ES2716142T3 - Derivados del alcohol de 1-fenil-2-piridinil alquilo como inhibidores de la fosfodiesterasa - Google Patents
Derivados del alcohol de 1-fenil-2-piridinil alquilo como inhibidores de la fosfodiesterasa Download PDFInfo
- Publication number
- ES2716142T3 ES2716142T3 ES13814850T ES13814850T ES2716142T3 ES 2716142 T3 ES2716142 T3 ES 2716142T3 ES 13814850 T ES13814850 T ES 13814850T ES 13814850 T ES13814850 T ES 13814850T ES 2716142 T3 ES2716142 T3 ES 2716142T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- methylsulfonamido
- ethyl
- methyl
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *c(ccc(C(Cc1ccncc1)=O)c1)c1O* Chemical compound *c(ccc(C(Cc1ccncc1)=O)c1)c1O* 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/155—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
- A61M15/0065—Inhalators with dosage or measuring devices
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
- A61M15/009—Inhalators using medicine packages with incorporated spraying means, e.g. aerosol cans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M16/00—Devices for influencing the respiratory system of patients by gas treatment, e.g. ventilators; Tracheal tubes
- A61M16/10—Preparation of respiratory gases or vapours
- A61M16/14—Preparation of respiratory gases or vapours by mixing different fluids, one of them being in a liquid phase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Pulmonology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Anesthesiology (AREA)
- Biomedical Technology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Biophysics (AREA)
- Emergency Medicine (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
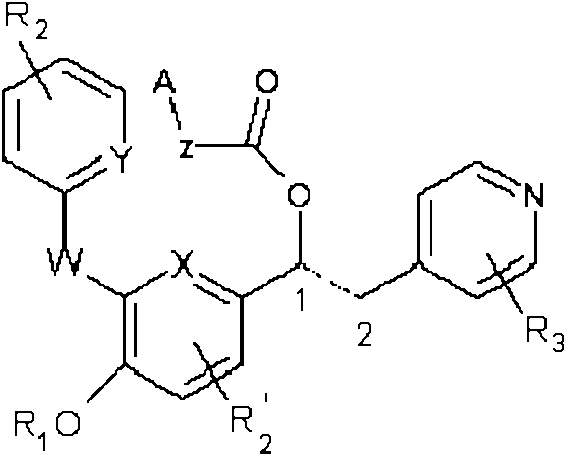
Un compuesto de la Fórmula general (I)**Fórmula** en donde: R1 y R2 son iguales o diferentes y se seleccionan independientemente del grupo que consiste en hidrógeno, metilo, etilo, difluorometilo, ciclopropilmetilo o ciclopropilo; R3 son los mismos dos átomos de cloro en la posición 3 y 5 del anillo piridilo; Z es un grupo (CH2)m en donde m = 0; A es un anillo de fenilo, que está opcionalmente sustituido con uno o más sustituyentes R4, que pueden ser iguales o diferentes y se seleccionan independientemente del grupo que consiste en: (C1-C2)alquilo lineal o ramificado opcionalmente sustituido por uno o más (C3-C7)heterocicloalquilo y preferentemente 4-morfolinilo; trifluorometilo; metiltio; halógeno seleccionado de fluoro o cloro; y OR7 en donde R7 se selecciona del grupo que consiste en -H; - (C1-C4) alquilo opcionalmente sustituido por un radical seleccionado de OH, ciclopropilo, 4-morfolinilo, 1- o 4- piperidinilo, 4-piridinilo y fenilo; - trifluorometilo o difluorometilo; y W es el grupo -NR9SO2R10 en donde R9 se selecciona del grupo que consiste en (C1-C4) alquilo opcionalmente sustituido por 4-morfolinilo, 1-piperidinilo, 1-pirrolidinilo, piperazinilo, 1,2- tiazolidin-3-ilo, piperazin-1-ilcarbonilo en donde cualquiera de dichos anillos puede estar opcionalmente sustituido con uno o más metilo, etilo u OH; y R11R12N-(C1-C3)alquileno en donde R11 y R12 son independientemente H o metilo opcionalmente sustituidos con un grupo OH; y R10 se selecciona del grupo que consiste en (C1-C4) alquilo opcionalmente sustituido por (C3-C7) cicloalquilo y (C3-C7) cicloalquilo; sus N-óxidos en el anillo de piridina, y sales farmacéuticamente aceptables de estos.
Description
DESCRIPCIÓN
Derivados del alcohol de 1-fenil-2-piridinil alquilo como inhibidores de lafosfodiesterasa
Campo de la invención
La presente invención se refiere a inhibidores de la enzima fosfodiesterasa 4 (PDE4). Más particularmente, la invención se refiere a compuestos que son derivados de alcoholes de 1-fenil-2-piridinil alquilo, métodos para preparar tales compuestos, composiciones que los contienen y uso terapéutico de los mismos.
Antecedentes de la invención
La obstrucción de las vías respiratorias caracteriza una serie de enfermedades respiratorias graves, que incluyen el asma y la enfermedad pulmonar obstructiva crónica (EPOC). Los eventos que conducen a la obstrucción de las vías respiratorias incluyen edema de las paredes de las vías respiratorias, aumento de la producción de moco e inflamación.
Los fármacos para tratar las enfermedades respiratorias tales como el asma y la EPOC se administran actualmente por inhalación. Una de las ventajas de la vía inhalatoria sobre la sistémica es la posibilidad de administrar el fármaco directamente en el sitio de acción, reduciendo los efectos secundarios sistémicos, lo que da como resultado una respuesta clínica más rápida y una mayor proporción terapéutica.
Los corticosteroides inhalados son la terapia de mantenimiento actual de elección para el asma y junto con el broncodilatador agonistas beta2 para el alivio de los síntomas agudos, forman el pilar de la terapia actual para la enfermedad. El tratamiento actual de la EPOC es en gran medida sintomático mediante la terapia broncodilatadora con anticolinérgicos inhalados y agonistas del adrenoceptor beta-2. Sin embargo, los corticosteroides no reducen la respuesta inflamatoria en la EPOC como lo hacen en el asma.
Otra clase de agentes terapéuticos que ha sido ampliamente investigada en vista de sus efectos antiinflamatorios para el tratamiento de enfermedades inflamatorias respiratorias tales como el asma y la EPOC está representada por los inhibidores de las enzimas fosfodiesterasas (PDE), en particular de la fosfodiesterasa tipo 4 (en lo sucesivo denominada PDE4).
En la técnica anterior se han descrito varios compuestos que actúan como inhibidores de la PDE4. Sin embargo, la utilidad de muchos inhibidores de la PDE4 de la primera generación, tal como el rolipram y el piclamilast, ha sido limitada debido a sus efectos secundarios indeseables. Dichos efectos incluyen náuseas y emesis debido a su acción sobre la PDE4 en el sistema nervioso central y la secreción del ácido gástrico debido a la acción sobre la PDE4 en células parietales en el intestino.
La causa de dichos efectos secundarios ha sido ampliamente investigada.
Se ha encontrado que la PDE4 existe en dos formas distintas que representan conformaciones diferentes, que se designaron como sitio de unión a rolipram de alta afinidad o HPDE4, especialmente presentes en el sistema nervioso central y en las células parietales y sitio de unión a rolipram de baja afinidad o LPDE4 (Jacobitz, S y otros Mol. Pharmacol, 1996, 50, 891-899.), que se encuentra en las células inmunes e inflamatorias. Si bien ambas formas parecen mostrar actividad catalítica, difieren con respecto a su sensibilidad a los inhibidores. En particular, los compuestos con mayor afinidad por LPDE4 parecen ser menos propensos a inducir efectos secundarios como náuseas, emesis y aumento de la secreción gástrica.
El esfuerzo de apuntar a LPDE4 ha resultado en una ligera mejora en la selectividad para los inhibidores de la PDE4 de segunda generación, tal como el roflumilast. No obstante, el roflumilast está bajo dosificación para lograr un perfil de efectos secundarios aceptable.
Otras clases de compuestos que actúan como inhibidores de la PDE4 se han descrito en la técnica anterior.
Por ejemplo, EP 1634606describe, entre otros, derivados de cetona como benzofurano o derivados de 1,3-benzodioxol.
WO 9402465describe, entre otros, derivados de cetona de la Fórmula general

en donde Ri es alquilo inferior y R2 puede ser alquilo, alquenilo, cicloalquilo, cicloalquilo, cicloalquenilo, ciclotioalquilo o ciclotioalquenilo.
WO 9535281a nombre de Celltech Therapeutics se refiere a derivados de fenilo trisustituidos.
WO2009/018909 describe derivados de alcoholes de 1-fenil-2-piridinil-alquilo que tienen la fórmula general informada a continuación

como inhibidores de la enzima fosfodiesterasa 4 (PDE4).
WO2009/077068 describe otros derivados de alcoholes de 1 -fenil-2-piridinil alquilo que tienen la fórmula general informada a continuación.
WO2010/089107 describe otros derivados de alcoholes de 1 -fenil-2-piridinil alquilo que tienen la fórmula general informada a continuación.

como inhibidores de la enzima fosfodiesterasa 4 (PDE4).
Aunque se han descrito varios inhibidores de la PDE4 como se informó anteriormente, todavía existe la necesidad de inhibidores adicionales de la PDE4. Particularmente, todavía existe la necesidad de inhibidores de la PDE4 adicionales dotados de una alta afinidad por la enzima PDE4.
Particularmente ventajosa también sería la identificación de otros inhibidores de la PDE4 dotados de una alta afinidad por la enzima PDE4 y de una buena solubilidad en agua y/o en un sistema acuoso.
Como tal, además de ser usado para las preparaciones inhalables en forma de inhalador de polvo seco, inhalador de dosis medida presurizado o formulación nebulizada sin propelente, dichos compuestos, debido a su capacidad de absorción y facilidad de formulación, también podrían administrarse por otras vías, tal como la vía oral o transdérmica, pero también en cualquier otra solución farmacéutica tal como, por ejemplo, aquellas para inyección, infusión u administración ocular.
La presente invención se dirige a la necesidad mencionada anteriormente proporcionando los compuestos de la invención.
Resumen de la invención
La invención se refiere a compuestos de la Fórmula general (I) que actúan como inhibidores de la enzima fosfodiesterasa 4 (PDE4), a los procesos para su preparación, a las composiciones que los comprenden, a los usos terapéuticos y combinaciones con otros ingredientes farmacéuticamente activos entre los que están, por ejemplo, los usados actualmente en el tratamiento de trastornos respiratorios, por ejemplo, antagonistas-beta2, corticosteroides, inhibidores de la P38 MAP quinasa, IKK2, inhibidores de HNE, antagonistas de M3, moduladores de leucotrienos, AINE y reguladores de moco.
Descripción detallada de la invención
La invención se dirige a los compuestos de la Fórmula general (I), que actúan como inhibidores de la enzima fosfodiesterasa 4 (PDE4), a procesos para la preparación de los mismos, composiciones que los comprenden y usos terapéuticos de los mismos.
La invención proporciona compuestos de la Fórmula (I)

en donde:
Ri y R2 son iguales o diferentes y se seleccionan independientemente del grupo que consiste en: hidrógeno, metilo, etilo, difluorometilo, ciclopropilmetilo o ciclopropilo;
R3 son los mismos dos átomos de cloro en la posición 3 y 5 del anillo piridilo;
Z es un grupo (CH2)m en donde m = 0;
A es un anillo de fenilo, que está opcionalmente sustituido con uno o más sustituyentes R4, que pueden ser iguales o diferentes y se seleccionan independientemente del grupo que consiste en:
(C1C2) alquilo lineal o ramificado opcionalmente sustituido con uno o más (C3-C7) heterocicloalquilo;
trifluorometilo; metiltio; halógeno seleccionado de flúor o cloro; y
OR7 en donde R7 se selecciona del grupo que consiste en
- H;
- (C1-C4) alquilo opcionalmente sustituido con un radical seleccionado de OH, ciclopropilo, 4-morfolinilo, 1- o 4 piperidinilo y fenilo;
- trifluorometilo o difluorometilo; y
Wes
- -NR9SO2R10 en donde
R9 se selecciona del grupo que consiste en (C1-C10) alquilo opcionalmente sustituido con 4-morfolinilo, 1 -piperidinilo, 1-pirrolidinilo, piperazinilo, 1,2-tiazolidin-3-ilo, piperazin-1-ilcarbonilo en donde cualquiera de dichos anillos puede estar opcionalmente sustituido con uno o más metilo, etilo o OH; y
RnR12N-(C1-C10) alquileno en donde Ruy R12 son independientemente H o metilo opcionalmente sustituido con un grupo OH; y
R10 se selecciona del grupo que consiste en (C1-C4) alquilo opcionalmente sustituido por (C3-C7) cicloalquilo y; sus N-óxidos en el anillo de piridina, y sales farmacéuticamente aceptables de estos.
La invención también abarca las sales y/o solvatos farmacéuticamente aceptables de estos.
El término "sales farmacéuticamente aceptables”, como se usa en la presente descripción, se refiere a derivados de los compuestos de la Fórmula (I) o de sus N-óxidos correspondientes en el anillo de piridina en donde el compuesto principal se modifica adecuadamente mediante la conversión de cualquiera de los ácidos libres o grupos básicos, si están presentes, en la sal de adición correspondiente con cualquier base o ácido convencionalmente indicado como farmacéuticamente aceptable.
Los ejemplos adecuados de dichas sales pueden incluir sales de adición de ácidos minerales u orgánicos de residuos básicos tales como grupos amino, así como residuos del ácidos minerales u orgánicos tales como grupos carboxílicos. Los cationes de bases inorgánicas que pueden usarse adecuadamente para preparar sales dentro de la invención comprenden iones de metales alcalinos o alcalinotérreos tales como potasio, sodio, calcio o magnesio.
Los obtenidos por reacción del compuesto principal, que funciona como una base, con un ácido inorgánico u orgánico para formar una sal, comprenden, por ejemplo, sales del ácido clorhídrico, ácido sulfúrico, ácido fosfórico, ácido metanosulfónico, ácido alcanforsulfónico, ácido oxálico, ácido maleico, ácido succínico y ácido cítrico.
Los expertos en la técnica de química orgánica apreciarán que muchos compuestos orgánicos pueden formar complejos con solventes en los que ellos reaccionan o a partir de los cuales precipitan o cristalizan. Estos complejos son conocidos como "solvatos". Los solvatos farmacéuticamente aceptables del compuesto de la invención están dentro del alcance de la invención.
Dentro del alcance de la presente invención también se incluyen los polimorfos y formas cristalinas de los compuestos de la Fórmula (I), de sus N-óxidos en el anillo de piridina, o de sales farmacéuticamente aceptables, o solvatos de estos. En lo adelante, los compuestos de la Fórmula (I), los N-óxidos correspondientes en el anillo de piridina, variaciones, enantiómeros, diastereoisómeros de estos, sus sales y solvatos farmacéuticamente aceptables y los polimorfos o formas cristalinas de estos definidos en cualquier aspecto de la invención (excepto los compuestos intermedios descritos en los procesos químicos) se denominan "compuestos de la invención".
La invención comprende, además, un proceso para la preparación de los compuestos de la invención.
La presente invención también proporciona composiciones farmacéuticas de compuestos de la invención solos o en combinación, en mezcla con uno o más portadores farmacéuticamente aceptables.
En un aspecto adicional, la presente invención proporciona el uso de los compuestos de la invención como un medicamento.
En un aspecto, la presente invención proporciona el uso de los compuestos de la invención para la fabricación de un medicamento.
En particular, la presente invención proporciona el uso de los compuestos de la invención para la prevención y/o el tratamiento de cualquier enfermedad caracterizada por la hiperactividad de la fosfodiesterasa 4 (PDE4) y/o en donde es deseable una inhibición de la actividad de la PDE4.
En particular, los compuestos de la invención solos o combinados con otros ingredientes activos pueden administrarse para la prevención y/o el tratamiento de una enfermedad del tracto respiratorio caracterizada por una obstrucción de las vías respiratorias tal como el asma y la EPOC.
En un aspecto particular, la presente invención proporciona el uso de los compuestos de la invención para la preparación de un medicamento para la prevención y/o el tratamiento de cualquier enfermedad caracterizada por la hiperactividad de la fosfodiesterasa 4 (PDE4) y/o en donde es deseable una inhibición de la actividad de la PDE4. Además, la presente invención proporciona un método para la prevención y/o el tratamiento de cualquier enfermedad en donde la inhibición de la PDE4 es deseable, dicho método comprende administrar a un paciente que necesita dicho tratamiento una cantidad terapéuticamente eficaz de un compuesto de la invención.
Definiciones
El término "átomos de halógeno" como se usa en la presente descripción incluye flúor, cloro, bromo y yodo, preferentemente cloro.
Como se usa en la presente descripción, el término "(Ci -Cx ) alquilo" donde x es un número entero mayor que 1, se refiere a grupos alquilo de cadena lineal y ramificada en donde el número de átomos de carbono constituyentes está en el intervalo de 1 a x. Los grupos alquilo particulares son metilo, etilo, n-propilo, isopropilo y t-butilo.
Por analogía, el término "(Ci -Cx)alquileno", se refiere a un radical (C-i-Cx) alquilo divalente, en donde (C-i-Cx) alquilo es como se definió anteriormente.
El término "(Ci -Cx) alcoxi" donde x es un número entero mayor que 1, se refiere a grupos alcoxi de cadena lineal y ramificada en donde el número de átomos de carbono constituyentes está en el intervalo de 1 a x. Los grupos alcoxi particulares son metoxilo, etoxilo, n-propoxilo, isopropoxilo y t-butoxilo.
La expresión "(Ci -Cx)haloalquilo" se refiere a los grupos (Ci -Cx)alquilo" definidos anteriormente en donde uno o más átomos de hidrógeno se reemplazan por uno o más átomos de halógeno, que pueden ser iguales o diferentes entre sí. Los ejemplos de dichos grupos (Ci -C6)haloalquilo pueden incluir grupos alquilo halogenados, polihalogenados y
completamente halogenados en donde todos los átomos de hidrógeno se reemplazan por átomos de halógeno, por ejemplo, grupos trifluorometilo o difluoro metilo.
El término "(C1-Cx)alquiltio" donde x es un número entero mayor que 1, se refiere a grupos alquil-S de cadena lineal y ramificada en donde el número de átomos de carbono constituyentes está en el intervalo de 1 a x. Los grupos alquiltio particulares son metiltio, etiltio, n-propiltio, isopropiltio y t-butiltio.
El término "(C3-Cy)cicloalquilo", donde y es un número entero mayor o igual a 3, se refiere a grupos hidrocarburos cíclicos saturados que contienen de 3 a y átomos de carbono en el anillo. Los ejemplos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y cicloheptilo.
La expresión derivada "(C3-Cy) heterocicloalquilo "se refiere a grupos (C3-Cy)cicloalquilo monocíclicos, en donde al menos un átomo de carbono del anillo se reemplaza por un heteroátomo (por ejemplo, N, NH, S u O). Los ejemplos no limitantes de (C3-Cy)heterocicloalquilo están representados por: pirrolidinilo, imidazolidina, tiazolidinilo, piperazinilo, piperidinilo, morfolinilo, tiomorfolinilo, azetidinilo y monóxido o dióxido de los mismos.
Por analogía, el término "(C3-Cy)heterocicloalquileno", se refiere a un radical (C3-Cy)heterocicloalquilo divalente, e donde (C3-Cy)heterocicloalquilo es como se definió anteriormente.
La expresión "(C3-Cy)cicloalquilcarbonilo" se refiere a grupos (C3-Cy)cicloalquilCO en donde el grupo "(C3-Cy)cicloalquilo" tiene el significado definido anteriormente.
El término "(C2-Cz)alquenilo" se refiere a cadenas de carbono lineales o ramificadas, conjugadas o no conjugadas con uno o más enlaces dobles, en configuración cis o trans, en donde el número de átomos está en el intervalo de 2 a 6. El término "(Cs-Cz) cicloalquenilo", donde z es un número entero mayor o igual a 5, se refiere a grupos hidrocarburos cíclicos que contienen de 5 a z átomos de carbono y uno o más enlaces dobles.
El término "(C2-C6)alquinilo" se refiere a cadenas de carbono lineales o ramificadas con uno o más enlaces triples en donde el número de átomos está en el intervalo de 2 a 6.
El término "(C3-Cy)heterocicloalquilo(C1-Cx)alquileno" se refiere al grupo "(C1-Cx)alquileno" anterior en donde un átomo de carbono terminal se une a un grupo "(C3-Cy)heterocicloalquilo".
Como se usa en la presente, la expresión "sistema de anillo" se refiere a sistemas de anillo mono- o bicíclicos que pueden estar saturados, parcialmente insaturados o insaturados, tales como arilo, (C3-C8) cicloalquilo, (C3-C7) heterocicloalquilo o heteroarilo, con 5 a 11 átomos en el anillo en el que al menos un átomo del anillo es un heteroátomo (por ejemplo, N, S u O).
La expresión "arilo" se refiere a sistemas anulares mono o bicíclicos que tienen 6 a 10 átomos del anillo, en donde al menos un anillo es aromático.
La expresión "heteroarilo" se refiere a sistemas anulares mono o bicíclicos con 5 a 11 átomos del anillo, en el cual al menos un anillo es aromático y en el cual al menos un átomo del anillo es un heteroátomo (por ejemplo N, NH, S u O). Los ejemplos de sistemas monocíclicos de heteroarilo de 5, 6 miembros adecuados incluyen, por ejemplo, benceno, tiofeno, pirrol, pirazol, imidazol, isoxazol, oxazol, isotiazole, tiazol, piridina, radicales derivados defuranoy similares. Los ejemplos de sistemas arilo o heteroarilo bicíclicos adecuados incluyen radicales naftaleno, bifenileno, purina, pteridina, benzotriazol, quinoleina, isoquinoleína, indol, isoindol, benzotiofeno, dihidrobenzo dioxina, dihidrobenzo dioxepina, benzo oxazina y similares.
Resultará evidente para los expertos en la técnica que los compuestos de la Fórmula general (I) al menos contienen un centro estereogénico, es decir, representado por el átomo de carbono (1) con un asterisco abajo y por lo tanto existen como estereoisómeros ópticos.

Cuando los compuestos de acuerdo con la invención tienen al menos un centro estereogénico, pueden existir por consiguiente como enantiómeros. Cuando los compuestos de acuerdo con la invención poseen dos o más centros estereogénicos, pueden existir adicionalmente como diastereoisómeros. Debe entenderse que todos estos isómeros y mezclas de estos en cualquier proporción están abarcados dentro del alcance de la presente invención.
En una modalidad preferida, la presente invención se dirige a compuestos de la Fórmula (I)', que son compuestos de la Fórmula (I), sus N-óxidos en el anillo de piridina, y sales farmacéuticamente aceptables de estos como se definió anteriormente, donde la configuración absoluta del carbono (1) es la que se muestra a continuación:

La configuración absoluta para el carbono (1) se asigna sobre la base de la nomenclatura Cahn-Ingold-Prelog en función de las prioridades de los grupos.
En una modalidad preferida, para los compuestos de la Fórmula (I), la configuración absoluta en el carbono (1) es (S).
Los compuestos de la invención están dotados de una alta afinidad por la enzima PDE4 y de una buena solubilidad en agua y/o en un sistema acuoso que puede proporcionarles una capacidad de absorción y/o facilidad de formulación oral o transdérmica favorable en cualquier solución farmacéutica, tal como para administración inyectable, por infusión u ocular.
Los compuestos de la invención se pueden preparar de acuerdo con la adaptación apropiada de los enfoques sintéticos que se describen a continuación en la Sección Experimental, Ejemplos 1 a 39.
Los procesos descritos a continuación no deben considerarse limitantes del alcance de los métodos sintéticos disponibles para la preparación de los compuestos de la invención.
El proceso descrito es particularmente ventajoso ya que es susceptible de ser modulado apropiadamente a través de cualquier variante apropiada conocida por el experto, para obtener cualquiera de los compuestos deseados de la invención. Tales variantes están comprendidas dentro del alcance de la presente invención.
De todo lo anterior, debe quedar claro para el experto que cualquiera de los grupos descritos puede estar presente como tal o en cualquier forma debidamente protegida.
En particular, los grupos funcionales presentes en los compuestos de la invención o productos intermedios de estos que podrían generar reacciones secundarias y subproductos no deseados, deben protegerse adecuadamente antes de que tenga lugar la alquilación, acilación, acoplamiento, oxidación o sulfonilación. Igualmente, la desprotección posterior de esos mismos grupos protegidos puede seguir tras el completamiento de dichas reacciones.
En la presente invención, a menos que se indique de cualquier otra forma, el término "grupo protector", designa un grupo protector adaptado para preservar la función del grupo al cual está unido. Típicamente, los grupos protectores se usan para preservar las funciones amino, hidroxilo o carboxilo. Los grupos protectores apropiados pueden incluir, por ejemplo, bencilo, benciloxicarbonilo, t-butoxicarbonilo, alquilo o ésteres de bencilo o similares, que son bien conocidos por los expertos en la técnica [ver, para una referencia general, T.W. Green; Protective Groups in Organic Síntesis (Wiley, N.Y. 1999)].
Del mismo modo, la protección y la desprotección selectiva de cualquiera de dichos grupos, por ejemplo, incluyendo los grupos carbonilo, hidroxilo o amino, pueden llevarse a cabo de acuerdo con métodos muy bien conocidos empleados comúnmente en química orgánica sintética.
Los N-óxidos en el anillo de 4-piridinilo de los compuestos de la Fórmula general (I) y sus variantes pueden prepararse de acuerdo con los métodos disponibles en la literatura y bien conocidos por los expertos. Por ejemplo, pueden prepararse disolviendo el compuesto de la Fórmula general (I) o sus variantes en CH2G 2 o CHCl3, y después agregando un agente oxidante como el ácido m-cloro perbenzoico (mCPBA) a la solución resultante. Otros agentes oxidantes que pueden usarse son el peróxido de hidrógeno, el ácido perbenzoico y el ácido peracético.
Alternativamente, en particular para aquellos compuestos que comprenden grupos funcionales sensibles a la oxidación,
los N-óxidos correspondientes se preparan llevando a cabo la etapa de oxidación antes de que se introduzcan otros grupos funcionales.
En una modalidad, el proceso para la preparación de compuestos de la Fórmula (I) o variantes de estos se realiza a partir del N-óxido en el anillo de piridina de los compuestos intermedios, lo que permite la preparación de compuestos de la Fórmula (I) o variantes de estos en forma de N-óxidos en el anillo de piridina.
La salificación opcional de los compuestos de la Fórmula (I) o N-óxidos en el anillo de piridina de estos puede llevarse a cabo convirtiendo adecuadamente cualquiera de los grupos ácido o amino libres en las sales farmacéuticamente aceptables correspondientes. En este caso también, las condiciones operativas empleadas para la salificación opcional de los compuestos de la invención están dentro del conocimiento ordinario de la persona experta.
A partir de lo anterior, resultará evidente para los expertos que el proceso anterior, incluyendo cualquier variante de este para la preparación de compuestos adecuados de la invención, puede modificarse convenientemente para adaptar las condiciones de reacción a las necesidades específicas, por ejemplo, por la elección de agentes de condensación, solventes, y grupos protectores adecuados, según el caso.
La presente invención proporciona, además, composiciones farmacéuticas de los compuestos de la invención en mezcla con uno o más portadores farmacéuticamente aceptables, por ejemplo, los descritos en Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., Estados Unidos.
La administración de los compuestos de la presente invención puede llevarse a cabo de acuerdo con las necesidades del paciente, por ejemplo, por vía oral, nasal, ocular administración parenteral (subcutánea, intravenosa, intramuscular, intraesternal y por infusión), por inhalación, rectal, vaginal, tópica, local, transdérmica, y por administración ocular. Diversas formas de dosificación orales sólidas se pueden usar para administrar los compuestos de la invención incluyendo formas sólidas tales como tabletas, cápsulas de gel, cápsulas, comprimidos oblongos, gránulos, pastillas y polvos a granel. Los compuestos de la presente invención o los compuestos de la Fórmula (II) pueden administrarse solos o combinados con varios portadores farmacéuticamente aceptables, diluyentes (tales como sacarosa, manitol, lactosa, almidones) y excipientes conocidos en la técnica que incluyen pero sin limitarse a agentes de suspensión, solubilizantes, agentes tamponantes, aglutinantes, disgregantes, conservantes, colorantes, saborizantes, lubricantes y similares. Las cápsulas, tabletas y geles de liberación en el tiempo también son ventajosos en la administración de los compuestos de la presente invención o los compuestos de la Fórmula (I).
También se pueden usar diversas formas de dosificación líquidas orales para administrar compuestos de la invención, que incluyen soluciones, emulsiones, suspensiones, jarabes y elixires acuosos y no acuosos. Tales formas de dosificación pueden contener además diluyentes inertes adecuados conocidos en la técnica tales como agua y excipientes adecuados conocidos en la técnica tales como conservantes, agentes humectantes, edulcorantes, saborizantes, así como también agentes para la emulsificación y/o suspensión de los compuestos de la invención o los compuestos de la Fórmula (I). Los compuestos de la presente invención pueden inyectarse, por ejemplo, por vía intravenosa, en forma de una solución estéril isotónica. Otras preparaciones también son posibles.
Los supositorios para administración rectal de los compuestos de la invención pueden prepararse mediante la mezcla del compuesto con un excipiente adecuado tal como manteca de cacao, salicilatos y polietilenglicoles.
Las formulaciones para administración vaginal pueden estar en forma de crema, gel, pasta, espuma, o fórmula de aerosol que contiene, además del ingrediente activo, dichos portadores adecuados como se conocen en la técnica. Para la administración tópica, la composición farmacéutica puede estar en forma de cremas, ungüentos, linimentos, lociones, emulsiones, suspensiones, geles, soluciones, pastas, polvos, aerosoles, y gotas adecuadas para la administración a la piel, ojo, oído o nariz. La administración tópica también puede implicar la administración transdérmica a través de medios tales como parches transdérmicos.
Para el tratamiento de las enfermedades del tracto respiratorio, los compuestos de acuerdo con la invención se administran preferentemente por inhalación.
Las composiciones inhalables incluyen polvos inhalables, aerosoles dosificadores que contienen gas propelente o formulaciones inhalables sin propelentes.
Para la administración como un polvo seco, se pueden usar los inhaladores de una o de múltiples dosis conocidos de la técnica anterior. En ese caso el polvo puede rellenarse en cápsulas de gelatina, de plástico o de otras cápsulas, cartuchos o envases de burbuja o en un reservorio.
Un diluyente o portador, generalmente no tóxico y químicamente inerte para los compuestos de la invención, por ejemplo, lactosa o cualquier otro aditivo adecuado para mejorar la fracción respirable puede ser añadido a los compuestos en polvo de la invención.
Los aerosoles de inhalación que contienen gas propelente tales como hidrofluoroalcanos pueden contener los compuestos de la invención ya sea en solución o en forma dispersa o partículas micronizadas suspendidas. Las formulaciones que contienen propelente también pueden contener otros ingredientes tales como cosolventes, estabilizadores y opcionalmente otros excipientes.
Las formulaciones inhalables sin propulsores que comprenden los compuestos de la invención pueden estar en forma de soluciones o suspensiones en un medio acuoso, alcohólico o hidroalcohólico y pueden ser entregados por nebulizadores de chorro o ultrasónicos conocidos de la técnica anterior o por nebulizadores de niebla suave tales como como Respimat®.
Los compuestos de la invención se pueden administrar como único agente activo o en combinación con otros ingredientes farmacéuticos activos, incluyendo los usados actualmente en el tratamiento de trastornos respiratorios, por ejemplo, agonistas beta2, agentes antimuscarínicos, corticoides, inhibidores de proteína quinasas activados por mitógeno (P38 MAP quinasa), inhibidores de la subunidad beta del factor nuclear kappa-B quinasa (IKK2), inhibidores de la elastasa neutrofílica humana (HNE), inhibidores de la fosfodiesterasa 4 (PDE4), moduladores del leucotrieno, agentes antiinflamatorios no esteroideos (AINE) y reguladores de moco.
La presente invención también proporciona combinaciones de un compuesto de la invención, con un agonista p2 seleccionado del grupo que consiste en carmoterol, vilanterol (GSK-642444), indacaterol, milveterol, arformoterol, formoterol, salbutamol, levalbuterol, terbutalina, AZD-3199, olodaterol (BI-1744-CL), abediterol (LAS-100977), bambuterol, isoproterenol, procaterol, clenbuterol, reproterol, fenoterol y ASF-1020 y sus sales.
La presente invención también proporciona combinaciones de un compuesto de la invención, con un corticosteroide seleccionado del grupo que consiste en propionato de fluticasona, furoato de fluticasona, furoato de mometasona, dipropionato de beclometasona, ciclesonida, budesonida, GSK 685698, GSK 870086.
La presente invención también proporciona combinaciones de un compuesto de la invención con un agente antimuscarínico seleccionado del grupo que consiste en sales de aclidinio, tiotropio, ipratropio, trospio, glicopirronio y oxitropio.
La presente invención proporciona además combinaciones de un compuesto de la invención con un inhibidor de PDE4 seleccionado del grupo que consiste en AN-2728, AN-2898, CBS-3595, apremilast, ELB-353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, cipamfilina, cilomilast, roflumilast, BAY19-8004 y SCH-351591, AN-6415, indus-82010, TPI-PD3, ELB-353, CC-11050, GSK-256066, oglemilast, OX-914, tetomilast, MEM-1414 y RPL-554.
La presente invención proporciona además combinaciones de un compuesto de la invención con un inhibidor de MAP quinasa P38 seleccionado del grupo que consiste en semapimod, talmapimod, pirfenidona, PH-797804, GSK-725, minokina y losmapimod y sales de estos.
En una modalidad preferida, la presente invención proporciona combinaciones de un compuesto de la invención con un inhibidor de IKK2.
La presente invención proporciona además combinaciones de un compuesto de la invención con un inhibidor de HNE seleccionado del grupo que consiste en AAT, ADC-7828, Aeriva, TAP-I, AE-3763, KRP-109, AX-9657, POL-6014, AER-002, AGTC-0106, respriva, AZD-9668, zemaira, AAT IV, PGX-100, elafin, SPHD-400, prolastina C y prolastina inhalada. La invención proporciona además combinaciones de un compuesto de la invención con un modulator de leukotrienos seleccionado del grupo que consiste en montelukast, zafirlukast y pranlukast.
La invención proporciona además combinaciones de un compuesto de la invención con un AINE seleccionado del grupo que consiste en ibuprofeno y ketoprofeno.
La presente invención proporciona además combinaciones de un compuesto de la invención con un regulador de moco seleccionado del grupo que consiste en INS-37217, diquafosol, sibenadet, CS-003, talnetant, DNK-333, MSI-1956 y gefitinib.
Las dosificaciones de los compuestos de la presente invención dependen de una variedad de factores que incluyen la enfermedad particular a tratar, la gravedad de los síntomas, la vía de administración, la frecuencia del intervalo de dosificación, el compuesto particular utilizado, la eficacia, perfil toxicológico, y perfil farmacocinético del compuesto. Ventajosamente, los compuestos de la invención pueden administrarse por ejemplo, a una dosificación comprendida entre 0.001 y 1000 mg/día, preferentemente entre 0.1 y 500 mg/día.
Cuando se administran por vía de inhalación, la dosis de los compuestos de la invención está comprendida ventajosamente entre 0.01 y 20 mg/día, preferentemente entre 0.1 y 10 mg/día.
Preferentemente, los compuestos de la invención solos o combinados con otros ingredientes activos pueden administrarse para la prevención y/o tratamiento de cualquier enfermedad respiratoria obstructiva tal como asma, bronquitis crónica y enfermedad pulmonar obstructiva crónica (EPOC).
Sin embargo, los compuestos de la invención pueden administrarse para la prevención y/o tratamiento de cualquier enfermedad en la que se requiera la inhibición de la PDE4. Dicha enfermedad incluye: estados de enfermedad alérgica tales como dermatitis atópica, urticaria, rinitis alérgica, conjuntivitis alérgica, conjuntivitis vernal, granuloma eosinofílico, psoriasis, artritis inflamatoria, artritis reumatoide, choque séptico, colitis ulcerosa, enfermedad de Crohn, lesión por reperfusión del miocardio y cerebro, glomerulonefritis crónica, choque endotóxico, fibrosis quística, reestenosis arterial, arterosclerosis, queratosis, espondilitis reumatoide, osteoartritis, piresis, diabetes mellitus, neumoconiosis, eczema por contacto tóxico y alérgico, eczema atópico, eczema seborreico, liquen simple, quemadura solar, prurito en el área anogenital, alopecia areata, cicatrices hipertróficas, lupus eritematoso discoide, lupus eritematoso sistémico, piodermias foliculares y de área amplia, acné endógeno y exógeno, acné rosácea, enfermedad de Behget, nefritis púrpura anafilactoide, enfermedad inflamatoria intestinal, leucemia, esclerosis múltiple, enfermedades gastrointestinales, enfermedades autoinmunes y similares.
También incluyen trastornos neurológicos y psiquiátricos, como la enfermedad de Alzheimer, esclerosis múltiple, esclerosis lateral amiotrófica (ELA), atrofia de sistemas múltiples (ASM), esquizofrenia, enfermedad de Parkinson, enfermedad de Huntington, enfermedad de Pick, depresión, accidente cerebrovascular y lesión de la médula espinal. La presente invención se describirá ahora más detalladamente por medio de los siguientes ejemplos no limitantes. Parte experimental
Los nombres químicos de los compuestos se generaron con el software Structure To Name Enterprise Cambridge 10.0. La purificación mediante cartucho SCX preempaquetado se refiere a Isolute SCX, un potente absorbente de intercambio catiónico.
Procedimientos para la formación de sal.
A menos que se indique lo contrario, las sales de trifluoroacetato y sales del ácido fórmico descritas en la sección experimental se obtuvieron de acuerdo con el siguiente procedimiento: Los compuestos que contenían uno o más centros básicos y purificados por HPLC preparativa se obtuvieron como sal del ácido fórmico (Método 1) o sal del ácido trifluoroacético (Método 2), una vez que las fracciones limpias recogidas de la cromatografía se evaporaron a presión reducida sin ningún tratamiento básico adicional.
Si no se indica lo contrario, cualquier otra sal se obtuvo al tratar la base con una solución del ácido correspondiente en condiciones conocidas por el experto.
La estequiometría de la sal se determinó, si era necesario, por RMN.
Caracterización por RMN
Los espectros de RMN se registraron con:
Los espectros de 1H-RMN se registraron en un espectrómetro Varian AS400 de 400 MHz. Los cambios químicos se reportan como valores de 8 en ppm con respecto al trimetil silano (TMS) como un estándar interno. Las constantes de acoplamiento (valores J) se dan en hercios (Hz) y las multiplicidades se informan usando la siguiente abreviatura (s=singlete, d=doblete, t=triplete, q=cuartete, m=multiplete, br=ancho, nd=no determinado).
O
Los espectros de 1H-RMN se registraron en un espectrómetro Bruker ARX300 a 300.13 MHz (1H) usando solventes deuterados, como dimetilsulfóxido deuterado (DMSO-d6) o cloroformo deuterado (CDC13). El instrumento estaba equipado con una sonda inversa multinuclear y un controlador de temperatura. Los desplazamientos químicos se expresan en partes por millon (ppm) a campo abajo de tetrametilsilano (unidades d). La multiplicidad se indica de la siguiente manera: (s) singlete, (d) doblete, (dd) doble doblete, (ddd) triple doblete, (t) triplete, (dt) doble triplete, (q) cuartete, (m) multiplete, (br s) señal amplia. Las constantes de acoplamiento J se expresan en unidades de hercios (Hz). HPLC Preparativo - Método 1
Columna: Waters Symmetry Prep C1817um 19x300
Flujo: 20 ml/min
Fase móvil: 90 % H2O, 10 % acetonitrilo, 0.05 % TFA (A), 10 % H2O, 90 % acetonitrilo, 0.05 % TFA (B)
Gradiente:

Se usó el mismo gradiente sin TFA en la fase móvil para HPLC preparativa en condiciones neutras.
HPLC Preparativo - Método 2
Waters Micromass ZQ; Gestor de muestras 2767; Detector de matriz de fotodiodos 2996;
Columna XTerra Prep MS C18 Columna (5 |jm, 19 x 150 mm, Waters); tasa de flujo de 20 ml/min con detección MS o UV fijada a 254 nm.
Gradiente:

Eluyente
Solvente A (agua:MeCN:HCOOH 95:5:0.05)
Solvente B (agua:MeCN:HCOOH 5:95:0.05)
HPLC Preparativa -Método 3
Waters Micromass ZQ / gestor de muestras 2767
Detector de matriz de fotodiodos: 2996
Columna: XTERRA Prep MS C1810um 19x300
Flujo: 20 ml/min
Fases Móviles: H2O, 0.1 % TFA (A); acetonitrilo, 0.1 % TFA (B)
Gradiente:

Acondicionamiento:

HLPC quiral:
La pureza enantiomérica se determinó en el sistema de HPLC Hewlett Packard 1050 usando una columna Chiracel OD (5|j 4.6X250 mm), eluyendo con el uso de una mezcla isocrática de hexano e isopropanol en diferentes proporciones como se indica en cada ejemplo específico.
Flujo = 0.8 ml/min
detección UV = 230 nm.
Determinación de la rotación óptica (actividad)
Las rotaciones específicas de los compuestos se midieron con un polarímetro Perkin Elmer modelo 241 o 341.

Los valores MS/ESI+ [MH]+ informados en el texto a continuación pueden obtenerse por MS con el instrumento Waters ZQ (o equivalente) o por UPLC con el instrumento Waters:
Instrumento de MS: Waters ZQ (o equivalente)
Polaridad ES+
Polaridad ES-

Instrumento para LC: Acquity Waters UPLC
Instrumento: UPLC Waters acoplado con micromasa ZQ e interconectado con detector PDA 2996
Columna: Acquity UPLC BEH C181.7 um 50x2.1 mm
Método: TFA largo
Condiciones: ESI+, 3.2KV, 25V, 350°C
Longitud de onda: PBI

Las rutas y procedimientos sintéticos detallados para ejemplos específicos se describen en los Ejemplos 1 a 39.
En los procedimientos que siguen, después de cada material de partida, a veces se proporciona una referencia a un número compuesto. Esto se proporciona simplemente para ayudar al químico experto. El material de partida no necesariamente pueden haberse preparado del lote referido.
Cuando se hace referencia al uso de un procedimiento "similar" o "análogo", como apreciarán los expertos en la técnica, dicho procedimiento puede implicar variaciones menores, por ejemplo, temperatura de reacción, cantidad de reactivo/solvente, tiempo de reacción, condiciones de tratamiento o condiciones de purificación cromatográfica.
Algunos compuestos o intermediarios mencionados en la presente patente y usados para la síntesis del compuesto de la presente invención se han descrito en solicitudes de patente anteriores como las enumeradas en la Tabla 1.
Tabla 1


Ejemplo 1
Síntesis de (S)-3,5-dicloro-4-(2-(3,4-dimetoxifenil)-2-hidroxietil)piridina 1-óxido (Compuesto 1)
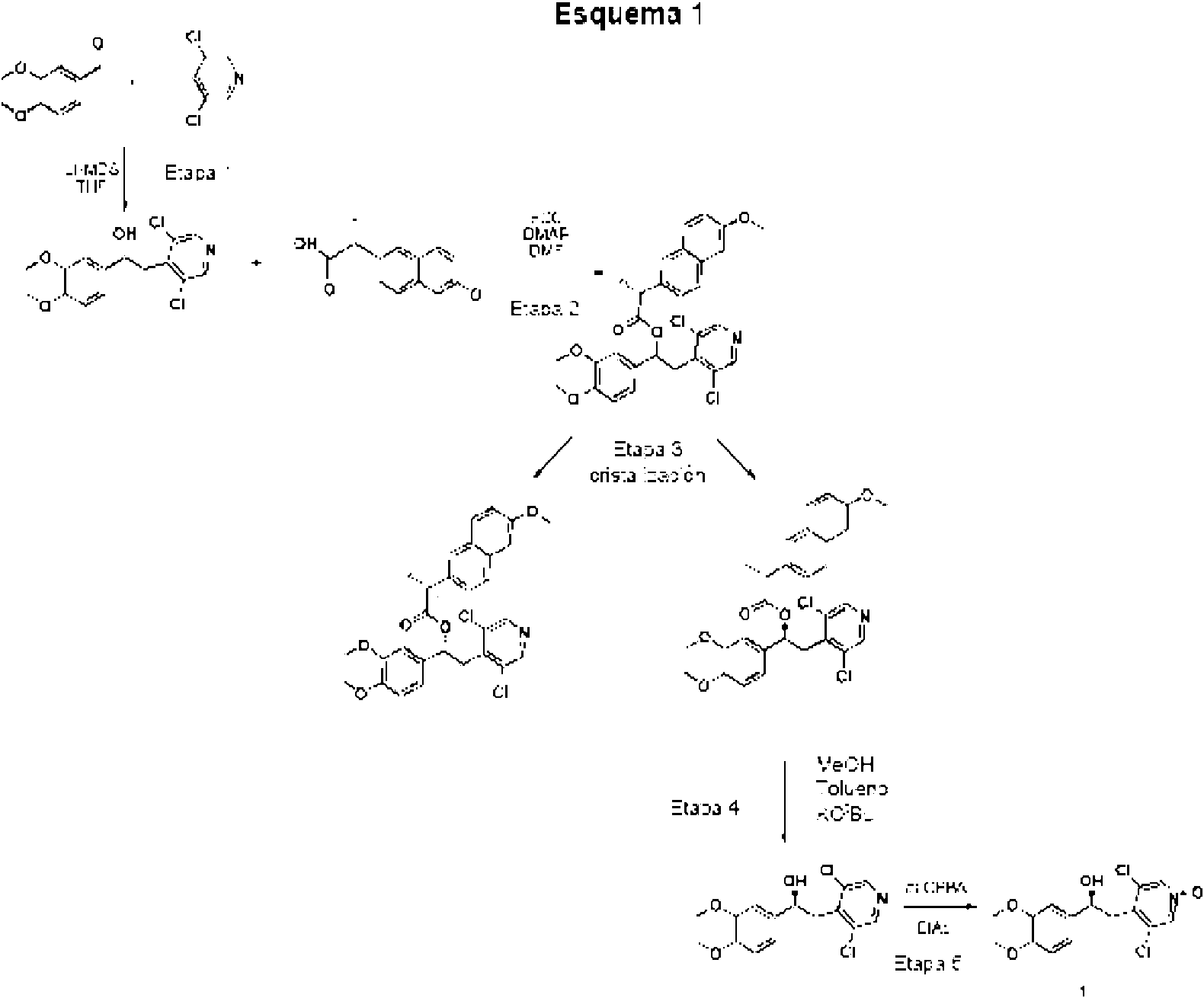
Etapa 1: Síntesis de 2-(3,5-didoropiridin-4-il)-1-(3,4-dimetoxifenil)etanol (2)
3,5-Didoro-4-metilpiridina (54 g, 331 mmol) se disolvió en THF seco (480 mL) bajo una atmósfera de argón y se enfrió a -78°C en un baño de hielo seco/acetona. Solución de LHMDS en THF (331ml, 331 mmol) se añadió en forma de gotas manteniendo la temperatura a -78°. La mezcla se agitó a -78° por 1 h. Después de eso, una solución de 3,4-dimetoxibenzaldehído (50 g, 301 mmol) en THF seco (120 ml) se añadió en forma de gotas manteniendo la temperatura a -78°C. Cuando se completó la adición, la mezcla se dejó calentar a temperatura ambiente.
La reacción se vertió en hielo y agua (1L) y la mezcla se agitó hasta que se formó un precipitado copioso. El sólido se filtró, y se disolvió en acetato de etilo (500 ml), se secó sobre Na2SO4 y el solvente se evaporó al vacío. El producto crudo se cristalizó en CHCl3/hexano. El precipitado se filtró, se lavó con hexano y se secó al vacío a 40°C por 8 h para dar 55 g (rendimiento 45 %). La solución del licor madre se evaporó al vacío a 40°C, se disolvió en acetato de etilo (200 ml) y se extrajo con 200 ml de agua. La solución orgánica se secó sobre Na2SO4 y el solvente se evaporó al vacío a
40°C. El producto crudo se cristalizó en CHCl3/hexano, y se obtuvieron 15 g adicionales del producto deseado (rendimiento general 70 %).
Etapa 2: Síntesis de ((S)-2-(3,5-didoropiridin-4-il)-1-(3,4-dimetoxifenil)etil) 2-(6-metoxinaftalen-2-il)propanoato (11) 2-(3,5-Didoropiridin-4-il)-1-(3,4-dimetoxifenil)etanol (compuesto 2, 50 g, 152 mmol) y ácido (R)-2-(6metoxinaftalen-2-il)propanoico comercialmente disponibles (compuesto 10, 38.6 g, 168 mmol), DMAP (20.5 g, 168 mmol) y EDC (43.8 g, 229 mmol) se disolvieron en DMF (300 ml) y la mezcla de reacción se agitó a temperatura ambiente por 2 h. Después de ese tiempo se añadió agua (500 ml), y la solución se agitó cuando se produjo la precipitación. El sólido se filtró y se disolvió en DCM (500 ml). La solución orgánica se lavó con HCl 1N acuoso (2x500 ml), solución de NaHCO3 saturado acuoso (500 ml) y se secó sobre Na2SO4. El solvente se evaporó al vacío y el residuo sólido se sonicó en EtOH (300 ml) y se trituró por 1 h. El precipitado resultante se recogió por filtración y se secó al vacío a 40°C por 4 h para dar el compuesto del título (79 g; rendimiento 99 %) como una mezcla diastereoisomérica.
Etapa 3: Síntesis de (R)-((S)-2-(3,5-dicloropiridin-4-il)-1-(3,4-dimetoxifenil)etil) 2-(6-metoxinaftalen-2-il)propanoato (12): ((S)-2-(3,5-Dicloropiridin-4-il)-1-(3,4-dimetoxifenil)etil) 2-(6-metoxi naftalen-2-il)propanoato (79 g, 146 mmol) se disolvió en CHCl3(100 ml) y MeOH (30 ml) se añadió lentamente hasta una opalescencia persistente y la mezcla se dejó a temperatura ambiente por 2 h. El sólido formado se recogió por filtración y se recristalizó por un sistema de solventes CHCh/MeOH (70 ml/20 ml) para obtener 35 g del compuesto deseado (rendimiento 88 %, ee 98 %).
Análisis HPLC quiral Rt= 42.33 min (isómero rápido); eluyente: hexano:isopropanol 97:3 1H RMN (600 MHz, CLOROFORMO-d) 8 ppm 8.04 (s, 2 H), 7.67 (d, J=8.79 Hz, 1 H), 7.58 (d, J=8.52 Hz, 1 H), 7.53 (m, 1 H), 7.12 - 7.20 (m, 3 H), 6.95 (dd, J=8.24, 1.92 Hz, 1 H), 6.78 - 6.88 (m, 2 H), 6.14 (dd, J=10.44, 4.12 Hz, 1 H), 3.95 (s, 3 H), 3.88 (s, 3 H), 3.78 - 3.81 (m, 4 H), 3.55 (dd, J=13.73, 10.44 Hz, 1 H), 3.14 (dd, J=13.60, 4.26 Hz, 1 H), 1.44 (d, J=7.14 Hz, 3 H).
Etapa 4: Síntesis de (S)-2-(3,5-dicloropiridin-4-il)-1-(3,4-dimetoxifenil)etanol (13)
(R) -((S)-2-(3,5-Dicloropiridin-4-il)-1-(3,4-dimetoxifenil)etil) 2-(6-metoxinaftalen-2-il)propanoato (30 g, 56 mmol) se disolvió en MeOH y se añadió tolueno lentamente. terc-Butóxido de potasio se añadió lentamente a la suspensión. La mezcla se agitó por 24 h a temperatura ambiente. La reacción se diluyó con agua (500 ml) y la mezcla acuosa se extrajo con CHCl3 (500 ml). La capa orgánica se secó sobre Na2SO4 y el solvente se evaporó al vacío. El residuo se cristalizó a partir de CHCh(100 ml) y hexano (20 ml, hasta una opalescencia persitente). El licor madre se concentró y se recristalizó de la misma forma dando una segunda cosecha del compuesto deseado. Se obtuvieron en total 16 g del compuesto deseado (rendimiento 87 %).
Análisis HPLC quiral Rt= 58.03 min; eluyente: hexano:isopropanol 95:5. [ Lal J2a0 = 10.21 (c=0.506, Metanol)
1H RMN (400 MHz, acetona) 8 ppm 8.47 (s, 2 H), 6.96 - 7.15 (m, 1 H), 6.87 (m, 2 H), 4.93 - 5.21 (m, 1 H), 4.50 (d, J=3.97 Hz, 1 H), 3.78 (s, 6 H), 3.44 (dd, J=12.79, 8.38 Hz, 1 H), 3.22 (dd, J=13.01, 5.51 Hz, 1 H).
MS/ESI+ [MH]+: 328.19
Etapa 5: Síntesis de (S)-3,5-dicloro-4-(2-(3,4-dimetoxifenil)-2-hidroxietil)piridina 1-óxido (1)
(S) -2-(3,5-Dicloropiridin-4-il)-1-(3,4-dimetoxifenil)etanol (4 g, 12 mmol) se disolvió en acetato de etilo y a la solución se añadió ácido m-CPB. La mezcla se agitó a temperatura ambiente por 5 h. El sólido formado se recogió por filtración, se lavó con acetato de etilo y se secó al vacío para dar 1.72 g del compuesto del título (rendimiento 41 %). Análisis HPLC quiral Rt= 22.16 min; eluyente: hexano:isopropanol 6:4. [«L = 68-91(c = 0.253, Metanol/CHCI31:1). MS/ESI+[MH]+: 344.19
1H RMN (400 MHz, CLOROFORMO-d) 8 ppm 8.15 (s, 2 H), 6.99 (m, 1 H), 6.79 - 6.88 (m, 2 H), 5.03 (dd, J=8.50, 5.32 Hz, 1 H), 3.75-3.98 (m, 6 H), 3.42 (dd, J=13.57, 8.56 Hz, 1 H), 3.19 (dd, J=13.51, 5.32 Hz, 1 H), 2.06-2.15 (m, 1 H). Ejemplo 2
Síntesis de clorhidrato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(N-(2-morfolinoetil)metilsulfonamido)benzoiloxi)-2-(3,4-dimetoxifenil)etil)piridina 1-óxido (Compuesto 14)

Etapa 1: Síntesis de (S)-4-(2-(4-(N-(terc-butoxicarbonN)metNsulfonamido)-3-(cidopropNmetoxi)benzoNoxi)-2-(3,4-dimetoxifenil)etil)-3,5-dicloropiridina 1-óxido (15)
(S)-3,5-Didoro-4-(2-(3,4-dimetoxifenil)-2-hidroxietil)piridina 1-óxido (734 mg 2.13 mmol) se disolvió en DMF (5,5 ml). Se añadieron EDAC (840 mg, 4.36 mmol), DMAP (390 mg, 3.2 mmol) y el compuesto 9 (1.23 g, 3.2 mmol). La mezcla se agitó a temperatura ambiente por 1h, después se añadió agua y la fase acuosa se extrajo con AcOEt dos veces. La fase orgánica combinada se lavó con HCl 1N, se secó sobre Na2SO4 y se evaporó hasta secarse. El producto crudo se trituró con n-hexano para dar 1.87 g del compuesto deseado (rendimiento 90 %). MS/ESI+ 710.15 [MH]+
Etapa 2: Síntesis de (S)-3,5-didoro-4-(2-(3-(cidopropNmetoxi)-4-(metNsulfonamido)benzoNoxi)-2-(3,4-dimetoxifenil)etil)piridina 1-óxido (16)
(S)-4-(2-(4-(N-(terc-Butoxicarbonil)metilsulfonamido)-3-(ciclopropilmetoxi)benzoiloxi)-2-(3,4-dimetoxifenil)etil)-3,5-dicloropiridina 1-óxido (240 mg, 0,34 mmol) se disolvió en DmF (4,5 ml) y la solución se agitó a 100 grados por 5 días hasta completarse. Después la mezcla se dejó enfriar hasta la temperatura ambiente y se añadió agua. La fase acuosa se extrajo con EtOAc dos veces. La fase orgánica se secó sobre Na2SO4 y se evaporó hasta secarse. El producto crudo se trituró con Et2O para dar el compuesto del título (160 mg, 80 % de rendimiento). MS/ESI+610.09 [MH]+ 1H RMN (400 MHz, cloroformo-d) 8 ppm 8.15 (s, 2 H), 7.70 (dd, J=8.38, 1.65 Hz, 1 H), 7.59 (d, J=8.44 Hz, 1 H), 7.47 (d, J=1.59 Hz, 1 H), 7.21 (m, 1 H), 6.95 - 7.08 (m, 2 H), 6.87 (d, J=8.31 Hz, 1 H), 6.29 (dd, J=10.15, 4.28 Hz, 1 H), 3.82 y 4.02 (2s, 6 H, 3H cada uno), 3.72 (dd, J=14.00, 10.09 Hz, 1 H), 3.34 (dd, J=14.06, 4.28 Hz, 1 H), 3.06 (s, 3 H), 1.22 -1.36 (m, 1 H), 0.60 - 0.77 (m, 2 H), 0.35 (q, J=5.01 Hz, 2 H)
Etapa 3: Síntesis de clorhidrato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(N-(2-morfolinoetil)metilsulfonamido) benzoiloxi)-2-(3,4-dimetoxifenil)etil)piridina 1-óxido (14)
(S)-3,5-Dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(metilsulfonamido)-benzoiloxi)-2-(3,4-dimetoxifenil)etil)piridina 1-óxido (40 mg, 0.065 mmol) se disolvió en DMF (1 ml). Se añadieron K2CO3 (25 mg, 0.18 mmol) y 4-(2-cloroetil)morfolina (20 mg, 0.133 mmol) y la mezcla se agitó a 45 grados toda la noche. La reacción se dejó después enfriar hasta la temperatura ambiente y se añadió agua. La fase acuosa se extrajo con AcOEt dos veces y la capa orgánica se secó sobre Na2SO4. El solvente se evaporó al vacío para dar el producto crudo, que se purificó por HPLC preparativa (Método 2) y se cristalizó a partir de éter de petróleo/éter de dietilo 1/1. La salificación se logró después del tratamiento con HCl/AcOEt para dar clorhidrato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(N-(2-morfolinoetil)metilsulfonamido)benzoiloxi)-2-(3,4-dimetoxifenil)etil)-piridina 1-óxido (10 mg, 0.013 mmol, rendimiento 20 %). MS/ESI+ 724.2 [MH]+
1H RMN (400 MHz, cloroformo-d) 8 ppm 8.16 (s, 2 H), 7.61 - 7.71 (m, 1 H), 7.53 (m, 1 H), 7.33 - 7.45 (m, 1 H), 6.92 -7.06 (m, 2 H), 6.87 (d, J=8.38 Hz, 1 H), 6.22 - 6.32 (m, 1 H), 3.84 - 3.96 (m, 12 H), 3.65 - 3.78 (m, 1 H), 3.28 - 3.41 (m, 1 H), 3.00 (s, 3 H), 2.4 -1.5 (m, 6 H), 1.25 (m, 1 H), 0.69 (d, J=7.94 Hz, 2 H), 0.37 (d, J=4.41 Hz, 2 H).
Los compuestos enumerados en la Tabla 2 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 2, al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados. Las variaciones específicas en los métodos experimentales o de purificación se indican en la tabla.
El ácido carboxílico se sintetizó siguiendo los procedimientos análogos a aquellos para el compuesto 9 (WO2010/089107 Ejemplo 18, Etapa 1-6).













Ċ

Los compuestos enumerados en la Tabla 3 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 2, al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados. Las variaciones específicas en los métodos experimentales o de purificación se indican en la tabla. Los ejemplos de referencia se indican por un asterisco (*) en la columna 2.
Las referencias a los procedimientos para la síntesis de diferentes alcoholes se enumeran en la Tabla 1.
El ácido carboxílico se sintetizó siguiendo el mismo procedimiento que para el compuesto 9 (WO2010/089107 Ejemplo 18, Esquema 2, Etapa 4-6)


Ejemplo 3 (Referencia)
Síntesis de (S)-3,5-didoro-4-(2-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(1,1-dioxotiomorfolinilmetil)benzoiloxi)-etil)piridina 1-óxido (Compuesto 37)

(S)-3,5-Didoro-4-(2-(3-(cidopropNmetoxi)-4-(difluorometoxi)fenN)-2-hidroxietil)piridina 1-óxido (compuesto 3, 40 mg, 0.095 mmol) se disolvió en DMF (1.5 ml). EDAC (55 mg, 0.285 mmol), DMAP (14 mg, 0.114 mmol) y ácido 1,1-dioxotiomorfolinilmetil)benzoico (39 mg, 0.143 mmol) se añadieron. La mezcla se agitó a temperatura ambiente por 1h, después se añadió agua y la fase acuosa se extrajo con AcOEt dos veces. La fase orgánica combinada se lavó con HCl 1N, se secó sobre Na2SO4 y se evaporó hasta secarse para dar 40 mg del compuesto deseado (rendimiento 65 %). MS/ESI+ 671.1 [MH]+
1H RMN (400 MHz, acetona) 8 ppm 8.27 (s, 2 H), 8.05 (d, J=7.06 Hz, 2 H), 7.54 (d, J=7.94 Hz, 2 H), 7.31 (m, 1 H), 7.20 (q, J=8.38 Hz, 2 H), 6.92 (t, J=75.00 Hz, 1 H), 6.34 (dd, J=9.26, 4.41 Hz, 1 H), 3.98 (d, J=7.06 Hz, 2 H), 3.70 - 3.83 (m, 3 H), 3.41 - 3.50 (m, 1 H), 3.07 (d, J=3.97 Hz, 4 H), 2.99 (d, J=4.41 Hz, 4 H), 1.22- 1.35 (m, 1 H), 1.11 (t, J=7.06 Hz, 1 H), 0.55 - 0.64 (m, 2 H), 0.33 - 0.42 (m, 2 H).
Ejemplo 4
Síntesis de clorhidrato de (S)-3,5-didoro-4-(2-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenN)-2-(4-hidroxi-3-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 42)

Etapa 1: Síntesis de metil 4-(benciloxi)-3-(metilsulfonamido)benzoato (47)
Metil 3-amino-4-(benciloxi)benzoato (1.7 g, 6.61 mmol) se disolvió en piridina (5 ml), después se añadió cloruro de metanosulfonilo (0.62 ml, 7.93 mmol) a 0°C, y la mezcla se agitó a temperatura ambiente por 2 h. La reacción se apagó con HCl 1N, y el producto se extrajo con acetato de etilo. La fase orgánica se lavó con HCl 1N (2x30ml) y salmuera, después se secó sobre Na2SO4. El solvente se eliminó para producir 1.7 g del intermediario deseado (rendimiento: 77 %).
Etapa 2: Síntesis de metil 4-(benciloxi)-3-(N-(2-morfolinoetil) metilsulfonamido)benzoato (46)
Metil 4-(benciloxi)-3-(metilsulfonamido)benzoato (700 mg, 2.1 mmol) se disolvió en DMF (5 ml). Se añadieron K2CO3 (580 mg, 4.2 mmol) y 4-(2 cloroetil) morfolina (628 mg, 4.2 mmol). La mezcla se agitó a 60 °C por 3 h. La reacción se apagó con agua, y el producto se extrajo con acetato de etilo. La capa orgánica se lavó con H2O (2x) y solución saturada de NaCl, se secó sobre Na2SO4 y se evaporó al vacío para producir 750 mg del compuesto deseado. (rendimiento 80 %).
Etapa 3: Síntesis de ácido 4-(benciloxi)-3-(N-(2-morfolinoetil) metilsulfonamido)benzoico (45)
Metil 4-(benciloxi)-3-(N-(2-morfolinoetil)metilsulfonamido)benzoato (750 mg, 1.7 mmol) se disolvió en THF (10 ml). Se añadió LiOH 1N (1.5 ml) y la mezcla resultante se agitó a temperatura ambiente por 5 h. El pH se ajustó hasta ser neutro añadiendo HCl 1N y después los volátiles se eliminaron al vacío para producir 650 mg del producto crudo.
Etapa 4: Síntesis de (S)-4-(2-(4-(benciloxi)-3-(N-(2-morfolinoetil) metilsulfonamido)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi) fenil)etil)-3,5-dicloropiridina 1-óxido (44)
El Compuesto 3 (200 mg, 0.5 mmol) y ácido 4-(benciloxi)-3-(N-(2-morfolinoetil)metilsulfonamido)benzoico (652 mg, 1.5 mmol) se disolvieron en DMF (5 ml), después se añadió EDC (767 mg, 4 mmol) y DMAP (122 mg, 1 mmol). La mezcla se agitó a temperatura ambiente por 24 h, después se apagó añadiendo agua y el producto se extrajo con acetato de etilo. La fase orgánica se lavó con solución saturada de K2CO3 (2x), agua (2x) y salmuera, se secó sobre Na2SO4 y se evaporó al vacío para producir 350 mg del producto crudo que se usó en la próxima etapa sin ninguna purificación adicional.
1H RMN (400 MHz, acetona) 8 ppm 8.25 (s, 2 H), 8.00 - 8.14 (m, 2 H), 7.58 (d, J=7.06 Hz, 2 H), 7.27 - 7.48 (m, 5 H), 7.13 - 7.24 (m, 2 H), 6.69-6.91-7.11 (t, 1 H, CHF2), 6.32 (dd, J=9.70, 4.85 Hz, 1 H), 5.35 (s, 2 H), 3.91 - 4.05 (m, 2 H), 3.74 (dd, J=14.11, 9.70 Hz, 3 H), 3.33- 3.54 (m, 5 H), 2.91 (s, 3 H), 2.19-2.50 (m, 6 H), 1.18-1.36 (m, 1 H), 0.54-0.66 (m, 2 H), 0.39 (q, J=4.85 Hz, 2 H).
MS/EST 836.2 [MH]+
Etapa 5: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-difluorometoxi)fenil)-2-(4-hidroxi-3-(N-(2-morfolinoetil)-metilsulfonamido) benzoiloxi)etil)piridina 1-óxido (43)
(S)-4-(2-(4-(Benciloxi)-3-(N-(2-morfolinoetil)metilsulfonamido)-benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (120 mg, 0.14 mmol) se disolvió en acetona (10 ml), después 5 % Pd/Ba2SO4 (149 mg, 0.07 mmol) se añadió y la mezcla se hidrogenó mediante agitación de la mezcla en un aparato de Parr (H2: 30 psi) por 30 min. Se añadió más 5 % Pd/Ba2SO4 (149 mg, 0.07 mmol) y la agitación continuó por 30'. El catalizador se filtró a través de una almohadilla de celita y el solvente se evaporó al vacío. El producto crudo se purificó por HPLC preparativa (Método 1) para producir 50 mg del compuesto del título.
MS/ESI+ 712.15 [MH]+1H RMN (400 MHz, acetona) 8 ppm 8.22 (m, 1 H), 8.07 (d, J=2.21 Hz, 1 H), 7.98 (d, J=6.62 Hz, 2 H), 7.40 (d, J=6.62 Hz, 1 H), 7.28 (m, 1 H), 7.04 - 7.22 (m, 2 H), 6.65 - 7.04 (m, 2 H), 6.20 - 6.28 (m, 1 H), 3.95 (d, J=5.73 Hz, 2 H), 3.73 (t, J=4.41 Hz, 6 H), 3.48 - 3.60 (m, 1 H), 3.31 - 3.43 (m, 1 H), 3.04 (s, 3 H), 2.75 - 2.62 (m, 6 H), 1.19 -1.37 (m, 1 H), 0.60 (d, J=7.06 Hz, 2 H), 0.37 (d, J=4.85 Hz, 2 H).
Etapa 6: Síntesis de clorhidrato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-hidroxi-3-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (42)
(S)-3,5-Dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-hidroxi-3-(N-(2-morfolinoetil)metilsulfonamido) benzoiloxi)etil)-piridina 1-óxido (50 mg, 0.07 mmol) se disolvió en acetato de etilo (1.5 ml) y HCl/Et2O 2N (100 pl) se añadió. Et2O (5 ml) se añadió hasta que se formó un precipitado copioso. El compuesto del título (35 mg) se obtuvo por filtración (rendimiento 67 %). MS/ESI+ 746.2 [MH]+1H RMN (400 MHz, acetona) 8 ppm 10.33 - 10.77 (m, bs H), 8.31 (s, 2 H), 7.99 - 8.12 (m, 1 H), 7.79 - 7.98 (m, 1 H), 7.27 - 7.36 (m, 1 H), 7.05 - 7.27 (m, 3 H), 6.90 (t, J=75.00 Hz, 1 H), 6.20 -6.38 (m, 1 H), 5.60 - 5.90 (m, bs H), 3.85 - 4.41 (m, 8 H), 3.69 - 3.81 (m, 1 H), 3.22 - 3.63 (m, 7 H), 3.14 (s, 3 H), 1.16 -1.39 (m, 1 H), 0.53 - 0.68 (m, 2 H), 0.29 - 0.48 (m, 2 H).
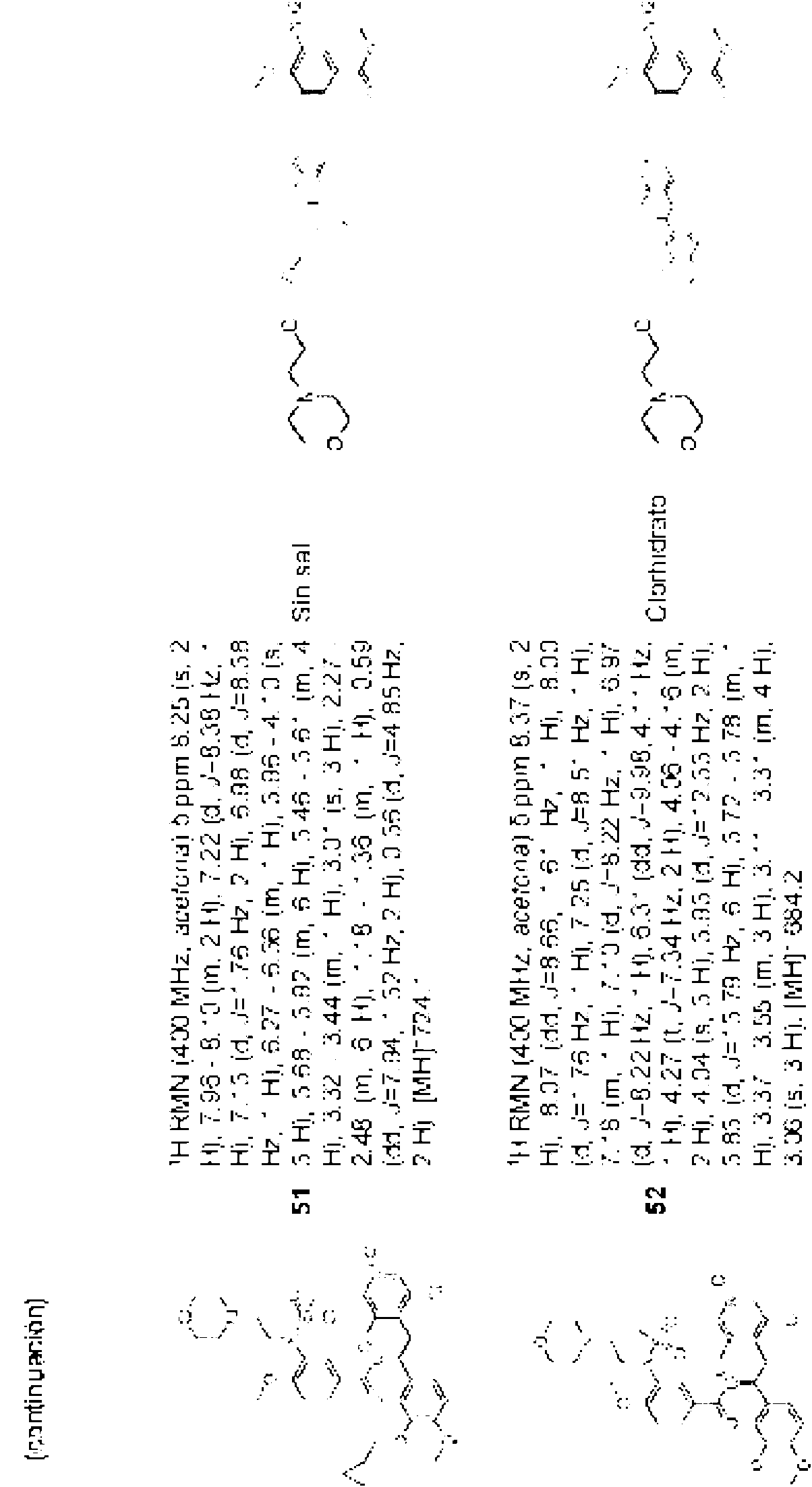
Los compuestos enumerados en la Tabla 5 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 4, Etapa 1-4, al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados.

Ejemplo 5
Síntesis de (S)-3,5-didoro-4-(2-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-fluoro-3-(N-(2-morfolinoetil)-metil sulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 53):

Etapa 1: Preparación de metil 3-amino-4-fluorobenzoato (54):
A una mezcla de ácido 3-amino-4-fluorobenzoico (1 g, 6.45 mmol) en MeOH (20 ml), se añadió H2SO4 conc. (0.687 ml, 12.89 mmol) en forma de gotas y la reacción se sometió a reflujo por 20 horas. Después de enfriar hasta la temperatura ambiente el solvente se evaporó y el residuo se particionó entre sol. sat. de NaHCO3 ac. y acetato de etilo; la fase orgánica se lavó con salmuera, se secó sobre Na2SO4 y se evaporó hasta secarse proporcionando metil 3-amino-4-fluorobenzoato como un sólido marrón (0.970 g, 5.73 mmol, 89 % de rendimiento, MS/ESI+ 170.0 [MH]+) (43) Etapa 2: Preparación de metil 4-fluoro-3-(metilsulfonamido)benzoato (55):
Una solución de metil 3-amino-4-fluorobenzoato (0.970 g, 5.73 mmol) en piridina seca (12 ml), se enfrió hasta 0°C y se añadió cloruro de metanosulfonilo (0.577 ml, 7.45 mmol) en forma de gotas. La mezcla resultante se dejó calentar hasta la temperatura ambiente y se agitó por 3 horas. El solvente se evaporó y el residuo se particionó entre DCM y sol. sat. de NaHCO3 ac. La capa orgánica se lavó con salmuera y se secó sobre Na2SO4; el solvente se evaporó y el residuo se purificó por cromatografía rápida en columna de gel de sílice (acetato de etilo : éter de petróleo : 35 : 65) proporcionando metil 4-fluoro-3-(metilsulfonamido) benzoato como un polvo blancuzco (0.400 g, 1.618 mmol, 28.2 % de rendimiento, MS/ESI+248.1 [MH]+).
Etapa 3: Preparación de metil 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido) benzoato (56):
A una solución de metil 4-fluoro-3-(metilsulfonamido)benzoato (0.400 g, 1.618 mmol) en DMF seco (16.200 ml) bajo una atmósfera de N2, K2CO3 (0.492 g, 3.56 mmol) se añadió seguido por 4-(2-cloroetil)morfolina clorhidrato (0.361 g, 1.941 mmol). La suspensión blanca resultante se calentó hasta 70°C por 1 hora, se agitó a temperatura ambiente toda la noche y se calentó hasta 70°C por 3.5 horas adicionales. Se añadió agua (20 ml) y la mezcla se extrajo varias veces con éter de etilo (25 ml x 5). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se evaporaron hasta secarse. El residuo se purificó por cromatografía rápida en columna de gel de sílice (acetato de etilo : éter de petróleo = 85 : 15) para dar metil 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoato como un aceite incoloro (0.512 g, 1.421 mmol, 88 % de rendimiento, MS/ESI+ 361.0 [MH]+).
Etapa 4: Preparación de clorhidrato de ácido 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoico (57):
Metil 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoato (0.504 g, 1.398 mmol) se disolvió en dioxano (14.000 ml) y HCl 6N ac. (1.398 ml, 8.39 mmol) se añadió. La mezcla se calentó hasta 70°C por 24 horas. HCl 6N ac. adicional (2.80 ml, 16.78 mmol) se añadió en tres porciones calentando hasta 100°C por 4 horas. Los volátiles se eliminaron al vacío proporcionando clorhidrato de ácido 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoico como un sólido amarillo pálido (0.518 g, 1.353 mmol, 97 % de rendimiento, MS/ESI+ 347.0 [MH]+). Este producto se usó sin ninguna purificación adicional.
Etapa 5: Preparación de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-fluoro-3-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (53):
Ácido 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoico clorhidrato (0.180 g, 0.470 mmol) se suspendió en DCM seco (13.100 ml); (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (0.165 g, 0.392 mmol), EDC (0.225 g, 1.175 mmol) y DMAP (0.096 g, 0.784 mmol) se añadieron secuencialmente y la mezcla se agitó a temperatura ambiente por 5 horas. Una segunda porción de clorhidrato de ácido 4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoico (0.030 g, 0.078 mmol) se añadió y la reacción se agitó a temperatura ambiente toda la noche. La mezcla se diluyó con DCM (20 ml) y se lavó con HCl 1N ac. (15 ml x 3). La fase orgánica se secó sobre Na2SO4 y se evaporó hasta secarse. El producto crudo se purificó por cromatografía rápida en columna de gel de sílice (DCM : MeOH = 97 : 3) proporcionando (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)-fenil)-2-(4-fluoro-3-(N-(2-morfolinoetil)metilsulfonamido)benzoiloxi)-etil)piridina 1-óxido como un sólido esponjoso blanco (0.202 g, 0.270 mmol, 69 % de rendimiento, MS/ESI+748.52 [MH]+, [ao]= -30.52, c= 0.5, MeOH).
1H RMN (300 MHz, DMSO-d6) d ppm 8.53 (s, 2 H), 7.97 - 8.14 (m, 2 H), 7.49 (t, 1 H), 7.23 (d, 1 H), 7.20 (d, 1 H), 7.09 (dd, 1 H), 7.06 (t, 1 H), 6.19 (dd, 1 H), 3.95 (dd, 1 H), 3.90 (dd, 1 H), 3.66 - 3.81 (m, 2 H), 3.64 (dd, 1 H), 3.31 - 3.44 (m, 5 H), 3.13 (s, 3 H), 2.32-2.45 (m, 2 H), 2.11 -2.31 (m, 4 H), 1.05 - 1.39 (m, 1 H), 0.46-0.67 (m, 2 H), 0.20-0.44 (m, 2 H) Los compuestos enumerados en la Tabla 6 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 5 al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados.
Las variaciones específicas en los métodos experimentales o de purificación se indican en la tabla.


Ejemplo 6
Síntesis de (S)-3,5-didoro-4-(2-(3-(cidopropilmetoxi)-4-(difluorometoxi) fenil)-2-(5-(N-(ciclopropilmetil)metilsulfonamido)-2-(2-morfolinoetoxi) benzoiloxi)etil)piridina 1-óxido (62)
Esquema 6

Etapa 1: Síntesis de metil 5-amino-2-hidroxibenzoato (63)
Ácido 5-amino-2-hidroxibenzoico (10 g, 65.3 mmol) se suspendió en MeOH (150 ml) y 96 % ácido sulfúrico (12 ml, 225 mmol) se añadió en forma de gotas. La mezcla se calentó a reflujo por 96h, después se enfrió hasta la temperatura ambiente: parte del solvente se evaporó (quedaron 90 ml) y se añadió solución de NaHCO3 al 5 % (pH básico). La mezcla se extrajo con AcOEt. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se evaporaron. El producto deseado se obtuvo como un sólido (9.68 g, 89 % de rendimiento) y se usó en la siguiente reacción sin purificación adicional.
MS/ESI+ 168 [MH]+
Etapa 2: Síntesis de metil 2-hidroxi-5-(metilsulfonamido)benzoato (64)
Metil 5-amino-2-hidroxibenzoato (9.58 g, 57.3 mmol) se disolvió en DCM (150 ml). Se añadieron piridina (9.27 ml, 115 mmol) y cloruro de metanosulfonilo (4.47 ml, 57.3 mmol). La mezcla de reacción se agitó a temperatura ambiente por 4h y después agua (50 ml) y 6M HCl (15 ml) se añadieron. Un sólido rosa comenzó a cristalizarse. El sólido se recuperó por filtración y se lavó con agua. El compuesto del título se obtuvo como un sólido rosa (13 g, 92 % de rendimiento) y se usó en la siguiente reacción sin purificación adicional.
MS/ESI+ 246 [MH]+
Etapa 3: Síntesis de metil 5-(N-(ciclopropilmetil)metilsulfonamido)-2-hidroxibenzoato (65)
Metil 2-hidroxi-5-(metilsulfonamido)benzoato (5.565 g, 22.69 mmol) se disolvió en acetonitrilo (250 ml). Bromometil ciclopropano (4.59 g, 34 mmol) y carbonato de potasio (6.27 g, 45.4 mmol) se añadieron y la mezcla se agitó a temperatura ambiente por 4 días. La mezcla de reacción se vertió en agua helada (200 ml) y 36 % HCl se añadió hasta pH=1. La mezcla se extrajo después con AcOEt, la capa orgánica se secó sobre Na2SO4y el solvente se evaporó. El producto resultante crudo se purificó por cromatografía rápida en gel de sílice (éter de petróleo : AcOEt 4:1) proporcionando 4.75 g del producto deseado (70 % de rendimiento)
MS/ESI+ 300 [MH]+
Etapa 4: Síntesis de metil 5-(N-(ciclopropilmetil)metilsulfonamido)-2-(2-morfolinoetoxi)benzoato (66)
Una mezcla de metil 5-(N-(cidopropilmetil)metilsulfonamido)-2-hidroxibenzoato (200 mg, 0.668 mmol), 4-(2-doroetil)morfolina clorhidrato (149 mg, 0.802 mmol) y K2CO3 (231 mg, 1.670 mmol) en 10 ml de DMF se agitó por 4h a 80°C. La mezcla se vertió en agua y se extrajo dos veces con AcOEt, la cual se lavó después con salmuera, se secó y se evaporó para obtener el producto deseado (259 mg, 94 % de rendimiento) el cual se usó en la próxima etapa sin purificación adicional.
MS/ESI+ 413.0 [MH]+
Etapa 5: Síntesis de clorhidrato del ácido 5-(N-(ciclopropilmetil)metilsulfonamido)-2-(2-morfolinoetoxi)benzoico (67)
Una mezcla de metil 5-(N-(ciclopropilmetil)metilsulfonamido)-2-(2-morfolinoetoxi)benzoato (260 mg, 0.630 mmol) y HCl conc. (1.5 ml, 18.00 mmol) en 3 ml de dioxano se calentó a 100°C por 1h bajo irradiación con microondas. La mezcla se evaporó para obtener el compuesto del título (303 mg, rendimiento cuantitativo) el cual se usó en la próxima etapa sin purificación adicional.
MS/ESI+ 434.9 [MH]+
Etapa 6 : Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(5-(N-(ciclopropilmetil) metilsulfonamido)-2-(2-morfolinoetoxi)benzoiloxi)etil)piridina 1-óxido (62)
Una mezcla de clorhidrato de ácido 5-(N-(ciclopropilmetil)metilsulfonamido)-2-(2-morfolinoetoxi)benzoico (303 mg, 0.627 mmol), el compuesto 3 (220 mg, 0.522 mmol), DMAP (128 mg, 1.045 mmol) y EDC (250 mg, 1.306 mmol) en 25 ml de DCM seco se agitó toda la noche a temperatura ambiente. La mezcla se lavó con HCl diluido y salmuera, después se secó y se evaporó. El producto crudo se purificó por HPLC prep.; las fracciones que contenían el producto se basificaron con NaHCO3 y se extrajeron con AcOEt, el cual se lavó después con salmuera, se secó y se evaporó para obtener (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(5-(N-(ciclopropilmetil)metilsulfonamido)-2-(2-morfolinoetoxi)benzoiloxi)etil)piridina 1-óxido (31 mg, 7.41 % de rendimiento).
MS/ESI+ 800.27 [MH]+,
[a£° = - 25.21 (c=0.365, MeOH).
1H RMN (300 MHz, DMSO-d6) 5 ppm 8.53 (s, 2 H), 7.64 (d, 1 H), 7.58 (dd, 1 H), 7.21 (d, 1 H), 7.20 (d, 1 H), 7.16 (d, 1 H), 7.06 (dd, 1 H), 7.06 (t, 1 H), 6.17 (dd, 1 H), 4.15 (t, 2 H), 3.93 (dd, 1 H), 3.87 (dd, 1 H), 3.56 (dd, 1 H), 3.49 - 3.53 (m, 4 H), 3.47 (d, 2 H), 3.32 (dd, 1 H), 2.97 (s, 3 H), 2.56 - 2.69 (m, 2 H), 2.33 - 2.45 (m, 4 H), 1.11 - 1.23 (m, 1 H), 0.72 -0.92 (m, 1 H), 0.48 - 0.63 (m, 2 H), 0.25 - 0.47 (m, 4 H), -0.07 - 0.19 (m, 2 H)
MS/ESI+ 800.27 [MH]+
Ejemplo 7
Síntesis de trifluoroacetato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)etil) metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (68)

Etapa 1: Preparación de metil 3-amino-4-metoxibenzoato (69):
Una mezcla de ácido 3-amino-4-metoxibenzoico (2.00 g, 11.96 mmol) y ácido sulfúrico (1.275 ml, 23.93 mmol) en MeOH (25 ml) se calentó hasta reflujo por 24 horas. Después de enfriar hasta la temperatura ambiente el solvente se eliminó al vacío y el residuo se particionó entre sol. sat. de NaHCO3 ac. y acetato de etilo. La capa orgánica se secó sobre Na2SO4 y se evaporó hasta secarse proporcionando metil 3-amino-4-metoxibenzoato como un polvo blancuzco (2.00 g, 11.04 mmol, 92 % de rendimiento, MS/ESI+182.1 [MH]+).
Etapa 2: Preparación de metil 4-metoxi-3-(metilsulfonamido)benzoato (70):
Una solución de metil 3-amino-4-metoxibenzoato (2.00 g, 11.04 mmol) en piridina seca (25 ml) se enfrió hasta 0°C y se añadió cloruro de metanosulfonilo (1.115 ml, 14.35 mmol) en forma de gotas. La reacción se dejó calentar hasta la temperatura ambiente y se agitó por 3 horas. El solvente se evaporó hasta secarse y el residuo se particionó entre DCM y HCl 2N ac.; la capa orgánica se lavó con salmuera, se secó sobre Na2SO4 y el solvente se eliminó al vacío. El residuo se trituró con iPr2O y el sólido resultante se recogió por filtración para dar metil 4-metoxi-3-(metilsulfonamido)benzoato como un polvo blancuzco (2.6 g, 10.03 mmol, 91 % de rendimiento, MS/ESI+ 260.1 [MH]+).
Etapa 3: Preparación de metil 3-(N-(2-hidroxietil)metilsulfonamido)-4-metoxibenzoato (71):
Una mezcla de metil 4-metoxi-3-(metilsulfonamido)benzoato (0.300 g, 1.157 mmol), 2-bromoetanol (0.434 g, 3.47 mmol) y K2CO3(0.320 g, 2.314 mmol) en acetonitrilo (5 ml) se calentó bajo irradiación con microondas a 110°C por 5 horas. El solvente se evaporó y el residuo se trató con acetato de etilo. Las sales inorgánicas se filtraron y el filtrado se evaporó hasta secarse. El producto crudo se purificó por cromatografía rápida en columna de gel de sílice (éter de petróleo : acetato de etilo = 1 : 9 ) proporcionando metil 3-(N-(2-hidroxietil) metilsulfonamido)-4-metoxibenzoato como un aceite marrón claro (0.200 g, 0.659 mmol, 57.0 % de rendimiento, MS/eSi+ 326.3 [MNa]+).
Etapa 4: Preparación de metil 3-(N-(2-bromoetil)metilsulfonamido)-4-metoxibenzoato (72):
A una solución de metil 3-(N-(2-hidroxietil)metilsulfonamido)-4-metoxibenzoato (0.200 g, 0.659 mmol) y trifenilfosfina (0.173 g, 0.659 mmol) en DCM seco (3 ml), CBr4 (219 mg, 0.659 mmol) se añadió en forma de porciones a temperatura ambiente bajo una atmósfera de nitrógeno y la mezcla se agitó por 30 minutos. El solvente se eliminó al vacío y el residuo se purificó por cromatografía rápida en columna de gel de sílice (acetato de etilo : éter de petróleo = 9 : 1 ) proporcionando metil 3-(N-(2-bromoetil)metilsulfonamido)-4-metoxibenzoato como un sólido blanco (0.150 g, 0.410 mmol, 62.1 % de rendimiento, MS/ESI+ 365.9-367.9 [MH]+).
Etapa 5: Preparación de metil 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)etil) metilsulfonamido)benzoato (73):
Una mezcla de metil 3-(N-(2-bromoetil)metilsulfonamido)-4-metoxibenzoato (0.150 g, 0.410 mmol), 1-metilpiperazina (0.041 g, 0.410 mmol) y K2CO3 (0.085 g, 0.614 mmol) en acetonitrilo (3 ml) se calentó bajo irradiación con microondas a 130°C por 3 horas. El residuo insoluble se filtró y el filtrado se evaporó hasta secarse. El producto crudo se purificó por cromatografía rápida en columna de gel de sílice (DCM : MeOH : ac. 32% NH4OH = 95 : 5 : 0.5) proporcionando metil 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)-benzoato como un aceite marrón claro (0.140 g, 0.363 mmol, 89 % de rendimiento, MS/ESI+ 386.4 [MH]+).
Etapa 6: Preparación de trifluoroacetato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)etil) metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (68):
A una solución de metil 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido) benzoato (0.140 g, 0.363 mmol) en THF (5 ml), una solución de LiOH*H20 (0.183 g, 0.436 mmol) en agua (0.5 ml) se añadió y la mezcla se agitó a temperatura ambiente por 24 horas. El solvente se evaporó al vacío. El producto crudo sólido seco se suspendió en Dc M/DMF = 1/1 con EDC (0.209 g, 1.089 mmol), (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (0.137 g, 0.327 mmol) y DMAP (0.44 g, 0.363 mmol) y la solución resultante se agitó a temperatura ambiente toda la noche. El solvente se evaporó y el residuo se particionó entre acetato de etilo y sol. sat. de NaHCO3 ac. La fase orgánica se lavó con salmuera y se secó sobre Na2SO4; el solvente se eliminó al vacío y el residuo se purificó por cromatografía rápida en columna de gel de sílice (DCM : MeOH : 32% NH4OH ac. = 95 : 5 : 0.5) proporcionando 0.150 g del producto deseado. Se necesitó una purificación adicional por HPLC preparativa (Método 3) para obtener (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)benzoiloxi)etil)-piridina 1-óxido como sal de trifluoroacetato (polvo blanco) (0.068 g, 0.077 mmol, 21.10 % de rendimiento, pureza LC-MS (BPI): 99.2%, MS/ESI+ 773.36 [MH]+, [ao]= -30.48, c= 0.5, DCM).
Ejemplo 8
Síntesis de (3,5-dicloro-4-((S)-2-(3-(ciclopropilmetoxi)-4-(N-(3-((2S,5R)-2,5-dimetilpiperazin-1-il)propil) metilsulfonamido)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)piridina 1-óxido (74)

Etapa 1: Síntesis de (S)-4-(2-(4-(N-(3-bromopropil)metilsulfonamido)-3-(ciclopropilmetoxi)benzoiloxi)-2-(3-(cidopropilmetoxi)-4-(difluoro-metoxi)fenil)etil)-3,5-didoropiridina 1-óxido (75)
A una solución de (S)-3,5-didoro-4-(2-(3-(ddopropNmetoxi)-4-(difluoroiTietoxi)fenN)-2-(3-(ddopropNiTietoxi)-4-(metilsulfonamido) benzoiloxi)etil)piridina 1-óxido, (comp. 7, 1 g, 1.5 mmol) se disolvió en DMF (10 ml), se añadieron 1,3-dibromopropano (1.52 ml, 15 mmol) y K2CO3(414 mg, 3 mmol) y la mezcla se agitó a 40 °C por 6 h. La reacción se diluyó con agua y después se extrajo con acetato de etilo. La fase orgánica se lavó con agua (3x 30 ml), se secó sobre Na2SO4 y se evaporó hasta secarse. El sólido se trituró con éter de petróleo y se filtró, para producir 700 mg del producto deseado (rendimiento: 58%).
MS/EST 808.51 [MH]+
1H RMN (400 MHz, acetona) 8 ppm 8.28 (s, 2 H), 7.68 (dd, J=7.94, 1.76 Hz, 1 H), 7.63 (d, J=1.76 Hz, 1 H), 7.44 (d, J=8.38 Hz, 1 H), 7.32 (d, J=1.76 Hz, 1 H), 7.15 - 7.25 (m, 2 H), 6.92 (t, J=75.00 Hz, 1 H), 6.31 (dd, J=9.92, 4.63 Hz, 1 H), 4.07 (d, J=7.06 Hz, 2 H), 3.99 (d, J=7.06 Hz, 2 H), 3.86 (t, J=6.62 Hz, 2 H), 3.77 (dd, J=14.33, 9.92 Hz, 1 H), 3.56 (t, J=6.62 Hz, 2 H), 3.44 (dd, J=14.11, 4.41 Hz, 1 H), 3.09 (s, 3 H), 1.96 -2.03 (m, 2 H), 1.34 - 1.46 (m, 1 H), 1.21 - 1.33 (m, 1 H), 0.64 - 0.75 (m, 2 H), 0.54 - 0.63 (m, 2 H), 0.42 - 0.51 (m, 2 H), 0.34 - 0.41 (m, 2 H).
Etapa 2: Síntesis de 4-((S)-2-(4-(N-(3-((2S,5R)-4-(terc-butoxicarbonil)-2,5-dimetilpiperazin-1-il)propil) metilsulfonamido)-3- (ciclopropilmetoxi)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridinal-óxido (76) A una solución de (S)-4-(2-(4-(N-(3-bromopropil)metilsulfonamido)-3-(ciclopropilmetoxi)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina1-óxido (200 mg, 0.21 mmol) en DMF (4ml) se añadieron (2R,5S)-terc-butil 2,5-dimetilpiperazina-1-carboxilato (135 mg, 0.63 mmol) y K2CO3 (58 mg, 0.42 mmol) y la mezcla se agitó a temperatura ambiente toda la noche. La reacción se diluyó con agua y se extrajo con acetato de etilo. La fase orgánica se lavó con agua, se secó sobre Na2SO4 y se evaporó hasta secarse, para producir 250 mg del producto crudo que se usó en la próxima etapa sin ninguna purificación adicional.
Etapa 3: Síntesis de 3,5-dicloro-4-((S)-2-(3-(ciclopropilmetoxi)-4-(N-(3-((2S,5R)-2,5-dimetilpiperazin-1-il)propil) metilsulfonamido)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)piridina 1-óxido (74)
4- ((S)-2-(4-(N-(3-((2S,5R)-4-(terc-Butoxicarbonil)-2,5-dimetilpiperazin-1-il)propil)metilsulfonamido)-3-(ciclopropilmetoxi)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (250 mg, 0.26 mmol) se disolvió en HCl/dioxano 4M y la reacción se agitó a temperatura ambiente por 6 h. El solvente se evaporó al vacío y el residuo se particionó entre solución saturada de NaHCO3 y acetato de etilo. La fase orgánica se secó sobre Na2SO4y se evaporó al vacío. El producto crudo obtenido se purificó por HPLC semi-preparativa para producir 150 mg del compuesto final (rendimiento: 68 %)
MS/ESI+ 841.8 [MH]+1H RMN (400 MHz, acetona) 5 ppm 8.23 (s, 2 H), 7.67 (d, J=7.94 Hz, 1 H), 7.61 (d, J=1.32 Hz, 1 H), 7.40 (d, J=7.94 Hz, 1 H), 7.32 (d, J=1.32 Hz, 1 H), 7.13 - 7.24 (m, 2 H), 6.91 (t, J=75.00 Hz, 1 H), 6.31 (dd, J=9.70, 4.41 Hz, 1 H), 4.05 (d, J=7.06 Hz, 2 H), 3.98 (d, J=6.62 Hz, 2 H), 3.65 - 3.87 (m, 4 H), 3.43 (dd, J=14.11, 4.41 Hz, 2 H), 2.93 - 3.07 (d, 3 H), 2.79 (d, J=12.79 Hz, 2 H), 2.39 (m, 1 H), 2.10 (s, 3 H), 1.72 - 1.84 (m, 1 H), 1.57 (m, 2 H), 1.32 -1.47 (m, 1 H), 1.21 - 1.31 (m, 1 H), 1.11 (dd, J=6.84, 4.19 Hz, 1 H), 1.01 (d, J=6.62 Hz, 1 H), 0.91 (d, J=5.73 Hz, 3 H), 0.64 - 0.73 (m, 2 H), 0.55 - 0.63 (m, 2 H), 0.45 (d, J=4.41 Hz, 2 H), 0.37 (d, J=5.73 Hz, 2 H).
Los compuestos enumerados en la Tabla 7 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 8, Esquema 8, Etapa 1-2 al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados. Las variaciones específicas en los métodos experimentales o de purificación se indican en la tabla.
Tabla 7

Ejemplo 9
Síntesis de 2,2,2-trifluoroacetato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(2-hidroxi-4-(N-(2-(morfolino-4-io)etil)metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 81):
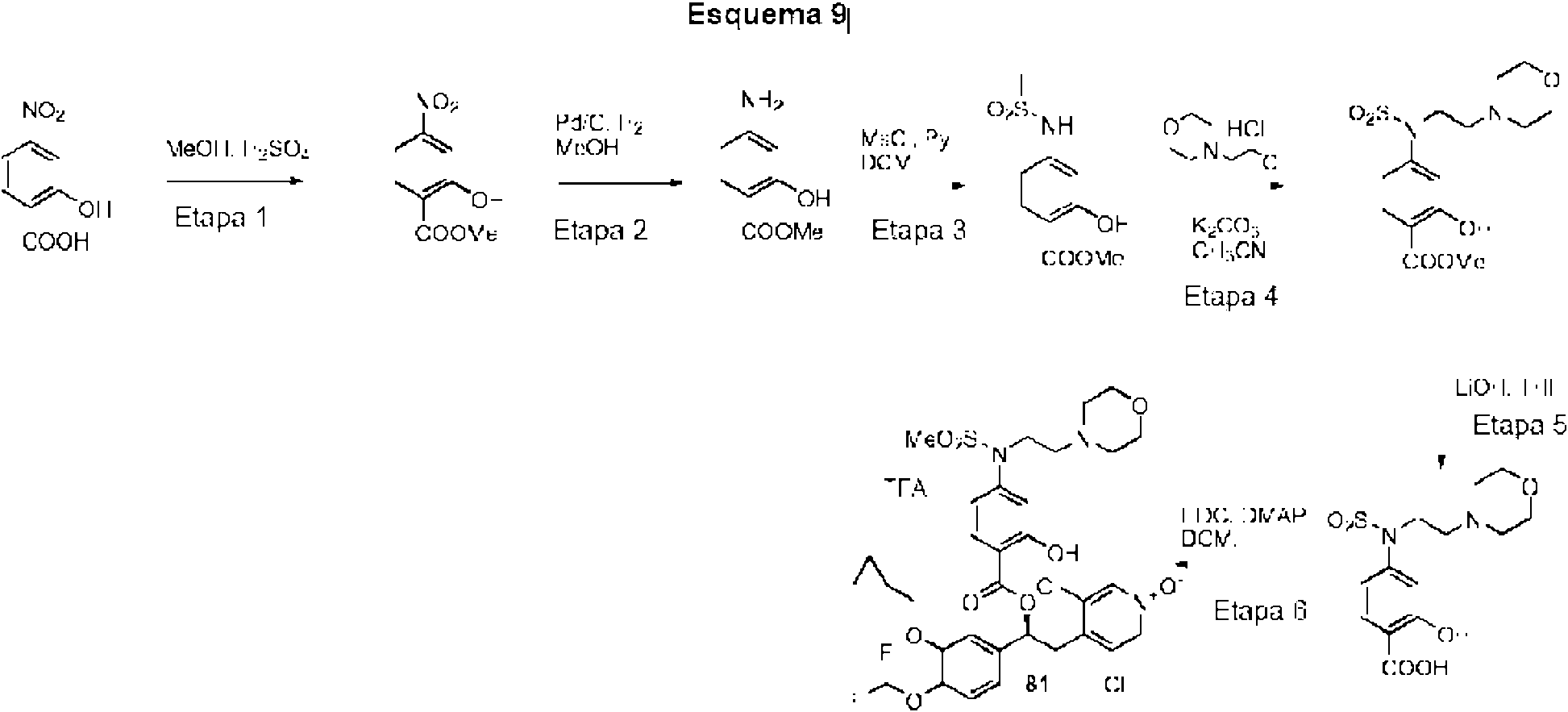
Etapa 1: Preparación de metil 2-hidroxi-4-nitrobenzoato (82):
A una solución de ácido 2-hidroxi-4-nitrobenzoico (3 g, 16.38 mmol) en MeOH (150 ml) enfriado a0°C (baño hielo/agua) se añadió ácido sulfúrico concentrado (15 ml) en forma de gotas. La reacción se calentó hasta la temperatura ambiente después se calentó a 80 °C por 16h. El MeOH se eliminó parcialmente al vacío y se añadió agua a la mezcla. El precipitado amarillo pálido que se formó se filtró y se lavó con mucha agua. Después de secar se obtuvo metil 2-hidroxi-4-nitrobenzoato como un sólido amarillo pálido (3.03 g, 15.37 mmol, 94 % de rendimiento, pureza UPLC-MS: 98 %, MS/ESI+ no detectable [MH]+).
Etapa 2: Preparación de metil 4-amino-2-hidroxibenzoato (83):
En una botella de Parrde 500 ml se añadió metil 2-hidroxi-4-nitrobenzoato (1.3 g, 6.59 mmol) a una suspensión de 10 % Pd/C (100 mg) en MeOH (40 ml). La reacción se agitó bajo una atmósfera de H2 (30 psi) por 1 h. El catalizador se filtró a través de una almohadilla de celita y los licores madre se evaporaron al vacío. Se obtuvo metil 4-amino-2-hidroxibenzoato como un sólido amarillo (1.08 g, 6.46 mmol, 98 % de rendimiento, pureza UPLC-MS: 100 %, MS/ESI+ 167.9 [MH]+).
Etapa 3: Preparación de metil 2-hidroxi-4-(metilsulfonamido)benzoato (84):
A una solución de metil 4-amino-2-hidroxibenzoato (1.08 g, 6.46 mmol) en DCM (100 ml) se añadió piridina (1.043 ml, 12.92 mmol) y cloruro de metanosulfonilo (0.600 ml, 7.75 mmol) a temperatura ambiente. La reacción se agitó a la misma temperatura por 48 h. HCl 2N se añadió a la mezcla, la fase orgánica se separó, se secó (sulfato de sodio), se filtró y se evaporó al vacío. Se obtuvo metil 2-hidroxi-4-(metilsulfonamido)benzoato como un sólido rosa (0.87 g, 3.55 mmol, 54.9 % de rendimiento, pureza UPLC-MS: 95 %, MS/ESI+ 245.8, 167.9 [MH]+) y se usó como tal en la siguiente reacción sin purificación adicional.
Etapa 4: Preparación de metil 2-hidroxi-4-(N-(2-morfolinoetil)metilsulfonamido)benzoato (85):
A una solución de metil 2-hidroxi-4-(metilsulfonamido)benzoato (0.435 g, 1.774 mmol) en CH3CN (50 ml), K2CO3 (0.368 g, 2.66 mmol) se añadió a temperatura ambiente. La reacción se agitó a la misma temperatura por 10 min, después se añadió clorhidrato de 4-(2-cloroetil)morfolina (0.330 g, 1.774 mmol) en una porción. La suspensión se agitó a 80 °C toda la noche. CH3CN se eliminó al vacío y el producto crudo restante se particionó entre una solución saturada acuosa de NH4Cl y DCM. La fase orgánica se separó y la fase acuosa se neutralizó y se extrajo con DCM. Los productos orgánicos combinados se secaron (sulfato de sodio), se filtraron y se evaporaron al vacío. La purificación por cromatografía rápida
en gel de sílice (DCM:MeOH 95:5) proporcionó metil 2-hidroxi-4-(N-(2-morfolinoetil)metilsulfonamido)benzoato como un aceite amarillo pálido (0.21 g, 0.586 mmol, 33.0 % de rendimiento, pureza UPLC-m S: 95 %, MS/ESI+ 358.9 [MH]+). Etapa 5: Preparación de 2,2,2-trifluoroacetato de 4-(2-(N-(4-carboxi-3-hidroxifenil)metilsulfonamido)etil)morfolin-4-io (86): 1M de hidróxido de litio (1.172 ml, 1.172 mmol) se añadió a una solución de metil 2-hidroxi-4-(N-(2-morfolinoetil)metilsulfonamido)benzoato (0.21 g, 0.586 mmol) en una mezcla de THF:MeOH (4:1) (20 ml). La reacción se agitó a 80 °C por 16 h. Los solventes se evaporaron al vacío. La purificación por HPLC preparativa de fase inversa proporcionó 2,2,2-trifluoroacetato de 4-(2-(N-(4-carboxi-3-hidroxifenil)metilsulfonamido)etil)morfolin-4-io como un sólido blanco (0.1 g, 0.218 mmol, 37.2 % de rendimiento, pureza UPLC-MS: 100 %, MS/ESI+ 345.0 [MH]+).
Etapa 6: Preparación de 2,2,2-trifluoroacetato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(2-hidroxi-4-(N-(2-(morfolino-4-io)etil)metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (81):
A una solución de 4-(2-(N-(4-carboxi-3-hidroxifenil)metilsulfonamido)-etil)morfolin-4-io 2,2,2-trifluoroacetato (0.1 g, 0.218 mmol) en DCM (10 ml), se añadió el Compuesto 3(0.092 g, 0.218 mmol), EDC (0.042 g, 0.218 mmol) y DMAP (0.053 g, 0.436 mmol) a temperatura ambiente. La reacción se agitó a la misma temperatura toda la noche. HCl 2N se añadió a la reacción y la fase orgánica se separó, se secó (sulfato de sodio), se filtró y se evaporó al vacío. El producto crudo obtenido se purificó por HPLC preparativa de fase inversa. 2,2,2-Trifluoroacetato de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(2-hidroxi-4-(N-(2-(morfolino-4-io)etil)metilsulfonamido)benzoiloxi)etil)piridina 1-óxido se obtuvo como un sólido blancuzco (0.032 g, 0.037 mmol, 17.04 % de rendimiento, pureza LC-MS: 100 %, MS/ESI+ 746.22 [MH]+, M = -37.84, c=0.5, MeOH).
Los compuestos enumerados en la Tabla 8 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 9, Esquema 9 al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados.

Ċ

Ejemplo 10
Preparación de (S)-3,5-didoro-4-(2-(2-(cidopropilmetoxi)-4-(N-(2-(morfolino-4-io)etil)metilsulfonamido) benzoiloxi)-2-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)etil)piridina 1-óxido (Compuesto 91):
Esquema 10

Etapa 1: Preparación de metil 2-(ciclopropilmetoxi)-4-nitrobenzoato (92):
A una solución de metil 2-hidroxi-4-nitrobenzoato (0.5 g, 2.54 mmol) en CH3CN (30 ml), K2CO3 (0.701 g, 5.07 mmol) se añadió a temperatura ambiente. La reacción se agitó a la misma temperatura por 15 minutos, después se añadió (bromometil)ciclopropano (0.372 ml, 3.80 mmol). La reacción se calentó hasta 80 °C y se agitó por 48h. El solvente se eliminó al vacío y el producto crudo se particionó entre una solución saturada de NH4Cl y DCM. La fase orgánica se separó, se secó (sulfato de sodio), se filtró y se evaporó al vacío. Metil 2-(ciclopropilmetoxi)-4-nitrobenzoato se obtuvo como un sólido amarillo pálido (0.49 g, 1.950 mmol, 77 % de rendimiento, MS/ESI+ no detectable [MH]+) y se usó en la próxima etapa sin purificación adicional.
Etapa 2: Preparación de metil 4-amino-2-(ciclopropilmetoxi)benzoato (93):
En una botella de Parr de 250 ml, se añadió metil 2-(ciclopropilmetoxi)-4-nitrobenzoato (0.49 g, 1.950 mmol) a una suspensión de 10 % Pd/C (0.05 g, 2.070 mmol) en MeOH (40 ml). La reacción se agitó bajo una atmósfera de hidrógeno (30 psi) por 1hr. El catalizador se eliminó por filtración y la solución se evaporó al vacío. Se obtuvo metil 4-amino-2-(ciclopropilmetoxi)benzoato como un aceite incoloro (0.27 g, 1.220 mmol, 62.6 % de rendimiento, MS/ESI+221.9, 243.9 [MH]+) y se usó en la próxima etapa sin purificación adicional.
Etapa 3: Preparación de metil 2-(ciclopropilmetoxi)-4-(metilsulfonamido)benzoato (94):
A una solución de metil 4-amino-2-(ciclopropilmetoxi)benzoato (0.27 g, 1.220 mmol) en DCM (20 ml) se añadió piridina (0.197 ml, 2.441 mmol) y cloruro de metanosulfonilo (0.113 ml, 1.464 mmol) a temperatura ambiente. La reacción se agitó a la misma temperatura y bajo nitrógeno por 3 días. HCl 2N se añadió a la mezcla, la fase orgánica se separó, se secó (sulfato de sodio), se filtró y se evaporó hasta secarse. Se obtuvo metil 2-(ciclopropilmetoxi)-4-(metilsulfonamido)benzoato como un aceite amarillo pálido (0.35 g, 1.169 mmol, 96 % de rendimiento, MS/ESI+ 300.0 [MH]+) y se usó en la próxima etapa sin purificación adicional.
Etapa 4: Preparación de metil 2-(ciclopropilmetoxi)-4-(N-(2-morfolinoetil)metilsulfonamido)benzoato (95):
A una solución de metil 2-(ciclopropilmetoxi)-4-(metilsulfonamido)-benzoato (0.35 g, 1.169 mmol) en CH3CN (20 ml), K2CO3 (0.323 g, 2.338 mmol) se añadió a temperatura ambiente y la suspensión así obtenida se agitó a la misma temperatura por 10 minutos. clorhidrato de 4-(2-cloroetil)morfolina (0.326 g, 1.754 mmol) se añadió y la mezcla se calentó a 80 °C por 20 h. El solvente se eliminó a presión reducida y el compuesto crudo se particionó entre agua y DCM. La capa orgánica se separó, se secó (sulfato de sodio), se filtró y se evaporó hasta secarse. Metil 2
(cidopropilmetoxi)-4-(N-(2-morfolinoetil)-metilsulfonamido)benzoato se obtuvo como un aceite amarillo pálido (0.42 g, 1.018 mmol, 87 % de rendimiento, MS/ESI+ 412.9 [MH]+) y se usó en la próxima etapa sin purificación adicional.
Etapa 5: Preparación de 2,2,2-trifluoroacetato de 4-(2-(N-(4-carboxi-3-(ciclopropilmetoxi)fenil)-metilsulfonamido) etil) morfolin-4-io (96):
A una solución de metil 2-(ciclopropilmetoxi)-4-(N-(2-morfolinoetil)metilsulfonamido)benzoato (0.42 g, 1.018 mmol) en THF (30 ml), se añadió 1M de hidróxido de litio (2.036 ml, 2.036 mmol). La reacción se agitó a temperatura ambiente toda la noche. Los solventes se eliminaron al vacío y el producto crudo restante se purificó por HPLC preparativa de fase inversa. 2,2,2-Trifluoroacetato de 4-(2-(N-(4-Carboxi-3-(ciclopropilmetoxi)fenil)metilsulfonamido)etil)morfolin-4-io se obtuvo como un aceite pegajoso incoloro(0.19 g, 0.371 mmol, 36.4 % de rendimiento, MS/ESI+ 398.9 [MH]+).
Etapa 6 : Preparación de (S)-3,5-dicloro-4-(2-(2-(ciclopropilmetoxi)-4-(N-(2-(morfolino-4-io)etil) metilsulfonamido) benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)piridina 1-óxido (91):
A una solución de 4-(2-(N-(4-carboxi-3-(ciclopropilmetoxi)fenil)-metilsulfonamido)etil)morfolin-4-io 2,2,2-trifluoroacetato (0.19 g, 0.371 mmol) en DCM (20 ml) se añadieron (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (0.171 g, 0.408 mmol), EDC (0.071 g, 0.371 mmol) y DMAP (0.091 g, 0.741 mmol) a temperatura ambiente. La reacción se agitó a la misma temperatura por 16 h. HCl 2N se añadió, la fase orgánica se separó, se secó (sulfato de sodio), se filtró y se evaporó al vacío. El producto crudo se purificó por HPLC preparativa. (Método 1) 2,2,2-trifluoroacetato de (S)-3,5-dicloro-4-(2-(2-(ciclopropilmetoxi)-4-(N-(2-(morfolino-4-io)etil) metilsulfonamido)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)piridina 1-óxido se obtuvo como un sólido blancuzco (0.04 g, 0.044 mmol, 11.80 % de rendimiento, MS/ESI+ 800.23 [MH]+, [ao]=-8.8, c=0.5, MeOH).
Los compuestos enumerados en la Tabla 9 se prepararon con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 10, Esquema 10 al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados.
Tabla 9

Ejemplo 11 (Referencia)
Síntesis de (S)-4-(2-(3-(2-(bis(2-hidroxietil)amino)etilsulfoniloxi)-4-metoxibenzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (99)

Etapa 1: Síntesis de (S)-l-terc-butil 2-(5-formil-2-metoxifenil) pirrolidina-1,2-dicarboxilato (105)
3-Hidroxi-4-metoxibenzaldehído (200 mg, 1.315 mmol) se disolvió en DMF (5 ml) seguido por (S)-1-(tercbutoxicarbonil)pirrolidina-2-ácido carboxílico (290 mg, 1.347 mmol), y DMAP (50 mg, 0.410 mmol). La reacción se sonicó por 30 min y se agitó por aproximadamente 2 h a temperatura ambiente. Después de ese tiempo, la reacción se diluyó con K2CO3 (50 ml) y se extrajo con diisopropil éter (50 ml). El extracto orgánico se lavó con sol. de K2CO3 conc. (6 x 50 ml). La solución se secó sobre Na2SO4 y el solvente se eliminó a presión reducida para proporcionar 400 mg del producto deseado (rendimiento = 87 %).
Etapa 2: Síntesis del ácido (S)-3-(1-(terc-butoxicarbonil)pirrolidina-2-carboniloxi)-4-metoxibenzoico (104)
(S)-1 -terc-butil 2-(5-formil-2-metoxifenil) pirrolidina-1,2-dicarboxilato (380 mg, 1.088 mmol) se disolvió en ácido acético y se enfrió hasta 0°C en un baño de hielo. Ácido sulfámico (106 mg, 1.088 mmol) se añadió seguido por la adición en forma de gotas de clorito de sodio (180 mg, 1.990 mmol) pre-disuelto en agua (0.5 mL). La reacción se agitó a temperatura ambiente toda la noche, se diluyó con agua (30 mL) y se extrajo con acetato de etilo (40 mL). El solvente se eliminó a presión reducida y el residuo aceitoso se trituró con un sistema de solventes cloroformo y hexano (10:90) dando lugar a un sólido blanco. El sólido se filtró y se lavó con agua (100 mL) antes de secarse en un horno al vacío toda la noche para dar 300 mg del compuesto del título (rendimiento 75 %).
Etapa 3: Síntesis de 4-((S)-2-(3-((S)-1-(terc-butoxicarbonil)pirrolidina-2-carboniloxi)-4-metoxibenzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil) etil)-3,5-dicloropiridina 1-óxido (103)
El acoplamiento de 3 a ácido (S)-3-(1-(terc-butoxicarbonil)pirrolidina-2-carboniloxi)-4-metoxibenzoico, bajo las mismas condiciones y procedimiento experimental mostrado en el Ejemplo 4, Esquema 4, Etapa 4, dio el derivado éster correspondiente en rendimiento cuantitativo.
Etapa 4: Síntesis de clorhidrato de 3,5-dicloro-4-((S)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-((S)-pirrolidina-2-carboniloxi)benzoiloxi)etil)piridina 1-óxido (102)
4-((S)-2-(3-((S)-1-(terc-butoxicarbonil)pirrolidina-2-carboniloxi)-4-metoxibenzoiloxi)-2-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5 dicloropiridina 1-óxido (620 mg; 0.808 mmol) se disolvió en HCl/AcOEt 4M (2.0 ml) y se agitó por 2 h a temperatura ambiente. El solvente se eliminó a presión reducida para dar el producto crudo que se recristalizó a partir de AcOEt:Hexano (1:2). El sólido se filtró para obtener el compuesto del título (480 mg; rendimiento = 89 %). MS/ESI+ 667.1 [MH]+.
1H RMN (400 MHz, CLOROFORMO-d) 8 ppm 8.29 (s, 2 H), 7.90 (d, J=9.00 Hz, 1 H), 7.73 (m, 1 H), 7.12 - 7.23 (m, 1 H), 6.93 - 7.10 (m, 3 H), 6.39 - 6.85 (m, 1 H, CHF2), 6.20 (dd, J=9.78, 3.91 Hz, 1 H), 4.83 (br. s., 1 H), 3.77 - 3.96 (m, 5 H), 3.53 - 3.76 (m, 2 H), 3.34 (d, J=11.35 Hz, 1 H), 1.80 - 2.70 (m, 6 H), 1.27 (d, J=4.30 Hz, 1 H), 0.58 - 0.75 (m, 2 H), 0.37 (q, J=5.09 Hz, 2 H).
Etapa 5: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-hidroxi-4-metoxibenzoiloxi) etil)piridina 1-óxido (101)
Clorhidrato de 3,5-dicloro-4-((S)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-((S)-pirrolidina-2-carboniloxi)benzoiloxi)etil)piridina 1-óxido (150.0 mg; 0.2 mmol) se disolvió en MeOH (2.0 ml) seguido por adición de sol. de NaHCO3 sat. (2.0 ml). La solución se agitó a temperatura ambiente por 5 h. MeOH se evaporó al vacío y la fase acuosa se acidificó con HCl 1M (50 ml) y después se extrajo con AcOEt (50 ml; x3). El extracto orgánico se secó sobre Na2SO4 (1.0 g) y el solvente se eliminó a presión reducida para producir 100.0 mg del producto deseado como polvo blanco (rendimiento = 78.0 %).
MS/ESI+ 570.366 [MH]+
Etapa 6: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(vinilsulfoniloxi) benzoiloxi)-etil)piridina 1-óxido (100)
(S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-hidroxi-4-metoxibenzoiloxi)etil)piridina 1-óxido (30 mg, 0.05 mmol) se disolvió en DCM (2.5 ml), después TEA (8.4 pl, 0.06 mmol) y 2-cloroetanesulfonil cloruro (6.3 pl, 0.06 mmol) se añadieron, y la mezcla se agitó a temperatura ambiente por 30'. La reacción se diluyó con DCM y se lavó con HCl 1N (2x 10 ml). La fase orgánica se secó sobre Na2SO4 y se evaporó al vacío para producir 30 mg del producto crudo que se usó en la próxima etapa sin ninguna purificación adicional.
Etapa 7: Síntesis de (S)-4-(2-(3-(2-(bis(2-hidroxietil)amino)-etilsulfoniloxi)-4-metoxibenzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (99)
(S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(vinilsulfoniloxi) benzoiloxi)etil)piridina 1-óxido (30 mg, 0.045 mmol) se disolvió en EtOH (1.5 ml), después dietanolamina (9.5 pl, 0.09 mmol) se añadió, y la reacción se agitó a temperatura ambiente por 30'. El solvente se evaporó, y el producto crudo se purificó por HPLC semipreparativa para producir 20 mg del producto final. (rendimiento = 59 %) Ms/ESI+ 765.3 [MH]+, 787.2 [MNa]+ 1H RMN (400 MHz, acetona) 8 ppm 8.27 (s, 2 H), 7.93 - 8.07 (m, 2 H), 7.27 - 7.35 (m, 2 H), 7.18 - 7.24 (m, 1 H), 7.13 -7.18 (m, 1 H), 6.91 (t, J=75.00 Hz, 1 H), 6.31 (dd, J=9.70, 4.41 Hz, 1 H), 4.03 (s, 3 H), 3.98 (dd, J=6.62, 3.97 Hz, 2 H), 3.65 - 3.82 (m, 3 H), 3.53 - 3.62 (m, 4 H), 3.43 (dd, J=14.11, 4.41 Hz, 1 H), 3.20 - 3.29 (m, 2 H), 2.72 - 2.78 (m, 4 H), 1.27 (m, 1 H), 0.60 (d, J=6.62 Hz, 2 H), 0.34 - 0.42 (m, 2 H).
Ejemplo 27
Síntesis de S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(2-hidroxietoxi)-4-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 225)
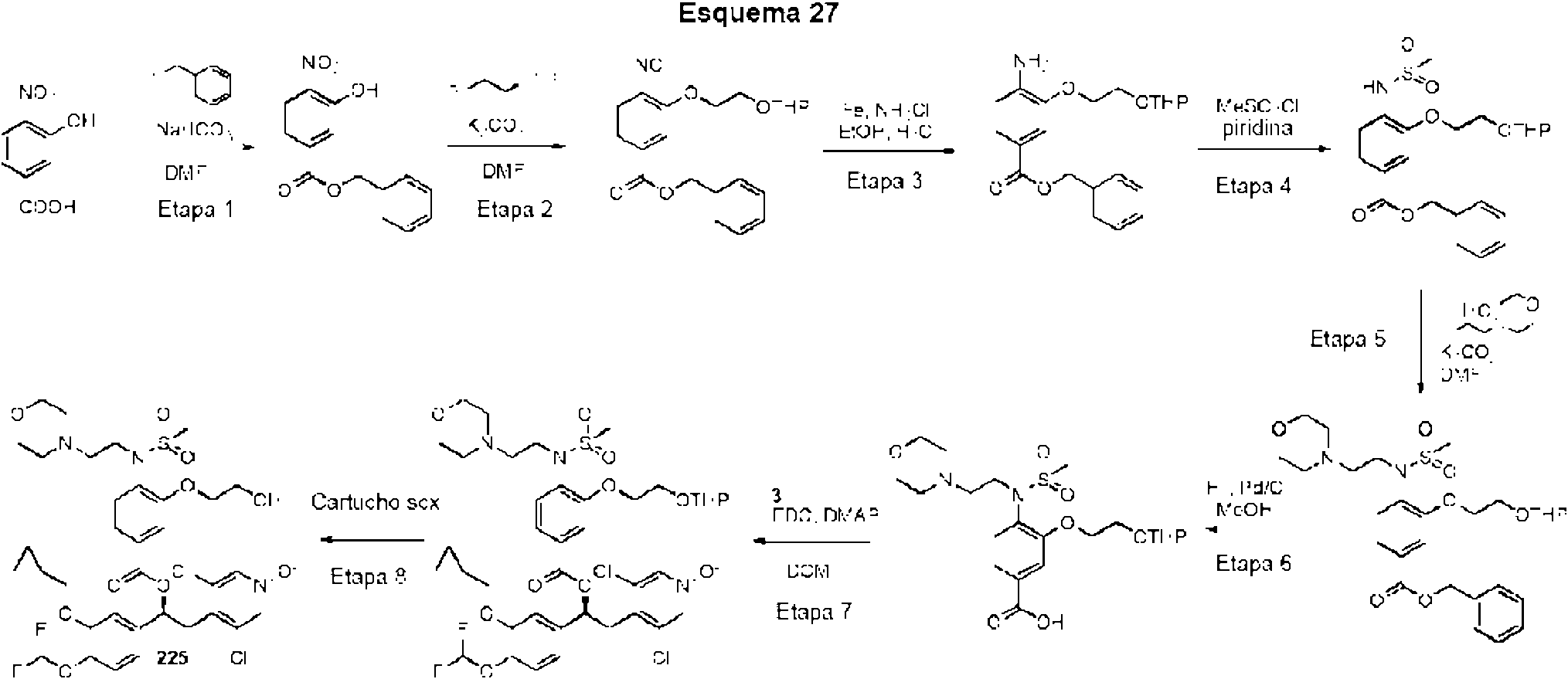
Etapa 1: Síntesis de bencil 3-hidroxi-4-nitrobenzoato (232)
A una solución de 3-hidroxi-4-nitrobenzoico ácido (1.1 g, 5.976 mmol) en DMF seco (55 ml), bicarbonato de sodio (0.606 g, 7.205 mmol) se añadió y la mezcla se agitó a temperatura ambiente por 45 minutos. Bromuro de bencilo (0.857 ml, 7.22205 mmol) se añadió y la reacción se calentó hasta 50°C por 5 h. Una segunda porción de bicarbonato de sodio (0.115 g, 1.365 mmol) y bromuro de bencilo (0.162 ml, 1.365 mmol) se añadió y la reacción se calentó hasta 50°C por 1 h adicional y se deja a temperatura ambiente toda la noche. Se añadió HCl 1N se añadió y la mezcla se extrajo dos veces con acetato de etilo. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se evaporó hasta secarse. El producto crudo se purificó por cromatografía rápida en columna de gel de sílice (éter de petróleo : acetato de etilo = 98 : 2) proporcionando bencil 3-hidroxi-4-nitrobenzoato como un sólido amarillo (1.600 g, 100 % de rendimiento).
Etapa 2: Síntesis de bencil 4-nitro-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato (231)
A una solución de bencil 3-hidroxi-4-nitrobenzoato (1.6 g, 5.86 mmol) en DMF seco (50 ml), 2-(2-bromoetoxi)tetrahidro-2H-piran (1.238 ml, 8.20 mmol) y K2CO3 (1.133 g, 8.20 mmol) se añadieron y la reacción se calentó hasta 100°C por 48 h. Una segunda porción de 2-(2-bromoetoxi)tetrahidro-2H-piran (0.442 ml, 2.93 mmol) y K2CO3(0.405 g, 2.93 mmol) se añadió y la mezcla se agitó a 100°C por 6 h adicionales. La mezcla de reacción se diluyó con HCl 1N y se extrajo varias veces con acetato de etilo. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se evaporó hasta secarse. El residuo se purificó por cromatografía rápida en columna de gel de sílice (éter de petróleo : acetato de etilo = 95 : 5 to 90 : 10) proporcionando bencil 4-nitro-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato con un aceite amarillo (0.7884 g, 33.5 % de rendimiento). MS/ESI+ 424.0 [MNa]+
Etapa 3: Síntesis de bencil 4-amino-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato (230)
A una suspensión de bencil 4-nitro-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato (0.707 g, 1.761 mmol) en etanol (15 ml) y agua (7 ml), cloruro de amonio (0.066 g, 1.233 mmol) se añadió seguido por polvo de hierro (0.590 g, 10.57 mmol). La mezcla resultante se calentó hasta 80°C por 2 h, se enfrió hasta la temperatura ambiente y se filtró. El filtrado se evaporó hasta secarse y el residuo se disolvió en acetato de etilo y se lavó con agua y salmuera. La fase orgánica se secó sobre Na2SO4 y el solvente se eliminó al vacío proporcionando bencil 4-amino-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato con un aceite amarillo (0.652 g, 100 % de rendimiento). Este producto se usó sin ninguna purificación adicional. MS/ESI+ 372.1 [MNa]+
Etapa 4: Síntesis de bencil 4-(metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato (229)
Una solución de bencil 4-amino-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato (0.652 g, 1.755 mmol) en piridina (20 ml) se enfrió a 0°C y se añadió cloruro de metanosulfonilo (0.150 ml, 1.931 mmol). La reacción se agitó a temperatura ambiente toda la noche y se calentó hasta 50°C por 2 h. Después de enfriar hasta temperatur ambiente, se añadió cloruro de metanosulfonilo adicional (0.232 ml, 2.984 mmol) durante 24 h. El solvente se eliminó al vacío y el residuo se disolvió en acetato de etilo y se lavó con HCl 1N ac. y después con salmuera. La capa orgánica se secó sobre Na2SO4 y se evaporó hasta secarse. El producto crudo se purificó por cromatografía rápida en columna de gel de sílice (éter de
petróleo : acetato de etilo = 75 : 25) proporcionando bencil 4-(metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi) benzoato con un aceite amarillo (0.720 g, 91 % de rendimiento). MS/ESI+ 472.0 [MNa]+
Etapa 5: Síntesis de bencil 4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi) etoxi)benzoato (228) A una solución de bencil 4-(metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi) benzoato (0.720 g, 1.602 mmol) en DMF (25 ml) se añadieron K2CO3 (0.531 g, 3.84 mmol) e clorhidrato de 4-(2-cloroetil)morfolina (0.358 g, 1.922 mmol). La mezcla de reacción se calentó hasta 70°C por 5 h y hasta 80°C por 1 h. La mezcla se particionó entre acetato de etilo y agua; la fase orgánica se lavó con salmuera, se secó sobre Na2SO3 y se evaporó hasta secarse. El producto crudo se purificó por cromatografía rápida en columna de gel de sílice (100 % acetato de etilo) proporcionando bencil 4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato con un aceite amarillo (0.680 g, 75 % de rendimiento). MS/ESI+563.2 [MH]+
Etapa 6: Síntesis de ácido 4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoico (227) Una mezcla de bencil 4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoato (0.680 g, 1.209 mmol) y una suspensión en agua (2 ml) de 10 % p/p Pd/C (0.643 g, 0.604 mmol) en metanol (15 ml) se hidrogenó en un aparato de Parr a 30 psi por 3.5 h. El catalizador se filtró y el filtrado se evaporó hasta secarse proporcionando ácido 4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi) etoxi)benzoico como un aceite rojo pálido (0.428 g, 74.9 % de rendimiento). Este producto se usó sin ninguna purificación adicional. MS/ESI+ 473.1 [MH]+ Etapa 7: Síntesis de 3,5-dicloro-4-((2S)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoiloxi)etil)piridina 1-óxido (226)
Una mezcla de ácido 4-(N-(2-morfolinoetil)metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoico (0.428 g, 0.906 mmol), (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (0.346 g, 0.823 mmol), EdC (0.474 g, 2.470 mmol) y DMAP (0.151 g, 1.235 mmol) en DCM (20 ml) se agitó a temperatura ambiente por 4 h. La mezcla de reacción se lavó con sol. de NH4Cl sat. ac., NaHCO3 ac. y salmuera; la fase orgánica se secó sobre Na2SO4 y el solvente se eliminó al vacío. Este producto crudo se filtró a través de un cartucho de gel de sílice (DCM : MeOH = 99 : 1) proporcionando el compuesto del título como una espuma blanca (0.566 g, 79 % de rendimiento). MS/ESI+ 874.2 [MH]+
Etapa 8: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(2-hidroxietoxi)-4-(N-(2-morfolinoetil) metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (225)
3,5-Dicloro-4-((2S)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)-fenil)-2-(4-(N-(2-morfolinoetil) metilsulfonamido)-3-(2-(tetrahidro-2H-piran-2-iloxi)etoxi)benzoiloxi) etil)piridina 1-óxido (0.250 g, 0.286 mmol) se cargó en un cartucho scx eluyendo primero con DCM : MeOH = 1 : 1 y después con MeOH : ac. 32% NH4OH = 9:1. Los volátiles se eliminaron al vacío y el residuo se purificó por cromatografía rápida en columna de gel de sílice (DCM : MeOH = 99 : 1 a 95 : 5) proporcionando (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(2-hidroxietoxi)-4-(N-(2-morfol¡noet¡l)met¡lsulfonam¡do)benzo¡lox¡)etil)p¡r¡d¡na 1-óxido como una espuma blanca (0.113 g, 50.0 % de rendimiento).MS/ESI+ 790.53 [MH]+. ía l20= -44 •O 63J’ c= 0.32, MeOH
1H RMN (300 MHz, DMSO-Ó6) ó ppm 8.56 (s, 2 H), 7.58 (d, 1 H), 7.58 (dd, 1 H), 7.40 (d, 1 H), 7.24 (d, 1 H), 7.20 (d, 1 H), 7.09 (dd, 1 H), 7.06 (t, 1 H), 6.19 (dd, 1 H), 4.88 (t, 1 H), 4.05 - 4.26 (m, 2 H), 3.94 (d, 2 H), 3.79 (q, 2 H), 3.69 (t, 2 H), 3.64 (dd, 1 H), 3.39 - 3.46 (m, 4 H), 3.35 (dd, 1 H), 3.08 (s, 3 H), 2.32 (t, 2 H), 2.16 - 2.29 (m, 4 H), 1.10 - 1.32 (m, 1 H), 0.48 - 0.66 (m, 2 H), 0.23 - 0.46 (m, 2 H)
Ejemplo 31
Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil) metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 259):

Etapa 1: Síntesis de bencil 4-metoxi-3-nitrobenzoato (264):
A una solución de ácido 4-metoxi-3-nitrobenzoico (1 g, 5.07 mmol) en DMF seco (50 ml), se añadieron bicarbonato de sodio (0.511 g, 6.09 mmol) y bromuro de bencilo (0.724 ml, 6.09 mmol) y la reacción se agitó a 50°C por 4 h. La mezcla se trató con HCl 1N y se extrajo dos veces con AcOEt; las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se evaporaron hasta secarse. El producto crudo se purificó por cromatografía rápida en gel de sílice (éter de petróleo/AcOEt = 9/1) para proporcionar bencil 4-metoxi-3-nitrobenzoato (1.2539 g, 4.36 mmol, 86 % de rendimiento, MS/ESI+ 288.1 [MH]+).
Etapa 2: Síntesis de bencil 3-amino-4-metoxibenzoato (263):
A una suspensión de bencil 4-metoxi-3-nitrobenzoato (1.2539 g, 4.36 mmol) en EtOH (25 ml) y agua (12 ml) se añadió cloruro de amonio (0.163 g, 3.06 mmol) seguido por polvo de hierro (1.463 g, 26.2 mmol) y la reacción se calentó a 80°C por 2 h. La parte insoluble se filtró y el filtrado se evaporó hasta secarse. El residuo se disolvió en EtOAc y se lavó con agua y salmuera; la fase orgánica se secó sobre Na2SO4y el solvente se eliminó proporcionando bencil 3-amino-4-metoxibenzoato (1.035 g, 4.02 mmol, 92 % de rendimiento, MS/ESI+257.9 [MH]+). Este producto se usó sin ninguna purificación adicional.
Etapa 3: Síntesis de bencil 4-metoxi-3-(metilsulfonamido)benzoato (262):
A una solución de bencil 3-amino-4-metoxibenzoato (1.035 g, 4.02 mmol) en piridina (40 ml) enfriada a 0°C, se añadió cloruro de metanosulfonilo (0.376 ml, 4.83 mmol) y la mezcla se agitó a temperatura ambiente toda la noche. El solvente se evaporó y el residuo se particionó entre EtOAc y HCl 1N; la fase orgánica se lavó con salmuera y se secó sobre Na2SO4. El solvente se eliminó al vacío y el producto crudo se purificó por trituración con éter de dietilo produciendo bencil 4-metoxi-3-(metilsulfonamido)benzoato (0.920 g, 2.74 mmol, 68.2 % de rendimiento, MS/ESI+ 336.0 [Mh ]+). Etapa 4: Síntesis de bencil 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)metilsulfonamido)benzoato (261):
Una mezcla de bencil 4-metoxi-3-(metilsulfonamido)benzoato (0.500 g, 1.491 mmol), 2-cloro-1-(4-metilpiperazin-1-il)etanona clorhidrato (0.381 g, 1.789 mmol) y carbonato de potasio (0.495 g, 3.58 mmol) en DMF (15 ml) se calentó a 70°C por 1 h. La mezcla se diluyó con EtOAc y se lavó con agua y varias veces con salmuera. La fase orgánica se secó
sobre sulfato de sodio, el solvente se eliminó y el producto crudo se purificó por cromatografía rápida en columna de gel de sílice (EtOAc : MeOH = 90 : 10 a 80 : 20) proporcionando bencil 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)metilsulfonamido)benzoato (0.561 g, 1.180 mmol, 79 % de rendimiento, MS/ESI+ 475.9 [MH]+).
Etapa 5: Síntesis de ácido 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)metilsulfonamido)benzoico (260):
Bencil 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)-metilsulfonamido)benzoato (0.555 g, 1.167 mmol) se disolvió en MeOH (22.200 ml) y se añadió Pd/C (0.248 g, 0.233 mmol) 10 % p/p. La mezcla resultante se hidrogenó en un aparato de Parr a 30 psi por 1 h. El catalizador se filtró y se lavó con MeOH, EtOAc y DCM. Los filtrados combinados se evaporaron hasta secarse produciendo ácido 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)-metilsulfonamido)benzoico (0.339 g, 0.880 mmol, 75 % de rendimiento, MS/ESI+ 386.1 [MH]+).
Etapa 5: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil) metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (259)
Ácido 4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)-metilsulfonamido)benzoico (0.200 g, 0.519 mmol) se suspendió en DCM seco (3.459 ml) y, agitando a temperatura ambiente, (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (0.145 g, 0.346 mmol), EDC (0.199 g, 1.038 mmol) y DMAP (0.0423 g, 0.346 mmol) se añadieron secuencialmente. La suspensión reaccionó por 24 h a temperatura ambiente. La mezcla se evaporó y el residuo se purificó por cromatografía rápida en columna s Na P de gel de sílice (DCM/MeOH = 95/5 a 93/7) para proporcionar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-metoxi-3-(N-(2-(4-metilpiperazin-1-il)-2-oxoetil)metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (197 mg, 0.250 mmol, 72.3 % de rendimiento, MS/ESI+ 787.24 [MH]+, [ap]= -41.60, c = 0.5, MeOH).
1H RMN (300 MHz, DMSO-cfe) 8 ppm 8.52 (s, 2 H), 8.15 (d, 1 H), 7.99 (dd, 1 H), 7.16 - 7.26 (m, 3 H), 7.06 (dd, 1 H), 7.06 (t, 1 H), 6.19 (dd, 1 H), 4.43 (s, 2 H), 3.94 (d, 2 H), 3.93 (s, 3 H), 3.62 (dd, 1 H), 3.33 - 3.45 (m, 4 H), 3.33 (dd, 1 H), 3.08 (s, 3 H), 2.20-2.36 (m, 4 H), 2.17 (s, 3 H), 1.07- 1.39 (m, 1 H), 0.49-0.66 (m, 2 H), 0.22 - 0.46 (m, 2 H)
Ejemplo 32
Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoiloxi)etil)piridina 1-óxido (Compuesto 265):

Etapa 1: Síntesis de ácido 3-nitro-4-(trifluorometoxi)benzoico (270):
A una solución de ácido 4-(trifluorometoxi)benzoico (900 mg, 4.37 mmol) en H2SO4 conc. (9 ml) enfriada a 0°C, se añadió una solución de nitrato de potasio (486 mg, 4.80 mmol) en H2SO4 conc. (4.5 ml) y la reacción se agitó a la misma temperatura por 2 h. La mezcla se trató con H2O (50 ml) y se extrajo con EtOAc (150 ml). La fase orgánica se lavó con salmuera (2 x 100 ml), se secó sobre Na2SO4y se evaporó hasta secarse proporcionando ácido 3-nitro-4-(trifluorometoxi)benzoico (2) (1.05 g, 4.18 mmol, 96 % de rendimiento, MS/ESI+252.1 [MH]+). Este producto se usósin purificación.
Etapa 2: Síntesis de metil 3-nitro-4-(trifluorometoxi)benzoato (269):
A una solución de ácido 3-nitro-4-(trifluorometoxi)benzoico (1.05 g, 4.18 mmol) en MeOH (40 ml), H2SO4 conc. (0.446 ml) se añadió y la mezcla resultante se calentó hasta reflujo toda la noche. El solvente se eliminó al vacío y NaHCO3 (60 ml) sat. acuoso se añadió al residuo. La mezcla se extrajo con EtOAc (2 x 100 ml) y las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se evaporaron para proporcionar metil 3-nitro-4-(trifluorometoxi)benzoato (1.05 g, 3.96 mmol, 95 % de rendimiento, MS/ESI+ 265.9 [MH]+).
Etapa 3: Síntesis de metil 3-amino-4-(trifluorometoxi)benzoato (268):
A una solución de etil 3-nitro-4-(trifluorometoxi)benzoato (1.0 g, 3.77 mmol) en MeOH (20 ml), una cantidad catalítica de Pd/C (10 % p/p) se añadió y la mezcla se hidrogenó en un aparato de Parr a 20 psi por 2 h. El catalizador se filtró y el filtrado se evaporó hasta secarse produciendo metil 3-amino-4-(trifluorometoxi)benzoato (730 mg, 3.10 mmol, 82 % de rendimiento, MS/ESI+ 235.9 [MH]+).
Etapa 4: Síntesis de metil 3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoato (267):
A una solución de metil 3-amino-4-(trifluorometoxi)benzoato (730 mg, 3.10 mmol) en piridina seca (8 ml) enfriada a 0 °C, se añadió cloruro de metanosulfonilo (0.721 ml, 9.31 mmol) y la mezcla se dejó calentar hasta la temperatura ambiente y se agitó toda la noche. El solvente se eliminó al vacío y el residuo se trató con NaHCO3 (50 ml) sat. acuoso y se extrajo con EtOAc (2 x 150 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se evaporaron a presión reducida. El residuo se trituró con Et2O (20 ml) para dar 1.00 g del producto crudo. 500 mg de este producto crudo se disolvieron en DMF seco (10 ml), 4-(2-cloroetil)morfolina clorhidrato (401 mg, 2.156 mmol) se añadió seguido por K2CO3 (496 mg, 3.59 mmol) y la mezcla de reacción se calentó a 100 °C toda la noche. El solvente se eliminó al vacío y el residuo se particionó entre EtOAc y agua; la fase orgánica se lavó con salmuera y se secó sobre sulfato de sodio. El solvente se eliminó y el producto crudo se purificó por cromatografía rápida en gel de sílice (DCM : EtOAc = 90 : 10 a 100 % EtOAc) proporcionando metil 3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoato (600 mg, 1.407 mmol, 91 % de rendimiento por encima del 50 % del material de partida, MS/ESI+ 426.9 [MH]+).
Etapa 5: Síntesis de clorhidrato de ácido 3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoico (266):
A una solución de metil 3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoato (600 mg, 1.407 mmol) en dioxano (10 ml), 6M HCl acuoso (2.814 ml, 16.89 mmol) se añadió y la mezcla se calentó a 100°C por 4 h. Los volátiles se eliminaron al vacío para obtener clorhidrato de ácido 3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoico (600 mg, 1.337 mmol, 95 % de rendimiento, MS/ESI+ 412.9 [MH]+).
Etapa 4: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(N-(2-morfolinoetil) metilsulfonamido)-4-(trifluorometoxi)benzoiloxi)etil)piridina 1-óxido (265):
Una mezcla de ácido 3-(N-(2-morfolinoetil)metilsulfonamido)-4-(trifluorometoxi)benzoico (236 mg, 0.571 mmol), (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (200 mg, 0.476 mmol), EdC (128 mg, 0.666 mmol) y DMAP (116 mg, 0.952 mmol) en DCM seco (10 ml) se agitó a temperatura ambiente por 3 días. La mezcla de reacción se trató con 1M HCl y se extrajo dos veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se evaporaron. El producto crudo se purificó por cromatografía rápida en gel de sílice (DCM/MeOH = 98/2 a 90/10). Una purificación adicional por cromatografía rápida en cartucho de gel de sílice (DCM/MeOH 99/1) se necesitó para proporcionar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(N-(2-morfolinoetil) metilsulfonamido)-4-(trifluorometoxi)benzoiloxi)etil)piridina 1-óxido (28 mg, 0.034 mmol, 7.22 % de rendimiento, MS/ESI+ 814.06 [MH]+, [ao]= -20.55, c=0.510, DCM).
1H RMN (300 MHz, DMSO-cfe) 8 ppm 8.54 (s, 2 H), 8.11 (d, 1 H), 8.11 (dd, 1 H), 7.55 - 7.66 (m, 1 H), 7.23 (d, 1 H), 7.20 (d, 1 H), 7.09 (dd, 1 H), 7.06 (t, 1 H), 6.20 (dd, 1 H), 3.83 - 4.03 (m, 2 H), 3.55 - 3.80 (m, 3 H), 3.31 - 3.42 (m, 5 H), 3.15 (s, 3 H), 2.09 - 2.46 (m, 6 H), 1.06 - 1.38 (m, 1 H), 0.45 - 0.69 (m, 2 H), 0.21 - 0.45 (m, 2 H)
Ejemplo 33
Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(N-(2-(4-metilpiperazin-1-il)etil)-metilsulfonamido)-4-(trifluorometil)benzoiloxi)etil)piridina 1-óxido (Compuesto 271):

Etapa 1: Síntesis de metil 3-(N-(2-hidroxietil)metilsulfonamido)-4-(trifluorometil)benzoato (275):
Una mezcla de metil 3-(N-(metilsulfonil)metilsulfonamido)-4-(trifluorometil)benzoato (1.12 g, 2.98 mmol), 2-bromoetanol (0.847 ml, 11.94 mmol) y K2CO3 (825 mg, 5.97 mmol) en CH3CN (20 ml) se dividió en dos recipientes y se calentó bajo irradiación con microondas a 110°C por 2 h. El solvente se eliminó a presión reducida y el residuo se purificó por cromatografía rápida en gel de sílice (éter de petróleo/EtOAc = 80/20) para dar metil 3-(N-(2-hidroxietil)metilsulfonamido)-4-(trifluorometil)benzoato (700 mg, 2.051 mmol, 69 % de rendimiento, MS/ESI+342.0 [Mh ]+). Etapa 2: Síntesis de metil 3-(N-(2-bromoetil)metilsulfonamido)-4-(trifluorometil)benzoato (274):
Una suspensión de PS-PPh3 (carga: 1.88 mmol/g, 2.3 g, 1.324 mmol) en DCM seco (40 ml) se agitó por 30 minutos a temperatura ambiente; CBr4 (874 mg, 2.64 mmol) y metil 3-(N-(2-hidroxietil)metilsulfonamido)-4-(trifluorometil)benzoato (500 mg, 1.465 mmol) se añadieron y la mezcla resultante se agitó a temperatura ambiente por 1.5 h. La resina se filtró y el filtrado se evaporó hasta secarse proporcionando metil 3-(N-(2-bromoetil)metilsulfonamido)-4-(trifluorometil)benzoato (590 mg, 1.460 mmol, rendimiento cuantitativo, MS/ESI+ 403.8-405.7 [MH]+). Este intermediario se usó sin ninguna purificación adicional.
Etapa 3: Síntesis de metil 3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)-4-(trifluorometil)benzoato (273):
Una mezcla de metil 3-(N-(2-bromoetil)metilsulfonamido)-4-(trifluorometil)benzoato (780 mg, 1.930 mmol), K2CO3 (400 mg, 2.89 mmol) y 1-metilpiperazina (0.536 ml, 4.82 mmol) en CH3CN (40 ml) se dividió en dos recipientes y se calentó bajo irradiación con microondas a 130°C por 1 h. El solvente se eliminó a presión reducida y el residuo se mezcló con el producto crudo obtenido realizando la misma reacción en 58 mg (0.143 mmol) del compuesto de partida. Los productos crudos combinados se purificaron por cromatografía rápida en gel de sílice (DMC/MeOH = 97/3 to 95/5) para proporcionar metil 3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)-4-(trifluorometil)benzoato (600 mg, 1.418 mmol, 47 % de rendimiento, MS/ESI+ 424.0 [MH]+).
Etapa 4: Síntesis de ácido 3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)-4-(trifluorometil)benzoico (272):
A una solución de metil 3-(N-(2-(4-metilpiperazin-1-il)etil)-metilsulfonamido)-4-(trifluorometil)benzoato (460 mg, 1.086 mmol) en dioxano (11 ml), HCl 6N acuoso (2.173 ml, 13.04 mmol) se añadió y la mezcla de reacción se calentó a 100 °C por 2 h. Los volátiles se eliminaron al vacío para proporcionar clorhidrato de ácido 3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)-4-(trifluorometil)benzoico (440 mg, 0.988 mmol, 92 % de rendimiento, MS/ESI+ 409.8 [Mh ]+).
Etapa 5: Síntesis de (S)-3,5-didoro-4-(2-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(N-(2-(4-metilpiperazin-1-il)etil)-metilsulfonamido)-4-(trifluorometil)benzoiloxi)etil)piridina 1-óxido (271):
Una mezcla de clorhidrato de ácido 3-(N-(2-(4-metilpiperazin-1-il)etil)metilsulfonamido)-4-(trifluorometil)benzoico (234 mg, 0.525 mmol), (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (200 mg, 0.476 mmol), EDC (128 mg, 0.666 mmol) y DMAP (116 mg, 0.952 mmol) en DCM seco (8 ml) se agitó a temperatura ambiente toda la noche. La mezcla de reacción se trató con 1M HCl y la fase acuosa se extrajo dos veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se evaporaron. El producto crudo se purificó por cromatografía rápida en gel de sílice (DCM/MeOH = 97/3 a 90/10) para proporcionar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(N-(2-(4-metilpiperazin-1-il)etil) metilsulfonamido)-4-(trifluorometil)benzoiloxi)etil)piridina 1-óxido (186 mg, 0.229 mmol, 48.2 % de rendimiento, MS/ESI+ 811.19 [MH]+, [aD]= -19.36, c = 0.500,DCM).
1H RMN (300 MHz, DMSO-d6) 8 ppm 8.54 y 8.55 (s, 2 H), 8.17 (d, 1 H), 8.10 (s, 1 H), 7.96 (dd, 1 H), 7.18 - 7.27 (m, 2 H), 7.06 - 7.13 (m, 1 H), 7.06 y 7.07 (t, 1 H), 6.10 - 6.32 (m, 1 H), 3.92 (t, 2 H), 3.75 - 4.07 (m, 1 H), 3.45 - 3.75 (m, 2 H), 3.37 (dt, 1 H), 3.19 (s, 3 H), 2.30 - 2.43 (m, 4 H), 2.06 - 2.24 (m, 4 H), 1.92 - 2.05 (m, 2 H), 1.98 y 2.00 (s, 3 H), 1.14 -1.32 (m, 1 H), 0.44- 0.70 (m, 2 H), 0.12-0.44 (m, 2 H)
Ejemplo 34
Síntesis de la sal del ácido 2,2,2-trifluoroacetático (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoiloxi)etil)piridina 1-óxido (Compuesto 276):
Esquema 34
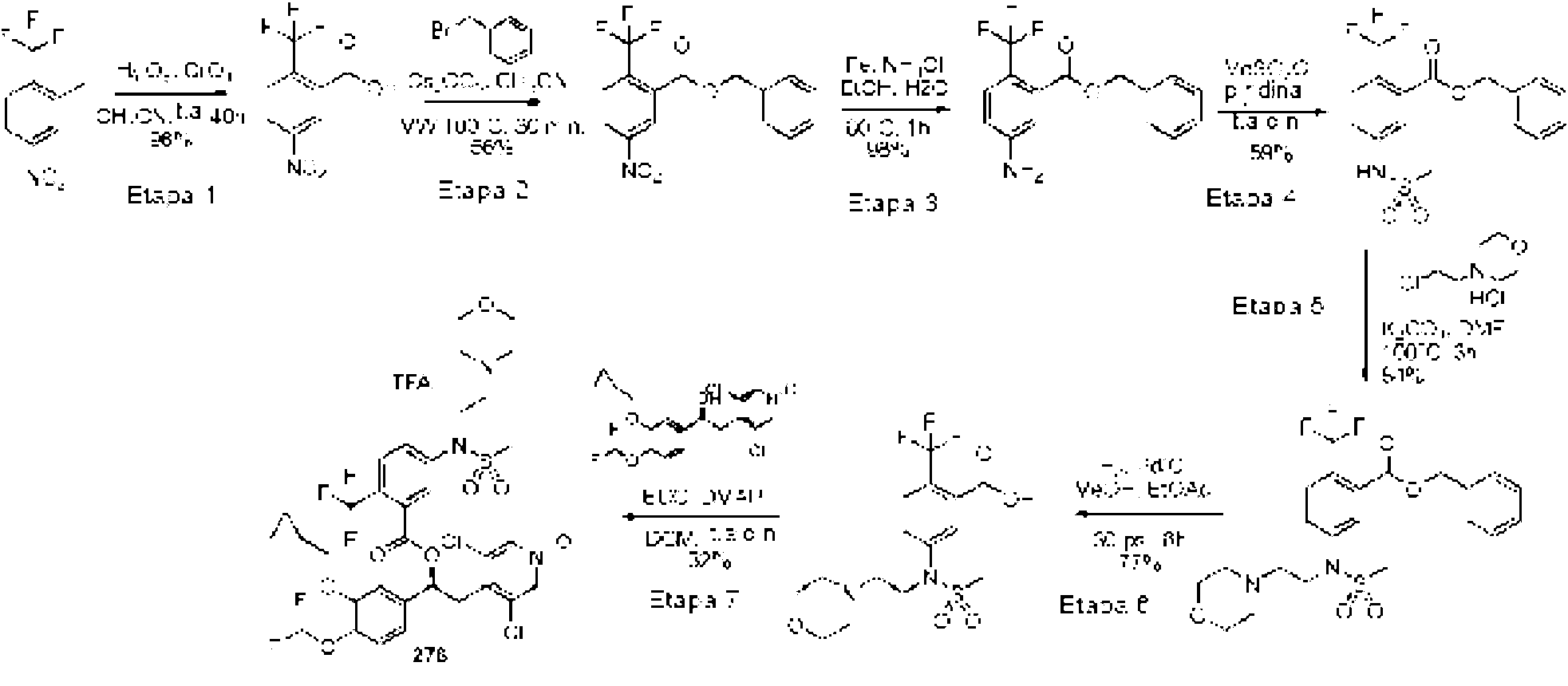
Etapa 1: Síntesis de ácido 5-nitro-2-(trifluorometil)benzoico (282):
A una solución de 2-metil-4-nitro-1-(trifluorometil)benceno (1 g, 4.87 mmol) en CH3CN (30 ml), H5IO6 (5.56 g, 24.37 mmol) y CrO3(0.126 ml, 3.40 mmol) se añadieron y la mezcla de reacción se agitó a temperatura ambiente por 2 h. Se añadió más CrO3 (0.0722 ml, 1.950 mmol) y la mezcla se agitó a temperatura ambiente toda la noche. iPrOH (150 ml) se añadió a la mezcla de reacción y la suspensión se volvió verde. El precipitado se filtró y el filtrado se evaporó hasta secarse; el residuo sólido se trituró con AcOEt (40 ml) y se filtró para proporcionar ácido 5-nitro-2-(trifluorometil)benzoico (1.1 g, 4.68 mmol, 96 % de rendimiento).
Etapa 2: Síntesis de bencil 5-nitro-2-(trifluorometil)benzoato (281):
A una solución de ácido 5-nitro-2-(trifluorometil)benzoico (1.0 g, 4.25 mmol) y bromuro de bencilo (0.506 ml, 4.25 mmol) en CH3CN (8 ml), CS2CO3 (416 mg, 1.276 mmol) se añadió y la mezcla resultante se calentó a 100 °C bajo irradiación de microondas por 30 minutos. La mezcla de reacción se concentró al vacío y el residuo se particionó entre agua y EtOAc. La fase orgánica se secó sobre Na2SO4, el solvente se eliminó a presión reducida y el producto crudo se purificó
por cromatografía rápida en gel de sílice (éter de petróleo/DCM = 95/5) para proporcionar bencil 5-nitro-2-(trifluorometil)benzoato (770 mg, 2.367 mmol, 56 % de rendimiento).
Etapa 3: Síntesis de bencil 5-amino-2-(trifluorometil)benzoato (280):
A una suspensión de bencil 5-nitro-2-(trifluorometil)benzoato (770 mg, 2.367 mmol) en etanol (8 ml) y agua (4 ml), cloruro de amonio (253 mg, 4.73 mmol) se añadió en una porción. La mezcla se calentó a 80°C, polvo de hierro (793 mg, 14.20 mmol) se añadió y la reacción se agitó a la misma temperatura por 1 h. La mezcla se filtró a través de una almohadilla de celita y el filtrado se evaporó hasta secarse. El residuo se disolvió en EtOAc y se lavó con agua y salmuera; la fase orgánica se secó sobre Na2SO4 y el solvente se eliminó proporcionando bencil 5-amino-2-(trifluorometil)benzoato (688 mg, 2.330 mmol, 98 % de rendimiento, MS/ESI+ 296.0 [Mh ]+). Este intermediario se usó sin purificación.
Etapa 4: Síntesis de bencil 5-(metilsulfonamido)-2-(trifluorometil)benzoato (279):
A una solución de bencil 5-amino-2-(trifluorometil)benzoato (688 mg, 2.330 mmol) en piridina seca (6 ml) enfriada a 0 °C, se añadió cloruro de metanosulfonilo (0.198 ml, 2.56 mmol) en forma de gotas y la mezcla de reacción se agitó a temperatura ambiente toda la noche. Los volátiles se eliminaron al vacío y el residuo se trató con NaHCO3 sat. acuoso y se extrajo dos veces con EtOAc; las capas orgánicas combinadas se secaron sobre Na2SO4, el solvente se eliminó y el producto crudo se purificó por trituración con Et2O (100 ml) para producir bencil 5-(metilsulfonamido)-2-(trifluorometil)benzoato (510 mg, 1.366 mmol, 59 % de rendimiento).
Etapa 5: Síntesis de bencil 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoato (278):
Bencil 5-(metilsulfonamido)-2-(trifluorometil)benzoato (510 mg, 1.366 mmol) se disolvió en DMF seco (6 ml) y se añadió clorhidrato de 4-(2-cloroetil)morfolina (381 mg, 2.049 mmol) seguido por K2CO3 (529 mg, 3.82 mmol). La reacción se calentó a 100 °C por 3 h. La mezcla se particionó entre acetato de etilo y agua; la capa orgánica se lavó varias veces con salmuera y se secó sobre sulfato de sodio. El solvente se eliminó a presión reducida y el producto crudo se purificó por cromatografía rápida en gel de sílice (EtOAc/éter de petróleo = 80/20 to AcOEt) para dar bencil 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoato (560 mg, 1.151 mmol, 84 % de rendimiento, MS/ESI+487.1 [MH]+).
Etapa 6: Síntesis de ácido 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoico (277):
Una mezcla de bencil 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoato (560 mg, 1.151 mmol) y una cantidad catalítica de 10 % Pd/C en AcOEt (5 ml) y MeOH (15 ml) se hidrogenó en un aparato de Parr a 30 psi por 6 h. El catalizador se filtró y el solvente se eliminó al vacío; el residuo crudo se trituró con MeOH (5 ml) para proporcionar después de la filtración ácido 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoico (350 mg, 0.883 mmol, 77 % de rendimiento, MS/ESI+396.9 [MH]+).
Etapa 5: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(5-(N-(2-morfolinoetil) metilsulfonamido)-2-(trifluorometil)benzoiloxi)etil)piridina 1-óxido, sal de 2,2,2-trifluoroacetato (276):
Una mezcla de ácido 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoico (200 mg, 0.505 mmol), (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (177 mg, 0.420 mmol), EDC (121 mg, 0.631 mmol) y DmAp (128 mg, 1.051 mmol) en DCM seco se agitó a temperatura ambiente por 3 h. Se añadió más ácido 5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoico (7) (167 mg, 0.420 mmol), EDC (81 mg, 0.420 mmol) y DMAP (25.7 mg, 0.210 mmol) y la reacción se agitó a temperatura ambiente toda la noche. La mezcla se lavó con agua y salmuera; la fase orgánica se secó sobre Na2SO4, se filtró y se evaporó. El producto crudo se purificó por cromatografía rápida en gel de sílice (DCM/MeOH = 95/05) y después por una cromatografía en gel de sílice adicional (DCM/AcOEt 60/40, después DCM/MeOH 90/10). El producto obtenido se trituró con EtOH (3 ml) y finalmente se purificó por HPLC preparativa produciendo (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(5-(N-(2-morfolinoetil)metilsulfonamido)-2-(trifluorometil)benzoiloxi)etil)piridina 1-óxido sal de 2,2,2-trifluoroacetato (121 mg, 0.133 mmol, 31.6 % de rendimiento, MS/ESI+ 798.1 [MH]+, [ao]= -2.513, c = 0.390, DCM).
1H RMN (300 MHz, DMSO-cfe Na2CO3) d ppm 8.56 (s, 2 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.78 (d, 1 H), 7.21 (d, 1 H), 7.17 (d, 1 H), 7.03 (dd, 1 H), 7.09 (t, 1 H), 6.20 (dd, 1 H), 3.94 (dd, 1 H), 3.87 (dd, 1 H), 3.85 (t, 2 H), 3.60 (dd, 1 H), 3.36 - 3.47 (m, 4 H), 3.40 (dd, 1 H), 3.13 (s, 3 H), 2.39 (t, 2 H), 2.18 - 2.32 (m, 4 H), 1.12 - 1.31 (m, 1 H), 0.48 - 0.69 (m, 2 H), 0.25-0.48 (m, 2 H)
Ejemplo 36
Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-hidroxi-5-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 287)

Etapa 1: Síntesis de metil 3-(cidopropilmetoxi)-5-(N-(2-morfolinoetil)metilsulfonamido)benzoato (292)
Metil 3-(cidopropilmetoxi)-5-(metilsulfonamido)benzoato (202 mg, 0.675 mmol) (obtenido siguiendo el mismo procedimiento descrito en WO2010/089107, Esquema 2, Ejemplo 16, Etapa 1-4, comenzando a partir de metil éster del ácido 3-hidroxi-5-nitro-benzoico), reacció como se describió en el Ejemplo 5. Esquema 5, Etapa 3, para dar el producto deseado (152 mg, 0.368 mmol, 55 % de rendimiento). MS/ESI+ 299.08 [MH]+.
Etapa 2: Síntesis de metil 3-hidroxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoato (291)
Metil 3-(ciclopropilmetoxi)-5-(N-(2-morfolinoetil) metilsulfonamido)benzoato (50 mg, 0.121 mmol) se disolvió en TFA (1.5 ml, 19.47 mmol). La reacción se agitó a temperatura ambiente por 4 h hasta lograr el completamiento. El solvente se concentró al vacío para dar metil 3-hidroxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoato (40 mg, 0.112 mmol, 92 % de rendimiento). MS/ESI+ 359.12 [MH]+
Etapa 3: Síntesis de ácido 3-hidroxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoico (290)
Metil 3-hidroxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoato (40 mg, 0.112 mmol) se disolvió en MeOH (1.5ml). NaOH 1M (500microl) se añadió, y la reacción se agitó a 50 Celsius grados por 2 h hasta lograr el completamiento. La mezcla de reacción se neutralizó con HCl 1N y se concentró para dar ácido 3-hidroxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoico (60 mg, 0.174 mmol, 156 % de rendimiento). MS/ESI+ 345.11 [MH]+
Etapa 4: Síntesis de ácido 3-acetoxi-5-(N-(2-morfolinoetil)-metilsulfonamido)benzoico (289)
Ácido 3-hidroxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoico (200 pl, 0.764 mmol) se disolvió en DCM (2ml). Se añadieron cloruro de acetilo (60 mg, 0.764 mmol) y DMAP (100 mg, 0.819 mmol) y la reacción se agitó a temperatura ambiente por 2 h hasta lograr el completamiento. La mezcla de reacción se concentró y el producto crudo se trituró en hexano, se filtró y se secó en un horno de vacío para dar ácido 3-acetoxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoico (60 mg, 0.155 mmol, 20.31 % de rendimiento). MS/ESI+ 387.42 [MH]+ Etapa 5: Síntesis de (S)-4-(2-(3-acetoxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (288)
(S)-3,5-Dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (20 mg, 0.048 mmol), ácido 3-acetoxi-5-(N-(2-morfolinoetil)metilsulfonamido)benzoico (60 mg, 0.155 mmol), DMAP (20 mg, 0.164 mmol) y EDC (100
mg, 0.522 mmol) se disolvieron en DMF (1.5ml). La reacción se agitó a 40 grados Celsius por 8 h hasta lograr el completamiento. La mezcla de reacción se diluyó con agua y el precipitado se disolvió en acetato de etilo. La fase orgánica se secó sobre Na2SO4 y se concentró al vacío para dar (S)-4-(2-(3-acetoxi-5-(N-(2-morfolinoetil)metilsulfonamido)-benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (30 mg, 0.038 mmol, 80 % de rendimiento). MS/ESI+788.64 [MH]+
Etapa 6: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-hidroxi-5-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (287)
(S)-4-(2-(3-Acetoxi-5-(N-(2-morfolinoetil)metilsulfonamido)-benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido se disolvió en MeOH (1.5ml). Se añadió una sol. sat de bicarbonato de sodio (500 j L), y la reacción se agitó a temperatura ambiente por 2 h hasta lograr el completamiento. La mezcla de reacción se diluyó con agua, se extrajo con acetato de etilo y se concentró al vacío. El producto crudo se purificó por HPLC preparativa para dar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-hidroxi-5-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (10 mg, 0.013 mmol). MS/eS|+ 745.8 [MH]+ 1H RMN (400 MHz, DMSOd) 8 ppm 10.03 - 10.22 (s, 1 H), 8.43 - 8.69 (s, 2 H), 7.38 - 7.45 (m, 1 H), 7.31 - 7.38 (m, 1 H), 7.16 - 7.28 (m, 2 H), 7.00 - 7.12 (m, 3 H), 6.05 - 6.25 (m, 1 H), 3.84 - 3.97 (m, 2 H), 3.66 - 3.78 (m, 2 H), 3.55 - 3.65 (m, 1 H), 3.47 (m, 5 H), 3.02 (s, 3 H), 2.23 - 2.38 (m, 6 H), 1.08 - 1.33 (m, 1 H), 0.49 - 0.67 (m, 2 H), 0.24 - 0.45 (m, 2 H). Ejemplo 37
Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(metiltio)-3-(N-(2-morfolinoetil)-metilsulfonamido)benzoiloxi)etil)piridina 1-óxido (Compuesto 293)

Etapa 1: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(metiltio)-3-nitrobenzoiloxi) etil)piridina1-óxido (296)
(S)-3,5-Dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-hidroxietil)piridina 1-óxido (240 mg, 0.571 mmol), N,N-dimetilpiridin-4-amina (84 mg, 0.685 mmol), clorhidrato de N1-((etilimino)metileno)-N3,N3-dimetilpropano-1,3-diamina (328 mg, 1.713 mmol) se disolvieron en DMF (2ml). La reacción se agitó a temperatura ambiente toda la noche. La mezcla de reacción se diluyó con HCl 1N, y el precipitado se filtró, se lavó con HCl 1N, se disolvió en acetato de etilo y se extrajo con HCl 1N, NaHcO35 % y salmuera. La fase orgánica se secó sobre Na2SO4 y se concentró al
vacío para dar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(metiltio)-3-nitrobenzoiloxi) etil)piridina 1-óxido (300 mg, 0.487 mmol, 85 % de rendimiento). MS/ESI+ 615.43 [Mh ]+
Etapa 2: Síntesis de (S)-4-(2-(3-amino-4-(metiltio)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi) fenil)etM)-3,5-didoropiridina 1-óxido (295)
(S)-3,5-Dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(metiltio)-3-nitrobenzoiloxi)etil)piridina 1-óxido (320 mg, 0.520 mmol) se disolvió en THF (5ml). CLORURO DE ESTAÑO(II) DIHIDRATADO (587 mg, 2.60 mmol) se añadió, y la mezcla se agitó a temperatura ambiente toda la noche. El solvente se concentró al vacío y el producto crudo se disolvió en acetato de etilo y se lavó con HCl 1N, salmuera, se secó sobre Na2SO4 y se concentró al vacío. 20 mg del producto crudo se purificaron por HPLC preparativa para dar (S)-4-(2-(3-amino-4-(metiltio)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (10 mg, 0.017 mmol, 3.29 % de rendimiento). MS/ESI+584.8 [MH]+
1H RMN (400 MHz, DMSOd) 5 ppm 8.57 (s, 2 H), 7.27 - 7.31 (m, 1 H), 7.19 (m, 4 H), 7.01 - 7.10 (m, 2 H), 6.06 - 6.27 (m, 1 H), 5.29 (s, 2 H), 3.91 (d, J=7.06 Hz, 2 H), 3.51 - 3.64 (m, 1 H), 3.29 (m, 1 H), 2.43 (s, 3 H), 1.12 - 1.30 (m, 1 H), 0.48 - 0.62 (m, 2 H), 0.26 - 0.41 (m, 2 H)
Etapa 3: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(metilsulfonamido)-4-(metiltio)benzoiloxi)etil)piridina 1-óxido (294)
(S)-4-(2-(3-Amino-4-(metiltio)benzoiloxi)-2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)etil)-3,5-dicloropiridina 1-óxido (200 mg, 0.342 mmol) se disolvió en DCM (3ml). Se añadieron cloruro de metanosulfonilo (0.053 ml, 0.683 mmol) y piridina (0.028 ml, 0.342 mmol) y la reacción se agitó a temperatura ambiente por 8 hr hasta lograr el completamiento. La mezcla de reacción se lavó con HCl 1N y salmuera, se secó sobre Na2SO4 y se concentró al vacío. 30 mg del producto crudo se purificaron por Hp Lc preparativa para dar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(metilsulfonamido)-4-(metiltio)benzoiloxi)etil)piridina 1-óxido (20 mg, 0.030 mmol, 8.82 % de rendimiento). MS/ESI+ 663.0 [MH]+
1H RMN (400 MHz, DMSOd) 5 ppm 9.32 - 9.56 (bs, 1 H), 8.55 (s, 2 H), 7.83 - 7.93 (m, 1 H), 7.76 - 7.82 (m, 1 H), 7.33 -7.44 (m, 1 H), 7.17 - 7.24 (m, 2 H), 7.00 - 7.10 (m, 2 H), 6.12 - 6.27 (m, 1 H), 3.85 - 3.97 (m, 2 H), 3.52 - 3.66 (m, 1 H), 3.36-3.42 (m, 1 H), 3.01 (s, 3 H), 2.49 (s, 3 H), 1.18-1.27 (m, 1 H), 0.50-0.61 (m, 2 H), 0.31 -0.40 (m, 2 H).
Etapa 4: Síntesis de (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(4-(metiltio)-3-(N-(2-morfolinoetil)metilsulfonamido)benzoiloxi)etil)piridina 1 -óxido (293)
(S)-3,5-Dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3-(metilsulfonamido)-4-(metiltio) benzoiloxi)etil)piridina 1-óxido (50 mg, 0.075 mmol) se disolvió en DMF. Se añadieron 4-(2-cloroetil)morfolina (56.4 mg, 0.377 mmol) y K2CO3 (20.83 mg, 0.151 mmol) y la reacción se agitó a 45 grados Celsius por 6 h hasta lograr el completamiento. La mezcla de reacción se diluyó con agua y se extrajo con AcOEt. La fase orgánica se secó sobre Na2SO4 y se concentró al vacío. El producto crudo se purificó por HPLC preparativa para dar (S)-3,5-dicloro-4-(2-(3-(ciclopropilmetoxi)-4-(difluorometoxi)-fenil)-2-(4-(metiltio)-3-(N-(2-morfolinoetil)metilsulfonamido)-benzoiloxi)etil)piridina 1-óxido (30 mg, 0.039 mmol, 51.3 % de rendimiento). MS/ESI+ 776.3 [MH]+
1H RMN (400 MHz, DMSOd) 5 ppm 8.55 (d, J=10.14 Hz, 2 H), 7.92 - 8.00 (m, 1 H), 7.87 (t, J=2.21 Hz, 1 H), 7.37 - 7.45 (m, 1 H), 7.21 (d, J=7.94 Hz, 2 H), 7.07 (m, 2 H), 6.13 - 6.24 (m, 1 H), 3.93 (d, J=7.06 Hz, 2 H), 3.70 - 3.84 (m, 1 H), 3.48 -3.67 (m, 2 H), 3.39 (d, J=4.41 Hz, 4 H), 3.18 (m, 1 H), 3.18 (s, 3 H), 2.50 (s, 3 H), 2.07-2.45 (m, 6 H), 1.14-1.29 (m, 1 H), 0.50 - 0.63 (m, 2 H), 0.27 - 0.42 (m, 2 H).
El compuesto enumerado en la Tabla 22 se preparó con etapas de síntesis y procedimientos análogos a los descritos en el Ejemplo 37 al hacer reaccionar los precursores apropiados (comercialmente disponibles o sintetizados por una persona experta en la técnica) con reactivos adecuados.
Tabla 22

Actividad farmacológica de los compuestos de la invención.
Determinación in vitro de la actividad inhibitoria de PDE4 en el ensayo sin células.
La actividad de PDE4 se determinó en el lisado de células sobrenadantes monocíticas humanas U937. Las células se cultivaron, se recogieron y la fracción de sobrenadante se preparó esencialmente como se describe en Torphy TJ y otros J. Pharmacol. Exp. Ther. 1992; 263:1195-1205.
Las células U937 (Cell Bank, Interlab Cell Line Collection, ICLC HTL94002) se cultivaron a 37°C, 5 % CO2 en RPMI 1640 con medio GlutaMAX™-I suplementado con 10 % de suero fetal bovino y 100 pg/ml de penicilina-estreptomicina (Gibco).
Las células se cosecharon y se lavaron dos veces por centrifugación (150 x g, 8 min) en PBS frío. Las células lavadas se resuspendieron en tampón Krebs-Ringer-Henseleit frío a una concentración final de 20 X 106 células/ml y se sonicaron. Después de la centrifugación a 15000 x g por 20 min, los los sobrenadantes se agruparon, se dividieron en alícuotas y se almacenaron - 80°C.
La actividad de PDE4 se determinó en células sobrenadantes mediante el ensayo de la desaparición de AMPc de las mezclas de incubación.
La concentración de los compuestos de ensayo osciló entre 10'12M y 10'6 M. Las reacciones fueron detenidas por la inactivación por calor de las enzimas (2.5 minutos a 100°C) y el contenido de AMPc residual se determinó con el uso del ensayo AMPc de LANCE de Perkin Elmer siguiendo las instrucciones de los proveedores.
Los resultados de los compuestos probados, representativos de la invención, expresados como media de la concentración nM del compuesto de prueba que produce 50 % de inhibición de desaparición de AMPc (IC50), son < 1 nM.
El porcentaje de inhibición de la actividad de PDE4 se calculó, asumiendo que la desaparición de AMPc en ausencia de inhibidores como 100 % y la desaparición de AMPc en muestras inactivadas con calor como 0 %.
Claims (13)
1. Un compuesto de la Fórmula general (I)

en donde:
Ri y R2 son iguales o diferentes y se seleccionan independientemente del grupo que consiste en hidrógeno, metilo, etilo, difluorometilo, ciclopropilmetilo o ciclopropilo;
R3 son los mismos dos átomos de cloro en la posición 3 y 5 del anillo piridilo;
Z es un grupo (CH2)m en donde m = 0;
A es un anillo de fenilo, que está opcionalmente sustituido con uno o más sustituyentes R4, que pueden ser iguales o diferentes y se seleccionan independientemente del grupo que consiste en:
(Ci-C2)alquilo lineal o ramificado opcionalmente sustituido por uno o más (C3-C7)heterocicloalquilo y preferentemente 4-morfolinilo;
trifluorometilo;
metiltio;
halógeno seleccionado de fluoro o cloro; y
OR7 en donde R7 se selecciona del grupo que consiste en
-H;
- (C1-C4) alquilo opcionalmente sustituido por un radical seleccionado de OH, ciclopropilo, 4-morfolinilo, 1- o 4-piperidinilo, 4-piridinilo y fenilo;
- trifluorometilo o difluorometilo; y
W es el grupo -NR9SO2R10 en donde R9 se selecciona del grupo que consiste en (C1-C4) alquilo opcionalmente sustituido por 4-morfolinilo, 1-piperidinilo, 1-pirrolidinilo, piperazinilo, 1,2-tiazolidin-3-ilo, piperazin-1-ilcarbonilo en donde cualquiera de dichos anillos puede estar opcionalmente sustituido con uno o más metilo, etilo u OH; y
RnRi2N-(Ci-C3)alquileno en donde R uy Ri2 son independientemente H o metilo opcionalmente sustituidos con un grupo OH; y
R10 se selecciona del grupo que consiste en (C1-C4) alquilo opcionalmente sustituido por (C3-C7) cicloalquilo y (C3-C7) cicloalquilo;
sus N-óxidos en el anillo de piridina, y sales farmacéuticamente aceptables de estos.
2. Un compuesto de acuerdo con la reivindicación 1 que son N-óxidos en el anillo de piridina, representado por la Fórmula (Ia)

en donde R1, R2, R3, Z, A y W son como se describen en la reivindicación 1.
3. Un compuesto de acuerdo con la reivindicación 1 que se representa por la Fórmula (I)' en donde la configuración absoluta del carbono (1) es la que se muestra a continuación:

en donde Ri, R2, R3, Z, A y W son como se definieron anteriormente para los compuestos de la Fórmula (I) y sus N-óxidos en el anillo de piridina, o sales farmacéuticamente aceptables de estos.
4. Una composición farmacéutica que comprende un compuesto de la Fórmula (I) como se define en cualquiera de las reivindicaciones 1 a 3 y uno o más vehículos y/o excipientes farmacéuticamente aceptables.
5. Un compuesto de la Fórmula (I) como se define en cualquiera de las reivindicaciones 1 a 3, para usar como medicamento.
6. Un compuesto de la Fórmula (I) como se define en cualquiera de las reivindicaciones 1 a 3, para usar en la prevención y/o el tratamiento de un estado de enfermedad alérgica o una enfermedad del tracto respiratorio caracterizada por la obstrucción de las vías aéreas.
7. Un compuesto para usar de acuerdo con la reivindicación 6 en donde la enfermedad del tracto respiratorio caracterizada por la obstrucción de las vías aéreas se selecciona de asma y EPOC.
8. Un compuesto para usar de acuerdo con la reivindicación 6 en donde el estado de enfermedad alérgica se selecciona de dermatitis atópica, urticaria y rinitis alérgica.
9. Uso de un compuesto de la Fórmula (I) como se define en cualquiera de las reivindicaciones 1 a 3 para la preparación de un medicamento para la prevención y/o el tratamiento de una enfermedad alérgica o una enfermedad de las vías respiratorias caracterizada por la obstrucción de las vías aéreas.
10. Uso de acuerdo con la reivindicación 9, en donde la enfermedad del tracto respiratorio caracterizada por la obstrucción de las vías aéreas se selecciona de asma y EPOC.
11. Uso de acuerdo con la reivindicación 9, en donde el estado de enfermedad alérgica se selecciona de dermatitis atópica, urticaria y rinitis alérgica.
12. Un dispositivo que comprende una composición farmacéutica de acuerdo con la reivindicación 4.
13. Un kit que comprende las composiciones farmacéuticas de la reivindicación 4 y un dispositivo que es un inhalador de polvo seco de dosis única o múltiple, un inhalador de dosis medida o un nebulizador de nebulización suave.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12195738 | 2012-12-05 | ||
| PCT/EP2013/075540 WO2014086865A1 (en) | 2012-12-05 | 2013-12-04 | 1-phenyl-2-pyridinyl alkyl alcohol derivatives as phosphodiesterase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2716142T3 true ES2716142T3 (es) | 2019-06-10 |
Family
ID=47325924
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13814850T Active ES2716142T3 (es) | 2012-12-05 | 2013-12-04 | Derivados del alcohol de 1-fenil-2-piridinil alquilo como inhibidores de la fosfodiesterasa |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US9944612B2 (es) |
| EP (1) | EP2928869B1 (es) |
| KR (1) | KR102192739B1 (es) |
| CN (2) | CN104822659A (es) |
| AR (1) | AR093796A1 (es) |
| BR (1) | BR112015012724A2 (es) |
| CA (1) | CA2893628C (es) |
| ES (1) | ES2716142T3 (es) |
| HK (1) | HK1213551A1 (es) |
| PL (1) | PL2928869T3 (es) |
| RU (1) | RU2655170C2 (es) |
| TR (1) | TR201907092T4 (es) |
| TW (1) | TWI628166B (es) |
| WO (1) | WO2014086865A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2216327A1 (en) | 2009-02-06 | 2010-08-11 | CHIESI FARMACEUTICI S.p.A. | Benzoic acid (1-phenyl-2-pyridin-4-yl)ethyl esters as phosphodiesterase inhibitors |
| RS61558B1 (sr) | 2013-10-22 | 2021-04-29 | Chiesi Farm Spa | Kristalni oblik pde4 inhibitora |
| AR104822A1 (es) * | 2015-06-01 | 2017-08-16 | Chiesi Farm Spa | Derivado de aminoésteres |
| AR104829A1 (es) * | 2015-06-01 | 2017-08-16 | Chiesi Farm Spa | Derivados de aminoésteres |
| EP3732164A1 (en) * | 2017-12-28 | 2020-11-04 | Chiesi Farmaceutici S.p.A. | Use of 1-phenyl-2-pyridinyl alkyl alcohol derivatives for treating cystic fibrosis |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3775684B2 (ja) | 1992-07-28 | 2006-05-17 | ローン−プーラン・ロレ・リミテツド | 脂肪族−またはヘテロ原子−含有連結基によりアリールまたはヘテロアリールに連結されたフェニル含有化合物 |
| US5786354A (en) | 1994-06-21 | 1998-07-28 | Celltech Therapeutics, Limited | Tri-substituted phenyl derivatives and processes for their preparation |
| ID27509A (id) * | 1998-10-06 | 2001-04-12 | Dainippon Pharmaceutical Co | Turunan piridin tersubstitusi-2,3 proses pembuatannya, komposisi farmasi yang mengandung senyawa tersebut, dan senyawa-antaranya |
| SE9903995D0 (sv) * | 1999-11-03 | 1999-11-03 | Astra Ab | New combination |
| US20060239927A1 (en) | 2003-03-31 | 2006-10-26 | Kyowa Hakko Kogyo Co., | Drug for airway administration |
| EP2044023B1 (en) * | 2006-07-14 | 2011-01-19 | CHIESI FARMACEUTICI S.p.A. | Derivatives of 1-phenyl-2-pyridynyl alkylene alcohols as phosphodiesterase inhibitors |
| JP2008174213A (ja) * | 2006-12-20 | 2008-07-31 | Nsk Ltd | 舵角可変式ステアリング装置 |
| EP2022783A1 (en) * | 2007-08-08 | 2009-02-11 | CHIESI FARMACEUTICI S.p.A. | "Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors" |
| EP2070913A1 (en) | 2007-12-14 | 2009-06-17 | CHIESI FARMACEUTICI S.p.A. | Ester derivatives as phosphodiesterase inhibitors |
| EP2110375A1 (en) | 2008-04-14 | 2009-10-21 | CHIESI FARMACEUTICI S.p.A. | Phosphodiesterase-4 inhibitors belonging to the tertiary amine class |
| EP2216327A1 (en) * | 2009-02-06 | 2010-08-11 | CHIESI FARMACEUTICI S.p.A. | Benzoic acid (1-phenyl-2-pyridin-4-yl)ethyl esters as phosphodiesterase inhibitors |
| CN103562185A (zh) | 2011-06-06 | 2014-02-05 | 奇斯药制品公司 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 |
| EP2760838B1 (en) | 2011-09-26 | 2017-05-10 | Chiesi Farmaceutici S.p.A. | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors |
| KR20150022789A (ko) | 2012-06-04 | 2015-03-04 | 키에시 파르마슈티시 엣스. 피. 에이. | 포스포디에스테라제 억제제로서 1-페닐-2-피리디닐 알킬 알코올의 유도체 |
-
2013
- 2013-12-04 ES ES13814850T patent/ES2716142T3/es active Active
- 2013-12-04 CA CA2893628A patent/CA2893628C/en active Active
- 2013-12-04 CN CN201380063353.XA patent/CN104822659A/zh active Pending
- 2013-12-04 RU RU2015121045A patent/RU2655170C2/ru active
- 2013-12-04 TR TR2019/07092T patent/TR201907092T4/tr unknown
- 2013-12-04 TW TW102144352A patent/TWI628166B/zh active
- 2013-12-04 BR BR112015012724A patent/BR112015012724A2/pt not_active Application Discontinuation
- 2013-12-04 AR ARP130104493A patent/AR093796A1/es active IP Right Grant
- 2013-12-04 HK HK16101442.4A patent/HK1213551A1/zh unknown
- 2013-12-04 PL PL13814850T patent/PL2928869T3/pl unknown
- 2013-12-04 CN CN201911008774.8A patent/CN110698459B/zh active Active
- 2013-12-04 EP EP13814850.7A patent/EP2928869B1/en active Active
- 2013-12-04 WO PCT/EP2013/075540 patent/WO2014086865A1/en not_active Ceased
- 2013-12-04 KR KR1020157017275A patent/KR102192739B1/ko active Active
- 2013-12-05 US US14/097,445 patent/US9944612B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CA2893628C (en) | 2021-11-30 |
| WO2014086865A1 (en) | 2014-06-12 |
| BR112015012724A2 (pt) | 2017-07-11 |
| TW201439067A (zh) | 2014-10-16 |
| RU2015121045A (ru) | 2017-01-11 |
| US9944612B2 (en) | 2018-04-17 |
| US20140155391A1 (en) | 2014-06-05 |
| HK1213551A1 (zh) | 2016-07-08 |
| KR20150092214A (ko) | 2015-08-12 |
| EP2928869B1 (en) | 2019-02-20 |
| CN110698459A (zh) | 2020-01-17 |
| KR102192739B1 (ko) | 2020-12-18 |
| CA2893628A1 (en) | 2014-06-12 |
| PL2928869T3 (pl) | 2019-08-30 |
| EP2928869A1 (en) | 2015-10-14 |
| AR093796A1 (es) | 2015-06-24 |
| CN104822659A (zh) | 2015-08-05 |
| TR201907092T4 (tr) | 2019-06-21 |
| CN110698459B (zh) | 2023-05-05 |
| RU2655170C2 (ru) | 2018-05-24 |
| TWI628166B (zh) | 2018-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2395528T3 (es) | Ésteres de (1-fenil-2-piridin-4-il)etilo del ácido benzoico como inhibidores de fosfodiesterasa | |
| CA2893627C (en) | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity | |
| WO2012168226A1 (en) | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors | |
| EP2928879B1 (en) | Phenylethylpyridine derivatives as pde4-inhibitors and muscarinic receptor antagonists | |
| ES2716142T3 (es) | Derivados del alcohol de 1-fenil-2-piridinil alquilo como inhibidores de la fosfodiesterasa | |
| EP2760838A1 (en) | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors | |
| CN104822671B (zh) | 作为pde4抑制剂的苯乙基吡啶衍生物 | |
| CN104837828A (zh) | 作为pde4-抑制剂的苯基乙基吡啶衍生物 | |
| ES2848699T3 (es) | Compuestos de 3-oxo-1,4-diazepinilo como activadores de NRF2 | |
| CN102947300B (zh) | 生物碱氨基酯衍生物及其药物组合物 | |
| EP3152204B1 (en) | Carbamate derivatives which are both phosphodiesterase 4 (pde4) enzyme inhibitors and muscarinic m3 receptor antagonists | |
| HK40018257B (en) | 1-phenyl-2-pyridinyl alkyl alcohol derivatives as phosphodiesterase inhibitors | |
| HK40018257A (en) | 1-phenyl-2-pyridinyl alkyl alcohol derivatives as phosphodiesterase inhibitors | |
| HK1182093B (en) | Alkaloid aminoester derivatives and medicinal compositions thereof |