EP4461356A2 - Methods of treating an ischemic disease - Google Patents
Methods of treating an ischemic disease Download PDFInfo
- Publication number
- EP4461356A2 EP4461356A2 EP24200691.4A EP24200691A EP4461356A2 EP 4461356 A2 EP4461356 A2 EP 4461356A2 EP 24200691 A EP24200691 A EP 24200691A EP 4461356 A2 EP4461356 A2 EP 4461356A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- cells
- mnbmcs
- tnfr1
- expression
- ischemic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 208000023589 ischemic disease Diseases 0.000 title claims abstract description 68
- 238000000034 method Methods 0.000 title abstract description 159
- 102100033732 Tumor necrosis factor receptor superfamily member 1A Human genes 0.000 claims abstract description 136
- 101710187743 Tumor necrosis factor receptor superfamily member 1A Proteins 0.000 claims abstract description 135
- 230000014509 gene expression Effects 0.000 claims abstract description 127
- 108060008682 Tumor Necrosis Factor Proteins 0.000 claims abstract description 77
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 claims abstract description 77
- 230000002829 reductive effect Effects 0.000 claims abstract description 41
- 210000004027 cell Anatomy 0.000 claims description 326
- 238000010453 CRISPR/Cas method Methods 0.000 claims description 46
- 230000002222 downregulating effect Effects 0.000 claims description 42
- 210000001185 bone marrow Anatomy 0.000 claims description 34
- 210000002901 mesenchymal stem cell Anatomy 0.000 claims description 28
- 102100033733 Tumor necrosis factor receptor superfamily member 1B Human genes 0.000 claims description 27
- 101710187830 Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 claims description 27
- 108020005004 Guide RNA Proteins 0.000 claims description 26
- 210000002798 bone marrow cell Anatomy 0.000 claims description 26
- 210000004698 lymphocyte Anatomy 0.000 claims description 23
- 208000031225 myocardial ischemia Diseases 0.000 claims description 21
- 230000000302 ischemic effect Effects 0.000 claims description 12
- 238000000432 density-gradient centrifugation Methods 0.000 claims description 8
- 239000000017 hydrogel Substances 0.000 claims description 6
- 210000004700 fetal blood Anatomy 0.000 claims description 5
- 210000005259 peripheral blood Anatomy 0.000 claims description 5
- 239000011886 peripheral blood Substances 0.000 claims description 5
- 229920001917 Ficoll Polymers 0.000 claims description 4
- 208000026106 cerebrovascular disease Diseases 0.000 claims description 3
- 208000019693 Lung disease Diseases 0.000 claims description 2
- 208000017169 kidney disease Diseases 0.000 claims description 2
- 230000000694 effects Effects 0.000 abstract description 96
- 239000002679 microRNA Substances 0.000 description 89
- 239000003795 chemical substances by application Substances 0.000 description 83
- 108091070501 miRNA Proteins 0.000 description 67
- 108090000623 proteins and genes Proteins 0.000 description 66
- 210000000130 stem cell Anatomy 0.000 description 65
- 210000001519 tissue Anatomy 0.000 description 51
- 206010061216 Infarction Diseases 0.000 description 46
- 230000007574 infarction Effects 0.000 description 46
- 241000282414 Homo sapiens Species 0.000 description 40
- 230000035772 mutation Effects 0.000 description 39
- 125000003729 nucleotide group Chemical group 0.000 description 39
- 239000008194 pharmaceutical composition Substances 0.000 description 39
- 239000002773 nucleotide Substances 0.000 description 38
- 108020004414 DNA Proteins 0.000 description 35
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 34
- 241000699670 Mus sp. Species 0.000 description 33
- 210000001671 embryonic stem cell Anatomy 0.000 description 31
- 241000699666 Mus <mouse, genus> Species 0.000 description 28
- 108020004459 Small interfering RNA Proteins 0.000 description 28
- 230000009368 gene silencing by RNA Effects 0.000 description 26
- 238000010459 TALEN Methods 0.000 description 25
- 239000000203 mixture Substances 0.000 description 25
- 108091033409 CRISPR Proteins 0.000 description 23
- 101710163270 Nuclease Proteins 0.000 description 23
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 23
- 239000004480 active ingredient Substances 0.000 description 23
- 150000007523 nucleic acids Chemical group 0.000 description 23
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 22
- 102000004169 proteins and genes Human genes 0.000 description 22
- 238000002347 injection Methods 0.000 description 21
- 239000007924 injection Substances 0.000 description 21
- 108020004999 messenger RNA Proteins 0.000 description 21
- 102000039446 nucleic acids Human genes 0.000 description 21
- 108020004707 nucleic acids Proteins 0.000 description 21
- 235000018102 proteins Nutrition 0.000 description 21
- 230000027455 binding Effects 0.000 description 20
- 238000011282 treatment Methods 0.000 description 20
- 210000002216 heart Anatomy 0.000 description 19
- 238000000338 in vitro Methods 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 16
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 16
- 239000002609 medium Substances 0.000 description 16
- 230000004075 alteration Effects 0.000 description 15
- 210000002966 serum Anatomy 0.000 description 15
- 230000000692 anti-sense effect Effects 0.000 description 14
- 210000002459 blastocyst Anatomy 0.000 description 14
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 14
- 239000002953 phosphate buffered saline Substances 0.000 description 14
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 13
- 238000003556 assay Methods 0.000 description 13
- 230000001413 cellular effect Effects 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 108700028369 Alleles Proteins 0.000 description 12
- 102000000574 RNA-Induced Silencing Complex Human genes 0.000 description 12
- 108010016790 RNA-Induced Silencing Complex Proteins 0.000 description 12
- 210000002540 macrophage Anatomy 0.000 description 12
- 208000010125 myocardial infarction Diseases 0.000 description 12
- 238000002054 transplantation Methods 0.000 description 12
- 102000004190 Enzymes Human genes 0.000 description 11
- 108090000790 Enzymes Proteins 0.000 description 11
- 241001465754 Metazoa Species 0.000 description 11
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 11
- 238000013459 approach Methods 0.000 description 11
- 229940088598 enzyme Drugs 0.000 description 11
- 238000010362 genome editing Methods 0.000 description 11
- 230000008595 infiltration Effects 0.000 description 11
- 238000001764 infiltration Methods 0.000 description 11
- 210000004165 myocardium Anatomy 0.000 description 11
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 11
- 108091007428 primary miRNA Proteins 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 11
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 10
- 108091034117 Oligonucleotide Proteins 0.000 description 10
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 10
- 238000012217 deletion Methods 0.000 description 10
- 230000037430 deletion Effects 0.000 description 10
- 230000003902 lesion Effects 0.000 description 10
- 230000037361 pathway Effects 0.000 description 10
- 102100023387 Endoribonuclease Dicer Human genes 0.000 description 9
- 101000907904 Homo sapiens Endoribonuclease Dicer Proteins 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 238000003776 cleavage reaction Methods 0.000 description 9
- 230000007423 decrease Effects 0.000 description 9
- 230000002255 enzymatic effect Effects 0.000 description 9
- 238000003780 insertion Methods 0.000 description 9
- 230000037431 insertion Effects 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 230000007017 scission Effects 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 239000013598 vector Substances 0.000 description 9
- 108020004705 Codon Proteins 0.000 description 8
- 210000001367 artery Anatomy 0.000 description 8
- 150000001875 compounds Chemical class 0.000 description 8
- 238000013461 design Methods 0.000 description 8
- 239000012997 ficoll-paque Substances 0.000 description 8
- 238000009472 formulation Methods 0.000 description 8
- 238000002955 isolation Methods 0.000 description 8
- 239000003550 marker Substances 0.000 description 8
- 210000000440 neutrophil Anatomy 0.000 description 8
- 230000006780 non-homologous end joining Effects 0.000 description 8
- 241000894007 species Species 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 102000004533 Endonucleases Human genes 0.000 description 7
- 108010042407 Endonucleases Proteins 0.000 description 7
- 206010061218 Inflammation Diseases 0.000 description 7
- 108091028043 Nucleic acid sequence Proteins 0.000 description 7
- 108010020764 Transposases Proteins 0.000 description 7
- 102000008579 Transposases Human genes 0.000 description 7
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 238000010494 dissociation reaction Methods 0.000 description 7
- 230000005593 dissociations Effects 0.000 description 7
- 230000034431 double-strand break repair via homologous recombination Effects 0.000 description 7
- 230000003828 downregulation Effects 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 239000012091 fetal bovine serum Substances 0.000 description 7
- 238000000684 flow cytometry Methods 0.000 description 7
- 238000002744 homologous recombination Methods 0.000 description 7
- 230000006801 homologous recombination Effects 0.000 description 7
- 230000001939 inductive effect Effects 0.000 description 7
- 230000004054 inflammatory process Effects 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 238000010172 mouse model Methods 0.000 description 7
- 230000007170 pathology Effects 0.000 description 7
- 239000013612 plasmid Substances 0.000 description 7
- 230000006798 recombination Effects 0.000 description 7
- 238000005215 recombination Methods 0.000 description 7
- 230000011664 signaling Effects 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 230000008685 targeting Effects 0.000 description 7
- 238000001262 western blot Methods 0.000 description 7
- 229910052725 zinc Inorganic materials 0.000 description 7
- 239000011701 zinc Substances 0.000 description 7
- 102000053602 DNA Human genes 0.000 description 6
- 238000002965 ELISA Methods 0.000 description 6
- 206010019280 Heart failures Diseases 0.000 description 6
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 6
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 6
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 6
- 206010028851 Necrosis Diseases 0.000 description 6
- 108091027967 Small hairpin RNA Proteins 0.000 description 6
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 6
- 150000001413 amino acids Chemical group 0.000 description 6
- 238000010171 animal model Methods 0.000 description 6
- 238000004113 cell culture Methods 0.000 description 6
- 230000004069 differentiation Effects 0.000 description 6
- 210000003743 erythrocyte Anatomy 0.000 description 6
- 210000000630 fibrocyte Anatomy 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- 230000017074 necrotic cell death Effects 0.000 description 6
- 230000001338 necrotic effect Effects 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- 102000004196 processed proteins & peptides Human genes 0.000 description 6
- 238000012545 processing Methods 0.000 description 6
- 108091008146 restriction endonucleases Proteins 0.000 description 6
- 238000001890 transfection Methods 0.000 description 6
- 238000013519 translation Methods 0.000 description 6
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- -1 Oct-3/4 Proteins 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 230000004071 biological effect Effects 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- 238000012258 culturing Methods 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 210000003754 fetus Anatomy 0.000 description 5
- 230000037433 frameshift Effects 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 229920002674 hyaluronan Polymers 0.000 description 5
- 229960003160 hyaluronic acid Drugs 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 230000002530 ischemic preconditioning effect Effects 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 210000001161 mammalian embryo Anatomy 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 239000003471 mutagenic agent Substances 0.000 description 5
- 231100000707 mutagenic chemical Toxicity 0.000 description 5
- 230000009437 off-target effect Effects 0.000 description 5
- 210000001778 pluripotent stem cell Anatomy 0.000 description 5
- 229920001184 polypeptide Polymers 0.000 description 5
- 102000005962 receptors Human genes 0.000 description 5
- 108020003175 receptors Proteins 0.000 description 5
- 239000004055 small Interfering RNA Substances 0.000 description 5
- 210000001082 somatic cell Anatomy 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 108091026890 Coding region Proteins 0.000 description 4
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 4
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 4
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 4
- 239000012981 Hank's balanced salt solution Substances 0.000 description 4
- 101100425753 Homo sapiens TNFRSF1A gene Proteins 0.000 description 4
- 101100425754 Mus musculus Tnfrsf1a gene Proteins 0.000 description 4
- 108010057163 Ribonuclease III Proteins 0.000 description 4
- 102000003661 Ribonuclease III Human genes 0.000 description 4
- 108091028113 Trans-activating crRNA Proteins 0.000 description 4
- 102000004142 Trypsin Human genes 0.000 description 4
- 108090000631 Trypsin Proteins 0.000 description 4
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 4
- 206010000891 acute myocardial infarction Diseases 0.000 description 4
- 210000004504 adult stem cell Anatomy 0.000 description 4
- 230000000735 allogeneic effect Effects 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 210000004271 bone marrow stromal cell Anatomy 0.000 description 4
- 210000004413 cardiac myocyte Anatomy 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 239000006285 cell suspension Substances 0.000 description 4
- 238000005520 cutting process Methods 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 239000008298 dragée Substances 0.000 description 4
- 230000030279 gene silencing Effects 0.000 description 4
- 239000008103 glucose Substances 0.000 description 4
- 238000001114 immunoprecipitation Methods 0.000 description 4
- 238000012744 immunostaining Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 210000003716 mesoderm Anatomy 0.000 description 4
- 210000004681 ovum Anatomy 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 102000040430 polynucleotide Human genes 0.000 description 4
- 108091033319 polynucleotide Proteins 0.000 description 4
- 239000002157 polynucleotide Substances 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 230000032361 posttranscriptional gene silencing Effects 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 230000010474 transient expression Effects 0.000 description 4
- 239000012588 trypsin Substances 0.000 description 4
- 238000011740 C57BL/6 mouse Methods 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 102000029816 Collagenase Human genes 0.000 description 3
- 108060005980 Collagenase Proteins 0.000 description 3
- 108010051219 Cre recombinase Proteins 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 108010046276 FLP recombinase Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 102000014150 Interferons Human genes 0.000 description 3
- 108010050904 Interferons Proteins 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 108700011259 MicroRNAs Proteins 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 101000648740 Mus musculus Tumor necrosis factor Proteins 0.000 description 3
- 240000007019 Oxalis corniculata Species 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 3
- 108010052160 Site-specific recombinase Proteins 0.000 description 3
- 108091023045 Untranslated Region Proteins 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 230000001464 adherent effect Effects 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 238000002659 cell therapy Methods 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 229960002424 collagenase Drugs 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 210000000805 cytoplasm Anatomy 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 230000005782 double-strand break Effects 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 210000003981 ectoderm Anatomy 0.000 description 3
- 238000004520 electroporation Methods 0.000 description 3
- 210000002257 embryonic structure Anatomy 0.000 description 3
- 210000001900 endoderm Anatomy 0.000 description 3
- 239000012894 fetal calf serum Substances 0.000 description 3
- 230000007045 gastrulation Effects 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 229940079322 interferon Drugs 0.000 description 3
- 208000028867 ischemia Diseases 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 230000005291 magnetic effect Effects 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 230000003278 mimic effect Effects 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 210000002894 multi-fate stem cell Anatomy 0.000 description 3
- 230000003505 mutagenic effect Effects 0.000 description 3
- 108091027963 non-coding RNA Proteins 0.000 description 3
- 102000042567 non-coding RNA Human genes 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 230000008186 parthenogenesis Effects 0.000 description 3
- 231100000915 pathological change Toxicity 0.000 description 3
- 230000036285 pathological change Effects 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000001172 regenerating effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 230000000392 somatic effect Effects 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 210000002303 tibia Anatomy 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 239000012096 transfection reagent Substances 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 210000000689 upper leg Anatomy 0.000 description 3
- 230000035899 viability Effects 0.000 description 3
- NUKQEEMKQGMUQH-UHFFFAOYSA-N 1-methyl-1-nitrosoguanidine Chemical compound O=NN(C)C(N)=N NUKQEEMKQGMUQH-UHFFFAOYSA-N 0.000 description 2
- 108020005345 3' Untranslated Regions Proteins 0.000 description 2
- 108020003589 5' Untranslated Regions Proteins 0.000 description 2
- 108091079001 CRISPR RNA Proteins 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 208000020446 Cardiac disease Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 230000004568 DNA-binding Effects 0.000 description 2
- PLUBXMRUUVWRLT-UHFFFAOYSA-N Ethyl methanesulfonate Chemical compound CCOS(C)(=O)=O PLUBXMRUUVWRLT-UHFFFAOYSA-N 0.000 description 2
- 108700024394 Exon Proteins 0.000 description 2
- 102000009123 Fibrin Human genes 0.000 description 2
- 108010073385 Fibrin Proteins 0.000 description 2
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 2
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 2
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 2
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 2
- 101000800116 Homo sapiens Thy-1 membrane glycoprotein Proteins 0.000 description 2
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 2
- 102000018251 Hypoxanthine Phosphoribosyltransferase Human genes 0.000 description 2
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 2
- 102000043136 MAP kinase family Human genes 0.000 description 2
- 108091054455 MAP kinase family Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 2
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 241000219061 Rheum Species 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 108010022394 Threonine synthase Proteins 0.000 description 2
- 102100033523 Thy-1 membrane glycoprotein Human genes 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 2
- 241000255993 Trichoplusia ni Species 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 101150063416 add gene Proteins 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 206010003119 arrhythmia Diseases 0.000 description 2
- 230000006793 arrhythmia Effects 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 230000003293 cardioprotective effect Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 230000019113 chromatin silencing Effects 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 210000004351 coronary vessel Anatomy 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 102000004419 dihydrofolate reductase Human genes 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 108010007093 dispase Proteins 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- 210000002308 embryonic cell Anatomy 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 230000004720 fertilization Effects 0.000 description 2
- 229950003499 fibrin Drugs 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 229960002963 ganciclovir Drugs 0.000 description 2
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 210000004392 genitalia Anatomy 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 210000001654 germ layer Anatomy 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 238000007901 in situ hybridization Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 210000005240 left ventricle Anatomy 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000009126 molecular therapy Methods 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 238000007481 next generation sequencing Methods 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 210000002826 placenta Anatomy 0.000 description 2
- 230000034190 positive regulation of NF-kappaB transcription factor activity Effects 0.000 description 2
- 230000000270 postfertilization Effects 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 230000035935 pregnancy Effects 0.000 description 2
- 238000004393 prognosis Methods 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- ZCCUUQDIBDJBTK-UHFFFAOYSA-N psoralen Chemical compound C1=C2OC(=O)C=CC2=CC2=C1OC=C2 ZCCUUQDIBDJBTK-UHFFFAOYSA-N 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 239000013608 rAAV vector Substances 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000008439 repair process Effects 0.000 description 2
- 238000003757 reverse transcription PCR Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 231100000241 scar Toxicity 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000035939 shock Effects 0.000 description 2
- 230000005783 single-strand break Effects 0.000 description 2
- 229940054269 sodium pyruvate Drugs 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 238000012033 transcriptional gene silencing Methods 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 230000036266 weeks of gestation Effects 0.000 description 2
- 210000004340 zona pellucida Anatomy 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- CXNPLSGKWMLZPZ-GIFSMMMISA-N (2r,3r,6s)-3-[[(3s)-3-amino-5-[carbamimidoyl(methyl)amino]pentanoyl]amino]-6-(4-amino-2-oxopyrimidin-1-yl)-3,6-dihydro-2h-pyran-2-carboxylic acid Chemical compound O1[C@@H](C(O)=O)[C@H](NC(=O)C[C@@H](N)CCN(C)C(N)=N)C=C[C@H]1N1C(=O)N=C(N)C=C1 CXNPLSGKWMLZPZ-GIFSMMMISA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- LFKLPJRVSHJZPL-UHFFFAOYSA-N 1,2:7,8-diepoxyoctane Chemical compound C1OC1CCCCC1CO1 LFKLPJRVSHJZPL-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- VXGRJERITKFWPL-UHFFFAOYSA-N 4',5'-Dihydropsoralen Natural products C1=C2OC(=O)C=CC2=CC2=C1OCC2 VXGRJERITKFWPL-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical compound BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 102000010565 Apoptosis Regulatory Proteins Human genes 0.000 description 1
- 108010063104 Apoptosis Regulatory Proteins Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 241000203069 Archaea Species 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108091007065 BIRCs Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 108020004513 Bacterial RNA Proteins 0.000 description 1
- 108700003785 Baculoviral IAP Repeat-Containing 3 Proteins 0.000 description 1
- 102100021677 Baculoviral IAP repeat-containing protein 2 Human genes 0.000 description 1
- 102100021662 Baculoviral IAP repeat-containing protein 3 Human genes 0.000 description 1
- 101100454433 Biomphalaria glabrata BG01 gene Proteins 0.000 description 1
- 101100454434 Biomphalaria glabrata BG04 gene Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 230000033616 DNA repair Effects 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 230000010558 Gene Alterations Effects 0.000 description 1
- 229940123611 Genome editing Drugs 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- MAJYPBAJPNUFPV-BQBZGAKWSA-N His-Cys Chemical compound SC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CN=CN1 MAJYPBAJPNUFPV-BQBZGAKWSA-N 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 1
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 1
- 101000608935 Homo sapiens Leukosialin Proteins 0.000 description 1
- 101000979342 Homo sapiens Nuclear factor NF-kappa-B p105 subunit Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 101000574648 Homo sapiens Retinoid-inducible serine carboxypeptidase Proteins 0.000 description 1
- 101100425757 Homo sapiens TNFRSF1B gene Proteins 0.000 description 1
- 101000801228 Homo sapiens Tumor necrosis factor receptor superfamily member 1A Proteins 0.000 description 1
- 101000801232 Homo sapiens Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 229920001612 Hydroxyethyl starch Polymers 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102100022338 Integrin alpha-M Human genes 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- 101150008942 J gene Proteins 0.000 description 1
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 102100030931 Ladinin-1 Human genes 0.000 description 1
- 101710177601 Ladinin-1 Proteins 0.000 description 1
- 102100039564 Leukosialin Human genes 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 241000699673 Mesocricetus auratus Species 0.000 description 1
- 101100095719 Mus musculus Sh2d2a gene Proteins 0.000 description 1
- 101100425758 Mus musculus Tnfrsf1b gene Proteins 0.000 description 1
- 208000029549 Muscle injury Diseases 0.000 description 1
- 201000002481 Myositis Diseases 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 1
- 206010067482 No adverse event Diseases 0.000 description 1
- 108020004485 Nonsense Codon Proteins 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 1
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 241000577979 Peromyscus spicilegus Species 0.000 description 1
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 1
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 1
- 102000009572 RNA Polymerase II Human genes 0.000 description 1
- 108010009460 RNA Polymerase II Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- 108010091086 Recombinases Proteins 0.000 description 1
- 102000018120 Recombinases Human genes 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- 102100025483 Retinoid-inducible serine carboxypeptidase Human genes 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 102000004389 Ribonucleoproteins Human genes 0.000 description 1
- 108010081734 Ribonucleoproteins Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 238000011579 SCID mouse model Methods 0.000 description 1
- 208000006117 ST-elevation myocardial infarction Diseases 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 241000193996 Streptococcus pyogenes Species 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 206010049418 Sudden Cardiac Death Diseases 0.000 description 1
- 101150052863 THY1 gene Proteins 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108700009124 Transcription Initiation Site Proteins 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 208000009982 Ventricular Dysfunction Diseases 0.000 description 1
- 208000033774 Ventricular Remodeling Diseases 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 210000004381 amniotic fluid Anatomy 0.000 description 1
- 230000003092 anti-cytokine Effects 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000037429 base substitution Effects 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- CXNPLSGKWMLZPZ-UHFFFAOYSA-N blasticidin-S Natural products O1C(C(O)=O)C(NC(=O)CC(N)CCN(C)C(N)=N)C=CC1N1C(=O)N=C(N)C=C1 CXNPLSGKWMLZPZ-UHFFFAOYSA-N 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000006931 brain damage Effects 0.000 description 1
- 231100000874 brain damage Toxicity 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 210000001715 carotid artery Anatomy 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 108020001778 catalytic domains Proteins 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 239000002962 chemical mutagen Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 230000002079 cooperative effect Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- CVSVTCORWBXHQV-UHFFFAOYSA-N creatine Chemical compound NC(=[NH2+])N(C)CC([O-])=O CVSVTCORWBXHQV-UHFFFAOYSA-N 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 101150047356 dec-1 gene Proteins 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 210000001840 diploid cell Anatomy 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000010573 double replacement reaction Methods 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000003090 exacerbative effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 210000000646 extraembryonic cell Anatomy 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 210000000604 fetal stem cell Anatomy 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 210000003953 foreskin Anatomy 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 230000005021 gait Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 230000037440 gene silencing effect Effects 0.000 description 1
- 238000012226 gene silencing method Methods 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 230000037442 genomic alteration Effects 0.000 description 1
- 102000005396 glutamine synthetase Human genes 0.000 description 1
- 108020002326 glutamine synthetase Proteins 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 229940064366 hespan Drugs 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 102000057041 human TNF Human genes 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 210000001822 immobilized cell Anatomy 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 210000004692 intercellular junction Anatomy 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000010468 interferon response Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 238000012977 invasive surgical procedure Methods 0.000 description 1
- 230000005865 ionizing radiation Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 208000037906 ischaemic injury Diseases 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 210000003127 knee Anatomy 0.000 description 1
- 238000011005 laboratory method Methods 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 201000002818 limb ischemia Diseases 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 210000003098 myoblast Anatomy 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000003061 neural cell Anatomy 0.000 description 1
- 230000037434 nonsense mutation Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 238000010397 one-hybrid screening Methods 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 230000002188 osteogenic effect Effects 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000001776 parthenogenetic effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000003169 placental effect Effects 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000000955 prescription drug Substances 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 238000012514 protein characterization Methods 0.000 description 1
- 235000004252 protein component Nutrition 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 108091007054 readthrough proteins Proteins 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 230000008672 reprogramming Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000003938 response to stress Effects 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 230000036573 scar formation Effects 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 210000001988 somatic stem cell Anatomy 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000035924 thermogenesis Effects 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 210000003014 totipotent stem cell Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 108091008023 transcriptional regulators Proteins 0.000 description 1
- 230000009752 translational inhibition Effects 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 210000000143 trophectoderm cell Anatomy 0.000 description 1
- 238000012920 two-step selection procedure Methods 0.000 description 1
- 210000003954 umbilical cord Anatomy 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 230000006815 ventricular dysfunction Effects 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 238000012070 whole genome sequencing analysis Methods 0.000 description 1
- 210000001325 yolk sac Anatomy 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Images











Classifications
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N1/00—Preservation of bodies of humans or animals, or parts thereof
- A01N1/10—Preservation of living parts
- A01N1/16—Physical preservation processes
- A01N1/162—Temperature processes, e.g. following predefined temperature changes over time
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/10—Cellular immunotherapy characterised by the cell type used
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/40—Cellular immunotherapy characterised by antigens that are targeted or presented by cells of the immune system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
- C07K14/7151—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons for tumor necrosis factor [TNF], for lymphotoxin [LT]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0663—Bone marrow mesenchymal stem cells (BM-MSC)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0669—Bone marrow stromal cells; Whole bone marrow
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K40/00
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K40/00 characterized by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K40/00
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K40/00 characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/80—Vectors containing sites for inducing double-stranded breaks, e.g. meganuclease restriction sites
Definitions
- the present invention in some embodiments thereof, relates to methods of treating an ischemic disease.
- Ischemic heart disease such as acute myocardial infarctions, congestive heart failure, arrhythmias, and sudden cardiac death, is the leading cause of morbidity and mortality in all industrialized nations.
- ischemic heart disease causes nearly 20 % of all deaths ( ⁇ 600,000 deaths each year).
- conventional medicine and surgery have offered many breakthroughs, resulting in a dramatic decline in mortality [ Mozaffarian et al. Circulation. (2015) 131(4):e29-322 .)
- the prognosis for patients who are admitted to hospital with heart failure remains poor, with a 5 year survival of about 50% and a 10 year survival of about 10 %.
- Tumor necrosis factor alpha is a pro-inflammatory cytokine that has been implicated in mediating or exacerbating various mammalian conditions such as myocardial infarction, heart failure, septic shock, inflammatory disease, HIV infection and tissue transplant.
- TNF ⁇ Tumor necrosis factor alpha
- soluble TNF receptor or an anti-TNF antibody was not beneficial to patients with heart failure.
- treatment with soluble TNF receptors significantly exacerbated ventricular dysfunction and remodeling with enhanced interstitial fibrosis after myocardial infarction (Monden et al [34]).
- TNF ⁇ initiates its biological effects by binding to two distinct cell surface receptors expressed in most cell types: TNFR1 (TNF ⁇ receptor type 1, with approximate mass of 55 kDa) and TNFR2 (TNF- ⁇ receptor type 2, with approximate mass of 75 kDa).
- TNFR1 TNF ⁇ receptor type 1
- TNFR2 TNF- ⁇ receptor type 2
- the specific roles of TNFR1 and TNFR2 signaling in ischemic damage remain unclear with several studies indicating that signaling via TNFR1 is deleterious and signaling via TNFR2 is protective against ischemic damage while others suggesting no difference in the function of the two receptors in protecting the heart from ischemia (see e.g. Kishore et al. [33], Monden et al [34], Moe et al 2004 [35] and Kurrelmeyer et al [37]).
- a method of treating an ischemic disease in a subject in need thereof comprising administering to the subject a therapeutically effective amount of mononuclear bone marrow cells (mnBMCs) comprising mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of the TNFR1, thereby treating the ischemic disease in the subject.
- mnBMCs mononuclear bone marrow cells
- MSCs mesenchymal stem cells
- a method of treating an ischemic disease in a subject in need thereof comprising administering to the subject a therapeutically effective amount of differentiated cells with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of the TNFR1, wherein the differentiated cells are of a same type of tissue affected in the ischemic disease, thereby treating the ischemic disease in the subject.
- the method further comprising treating the cells with reduced expression and/or activity of TNFR1 with TNF ⁇ prior to the administering.
- the method comprising cryopreserving the cells prior to the treating with the TNF ⁇ .
- a method of treating an ischemic disease in a subject in need thereof comprising:
- the cells comprise differentiated cells of a same type of tissue affected in the ischemic disease.
- the cells with reduced level of expression and/or activity of TNFR1 have the same level of expression and/or activity of TNFR2 as compared to the control stem cells.
- the cells with reduced expression and/or activity of TNFR1 are genetically modified cells.
- the genetically modified comprises genetically modified with a CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN) or meganuclease for downregulating expression of the TNFR1.
- ZFN Zinc finger nuclease
- TALEN transcription-activator like effector nuclease
- meganuclease for downregulating expression of the TNFR1.
- the genetically modified comprises genetically modified with a CRISPR/Cas system for downregulating expression of the TNFR1.
- a method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells comprising:
- the (i) is effected prior to the (ii).
- the agent does not downregulate expression and/or activity of TNFR2.
- the agent is selected from the group consisting of CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN), meganuclease, antisense and siRNA.
- ZFN Zinc finger nuclease
- TALEN transcription-activator like effector nuclease
- siRNA siRNA
- the agent comprises a CRISPR/Cas system.
- a method of treating an ischemic disease in a subject in need thereof comprising:
- the differentiated cells are of a same type of tissue affected in the ischemic disease.
- the method comprising cryopreserving the cells following the (i) and prior to the (ii).
- the cells are non-autologous to the subject.
- a method of treating an ischemic disease in a subject in need thereof comprising administering to the subject a therapeutically effective amount of a CRISPR/Cas system for downregulating expression of TNFR1, thereby treating the ischemic disease in the subject.
- a pharmaceutical composition comprising as an active ingredient differentiated cells or bone marrow stem cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1.
- a pharmaceutical composition comprising as active ingredients cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1 and at least 2 ng/ml TNF ⁇ .
- the CRISPR/Cas system does not downregulate expression of TNFR2.
- the cells comprise differentiated cells.
- the differentiated cells are cardiomyocytes.
- the cells comprise stem cells.
- the stem cells are selected from the group consisting of embryonic stem cells (ESCs), induced pluripotent stem cells (iPS), hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
- ESCs embryonic stem cells
- iPS induced pluripotent stem cells
- HSCs hematopoietic stem cells
- MSCs mesenchymal stem cells
- the stem cells comprise hematopoietic stem cells (HSCs).
- HSCs hematopoietic stem cells
- the cells are comprised in mononuclear bone marrow cells (mnBMCs).
- mnBMCs mononuclear bone marrow cells
- the mnBMCs comprise lymphocytes or progenitors thereof.
- the cells are obtained by density gradient centrifugation of bone marrow cells.
- the method comprising obtaining the cells by density gradient centrifugation of bone marrow cells.
- the cells are human cells.
- the cells are cryopreserved cells.
- the ischemic disease is ischemic heart disease.
- the ischemic heart disease is myocardial infarction.
- the ischemic heart disease is ischemic cardiomyopathy.
- the subject is not treated with TNF ⁇ .
- the present invention in some embodiments thereof, relates to methods of treating an ischemic disease.
- Ischemic heart disease is the leading cause of morbidity and mortality in all industrialized nations. Over the past half a century conventional medicine and surgery have offered many breakthroughs, resulting in a dramatic decline in mortality; however, despite the major advances in treatment, the prognosis for patients who are admitted to hospital with heart failure remains poor.
- the approach of using stem or precursor cells has emerged in the last decade as a regenerative strategy to address cardiac disease, with pre-clinical and clinical trials showing beneficial effects of progenitor cell therapy in acute myocardial infarction and ischemic cardiomyopathy.
- LAD left anterior descending artery
- some embodiments of the present teachings suggest that therapeutic cells with reduced level of expression and/or activity of TNFR1 can be used for treating an ischemic disease.
- a method of treating an ischemic disease in a subject in need thereof comprising administering to the subject a therapeutically effective amount of mononuclear bone marrow cells (mnBMCs) comprising mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, thereby treating the ischemic disease in the subject.
- mnBMCs mononuclear bone marrow cells
- MSCs mesenchymal stem cells
- lymphocytes or progenitors thereof with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, thereby treating the ischemic disease in the subject.
- a method of treating an ischemic disease in a subject in need thereof comprising administering to the subject a therapeutically effective amount of differentiated cells with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, wherein said differentiated cells are of a same type of tissue affected in said ischemic disease, thereby treating the ischemic disease in the subject.
- the method further comprising treating said cells with reduced expression and/or activity of TNFR1 with TNF ⁇ prior to said administering.
- a method of treating an ischemic disease in a subject in need thereof comprising:
- treating refers to inhibiting, preventing or arresting the development of a pathology (disease, disorder or medical condition e.g. ischemic disease e.g. ischemic heart disease) and/or causing the reduction, remission, or regression of a pathology or a symptom of a pathology.
- ischemic disease e.g. ischemic heart disease
- a pathology disease, disorder or medical condition e.g. ischemic disease e.g. ischemic heart disease
- ischemic disease e.g. ischemic heart disease
- ischemic disease refers to a disease characterized by reduced blood (and hence oxygen) supply to the diseased tissue/organ.
- the ischemic disease is an acute ischemic disease.
- the ischemic disease is a chronic ischemic disease.
- ischemic diseases include trauma, ischemic cerebrovascular disorder (such as apoplexy or cerebral infarction), ischemic renal disease, ischemic pulmonary disease, infection-related ischemic disease, ischemic disease of limbs, and ischemic heart disease.
- the ischemic disease is not a rejection reaction following transplantation (e.g. GVHD).
- the ischemic disease is ischemic heart disease.
- ischemic heart disease refers to a disease in which heart muscle is damaged or works inefficiently because of reduced blood (and hence oxygen) supply to the heart.
- Non-limiting Examples of ischemic heart diseases include ischemic cardiomyopathy, myocardial infarction or ischemic heart failure and chronic ischemic heart disease.
- the ischemic disease is myocardial infarction (also known as heart attack).
- the myocardial infarction is ST-segment elevation MI (STEMI) myocardial infarction (i.e. the coronary artery is completely blocked, thus virtually all of the heart muscle supplied by the affected artery becomes infarcted), as determined by ECG.
- ST-segment elevation MI STEMI
- ECG ECG
- the myocardial infarction is non-ST-segment elevation MI (NSTEMI) myocardial infarction (i.e. the artery is only partially occluded thus only a portion of the heart muscle supplied by the artery becomes infarcted), as determined by ECG.
- NSTEMI non-ST-segment elevation MI
- the ischemic disease is ischemic cardiomyopathy.
- the term "subject” includes mammals, e.g., human beings at any age and of any gender who suffer from the pathology (medical condition). According to specific embodiments, this term encompasses individuals who are at risk to develop the pathology.
- the subject is not treated with TNF ⁇ .
- TNF ⁇ a cytokine also known as Tumor necrosis factor alpha.
- TNF ⁇ can bind TNFR1 and TNFR2.
- the TNF ⁇ protein refers to the human protein, such as provided in the following GenBank Number NP_000585 (SEQ ID NO: 1).
- the term also encompasses functional homologues (naturally occurring or synthetically/recombinantly produced), orthologs (from other species) which exhibit the desired activity (i.e., binding TNFR1, binding TNFR2, activation of NF ⁇ B, activation of the MAPK pathway).
- Such homologues can be, for example, at least 80 %, at least 81 %, at least 82 %, at least 83 %, at least 84 %, at least 85 %, at least 86 %, at least 87 %, at least 88 %, at least 89 %, at least 90 %, at least 91 %, at least 92 %, at least 93 %, at least 94 %, at least 95 %, at least 96 %, at least 97 %, at least 98 %, at least 99 % or 100 % identical or homologous to the polypeptide SEQ ID NO: 1 or 80 %, at least 81 %, at least 82 %, at least 83 %, at least 84 %, at least 85 %, at least 86 %, at least 87 %, at least 88 %, at least 89 %, at least 90 %, at least 91 %, at least 92 %, at least
- Sequence identity or homology can be determined using any protein or nucleic acid sequence alignment algorithm such as Blast, ClustalW, and MUSCLE.
- the functional homologs also refer to functional portions of TNF ⁇ which maintain the activity of the full length protein (i.e. binding TNFR1, binding TNFR2, activation of NF ⁇ B, activation of the MAPK pathway).
- the TNF ⁇ is a recombinant TNF ⁇ , which can be prepared by standard recombinant expression methods or purchased commercially.
- TNF ⁇ can be commercially obtained from e.g. R&D Systems.
- the cells are treated (or contacted) with TNF ⁇ .
- the effect of treatment with TNF ⁇ is additive.
- the effect of treatment with TNF ⁇ is synergistic.
- treatment (or contacting) with TNF ⁇ is effected at a concentration of at least 2 ng/ml, at least 5 ng/ml, at least 10 ng/ml, at least 20 ng/ml, at least 30 ng/ml, at least 40 ng/ml, at least 50 ng/ml or at least 60 ng/ml, each possibility represents a separate embodiment of the present invention.
- treatment (or contacting) with TNF ⁇ is effected at a concentration of 2 - 200 ng/ml or 20 - 200 ng/ml.
- treatment (or contacting) with TNF ⁇ is effected 1-72 hours, 1-48, 1-24, 1-10 or 1-5 hours prior to the administering, each possibility represents a separate embodiment of the present invention.
- treatment (or contacting) with TNF ⁇ comprises a single TNF ⁇ treatment.
- treatment (or contacting) with TNF ⁇ comprises a plurality of TNF ⁇ treatments.
- the cells comprise differentiated cells.
- Non-limiting examples of differentiated cells that can be used with some embodiments of the present invention include differentiated cells derived from heart, kidney, liver, lung and brain.
- the differentiated cells comprise differentiated cells of a same type of tissue affected in the ischemic disease.
- a cell suspension may be obtained by any mechanical or chemical (e.g. enzymatic) means.
- Several methods exist for dissociating cell clusters to form cell suspensions e.g. single cell suspension
- cell suspensions e.g. single cell suspension
- aggregates e.g., physical forces (mechanical dissociation such as cell scraper, trituration through a narrow bore pipette, fine needle aspiration, vortex disaggregation and forced filtration through a fine nylon or stainless steel mesh), enzymes (enzymatic dissociation such as trypsin, collagenase, Acutase and the like ) or a combination of both.
- enzymatic digestion of tissue/organ into isolate cells can be performed by subjecting the tissue to an enzyme such as type IV Collagenase (Worthington biochemical corporation, Lakewood, NJ, USA) and/or Dispase (Invitrogen Corporation products, Grand Island NY, USA).
- the tissue may be enzyme digested by finely mincing tissue with a razor blade in the presence of e.g. collagenase, dispase and CaCl2 at 37 °C for about 1 hour.
- the method may further comprise removal of nonspecific debris from the resultant cell suspension by, for example, sequential filtration through filters (e.g. 70- and 40- ⁇ m filters), essentially as described under "General Materials and Experimental Methods" of the Examples section which follows.
- mechanical dissociation of tissue into isolated cells can be performed using a device designed to break the tissue to a predetermined size.
- a device designed to break the tissue to a predetermined size.
- mechanical dissociation can be manually performed using a needle such as a 27g needle (BD Microlance, Drogheda, Ireland) while viewing the tissue/cells under an inverted microscope.
- the dissociated cells are further broken to small clumps using 200 ⁇ l Gilson pipette tips (e.g., by pipetting up and down the cells).
- such differentiated cells can be obtained by differentiation of stem cell.
- the differentiated cells have been ex-vivo differentiated.
- the differentiated cells comprise cardiomyocytes.
- the differentiated cells comprise lymphocytes.
- the cells comprise bone marrow cells.
- bone marrow can be obtained from iliac crest, femora, tibiae, spine, rib or other medullar spaces.
- the desired cellular fraction can be purified from the bone marrow aspirates.
- the cells comprise mononuclear bone marrow cells (mnBMCs).
- the cells are comprised in mononuclear bone marrow cells (mnBMCs).
- mnBMCs mononuclear bone marrow cells
- the cells comprise at least 50 % at least 60 %, at least 70 %, at least 80 %, at least 90 % or at least 95 % mnBMCs.
- the mononuclear fraction can be purified from the bone marrow aspirates.
- methods and reagents known to those skilled in the art for purifying mnBMCs from bone marrow such as, density gradient centrifugation (e.g. ficoll), sedimentation (e.g. Hespan), centrifugal elutriation, fractionation, automated processes (e.g. Sepax ® from Biosafe SA, Eysins, Switzerland and the AutoXpress Platform ® (AXP) from Thermogenesis, Collinso Cordova, CA chemical lysis of e.g. red blood cells (e.g. by ACK), selection of specific cell types using cell surface markers (using e.g.
- FACS sorter or magnetic cell separation techniques such as are commercially available e.g. from Invitrogen, Stemcell Technologies, Cellpro, Advanced Magnetics, or Miltenyi Biotec.), and depletion of specific cell types by methods such as eradication (e.g. killing) with specific antibodies or by affinity based purification based on negative selection (using e.g. magnetic cell separation techniques, FACS sorter and/or capture ELISA labeling).
- eradication e.g. killing
- affinity based purification based on negative selection using e.g. magnetic cell separation techniques, FACS sorter and/or capture ELISA labeling.
- Such methods are described for example in THE HANDBOOK OF EXPERIMENTAL IMMUNOLOGY, Volumes 1 to 4, (D.N. Weir, edit or) and FLOW CYTOMETRY AND CELL SORTING (A. Radbruch, editor, Springer Verlag, 2000 ).
- the bone marrow cells are obtained by density gradient centrifugation (e.g. ficoll) of bone marrow cells.
- the bone marrow cells comprise stem cells (e.g. hematopoietic stem cells, mesenchymal stem cells). Methods of obtaining bone marrow stem cells are well known in the art and are further described hereinbelow.
- the bone marrow cells comprise mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof.
- the bone marrow cells comprise lymphocytes or progenitors thereof.
- lymphocytes progenitors refers to hematopoietic progenitor cells that can differentiate to lymphocytes.
- lymphocytes progenitor cells are mobilized to the peripheral blood by agents such as G-CSF with or without chemotherapy.
- the cells comprise stem cells.
- stem cells refers to cells which are capable of remaining in an undifferentiated state (e.g., totipotent, pluripotent or multipotent stem cells) for extended periods of time in culture until induced to differentiate into other cell types having a particular, specialized function (e.g., fully differentiated cells).
- Totipotent cells such as embryonic cells within the first couple of cell divisions after fertilization are the only cells that can differentiate into embryonic and extra-embryonic cells and are able to develop into a viable human being.
- pluripotent stem cells refers to cells which can differentiate into all three embryonic germ layers, i.e., ectoderm, endoderm and mesoderm or remaining in an undifferentiated state.
- the pluripotent stem cells include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPS).
- the multipotent stem cells include e.g. adult stem cells, hematopoietic stem cells and mesenchymal stem cells.
- undifferentiated stem cells are of a distinct morphology, which is clearly distinguishable from differentiated cells of embryo or adult origin by the skilled in the art. Typically, undifferentiated stem cells have high nuclear/cytoplasmic ratios, prominent nucleoli and compact colony formation with poorly discernable cell junctions. Additional features of undifferentiated stem cells are further described hereinunder.
- the stem cells are selected from the group consisting of embryonic stem cells (ESCs), induced pluripotent stem cells (iPS), hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
- ESCs embryonic stem cells
- iPS induced pluripotent stem cells
- HSCs hematopoietic stem cells
- MSCs mesenchymal stem cells
- the cells are embryonic stem cells (ESCs).
- ESCs embryonic stem cells
- embryonic stem cells refers to ESCs which are capable of differentiating into cells of all three embryonic germ layers (i.e., endoderm, ectoderm and mesoderm), or remaining in an undifferentiated state.
- embryonic stem cells may comprise cells which are obtained from the embryonic tissue formed after gestation (e.g., blastocyst) before implantation of the embryo (i.e., a pre-implantation blastocyst), extended blastocyst cells (EBCs) which are obtained from a post-implantation/pre-gastrulation stage blastocyst (see WO2006/040763 ), embryonic germ (EG) cells which are obtained from the genital tissue of a fetus any time during gestation, preferably before 10 weeks of gestation, and cells originating from an unfertilized ova which are stimulated by parthenogenesis (parthenotes).
- gestation e.g., blastocyst
- EBCs extended blastocyst cells
- EG embryonic germ
- the ESCs of some embodiments of the invention can be obtained using well-known cell-culture methods.
- human embryonic stem cells can be isolated from human blastocysts.
- Human blastocysts are typically obtained from human in vivo preimplantation embryos or from in vitro fertilized (IVF) embryos.
- IVF in vitro fertilized
- a single cell human embryo can be expanded to the blastocyst stage.
- the zona pellucida is removed from the blastocyst and the inner cell mass (ICM) is isolated by immunosurgery, in which the trophectoderm cells are lysed and removed from the intact ICM by gentle pipetting.
- ICM inner cell mass
- the ICM is then plated in a tissue culture flask containing the appropriate medium which enables its outgrowth. Following 9 to 15 days, the ICM derived outgrowth is dissociated into clumps either by a mechanical dissociation or by an enzymatic degradation and the cells are then re-plated on a fresh tissue culture medium. Colonies demonstrating undifferentiated morphology are individually selected by micropipette, mechanically dissociated into clumps, and re-plated. Resulting ES cells are then routinely split every 4-7 days. For further details on methods of preparation human ES cells see Thomson et al., [U.S. Pat. No. 5,843,780 ; Science 282: 1145, 1998 ; Curr. Top. Dev.
- Human ESCs can be purchased from the NIH human embryonic stem cells registry [Hypertext Transfer Protocol://grants (dot) nih (dot) gov/stem_cells/registry/current (dot) htm].
- Non-limiting examples of commercially available embryonic stem cell lines are BG01, BG02, BG03, BG04, CY12, CY30, CY92, CY10, TE03, TE32, CHB-4, CHB-5, CHB-6, CHB-8, CHB-9, CHB-10, CHB-11, CHB-12, HUES 1, HUES 2, HUES 3, HUES 4, HUES 5, HUES 6, HUES 7, HUES 8, HUES 9, HUES 10, HUES 11, HUES 12, HUES 13, HUES 14, HUES 15, HUES 16, HUES 17, HUES 18, HUES 19, HUES 20, HUES 21, HUES 22, HUES 23, HUES 24, HUES 25, HUES 26, HUES 27, HUES 28, CyT49, RUES3, WA01, UCSF4, NYUES1, NYUES2, NYUES3, NYUES4, NYUES5, NYUES6, NYUES7, UCLA 1, UCLA 2, UCLA 3, WA077 (H7)
- ESCs can be obtained from other species as well, including mouse (Mills and Bradley, 2001), golden hamster [ Doetschman et al., 1988, Dev Biol. 127: 224-7 ], rat [ Iannaccone et al., 1994, Dev Biol. 163: 288-92 ] rabbit [ Giles et al. 1993, Mol Reprod Dev. 36: 130-8 ; Graves & Moreadith, 1993, Mol Reprod Dev. 1993, 36: 424-33 ], several domestic animal species [ Notarianni et al., 1991, J Reprod Fertil Suppl. 43: 255-60 ; Wheeler 1994, Reprod Fertil Dev.
- EBCs Extended blastocyst cells
- EBCs can be obtained from a blastocyst of at least nine days post fertilization at a stage prior to gastrulation.
- the zona pellucida Prior to culturing the blastocyst, the zona pellucida is digested [for example by Tyrode's acidic solution (Sigma Aldrich, St Louis, MO, USA)] so as to expose the inner cell mass.
- the blastocysts are then cultured as whole embryos for at least nine and no more than fourteen days post fertilization ( i.e., prior to the gastrulation event) in vitro using standard embryonic stem cell culturing methods.
- EG cells are prepared from the primordial germ cells obtained from fetuses of about 8-11 weeks of gestation (in the case of a human fetus) using laboratory techniques known to anyone skilled in the arts.
- the genital ridges are dissociated and cut into small chunks which are thereafter disaggregated into cells by mechanical dissociation.
- the EG cells are then grown in tissue culture flasks with the appropriate medium.
- the cells are cultured with daily replacement of medium until a cell morphology consistent with EG cells is observed, typically after 7-30 days or 1-4 passages.
- Shamblott et al. [Proc. Natl. Acad. Sci. USA 95: 13726, 1998 ] and U.S. Pat. No. 6,090,622 .
- ESCs e.g., human ESCs
- parthenogenesis e.g., Zhenyu Lu et al., 2010. J. Assist Reprod. Genet. 27:285-291 ; "Derivation and long-term culture of human parthenogenetic embryonic stem cells using human foreskin feeders", which is fully incorporated herein by reference).
- Parthenogenesis refers to the initiation of cell division by activation of ova in the absence of sperm cells, for example using electrical or chemical stimulation.
- the activated ovum (parthenote) is capable of developing into a primitive embryonic structure (called a blastocyst) but cannot develop to term as the cells are pluripotent, meaning that they cannot develop the necessary extra-embryonic tissues (such as amniotic fluid) needed for a viable human fetus.
- the cells are embryonic stem cells Induced pluripotent stem cells (iPS).
- iPS embryonic stem cells Induced pluripotent stem cells
- Induced pluripotent stem cells are cells obtained by de-differentiation of adult somatic cells which are endowed with pluripotency (i.e., being capable of differentiating into the three embryonic germ cell layers, i.e., endoderm, ectoderm and mesoderm).
- pluripotency i.e., being capable of differentiating into the three embryonic germ cell layers, i.e., endoderm, ectoderm and mesoderm.
- such cells are obtained from a differentiated tissue (e.g., a somatic tissue such as skin) and undergo de-differentiation by genetic manipulation which re-program the cell to acquire embryonic stem cells characteristics.
- the induced pluripotent stem cells are formed by inducing the expression of Oct-4, Sox2, Kfl4 and c-Myc in a somatic stem cell.
- iPS Induced pluripotent stem cells
- somatic cells can be generated from somatic cells by genetic manipulation of somatic cells, e.g., by retroviral transduction of somatic cells such as fibroblasts, hepatocytes, gastric epithelial cells with transcription factors such as Oct-3/4, Sox2, c-Myc, and KLF4
- somatic cells such as fibroblasts, hepatocytes, gastric epithelial cells with transcription factors such as Oct-3/4, Sox2, c-Myc, and KLF4
- adult stem cells also called “tissue stem cells” or a stem cell from a somatic tissue refers to any stem cell derived from a somatic tissue [of either a postnatal or prenatal animal (especially the human)].
- the adult stem cell is generally thought to be a multipotent stem cell, capable of differentiation into multiple cell types.
- Adult stem cells can be derived from any adult, neonatal or fetal tissue such as adipose tissue, skin, kidney, liver, prostate, pancreas, intestine, bone marrow and placenta.
- isolation of adult tissue stem cells is based on the discrete location (or niche) of each cell type included in the adult tissue, i.e., the stem cells, the transit amplifying cells and the terminally differentiated cells [ Potten, C. S. and Morris, R. J. (1988). Epithelial stem cells in vivo. J. Cell Sci. Suppl. 10, 45-62 ].
- HSCs Hematopoietic stem cells
- adult tissue stem cells include stem cells obtained from blood or bone marrow tissue of an individual at any age or from cord blood of a newborn individual.
- the cells comprise HSCs.
- hematopoietic stem cell is a CD34+ cell.
- CD34+ cell refers to a hematopoietic stem cell positive for the CD34 marker that can differentiate to each of the cell types in the blood, i.e.; the myeloid (monocyte, macrophage, neutrophil, basophil, eosinophil, erythrocyte, megakaryocyte, dendritic cell) or lymphoid (T cell, B cell, NK cell) lineages.
- myeloid monocyte, macrophage, neutrophil, basophil, eosinophil, erythrocyte, megakaryocyte, dendritic cell
- T cell B cell, NK cell
- the HSCs may be a specific cell line, alternatively may be generated from iPS or embryonic stem cells [see for example Pick M et al. (2007) Stem Cells, 25(9): 2206-14 ; and Pick M et al. (2013) PLoS One, 8(2): e55530 ] or alternatively may be isolated using various methods known in the arts such as those disclosed by " Handbook of Stem Cells" edit by Robert Lanze, Elsevier Academic Press, 2004, Chapter 54, pp609-614 , isolation and characterization of hematopoietic stem cells, by Gerald J Spangrude and William B Stayton.
- HSCs can be isolated from cord blood, peripheral blood or BM samples by means of density gradient centrifugation using for example Ficoll-Paque (can be obtained from GE Healthcare Bio-Science AB) followed by immunomagnetic or immunofluorescent methods (such as Diamond or Microbeads CD34+ isolation kit obtained from Miltenyi Biotech). Purity of the purified fraction can be assessed by flow cytometry for the specified markers (for example CD34).
- Ficoll-Paque can be obtained from GE Healthcare Bio-Science AB
- immunomagnetic or immunofluorescent methods such as Diamond or Microbeads CD34+ isolation kit obtained from Miltenyi Biotech.
- Purity of the purified fraction can be assessed by flow cytometry for the specified markers (for example CD34).
- the HSCs are mobilized to the peripheral blood by agents such as G-CSF with or without chemotherapy.
- Placental and cord blood stem cells may also be referred to as "young stem cells”.
- Fetal stem cells can be isolated using various methods known in the art such as those disclosed by Eventov-Friedman S, et al., PLoS Med. 2006, 3: e215 ; Eventov-Friedman S, et al., Proc Natl Acad Sci USA. 2005, 102: 2928-33 ; Dekel B, et al., 2003, Nat Med. 9: 53-60 ; and Dekel B, et al., 2002, J. Am. Soc. Nephrol. 13: 977-90 .
- the cells comprise mesenchymal stem cells (MSCs).
- MSCs mesenchymal stem cells
- Mesenchymal stem cells give rise to one or more mesenchymal tissues (e.g., adipose, osseous, cartilaginous, elastic and fibrous connective tissues, myoblasts) as well as to tissues other than those originating in the embryonic mesoderm (e.g., neural cells) depending upon various influences from bioactive factors such as cytokines.
- mesenchymal tissues e.g., adipose, osseous, cartilaginous, elastic and fibrous connective tissues, myoblasts
- tissues other than those originating in the embryonic mesoderm e.g., neural cells
- bioactive factors such as cytokines.
- Methods of isolating, purifying and expanding MSCs are known in the art and include, for example, those disclosed by Caplan and Haynesworth in U.S. Pat. No. 5,486,359 and Jones E.A. et al., 2002, Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells, Arthritis Rheum. 46(12): 3349-60 .
- MSC cultures are generated by diluting bone marrow aspirates (usually 20 ml) with equal volumes of Hank's balanced salt solution (HBSS; GIBCO Laboratories, Grand Island, NY, USA) and layering the diluted cells over about 10 ml of a Ficoll column (Ficoll-Paque; Pharmacia, Piscataway, NJ, USA). Following 30 minutes of centrifugation at 2,500 x g, the mononuclear cell layer is removed from the interface and suspended in HBSS.
- HBSS Hank's balanced salt solution
- MSCs are then centrifuged at 1,500 x g for 15 minutes and resuspended in a complete medium (MEM, ⁇ medium without deoxyribonucleotides or ribonucleotides; GIBCO); 2-20 % fetal calf serum (FCS) derived from a lot selected for rapid growth of MSCs (Atlanta Biologicals, Norcross, GA); 100 units/ml penicillin (GIBCO), 100 ⁇ g/ml streptomycin (GIBCO); and 2 mM L-glutamine (GIBCO).
- MEM complete medium
- FCS fetal calf serum
- Resuspended cells are plated in about 25 ml of medium in a 10 cm culture dish (Corning Glass Works, Corning, NY) and incubated at 37 °C with 5 % humidified CO 2 . Following 24 hours in culture, nonadherent cells are discarded, and the adherent cells are thoroughly washed twice with phosphate buffered saline (PBS). The medium is replaced with a fresh complete medium every 3 or 4 days for about 14 days. Adherent cells are then harvested with 0.25 % trypsin and 1 mM EDTA (Trypsin/EDTA, GIBCO) for 5 min at 37 °C, replated in a 6-cm plate and cultured for another 14 days.
- Trypsin/EDTA GIBCO
- Cells are then trypsinized and counted using a cell counting device such as for example, a hemocytometer (Hausser Scientific, Horsham, PA). Cultured cells are recovered by centrifugation and resuspended with 5 % DMSO and 30 % FCS at a concentration of 1 to 2 ⁇ 10 6 cells per ml. Aliquots of about 1 ml each are slowly frozen and stored in liquid nitrogen.
- a cell counting device such as for example, a hemocytometer (Hausser Scientific, Horsham, PA).
- MSC cultures are resuspended in a complete medium and plated at a concentration of about 5,000 cells/cm 2 . Following 24 hours in culture, nonadherent cells are removed and the adherent cells are harvested using Trypsin/EDTA, dissociated by passage through a narrowed Pasteur pipette, and preferably replated at a density of about 1.5 to about 3.0 cells/cm 2 . Under these conditions, MSC cultures can grow for about 50 population doublings and be expanded for about 2000 fold [ Colter DC., et al. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc Natl Acad Sci USA. 97: 3213-3218, 2000 ].
- MSC cultures utilized by some embodiments of the invention include three groups of cells which are defined by their morphological features: small and agranular cells (referred to as RS-1, hereinbelow), small and granular cells (referred to as RS-2, hereinbelow) and large and moderately granular cells (referred to as mature MSCs, hereinbelow).
- RS-1 small and agranular cells
- RS-2 small and granular cells
- mature MSCs large and moderately granular cells
- MSCs When MSCs are cultured under the culturing conditions of some embodiments of the invention they exhibit negative staining for the HSC markers CD34, CD11B, CD43 and CD45. A small fraction of cells (less than 10 %) are dimly positive for CD31 and/or CD38 markers. In addition, mature MSCs are dimly positive for the HSC marker, CD117 (c-Kit), moderately positive for the osteogenic MSCs marker, Stro-1 [ Simmons, P. J. & Torok-Storb, B. (1991). Blood 78, 5562 ] and positive for the thymocytes and peripheral T lymphocytes marker, CD90 (Thy-1). On the other hand, the RS-1 cells are negative for the CD117 and Stro1 markers and are dimly positive for the CD90 marker, and the RS-2 cells are negative for all of these markers.
- the cells of some embodiments of the present invention can be a primary cell (non-cultured and alternatively or additionally non-immortalized cell) or a cell-line.
- the cells used and/or obtained according to the present invention can be freshly isolated, stored e.g., cryopreserved (i.e. frozen) at e.g. liquid nitrogen temperature at any stage (e.g. following their retrieval, following contacting with the agent, following treatment with TNF ⁇ ) for long periods of time (e.g., months, years) for future use; and cell lines.
- cryopreserved i.e. frozen
- liquid nitrogen temperature e.g. liquid nitrogen temperature
- any stage e.g. following their retrieval, following contacting with the agent, following treatment with TNF ⁇
- long periods of time e.g., months, years
- the cells are cryopreserved cells.
- the cells are cryopreserved prior to contacting with the agent which downregulates expression and/or activity of TNFR1.
- the cells are cryopreserved following contacting with the agent which downregulates expression and/or activity of TNFR1.
- the cells are cryopreserved prior to treatment with TNF ⁇ .
- the cells are cryopreserved following treatment with TNF ⁇ .
- the cells are cryopreserved prior to administering to the subject.
- the cells obtained according to the present invention can be stored in a cell bank or a depository or storage facility.
- the cells are freshly isolated (i.e., not subjected to preservation processes).
- the cells used according to specific embodiments of the present invention may be autologous or non-autologous; they can be syngeneic or non-synegeneic: allogeneic or xenogeneic to the subject; each possibility represents a separate embodiment of the present invention.
- the cells are autologous to said subject.
- autologous means that the donor subject is the recipient subject.
- the cells have been removed and re-introduced e.g., re-infused to the subject.
- the cells are non-autologous to said subject.
- non-autologous means that the donor subject is not the recipient subject.
- syngeneic means that the donor subject is essentially genetically identical with the recipient subject.
- Examples of syngeneic transplantation include transplantation of cells derived from the subject (also referred to in the art as “autologous”), a clone of the subject, or a homozygotic twin of the subject
- allogeneic means that the donor is of the same species as the recipient, but which is substantially non-clonal with the recipient. Typically, outbred, non-zygotic twin mammals of the same species are allogeneic with each other. It will be appreciated that an allogeneic donor may be HLA identical or HLA non-identical with respect to the subject
- xenogeneic means that the donor subject is from a different species relative to the recipient subject.
- the cells are mammalian cells.
- the cells are primate cells.
- the cells are human cells.
- the cells are rodent cells (e.g. mouse, rat).
- the cells of some embodiments of the present invention are characterized by reduced level of expression and/or activity of TNFR1.
- TNFR1 Tumor necrosis factor receptor 1
- TNFRSF1A tumor necrosis factor receptor superfamily member 1A
- CD120a and p55 refers to the TNFR1 gene and to its polynucleotide or polypeptide expression product.
- the TNFR1 refers to the human TNFR1, such as provided in Gene ID: 7132 (SEQ ID NO: 2) and the following Accession Numbers: NM_001065 (SEQ ID NO: 3), NM_001346091 (SEQ ID NO: 4), NM_001346092 (SEQ ID NO: 5), NP_001056 (SEQ ID NO: 6), NP_001333020 (SEQ ID NO: 7) and NP_001333021 (SEQ ID NO: 8).
- the TNFR1 refers to the mouse TNFR1, such as provided in Gene ID: 21937 (SEQ ID NO: 9) and in the following Accession Numbers: NM_011609 (SEQ ID NO: 10) and NP_035739 (SEQ ID NO: 11).
- TNFR1 activity is at least one of (or two of or all of): binding TNF ⁇ , forming a trimer, activating the transcription factor NF ⁇ B and/or mediating apoptosis.
- reduced level of expression and/or activity refers to a decrease of at least 10 % in TNFR1 expression or activity in comparison to a control cell of the same origin which was not contacted with an agent which downregulates expression and/or activity of TNFR1, as may be determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNF ⁇ signaling assays.
- the decrease is in at least 20 %, 30 %, 40 % or even higher say, 50 %, 60 %, 70 %, 80 %, 90 % or even 100 %.
- the cell does not express TNFR1.
- the cells with reduced expression and/or activity of TNFR1 are genetically modified cells.
- Methods and agents for genetically modifying cells are well known in the art and are further described in details hereinbelow.
- the cells are genetically modified with a CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN) or meganuclease for downregulating expression of said TNFR1.
- ZFN Zinc finger nuclease
- TALEN transcription-activator like effector nuclease
- meganuclease for downregulating expression of said TNFR1.
- the cells are genetically modified with a CRISPR/Cas system for downregulating expression of said TNFR1.
- the cells with reduced level of expression and/or activity of TNFR1 express similar levels and/or activity of TNFR2 in comparison to a control cell of the same origin which was not contacted with an agent which downregulates expression and/or activity of TNFR1, as may be determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNF ⁇ signaling assays.
- TNFR2 Tumor necrosis factor receptor 2
- TNFRSF1B tumor necrosis factor receptor superfamily member 1B
- CD120b and p75 refers to the TNFR2 gene and to its polynucleotide or polypeptide expression product.
- the TNFR2 refers to the human TNFR2, such as provided in Gene ID: 7133 (SEQ ID NO: 12) and the following Accession Numbers: NM_001066 (SEQ ID NO: 13) and NP_001057 (SEQ ID NO: 14).
- the TNFR1 refers to the mouse TNFR2, such as provided in Gene ID: 21938 (SEQ ID NO: 15) and in the following Accession Numbers: NM_011610 (SEQ ID NO: 16) and NP_035740 (SEQ ID NO: 17).
- TNFR2 activity is at least one of (or two of or all of): binding TNF ⁇ and/or mediating the recruitment of two anti-apoptotic proteins, c-IAP1 and c-IAP2.
- the present invention also contemplates methods of obtaining such cells.
- a method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells comprising:
- a method of downregulating expression and/or activity of TNFR1 in differentiated cells comprising:
- inducing differentiation of the stem cells is effected prior to contacting with the agent.
- Methods of inducing differentiation of stem cells are known in the art and are further described hereinabove.
- a method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells comprising genetically modifying ex-vivo or in-vitro differentiated cells or bone marrow stem cells with an agent which downregulates expression and/or activity of TNFR1.
- the method further comprising contacting the cells ex-vivo or in-vitro with TNF ⁇ .
- contacting the cells with the agent is effected prior to contacting the cells with the TNF ⁇ .
- contacting the cells with the agent is effected following contacting the cells with the TNF ⁇ .
- Specific embodiments of the present invention contemplates a method of treating an ischemic disease in a subject in need thereof, the method comprising:
- downstreamregulating expression and/or activity and “downregulates expression and/or activity” refer to a decrease of at least 5 % in expression and/or biological function in the presence of the agent in comparison to same in the absence of the agent, as determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNF ⁇ signaling assays.
- the decrease is in at least 10 %, 30 %, 40 % or even higher say, 50 %, 60 %, 70 %, 80 %, 90 % or even 100 %.
- the decrease is at least 1.5 fold, at least 2 fold, at least 3 fold, at least 5 fold, at least 10 fold, or at least 20 fold as compared to same in the absence of the agent.
- downregulating expression refers to the absence of mRNA and/or protein, as detected by RT-PCR or Western blot, respectively.
- downregulating expression and/or activity of TNFR1 and “downregulates expression and/or activity of TNFR1 "refer to the ability to specifically downregulate the expression and/or activity of TNFR1 and not to downregulate the expression and/or activity of TNFR2, as determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNF ⁇ signaling assays.
- This selective inhibition can be manifested as higher affinity (e.g., K d ) of the agent to TNFR1 than to TNFR2.
- Increased affinity can be of at least 5, 10, 100, 1000 or 10000 fold.
- the agent does not downregulate expression and/or activity of TNFR2.
- Downregulating expression and/or activity can be effected at the genomic (e.g. homologous recombination and site specific endonucleases) and/or the transcript level using a variety of molecules which interfere with transcription and/or translation (e.g., RNA silencing agents) of a target expression product described herein, but may also be effected at the protein level (e.g., antibodies, small molecules, inhibitory peptides, enzymes that cleave the polypeptide, aptamers and the like).
- the agent directly binds TNFR1.
- the agent indirectly binds TNFR1 by acting through an intermediary molecule, for example the agent binds to or modulates a molecule that in turn binds to or modulates the target.
- the agent does not bind TNFR2.
- Down-regulation at the nucleic acid level is typically effected using a nucleic acid agent, having a nucleic acid backbone, DNA, RNA, mimetics thereof or a combination of same.
- the nucleic acid agent may be encoded from a DNA molecule or provided to the cell per se.
- Downregulation can be achieved by inactivating the gene (i.e. the TNFR1 gene) via introducing targeted mutations involving loss-of function alterations (e.g. point mutations, deletions and insertions) in the gene structure.
- the gene i.e. the TNFR1 gene
- targeted mutations involving loss-of function alterations e.g. point mutations, deletions and insertions
- loss-of-function alterations refers to any mutation in the DNA sequence of a gene which results in downregulation of the expression level and/or activity of the expressed product, i.e., the mRNA transcript and/or the translated protein.
- Non-limiting examples of such loss-of-function alterations include a missense mutation, i.e., a mutation which changes an amino acid residue in the protein with another amino acid residue and thereby abolishes the enzymatic activity of the protein; a nonsense mutation, i.e., a mutation which introduces a stop codon in a protein, e.g., an early stop codon which results in a shorter protein devoid of the enzymatic activity; a frame-shift mutation, i.e ., a mutation, usually, deletion or insertion of nucleic acid(s) which changes the reading frame of the protein, and may result in an early termination by introducing a stop codon into a reading frame (e.g., a truncated protein, devoid of the enzymatic activity), or in a longer amino acid sequence (e.g., a readthrough protein) which affects the secondary or tertiary structure of the protein and results in a non-functional protein, devoid of the enzymatic
- loss-of-function alteration of a gene may comprise at least one allele of the gene.
- allele refers to any of one or more alternative forms of a gene locus, all of which alleles relate to a trait or characteristic. In a diploid cell or organism, the two alleles of a given gene occupy corresponding loci on a pair of homologous chromosomes.
- loss-of-function alteration of a gene comprises both alleles of the gene.
- the TNFR1 may be in a homozygous form or in a heterozygous form.
- Genome Editing using engineered endonucleases - this approach refers to a reverse genetics method using artificially engineered nucleases to cut and create specific double-stranded breaks at a desired location(s) in the genome, which are then repaired by cellular endogenous processes such as, homology directed repair (HDR) and non-homologous end-joining (NHEJ).
- HDR homology directed repair
- NHEJ directly joins the DNA ends in a double-stranded break
- HDR utilizes a homologous sequence as a template for regenerating the missing DNA sequence at the break point.
- a DNA repair template containing the desired sequence must be present during HDR.
- Genome editing cannot be performed using traditional restriction endonucleases since most restriction enzymes recognize a few base pairs on the DNA as their target and the probability is very high that the recognized base pair combination will be found in many locations across the genome resulting in multiple cuts not limited to a desired location.
- restriction enzymes recognize a few base pairs on the DNA as their target and the probability is very high that the recognized base pair combination will be found in many locations across the genome resulting in multiple cuts not limited to a desired location.
- ZFNs Zinc finger nucleases
- TALENs transcription-activator like effector nucleases
- CRISPR/Cas system CRISPR/Cas system.
- Meganucleases are commonly grouped into four families: the LAGLIDADG family, the GIY-YIG family, the His-Cys box family and the HNH family. These families are characterized by structural motifs, which affect catalytic activity and recognition sequence. For instance, members of the LAGLIDADG family are characterized by having either one or two copies of the conserved LAGLIDADG motif. The four families of meganucleases are widely separated from one another with respect to conserved structural elements and, consequently, DNA recognition sequence specificity and catalytic activity. Meganucleases are found commonly in microbial species and have the unique property of having very long recognition sequences (>14bp) thus making them naturally very specific for cutting at a desired location.
- Meganucleases can be designed using the methods described in e.g., Certo, MT et al. Nature Methods (2012) 9:073-975 ; U.S. Patent Nos. 8,304,222 ; 8,021,867 ; 8, 119,381 ; 8, 124,369 ; 8, 129,134 ; 8,133,697 ; 8,143,015 ; 8,143,016 ; 8, 148,098 ; or 8, 163,514 , the contents of each are incorporated herein by reference in their entirety.
- meganucleases with site specific cutting characteristics can be obtained using commercially available technologies e.g., Precision Biosciences' Directed Nuclease Editor TM genome editing technology.
- ZFNs and TALENs Two distinct classes of engineered nucleases, zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), have both proven to be effective at producing targeted double-stranded breaks (Christian et al., 2010; Kim et al., 1996; Li et al., 2011; Mahfouz et al., 2011; Miller et al., 2010).
- ZFNs and TALENs restriction endonuclease technology utilizes a non-specific DNA cutting enzyme which is linked to a specific DNA binding domain (either a series of zinc finger domains or TALE repeats, respectively).
- a restriction enzyme whose DNA recognition site and cleaving site are separate from each other is selected. The cleaving portion is separated and then linked to a DNA binding domain, thereby yielding an endonuclease with very high specificity for a desired sequence.
- An exemplary restriction enzyme with such properties is Fokl. Additionally Fokl has the advantage of requiring dimerization to have nuclease activity and this means the specificity increases dramatically as each nuclease partner recognizes a unique DNA sequence.
- Fokl nucleases have been engineered that can only function as heterodimers and have increased catalytic activity.
- the heterodimer functioning nucleases avoid the possibility of unwanted homodimer activity and thus increase specificity of the double-stranded break.
- ZFNs and TALENs are constructed as nuclease pairs, with each member of the pair designed to bind adjacent sequences at the targeted site.
- the nucleases bind to their target sites and the FokI domains heterodimerize to create a double-stranded break. Repair of these double-stranded breaks through the non-homologous end-joining (NHEJ) pathway most often results in small deletions or small sequence insertions. Since each repair made by NHEJ is unique, the use of a single nuclease pair can produce an allelic series with a range of different deletions at the target site.
- NHEJ non-homologous end-joining
- deletions typically range anywhere from a few base pairs to a few hundred base pairs in length, but larger deletions have successfully been generated in cell culture by using two pairs of nucleases simultaneously (Carlson et al., 2012; Lee et al., 2010).
- the double-stranded break can be repaired via homology directed repair to generate specific modifications (Li et al., 2011; Miller et al., 2010; Urnov et al., 2005).
- ZFNs rely on Cys2- His2 zinc fingers and TALENs on TALEs. Both of these DNA recognizing peptide domains have the characteristic that they are naturally found in combinations in their proteins. Cys2-His2 Zinc fingers typically found in repeats that are 3 bp apart and are found in diverse combinations in a variety of nucleic acid interacting proteins. TALEs on the other hand are found in repeats with a one-to-one recognition ratio between the amino acids and the recognized nucleotide pairs.
- Zinc fingers correlated with a triplet sequence are attached in a row to cover the required sequence
- OPEN low-stringency selection of peptide domains vs. triplet nucleotides followed by high-stringency selections of peptide combination vs. the final target in bacterial systems
- ZFNs can also be designed and obtained commercially from e.g., Sangamo Biosciences TM (Richmond, CA).
- TALEN Method for designing and obtaining TALENs are described in e.g. Reyon et al. Nature Biotechnology 2012 May;30(5):460-5 ; Miller et al. Nat Biotechnol. (2011) 29: 143-148 ; Cermak et al. Nucleic Acids Research (2011) 39 (12): e82 and Zhang et al. Nature Biotechnology (2011) 29 (2): 149-53 .
- a recently developed web-based program named Mojo Hand was introduced by Mayo Clinic for designing TAL and TALEN constructs for genome editing applications (can be accessed through www(dot)talendesign(dot)org).
- TALEN can also be designed and obtained commercially from e.g., Sangamo Biosciences TM (Richmond, CA).
- CRISPR-Cas system Many bacteria and archaea contain endogenous RNA-based adaptive immune systems that can degrade nucleic acids of invading phages and plasmids. These systems consist of clustered regularly interspaced short palindromic repeat (CRISPR) genes that produce RNA components and CRISPR associated (Cas) genes that encode protein components.
- CRISPR RNAs crRNAs
- crRNAs contain short stretches of homology to specific viruses and plasmids and act as guides to direct Cas nucleases to degrade the complementary nucleic acids of the corresponding pathogen.
- RNA/protein complex RNA/protein complex and together are sufficient for sequence-specific nuclease activity: the Cas9 nuclease, a crRNA containing 20 base pairs of homology to the target sequence, and a trans-activating crRNA (tracrRNA) ( Jinek et al. Science (2012) 337: 816-821 .). It was further demonstrated that a synthetic chimeric guide RNA (gRNA) composed of a fusion between crRNA and tracrRNA could direct Cas9 to cleave DNA targets that are complementary to the crRNA in vitro.
- gRNA synthetic chimeric guide RNA
- transient expression of Cas9 in conjunction with synthetic gRNAs can be used to produce targeted double-stranded brakes in a variety of different species (Cho et al., 2013; Cong et al., 2013; DiCarlo et al., 2013; Hwang et al., 2013a,b; Jinek et al., 2013; Mali et al., 2013).
- the CRIPSR/Cas system for genome editing contains two distinct components: a gRNA and an endonuclease e.g. Cas9.
- the gRNA is typically a 20 nucleotide sequence encoding a combination of the target homologous sequence (crRNA) and the endogenous bacterial RNA that links the crRNA to the Cas9 nuclease (tracrRNA) in a single chimeric transcript.
- the gRNA/Cas9 complex is recruited to the target sequence by the base-pairing between the gRNA sequence and the complement genomic DNA.
- the genomic target sequence must also contain the correct Protospacer Adjacent Motif (PAM) sequence immediately following the target sequence.
- PAM Protospacer Adjacent Motif
- the binding of the gRNA/Cas9 complex localizes the Cas9 to the genomic target sequence so that the Cas9 can cut both strands of the DNA causing a double-strand break.
- the double-stranded brakes produced by CRISPR/Cas can undergo homologous recombination or NHEJ.
- the Cas9 nuclease has two functional domains: RuvC and HNH, each cutting a different DNA strand. When both of these domains are active, the Cas9 causes double strand breaks in the genomic DNA.
- CRISPR/Cas A significant advantage of CRISPR/Cas is that the high efficiency of this system coupled with the ability to easily create synthetic gRNAs enables multiple genes to be targeted simultaneously. In addition, the majority of cells carrying the mutation present biallelic mutations in the targeted genes.
- 'nickases Modified versions of the Cas9 enzyme containing a single inactive catalytic domain, either RuvC- or HNH-, are called 'nickases'. With only one active nuclease domain, the Cas9 nickase cuts only one strand of the target DNA, creating a single-strand break or 'nick'. A single-strand break, or nick, is normally quickly repaired through the HDR pathway, using the intact complementary DNA strand as the template. However, two proximal, opposite strand nicks introduced by a Cas9 nickase are treated as a double-strand break, in what is often referred to as a 'double nick' CRISPR system.
- a double-nick can be repaired by either NHEJ or HDR depending on the desired effect on the gene target.
- using the Cas9 nickase to create a double-nick by designing two gRNAs with target sequences in close proximity and on opposite strands of the genomic DNA would decrease off-target effect as either gRNA alone will result in nicks that will not change the genomic DNA.
- dCas9 Modified versions of the Cas9 enzyme containing two inactive catalytic domains
- dCas9 can be utilized as a platform for DNA transcriptional regulators to activate or repress gene expression by fusing the inactive enzyme to known regulatory domains.
- the binding of dCas9 alone to a target sequence in genomic DNA can interfere with gene transcription.
- Non-limiting examples of gRNA sequences that can be used with some embodiments of the present invention are described in Tables 1A and 1B hereinbelow.
- the gRNA sequence does not have a significant off target effect.
- Methods of determining off target effect are well known in the art, such as BGI Human Whole Genome Sequencing (described in Nature;491:65-56.2012 ), next generation sequencing (NGS) using e.g. commercially available kits such as Alt-R-Genom Editing (IDT detection kit) or Sure select target enrich ⁇ 1% variant allele frequency (Agilent).
- both gRNA and Cas9 should be expressed in a target cell.
- the insertion vector can contain both cassettes on a single plasmid or the cassettes are expressed from two separate plasmids.
- CRISPR plasmids are commercially available such as the px330 plasmid from Addgene.
- the target cell can be transfected with both gRNA and Cas9 without plasmid using e.g. a transfection reagent such as CRISPRMAX [see e.g. Yu et al. (2016) JDlBiotechnol Lett. 38(6):919-29 ].
- Table 1A TNFR1 guide RNA sequences designed according to the mouse TNFR1 Sequence of gRNA Forward Reverse GCD Band sizes CRISPR1 604 378bp 226bp CRISPR2 600 357bp 243bp CRISPR3 608 417bp 191bp * Mouse tumor necrosis factor receptor superfamily, member 1a mRNA: NM_011609 (SEQ ID NO: 10) Total No.
- Table 1b TNFR1 guide RNA sequences designed according to the human TNFR1 Sequence of gRNA Forward Reverse GCD Band sizes CRISPR1 625 415bp 210bp CRISPR2 648 420bp 228bp CRISPR3 484 360bp 124bp CRISPR4 CRISPR5 CRISPR6 CRISPR7 * Human tumor necrosis factor receptor superfamily, member 1a mRNA: NM_001065 (SEQ ID NO: 3)
- “Hit and run” or “in-out” - involves a two-step recombination procedure.
- an insertion-type vector containing a dual positive/negative selectable marker cassette is used to introduce the desired sequence alteration.
- the insertion vector contains a single continuous region of homology to the targeted locus and is modified to carry the mutation of interest.
- This targeting construct is linearized with a restriction enzyme at a one site within the region of homology, electroporated into the cells, and positive selection is performed to isolate homologous recombinants. These homologous recombinants contain a local duplication that is separated by intervening vector sequence, including the selection cassette.
- targeted clones are subjected to negative selection to identify cells that have lost the selection cassette via intrachromosomal recombination between the duplicated sequences.
- the local recombination event removes the duplication and, depending on the site of recombination, the allele either retains the introduced mutation or reverts to wild type. The end result is the introduction of the desired modification without the retention of any exogenous sequences.
- the "double-replacement" or “tag and exchange” strategy - involves a two-step selection procedure similar to the hit and run approach, but requires the use of two different targeting constructs.
- a standard targeting vector with 3' and 5' homology arms is used to insert a dual positive/negative selectable cassette near the location where the mutation is to be introduced.
- homologously targeted clones are identified.
- a second targeting vector that contains a region of homology with the desired mutation is electroporated into targeted clones, and negative selection is applied to remove the selection cassette and introduce the mutation.
- the final allele contains the desired mutation while eliminating unwanted exogenous sequences.
- Site-Specific Recombinases The Cre recombinase derived from the P1 bacteriophage and Flp recombinase derived from the yeast Saccharomyces cerevisiae are site-specific DNA recombinases each recognizing a unique 34 base pair DNA sequence (termed “Lox” and "FRT", respectively) and sequences that are flanked with either Lox sites or FRT sites can be readily removed via site-specific recombination upon expression of Cre or Flp recombinase, respectively.
- the Lox sequence is composed of an asymmetric eight base pair spacer region flanked by 13 base pair inverted repeats.
- Cre recombines the 34 base pair lox DNA sequence by binding to the 13 base pair inverted repeats and catalyzing strand cleavage and religation within the spacer region.
- the staggered DNA cuts made by Cre in the spacer region are separated by 6 base pairs to give an overlap region that acts as a homology sensor to ensure that only recombination sites having the same overlap region recombine.
- the site specific recombinase system offers means for the removal of selection cassettes after homologous recombination. This system also allows for the generation of conditional altered alleles that can be inactivated or activated in a temporal or tissue-specific manner.
- the Cre and Flp recombinases leave behind a Lox or FRT "scar" of 34 base pairs. The Lox or FRT sites that remain are typically left behind in an intron or 3' UTR of the modified locus, and current evidence suggests that these sites usually do not interfere significantly with gene function.
- Cre/Lox and Flp/FRT recombination involves introduction of a targeting vector with 3' and 5' homology arms containing the mutation of interest, two Lox or FRT sequences and typically a selectable cassette placed between the two Lox or FRT sequences. Positive selection is applied and homologous recombinants that contain targeted mutation are identified. Transient expression of Cre or Flp in conjunction with negative selection results in the excision of the selection cassette and selects for cells where the cassette has been lost. The final targeted allele contains the Lox or FRT scar of exogenous sequences.
- Transposases refers to an enzyme that binds to the ends of a transposon and catalyzes the movement of the transposon to another part of the genome.
- transposon refers to a mobile genetic element comprising a nucleotide sequence which can move around to different positions within the genome of a single cell. In the process the transposon can cause mutations and/or change the amount of a DNA in the genome of the cell.
- transposon systems that are able to also transpose in cells e.g. vertebrates have been isolated or designed, such as Sleeping Beauty [ Izsvák and Ivics Molecular Therapy (2004) 9, 147-156 ], piggyBac [ Wilson et al. Molecular Therapy (2007) 15, 139-145 ], Tol2 [ Kawakami et al. PNAS (2000) 97 (21): 11403-11408 ] or Frog Prince [ Miskey et al. Nucleic Acids Res. Dec 1, (2003) 31(23): 6873-6881 ].
- DNA transposons translocate from one DNA site to another in a simple, cut-and-paste manner.
- PB is a 2.5 kb insect transposon originally isolated from the cabbage looper moth, Trichoplusia ni.
- the PB transposon consists of asymmetric terminal repeat sequences that flank a transposase, PBase.
- PBase recognizes the terminal repeats and induces transposition via a "cut-and-paste" based mechanism, and preferentially transposes into the host genome at the tetranucleotide sequence TTAA.
- the TTAA target site is duplicated such that the PB transposon is flanked by this tetranucleotide sequence.
- PB When mobilized, PB typically excises itself precisely to reestablish a single TTAA site, thereby restoring the host sequence to its pretransposon state. After excision, PB can transpose into a new location or be permanently lost from the genome.
- the transposase system offers an alternative means for the removal of selection cassettes after homologous recombination quit similar to the use Cre/Lox or Flp/FRT.
- the PB transposase system involves introduction of a targeting vector with 3' and 5' homology arms containing the mutation of interest, two PB terminal repeat sequences at the site of an endogenous TTAA sequence and a selection cassette placed between PB terminal repeat sequences. Positive selection is applied and homologous recombinants that contain targeted mutation are identified.
- Transient expression of PBase removes in conjunction with negative selection results in the excision of the selection cassette and selects for cells where the cassette has been lost.
- the final targeted allele contains the introduced mutation with no exogenous sequences.
- Genome editing using recombinant adeno-associated virus (rAAV) platform is based on rAAV vectors which enable insertion, deletion or substitution of DNA sequences in the genomes of live mammalian cells.
- the rAAV genome is a single-stranded deoxyribonucleic acid (ssDNA) molecule, either positive- or negative-sensed, which is about 4.7 kb long.
- ssDNA deoxyribonucleic acid
- These single-stranded DNA viral vectors have high transduction rates and have a unique property of stimulating endogenous homologous recombination in the absence of double-strand DNA breaks in the genome.
- rAAV genome editing has the advantage in that it targets a single allele and does not result in any off-target genomic alterations.
- rAAV genome editing technology is commercially available, for example, the rAAV GENESIS TM system from Horizon TM (Cambridge, UK).
- the agent can be a mutagen that causes random mutations and the cells exhibiting downregulation of the expression level and/or activity of the target may be selected.
- the mutagens may be, but are not limited to, genetic, chemical or radiation agents.
- the mutagen may be ionizing radiation, such as, but not limited to, ultraviolet light, gamma rays or alpha particles.
- Other mutagens may include, but not be limited to, base analogs, which can cause copying errors; deaminating agents, such as nitrous acid; intercalating agents, such as ethidium bromide; alkylating agents, such as bromouracil; transposons; natural and synthetic alkaloids; bromine and derivatives thereof; sodium azide; psoralen (for example, combined with ultraviolet radiation).
- the mutagen may be a chemical mutagen such as, but not limited to, ICR191, 1,2,7,8-diepoxy-octane (DEO), 5-azaC, N-methyl-N-nitrosoguanidine (MNNG) or ethyl methane sulfonate (EMS).
- DEO 1,2,7,8-diepoxy-octane
- MNNG N-methyl-N-nitrosoguanidine
- EMS ethyl methane sulfonate
- Methods for qualifying efficacy and detecting sequence alteration include, but not limited to, DNA sequencing, electrophoresis, an enzyme-based mismatch detection assay and a hybridization assay such as PCR, RT-PCR, RNase protection, in-situ hybridization, primer extension, Southern blot, Northern Blot and dot blot analysis.
- Sequence alterations in a specific gene can also be determined at the protein level using e.g. chromatography, electrophoretic methods, immunodetection assays such as ELISA and western blot analysis and immunohistochemistry.
- knock-in/knock-out construct including positive and/or negative selection markers for efficiently selecting transformed cells that underwent a homologous recombination event with the construct.
- Positive selection provides a means to enrich the population of clones that have taken up foreign DNA.
- positive markers include glutamine synthetase, dihydrofolate reductase (DHFR), markers that confer antibiotic resistance, such as neomycin, hygromycin, puromycin, and blasticidin S resistance cassettes.
- Negative selection markers are necessary to select against random integrations and/or elimination of a marker sequence (e.g. positive marker).
- Non-limiting examples of such negative markers include the herpes simplex-thymidine kinase (HSV-TK) which converts ganciclovir (GCV) into a cytotoxic nucleoside analog, hypoxanthine phosphoribosyltransferase (HPRT) and adenine phosphoribosytransferase (ARPT).
- HSV-TK herpes simplex-thymidine kinase
- GCV ganciclovir
- HPRT hypoxanthine phosphoribosyltransferase
- ARPT adenine phosphoribosytransferase
- RNA silencing refers to a group of regulatory mechanisms [e.g. RNA interference (RNAi), transcriptional gene silencing (TGS), post-transcriptional gene silencing (PTGS), quelling, co-suppression, and translational repression] mediated by RNA molecules which result in the inhibition or "silencing" of the expression of a corresponding protein-coding gene.
- RNA silencing has been observed in many types of organisms, including plants, animals, and fungi.
- RNA silencing agent refers to an RNA which is capable of specifically inhibiting or “silencing" the expression of a target gene.
- the RNA silencing agent is capable of preventing complete processing (e.g., the full translation and/or expression) of an mRNA molecule through a post-transcriptional silencing mechanism.
- RNA silencing agents include non-coding RNA molecules, for example RNA duplexes comprising paired strands, as well as precursor RNAs from which such small non-coding RNAs can be generated.
- Exemplary RNA silencing agents include dsRNAs such as siRNAs, miRNAs and shRNAs.
- the RNA silencing agent is capable of inducing RNA interference.
- the RNA silencing agent is capable of mediating translational repression.
- the RNA silencing agent is specific to the target RNA (i.e. TNFR1) and does not cross inhibit or silence other targets or a splice variant which exhibits 99% or less global homology to the target gene, e.g., less than 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81% global homology to the target gene; as determined by PCR, Western blot, Immunohistochemistry and/or flow cytometry.
- RNA interference refers to the process of sequence-specific post-transcriptional gene silencing in animals mediated by short interfering RNAs (siRNAs).
- RNA silencing agents that can be used according to specific embodiments of the present invention.
- DsRNA, siRNA and shRNA - The presence of long dsRNAs in cells stimulates the activity of a ribonuclease III enzyme referred to as dicer.
- Dicer is involved in the processing of the dsRNA into short pieces of dsRNA known as short interfering RNAs (siRNAs).
- Short interfering RNAs derived from dicer activity are typically about 21 to about 23 nucleotides in length and comprise about 19 base pair duplexes.
- the RNAi response also features an endonuclease complex, commonly referred to as an RNA-induced silencing complex (RISC), which mediates cleavage of single-stranded RNA having sequence complementary to the antisense strand of the siRNA duplex. Cleavage of the target RNA takes place in the middle of the region complementary to the antisense strand of the siRNA duplex.
- RISC RNA-induced silencing complex
- some embodiments of the invention contemplate use of dsRNA to downregulate protein expression from mRNA.
- dsRNA longer than 30 bp are used.
- dsRNA is provided in cells where the interferon pathway is not activated, see for example Billy et al., PNAS 2001, Vol 98, pages 14428-14433 ; and Diallo et al, Oligonucleotides, October 1, 2003, 13(5): 381-392. doi: 10.1089/154545703322617069 .
- the long dsRNA are specifically designed not to induce the interferon and PKR pathways for down-regulating gene expression.
- Shinagwa and Ishii [Genes & Dev. 17 (11): 1340-1345, 2003 ] have developed a vector, named pDECAP, to express long double-strand RNA from an RNA polymerase II (Pol II) promoter. Because the transcripts from pDECAP lack both the 5'-cap structure and the 3'-poly(A) tail that facilitate ds-RNA export to the cytoplasm, long ds-RNA from pDECAP does not induce the interferon response.
- siRNAs small inhibitory RNAs
- siRNA refers to small inhibitory RNA duplexes (generally between 18-30 base pairs) that induce the RNA interference (RNAi) pathway.
- RNAi RNA interference
- siRNAs are chemically synthesized as 21mers with a central 19 bp duplex region and symmetric 2-base 3'-overhangs on the termini, although it has been recently described that chemically synthesized RNA duplexes of 25-30 base length can have as much as a 100-fold increase in potency compared with 21mers at the same location.
- RNA silencing agent of some embodiments of the invention may also be a short hairpin RNA (shRNA).
- RNA agent refers to an RNA agent having a stem-loop structure, comprising a first and second region of complementary sequence, the degree of complementarity and orientation of the regions being sufficient such that base pairing occurs between the regions, the first and second regions being joined by a loop region, the loop resulting from a lack of base pairing between nucleotides (or nucleotide analogs) within the loop region.
- the number of nucleotides in the loop is a number between and including 3 to 23, or 5 to 15, or 7 to 13, or 4 to 9, or 9 to 11. Some of the nucleotides in the loop can be involved in base-pair interactions with other nucleotides in the loop.
- RNA silencing agents suitable for use with some embodiments of the invention can be effected as follows. First, the mRNA sequence is scanned downstream of the AUG start codon for AA dinucleotide sequences. Occurrence of each AA and the 3' adjacent 19 nucleotides is recorded as potential siRNA target sites. Preferably, siRNA target sites are selected from the open reading frame, as untranslated regions (UTRs) are richer in regulatory protein binding sites. UTR-binding proteins and/or translation initiation complexes may interfere with binding of the siRNA endonuclease complex [ Tuschl ChemBiochem. 2:239-245 ].
- UTRs untranslated regions
- siRNAs directed at untranslated regions may also be effective, as demonstrated for GAPDH wherein siRNA directed at the 5' UTR mediated about 90 % decrease in cellular GAPDH mRNA and completely abolished protein level (www(dot)ambion(dot)com/techlib/tn/91/912(dot)html).
- potential target sites are compared to an appropriate genomic database (e.g., human, mouse, rat etc.) using any sequence alignment software, such as the BLAST software available from the NCBI server (www(dot)ncbi(dot)nlm(dot)nih(dot)gov/BLAST/). Putative target sites which exhibit significant homology to other coding sequences are filtered out.
- sequence alignment software such as the BLAST software available from the NCBI server (www(dot)ncbi(dot)nlm(dot)nih(dot)gov/BLAST/).
- Qualifying target sequences are selected as template for siRNA synthesis.
- Preferred sequences are those including low G/C content as these have proven to be more effective in mediating gene silencing as compared to those with G/C content higher than 55 %.
- Several target sites are preferably selected along the length of the target gene for evaluation.
- a negative control is preferably used in conjunction.
- Negative control siRNA preferably include the same nucleotide composition as the siRNAs but lack significant homology to the genome.
- a scrambled nucleotide sequence of the siRNA is preferably used, provided it does not display any significant homology to any other gene.
- RNA silencing agent of some embodiments of the invention need not be limited to those molecules containing only RNA, but further encompasses chemically-modified nucleotides and non-nucleotides.
- RNA silencing agent may be a miRNA.
- miRNA refers to a collection of non-coding single-stranded RNA molecules of about 19-28 nucleotides in length, which regulate gene expression. miRNAs are found in a wide range of organisms (viruses.fwdarw.humans) and have been shown to play a role in development, homeostasis, and disease etiology.
- the pri-miRNA is typically part of a polycistronic RNA comprising multiple pri-miRNAs.
- the pri-miRNA may form a hairpin with a stem and loop.
- the stem may comprise mismatched bases.
- the hairpin structure of the pri-miRNA is recognized by Drosha, which is an RNase III endonuclease. Drosha typically recognizes terminal loops in the pri-miRNA and cleaves approximately two helical turns into the stem to produce a 60-70 nucleotide precursor known as the pre-miRNA. Drosha cleaves the pri-miRNA with a staggered cut typical of RNase III endonucleases yielding a pre-miRNA stem loop with a 5' phosphate and ⁇ 2 nucleotide 3' overhang. It is estimated that approximately one helical turn of stem ( ⁇ 10 nucleotides) extending beyond the Drosha cleavage site is essential for efficient processing. The pre-miRNA is then actively transported from the nucleus to the cytoplasm by Ran-GTP and the export receptor Ex-portin-5.
- the double-stranded stem of the pre-miRNA is then recognized by Dicer, which is also an RNase III endonuclease. Dicer may also recognize the 5' phosphate and 3' overhang at the base of the stem loop. Dicer then cleaves off the terminal loop two helical turns away from the base of the stem loop leaving an additional 5' phosphate and ⁇ 2 nucleotide 3' overhang.
- the resulting siRNA-like duplex which may comprise mismatches, comprises the mature miRNA and a similar-sized fragment known as the miRNA*.
- the miRNA and miRNA* may be derived from opposing arms of the pri-miRNA and pre-miRNA. miRNA* sequences may be found in libraries of cloned miRNAs but typically at lower frequency than the miRNAs.
- RISC RNA-induced silencing complex
- the miRNA strand of the miRNA:miRNA* duplex When the miRNA strand of the miRNA:miRNA* duplex is loaded into the RISC, the miRNA* is removed and degraded.
- the strand of the miRNA:miRNA* duplex that is loaded into the RISC is the strand whose 5' end is less tightly paired. In cases where both ends of the miRNA:miRNA* have roughly equivalent 5' pairing, both miRNA and miRNA* may have gene silencing activity.
- the RISC identifies target nucleic acids based on high levels of complementarity between the miRNA and the mRNA, especially by nucleotides 2-7 of the miRNA.
- the target sites in the mRNA may be in the 5' UTR, the 3' UTR or in the coding region.
- multiple miRNAs may regulate the same mRNA target by recognizing the same or multiple sites.
- the presence of multiple miRNA binding sites in most genetically identified targets may indicate that the cooperative action of multiple RISCs provides the most efficient translational inhibition.
- miRNAs may direct the RISC to downregulate gene expression by either of two mechanisms: mRNA cleavage or translational repression.
- the miRNA may specify cleavage of the mRNA if the mRNA has a certain degree of complementarity to the miRNA. When a miRNA guides cleavage, the cut is typically between the nucleotides pairing to residues 10 and 11 of the miRNA.
- the miRNA may repress translation if the miRNA does not have the requisite degree of complementarity to the miRNA. Translational repression may be more prevalent in animals since animals may have a lower degree of complementarity between the miRNA and binding site.
- any pair of miRNA and miRNA* there may be variability in the 5' and 3' ends of any pair of miRNA and miRNA*. This variability may be due to variability in the enzymatic processing of Drosha and Dicer with respect to the site of cleavage. Variability at the 5' and 3' ends of miRNA and miRNA* may also be due to mismatches in the stem structures of the pri-miRNA and pre-miRNA. The mismatches of the stem strands may lead to a population of different hairpin structures. Variability in the stem structures may also lead to variability in the products of cleavage by Drosha and Dicer.
- miRNA mimic refers to synthetic non-coding RNAs that are capable of entering the RNAi pathway and regulating gene expression. miRNA mimics imitate the function of endogenous miRNAs and can be designed as mature, double stranded molecules or mimic precursors (e.g., or pre-miRNAs). miRNA mimics can be comprised of modified or unmodified RNA, DNA, RNA-DNA hybrids, or alternative nucleic acid chemistries (e.g., LNAs or 2'-O,4'-C-ethylene-bridged nucleic acids (ENA)).
- nucleic acid chemistries e.g., LNAs or 2'-O,4'-C-ethylene-bridged nucleic acids (ENA)
- the length of the duplex region can vary between 13-33, 18-24 or 21-23 nucleotides.
- the miRNA may also comprise a total of at least 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 or 40 nucleotides.
- the sequence of the miRNA may be the first 13-33 nucleotides of the pre-miRNA.
- the sequence of the miRNA may also be the last 13-33 nucleotides of the pre-miRNA.
- Preparation of miRNAs mimics can be effected by any method known in the art such as chemical synthesis or recombinant methods.
- contacting cells with a miRNA may be effected by transfecting the cells with e.g. the mature double stranded miRNA, the pre-miRNA or the pri-miRNA.
- the pre-miRNA sequence may comprise from 45-90, 60-80 or 60-70 nucleotides.
- the pri-miRNA sequence may comprise from 45-30,000, 50-25,000, 100-20,000, 1,000-1,500 or 80-100 nucleotides.
- Antisense - Antisense is a single stranded RNA designed to prevent or inhibit expression of a gene by specifically hybridizing to its mRNA. Downregulation can be effected using an antisense polynucleotide capable of specifically hybridizing with an mRNA transcript encoding the target (i.e. TNFR1).
- the first aspect is delivery of the oligonucleotide into the cytoplasm of the appropriate cells, while the second aspect is design of an oligonucleotide which specifically binds the designated mRNA within cells in a way which inhibits translation thereof.
- the agent is selected from the group consisting of CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN), meganuclease, antisense and siRNA.
- ZFN Zinc finger nuclease
- TALEN transcription-activator like effector nuclease
- siRNA siRNA
- down-regulating expression and/or activity is effected at the genomic level.
- the agent comprises a CRISPR/Cas system.
- Embodiments of the invention further contemplate the use of the CRISPR/Cas system per se (as the therapeutic agent and not as a cell therapy) for downregulating expression of TNFR1 for treating an ischemic disease.
- a method of treating an ischemic disease in a subject in need thereof comprising administering to the subject a therapeutically effective amount of a CRISPR/Cas system for downregulating expression of TNFR1, thereby treating the ischemic disease in the subject.
- the CRISPR/Cas system does not downregulate expression of TNFR2.
- the cells or the agents e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- a "pharmaceutical composition” refers to a preparation of one or more of the active ingredients described herein with other chemical components such as physiologically suitable carriers and excipients.
- the purpose of a pharmaceutical composition is to facilitate administration of a compound to an organism.
- active ingredient refers to the cell or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) accountable for the biological effect.
- physiologically acceptable carrier and “pharmaceutically acceptable carrier” which may be interchangeably used refer to a carrier or a diluent that does not cause significant irritation to an organism and does not abrogate the biological activity and properties of the administered compound.
- An adjuvant is included under these phrases.
- excipient refers to an inert substance added to a pharmaceutical composition to further facilitate administration of an active ingredient.
- excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
- Suitable routes of administration may, for example, include oral, rectal, transmucosal, especially transnasal, intestinal or parenteral delivery, including intramuscular, subcutaneous and intramedullary injections as well as intrathecal, direct intraventricular, intracardiac, e.g., into the right or left ventricular cavity, into the common coronary artery, intravenous, intraperitoneal, intranasal, or intraocular injections.
- the cells or the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- the pharmaceutical composition comprising same is administered intravenously.
- neurosurgical strategies e.g., intracerebral injection or intracerebroventricular infusion
- molecular manipulation of the agent e.g., production of a chimeric fusion protein that comprises a transport peptide that has an affinity for an endothelial cell surface molecule in combination with an agent that is itself incapable of crossing the BBB
- pharmacological strategies designed to increase the lipid solubility of an agent (e.g., conjugation of water-soluble agents to lipid or cholesterol carriers)
- the transitory disruption of the integrity of the BBB by hyperosmotic disruption resulting from the infusion of a mannitol solution into the carotid artery or the use of a biologically active agent such as an angiotensin peptide).
- each of these strategies has limitations, such as the inherent risks associated with an invasive surgical procedure, a size limitation imposed by a limitation inherent in the endogenous transport systems, potentially undesirable biological side effects associated with the systemic administration of a chimeric molecule comprised of a carrier motif that could be active outside of the CNS, and the possible risk of brain damage within regions of the brain where the BBB is disrupted, which renders it a suboptimal delivery method.
- the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- the pharmaceutical composition comprising same in a local rather than systemic manner, for example, via injection of the pharmaceutical composition directly into a tissue region of a patient.
- the cells or the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- the pharmaceutical composition comprising same is administered into the ischemic tissue.
- the cells or the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- the pharmaceutical composition comprising same is administered by transendocardial injection.
- the cells or the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- the pharmaceutical composition comprising same is administered intracoronary or intramyocardialy.
- the cells or the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- an implantable device such as a stent, a valve, an intravascular device and a pacemaker.
- the cells of the present invention are administered in a hydrated gel (hydrogel) such as a hyaluronic acid-based hydrogels (see the Examples section which follows).
- a hydrated gel such as a hyaluronic acid-based hydrogels (see the Examples section which follows).
- hydrogels are known in the art and disclosed e.g. in Xu et al. (2012) Soft Matter. 8(12):3280-3294 ; Kim et al. (2012) Knee Surg Relat Res. Sep; 24(3): 164-172 , the contents of which are fully incorporated herein by reference.
- the hyaluronic acid is provided at a concentration range of about 0.1-10 %, e.g., about 0.5-10 %, e.g., about 0.5-5, e.g., about 1-5 %, e.g., about 2-5 %, e.g., about 3-5 %, e.g., about 3-4 % in the composition e.g. hydrogel.
- the hyaluronic acid is provided at a concentration range of about 3-4 % in the composition e.g. hydrogel.
- the cells of the present invention are administered in a biodegradable co-polymer or scaffold.
- a biodegradable co-polymer or scaffold Such scaffolds are known in the art and disclosed e.g. in Florian Weinbergeret al. (2017) Circulation Research. 120:1487-1500 ; Rochkind S et al (2004) Neurol Res. 26(2):161-6 ; Rochkind S. et al. (2006) Eur Spine J. 15(2):234-45 ; and FSY Wong, ACY Lo (2015) J Stem Cell Res Ther 5:267 , the contents of which are fully incorporated herein by reference.
- compositions of some embodiments of the invention may be manufactured by processes well known in the art, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes.
- compositions for use in accordance with some embodiments of the invention thus may be formulated in conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active ingredients into preparations which, can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen.
- the active ingredients of the pharmaceutical composition may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological salt buffer.
- physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological salt buffer.
- penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
- the pharmaceutical composition can be formulated readily by combining the active compounds with pharmaceutically acceptable carriers well known in the art.
- Such carriers enable the pharmaceutical composition to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like, for oral ingestion by a patient.
- Pharmacological preparations for oral use can be made using a solid excipient, optionally grinding the resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries if desired, to obtain tablets or dragee cores.
- Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carbomethylcellulose; and/or physiologically acceptable polymers such as polyvinylpyrrolidone (PVP).
- disintegrating agents may be added, such as cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- Dragee cores are provided with suitable coatings.
- suitable coatings For this purpose, concentrated sugar solutions may be used which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, titanium dioxide, lacquer solutions and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
- compositions which can be used orally include push-fit capsules made of gelatin as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- the push-fit capsules may contain the active ingredients in admixture with filler such as lactose, binders such as starches, lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active ingredients may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added. All formulations for oral administration should be in dosages suitable for the chosen route of administration.
- compositions may take the form of tablets or lozenges formulated in conventional manner.
- the active ingredients for use according to some embodiments of the invention are conveniently delivered in the form of an aerosol spray presentation from a pressurized pack or a nebulizer with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichloro-tetrafluoroethane or carbon dioxide.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichloro-tetrafluoroethane or carbon dioxide.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- Capsules and cartridges of, e.g., gelatin for use in a dispenser may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
- compositions described herein may be formulated for parenteral administration, e.g., by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multidose containers with optionally, an added preservative.
- the compositions may be suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- compositions for parenteral administration include aqueous solutions of the active preparation in water-soluble form. Additionally, suspensions of the active ingredients may be prepared as appropriate oily or water based injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acids esters such as ethyl oleate, triglycerides or liposomes. Aqueous injection suspensions may contain substances, which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the active ingredients to allow for the preparation of highly concentrated solutions.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile, pyrogen-free water based solution, before use.
- a suitable vehicle e.g., sterile, pyrogen-free water based solution
- compositions of some embodiments of the invention may also be formulated in rectal compositions such as suppositories or retention enemas, using, e.g., conventional suppository bases such as cocoa butter or other glycerides.
- Alternative embodiments include depots providing sustained release or prolonged duration of activity of the active ingredient in the subject, as are well known in the art.
- compositions suitable for use in context of some embodiments of the invention include compositions wherein the active ingredients are contained in an amount effective to achieve the intended purpose. More specifically, a therapeutically effective amount means an amount of active ingredients (the cells, the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) effective to prevent, alleviate or ameliorate symptoms of a disorder (e.g., ischemic disease, e.g., ischemic heart disease) or prolong the survival of the subject being treated.
- a therapeutically effective amount means an amount of active ingredients (the cells, the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) effective to prevent, alleviate or ameliorate symptoms of a disorder (e.g., ischemic disease, e.g., ischemic heart disease) or prolong the survival of the subject being treated.
- a disorder e.g., ischemic disease, e.g., ischemic heart disease
- the therapeutically effective amount or dose can be estimated initially from in vitro and cell culture assays.
- a dose can be formulated in animal models to achieve a desired concentration or titer. Such information can be used to more accurately determine useful doses in humans.
- Toxicity and therapeutic efficacy of the active ingredients described herein can be determined by standard pharmaceutical procedures in vitro, in cell cultures or experimental animals.
- the data obtained from these in vitro and cell culture assays and animal studies can be used in formulating a range of dosage for use in human.
- the dosage may vary depending upon the dosage form employed and the route of administration utilized.
- the exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition. (See e.g., Fingl, et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p.1 ).
- Dosage amount and interval may be adjusted individually to provide the active ingredient in levels sufficient to induce or suppress the biological effect (minimal effective concentration, MEC).
- MEC minimum effective concentration
- the MEC will vary for each preparation, but can be estimated from in vitro data. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. Detection assays can be used to determine plasma concentrations.
- dosing can be of a single or a plurality of administrations, with course of treatment lasting from several days to several weeks or until cure is effected or diminution of the disease state is achieved.
- the cells, the agent e.g. a CRISPR/Cas system for downregulating expression of TNFR1
- the pharmaceutical composition comprising same are administered within 1.5-24 hours following diagnosis of said ischemic disease.
- compositions to be administered will, of course, be dependent on the subject being treated, the severity of the affliction, the manner of administration, the judgment of the prescribing physician, etc.
- compositions of some embodiments of the invention may, if desired, be presented in a pack or dispenser device, such as an FDA approved kit, which may contain one or more unit dosage forms containing the active ingredient.
- the pack may, for example, comprise metal or plastic foil, such as a blister pack.
- the pack or dispenser device may be accompanied by instructions for administration.
- the pack or dispenser may also be accommodated by a notice associated with the container in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the compositions or human or veterinary administration. Such notice, for example, may be of labeling approved by the U.S. Food and Drug Administration for prescription drugs or of an approved product insert.
- Compositions comprising a preparation of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition, as is further detailed above.
- a pharmaceutical composition comprising as an active ingredient differentiated cells or bone marrow stem cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1.
- a pharmaceutical composition comprising as active ingredients cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1 and at least 2 ng/ml TNF ⁇ .
- an article of manufacture comprising TNF ⁇ and the cells disclosed herein with reduced expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1.
- the TNF ⁇ and the cells are in a co-formulation.
- the TNF ⁇ and the cells are in separate containers.
- an article of manufacture comprising TNF ⁇ and a CRISPR/Cas system for downregulating expression of TNFR1.
- the TNF ⁇ and a CRISPR/Cas system are in a co-formulation.
- the TNF ⁇ and a CRISPR/Cas system are in separate containers.
- TNF ⁇ is provided at a concentration above its physiological concentration in the cells.
- TNF ⁇ is provided at a concentration of at least 2 ng/ml, at least 5 ng/ml, at least 10 ng/ml, at least 20 ng/ml, at least 30 ng/ml, at least 40 ng/ml or at least 50 ng/ml, each possibility represents a separate embodiment of the present invention.
- TNF ⁇ is provided at a concentration of 2 - 200 ng/ml or 20 - 200 ng/ml.
- compositions, method or structure may include additional ingredients, steps and/or parts, but only if the additional ingredients, steps and/or parts do not materially alter the basic and novel characteristics of the claimed composition, method or structure.
- a compound or “at least one compound” may include a plurality of compounds, including mixtures thereof.
- range format is merely for convenience and brevity and should not be construed as an inflexible limitation on the scope of the invention. Accordingly, the description of a range should be considered to have specifically disclosed all the possible subranges as well as individual numerical values within that range. For example, description of a range such as from 1 to 6 should be considered to have specifically disclosed subranges such as from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from 3 to 6 etc., as well as individual numbers within that range, for example, 1, 2, 3, 4, 5, and 6. This applies regardless of the breadth of the range.
- method refers to manners, means, techniques and procedures for accomplishing a given task including, but not limited to, those manners, means, techniques and procedures either known to, or readily developed from known manners, means, techniques and procedures by practitioners of the chemical, pharmacological, biological, biochemical and medical arts.
- sequences that substantially correspond to its complementary sequence as including minor sequence variations, resulting from, e.g., sequencing errors, cloning errors, or other alterations resulting in base substitution, base deletion or base addition, provided that the frequency of such variations is less than 1 in 50 nucleotides, alternatively, less than 1 in 100 nucleotides, alternatively, less than 1 in 200 nucleotides, alternatively, less than 1 in 500 nucleotides, alternatively, less than 1 in 1000 nucleotides, alternatively, less than 1 in 5,000 nucleotides, alternatively, less than 1 in 10,000 nucleotides.
- red blood cell (RBC) lysis buffer (Roche) was added and the sample was incubated for 3 minutes at room temperature.
- Mononuclear bone marrow cells (mnBMCs) were separated from the BM sample by Ficoll-Paque (Histopaque1077, Sigma), with 3 technical replicates.
- BM cells were resuspended in 2 mL PBS supplemented with 2 % murine serum were layered on 1.5 ml Ficoll-Paque. The Ficoll-Paque gradients were centrifuged for 40 minutes at 400 g without brake. The mnBM layer was then collected, washed in PBS supplemented with 2 % FBS. Viability and cell count was determined by trypan blue exclusion method using hemocytometer. A total of 100 ⁇ 10 6 mnBMCs was counted. 60 ⁇ 10 6 mnBMCs were frozen at -80 °C.
- 40 ⁇ 10 6 cells were seeded in 24 wells plates (120,000 mnBMCs per well) in 1 mL / well DMEM medium containing- glutamine, sodium pyruvate, pen/strep and 3 % mouse serum and cultured under standard culture conditions. Following 24 hours of incubation, the medium was replaced with optimem medium (Invitrogen) and cells were transfected with TNFR1 CRISPR for 2 days. Control cells, which were not transfected with TNFR1 CRISPR were cultured under the same culturing conditions: 120,000 mnBMCs per well in 24 wells plates in DMEM medium containing 2 % FBS.
- TNFR1 CRISPR transfection For CRISPR transfection three different mouse TNFR1 guide RNA were designed and synthesized (Table 1A hereinabove) and mnBMCs were transfected with case 9 protein with the lipophilic transfection reagent CRISPRMAX as described before (Yu et al 2016).
- mnBM Transplantation 750,000 cultured mnBMCs were treated with 30 ng / ml mouse TNF ⁇ (PeproTech) for 20 minutes and added to 50 ⁇ L gel containing 3.75 % hyaluronic acid (HA), 20 % non-activated murine serum (serum obtained from C57BL/6 mice and inactivated in 56 °C) in DMEM.
- 50 ⁇ L gel containing mnBMCs were injected to the heart as described in Toma et al 2002 and Kawada et al 2016.
- mice Animal model - Animal experiments were conducted according to the guidelines of the Animal Care and Use Committee of Israel, and the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health. 18 C57BL/6 mice were purchased at 8 weeks (20-23 gr) from Ex-Vivo Israel. Following 1 week of acclimation the mice were intubated and anesthetized with 0.5 % isoflurane gas. Permanent ligation of the left anterior descending artery (LAD) was performed as described in Kolk et al. (2009) JoVE. 32 (ref 41). The mice were randomly assigned into three groups, see table 2 hereinbelow:
- Injected mnBMCs in all groups (1M, 2M, 3M) were treated for 20 minutes prior to injection with 30 ng/mL mouse TNF ⁇ (PeproTech) containing 1 mg / ml murine serum.
- mice were sacrificed 24 hours or 4 days following LAD occlusion and ⁇ 200 ⁇ l whole blood was collected from the retro-orbital sinus into yellow cap serum tubes. The collected blood was centrifuged (at 3000 rpm for 10 minutes) for serum preparation. Samples were frozen at (-60 °C) - (-90 °C) and stored in appropriately labeled test-tubes for further Creatin Kinase (CK) detection (Sigma). From each sacrificed mouse heart was dissected, weighed, fixated and sent for histological evaluation.
- Table 2 Experimental design: Group Mouse No.
- Histology - Hearts were harvested and fixed in 10 % formaldehyde at Pharmaseed and transferred to Patho-Logica. All tissues were trimmed in the same manner into block cassettes. Transverse cross sections were performed in each heart producing four equal cross sections per organ. The tissues were embedded in paraffin, sectioned at no more than 5 micron thickness, and stained with Hematoxylin & Eosin (H&E) and Masson Trichrome (MT), to trace the injected cells, pathological changes and to evaluate the volume of the induced infarcts. Furthermore, to evaluated viability an apoptosis TUNEL kit stain was performed. The slides were photographed using a microscope (Olympus BX60), at magnifications of X10, X20 and X40, equipped with an Olympus DP-73 camera.
- H&E Hematoxylin & Eosin
- MT Masson Trichrome
- mnBMCs transfected with TNFR1 CRISPR and treated with TNF ⁇ were injected to the LAD area and their effect on infract volume and cellular response was compared to the effect of injection of control mnBMCs (non-transfected and treated with TNF ⁇ ).
- Ischemic preconditioning has been recognized as one of the most potent mechanisms to protect against myocardial ischemic injury.
- ischemia In experimental animals and humans, a brief period of ischemia has been shown to protect the heart from more prolonged episodes of ischemia, reducing infarct size, attenuating the incidence, and severity of reperfusion-induced arrhythmias, and preventing endothelial cell dysfunction (e.g. 6). It has been shown that classical preconditioning can be mimicked by administration of TNF ⁇ [9-13].
- CK were determined in the serum of the transplanted mice.
- CK made up of three enzyme forms, including CK-MB
- levels can rise following heart attack, skeletal muscle injury and strenuous exercise.
- detection of CK levels in the serum indicated that injection of the mnBMCs (either transfected or not transfected with TNFR1 CRISPR) did not have an effect of CK levels 4 days following LAD occlusion ( Figure 10 ).
- mnBMCs transfected with human TNFR1 CRISPR and treated with TNF ⁇ are injected to the LAD area and their effect on infract volume and cellular response is compared to the effect of injection of control human mnBMCs (non-transfected and treated with TNF ⁇ ).
- BM Bone marrow
- the Ficoll-Paque gradients are centrifuged for 40 minutes at 400 g without brake. Following, the mnBM layer is collected, washed in PBS supplemented with 2 % human serum. Viability and cell count is determined by trypan blue exclusion method using hemocytometer. A fraction of the mnBMCs are frozen at -80 °C. Another fraction is seeded in 24 wells plates (120,000 mnBMCs per well) in 1 mL / well DMEM medium containing- glutamine, sodium pyruvate, pen/strep and 3 % human serum and cultured under standard culture conditions.
- TNFR1 CRISPR transfection For CRISPR transfection seven different human TNFR1 guide RNA were designed and synthesized (Table 1B hereinabove). mnBMCs are transfected with case 9 protein with the lipophilic transfection reagent CRISPRMAX as described before (Yu et al 2016) or transfected immediately by electroporation.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Cell Biology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Developmental Biology & Embryology (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Microbiology (AREA)
- Hematology (AREA)
- Biophysics (AREA)
- Virology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Rheumatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Physics & Mathematics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Plant Pathology (AREA)
- Mycology (AREA)
- Toxicology (AREA)
Abstract
Description
- The present invention, in some embodiments thereof, relates to methods of treating an ischemic disease.
- Ischemic heart disease, such as acute myocardial infarctions, congestive heart failure, arrhythmias, and sudden cardiac death, is the leading cause of morbidity and mortality in all industrialized nations. In the United States, ischemic heart disease causes nearly 20 % of all deaths (≈600,000 deaths each year). Over the past half a century conventional medicine and surgery have offered many breakthroughs, resulting in a dramatic decline in mortality [Mozaffarian et al. Circulation. (2015) 131(4):e29-322.) Despite the major advances in treatment, the prognosis for patients who are admitted to hospital with heart failure remains poor, with a 5 year survival of about 50% and a 10 year survival of about 10 %. The approach of using stem or precursor cells has emerged in the last decade as a regenerative strategy to address cardiac disease, with pre-clinical and clinical trials showing beneficial effects of progenitor cell therapy in acute myocardial infarction and ischemic cardiomyopathy [e.g. Choudhury et al. Eur J Heart Fail. (2017) 19(1):138-147; and Karantalis et al. Circ Res. (2015) 116(8):1413-30].
- Tumor necrosis factor alpha (TNFα) is a pro-inflammatory cytokine that has been implicated in mediating or exacerbating various mammalian conditions such as myocardial infarction, heart failure, septic shock, inflammatory disease, HIV infection and tissue transplant. However, in anti-cytokine clinical trials, the use of either a soluble TNF receptor, or an anti-TNF antibody was not beneficial to patients with heart failure. In addition, treatment with soluble TNF receptors significantly exacerbated ventricular dysfunction and remodeling with enhanced interstitial fibrosis after myocardial infarction (Monden et al [34]). These findings suggest that TNFα may not be exclusively toxic but may be partially protective in cardiovascular diseases. TNFα initiates its biological effects by binding to two distinct cell surface receptors expressed in most cell types: TNFR1 (
TNFα receptor type 1, with approximate mass of 55 kDa) and TNFR2 (TNF-α receptor type 2, with approximate mass of 75 kDa). The specific roles of TNFR1 and TNFR2 signaling in ischemic damage remain unclear with several studies indicating that signaling via TNFR1 is deleterious and signaling via TNFR2 is protective against ischemic damage while others suggesting no difference in the function of the two receptors in protecting the heart from ischemia (see e.g. Kishore et al. [33], Monden et al [34], Moe et al 2004 [35] and Kurrelmeyer et al [37]). - Additional background art includes:
- International Patent Application Publication Nos.
WO2000000504 ,WO2015104322 andWO2011006914 ; - US Patent Application Publication Nos:
US20120014879 andUS20110189768 ; - European Patent No.
EP2746396 ; - Chinese Patent No.
CN101401930 ; - Bao et al. (2008) Scand. Cardiovasc. J. 42, 56-62;
- Farhang et al. (2017) Tissue Eng Part A. 23(15-16):738-749;
- Kelly et al. (2010) Shock. 33(6): 602-607; and
- Tan et al. (2010) Shock. 34(3):236-42.
- According to an aspect of some embodiments of the present invention there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of mononuclear bone marrow cells (mnBMCs) comprising mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of the TNFR1, thereby treating the ischemic disease in the subject.
- According to an aspect of some embodiments of the present invention there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of differentiated cells with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of the TNFR1, wherein the differentiated cells are of a same type of tissue affected in the ischemic disease, thereby treating the ischemic disease in the subject.
- According to some embodiments of the invention, the method further comprising treating the cells with reduced expression and/or activity of TNFR1 with TNFα prior to the administering.
- According to some embodiments of the invention, the method comprising cryopreserving the cells prior to the treating with the TNFα.
- According to an aspect of some embodiments of the present invention there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising:
- (i) treating with TNFα cells with reduced expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of the TNFR1, wherein the cells are selected from the group consisting of mononuclear bone marrow cells (mnBMCs), stem cells and differentiated cells of a same type of tissue affected in the ischemic disease; and
- (ii) administering to the subject a therapeutically effective amount of the cells with reduced level of expression and/or activity of TNFR1 following the (i), thereby treating the ischemic disease in the subject.
- According to some embodiments of the invention, the cells comprise differentiated cells of a same type of tissue affected in the ischemic disease.
- According to some embodiments of the invention, the cells with reduced level of expression and/or activity of TNFR1 have the same level of expression and/or activity of TNFR2 as compared to the control stem cells.
- According to some embodiments of the invention, the cells with reduced expression and/or activity of TNFR1 are genetically modified cells.
- According to some embodiments of the invention, the genetically modified comprises genetically modified with a CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN) or meganuclease for downregulating expression of the TNFR1.
- According to some embodiments of the invention, the genetically modified comprises genetically modified with a CRISPR/Cas system for downregulating expression of the TNFR1.
- According to an aspect of some embodiments of the present invention there is provided a method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells, the method comprising:
- (i) contacting ex-vivo or in-vitro differentiated cells or bone marrow stem cells with an agent which downregulates expression and/or activity of TNFR1; and
- (ii) contacting ex-vivo or in-vitro the cells with TNFα.
- According to some embodiments of the invention, the (i) is effected prior to the (ii).
- According to some embodiments of the invention, the agent does not downregulate expression and/or activity of TNFR2.
- According to some embodiments of the invention, the agent is selected from the group consisting of CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN), meganuclease, antisense and siRNA.
- According to some embodiments of the invention, the agent comprises a CRISPR/Cas system.
- According to some embodiments of the invention, there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising:
- (i) obtaining cells according to the method of the present invention; and
- (ii) administering to the subject a therapeutically effective amount of the cells, thereby treating the ischemic disease in the subject.
- According to some embodiments of the invention, the differentiated cells are of a same type of tissue affected in the ischemic disease.
- According to some embodiments of the invention, the method comprising cryopreserving the cells following the (i) and prior to the (ii).
- According to some embodiments of the invention, the cells are non-autologous to the subject.
- According to an aspect of some embodiments of the present invention there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a CRISPR/Cas system for downregulating expression of TNFR1, thereby treating the ischemic disease in the subject.
- According to an aspect of some embodiments of the present invention there is provided a pharmaceutical composition comprising as an active ingredient differentiated cells or bone marrow stem cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1.
- According to an aspect of some embodiments of the present invention there is provided a pharmaceutical composition comprising as active ingredients cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1 and at least 2 ng/ml TNFα.
- According to some embodiments of the invention, the CRISPR/Cas system does not downregulate expression of TNFR2.
- According to some embodiments of the invention, the cells comprise differentiated cells.
- According to some embodiments of the invention, the differentiated cells are cardiomyocytes.
- According to some embodiments of the invention, the cells comprise stem cells.
- According to some embodiments of the invention, the stem cells are selected from the group consisting of embryonic stem cells (ESCs), induced pluripotent stem cells (iPS), hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
- According to some embodiments of the invention, the stem cells comprise hematopoietic stem cells (HSCs).
- According to some embodiments of the invention, the cells are comprised in mononuclear bone marrow cells (mnBMCs).
- According to some embodiments of the invention, the mnBMCs comprise lymphocytes or progenitors thereof.
- According to some embodiments of the invention, the cells are obtained by density gradient centrifugation of bone marrow cells.
- According to some embodiments of the invention, the method comprising obtaining the cells by density gradient centrifugation of bone marrow cells.
- According to some embodiments of the invention, the cells are human cells.
- According to some embodiments of the invention, the cells are cryopreserved cells.
- According to some embodiments of the invention, the ischemic disease is ischemic heart disease.
- According to some embodiments of the invention, the ischemic heart disease is myocardial infarction.
- According to some embodiments of the invention, the ischemic heart disease is ischemic cardiomyopathy.
- According to some embodiments of the invention, the subject is not treated with TNFα.
- Unless otherwise defined, all technical and/or scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which the invention pertains. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of embodiments of the invention, exemplary methods and/or materials are described below. In case of conflict, the patent specification, including definitions, will control. In addition, the materials, methods, and examples are illustrative only and are not intended to be necessarily limiting.
- Some embodiments of the invention are herein described, by way of example only, with reference to the accompanying drawings. With specific reference now to the drawings in detail, it is stressed that the particulars shown are by way of example and for purposes of illustrative discussion of embodiments of the invention. In this regard, the description taken with the drawings makes apparent to those skilled in the art how embodiments of the invention may be practiced.
- In the drawings:
-
Figures 1A-F show representative photomicrographs of histological sections of hearts obtained from Control mice (Group 3M, see Table 2 hereinbelow) 24 hours following left anterior descending artery (LAD) occlusion and stained with Masson Trichrome (MT,Figures 1A, 1C, 1E ) or Hematoxylin & Eosin (H&E,Figures 1B, 1D, 1F). Figures 1A-B demonstrate a middle sized infarct volume (Figure 1A ) and cellular infiltration surrounding injected round clear cells (arrow,Figure 1B ) in mouse no. 15.Figures 1C-D demonstrate middle sized infarct volume (Figure 1C ) and cellular infiltration surrounding injected round clear cells (arrow) and some adipocytes (Figure 1D ) in mouse no. 16.Figures 1E-F demonstrate middle sized infarct volume (Figure 1E ) and marked cellular infiltration in and few injected cells (arrow,Figure 1F ) in mouse no. 18. -
Figures 2A-F show representative photomicrographs of histological sections of hearts obtained from Group 2M mice (see Table 2 hereinbelow) 24 hours following LAD occlusion and stained with MT (Figures 2A, 2C, 2E ) or H&E (Figures 2B, 2D, 2F). Figures 2A-B demonstrate a small infarct volume (Figure 2A ) and mild cellular infiltration (Figure 2B ) in mouse no. 7.Figures 2C-D demonstrate middle sized infarct volume (Figure 2C ) and focal extensive infiltration with some cellular debris of the injected cells (arrow,Figure 2D ) in mouse no. 9.Figures 2E-F a transmural infarct with still intact left ventricle wall (Figure 2E ) and marked cellular infiltration in the inner part of the ventricle wall (Figure 2F ) in mouse no. 10. -
Figures 3A-F show representative photomicrographs of histological sections of hearts obtained from Group 1M mice (see Table 2 hereinbelow) 24 hours following LAD occlusion and stained with MT (Figures 3A, 3C, 3E ) or H&E (Figures 3B, 3D, 3F). Figures 3A-B demonstrate a transmural infarct with a marked reduced myocardium tissue (Figure 3A ) and minimal cellular infiltration (Figure 3B ) in mouse no. 3.Figures 3C-D , demonstrate a relative small infarct volume in the wall of the left ventricle (Figure 3C ) and loss of tissue, inflammatory reaction and some fibrin material in the epicardium (arrow,Figure 3D ) in mouse no. 5.Figures 3E-F demonstrate a transmural infarct with a marked reduced myocardium tissue (Figure 3E ) and mild infiltration of inflammatory cells (Figure 3F ) in mouse no. 3. -
Figures 4A-D show representative photomicrographs of histological sections of hearts obtained from Control mice (Group 3M, see Table 2 hereinbelow) 4 days following LAD occlusion and stained with MT (Figures 4A, 4C ) or H&E (Figures 4B, 4D). Figures 4A-B demonstrate a very large infarct volume (Figure 4A ) and severe inflammation and necrosis (N) in the affected myocardium (Figure 4B ) in mouse no. 13.Figures 4C-D demonstrate a very large infarct volume (Figure 4C ) and severe inflammation and necrosis (N) in the affected myocardium (Figure 4D ) in mouse no. 14. -
Figures 5A-D show representative photomicrographs of histological sections of hearts obtained from Group 2M mice (see Table 2 hereinbelow) 4 days following LAD occlusion and stained with MT (Figures 5A, 5C ) or H&E (Figures 5B, 5D). Figures 5A-B demonstrate a very large infarct volume (Figure 5A ) and severe inflammation and necrosis in the affected myocardium (Figure 5B ) in mouse no. 11.Figures 5C-D demonstrate a very large infarct volume (Figure 5C ) and severe inflammation and necrosis (N) in the affected myocardium (Figure 5D ) in mouse no. 12. -
Figures 6A-D show representative photomicrographs of histological sections of hearts obtained from Group 1M mice (see Table 2 hereinbelow) 4 days following LAD occlusion and stained with MT (Figures 6A, 6C ) or H&E (Figures 6B, 6D). Figures 6A-B demonstrate a very large infarct volume (Figure 6A ) and severe inflammation and necrosis in the affected myocardium (Figure 6B ) in mouse no. 1.Figures 6C-D demonstrate a very large infarct volume (Figure 6C ) and severe inflammation and necrosis in the affected myocardium (Figure 6D ) in mouse no. 2. -
Figures 7A-F show representative photomicrographs of histological sections of hearts obtained from Control mice (Group 3M), Group 2M mice, and Group 1M mice (see Table 2 hereinbelow) 24 hours following LAD occlusion and stained with TUNEL.Figure 7A demonstrate a mild to moderate TUNEL reaction in the infarct lesion in mouse no. 6 (Group 1M).Figures 7B-D demonstrate high TUNEL reaction in the infarct lesion in mice no. 7 (Figure 7B ), 9 (Figure 7C ) and10 (Figure 7D ) (Group 2M).Figures 7E-F demonstrate moderate to high TUNEL reaction in the infarct lesion in mice no. 15 (Figure 7E ) and 16 (Figure 7F ) (Control mice, Group 3M). -
Figures 8A-F show representative photomicrographs of histological sections of hearts obtained from Control mice (Group 3M), Group 2M mice, and Group 1M mice (see Table 2 hereinbelow) 4 days following LAD occlusion and stained with TUNEL.Figures 8A-B demonstrate high TUNEL reaction in the infarct lesion in mouse no. 1 and mild to moderate TUNEL reaction in the infarct lesion in mouse no. 2 (Group 1M).Figures 8C-D demonstrates mild TUNEL reaction in the infarct lesion in mice no. 11 and 12 (Group 2M).Figures 8E-F demonstrate high TUNEL reaction in the infarct lesion in mouse no. 13 and moderate TUNEL reaction in the infarct lesion in mouse no. 14 (Control mice, Group 3M). -
Figures 9A-B are bar graphs demonstrating that transplantation of mono-nuclear bone marrow cells (mnBMCs) transfected with TNFR1 CRISPR reduced infract size in the LAD occlusion mouse model compared to control mice transplanted with non-transfected mnBMCs, as determined byhistological evaluation 24 hours (Figure 9A ) and4 days (Figure 9B ) following LAD occlusion. -
Figures 10 is a bar graph demonstrating that transplantation of mnBMCs transfected with TNFR1 CRISPR had no toxic effect (such as severe muscle breakdown, acute kidney injury or autoimmune myositis) in the LAD occlusion mouse model compared to control mice transplanted with non-transfected mnBMCs, as determined byCK levels 4 days following LAD occlusion. - The present invention, in some embodiments thereof, relates to methods of treating an ischemic disease.
- Before explaining at least one embodiment of the invention in detail, it is to be understood that the invention is not necessarily limited in its application to the details set forth in the following description or exemplified by the Examples. The invention is capable of other embodiments or of being practiced or carried out in various ways.
- Ischemic heart disease is the leading cause of morbidity and mortality in all industrialized nations. Over the past half a century conventional medicine and surgery have offered many breakthroughs, resulting in a dramatic decline in mortality; however, despite the major advances in treatment, the prognosis for patients who are admitted to hospital with heart failure remains poor. The approach of using stem or precursor cells has emerged in the last decade as a regenerative strategy to address cardiac disease, with pre-clinical and clinical trials showing beneficial effects of progenitor cell therapy in acute myocardial infarction and ischemic cardiomyopathy.
- Whilst reducing the present invention to practice, the present inventors have now uncovered in a left anterior descending artery (LAD) mouse model that transplantation of mononuclear MB cells (mnBMCs) transfected with TNFR1 CRISPR/Cas system and treated with TNFα provides a cardio-protective effect from ischemic damage, as demonstrated by reduced infarct depth and width (Examples section and
Figures 1A-10 ). - Consequently, some embodiments of the present teachings suggest that therapeutic cells with reduced level of expression and/or activity of TNFR1 can be used for treating an ischemic disease.
- Thus, according to a first aspect of the present invention, there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of mononuclear bone marrow cells (mnBMCs) comprising mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, thereby treating the ischemic disease in the subject.
- According to another aspect of the present invention, there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of differentiated cells with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, wherein said differentiated cells are of a same type of tissue affected in said ischemic disease, thereby treating the ischemic disease in the subject.
- According to specific embodiments, the method further comprising treating said cells with reduced expression and/or activity of TNFR1 with TNFα prior to said administering.
- According to another aspect of the present invention, there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising:
- (i) treating with TNFα cells with reduced expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, wherein said cells are selected from the group consisting of mononuclear bone marrow cells (mnBMCs), stem cells and differentiated cells of a same type of tissue affected in said ischemic disease; and
- (ii) administering to the subject a therapeutically effective amount of said cells with reduced level of expression and/or activity of TNFR1 following said (i), thereby treating the ischemic disease in the subject.
- The term "treating" or "treatment" refers to inhibiting, preventing or arresting the development of a pathology (disease, disorder or medical condition e.g. ischemic disease e.g. ischemic heart disease) and/or causing the reduction, remission, or regression of a pathology or a symptom of a pathology. Those of skill in the art will understand that various methodologies and assays can be used to assess the development of a pathology, and similarly, various methodologies and assays may be used to assess the reduction, remission or regression of a pathology.
- As used herein, the term "ischemic disease" refers to a disease characterized by reduced blood (and hence oxygen) supply to the diseased tissue/organ. According to specific embodiments, the ischemic disease is an acute ischemic disease. According to other specific embodiments, the ischemic disease is a chronic ischemic disease. Non-limiting Examples of ischemic diseases include trauma, ischemic cerebrovascular disorder (such as apoplexy or cerebral infarction), ischemic renal disease, ischemic pulmonary disease, infection-related ischemic disease, ischemic disease of limbs, and ischemic heart disease.
- According to specific embodiments, the ischemic disease is not a rejection reaction following transplantation (e.g. GVHD).
- According to specific embodiments, the ischemic disease is ischemic heart disease.
- As used herein the term "ischemic heart disease" refers to a disease in which heart muscle is damaged or works inefficiently because of reduced blood (and hence oxygen) supply to the heart. Non-limiting Examples of ischemic heart diseases include ischemic cardiomyopathy, myocardial infarction or ischemic heart failure and chronic ischemic heart disease.
- According to specific embodiments, the ischemic disease is myocardial infarction (also known as heart attack).
- According to specific embodiments, the myocardial infarction is ST-segment elevation MI (STEMI) myocardial infarction (i.e. the coronary artery is completely blocked, thus virtually all of the heart muscle supplied by the affected artery becomes infarcted), as determined by ECG.
- According to other specific embodiments, the myocardial infarction is non-ST-segment elevation MI (NSTEMI) myocardial infarction (i.e. the artery is only partially occluded thus only a portion of the heart muscle supplied by the artery becomes infarcted), as determined by ECG.
- According to specific embodiments, the ischemic disease is ischemic cardiomyopathy.
- As used herein, the term "subject" includes mammals, e.g., human beings at any age and of any gender who suffer from the pathology (medical condition). According to specific embodiments, this term encompasses individuals who are at risk to develop the pathology.
- According to specific embodiments, the subject is not treated with TNFα.
- "TNFα", a cytokine also known as Tumor necrosis factor alpha. TNFα can bind TNFR1 and TNFR2. According to specific embodiments, the TNFα protein refers to the human protein, such as provided in the following GenBank Number NP_000585 (SEQ ID NO: 1).
- The term also encompasses functional homologues (naturally occurring or synthetically/recombinantly produced), orthologs (from other species) which exhibit the desired activity (i.e., binding TNFR1, binding TNFR2, activation of NFκB, activation of the MAPK pathway). Such homologues can be, for example, at least 80 %, at least 81 %, at least 82 %, at least 83 %, at least 84 %, at least 85 %, at least 86 %, at least 87 %, at least 88 %, at least 89 %, at least 90 %, at least 91 %, at least 92 %, at least 93 %, at least 94 %, at least 95 %, at least 96 %, at least 97 %, at least 98 %, at least 99 % or 100 % identical or homologous to the polypeptide SEQ ID NO: 1 or 80 %, at least 81 %, at least 82 %, at least 83 %, at least 84 %, at least 85 %, at least 86 %, at least 87 %, at least 88 %, at least 89 %, at least 90 %, at least 91 %, at least 92 %, at least 93 %, at least 94 %, at least 95 %, at least 96 %, at least 97 %, at least 98 %, at least 99 % or 100 % identical to the polynucleotide sequence encoding same (as further described hereinbelow).
- Sequence identity or homology can be determined using any protein or nucleic acid sequence alignment algorithm such as Blast, ClustalW, and MUSCLE.
- The functional homologs also refer to functional portions of TNFα which maintain the activity of the full length protein (i.e. binding TNFR1, binding TNFR2, activation of NFκB, activation of the MAPK pathway).
- According to specific embodiments, the TNFα is a recombinant TNFα, which can be prepared by standard recombinant expression methods or purchased commercially.
- TNFα can be commercially obtained from e.g. R&D Systems.
- According to specific embodiments, the cells are treated (or contacted) with TNFα.
- According to specific embodiments, the effect of treatment with TNFα is additive.
- According to other specific embodiments, the effect of treatment with TNFα is synergistic.
- According to specific embodiments, treatment (or contacting) with TNFα is effected at a concentration of at least 2 ng/ml, at least 5 ng/ml, at least 10 ng/ml, at least 20 ng/ml, at least 30 ng/ml, at least 40 ng/ml, at least 50 ng/ml or at least 60 ng/ml, each possibility represents a separate embodiment of the present invention.
- According to specific embodiments, treatment (or contacting) with TNFα is effected at a concentration of 2 - 200 ng/ml or 20 - 200 ng/ml.
- According to specific embodiments, treatment (or contacting) with TNFα is effected 1-72 hours, 1-48, 1-24, 1-10 or 1-5 hours prior to the administering, each possibility represents a separate embodiment of the present invention.
- According to specific embodiments, treatment (or contacting) with TNFα comprises a single TNFα treatment.
- According to other specific embodiments, treatment (or contacting) with TNFα comprises a plurality of TNFα treatments.
- Several types of cells can be used and/or obtained according to specific embodiments of the present invention.
- According to specific embodiments, the cells comprise differentiated cells.
- Non-limiting examples of differentiated cells that can be used with some embodiments of the present invention include differentiated cells derived from heart, kidney, liver, lung and brain.
- According to specific embodiments, the differentiated cells comprise differentiated cells of a same type of tissue affected in the ischemic disease.
- Methods of obtaining such differentiated cells are well known in the art. For example a cell suspension may be obtained by any mechanical or chemical (e.g. enzymatic) means. Several methods exist for dissociating cell clusters to form cell suspensions (e.g. single cell suspension) from primary tissues, attached cells in culture, and aggregates, e.g., physical forces (mechanical dissociation such as cell scraper, trituration through a narrow bore pipette, fine needle aspiration, vortex disaggregation and forced filtration through a fine nylon or stainless steel mesh), enzymes (enzymatic dissociation such as trypsin, collagenase, Acutase and the like ) or a combination of both. Thus, for example, enzymatic digestion of tissue/organ into isolate cells can be performed by subjecting the tissue to an enzyme such as type IV Collagenase (Worthington biochemical corporation, Lakewood, NJ, USA) and/or Dispase (Invitrogen Corporation products, Grand Island NY, USA). For example, the tissue may be enzyme digested by finely mincing tissue with a razor blade in the presence of e.g. collagenase, dispase and CaCl2 at 37 °C for about 1 hour. The method may further comprise removal of nonspecific debris from the resultant cell suspension by, for example, sequential filtration through filters (e.g. 70- and 40-µm filters), essentially as described under "General Materials and Experimental Methods" of the Examples section which follows. Furthermore, mechanical dissociation of tissue into isolated cells can be performed using a device designed to break the tissue to a predetermined size. Such a device can be obtained from CellArtis Goteborg, Sweden. Additionally or alternatively, mechanical dissociation can be manually performed using a needle such as a 27g needle (BD Microlance, Drogheda, Ireland) while viewing the tissue/cells under an inverted microscope. Following enzymatic or mechanical dissociation of the tissue, the dissociated cells are further broken to small clumps using 200 µl Gilson pipette tips (e.g., by pipetting up and down the cells). Alternatively, such differentiated cells can be obtained by differentiation of stem cell. Hence, according to specific embodiments, the differentiated cells have been ex-vivo differentiated. Methods of inducing differentiation of stem cells are well known to the skilled in the art. Thus, for example approaches to differentiate pluripotent cells into cardiomyocytes are disclosed in e.g. Burridge et al. (2012) Cell Stem Cell 10:16-28; Mummery et al. Circ Res. 2012 ;
WO2014200339 ,WO2011056416 ,WO2015004539 ,WO201202649 WO2014078414 andUS7534607 , the contents of which are fully incorporated herein by reference. - It will be appreciated that commercially available differentiated cells can also be used according to some embodiments of the invention.
- According to specific embodiments, the differentiated cells comprise cardiomyocytes.
- According to specific embodiments, the differentiated cells comprise lymphocytes.
- According to specific embodiments, the cells comprise bone marrow cells.
- Methods of obtaining bone marrow cells are well known in the art. Thus, for example, bone marrow can be obtained from iliac crest, femora, tibiae, spine, rib or other medullar spaces. Following, the desired cellular fraction can be purified from the bone marrow aspirates.
- According to specific embodiments, the cells comprise mononuclear bone marrow cells (mnBMCs).
- According to specific embodiments, the cells are comprised in mononuclear bone marrow cells (mnBMCs).
- According to specific embodiment, the cells comprise at least 50 % at least 60 %, at least 70 %, at least 80 %, at least 90 % or at least 95 % mnBMCs.
- Thus, for example, the mononuclear fraction can be purified from the bone marrow aspirates. There are several methods and reagents known to those skilled in the art for purifying mnBMCs from bone marrow such as, density gradient centrifugation (e.g. ficoll), sedimentation (e.g. Hespan), centrifugal elutriation, fractionation, automated processes (e.g. Sepax® from Biosafe SA, Eysins, Switzerland and the AutoXpress Platform® (AXP) from Thermogenesis, Rancho Cordova, CA chemical lysis of e.g. red blood cells (e.g. by ACK), selection of specific cell types using cell surface markers (using e.g. FACS sorter or magnetic cell separation techniques such as are commercially available e.g. from Invitrogen, Stemcell Technologies, Cellpro, Advanced Magnetics, or Miltenyi Biotec.), and depletion of specific cell types by methods such as eradication (e.g. killing) with specific antibodies or by affinity based purification based on negative selection (using e.g. magnetic cell separation techniques, FACS sorter and/or capture ELISA labeling). Such methods are described for example in THE HANDBOOK OF EXPERIMENTAL IMMUNOLOGY, or) and FLOW CYTOMETRY AND CELL SORTING (A. Radbruch, editor, Springer Verlag, 2000).
- According to specific embodiments, the bone marrow cells are obtained by density gradient centrifugation (e.g. ficoll) of bone marrow cells.
- According to specific embodiments, the bone marrow cells comprise stem cells (e.g. hematopoietic stem cells, mesenchymal stem cells). Methods of obtaining bone marrow stem cells are well known in the art and are further described hereinbelow.
- According to specific embodiments, the bone marrow cells comprise mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof.
- According to specific embodiments, the bone marrow cells comprise lymphocytes or progenitors thereof.
- As used herein the term "lymphocytes progenitors" refers to hematopoietic progenitor cells that can differentiate to lymphocytes.
- According to a specific embodiment the lymphocytes progenitor cells are mobilized to the peripheral blood by agents such as G-CSF with or without chemotherapy.
- According to specific embodiments, the cells comprise stem cells.
- As used herein, the phrase "stem cells" refers to cells which are capable of remaining in an undifferentiated state (e.g., totipotent, pluripotent or multipotent stem cells) for extended periods of time in culture until induced to differentiate into other cell types having a particular, specialized function (e.g., fully differentiated cells). Totipotent cells, such as embryonic cells within the first couple of cell divisions after fertilization are the only cells that can differentiate into embryonic and extra-embryonic cells and are able to develop into a viable human being. Preferably, the phrase "pluripotent stem cells" refers to cells which can differentiate into all three embryonic germ layers, i.e., ectoderm, endoderm and mesoderm or remaining in an undifferentiated state. The pluripotent stem cells include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPS). The multipotent stem cells include e.g. adult stem cells, hematopoietic stem cells and mesenchymal stem cells.
- It will be appreciated that undifferentiated stem cells are of a distinct morphology, which is clearly distinguishable from differentiated cells of embryo or adult origin by the skilled in the art. Typically, undifferentiated stem cells have high nuclear/cytoplasmic ratios, prominent nucleoli and compact colony formation with poorly discernable cell junctions. Additional features of undifferentiated stem cells are further described hereinunder.
- According to specific embodiments, the stem cells are selected from the group consisting of embryonic stem cells (ESCs), induced pluripotent stem cells (iPS), hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
- According to specific embodiments, the cells are embryonic stem cells (ESCs).
- The phrase "embryonic stem cells (ESCs)" refers to ESCs which are capable of differentiating into cells of all three embryonic germ layers (i.e., endoderm, ectoderm and mesoderm), or remaining in an undifferentiated state. The phrase "embryonic stem cells (ESCs)" may comprise cells which are obtained from the embryonic tissue formed after gestation (e.g., blastocyst) before implantation of the embryo (i.e., a pre-implantation blastocyst), extended blastocyst cells (EBCs) which are obtained from a post-implantation/pre-gastrulation stage blastocyst (see
WO2006/040763 ), embryonic germ (EG) cells which are obtained from the genital tissue of a fetus any time during gestation, preferably before 10 weeks of gestation, and cells originating from an unfertilized ova which are stimulated by parthenogenesis (parthenotes). - The ESCs of some embodiments of the invention can be obtained using well-known cell-culture methods. For example, human embryonic stem cells can be isolated from human blastocysts. Human blastocysts are typically obtained from human in vivo preimplantation embryos or from in vitro fertilized (IVF) embryos. Alternatively, a single cell human embryo can be expanded to the blastocyst stage. For the isolation of human ESCs the zona pellucida is removed from the blastocyst and the inner cell mass (ICM) is isolated by immunosurgery, in which the trophectoderm cells are lysed and removed from the intact ICM by gentle pipetting. The ICM is then plated in a tissue culture flask containing the appropriate medium which enables its outgrowth. Following 9 to 15 days, the ICM derived outgrowth is dissociated into clumps either by a mechanical dissociation or by an enzymatic degradation and the cells are then re-plated on a fresh tissue culture medium. Colonies demonstrating undifferentiated morphology are individually selected by micropipette, mechanically dissociated into clumps, and re-plated. Resulting ES cells are then routinely split every 4-7 days. For further details on methods of preparation human ES cells see
Thomson et al., [U.S. Pat. No. 5,843,780 ; Science 282: 1145, 1998; Curr. Top. Dev. Biol. 38: 133, 1998; Proc. Natl. Acad. Sci. USA 92: 7844, 1995]; Bongso et al., [Hum Reprod 4: 706, 1989]; and Gardner et al., [Fertil. Steril. 69: 84, 1998]. - It will be appreciated that commercially available stem cells can also be used according to some embodiments of the invention. Human ESCs can be purchased from the NIH human embryonic stem cells registry [Hypertext Transfer Protocol://grants (dot) nih (dot) gov/stem_cells/registry/current (dot) htm]. Non-limiting examples of commercially available embryonic stem cell lines are BG01, BG02, BG03, BG04, CY12, CY30, CY92, CY10, TE03, TE32, CHB-4, CHB-5, CHB-6, CHB-8, CHB-9, CHB-10, CHB-11, CHB-12,
HUES 1,HUES 2, HUES 3,HUES 4, HUES 5,HUES 6, HUES 7, HUES 8, HUES 9, HUES 10, HUES 11, HUES 12,HUES 13, HUES 14, HUES 15, HUES 16, HUES 17, HUES 18, HUES 19,HUES 20, HUES 21, HUES 22, HUES 23,HUES 24, HUES 25, HUES 26, HUES 27, HUES 28, CyT49, RUES3, WA01, UCSF4, NYUES1, NYUES2, NYUES3, NYUES4, NYUES5, NYUES6, NYUES7,UCLA 1,UCLA 2, UCLA 3, WA077 (H7), WA09 (H9), WA13 (H13), WA14 (H14), HUES 62, HUES 63, HUES 64, CT1, CT2, CT3, CT4, MA135, Eneavour-2, WIBR1, WIBR2, WIBR3, WIBR4, WIBR5, WIBR6, HUES 45, Shef 3,Shef 6, BJNhem19, BJNhem20, SA001, SA001. - In addition, ESCs can be obtained from other species as well, including mouse (Mills and Bradley, 2001), golden hamster [Doetschman et al., 1988, Dev Biol. 127: 224-7], rat [Iannaccone et al., 1994, Dev Biol. 163: 288-92] rabbit [Giles et al. 1993, Mol Reprod Dev. 36: 130-8; Graves & Moreadith, 1993, Mol Reprod Dev. 1993, 36: 424-33], several domestic animal species [Notarianni et al., 1991, J Reprod Fertil Suppl. 43: 255-60; Wheeler 1994, Reprod Fertil Dev. 6: 563-8; Mitalipova et al., 2001, Cloning. 3: 59-67] and non-human primate species (Rhesus monkey and marmoset) [Thomson et al., 1995, Proc Natl Acad Sci USA. 92: 7844-8; Thomson et al., 1996, Biol Reprod. 55: 254-9].
- Extended blastocyst cells (EBCs) can be obtained from a blastocyst of at least nine days post fertilization at a stage prior to gastrulation. Prior to culturing the blastocyst, the zona pellucida is digested [for example by Tyrode's acidic solution (Sigma Aldrich, St Louis, MO, USA)] so as to expose the inner cell mass. The blastocysts are then cultured as whole embryos for at least nine and no more than fourteen days post fertilization (i.e., prior to the gastrulation event) in vitro using standard embryonic stem cell culturing methods.
- Another method for preparing ESCs is described in Chung et al., Cell Stem Cell, . This method comprises removing a single cell from an embryo during an in vitro fertilization process. The embryo is not destroyed in this process.
- EG cells are prepared from the primordial germ cells obtained from fetuses of about 8-11 weeks of gestation (in the case of a human fetus) using laboratory techniques known to anyone skilled in the arts. The genital ridges are dissociated and cut into small chunks which are thereafter disaggregated into cells by mechanical dissociation. The EG cells are then grown in tissue culture flasks with the appropriate medium. The cells are cultured with daily replacement of medium until a cell morphology consistent with EG cells is observed, typically after 7-30 days or 1-4 passages. For additional details on methods of preparation human EG cells see Shamblott et al., [Proc. Natl. Acad. Sci. USA 95: 13726, 1998] and
U.S. Pat. No. 6,090,622 . - ESCs (e.g., human ESCs) originating from an unfertilized ova stimulated by parthenogenesis (parthenotes) are known in the art (e.g., Zhenyu Lu et al., 2010. J. Assist Reprod. Genet. 27:285-291; "Derivation and long-term culture of human parthenogenetic embryonic stem cells using human foreskin feeders", which is fully incorporated herein by reference). Parthenogenesis refers to the initiation of cell division by activation of ova in the absence of sperm cells, for example using electrical or chemical stimulation. The activated ovum (parthenote) is capable of developing into a primitive embryonic structure (called a blastocyst) but cannot develop to term as the cells are pluripotent, meaning that they cannot develop the necessary extra-embryonic tissues (such as amniotic fluid) needed for a viable human fetus.
- According to specific embodiments, the cells are embryonic stem cells Induced pluripotent stem cells (iPS).
- Induced pluripotent stem cells (iPS; embryonic-like stem cells), are cells obtained by de-differentiation of adult somatic cells which are endowed with pluripotency (i.e., being capable of differentiating into the three embryonic germ cell layers, i.e., endoderm, ectoderm and mesoderm). According to some embodiments of the invention, such cells are obtained from a differentiated tissue (e.g., a somatic tissue such as skin) and undergo de-differentiation by genetic manipulation which re-program the cell to acquire embryonic stem cells characteristics. According to some embodiments of the invention, the induced pluripotent stem cells are formed by inducing the expression of Oct-4, Sox2, Kfl4 and c-Myc in a somatic stem cell.
- Induced pluripotent stem cells (iPS) (embryonic-like stem cells) can be generated from somatic cells by genetic manipulation of somatic cells, e.g., by retroviral transduction of somatic cells such as fibroblasts, hepatocytes, gastric epithelial cells with transcription factors such as Oct-3/4, Sox2, c-Myc, and KLF4 [Yamanaka S, Cell Stem Cell. 2007, 1(1):39-49; Aoi T, et al., Generation of Pluripotent Stem Cells from Adult Mouse Liver and Stomach Cells. Science. 2008 Feb 14. (Epub ahead of print); IH Park, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008;451:141-146; K Takahashi, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131:861-872]. Other embryonic-like stem cells can be generated by nuclear transfer to oocytes, fusion with embryonic stem cells or nuclear transfer into zygotes if the recipient cells are arrested in mitosis.
- The phrase "adult stem cells" (also called "tissue stem cells" or a stem cell from a somatic tissue) refers to any stem cell derived from a somatic tissue [of either a postnatal or prenatal animal (especially the human)]. The adult stem cell is generally thought to be a multipotent stem cell, capable of differentiation into multiple cell types. Adult stem cells can be derived from any adult, neonatal or fetal tissue such as adipose tissue, skin, kidney, liver, prostate, pancreas, intestine, bone marrow and placenta.
- Adult tissue stem cells can be isolated using various methods known in the art such as those disclosed by Alison, M.R. [J Pathol. (2003) 200(5): 547-50], Cai, J. et al., [Blood Cells Mol Dis. 2003 31(1): 18-27], Collins, A.T. et al., [J Cell Sci. 2001; 114(Pt 21): 3865-72], Potten, C. S. and Morris, R. J. [Epithelial stem cells in vivo. 1988. J. Cell Sci. Suppl. 10, 45-62], Dominici, M et al., [J. Biol. Regul. Homeost. Agents. 2001, 15: 28-37],
Caplan and Haynesworth [U.S. Pat. No. 5,486,359 ] Jones E.A. et al., [Arthritis Rheum. 2002, 46(12): 3349-60]. Generally, isolation of adult tissue stem cells is based on the discrete location (or niche) of each cell type included in the adult tissue, i.e., the stem cells, the transit amplifying cells and the terminally differentiated cells [Potten, C. S. and Morris, R. J. (1988). Epithelial stem cells in vivo. J. Cell Sci. Suppl. 10, 45-62]. - Hematopoietic stem cells (HSCs), which may also referred to as adult tissue stem cells, include stem cells obtained from blood or bone marrow tissue of an individual at any age or from cord blood of a newborn individual. According to specific embodiments, the cells comprise HSCs.
- According to a specific embodiment, hematopoietic stem cell is a CD34+ cell.
- As used herein the term "CD34+ cell" refers to a hematopoietic stem cell positive for the CD34 marker that can differentiate to each of the cell types in the blood, i.e.; the myeloid (monocyte, macrophage, neutrophil, basophil, eosinophil, erythrocyte, megakaryocyte, dendritic cell) or lymphoid (T cell, B cell, NK cell) lineages.
- The HSCs may be a specific cell line, alternatively may be generated from iPS or embryonic stem cells [see for example Pick M et al. (2007) Stem Cells, 25(9): 2206-14; and Pick M et al. (2013) PLoS One, 8(2): e55530] or alternatively may be isolated using various methods known in the arts such as those disclosed by "Handbook of Stem Cells" edit by Robert Lanze, Elsevier Academic Press, 2004, Chapter 54, pp609-614, isolation and characterization of hematopoietic stem cells, by Gerald J Spangrude and William B Stayton. Thus, for example, HSCs can be isolated from cord blood, peripheral blood or BM samples by means of density gradient centrifugation using for example Ficoll-Paque (can be obtained from GE Healthcare Bio-Science AB) followed by immunomagnetic or immunofluorescent methods (such as Diamond or Microbeads CD34+ isolation kit obtained from Miltenyi Biotech). Purity of the purified fraction can be assessed by flow cytometry for the specified markers (for example CD34).
- According to a specific embodiment the HSCs are mobilized to the peripheral blood by agents such as G-CSF with or without chemotherapy.
- Placental and cord blood stem cells may also be referred to as "young stem cells".
- Fetal stem cells can be isolated using various methods known in the art such as those disclosed by Eventov-Friedman S, et al., PLoS Med. 2006, 3: e215; Eventov-Friedman S, et al., Proc Natl Acad Sci USA. 2005, 102: 2928-33; Dekel B, et al., 2003, Nat Med. 9: 53-60; and Dekel B, et al., 2002, J. Am. Soc. Nephrol. 13: 977-90.
- According to specific embodiments, the cells comprise mesenchymal stem cells (MSCs).
- Mesenchymal stem cells give rise to one or more mesenchymal tissues (e.g., adipose, osseous, cartilaginous, elastic and fibrous connective tissues, myoblasts) as well as to tissues other than those originating in the embryonic mesoderm (e.g., neural cells) depending upon various influences from bioactive factors such as cytokines. Although such cells can be isolated from embryonic yolk sac, placenta, umbilical cord, fetal and adolescent skin, blood and other tissues, their abundance in the bone marrow far exceeds their abundance in other tissues and as such isolation from bone marrow is presently preferred.
- Methods of isolating, purifying and expanding MSCs are known in the art and include, for example, those disclosed by
Caplan and Haynesworth in U.S. Pat. No. 5,486,359 and Jones E.A. et al., 2002, Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells, Arthritis Rheum. 46(12): 3349-60. - According to specific embodiments, MSC cultures are generated by diluting bone marrow aspirates (usually 20 ml) with equal volumes of Hank's balanced salt solution (HBSS; GIBCO Laboratories, Grand Island, NY, USA) and layering the diluted cells over about 10 ml of a Ficoll column (Ficoll-Paque; Pharmacia, Piscataway, NJ, USA). Following 30 minutes of centrifugation at 2,500 x g, the mononuclear cell layer is removed from the interface and suspended in HBSS. Cells are then centrifuged at 1,500 x g for 15 minutes and resuspended in a complete medium (MEM, α medium without deoxyribonucleotides or ribonucleotides; GIBCO); 2-20 % fetal calf serum (FCS) derived from a lot selected for rapid growth of MSCs (Atlanta Biologicals, Norcross, GA); 100 units/ml penicillin (GIBCO), 100 µg/ml streptomycin (GIBCO); and 2 mM L-glutamine (GIBCO). Resuspended cells are plated in about 25 ml of medium in a 10 cm culture dish (Corning Glass Works, Corning, NY) and incubated at 37 °C with 5 % humidified CO2. Following 24 hours in culture, nonadherent cells are discarded, and the adherent cells are thoroughly washed twice with phosphate buffered saline (PBS). The medium is replaced with a fresh complete medium every 3 or 4 days for about 14 days. Adherent cells are then harvested with 0.25 % trypsin and 1 mM EDTA (Trypsin/EDTA, GIBCO) for 5 min at 37 °C, replated in a 6-cm plate and cultured for another 14 days. Cells are then trypsinized and counted using a cell counting device such as for example, a hemocytometer (Hausser Scientific, Horsham, PA). Cultured cells are recovered by centrifugation and resuspended with 5 % DMSO and 30 % FCS at a concentration of 1 to 2 × 106 cells per ml. Aliquots of about 1 ml each are slowly frozen and stored in liquid nitrogen.
- To expand the MSC fraction, cells are resuspended in a complete medium and plated at a concentration of about 5,000 cells/cm2. Following 24 hours in culture, nonadherent cells are removed and the adherent cells are harvested using Trypsin/EDTA, dissociated by passage through a narrowed Pasteur pipette, and preferably replated at a density of about 1.5 to about 3.0 cells/cm2. Under these conditions, MSC cultures can grow for about 50 population doublings and be expanded for about 2000 fold [Colter DC., et al. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc Natl Acad Sci USA. 97: 3213-3218, 2000].
- MSC cultures utilized by some embodiments of the invention include three groups of cells which are defined by their morphological features: small and agranular cells (referred to as RS-1, hereinbelow), small and granular cells (referred to as RS-2, hereinbelow) and large and moderately granular cells (referred to as mature MSCs, hereinbelow). The presence and concentration of such cells in culture can be assayed by identifying a presence or absence of various cell surface markers, by using, for example, immunofluorescence, in situ hybridization, and activity assays.
- When MSCs are cultured under the culturing conditions of some embodiments of the invention they exhibit negative staining for the HSC markers CD34, CD11B, CD43 and CD45. A small fraction of cells (less than 10 %) are dimly positive for CD31 and/or CD38 markers. In addition, mature MSCs are dimly positive for the HSC marker, CD117 (c-Kit), moderately positive for the osteogenic MSCs marker, Stro-1 [Simmons, P. J. & Torok-Storb, B. (1991). Blood 78, 5562] and positive for the thymocytes and peripheral T lymphocytes marker, CD90 (Thy-1). On the other hand, the RS-1 cells are negative for the CD117 and Stro1 markers and are dimly positive for the CD90 marker, and the RS-2 cells are negative for all of these markers.
- The cells of some embodiments of the present invention can be a primary cell (non-cultured and alternatively or additionally non-immortalized cell) or a cell-line.
- According to specific embodiments, the cells used and/or obtained according to the present invention can be freshly isolated, stored e.g., cryopreserved (i.e. frozen) at e.g. liquid nitrogen temperature at any stage (e.g. following their retrieval, following contacting with the agent, following treatment with TNFα) for long periods of time (e.g., months, years) for future use; and cell lines.
- Thus, according to specific embodiments, the cells are cryopreserved cells.
- According to specific embodiments, the cells are cryopreserved prior to contacting with the agent which downregulates expression and/or activity of TNFR1.
- According to specific embodiments, the cells are cryopreserved following contacting with the agent which downregulates expression and/or activity of TNFR1.
- According to specific embodiments, the cells are cryopreserved prior to treatment with TNFα.
- According to specific embodiments, the cells are cryopreserved following treatment with TNFα.
- According to specific embodiments, the cells are cryopreserved prior to administering to the subject.
- Methods of cryopreservation are commonly known by one of ordinary skill in the art and are disclosed e.g. in International Patent Application Publication Nos.
WO2007054160 andWO 2001039594 and US Patent Application Publication No.US20120149108 . - According to specific embodiments, the cells obtained according to the present invention can be stored in a cell bank or a depository or storage facility.
- According to specific embodiments, the cells are freshly isolated (i.e., not subjected to preservation processes).
- The cells used according to specific embodiments of the present invention may be autologous or non-autologous; they can be syngeneic or non-synegeneic: allogeneic or xenogeneic to the subject; each possibility represents a separate embodiment of the present invention.
- According to specific embodiments, the cells are autologous to said subject.
- As used herein, the term "autologous" means that the donor subject is the recipient subject. Thus, in autologous transplantation the cells have been removed and re-introduced e.g., re-infused to the subject.
- According to specific embodiments, the cells are non-autologous to said subject.
- As used herein, the term "non-autologous" means that the donor subject is not the recipient subject.
- As used herein, the term "syngeneic" means that the donor subject is essentially genetically identical with the recipient subject. Examples of syngeneic transplantation include transplantation of cells derived from the subject (also referred to in the art as "autologous"), a clone of the subject, or a homozygotic twin of the subject
- As used herein, the term "allogeneic" means that the donor is of the same species as the recipient, but which is substantially non-clonal with the recipient. Typically, outbred, non-zygotic twin mammals of the same species are allogeneic with each other. It will be appreciated that an allogeneic donor may be HLA identical or HLA non-identical with respect to the subject
- As used herein, the term "xenogeneic" means that the donor subject is from a different species relative to the recipient subject.
- According to specific embodiments, the cells are mammalian cells.
- According to specific embodiments, the cells are primate cells.
- According to specific embodiments, the cells are human cells.
- According to other specific embodiments, the cells are rodent cells (e.g. mouse, rat).
- The cells of some embodiments of the present invention are characterized by reduced level of expression and/or activity of TNFR1.
- As used herein, the term "TNFR1 (Tumor necrosis factor receptor 1)", also known as tumor necrosis factor receptor superfamily member 1A (TNFRSF1A), CD120a and p55 refers to the TNFR1 gene and to its polynucleotide or polypeptide expression product. According to specific embodiments, the TNFR1 refers to the human TNFR1, such as provided in Gene ID: 7132 (SEQ ID NO: 2) and the following Accession Numbers: NM_001065 (SEQ ID NO: 3), NM_001346091 (SEQ ID NO: 4), NM_001346092 (SEQ ID NO: 5), NP_001056 (SEQ ID NO: 6), NP_001333020 (SEQ ID NO: 7) and NP_001333021 (SEQ ID NO: 8). According to specific embodiments, the TNFR1 refers to the mouse TNFR1, such as provided in Gene ID: 21937 (SEQ ID NO: 9) and in the following Accession Numbers: NM_011609 (SEQ ID NO: 10) and NP_035739 (SEQ ID NO: 11).
- According to specific embodiments, TNFR1 activity is at least one of (or two of or all of): binding TNFα, forming a trimer, activating the transcription factor NFκB and/or mediating apoptosis.
- As used herein, "reduced level of expression and/or activity" refers to a decrease of at least 10 % in TNFR1 expression or activity in comparison to a control cell of the same origin which was not contacted with an agent which downregulates expression and/or activity of TNFR1, as may be determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNFα signaling assays. According to a specific embodiment, the decrease is in at least 20 %, 30 %, 40 % or even higher say, 50 %, 60 %, 70 %, 80 %, 90 % or even 100 %.
- According to specific embodiments, the cell does not express TNFR1.
- According to specific embodiments the cells with reduced expression and/or activity of TNFR1 are genetically modified cells. Methods and agents for genetically modifying cells are well known in the art and are further described in details hereinbelow.
- According to specific embodiments, the cells are genetically modified with a CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN) or meganuclease for downregulating expression of said TNFR1.
- According to specific embodiments, the cells are genetically modified with a CRISPR/Cas system for downregulating expression of said TNFR1.
- According to specific embodiments, the cells with reduced level of expression and/or activity of TNFR1 express similar levels and/or activity of TNFR2 in comparison to a control cell of the same origin which was not contacted with an agent which downregulates expression and/or activity of TNFR1, as may be determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNFα signaling assays.
- As used herein, the term "TNFR2 (Tumor necrosis factor receptor 2)", also known as tumor necrosis factor receptor superfamily member 1B (TNFRSF1B) and CD120b and p75 refers to the TNFR2 gene and to its polynucleotide or polypeptide expression product. According to specific embodiments, the TNFR2 refers to the human TNFR2, such as provided in Gene ID: 7133 (SEQ ID NO: 12) and the following Accession Numbers: NM_001066 (SEQ ID NO: 13) and NP_001057 (SEQ ID NO: 14). According to specific embodiments, the TNFR1 refers to the mouse TNFR2, such as provided in Gene ID: 21938 (SEQ ID NO: 15) and in the following Accession Numbers: NM_011610 (SEQ ID NO: 16) and NP_035740 (SEQ ID NO: 17).
- According to specific embodiments, TNFR2 activity is at least one of (or two of or all of): binding TNFα and/or mediating the recruitment of two anti-apoptotic proteins, c-IAP1 and c-IAP2.
- As the cells of some embodiments of the present invention are characterized by reduced level of expression and/or activity of TNFR1, the present invention also contemplates methods of obtaining such cells.
- Thus, according to an aspect of the present invention there is provided a method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells, the method comprising:
- (i) contacting ex-vivo or in-vitro differentiated cells or bone marrow stem cells with an agent which downregulates expression and/or activity of TNFR1; and
- (ii) contacting ex-vivo or in-vitro said cells with TNFα.
- According to another aspect of the present invention there is provided a method of downregulating expression and/or activity of TNFR1 in differentiated cells, the method comprising:
- (i) inducing ex-vivo or in-vivo differentiation of stem cells;
- (ii) contacting ex-vivo or in-vitro said cells with an agent which downregulates expression and/or activity of TNFR1; and
- (iii) contacting ex-vivo or in-vitro said cells with TNFα.
- According to specific embodiments, inducing differentiation of the stem cells is effected prior to contacting with the agent. Methods of inducing differentiation of stem cells are known in the art and are further described hereinabove.
- According to another aspect of the present invention there is provided a method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells, the method comprising genetically modifying ex-vivo or in-vitro differentiated cells or bone marrow stem cells with an agent which downregulates expression and/or activity of TNFR1.
- According to specific embodiments, the method further comprising contacting the cells ex-vivo or in-vitro with TNFα.
- According to specific embodiments, contacting the cells with the agent is effected prior to contacting the cells with the TNFα.
- According to specific embodiments, contacting the cells with the agent is effected following contacting the cells with the TNFα.
- Specific embodiments of the present invention contemplates a method of treating an ischemic disease in a subject in need thereof, the method comprising:
- (i) obtaining cells according to the methods of downregulating expression and/or activity of TNFR1 described herein; and
- (ii) administering to the subject a therapeutically effective amount of said cells, thereby treating the ischemic disease in the subject.
- As used herein, "downregulating expression and/or activity" and "downregulates expression and/or activity" refer to a decrease of at least 5 % in expression and/or biological function in the presence of the agent in comparison to same in the absence of the agent, as determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNFα signaling assays. According to a specific embodiment, the decrease is in at least 10 %, 30 %, 40 % or even higher say, 50 %, 60 %, 70 %, 80 %, 90 % or even 100 %. According to specific embodiments, the decrease is at least 1.5 fold, at least 2 fold, at least 3 fold, at least 5 fold, at least 10 fold, or at least 20 fold as compared to same in the absence of the agent.
- According to specific embodiments, downregulating expression refers to the absence of mRNA and/or protein, as detected by RT-PCR or Western blot, respectively.
- According to specific embodiments, "downregulating expression and/or activity of TNFR1" and "downregulates expression and/or activity of TNFR1 "refer to the ability to specifically downregulate the expression and/or activity of TNFR1 and not to downregulate the expression and/or activity of TNFR2, as determined by e.g. PCR, ELISA, Western blot analysis, immunoprecipitation, flow cytometry, immuno-staining, TNFα signaling assays. This selective inhibition can be manifested as higher affinity (e.g., Kd) of the agent to TNFR1 than to TNFR2. Increased affinity can be of at least 5, 10, 100, 1000 or 10000 fold.
- Hence, according to specific embodiments, the agent does not downregulate expression and/or activity of TNFR2.
- Downregulating expression and/or activity can be effected at the genomic (e.g. homologous recombination and site specific endonucleases) and/or the transcript level using a variety of molecules which interfere with transcription and/or translation (e.g., RNA silencing agents) of a target expression product described herein, but may also be effected at the protein level (e.g., antibodies, small molecules, inhibitory peptides, enzymes that cleave the polypeptide, aptamers and the like).
- According to specific embodiments, the agent directly binds TNFR1.
- According to other specific embodiments, the agent indirectly binds TNFR1 by acting through an intermediary molecule, for example the agent binds to or modulates a molecule that in turn binds to or modulates the target.
- According to specific embodiments, the agent does not bind TNFR2.
- Non-limiting examples of down-regulating agents at the nucleic acid level are described in details hereinbelow.
- Down-regulation at the nucleic acid level is typically effected using a nucleic acid agent, having a nucleic acid backbone, DNA, RNA, mimetics thereof or a combination of same. The nucleic acid agent may be encoded from a DNA molecule or provided to the cell per se.
- Downregulation can be achieved by inactivating the gene (i.e. the TNFR1 gene) via introducing targeted mutations involving loss-of function alterations (e.g. point mutations, deletions and insertions) in the gene structure.
- As used herein, the phrase "loss-of-function alterations" refers to any mutation in the DNA sequence of a gene which results in downregulation of the expression level and/or activity of the expressed product, i.e., the mRNA transcript and/or the translated protein. Non-limiting examples of such loss-of-function alterations include a missense mutation, i.e., a mutation which changes an amino acid residue in the protein with another amino acid residue and thereby abolishes the enzymatic activity of the protein; a nonsense mutation, i.e., a mutation which introduces a stop codon in a protein, e.g., an early stop codon which results in a shorter protein devoid of the enzymatic activity; a frame-shift mutation, i.e., a mutation, usually, deletion or insertion of nucleic acid(s) which changes the reading frame of the protein, and may result in an early termination by introducing a stop codon into a reading frame (e.g., a truncated protein, devoid of the enzymatic activity), or in a longer amino acid sequence (e.g., a readthrough protein) which affects the secondary or tertiary structure of the protein and results in a non-functional protein, devoid of the enzymatic activity of the non-mutated polypeptide; a readthrough mutation due to a frame-shift mutation or a modified stop codon mutation (i.e., when the stop codon is mutated into an amino acid codon), with an abolished enzymatic activity; a promoter mutation, i.e., a mutation in a promoter sequence, usually 5' to the transcription start site of a gene, which results in down-regulation of a specific gene product; a regulatory mutation, i.e., a mutation in a region upstream or downstream, or within a gene, which affects the expression of the gene product; a deletion mutation, i.e., a mutation which deletes coding nucleic acids in a gene sequence and which may result in a frame-shift mutation or an in-frame mutation (within the coding sequence, deletion of one or more amino acid codons); an insertion mutation, i.e., a mutation which inserts coding or non-coding nucleic acids into a gene sequence, and which may result in a frame-shift mutation or an in-frame insertion of one or more amino acid codons; an inversion, i.e., a mutation which results in an inverted coding or non-coding sequence; a splice mutation i.e., a mutation which results in abnormal splicing or poor splicing; and a duplication mutation, i.e., a mutation which results in a duplicated coding or non-coding sequence, which can be in-frame or can cause a frame-shift.
- According to specific embodiments loss-of-function alteration of a gene may comprise at least one allele of the gene.
- The term "allele" as used herein, refers to any of one or more alternative forms of a gene locus, all of which alleles relate to a trait or characteristic. In a diploid cell or organism, the two alleles of a given gene occupy corresponding loci on a pair of homologous chromosomes.
- According to other specific embodiments loss-of-function alteration of a gene comprises both alleles of the gene. In such instances the TNFR1 may be in a homozygous form or in a heterozygous form.
- Methods of introducing nucleic acid alterations to a gene of interest are well known in the art [see for example Menke D. Genesis (2013) 51: - 618; Capecchi, Science (1989) 244:1288-1292; Santiago et al. Proc Natl Acad Sci USA (2008) 105:5809-5814; International Patent Application Nos.
WO 2014085593 ,WO 2009071334 andWO 2011146121 ;US Patent Nos. 8771945 ,8586526 ,6774279 and UP Patent Application Publication Nos.20030232410 ,20050026157 ,US20060014264 ; the contents of which are incorporated by reference in their entireties] and include targeted homologous recombination, site specific recombinases, PB transposases and genome editing by engineered nucleases. Agents for introducing nucleic acid alterations to a gene of interest can be designed publically available sources or obtained commercially from Transposagen, Addgene and Sangamo Biosciences. - Following is a description of various exemplary methods used to introduce nucleic acid alterations to a gene of interest and agents for implementing same that can be used according to specific embodiments of the present invention.
- Genome Editing using engineered endonucleases - this approach refers to a reverse genetics method using artificially engineered nucleases to cut and create specific double-stranded breaks at a desired location(s) in the genome, which are then repaired by cellular endogenous processes such as, homology directed repair (HDR) and non-homologous end-joining (NHEJ). NHEJ directly joins the DNA ends in a double-stranded break, while HDR utilizes a homologous sequence as a template for regenerating the missing DNA sequence at the break point. In order to introduce specific nucleotide modifications to the genomic DNA, a DNA repair template containing the desired sequence must be present during HDR. Genome editing cannot be performed using traditional restriction endonucleases since most restriction enzymes recognize a few base pairs on the DNA as their target and the probability is very high that the recognized base pair combination will be found in many locations across the genome resulting in multiple cuts not limited to a desired location. To overcome this challenge and create site-specific single- or double-stranded breaks, several distinct classes of nucleases have been discovered and bioengineered to date. These include the meganucleases, Zinc finger nucleases (ZFNs), transcription-activator like effector nucleases (TALENs) and CRISPR/Cas system.
- Meganucleases - Meganucleases are commonly grouped into four families: the LAGLIDADG family, the GIY-YIG family, the His-Cys box family and the HNH family. These families are characterized by structural motifs, which affect catalytic activity and recognition sequence. For instance, members of the LAGLIDADG family are characterized by having either one or two copies of the conserved LAGLIDADG motif. The four families of meganucleases are widely separated from one another with respect to conserved structural elements and, consequently, DNA recognition sequence specificity and catalytic activity. Meganucleases are found commonly in microbial species and have the unique property of having very long recognition sequences (>14bp) thus making them naturally very specific for cutting at a desired location. This can be exploited to make site-specific double-stranded breaks in genome editing. One of skill in the art can use these naturally occurring meganucleases, however the number of such naturally occurring meganucleases is limited. To overcome this challenge, mutagenesis and high throughput screening methods have been used to create meganuclease variants that recognize unique sequences. For example, various meganucleases have been fused to create hybrid enzymes that recognize a new sequence. Alternatively, DNA interacting amino acids of the meganuclease can be altered to design sequence specific meganucleases (see e.g.,
US Patent 8,021,867 ). Meganucleases can be designed using the methods described in e.g., Certo, MT et al. Nature Methods (2012) 9:073-975;U.S. Patent Nos. 8,304,222 ;8,021,867 ;8, 119,381 ;8, 124,369 ;8, 129,134 ;8,133,697 ;8,143,015 ;8,143,016 ;8, 148,098 ; or8, 163,514 , the contents of each are incorporated herein by reference in their entirety. Alternatively, meganucleases with site specific cutting characteristics can be obtained using commercially available technologies e.g., Precision Biosciences' Directed Nuclease Editor™ genome editing technology. - ZFNs and TALENs - Two distinct classes of engineered nucleases, zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), have both proven to be effective at producing targeted double-stranded breaks (Christian et al., 2010; Kim et al., 1996; Li et al., 2011; Mahfouz et al., 2011; Miller et al., 2010).
- Basically, ZFNs and TALENs restriction endonuclease technology utilizes a non-specific DNA cutting enzyme which is linked to a specific DNA binding domain (either a series of zinc finger domains or TALE repeats, respectively). Typically a restriction enzyme whose DNA recognition site and cleaving site are separate from each other is selected. The cleaving portion is separated and then linked to a DNA binding domain, thereby yielding an endonuclease with very high specificity for a desired sequence. An exemplary restriction enzyme with such properties is Fokl. Additionally Fokl has the advantage of requiring dimerization to have nuclease activity and this means the specificity increases dramatically as each nuclease partner recognizes a unique DNA sequence. To enhance this effect, Fokl nucleases have been engineered that can only function as heterodimers and have increased catalytic activity. The heterodimer functioning nucleases avoid the possibility of unwanted homodimer activity and thus increase specificity of the double-stranded break.
- Thus, for example to target a specific site, ZFNs and TALENs are constructed as nuclease pairs, with each member of the pair designed to bind adjacent sequences at the targeted site. Upon transient expression in cells, the nucleases bind to their target sites and the FokI domains heterodimerize to create a double-stranded break. Repair of these double-stranded breaks through the non-homologous end-joining (NHEJ) pathway most often results in small deletions or small sequence insertions. Since each repair made by NHEJ is unique, the use of a single nuclease pair can produce an allelic series with a range of different deletions at the target site. The deletions typically range anywhere from a few base pairs to a few hundred base pairs in length, but larger deletions have successfully been generated in cell culture by using two pairs of nucleases simultaneously (Carlson et al., 2012; Lee et al., 2010). In addition, when a fragment of DNA with homology to the targeted region is introduced in conjunction with the nuclease pair, the double-stranded break can be repaired via homology directed repair to generate specific modifications (Li et al., 2011; Miller et al., 2010; Urnov et al., 2005).
- Although the nuclease portions of both ZFNs and TALENs have similar properties, the difference between these engineered nucleases is in their DNA recognition peptide. ZFNs rely on Cys2- His2 zinc fingers and TALENs on TALEs. Both of these DNA recognizing peptide domains have the characteristic that they are naturally found in combinations in their proteins. Cys2-His2 Zinc fingers typically found in repeats that are 3 bp apart and are found in diverse combinations in a variety of nucleic acid interacting proteins. TALEs on the other hand are found in repeats with a one-to-one recognition ratio between the amino acids and the recognized nucleotide pairs. Because both zinc fingers and TALEs happen in repeated patterns, different combinations can be tried to create a wide variety of sequence specificities. Approaches for making site-specific zinc finger endonucleases include, e.g., modular assembly (where Zinc fingers correlated with a triplet sequence are attached in a row to cover the required sequence), OPEN (low-stringency selection of peptide domains vs. triplet nucleotides followed by high-stringency selections of peptide combination vs. the final target in bacterial systems), and bacterial one-hybrid screening of zinc finger libraries, among others. ZFNs can also be designed and obtained commercially from e.g., Sangamo Biosciences™ (Richmond, CA).
- Method for designing and obtaining TALENs are described in e.g. Reyon et al. Nature Biotechnology 2012 May;30(5):460-5; Miller et al. Nat Biotechnol. (2011) 29: 143-148; Cermak et al. Nucleic Acids Research (2011) 39 (12): e82 and Zhang et al. Nature Biotechnology (2011) 29 (2): 149-53. A recently developed web-based program named Mojo Hand was introduced by Mayo Clinic for designing TAL and TALEN constructs for genome editing applications (can be accessed through www(dot)talendesign(dot)org). TALEN can also be designed and obtained commercially from e.g., Sangamo Biosciences™ (Richmond, CA).
- CRISPR-Cas system - Many bacteria and archaea contain endogenous RNA-based adaptive immune systems that can degrade nucleic acids of invading phages and plasmids. These systems consist of clustered regularly interspaced short palindromic repeat (CRISPR) genes that produce RNA components and CRISPR associated (Cas) genes that encode protein components. The CRISPR RNAs (crRNAs) contain short stretches of homology to specific viruses and plasmids and act as guides to direct Cas nucleases to degrade the complementary nucleic acids of the corresponding pathogen. Studies of the type II CRISPR/Cas system of Streptococcus pyogenes have shown that three components form an RNA/protein complex and together are sufficient for sequence-specific nuclease activity: the Cas9 nuclease, a crRNA containing 20 base pairs of homology to the target sequence, and a trans-activating crRNA (tracrRNA) (Jinek et al. Science (2012) 337: 816-821.). It was further demonstrated that a synthetic chimeric guide RNA (gRNA) composed of a fusion between crRNA and tracrRNA could direct Cas9 to cleave DNA targets that are complementary to the crRNA in vitro. It was also demonstrated that transient expression of Cas9 in conjunction with synthetic gRNAs can be used to produce targeted double-stranded brakes in a variety of different species (Cho et al., 2013; Cong et al., 2013; DiCarlo et al., 2013; Hwang et al., 2013a,b; Jinek et al., 2013; Mali et al., 2013).
- The CRIPSR/Cas system for genome editing contains two distinct components: a gRNA and an endonuclease e.g. Cas9.
- The gRNA is typically a 20 nucleotide sequence encoding a combination of the target homologous sequence (crRNA) and the endogenous bacterial RNA that links the crRNA to the Cas9 nuclease (tracrRNA) in a single chimeric transcript. The gRNA/Cas9 complex is recruited to the target sequence by the base-pairing between the gRNA sequence and the complement genomic DNA. For successful binding of Cas9, the genomic target sequence must also contain the correct Protospacer Adjacent Motif (PAM) sequence immediately following the target sequence. The binding of the gRNA/Cas9 complex localizes the Cas9 to the genomic target sequence so that the Cas9 can cut both strands of the DNA causing a double-strand break. Just as with ZFNs and TALENs, the double-stranded brakes produced by CRISPR/Cas can undergo homologous recombination or NHEJ.
- The Cas9 nuclease has two functional domains: RuvC and HNH, each cutting a different DNA strand. When both of these domains are active, the Cas9 causes double strand breaks in the genomic DNA.
- A significant advantage of CRISPR/Cas is that the high efficiency of this system coupled with the ability to easily create synthetic gRNAs enables multiple genes to be targeted simultaneously. In addition, the majority of cells carrying the mutation present biallelic mutations in the targeted genes.
- However, apparent flexibility in the base-pairing interactions between the gRNA sequence and the genomic DNA target sequence allows imperfect matches to the target sequence to be cut by Cas9.
- Modified versions of the Cas9 enzyme containing a single inactive catalytic domain, either RuvC- or HNH-, are called 'nickases'. With only one active nuclease domain, the Cas9 nickase cuts only one strand of the target DNA, creating a single-strand break or 'nick'. A single-strand break, or nick, is normally quickly repaired through the HDR pathway, using the intact complementary DNA strand as the template. However, two proximal, opposite strand nicks introduced by a Cas9 nickase are treated as a double-strand break, in what is often referred to as a 'double nick' CRISPR system. A double-nick can be repaired by either NHEJ or HDR depending on the desired effect on the gene target. Thus, if specificity and reduced off-target effects are crucial, using the Cas9 nickase to create a double-nick by designing two gRNAs with target sequences in close proximity and on opposite strands of the genomic DNA would decrease off-target effect as either gRNA alone will result in nicks that will not change the genomic DNA.
- Modified versions of the Cas9 enzyme containing two inactive catalytic domains (dead Cas9, or dCas9) have no nuclease activity while still able to bind to DNA based on gRNA specificity. The dCas9 can be utilized as a platform for DNA transcriptional regulators to activate or repress gene expression by fusing the inactive enzyme to known regulatory domains. For example, the binding of dCas9 alone to a target sequence in genomic DNA can interfere with gene transcription.
- There are a number of publically available tools available to help choose and/or design target sequences as well as lists of bioinformatically determined unique gRNAs for different genes in different species such as the Feng Zhang lab's Target Finder, the Michael Boutros lab's Target Finder (E-CRISP), the RGEN Tools: Cas-OFFinder, the CasFinder: Flexible algorithm for identifying specific Cas9 targets in genomes and the CRISPR Optimal Target Finder.
- Non-limiting examples of gRNA sequences that can be used with some embodiments of the present invention are described in Tables 1A and 1B hereinbelow.
- According to specific embodiments, the gRNA sequence does not have a significant off target effect. Methods of determining off target effect are well known in the art, such as BGI Human Whole Genome Sequencing (described in Nature;491:65-56.2012), next generation sequencing (NGS) using e.g. commercially available kits such as Alt-R-Genom Editing (IDT detection kit) or Sure select target enrich <1% variant allele frequency (Agilent).
- In order to use the CRISPR system, both gRNA and Cas9 should be expressed in a target cell. The insertion vector can contain both cassettes on a single plasmid or the cassettes are expressed from two separate plasmids. CRISPR plasmids are commercially available such as the px330 plasmid from Addgene. Alternatively, the target cell can be transfected with both gRNA and Cas9 without plasmid using e.g. a transfection reagent such as CRISPRMAX [see e.g. Yu et al. (2016) JDlBiotechnol Lett. 38(6):919-29]. In some cells electroporation can improve the transfection of the gRNA and the Cas9 [see e.g. Liang et al. (2015) Journal of Biotechnology 208, 2015, Pages 44-53; and Liang et al. (2017) Journal of Biotechnology, Volume 241, 2017, pp. 136-146].
Table 1A: TNFR1 guide RNA sequences designed according to the mouse TNFR1 Sequence of gRNA Forward Reverse GCD Band sizes CRISPR1 


604 378bp 226bp CRISPR2 


600 357bp 243bp CRISPR3 


608 417bp 191bp * Mouse tumor necrosis factor receptor superfamily, member 1a mRNA: NM_011609 (SEQ ID NO: 10) Total No. of Exons: 10, Targeted Exons: 2 (2, 3) Table 1b: TNFR1 guide RNA sequences designed according to the human TNFR1 Sequence of gRNA Forward Reverse GCD Band sizes CRISPR1 

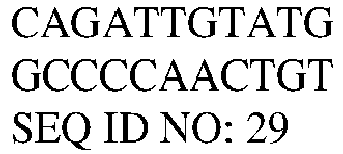
625 415bp 210bp CRISPR2 


648 420bp 228bp CRISPR3 


484 360bp 124bp CRISPR4 


CRISPR5 


CRISPR6 


CRISPR7 


* Human tumor necrosis factor receptor superfamily, member 1a mRNA: NM_001065 (SEQ ID NO: 3) - "Hit and run" or "in-out" - involves a two-step recombination procedure. In the first step, an insertion-type vector containing a dual positive/negative selectable marker cassette is used to introduce the desired sequence alteration. The insertion vector contains a single continuous region of homology to the targeted locus and is modified to carry the mutation of interest. This targeting construct is linearized with a restriction enzyme at a one site within the region of homology, electroporated into the cells, and positive selection is performed to isolate homologous recombinants. These homologous recombinants contain a local duplication that is separated by intervening vector sequence, including the selection cassette. In the second step, targeted clones are subjected to negative selection to identify cells that have lost the selection cassette via intrachromosomal recombination between the duplicated sequences. The local recombination event removes the duplication and, depending on the site of recombination, the allele either retains the introduced mutation or reverts to wild type. The end result is the introduction of the desired modification without the retention of any exogenous sequences.
- The "double-replacement" or "tag and exchange" strategy - involves a two-step selection procedure similar to the hit and run approach, but requires the use of two different targeting constructs. In the first step, a standard targeting vector with 3' and 5' homology arms is used to insert a dual positive/negative selectable cassette near the location where the mutation is to be introduced. After electroporation and positive selection, homologously targeted clones are identified. Next, a second targeting vector that contains a region of homology with the desired mutation is electroporated into targeted clones, and negative selection is applied to remove the selection cassette and introduce the mutation. The final allele contains the desired mutation while eliminating unwanted exogenous sequences.
- Site-Specific Recombinases - The Cre recombinase derived from the P1 bacteriophage and Flp recombinase derived from the yeast Saccharomyces cerevisiae are site-specific DNA recombinases each recognizing a unique 34 base pair DNA sequence (termed "Lox" and "FRT", respectively) and sequences that are flanked with either Lox sites or FRT sites can be readily removed via site-specific recombination upon expression of Cre or Flp recombinase, respectively. For example, the Lox sequence is composed of an asymmetric eight base pair spacer region flanked by 13 base pair inverted repeats. Cre recombines the 34 base pair lox DNA sequence by binding to the 13 base pair inverted repeats and catalyzing strand cleavage and religation within the spacer region. The staggered DNA cuts made by Cre in the spacer region are separated by 6 base pairs to give an overlap region that acts as a homology sensor to ensure that only recombination sites having the same overlap region recombine.
- Basically, the site specific recombinase system offers means for the removal of selection cassettes after homologous recombination. This system also allows for the generation of conditional altered alleles that can be inactivated or activated in a temporal or tissue-specific manner. Of note, the Cre and Flp recombinases leave behind a Lox or FRT "scar" of 34 base pairs. The Lox or FRT sites that remain are typically left behind in an intron or 3' UTR of the modified locus, and current evidence suggests that these sites usually do not interfere significantly with gene function.
- Thus, Cre/Lox and Flp/FRT recombination involves introduction of a targeting vector with 3' and 5' homology arms containing the mutation of interest, two Lox or FRT sequences and typically a selectable cassette placed between the two Lox or FRT sequences. Positive selection is applied and homologous recombinants that contain targeted mutation are identified. Transient expression of Cre or Flp in conjunction with negative selection results in the excision of the selection cassette and selects for cells where the cassette has been lost. The final targeted allele contains the Lox or FRT scar of exogenous sequences.
- Transposases - As used herein, the term "transposase" refers to an enzyme that binds to the ends of a transposon and catalyzes the movement of the transposon to another part of the genome.
- As used herein the term "transposon" refers to a mobile genetic element comprising a nucleotide sequence which can move around to different positions within the genome of a single cell. In the process the transposon can cause mutations and/or change the amount of a DNA in the genome of the cell.
- A number of transposon systems that are able to also transpose in cells e.g. vertebrates have been isolated or designed, such as Sleeping Beauty [Izsvák and Ivics Molecular Therapy (2004) 9, 147-156], piggyBac [Wilson et al. Molecular Therapy (2007) 15, 139-145], Tol2 [Kawakami et al. PNAS (2000) 97 (21): 11403-11408] or Frog Prince [Miskey et al. Nucleic Acids Res. ]. Generally, DNA transposons translocate from one DNA site to another in a simple, cut-and-paste manner. Each of these elements has their own advantages, for example, Sleeping Beauty is particularly useful in region-specific mutagenesis, whereas Tol2 has the highest tendency to integrate into expressed genes. Hyperactive systems are available for Sleeping Beauty and piggyBac. Most importantly, these transposons have distinct target site preferences, and can therefore introduce sequence alterations in overlapping, but distinct sets of genes. Therefore, to achieve the best possible coverage of genes, the use of more than one element is particularly preferred. The basic mechanism is shared between the different transposases, therefore we will describe piggyBac (PB) as an example.
- PB is a 2.5 kb insect transposon originally isolated from the cabbage looper moth, Trichoplusia ni. The PB transposon consists of asymmetric terminal repeat sequences that flank a transposase, PBase. PBase recognizes the terminal repeats and induces transposition via a "cut-and-paste" based mechanism, and preferentially transposes into the host genome at the tetranucleotide sequence TTAA. Upon insertion, the TTAA target site is duplicated such that the PB transposon is flanked by this tetranucleotide sequence. When mobilized, PB typically excises itself precisely to reestablish a single TTAA site, thereby restoring the host sequence to its pretransposon state. After excision, PB can transpose into a new location or be permanently lost from the genome.
- Typically, the transposase system offers an alternative means for the removal of selection cassettes after homologous recombination quit similar to the use Cre/Lox or Flp/FRT. Thus, for example, the PB transposase system involves introduction of a targeting vector with 3' and 5' homology arms containing the mutation of interest, two PB terminal repeat sequences at the site of an endogenous TTAA sequence and a selection cassette placed between PB terminal repeat sequences. Positive selection is applied and homologous recombinants that contain targeted mutation are identified. Transient expression of PBase removes in conjunction with negative selection results in the excision of the selection cassette and selects for cells where the cassette has been lost. The final targeted allele contains the introduced mutation with no exogenous sequences.
- For PB to be useful for the introduction of sequence alterations, there must be a native TTAA site in relatively close proximity to the location where a particular mutation is to be inserted.
- Genome editing using recombinant adeno-associated virus (rAAV) platform - this genome-editing platform is based on rAAV vectors which enable insertion, deletion or substitution of DNA sequences in the genomes of live mammalian cells. The rAAV genome is a single-stranded deoxyribonucleic acid (ssDNA) molecule, either positive- or negative-sensed, which is about 4.7 kb long. These single-stranded DNA viral vectors have high transduction rates and have a unique property of stimulating endogenous homologous recombination in the absence of double-strand DNA breaks in the genome. One of skill in the art can design a rAAV vector to target a desired genomic locus and perform both gross and/or subtle endogenous gene alterations in a cell. rAAV genome editing has the advantage in that it targets a single allele and does not result in any off-target genomic alterations. rAAV genome editing technology is commercially available, for example, the rAAV GENESIS™ system from Horizon™ (Cambridge, UK).
- It will be appreciated that the agent can be a mutagen that causes random mutations and the cells exhibiting downregulation of the expression level and/or activity of the target may be selected.
- The mutagens may be, but are not limited to, genetic, chemical or radiation agents. For example, the mutagen may be ionizing radiation, such as, but not limited to, ultraviolet light, gamma rays or alpha particles. Other mutagens may include, but not be limited to, base analogs, which can cause copying errors; deaminating agents, such as nitrous acid; intercalating agents, such as ethidium bromide; alkylating agents, such as bromouracil; transposons; natural and synthetic alkaloids; bromine and derivatives thereof; sodium azide; psoralen (for example, combined with ultraviolet radiation). The mutagen may be a chemical mutagen such as, but not limited to, ICR191, 1,2,7,8-diepoxy-octane (DEO), 5-azaC, N-methyl-N-nitrosoguanidine (MNNG) or ethyl methane sulfonate (EMS).
- Methods for qualifying efficacy and detecting sequence alteration are well known in the art and include, but not limited to, DNA sequencing, electrophoresis, an enzyme-based mismatch detection assay and a hybridization assay such as PCR, RT-PCR, RNase protection, in-situ hybridization, primer extension, Southern blot, Northern Blot and dot blot analysis.
- Sequence alterations in a specific gene can also be determined at the protein level using e.g. chromatography, electrophoretic methods, immunodetection assays such as ELISA and western blot analysis and immunohistochemistry.
- In addition, one ordinarily skilled in the art can readily design a knock-in/knock-out construct including positive and/or negative selection markers for efficiently selecting transformed cells that underwent a homologous recombination event with the construct. Positive selection provides a means to enrich the population of clones that have taken up foreign DNA. Non-limiting examples of such positive markers include glutamine synthetase, dihydrofolate reductase (DHFR), markers that confer antibiotic resistance, such as neomycin, hygromycin, puromycin, and blasticidin S resistance cassettes. Negative selection markers are necessary to select against random integrations and/or elimination of a marker sequence (e.g. positive marker). Non-limiting examples of such negative markers include the herpes simplex-thymidine kinase (HSV-TK) which converts ganciclovir (GCV) into a cytotoxic nucleoside analog, hypoxanthine phosphoribosyltransferase (HPRT) and adenine phosphoribosytransferase (ARPT).
- Downregulation can also be achieved by RNA silencing. As used herein, the phrase "RNA silencing" refers to a group of regulatory mechanisms [e.g. RNA interference (RNAi), transcriptional gene silencing (TGS), post-transcriptional gene silencing (PTGS), quelling, co-suppression, and translational repression] mediated by RNA molecules which result in the inhibition or "silencing" of the expression of a corresponding protein-coding gene. RNA silencing has been observed in many types of organisms, including plants, animals, and fungi.
- As used herein, the term "RNA silencing agent" refers to an RNA which is capable of specifically inhibiting or "silencing" the expression of a target gene. In certain embodiments, the RNA silencing agent is capable of preventing complete processing (e.g., the full translation and/or expression) of an mRNA molecule through a post-transcriptional silencing mechanism. RNA silencing agents include non-coding RNA molecules, for example RNA duplexes comprising paired strands, as well as precursor RNAs from which such small non-coding RNAs can be generated. Exemplary RNA silencing agents include dsRNAs such as siRNAs, miRNAs and shRNAs.
- In one embodiment, the RNA silencing agent is capable of inducing RNA interference.
- In another embodiment, the RNA silencing agent is capable of mediating translational repression.
- According to an embodiment of the invention, the RNA silencing agent is specific to the target RNA (i.e. TNFR1) and does not cross inhibit or silence other targets or a splice variant which exhibits 99% or less global homology to the target gene, e.g., less than 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81% global homology to the target gene; as determined by PCR, Western blot, Immunohistochemistry and/or flow cytometry.
- RNA interference refers to the process of sequence-specific post-transcriptional gene silencing in animals mediated by short interfering RNAs (siRNAs).
- Following is a detailed description on RNA silencing agents that can be used according to specific embodiments of the present invention.
- DsRNA, siRNA and shRNA - The presence of long dsRNAs in cells stimulates the activity of a ribonuclease III enzyme referred to as dicer. Dicer is involved in the processing of the dsRNA into short pieces of dsRNA known as short interfering RNAs (siRNAs). Short interfering RNAs derived from dicer activity are typically about 21 to about 23 nucleotides in length and comprise about 19 base pair duplexes. The RNAi response also features an endonuclease complex, commonly referred to as an RNA-induced silencing complex (RISC), which mediates cleavage of single-stranded RNA having sequence complementary to the antisense strand of the siRNA duplex. Cleavage of the target RNA takes place in the middle of the region complementary to the antisense strand of the siRNA duplex.
- Accordingly, some embodiments of the invention contemplate use of dsRNA to downregulate protein expression from mRNA.
- According to one embodiment dsRNA longer than 30 bp are used. Various studies demonstrate that long dsRNAs can be used to silence gene expression without inducing the stress response or causing significant off-target effects - see for example [Strat et al., Nucleic Acids Research, 2006, Vol. 34, No. 13 3803-3810; Bhargava A et al. Brain Res. Protoc. 2004;13:115-125; Diallo M., et al., Oligonucleotides. 2003;13:381-392; Paddison P.J., et al., Proc. Natl Acad. Sci. USA. 2002;99:1443-1448; Tran N., et al., FEBS Lett. 2004;573:127-134].
- According to some embodiments of the invention, dsRNA is provided in cells where the interferon pathway is not activated, see for example Billy et al., PNAS 2001, Vol 98, pages 14428-14433; and Diallo et al, Oligonucleotides, October 1, 2003, 13(5): 381-392. doi: 10.1089/154545703322617069.
- According to an embodiment of the invention, the long dsRNA are specifically designed not to induce the interferon and PKR pathways for down-regulating gene expression. For example, Shinagwa and Ishii [Genes & Dev. 17 (11): 1340-1345, 2003] have developed a vector, named pDECAP, to express long double-strand RNA from an RNA polymerase II (Pol II) promoter. Because the transcripts from pDECAP lack both the 5'-cap structure and the 3'-poly(A) tail that facilitate ds-RNA export to the cytoplasm, long ds-RNA from pDECAP does not induce the interferon response.
- Another method of evading the interferon and PKR pathways in mammalian systems is by introduction of small inhibitory RNAs (siRNAs) either via transfection or endogenous expression.
- The term "siRNA" refers to small inhibitory RNA duplexes (generally between 18-30 base pairs) that induce the RNA interference (RNAi) pathway. Typically, siRNAs are chemically synthesized as 21mers with a central 19 bp duplex region and symmetric 2-base 3'-overhangs on the termini, although it has been recently described that chemically synthesized RNA duplexes of 25-30 base length can have as much as a 100-fold increase in potency compared with 21mers at the same location. The observed increased potency obtained using longer RNAs in triggering RNAi is suggested to result from providing Dicer with a substrate (27mer) instead of a product (21mer) and that this improves the rate or efficiency of entry of the siRNA duplex into RISC.
- It has been found that position of the 3'-overhang influences potency of an siRNA and asymmetric duplexes having a 3'-overhang on the antisense strand are generally more potent than those with the 3'-overhang on the sense strand (Rose et al., 2005). This can be attributed to asymmetrical strand loading into RISC, as the opposite efficacy patterns are observed when targeting the antisense transcript.
- The strands of a double-stranded interfering RNA (e.g., a siRNA) may be connected to form a hairpin or stem-loop structure (e.g., a shRNA). Thus, as mentioned, the RNA silencing agent of some embodiments of the invention may also be a short hairpin RNA (shRNA).
- The term "shRNA", as used herein, refers to an RNA agent having a stem-loop structure, comprising a first and second region of complementary sequence, the degree of complementarity and orientation of the regions being sufficient such that base pairing occurs between the regions, the first and second regions being joined by a loop region, the loop resulting from a lack of base pairing between nucleotides (or nucleotide analogs) within the loop region. The number of nucleotides in the loop is a number between and including 3 to 23, or 5 to 15, or 7 to 13, or 4 to 9, or 9 to 11. Some of the nucleotides in the loop can be involved in base-pair interactions with other nucleotides in the loop. Examples of oligonucleotide sequences that can be used to form the loop include 5'-CAAGAGA-3' and 5'-UUACAA-3' (International Patent Application Nos.
WO2013126963 andWO2014107763 ). It will be recognized by one of skill in the art that the resulting single chain oligonucleotide forms a stem-loop or hairpin structure comprising a double-stranded region capable of interacting with the RNAi machinery. - Synthesis of RNA silencing agents suitable for use with some embodiments of the invention can be effected as follows. First, the mRNA sequence is scanned downstream of the AUG start codon for AA dinucleotide sequences. Occurrence of each AA and the 3' adjacent 19 nucleotides is recorded as potential siRNA target sites. Preferably, siRNA target sites are selected from the open reading frame, as untranslated regions (UTRs) are richer in regulatory protein binding sites. UTR-binding proteins and/or translation initiation complexes may interfere with binding of the siRNA endonuclease complex [Tuschl ChemBiochem. 2:239-245]. It will be appreciated though, that siRNAs directed at untranslated regions may also be effective, as demonstrated for GAPDH wherein siRNA directed at the 5' UTR mediated about 90 % decrease in cellular GAPDH mRNA and completely abolished protein level (www(dot)ambion(dot)com/techlib/tn/91/912(dot)html).
- Second, potential target sites are compared to an appropriate genomic database (e.g., human, mouse, rat etc.) using any sequence alignment software, such as the BLAST software available from the NCBI server (www(dot)ncbi(dot)nlm(dot)nih(dot)gov/BLAST/). Putative target sites which exhibit significant homology to other coding sequences are filtered out.
- Qualifying target sequences are selected as template for siRNA synthesis. Preferred sequences are those including low G/C content as these have proven to be more effective in mediating gene silencing as compared to those with G/C content higher than 55 %. Several target sites are preferably selected along the length of the target gene for evaluation. For better evaluation of the selected siRNAs, a negative control is preferably used in conjunction. Negative control siRNA preferably include the same nucleotide composition as the siRNAs but lack significant homology to the genome. Thus, a scrambled nucleotide sequence of the siRNA is preferably used, provided it does not display any significant homology to any other gene.
- It will be appreciated that, and as mentioned hereinabove, the RNA silencing agent of some embodiments of the invention need not be limited to those molecules containing only RNA, but further encompasses chemically-modified nucleotides and non-nucleotides.
- miRNA and miRNA mimics - According to another embodiment the RNA silencing agent may be a miRNA.
- The term "microRNA", "miRNA", and "miR" are synonymous and refer to a collection of non-coding single-stranded RNA molecules of about 19-28 nucleotides in length, which regulate gene expression. miRNAs are found in a wide range of organisms (viruses.fwdarw.humans) and have been shown to play a role in development, homeostasis, and disease etiology.
- Below is a brief description of the mechanism of miRNA activity.
- Genes coding for miRNAs are transcribed leading to production of an miRNA precursor known as the pri-miRNA. The pri-miRNA is typically part of a polycistronic RNA comprising multiple pri-miRNAs. The pri-miRNA may form a hairpin with a stem and loop. The stem may comprise mismatched bases.
- The hairpin structure of the pri-miRNA is recognized by Drosha, which is an RNase III endonuclease. Drosha typically recognizes terminal loops in the pri-miRNA and cleaves approximately two helical turns into the stem to produce a 60-70 nucleotide precursor known as the pre-miRNA. Drosha cleaves the pri-miRNA with a staggered cut typical of RNase III endonucleases yielding a pre-miRNA stem loop with a 5' phosphate and ~2 nucleotide 3' overhang. It is estimated that approximately one helical turn of stem (~10 nucleotides) extending beyond the Drosha cleavage site is essential for efficient processing. The pre-miRNA is then actively transported from the nucleus to the cytoplasm by Ran-GTP and the export receptor Ex-portin-5.
- The double-stranded stem of the pre-miRNA is then recognized by Dicer, which is also an RNase III endonuclease. Dicer may also recognize the 5' phosphate and 3' overhang at the base of the stem loop. Dicer then cleaves off the terminal loop two helical turns away from the base of the stem loop leaving an additional 5' phosphate and ~2 nucleotide 3' overhang. The resulting siRNA-like duplex, which may comprise mismatches, comprises the mature miRNA and a similar-sized fragment known as the miRNA*. The miRNA and miRNA* may be derived from opposing arms of the pri-miRNA and pre-miRNA. miRNA* sequences may be found in libraries of cloned miRNAs but typically at lower frequency than the miRNAs.
- Although initially present as a double-stranded species with miRNA*, the miRNA eventually becomes incorporated as a single-stranded RNA into a ribonucleoprotein complex known as the RNA-induced silencing complex (RISC). Various proteins can form the RISC, which can lead to variability in specificity for miRNA/miRNA* duplexes, binding site of the target gene, activity of miRNA (repress or activate), and which strand of the miRNA/miRNA* duplex is loaded in to the RISC.
- When the miRNA strand of the miRNA:miRNA* duplex is loaded into the RISC, the miRNA* is removed and degraded. The strand of the miRNA:miRNA* duplex that is loaded into the RISC is the strand whose 5' end is less tightly paired. In cases where both ends of the miRNA:miRNA* have roughly equivalent 5' pairing, both miRNA and miRNA* may have gene silencing activity.
- The RISC identifies target nucleic acids based on high levels of complementarity between the miRNA and the mRNA, especially by nucleotides 2-7 of the miRNA.
- A number of studies have looked at the base-pairing requirement between miRNA and its mRNA target for achieving efficient inhibition of translation (reviewed by Bartel 2004, Cell 116-281). In mammalian cells, the first 8 nucleotides of the miRNA may be important (Doench & Sharp 2004 GenesDev 2004-504). However, other parts of the microRNA may also participate in mRNA binding. Moreover, sufficient base pairing at the 3' can compensate for insufficient pairing at the 5' (Brennecke et al, 2005 PLoS 3-e85). Computation studies, analyzing miRNA binding on whole genomes have suggested a specific role for bases 2-7 at the 5' of the miRNA in target binding but the role of the first nucleotide, found usually to be "A" was also recognized (Lewis et at 2005 Cell 120-15). Similarly, nucleotides 1-7 or 2-8 were used to identify and validate targets by Krek et al. (2005, Nat Genet 37-495).
- The target sites in the mRNA may be in the 5' UTR, the 3' UTR or in the coding region. Interestingly, multiple miRNAs may regulate the same mRNA target by recognizing the same or multiple sites. The presence of multiple miRNA binding sites in most genetically identified targets may indicate that the cooperative action of multiple RISCs provides the most efficient translational inhibition.
- miRNAs may direct the RISC to downregulate gene expression by either of two mechanisms: mRNA cleavage or translational repression. The miRNA may specify cleavage of the mRNA if the mRNA has a certain degree of complementarity to the miRNA. When a miRNA guides cleavage, the cut is typically between the nucleotides pairing to residues 10 and 11 of the miRNA. Alternatively, the miRNA may repress translation if the miRNA does not have the requisite degree of complementarity to the miRNA. Translational repression may be more prevalent in animals since animals may have a lower degree of complementarity between the miRNA and binding site.
- It should be noted that there may be variability in the 5' and 3' ends of any pair of miRNA and miRNA*. This variability may be due to variability in the enzymatic processing of Drosha and Dicer with respect to the site of cleavage. Variability at the 5' and 3' ends of miRNA and miRNA* may also be due to mismatches in the stem structures of the pri-miRNA and pre-miRNA. The mismatches of the stem strands may lead to a population of different hairpin structures. Variability in the stem structures may also lead to variability in the products of cleavage by Drosha and Dicer.
- The term "microRNA mimic" or "miRNA mimic" refers to synthetic non-coding RNAs that are capable of entering the RNAi pathway and regulating gene expression. miRNA mimics imitate the function of endogenous miRNAs and can be designed as mature, double stranded molecules or mimic precursors (e.g., or pre-miRNAs). miRNA mimics can be comprised of modified or unmodified RNA, DNA, RNA-DNA hybrids, or alternative nucleic acid chemistries (e.g., LNAs or 2'-O,4'-C-ethylene-bridged nucleic acids (ENA)). For mature, double stranded miRNA mimics, the length of the duplex region can vary between 13-33, 18-24 or 21-23 nucleotides. The miRNA may also comprise a total of at least 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 or 40 nucleotides. The sequence of the miRNA may be the first 13-33 nucleotides of the pre-miRNA. The sequence of the miRNA may also be the last 13-33 nucleotides of the pre-miRNA.
- Preparation of miRNAs mimics can be effected by any method known in the art such as chemical synthesis or recombinant methods.
- It will be appreciated from the description provided herein above that contacting cells with a miRNA may be effected by transfecting the cells with e.g. the mature double stranded miRNA, the pre-miRNA or the pri-miRNA.
- The pre-miRNA sequence may comprise from 45-90, 60-80 or 60-70 nucleotides.
- The pri-miRNA sequence may comprise from 45-30,000, 50-25,000, 100-20,000, 1,000-1,500 or 80-100 nucleotides.
- Antisense - Antisense is a single stranded RNA designed to prevent or inhibit expression of a gene by specifically hybridizing to its mRNA. Downregulation can be effected using an antisense polynucleotide capable of specifically hybridizing with an mRNA transcript encoding the target (i.e. TNFR1).
- Design of antisense molecules which can be used to efficiently downregulate a target must be effected while considering two aspects important to the antisense approach. The first aspect is delivery of the oligonucleotide into the cytoplasm of the appropriate cells, while the second aspect is design of an oligonucleotide which specifically binds the designated mRNA within cells in a way which inhibits translation thereof.
- The prior art teaches of a number of delivery strategies which can be used to efficiently deliver oligonucleotides into a wide variety of cell types [see, for example, Jääskeläinen et al. Cell Mol Biol Lett. (2002) 7(2):236-7; Gait, Cell Mol Life Sci. (2003) 60(5):844-53; Martino et al. J Biomed Biotechnol. (2009) 2009:410260; Grijalvo et al. Expert Opin Ther Pat. (2014) 24(7):801-19; Falzarano et al, Nucleic Acid Ther. (2014) 24(1):87-100; Shilakari et al. Biomed Res Int. (2014) 2014: 526391; Prakash et al. Nucleic Acids Res. (2014) 42(13):8796-807 and Asseline et al. J Gene Med. (2014) 16(7-8):157-65].
- In addition, algorithms for identifying those sequences with the highest predicted binding affinity for their target mRNA based on a thermodynamic cycle that accounts for the energetics of structural alterations in both the target mRNA and the oligonucleotide are also available [see, for example, Walton et al. Biotechnol Bioeng 65: 1-9 (1999)]. Such algorithms have been successfully used to implement an antisense approach in cells.
- In addition, several approaches for designing and predicting efficiency of specific oligonucleotides using an in vitro system were also published (Matveeva et al., Nature Biotechnology 16: 1374 - 1375 (1998)].
- Thus, the generation of highly accurate antisense design algorithms and a wide variety of oligonucleotide delivery systems, enable an ordinarily skilled artisan to design and implement antisense approaches suitable for downregulating expression of known sequences without having to resort to undue trial and error experimentation.
- According to specific embodiments, the agent is selected from the group consisting of CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN), meganuclease, antisense and siRNA.
- According to specific embodiments, down-regulating expression and/or activity is effected at the genomic level.
- According to specific embodiments, the agent comprises a CRISPR/Cas system.
- Embodiments of the invention further contemplate the use of the CRISPR/Cas system per se (as the therapeutic agent and not as a cell therapy) for downregulating expression of TNFR1 for treating an ischemic disease.
- Thus, according to an aspect of the present invention, there is provided a method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a CRISPR/Cas system for downregulating expression of TNFR1, thereby treating the ischemic disease in the subject.
- Such CRISPR/Cas systems are described in details hereinabove.
- According to specific embodiments, the CRISPR/Cas system does not downregulate expression of TNFR2.
- The cells or the agents (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) of some embodiments of the invention can be administered to an organism per se, or in a pharmaceutical composition where it is mixed with suitable carriers or excipients.
- As used herein a "pharmaceutical composition" refers to a preparation of one or more of the active ingredients described herein with other chemical components such as physiologically suitable carriers and excipients. The purpose of a pharmaceutical composition is to facilitate administration of a compound to an organism.
- Herein the term "active ingredient" refers to the cell or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) accountable for the biological effect.
- Hereinafter, the phrases "physiologically acceptable carrier" and "pharmaceutically acceptable carrier" which may be interchangeably used refer to a carrier or a diluent that does not cause significant irritation to an organism and does not abrogate the biological activity and properties of the administered compound. An adjuvant is included under these phrases.
- Herein the term "excipient" refers to an inert substance added to a pharmaceutical composition to further facilitate administration of an active ingredient. Examples, without limitation, of excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
- Techniques for formulation and administration of drugs may be found in "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, latest edition, which is incorporated herein by reference.
- Suitable routes of administration may, for example, include oral, rectal, transmucosal, especially transnasal, intestinal or parenteral delivery, including intramuscular, subcutaneous and intramedullary injections as well as intrathecal, direct intraventricular, intracardiac, e.g., into the right or left ventricular cavity, into the common coronary artery, intravenous, intraperitoneal, intranasal, or intraocular injections.
- According to specific embodiments, the cells or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same is administered intravenously.
- Conventional approaches for drug delivery to the central nervous system (CNS) include: neurosurgical strategies (e.g., intracerebral injection or intracerebroventricular infusion); molecular manipulation of the agent (e.g., production of a chimeric fusion protein that comprises a transport peptide that has an affinity for an endothelial cell surface molecule in combination with an agent that is itself incapable of crossing the BBB) in an attempt to exploit one of the endogenous transport pathways of the BBB; pharmacological strategies designed to increase the lipid solubility of an agent (e.g., conjugation of water-soluble agents to lipid or cholesterol carriers); and the transitory disruption of the integrity of the BBB by hyperosmotic disruption (resulting from the infusion of a mannitol solution into the carotid artery or the use of a biologically active agent such as an angiotensin peptide). However, each of these strategies has limitations, such as the inherent risks associated with an invasive surgical procedure, a size limitation imposed by a limitation inherent in the endogenous transport systems, potentially undesirable biological side effects associated with the systemic administration of a chimeric molecule comprised of a carrier motif that could be active outside of the CNS, and the possible risk of brain damage within regions of the brain where the BBB is disrupted, which renders it a suboptimal delivery method.
- Alternately, one may administer the cells or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same in a local rather than systemic manner, for example, via injection of the pharmaceutical composition directly into a tissue region of a patient.
- According to specific embodiments, the cells or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same is administered into the ischemic tissue.
- According to specific embodiments, the cells or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same is administered by transendocardial injection.
- According to specific embodiments, the cells or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same is administered intracoronary or intramyocardialy.
- According to specific embodiments, the cells or the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same is attached to an implantable device such as a stent, a valve, an intravascular device and a pacemaker.
- According to some embodiments, the cells of the present invention are administered in a hydrated gel (hydrogel) such as a hyaluronic acid-based hydrogels (see the Examples section which follows). Such hydrogels are known in the art and disclosed e.g. in Xu et al. (2012) Soft Matter. 8(12):3280-3294; Kim et al. (2012) Knee Surg Relat Res. Sep; 24(3): 164-172, the contents of which are fully incorporated herein by reference.
- According to specific embodiments, the hyaluronic acid is provided at a concentration range of about 0.1-10 %, e.g., about 0.5-10 %, e.g., about 0.5-5, e.g., about 1-5 %, e.g., about 2-5 %, e.g., about 3-5 %, e.g., about 3-4 % in the composition e.g. hydrogel.
- According to a specific embodiment, the hyaluronic acid is provided at a concentration range of about 3-4 % in the composition e.g. hydrogel.
- According to some embodiments, the cells of the present invention are administered in a biodegradable co-polymer or scaffold. Such scaffolds are known in the art and disclosed e.g. in Florian Weinbergeret al. (2017) Circulation Research. 120:1487-1500; Rochkind S et al (2004) Neurol Res. 26(2):161-6; Rochkind S. et al. (2006) Eur Spine J. 15(2):234-45; and FSY Wong, ACY Lo (2015) J Stem Cell Res Ther 5:267, the contents of which are fully incorporated herein by reference.
- Pharmaceutical compositions of some embodiments of the invention may be manufactured by processes well known in the art, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes.
- Pharmaceutical compositions for use in accordance with some embodiments of the invention thus may be formulated in conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active ingredients into preparations which, can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen.
- For injection, the active ingredients of the pharmaceutical composition may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological salt buffer. For transmucosal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
- For oral administration, the pharmaceutical composition can be formulated readily by combining the active compounds with pharmaceutically acceptable carriers well known in the art. Such carriers enable the pharmaceutical composition to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like, for oral ingestion by a patient. Pharmacological preparations for oral use can be made using a solid excipient, optionally grinding the resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries if desired, to obtain tablets or dragee cores. Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carbomethylcellulose; and/or physiologically acceptable polymers such as polyvinylpyrrolidone (PVP). If desired, disintegrating agents may be added, such as cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, titanium dioxide, lacquer solutions and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
- Pharmaceutical compositions which can be used orally, include push-fit capsules made of gelatin as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. The push-fit capsules may contain the active ingredients in admixture with filler such as lactose, binders such as starches, lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active ingredients may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition, stabilizers may be added. All formulations for oral administration should be in dosages suitable for the chosen route of administration.
- For buccal administration, the compositions may take the form of tablets or lozenges formulated in conventional manner.
- For administration by nasal inhalation, the active ingredients for use according to some embodiments of the invention are conveniently delivered in the form of an aerosol spray presentation from a pressurized pack or a nebulizer with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichloro-tetrafluoroethane or carbon dioxide. In the case of a pressurized aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount. Capsules and cartridges of, e.g., gelatin for use in a dispenser may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
- The pharmaceutical composition described herein may be formulated for parenteral administration, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multidose containers with optionally, an added preservative. The compositions may be suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- Pharmaceutical compositions for parenteral administration include aqueous solutions of the active preparation in water-soluble form. Additionally, suspensions of the active ingredients may be prepared as appropriate oily or water based injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acids esters such as ethyl oleate, triglycerides or liposomes. Aqueous injection suspensions may contain substances, which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the active ingredients to allow for the preparation of highly concentrated solutions.
- Alternatively, the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile, pyrogen-free water based solution, before use.
- The pharmaceutical composition of some embodiments of the invention may also be formulated in rectal compositions such as suppositories or retention enemas, using, e.g., conventional suppository bases such as cocoa butter or other glycerides.
- Alternative embodiments include depots providing sustained release or prolonged duration of activity of the active ingredient in the subject, as are well known in the art.
- Pharmaceutical compositions suitable for use in context of some embodiments of the invention include compositions wherein the active ingredients are contained in an amount effective to achieve the intended purpose. More specifically, a therapeutically effective amount means an amount of active ingredients (the cells, the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) effective to prevent, alleviate or ameliorate symptoms of a disorder (e.g., ischemic disease, e.g., ischemic heart disease) or prolong the survival of the subject being treated.
- Determination of a therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- For any preparation used in the methods of the invention, the therapeutically effective amount or dose can be estimated initially from in vitro and cell culture assays. For example, a dose can be formulated in animal models to achieve a desired concentration or titer. Such information can be used to more accurately determine useful doses in humans.
- Toxicity and therapeutic efficacy of the active ingredients described herein can be determined by standard pharmaceutical procedures in vitro, in cell cultures or experimental animals. The data obtained from these in vitro and cell culture assays and animal studies can be used in formulating a range of dosage for use in human. The dosage may vary depending upon the dosage form employed and the route of administration utilized. The exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition. (See e.g., Fingl, et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p.1).
- Dosage amount and interval may be adjusted individually to provide the active ingredient in levels sufficient to induce or suppress the biological effect (minimal effective concentration, MEC). The MEC will vary for each preparation, but can be estimated from in vitro data. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. Detection assays can be used to determine plasma concentrations.
- Depending on the severity and responsiveness of the condition to be treated, dosing can be of a single or a plurality of administrations, with course of treatment lasting from several days to several weeks or until cure is effected or diminution of the disease state is achieved.
- According to specific embodiments, the cells, the agent (e.g. a CRISPR/Cas system for downregulating expression of TNFR1) or the pharmaceutical composition comprising same are administered within 1.5-24 hours following diagnosis of said ischemic disease.
- The amount of a composition to be administered will, of course, be dependent on the subject being treated, the severity of the affliction, the manner of administration, the judgment of the prescribing physician, etc.
- Compositions of some embodiments of the invention may, if desired, be presented in a pack or dispenser device, such as an FDA approved kit, which may contain one or more unit dosage forms containing the active ingredient. The pack may, for example, comprise metal or plastic foil, such as a blister pack. The pack or dispenser device may be accompanied by instructions for administration. The pack or dispenser may also be accommodated by a notice associated with the container in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the compositions or human or veterinary administration. Such notice, for example, may be of labeling approved by the U.S. Food and Drug Administration for prescription drugs or of an approved product insert. Compositions comprising a preparation of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition, as is further detailed above.
- According to an aspect of the present invention, there is provided a pharmaceutical composition comprising as an active ingredient differentiated cells or bone marrow stem cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1.
- According to another aspect of the present invention, there is provided a pharmaceutical composition comprising as active ingredients cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1 and at least 2 ng/ml TNFα.
- According to another aspect of the present invention there is provided an article of manufacture comprising TNFα and the cells disclosed herein with reduced expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1.
- According to specific embodiments, the TNFα and the cells are in a co-formulation.
- According to specific embodiments, the TNFα and the cells are in separate containers.
- According to another aspect of the present invention there is provided an article of manufacture comprising TNFα and a CRISPR/Cas system for downregulating expression of TNFR1.
- According to specific embodiments, the TNFα and a CRISPR/Cas system are in a co-formulation.
- According to specific embodiments, the TNFα and a CRISPR/Cas system are in separate containers.
- According to specific embodiments, TNFα is provided at a concentration above its physiological concentration in the cells.
- According to specific embodiments, TNFα is provided at a concentration of at least 2 ng/ml, at least 5 ng/ml, at least 10 ng/ml, at least 20 ng/ml, at least 30 ng/ml, at least 40 ng/ml or at least 50 ng/ml, each possibility represents a separate embodiment of the present invention.
- According to specific embodiments, TNFα is provided at a concentration of 2 - 200 ng/ml or 20 - 200 ng/ml.
- As used herein the term "about" refers to ± 10 %
- The terms "comprises", "comprising", "includes", "including", "having" and their conjugates mean "including but not limited to".
- The term "consisting of" means "including and limited to".
- The term "consisting essentially of" means that the composition, method or structure may include additional ingredients, steps and/or parts, but only if the additional ingredients, steps and/or parts do not materially alter the basic and novel characteristics of the claimed composition, method or structure.
- As used herein, the singular form "a", "an" and "the" include plural references unless the context clearly dictates otherwise. For example, the term "a compound" or "at least one compound" may include a plurality of compounds, including mixtures thereof.
- Throughout this application, various embodiments of this invention may be presented in a range format. It should be understood that the description in range format is merely for convenience and brevity and should not be construed as an inflexible limitation on the scope of the invention. Accordingly, the description of a range should be considered to have specifically disclosed all the possible subranges as well as individual numerical values within that range. For example, description of a range such as from 1 to 6 should be considered to have specifically disclosed subranges such as from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from 3 to 6 etc., as well as individual numbers within that range, for example, 1, 2, 3, 4, 5, and 6. This applies regardless of the breadth of the range.
- Whenever a numerical range is indicated herein, it is meant to include any cited numeral (fractional or integral) within the indicated range. The phrases "ranging/ranges between" a first indicate number and a second indicate number and "ranging/ranges from" a first indicate number "to" a second indicate number are used herein interchangeably and are meant to include the first and second indicated numbers and all the fractional and integral numerals therebetween.
- As used herein the term "method" refers to manners, means, techniques and procedures for accomplishing a given task including, but not limited to, those manners, means, techniques and procedures either known to, or readily developed from known manners, means, techniques and procedures by practitioners of the chemical, pharmacological, biological, biochemical and medical arts.
- When reference is made to particular sequence listings, such reference is to be understood to also encompass sequences that substantially correspond to its complementary sequence as including minor sequence variations, resulting from, e.g., sequencing errors, cloning errors, or other alterations resulting in base substitution, base deletion or base addition, provided that the frequency of such variations is less than 1 in 50 nucleotides, alternatively, less than 1 in 100 nucleotides, alternatively, less than 1 in 200 nucleotides, alternatively, less than 1 in 500 nucleotides, alternatively, less than 1 in 1000 nucleotides, alternatively, less than 1 in 5,000 nucleotides, alternatively, less than 1 in 10,000 nucleotides.
- It is appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the invention, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable subcombination or as suitable in any other described embodiment of the invention. Certain features described in the context of various embodiments are not to be considered essential features of those embodiments, unless the embodiment is inoperative without those elements.
- Various embodiments and aspects of the present invention as delineated hereinabove and as claimed in the claims section below find experimental support in the following examples.
- Reference is now made to the following examples, which together with the above descriptions illustrate some embodiments of the invention in a non limiting fashion.
- Generally, the nomenclature used herein and the laboratory procedures utilized in the present invention include molecular, biochemical, microbiological and recombinant DNA techniques. Such techniques are thoroughly explained in the literature. See, for example, "Molecular Cloning: A laboratory Manual" Sambrook et al., (1989); "Current Protocols in Molecular Biology" Volumes I-III Ausubel, R. M., ed. (1994); Ausubel et al., "Current Protocols in Molecular Biology", John Wiley and Sons, Baltimore, Maryland (1989); Perbal, "A Practical Guide to Molecular Cloning", John Wiley & Sons, New York (1988); Watson et al., "Recombinant DNA", Scientific American Books, New York; Birren et al. (eds) "Genome Analysis: A Laboratory Manual Series", Vols. 1-4, Cold Spring Harbor Laboratory Press, New York (1998); methodologies as set forth in
U.S. Pat. Nos. 4,666,828 ;4,683,202 ;4,801,531 ;5,192,659 and5,272,057 ; "Cell Biology: A Laboratory Handbook", Volumes I-III Cellis, J. E., ed. (1994); "Culture of Animal Cells - A Manual of Basic Technique" by Freshney, Wiley-Liss, N. Y. (1994), Third Edition; "Current Protocols in Immunology" Volumes I-III Coligan J. E., ed. (1994); Stites et al. (eds), "Basic and Clinical Immunology" (8th Edition), Appleton & Lange, Norwalk, CT (1994); Mishell and Shiigi (eds), "Selected Methods in Cellular Immunology", W. H. Freeman and Co., New York (1980); available immunoassays are extensively described in the patent and scientific literature, see, for example,U.S. Pat. Nos. 3,791,932 ;3,839,153 ;3,850,752 ;3,850,578 ;3,853,987 ;3,867,517 ;3,879,262 ;3,901,654 ;3,935,074 ;3,984,533 ;3,996,345 ;4,034,074 ;4,098,876 ;4,879,219 ;5,011,771 and5,281,521 ; "Oligonucleotide Synthesis" Gait, M. J., ed. (1984); "Nucleic Acid Hybridization" Hames, B. D., and Higgins S. J., eds. (1985); "Transcription and Translation" Hames, B. D., and Higgins S. J., eds. (1984); "Animal Cell Culture" Freshney, R. I., ed. (1986); "Immobilized Cells and Enzymes" IRL Press, (1986); "A Practical Guide to Molecular Cloning" Perbal, B., (1984) and "Methods in Enzymology" Vol. 1-317, Academic Press; "PCR Protocols: A Guide To Methods And Applications", Academic Press, San Diego, CA (1990); Marshak et al., "Strategies for Protein Purification and Characterization - A Laboratory Course Manual" CSHL Press (1996); all of which are incorporated by reference as if fully set forth herein. Other general references are provided throughout this document. The procedures therein are believed to be well known in the art and are provided for the convenience of the reader. All the information contained therein is incorporated herein by reference. - Isolation of mononuclear bone marrow cells - Seven 8 weeks old C57B1/6 (OldHsd) male mice weighing 20-23 gram were purchased from Envigo RMS (Israel) LTD. Bone marrow (BM) was flushed from tibiae and femurs of each mouse using 2 mL PBS supplemented with 2 % fetal bovine serum (FBS). The suspension was resuspended and mashed through a 40-100 µM cell strainer. The sample was centrifuged at 300 g for 5 minutes and resuspended in 100 µL PBS. Following, 1 mL of red blood cell (RBC) lysis buffer (Roche) was added and the sample was incubated for 3 minutes at room temperature. 9 mL PBS (3 g/L glucose) supplemented with 2 % FBS (3 g/L glucose) was added to stop the reaction and the sample was centrifuged at 300 g for 5 minutes and immediately resuspended in 1 mL of ice cold PBS supplemented with 2 % fetal bovine serum (FBS). Mononuclear bone marrow cells (mnBMCs) were separated from the BM sample by Ficoll-Paque (Histopaque1077, Sigma), with 3 technical replicates. BM cells were resuspended in 2 mL PBS supplemented with 2 % murine serum were layered on 1.5 ml Ficoll-Paque. The Ficoll-Paque gradients were centrifuged for 40 minutes at 400 g without brake. The mnBM layer was then collected, washed in PBS supplemented with 2 % FBS. Viability and cell count was determined by trypan blue exclusion method using hemocytometer. A total of 100×106 mnBMCs was counted. 60 ×106 mnBMCs were frozen at -80 °C. 40 ×106 cells were seeded in 24 wells plates (120,000 mnBMCs per well) in 1 mL / well DMEM medium containing- glutamine, sodium pyruvate, pen/strep and 3 % mouse serum and cultured under standard culture conditions. Following 24 hours of incubation, the medium was replaced with optimem medium (Invitrogen) and cells were transfected with TNFR1 CRISPR for 2 days. Control cells, which were not transfected with TNFR1 CRISPR were cultured under the same culturing conditions: 120,000 mnBMCs per well in 24 wells plates in DMEM medium containing 2 % FBS.
- TNFR1 CRISPR transfection - For CRISPR transfection three different mouse TNFR1 guide RNA were designed and synthesized (Table 1A hereinabove) and mnBMCs were transfected with case 9 protein with the lipophilic transfection reagent CRISPRMAX as described before (Yu et al 2016).
- mnBM Transplantation - 750,000 cultured mnBMCs were treated with 30 ng / ml mouse TNFα (PeproTech) for 20 minutes and added to 50 µL gel containing 3.75 % hyaluronic acid (HA), 20 % non-activated murine serum (serum obtained from C57BL/6 mice and inactivated in 56 °C) in DMEM. Following ligation of the left anterior descending artery (LAD), 50 µL gel containing mnBMCs were injected to the heart as described in Toma et al 2002 and Kawada et al 2016.
- Animal model - Animal experiments were conducted according to the guidelines of the Animal Care and Use Committee of Israel, and the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health. 18 C57BL/6 mice were purchased at 8 weeks (20-23 gr) from Ex-Vivo Israel. Following 1 week of acclimation the mice were intubated and anesthetized with 0.5 % isoflurane gas. Permanent ligation of the left anterior descending artery (LAD) was performed as described in Kolk et al. (2009) JoVE. 32 (ref 41). The mice were randomly assigned into three groups, see table 2 hereinbelow:
- Group 1M - on
day - Group 2M - on
day 1, following LAD, mice were injected to the LAD area with 750,000 mnBMCs transfected with TNFR1 CRISPR, n = 12; - Group 3M - on
day 1, following LAD, mice were injected to the LAD area with 750,000 mnBMCs that were not transfected with TNFR1 CRISPR, n = 6. - Injected mnBMCs in all groups (1M, 2M, 3M) were treated for 20 minutes prior to injection with 30 ng/mL mouse TNFα (PeproTech) containing 1 mg / ml murine serum.
- Body weight, mortality and morbidity were evaluated once a day. Mice were sacrificed 24 hours or 4 days following LAD occlusion and ~200 µl whole blood was collected from the retro-orbital sinus into yellow cap serum tubes. The collected blood was centrifuged (at 3000 rpm for 10 minutes) for serum preparation. Samples were frozen at (-60 °C) - (-90 °C) and stored in appropriately labeled test-tubes for further Creatin Kinase (CK) detection (Sigma). From each sacrificed mouse heart was dissected, weighed, fixated and sent for histological evaluation.
Table 2: Experimental design: Group Mouse No. Time Treatment 1M 3 24 hours TNFα IP + Injection of mnBMCs transfected with TNFR1 CRISPR and treated with TNFCI, 5 6 2M 7 Injection of mnBMCs transfected with TNFR1 CRISPR and treated with TNFCI, 9 10 3M 15 Control: Injection of non-transfected mnBMCs treated with TNFα 16 18 1M 1 4 days TNF IP + Injection of mnBMCs transfected with TNFR1 CRISPR and treated with TNFCI, 2 2M 11 Injection of mnBMCs transfected with TNFR1 CRISPR and treated with TNFCI, 12 3M 13 Control: Injection of non-transfected mnBMCs treated with TNFα 14 - Histology - Hearts were harvested and fixed in 10 % formaldehyde at Pharmaseed and transferred to Patho-Logica. All tissues were trimmed in the same manner into block cassettes. Transverse cross sections were performed in each heart producing four equal cross sections per organ. The tissues were embedded in paraffin, sectioned at no more than 5 micron thickness, and stained with Hematoxylin & Eosin (H&E) and Masson Trichrome (MT), to trace the injected cells, pathological changes and to evaluate the volume of the induced infarcts. Furthermore, to evaluated viability an apoptosis TUNEL kit stain was performed. The slides were photographed using a microscope (Olympus BX60), at magnifications of X10, X20 and X40, equipped with an Olympus DP-73 camera.
-
- Infarct diameter (mm);
- Presence of injected mnBMCs (-, +, ++, +++);
- Additional pathological changes
-
- Grade: -, no signs of positive TUNEL expression in all tissues and cells.
- Grade: +, Only few cells (< 10) per cross section are positive.
- Grade ++, More cells (> 10, >50) per cross section are positive.
- Grade +++, Many cells (>50) per cross section are positive.
- The effect of transplantation of stem cells with reduced expression of TNFR1 on ischemic heart disease was assessed in a permanent ligation of the left anterior descending artery (LAD) mouse model, a known model of infarction and myocardial ischemia [Kolk et al. (2009) JoVE. 32]. To this end, mnBMCs transfected with TNFR1 CRISPR and treated with TNFα were injected to the LAD area and their effect on infract volume and cellular response was compared to the effect of injection of control mnBMCs (non-transfected and treated with TNFα).
- Histological evaluation of hearts extracted from LAD occluded mice injected with mnBMCs transfected with TNFR1 CRISPR demonstrated reduced infarct size (both width and depth), as determined 24 hours and 4 days following LAD occlusion (Group 2M) compared to control mice that were injected with non-transfected mnBMCs (Group 3M) (
Figures 1A-2F ,4A-5D and9A-9B and Table 3 hereinbelow). Interestingly, moderate to high TUNEL reaction in the infarct lesion was demonstrated in these mice (Figures 7A-8F ), mainly localized in the inner part of the ventricle wall. - Ischemic preconditioning (IP) has been recognized as one of the most potent mechanisms to protect against myocardial ischemic injury. In experimental animals and humans, a brief period of ischemia has been shown to protect the heart from more prolonged episodes of ischemia, reducing infarct size, attenuating the incidence, and severity of reperfusion-induced arrhythmias, and preventing endothelial cell dysfunction (e.g. 6). It has been shown that classical preconditioning can be mimicked by administration of TNFα [9-13].
- In the present model, treatment of the mice with
TNFα 2 hours prior to LAD occlusion followed by injection of the TNFR1 CRISPR transfected mnBMCs (Group 1M) did not have a beneficial effect on infarct size. On the contrary, histological evaluation of hearts extracted from these mice revealed a large infarct volume with a relative low cellular reaction and mild to moderate TUNEL reaction in the infarct lesion, as detected 24 hours and 4 days following LAD (Figures 3A-F ,6A-D ,7A-8F and Table 3 hereinbelow). - To evaluate toxicity, CK were determined in the serum of the transplanted mice. CK (made up of three enzyme forms, including CK-MB) levels can rise following heart attack, skeletal muscle injury and strenuous exercise. Importantly, detection of CK levels in the serum indicated that injection of the mnBMCs (either transfected or not transfected with TNFR1 CRISPR) did not have an effect of
CK levels 4 days following LAD occlusion (Figure 10 ). - Taken together, the results indicated a cardio-protective effect of transplantation of mnMB cells transfected with TNFR1 CRISPR against ischemic damage as demonstrated by reduced infarct depth and width in the mouse LAD model.
Table 3: Histological results of cardiac infarct parameters Animal # Group Infarct volume mm (MT) Depth / width BM cells in infarct TUNNEL reaction Additional pathological changes 24 hours post LAD 3 1M Transmural / 1.57 - - Few neutrophils and lymphocytes 5 1M 0.85 / 0.45 - - Moderate infiltration of lymphocytes and few neutrophils. Fibrin on top of the infarct. 6 1M Transmural / 1.12 - ++ Almost no reaction 7 2M 0.22 / 1.76 - +++ Mild infiltration of mainly lymphocytes and macrophages. 9 2M 0.59 / 1.37 + +++ Macrophages and neutrophils focally infiltrated next to injected cells debris 10 2M Transmural / 1.49 - +++ Cellular infiltration of neutrophils and lymphocytes in the inner part of the ventricle wall 15 3M 0.37 / 0.71 ++ ++ Macrophages and neutrophils focally infiltrated next to injected cells debris 16 3M 0.44 / 1.58 ++ +++ Macrophages and neutrophils focally infiltrated next to injected cells debris and some adipocytes 18 3M 0.32 / 0.88 + +++ Macrophages and neutrophils focally infiltrated next to injected cells debris 4 days post LAD 1 1M Transmural (2.02) / 3.29 - +++ A very large infarct with a central necrotic core, surrounded by many macrophages, lymphocytes and some fibrocytes. 2 1M Transmural (1.06) / 4.74 - ++ A very large infarct with a central necrotic core, surrounded by many macrophages, lymphocytes and some fibrocytes. 11 2M Transmural (1.14) / 2.74 - ++ A very large infarct with a central necrotic core, surrounded by many macrophages, lymphocytes and some fibrocytes. 12 2M Transmural (1.01) / 4.14 - ++ A very large infarct with a central necrotic core, surrounded by many macrophages, lymphocytes and some fibrocytes. 13 3M 1.77 / 4.36 - +++ A very large infarct with a central necrotic core, surrounded by many macrophages, lymphocytes and some fibrocytes. 14 3M 2.02 / 3.99 - ++ A very large infarct with a central necrotic core, surrounded by many macrophages, lymphocytes and some fibrocytes. - The effect of transplantation of human stem cells with reduced expression of TNFR1 on ischemic heart disease is assessed in a LAD occluded immune-deficient mouse model. To this end, human mnBMCs transfected with human TNFR1 CRISPR and treated with TNFα are injected to the LAD area and their effect on infract volume and cellular response is compared to the effect of injection of control human mnBMCs (non-transfected and treated with TNFα).
- Isolation of human mnBMCs - Bone marrow (BM) is flushed from tibiae, femurs or from peripheral blood or taken from human blood bank or human umbilical cord blood bank. Each sample is handled using 2 mL PBS supplemented with 2 % human serum. The suspension is resuspended and mashed through a 40-100 µM cell strainer. The sample is centrifuged at 300 g for 5 minutes and re-suspended in 100 µL PBS. Following, 1 mL of red blood cell (RBC) lysis buffer (Roche) is added and the sample is incubated for 3 minutes at room temperature. 9 mL PBS (3 g / L glucose) supplemented with 2 % human serum (3 g / L glucose) is added to stop the reaction and the sample is centrifuged at 300 g for 5 minutes and immediately re-suspended in 1 mL of ice cold PBS supplemented with 2 % human serum. Mononuclear bone marrow cells (mnBMCs) are separated from the BM sample by Ficoll-Paque (Histopaque1077, Sigma), with 3 technical replicates, as follows: BM cells re-suspended in 2 mL PBS supplemented with 2 % human serum are ayered on 1.5 ml Ficoll-Paque. The Ficoll-Paque gradients are centrifuged for 40 minutes at 400 g without brake. Following, the mnBM layer is collected, washed in PBS supplemented with 2 % human serum. Viability and cell count is determined by trypan blue exclusion method using hemocytometer. A fraction of the mnBMCs are frozen at -80 °C. Another fraction is seeded in 24 wells plates (120,000 mnBMCs per well) in 1 mL / well DMEM medium containing- glutamine, sodium pyruvate, pen/strep and 3 % human serum and cultured under standard culture conditions. Following 1-24 hours of incubation, the medium is replaced with optimem medium (Invitrogen) and cells are transfected with TNFR1 CRISPR for 5 minutes up to 2 days or immediately electroporated. Control cells, which are not transfected with TNFR1 CRISPR are cultured under the same culturing conditions.
- TNFR1 CRISPR transfection - For CRISPR transfection seven different human TNFR1 guide RNA were designed and synthesized (Table 1B hereinabove). mnBMCs are transfected with case 9 protein with the lipophilic transfection reagent CRISPRMAX as described before (Yu et al 2016) or transfected immediately by electroporation.
- mnBM Transplantation - As described in Example 1 hereinabove.
- Animal model - As described in Example 1 hereinabove with Nude and/or SCID mice instead of C57BL/6 mice.
- Histology - As described in Example 1 hereinabove
- Although the invention has been described in conjunction with specific embodiments thereof, it is evident that many alternatives, modifications and variations will be apparent to those skilled in the art. Accordingly, it is intended to embrace all such alternatives, modifications and variations that fall within the spirit and broad scope of the appended claims.
- All publications, patents and patent applications mentioned in this specification are herein incorporated in their entirety by reference into the specification, to the same extent as if each individual publication, patent or patent application was specifically and individually indicated to be incorporated herein by reference. In addition, citation or identification of any reference in this application shall not be construed as an admission that such reference is available as prior art to the present invention. To the extent that section headings are used, they should not be construed as necessarily limiting.
-
- 1 Beutler, B. and van Huffel, C. (1994) Unraveling function in the TNF ligand and receptor families. Science. 264, 667-668
- 2 Kolesnick, R. and Golde, D. W. (1994) The sphingomyelin pathway in tumor-necrosis-factor and interleukin-1 signaling. Cell. 77, 325-328
- 3 Beyaert, R. and Fiers, W. (1994) Molecular mechanisms of tumor necrosis factor-induced cytotoxicity - What we do understand and what we do not. FEBS Lett. 340, 9-16
- 4 Valgimigli, M., Ceconi, C., Malagutti, P., Merli, E., Soukhomovskaia, O., Francolini, G., Cicchitelli, G., Olivares, A., Parrinello, G., Percoco, G., Guardigli, G., Mele, D., Pirani, R. and Ferrari, R. (2005) Tumor necrosis factor-
- 5 Kleinbongard, P., Schulz, R. and Heusch, G. (2010) TNFalpha in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 16, 49-69
- 6 Murry, C. E., Jennings, R. B. and Reimer, K. A. (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 74, 1124-1136
- 7 Marber, M. S., Latchman, D. S., Walker, J. M. and Yellon, D. M. (1993) Cardiac
- 8 Schulz, R., Gres, P., Konietzka, I. and Heusch, G. (2005) Regional differences of myocardial infarct development and ischemic preconditioning. Basic Res. Cardiol. 100, 48-56
- 9 Smith, R. M., Suleman, N., McCarthy, J. and Sack, M. N. (2002) Classic ischemic but not pharmacologic preconditioning is abrogated following genetic ablation of the TNF alpha gene. Cardiovasc. Res. 55, 553-560
- 10 Ran, K., Duan, K. M., Zou, D. Q., Li, Z. J., Jin, L. Y. and Chang, Y. T. (2008) Effect of isoflurane delayed preconditioning on myocardial ischemia reperfusion injury in rabbits. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 33, 146-150
- 11 Lecour, S., Smith, R. M., Woodward, B., Opie, L. H., Rochette, L. and Sack, M. N. (2002) Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J. Mol. Cell. Cardiol. 34, 509-518
- 12 Lecour, S., Rochette, L. and Opie, L. (2005) Free radicals trigger TNF alpha-induced cardioprotection. Cardiovasc. Res. 65, 239-243
- 13 El-Ani, D., Zimlichman, R., Mashiach, Y. and Shainberg, A. (2007) Adenosine and TNF-alpha exert similar inotropic effect on heart cultures, suggesting a cardioprotective mechanism against hypoxia. Life Sci. 81, 803-813
- 14 Dorge, H., Schulz, R., Belosjorow, S., Post, H., van de Sand, A., Konietzka, I., Frede, S., Hartung, T., Vinten-Johansen, J., Youker, K. A., Entman, M. L., Erbel, R. and Heusch, G. (2002) Coronary microembolization: The role of TNF-alpha in contractile dysfunction. J. Mol. Cell. Cardiol. 34, 51-62
- 15 Reil, J. C., Gilles, S., Zahler, S., Brandl, A., Drexler, H., Hultner, L., Matrisian, L. M., Welsch, U. and Becker, B. F. (2007) Insights from knock-out models concerning postischemic release of TNF alpha from isolated mouse hearts. J. Mol. Cell. Cardiol. 42, 133-141
- 16 Skyschally, A., Gres, P., Hoffmann, S., Haude, M., Erbel, R., Schulz, R. and Heusch, G. (2007) Bidirectional role of tumor necrosis factor-alpha in coronary microembolization - Progressive contractile dysfunction versus delayed protection against infarction. Circul. Res. 100, 140-146
- 17 Heusch, P., Skyschally, A., Leineweber, K., Haude, M., Erbel, R. and Heusch, G. (2007) The interaction of coronary microembolization and ischemic preconditioning: A third window of cardioprotection through TNF-alpha. Archives of Medical Science. 3, 83-92
- 18 Thielmann, M., Dorge, H., Martin, C., Belosjorow, S., Schwanke, U., van de Sand, A., Konietzka, I., Buchert, A., Kruger, A., Schulz, R. and Heusch, G. (2002) Myocardial dysfunction with coronary microembolization - Signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine. Circul. Res. 90, 807-813
- 19 El-Ani, D. and Zimlichman, R. (2003) TNFalpha stimulated ATP-sensitive potassium channels and attenuated deoxyglucose and Ca uptake of H9c2 cardiomyocytes. Ann. N. Y. Acad. Sci. 1010, 716-720
- 20 Meldrum, D. R., Dinarello, C. A., Shames, B. D., Cleveland, J. C., Cain, B. S., Banerjee, A., Meng, X. Z. and Harken, A. H. (1998) Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-alpha production - Potential ultimate effector mechanism of preconditioning. Circulation. 98, II214-II218
- 21 Mubagwa, K. and Flameng, W. (2001) Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc. Res. 52, 25-39
- 22 Hutchinson, S. A. and Scammells, P. J. (2004) A(1) adenosine receptor agonists: medicinal chemistry and therapeutic potential. Curr.Pharm.Des. 10, 2021-2039
- 23 Lotz, C., Liem, D. and Ping, P. P. (2011) New frontiers in myocardial protection: A systems biology approach. Journal of Cardiovascular Pharmacology and Therapeutics. 16, 285-289
- 24 Hinkel, R., Trenkwalder, T. and Kupatt, C. (2011) Gene therapy for ischemic heart disease. Expert Opinion on Biological Therapy. 11, 723-737
- 25 Marais, E., Genade, S. and Lochner, A. (2008) CREB activation and ischaemic preconditioning. Cardiovasc. Drugs Ther. 22, 3-17
- 26 Qu, S., Zhu, H., Wei, X., Zhang, C., Jiang, L., Liu, Y. and Xiao, X. (2010) Oxidative stress-mediated up-regulation of myocardial ischemic preconditioning up-regulated .
- 27 Muller, B. A. and Dhalla, N. S. (2010) Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-referfused heart. Curr. Cardiol. Rev. 6, 255-264
- 28 Schmitt, J. P., Ahmad, F., Lorenz, K., Hein, L., Schulz, S., Asahi, M., MacLennan, D. H., Seidman, C. E., Seidman, J. G. and Lohse, M. J. (2009) Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+-ATPase inhibition. Circulation. 119, 436-444
- 29 Cerra, M. C. and Imbrogno, S. (2012) Phospholamban and cardiac function: a comparative perspective in vertebrates. Acta Physiologica. 205, 9-25
- 30 Kranias, E. G. and Hajjar, R. J. (2012) Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circul. Res. 110, 1646-1660
- 31 Louch, W. E., Vangheluwe, P., Bito, V., Raeymaekers, L., Wuytack, F. and Sipido, K. R. (2012) Phospholamban ablation in hearts expressing the high affinity SERCA2b isoform normalizes global Ca2+ homeostasis but not Ca2+-dependent hypertrophic signaling. American Journal of Physiology-Heart and Circulatory Physiology. 302, H2574-H2582
- 32 MacLennan, D. H. and Kranias, E. G. (2003) Phospholamban: A crucial regulator of cardiac contractility. Nature Reviews Molecular Cell Biology. 4, 566-577
- 33 Kishore, R., Tkebuchava, T., Sasi, S. P., Silver, M., Gilbert, H. Y., Yoon, Y. S., Park, H. Y, Thorne, T., Losordo, D. W. and Goukassian, D. A. (2011) Tumor necrosis factor-alpha signaling via TNFR1/p55 is deleterious whereas TNFR2/p75 signaling is protective in adult infarct myocardium. Advances in TNF Family Research. 691, 433-448
- 34 Monden, Y, Kubota, T., Inoue, T., Tsutsumi, T., Kawano, S., Ide, T., Tsutsui, H. and Sunagawa, K. (2007) Tumor necrosis factor-alpha is toxic via
- 35 Moe, G. W., Marin-Garcia, J., Konig, A., Goldenthal, M., Lu, X. and Feng, Q. (2004) In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am. J. Physiol. Heart Circ. Physiol. 287, H1813-1820
- 36 Bao, C., Guo, J., Lin, G., Hu, M. and Hu, Z. (2008) TNFR gene-modified mesenchymal stem cells attenuate inflammation and cardiac dysfunction following MI. Scand. Cardiovasc. J. 42, 56-62
- 37 Kurrelmeyer, K. M., Michael, L. H., Baumgarten, G., Taffet, G. E., Peschon, J. J., Sivasubramanian, N., Entman, M. L. and Mann, D. L. (2000) Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc. Nat. Acad. Sci. U.S.A. 97, 5456-5461
- 38 Wang, M., Crisostomo, P. R., Markel, T. A., Wang, Y. and Meldrum, D. R. (2008) Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation. 118, S38-45
- 39 El-Ani, D., Stav, H., Guetta, V., Arad, M. and Shainberg, A. (2011) Rapamycin (sirolimus) protects against hypoxic damage in primary heart cultures via Na(+)/Ca(2+) exchanger activation. Life Sci. 89.
- 40 El-Ani D, Philipchik I, Stav H, Levi M, Zerbib J and Shainberg A. TNF alpha protects heart rat cultures against hypoxic damage via activation of PKA and phospholamban to prevent calcium overload.2014. Can J Physiol Pharmacol 92(11):917-25.2014.
- 41 Kolk M.V., Meyberg D., Deuse T., Tang-Quan K.R., Robbins R.C., Reichenspurner H., Schrepfer S. (2009). LAD-Ligation: A Murine Model of Myocardial Infarction. JoVE. 32.
- The present invention also comprises the following embodiments:
- 1. A method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of mononuclear bone marrow cells (mnBMCs) comprising mesenchymal stem cells (MSCs) and lymphocytes or progenitors thereof with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, thereby treating the ischemic disease in the subject.
- 2. A method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of differentiated cells with reduced level of expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, wherein said differentiated cells are of a same type of tissue affected in said ischemic disease, thereby treating the ischemic disease in the subject.
- 3. The method of any one of embodiments 1-2, further comprising treating said cells with reduced expression and/or activity of TNFR1 with TNFα prior to said administering.
- 4. The method of embodiment 3, comprising cryopreserving said cells prior to said treating with said TNFα.
- 5. A method of treating an ischemic disease in a subject in need thereof, the method comprising:
- (i) treating with TNFα cells with reduced expression and/or activity of TNFR1 as compared to control cells of the same origin not contacted with an agent which downregulates expression and/or activity of said TNFR1, wherein said cells are selected from the group consisting of mononuclear bone marrow cells (mnBMCs), stem cells and differentiated cells of a same type of tissue affected in said ischemic disease; and
- (ii) administering to the subject a therapeutically effective amount of said cells with reduced level of expression and/or activity of TNFR1 following said (i), thereby treating the ischemic disease in the subject.
- 6. The method of embodiment 5, wherein said cells comprise differentiated cells of a same type of tissue affected in said ischemic disease.
- 7. The method of any one of embodiments 1-6, wherein said cells with reduced level of expression and/or activity of TNFR1 have the same level of expression and/or activity of TNFR2 as compared to said control stem cells.
- 8. The method of any one of embodiments 1-7, wherein said cells with reduced expression and/or activity of TNFR1 are genetically modified cells.
- 9. The method of embodiment 8, wherein said genetically modified comprises genetically modified with a CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN) or meganuclease for downregulating expression of said TNFR1.
- 10. The method of embodiment 8, wherein said genetically modified comprises genetically modified with a CRISPR/Cas system for downregulating expression of said TNFR1.
- 11. A method of downregulating expression and/or activity of TNFR1 in differentiated cells or bone marrow stem cells, the method comprising:
- (i) contacting ex-vivo or in-vitro differentiated cells or bone marrow stem cells with an agent which downregulates expression and/or activity of TNFR1; and
- (ii) contacting ex-vivo or in-vitro said cells with TNFα.
- 12. The method of embodiment 11, wherein said (i) is effected prior to said (ii).
- 13. The method of any one of embodiments 11-12, wherein said agent does not downregulate expression and/or activity of TNFR2.
- 14. The method of any one of embodiments 11-13, wherein said agent is selected from the group consisting of CRISPR/Cas system, a Zinc finger nuclease (ZFN), transcription-activator like effector nuclease (TALEN), meganuclease, antisense and siRNA.
- 15. The method of any one of embodiments 11-13, wherein said agent comprises a CRISPR/Cas system.
- 16. A method of treating an ischemic disease in a subject in need thereof, the method comprising:
- (i) obtaining cells according to the method of any one of embodiments 11-14; and
- (ii) administering to the subject a therapeutically effective amount of said cells, thereby treating the ischemic disease in the subject.
- 17. The method of embodiment 16, wherein said differentiated cells are of a same type of tissue affected in said ischemic disease.
- 18. The method of any one of embodiments 5-10 and 16-17, comprising cryopreserving said cells following said (i) and prior to said (ii).
- 19. The method of any one of embodiments 1-10 and 16-18, wherein said cells are non-autologous to said subject.
- 20. A method of treating an ischemic disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a CRISPR/Cas system for downregulating expression of TNFR1, thereby treating the ischemic disease in the subject.
- 21. A pharmaceutical composition comprising as an active ingredient differentiated cells or bone marrow stem cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1.
- 22. A pharmaceutical composition comprising as active ingredients cells genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1 and at least 2 ng/ml TNFα.
- 23. The method of
embodiment 20 or the pharmaceutical composition of any one of embodiments 21-22, wherein said CRISPR/Cas system does not downregulate expression of TNFR2. - 24. The pharmaceutical composition of any one of embodiments 21-22, wherein said cells comprise differentiated cells.
- 25. The method of any one of embodiments 2-3 and 6-19 or the pharmaceutical composition of any one of embodiments 21 and 23-24, wherein said differentiated cells are cardiomyocytes.
- 26. The method of any one of embodiments 5, 7-10 and 18-19 or the pharmaceutical composition of any one of embodiments 22-23, wherein said cells comprise stem cells.
- 27. The method or the pharmaceutical composition of embodiment 26, wherein said stem cells are selected from the group consisting of embryonic stem cells (ESCs), induced pluripotent stem cells (iPS), hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).
- 28. The method or the pharmaceutical composition of embodiment 26, wherein said stem cells comprise hematopoietic stem cells (HSCs).
- 29. The method or the pharmaceutical composition of any one of embodiments 5, 7-16, 18-19 and 21-28, wherein said cells are comprised in mononuclear bone marrow cells (mnBMCs).
- 30. The method or the pharmaceutical composition of embodiment 29, wherein said mnBMCs comprise lymphocytes or progenitors thereof.
- 31. The method or the pharmaceutical composition of any one of
embodiments 1, 3-5, 7-16, 18-19, 21-23 and 29-30, wherein said cells are obtained by density gradient centrifugation of bone marrow cells. - 32. The method of any one of
embodiments 1, 3-5, 7-16, 18-19, 23 and 29-30, comprising obtaining said cells by density gradient centrifugation of bone marrow cells. - 33. The method or the pharmaceutical composition of any one of embodiments 1-19 and 21-32, wherein said cells are human cells.
- 34. The method of any one of embodiments 1-18 or the pharmaceutical composition of any one of embodiments 21-32, wherein said cells are cryopreserved cells.
- 35. The method of any one of embodiments 1-10, 16-20 and 25-33, wherein said ischemic disease is ischemic heart disease.
- 36. The method of embodiment 35, wherein said ischemic heart disease is myocardial infarction.
- 37. The method of embodiment 35, wherein said ischemic heart disease is ischemic cardiomyopathy.
- 38. The method of any one of embodiments 1-10, 16-20 and 25-37, wherein said subject is not treated with TNFα.
Claims (14)
- Mononuclear bone marrow cells (mnBMCs) comprising (i) mesenchymal stem cells (MSCs) and (ii) lymphocytes or lymphocytes progenitors, the mnBMCs being genetically modified with a CRISPR/Cas system for downregulating expression of TNFR1 and having at least 20 % reduced level of expression of said TNFR1 as compared to non-genetically modified control cells of the same origin as determined by PCR, for use in treating an ischemic disease in a subject in need thereof.
- The mnBMCs for use of claim 1, wherein said genetically modified mnBMCs have the same level of expression of TNFR2 as compared to said non-genetically modified control cells as determined by PCR.
- The mnBMCs for use of any one of claims 1-2, wherein said at least 20 % reduced level of expression of said TNFR1 is at least 50 %.
- The mnBMCs for use of any one of claims 1-3, wherein CRISPR/Cas system comprises a guide RNA sequence selected from the group consisting of SEQ ID NO: 18, 21, 24, 27, 30, 33, 36, 39, 42 and 45.
- The mnBMCs for use of any one of claims 1-4, being obtained from bone marrow, peripheral blood or umbilical cord blood by ficoll density gradient centrifugation.
- The mnBMCs for use of any one of claims 1-5, being cryopreserved cells.
- The mnBMCs for use of any one of claims 1-6, being formulated in a hydrogel.
- The mnBMCs for use of any one of claims 1-7, being formulated for intracardial administration.
- The mnBMCs for use of any one of claims 1-8, being treated with TNFα prior to administration to said subject.
- The mnBMCs for use of any one of claims 1-9, wherein said subject is not treated with TNFα.
- The mnBMCs for use of any one of claims 1-10, being non-autologous to said subject.
- The mnBMCs for use of any one of claims 1-10, being autologous to said subject.
- The mnBMCs for use of any one of claims 1-12, wherein said ischemic disease is selected from the group consisting of ischemic cerebrovascular disorder, ischemic renal disease, ischemic pulmonary disease and ischemic heart disease
- The mnBMCs for use of any one of claims 1-13, wherein said ischemic disease is ischemic heart disease.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762576094P | 2017-10-24 | 2017-10-24 | |
| EP18871526.2A EP3700543A4 (en) | 2017-10-24 | 2018-10-24 | METHOD OF TREATMENT OF ISCHEMIC DISEASE |
| PCT/IL2018/051139 WO2019082184A1 (en) | 2017-10-24 | 2018-10-24 | Methods of treating an ischemic disease |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP18871526.2A Division EP3700543A4 (en) | 2017-10-24 | 2018-10-24 | METHOD OF TREATMENT OF ISCHEMIC DISEASE |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP4461356A2 true EP4461356A2 (en) | 2024-11-13 |
| EP4461356A3 EP4461356A3 (en) | 2025-01-22 |
Family
ID=66247200
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP18871526.2A Withdrawn EP3700543A4 (en) | 2017-10-24 | 2018-10-24 | METHOD OF TREATMENT OF ISCHEMIC DISEASE |
| EP24200691.4A Pending EP4461356A3 (en) | 2017-10-24 | 2018-10-24 | Methods of treating an ischemic disease |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP18871526.2A Withdrawn EP3700543A4 (en) | 2017-10-24 | 2018-10-24 | METHOD OF TREATMENT OF ISCHEMIC DISEASE |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US11701391B2 (en) |
| EP (2) | EP3700543A4 (en) |
| CN (1) | CN111565731B (en) |
| IL (1) | IL274190A (en) |
| WO (1) | WO2019082184A1 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11701391B2 (en) | 2017-10-24 | 2023-07-18 | Dalia ELANI | Methods of treating an ischemic disease |
| CN116492464B (en) * | 2023-04-12 | 2025-08-19 | 天津医科大学总医院 | Application of TNFRSF1A inhibitor miR-3059-5p in treating cerebral apoplexy |
Citations (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3791932A (en) | 1971-02-10 | 1974-02-12 | Akzona Inc | Process for the demonstration and determination of reaction components having specific binding affinity for each other |
| US3839153A (en) | 1970-12-28 | 1974-10-01 | Akzona Inc | Process for the detection and determination of specific binding proteins and their corresponding bindable substances |
| US3850578A (en) | 1973-03-12 | 1974-11-26 | H Mcconnell | Process for assaying for biologically active molecules |
| US3850752A (en) | 1970-11-10 | 1974-11-26 | Akzona Inc | Process for the demonstration and determination of low molecular compounds and of proteins capable of binding these compounds specifically |
| US3853987A (en) | 1971-09-01 | 1974-12-10 | W Dreyer | Immunological reagent and radioimmuno assay |
| US3867517A (en) | 1971-12-21 | 1975-02-18 | Abbott Lab | Direct radioimmunoassay for antigens and their antibodies |
| US3879262A (en) | 1972-05-11 | 1975-04-22 | Akzona Inc | Detection and determination of haptens |
| US3901654A (en) | 1971-06-21 | 1975-08-26 | Biological Developments | Receptor assays of biologically active compounds employing biologically specific receptors |
| US3935074A (en) | 1973-12-17 | 1976-01-27 | Syva Company | Antibody steric hindrance immunoassay with two antibodies |
| US3984533A (en) | 1975-11-13 | 1976-10-05 | General Electric Company | Electrophoretic method of detecting antigen-antibody reaction |
| US3996345A (en) | 1974-08-12 | 1976-12-07 | Syva Company | Fluorescence quenching with immunological pairs in immunoassays |
| US4034074A (en) | 1974-09-19 | 1977-07-05 | The Board Of Trustees Of Leland Stanford Junior University | Universal reagent 2-site immunoradiometric assay using labelled anti (IgG) |
| US4098876A (en) | 1976-10-26 | 1978-07-04 | Corning Glass Works | Reverse sandwich immunoassay |
| US4666828A (en) | 1984-08-15 | 1987-05-19 | The General Hospital Corporation | Test for Huntington's disease |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4801531A (en) | 1985-04-17 | 1989-01-31 | Biotechnology Research Partners, Ltd. | Apo AI/CIII genomic polymorphisms predictive of atherosclerosis |
| US4879219A (en) | 1980-09-19 | 1989-11-07 | General Hospital Corporation | Immunoassay utilizing monoclonal high affinity IgM antibodies |
| US5011771A (en) | 1984-04-12 | 1991-04-30 | The General Hospital Corporation | Multiepitopic immunometric assay |
| US5192659A (en) | 1989-08-25 | 1993-03-09 | Genetype Ag | Intron sequence analysis method for detection of adjacent and remote locus alleles as haplotypes |
| US5272057A (en) | 1988-10-14 | 1993-12-21 | Georgetown University | Method of detecting a predisposition to cancer by the use of restriction fragment length polymorphism of the gene for human poly (ADP-ribose) polymerase |
| US5281521A (en) | 1992-07-20 | 1994-01-25 | The Trustees Of The University Of Pennsylvania | Modified avidin-biotin technique |
| US5486359A (en) | 1990-11-16 | 1996-01-23 | Osiris Therapeutics, Inc. | Human mesenchymal stem cells |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| WO2000000504A1 (en) | 1998-06-26 | 2000-01-06 | Isis Pharmaceuticals, Inc. | Antisense modulation of tnfr1 expression |
| US6090622A (en) | 1997-03-31 | 2000-07-18 | The Johns Hopkins School Of Medicine | Human embryonic pluripotent germ cells |
| WO2001039594A2 (en) | 1999-12-03 | 2001-06-07 | Dendreon Corporation | Cryopreservation of antigen-loaded dendritic cells and their precursors in serum-free media |
| US20030232410A1 (en) | 2002-03-21 | 2003-12-18 | Monika Liljedahl | Methods and compositions for using zinc finger endonucleases to enhance homologous recombination |
| US6774279B2 (en) | 1997-05-30 | 2004-08-10 | Carnegie Institution Of Washington | Use of FLP recombinase in mice |
| US20050026157A1 (en) | 2002-09-05 | 2005-02-03 | David Baltimore | Use of chimeric nucleases to stimulate gene targeting |
| US20060014264A1 (en) | 2004-07-13 | 2006-01-19 | Stowers Institute For Medical Research | Cre/lox system with lox sites having an extended spacer region |
| WO2006040763A2 (en) | 2004-10-12 | 2006-04-20 | Technion Research & Development Foundation Ltd. | Isolated primate embryonic cells and methods of generating and using same |
| WO2007054160A2 (en) | 2005-11-09 | 2007-05-18 | Paul-Ehrlich-Institut Bundesamt Für Sera Und Impfstoffe | Method for the cryopreservation of human blood |
| CN101401930A (en) | 2008-10-29 | 2009-04-08 | 中山大学 | Polypeptide and uses in preparing anti-heart failure and anti-inflammatory reaction medicament |
| US7534607B1 (en) | 2005-12-27 | 2009-05-19 | Industrial Technology Research Institute | Method of producing cardiomyocytes from mesenchymal stem cells |
| WO2009071334A2 (en) | 2007-12-07 | 2009-06-11 | Max-Delbrück-Centrum Für Molekulare Medizin (Mdc) | Transposon-mediated mutagenesis in spermatogonial stem cells |
| WO2011006914A2 (en) | 2009-07-16 | 2011-01-20 | Glaxo Group Limited | Antagonists, uses & methods for partially inhibiting tnfr1 |
| WO2011056416A2 (en) | 2009-10-19 | 2011-05-12 | Cellular Dynamics International, Inc. | Cardiomyocyte production |
| US20110189768A1 (en) | 2006-01-13 | 2011-08-04 | Alla Danilkovitch | Mesenchymal Stem Cells Expressing TNF-alpha Receptors |
| US8021867B2 (en) | 2005-10-18 | 2011-09-20 | Duke University | Rationally-designed meganucleases with altered sequence specificity and DNA-binding affinity |
| WO2011146121A1 (en) | 2010-05-17 | 2011-11-24 | Sangamo Biosciences, Inc. | Novel dna-binding proteins and uses thereof |
| WO2012002649A2 (en) | 2010-06-30 | 2012-01-05 | 삼성정밀화학(주) | Method for preparing chlorohydrins and method for preparing epichlorohydrin using chlorohydrins prepared thereby |
| US20120014879A1 (en) | 2005-10-24 | 2012-01-19 | Caritas St. Elizabeth Medical Center of Boston Inc | USE OF TUMOR NECROSIS FACTOR-alpha RECEPTOR p75 FOR TREATMENT OF ISCHEMIA-INDUCED NEOVASCULARIZATION |
| US20120149108A1 (en) | 2009-08-19 | 2012-06-14 | Masashige Tanabe | Cell preservation method |
| WO2013126963A1 (en) | 2012-02-29 | 2013-09-06 | Benitec Biopharma Limited | Pain treatment |
| WO2014078414A1 (en) | 2012-11-13 | 2014-05-22 | Joseph Wu | Chemically defined production of cardiomyocytes from pluripotent stem cells |
| WO2014085593A1 (en) | 2012-11-27 | 2014-06-05 | Children's Medical Center Corporation | Targeting bcl11a distal regulatory elements for fetal hemoglobin reinduction |
| EP2746396A1 (en) | 2012-12-20 | 2014-06-25 | PLS-Design GmbH | Selective local inhibition of TNFR1-mediated functions at the site of antigen/allergen presentation |
| US8771945B1 (en) | 2012-12-12 | 2014-07-08 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| WO2014107763A1 (en) | 2013-01-08 | 2014-07-17 | Benitec Biopharma Limited | Age-related macular degeneration treatment |
| WO2014200339A1 (en) | 2013-06-11 | 2014-12-18 | Pluriomics B.V. | Culture medium compositions for maturating cardiomyocytes derived from pluripotent mammalian stem cells |
| WO2015004539A2 (en) | 2013-07-02 | 2015-01-15 | National University Of Singapore | Differentiation of pluripotent stem cells and cardiac progenitor cells into striated cardiomyocyte fibers using laminins ln-511, ln-521 and ln-221 |
| WO2015104322A1 (en) | 2014-01-09 | 2015-07-16 | Glaxosmithkline Intellectual Property Development Limited | Treatment of inflammatory diseases with non-competitive tnfr1 antagonists |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004113387A2 (en) * | 2003-06-24 | 2004-12-29 | Merck Patent Gmbh | Tumour necrosis factor receptor molecules with reduced immunogenicity |
| WO2015191545A1 (en) | 2014-06-09 | 2015-12-17 | University Of Washington | Methods of protection against ischemia reperfusion injury |
| US11319555B2 (en) | 2014-11-20 | 2022-05-03 | Duke University | Compositions, systems and methods for cell therapy |
| US9675847B2 (en) | 2014-11-21 | 2017-06-13 | One World Play Project LLC | Sports ball and method of manufacture |
| US11280790B2 (en) * | 2016-10-04 | 2022-03-22 | Svar Life Science Ab | System and products for improved quantification of ADCC activity |
| US11701391B2 (en) | 2017-10-24 | 2023-07-18 | Dalia ELANI | Methods of treating an ischemic disease |
-
2018
- 2018-10-24 US US16/758,457 patent/US11701391B2/en active Active
- 2018-10-24 EP EP18871526.2A patent/EP3700543A4/en not_active Withdrawn
- 2018-10-24 CN CN201880082166.9A patent/CN111565731B/en active Active
- 2018-10-24 WO PCT/IL2018/051139 patent/WO2019082184A1/en not_active Ceased
- 2018-10-24 EP EP24200691.4A patent/EP4461356A3/en active Pending
-
2020
- 2020-04-23 IL IL274190A patent/IL274190A/en unknown
-
2023
- 2023-05-23 US US18/200,595 patent/US20230330148A1/en not_active Abandoned
Patent Citations (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3850752A (en) | 1970-11-10 | 1974-11-26 | Akzona Inc | Process for the demonstration and determination of low molecular compounds and of proteins capable of binding these compounds specifically |
| US3839153A (en) | 1970-12-28 | 1974-10-01 | Akzona Inc | Process for the detection and determination of specific binding proteins and their corresponding bindable substances |
| US3791932A (en) | 1971-02-10 | 1974-02-12 | Akzona Inc | Process for the demonstration and determination of reaction components having specific binding affinity for each other |
| US3901654A (en) | 1971-06-21 | 1975-08-26 | Biological Developments | Receptor assays of biologically active compounds employing biologically specific receptors |
| US3853987A (en) | 1971-09-01 | 1974-12-10 | W Dreyer | Immunological reagent and radioimmuno assay |
| US3867517A (en) | 1971-12-21 | 1975-02-18 | Abbott Lab | Direct radioimmunoassay for antigens and their antibodies |
| US3879262A (en) | 1972-05-11 | 1975-04-22 | Akzona Inc | Detection and determination of haptens |
| US3850578A (en) | 1973-03-12 | 1974-11-26 | H Mcconnell | Process for assaying for biologically active molecules |
| US3935074A (en) | 1973-12-17 | 1976-01-27 | Syva Company | Antibody steric hindrance immunoassay with two antibodies |
| US3996345A (en) | 1974-08-12 | 1976-12-07 | Syva Company | Fluorescence quenching with immunological pairs in immunoassays |
| US4034074A (en) | 1974-09-19 | 1977-07-05 | The Board Of Trustees Of Leland Stanford Junior University | Universal reagent 2-site immunoradiometric assay using labelled anti (IgG) |
| US3984533A (en) | 1975-11-13 | 1976-10-05 | General Electric Company | Electrophoretic method of detecting antigen-antibody reaction |
| US4098876A (en) | 1976-10-26 | 1978-07-04 | Corning Glass Works | Reverse sandwich immunoassay |
| US4879219A (en) | 1980-09-19 | 1989-11-07 | General Hospital Corporation | Immunoassay utilizing monoclonal high affinity IgM antibodies |
| US5011771A (en) | 1984-04-12 | 1991-04-30 | The General Hospital Corporation | Multiepitopic immunometric assay |
| US4666828A (en) | 1984-08-15 | 1987-05-19 | The General Hospital Corporation | Test for Huntington's disease |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683202B1 (en) | 1985-03-28 | 1990-11-27 | Cetus Corp | |
| US4801531A (en) | 1985-04-17 | 1989-01-31 | Biotechnology Research Partners, Ltd. | Apo AI/CIII genomic polymorphisms predictive of atherosclerosis |
| US5272057A (en) | 1988-10-14 | 1993-12-21 | Georgetown University | Method of detecting a predisposition to cancer by the use of restriction fragment length polymorphism of the gene for human poly (ADP-ribose) polymerase |
| US5192659A (en) | 1989-08-25 | 1993-03-09 | Genetype Ag | Intron sequence analysis method for detection of adjacent and remote locus alleles as haplotypes |
| US5486359A (en) | 1990-11-16 | 1996-01-23 | Osiris Therapeutics, Inc. | Human mesenchymal stem cells |
| US5281521A (en) | 1992-07-20 | 1994-01-25 | The Trustees Of The University Of Pennsylvania | Modified avidin-biotin technique |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US6090622A (en) | 1997-03-31 | 2000-07-18 | The Johns Hopkins School Of Medicine | Human embryonic pluripotent germ cells |
| US6774279B2 (en) | 1997-05-30 | 2004-08-10 | Carnegie Institution Of Washington | Use of FLP recombinase in mice |
| WO2000000504A1 (en) | 1998-06-26 | 2000-01-06 | Isis Pharmaceuticals, Inc. | Antisense modulation of tnfr1 expression |
| WO2001039594A2 (en) | 1999-12-03 | 2001-06-07 | Dendreon Corporation | Cryopreservation of antigen-loaded dendritic cells and their precursors in serum-free media |
| US20030232410A1 (en) | 2002-03-21 | 2003-12-18 | Monika Liljedahl | Methods and compositions for using zinc finger endonucleases to enhance homologous recombination |
| US20050026157A1 (en) | 2002-09-05 | 2005-02-03 | David Baltimore | Use of chimeric nucleases to stimulate gene targeting |
| US20060014264A1 (en) | 2004-07-13 | 2006-01-19 | Stowers Institute For Medical Research | Cre/lox system with lox sites having an extended spacer region |
| WO2006040763A2 (en) | 2004-10-12 | 2006-04-20 | Technion Research & Development Foundation Ltd. | Isolated primate embryonic cells and methods of generating and using same |
| US8143015B2 (en) | 2005-10-18 | 2012-03-27 | Duke University | Methods of cleaving DNA with rationally-designed meganucleases |
| US8129134B2 (en) | 2005-10-18 | 2012-03-06 | Duke University | Methods of cleaving DNA with rationally-designed meganucleases |
| US8148098B2 (en) | 2005-10-18 | 2012-04-03 | Duke University | Methods of cleaving DNA with rationally-designed meganucleases |
| US8163514B2 (en) | 2005-10-18 | 2012-04-24 | Duke University | Methods of cleaving DNA with rationally-designed meganucleases |
| US8143016B2 (en) | 2005-10-18 | 2012-03-27 | Duke University | Methods of cleaving DNA with rationally-designed meganucleases |
| US8124369B2 (en) | 2005-10-18 | 2012-02-28 | Duke University | Method of cleaving DNA with rationally-designed meganucleases |
| US8119381B2 (en) | 2005-10-18 | 2012-02-21 | Duke University | Rationally-designed meganucleases with altered sequence specificity and DNA-binding affinity |
| US8021867B2 (en) | 2005-10-18 | 2011-09-20 | Duke University | Rationally-designed meganucleases with altered sequence specificity and DNA-binding affinity |
| US8133697B2 (en) | 2005-10-18 | 2012-03-13 | Duke University | Methods of cleaving DNA with rationally-designed meganucleases |
| US8304222B1 (en) | 2005-10-18 | 2012-11-06 | Duke University | Rationally-designed meganucleases with altered sequence specificity and heterodimer formation |
| US20120014879A1 (en) | 2005-10-24 | 2012-01-19 | Caritas St. Elizabeth Medical Center of Boston Inc | USE OF TUMOR NECROSIS FACTOR-alpha RECEPTOR p75 FOR TREATMENT OF ISCHEMIA-INDUCED NEOVASCULARIZATION |
| WO2007054160A2 (en) | 2005-11-09 | 2007-05-18 | Paul-Ehrlich-Institut Bundesamt Für Sera Und Impfstoffe | Method for the cryopreservation of human blood |
| US7534607B1 (en) | 2005-12-27 | 2009-05-19 | Industrial Technology Research Institute | Method of producing cardiomyocytes from mesenchymal stem cells |
| US20110189768A1 (en) | 2006-01-13 | 2011-08-04 | Alla Danilkovitch | Mesenchymal Stem Cells Expressing TNF-alpha Receptors |
| WO2009071334A2 (en) | 2007-12-07 | 2009-06-11 | Max-Delbrück-Centrum Für Molekulare Medizin (Mdc) | Transposon-mediated mutagenesis in spermatogonial stem cells |
| CN101401930A (en) | 2008-10-29 | 2009-04-08 | 中山大学 | Polypeptide and uses in preparing anti-heart failure and anti-inflammatory reaction medicament |
| WO2011006914A2 (en) | 2009-07-16 | 2011-01-20 | Glaxo Group Limited | Antagonists, uses & methods for partially inhibiting tnfr1 |
| US20120149108A1 (en) | 2009-08-19 | 2012-06-14 | Masashige Tanabe | Cell preservation method |
| WO2011056416A2 (en) | 2009-10-19 | 2011-05-12 | Cellular Dynamics International, Inc. | Cardiomyocyte production |
| WO2011146121A1 (en) | 2010-05-17 | 2011-11-24 | Sangamo Biosciences, Inc. | Novel dna-binding proteins and uses thereof |
| US8586526B2 (en) | 2010-05-17 | 2013-11-19 | Sangamo Biosciences, Inc. | DNA-binding proteins and uses thereof |
| WO2012002649A2 (en) | 2010-06-30 | 2012-01-05 | 삼성정밀화학(주) | Method for preparing chlorohydrins and method for preparing epichlorohydrin using chlorohydrins prepared thereby |
| WO2013126963A1 (en) | 2012-02-29 | 2013-09-06 | Benitec Biopharma Limited | Pain treatment |
| WO2014078414A1 (en) | 2012-11-13 | 2014-05-22 | Joseph Wu | Chemically defined production of cardiomyocytes from pluripotent stem cells |
| WO2014085593A1 (en) | 2012-11-27 | 2014-06-05 | Children's Medical Center Corporation | Targeting bcl11a distal regulatory elements for fetal hemoglobin reinduction |
| US8771945B1 (en) | 2012-12-12 | 2014-07-08 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| EP2746396A1 (en) | 2012-12-20 | 2014-06-25 | PLS-Design GmbH | Selective local inhibition of TNFR1-mediated functions at the site of antigen/allergen presentation |
| WO2014107763A1 (en) | 2013-01-08 | 2014-07-17 | Benitec Biopharma Limited | Age-related macular degeneration treatment |
| WO2014200339A1 (en) | 2013-06-11 | 2014-12-18 | Pluriomics B.V. | Culture medium compositions for maturating cardiomyocytes derived from pluripotent mammalian stem cells |
| WO2015004539A2 (en) | 2013-07-02 | 2015-01-15 | National University Of Singapore | Differentiation of pluripotent stem cells and cardiac progenitor cells into striated cardiomyocyte fibers using laminins ln-511, ln-521 and ln-221 |
| WO2015104322A1 (en) | 2014-01-09 | 2015-07-16 | Glaxosmithkline Intellectual Property Development Limited | Treatment of inflammatory diseases with non-competitive tnfr1 antagonists |
Non-Patent Citations (138)
| Title |
|---|
| "Gene", Database accession no. 21938 |
| "Nucleic Acid Hybridization", 1985 |
| "PCR Protocols: A Guide To Methods And Applications", vol. 1, 1990, ACADEMIC PRESS, pages: 317 |
| "Phospholamban ablation in hearts expressing the high affinity SERCA2b isoform normalizes global Ca2+ homeostasis but not Ca2+-dependent hypertrophic signaling", AMERICAN JOURNAL OF PHYSIOLOGY-HEART AND CIRCULATORY PHYSIOLOGY, vol. 302, 2012, pages 2574 - 2582 |
| "Selected Methods in Cellular Immunology", 1980, W. H. FREEMAN AND CO. |
| "Transcription and Translation", 1984 |
| ALISON, M.R, J PATHOL, vol. 200, no. 5, 2003, pages 547 - 50 |
| AOI T ET AL.: "Generation of Pluripotent Stem Cells from Adult Mouse Liver and Stomach Cells", SCIENCE, 14 February 2008 (2008-02-14) |
| ASSELINE ET AL., J GENE MED, vol. 16, no. 7-8, 2014, pages 157 - 65 |
| BAO, C.GUO, J.LIN, G.HU, MHU, Z.: "TNFR gene-modified mesenchymal stem cells attenuate inflammation and cardiac dysfunction following MI", SCAND. CARDIOVASC. J, vol. 42, 2008, pages 56 - 62 |
| BARTEL, CELL, vol. 116, 2004, pages 281 |
| BEUTLER, BVAN HUFFEL, C: "Unraveling function in the TNF ligand and receptor families", SCIENCE, vol. 264, 1994, pages 667 - 668, XP001247252, DOI: 10.1126/science.8171316 |
| BEYAERT, RFIERS, W: "Molecular mechanisms of tumor necrosis factor-induced cytotoxicity - What we do understand and what we do not", FEBS LETT, vol. 340, 1994, pages 9 - 16 |
| BHARGAVA A ET AL., BRAIN RES. PROTOC, vol. 13, 2004, pages 115 - 125 |
| BILLY ET AL., PNAS, vol. 98, 2001, pages 14428 - 14433 |
| BONGSO ET AL., HUM REPROD, vol. 4, 1989, pages 706 |
| BRENNECKE ET AL., PLOS, vol. 3, 2005, pages e85 |
| BURRIDGE ET AL., CELL STEM CELL, vol. 10, 2012, pages 16 - 28 |
| CAI, J ET AL., BLOOD CELLS MOL DIS, vol. 31, no. 1, 2003, pages 18 - 27 |
| CAPECCHI, SCIENCE, vol. 244, 1989, pages 1288 - 1292 |
| CERMAK ET AL., NUCLEIC ACIDS RESEARCH, vol. 39, no. 12, 2011, pages e82 |
| CERRA, M. CIMBROGNO, S: "Phospholamban and cardiac function: a comparative perspective in vertebrates", ACTA PHYSIOLOGICA, vol. 205, 2012, pages 9 - 25, XP072378072, DOI: 10.1111/j.1748-1716.2012.02389.x |
| CERTO, MT ET AL., NATURE METHODS, vol. 9, 2012, pages 073 - 975 |
| CHOUDHURY ET AL., EUR J HEART FAIL., vol. 19, no. 1, 2017, pages 138 - 147 |
| CHUNG ET AL., CELL STEM CELL, vol. 2, 7 February 2008 (2008-02-07), pages 113 - 117 |
| COLLINS, A.T ET AL., J CELL SCI, vol. 114, 2001, pages 3865 - 72 |
| COLTER DC. ET AL.: "Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow", PROC NATL ACAD SCI USA., vol. 97, 2000, pages 3213 - 3218, XP002205331, DOI: 10.1073/pnas.070034097 |
| CURR. TOP. DEV. BIOL, vol. 38, 1998, pages 133 |
| DEKEL B ET AL., J. AM. SOC. NEPHROL, vol. 13, 2002, pages 977 - 90 |
| DEKEL B ET AL., NAT MED, vol. 9, 2003, pages 53 - 60 |
| DIALLO ET AL., OLIGONUCLEOTIDES, vol. 13, no. 5, 1 October 2003 (2003-10-01), pages 381 - 392 |
| DIALLO M. ET AL., OLIGONUCLEOTIDES, vol. 13, 2003, pages 381 - 392 |
| DOENCHSHARP, GENESDEV, vol. 2004, 2004, pages 504 |
| DOETSCHMAN ET AL., DEV BIOL, vol. 127, 1988, pages 224 - 7 |
| DOMINICI, M ET AL., J. BIOL. REGUL. HOMEOST. AGENTS, vol. 15, 2001, pages 28 - 37 |
| DORGE, H., SCHULZ, R., BELOSJOROW, S., POST, H., VAN DE SAND, A., KONIETZKA, I., FREDE, S.,HARTUNG, T., VINTEN-JOHANSEN, J., YOUKE: "Coronary microembolization: The role of TNF-alpha in contractile dysfunction.", MOL. CELL. CARDIOL, vol. 34, 2002, pages 51 - 62 |
| EL-ANI DPHILIPCHIK ISTAV HLEVI MZERBIB JSHAINBERG A: "TNF alpha protects heart rat cultures against hypoxic damage via activation of PKA and phospholamban to prevent calcium overload.2014", CAN J PHYSIOL PHARMACOL, vol. 92, no. 11, 2014, pages 917 - 25 |
| EL-ANI, D.STAV, H.GUETTA, V.ARAD, MSHAINBERG, A: "Rapamycin (sirolimus) protects against hypoxic damage in primary heart cultures via Na(+)/Ca(2+) exchanger activation", LIFE SCI, vol. 89, 2011 |
| EL-ANI, D.ZIMLICHMAN, R.MASHIACH, YSHAINBERG, A: "Adenosine and TNF-alpha exert similar inotropic effect on heart cultures, suggesting a cardioprotective mechanism against hypoxia", LIFE SCI, vol. 81, 2007, pages 803 - 813, XP022227403, DOI: 10.1016/j.lfs.2007.06.030 |
| EL-ANI, DZIMLICHMAN, R: "TNFalpha stimulated ATP-sensitive potassium channels and attenuated deoxyglucose and Ca uptake of H9c2 cardiomyocytes", ANN. N. Y. ACAD. SCI, vol. 1010, 2003, pages 716 - 720 |
| EVENTOV-FRIEDMAN S ET AL., PLOS MED, vol. 3, 2006, pages e215 |
| EVENTOV-FRIEDMAN S ET AL., PROC NATL ACAD SCI U S A., vol. 102, 2005, pages 2928 - 33 |
| FALZARANO ET AL., NUCLEIC ACID THER, vol. 24, no. 1, 2014, pages 87 - 100 |
| FARHANG ET AL., TISSUE ENG PART A, vol. 23, no. 15-16, 2017, pages 738 - 749 |
| FINGL ET AL., THE PHARMACOLOGICAL BASIS OF THERAPEUTICS, 1975, pages 1 |
| FLORIAN WEINBERGERET, CIRCULATION RESEARCH, vol. 120, 2017, pages 1487 - 1500 |
| FSY WONGACY LO, J STEM CELL RES THER, vol. 5, 2015, pages 267 |
| GAIT, CELL MOL LIFE SCI., vol. 60, no. 5, 2003, pages 844 - 53 |
| GARDNER ET AL., FERTIL. STERIL, vol. 69, 1998, pages 84 |
| GRAVESMOREADITH, MOL REPROD DEV, vol. 36, 1993, pages 424 - 33 |
| GRIJALVO ET AL., EXPERT OPIN THER PAT, vol. 24, no. 7, 2014, pages 801 - 19 |
| HEUSCH, P., SKYSCHALLY, A., LEINEWEBER, K., HAUDE, M., ERBEL, R. AND HEUSCH, G.: "The interaction of coronary microembolization and ischemic preconditioning: A third window of cardioprotection through TNF-alpha.", ARCHIVES OF MEDICAL SCIENCE, vol. 3, 2007, pages 83 - 92 |
| HINKEL, R., TRENKWALDER, T. AND KUPATT, C.: "Gene therapy for ischemic heart disease.", EXPERT OPINION ON BIOLOGICAL THERAPY, vol. 11, 2011, pages 723 - 737 |
| HUTCHINSON, S. ASCAMMELLS, P. J: "A(1) adenosine receptor agonists: medicinal chemistry and therapeutic potential", CURR.PHARM.DES, vol. 10, 2004, pages 2021 - 2039 |
| IANNACCONE ET AL., DEV BIOL, vol. 163, 1994, pages 288 - 92 |
| IH PARKZHAO RWEST JA ET AL.: "Reprogramming of human somatic cells to pluripotency with defined factors", NATURE, vol. 451, 2008, pages 141 - 146 |
| IZSVÁKIVICS, MOLECULAR THERAPY, vol. 54, 2004, pages 147 - 156 |
| JÄÄSKELÄINEN ET AL., CELL MOL BIOL LETT., vol. 7, no. 2, 2002, pages 236 - 7 |
| JINEK ET AL., SCIENCE, vol. 337, 2012, pages 816 - 821 |
| JONES E.A. ET AL.: "Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells", ARTHRITIS RHEUM, vol. 46, no. 12, 2002, pages 3349 - 60, XP009083397, DOI: 10.1002/art.10696 |
| K TAKAHASHITANABE KOHNUKI M ET AL.: "Induction of pluripotent stem cells from adult human fibroblasts by defined factors", CELL, vol. 131, 2007, pages 861 - 872 |
| KARANTALIS ET AL., CIRC RES., vol. 116, no. 8, 2015, pages 1413 - 30 |
| KAWAKAMI ET AL., PNAS, vol. 97, no. 21, 2000, pages 11403 - 11408 |
| KELLY ET AL., SHOCK, vol. 34, no. 3, 2010, pages 236 - 607 |
| KIM ET AL., KNEE SURG RELAT RES, vol. 24, no. 3, 2012, pages 164 - 172 |
| KISHORE, R.TKEBUCHAVA, T.SASI, S. P.SILVER, M.GILBERT, H. Y.YOON, Y. S.PARK, H. Y.THORNE, T.LOSORDO, D. WGOUKASSIAN, D. A: "Tumor necrosis factor-alpha signaling via TNFR1/p55 is deleterious whereas TNFR2/p75 signaling is protective in adult infarct myocardium", ADVANCES IN TNF FAMILY RESEARCH, vol. 691, 2011, pages 433 - 448 |
| KLEINBONGARD, P.SCHULZ, RHEUSCH, G: "TNFalpha in myocardial ischemia/reperfusion, remodeling and heart failure", HEART FAIL REV, vol. 16, 2010, pages 49 - 69, XP019869217, DOI: 10.1007/s10741-010-9180-8 |
| KOLESNICK, RGOLDE, D. W: "The sphingomyelin pathway in tumor-necrosis-factor and interleukin-1 signaling", CELL, vol. 77, 1994, pages 325 - 328, XP024244732, DOI: 10.1016/0092-8674(94)90147-3 |
| KOLK M.V.MEYBERG D.DEUSE T.TANG-QUAN K.R.ROBBINS R.C.REICHENSPURNER H.SCHREPFER S: "LAD-Ligation: A Murine Model of Myocardial Infarction", JOVE, vol. 32, 2009 |
| KRANIAS, E. GAND HAJJAR, R. J: "Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome", CIRCUL. RES, vol. 110, 2012, pages 1646 - 1660 |
| KREK ET AL., NAT GENET, vol. 37, 2005, pages 495 |
| KURRELMEYER, K. M.MICHAEL, L. H.BAUMGARTEN, G.TAFFET, G. E.PESCHON, J. J.SIVASUBRAMANIAN, NENTMAN, M. LMANN, D. L: "Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction", PROC. NAT. ACAD. SCI. U.S.A., vol. 97, 2000, pages 5456 - 5461 |
| LECOUR, S.ROCHETTE, LOPIE, L: "Free radicals trigger TNF alpha-induced cardioprotection", CARDIOVASC. RES, vol. 65, 2005, pages 239 - 243, XP004696923, DOI: 10.1016/j.cardiores.2004.10.003 |
| LECOUR, S.SMITH, R. M.WOODWARD, B.OPIE, L. H.ROCHETTE, LSACK, M. N: "Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection", J. MOL. CELL. CARDIOL, vol. 34, 2002, pages 509 - 518 |
| LEWIS, CELL, vol. 120, 2005, pages 15 |
| LIANG ET AL., JOURNAL OF BIOTECHNOLOGY, vol. 208, 2015, pages 44 - 53 |
| LIANG ET AL., JOURNAL OF BIOTECHNOLOGY, vol. 241, 2017, pages 136 - 146 |
| LOTZ, C.LIEM, DPING, P. P: "New frontiers in myocardial protection: A systems biology approach", JOURNAL OF CARDIOVASCULAR PHARMACOLOGY AND THERAPEUTICS, vol. 16, 2011, pages 285 - 289 |
| MACLENNAN, D. HKRANIAS, E. G: "Phospholamban: A crucial regulator of cardiac contractility", NATURE REVIEWS MOLECULAR CELL BIOLOGY, vol. 4, 2003, pages 566 - 577, XP009052993 |
| MARAIS, E.GENADE, SLOCHNER, A: "CREB activation and ischaemic preconditioning. Cardiovasc", DRUGS THER, vol. 22, 2008, pages 3 - 17, XP019570707 |
| MARBER, M. S.LATCHMAN, D. S.WALKER, J. MYELLON, D. M: "Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction", CIRCULATION, vol. 88, 1993, pages 1264 - 1272, XP001117531 |
| MARSHAK ET AL.: "Strategies for Protein Purification and Characterization - A Laboratory Course Manual", 1996, CSHL PRESS |
| MARTINO ET AL., J BIOMED BIOTECHNOL, vol. 2009, 2009, pages 410260 |
| MATVEEVA ET AL., NATURE BIOTECHNOLOGY, vol. 16, 1998, pages 1374 - 1375 |
| MELDRUM, D. RDINARELLO, C. A.SHAMES, B. D.CLEVELAND, J. C.CAIN, B. SBANERJEE, A.MENG, X. ZHARKEN, A. H: "Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-alpha production - Potential ultimate effector mechanism of preconditioning", CIRCULATION, vol. 1-4, 1998, pages 11214 - 11218 |
| MENKE D, GENESIS, vol. 51, 2013, pages 618 |
| MILLER ET AL., NAT BIOTECHNOL., vol. 29, 2011, pages 143 - 148 |
| MISKEY ET AL., NUCLEIC ACIDS RES, vol. 31, no. 23, pages 6873 - 6881 |
| MITALIPOVA ET AL., CLONING, vol. 3, 2001, pages 59 - 67 |
| MOE, G. W.MARIN-GARCIA, J.KONIG, A.GOLDENTHAL, M.LU, XFENG, Q: "In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure", AM. J. PHYSIOL. HEART CIRC. PHYSIOL, vol. 287, 2004, pages 1813 - 1820 |
| MONDEN, Y.KUBOTA, T.INOUE, T.TSUTSUMI, T.KAWANO, S.IDE, TTSUTSUI, HSUNAGAWA, K: "Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction", AM. J. PHYSIOL. HEART CIRC. PHYSIOL, 2007 |
| MOZAFFARIAN ET AL., CIRCULATION, vol. 131, no. 4, 2015, pages e29 - 322 |
| MUBAGWA, KFLAMENG, W: "Adenosine, adenosine receptors and myocardial protection: an updated overview", CARDIOVASC. RES, vol. 52, 2001, pages 25 - 39 |
| MULLER, B. ADHALLA, N. S: "Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-referfused heart", CURR. CARDIOL. REV, vol. 6, 2010, pages 255 - 264 |
| MUMMERY ET AL., CIRC RES, vol. 111, no. 3, 20 July 2012 (2012-07-20), pages 344 - 58 |
| MURRY, C. E.ENNINGS, R. BREIMER, K. A: "Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium", CIRCULATION, vol. 74, 1986, pages 1124 - 1136 |
| NATURE, vol. 491, 2012, pages 65 - 56 |
| NOTARIANNI ET AL., J REPROD FERTIL SUPPL, vol. 43, 1991, pages 255 - 60 |
| PADDISON P.J. ET AL., PROC. NATL ACAD. SCI. USA., vol. 99, 2002, pages 1443 - 1448 |
| PERBAL, B.: "A Practical Guide to Molecular Cloning", 1984, JOHN WILEY & SONS |
| PICK M ET AL., PLOS ONE, vol. 8, no. 2, 2013, pages e55530 |
| PICK M ET AL., STEM CELLS, vol. 25, no. 9, 2007, pages 2206 - 14 |
| POTTEN, C. SMORRIS, R. J, J. CELL SCI, vol. 10, 1988, pages 45 - 62 |
| PRAKASH ET AL., NUCLEIC ACIDS RES., vol. 42, no. 13, 2014, pages 8796 - 807 |
| PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 7844 |
| QU, S.ZHU, H.WEI, X.ZHANG, C.JIANG, L.LIU, YXIAO, X: "Oxidative stress-mediated up-regulation of myocardial ischemic preconditioning up-regulated protein 1 gene expression in H9c2 cardiomyocytes is regulated by cyclic AMP-response element binding protein", FREE RADIC. BIOL. MED, vol. 49, 2010, pages 580 - 586, XP027136595 |
| RAN, K.DUAN, K. M.ZOU, D. Q.LI, Z. J.JIN, L. YCHANG, Y. T: "Effect of isoflurane delayed preconditioning on myocardial ischemia reperfusion injury in rabbits", ZHONG NAN DA XUE XUE BAO YI XUE BAN, vol. 33, 2008, pages 146 - 150 |
| REIL, J. C.GILLES, S.ZAHLER, S.BRANDL, A.DREXLER, H.HULTNER, LMATRISIAN, L. M.WELSCH, UBECKER, B. F: "Insights from knock-out models concerning postischemic release of TNF alpha from isolated mouse hearts", J. MOL. CELL. CARDIOL, vol. 42, 2007, pages 133 - 141 |
| REYON ET AL., NATURE BIOTECHNOLOGY, vol. 30, no. 5, May 2012 (2012-05-01), pages 460 - 5 |
| ROCHKIND S ET AL., EUR SPINE J, vol. 15, no. 2, 2006, pages 234 - 45 |
| ROCHKIND S ET AL., NEUROL RES, vol. 26, no. 2, 2004, pages 161 - 6 |
| SAMBROOK ET AL.: "Current Protocols in Molecular Biology", 1989, JOHN WILEY AND SONS |
| SANTIAGO ET AL., PROC NATL ACAD SCI USA, vol. 105, 2008, pages 5809 - 5814 |
| SCHMITT, J. P.AHMAD, F.LORENZ, K.HEIN, L.SCHULZ, S.ASAHI, M.MACLENNAN, D. H.SEIDMAN, C. E.SEIDMAN, J. GLOHSE, M. J: "Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+-ATPase inhibition", CIRCULATION, vol. 119, 2009, pages 436 - 444 |
| SCHULZ, R.GRES, P.KONIETZKA, IHEUSCH, G: "Regional differences of myocardial infarct development and ischemic preconditioning", BASIC RES. CARDIOL, vol. 100, 2005, pages 48 - 56 |
| SCIENCE, vol. 282, 1998, pages 1145 |
| SHAMBLOTT ET AL., PROC. NATL. ACAD. SCI. USA, vol. 95, 1998, pages 13726 |
| SHILAKARI ET AL., BIOMED RES INT, vol. 2014, 2014, pages 526391 |
| SHINAGWAISHII, GENES & DEV, vol. 17, no. 11, 2003, pages 1340 - 1345 |
| SIMMONS, P. JTOROK-STORB, B, BLOOD, vol. 78, 1991, pages 5562 |
| SKYSCHALLY, A., GRES, P., HOFFMANN, S., HAUDE, M., ERBEL, R., SCHULZ, R. AND HEUSCH, G.: "Bidirectional role of tumor necrosis factor-alpha in coronary microembolization Progressive contractile dysfunction versus delayed protection against infarction.-", CIRCUL. RES, vol. 100, 2007, pages 140 - 146 |
| SMITH, R. M.SULEMAN, N.MCCARTHY, JSACK, M. N: "Classic ischemic but not pharmacologic preconditioning is abrogated following genetic ablation of the TNF alpha gene", CARDIOVASC. RES, vol. 55, 2002, pages 553 - 560 |
| STRAT ET AL., NUCLEIC ACIDS RESEARCH, vol. 34, no. 13, 2006, pages 3803 - 3810 |
| THIELMANN, M., DORGE, H., MARTIN, C., BELOSJOROW, S., SCHWANKE, U., VAN DE SAND, A.,KONIETZKA, I., BUCHERT, A., KRUGER, A., SCHULZ: "Myocardial dysfunction with coronary microembolization - Signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine.", CIRCUL. RES, vol. 90, 2002, pages 807 - 813 |
| THOMSON ET AL., BIOL REPROD, vol. 55, 1996, pages 254 - 9 |
| THOMSON ET AL., PROC NATL ACAD SCI U S A., vol. 92, 1995, pages 7844 - 8 |
| TRAN N. ET AL., FEBS LETT, vol. 573, 2004, pages 127 - 134 |
| TUSCHL, CHEMBIOCHEM, vol. 2, pages 239 - 245 |
| VALGIMIGLI, M.CECONI, C.MALAGUTTI, P.MERLI, E.SOUKHOMOVSKAIA, OFRANCOLINI, G.CICCHITELLI, G.OLIVARES, A.PARRINELLO, G.PERCOCO, G: "Tumor necrosis factor-alpha receptor 1 is a major predictor of mortality and new-onset heart failure in patients with acute myocardial infarction - The cytokine-activation and long-term prognosis in myocardial infarction (C-ALPHA) stud", CIRCULATION, vol. 111, 2005, pages 863 - 870 |
| WALTON ET AL., BIOTECHNOL BIOENG, vol. 65, 1999, pages 1 - 9 |
| WANG, M.CRISOSTOMO, P. R.MARKEL, T. A.WANG, YMELDRUM, D. R.: "Mechanisms of sex differences in TNFR2-mediated cardioprotection", CIRCULATION, vol. 118, 2008, pages 38 - 45 |
| WHEELER, REPROD FERTIL DEV, vol. 6, 1994, pages 563 - 8 |
| WILSON ET AL., MOLECULAR THERAPY, vol. 15, 2007, pages 139 - 145 |
| XU ET AL., SOFT MATTER, vol. 8, no. 12, 2012, pages 3280 - 3294 |
| YAMANAKA S, CELL STEM CELL, vol. 1, no. 1, 2007, pages 39 - 49 |
| YU ET AL., JD1BIOTECHNOL LETT, vol. 38, no. 6, 2016, pages 919 - 29 |
| ZHANG ET AL., NATURE BIOTECHNOLOGY, vol. 29, no. 2, 2011, pages 149 - 53 |
| ZHENYU LU ET AL., J. ASSIST REPROD. GENET, vol. 27, 2010, pages 285 - 291 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019082184A1 (en) | 2019-05-02 |
| US20230330148A1 (en) | 2023-10-19 |
| EP4461356A3 (en) | 2025-01-22 |
| US20200254025A1 (en) | 2020-08-13 |
| IL274190A (en) | 2020-06-30 |
| EP3700543A1 (en) | 2020-09-02 |
| CN111565731A (en) | 2020-08-21 |
| US11701391B2 (en) | 2023-07-18 |
| CN111565731B (en) | 2024-03-08 |
| EP3700543A4 (en) | 2021-08-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP4176048B1 (en) | Genetically engineered t cells with regnase-1 and/or tgfbrii disruption have improved functionality and persistence | |
| US20230330148A1 (en) | Methods of treating an ischemic disease | |
| US20230346836A1 (en) | Genetically engineered t cells with disrupted casitas b-lineage lymphoma proto-oncogene-b (cblb) and uses thereof | |
| WO2017153982A1 (en) | Method for modulating myelination | |
| KR20250068649A (en) | Immune cells with co-expressed TGFBR SHRNA | |
| KR20200141470A (en) | Composition and method for adjusting somatic cell reprogramming and imprinting | |
| US20250288673A1 (en) | Gene therapy for the treatment of activated pi3kinase delta syndrome type 1 (apds1) | |
| TW202409271A (en) | Compositions and methods for reducing mhc class i in a cell | |
| WO2022189967A1 (en) | Genetically engineered t cells with ptpn2 knockout have improved functionality and anti-tumor activity | |
| US20250222103A1 (en) | Immune cells with combination gene perturbations | |
| US12109224B2 (en) | USB1 directed regulation of miRNAs related hematopoietic differentiation and affected in multiple forms of leukemia | |
| US20240318154A1 (en) | Gene therapy for the treatment of severe combined immunodeficiency (scid) related to rag1 | |
| CN120898000A (en) | New polynucleotides, cells and methods | |
| Yang | Revealing a Non-canonical Role of Anti-apoptotic MCL-1 in Early Embryonic Development |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20240926 |
|
| AC | Divisional application: reference to earlier application |
Ref document number: 3700543 Country of ref document: EP Kind code of ref document: P |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R079 Free format text: PREVIOUS MAIN CLASS: A61P0009100000 Ipc: A61K0035280000 |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: A61P 9/10 20060101ALI20241213BHEP Ipc: C12N 15/90 20060101ALI20241213BHEP Ipc: C12N 5/0775 20100101ALI20241213BHEP Ipc: C07K 14/715 20060101ALI20241213BHEP Ipc: A61K 48/00 20060101ALI20241213BHEP Ipc: A61K 39/00 20060101ALI20241213BHEP Ipc: C12N 15/85 20060101ALI20241213BHEP Ipc: C12N 15/63 20060101ALI20241213BHEP Ipc: A61K 35/28 20150101AFI20241213BHEP |