capsules, gel capsules, sacks, tablets or microtablets. Thickeners, flavorings, diluents, emulsifiers, dispersion aids or binders may be desirable. A preferred oral formulation is one in which the oligonucleotide of the invention is mixed with one or more penetration enhancer surfactants and chelating agents. Typical surfactants include fatty acids and / or esters or salts thereof, bile acids and / or salts thereof. Typical bile acids / salts include chenodeoxycholic acid (CDCA) and ursodeoxy chenedodecholic acid (UDCA), cholic acid, dehydrocholic acid, deoxycholic acid, glucholic acid, glycolic acid, glycodeoxycholic acid, taurochol Examples include acids, taurodeoxycholic acid, sodium tauro-24,25-dihydro-fusidate, sodium glycodihydrofusidate. Typical fatty acids include arachidonic acid, undecanoic acid, oleic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin Glyceryl 1-monocaprate, 1-dodecylazacycloheptan-2-one, acrylic carnitine, acrylic choline, monoglyceride, diglyceride or a pharmaceutically acceptable salt thereof (for example, sodium). Also preferred are combinations of penetration enhancers, such as fatty acids / salts used in combination with bile acids / salts. Particularly preferred combinations are lauric acid, capric acid and UDCA sodium salt. Further typical penetration enhancers include polyoxyethylene-9-lauryl ether, polyoxyethylene-20-cetyl ether. The oligonucleotides of the invention may be delivered orally in the form of granules containing spray-dried particles or may be conjugated to form microparticles or nanoparticles. Oligonucleotide complexing agents include polyamino acids; polyimines; polyacrylates; polyalkyl acrylates, polyoxyethanes, polyalkyl cyanoacrylates; cationized gelatin, albumin, starch, acrylates, polyethylene glycol (PEG) And polyalkylcyanoacrylates; DEAE-derivatized polyimines, porlans, cellulose and starch. Particularly advantageous complexing agents include chitosan, N-trimethylchitosan, poly-L-lysine, polyhistidine, polyornithine, polyspermine, protamine, polyvinylpyridine, polythiodiethylaminomethylethylene P (TDAE), polyaminostyrene (for example , P-amino), poly (methyl cyanoacrylate), poly (ethyl cyanoacrylate), poly (butyl cyanoacrylate), poly (isobutyl cyanoacrylate), poly (isohexyl cyanoacrylate), DEAE-methacrylate, DEAE-hexyl acrylate , DEAE-acrylamide, DEAE-albumin and DEAE-dextran, polymethyl acrylate, polyhexyl acrylate, poly (D, L-lactic acid), poly (DL-lactic acid-co-glycolic acid (PLGA), alginate and polyethylene glycol (PEG) ) It is.
経膣送達のための組成物は、例えば、ジェル、クリーム、錠剤、丸剤、カプセル、坐薬、フィルム、または粘膜に接着し容易に洗浄除去されない任意の他の薬学的に許容される形態を含む様々な形態を有し得る。更に経膣送達のための非常に多様な異なる製剤が、当該技術分野にて、例えば米国特許第4,615,697号および米国特許第6,699,494号に記載されており、これらはその全体が参照により本明細書に組み入れられる。 Compositions for vaginal delivery include, for example, gels, creams, tablets, pills, capsules, suppositories, films, or any other pharmaceutically acceptable form that adheres to the mucosa and is not easily washed away. It can have various forms. In addition, a great variety of different formulations for vaginal delivery are described in the art, for example in US Pat. No. 4,615,697 and US Pat. No. 6,699,494, which are hereby incorporated by reference in their entirety. Be incorporated.
加えて、添加物(特許第4,615,697号に記載されているような)を、送達系の最大のもしくは所望の効力のために、または患者の心地よさのために、製剤中に組み合わせてもよい。そのような添加物は、例えば、滑沢剤、可塑化剤、保存剤、ゲル形成剤、錠剤形成剤、丸剤形成剤、坐薬形成剤、フィルム形成剤、クリーム形成剤、錠剤崩壊剤、コーティング、結合剤、ビヒクル、着色剤、味覚および/または香り調整剤、湿潤剤、粘度調節剤、pH調整剤、ならびに同様の薬剤を含む。 In addition, additives (such as those described in US Pat. No. 4,615,697) may be combined in the formulation for maximum or desired efficacy of the delivery system or for patient comfort. Such additives include, for example, lubricants, plasticizers, preservatives, gel forming agents, tablet forming agents, pill forming agents, suppository forming agents, film forming agents, cream forming agents, tablet disintegrating agents, coatings. , Binders, vehicles, colorants, taste and / or fragrance modifiers, wetting agents, viscosity modifiers, pH modifiers, and similar agents.
所定の態様において、組成物は、一般に米国特許第4,615,697号に記載されている架橋ポリカルボン酸ポリマー調合物を含み得る。一般に、このような態様において、そのような調合物中のポリマーの単量体の少なくとも80パーセントは、少なくとも1個のカルボキシル官能基を含んでいるはずである。架橋剤は、十分な生体接着を提供して、系が十分な時間、標的上皮表面に取り付けられた状態を保ち、所望の投薬が行われる量で存在する必要がある。 In certain embodiments, the composition may comprise a crosslinked polycarboxylic acid polymer formulation generally described in US Pat. No. 4,615,697. In general, in such embodiments, at least 80 percent of the monomers of the polymer in such formulations should contain at least one carboxyl functional group. The cross-linking agent must be present in an amount that provides sufficient bioadhesion to keep the system attached to the target epithelial surface for a sufficient amount of time and to achieve the desired dosage.
経膣投与に関して、このような製剤は、上皮表面に少なくとも約24〜48時間、取り付けられた状態を保つ。これらの結果は、ポリマーの連続的存在による膣からのサンプルのpH低下を試験することにより、様々な期間を通して臨床的に測定してもよい。この生体接着の好ましいレベルは、通常、架橋剤がポリマーの約0.1〜6.0重量パーセントにて存在する場合に得られ、適切なレベルの生体接着が得られる限り、約1.0〜2.0重量パーセントが最も好ましい。生体接着は、接着強度の測定に使用される市販の表面張力計を用いても測定することができる。 For vaginal administration, such formulations remain attached to the epithelial surface for at least about 24-48 hours. These results may be measured clinically over various time periods by testing the pH drop of the sample from the vagina due to the continuous presence of the polymer. This preferred level of bioadhesion is usually obtained when the crosslinker is present at about 0.1 to 6.0 weight percent of the polymer, with about 1.0 to 2.0 weight percent being most preferred as long as an adequate level of bioadhesion is obtained. . Bioadhesion can also be measured using a commercially available surface tensiometer used for measuring adhesive strength.
ポリマー調合物は、ポリマー中の架橋剤の量を変動させることにより、放出速度を制御するよう調整することができる。適切な架橋剤は、ジビニルグリコール、ジビニルベンゼン、N,N-ジアリルアクリルアミド、3,4-ジヒドロキシ-1,5-ヘキサジエン、2,5-ジメチル-1,5-ヘキサジエンおよび同様の薬剤を含む。 The polymer formulation can be adjusted to control the release rate by varying the amount of crosslinker in the polymer. Suitable crosslinkers include divinyl glycol, divinyl benzene, N, N-diallylacrylamide, 3,4-dihydroxy-1,5-hexadiene, 2,5-dimethyl-1,5-hexadiene and similar agents.
このような調合物に使用する好ましいポリマーは、商品名NOVEON.RTM.-AA1の下でB. F. Goodrich Speciality Polymers of Cleveland, Ohioから市販されているポリカルボフィル(Polycarbophil)、U.S.P.である。米国薬局方、1995版、United States Pharmacopeial Convention、Inc., Rockville, Md.の1240〜41ページには、ポリカルボフィルは、ジビニルグリコールと架橋したポリアクリル酸であることが示されている。 A preferred polymer for use in such formulations is Polycarbophil, U.S.P., commercially available from BF Goodrich Specialty Polymers of Cleveland, Ohio under the trade name NOVEON.RTM.-AA1. In the United States Pharmacopeia, 1995, United States Pharmacopeial Convention, Inc., Rockville, Md., Pages 1240-41, it is shown that polycarbophil is polyacrylic acid crosslinked with divinyl glycol.
このような薬物送達系の製剤に使用できる他の有用な生体接着性ポリマーは、特許第4,615,697号に言及されている。例えば、これらは、例えば3,4-ジヒドロキシ-1,5-ヘキサジエンと架橋したポリアクリル酸ポリマー、および例えば、ジビニルベンゼンと架橋したポリメタクリルポリマーを含む。典型的には、これらのポリマーは、生体接着能力を低下させることから、その塩形態では使用されないであろう。このような生体接着性ポリマーは、例えば過酸化ベンゾイル、アゾビスイソブチロニトリル等のような開始剤を用いた、従来の遊離ラジカル重合法により製造され得る。有用な生体接着剤の例示的な製剤は、特許第4,615,697号に提供されている。 Other useful bioadhesive polymers that can be used in such drug delivery system formulations are mentioned in US Pat. No. 4,615,697. For example, these include polyacrylic acid polymers crosslinked with, for example, 3,4-dihydroxy-1,5-hexadiene, and polymethacrylic polymers crosslinked with, for example, divinylbenzene. Typically, these polymers will not be used in their salt form because they reduce bioadhesive ability. Such bioadhesive polymers can be produced by conventional free radical polymerization methods using initiators such as benzoyl peroxide, azobisisobutyronitrile, and the like. An exemplary formulation of a useful bioadhesive is provided in US Pat. No. 4,615,697.
非経口、くも膜下又は脳室内の投与のための組成物及び製剤は、緩衝液、希釈液及び、たとえば、浸透増強剤、担体化合物及びそのほかの薬学的に許容される担体又は賦形剤に限定されない添加物も含有してもよい、無菌の水溶液を包含してもよい。 Compositions and formulations for parenteral, intrathecal or intraventricular administration are limited to buffers, diluents and, for example, penetration enhancers, carrier compounds and other pharmaceutically acceptable carriers or excipients. Sterile aqueous solutions may also be included that may also contain additives that are not.
本発明の薬学的組成物は、溶液、エマルション、及びリポソーム含有製剤を包含してもよいが、これらに限定されない。これらの組成物は、予備形成された液体、自己乳化する固体及び自己乳化する半固体を含むが、これらに限定されない種々の成分から生成してもよい。 The pharmaceutical compositions of the present invention may include, but are not limited to, solutions, emulsions, and liposome-containing formulations. These compositions may be formed from a variety of ingredients including, but not limited to, preformed liquids, self-emulsifying solids and self-emulsifying semisolids.
都合良く、単位投与形態で提示してもよい本発明の薬学的製剤は、製薬業界で周知の従来技術に従って調製してもよい。そのような技術は、薬学的担体又は賦形剤と共に有効成分を会合させる工程を包含する。一般に、製剤は、有効成分を液体担体又は微細に分割した固体担体又はその双方と均一且つ密接に会合させ、必要に応じて生成物を振盪することによって調製される。 Conveniently, the pharmaceutical formulations of the invention that may be presented in unit dosage form may be prepared according to conventional techniques well known in the pharmaceutical industry. Such techniques include the step of bringing into association the active ingredient with the pharmaceutical carrier or excipient. In general, the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and, if necessary, shaking the product.
本発明の組成物は、錠剤、カプセル、ジェルカプセル、液体シロップ、軟ジェル、座薬及び浣腸を含むが、これらに限定されない多数の可能な投与形態に製剤化してもよい。本発明の組成物はまた、水性、非水性又はその混合の媒体中での懸濁液として製剤化されてもよい。水性懸濁液はさらに、たとえば、カルボキシメチルセルロースナトリウム、ソルビトール及び/又はデキストランを含む、懸濁液の粘度を高める物質を含有してもよい。懸濁液はまた安定剤も含有してもよい。 The compositions of the invention may be formulated into a number of possible dosage forms including, but not limited to, tablets, capsules, gel capsules, liquid syrups, soft gels, suppositories, and enemas. The compositions of the present invention may also be formulated as suspensions in aqueous, non-aqueous or mixed media. Aqueous suspensions may further contain substances that increase the viscosity of the suspension, including, for example, sodium carboxymethylcellulose, sorbitol and / or dextran. The suspension may also contain stabilizers.
本発明の態様の1つでは、薬学的組成物は発泡体として製剤化し、使用する。薬学的発泡体には、エマルション、微細エマルション、クリーム、ゼリー及びリポソームのような剤形が挙げられるが、これらに限定されない。性質では基本的に類似しているものの、これらの剤形は、最終産物の成分及び濃度において異なる。そのような組成物及び製剤の調製は一般に、製薬及び製剤技術の当業者に知られており、本発明の組成物の製剤に適用してもよい。 In one embodiment of the invention, the pharmaceutical composition is formulated and used as a foam. Pharmaceutical foams include, but are not limited to, dosage forms such as emulsions, microemulsions, creams, jellies and liposomes. Although fundamentally similar in nature, these dosage forms differ in the components and concentrations of the final product. The preparation of such compositions and formulations is generally known to those skilled in the pharmaceutical and formulation arts and may be applied to the formulation of the compositions of the present invention.
エマルション
本発明の製剤及び組成物は、エマルションとして調製し、製剤化してもよい。エマルションは、普通、直径0.1μmを超える液滴の形態で、一方の液体が他方に分散された通常、不均質システムである。(Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (lids.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199; Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., Volume 1, p. 245; Block in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 2, p. 335; Higuchi et at., in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 301)。エマルションは、互いに密接に混合され分散された非混和性の液相を含む二相性システムであることが多い。一般に、エマルションは、油中水型(w/o)又は水中油型(o/w)型のいずれかであってもよい。水性相が微細な液滴として大量の油相に細かく分割され、分散されている場合、得られる組成物は油中水(w/o)エマルションと呼ばれる。又は、油相が微細な液滴として大量の水性相に細かく分割され、分散されている場合、得られる組成物は水中油(o/w)エマルションと呼ばれる。エマルションは、分散相及び、水性相、油相に液体として存在する有効薬剤又は分離相としてのそれ自体に加えて、追加の成分を含有してもよい。乳化剤、安定剤、染料及び抗酸化剤のような薬学的な賦形剤も、必要に応じてエマルションに存在してもよい。薬学的エマルションは、たとえば、油中水中油(o/w/o)及び水中油中水(w/o/w)のような二相より多くから構成される複数のエマルションであってもよい。そのような複雑な製剤は、単純な二相性エマルションが提供しない、特定の利点を提供することが多い。o/wエマルションの個々の油性液滴が小さな水性液滴を内包する複数のエマルションがw/o/wエマルションを構成する。同様に、油性連続相で安定化された水の小球に内包された油性液滴のシステムがo/w/oエマルションを提供する。
Emulsion The formulations and compositions of the present invention may be prepared as an emulsion and formulated. Emulsions are usually heterogeneous systems, usually in the form of droplets with a diameter greater than 0.1 μm, with one liquid dispersed in the other. (Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (lids.), 1988, Marcel Dekker, Inc., New York, NY, volume 1, p. 199; Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, NY, Volume 1, p. 245; Block in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, NY, volume 2, p. 335; Higuchi et at., In Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 301). Emulsions are often biphasic systems that contain immiscible liquid phases that are intimately mixed and dispersed with each other. In general, emulsions may be either water-in-oil (w / o) or oil-in-water (o / w) types. When the aqueous phase is finely divided and dispersed into a large amount of oil phase as fine droplets, the resulting composition is called a water-in-oil (w / o) emulsion. Alternatively, if the oil phase is finely divided and dispersed into a large amount of aqueous phase as fine droplets, the resulting composition is called an oil-in-water (o / w) emulsion. Emulsions may contain additional components in addition to the dispersed phase and the active agent present as a liquid in the aqueous phase, oil phase or itself as a separate phase. Pharmaceutical excipients such as emulsifiers, stabilizers, dyes and antioxidants may also be present in the emulsion if desired. The pharmaceutical emulsion may be a plurality of emulsions composed of more than two phases such as, for example, oil-in-water-in-oil (o / w / o) and water-in-oil-in-water (w / o / w). Such complex formulations often offer certain advantages that simple biphasic emulsions do not provide. Multiple emulsions in which individual oil droplets of an o / w emulsion enclose small aqueous droplets constitute a w / o / w emulsion. Similarly, a system of oily droplets encapsulated in water globules stabilized with an oily continuous phase provides an o / w / o emulsion.
エマルションは、熱力学的安定性がほとんどないか、全くないことを特徴とする。エマルションの分散された又は不連続の相が、製剤の乳化剤又は粘度の手段を介して、外部の又は連続した相に上手く分散され、この形態で維持されることが多い。エマルション型の軟膏基剤やクリ-ムの場合のように、エマルションのいずれかの相が半固体又は固体であってもよい。エマルションを安定化するそのほかの手段は、エマルションのいずれかの相に組み入れられてもよい乳化剤の使用を伴う。乳化剤は、おおまかに4つのカテゴリー:合成の界面活性剤、天然に存在する乳化剤、吸収基剤及び微細に分散された固体に分類してもよい(Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, N.Y., volume 1 p.199)。 Emulsions are characterized by little or no thermodynamic stability. Often the dispersed or discontinuous phase of the emulsion is well dispersed and maintained in this form via the emulsifier or viscosity means of the formulation to the external or continuous phase. As in the case of an emulsion type ointment base or cream, any phase of the emulsion may be semi-solid or solid. Another means of stabilizing the emulsion involves the use of emulsifiers that may be incorporated into any phase of the emulsion. Emulsifiers may be broadly classified into four categories: synthetic surfactants, naturally occurring emulsifiers, absorbent bases and finely dispersed solids (Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker ( Eds.), 1988, Marcel Dekker Inc., New York, NY, volume 1 p.199).
表面活性化剤としても知られる合成界面活性剤は、エマルションの配合において広い適用性が見い出されており、文献(Rieger, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 285; Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), Marcel Dekker, Inc., New York, N.Y., 1988, volume 1, p. 199)で概説されている。界面活性剤は、通常、両親媒性であり、親水性部分及び疎水性部分を含む。界面活性剤の親水性の性質と疎水性の性質の比は、親水性/親油性バランス(HLB)と呼ばれ、製剤の調製で界面活性剤を分類し、選択する有益なツールである。界面活性剤は、親水性基の性質:非イオン性、陰イオン性、陽イオン性及び両性イオン性に基づいて異なる部類に分類してもよい(Rieger, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, N.Y., Vol.1 p.285)。 Synthetic surfactants, also known as surface activators, have found wide applicability in emulsion formulations and have been described in the literature (Rieger, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, NY, volume 1, p. 285; Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), Marcel Dekker, Inc., New York, NY, 1988, volume 1, p. 199 ). Surfactants are usually amphiphilic and include a hydrophilic portion and a hydrophobic portion. The ratio between the hydrophilic and hydrophobic properties of a surfactant is called the hydrophilic / lipophilic balance (HLB) and is a valuable tool for classifying and selecting surfactants in the preparation of a formulation. Surfactants may be classified into different classes based on the nature of the hydrophilic group: nonionic, anionic, cationic and zwitterionic (Rieger, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker ( Eds.), 1988, Marcel Dekker Inc., New York, NY, Vol. 1 p.285).
エマルション配合で使用される天然に存在する乳化剤には、ラノリン、ハチミツ、ホスファチド、レシチン及びアカシアが挙げられる。吸収基剤は、無水ラノリンや親水性ワセリンのような半固体の一貫性を保持するけれども、水を吸収してw/oエマルションを形成するように親水性特性を持つ。微細に分割された固体も特に、界面活性剤との併用で、及び種々の調製物中で良好な乳化剤として使用されている。これらには、重金属水酸化物のような極性無機固体、ベントナイト、アタパルガイト、ヘクトライト、カオリン、モントモリロナイトのような非膨張粘土、コロイド状珪酸アルミニウム及びコロイド状珪酸マグネシウムアルミニウム、色素、及び炭素又はトリステアリン酸グリセリルのような非極性固体が挙げられる。 Naturally occurring emulsifiers used in emulsion formulations include lanolin, honey, phosphatide, lecithin and acacia. Absorbent bases retain the properties of semisolids such as anhydrous lanolin and hydrophilic petrolatum, but have hydrophilic properties to absorb water and form w / o emulsions. Finely divided solids are also used in particular in combination with surfactants and as good emulsifiers in various preparations. These include polar inorganic solids such as heavy metal hydroxides, non-expanded clays such as bentonite, attapulgite, hectorite, kaolin, montmorillonite, colloidal aluminum silicate and colloidal magnesium aluminum silicate, pigments, and carbon Or a non-polar solid such as glyceryl tristearate.
多種多様な非乳化材料がエマルション製剤に包含され、エマルションの特性に寄与する。これらには、脂肪、油、ワックス、脂肪酸、脂肪アルコール類、脂肪エステル類、湿潤剤、親水性コロイド類、防腐剤及び抗酸化剤が挙げられる(Block, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 335; Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199)。 A wide variety of non-emulsifying materials are included in the emulsion formulation and contribute to the properties of the emulsion. These include fats, oils, waxes, fatty acids, fatty alcohols, fatty esters, wetting agents, hydrophilic colloids, preservatives and antioxidants (Block, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker ( Eds.), 1988, Marcel Dekker, Inc., New York, NY, volume 1, p. 335; Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York , NY, volume 1, p. 199).
親水性コロイド類又は親水コロイド類には、天然に存在するゴム及び、多糖類のような合成ポリマー類(たとえば、アカシア、寒天、アルギン酸、カラギーナン、グアーゴム、カラヤゴム及びトラガカント)、セルロース誘導体(たとえば、カルボキシメチルセルロース及びカルボキシプロピルセルロース)、及び合成ポリマー類(たとえば、カルボマー類、セルロースエーテル類及びカルボキシビニルポリマー類)が挙げられる。これらは水に分散し、又は水中で膨潤し、分散相液滴の回りに強力な表面間の被膜を形成し、内部相の粘度を高めることによってエマルションを安定化するコロイド溶液を形成する。 Hydrophilic colloids or hydrocolloids include naturally occurring rubbers and synthetic polymers such as polysaccharides (eg acacia, agar, alginic acid, carrageenan, guar gum, karaya gum and tragacanth), cellulose derivatives (eg carboxy Methylcellulose and carboxypropylcellulose), and synthetic polymers such as carbomers, cellulose ethers and carboxyvinyl polymers. They disperse in water or swell in water, forming a strong inter-surface coating around the dispersed phase droplets and forming a colloidal solution that stabilizes the emulsion by increasing the viscosity of the internal phase.
エマルションは、微生物の増殖を容易に支える炭水化物、タンパク質、ステロール及びホスファチドのような多数の成分を含有することが多いので、これらエマルションには防腐剤を組み入れることが多い。エマルション製剤に包含され一般に使用される防腐剤には、メチルパラベン、プロピルパラベン、四級アンモニウム塩、塩化ベンザルコニウム、p-ヒドロキシ安息香酸のエステル類、及びホウ酸が挙げられる。抗酸化剤も一般にエマルション製剤に添加され、製剤の劣化を防止する。使用される抗酸化剤は、トコフェロール類、没食子酸アルキル、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエンのような遊離のラジカルスカベンジャー、又はアスコルビン酸及びメタ重亜硫酸ナトリウムのような還元剤、並びにクエン酸、酒石酸及びレシチンのような抗酸化共力剤であってもよい。 Because emulsions often contain a number of ingredients such as carbohydrates, proteins, sterols and phosphatides that readily support microbial growth, these emulsions often incorporate preservatives. Preservatives that are included and commonly used in emulsion formulations include methylparaben, propylparaben, quaternary ammonium salts, benzalkonium chloride, esters of p-hydroxybenzoic acid, and boric acid. Antioxidants are also commonly added to emulsion formulations to prevent degradation of the formulation. Antioxidants used include tocopherols, alkyl gallate, butylated hydroxyanisole, free radical scavengers such as butylated hydroxytoluene, or reducing agents such as ascorbic acid and sodium metabisulfite, and citric acid, Antioxidant synergists such as tartaric acid and lecithin may also be used.
皮膚、経口及び非経口の経路を介したエマルション製剤の適用並びにその製造方法は、文献に概説されている(Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, N.Y., Vol.1 p.199)。容易な製剤化、吸収及び生物利用性の観点からの有効性のために経口送達のためのエマルション製剤は非常に広く使用されている(Rosoff, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1 , p. 245; Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1 , p. 199)。鉱物油系の下剤、油溶性ビタミン及び高脂肪栄養剤は、o/wエマルションとして一般に経口で投与される物質のうちである。 The application of emulsion formulations via the dermal, oral and parenteral routes and methods for their preparation have been reviewed in the literature (Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc. , New York, NY, Vol.1 p.199). Emulsion formulations for oral delivery are very widely used because of their ease of formulation, absorption and bioavailability (Rosoff, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.) , 1988, Marcel Dekker, Inc., New York, NY, volume 1, p. 245; Idson, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, NY, volume 1, p. 199). Mineral oil-based laxatives, oil-soluble vitamins and high fat nutrients are among the substances commonly administered orally as o / w emulsions.
本発明の態様の1つでは、オリゴヌクレオチドの組成物がマイクロエマルションとして製剤化される。マイクロエマルションは、光学的に単一の等方性で、熱力学的に安定な溶液である、水、油及び両親媒性物質の系として定義されてもよい(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, N.Y., volume 1, p245)。通常、先ず、水性の界面活性剤溶液に油を分散し、次いで、十分量の第4の成分、一般的には中間鎖長のアルコールを加えて透明な系を形成することによりマイクロエマルションを調製する。従って、マイクロエマルションは、表面活性化分子の表面間被膜により安定化される2つの非混和性の液の熱力学的に安定な、等方的に透明な分散液としても記載されている(Leung and Shah, in Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., Ed., VCH Publishers, New York, p.185-215, 1989)。一般に、油、水、界面活性剤、界面活性剤共力剤及び電解質を含む3〜5つの成分の組み合わせによってマイクロエマルションを調製する。マイクロエマルションが油中水(w/o)型であるか、水中油(o/w)型であるのかは、使用する油及び界面活性剤の特性、及び界面活性剤分子の極性頭部及び炭化水素の尾部の構造及び幾何的詰め込みに依存する(Schott, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., p.271, 1985)。 In one embodiment of the invention, the oligonucleotide composition is formulated as a microemulsion. A microemulsion may be defined as a system of water, oil and amphiphile, which is an optically single isotropic, thermodynamically stable solution (Rosoff, in Pharmaceutical Dosage Forms, Lieberman Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, NY, volume 1, p245). Usually, a microemulsion is prepared by first dispersing the oil in an aqueous surfactant solution and then adding a sufficient amount of a fourth component, typically an intermediate chain length alcohol, to form a transparent system. To do. Thus, microemulsions are also described as thermodynamically stable, isotropically transparent dispersions of two immiscible liquids that are stabilized by an intersurface coating of surface-activating molecules (Leung and Shah, in Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., Ed., VCH Publishers, New York, p.185-215, 1989). In general, microemulsions are prepared by a combination of 3 to 5 components including oil, water, surfactant, surfactant synergist and electrolyte. Whether the microemulsion is a water-in-oil (w / o) or oil-in-water (o / w) type depends on the properties of the oil and surfactant used and the polar head and carbonization of the surfactant molecule. Depends on the structure and geometric packing of the hydrogen tail (Schott, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., P.271, 1985).
位相図を利用した現象論的アプローチが鋭意検討され、マイクロエマルションの製剤化方法の包括的な知識が当業者に与えられてきた(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245; Block, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 335)。従来のエマルションに比べて、マイクロエマルションは、自然に形成される熱力学的に安定な液滴形成物における水不溶性薬剤の可溶化という利点を提供する。 A phenomenological approach using phase diagrams has been extensively studied and comprehensive knowledge of microemulsion formulation methods has been given to those skilled in the art (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.) , 1988, Marcel Dekker, Inc., New York, NY, volume 1, p. 245; Block, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, NY , volume 1, p. 335). Compared to conventional emulsions, microemulsions offer the advantage of solubilization of water-insoluble drugs in naturally formed thermodynamically stable droplet formers.
マイクロエマルションの調製で使用される界面活性剤には、イオン性界面活性剤、非イオン性界面活性剤、Brij96、ポリオキシエチレンオレイルエーテル類、ポリグリセロール脂肪酸エステル類、単独又は界面活性剤共力剤との組み合わせで、テトラグリセロールモノラウレート(ML310)、テトラグリセロールモノオレート(MO310)、ヘキサグリセロールモノオレート(PO310)、ヘキサグリセロールペンタオレート(PO500)、デカグリセロールモノカプレート(MCA750)、デカグリセロールモノオレート(MO750)、デカグリセロールセスキオレート(SO750)デカグリセロールデカオレート(DA0750)が挙げられるが、これらに限定されない。界面活性剤共力剤は、通常、エタノール、1-プロパノール及び1-ブタノールのような短鎖アルコールであり、界面活性剤分子の間で生じる空隙のために、界面活性剤被膜に浸透し、その結果、無秩序の被膜を創製することによって表面間の流動性を高めるように作用する。しかしながら、マイクロエマルションは、界面活性剤共力剤を使用せずに調製されてもよく、アルコールを含まない自己乳化エマルションの系は当該技術で既知である。水性相は、通常、水、薬剤の水溶液、グリセロール、PEG300、PEG400、ポリグリセロール類、プロピレングリコール、及びエチレングリコール誘導体であってもよい。油相は、カプテックス300、カプテックス355、カプムールMCM、脂肪酸エステル、中鎖(C8〜C12)モノ、ジ、及びトリグリセリド、ポリオキシエチル化グリセリル脂肪酸エステル、脂肪アルコール、ポリグリコール化グリセリド、飽和ポリグリコール化C8〜C10のグリセリド、植物油及びシリコーン油を挙げてもよいが、これらに限定されない。 Surfactants used in the preparation of microemulsions include ionic surfactants, nonionic surfactants, Brij96, polyoxyethylene oleyl ethers, polyglycerol fatty acid esters, alone or surfactant synergists In combination with tetraglycerol monolaurate (ML310), tetraglycerol monooleate (MO310), hexaglycerol monooleate (PO310), hexaglycerol pentaoleate (PO500), decaglycerol monocaprate (MCA750), decaglycerol monooleate (MO750), decaglycerol sesquiolate (SO750) decaglycerol dekaorate (DA0750), but not limited thereto. Surfactant synergists are usually short chain alcohols such as ethanol, 1-propanol and 1-butanol, which penetrate the surfactant coating due to voids created between the surfactant molecules, As a result, it acts to increase the fluidity between the surfaces by creating a disordered coating. However, microemulsions may be prepared without the use of surfactant synergists, and alcohol-free self-emulsifying emulsion systems are known in the art. The aqueous phase may usually be water, aqueous drug solutions, glycerol, PEG300, PEG400, polyglycerols, propylene glycol, and ethylene glycol derivatives. The oil phase consists of Captex 300, Captex 355, Capmul MCM, fatty acid esters, medium chain (C8-C12) mono, di and triglycerides, polyoxyethylated glyceryl fatty acid esters, fatty alcohols, polyglycolized glycerides, saturated poly Examples may include, but are not limited to, glycolated C8-C10 glycerides, vegetable oils and silicone oils.
マイクロエマルションは、薬剤の可溶化及び薬剤の吸収の向上の観点から特に興味深い。脂質系マイクロエマルション(o/w及びw/oの双方)は、ペプチドを含む薬剤の経口の生物利用性を高めることが提案されている(Constantinides et al., Pharmaceutical Research, 11:1385-1390, 1994; Ritschet, Met/i. Find. Exp. Clin. Pharmacol., 13:205, 1993)。マイクロエマルションは、薬剤可溶化の改善、酵素的加水分解からの薬剤の保護、膜の流動性及び透過性における界面活性剤が誘発する変化による薬剤吸収の向上の可能性、容易な調製、固形投与形態での容易な経口投与、臨床的効能の改善及び毒性の低下という利点を提供する(Constantinides et al., Pharmaceutical Research, 11:1385, 1994; Ho et al., J. Pharm. Set., 85:138-143, 1996)。その成分を常温で一緒にするとマイクロエマルションが自然に形成してもよいことが多い。熱不安定性の薬剤、ペプチド又はオリゴヌクレオチドを配合する際、これは特に有利であってもよい。マイクロエマルションは、化粧品への適用及び薬学的な適用の双方において有効成分の経皮送達にも有効である。本発明のマイクロエマルション組成物及び製剤は、消化管、膣、口腔及び投与のそのほかの場所におけるオリゴヌクレオチド及び核酸の局所の細胞取り込みを改善すると共に、消化管からのオリゴヌクレオチド及び核酸の全身性の吸収を促進することが期待される。 Microemulsions are of particular interest in terms of drug solubilization and improved drug absorption. Lipid-based microemulsions (both o / w and w / o) have been proposed to enhance the oral bioavailability of drugs containing peptides (Constantinides et al., Pharmaceutical Research, 11: 1385-1390, 1994; Ritschet, Met / i. Find. Exp. Clin. Pharmacol., 13: 205, 1993). Microemulsions improve drug solubilization, protect drugs from enzymatic hydrolysis, potentially improve drug absorption by surfactant-induced changes in membrane fluidity and permeability, easy preparation, solid administration Provides the benefits of easy oral administration in form, improved clinical efficacy and reduced toxicity (Constantinides et al., Pharmaceutical Research, 11: 1385, 1994; Ho et al., J. Pharm. Set., 85 : 138-143, 1996). Often the microemulsions may form spontaneously when the ingredients are brought together at room temperature. This may be particularly advantageous when formulating thermolabile drugs, peptides or oligonucleotides. Microemulsions are also effective for transdermal delivery of active ingredients in both cosmetic and pharmaceutical applications. The microemulsion compositions and formulations of the present invention improve the local cellular uptake of oligonucleotides and nucleic acids in the gastrointestinal tract, vagina, buccal cavity and other places of administration, as well as systemic oligonucleotide and nucleic acids from the gastrointestinal tract. It is expected to promote absorption.
本発明のマイクロエマルションはまた、製剤の特性を改善し、本発明のオリゴヌクレオチド及び核酸の吸収を高めるソルビタンモノステアレート(グリル3)、ラブラソール、及び浸透増強剤のような追加成分及び添加剤を含有してもよい。本発明の浸透増強剤は、5つの大まかなカテゴリー、界面活性剤、脂肪酸、胆汁塩、キレート剤及び非界面活性剤の1つに属するとして分類することができる(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92)。 The microemulsions of the present invention also contain additional ingredients and additives such as sorbitan monostearate (Grill 3), labrasol, and penetration enhancers that improve the properties of the formulation and enhance the absorption of the oligonucleotides and nucleic acids of the present invention. You may contain. The penetration enhancers of the present invention can be classified as belonging to one of five broad categories, surfactants, fatty acids, bile salts, chelating agents and non-surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92).
リポソーム
薬剤の製剤化に検討され、使用されているマイクロエマルションに加えて、整理された界面活性剤の構造体が多数ある。これらには、単層、ミセル、二重層及び小胞が挙げられる。小胞は、薬剤送達の作用の特異性及び延長した持続時間を提供する。従って、本明細書で使用されるとき、用語「リポソーム」は、球状の二重層に配置された両親媒性脂質から構成される小胞を言い、すなわち、リポソームは、親油性物質から形成される膜及び水性の内部を有する単層状体又は複層状体である。水性部分は通常、送達されるべき組成物を含有する。無傷の哺乳類の皮膚を横切るために、脂質小胞は、好適な経皮勾配の影響下、それぞれ直径50nm未満の一連の微細孔を通過するべきである。従って、極めて変形しやすい、且つ、そのような微細孔を通過できるリポソームを使用することが望ましい。リポソームの追加の因子には、脂質表面の変化及びリポソームの水性容積が挙げられる。
Liposome In addition to the microemulsions that have been investigated and used for drug formulation, there are a number of organized surfactant structures. These include monolayers, micelles, bilayers and vesicles. Vesicles provide the specificity and extended duration of action for drug delivery. Thus, as used herein, the term “liposomes” refers to vesicles composed of amphiphilic lipids arranged in a spherical bilayer, ie liposomes are formed from lipophilic substances. A monolayer or multilayer having a membrane and an aqueous interior. The aqueous portion usually contains the composition to be delivered. In order to cross intact mammalian skin, lipid vesicles should pass through a series of micropores, each less than 50 nm in diameter, under the influence of a suitable transdermal gradient. Therefore, it is desirable to use liposomes that are very easily deformable and that can pass through such micropores. Additional factors for liposomes include changes in lipid surface and aqueous volume of the liposomes.
リポソームのさらなる利点には、天然のリン脂質から得られるリポソームは生体適合性であり、生分解性であること;リポソームは広範囲の水溶性及び脂溶性の薬剤を組み入れることができること;リポソームはその内部区画にて内包された薬剤を代謝及び分解から保護できること(Rosoff, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, N.Y., volume 1, p245)が挙げられる。 A further advantage of liposomes is that liposomes derived from natural phospholipids are biocompatible and biodegradable; liposomes can incorporate a wide range of water-soluble and lipid-soluble drugs; The ability to protect the drugs contained in the compartment from metabolism and degradation (Rosoff, Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker Inc., New York, NY, volume 1, p245) It is done.
局所投与については、リポソームはそのほかの製剤に対して幾つかの利点を提示するという証拠がある。そのような利点には、投与された薬剤の高い全身性の吸収に関連する副作用を減らすこと、所望の標的にて投与された薬剤の蓄積を増やすこと、皮膚に対して親水性及び疎水性の両方であることが挙げられる。鎮痛剤、抗体、ホルモン及び高分子量のDNAを含む化合物が皮膚に投与され、一般に上部上皮が標的とされる。 For topical administration, there is evidence that liposomes present several advantages over other formulations. Such benefits include reducing side effects associated with high systemic absorption of the administered drug, increasing the accumulation of the drug administered at the desired target, hydrophilic and hydrophobic to the skin. It is mentioned that it is both. Compounds containing analgesics, antibodies, hormones, and high molecular weight DNA are administered to the skin, generally targeting the upper epithelium.
リポソームは2つに大別される。陽イオンリポソームは、正に荷電したリポソームであり、負に荷電したDNAと相互作用して安定な複合体を形成する。正に荷電したDNA/リポソーム複合体は、負に荷電した細胞表面に結合し、エンドソームに内部移行する。エンドソーム内の酸性pHのために、リポソームは破裂し、細胞の細胞質にその内容物を放出する(Wang et al., Biochem. Biophys. Res. Commun., 1987, 147,980-985)。 Liposomes are roughly divided into two types. Cationic liposomes are positively charged liposomes that interact with negatively charged DNA to form stable complexes. The positively charged DNA / liposome complex binds to the negatively charged cell surface and is internalized into the endosome. Due to the acidic pH within the endosome, the liposomes rupture and release their contents into the cell cytoplasm (Wang et al., Biochem. Biophys. Res. Commun., 1987, 147, 980-985).
pH感受性又は負に荷電したリポソームは、DNAと複合するよりもDNAを捕捉する。DNA及び脂質は似たように荷電しているので、複合体形成ではなく相反が生じる。従って、DNAは、これらリポソームの水性内部に捕捉される。pH感受性のリポソームは、たとえば、チミジンキナーゼ遺伝子をコードするDNAを培養における細胞の単層に送達するのに使用されている(Zhou et al., Journal of Controlled Release, 1992, 19,269-274)。 Liposomes that are pH sensitive or negatively charged capture DNA rather than complex with DNA. Since DNA and lipids are similarly charged, a reciprocal rather than complex formation occurs. Thus, DNA is entrapped within the aqueous interior of these liposomes. pH-sensitive liposomes have been used, for example, to deliver DNA encoding the thymidine kinase gene to cell monolayers in culture (Zhou et al., Journal of Controlled Release, 1992, 19,269-274).
リポソーム組成物の主要な種類の1つに天然由来のホスファチジルコリン以外のリン脂質が挙げられる。中性のリポソーム組成物は、たとえば、ジミリストイルホスファチジルコリン(DMPC)又はジパルミトイルホスファチジルコリン(DPPC)から形成することができる。陰イオンリポソーム組成物は一般に、ジミリストイルホスファチジルグリセロールから形成されるが、陰イオン膜融合性のリポソームはジオレイルホスファチジルエタノールアミン(DOPE)から主として形成される。もう1つの種類のリポソームは、たとえば、大豆PC及び卵PCのようなホスファチジルコリン(PC)から形成される。もう1つの種類は、リン脂質及び/又はホスファチジルコリン及び/又はコレステロールから形成される。 One of the main types of liposome compositions includes phospholipids other than naturally derived phosphatidylcholine. Neutral liposome compositions can be formed, for example, from dimyristoyl phosphatidylcholine (DMPC) or dipalmitoyl phosphatidylcholine (DPPC). Anionic liposome compositions are generally formed from dimyristoyl phosphatidylglycerol, whereas anionic membrane fusogenic liposomes are formed primarily from dioleyl phosphatidylethanolamine (DOPE). Another type of liposome is formed from phosphatidylcholine (PC) such as, for example, soy PC and egg PC. Another type is formed from phospholipids and / or phosphatidylcholines and / or cholesterol.
幾つかの研究によってリポソーム薬剤製剤の皮膚への局所送達が評価されている。インターフェロンを含有するリポソームのモルモット皮膚への適用により、皮膚のヘルペスによる傷を軽減したが、他の手段を介した送達(たとえば、溶液として、又はエマルションとして)は効果がなかった(Weiner et al., Journal of Drug Targeting, 1992, 2,405-410)。追加の研究により、水性系を用いたインターフェロンの投与に対する、リポソーム製剤の一部として投与されたインターフェロンの有効性を調べ、リポソーム製剤は水性投与より優れていると結論付けた(du Plessis et al., Antiviral Research, 1992, 18,259-265)。 Several studies have evaluated the topical delivery of liposomal drug formulations to the skin. Application of liposomes containing interferon to guinea pig skin alleviated skin herpes wounds, but delivery via other means (eg, as a solution or as an emulsion) was ineffective (Weiner et al. , Journal of Drug Targeting, 1992, 2,405-410). Additional studies investigated the effectiveness of interferon administered as part of a liposomal formulation for interferon administration using an aqueous system and concluded that the liposomal formulation is superior to aqueous administration (du Plessis et al. , Antiviral Research, 1992, 18, 259-265).
非イオン性のリポソーム系も調べられ、特に非イオン性界面活性剤及びコレステロールを含む系で、皮膚への薬剤送達における利便性が確定している。ノバソン(商標)I(グリセリルジラウレート/コレステロール/ポリオキシエチレン-10-ステアリルエーテル)及びノバソーム(商標)II(グリセリルジステアレート/コレステロール/ポリオキシエチレン-10-ステアリルエーテル)を含む非イオン性リポソーム製剤を用いて、マウスの皮膚の真皮にシクロスポリンAを送達した。結果は、そのような非イオン性リポソーム系が皮膚の異なる層へのシクロスポリンAの沈着を促進する上で有効であることを示した(Hu et al., S.T.P. Pharma Sci., 1994, 4,6,466)。 Non-ionic liposome systems have also been investigated, particularly in systems containing non-ionic surfactants and cholesterol, which have established convenience in drug delivery to the skin. Nonionic liposome formulation comprising Novason ™ I (glyceryl dilaurate / cholesterol / polyoxyethylene-10-stearyl ether) and Novasome ™ II (glyceryl distearate / cholesterol / polyoxyethylene-10-stearyl ether) Was used to deliver cyclosporin A into the dermis of mouse skin. The results showed that such nonionic liposome systems are effective in promoting the deposition of cyclosporin A in different layers of the skin (Hu et al., STP Pharma Sci., 1994, 4,6,466 ).
リポソームはまた、「立体的に安定化された」リポソームを包含し、その用語は、本明細書で使用されるとき、1以上の特殊化された脂質を含むリポソームを言い、特殊化された脂質はリポソームに組み入れられると、そのような特殊化された脂質を欠くリポソームに比べて循環寿命が長くなる。立体的に安定化されたリポソームの例は、小胞形成する脂質部分の一部がモノシアロガングリオシドGM1のような糖脂質を1以上包含するもの、又はポリエチレングリコール(PEG)部分のような親水性ポリマーの1以上で誘導体化されるものである。特定の理論に束縛されないで、ガングリオシド、スフィンゴミエリン、又はPEGで誘導体化された脂質を含有する立体的に安定化されたリポソームについては、これら立体的に安定化されたリポソームの循環寿命の延長は、網内系(RES)の細胞への取り込みの低下によると考えられている(Allen et al., FEBS Lett., 1987, 223,42; Wu et al., Cancer Research 1993, 53,3765)。 Liposomes also encompass “sterically stabilized” liposomes, the term as used herein refers to liposomes containing one or more specialized lipids, and specialized lipids. When incorporated into liposomes, it has a longer circulation life than liposomes lacking such specialized lipids. Examples of sterically stabilized liposomes are those in which part of the lipid portion of the vesicle formation include glycolipids, such as monosialoganglioside G M1 1 or more, or hydrophilic, such as polyethylene glycol (PEG) moiety Derivatized with one or more of the functional polymers. Without being bound by a particular theory, for sterically stabilized liposomes containing lipids derivatized with gangliosides, sphingomyelin, or PEG, the circulation life extension of these sterically stabilized liposomes is This is thought to be due to a decrease in reticuloendothelial (RES) uptake into cells (Allen et al., FEBS Lett., 1987, 223,42; Wu et al., Cancer Research 1993, 53, 3765).
1以上の糖脂質を含む種々のリポソームは、Papahadjopoulos et al., Ann. N. Y. Acad. Sci., 1987, 507:64, (モノシアロガングリオシドGM1、硫酸ガラクトセレブロシド及びホスファチジルイノシトール);Gabizon et al., Proc. Natl. Acad. Sci. USA, 1988, 85,6949; Allen et al., 米国特許第4,837,028号及び国際出願公報、国際公開公報第88/04924号(スフィンゴミエリン及びガングリオシドGM1又は硫酸ガラクトセレブロシドエステル);Webb et al., 米国特許第5,543,152号(スフィンゴミエリン);Lim et al., 国際公開公報第97/13499号(1,2-sn-ジミリストイルホスファチジルコリン)に報告されている。 Various liposomes containing one or more glycolipids are described in Papahadjopoulos et al., Ann. NY Acad. Sci., 1987, 507: 64, (monosialoganglioside G M1 , galactocerebroside sulfate and phosphatidylinositol); Gabizon et al. , Proc Natl Acad Sci USA, 1988 , 85,6949;..... Allen et al, U.S. Patent No. 4,837,028 and International application Publication WO 88/04924 (sphingomyelin and gangliosides G M1 or sulphate galacto Cerebroside esters); Webb et al., US Pat. No. 5,543,152 (sphingomyelin); Lim et al., WO 97/13499 (1,2-sn-dimyristoylphosphatidylcholine).
1以上の親水性ポリマーで誘導体化された脂質を含むリポソーム、及び調製方法は、たとえば、Sunamoto et al., Bull. Chem. Soc. Jpn., 1980, 53, 2778(非イオン性界面活性剤、PEG部分を含む2C1215G);Illum et al., FEBS Lett., 1984, 167,79(ポリマーグリコールによるポリスチレン粒子の親水性被覆);Sears, 米国特許第4,426,330号及び同第4,534,899号(ポリアルキレングリコール(たとえば、PEG)のカルボキシル基の結合により修飾された合成リン脂質);Klibanov et al., FEBS Lett., 1990, 268, 235(PEG又はPEGステアレートで誘導体化されたホスファチジルエタノールアミン(PE);Blume et al., Biochimica et Biophysica Acta, 1990, 1029:91(PEGで誘導体化されたリン脂質、たとえば、ジステアロイルホスファチジルエタノールアミン(DSPE)とPEGとの組み合わせから形成されるDSPE-PEG);Fisher, 欧州特許第EP 0 445 131 B1号及び国際公開公報第90/04384号(リポソームの外側表面で共有結合したPEG部分);Woodle et al., 米国特許第5,013,556号及び同第5,356,633号、及びMartin et al., 米国特許第5,213,804号及び欧州特許第EP0496813B1(1〜20モルパーセントのPEGで誘導体化したPEを含有するリポソーム組成物);Martin et al., 国際公開公報第91/05545号及び米国特許第5,225,212号及びZalipsky et al., 国際公開公報第94/20073号(多数のそのほかの脂質-ポリマー抱合体を含有するリポソーム);Choi et al., 国際公開公報第96/10391号(PEGで修飾したセラミド脂質を含むリポソーム);Miyazaki et al., 米国特許第5,540,935号及びTagawa et al., 米国特許第5,556,948号(その表面で機能部分によりさらに誘導体化することができるPEGを含有するリポソーム)に記載されている。 Liposomes containing lipids derivatized with one or more hydrophilic polymers and methods of preparation are described, for example, in Sunamoto et al., Bull. Chem. Soc. Jpn., 1980, 53, 2778 (nonionic surfactants, 2C 12 15G including PEG moiety);.. Illum et al, FEBS Lett, 1984, a hydrophilic coating of polystyrene particles by 167,79 (polymer glycol); Sears, U.S. Patent No. 4,426,330 and ibid. No. 4,534,899 (polyalkylene Glycols (eg, synthetic phospholipids modified by attachment of the carboxyl group of PEG); Klibanov et al., FEBS Lett., 1990, 268, 235 (phosphatidylethanolamine derivatized with PEG or PEG stearate (PE) ); Blume et al., Biochimica et Biophysica Acta, 1990, 1029: 91 (phospholipids derivatized with PEG, eg, DSPE-PEG formed from a combination of distearoylphosphatidylethanolamine (DSPE) and PEG) ; Fisher, Europe State Patent Nos. EP 0 445 131 B1 and WO 90/04384 (PEG moieties covalently attached to the outer surface of liposomes); Woodle et al., US Pat. Nos. 5,013,556 and 5,356,633, and Martin et al. al., US Patent No. 5,213,804 and European Patent No. EP0496813B1 (liposome composition containing PE derivatized with 1-20 mole percent PEG); Martin et al., WO 91/05545 and US Patent 5,225,212 and Zalipsky et al., WO 94/20073 (liposomes containing a number of other lipid-polymer conjugates); Choi et al., WO 96/10391 (modified with PEG) Liposomes containing ceramide lipids); Miyazaki et al., US Pat. No. 5,540,935 and Tagawa et al., US Pat. No. 5,556,948 (liposomes containing PEG that can be further derivatized with functional moieties on their surface) Are listed.
核酸を含むリポソームは、たとえば、Thierry et al., 国際公開公報第96/40062(高分子量の核酸をリポソームに内包する方法);Tagawa et al., 米国特許第5,264,221号(タンパク質に結合した、RNAを含有するリポソーム);Rahman et al., 米国特許第5,665,710号(オリゴデオキシヌクレオチドをリポソームに内包する方法);Love et al., 国際公開公報第97/04787号(アンチセンスオリゴヌクレオチドを含むリポソーム)に記載されている。 Liposomes containing nucleic acids are described, for example, in Thierry et al., WO 96/40062 (method of encapsulating high molecular weight nucleic acids in liposomes); Tagawa et al., US Pat. No. 5,264,221 (protein-bound, RNA Rahman et al., US Pat. No. 5,665,710 (method of encapsulating oligodeoxynucleotides in liposomes); Love et al., WO 97/04787 (liposomes containing antisense oligonucleotides) It is described in.
もう1つの種類のリポソームである、トランスファーソームは極めて変形しやすい脂質の凝集体であり、薬剤送達ビヒクルに魅力的である(Cevc et al., 1998, Biochim. Biophys. Acta, 1368(2):201-15)。トランスファーソームは、液滴よりも小さい孔を介して浸透することができる極めて変形しやすい脂質液滴として記載されてもよい。トランスファーソームはそれが使用される環境に適用可能であり、たとえば、それらは、形状適用可能であり、自己修復性であり、分断することなく標的に到達し、自己負荷することが多い。トランスファーソームは、たとえば、標準のリポソーム組成物に表面端活性化剤、通常、界面活性剤を添加することにより作製することができる。 Another type of liposome, transfersomes, are highly deformable lipid aggregates that are attractive for drug delivery vehicles (Cevc et al., 1998, Biochim. Biophys. Acta, 1368 (2): 201-15). Transfersomes may be described as highly deformable lipid droplets that can penetrate through pores smaller than the droplets. Transfersomes are applicable to the environment in which they are used, for example, they are shape-applicable, self-repairing, often reach the target without disruption and self-load. Transfersomes can be made, for example, by adding a surface edge activator, usually a surfactant, to a standard liposome composition.
界面活性剤
界面活性剤は、エマルション(マイクロエマルションを含む)及びリポソームのような製剤で広く使用されている。天然の及び合成の多数の異なる種類の界面活性剤を分類し、ランク付けする最も共通した方法は、親水性/親油性比(HLB)の使用による。親水性基(「頭部」としても知られる)の性質は、製剤で使用される様々な界面活性剤を分類する最も有用な手段を提供する(Rieger, Pharmaceutical Dosage Forms, Marcel Dekker Inc., New York, N.Y., 1988 p.285)。
Surfactants Surfactants are widely used in formulations such as emulsions (including microemulsions) and liposomes. The most common way to classify and rank many different types of natural and synthetic surfactants is by using the hydrophilic / lipophilic ratio (HLB). The nature of the hydrophilic group (also known as the “head”) provides the most useful means of classifying the various surfactants used in the formulation (Rieger, Pharmaceutical Dosage Forms, Marcel Dekker Inc., New York, NY, 1988 p.285).
界面活性剤の分子がイオン化されていなければ、非イオン性界面活性剤として分類される。非イオン性界面活性剤は薬学的製品及び化粧品で広く使用されており、広い範囲のpH値にわたって使用でき、典型的なHLB値は、構造によって2〜約18である。非イオン性界面活性剤には、エチレングリコールエステル、プロピレングリコールエステル、グリセリルエステル、ポリグリセリルエステル、ソルビタンエステル、スクロースエステル、及びエトキシル化エステルのような非イオン性エステル;並びに脂肪アルコールエトキシレート、プロポキシル化アルコールのような非イオン性アルカノールアミドが挙げられ、エトキシル化/プロポキシル化ブロックポリマーもこの部類に包含される。ポリオキシエチレン界面活性剤は非イオン性界面活性剤の部類で最もよく使用されるものである。 If the surfactant molecule is not ionized, it is classified as a nonionic surfactant. Nonionic surfactants are widely used in pharmaceutical products and cosmetics and can be used over a wide range of pH values, with typical HLB values ranging from 2 to about 18 depending on the structure. Nonionic surfactants include nonionic esters such as ethylene glycol esters, propylene glycol esters, glyceryl esters, polyglyceryl esters, sorbitan esters, sucrose esters, and ethoxylated esters; and fatty alcohol ethoxylates, propoxylated Nonionic alkanolamides such as alcohols are included, and ethoxylated / propoxylated block polymers are also included in this class. Polyoxyethylene surfactants are those most commonly used in the class of nonionic surfactants.
水に溶解するか、又は分散したとき負の電荷を持つ界面活性剤分子は陰イオン性に分類される。陰イオン性界面活性剤には、石鹸のようなカルボキシレート、アシルラクチレート、アミノ酸のアシルアミド、アルキルサルフェート及びエトキシル化アルキルサルフェートのような硫酸のエステル、アルキルベンゼンスルホネートのようなスルホネート、アシルイソチオネート、アシルラウレート及びスルホスクシネート及びホスフェートが挙げられる。アルキルサルフェート及び石鹸が、最もよく使用される陰イオン性界面活性剤である。 Surfactant molecules that have a negative charge when dissolved or dispersed in water are classified as anionic. Anionic surfactants include carboxylates such as soaps, acyl lactylates, acylamides of amino acids, esters of sulfuric acids such as alkyl sulfates and ethoxylated alkyl sulfates, sulfonates such as alkyl benzene sulfonates, acyl isothionates, Examples include acyl laurate and sulfosuccinate and phosphate. Alkyl sulfates and soaps are the most commonly used anionic surfactants.
水に溶解するか、又は分散したとき正の電荷を持つ界面活性剤分子は陽イオン性に分類される。陽イオン性界面活性剤には、四級アンモニウム塩及びエトキシル化アミンが挙げられ、四級アンモニウム塩が最も頻繁に使用される。 Surfactant molecules that have a positive charge when dissolved or dispersed in water are classified as cationic. Cationic surfactants include quaternary ammonium salts and ethoxylated amines, with quaternary ammonium salts being most frequently used.
正の電荷又は負の電荷のいずれかを持つことができる界面活性剤分子は両性として分類される。両性界面活性剤には、アクリル酸誘導体、置換アルキルアミド、N-アルキルベタイン及びホスファチドが挙げられる。 Surfactant molecules that can have either a positive charge or a negative charge are classified as amphoteric. Amphoteric surfactants include acrylic acid derivatives, substituted alkylamides, N-alkylbetaines and phosphatides.
薬剤製品、製剤及びエマルションにおける界面活性剤の使用は、Rieger, in Pharmaceutical Dosage Forms, Marcel Dekker Inc., New York, N.Y., 1988, p.285に概説されている。 The use of surfactants in pharmaceutical products, formulations and emulsions is reviewed in Rieger, in Pharmaceutical Dosage Forms, Marcel Dekker Inc., New York, N.Y., 1988, p.285.
浸透増強剤
幾つかの態様では、組成物中で又は組成物と共に浸透増強剤を用いて核酸、特にオリゴヌクレオチドの動物皮膚へのまたは粘膜を通した送達を高める。ほとんどの薬剤はイオン化された形態及び非イオン化の形態の双方で溶液中に存在する。しかしながら、普通、脂溶性又は親油性の薬剤だけが細胞膜を容易に横切る。横切るべき膜が浸透増強剤で処理されていれば、非親油性薬剤であっても細胞膜を横切ることができることが発見されている。非親油性の薬剤が細胞膜を横切るのを助けるのに加えて、浸透増強剤は、親油性薬剤の透過性も高める。
Penetration enhancers In some embodiments, penetration enhancers are used in or with the composition to enhance delivery of nucleic acids, particularly oligonucleotides, to the animal skin or through mucosa. Most drugs exist in solution in both ionized and non-ionized forms. However, usually only fat-soluble or lipophilic drugs easily cross cell membranes. It has been discovered that even non-lipophilic drugs can cross cell membranes if the membrane to be crossed is treated with a penetration enhancer. In addition to helping non-lipophilic drugs cross cell membranes, penetration enhancers also increase the permeability of lipophilic drugs.
浸透増強剤は、5つの大まかなカテゴリー、すなわち、界面活性剤、脂肪酸、胆汁塩、キレート剤及び非キレート性非界面活性剤の1つに属するとして分類される(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92)。これらの部類の浸透増強剤のそれぞれを以下で詳細に説明する。 Penetration enhancers are classified as belonging to one of five broad categories: surfactants, fatty acids, bile salts, chelating agents and non-chelating non-surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92). Each of these classes of penetration enhancers is described in detail below.
界面活性剤:本発明に関連して、界面活性剤(又は「表面活性化剤」)は、水性溶液に溶解すると溶液の表面張力又は水性溶液と別の液体との界面張力を低下させ、その結果、粘膜を介したオリゴヌクレオチドの吸収が高められる化学構成要素である。これら浸透増強剤には、たとえば、ラウリル硫酸ナトリウム、ポリオキシエチレン-9-ラウリルエーテル及びポリオキシエチレン-20-セチルエーテル(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92);並びにFC-43のようなパーフルオロ化合物エマルション(Takahashi et al., J. Pharm. Pharmacol., 1988, 40,252)が挙げられ、それぞれその全体を参考として本明細書に組み入れる。 Surfactant: In the context of the present invention, a surfactant (or “surface activator”), when dissolved in an aqueous solution, reduces the surface tension of the solution or the interfacial tension between the aqueous solution and another liquid, The result is a chemical component that enhances the absorption of oligonucleotides through the mucosa. These penetration enhancers include, for example, sodium lauryl sulfate, polyoxyethylene-9-lauryl ether and polyoxyethylene-20-cetyl ether (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92). As well as perfluoro compound emulsions such as FC-43 (Takahashi et al., J. Pharm. Pharmacol., 1988, 40,252), each of which is incorporated herein by reference in its entirety.
脂肪酸:浸透増強剤として作用する種々の脂肪酸及びその誘導体には、たとえば、オレイン酸、ラウリル酸、カプリン酸(n-デカン酸)、ミリスチル酸、パルミチン酸、ステアリン酸、リノール酸、リノレン酸、ジカプレート、トリカプレート、モノオレイン(1-モノオレオイル-rac-グリセロール)、ジラウリン、カープリル酸、アラキドン酸、グリセロール1-モノカプレート、1-ドデシルアザシクロヘプタン-2-オン、アシルカルニチン、アシルコリン、それらのC1〜10アルキルエステル(たとえば、メチル、イソプロピル及びt-ブチル)並びにそれらのモノ-及びジグリセリド(すなわち、オレート、ラウレート、カプレート、ミリステート、パルミテート、ステアレート、リノレートなど)(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; El Hariri et al., J. Pharm. Pharmacol., 1992, 44, 651-654)が挙げられ、それぞれその全体を参考として本明細書に組み入れる。 Fatty acids: Various fatty acids and their derivatives that act as penetration enhancers include, for example, oleic acid, lauric acid, capric acid (n-decanoic acid), myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dica Plate, tricaplate, monoolein (1-monooleoyl-rac-glycerol), dilaurin, carprilic acid, arachidonic acid, glycerol 1-monocaprate, 1-dodecylazacycloheptan-2-one, acylcarnitine, acylcholine, them C 1-10 alkyl esters of (eg, methyl, isopropyl and t-butyl) and their mono- and diglycerides (ie, oleate, laurate, caprate, myristate, palmitate, stearate, linoleate, etc.) (Lee et al. , Critical Reviews in Therapeutic Drug Carrier Systems, 1991 , p.92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; El Hariri et al., J. Pharm. Pharmacol., 1992, 44, 651-654). The entirety is incorporated herein by reference.
胆汁塩:胆汁の生理学的役割には、脂質及び脂溶性ビタミンの分散及び吸収の促進が挙げられる(Brunton, Chapter 38, Goodman & Gilman's The Pharmacological Basis of Therpeutics, 9th Ed., Hardman et al., Eds., McGraw-Hill, New York, 1996, p.934-935)。種々の天然の胆汁塩及びその合成誘導体が浸透増強剤として作用する。従って、用語、「胆汁塩」は、胆汁の天然に存在するいかなる成分及びそのいかなる合成誘導体も包含する。本発明の胆汁塩には、たとえば、コール酸(又は薬学的に許容されるそのナトリウム塩、コール酸ナトリウム)、ジヒドロコール酸(ジヒドロコール酸ナトリウム)、デオキシコール酸(デオキシコール酸ナトリウム)、グルコール酸(グルコール酸ナトリウム)、グリコール酸(グリコール酸ナトリウム)、グリコデオキシコール酸(グリコデオキシコール酸ナトリウム)、タウロコール酸(タウロコール酸ナトリウム)、タウロデオキシコール酸(タウロデオキシコール酸ナトリウム)、ケノデオキシコール酸(ケノデオキシコール酸ナトリウム)、ウルソデオキシコール酸(UDCA)、タウロ-24,25-ジヒドロ-フシジン酸ナトリウム(STDHF)、グリコジヒドロフシジン酸ナトリウム及びポリオキシエチレン-9-ラウリルエーテル(POE)が挙げられる。(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991 , page 92; Swinyard, Chapter 39, Remington's Pharmaceutical Sciences, 18th Ed., Gennaro, ed., Mack Publishing Co., Easton, Pa., 1990, pages 782-783; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Yamamoto et al., J. Pharm. Exp. Ther, 1992, 263, 25; Yamashita et al., J. Pharm:. Sci., 1990, 79, 579-583)。 Bile salts: The physiological role of bile includes promoting the dispersion and absorption of lipids and fat-soluble vitamins (Brunton, Chapter 38, Goodman & Gilman's The Pharmacological Basis of Therpeutics, 9th Ed., Hardman et al., Eds , McGraw-Hill, New York, 1996, p.934-935). Various natural bile salts and synthetic derivatives thereof act as penetration enhancers. Thus, the term “bile salt” encompasses any naturally occurring component of bile and any synthetic derivatives thereof. Examples of the bile salt of the present invention include cholic acid (or a pharmaceutically acceptable sodium salt thereof, sodium cholate), dihydrocholic acid (sodium dihydrocholate), deoxycholic acid (sodium deoxycholate), glycol. Acid (sodium glycolate), glycolic acid (sodium glycolate), glycodeoxycholic acid (sodium glycodeoxycholate), taurocholic acid (sodium taurocholate), taurodeoxycholic acid (sodium taurodeoxycholate), chenodeoxycholic acid ( Examples include sodium chenodeoxycholic acid), ursodeoxycholic acid (UDCA), tauro-24,25-dihydro-fusidic acid sodium (STDHF), sodium glycodihydrofusidic acid and polyoxyethylene-9-lauryl ether (POE). That. (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Swinyard, Chapter 39, Remington's Pharmaceutical Sciences, 18th Ed., Gennaro, ed., Mack Publishing Co., Easton, Pa., 1990, pages 782-783; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Yamamoto et al., J. Pharm. Exp. Ther, 1992, 263, 25; Yamashita et al., J. Pharm: Sci., 1990, 79, 579-583).
キレート剤:本背景では、キレート剤は、それと錯体を形成することにより溶液から金属イオンを除き、その結果粘膜を介したオリゴヌクレオチドの吸収が高められる化合物とみなすことができる。本発明での浸透増強剤としてのその使用に関して、性状分析されたDNAヌクレアーゼのほとんどが触媒について二価の金属イオンを必要とし、キレート剤により阻害されるので、キレート剤はDNA分解酵素阻害剤として作用するという追加的な利点を有する(Jarrett, J. Chromatogr., 1993, 618,315-339)。限定することなく、キレート剤には、エチレンジアミン四酢酸二ナトリウム(EDTA)、クエン酸、サリチレート(たとえば、サリチル酸ナトリウム、5-メトキシサリチレート及びホモバニレート)、コラーゲンの N-アシル誘導体、ベータジケトンのラウレス-9及びN-アミノアシル誘導体(エナミン)(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7,1-33; Buur et al., J. Control Rel., 1990, 14,43-51)が挙げられる。 Chelating agent: In this context, a chelating agent can be considered a compound that forms a complex with it to remove metal ions from the solution, resulting in enhanced absorption of the oligonucleotide through the mucosa. With regard to its use as a penetration enhancer in the present invention, most of the DNA nucleases that have been characterized require a divalent metal ion for the catalyst and are inhibited by the chelating agent, so that the chelating agent is a DNA degrading enzyme inhibitor. Has the additional advantage of acting (Jarrett, J. Chromatogr., 1993, 618, 315-339). Without limitation, chelating agents include disodium ethylenediaminetetraacetate (EDTA), citric acid, salicylates (eg, sodium salicylate, 5-methoxysalicylate and homovanillate), N-acyl derivatives of collagen, laureth of beta diketone. -9 and N-aminoacyl derivatives (enamines) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7,1-33; Buur et al , J. Control Rel., 1990, 14, 43-51).
非キレート剤非界面活性剤:本明細書で使用されるとき、非キレート剤非界面活性剤の浸透を増強する化合物は、有意なキレート剤活性及び界面活性剤活性を有さず、消化器系粘膜を介したオリゴヌクレオチドの吸収を高める化合物である(Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7,1-33)。そのような浸透増強剤の例には、不飽和環状尿素1-アルキル及び1-アルケニルアザシクロ-アルカノン誘導体(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92)並びにジクロフェナックナトリウム、インドメタシン及びフェニルブタゾンのような非ステロイド抗炎症剤(Yamashita et al., J. Pharm. Pharmacol., 1987, 39,621-626)が挙げられる。 Non-chelating non-surfactant: As used herein, compounds that enhance the penetration of non-chelating non-surfactants do not have significant chelating and surfactant activity and are digestive system It is a compound that enhances the absorption of oligonucleotides through the mucosa (Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33). Examples of such penetration enhancers include unsaturated cyclic urea 1-alkyl and 1-alkenylazacyclo-alkanone derivatives (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92) and diclofenac sodium, Non-steroidal anti-inflammatory agents such as indomethacin and phenylbutazone (Yamashita et al., J. Pharm. Pharmacol., 1987, 39, 621-626).
細胞レベルでオリゴヌクレオチドの取り込みを高める作用剤を本発明の薬学的組成物及びそのほかの組成物及び製剤に添加してもよい。たとえば、リポフェクチン(Junichi et al., 米国特許第5,705,188号)のような陽イオン性脂質、陽イオン性グリセロール誘導体、及びポリリジン(Lollo et al., PCT出願、国際公開公報第97/30731号)のようなポリ陽イオン分子は、オリゴヌクレオチドの細胞内への取り込みを高めることも知られている。 Agents that enhance the uptake of oligonucleotides at the cellular level may be added to the pharmaceutical compositions and other compositions and formulations of the present invention. For example, cationic lipids such as lipofectin (Junichi et al., US Pat. No. 5,705,188), cationic glycerol derivatives, and polylysine (Lollo et al., PCT application, WO 97/30731) Such polycationic molecules are also known to enhance uptake of oligonucleotides into cells.
エチレングリコール及びプロピレングリコールのようなグリコール類、2-ピロールのようなピロール類、アゾン類(azones)、並びにリモネン及びメントンのようなテルペン類を含むそのほかの作用剤を利用して投与された核酸の浸透を高めてもよい。 Of nucleic acids administered using other agents including glycols such as ethylene glycol and propylene glycol, pyrroles such as 2-pyrrole, azones, and terpenes such as limonene and menthone. Penetration may be increased.
担体
本発明の特定の組成物はまた、製剤中に担体化合物を組み入れる。本明細書で使用されるとき、「担体化合物」又は「担体」は、核酸又はその類縁体を言い、それは不活性(すなわち、それ自体の生物活性を持たない)であるが、たとえば、生物学的に活性のある核酸を分解する又は循環からのその除去を促進することによって生物活性を有する核酸の生物利用性を低下させる生体内の過程によって核酸として認識される。核酸と担体化合物の同時投与は、後者の物質が過剰であることが多いが、肝臓、腎臓、又は循環外のそのほかの貯蔵場所で回収される核酸の量の実質的低下を招く。たとえば、肝臓組織におけるホスホロチオエートの回収は、それが、ポリイノシン酸、硫酸デキストラン、ポリシチジン酸又は4-アセトアミド-4'イソチオシアノ-スチルベン-2,2-ジスルホン酸と共に同時投与された場合、低下させることができ(Miyao et al., Antisense Res. Dev., 1995, 5,115-121; Takakura et al., Antisense & Nucl Acid Drug Dev., 1996, 6,177-183)、それぞれその全体を参考として本明細書に組み入れる。
Carrier Certain compositions of the present invention also incorporate a carrier compound in the formulation. As used herein, “carrier compound” or “carrier” refers to a nucleic acid or analog thereof, which is inactive (ie, does not have its own biological activity), eg, biology It is recognized as a nucleic acid by an in vivo process that degrades the bioavailability of a biologically active nucleic acid by degrading the active nucleic acid or promoting its removal from the circulation. Co-administration of nucleic acid and carrier compound often results in an excess of the latter, but results in a substantial reduction in the amount of nucleic acid recovered in the liver, kidney, or other storage location outside the circulation. For example, the recovery of phosphorothioate in liver tissue can be reduced when it is co-administered with polyinosinic acid, dextran sulfate, polycytidic acid or 4-acetamido-4'isothiocyano-stilbene-2,2-disulfonic acid. (Miyao et al., Antisense Res. Dev., 1995, 5,115-121; Takakura et al., Antisense & Nucl Acid Drug Dev., 1996, 6,177-183), each of which is incorporated herein by reference in its entirety.
賦形剤
担体化合物とは対照的に、「薬学的担体」又は「賦形剤」は1以上の核酸を動物に送達するための薬学的に許容される溶媒、懸濁剤又はそのほかの任意の薬学的に不活性のビヒクルであり、通常、液体又は固体である。薬学的担体は、所定の薬学的組成物の核酸及びそのほかの成分と組み合わせた場合、意図する投与方式の観点から一般的に選択され所望の嵩、濃度等を提供する。典型的な薬学的担体には、結合剤(たとえば、予備ゲル化したトウモロコシデンプン、ポリビニルピロリドン又はヒドロキシプロピルメチルセルロースなど);充填剤(たとえば、ラクトース及びそのほかの糖、微結晶セルロース、ペクチン、ゼラチン、硫酸カルシウム、エチルセルロース、ポリアクリレート又はリン酸水素カルシウムなど);潤滑剤(たとえば、ステアリン酸マグネシウム、タルク、シリカ、コロイド状二酸化珪素、ステアリン酸、金属ステアレート、水素添加植物油、コーンスターチ、ポリエチレングリコール、安息香酸ナトリウム、酢酸ナトリウムなど);崩壊剤(たとえば、デンプン、デンプングリコテートナトリウムなど);並びに湿潤剤(たとえば、ラウリル硫酸ナトリウムなど)が挙げられるが、これらに限定されない。
Excipient In contrast to a carrier compound, a “pharmaceutical carrier” or “excipient” is a pharmaceutically acceptable solvent, suspending agent or any other agent for delivering one or more nucleic acids to an animal. A pharmaceutically inert vehicle, usually a liquid or a solid. Pharmaceutical carriers are generally selected from the viewpoint of the intended mode of administration and provide the desired bulk, concentration, etc. when combined with nucleic acids and other components of a given pharmaceutical composition. Typical pharmaceutical carriers include binders (eg, pregelatinized corn starch, polyvinylpyrrolidone or hydroxypropylmethylcellulose); fillers (eg, lactose and other sugars, microcrystalline cellulose, pectin, gelatin, sulfuric acid) Calcium, ethyl cellulose, polyacrylate or calcium hydrogen phosphate, etc .; lubricants (eg magnesium stearate, talc, silica, colloidal silicon dioxide, stearic acid, metal stearate, hydrogenated vegetable oil, corn starch, polyethylene glycol, benzoic acid Sodium, sodium acetate, etc.); disintegrants (eg, starch, sodium starch glycolate, etc.); and wetting agents (eg, sodium lauryl sulfate, etc.). Not.
本発明の組成物を製剤化するのに、核酸と有害に反応しない非経口投与に好適な薬学的に許容される有機又は無機の賦形剤も使用することができる。好適な、薬学的に許容される担体には、水、塩溶液、アルコール類、ポリエチレングリコール類、ゼラチン、ラクトース、アミロース、ステアリン酸マグネシウム、タルク、珪酸、粘度のあるパラフィン、ヒドロキシメチルセルロース、ポリビニルピロリドンなどが挙げられるが、これらに限定されない。 Pharmaceutically acceptable organic or inorganic excipients suitable for parenteral administration that do not deleteriously react with nucleic acids can also be used to formulate the compositions of the present invention. Suitable pharmaceutically acceptable carriers include water, salt solutions, alcohols, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin, hydroxymethylcellulose, polyvinylpyrrolidone and the like However, it is not limited to these.
核酸の局所投与のための製剤には、無菌及び非無菌の水溶液、アルコールのような一般の溶媒中での非水性溶液、又は液体若しくは固体油の基剤における核酸の溶液が挙げられてもよい。溶液は、緩衝液、希釈液及びそのほかの好適な添加剤を含有してもよい。核酸と有害に反応しない非経口投与に好適な薬学的に許容される有機又は無機の賦形剤を使用することができる。 Formulations for topical administration of nucleic acids may include sterile and non-sterile aqueous solutions, non-aqueous solutions in common solvents such as alcohol, or solutions of nucleic acids in liquid or solid oil bases. . The solution may contain buffers, diluents and other suitable additives. Pharmaceutically acceptable organic or inorganic excipients suitable for parenteral administration that do not adversely react with nucleic acids can be used.
そのほかの薬学的組成物成分
本組成物は、薬学的組成物に従来見い出されるそのほかの成分を、技術で確立した用途レベルで追加的に含有してもよい。従って、たとえば、組成物は、たとえば、かゆみ止め剤、収斂剤、局所麻酔剤又は抗炎症剤のような追加的な、相溶性の、薬学的に活性のある物質を含有してもよく、又は、たとえば、染料、香味剤、防腐剤、抗酸化剤、乳白剤、濃厚剤及び安定剤のような、本発明の組成物の種々の投与形態を物理的に製剤化するのに有用な追加の物質を含有してもよい。しかしながら、そのような物質は、添加した際、本発明の組成物の成分の生物活性に過度に干渉すべきではない。製剤を滅菌し、所望であれば、製剤の核酸と有害に相互作用しない補助剤、たとえば、潤滑剤、防腐剤、安定剤、湿潤剤、乳化剤、浸透圧に影響を与える塩、緩衝液、着色剤、香味剤及び/又は芳香族物質などを混合する。
Other Pharmaceutical Composition Ingredients The composition may additionally contain other ingredients conventionally found in pharmaceutical compositions at the level of application established in the art. Thus, for example, the composition may contain additional, compatible, pharmaceutically active substances such as, for example, anti-itch agents, astringents, local anesthetics or anti-inflammatory agents, or Additional useful for physically formulating various dosage forms of the compositions of the invention, such as, for example, dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickeners and stabilizers. A substance may be contained. However, such materials should not unduly interfere with the biological activity of the components of the composition of the invention when added. Sterilizes the formulation and, if desired, adjuvants that do not adversely interact with the nucleic acid of the formulation, eg, lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts that affect osmotic pressure, buffers, coloring Mixing agents, flavoring agents and / or aromatic substances.
水性懸濁液は、たとえば、カルボキシメチルセルロースナトリウム、ソルビトール及び/又はデキストランを含む懸濁液の粘度を高める物質、及び/又は安定剤を含有してもよい。 Aqueous suspensions may contain substances that increase the viscosity of the suspension, including, for example, sodium carboxymethylcellulose, sorbitol and / or dextran, and / or stabilizers.
本発明の特定の態様は、(a)1以上の抗ウイルス性オリゴヌクレオチド及び(b)異なるメカニズムにより機能する1以上のそのほかの化学療法剤を含有する薬学的組成物を提供する。そのような化学療法剤の例には、ダウノルビシン、ダウノマイシン、ダクチノマイシン、ドキソルビシン、エピルビシン、イダルビシン、エソルビシン、ブレオマイシン、マフォスファミド、イフォスファミド、シトシンアラビノシド、ビスクロロエチルニトロソ尿素、ブスルファン、マイトマイシンC、アクチノマイシンD、ミトラマイシン、プレドニゾン、ヒドロキシプロゲステロン、テストステロン、タモキシフェン、ダカルバジン、プロカルバジン、ヘキサメチルメラミン、ペンタメチルメタミン、ミトキサントロン、アムサクリン、クロラムブシル、メチルシクロヘキシルニトロソ尿素、窒素マスタード、メラファラン、シクロホスファミド、6-メルカプトプリン、6-チオグアニン、シタラビン、5-アザシチジン、ヒドロキシ尿素、デオキシコフォマイシン、4-ヒドロキシパーオキシシクロホスホロアミド、5-フルオロウラシル(5-FU)、5-フルオロデオキシウリジン(5-FUdR)、メソトレキセート(MTX)、コルヒチン、タキソール、ビンクリスチン、ビンブラスチン、エトポシド(VP-16)、トリメトレキサート、イリノテカン、トポテカン、ゲムシタビン、テニポシド、シスプラチン及びジエチルスチルベストロール(DES)が挙げられるが、これらに限定されない。一般に、The Merck Manual of Diagnosis and Therapy, 15th Ed. 1987, pp. 1206-1228, Berkow et al., eds., Rahway, NJ.を参照のこと。本発明の化合物と共に使用する場合、個々に(たとえば、5-FUとオリゴヌクレオチド)、順に(たとえば、5-FUとオリゴヌクレオチドを一定期間、続いてMTXとオリゴヌクレオチド)又は1以上のそのような化学療法剤と組み合わせて(たとえば、5-EU、MTX及びオリゴヌクレオチド、又は5-FU、放射線療法及びオリゴヌクレオチド)そのような化学療法剤を使用すればよい。抗炎症薬には、非ステロイド性抗炎症薬及びコルチコステロイド類が挙げられるが、これらに限定されず、抗ウイルス剤には、リバビリン、シドフォビル、ビダラビン、アシクロビル及びガンシクロビルが挙げられるが、これらに限定されず、本発明の組成物で組み合わせてもよい。一般に、The Merck Manual of Diagnosis and Therapy, 15th Ed. Berkow et al., eds., 1987, Rahway, NJ., pages 2499-2506を参照のこと。そのほかの非オリゴヌクレオチドの化学療法剤も本発明の範囲内である2以上の化合物を一緒に又は順に用いてもよい。 Certain embodiments of the present invention provide pharmaceutical compositions comprising (a) one or more antiviral oligonucleotides and (b) one or more other chemotherapeutic agents that function by different mechanisms. Examples of such chemotherapeutic agents include daunorubicin, daunomycin, dactinomycin, doxorubicin, epirubicin, idarubicin, esorubicin, bleomycin, mafosfamide, ifosfamide, cytosine arabinoside, bischloroethylnitrosourea, busulfan, mitomycin C, actino Mycin D, mitramycin, prednisone, hydroxyprogesterone, testosterone, tamoxifen, dacarbazine, procarbazine, hexamethylmelamine, pentamethylmethamine, mitoxantrone, amsacrine, chlorambucil, methylcyclohexylnitrosourea, nitrogen mustard, melphalan, cyclophosphamide , 6-mercaptopurine, 6-thioguanine, cytarabine, 5-azacytidine, hydroxyurea, de Xycofomycin, 4-hydroxyperoxycyclophosphoramide, 5-fluorouracil (5-FU), 5-fluorodeoxyuridine (5-FUdR), methotrexate (MTX), colchicine, taxol, vincristine, vinblastine, etoposide (VP -16), trimethrexate, irinotecan, topotecan, gemcitabine, teniposide, cisplatin and diethylstilbestrol (DES), but are not limited thereto. See generally, The Merck Manual of Diagnosis and Therapy, 15th Ed. 1987, pp. 1206-1228, Berkow et al., Eds., Rahway, NJ. When used with a compound of the invention, individually (eg, 5-FU and oligonucleotide), sequentially (eg, 5-FU and oligonucleotide for a period, followed by MTX and oligonucleotide) or one or more such Such chemotherapeutic agents may be used in combination with chemotherapeutic agents (eg, 5-EU, MTX and oligonucleotides, or 5-FU, radiation therapy and oligonucleotides). Anti-inflammatory drugs include, but are not limited to, non-steroidal anti-inflammatory drugs and corticosteroids, and antiviral drugs include, but are not limited to, ribavirin, cidofovir, vidarabine, acyclovir and ganciclovir. It is not limited, You may combine with the composition of this invention. See generally, The Merck Manual of Diagnosis and Therapy, 15th Ed. Berkow et al., Eds., 1987, Rahway, NJ., Pages 2499-2506. Other non-oligonucleotide chemotherapeutic agents may also be used together or in sequence with two or more compounds within the scope of the present invention.
実施例
実施例1:単純ヘルペスウイルス
単純ヘルペスウイルス(HSV)はヒト集団の有意な割合を冒す。ランダムON又はONランダマーはHSVのようなウイルスの感染性を阻害することが本発明で見い出された。VERO細胞(HSV-1(KOS株)及びHSV-2(MS2株)の感染に感受性)におけるHSVの細胞性複製アッセイを用いて、HSVの複製開始点に相補的な一本鎖PS-ONがHSV-1及びHSV-2の複製を阻害した。驚くべきことに、ヒト(343ARS)及びプラスミド(pBR322/pUC)の開始点に相補的な対照のPS-ONもウイルスの感染性を阻害した。ランダム配列のPS-ON及びPS-ONランダマーによる実験は、ONのサイズの増加に伴ってウイルス感染の阻害が高まることを実証した。これらのデータは、ONは、ウイルス感染の治療的処置に有用な強力な抗ウイルス剤であることを示している。
Examples Example 1: Herpes simplex virus Herpes simplex virus (HSV) affects a significant proportion of the human population. It has been found in the present invention that random ON or ON randomers inhibit the infectivity of viruses such as HSV. Using HSV cellular replication assay in VERO cells (susceptible to HSV-1 (KOS strain) and HSV-2 (MS2 strain) infection), single-stranded PS-ON complementary to HSV replication origin HSV-1 and HSV-2 replication was inhibited. Surprisingly, the control PS-ON complementary to the start of human (343ARS) and plasmid (pBR322 / pUC) also inhibited viral infectivity. Experiments with random-sequence PS-ON and PS-ON randomers demonstrated that inhibition of viral infection increased with increasing ON size. These data indicate that ON is a powerful antiviral agent useful for the therapeutic treatment of viral infections.
本発明者らは、HHVのようなウイルスの広がりを阻止する可能性のあるメカニズムはそのDNAの複製を妨げることであると理論付けた。このことを念頭に置いて、HSV1及びHSV2の複製開始点に相補的なホスホロチオエートオリゴヌクレオチド(ON)を感染細胞に導入した。これらのONは、ウイルスの複製開始点にてDNAの三重体形成を生じ、必要なトランス作用因子の会合及びウイルスDNAの複製を阻止した。驚くべきことに、実験的パーラダイムにおいて、ウイルス感染を阻害するONの能力はそのサイズ(長さ)が増加するにつれて高まることを示す結果が本明細書で提示される。 The inventors theorized that a possible mechanism to prevent the spread of viruses such as HHV is to prevent its DNA replication. With this in mind, phosphorothioate oligonucleotides (ON) complementary to the HSV1 and HSV2 origins of replication were introduced into infected cells. These ONs produced DNA triplets at the viral origin of replication, preventing the association of necessary trans-acting factors and viral DNA replication. Surprisingly, results are presented herein that show that in an experimental paradigm, the ability of ON to inhibit viral infection increases as its size (length) increases.
HSV-1の阻害
プラーク減少アッセイ(PRA)にてHSV-1を阻害するPS-ONの能力を測定した。不死化したアフリカミドリザルの腎臓細胞(VERO)を10%のウシ胎児血清をプラスし、ゲンタマイシン、バンコマイシン及びアンフォテレシンBを補完したMEM(最低必須培地)中で、37℃、5% CO2にて培養した。4日間の増殖で集密な単層細胞が得られる密度にて細胞を12ウェルプレートに播いた。集密に達すると、培地を5%血清と上記の補完物だけを含有するものに代え、次いで、試験化合物の存在下、細胞を90分間、HSV-1(KOS株、総計およそ40〜60PFU)にさらした。ウイルスに暴露したのち、培地を、5%血清、1%ヒト免疫グロブリン、上記の補完物及び試験化合物を含有する新しい「重層する」培地に変更した。感染の3〜4日後、感染培養物のホルマリン固定及びクレシルバイオレット染色に続いて、プラークの計数を行った。
Inhibition of HSV-1 The ability of PS-ON to inhibit HSV-1 was measured in a plaque reduction assay (PRA). Cultured immortalized African green monkey kidney cells (VERO) in MEM (minimum essential medium) supplemented with gentamicin, vancomycin and amphotericin B plus 10% fetal bovine serum at 37 ° C, 5% CO 2 did. Cells were seeded in 12-well plates at a density that yielded confluent monolayer cells after 4 days of growth. When confluence is reached, the medium is replaced with one containing only 5% serum and the above complements, and then the cells are placed in the presence of the test compound for 90 minutes in HSV-1 (KOS strain, approximately 40-60 PFU total) Exposed to. After exposure to the virus, the medium was changed to a new “layered” medium containing 5% serum, 1% human immunoglobulin, the above complements and test compounds. Three to four days after infection, plaque counts were performed following formalin fixation and cresyl violet staining of infected cultures.
ONはすべて(別に言及したものは除いて)University of Calgary、コアDNAサービスラボで合成した1又は15μMの合成スケ-ルでON(表21を参照のこと)を調製し、50cmのセファデックスG-25カラムで脱保護し、脱塩した。UVシャドウゲル電気泳動により得られたONを分析し、約95%の完全長、n-1及びn-2オリゴ並びに5%までの短いオリゴ種(これらはランダム欠失を有すると思われる)を含有することを確定した。ランダムなオリゴ合成については、アデニン、グアノシン、シトシン及びチミジンのアミダイトを等モル量にて一緒に混合し、合成の間、ONの各位置での取り込みの無作為さをできるだけ大きくした。 All ONs (except as otherwise noted) were prepared at the University of Calgary, Core DNA Service Lab 1 or 15 μM synthesis scale (see Table 21) and 50 cm Sephadex G -25 column for deprotection and desalting. Analyzing the ON obtained by UV shadow gel electrophoresis, about 95% full length, n-1 and n-2 oligos and up to 5% short oligo species (these are likely to have random deletions) It was confirmed to contain. For random oligo synthesis, adenine, guanosine, cytosine and thymidine amidites were mixed together in equimolar amounts to maximize the randomness of incorporation at each ON position during synthesis.
PS-ONによるHSV-1阻害の能力を試験するために、HSV-1 PRAでREP 1001、2001および3007を試験する。PS-ONがHSV内の複製起点に指向されているため、REP 2001のみが任意の活性を示すであろうと想定された(他の2つは、ヒトおよびプラスミド内の複製起点に指向されている)。しかしながら、3つのPS-ON全部が、抗HSV-1活性を示した。試験は、HSV-1 (KOS株)を用いて、VERO細胞内で実施したプラーク減少アッセイにより行った。感染細胞は、濃度を増大させたREP 1001、REP 2001、またはREP 3007により処理した。アッセイ結果の直線回帰から計算したIC50値は、各々、2.76、0.77、および5.33マイクロモルであった。更に、抗HSV-1効果の有効性は、オリゴのサイズに依存することが見出された。 To test the ability of HSV-1 inhibition by PS-ON, REP 1001, 2001 and 3007 are tested with HSV-1 PRA. Since PS-ON is directed to the origin of replication in HSV, it was assumed that only REP 2001 would show any activity (the other two are directed to the origin of replication in humans and plasmids ). However, all three PS-ONs showed anti-HSV-1 activity. The test was performed by plaque reduction assay performed in VERO cells using HSV-1 (KOS strain). Infected cells were treated with increasing concentrations of REP 1001, REP 2001, or REP 3007. IC 50 values calculated from linear regression of the assay results were 2.76, 0.77, and 5.33 micromolar, respectively. Furthermore, the effectiveness of the anti-HSV-1 effect was found to depend on the size of the oligo.
PS-ONの抗HSV-1活性上のサイズ依存性および相対的な配列非依存性を確認するために、本発明者らは、様々なサイズのPS-ON (REP 2002、2003、2004、2005および2006)を、抗ウイルス薬アシクロビルと共に試験した。これらPS-ONは、各塩基を「ゆらぎ」(N)として合成して、各PS-ONが実際に同一サイズの異なるランダム配列を有する集団とすることにより、配列特異的な効果に関して不活性にされている;これらのPS-ONは、「ランダマー」と称される。HSV-1 (KOS株)を用いて、VERO細胞内でプラーク減少アッセイを実施した。感染細胞を、濃度を増大させたREP 2001、REP 2002またはREP 3003、REP 2004、REP 2005、REP 2006およびアシクロビルにより処理する。アッセイデータの直線回帰からIC50値を計算した。HSV-1に対するPS-ONのサイズとIC50との関係は、抗HSV-1活性を示した、試験した各PS-ONの特定のサイズに対してIC50値をプロットすることにより決定した。アシクロビルのIC50を、臨床的相関の参照として使用した。本発明者らは、10塩基またはそれ未満のオリゴは、検出可能な抗HSV-1活性を有さないが、PS-ONのサイズが10塩基を越えて増加するにつれ、有効性もまた上昇する(IC50低下)ことを見出した。本発明者らはまた、20塩基より大きいPS-ONは、臨床的に許容される抗HSV-1薬、アシクロビルと比較して有意に低いIC50値を有することにも注目した。 To confirm the size-dependence and relative sequence-independence on the anti-HSV-1 activity of PS-ON, we have determined that various sizes of PS-ON (REP 2002, 2003, 2004, 2005 And 2006) were tested with the antiviral drug acyclovir. These PS-ONs are inactive with respect to sequence-specific effects by synthesizing each base as a “fluctuation” (N) and making each PS-ON actually a group with different random sequences of the same size. These PS-ONs are referred to as “Randamers”. Plaque reduction assay was performed in VERO cells using HSV-1 (KOS strain). Infected cells are treated with increasing concentrations of REP 2001, REP 2002 or REP 3003, REP 2004, REP 2005, REP 2006 and acyclovir. IC 50 values were calculated from linear regression of assay data. The relationship between the size of PS-ON for HSV-1 and IC 50 was determined by plotting the IC 50 value against the specific size of each PS-ON tested that showed anti-HSV-1 activity. The IC 50 of acyclovir was used as a reference for clinical correlations. We have oligos of 10 bases or less that have no detectable anti-HSV-1 activity, but effectiveness also increases as the size of PS-ON increases beyond 10 bases. (IC 50 decreased). We also noted that PS-ONs greater than 20 bases have significantly lower IC 50 values compared to the clinically acceptable anti-HSV-1 drug, acyclovir.
抗HSV-1活性に関するPS-ONの有効なサイズ範囲を更に明確にするために、本発明者らは、より広範なサイズに亘る10〜120塩基のPS-ONランダマーを試験した。HSV-1 (KOS株)を用いて、VERO細胞内でプラーク減少アッセイを実施した。REP 2003、REP 2009、REP 2010、REP 2011、REP 2012、REP 2004、REP 2006、REP 2007およびREP 2008の濃度を上昇させながら、広範なサイズのPS-ONランダマーを試験した。直線回帰からIC50値を計算した。本発明者らは、12塩基およびそれより大きいオリゴが、検出可能な抗HSV-1活性を有し、HSV-1に対する効力は、PS-ONランダマー長が少なくとも120塩基迄上昇すると共に増大することも発見した。しかしながら、サイズにおける塩基の増加による効力の上昇は、40塩基より大きいPS-ONランダマーでより少ない。 To further define the effective size range of PS-ON for anti-HSV-1 activity, we tested 10-120 base PS-ON randomers over a wider range of sizes. Plaque reduction assay was performed in VERO cells using HSV-1 (KOS strain). A wide range of PS-ON randomers were tested with increasing concentrations of REP 2003, REP 2009, REP 2010, REP 2011, REP 2012, REP 2004, REP 2006, REP 2007 and REP 2008. IC 50 values were calculated from linear regression. We found that 12 bases and larger oligos have detectable anti-HSV-1 activity, and potency against HSV-1 increases with increasing PS-ON randomer length to at least 120 bases. Also found. However, the increase in potency with increasing bases in size is less with PS-ON randomers larger than 40 bases.
非PS-ONランダマー、ランダム配列PS-ONおよびHSV-1特異的配列PS-ONの効力を比較するために、本発明者らは、サイズ10、20および40塩基のONに対するこれらの3つの種類の修飾を試験した。HSV-1(KOS株)を用いて、VERO細胞内でプラーク減少アッセイを実施した。非修飾ON、ランダム配列を伴うPS-ON、およびHSV-1の開始コドンIE110を標的とするPS-ONを、濃度を上昇させて試験した。ONは、REP 2013、REP 2014、REP 2015、REP 2016、REP 2017、REP 2018、REP 2019、REP 2020およびREP 2021であった。直線回帰からIC50値を計算した。この系では、非修飾ONランダマーは、試験したサイズにおいて検出可能な抗HSV-1活性を有さない。ランダム配列およびHSV-1特異的配列PS-ONの双方は、サイズ依存性の抗HSV-1活性を示す(これらの修飾のいずれも10塩基では活性を認めない)。ランダム配列、HSV-1特異的配列およびランダマーPS-ONの比較により、20塩基長のPS-ONでは、HSV-1特異的配列による抗HSV-1活性の増大が存在したが、40塩基長では、全修飾、ランダマー、ランダム配列またはHSV-1特異的配列のいずれも、HSV-1に対して同等に有効であることが示された。 In order to compare the potency of non-PS-ON randomers, random sequence PS-ON and HSV-1 specific sequence PS-ON, we have identified these three types for ONs of size 10, 20 and 40 bases. The modification of was tested. Plaque reduction assay was performed in VERO cells using HSV-1 (KOS strain). Unmodified ON, PS-ON with random sequence, and PS-ON targeting HSV-1 start codon IE110 were tested at increasing concentrations. ON were REP 2013, REP 2014, REP 2015, REP 2016, REP 2017, REP 2018, REP 2019, REP 2020 and REP 2021. IC 50 values were calculated from linear regression. In this system, the unmodified ON randomer has no detectable anti-HSV-1 activity at the size tested. Both the random sequence and the HSV-1 specific sequence PS-ON show size-dependent anti-HSV-1 activity (none of these modifications are active at 10 bases). According to the comparison of random sequence, HSV-1 specific sequence and randomer PS-ON, there was an increase in anti-HSV-1 activity due to HSV-1 specific sequence in PS-ON of 20 base length, but in 40 base length All modifications, randomers, random sequences or HSV-1 specific sequences have been shown to be equally effective against HSV-1.
本発明者らが知る限り、PS-ONに関して、0.059μMおよび0.043μMもの低いHSV-1に関するIC50が報告されたことは、これが初めてである。 To the best of our knowledge, this is the first time that an IC 50 for HSV-1 as low as 0.059 μM and 0.043 μM has been reported for PS-ON.
実施例2:HSV-2の阻害
PS-ONによるHSV-2阻害の能力を、PRAにより測定する。不死化アフリカミドリザル腎臓(VERO)細胞を、37℃および5% CO2において、ゲンタマイシン、バンコマイシンおよびアンフォテリシンBで補充したMEMならびに10%ウシ胎児血清中で培養する。細胞を、増殖から4日後に細胞のコンフルエントな単層を得る密度で、12ウェルプレート内に播種する。コンフルエンシーに到達したら、培地を、5%血清および上記の補充物のみを含有するように交換し、次いで細胞を、試験化合物の存在下、HSV-2 (MS2株、合計約40〜60PFU)に90分間暴露する。ウイルス暴露後、培地を、5%血清、1%ヒト免疫グロブリン、上記の補充物および試験化合物を含有する新しい「オーバーレイ」培地で代替する。感染から3〜4日間後、感染培養物をホルマリン固定およびクレシルバイオレット染色した後、プラーク計数を実施する。
Example 2: Inhibition of HSV-2
The ability of HS--2 inhibition by PS-ON is measured by PRA. Immortalized African green monkey kidney (VERO) cells are cultured in MEM supplemented with gentamicin, vancomycin and amphotericin B and 10% fetal bovine serum at 37 ° C. and 5% CO 2 . Cells are seeded in 12-well plates at a density that gives a confluent monolayer of cells 4 days after growth. When confluency is reached, the medium is changed to contain only 5% serum and the above supplements, and the cells are then transferred to HSV-2 (MS2 strain, total about 40-60 PFU) in the presence of the test compound. Exposure for 90 minutes. After virus exposure, the medium is replaced with fresh “overlay” medium containing 5% serum, 1% human immunoglobulin, the above supplements and test compounds. Three to four days after infection, plaque counts are performed after infected cultures are formalin fixed and cresyl violet stained.
PS-ONによるHSV-2阻害の能力を試験するために、REP 1001、2001および3007を、HSV-2 PRAで試験する。HSV-2(MS2株)を用いて、感染した細胞を、濃度を増大させたREP 1001、REP 2001またはREP 3007により処理して、ヒト神経芽細胞内でプラーク減少アッセイを実施した。直線回帰からIC50値を計算した。阻害活性が、アンチセンスまたは他の配列相補的な機序によるものであれば、REP 2001のみ任意の活性を示すと予期され、それはこのPS-ONがHSV-1/2内の複製起点に指向されているためである(他の2つは、各々、ヒトおよびプラスミド内の複製起点に指向されている)。3つのPS-ON全部が、抗HSV-2活性を示した。更に、抗HSV-2効果の有効性は、PS-ONのサイズに依存し、配列に依存しない。 To test the ability of HSV-2 inhibition by PS-ON, REP 1001, 2001 and 3007 are tested with HSV-2 PRA. Using HSV-2 (MS2 strain), infected cells were treated with increasing concentrations of REP 1001, REP 2001 or REP 3007 to perform plaque reduction assays in human neuroblasts. IC 50 values were calculated from linear regression. If the inhibitory activity is due to antisense or other sequence-complementary mechanisms, only REP 2001 is expected to show any activity, which indicates that this PS-ON is directed to the origin of replication within HSV-1 / 2 (The other two are directed to the origin of replication in humans and plasmids, respectively). All three PS-ONs showed anti-HSV-2 activity. Furthermore, the effectiveness of the anti-HSV-2 effect depends on the size of the PS-ON and not on the sequence.
PS-ONの抗HSV-2活性上のサイズ依存性および配列非依存性を確認するために、本発明者らは、様々なサイズのPS-ON (REP 2001、2002、2003、2004、2005および2006)を試験した。これらPS-ONは、各塩基を「ゆらぎ」(N)として合成して、各PS-ONが実際に同一サイズの異なるランダム配列を有する集団とすることによって、配列特異的な効果に関して不活性にされており、これらのPS-ONは「ランダマー」と称される。これらのPS-ONをHSV-2PRAで試験する場合、本発明者らは、10塩基またはそれ未満のオリゴは、検出可能な抗HSV-2活性を有さないが、PS-ONのサイズが10塩基より大きく増加するにつれ、有効性もまた上昇する(IC50低下)ことを見い出す。本発明者らはまた、20塩基より大きいPS-ONは、臨床的に許容される抗HSV-2薬、アシクロビル(商標)と比較して有意に低いIC50値を有することにも注目した。 In order to confirm the size-dependence and sequence-independence on the anti-HSV-2 activity of PS-ON, we have determined that various sizes of PS-ON (REP 2001, 2002, 2003, 2004, 2005 and 2006). These PS-ONs are inactive with respect to sequence-specific effects by synthesizing each base as a “fluctuation” (N) and making each PS-ON actually a population with random sequences of the same size. These PS-ONs are called “Randamers”. When testing these PS-ONs with HSV-2PRA, we found that oligos of 10 bases or less had no detectable anti-HSV-2 activity, but the size of PS-ON was 10 We find that as we increase larger than the base, the effectiveness also increases (IC 50 reduction). We also noted that PS-ONs greater than 20 bases have significantly lower IC 50 values compared to the clinically acceptable anti-HSV-2 drug, Acyclovir ™.
本発明者らが知る限り、PS-ONに関して、0.012μMもの低いHSV-2に関するIC50が報告されたことは、これが初めてである。 To the best of our knowledge, this is the first time that an IC 50 for HSV-2 as low as 0.012 μM has been reported for PS-ON.
配列非特異的な組成が、ONの抗ウイルス活性上に効果を有するか否かを測定するために、等価なサイズを有するが、配列組成が異なる数種のPS-ONの抗HSV1活性を、HSV-1 PRAで試験した。試験したPS-ONは、REP 2006 (N20)、REP 2028 (G40)、REP 2029 (A40)、REP 2030 (T40)およびREP 2031 (C40)であった。HSV-1 PRAから生成したIC50値は、REP 2006 (N40)が、試験した全配列中最も活性が高かった一方で、REP 2029 (A40)は最も活性が低かったことを示す。本発明者らは、他の全PS-ONは、N40と比較して有意に低い活性を有し、効力の点でN40>C40>T40>A40>G40と序列されることにも注目する。 In order to determine whether a non-sequence specific composition has an effect on the antiviral activity of ON, the anti-HSV1 activity of several PS-ONs with equivalent size but different sequence composition Tested with HSV-1 PRA. The PS-ONs tested were REP 2006 (N20), REP 2028 (G40), REP 2029 (A40), REP 2030 (T40) and REP 2031 (C40). IC 50 values generated from HSV-1 PRA indicate that REP 2006 (N40) was the most active of all sequences tested, while REP 2029 (A40) was the least active. We also note that all other PS-ONs have significantly lower activity compared to N40 and are ordered N40>C40>T40>A40> G40 in terms of potency.
本発明者らはまた、2つの異なるヌクレオチドを用いて、様々な配列組成を有する異なるPS ONの効力を試験した。PS-ONランダマー(REP 2006)は、HSV-1に対して、AC20 (REP 2055)、TC20 (REP 2056)またはAG20 (REP 2057)と比較して有意に高い効力を有し、それらの効力は、以下のように序列された:N40>AG>AC>TC。このデータは、抗ウイルス効果が配列相補的ではなく、非特異的な配列組成(即ち、C40およびN40)は、より強力な抗ウイルス活性を有することを示唆する。本発明者らは、この現象が固有のタンパク質結合能を保持する一方で、C40、A40、T40およびG40のような配列が、おそらく、これらの配列内で形成されたいくつかの制限的な三次構造により、より少数のウイルスタンパク質と高い親和性で結合する事実により説明することができることを提案する。その一方、N40のランダムな性質により、N40は広範囲の抗ウイルスタンパク質に高い親和性で結合する能力を維持し、このことは、その強力な抗ウイルス活性に寄与する。 We also tested the efficacy of different PSONs with various sequence compositions using two different nucleotides. PS-ON randomer (REP 2006) has significantly higher potency against HSV-1 compared to AC20 (REP 2055), TC20 (REP 2056) or AG20 (REP 2057) , Ordered as follows: N40> AG> AC> TC. This data suggests that the antiviral effect is not sequence complementary and that the non-specific sequence composition (ie C40 and N40) has a stronger antiviral activity. We have observed that while this phenomenon retains the intrinsic protein binding ability, sequences such as C40, A40, T40 and G40 probably have some restricted tertiary formation formed within these sequences. We propose that the structure can be explained by the fact that it binds with a smaller number of viral proteins with high affinity. On the other hand, due to the random nature of N40, N40 maintains the ability to bind to a wide range of antiviral proteins with high affinity, which contributes to its strong antiviral activity.
実施例3:CMVの阻害
PS-ONによるCMV阻害の能力を、プラーク減少アッセイ(PRA)にて測定する。このアッセイは、CMV(AD169株)をウイルス接種材料として使用し、かつヒト神経芽繊維を細胞宿主として使用した以外は、抗HSV-1および抗HSV-2の試験に用いたアッセイと同一である。
Example 3: Inhibition of CMV
The ability of PS-ON to inhibit CMV is measured by plaque reduction assay (PRA). This assay is identical to the assay used for anti-HSV-1 and anti-HSV-2 tests, except that CMV (strain AD169) was used as the virus inoculum and human neuroblast was used as the cell host. .
PS-ONの抗CMV活性上のサイズ依存性および配列非依存性を試験するために、本発明者らは、様々なサイズのPS-ONランダマーを試験した。CMV (AD169株)を用いて、VERO細胞内でプラーク減少アッセイを実施した。感染細胞を、濃度を増大させたREP 2004 (a)またはREP 2006 (b)で処理した。直線回帰からIC50値を計算し、CMVに対するPS-ONサイズとIC50との関係を、試験した各PS-ONの特定のサイズに対してIC50値をプロットすることにより決定した。これらのPS-ONをCMV PRAにて試験する場合、本発明者らは、PS-ONのサイズが増大するにつれ、有効性もまた上昇する(IC50低下)ことを発見する。 To test the size-dependence and sequence-independence on the anti-CMV activity of PS-ON, we tested various sizes of PS-ON randomers. Plaque reduction assay was performed in VERO cells using CMV (AD169 strain). Infected cells were treated with increasing concentrations of REP 2004 (a) or REP 2006 (b). IC 50 values were calculated from linear regression and the relationship between PS-ON size and IC 50 versus CMV was determined by plotting IC 50 values against the specific size of each PS-ON tested. When testing these PS-ON at CMV PRA, the present inventors have found that as the size of the PS-ON is increased, it discovers that effectiveness also increases (IC 50 decreases).
抗CMV活性に関するPS-ONの有効なサイズ範囲をより明らかにするために、本発明者らは、より広範なサイズに亘る10〜80塩基のPS-ONランダマーを試験した。本発明者らはまた、臨床的に許容できる数種の小分子CMVによる処理(ガンシクロビル、ホスカルネットおよびシドフォビル)ならびにCMV網膜炎の2バージョンの市販のアンチセンス処理(Vitravene(商標);University of Calgaryにより合成および市販されている)も含めた。CMV(AD169株)を用いて、VERO細胞内でプラーク減少アッセイを実施した。3種の臨床的CMV治療を試験した:ガンシクロビル、ホスカルネットおよびシドフォビル。REP 2003、REP 2004、REP 2006、およびREP 2007の濃度を上昇させ、広範なPS-ONランダマーサイズも試験した。最後に、室内で合成したおよび市販されているREP 2036 (Vitravene)を試験した。直線回帰からIC50値を計算した。本発明者らは、PS-ONランダマーサイズの上昇が効力の増大をもたらす一方、この効果は、約40塩基にて飽和することを発見した。更に、20、40および80塩基のPS-ONランダマーは全て、試験した任意の小分子治療と比較して有意に高い効力を有する。加えて、40および80塩基のPS-ONランダマーは、Vitravene(商標)と比較して高い効力を有する。 To further clarify the effective size range of PS-ON for anti-CMV activity, we tested 10-80 base PS-ON randomers over a wider range of sizes. We also treated with several clinically acceptable small molecule CMVs (ganciclovir, foscarnet and cidofovir) and two versions of CMV retinitis commercially available antisense treatment (Vitravene ™; University of Also synthesized and marketed by Calgary). Plaque reduction assay was performed in VERO cells using CMV (AD169 strain). Three clinical CMV treatments were tested: ganciclovir, foscarnet and cidofovir. Increasing concentrations of REP 2003, REP 2004, REP 2006, and REP 2007 also tested a wide range of PS-ON randomer sizes. Finally, REP 2036 (Vitravene) synthesized indoors and commercially available was tested. IC 50 values were calculated from linear regression. We have found that an increase in PS-ON randomer size results in increased potency, while this effect saturates at about 40 bases. Furthermore, 20, 40 and 80 base PS-ON randomers all have significantly higher potency compared to any small molecule therapy tested. In addition, 40- and 80-base PS-ON randomers have higher potency compared to Vitravene ™.
本発明者らが知る限り、PS-ONに関して、0.067μMもの低いCMVに関するIC50が報告されたことは、これが初めてである。 To the best of our knowledge, this is the first time that an IC 50 for CMV as low as 0.067 μM has been reported for PS-ON.
実施例4:HIV-1の阻害
2つの異なるアッセイにより、PS-ONのHIV-1を阻害する能力を測定した。
Example 4: Inhibition of HIV-1
Two different assays measured the ability of PS-ON to inhibit HIV-1.
細胞変性効果(CPE)
細胞の代謝程度を報告するため、MTT色素を用いて細胞変性効果をモニターする。10%ウシ胎児血清を加え、抗生物質を補完したMEM中で、37℃、5% CO2にて不死化したヒトリンパ球(MT4)を培養する。96ウェルプレートにて適当な試験化合物を含有する培地に細胞を播き2時間インキュベートする。試験化合物との予備インキュベートの後、HIV-1(NL4-3株)をウエルに加えた(0.0002TCID50/細胞)。6日間の追加のインキュベートの後、MTT変換によりCPEをモニターする。ウイルス播種の非存在下で6日間薬剤をインキュベートすることにより細胞障害性を測定する。MTTの吸収値を%生存率に変換するには、非感染未処置細胞における吸収を100%に設定し、感染未処置細胞の吸収を0%に設定する。
Cytopathic effect (CPE)
To report the degree of cellular metabolism, the cytopathic effect is monitored using MTT dye. Human lymphocytes (MT4) immortalized at 37 ° C. and 5% CO 2 are cultured in MEM supplemented with antibiotics with 10% fetal bovine serum. Cells are seeded in medium containing the appropriate test compound in a 96 well plate and incubated for 2 hours. After preincubation with the test compound, HIV-1 (strain NL4-3) was added to the wells (0.0002TCID 50 / cell). After 6 days of additional incubation, CPE is monitored by MTT conversion. Cytotoxicity is measured by incubating the drug for 6 days in the absence of virus seeding. To convert the MTT absorption value to% viability, the absorption in uninfected untreated cells is set to 100% and the absorption of infected untreated cells is set to 0%.
複製アッセイ(RA)
不死化したヒト胎児腎臓細胞(293A)においてHIVの複製能力をモニターする。2種のプラスミドをこれらの細胞に同時形質移入する。一方のプラスミドはルシフェラーゼ発現カセットを介入させたenv遺伝子を有する組換え野生型HIV-1ゲノム(NL4-3)を含有し(CNDO株として同定される)、他方のプラスミドはマウス白血病ウイルス(MLV)に由来するenv遺伝子を含有する。これら2種のプラスミドは、MLVのenv遺伝子に由来するトランスで提供されるタンパク質産物を除いて、HIV-1に由来するすべての成分を有する成熟キメラウイルスを産生するあらゆるタンパク質因子をトランスで提供する。これらの細胞から産生されるビリオンは感染性であり複製するが、env遺伝子を欠くために感染性ビリオンの次世代を産生することはできない。
Replication assay (RA)
HIV replication capacity is monitored in immortalized human fetal kidney cells (293A). Two plasmids are co-transfected into these cells. One plasmid contains the recombinant wild-type HIV-1 genome (NL4-3) with the env gene intervening luciferase expression cassette (identified as CNDO strain), the other plasmid is murine leukemia virus (MLV) Contains the env gene derived from These two plasmids provide in trans any protein factor that produces mature chimeric virus with all components derived from HIV-1, except for the protein product provided in trans from the env gene of MLV . The virions produced from these cells are infectious and replicate, but they cannot produce the next generation of infectious virions due to the lack of the env gene.
形質移入の24時間後、これらの細胞をトリプシン処理し、96ウェルプレートに播く。細胞が付着した後、培地を洗浄し、試験化合物を含有する培地と交換する。さらなる24時間の間にウイルス産生が進む。次いで、上清を回収し、元のままの293A細胞を再び感染させるのに使用する。ルシフェラーゼ遺伝子産物によって感染させた元の細胞を同定する。ルシフェラーゼ陽性細胞の数が、組換えHIV-1による複製及び/又は感染の程度の測定値である。幾つかの異なる部類の抗HIV薬に耐性を誘導することが知られている幾つかの点突然変異を持つHIV-1ゲノムを用いることにより多数の臨床的に認可されている抗HIV-1薬に対する耐性を調べるのにも、このアッセイが適用される。パーセント阻害は、検出可能なルシフェラーゼ陽性細胞がない場合を100%、感染細胞における陽性細胞の数を0%、未処置細胞を対照に設定する。 Twenty-four hours after transfection, these cells are trypsinized and seeded in 96-well plates. After the cells attach, the medium is washed and replaced with medium containing the test compound. Virus production proceeds in another 24 hours. The supernatant is then collected and used to reinfect intact 293A cells. The original cells infected with the luciferase gene product are identified. The number of luciferase positive cells is a measure of the extent of replication and / or infection by recombinant HIV-1. Numerous clinically approved anti-HIV-1 drugs by using the HIV-1 genome with several point mutations known to induce resistance to several different classes of anti-HIV drugs This assay is also applied to examine resistance to. Percent inhibition is set as 100% when there are no detectable luciferase positive cells, 0% positive cells in infected cells, and untreated cells as controls.
PS-ONの抗HSV-1活性上のサイズ依存性および配列非依存性を試験するために、本発明者らは、様々なサイズのPS-ONランダマーを試験した。HIV-1 (NL4-3株)を用いて、MT4細胞内でCPEアッセイを実施した。感染した細胞を、濃度を増大させたREP 2004またはREP 2006で処理した。直線回帰からIC50値を計算した。REP 2004およびREP 2006に関する非感染MT4細胞内での細胞毒性のプロファイルを測定した。本発明者らは、PS-ONのサイズが増大するにつれ、有効性もまた上昇(IC50低下)することを見出した。本発明者らはまた、このアッセイにてPS-ONランダマーが宿主細胞に有意な毒性を示さないことにも注目した。 To test the size-dependence and sequence-independence on the anti-HSV-1 activity of PS-ON, we tested various sizes of PS-ON randomers. CPE assay was performed in MT4 cells using HIV-1 (NL4-3 strain). Infected cells were treated with increasing concentrations of REP 2004 or REP 2006. IC 50 values were calculated from linear regression. Cytotoxicity profiles in uninfected MT4 cells for REP 2004 and REP 2006 were measured. The inventors have found that as PS-ON size increases, the effectiveness also increases (IC 50 decreases). The inventors also noted that PS-ON randomers did not show significant toxicity to host cells in this assay.
本発明者らが知る限り、PS-ONに関して、0.011μMもの低いHIV-1に関するIC50が報告されたことは、これが初めてである。 To the best of our knowledge, this is the first time that an IC 50 for HIV-1 as low as 0.011 μM has been reported for PS-ON.
抗HIV-1活性に関するPS-ONの有効なサイズ範囲をより明らかにするために、本発明者らは、野生型HIV-I(組換えNL4-3 (CNDO))を用いたRAにより、より広範なサイズに亘る10〜80塩基のPS-ONランダマーを試験した。組換え野生型HIV-1 NL4-3 (CNDO株)を用いて、293A細胞内で複製アッセイを実施した。加えて、本発明者らは、現在臨床で使用されている4つのプロテアーゼ阻害剤(アンプレナビル(aprenavir)、インジナビル、ロピナビルおよびサキナビル)を試験した。感染した細胞を、濃度を増大させたアンプレナビル、インジナビル、ロピナビル、サキナビル、REP 2003、REP 2004、REP 2006およびREP 2007により処理した。本発明者らは、10塩基およびそれより大きいPS-ONランダマーは抗HIV-1活性を有し、およびHIV-1に対する効力は、PS-ONランダマーの長さの上昇と共に増大するが、約40塩基にて飽和することを発見した。更に、40および80塩基のPS-ONランダマーは、4つの臨床的対照による効力と殆ど等価であった。 To further elucidate the effective size range of PS-ON for anti-HIV-1 activity, we made more by RA with wild-type HIV-I (recombinant NL4-3 (CNDO)). PS-ON randomers of 10-80 bases over a wide range of sizes were tested. Replication assays were performed in 293A cells using recombinant wild type HIV-1 NL4-3 (CNDO strain). In addition, we tested four protease inhibitors currently in clinical use (amprenavir, indinavir, lopinavir and saquinavir). Infected cells were treated with increasing concentrations of amprenavir, indinavir, lopinavir, saquinavir, REP 2003, REP 2004, REP 2006 and REP 2007. We found that PS-ON randomers with 10 bases and larger have anti-HIV-1 activity, and potency against HIV-1 increases with increasing length of PS-ON randomers, but about 40 Found to be saturated with base. Furthermore, the 40- and 80-base PS-ON randomers were almost equivalent to the efficacy with the four clinical controls.
本発明者らが知る限り、PS-ONに関して、0.014μMもの低いHIV-1に関するIC50が報告されたことは、これが初めてである。 To the best of our knowledge, this is the first time that an IC 50 for HIV-1 as low as 0.014 μM has been reported for PS-ON.
PS-ONランダマーの、HIVの薬物耐性株阻害の能力を試験するために、本発明者らは、HIV-1の組換えMDRC4株を用いて、上記の試験を再現した。この組換え株は、臨床的に許容される薬物の全クラス:ヌクレオチドRT阻害剤、非ヌクレオチドRT阻害剤およびプロテアーゼ阻害剤のうち少なくとも16種に対して有意な耐性を示す。本発明者らは、試験した全部のPS-ONランダマーが、耐性株に対して、野生型株に対すると同様に機能することを見出した。しかしながら、4種のプロテアーゼ阻害剤のうち3種は、変異型株に対する効力が低下し、従って40および80塩基PS-ONランダマーは、これらの薬物と比較して、この耐性株に対してより強力であることが示された。 In order to test the ability of PS-ON randomers to inhibit HIV drug-resistant strains, we reproduced the above test using a recombinant MDRC4 strain of HIV-1. This recombinant strain exhibits significant resistance to at least 16 of all classes of clinically acceptable drugs: nucleotide RT inhibitors, non-nucleotide RT inhibitors and protease inhibitors. The inventors have found that all PS-ON randomers tested function similarly to resistant strains as to wild type strains. However, three of the four protease inhibitors have reduced potency against mutant strains, so 40- and 80-base PS-ON randomers are more potent against this resistant strain compared to these drugs. It was shown that.
実施例5:RSVの阻害
PS-ONランダマーによるRSV阻害の能力を、CPEをアラマーブルー(細胞代謝の間接的指標)で監視することにより測定する。ヒト喉頭癌(Hep2)細胞を、37℃および5% CO2において、5%ウシ胎児血清を加えたMEM中で培養する。細胞を、増殖から5〜6日後に細胞のコンフルエントな単層を得る密度で、96ウェルプレート内に播種する。プレーティングの翌日、細胞を、試験化合物の存在下、低下した容積にて、RSV (A2株、108.2TCID50/mL)で2時間感染させた。接種後、培地を交換して、試験化合物を補充した。感染から6日後、アラマーブルーの蛍光変換を測定することにより、CPEを監視した。Hep2細胞内の試験化合物の毒性を、非感染細胞の7日間の処理、およびこれら細胞内のアラマーブルー変換の測定により監視した。非感染、未処理細胞内のアラマーブルー測定値を100%生存に設定し、感染、未処理細胞内の測定値を0%生存に設定した。
Example 5: Inhibition of RSV
The ability of PS-ON randomers to inhibit RSV is measured by monitoring CPE with Alamar Blue (an indirect indicator of cellular metabolism). Human laryngeal carcinoma (Hep2) cells are cultured in MEM supplemented with 5% fetal calf serum at 37 ° C. and 5% CO 2 . Cells are seeded in 96-well plates at a density that yields a confluent monolayer of cells 5-6 days after growth. The day after plating, cells were infected with RSV (A2 strain, 10 8.2 TCID 50 / mL) in reduced volume in the presence of test compound for 2 hours. After inoculation, the medium was changed and supplemented with the test compound. Six days after infection, CPE was monitored by measuring the fluorescence conversion of alamar blue. The toxicity of test compounds in Hep2 cells was monitored by treatment of uninfected cells for 7 days and measurement of alamar blue conversion in these cells. The measurement value of alamar blue in uninfected and untreated cells was set to 100% survival, and the measurement value in infected and untreated cells was set to 0% survival.
PS-ONの抗RSV活性上のサイズ依存性および配列非依存性を確認するために、本発明者らは、様々なサイズのPS-ONランダマーを試験した。加えて、本発明者らは、RSV感染のための臨床的に許容される処置、リバビリン(Virazole(商標))を試験した。RSV (A2株)を用いて、Hep2細胞内でCPEアッセイを実施した。感染細胞を、濃度を増大させたREP 2004、REP 2006、REP 2007、またはリバビリンで処理する。直線回帰からIC50値を計算し、各グラフに報告する。REP 2004、REP 2006、REP 2007またはリバビリンに関する非感染Hep2細胞内での細胞毒性のプロファイルを測定した。本発明者らは、PS-ONランダマーのサイズが増大するにつれ、有効性もまた上昇するが、約40塩基のサイズで飽和することを見出した。本発明者らはまた、20、40および80塩基のPS-ONランダマーが、臨床的に許容される抗RSV薬、リバビリンと比較して有意に低いIC50値を有することにも注目した。リバビリンが有意な毒性(治療係数=2.08)を示した一方、PS-ONランダマーはHep2細胞内で毒性を示さなかった。 To confirm the size dependence and sequence independence on the anti-RSV activity of PS-ON, we tested various sizes of PS-ON randomers. In addition, we tested a clinically acceptable treatment for RSV infection, ribavirin (Virazole ™). CPE assay was performed in Hep2 cells using RSV (A2 strain). Infected cells are treated with increasing concentrations of REP 2004, REP 2006, REP 2007, or ribavirin. IC 50 values are calculated from linear regression and reported on each graph. The cytotoxicity profile in non-infected Hep2 cells for REP 2004, REP 2006, REP 2007 or ribavirin was measured. The inventors have found that as PS-ON randomer size increases, effectiveness also increases, but saturates at a size of about 40 bases. We also noted that the 20, 40 and 80 base PS-ON randomers have significantly lower IC 50 values compared to the clinically acceptable anti-RSV drug, ribavirin. Ribavirin showed significant toxicity (therapeutic index = 2.08), while PS-ON randomer showed no toxicity in Hep2 cells.
本発明者らが知る限り、PS-ONに関して、0.015μMもの低いRSV-1に関するIC50が報告されたことは、これが初めてである。 To the best of our knowledge, this is the first time that an IC 50 for RSV-1 as low as 0.015 μM has been reported for PS-ON.
実施例6:コクサッキーウイルスB2型の阻害
PS-ONランダマーによるCOX B2阻害の能力を、CPEをアラマーブルー(細胞代謝の間接的指標)で監視することにより測定する。アカゲザル腎臓(LLC-MK2)細胞を、37℃および5%CO2において、5%ウシ胎児血清を加えたMEM中で培養する。細胞を、増殖から5〜6日後に細胞のコンフルエントな単層を得る密度で、96ウェルプレート内に播種する。プレーティングの翌日、細胞を、試験化合物の存在下、低下した容積にて、COX B2 (Ohio-1株、107.8TCID50/mL)で2時間感染させた。接種後、培地を交換して、試験化合物を補充した。感染から6日後、アラマーブルーの蛍光変換を測定することにより、CPEを監視した。LLC-MK2細胞内の試験化合物の毒性を、非感染細胞の7日間の処理、およびこれら細胞内のアラマーブルー変換の測定により監視した。非感染、未処理細胞内のアラマーブルー測定値を100%生存に設定し、感染、未処理細胞内の測定値を0%生存に設定した。
Example 6: Inhibition of Coxsackievirus type B2
The ability of the PS-ON randomer to inhibit COX B2 is measured by monitoring CPE with Alamar Blue (an indirect indicator of cellular metabolism). Rhesus monkey kidney (LLC-MK2) cells are cultured in MEM supplemented with 5% fetal calf serum at 37 ° C. and 5% CO 2 . Cells are seeded in 96-well plates at a density that yields a confluent monolayer of cells 5-6 days after growth. The day after plating, cells were infected with COX B2 (Ohio-1 strain, 10 7.8 TCID 50 / mL) in reduced volume in the presence of test compound for 2 hours. After inoculation, the medium was changed and supplemented with the test compound. Six days after infection, CPE was monitored by measuring the fluorescence conversion of alamar blue. Toxicity of test compounds in LLC-MK2 cells was monitored by treatment of uninfected cells for 7 days and measurement of alamar blue conversion in these cells. The measurement value of alamar blue in uninfected and untreated cells was set to 100% survival, and the measurement value in infected and untreated cells was set to 0% survival.
本発明者らは、COX B2 CPEアッセイにおいて、REP 2006の抗COX B2活性を試験した。コクサッキーウイルスB2型(Ohio-1株)を用いて、LLC-MK2細胞内でCPEアッセイを実施した。感染細胞を、濃度を増大させたREP 2006により処理した。REP 2006に関するLLC-MK2細胞内での細胞毒性のプロファイルを測定した。本発明者らは、このPS-ONランダマーが、LLC-MK2細胞内で、ある僅かな毒性を示す一方、感染LLC-MK2細胞を、ある程度COX B2感染から救い得ることを見出した。 We tested the anti-COX B2 activity of REP 2006 in the COX B2 CPE assay. CPE assay was performed in LLC-MK2 cells using Coxsackievirus type B2 (Ohio-1 strain). Infected cells were treated with REP 2006 at increasing concentrations. The cytotoxicity profile in LLC-MK2 cells for REP 2006 was measured. The present inventors have found that this PS-ON randomer exhibits some slight toxicity in LLC-MK2 cells, while infecting LLC-MK2 cells can be rescued to some extent from COX B2 infection.
実施例7:ワクシニアウイルスの阻害
本発明者らは、天然痘ウイルスを含むポックスウイルスに対する本発明者らの化合物の効力の指標として、ワクシニア感染モデルを使用した。PS-ONランダマーによるワクシニア阻害の能力を、CPEをアラマーブルー(細胞代謝の間接的指標)で監視することにより測定する。Vero細胞を、37℃および5% CO2において、5%ウシ胎児血清を加えたMEM中で培養する。細胞を、増殖から5〜6日後に細胞のコンフルエントな単層を得る密度で、96ウェルプレート内に播種する。プレーティングの翌日、細胞を、試験化合物の存在下、低下した容積にて、ワクシニア(107.9TCID50/mL)で2時間感染させた。接種後、培地を交換して、試験化合物(50μMで使用したシドフォビルを除いて、全て10μM)を補充した。感染から5日後、上清を回収した。VERO細胞を1:100に希釈した上清で再感染させて、再感染から7日後、アラマーブルーの蛍光変換を測定することによりCPEを監視して、上清中のウイルス負荷を測定した。
Example 7: Inhibition of vaccinia virus We used a vaccinia infection model as an indicator of the efficacy of our compounds against poxviruses, including smallpox virus. The ability of PS-ON randomer to inhibit vaccinia is measured by monitoring CPE with Alamar Blue (an indirect indicator of cellular metabolism). Vero cells are cultured in MEM supplemented with 5% fetal calf serum at 37 ° C. and 5% CO 2 . Cells are seeded in 96-well plates at a density that yields a confluent monolayer of cells 5-6 days after growth. The day after plating, cells were infected with vaccinia (10 7.9 TCID 50 / mL) in reduced volume in the presence of test compound for 2 hours. After inoculation, the medium was changed and supplemented with test compounds (all 10 μM except for cidofovir used at 50 μM). Supernatant was collected 5 days after infection. VERO cells were reinfected with 1: 100 diluted supernatant and 7 days after reinfection, CPE was monitored by measuring fluorescence conversion of alamar blue to determine viral load in the supernatant.
本発明者らは、様々なサイズのPS-ONランダマー(REP 2004、2006および2007)を試験した。加えて、本発明者らは、ワクシニア感染の公知の有効な治療薬、シドフォビル(Vistide(商標))を試験した。ワクシニア感染VERO細胞からの感染上清中のウイルス負荷の間接的な測定は、これら上清により誘導されたナイーブ細胞内のCPEを測定することにより決定した。REP 2004、2006および2007は10μMで試験した一方、シドフォビルは50μMで試験した。ワクシニアCPEアッセイにて試験した場合、本発明者らは、REP 2004、2006および2007による処理が、全て抗ウイルス活性を示した(即ち、再感染により上清中のCPEの減少が示された)が、この活性は、シドフォビルに関して観察されたものと比較して弱いことを見出した。 We tested various sizes of PS-ON randomers (REP 2004, 2006 and 2007). In addition, we tested a known effective treatment for vaccinia infection, cidofovir (Vistide ™). Indirect measurement of viral load in infected supernatants from vaccinia infected VERO cells was determined by measuring CPE in naive cells induced by these supernatants. REP 2004, 2006 and 2007 were tested at 10 μM, while cidofovir was tested at 50 μM. When tested in the vaccinia CPE assay, we all treated with REP 2004, 2006 and 2007 showed antiviral activity (i.e., reinfection showed a decrease in CPE in the supernatant). However, this activity was found to be weak compared to that observed for cidofovir.
実施例8:DHBV、パラインフルエンザ3型ウイルスおよびハンタウイルスの阻害
DHBV、パラインフルエンザ3型ウイルスおよびハンタウイルスは、測定可能なプラークまたはCPEを容易に生成しないため、本発明者らは、蛍光焦点形成ユニット(FFFU)検出により、これらウイルス内のREP 2006の効力を試験した。このアッセイでは、REP 2006(最終濃度10μMにおける)をウイルスと混合し、次いでこれを細胞上に吸着させる。吸着後、感染した細胞を更に7〜14日間インキュベートし、その時点でこれらをメタノール中で固定する。ウイルス複製の領域を、適切なウイルス抗原に対する免疫蛍光顕微鏡により検出する。試験した3つのウイルスの各々に関する特定の実験条件および結果を、下の表1に示す。
Example 8: Inhibition of DHBV, parainfluenza 3 virus and hantavirus
Since DHBV, parainfluenza type 3 virus and hantavirus do not readily produce measurable plaques or CPE, we have demonstrated the efficacy of REP 2006 within these viruses by detecting fluorescence focus forming units (FFFU). Tested. In this assay, REP 2006 (at a final concentration of 10 μM) is mixed with the virus, which is then adsorbed onto the cells. After adsorption, the infected cells are further incubated for 7-14 days, at which time they are fixed in methanol. The region of viral replication is detected by immunofluorescence microscopy against the appropriate viral antigen. Specific experimental conditions and results for each of the three viruses tested are shown in Table 1 below.
(表1)DHBV、パラインフルエンザ3型ウイルスおよびハンタウイルスの阻害
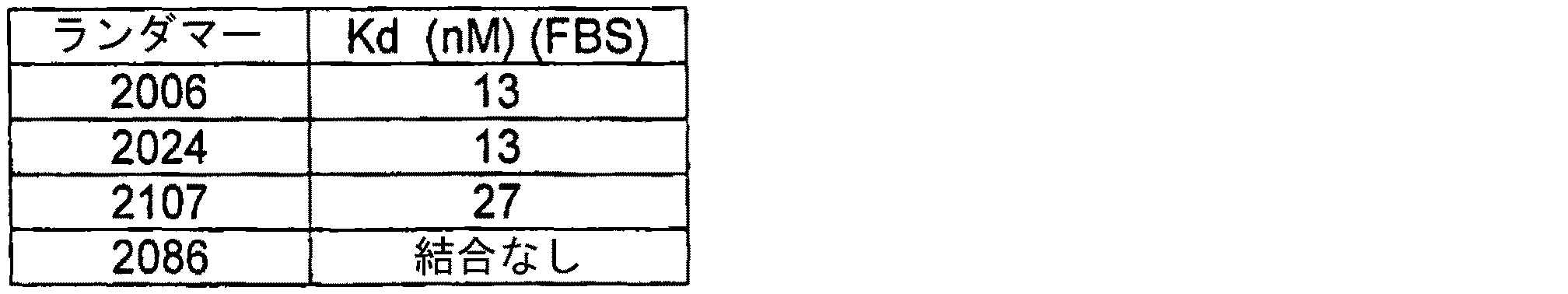
この最初のデータは、10μMにおいて、REP 2006は、DHBV、パラインフルエンザ2型およびハンタウイルスの阻害に有効であることを示す。本発明者らは、予備試験における所与の強力な応答により、IC50値がより低いであろうことを予想する。これらのデータは、ハンタウイルスおよびB型肝炎(DHBVと密接に関連する)のヒト感染の処置のためのPS-ONランダマーの効力を支持すると共に、処置を開始するための鑑別診断を必要とし得ない、RSVおよびパラインフルエンザ3型に起因する小児気管支炎の急性処置のための原理を提供する。 This initial data shows that at 10 μM, REP 2006 is effective at inhibiting DHBV, parainfluenza 2 and hantaviruses. The present inventors have found that by a given potent responses in the preliminary test, expect that an IC 50 value would be lower. These data support the efficacy of PS-ON randomers for treatment of human infections with hantavirus and hepatitis B (closely associated with DHBV) and may require differential diagnosis to initiate treatment. Provide principles for the acute treatment of childhood bronchitis due to RSV and parainfluenza type 3.
実施例9:現在の非応答性ウイルス
今日迄、本発明者らは、表2に示す以下のウイルス系において、送達系、薬物の組み合わせまたは化学的修飾を使用しない、PS-ONランダマー(10μM迄)の検出可能な抗ウイルス効力を観察していない。
Example 9: Current Non-Responsive Viruses To date, we have not used delivery systems, drug combinations or chemical modifications in the following viral systems shown in Table 2, PS-ON randomers (up to 10 μM). ) No detectable antiviral efficacy.
(表2)ウイルス系
現在の試験手順の下では、本発明者らは活性の実証に至らなかった。しかしながら、実証された抗ウイルス活性の欠如は、使用した特定のアッセイの限界によるものであり得る。これらのウイルスの効力を実証するための更なる試験が進行中である。 Under the current test procedure, we did not demonstrate activity. However, the lack of demonstrated antiviral activity may be due to the limitations of the particular assay used. Further studies are ongoing to demonstrate the efficacy of these viruses.
本発明者らの証明は、数種の異なるファミリーのウイルスを阻害する際に、PS-ONの電荷特性が重要であることを示しているため、本発明者らは、ウイルス活性阻害のためのこの電荷依存性機序が、カプシド形成した全ウイルスの活性を阻害する可能性を有することを期待する。これに対する推論は、実施例9に列挙したそれらのウイルスに対する抗ウイルス効力が検出されないことは、これらウイルスの複製中の、PS-ONとこれらのウイルスの構造タンパク質との間の相互作用が、ウイルスタンパク質の相互作用を阻止するに十分強力ではない可能性を示唆する。この場合、これらウイルスに対する効力を実現する一つの方法は、DNAまたは抗ウイルスポリマーの電荷特性を変更(例えば、DNAのホスホロチオエート結合をホスホロジチオエートで置換)して、ウイルスタンパク質に対するそれらの親和性を増大させることである。 Our evidence shows that the charge properties of PS-ON are important in inhibiting several different families of viruses, so we have We hope this charge-dependent mechanism has the potential to inhibit the activity of all encapsidated viruses. The reasoning for this is that the antiviral efficacy against those viruses listed in Example 9 is not detected, because the interaction between PS-ON and the structural proteins of these viruses during replication of these viruses This suggests that it may not be strong enough to prevent protein interactions. In this case, one way to achieve efficacy against these viruses is to alter the charge properties of the DNA or antiviral polymer (e.g., to replace the phosphorothioate linkage of the DNA with a phosphorodithioate) so that their affinity for viral proteins. Is to increase.
実施例10:A型インフルエンザの阻害
PS-ONによるA型インフルエンザウイルス(INF)阻害の能力を、プラーク減少アッセイ(PRA)により測定する。不死化犬腎臓(MDCK)細胞を、37℃および5% CO2において、ゲンタマイシン、バンコマイシンおよびアンフォテリシンBで補充した、10%ウシ胎児血清を加えたMEM中で培養する。細胞を、増殖から6日後に細胞のコンフルエントな単層を得る密度で、6ウェルプレート内に播種する。コンフルエンシーに到達したら、培地を、上記の補充物のみを含有するよう交換し、次いで細胞を、試験化合物の存在下、INF A (H3N2株、合計約35〜70PFU)に60分間暴露する。ウイルス暴露後、培地を、薬物のみを含有する新しい培地で代替する。感染から24時間後、培地を再度、4%アルブミン、0.025% DEAEデキストラン、2mg/mL TPCK-処理トリプシンおよび0.8%シープラークアガロース、上記の補充物を含有し、試験化合物を含有しないオーバーレイ培地で代替する。感染から2〜3日間後、感染培養物をホルマリン固定およびクレシルバイオレット染色した後、プラーク計数を実施する。
Example 10: Inhibition of influenza A
The ability of PS-ON to inhibit influenza A virus (INF) is measured by plaque reduction assay (PRA). Immortalized canine kidney (MDCK) cells are cultured in MEM with 10% fetal calf serum supplemented with gentamicin, vancomycin and amphotericin B at 37 ° C. and 5% CO 2 . Cells are seeded in 6-well plates at a density that yields a confluent monolayer of cells after 6 days of growth. When confluency is reached, the medium is changed to contain only the above supplements, and the cells are then exposed to INF A (H3N2 strain, about 35-70 PFU total) for 60 minutes in the presence of the test compound. After virus exposure, the medium is replaced with fresh medium containing only the drug. 24 hours after infection, the medium is again replaced with 4% albumin, 0.025% DEAE dextran, 2 mg / mL TPCK-treated trypsin and 0.8% sheep plaque agarose, overlay medium containing the above supplements and no test compound. To do. Two to three days after infection, infected cultures are formalin fixed and cresyl violet stained before plaque counting.
本発明者らは、INF A PRAアッセイにおいて、多様なPS-ONランダマーの抗INF A活性を試験した。本発明者らは、REP 2006のみが任意の測定可能な抗ウイルス活性を示すが、しかしこの活性は、有意であることを見出した(下の表3参照)。 We tested the anti-INF A activity of various PS-ON randomers in the INF A PRA assay. We have found that only REP 2006 shows any measurable antiviral activity, but this activity is significant (see Table 3 below).
(表3)INF A(H3N2)に対するPS-ONランダマーの活性
最大のランダマーのみが、幾分かの活性を有するように思われ、本発明者らは他の多数のウイルスにおけるランダマーの活性が、サイズ依存性であったことを理解しているため、本発明者らは、より幅広い希釈範囲を用いて、より大きいサイズに分布するランダマーの抗ウイルス活性を試験した。本発明者らは、本発明者らが試験した他のウイルスに関して、ランダマーの抗INF A活性は、それらの長さが増大するにつれより強力となるが、40塩基長を越えるランダマーに関しては、活性の有意な増大はないことを発見した。 Since only the largest randomer appears to have some activity, we understand that the activity of the randomer in many other viruses was size dependent. They tested the antiviral activity of randomers distributed over a larger size using a wider dilution range. We found that for other viruses we tested, the anti-INF A activity of randomers became stronger as their length increased, but for randomers over 40 bases in length, the activity We found that there was no significant increase.
(表4)PS-ONランダマーのサイズ依存的な抗INF A活性
REP 2006の作用機序を決定するため、本発明者らは、感染前、感染中および感染後の様々な時間におけるREP 2006 (IC99濃度)の添加の効果を測定する試みを行った。この実験において、本発明者らは、感染から5時間(300分)後でさえも、REP 2006の添加は、INF A活性を完全に阻害することを観察した(下の表を参照)。これら結果は、インフルエンザに対するREP 2006の作用の少なくとも有意な部分が、感染後に起こることを示す。PS-ONランダマーは細胞内に容易に進入しないため、PS-ONランダマーは宿主細胞から出芽するウイルスにも干渉するであろう。 In order to determine the mechanism of action of REP 2006, we attempted to measure the effect of adding REP 2006 (IC99 concentration) at various times before, during and after infection. In this experiment, we observed that the addition of REP 2006 completely inhibited INF A activity even after 5 hours (300 minutes) after infection (see table below). These results indicate that at least a significant portion of the effect of REP 2006 on influenza occurs after infection. Since PS-ON randomers do not enter cells easily, PS-ON randomers will also interfere with viruses that bud from host cells.
(表5)REP 2006の添加時間対INF A活性上の効果
実施例11:オリゴヌクレオチドが大部分は配列非依存的な作用様式により作用することを決定する試験
本発明者らは、本明細書において本発明のONの抗ウイルス活性が配列非依存的な作用様式により生じることを証明した。勿論、当業者は、配列特異的なON、例えば特定のウイルスのmRNAを標的とするアンチセンスONを調製し、ならびに全てのホスホロチオエートおよび2'-O-メチル修飾を組み込むことができる。しかしながら、そのようなONは、本発明者らが本明細書に記載したON修飾、および本発明者らが本明細書にて証明したこのような修飾されたONの活性が配列非依存的であるという事実から利益を受けることができると考えられる。従って、ONは、以下に概略する5つの試験の任意の一つの基準を満たす場合、即ち、その機能の実質的な部分が配列非依存的活性によるものである場合、配列非依存的活性を有するものと見なされる。以下の試験に使用するONは、PS-ONの合成に関して実施例12に記述する一般的な方法体系に従って調製し得る。
Example 11: Testing to Determine that Oligonucleotides Act Mostly in a Sequence Independent Mode of Action We hereby describe the effect that the antiviral activity of the ON of the invention is sequence independent. Proved to be caused by the style. Of course, one skilled in the art can prepare sequence-specific ONs, eg, antisense ONs that target specific viral mRNAs, and incorporate all phosphorothioate and 2′-O-methyl modifications. However, such ONs are sequence independent of the ON modifications we have described herein and the activity of such modified ONs we have demonstrated herein. It is thought that it can benefit from the fact that there is. Thus, ON has sequence-independent activity if it meets any one of the criteria of the five tests outlined below, ie if a substantial part of its function is due to sequence-independent activity. It is considered a thing. The ON used in the following tests can be prepared according to the general methodology described in Example 12 for the synthesis of PS-ON.
試験#1 - 候補ONの部分縮重のその抗ウイルス効力上の効果
この試験は、その配列の一部が縮重された場合の、候補ON配列の抗ウイルス活性を測定する役割を果たす。同一の化学構造を有する候補ONの縮重バージョンが、以下に記載するようにその活性を保持する場合、配列非依存的活性を有すると見なされる。候補ONは、以下の規則に従って縮重される。
Test # 1-Effect of Partial Degeneration of Candidate ON on Its Antiviral Efficacy This test serves to measure the antiviral activity of a candidate ON sequence when part of its sequence is degenerate. A degenerate version of a candidate ON having the same chemical structure is considered to have sequence-independent activity if it retains its activity as described below. Candidate ONs are degenerated according to the following rules.
L=候補ONの塩基数
X=(候補ONと同一の化学構造を有するが)縮重されたオリゴの各末端上の塩基数。
Lが偶数の場合、X=整数(L/4)である。
Lが奇数の場合、X=整数((L+1)/4)である。
Xは、4と等しいかまたはそれ以上でなければならない。
L = number of candidate ON bases
X = number of bases on each end of the degenerate oligo (although it has the same chemical structure as the candidate ON).
When L is an even number, X = integer (L / 4).
When L is an odd number, X = integer ((L + 1) / 4).
X must be greater than or equal to 4.
候補ONが、ヘルペスウイルス科、レトロウイルスまたはパラミクソウイルスファミリーの一メンバーに対する抗ウイルス活性を有すると断定された場合、そのウイルスファミリーに関して、好ましくは指示したウイルス株を使用して、本明細書に記載したアッセイによりIC50生成を実行する。候補ONが上記していない特定のウイルスファミリーの一メンバーに対する抗ウイルス活性を有すると断定された場合、医薬品産業により容認されている抗ウイルス効力試験によりIC50値を生成する。IC50値は、化合物の最低7種の濃度を使用して、用量応答曲線の直線範囲内の3つまたはそれ以上の点により生成する。これらの試験を用いて、候補ONのIC50をその縮重対応物と比較する。部分縮重ONのIC50が、元の候補ONの5倍よりも小さい場合(最小3回の測定に基づき、標準偏差が平均の15%を越えない)、ONは、大部分は配列非依存的作用様式により作用するものと見なす。 If a candidate ON is determined to have antiviral activity against a member of the herpesviridae, retrovirus or paramyxovirus family, the virus strain preferably indicated for that virus family is used herein, preferably IC 50 generation is performed by the described assay. If a candidate ON is determined to have antiviral activity against a member of a particular viral family not listed above, an IC 50 value is generated by an antiviral efficacy test accepted by the pharmaceutical industry. IC 50 values are generated by 3 or more points within the linear range of the dose response curve using a minimum of 7 concentrations of the compound. These tests are used to compare the candidate ON IC 50 to its degenerate counterpart. Partial degeneracy IC 50 of ON is 5 times smaller case than the original candidate ON (based on the measurement of minimum 3 times the standard deviation does not exceed 15% of the average), ON is largely sequence-independent It is considered to work according to the mode of action.
試験#2 - 候補ONとONランダマーの抗ウイルス活性の比較
この試験は、同一ウイルス内で候補ONの抗ウイルス効力を等価なサイズおよび化学構造のランダマーONの抗ウイルス効力と比較する役割を果たす。
Test # 2—Comparison of Antiviral Activity of Candidate ON and ON Randomer This test serves to compare the antiviral efficacy of candidate ON with that of a randomer ON of equivalent size and chemical structure within the same virus.
候補ONが、ヘルペスウイルス科、レトロウイルスまたはパラミクソウイルスファミリーの一メンバーに対する抗ウイルス活性を有すると断定された場合、そのウイルスファミリーに関して、好ましくは指示したウイルス株を使用して、本明細書に記載したアッセイよりIC50生成を実行する。候補ONが上記していない特定のウイルスファミリーの一メンバーに対する抗ウイルス活性を有すると断定された場合、医薬品産業により容認されている抗ウイルス効力試験によりIC50値を生成する。IC50値は、化合物の最低7種の濃度を使用して、用量応答曲線の直線範囲内の3つまたはそれ以上の点により生成する。この試験を用いて、候補ONのIC50を等価なサイズおよび化学的性質を有するONランダマーと比較する。ONランダマーのIC50が候補ONの5倍よりも小さい場合(最小3回の測定に基づき、標準偏差が平均の15%を越えない)、候補ONは、大部分は配列非依存的作用様式により作用するものと見なす。 If a candidate ON is determined to have antiviral activity against a member of the herpesviridae, retrovirus or paramyxovirus family, the virus strain preferably indicated for that virus family is used herein, preferably IC 50 generation is performed from the described assay. If a candidate ON is determined to have antiviral activity against a member of a particular viral family not listed above, an IC 50 value is generated by an antiviral efficacy test accepted by the pharmaceutical industry. IC 50 values are generated by 3 or more points within the linear range of the dose response curve using a minimum of 7 concentrations of the compound. This test is used to compare the candidate ON IC 50 to ON randomers with equivalent size and chemistry. If the ON randomer IC 50 is less than 5 times that of the candidate ON (based on a minimum of 3 measurements, the standard deviation does not exceed 15% of the mean), the candidate ON is largely due to a sequence-independent mode of action. It is considered to work.
試験#3 - 同一ウイルスファミリーの2つの非相同ウイルスにおける候補ONの抗ウイルス活性の比較
この試験は、ゲノムが候補ONと相同である標的ウイルスに対する候補ONの効力を、ゲノムが候補ONのゲノムと相同性を有さないが同一のウイルスファミリーに属する第二のウイルスに対する候補ONの効力と比較する役割を果たす。例えば、候補ONがHSVに対する活性を有すると報告された場合、そのHSVに対する活性をそのCMVまたはVZV等に対する活性と比較する。標的ウイルスおよび第二のウイルスにおける候補ONの相対的な活性の比較は、両方のウイルス内での同一の長さおよび化学構造のONランダマーの活性を使用して、2つのウイルス内で得られた候補ONのIC50値を正規化することにより遂行される。
Test # 3-Comparison of antiviral activity of candidate ONs in two non-homologous viruses of the same virus family It serves to compare the potency of the candidate ON against a second virus that has no homology but belongs to the same virus family. For example, if the candidate ON is reported to have activity against HSV, its activity against HSV is compared with its activity against CMV or VZV. A comparison of the relative activity of candidate ONs in the target virus and the second virus was obtained in the two viruses using the activity of the ON randomer of the same length and chemical structure in both viruses This is accomplished by normalizing the candidate ON IC 50 values.
従って、候補ONが、所定のウイルスに対して抗ウイルス活性を有すると断定された場合、ヘルペスウイルス科、レトロウイルスもしくはパラミクソウイルスファミリーに関して、本明細書に記載したアッセイの一つまたは当該技術分野で公知の他のアッセイを用いてこのウイルス内でのIC50生成を決定する。同様に、本明細書に記載したアッセイの一つまたは医薬品産業により容認されているアッセイを用いて、ゲノムが候補ONの配列に対して相同性を有さないが、同一ウイルスファミリーに属するウイルスに関して、第二のウイルスに対する候補ONに関するIC50生成を実行する。IC50はまた、各ウイルスに対する等価なサイズおよび化学的性質を有するランダマーに関しても生成する。2つのウイルスに対するONランダマーのIC50を使用して、以下のように2つのウイルスに対する候補ONのIC50値を正規化する。 Thus, if a candidate ON is determined to have antiviral activity against a given virus, one of the assays described herein or the art for the herpesviridae, retrovirus or paramyxovirus family Other assays known in the above are used to determine IC 50 production within this virus. Similarly, using one of the assays described herein or an assay accepted by the pharmaceutical industry, for viruses that do not have homology to the candidate ON sequence but whose genome belongs to the same virus family Perform IC 50 generation on candidate ON for the second virus. IC 50 is also generated for randomers with equivalent size and chemistry for each virus. The IC 50 of the ON randomer for the two viruses is used to normalize the IC 50 value of the candidate ON for the two viruses as follows:
第一の(相同)ウイルス内での候補ONおよびONランダマーのIC50に等価な代数的変換を適用して、ランダマーのIC50を1とする。 First (homologous) by applying the equivalent algebraic converted into IC 50 of candidate ON and ON randomers within virus, the IC 50 of randomer 1.
第二の(非相同)ウイルス内での候補ONおよびONランダマーのIC50に等価な代数的変換を適用して、ランダマーのIC50を1とする。 The second (non-homologous) by applying the equivalent algebraic converted into IC 50 of candidate ON and ON randomers within virus, the IC 50 of randomer 1.
相同ウイルス内での候補ONのIC50と、対非相同ウイルス内での候補ONのIC50の倍数の差異を、非相同ウイルス内での候補ONの変換IC50を相同ウイルス内での候補ONの変換IC50で割ることにより計算する。 Phase candidates IC 50 of ON at the same the virus pair differences multiples of IC 50 of candidate ON in heterologous in viruses, candidate ON of the conversion IC 50 of candidate ON in heterologous in viral phases at the same within the viral Calculate by dividing by the conversion IC 50 .
2つのウイルス間のIC50の倍数の差異が5未満の場合、候補ONは、大部分は配列非依存的な作用様式により作用するものと見なす。 If the difference in IC 50 fold difference between the two viruses is less than 5, the candidate ON is considered to act largely by a sequence-independent mode of action.
試験#4 - 異なるウイルスファミリー内での候補ONの抗ウイルス活性
この試験は、候補ONがウイルス内で薬物様活性を有するか否かを決定する役割を果たし、ここで該候補ONの配列は、ウイルスゲノムの任意の部分と相同ではなく、かつ該ウイルスは異なるファミリーに属する。従って、ヘルペスウイルス科、レトロウイルスまたはパラミクソウイルスファミリーに関して本明細書に記載したアッセイの一つを用いて、試験する候補ONの配列が、使用するウイルスのゲノムの任意の部分に対して相同ではないのような候補ONを試験する。候補ONの最低7種の濃度を使用して、用量応答曲線の直線範囲内の3つまたはそれ以上の点によりIC50値を生成する。得られた用量応答曲線が、薬物様活性を示す場合(典型的には、候補ONの濃度の低下に伴い抗ウイルス効力の低下を示す、減衰またはシグモイド曲線として出現し得る)、および曲線から生成したIC50が10μM未満の場合、候補ONは薬物様活性を有すると見なされる。候補ONが、該候補ONが相補的ではないために配列依存的なアンチセンス活性を有し得ない、異なるファミリーのウイルス内で薬物様活性を有すると見なされる場合、大部分は配列非依存的な作用様式で作用するものと見なす。
Test # 4-Antiviral Activity of Candidate ONs in Different Virus Families This test serves to determine whether the candidate ON has drug-like activity in the virus, where the sequence of the candidate ON is It is not homologous to any part of the viral genome and the virus belongs to a different family. Thus, using one of the assays described herein for the herpesviridae, retrovirus or paramyxovirus family, the sequence of the candidate ON to be tested is not homologous to any part of the viral genome used. Test candidate ON like no. A minimum of 7 concentrations of candidate ON is used to generate IC 50 values with 3 or more points within the linear range of the dose response curve. If the resulting dose response curve shows drug-like activity (typically appears as a decaying or sigmoid curve, showing a decrease in antiviral efficacy with decreasing concentrations of the candidate ON) and generated from the curve If the IC 50 is less than 10 μM, the candidate ON is considered to have drug-like activity. When a candidate ON is considered to have drug-like activity in a different family of viruses that cannot have sequence-dependent antisense activity because the candidate ON is not complementary, it is largely sequence-independent Are considered to work in different modes of action.
試験#5 - 候補ONの細胞外抗ウイルス活性
本発明者らが示す目下の結果により、ONの配列非依存的な抗ウイルス活性は、細胞外で生じることが示される。最先端のON技術は、ONが細胞を容易に浸透できないことから、それらは、アンチセンス活性を示すために適切な担体により細胞膜を貫通して送達される必要があることを教示している。従って、定義によるアンチセンスONの抗ウイルス活性は、活性のための細胞内送達に依存する。特定の配列特異的な候補ONが、インビトロで裸で使用された際に抗ウイルス活性を有する場合(従って細胞内浸透が乏しい場合)、本発明に記載するONの配列非依存的性質により利益を受けると思われる。
Test # 5-Extracellular Antiviral Activity of Candidate ON The current results we show indicate that the sequence-independent antiviral activity of ON occurs extracellularly. State-of-the-art ON technology teaches that since ON cannot easily penetrate cells, they need to be delivered across the cell membrane with a suitable carrier to exhibit antisense activity. Thus, by definition, the antiviral activity of antisense ON depends on intracellular delivery for activity. If a particular sequence-specific candidate ON has antiviral activity when used naked in vitro (and therefore has poor intracellular penetration), it may benefit from the sequence-independent nature of the ON described in the present invention. It seems to receive.
配列特異的な候補ONが、HSV-1、HIV-1またはRSVのゲノムの一部と相補的である場合、候補ONの配列非依存的抗ウイルス活性の存在は、以下に記載する適切なアッセイにて決定され得る。候補ONが、HSV-1、HIV-1またはRSVではないウイルスに対して相補的である場合、候補ONの抗ウイルス活性は、医薬品産業により容認されたアッセイを用いて決定される。 If the sequence-specific candidate ON is complementary to a portion of the HSV-1, HIV-1 or RSV genome, the presence of the sequence-independent antiviral activity of the candidate ON is determined by a suitable assay described below. Can be determined. If the candidate ON is complementary to a virus that is not HSV-1, HIV-1 or RSV, the antiviral activity of the candidate ON is determined using an assay accepted by the pharmaceutical industry.
適切なアッセイを用いて、裸の候補ONの抗ウイルス活性を、封入した(移入のために)候補ONの抗ウイルス活性と比較する(裸の状態および封入した状態の両方の候補ONの同一の濃度を用いて)。活性は、7種以上の濃度を用いた、そのうちの少なくとも3種は、ウイルス活性の50%阻害を含む直線範囲内に入る用量応答曲線により測定する。IC50(ウイルス活性を50%低下させる濃度)を、上記に定義した用量応答曲線の直線範囲の直線回帰により計算する。裸の候補ONのIC50が、封入した候補ONのIC50の5倍よりも小さい場合、候補ONの活性は、大部分は配列非依存的な作用様式により作用するものと見なす。 Using a suitable assay, compare the antiviral activity of the naked candidate ON with the antiviral activity of the encapsulated candidate ON (for transfer) (identical for both naked and encapsulated candidate ON). With concentration). Activity is measured by dose response curves using more than 7 concentrations, at least 3 of which fall within a linear range containing 50% inhibition of viral activity. IC 50 (concentration that reduces viral activity by 50%) is calculated by linear regression of the linear range of the dose response curve defined above. IC 50 of bare candidate ON is less than five times the IC 50 of encapsulated candidate ON, the candidate ON activity, mostly deemed to act by sequence-independent mode of action.
これらの試験で使用した閾値
これらの試験の目的は、ONが本明細書に記載したONの配列非依存的な抗ウイルス性質により利益を得るか、またはそれを利用するかおよび配列非依存的な活性で作用するかを合理的な分析により決定することである。勿論、任意の当業者は、試験の全方法体系、特に抗ウイルス試験方法の固有の変動性を想定して、2つの化合物間の抗ウイルス活性の差異の測定は、閾値が該化合物の活性の間で2倍または3倍の差異に設定されている場合、確実には結論付けられないことを認識するであろう。このことは、これらのアッセイにおいて同一化合物による実験ごとの変動が、この範囲内で変動するIC50を与え得る事実に起因する。従って、2種の化合物(例、2種のON)の活性間に実際に差異が存在することを合理的に確認するために、本発明者らは、該化合物のIC50間の少なくとも5倍の差異を閾値に設定する。この閾値は、上述した試験の評価の信頼性を保証する。
Thresholds used in these tests The purpose of these tests was to determine whether the ON would benefit from or utilize the sequence-independent antiviral properties of the ON described herein and be sequence-independent. It is determined by rational analysis whether it acts in an activity. Of course, any person skilled in the art will be able to measure the difference in antiviral activity between two compounds, assuming that the entire methodology of the test, in particular the inherent variability of the antiviral test method, is the threshold of the activity of the compound. You will recognize that it is not possible to conclude reliably if the difference is set to 2 or 3 times. This is due to the fact that variability from experiment to experiment with the same compound in these assays can give an IC 50 that varies within this range. Therefore, in order to reasonably confirm that there is indeed a difference between the activities of two compounds (e.g., two ONs), we have at least five-fold between the IC 50 of the compounds. Is set as a threshold value. This threshold ensures the reliability of the test evaluation described above.
上記の試験1〜3および5に記載した閾値は、初期設定の閾値である。特に示す場合、上記の比較試験1〜3および5に他の閾値を使用してもよい。従って例えば、特に示す場合、ONが配列非依存的な活性で作用しているか否かを決定する閾値は、10倍、8倍、6倍、5倍、4倍、3倍、2倍、1.5倍および同一のいずれかであり得る。上記の試験4に記載した閾値も、初期設定の閾値である。特に示す場合、試験4でONが配列非依存的活性を有するか否かを決定するための閾値は、10μM、5μM、1μM、0.8μM、0.6μM、0.5μM、0.4μM、0.3μM、0.2μMまたは0.1μM未満のIC50であり得る。 The threshold values described in tests 1 to 3 and 5 above are the default threshold values. Other thresholds may be used for the above comparative tests 1-3 and 5 if specifically indicated. Thus, for example, where indicated, thresholds that determine whether ON is acting with sequence-independent activity are 10 times, 8 times, 6 times, 5 times, 4 times, 3 times, 2 times, 1.5 times It can be either double and identical. The threshold values described in Test 4 above are also the default threshold values. In particular, the threshold for determining whether ON has sequence-independent activity in Test 4 is 10 μM, 5 μM, 1 μM, 0.8 μM, 0.6 μM, 0.5 μM, 0.4 μM, 0.3 μM, 0.2 μM Or an IC 50 of less than 0.1 μM.
同様に、初期設定が、上記の5試験の任意の一つを満たすに十分であっても、特に示す場合、ONは、任意の2つ(例、試験1および2、1および3、1および4、1および5、2および3、2および4、2および5、3および4、ならびに3および5)、任意の3つ(例、試験1および2および3、1および2および4、1および2および5、1および3および4、1および3および5、2および3および4、ならびに2および4および5)、試験の任意の4つ(例、1および2および3および4、1および2および3および5、ならびに2および3および4および5)を初期設定の閾値で、または特に示す場合、上記に示した別の一閾値で満たす必要があり得る。 Similarly, even if the initial settings are sufficient to satisfy any one of the five tests above, ON is specifically indicated if any two (e.g., tests 1 and 2, 1 and 3, 1 and 4, 1 and 5, 2 and 3, 2 and 4, 2 and 5, 3 and 4, and 3 and 5), any three (e.g., tests 1 and 2 and 3, 1 and 2 and 4, 1 and 2 and 5, 1 and 3 and 4, 1 and 3 and 5, 2 and 3 and 4, and 2 and 4 and 5), any four of the tests (eg, 1 and 2 and 3 and 4, 1 and 2) And 3 and 5, and 2 and 3, and 4 and 5) may need to be met with a default threshold or, if specifically indicated, with another threshold shown above.
実施例12:方法体系
以下の方法は、実施例11に記載した試験に適用するために提供する。
Example 12 Methodology The following method is provided for application to the test described in Example 11.
オリゴヌクレオチド合成
本発明のオリゴヌクレオチドは、当該技術分野で周知の方法により合成し得る。例えば、非置換および置換ホスホジエステル(P=O)オリゴヌクレオチドは、自動化DNA合成機(例、Applied Biosystems model 380BまたはAkta Oligopilot 100)上で、標準的なホスホロアミダイト化学構造を用いて、ヨウ素で酸化して合成し得る。ホスホロチオエート(P=S)は、標準的な酸化瓶を、アセトニトリル中の311-1,2-ベンゾジチオール-3-オン 1,1-ジオキシドの0.2M溶液で代替し、亜リン酸結合の段階的にチオエート化すること以外は、ホスホジエステルオリゴヌクレオチドと同様に合成し得る。チオエート化待機ステップは、68秒に延長してもよく、次にキャッピングのステップが続く。支持カラムから切断し、55℃にて濃縮水酸化アンモニウム中で非ブロック化(18時間)した後、オリゴヌクレオチドをエタノール2.5容積で0.5M NaCl溶液から2回沈殿させることにより精製し得る。
Oligonucleotide Synthesis The oligonucleotides of the present invention can be synthesized by methods well known in the art. For example, unsubstituted and substituted phosphodiester (P = O) oligonucleotides can be synthesized with iodine on an automated DNA synthesizer (e.g., Applied Biosystems model 380B or Akta Oligopilot 100) using standard phosphoramidite chemical structures. It can be synthesized by oxidation. Phosphorothioate (P = S) replaces the standard oxidation bottle with a 0.2M solution of 311-1,2-benzodithiol-3-one 1,1-dioxide in acetonitrile, stepwise for phosphite binding It can be synthesized in the same manner as a phosphodiester oligonucleotide, except that it is thioated. The thioate waiting step may be extended to 68 seconds, followed by a capping step. After cleaving from the support column and deblocking in concentrated ammonium hydroxide at 55 ° C. (18 hours), the oligonucleotide can be purified by precipitating twice from 0.5 M NaCl solution with 2.5 volumes of ethanol.
ヘルペスウイルス科に関する抗ウイルスアッセイ
ヘルペスウイルス科に関するプラーク減少を、以下のように実施する:
HSV-1またはHSV-2に関しては、VERO細胞(ATCC# CCL-81)を、12ウェルの組織培地プレート(NUNCまたは相当物)内で、37℃にておよび5% CO2において、10%熱不活性化ウシ胎児血清、ならびにゲンタマイシン、バンコマイシンおよびアンフォテリシンBで補充したMEMの存在下、コンフルエンス迄増殖させる。コンフルエンシーに到達したら、培地を、HSV-1 (KOS株、合計40〜60PFU)またはHSV-2 (MS2株、合計40〜60PFU)のいずれかで補充した5%ウシ胎児血清および上記に詳細した抗生物質を含有するよう交換する。ウイルス吸着を90分間進行させた後、細胞を洗浄し、5%ウシ胎児血清および1%ヒト免疫グロブリンを含有する新しい「オーバーレイ」培地で代替する。吸着から3〜4日間後、細胞をホルマリンで固定して、ホルマリン固定およびクレシルバイオレット染色後、プラークを計数する。
Antiviral Assay for Herpesviridae Plaque reduction for Herpesviridae is performed as follows:
For HSV-1 or HSV-2, VERO cells (ATCC # CCL-81) are heated in a 12-well tissue culture plate (NUNC or equivalent) at 37 ° C. and 5% CO 2 with 10% heat. Grow to confluence in the presence of inactivated fetal bovine serum and MEM supplemented with gentamicin, vancomycin and amphotericin B. Once confluency was reached, the medium was supplemented with either HSV-1 (KOS strain, total 40-60 PFU) or HSV-2 (MS2 strain, total 40-60 PFU) and 5% fetal bovine serum detailed above Change to contain antibiotics. After allowing the virus adsorption to proceed for 90 minutes, the cells are washed and replaced with fresh “overlay” medium containing 5% fetal bovine serum and 1% human immunoglobulin. Three to four days after adsorption, cells are fixed with formalin and plaques are counted after formalin fixation and cresyl violet staining.
CMVに関しては、ヒト神経芽細胞を、HSV-1/2アッセイでVERO細胞に関して詳細したように増殖させる。培地成分および吸着/オーバーレイ手順は、以下の例外を除き同一である:
1. 吸着中、細胞の感染にCMV(AD169株、合計40〜60PFU)を用いる。
2. オーバーレイ培地中、1%ヒト免疫グロブリンを4%シープラークアガロースで代替する。
For CMV, human neuroblasts are grown as detailed for VERO cells in the HSV-1 / 2 assay. Media components and adsorption / overlay procedures are the same with the following exceptions:
1. During adsorption, use CMV (AD169 strain, total 40-60 PFU) for cell infection.
2. Replace 1% human immunoglobulin with 4% sieve plaque agarose in overlay medium.
他のヘルペスウイルス科に関して、試験は、吸着中、適切な細胞宿主および40〜60PFUのウイルスを用いて上述したプラークアッセイにて実施される。 For other herpesviridae, tests are performed in the plaque assay described above using a suitable cellular host and 40-60 PFU virus during adsorption.
この試験は、試験の終盤に化合物の不在下で識別可能なプラークが存在する場合のみ有効である。 This test is only effective if there are plaques identifiable in the absence of the compound at the end of the test.
この試験において、IC50は、未処理対照と対比して50%のプラークが存在する濃度である。 In this test, the IC 50 is the concentration at which 50% plaque is present compared to the untreated control.
試験すべき化合物は、吸着中およびオーバーレイ中に存在する。 The compound to be tested is present during adsorption and overlay.
レトロウイルスに関する抗ウイルスアッセイ
レトロウイルスHIV-1に関するアッセイは、HIV-1感染細胞の上清中の総p24を、以下のように実施したELISAにより検出することにより実施する。
Antiviral Assay for Retrovirus The assay for retrovirus HIV-1 is performed by detecting total p24 in the supernatant of HIV-1-infected cells by ELISA performed as follows.
HIVおよびHBVに関して血清陰性である、スクリーニングを受けたドナーの新鮮なヒト血液からPBMCを単離した。末梢血細胞を低速遠心分離およびPBS中の再懸濁によりペレット化/2〜3回洗浄して、混入した血小板を除去した。次いで、洗浄した血液細胞をDulbeccoのリン酸緩衝生理食塩水(PBS)で1:1に希釈し、50mL遠心分離管内でリンパ球分離用溶液(Mediatech, Inc.のLSM; cellgro(登録商標); 密度1.078+/-0.002g/mL; Cat.# 85-072-CL) 14mL上に重ね、600×gで30分間遠心分離した。得られた界面から帯状PBMCを緩やかに吸引し、次に低速遠心分離によりPBSで2回洗浄した。最終洗浄後、細胞をトリパンブルー排除により計数し、15%ウシ胎児血清(FBS)、2mM L-グルタミン、4μg/mL PHA-Pで補充したRPMI 1640中に1×107細胞/mLにて再懸濁した。細胞を37℃で48〜72時間インキュベートした。インキュベーション後、PBMCを遠心分離にかけ、15% FBS、2mM L-グルタミン、100U/mLペニシリン、100μg/mLストレプトマイシン、10μg/mLゲンタマイシンおよび20U/mL組換えヒトIL-2を伴うRPMI 1640中に再懸濁した。PBMCをこの培地中で1〜2×106細胞/mLの濃度にて維持し、アッセイプロトコールに使用する迄、隔週で培地交換した。組織培養フラスコに吸着した結果として、単球は培養物から枯渇した。 PBMCs were isolated from fresh human blood of screened donors that were seronegative for HIV and HBV. Peripheral blood cells were pelleted / washed 2-3 times by low speed centrifugation and resuspension in PBS to remove contaminating platelets. The washed blood cells were then diluted 1: 1 with Dulbecco's phosphate buffered saline (PBS), and the lymphocyte separation solution (Mediatech, Inc. LSM; cellgro®; Density 1.078 +/- 0.002 g / mL; Cat. # 85-072-CL) Overlaid on 14 mL and centrifuged at 600 × g for 30 minutes. The band-shaped PBMC was gently sucked from the obtained interface, and then washed twice with PBS by low speed centrifugation. After the final wash, cells were counted by trypan blue exclusion and reconstituted at 1 × 10 7 cells / mL in RPMI 1640 supplemented with 15% fetal bovine serum (FBS), 2 mM L-glutamine, 4 μg / mL PHA-P. Suspended. Cells were incubated for 48-72 hours at 37 ° C. After incubation, the PBMCs are centrifuged and resuspended in RPMI 1640 with 15% FBS, 2 mM L-glutamine, 100 U / mL penicillin, 100 μg / mL streptomycin, 10 μg / mL gentamicin and 20 U / mL recombinant human IL-2. It became cloudy. PBMC were maintained at a concentration of in the medium 1 to 2 × 10 6 cells / mL, until use in assay protocol, the medium was replaced every two weeks. Monocytes were depleted from the culture as a result of adsorption to the tissue culture flask.
標準的なPBMCアッセイのために、少なくとも2人の正常なドナーからのPHA-P刺激細胞をプールして、新鮮な培地中で最終濃度1×106細胞/mLに希釈し、96ウェル丸底マイクロプレートのウェル内に、50μL/ウェル(5×104細胞/ウェル)にてプレーティングした。試験薬物の希釈物をマイクロタイターチューブ内にて2×濃度で調製し、各濃縮物100μLを標準的なフォーマットの適切なウェル内に配置した。2時間のプレインキュベーション期間(細胞+薬物)の後、ウイルスの既定の希釈物のストック50μLを、各試験ウェル内に配置した(最終MOI 0.1)。細胞およびウイルスのみを含むウェルを、ウイルス対照に使用した。MTSアッセイ系を用いて(以下に記載する)、薬物の細胞毒性試験のためにウイルスを除いた別個のプレートを同一に調製した。PBMC培養物を感染後7日間維持し、その時点で、細胞フリー上清サンプルを収集して、以下に記載するように逆転写酵素活性についてアッセイした。 For standard PBMC assays, PHA-P stimulated cells from at least 2 normal donors are pooled and diluted to a final concentration of 1 × 10 6 cells / mL in fresh medium, 96 well round bottom Plating was performed in a microplate well at 50 μL / well (5 × 10 4 cells / well). Test drug dilutions were prepared at 2 × concentration in microtiter tubes and 100 μL of each concentrate was placed in appropriate wells of a standard format. After a 2 hour preincubation period (cell + drug), 50 μL of a stock dilution of virus was placed in each test well (final MOI 0.1). Wells containing only cells and virus were used for virus control. Using the MTS assay system (described below), separate plates with the virus removed were prepared identically for drug cytotoxicity testing. PBMC cultures were maintained for 7 days post infection, at which time cell-free supernatant samples were collected and assayed for reverse transcriptase activity as described below.
p24 ELISAキットをCoulter Electronicsから購入した。製造業者の指示に従って、アッセイを実施する。各サンプル中のp24抗原の量を正確に定量するために、各アッセイにおいてコントロールカーブ(Control curves)を生成する。データは、Molecular Devices Vmaxプレートリーダーを用いた450nmでの分光光度分析により獲得する。最終濃度を光学密度の値から計算する。 The p24 ELISA kit was purchased from Coulter Electronics. The assay is performed according to the manufacturer's instructions. Control curves are generated in each assay to accurately quantify the amount of p24 antigen in each sample. Data are acquired by spectrophotometric analysis at 450 nm using a Molecular Devices Vmax plate reader. The final density is calculated from the optical density value.
この試験は、感染した未処理細胞の組織培養上清中に、p24の蓄積が存在する場合のみ有効である。 This test is valid only if p24 accumulation is present in the tissue culture supernatant of infected untreated cells.
この試験において、IC50は、検出可能なp24量が未処理対照内に存在するp24の50%である濃度である。 In this test, the IC 50 is the concentration at which the detectable amount of p24 is 50% of the p24 present in the untreated control.
試験するべき化合物は、吸着中および吸着後、培地中に存在する。 The compound to be tested is present in the medium during and after adsorption.
パラミクソウイルス科に関する抗ウイルスアッセイ
RSVに関して、CPEの測定を以下のように実施する。
Antiviral assay for Paramyxoviridae
For RSV, CPE measurement is performed as follows.
Hep2細胞を96ウェルプレート内にプレーティングして、5%ウシ胎児血清を加えたMEM中、5% CO2で37℃において一夜増殖させた。翌日、細胞を、吸着により2時間、RSV (100ul/ウェル内にA2株、108.2TCID50/mL)で感染させる。吸着後、培地を交換し、7日間の増殖後、CPEを、アラマーブルー色素の生細胞によるその蛍光付加体への変換により測定する。 Hep2 cells were plated in 96 well plates and grown overnight at 37 ° C. with 5% CO 2 in MEM with 5% fetal calf serum. The next day, cells are infected with RSV (A2 strain in 100 ul / well, 10 8.2 TCID50 / mL) for 2 hours by adsorption. After adsorption, the medium is changed and after 7 days of growth, CPE is measured by conversion of the alamar blue dye to its fluorescent adduct by living cells.
この試験は、化合物の不在下での感染細胞内のCPE測定値(アラマーブルー変換により測定)が、非感染細胞内で測定した変換の10%である場合にのみ有効である。 This test is only valid when the CPE measurement in infected cells in the absence of compound (measured by Alamar Blue conversion) is 10% of the conversion measured in uninfected cells.
IC50を比較するために、100% CPEは、感染細胞内で見られる変換レベルに設定し、0% CPEは、非感染細胞で見られる変換レベルに設定する。従ってIC50は、50% CPEを生成する化合物の濃度である。 To compare IC 50 s , 100% CPE is set to the conversion level found in infected cells and 0% CPE is set to the conversion level found in uninfected cells. IC 50 is therefore the concentration of compound that produces 50% CPE.
試験するべき化合物は、吸収中および吸収後、培地中に存在する。 The compound to be tested is present in the medium during and after absorption.
実施例13:2'-Oメチル化ホスホロチオエート化ランダマーは、pH耐性の増大および血清タンパク質結合の低下に伴う強力な抗ウイルス活性を示す
本発明者らは、本明細書において、PS-ONランダマーが配列特異的な機序を介して作用しない(即ち、それらの活性は、核酸に対する結合を必要とせず、および該活性は、配列特異的なアプタマー効果によるものではない)ことを示す。本発明者らは、更に、この実施例において、40塩基ランダマー上に非修飾結合、ホスホロチオエート結合、2'-Oメチル修飾リボースおよび非修飾リボヌクレオチドを組み合わせたオリゴヌクレオチドの、それらの抗ウイルス活性、血清タンパク質相互作用および化学的安定性に対する効果を示す。
Example 13: 2′-O methylated phosphorothioated randomers show potent antiviral activity with increased pH tolerance and decreased serum protein binding We have herein described that PS-ON randomers are Indicates that they do not act through sequence-specific mechanisms (ie, their activity does not require binding to nucleic acids and the activity is not due to sequence-specific aptamer effects). The inventors further in this example show their antiviral activity of oligonucleotides combining unmodified bonds, phosphorothioate bonds, 2'-O methyl modified ribose and unmodified ribonucleotides on a 40 base randomer, Shows effects on serum protein interactions and chemical stability.
全ランダマーは、University of Calgary Core DNA Services labにて、標準的な固相、バッチ合成を用いて、1または15 □mol合成規模で調製し、脱保護し、および50cm Sephadex G-25カラム上で脱塩した。 All randomers are prepared at the University of Calgary Core DNA Services lab using standard solid phase, batch synthesis, 1 or 15 □ mol synthesis scale, deprotected, and on a 50 cm Sephadex G-25 column. Desalted.
A型インフルエンザ(INF A)内での抗ウイルス活性試験のために、不死化犬腎臓(MDCK)細胞を、37℃および5% CO2において、ゲンタマイシン、バンコマイシンおよびアンフォテリシンBで補充した、10%ウシ胎児血清を加えたMEM中で培養する。細胞を増殖から6日後にコンフルエントな単層を得る密度で、6ウェルプレート内に播種する。コンフルエンシーに到達したら、培地を上記の補充物のみ含有するよう交換し、次いで細胞をINF A(H3N2株、合計約35〜70PFU)に60分間暴露する。ウイルス暴露後、培地を薬物のみ含有する新しい培地と交換する。感染から2〜3日後、感染培養物のホルマリン固定およびクレシルバイオレット染色後、プラーク計数を実施する。 Immortalized canine kidney (MDCK) cells supplemented with gentamicin, vancomycin and amphotericin B at 37 ° C. and 5% CO 2 for antiviral activity testing in influenza A (INF A), 10% bovine Incubate in MEM with fetal serum. Cells are seeded in 6-well plates at a density that yields a confluent monolayer 6 days after growth. Once confluency is reached, the medium is changed to contain only the above supplements, and the cells are then exposed to INF A (H3N2 strain, about 35-70 PFU total) for 60 minutes. After virus exposure, the medium is replaced with a new medium containing only the drug. Two to three days after infection, plaque counts are performed after formalin fixation and cresyl violet staining of infected cultures.
HSV内での抗ウイルス試験のために、不死化アフリカミドリザル腎臓(VERO)細胞を、37℃および5% CO2において、ゲンタマイシン、バンコマイシンおよびアンフォテリシンBで補充した、10%ウシ胎児血清を加えたMEM中で培養する。細胞を、増殖から4日後に細胞のコンフルエントな単層を得る密度で、12ウェルプレート内に播種する。コンフルエンシーに到達したら、培地を5%血清および上記の補充物のみを含有するよう交換し、次いで細胞を、試験化合物の存在下、HSV-1 (KOS株、合計約40〜60PFU)に90分間暴露する。ウイルス暴露後、培地を、5%血清、1%ヒト免疫グロブリン、上記の補充物および試験化合物を含有する新しい「オーバーレイ」培地で代替する。感染から3〜4日間後、感染培養物をホルマリン固定およびクレシルバイオレット染色後、プラーク計数を実施する。 For antiviral testing in HSV, immortalized African green monkey kidney (VERO) cells were supplemented with gentamicin, vancomycin and amphotericin B at 37 ° C. and 5% CO 2 with MEM supplemented with 10% fetal calf serum Incubate in. Cells are seeded in 12-well plates at a density that gives a confluent monolayer of cells 4 days after growth. When confluency is reached, the medium is changed to contain only 5% serum and the above supplements, and then the cells are placed in HSV-1 (KOS strain, total about 40-60 PFU) for 90 minutes in the presence of the test compound To expose. After virus exposure, the medium is replaced with fresh “overlay” medium containing 5% serum, 1% human immunoglobulin, the above supplements and test compounds. Three to four days after infection, infected cultures are formalin fixed and cresyl violet stained and plaque counts are performed.
血清タンパク質相互作用を測定するために、3'端においてFITC(ベイト)で標識したホスホロチオエートランダマーを、アッセイ緩衝液(10mM Tris、pH7.2、80mM NaCl、10mM EDTA、100mM b-メルカプトエタノールおよび1%トゥイーン20)中で2nMに希釈する。次いで、このオリゴを、熱不活性化されていない適量のFBSと混合する。ランダマー-FBS相互作用の後、複合体を様々な非標識ランダマーでチャレンジして、それらのベイトをその複合体から置換する能力について評価する。置換されたベイトを蛍光偏光により測定する。置換曲線を使用して、Kdを決定した。 To measure serum protein interactions, phosphorothioate randomers labeled with FITC (bait) at the 3 ′ end were added to assay buffer (10 mM Tris, pH 7.2, 80 mM NaCl, 10 mM EDTA, 100 mM b-mercaptoethanol and 1% Dilute to 2 nM in tween 20). The oligo is then mixed with an appropriate amount of FBS that has not been heat inactivated. Following the randomer-FBS interaction, the complexes are challenged with various unlabeled randomers to assess their ability to displace the baits from the complex. The displaced bait is measured by fluorescence polarization. A displacement curve was used to determine Kd.
pH耐性は、HClで適切なpHに調整したPBS中で、ランダマーをインキュベートすることにより測定した。インキュベーションから24時間後、サンプルを1M TRIS、pH7.4で中和して、変性アクリルアミドゲル上で移動させ、EtBr染色に次いで視覚化した。 The pH tolerance was measured by incubating the randomer in PBS adjusted to the appropriate pH with HCl. After 24 hours of incubation, the samples were neutralized with 1M TRIS, pH 7.4, run on a denaturing acrylamide gel and visualized following EtBr staining.
これらの実験に関して、本発明者らは、異なる修飾ランダマー:REP 2006、REP 2024、REP 2107、REP 2086およびREP 2060の挙動を比較した(この実施例の表6参照)。これらランダマーの抗ウイルス活性を、HSVおよびA型インフルエンザ内での抗ウイルス活性について、プラーク減少アッセイにより試験した(この実施例の表7参照)。これらの2種のウイルス内で、REP 2006、2024および2107は、同様の、かつ強力な抗ウイルス活性を有し、REP 2060は有意な抗HSV活性を示し、またREP 2086は、これらのアッセイ条件下で、HSV-1およびA型インフルエンザの両方内で、検出可能な抗ウイルス活性を有さなかった。 For these experiments, we compared the behavior of different modified randomers: REP 2006, REP 2024, REP 2107, REP 2086 and REP 2060 (see Table 6 in this example). The antiviral activity of these randomers was tested for antiviral activity within HSV and influenza A by plaque reduction assay (see Table 7 in this example). Within these two viruses, REP 2006, 2024, and 2107 have similar and potent antiviral activity, REP 2060 exhibits significant anti-HSV activity, and REP 2086 Below, it had no detectable antiviral activity within both HSV-1 and influenza A.
(表6)ランダマーの記述
N=非修飾デオキシリボヌクレオチド、ホスホロチオエート結合
N=2'-Oメチル修飾リボース、非修飾結合
N=2'-Oメチル修飾リボース+ホスホロチオエート結合
N=非修飾リボヌクレオチド+ホスホロチオエート結合
(Table 6) Description of randomer
N = unmodified deoxyribonucleotide, phosphorothioate linkage
N = 2'-O methyl modified ribose, unmodified bond
N = 2'-O methyl modified ribose + phosphorothioate linkage
N = unmodified ribonucleotide + phosphorothioate linkage
(表7)HSVおよびA型インフルエンザ内における様々なランダマーの抗ウイルス活性
血清タンパク質に対するこれらのランダマーの相対的な親和性を上述したように測定した。これらの実験の結果は、REP 2107が、REP 2006またはREP 2024よりも低い血清タンパク質に対する親和性を有し(この実施例の表8参照)、およびREP 2086と血清タンパク質との間に相互作用が検出されないことを示した。更に、競合の飽和において、REP 2107は、結合したベイトの置換において、REP 2006またはREP 2024と比較して低い有効性を有した(この実施例の表9参照)。 The relative affinity of these randomers for serum proteins was determined as described above. The results of these experiments show that REP 2107 has a lower affinity for serum proteins than REP 2006 or REP 2024 (see Table 8 in this example), and there is no interaction between REP 2086 and serum proteins. It was not detected. Furthermore, at competitive saturation, REP 2107 was less effective at replacing bound baits compared to REP 2006 or REP 2024 (see Table 9 in this example).
(表8)様々なランダマーの血清タンパク質親和性
(表9)飽和におけるベイトランダマーの置換
最後に、本発明者らは、これらのランダマーの、37℃で24時間のインキュベーションの間の、pH1〜pH7の範囲内におけるpH安定性を試験した。REP 2006およびREP 2024がpH3で有意な分解を示し、pH2.5で完全に分解した一方、REP 2107、2086および2060は、24時間のインキュベーション後、pH1で完全に安定であった。 Finally, we tested the pH stability of these randomers within the range of pH 1 to pH 7 during incubation for 24 hours at 37 ° C. REP 2006 and REP 2024 showed significant degradation at pH 3 and were completely degraded at pH 2.5, while REP 2107, 2086 and 2060 were completely stable at pH 1 after 24 hours of incubation.
これら結果は、ONランダマーのホスホロチオエート化が、それらの抗ウイルス活性において非常に有益であるとの本発明者らの以前の研究結果を再現する。本発明者らは、更に、PS-ONランダマー内の2'-Oメチル修飾の組み込みが、たとえPS-ONランダマー内の各リボースが2'-O-メチル修飾を含むとしても、これら分子の抗ウイルス活性に影響を与えないことを示す。更に、完全な2'-O-メチル化および完全なホスホロチオエート化されたランダマー(REP 2107)は、血清タンパク質との相互作用が比較的弱く、低pHにより誘導される加水分解に対する耐性が有意に増大されている。 These results reproduce the results of our previous study that phosphorothioation of ON randomers is highly beneficial in their antiviral activity. The inventors further noted that the incorporation of 2′-O methyl modifications within the PS-ON randomer may prevent the anti-reactivity of these molecules even though each ribose within the PS-ON randomer contains a 2′-O-methyl modification. Indicates that it does not affect virus activity. Furthermore, fully 2'-O-methylated and fully phosphorothioated randomer (REP 2107) has relatively weak interaction with serum proteins and significantly increased resistance to hydrolysis induced by low pH Has been.
実施例14:PS-ONは、大部分は細胞外作用様式により作用する
先行技術は、PS-ONの細胞内濃度を増大させる送達剤の使用が、それらの活性に有益であろうことを教示している。本発明者らは、本明細書において、PS-ONの抗ウイルス活性が大部分は細胞外で作用し、従って細胞内送達剤の移入促進により主な利益を受容しないであろうことを示す。
Example 14: PS-ON acts in large part by an extracellular mode of action Prior art teaches that the use of delivery agents that increase the intracellular concentration of PS-ON would be beneficial to their activity is doing. We show here that the antiviral activity of PS-ON acts largely extracellularly and therefore would not accept a major benefit by facilitating the transfer of intracellular delivery agents.
この実施例において、本発明者らは、例えばリボヌクレオチド(RNA)または2'-O-メチル修飾のような他の修飾を含まない、デオキシリボヌクレオチド(DNA)から作製されたPS-ONを使用する。これらの分子は、特にアンチセンス活性の場合、送達系またはトランスフェクション剤の補助なしでは、インビトロで細胞を容易に貫通しないことが当該技術分野にて周知であるため、このデータは、更なる修飾を支持するPS-ONに適用されると考えることが安全である。 In this example, we use PS-ONs made from deoxyribonucleotides (DNA) that do not contain other modifications such as ribonucleotides (RNA) or 2′-O-methyl modifications. . Since these molecules are well known in the art to not easily penetrate cells in vitro without the aid of a delivery system or transfection agent, especially in the case of antisense activity, this data represents further modification. It is safe to think that it applies to PS-ON that supports
細胞送達の測定のために、HeLa細胞を、標準的な条件下で培養し、次いで裸でまたは送達剤(この場合、DOTAP [1,2-ジオレオイル-3-トリメチルアンモニウム-プロパン]、カチオン性脂質)で封入して、蛍光標識したREP 2006 (FL-REP 2006、3'フルオレセインイソチオシアナート結合40塩基PS-ONランダマー)と共にインキュベートした。インキュベーションの様々な時間の後、細胞をPSBで徹底的に洗浄して、任意の非内在化ONを除去し、次いで細胞を溶解した。細胞溶解物中の細胞内ONのレベルを、蛍光プレートリーダーにより測定した。 For measurement of cell delivery, HeLa cells are cultured under standard conditions and then naked or with a delivery agent (in this case DOTAP [1,2-dioleoyl-3-trimethylammonium-propane], cationic lipid ) And incubated with fluorescently labeled REP 2006 (FL-REP 2006, 3 'fluorescein isothiocyanate-conjugated 40 base PS-ON randomer). After various times of incubation, the cells were washed thoroughly with PSB to remove any non-internalized ON and then the cells were lysed. The level of intracellular ON in the cell lysate was measured with a fluorescent plate reader.
HSV-1およびインフルエンザ内の裸のREP 2006、ならびにDOTAPおよびPEI(ポリエチレンイミン)で封入したREP 2006による抗ウイルス効力は、上述したように測定した。 Antiviral efficacy with HSV-1 and naked REP 2006 in influenza, and REP 2006 encapsulated with DOTAP and PEI (polyethyleneimine) was measured as described above.
HSV-1の感染サイクル中のREP 2006の作用時間を、上述したように測定したが、REP 2006を感染前、感染中および感染後の様々な時間において加えた。HIV-1内では、REP 2006を感染前、感染中および感染後の様々な時間において、HIV-LTR-β-gal HeLa細胞に加えることにより測定した。HIV-1感染は、吸収光度法を用いたβ-gal産生の比色分析アッセイにより監視した。 The duration of action of REP 2006 during the HSV-1 infection cycle was measured as described above, but REP 2006 was added at various times before, during and after infection. Within HIV-1, REP 2006 was measured by addition to HIV-LTR-β-gal HeLa cells at various times before, during and after infection. HIV-1 infection was monitored by a colorimetric assay for β-gal production using absorption photometry.
最初に、本発明者らは、DOTAPおよびPEIが蛍光REP 2006を細胞内に送達し得ることを測定した(表10参照)。このデータは、DOTAPおよびPEIの両方が、FL-2006(および推論によりREP 2006)を細胞内に送達できることを示した。 Initially, we measured that DOTAP and PEI can deliver fluorescent REP 2006 into cells (see Table 10). This data showed that both DOTAP and PEI can deliver FL-2006 (and REP 2006 by reasoning) into the cells.
(表10)送達を伴うおよび伴わないFL-REP 2006の細胞内濃度(pmol/細胞)
次に、本発明者らは、HSV-1、A型インフルエンザ内での封入(DOTAPまたはPEI) REP 2006の活性を測定した(この実施例の表11および表12参照)。これらの結果は、封入REP 2006が、HSV-1およびインフルエンザの両方内で、検出可能な抗ウイルス活性を有さないことを示した。 Next, we measured the activity of HSV-1, encapsulation within influenza A (DOTAP or PEI) REP 2006 (see Table 11 and Table 12 in this example). These results indicated that encapsulated REP 2006 did not have detectable antiviral activity in both HSV-1 and influenza.
(表11)HSV-1内における封入REP 2006の活性(IC50、μM)
![]()
![]()
(表12)A型インフルエンザ内におけるPEI封入REP 2006の活性(%プラーク形成の阻害)
最後に、HSV-1およびHIV-1内における添加時間の研究を行った。ここで、REP 2006は、感染前、感染中および感染後の様々な時間に加えられた。これらの結果は、REP 2006が、両方のウイルス内で、感染前または感染中に存在した場合に最も有効であることを示し、REP 2006がHSV-1およびHIV-1内で融合/進入阻害剤であることを示した。 Finally, a study of the addition time within HSV-1 and HIV-1 was conducted. Here, REP 2006 was added at various times before, during and after infection. These results indicate that REP 2006 is most effective in both viruses when present before or during infection, and REP 2006 is a fusion / entry inhibitor in HSV-1 and HIV-1. It showed that.
これらの結果は、REP 2006および例えばリボヌクレオチド(RNA)または2'-O-メチルのような、但しこれらに限定されない、更なる修飾を支持するPS-ONの抗ウイルス活性が、主として細胞外で生じることを示している。 These results indicate that the antiviral activity of PS-ON supporting additional modifications such as, but not limited to, REP 2006 and ribonucleotides (RNA) or 2'-O-methyl is primarily extracellular. It shows what happens.
実施例15:REP 2107は、優れたヌクレアーゼ耐性を示す
様々な化学構造の40merランダマーを、様々なヌクレアーゼによる分解に抵抗するそれらの能力について、37℃で4時間評価した(この実施例の表13参照)。殆どの化学構造が一種より多いヌクレアーゼに耐性を示したが、REP 2107のみが、試験した4種の全ヌクレアーゼに対して耐性であった。REP 2024(分子の各末端の4リボースに、2'-Oメチル修飾を有する)は、その親分子REP 2006と同一の耐性プロファイルを示し、S1ヌクレアーゼ分解に感受性であったが、2107(完全に2'-Oメチル修飾された)は、この酵素に耐性であったことに注目することが重要である。これらの結果は、試験したオリゴヌクレオチドのうち、REP 2107が、血液中でのヌクレアーゼ分解に対する耐性において最も有効であろうことを示唆している。
Example 15: REP 2107 exhibits excellent nuclease resistance 40mer randomers of various chemical structures were evaluated for 4 hours at 37 ° C for their ability to resist degradation by various nucleases (Table 13 of this example) reference). Although most chemical structures were resistant to more than one nuclease, only REP 2107 was resistant to all four nucleases tested. REP 2024 (with a 2'-O methyl modification at the 4 ribose at each end of the molecule) showed the same resistance profile as its parent molecule REP 2006 and was sensitive to S1 nuclease degradation, but 2107 (fully It is important to note that 2′-O methyl modified) was resistant to this enzyme. These results suggest that of the oligonucleotides tested, REP 2107 would be most effective in resistance to nuclease degradation in blood.
(表13)異なるランダマー化学構造による様々なヌクレアーゼに対する耐性
実施例16:ホスホロチオエート化ポリピリミジンONは、酸耐性およびヌクレアーゼ耐性を示す
ONの酸耐性のONの程度を測定するために、異なる化学構造および/または配列を有する様々な40塩基ONを、異なるpH値に緩衝したPBS中で、37℃で24時間インキュベートする。これらのONの分解を、尿素ポリアクリルアミドゲル電気泳動により評価した(表14参照)。
Example 16: Phosphorothioated polypyrimidine ON exhibits acid resistance and nuclease resistance
To measure the degree of ON of acid resistance of ON, various 40 base ONs with different chemical structures and / or sequences are incubated for 24 hours at 37 ° C. in PBS buffered to different pH values. The degradation of these ONs was evaluated by urea polyacrylamide gel electrophoresis (see Table 14).
これらの研究の結果は、ランダマーON(ピリミジンヌクレオチドおよびプリンヌクレオチドの両方を含む)が、それらが完全に2'-O-メチル化された場合にのみ、酸性のpHに耐性であることを示す。本発明者らのデータは、部分2'-O-メチル化ON(ギャップマー、REP 2024)でも、完全ホスホロチオエート化ONと比較して、酸耐性の有意な増大を全く示さないことを示した。完全ホスホロチオエート化ランダマーでさえも、非修飾ONと比較して、pH耐性の増大を示さない。対照的に、本発明者らは、ピリミジンヌクレオチドシトシン(ポリC、REP 2031)もしくはチミジン(ポリT、REP 2030)またはポリTCヘテロポリマー(REP 2056)のみを含むホスホロチオエート化40mer ONは、ホスホロチオエート化された(REP 2107)またはされていない(REP 2086)完全2'-O-メチル化ランダマーと等価な酸耐性を有することに気付いた。ポリピリミジンオリゴヌクレオチドの結果と対照的に、プリンヌクレオチドアデノシン(ポリA、REP 2029)のみを含むホスホロチオエート化オリゴヌクレオチドまたは任意のアデノシンもしくはグアノシンヌクレオチド(REP 2033、2055、2057)は、非修飾DNAと比較して高い酸耐性を全く示さなかった。 The results of these studies indicate that randomer ONs (including both pyrimidine and purine nucleotides) are resistant to acidic pH only when they are fully 2'-O-methylated. Our data showed that even the partial 2′-O-methylated ON (gapmer, REP 2024) did not show any significant increase in acid resistance compared to fully phosphorothioated ON. Even fully phosphorothioated randomers do not show increased pH tolerance compared to unmodified ON. In contrast, we have phosphorothioated 40mer ON containing only the pyrimidine nucleotide cytosine (poly C, REP 2031) or thymidine (poly T, REP 2030) or poly TC heteropolymer (REP 2056). (REP 2107) or not (REP 2086) was found to have acid resistance equivalent to the fully 2′-O-methylated randomer. In contrast to polypyrimidine oligonucleotide results, phosphorothioated oligonucleotides containing only the purine nucleotide adenosine (poly A, REP 2029) or any adenosine or guanosine nucleotide (REP 2033, 2055, 2057) compared to unmodified DNA And did not show any high acid resistance.
これらの結果は、ホスホロチオエート化ONと比較して高い酸耐性を達成するための、先行技術に記載された好ましい方法が、2'-O-メチル修飾(もしくは他の2'-リボース修飾)または他の修飾を加えることであることから有意である。本発明のデータは、ONがポリピリミジン(即ち、ピリミジンヌクレオチドのみを含む[例、シトシンもしくはチミジンのホモポリマー、またはシトシンおよびチミジンのヘテロポリマー])の場合、pH耐性およびヌクレアーゼ耐性を達成するために、2'-O-メチルリボース修飾または他の2'-リボース修飾を必要としないことを示す。ピリミジン存在下におけるプリン(AまたはG)の存在も、ONを酸に不安定なものとし得る。 These results indicate that the preferred method described in the prior art to achieve high acid resistance compared to phosphorothioated ON is a 2′-O-methyl modification (or other 2′-ribose modification) or other This is significant because of the modification. The data of the present invention show that when ON is polypyrimidine (i.e. containing only pyrimidine nucleotides [e.g. cytosine or thymidine homopolymer, or cytosine and thymidine heteropolymer]) to achieve pH resistance and nuclease resistance. , 2'-O-methyl ribose modification or other 2'-ribose modifications are not required. The presence of purines (A or G) in the presence of pyrimidines can also make ON acid labile.
(表14)様々な40mer ONの酸安定性
ヌクレアーゼ耐性に対するONヌクレオチドの組成および修飾の範囲を決定するために、異なるヌクレオチド組成および修飾を有する様々な40塩基ONを、様々なエンドヌクレアーゼおよびエキソヌクレアーゼの存在下、37℃で4時間インキュベートした。これらONの分解を、尿素ポリアクリルアミドゲル電気泳動により評価した。 To determine the range of ON nucleotide compositions and modifications for nuclease resistance, various 40 base ONs with different nucleotide compositions and modifications were incubated at 37 ° C. for 4 hours in the presence of various endonucleases and exonucleases. These ON degradations were evaluated by urea polyacrylamide gel electrophoresis.
これらの研究の結果は、ランダマーONが、それらが完全にホスホロチオエート化および完全に2'-O-メチル化された場合のみ、試験した4種の全酵素(ホスホジエステラーゼII[Sigma]、S1ヌクレアーゼ[Fermentas], Bal31 [New England Biolabs]およびエキソヌクレアーゼ1 [New England Biolabs])に耐性であることを示した(表15参照)。ランダマー内でのこれらのいずれかの修飾の脱落は、試験した1種または複数種のヌクレアーゼに対する感受性を増大させた。本発明者らは、完全ホスホロチオエート化、部分2'-O-メチル化ランダマー(REP 2024)が、ヌクレアーゼ耐性においてREP 2006と等価であり、最適なヌクレアーゼ耐性の達成のためには、ホスホロチオエート化ONの各ヌクレオチドに2'-O-メチル化が必要であり得ることに気付いた。しかしながら、本発明者らは、ホスホロチオエート化40merポリピリミジンポリシトシン(ポリC、REP 2031)が、完全ホスホロチオエート化、完全2'0メチル化ランダマー(REP 2107)と等価なヌクレアーゼ耐性を有することに気付いた。 The results of these studies show that Randamer ON is one of the four total enzymes tested (phosphodiesterase II [Sigma], S1 nuclease [Fermentas] only when they are fully phosphorothioated and fully 2'-O-methylated. ], Bal31 [New England Biolabs] and exonuclease 1 [New England Biolabs]) (see Table 15). Deletion of any of these modifications within the randomer increased sensitivity to one or more nucleases tested. We have fully phosphorothioated, the partial 2'-O-methylated randomer (REP 2024) is equivalent to REP 2006 in nuclease resistance, and for achieving optimal nuclease resistance, phosphorothioated ON It has been found that 2'-O-methylation may be required for each nucleotide. However, the inventors have found that phosphorothioated 40mer polypyrimidine polycytosine (Poly C, REP 2031) has nuclease resistance equivalent to fully phosphorothioated, fully 2'0 methylated randomer (REP 2107). .
これらの結果は、先行技術がホスホロチオエート化ONのヌクレアーゼ耐性を高める好ましい方法は、2'-O-メチル修飾、他の2'-リボース修飾または他の修飾を加えることであると教示していることから有意である。この新しいデータは、ONが完全にホスホロチオエート化されかつピリミジンのホモポリマーからなる場合、ヌクレアーゼ耐性を高めるために2'-O-メチル修飾もしくは他の2'-リボース修飾または任意の他の修飾を必要としないことを示す。 These results teach that the prior art is the preferred way to increase the nuclease resistance of phosphorothioated ON is to add 2'-O-methyl modifications, other 2'-ribose modifications or other modifications Is significant. This new data shows that when ON is fully phosphorothioated and consists of a pyrimidine homopolymer, 2'-O-methyl or other 2'-ribose modifications or any other modification is required to increase nuclease resistance It means not to.
(表15)様々な40mer ONのヌクレアーゼ耐性
これらの結果は、シトシンおよび/またはチミジンおよび/または他のピリミジンを含む、ピリミジンヌクレオチドのみを含有するホスホロチオエート化ONが低pHに耐性であり、かつシトシンヌクレオチドのみを含有するホスホロチオエート化ONが優れたヌクレアーゼ耐性を示し、抗ウイルスONの経口投与に重要な2つの特性を有することを示す。従って、抗ウイルスONの高いピリミジンヌクレオチド含有量は、低pHに対する耐性を提供するのに有利であり、高いシトシン含有量は、改善されたヌクレアーゼ耐性を提供するのに有利である。例えば、ある実施態様において、このようなオリゴヌクレオチドのピリミジン含有量は、50%より多く、60%より多く、もしくは70%より多く、もしくは80%より多く、もしくは90%より多くまたは100%である。更に、これらの結果は、ピリミジンヌクレオチドから構成された、治療有効量の少なくとも一種の薬学的に許容されるONの経口投与による処置方法の可能性を示す。これらの結果はまた、抗ウイルスON製剤の成分としてピリミジンヌクレオチドを高いレベルで含有するONの可能性も示す。 These results indicate that phosphorothioated ONs containing only pyrimidine nucleotides, including cytosine and / or thymidine and / or other pyrimidines, are resistant to low pH and phosphorothioated ON containing only cytosine nucleotides are superior nucleases It is resistant and shows two important properties for oral administration of antiviral ON. Thus, a high pyrimidine nucleotide content of antiviral ON is advantageous to provide resistance to low pH, and a high cytosine content is advantageous to provide improved nuclease resistance. For example, in certain embodiments, the pyrimidine content of such oligonucleotides is greater than 50%, greater than 60%, or greater than 70%, or greater than 80%, or greater than 90% or 100%. . Furthermore, these results indicate the possibility of a treatment method by oral administration of a therapeutically effective amount of at least one pharmaceutically acceptable ON composed of pyrimidine nucleotides. These results also indicate the possibility of ON containing high levels of pyrimidine nucleotides as a component of antiviral ON formulations.
実施例17:インビボでのONの配列非依存的な広域な活性
本発明者らは、本明細書において、40塩基の配列非依存的なPS-ONランダマーがウイルス感染した6種の異なる動物モデルにおいて強力な抗ウイルス活性を有することを示す(表16参照)。40塩基PS-ONランダマーを、皮下、腹腔内およびエアロゾル(吸入)を含む多数の投与経路により動物に導入した。これらデータは、配列非依存的なONの広域抗ウイルス薬としての治療的可能性を強力に支持する。
Example 17: Sequence-independent broad activity of ON in vivo We here describe six different animal models in which a 40 base sequence-independent PS-ON randomer was virally infected. It has a strong antiviral activity in (see Table 16). The 40 base PS-ON randomer was introduced into animals by multiple routes of administration including subcutaneous, intraperitoneal and aerosol (inhalation). These data strongly support the therapeutic potential of sequence-independent ON as a broad spectrum antiviral agent.
(表16)PS-ONランダマーは、インビボで強力な広域抗ウイルス活性を有する
実施例18:オリゴヌクレオチドは、広域なウイルスに抗ウイルス活性を有する
本発明者らは、本明細書において、40 PS-ONランダマーがインビトロで13のウイルスファミリーに対して抗ウイルス活性を有することを示す(表17参照)。
Example 18: Oligonucleotides have antiviral activity against a broad spectrum of viruses We have here demonstrated that 40 PS-ON randomers have antiviral activity against 13 viral families in vitro. Shown (see Table 17).
(表17)PS-ONランダマーは、インビトロで広域な抗ウイルス活性を有する
実施例19:PS-ONのインビボおよびインビトロでの抗インフルエンザ活性
ONの抗インフルエンザ活性を更に評価するために、赤血球凝集アッセイにより、REP 2006をインフルエンザの異なる株に対して試験した。表18に示すように、REP 2006は、広域な抗インフルエンザ活性を示した。
Example 19: In vivo and in vitro anti-influenza activity of PS-ON
To further evaluate the anti-influenza activity of ON, REP 2006 was tested against different strains of influenza by a hemagglutination assay. As shown in Table 18, REP 2006 showed a wide range of anti-influenza activity.
(表18)インフルエンザの多数の株に対するREP 2006の広域な抗ウイルス活性
ONのインフルエンザ処置のための薬物としての可能性を評価するために、REP 2006をインフルエンザ感染のマウスモデル内で試験した。REP 2006を注射用蒸留水中にて2つの濃度で調製し、噴霧によりエアロゾル化し、ここで排気口は、Andersonのカスケードチャンバ(cascade chamber)に接続されていた。エアロゾルチャンバ内の様々な濃度のREP 2006 10mLを用いて、20g Balb/cマウスをエアロゾル化ランダマー1に連日、30分間暴露した。マウスにA型インフルエンザ(H3N2、A/Hong Kong/68)約100TCIDを鼻腔内感染させ、感染から4日後、動物を屠殺にし、肺ウイルス力価を赤血球凝集アッセイにより測定した。表19に示すように、REP 2006は、インビボで強力な抗インフルエンザ活性を示した。 To evaluate the potential of ON as a drug for influenza treatment, REP 2006 was tested in a mouse model of influenza infection. REP 2006 was prepared in two concentrations in distilled water for injection and aerosolized by spraying, where the exhaust was connected to Anderson's cascade chamber. 20 g Balb / c mice were exposed to aerosolized randomer 1 daily for 30 minutes using 10 mL of various concentrations of REP 2006 in an aerosol chamber. Mice were infected intranasally with approximately 100 TCID of influenza A (H3N2, A / Hong Kong / 68), animals were sacrificed 4 days after infection, and pulmonary virus titers were measured by hemagglutination assay. As shown in Table 19, REP 2006 showed potent anti-influenza activity in vivo.
(表19)インビボでのREP 2006のA型インフルエンザに対する効力
実施例20:ホスホロチオエート化ポリピリミジンONは、インビボで酸性環境において改善された抗ウイルス活性を示す
ポリピリミジンONの低いpHに対する耐性と、それらがインビボにてより低pHで活性薬物となる能力を評価するために、REP 2031 (PS ポリC)をHSV-2膣内マウスモデルで試験した。雌のSwiss Websterのグループに、ウイルスチャレンジの7日前および1日前、酢酸メドロキシプロゲステロン3mgを含有する縣濁液0.1mLを、皮下注射により投与して、膣内HSV-2感染の感受性を増大させた。膣腔を綿棒で2回、最初はアルギン酸カルシウムを先端につけた湿潤タイプ1の綿棒で、次に乾燥綿棒で拭き取った。連続分注ピペットを使用して、候補溶液またはプラセボ対照のいずれか15μLで動物を処置した。5分後、動物を、HSV-2、186株104pfuを含有する縣濁液15μLの滴下注入により接種した。膣拭き取りサンプルを、接種から2日後に全動物から収集し、ウイルスの存在をアッセイする迄、培養液により冷凍貯蔵した(-80℃)。マウスを、接種から21日後迄、脱毛および会陰周辺の紅斑、慢性尿失禁、後肢麻痺および死亡を含み得る、症候性感染の兆候に関して連日評価した。症候を発現しなかった動物は、接種から2日後に収集した膣拭き取りサンプルからウイルスが単離された場合、感染したものと定義した。結果は、ポリピリミジンREP 2031が、例えばこの実施例では膣または胃のような酸性の環境内で抗ウイルス活性を有することを示した(表20)。
Example 20: Phosphorothioated polypyrimidine ON exhibits improved antiviral activity in an acidic environment in vivo Polypyrimidine ON's tolerance to low pH and their ability to become active drugs at lower pH in vivo To do so, REP 2031 (PS Poly C) was tested in the HSV-2 intravaginal mouse model. A group of female Swiss Websters were administered by subcutaneous injection 0.1 mL of a suspension containing 3 mg of medroxyprogesterone acetate 7 and 1 day before the viral challenge to increase the susceptibility of intravaginal HSV-2 infection. It was. The vaginal cavity was wiped twice with a cotton swab, first with a wet type 1 swab with a calcium alginate tip and then with a dry swab. Animals were treated with 15 μL of either the candidate solution or a placebo control using a continuous pipette. Five minutes later, the animals were inoculated by instillation of 15 μL of suspension containing HSV-2, 186 strain 10 4 pfu. Vaginal wipe samples were collected from all animals 2 days after inoculation and stored frozen (-80 ° C) in culture until assayed for the presence of virus. Mice were evaluated daily for signs of symptomatic infection until 21 days after inoculation, including hair loss and erythema around the perineum, chronic urinary incontinence, hindlimb paralysis and death. Animals that did not develop symptoms were defined as infected if the virus was isolated from a vaginal wipe sample collected 2 days after inoculation. The results showed that polypyrimidine REP 2031 has antiviral activity in an acidic environment such as the vagina or stomach in this example (Table 20).
(表20)HSV-2に対するONの膣内効力
明細書に引用された特許及びそのほかの参考文献はすべて、本発明が関係する当業者の技量のレベルを示しており、いかなる表及び図も含めて、それぞれの参考文献が個々にその全体を参考として本明細書に組み入れたのと同等に、それら全体を参考として組み入れる。 All patents and other references cited in the specification are indicative of the level of skill of those skilled in the art to which this invention pertains, and each reference, including any tables and figures, individually and entirely. As if incorporated herein by reference in their entirety.
当業者は、本発明が上手く適用されて、言及された結果及び利点並びにその中で固有のものを得ることを容易に十分に理解するであろう。好ましい態様の現在の代表として本明細書に記載された方法、変異及び組成物は典型的なものであって、本発明の範囲を限定することを意図するものではない。その中の変更及びそのほかの用途が当業者に生じるであろうが、それらは本発明の範囲内に包含され、請求の範囲によって定義されている。 Those skilled in the art will readily and fully appreciate that the present invention has been successfully applied to obtain the results and advantages mentioned, as well as those inherent therein. The methods, variations and compositions described herein as presently representative of preferred embodiments are exemplary and are not intended as limitations on the scope of the invention. Changes therein and other uses will occur to those skilled in the art, which are encompassed within the scope of the invention and are defined by the claims.
本発明の範囲と精神から逸脱することなく、本明細書で開示された発明に対して種々の置換及び修正を行ってもよいことは当業者には容易に明らかであろう。たとえば、合成条件及びオリゴヌクレオチドの組成物に対して変更を行うことができる。従って、そのような追加的態様は、本発明及び請求の範囲内である。 It will be readily apparent to those skilled in the art that various substitutions and modifications can be made to the invention disclosed herein without departing from the scope and spirit of the invention. For example, changes can be made to the synthesis conditions and the composition of the oligonucleotide. Accordingly, such additional embodiments are within the scope of the present invention and claims.
本明細書で好適に、説明的に記載された本発明を、本明細書で具体的に開示されていないいかなる要素、限定の非存在下で実践してもよい。従って、たとえば、本明細書の各例において、用語「含む」、「本質的に成る」及び「成る」のいかなるものも、ほかの2つの用語のいずれかで置き換えてもよい。採用された用語及び表現は、説明の用語として使用され、限定の用語ではなく、そのような用語及び表現の使用において、示された及び記載された特徴の同等物、又はその一部を排除する意図はないが、請求された発明の範囲内で様々な改変が可能であることが認識される。従って、本発明は好ましい態様及び任意の特徴によって具体的に開示されているが、本明細書に開示された概念の修正及び変更は当業者に頼ってもよく、そのような修正及び変更が、添付の特許請求の範囲によって定義される本発明の範囲内であるとみなされることが理解されるべきである。 The invention described in the description, suitably described herein, may be practiced in the absence of any elements, limitations not specifically disclosed herein. Thus, for example, in each example herein, any of the terms “comprising”, “consisting essentially of” and “consisting of” may be replaced with either of the other two terms. The terms and expressions employed are used as descriptive terms and are not limiting terms, and exclude the equivalents, or portions thereof, of the features shown and described in the use of such terms and expressions. Although not intended, it will be recognized that various modifications are possible within the scope of the claimed invention. Thus, although the invention has been specifically disclosed with preferred embodiments and optional features, modifications and changes to the concepts disclosed herein may be relied upon by those skilled in the art, and such modifications and changes may be It should be understood that it is deemed to be within the scope of the present invention as defined by the appended claims.
さらに、本発明の特徴及び局面がマーカッシュ群又は代替の他の群という点で記載される場合、当業者は、本発明がそれによって、マーカッシュ群又はそのほかの群の個々の一員又は亜群の一員という点で記載されることを理解するであろう。 Further, where features and aspects of the invention are described in terms of a Markush group or other alternative group, one of ordinary skill in the art will recognize that the invention is thereby a member of an individual or subgroup of the Markush group or other group. You will understand that
また、それとは反対に、記載していなければ、様々な数値が態様に対して提供される場合、範囲の端点として任意の2つの異なる値を利用することによって追加の態様が記載される。そのような範囲も記載された本発明の範囲内である。 Also, on the contrary, if not stated, additional aspects are described by utilizing any two different values as the endpoints of the range when various numerical values are provided for the aspects. Such ranges are also within the scope of the described invention.
従って、追加の態様も本発明の範囲内であり、特許請求の範囲の範囲内である。 Accordingly, additional embodiments are within the scope of the invention and are within the scope of the claims.
(表21) オリゴヌクレオチドの記述
Claims (76)
薬学的に許容される担体
を含有する、抗ウイルス薬学的組成物。 An antiviral pharmaceutical comprising a therapeutically effective amount of at least one pharmacologically acceptable antiviral oligonucleotide as defined in any one of claims 1-18; and a pharmaceutically acceptable carrier. Composition.
薬学的に許容される担体
を含有する、抗ウイルス薬学的組成物。 49. An antiviral pharmaceutical comprising a therapeutically effective amount of at least one pharmacologically acceptable antiviral oligonucleotide as defined in any one of claims 31 to 48; and a pharmaceutically acceptable carrier. Composition.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/969,812 US20050196382A1 (en) | 2002-09-13 | 2004-10-19 | Antiviral oligonucleotides targeting viral families |
| US66898305P | 2005-04-07 | 2005-04-07 | |
| PCT/CA2005/001623 WO2006042418A1 (en) | 2004-10-19 | 2005-10-19 | Antiviral oligonucleotides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008516996A true JP2008516996A (en) | 2008-05-22 |
| JP2008516996A5 JP2008516996A5 (en) | 2008-08-21 |
Family
ID=36202655
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007537092A Withdrawn JP2008516996A (en) | 2004-10-19 | 2005-10-19 | Antiviral oligonucleotide |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1802643A1 (en) |
| JP (1) | JP2008516996A (en) |
| AU (1) | AU2005297376A1 (en) |
| CA (1) | CA2584207A1 (en) |
| WO (1) | WO2006042418A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150013309A (en) * | 2012-05-18 | 2015-02-04 | 레플리코르 인코포레이티드 | Oligonucleotide chelate complex-polypeptide compositions and methods |
| JP2015517503A (en) * | 2012-05-18 | 2015-06-22 | レプリコール インコーポレーティッド | Oligonucleotide chelate complex method |
| JP2016520312A (en) * | 2013-05-22 | 2016-07-14 | アルナイラム ファーマシューティカルズ, インコーポレイテッドAlnylam Pharmaceuticals, Inc. | SERPINA1 iRNA composition and methods of use thereof |
| JP2024526189A (en) * | 2021-06-24 | 2024-07-17 | ヨハン ヴォルフガング ゲーテ-ウニヴェルズィテート フランクフルト | G-QUADRUCLES-CONTAINING OLIGONUCLEOTIDES FOR PROPHYLAXIS AND THERAPY - Patent application |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100129437A1 (en) | 2007-03-23 | 2010-05-27 | Bbb Holding B.V. | Targeted intracellular delivery of antiviral agents |
| WO2016030863A1 (en) * | 2014-08-29 | 2016-03-03 | Glaxosmithkline Intellectual Property Development Limited | Compounds and methods for treating viral infections |
| EP3184674A1 (en) * | 2015-12-23 | 2017-06-28 | Technische Universität Dortmund | Dna-encoded chemical library, use thereof and method to synthesize the library |
| TWI788312B (en) | 2016-11-23 | 2023-01-01 | 美商阿尼拉製藥公司 | SERPINA1 iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| US11166976B2 (en) | 2018-11-08 | 2021-11-09 | Aligos Therapeutics, Inc. | S-antigen transport inhibiting oligonucleotide polymers and methods |
| WO2021119325A1 (en) * | 2019-12-12 | 2021-06-17 | Aligos Therapeutics, Inc. | S-antigen transport inhibiting oligonucleotide polymers and methods |
| WO2021193965A1 (en) * | 2020-03-26 | 2021-09-30 | 国立研究開発法人国立循環器病研究センター | Antisense nucleic acid targeting apoc3 |
| WO2022109129A1 (en) * | 2020-11-20 | 2022-05-27 | Aligos Therapeutics, Inc. | Conjugates of s-antigen transport inhibiting oligonucleotide polymers having enhanced liver targeting |
| WO2022229350A2 (en) | 2021-04-30 | 2022-11-03 | Tirmed Pharma Ab | Single-stranded oligonucleotides for use in the medical treatment and/or prophylaxis of virus infections |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07501929A (en) * | 1991-08-23 | 1995-03-02 | アイシス・ファーマシューティカルス・インコーポレーテッド | Synthetic non-randomization of oligomeric fragments |
| US5523389A (en) * | 1992-09-29 | 1996-06-04 | Isis Pharmaceuticals, Inc. | Inhibitors of human immunodeficiency virus |
| BR0314236A (en) * | 2002-09-13 | 2005-08-09 | Replicor Inc | Oligonucleotide formulation, pharmaceutical composition, kit, antiviral compound, preparation of oligonucleotide and methods for selection of an antiviral oligonucleotide for use as an antiviral agent, for prophylaxis or treatment of a viral infection in a patient, for prophylactic treatment of cancer caused by oncoviruses. for identifying a compound that alters the binding of an oligonucleotide to at least one viral component, for purifying oligonucleotide binding to at least one viral component and for enriching oligonucleotides from an oligonucleotide cluster |
-
2005
- 2005-10-19 JP JP2007537092A patent/JP2008516996A/en not_active Withdrawn
- 2005-10-19 EP EP05797197A patent/EP1802643A1/en not_active Withdrawn
- 2005-10-19 CA CA002584207A patent/CA2584207A1/en not_active Abandoned
- 2005-10-19 AU AU2005297376A patent/AU2005297376A1/en not_active Abandoned
- 2005-10-19 WO PCT/CA2005/001623 patent/WO2006042418A1/en not_active Ceased
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150013309A (en) * | 2012-05-18 | 2015-02-04 | 레플리코르 인코포레이티드 | Oligonucleotide chelate complex-polypeptide compositions and methods |
| JP2015517504A (en) * | 2012-05-18 | 2015-06-22 | レプリコール インコーポレーティッド | Oligonucleotide chelate complexes-polypeptide compositions and methods |
| JP2015517503A (en) * | 2012-05-18 | 2015-06-22 | レプリコール インコーポレーティッド | Oligonucleotide chelate complex method |
| JP2018058853A (en) * | 2012-05-18 | 2018-04-12 | レプリコール インコーポレーティッド | Oligonucleotide chelate complex methods |
| KR102068109B1 (en) * | 2012-05-18 | 2020-01-21 | 레플리코르 인코포레이티드 | Oligonucleotide chelate complex-polypeptide compositions and methods |
| JP2016520312A (en) * | 2013-05-22 | 2016-07-14 | アルナイラム ファーマシューティカルズ, インコーポレイテッドAlnylam Pharmaceuticals, Inc. | SERPINA1 iRNA composition and methods of use thereof |
| JP2019107029A (en) * | 2013-05-22 | 2019-07-04 | アルナイラム ファーマシューティカルズ, インコーポレイテッドAlnylam Pharmaceuticals, Inc. | SERPINA 1 iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| JP2021191278A (en) * | 2013-05-22 | 2021-12-16 | アルナイラム ファーマシューティカルズ, インコーポレイテッドAlnylam Pharmaceuticals, Inc. | SERPINA1 iRNA COMPOSITION AND METHOD FOR USING THE SAME |
| JP2024526189A (en) * | 2021-06-24 | 2024-07-17 | ヨハン ヴォルフガング ゲーテ-ウニヴェルズィテート フランクフルト | G-QUADRUCLES-CONTAINING OLIGONUCLEOTIDES FOR PROPHYLAXIS AND THERAPY - Patent application |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2005297376A1 (en) | 2006-04-27 |
| CA2584207A1 (en) | 2006-04-27 |
| EP1802643A1 (en) | 2007-07-04 |
| WO2006042418A9 (en) | 2007-05-31 |
| WO2006042418A1 (en) | 2006-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5796024B2 (en) | Non-sequence complementary antiviral oligonucleotides | |
| JP2008516996A (en) | Antiviral oligonucleotide | |
| JP2011130772A (en) | Composition and method for controlling expression of jc virus gene | |
| US20100172965A1 (en) | Antiviral oligonucleotides targeting viral families | |
| CN101084232A (en) | Antiviral oligonucleotides | |
| EP3047023A2 (en) | Compositions and methods for inhibiting jc virus (jcv) | |
| CN101370818A (en) | RNAi suppresses replication of influenza virus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080702 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20081020 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20090724 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20090724 |