CN116287222A - Methylation marker for diagnosis of benign and malignant thyroid cancer nodules and application thereof - Google Patents
Methylation marker for diagnosis of benign and malignant thyroid cancer nodules and application thereof Download PDFInfo
- Publication number
- CN116287222A CN116287222A CN202111496935.XA CN202111496935A CN116287222A CN 116287222 A CN116287222 A CN 116287222A CN 202111496935 A CN202111496935 A CN 202111496935A CN 116287222 A CN116287222 A CN 116287222A
- Authority
- CN
- China
- Prior art keywords
- gene
- sequence
- genome
- methylation
- chr19
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 230000011987 methylation Effects 0.000 title claims abstract description 201
- 238000007069 methylation reaction Methods 0.000 title claims abstract description 201
- 230000003211 malignant effect Effects 0.000 title claims abstract description 94
- 208000024770 Thyroid neoplasm Diseases 0.000 title claims abstract description 45
- 239000003550 marker Substances 0.000 title abstract description 115
- 238000003745 diagnosis Methods 0.000 title abstract description 16
- 201000002510 thyroid cancer Diseases 0.000 title description 11
- 239000003153 chemical reaction reagent Substances 0.000 claims abstract description 83
- 108091029430 CpG site Proteins 0.000 claims abstract description 64
- 208000009453 Thyroid Nodule Diseases 0.000 claims abstract description 54
- 108700012457 TACSTD2 Proteins 0.000 claims abstract description 42
- 238000001514 detection method Methods 0.000 claims abstract description 22
- 238000009007 Diagnostic Kit Methods 0.000 claims abstract description 19
- 102100027212 Tumor-associated calcium signal transducer 2 Human genes 0.000 claims abstract description 18
- 101000686942 Homo sapiens Histone-lysine N-methyltransferase PRDM16 Proteins 0.000 claims abstract description 13
- 102100024594 Histone-lysine N-methyltransferase PRDM16 Human genes 0.000 claims abstract description 12
- 101000913913 Homo sapiens Calcium/calmodulin-dependent protein kinase II inhibitor 1 Proteins 0.000 claims abstract description 9
- 102100026252 Calcium/calmodulin-dependent protein kinase II inhibitor 1 Human genes 0.000 claims abstract description 8
- 238000002360 preparation method Methods 0.000 claims abstract description 8
- 108020004414 DNA Proteins 0.000 claims description 122
- 150000007523 nucleic acids Chemical class 0.000 claims description 85
- 206010028980 Neoplasm Diseases 0.000 claims description 78
- 108090000623 proteins and genes Proteins 0.000 claims description 76
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 72
- 102000039446 nucleic acids Human genes 0.000 claims description 71
- 108020004707 nucleic acids Proteins 0.000 claims description 71
- 201000011510 cancer Diseases 0.000 claims description 70
- 239000000523 sample Substances 0.000 claims description 70
- 230000036210 malignancy Effects 0.000 claims description 57
- 238000000034 method Methods 0.000 claims description 57
- 102000004190 Enzymes Human genes 0.000 claims description 55
- 108090000790 Enzymes Proteins 0.000 claims description 55
- 230000003321 amplification Effects 0.000 claims description 45
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 45
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 41
- 238000009396 hybridization Methods 0.000 claims description 39
- 101150000195 EGR3 gene Proteins 0.000 claims description 36
- 101000616757 Homo sapiens Small integral membrane protein 24 Proteins 0.000 claims description 35
- 238000006243 chemical reaction Methods 0.000 claims description 35
- 101100314148 Homo sapiens TNK1 gene Proteins 0.000 claims description 29
- 101150110588 TNK1 gene Proteins 0.000 claims description 29
- 239000012634 fragment Substances 0.000 claims description 29
- 101000970561 Homo sapiens Myc box-dependent-interacting protein 1 Proteins 0.000 claims description 28
- 125000003729 nucleotide group Chemical group 0.000 claims description 28
- 101150086895 Baiap2 gene Proteins 0.000 claims description 26
- 239000002773 nucleotide Substances 0.000 claims description 26
- 102100031149 Deoxyribonuclease gamma Human genes 0.000 claims description 25
- 101150112693 Dusp26 gene Proteins 0.000 claims description 25
- 102100021717 Early growth response protein 3 Human genes 0.000 claims description 25
- 101000845618 Homo sapiens Deoxyribonuclease gamma Proteins 0.000 claims description 25
- 101000896450 Homo sapiens Early growth response protein 3 Proteins 0.000 claims description 25
- 239000012472 biological sample Substances 0.000 claims description 25
- 108091008146 restriction endonucleases Proteins 0.000 claims description 25
- 101150041872 DNASE1L3 gene Proteins 0.000 claims description 24
- 101000663003 Homo sapiens Non-receptor tyrosine-protein kinase TNK1 Proteins 0.000 claims description 24
- 102100037669 Non-receptor tyrosine-protein kinase TNK1 Human genes 0.000 claims description 24
- 101150028074 2 gene Proteins 0.000 claims description 22
- 101100495925 Schizosaccharomyces pombe (strain 972 / ATCC 24843) chr3 gene Proteins 0.000 claims description 22
- 230000000295 complement effect Effects 0.000 claims description 21
- 238000011282 treatment Methods 0.000 claims description 21
- 101150049556 Bcr gene Proteins 0.000 claims description 20
- 102100026346 Brain-specific angiogenesis inhibitor 1-associated protein 2 Human genes 0.000 claims description 20
- 101150008702 CRABP2 gene Proteins 0.000 claims description 20
- 101000766212 Homo sapiens Brain-specific angiogenesis inhibitor 1-associated protein 2 Proteins 0.000 claims description 20
- 101150118850 Med16 gene Proteins 0.000 claims description 20
- 102100034127 Dual specificity protein phosphatase 26 Human genes 0.000 claims description 19
- 101001017423 Homo sapiens Dual specificity phosphatase 28 Proteins 0.000 claims description 19
- 101001017415 Homo sapiens Dual specificity protein phosphatase 26 Proteins 0.000 claims description 19
- 102100021845 Small integral membrane protein 24 Human genes 0.000 claims description 19
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 claims description 18
- 229940104302 cytosine Drugs 0.000 claims description 18
- 101150094832 Cep295nl gene Proteins 0.000 claims description 17
- 101001003233 Homo sapiens Immediate early response gene 2 protein Proteins 0.000 claims description 17
- 101100521059 Homo sapiens PRDM16 gene Proteins 0.000 claims description 17
- 101100537447 Homo sapiens TMC6 gene Proteins 0.000 claims description 17
- 101100321485 Homo sapiens ZNF219 gene Proteins 0.000 claims description 17
- 101150031424 Icam5 gene Proteins 0.000 claims description 17
- 101150081525 LIMK1 gene Proteins 0.000 claims description 17
- 101150089055 NAV2 gene Proteins 0.000 claims description 17
- 101150009380 PPIF gene Proteins 0.000 claims description 17
- 101150063753 PRDM16 gene Proteins 0.000 claims description 17
- 101150108294 Rtn4r gene Proteins 0.000 claims description 17
- 101150045328 SBNO2 gene Proteins 0.000 claims description 17
- 101150026328 TMC6 gene Proteins 0.000 claims description 17
- 101150024094 UACA gene Proteins 0.000 claims description 17
- 101150070562 CRTC1 gene Proteins 0.000 claims description 16
- 101000582813 Homo sapiens Mediator of RNA polymerase II transcription subunit 16 Proteins 0.000 claims description 16
- 102100030253 Mediator of RNA polymerase II transcription subunit 16 Human genes 0.000 claims description 16
- 101150092254 ASF1 gene Proteins 0.000 claims description 15
- 101150038182 CAMK2N1 gene Proteins 0.000 claims description 15
- 101001062353 Homo sapiens Hepatocyte nuclear factor 3-alpha Proteins 0.000 claims description 15
- 101100342331 Homo sapiens KLF16 gene Proteins 0.000 claims description 14
- 101100136964 Homo sapiens PLXNC1 gene Proteins 0.000 claims description 14
- 101001129467 Homo sapiens Pyroglutamyl-peptidase 1-like protein Proteins 0.000 claims description 14
- 101150050263 ICAM1 gene Proteins 0.000 claims description 14
- 101150097667 ICAM2 gene Proteins 0.000 claims description 14
- 101150104233 KLF16 gene Proteins 0.000 claims description 14
- 101150052935 LSG1 gene Proteins 0.000 claims description 14
- 101150058494 PLXNC1 gene Proteins 0.000 claims description 14
- 102100038504 Cellular retinoic acid-binding protein 2 Human genes 0.000 claims description 13
- 101001099851 Homo sapiens Cellular retinoic acid-binding protein 2 Proteins 0.000 claims description 13
- 101000960337 Homo sapiens Intercellular adhesion molecule 5 Proteins 0.000 claims description 13
- 102100039919 Intercellular adhesion molecule 5 Human genes 0.000 claims description 13
- 238000004458 analytical method Methods 0.000 claims description 13
- 239000000872 buffer Substances 0.000 claims description 13
- 101150054360 ADM gene Proteins 0.000 claims description 11
- 101150071235 ANO6 gene Proteins 0.000 claims description 11
- 101150044315 ASF1B gene Proteins 0.000 claims description 11
- 102100036379 CEP295 N-terminal-like protein Human genes 0.000 claims description 11
- 101150071384 CIRBP gene Proteins 0.000 claims description 11
- 101150114117 EGR1 gene Proteins 0.000 claims description 11
- 101150059401 EGR2 gene Proteins 0.000 claims description 11
- 101150063771 EHBP1L1 gene Proteins 0.000 claims description 11
- 101100190607 Homo sapiens AGPAT2 gene Proteins 0.000 claims description 11
- 101100269862 Homo sapiens ANO6 gene Proteins 0.000 claims description 11
- 101000714814 Homo sapiens CEP295 N-terminal-like protein Proteins 0.000 claims description 11
- 101100007687 Homo sapiens CREB5 gene Proteins 0.000 claims description 11
- 101100172996 Homo sapiens FAM20C gene Proteins 0.000 claims description 11
- 101100232471 Homo sapiens IER5 gene Proteins 0.000 claims description 11
- 101100510662 Homo sapiens LARP1 gene Proteins 0.000 claims description 11
- 101001005128 Homo sapiens LIM domain kinase 1 Proteins 0.000 claims description 11
- 101100518947 Homo sapiens PARP11 gene Proteins 0.000 claims description 11
- 101001091191 Homo sapiens Peptidyl-prolyl cis-trans isomerase F, mitochondrial Proteins 0.000 claims description 11
- 101100483042 Homo sapiens TTBK1 gene Proteins 0.000 claims description 11
- 101000608672 Homo sapiens Uveal autoantigen with coiled-coil domains and ankyrin repeats Proteins 0.000 claims description 11
- 101000782130 Homo sapiens Zinc finger protein 219 Proteins 0.000 claims description 11
- 101150065686 ITGB1BP1 gene Proteins 0.000 claims description 11
- 101150102269 ITPKB gene Proteins 0.000 claims description 11
- 102100026023 LIM domain kinase 1 Human genes 0.000 claims description 11
- 101150088720 MTHFD2 gene Proteins 0.000 claims description 11
- 102100021970 Myc box-dependent-interacting protein 1 Human genes 0.000 claims description 11
- 101150077147 NR2F1 gene Proteins 0.000 claims description 11
- 101150057903 NRARP gene Proteins 0.000 claims description 11
- 101150068134 PAPLN gene Proteins 0.000 claims description 11
- 101150091105 PARP11 gene Proteins 0.000 claims description 11
- 101150065640 PHLDB1 gene Proteins 0.000 claims description 11
- 101150035965 PRKAG2 gene Proteins 0.000 claims description 11
- 102100034943 Peptidyl-prolyl cis-trans isomerase F, mitochondrial Human genes 0.000 claims description 11
- 101150114601 RARS gene Proteins 0.000 claims description 11
- 101150057435 SH3BP2 gene Proteins 0.000 claims description 11
- 101150052128 SLC12A7 gene Proteins 0.000 claims description 11
- 101150098920 SLC39A14 gene Proteins 0.000 claims description 11
- 101150009693 TBCD gene Proteins 0.000 claims description 11
- 102100039543 Uveal autoantigen with coiled-coil domains and ankyrin repeats Human genes 0.000 claims description 11
- 102100036594 Zinc finger protein 219 Human genes 0.000 claims description 11
- 101150060483 chid1 gene Proteins 0.000 claims description 11
- 101150115803 metrnl gene Proteins 0.000 claims description 11
- 101150096543 rpl19 gene Proteins 0.000 claims description 11
- 239000000126 substance Substances 0.000 claims description 11
- 101000640056 Homo sapiens Protein strawberry notch homolog 2 Proteins 0.000 claims description 10
- 102100033980 Protein strawberry notch homolog 2 Human genes 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 10
- 238000012164 methylation sequencing Methods 0.000 claims description 10
- 101150033839 4 gene Proteins 0.000 claims description 9
- 229940035893 uracil Drugs 0.000 claims description 9
- 101100126930 Homo sapiens KCNK15 gene Proteins 0.000 claims description 8
- 101100129000 Homo sapiens LTBP4 gene Proteins 0.000 claims description 8
- 101100371695 Homo sapiens UCKL1 gene Proteins 0.000 claims description 8
- 101100107193 Homo sapiens ZNF536 gene Proteins 0.000 claims description 8
- 101000687648 Homo sapiens snRNA-activating protein complex subunit 2 Proteins 0.000 claims description 8
- 101150071810 Itpripl2 gene Proteins 0.000 claims description 8
- 101150043513 KCNK15 gene Proteins 0.000 claims description 8
- 101150078565 TEF gene Proteins 0.000 claims description 8
- 101150086775 ZNF536 gene Proteins 0.000 claims description 8
- 102100040775 CREB-regulated transcription coactivator 1 Human genes 0.000 claims description 7
- 101000891939 Homo sapiens CREB-regulated transcription coactivator 1 Proteins 0.000 claims description 7
- 101000599858 Homo sapiens Intercellular adhesion molecule 2 Proteins 0.000 claims description 7
- 101000984653 Homo sapiens Large subunit GTPase 1 homolog Proteins 0.000 claims description 7
- 102100037872 Intercellular adhesion molecule 2 Human genes 0.000 claims description 7
- 102100027113 Large subunit GTPase 1 homolog Human genes 0.000 claims description 7
- 102000000343 Nogo Receptor 1 Human genes 0.000 claims description 7
- 108010041199 Nogo Receptor 1 Proteins 0.000 claims description 7
- 230000000694 effects Effects 0.000 claims description 7
- 102100038369 1-acyl-sn-glycerol-3-phosphate acyltransferase beta Human genes 0.000 claims description 6
- 102100023774 Cold-inducible RNA-binding protein Human genes 0.000 claims description 6
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 claims description 6
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 claims description 6
- 101000605571 Homo sapiens 1-acyl-sn-glycerol-3-phosphate acyltransferase beta Proteins 0.000 claims description 6
- 101000906744 Homo sapiens Cold-inducible RNA-binding protein Proteins 0.000 claims description 6
- 101001046593 Homo sapiens Krueppel-like factor 16 Proteins 0.000 claims description 6
- 102100022324 Krueppel-like factor 16 Human genes 0.000 claims description 6
- 101100436059 Schizosaccharomyces pombe (strain 972 / ATCC 24843) cia1 gene Proteins 0.000 claims description 6
- 101100163864 Xenopus laevis asf1aa gene Proteins 0.000 claims description 6
- 239000007850 fluorescent dye Substances 0.000 claims description 6
- 238000004949 mass spectrometry Methods 0.000 claims description 6
- 238000003752 polymerase chain reaction Methods 0.000 claims description 6
- 108060002716 Exonuclease Proteins 0.000 claims description 5
- 230000029087 digestion Effects 0.000 claims description 5
- 102000013165 exonuclease Human genes 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 102100024626 5'-AMP-activated protein kinase subunit gamma-2 Human genes 0.000 claims description 4
- 102100021206 60S ribosomal protein L19 Human genes 0.000 claims description 4
- 102100036523 Anoctamin-6 Human genes 0.000 claims description 4
- 102100036131 Arginine-tRNA ligase, cytoplasmic Human genes 0.000 claims description 4
- 102100028228 COUP transcription factor 1 Human genes 0.000 claims description 4
- 102100030298 Chitinase domain-containing protein 1 Human genes 0.000 claims description 4
- 102100023227 E3 SUMO-protein ligase EGR2 Human genes 0.000 claims description 4
- 102100023226 Early growth response protein 1 Human genes 0.000 claims description 4
- 102100027300 Extracellular serine/threonine protein kinase FAM20C Human genes 0.000 claims description 4
- 102100029283 Hepatocyte nuclear factor 3-alpha Human genes 0.000 claims description 4
- 101000760987 Homo sapiens 5'-AMP-activated protein kinase subunit gamma-2 Proteins 0.000 claims description 4
- 101001105789 Homo sapiens 60S ribosomal protein L19 Proteins 0.000 claims description 4
- 101000928362 Homo sapiens Anoctamin-6 Proteins 0.000 claims description 4
- 101000874860 Homo sapiens Arginine-tRNA ligase, cytoplasmic Proteins 0.000 claims description 4
- 101000860854 Homo sapiens COUP transcription factor 1 Proteins 0.000 claims description 4
- 101000991102 Homo sapiens Chitinase domain-containing protein 1 Proteins 0.000 claims description 4
- 101001049692 Homo sapiens E3 SUMO-protein ligase EGR2 Proteins 0.000 claims description 4
- 101001049697 Homo sapiens Early growth response protein 1 Proteins 0.000 claims description 4
- 101000937709 Homo sapiens Extracellular serine/threonine protein kinase FAM20C Proteins 0.000 claims description 4
- 101001003310 Homo sapiens Immediate early response gene 5 protein Proteins 0.000 claims description 4
- 101001033889 Homo sapiens Inositol 1,4,5-trisphosphate receptor-interacting protein-like 2 Proteins 0.000 claims description 4
- 101000852593 Homo sapiens Inositol-trisphosphate 3-kinase B Proteins 0.000 claims description 4
- 101000997642 Homo sapiens Integrin beta-1-binding protein 1 Proteins 0.000 claims description 4
- 101001138022 Homo sapiens La-related protein 1 Proteins 0.000 claims description 4
- 101000985328 Homo sapiens Methenyltetrahydrofolate cyclohydrolase Proteins 0.000 claims description 4
- 101000582002 Homo sapiens Neuron navigator 2 Proteins 0.000 claims description 4
- 101000577309 Homo sapiens Notch-regulated ankyrin repeat-containing protein Proteins 0.000 claims description 4
- 101001091425 Homo sapiens Papilin Proteins 0.000 claims description 4
- 101000597240 Homo sapiens Pleckstrin homology-like domain family B member 1 Proteins 0.000 claims description 4
- 101001094872 Homo sapiens Plexin-C1 Proteins 0.000 claims description 4
- 101000613612 Homo sapiens Protein mono-ADP-ribosyltransferase PARP11 Proteins 0.000 claims description 4
- 101000761644 Homo sapiens SH3 domain-binding protein 2 Proteins 0.000 claims description 4
- 101000759314 Homo sapiens Tau-tubulin kinase 1 Proteins 0.000 claims description 4
- 101000851627 Homo sapiens Transmembrane channel-like protein 6 Proteins 0.000 claims description 4
- 101000652500 Homo sapiens Tubulin-specific chaperone D Proteins 0.000 claims description 4
- 102100020688 Immediate early response gene 5 protein Human genes 0.000 claims description 4
- 102100039741 Inositol 1,4,5-trisphosphate receptor-interacting protein-like 2 Human genes 0.000 claims description 4
- 102100036404 Inositol-trisphosphate 3-kinase B Human genes 0.000 claims description 4
- 102100033335 Integrin beta-1-binding protein 1 Human genes 0.000 claims description 4
- 102100020859 La-related protein 1 Human genes 0.000 claims description 4
- 102100032280 Metal cation symporter ZIP14 Human genes 0.000 claims description 4
- 102100028687 Methenyltetrahydrofolate cyclohydrolase Human genes 0.000 claims description 4
- 102100030465 Neuron navigator 2 Human genes 0.000 claims description 4
- 102100028809 Notch-regulated ankyrin repeat-containing protein Human genes 0.000 claims description 4
- 102100034934 Papilin Human genes 0.000 claims description 4
- 102100035150 Pleckstrin homology-like domain family B member 1 Human genes 0.000 claims description 4
- 102100035381 Plexin-C1 Human genes 0.000 claims description 4
- 102100040850 Protein mono-ADP-ribosyltransferase PARP11 Human genes 0.000 claims description 4
- 102100024865 SH3 domain-binding protein 2 Human genes 0.000 claims description 4
- 108091006626 SLC12A7 Proteins 0.000 claims description 4
- 108091006944 SLC39A14 Proteins 0.000 claims description 4
- 102100034252 Solute carrier family 12 member 7 Human genes 0.000 claims description 4
- 102100023277 Tau-tubulin kinase 1 Human genes 0.000 claims description 4
- 102100036810 Transmembrane channel-like protein 6 Human genes 0.000 claims description 4
- 102100030290 Tubulin-specific chaperone D Human genes 0.000 claims description 4
- 238000004590 computer program Methods 0.000 claims description 4
- 238000002844 melting Methods 0.000 claims description 4
- 230000008018 melting Effects 0.000 claims description 4
- 101150084750 1 gene Proteins 0.000 claims description 3
- 102100027309 Cyclic AMP-responsive element-binding protein 5 Human genes 0.000 claims description 3
- 101000726193 Homo sapiens Cyclic AMP-responsive element-binding protein 5 Proteins 0.000 claims description 3
- 102100020702 Immediate early response gene 2 protein Human genes 0.000 claims description 3
- 102100031114 Pyroglutamyl-peptidase 1-like protein Human genes 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 3
- 102000002260 Alkaline Phosphatase Human genes 0.000 claims description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 claims description 2
- 102100022789 Calcium/calmodulin-dependent protein kinase type IV Human genes 0.000 claims description 2
- 238000001712 DNA sequencing Methods 0.000 claims description 2
- 101150063735 DNASE1 gene Proteins 0.000 claims description 2
- 101100287682 Homo sapiens CAMK2G gene Proteins 0.000 claims description 2
- 101100126883 Homo sapiens CAMK4 gene Proteins 0.000 claims description 2
- 101000727472 Homo sapiens Reticulon-4 Proteins 0.000 claims description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 claims description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical class OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 claims description 2
- 238000009585 enzyme analysis Methods 0.000 claims description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 claims description 2
- 238000010200 validation analysis Methods 0.000 description 52
- 238000012549 training Methods 0.000 description 27
- 230000035945 sensitivity Effects 0.000 description 26
- 238000007477 logistic regression Methods 0.000 description 21
- 206010054107 Nodule Diseases 0.000 description 18
- 210000001685 thyroid gland Anatomy 0.000 description 18
- 108091028043 Nucleic acid sequence Proteins 0.000 description 17
- 238000004445 quantitative analysis Methods 0.000 description 17
- 239000000243 solution Substances 0.000 description 17
- 230000007067 DNA methylation Effects 0.000 description 16
- 230000002380 cytological effect Effects 0.000 description 16
- 238000012163 sequencing technique Methods 0.000 description 14
- 210000004369 blood Anatomy 0.000 description 13
- 239000008280 blood Substances 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- 239000000203 mixture Substances 0.000 description 13
- 238000012795 verification Methods 0.000 description 12
- 210000002381 plasma Anatomy 0.000 description 11
- 230000002255 enzymatic effect Effects 0.000 description 10
- 210000001519 tissue Anatomy 0.000 description 10
- 238000003556 assay Methods 0.000 description 9
- 238000003753 real-time PCR Methods 0.000 description 9
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 9
- -1 AGTAT 2 Proteins 0.000 description 8
- 108091034117 Oligonucleotide Proteins 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 108091033319 polynucleotide Proteins 0.000 description 8
- 102000040430 polynucleotide Human genes 0.000 description 8
- 239000002157 polynucleotide Substances 0.000 description 8
- 238000007855 methylation-specific PCR Methods 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- 238000003776 cleavage reaction Methods 0.000 description 6
- 230000003325 follicular Effects 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 230000007017 scission Effects 0.000 description 6
- 102000053602 DNA Human genes 0.000 description 5
- 108010006785 Taq Polymerase Proteins 0.000 description 5
- 210000001124 body fluid Anatomy 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 238000002493 microarray Methods 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- 208000037065 Subacute sclerosing leukoencephalitis Diseases 0.000 description 4
- 206010042297 Subacute sclerosing panencephalitis Diseases 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 230000006399 behavior Effects 0.000 description 4
- 239000010839 body fluid Substances 0.000 description 4
- 108091092356 cellular DNA Proteins 0.000 description 4
- 239000000975 dye Substances 0.000 description 4
- 238000001962 electrophoresis Methods 0.000 description 4
- 238000006911 enzymatic reaction Methods 0.000 description 4
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 238000013178 mathematical model Methods 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 238000012706 support-vector machine Methods 0.000 description 4
- 229940113082 thymine Drugs 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 108700028369 Alleles Proteins 0.000 description 3
- 108091093088 Amplicon Proteins 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 238000012408 PCR amplification Methods 0.000 description 3
- 101001074199 Rattus norvegicus Glycerol kinase Proteins 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 238000001574 biopsy Methods 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 230000009615 deamination Effects 0.000 description 3
- 238000006481 deamination reaction Methods 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 238000002405 diagnostic procedure Methods 0.000 description 3
- 238000013399 early diagnosis Methods 0.000 description 3
- 230000001605 fetal effect Effects 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000003780 insertion Methods 0.000 description 3
- 230000037431 insertion Effects 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 238000007857 nested PCR Methods 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000012175 pyrosequencing Methods 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 239000001488 sodium phosphate Substances 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- JTNCEQNHURODLX-UHFFFAOYSA-N 2-phenylethanimidamide Chemical compound NC(=N)CC1=CC=CC=C1 JTNCEQNHURODLX-UHFFFAOYSA-N 0.000 description 2
- 101100460704 Aspergillus sp. (strain MF297-2) notI gene Proteins 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 208000004434 Calcinosis Diseases 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 2
- 206010033701 Papillary thyroid cancer Diseases 0.000 description 2
- 101100006527 Penicillium crustosum claI gene Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical group [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- BIGPRXCJEDHCLP-UHFFFAOYSA-N ammonium bisulfate Chemical compound [NH4+].OS([O-])(=O)=O BIGPRXCJEDHCLP-UHFFFAOYSA-N 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- JXRVKYBCWUJJBP-UHFFFAOYSA-L calcium;hydrogen sulfate Chemical compound [Ca+2].OS([O-])(=O)=O.OS([O-])(=O)=O JXRVKYBCWUJJBP-UHFFFAOYSA-L 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- CTMZLDSMFCVUNX-VMIOUTBZSA-N cytidylyl-(3'->5')-guanosine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=C(C(N=C(N)N3)=O)N=C2)O)[C@@H](CO)O1 CTMZLDSMFCVUNX-VMIOUTBZSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000006326 desulfonation Effects 0.000 description 2
- 238000005869 desulfonation reaction Methods 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- APGUSRKQFBWUPZ-UHFFFAOYSA-K disulfooxyalumanyl hydrogen sulfate Chemical compound [Al+3].OS([O-])(=O)=O.OS([O-])(=O)=O.OS([O-])(=O)=O APGUSRKQFBWUPZ-UHFFFAOYSA-K 0.000 description 2
- 230000001973 epigenetic effect Effects 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000009830 intercalation Methods 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- 238000007834 ligase chain reaction Methods 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 238000001471 micro-filtration Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 101150067874 narI gene Proteins 0.000 description 2
- 238000007899 nucleic acid hybridization Methods 0.000 description 2
- 239000002751 oligonucleotide probe Substances 0.000 description 2
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 2
- 238000004321 preservation Methods 0.000 description 2
- 238000004393 prognosis Methods 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 2
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 2
- 239000004289 sodium hydrogen sulphite Substances 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 description 2
- 238000002604 ultrasonography Methods 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 1
- 102100026008 Breakpoint cluster region protein Human genes 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 102100027473 Cartilage oligomeric matrix protein Human genes 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108010045171 Cyclic AMP Response Element-Binding Protein Proteins 0.000 description 1
- 102000005636 Cyclic AMP Response Element-Binding Protein Human genes 0.000 description 1
- 238000007400 DNA extraction Methods 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 102100030012 Deoxyribonuclease-1 Human genes 0.000 description 1
- 102100029650 EH domain-binding protein 1-like protein 1 Human genes 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- 101000725508 Homo sapiens Cartilage oligomeric matrix protein Proteins 0.000 description 1
- 101000919645 Homo sapiens Collagen alpha-2(IX) chain Proteins 0.000 description 1
- 101000919644 Homo sapiens Collagen alpha-3(IX) chain Proteins 0.000 description 1
- 101000863721 Homo sapiens Deoxyribonuclease-1 Proteins 0.000 description 1
- 101001012961 Homo sapiens EH domain-binding protein 1-like protein 1 Proteins 0.000 description 1
- 101001034286 Homo sapiens Meteorin-like protein Proteins 0.000 description 1
- 101000690940 Homo sapiens Pro-adrenomedullin Proteins 0.000 description 1
- 101000654381 Homo sapiens Sodium channel protein type 8 subunit alpha Proteins 0.000 description 1
- 101150042441 K gene Proteins 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102100039669 Meteorin-like protein Human genes 0.000 description 1
- 102000016397 Methyltransferase Human genes 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 101150073096 NRAS gene Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 108091005461 Nucleic proteins Chemical group 0.000 description 1
- 108010047956 Nucleosomes Proteins 0.000 description 1
- 239000012807 PCR reagent Substances 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102100026651 Pro-adrenomedullin Human genes 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 1
- 101150077555 Ret gene Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 208000033781 Thyroid carcinoma Diseases 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 125000003275 alpha amino acid group Chemical group 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 210000004883 areola Anatomy 0.000 description 1
- 238000010420 art technique Methods 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 230000002308 calcification Effects 0.000 description 1
- 238000004422 calculation algorithm Methods 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 108091092328 cellular RNA Proteins 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 230000003196 chaotropic effect Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 238000013499 data model Methods 0.000 description 1
- 238000012350 deep sequencing Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000003935 denaturing gradient gel electrophoresis Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000007614 genetic variation Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000011544 gradient gel Substances 0.000 description 1
- 239000005337 ground glass Substances 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000004660 morphological change Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 210000001623 nucleosome Anatomy 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 210000004910 pleural fluid Anatomy 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000012257 pre-denaturation Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000013615 primer Substances 0.000 description 1
- 239000002987 primer (paints) Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 235000004252 protein component Nutrition 0.000 description 1
- 101150103120 ptc gene Proteins 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 102000016914 ras Proteins Human genes 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 238000012502 risk assessment Methods 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 238000000528 statistical test Methods 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 208000013077 thyroid gland carcinoma Diseases 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6834—Enzymatic or biochemical coupling of nucleic acids to a solid phase
- C12Q1/6837—Enzymatic or biochemical coupling of nucleic acids to a solid phase using probe arrays or probe chips
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
- G16B25/10—Gene or protein expression profiling; Expression-ratio estimation or normalisation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/112—Disease subtyping, staging or classification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/166—Oligonucleotides used as internal standards, controls or normalisation probes
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Analytical Chemistry (AREA)
- Molecular Biology (AREA)
- Physics & Mathematics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Pathology (AREA)
- Biomedical Technology (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Plant Pathology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Evolutionary Biology (AREA)
- Medical Informatics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Theoretical Computer Science (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
The invention relates to a methylation marker for diagnosing benign and malignant thyroid nodules and application thereof, in particular to application of a reagent for detecting methylation state or level of at least one CpG dinucleotide of one or more target markers in preparation of a detection reagent or a diagnosis reagent kit for diagnosing benign and malignant thyroid nodules of an individual, and application of a device for determining methylation state or level of at least one CpG dinucleotide of one or more target markers in preparation of a diagnosis reagent kit for diagnosing benign and malignant thyroid nodules of the individual, wherein the target markers comprise PRDM16 sequences of PRDM16 genes or genomes, CAMK2N1 sequences of CAMK2N1 genes or genomes, TACSTD2 sequences of TACSTD2 genes or genomes and the like. The invention also includes a diagnostic reagent or diagnostic kit for detecting the methylation state or methylation level of at least one CpG dinucleotide in the target marker to diagnose benign and malignant thyroid nodule.
Description
Technical Field
The invention relates to a methylation marker for diagnosing benign and malignant thyroid cancer nodules and application thereof.
Background
Thyroid cancer is a malignancy that originates in the epithelium of thyroid follicles. The female morbidity is more, and the male and female morbidity proportion is 1: (2-4), the age of onset is generally 21-40 years. Papillary thyroid carcinoma (Papillary thyroid cancer, PTC) is the most common thyroid carcinoma, accounting for approximately 80% of all thyroid carcinomas. In recent years, the incidence of domestic thyroid cancer is on the rise. The thyroid cancer is discovered early and treated in time, the prognosis is good, and the survival rate of 10 years can reach more than 90 percent; however, if the patients are in early stage and leak diagnosis, the patients develop to local advanced stage, the patients lose the opportunity of operation and cannot be cured, and the survival rate of 5 years is obviously reduced.
The clinical routine diagnostic method is an imaging examination. Ultrasound examination is highly suspected of malignant thyroid nodules and requires further fine needle puncture cytology (fine needle aspiration, FNA) examination to confirm diagnosis. Malignant and benign nodules present some difficulty in diagnosing PTC due to approximate cytologic characteristics, and up to 40% of thyroid nodules are difficult to accurately diagnose by cytologic characteristics. Current molecular diagnostic methods improve the accuracy of identification, but the sensitivity of these methods remains to be improved. Gene Expression Classifier is commonly used, but the positive predictive value (positive predictive value, PPV) is only 47%, and the detection of fresh puncture tissue is only performed, so that the wide application of some samples is limited. ThyroSeqv2 detects H/K/NRAS gene mutations and RET/PTC gene rearrangements frequently carried by benign nodules, with PPV of only 42-77%. Furthermore, diagnostic DNA Methylation Signature approach (DDMS) is a diagnostic method based on DNA methylation characteristics for the identification of benign and malignant thyroid cancer tissue. Although the method is highly accurate, some samples cannot be detected by the method for technical reasons [ John H YIm, audrey H Choi, arthur X Li et al Identification of Tissue-Specific DNA Methylation Signatures for Thyroid Nodule Diagnostics, clin Cancer Res,2019Jan 1 ] 5;25(2):544-551〕。
Gene Expression Classifier is commonly used, but the positive predictive value (positive predictive value, PPV) is only 47%, and the detection of fresh puncture tissue is only performed, so that the wide application of some samples is limited. ThyroSeqv2 detects H/K/NRAS gene mutations and RET/PTC gene rearrangements frequently carried by benign nodules, with PPV of only 42-77%. Furthermore, diagnostic DNA Methylation Signature approach (DDMS) is a diagnostic method based on DNA methylation characteristics for the identification of benign and malignant thyroid cancer tissue. Although the method is highly accurate, some samples cannot be detected by the method for technical reasons [ John H YIm, audrey H Choi, arthur X Li et al Identification of Tissue-Specific DNA Methylation Signatures for Thyroid Nodule Diagnostics, clin Cancer Res,2019Jan 1 ] 5;25(2):544-551〕。
Disclosure of Invention
In a first aspect, the present invention provides the use of a reagent for detecting the methylation status or level of at least one CpG dinucleotide of one or more markers of interest for the manufacture of a detection reagent or a diagnostic kit for diagnosing benign and malignant thyroid nodules in an individual, and the use of a device for determining the methylation status or level of at least one CpG dinucleotide of one or more markers of interest for the manufacture of a diagnostic kit for diagnosing benign and malignant thyroid nodules in an individual, wherein the one or more markers of interest are selected from the group consisting of: PRDM16 gene or genome PRDM16 sequence, CAMK2N1 gene or genome CAMK2N1 sequence, TACSTD2 gene or genome TACSTD2 sequence, CRABP2 gene or genome CRABP2 sequence, IER5 gene or genome IER5 sequence, ITPKB gene or genome ITPKB sequence, ITGB1BP1 gene or genome ITGB1BP1 sequence, MTHFD2 gene or genome MTHFD2 sequence, BIN1 gene or genome BIN1 sequence, DNASE1L3 gene or genome DNASE1L3 sequence, DNASE1L3 sequence LSG1 sequence of LSG1 gene or genome, SH3BP2 sequence of SH3BP2 gene or genome, SLC12A7 sequence of SLC12A7 gene or genome, NR2F1 sequence of NR2F1 gene or genome, EGR1 sequence of EGR1 gene or genome, LARP1 sequence of LARP1 gene or genome, RARS sequence of RARS gene or genome, TTBK1 sequence of TTBK1 gene or genome, FAM20C sequence of FAM20C gene or genome, CREB5 sequence of CREB5 gene or genome, LIMK1 sequence of LIMK1 gene or genome PRKAG2 sequence of PRKAG2 gene or genome, SLC39A14 sequence of SLC39A14 gene or genome, EGR3 sequence of EGR3 gene or genome, DUSP26 sequence of DUSP26 gene or genome, AGPAT2 sequence of AGPAT2 gene or genome, AGPAT2 sequence of gene or genome, gene, or genome, or sequence, or gene, or NRARP sequence of NRARP gene or genome, EGR2 sequence of EGR2 gene or genome, PPIF sequence of PPIF gene or genome, CHID1 sequence of CHID1 gene or genome, ADM sequence of ADM gene or genome, NAV2 sequence of NAV2 gene or genome, and/or its/their/EHBP 1L1 sequence of the EHBP1L1 gene or genome, PHLDB1 sequence of the PHLDB1 gene or genome, PARP11 sequence of the PARP11 gene or genome, ANO6 sequence of the ANO6 gene or genome, PLXNC1 sequence of the PLXNC1 gene or genome, ZNF219 sequence of the ZNF219 gene or genome, FOXA1 sequence of the FOXA1 gene or genome, PAPLN sequence of the PAPLN gene or genome, UACA sequence of the UACA gene or genome, PGPEP1L sequence of the PGPEP1L gene or genome, ITPRIPL2 sequence of the PLXNC1 gene or genome, TNK1 gene or genome's TNK1 sequence, RPL19 gene or genome's RPL19 sequence, ICAM2 gene or genome's ICAM2 sequence, TMC6 gene or genome's TMC6 sequence, CEP295NL gene or genome's CEP295NL sequence, BAIAP2 gene or genome's BAIAP2 sequence, TBCD gene or genome's TBCD sequence, METRNL gene or genome's METRL sequence, MED16 gene or genome's MED16 sequence, SBNO2 gene or genome's SBNO2 sequence, CIRBP gene or genome's CIRBP sequence, KLF16 gene or genome's KLF16 sequence, C19orf77 gene or genome's C19orf77 sequence, SNNK 2 gene or genome's SNN 2 sequence, ICAM1 gene or genome's ICAM5 sequence, IER2 gene or genome's IER2 sequence, ASF1B gene or genome's MED16 sequence, ASF1 gene or genome's CIRBP sequence, KLF16 gene or genome's KLF16 sequence, C19orf77 sequence, SNNK 2 gene or genome's SNNK 2 sequence, ICAM1 gene or genome's ICAM5 sequence, ICAM5 gene or genome's TC 2 gene's IER2 gene or genome's IEF 1, ASF1 gene or ASF1 gene's ASF1 or genome's ASF1, ASF 4 or gene's BCF 4 gene's 4 sequence, or its BCF 4 gene or genome's 4 sequence, and its KCR 4 gene or its 4 sequence.
In one or more embodiments, the one or more markers of interest are selected from the group consisting of: the PRDM16 gene or genome PRDM16 sequence, BIN1 sequence of a BIN1 gene or genome, LIMK1 sequence of a LIMK1 gene or genome, EGR3 sequence of a CRTC1 gene or genome, PPIF sequence of a PPIF gene or genome, ZNF219 sequence of a ZNF219 gene or genome, UACA sequence of a UACA gene or genome, TNK1 sequence of a TNK1 gene or genome, CEP295NL sequence of a CEP295NL gene or genome, SBNO2 sequence of a SBNO2 gene or genome, C19orf77 sequence of a C19orf77 gene or genome, ICAM5 sequence of a ICAM5 gene or genome, CRTC1 sequence of a CRTC1 gene or genome, RTN4R sequence of a RTN4 gene or genome, CAMK2N1 sequence of a CAMK 1 gene or genome, DNASE1L3 sequence of a DNASE1 gene or genome, DUSP26 sequence of a DUSP26 gene or genome, a cstr 2 sequence of a csag 2 gene or genome, a cstr 2 sequence of a cstr 2 gene or genome, a cstr 2 or a cstr 2 gene or genome.
In one or more embodiments, the one or more target markers include at least one or more of the following target markers: the EGR3 sequence of the EGR3 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, the TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the BCR sequence of the BCR gene or genome.
In one or more embodiments, the one or more target markers comprise: the PRDM16 gene or genome PRDM16 sequence, BIN1 sequence of a BIN1 gene or genome, LIMK1 sequence of a LIMK1 gene or genome, EGR3 sequence of an EGR3 gene or genome, PPIF sequence of a PPIF gene or genome, ZNF219 sequence of a ZNF219 gene or genome, UACA sequence of a UACA gene or genome, TNK1 sequence of a TNK1 gene or genome, CEP295NL sequence of a CEP295NL gene or genome, SBNO2 sequence of a SBNO2 gene or genome, C19orf77 sequence of a C19orf77 gene or genome, ICAM5 sequence of an ICAM5 gene or genome, CRTC1 sequence of a CRTC1 gene or genome, and RTN4R sequence of an RTN4R gene or genome.
In one or more embodiments, the CAMK2N1 sequence of the CAMK2N1 gene or genome, the DNASE1L3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the ICAM2 sequence of the ICAM2 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome, and the non 4L-DT sequence of the non 4L-DT gene or genome.
In one or more embodiments, the TACSTD2 sequence of a TACSTD2 gene or genome, the CRABP2 sequence of a CRABP2 gene or genome, the DNASE1L3 sequence of a DNASE1L3 gene or genome, the LSG1 sequence of a LSG1 gene or genome, the EGR3 sequence of an EGR3 gene or genome, the TNK1 sequence of a TNK1 gene or genome, the BAIAP2 sequence of a BAIAP2 gene or genome, the NOL4L-DT sequence of a NOL4L-DT gene or genome, and the BCR sequence of a BCR gene or genome.
In one or more embodiments, the TACSTD2 sequence of a TACSTD2 gene or genome, the CRABP2 sequence of a CRABP2 gene or genome, the DNASE1L3 sequence of a DNASE1L3 gene or genome, the EGR3 sequence of an EGR3 gene or genome, the DUSP26 sequence of a DUSP26 gene or genome, the TNK1 sequence of a TNK1 gene or genome, the BAIAP2 sequence of a BAIAP2 gene or genome, the NOL4L-DT sequence of a NOL4L-DT gene or genome, and the BCR sequence of a BCR gene or genome.
In one or more embodiments, the TACSTD2 sequence of a TACSTD2 gene or genome, the DNASE1L3 sequence of a DNASE1L3 gene or genome, the EGR3 sequence of an EGR3 gene or genome, the DUSP26 sequence of a DUSP26 gene or genome, the TNK1 sequence of a TNK1 gene or genome, the BAIAP2 sequence of a BAIAP2 gene or genome, the MED16 sequence of a MED16 gene or genome, the NOL4L-DT sequence of a NOL4L-DT gene or genome, and the BCR sequence of a BCR gene or genome.
In one or more embodiments, the Hg19 coordinates of the one or more target markers are as follows: PRDM16 gene: chr1:3155051:3155760; CAMK2N1 gene: chr1:20813203:20813902; TACSTD2 Gene: chr 1:5904615:59042314; CRABP2 Gene: chr1:15667274:156676773; IER5 gene: chr1:181074539:181075238; ITPKB gene: chr1:226924700:226925399; ITGB1BP1 gene: chr2:9526804:9527503; MTHFD2 gene: chr2:74453839:74454538; BIN1 gene: chr2, 127822196, 127822895; DNASE1L3 gene: chr3:58153211:58153910; LSG1 gene: chr3:194408527:194409226; SH3BP2 gene: chr 4:2795932:2795331; SLC12A7 gene: chr5:1117661:1118360; NR2F1 gene: chr5:92914797:92915496; EGR1 gene: chr5:137802399:137803098; LARP1 gene: chr5:154133955:154134654; RARS gene: chr 5:167837780:167838499; TTBK1 gene: chr6:43215063:43215762; FAM20C gene: chr7:193512:194211; CREB5 gene: chr7:28449041:28449740; LIMK1 gene: chr7:73508743:73509442; PRKAG2 gene: chr7:151424814:151425513; SLC39a14 gene: chr8:22236914:22237613; EGR3 gene: chr8:22547976:22549090; DUSP26 gene: chr8:34104888:34105587; AGPAT2 gene: chr9:139581855:139582554; NRARP gene: chr9:140205734:140206433; EGR2 gene: chr10:64578269:64578968; PPIF gene: chr10:81001706:81002405; CHID1 gene: chr11:911289:911988; ADM gene: chr11:10328946:10329645; NAV2 gene: chr11:19734801:19736359; EHBP1L1 gene: chr11:65343387:65344086; PHLDB1 gene: chr11:118479144:118479843; PARP11 gene: chr12:4139935:4140634; ANO6 gene: chr12:45610331:45611030; PLXNC1 gene: chr12:94544076:94544775; ZNF219 gene: chr14:21559748:21560447; FOXA1 gene: chr14:3806876:38065575; PAPLN gene: chr14:73704629:73705328; UACA gene: chr15:70766881:70767580; PGPEP1L gene: chr15:99466242:99466941; ITPRIPL2 gene: chr16:19125694:19126393; TNK1 gene: chr17:7286958:7287657; RPL19 gene: chr17:37366033:37366732; ICAM2 gene: chr 17:6207008:6207677; TMC6 gene: chr 17:7613226:7624091; CEP295NL gene: chr17:7689761:768880460; the BAIAP2 gene: chr17:79060865:79061564; TBCD gene: chr17:80744791:80745490; METRNL gene: chr17:81083812:81084511; MED16 gene: chr19:883793:884492; SBNO2 gene: chr19:1177275:1177974; CIRBP gene: chr19:1265690:1266389; KLF16 gene: chr19:1860343:1861042; c19orf77 gene: chr19:34666:3435687; SNAPC2 gene: chr 19:7985709:7986108; ICAM1 gene: chr19:10381317:10382016; ICAM5 gene: chr19:10404832:10405531; IER2 gene: chr19:13266647:13267346; ASF1B gene: chr 19:14248133:14248172; CRTC1 gene: chr19:18770961:18771660; ZNF536 gene: chr19:31039247:31039946; LTBP4 gene: chr19:41105706:41106405; NOL4L-DT gene: chr20:31162101:31162800; KCNK15 gene: chr20:43374048:43374747; UCKL1 gene: chr20:62588113:62588812; RTN4R gene: chr22:20226373:20227274; BCR gene: chr22:23624092:23624791; TEF gene: chr22:41771229:41771928.
In one or more embodiments, the Hg19 coordinates of the one or more target markers are as follows: PRDM16 gene: chr1:3155311:3155510; CAMK2N1 gene: chr1:20813453:20813652; TACSTD2 Gene: chr 1:59041685:59042064; CRABP2 Gene: chr1:15667684:156676723; IER5 gene: chr1:181074789:181074988; ITPKB gene: chr1:226924950:226925149; ITGB1BP1 gene: chr2:9527054:9527253; MTHFD2 gene: chr2:74454089:74454288; BIN1 gene: chr2, 127822446, 127822645; DNASE1L3 gene: chr3:58153461:58153660; LSG1 gene: chr3:194408777:194408976; SH3BP2 gene: chr 4:279282:2795581; SLC12A7 gene: chr5:1117911:1118110; NR2F1 gene: chr5:92915047:92915246; EGR1 gene: chr5:137802649:137802848; LARP1 gene: chr5:154134205:154134404; RARS gene: chr 5:167838020:167838129; TTBK1 gene: chr6:43215313:43215512; FAM20C gene: chr7:193762:193961; CREB5 gene: chr7:28449291:28449490; LIMK1 gene: chr7:73508993:73509192; PRKAG2 gene: chr7:151425064:151425263; SLC39a14 gene: chr8:22237164:22237363; EGR3 gene: chr8:22548226:22548425; EGR3 gene: chr8:22548641:22548840; DUSP26 gene: chr 8:3405138:34105337; AGPAT2 gene: chr9:139582105:13958234; NRARP gene: chr9:140205984:140206183; EGR2 gene: chr 10:64578519:6457878; PPIF gene: chr10:81001956:81002155; CHID1 gene: chr11:911539:911738; ADM gene: chr 11:1032996:10329395; NAV2 gene: chr11:19735051:19735250; NAV2 gene: chr11:19735910:19736109; EHBP1L1 gene: chr11:65343637:65343836; PHLDB1 gene: chr11:118479394:118479593; PARP11 gene: chr12, 4140185:4140384; ANO6 gene: chr12:45610581:45610780; PLXNC1 gene: chr12:94544326:94544525; ZNF219 gene: chr 14:21559998:2156097; FOXA1 gene: chr14:380665126:380565325; PAPLN gene: chr14:73704879:73705078; UACA gene: chr15:70767131:70767330; PGPEP1L gene: chr 15:99466492:466691; ITPRIPL2 gene: chr16:19125944:19126143; TNK1 gene: chr17:7287208:7287407; RPL19 gene: chr17:37366283:37366482; ICAM2 gene: chr 17:62076858:62057657; TMC6 gene: chr 17:7613476:7613675; TMC6 gene: chr 17:7623642:7623841; CEP295NL gene: chr 17:768880011:768880210; the BAIAP2 gene: chr17:79061115:79061314; TBCD gene: chr17, 80745041, 80745240; METRNL gene: chr17:81084062:81084261; MED16 gene: chr19:884043:884242; SBNO2 gene: chr19:1177525:1177724; CIRBP gene: chr19:1265940:1266139; KLF16 gene: chr19:1860593:1860792; c19orf77 gene: chr19:34916:3435115; c19orf77 gene: chr19:3435238:3435437; SNAPC2 gene: chr19:7985959:7986158; ICAM1 gene: chr19:10381567:10381766; ICAM5 gene: chr19:10405082:10405281; IER2 gene: chr19:13266897:13267096; ASF1B gene: chr19:14248383:14248582; CRTC1 gene: chr19:18771211:18771410; ZNF536 gene: chr19:31039497:31039696; LTBP4 gene: chr19:41105956:4106155; NOL4L-DT gene: chr20:31162351:31162550; KCNK15 gene: chr20:43374298:43374497; UCKL1 gene: chr20:62588863:6258562; RTN4R gene: chr22:20226623:20226822; RTN4R gene: chr22:20226825:20227024; BCR gene: chr22:23624342:23624541; TEF gene: chr22:41771479:41771678.
In one or more embodiments, the reagents include primer and/or probe molecules; preferably, the primer molecule is identical, complementary or hybridizes under stringent conditions to the one or more target markers and comprises at least 9 consecutive nucleotides, and the probe molecule hybridizes under stringent conditions to the amplification product of the one or more target markers.
In one or more embodiments, the reagents are required to implement genome-simplified methylation sequencing techniques. In one or more embodiments, the reagents required to implement the genome-simplified methylation sequencing technology include reagents required for cleavage, reagents required for library construction (e.g., end repair, addition of a-tails and adaptors, etc.), reagents required for cytosine conversion, reagents required for PCR amplification, and the like. One or more of the above-described reagents may be included in the detection reagent or diagnostic kit of the present invention.
In a second aspect, the invention provides a diagnostic reagent or diagnostic kit for detecting the methylation state or methylation level of at least one CpG dinucleotide of one or more markers of interest according to any of the embodiments herein, for diagnosing a benign or malignant thyroid nodule, comprising a reagent for detecting the methylation state or level of at least one CpG dinucleotide of one or more markers of interest.
In one or more embodiments, the diagnostic reagent or diagnostic kit comprises a primer and/or probe molecule, wherein the primer molecule is identical, complementary or hybridizes under stringent conditions to the one or more target markers and comprises at least 9 consecutive nucleotides; hybridizing the probe molecules to the amplified products of the one or more target markers under stringent conditions; optionally, the diagnostic reagent or diagnostic kit further comprises a primer molecule and/or a probe molecule for detecting the reference gene ACTB.
In one or more embodiments, the diagnostic reagent or diagnostic kit further comprises one or more substances selected from the group consisting of: PCR buffer, polymerase, dNTP, restriction endonuclease, digestion buffer, fluorescent dye, fluorescence quencher, fluorescent reporter, exonuclease, alkaline phosphatase, internal standard, control, KCl, mgCl 2 And (NH) 4 ) 2 SO 4 。
In one or more embodiments, the reagent for detecting methylation further comprises reagents used in one or more of the following methods: bisulfite conversion-based PCR, DNA sequencing, methylation-sensitive restriction enzyme analysis, fluorescent quantitation, methylation-sensitive high resolution melting curve, chip-based methylation profile analysis, and mass spectrometry.
In one or more embodiments, the agent is selected from one or more of the following: bisulfite and derivatives thereof, fluorescent dyes, fluorescence quenchers, fluorescence reporters, internal standards and controls.
In a third aspect the invention provides the use of at least one reagent or set of reagents for distinguishing between methylated and unmethylated CpG dinucleotides in at least one target region of genomic DNA, wherein the method comprises contacting genomic DNA isolated from a biological sample of the individual with the at least one reagent or set of reagents, wherein the target region is identical or complementary to a sequence of at least 16 consecutive nucleotides of one or more markers of interest as described in any of the embodiments herein, wherein the consecutive nucleotides comprise at least one CpG dinucleotide sequence, thereby at least partially providing for the detection and/or classification of benign and malignant thyroid nodules in an individual.
In a fourth aspect the invention provides the use of one or more reagents for converting an unmethylated cytosine base at position 5 to uracil or other bases detectably different from cytosine in terms of hybridization properties, an amplification enzyme and at least one primer comprising at least 9 consecutive nucleotides in the preparation of a kit for use in a method of detecting and/or classifying a benign and malignant thyroid nodule in an individual, wherein the method comprises: a) Isolating genomic DNA from the individual biological sample; b) Treating the genomic DNA of a) or fragment thereof with the one or more reagents; c) Contacting the treated genomic DNA or a treated fragment thereof with the amplification enzyme and the at least one primer that is identical, complementary or hybridizes under stringent conditions to one or more markers of interest as described in any of the embodiments herein, wherein the treated genomic DNA or fragment thereof is amplified to produce at least one amplification product or is not amplified; and d) determining the methylation status or level of at least one CpG dinucleotide of the one or more markers of interest, or a mean or value reflecting the mean methylation status or level of a plurality of CpG dinucleotides of the one or more markers of interest, based on the presence or nature of the amplificate, thereby at least partially detecting and/or classifying a benign malignancy of a thyroid nodule in an individual.
In one or more embodiments, in step b), the genomic DNA or fragment thereof is treated with an agent selected from the group consisting of bisulfites, acid sulfites, metabisulfites, and combinations thereof.
In one or more embodiments, c) contacting or amplifying the nucleic acid molecule is performed by using a thermostable DNA polymerase as the amplification enzyme, using a polymerase lacking 5'-3' exonuclease activity, using a polymerase chain reaction and/or producing an amplification product with a detectable label.
In one or more embodiments, the contacting or amplifying in c) comprises using methylation specific primers.
In a fifth aspect the present invention provides the use of one or more methylation sensitive restriction and amplification enzymes and at least one primer comprising at least 9 consecutive nucleotides, wherein the primer is identical, complementary or hybridizes under stringent conditions to one or more of the markers of interest described in any of the embodiments herein, for the preparation of a kit for use in a method of detecting and/or classifying a benign and malignant thyroid nodule in an individual; the method comprises the following steps: a) Isolating genomic DNA from the individual biological sample; b) Digesting the genomic DNA of a) or a fragment thereof with the one or more methylation sensitive restriction enzymes, and contacting the resulting digestion product with the amplification enzyme and the at least one primer; and c) determining the methylation status or level of at least one CpG dinucleotide of the one or more markers of interest based on the presence or the nature of the amplificate, thereby at least partially detecting and/or classifying a benign malignancy of a thyroid nodule in an individual.
In one or more embodiments, the presence or absence of an amplification product is determined by hybridizing at least one nucleic acid or peptide nucleic acid that is identical or complementary to at least a 16 base long fragment of a sequence selected from the one or more markers of interest.
In a sixth aspect the invention provides the use of a treated nucleic acid derived from one or more markers of interest as described in any of the embodiments herein, in the manufacture of a kit for diagnosing benign or malignant thyroid nodule, wherein the treatment is suitable for converting at least one unmethylated cytosine base of the one or more markers of interest to uracil or other bases detectably different from cytosine on hybridization.
A seventh aspect of the present invention provides an apparatus for detecting and diagnosing benign and malignant thyroid nodules in an individual, the apparatus comprising a memory, a processor and a computer program stored on the memory and executable on the processor, the processor executing the program to effect the steps of: (1) Obtaining the methylation level or methylation state of at least one CpG dinucleotide of one or more of the markers of interest described in any of the embodiments herein in the sample, and (2) interpreting the benign and malignant thyroid nodule according to the methylation level or methylation state of (1).
Drawings
Fig. 1: example 1 model constructed with marker combinations ROC curves for malignant nodules in training set and two sets of validation set samples
Fig. 2: example 2 model constructed with marker combinations ROC curves for malignant nodules were diagnosed in training set and two sets of validation set samples.
Fig. 3: example 3 model constructed with marker combinations ROC curves for malignant nodules were diagnosed in training set and two sets of validation set samples.
Fig. 4: example 4 model constructed with marker combinations ROC curves for malignant nodules were diagnosed in training set and two sets of validation set samples.
Fig. 5: example 5 model constructed with marker combinations ROC curves for malignant nodules were diagnosed in training set and two sets of validation set samples.
Detailed Description
While various aspects and embodiments of the present application have been disclosed, various equivalent changes or modifications can be made by those skilled in the art without departing from the spirit and scope of the present application. The various aspects and embodiments disclosed herein are illustrative and not intended to limit the scope of the application, which is defined in the claims. Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art. All references, patents and patent applications cited in this application are incorporated by reference into this application.
It is noted that, in the specification and claims of this application, the singular forms "a," "an," and "the" include plural referents unless the context clearly dictates otherwise. Thus, for example, reference to "an agent" includes a plurality of agents.
In the description and claims of this application, unless otherwise indicated, the terms "comprising," "including," or "containing" are intended to include the listed values, steps, or components, but do not exclude the presence of other values, steps, or components.
As a result of intensive studies, the present inventors have found some target markers associated with malignant thyroid nodules, including: PRDM16 gene or genome PRDM16 sequence, CAMK2N1 gene or genome CAMK2N1 sequence, TACSTD2 gene or genome TACSTD2 sequence, CRABP2 gene or genome CRABP2 sequence, IER5 gene or genome IER5 sequence, ITPKB gene or genome ITPKB sequence, ITGB1BP1 gene or genome ITGB1BP1 sequence, MTHFD2 gene or genome MTHFD2 sequence, BIN1 gene or genome BIN1 sequence, DNASE1L3 gene or genome DNASE1L3 sequence, DNASE1L3 sequence LSG1 sequence of LSG1 gene or genome, SH3BP2 sequence of SH3BP2 gene or genome, SLC12A7 sequence of SLC12A7 gene or genome, NR2F1 sequence of NR2F1 gene or genome, EGR1 sequence of EGR1 gene or genome, LARP1 sequence of LARP1 gene or genome, RARS sequence of RARS gene or genome, TTBK1 sequence of TTBK1 gene or genome, FAM20C sequence of FAM20C gene or genome, CREB5 sequence of CREB5 gene or genome, LIMK1 sequence of LIMK1 gene or genome PRKAG2 sequence of PRKAG2 gene or genome, SLC39A14 sequence of SLC39A14 gene or genome, EGR3 sequence of EGR3 gene or genome, DUSP26 sequence of DUSP26 gene or genome, AGPAT2 sequence of AGPAT2 gene or genome, AGPAT2 sequence of gene or genome, gene, or genome, or sequence, or gene, or NRARP sequence of NRARP gene or genome, EGR2 sequence of EGR2 gene or genome, PPIF sequence of PPIF gene or genome, CHID1 sequence of CHID1 gene or genome, ADM sequence of ADM gene or genome, NAV2 sequence of NAV2 gene or genome, and/or its/their/EHBP 1L1 sequence of the EHBP1L1 gene or genome, PHLDB1 sequence of the PHLDB1 gene or genome, PARP11 sequence of the PARP11 gene or genome, ANO6 sequence of the ANO6 gene or genome, PLXNC1 sequence of the PLXNC1 gene or genome, ZNF219 sequence of the ZNF219 gene or genome, FOXA1 sequence of the FOXA1 gene or genome, PAPLN sequence of the PAPLN gene or genome, UACA sequence of the UACA gene or genome, PGPEP1L sequence of the PGPEP1L gene or genome, ITPRIPL2 sequence of the PLXNC1 gene or genome, TNK1 gene or genome's TNK1 sequence, RPL19 gene or genome's RPL19 sequence, ICAM2 gene or genome's ICAM2 sequence, TMC6 gene or genome's TMC6 sequence, CEP295NL gene or genome's CEP295NL sequence, BAIAP2 gene or genome's BAIAP2 sequence, TBCD gene or genome's TBCD sequence, METRNL gene or genome's METRL sequence, MED16 gene or genome's MED16 sequence, SBNO2 gene or genome's SBNO2 sequence, CIRBP gene or genome's CIRBP sequence, KLF16 gene or genome's KLF16 sequence, C19orf77 gene or genome's C19orf77 sequence, SNNK 2 gene or genome's SNN 2 sequence, ICAM1 gene or genome's ICAM5 sequence, IER2 gene or genome's IER2 sequence, ASF1B gene or genome's MED16 sequence, ASF1 gene or genome's CIRBP sequence, KLF16 gene or genome's KLF16 sequence, C19orf77 sequence, SNNK 2 gene or genome's SNNK 2 sequence, ICAM1 gene or genome's ICAM5 sequence, ICAM5 gene or genome's TC 2 gene's IER2 gene or genome's IEF 1, ASF1 gene or ASF1 gene's ASF1 or genome's ASF1, ASF 4 or gene's BCF 4 gene's 4 sequence, or its BCF 4 gene or genome's 4 sequence, and its KCR 4 gene or its 4 sequence. The benign and malignant thyroid nodules can be identified by detecting the methylation level of one or more of the target markers in a biological sample from the individual comprising DNA.
I. Target marker and target region thereof
As used herein, the term "target marker" refers to a nucleic acid or gene region of interest that: its methylation level indicates benign and malignant thyroid nodule. The term "target marker" shall be considered to include all transcriptional variants of the genes described herein and all promoter and regulatory elements thereof. As will be appreciated by those skilled in the art, certain genes are known to exhibit allelic variation or single nucleotide polymorphism ("SNP") between individuals. SNPs include insertions and deletions of simple repeat sequences of different lengths (e.g., dinucleotide and trinucleotide repeats). Thus, the present application should be understood to extend to all forms of markers/genes arising from any other mutation, polymorphism or allelic variation. In addition, it is understood that the term "target marker" shall include both the sense strand sequence of a marker or gene and the antisense strand sequence of a marker or gene.
The term "target marker" as used herein is to be construed broadly to include both 1) the original marker found in a biological sample or genomic DNA (in a specific methylation state) and 2) its treated sequence (e.g., the corresponding region after bisulfite conversion or the corresponding region after MSRE treatment). The corresponding region after bisulfite conversion differs from the target marker in the genomic sequence in that one or more unmethylated cytosine residues are converted to uracil bases, thymine bases, or other bases that differ from cytosine in hybridization behavior. The MSRE treated corresponding region differs from the target marker in the genomic sequence in that the sequence is cleaved at one or more MSRE cleavage sites.
The molecular diagnosis in the invention comprises early diagnosis of thyroid malignant tumor, late diagnosis of thyroid malignant tumor, and also comprises thyroid malignant tumor screening, risk assessment, prognosis and disease recognition. Early diagnosis refers to the likelihood of finding cancer prior to metastasis, preferably before morphological changes in tissue or cells can be observed.
In this context, it should be understood that the target markers PRDM16, CAMK2N1, TACSTD2, CRABP2, IER5, ITPKB, ITGB1BP1, MTHFD2, BIN1, DNASE1L3, LSG1, SH3BP2, SLC12A7, NR2F1, EGR1, LARP1, RARS, TTBK1, FAM20C, CREB, LIMK1, PRKAG2, SLC39A14, EGR3, DUSP26, AGTAT 2, NRARP, EGR2, PPIF, CHID1, ADM, NAV2, EHBP1L1, PHLDB1, PARP11, ANO6, PLXNC1, ZNF219, FOXA1, PAPLN, UACA, PGPEP L, ITPRIPL2, TNK1, RPL19, ICAM2, TMC6, CEP295NL, BAIAP2, TBCD, METRNL, MED, NO2, RBP 16, CIP 16, CIF 19, ASRP 2, ASRP 4, RBF 4, and the like, may be described by their names as being equal to or more than two, or equal to, depending on the respective products, uses and methods described herein. The chromosome coordinates are consistent with the Hg19 version of the human genome database published in month 2 2009 (referred to herein as "Hg19 coordinates"). It is understood that the sequences of a certain gene and its genome as described herein also include fragments of each gene containing at least one CpG dinucleotide sequence. In some embodiments, the fragment is a region of interest of each gene described herein.
In some embodiments, the Hg19 coordinates of each gene mentioned herein are as follows: PRDM16 gene: chr1:3155051:3155760; CAMK2N1 gene: chr1:20813203:20813902; TACSTD2 Gene: chr 1:5904615:59042314; CRABP2 Gene: chr1:15667274:156676773; IER5 gene: chr1:181074539:181075238; ITPKB gene: chr1:226924700:226925399; ITGB1BP1 gene: chr2:9526804:9527503; MTHFD2 gene: chr2:74453839:74454538; BIN1 gene: chr2, 127822196, 127822895; DNASE1L3 gene: chr3:58153211:58153910; LSG1 gene: chr3:194408527:194409226; SH3BP2 gene: chr 4:2795932:2795331; SLC12A7 gene: chr5:1117661:1118360; NR2F1 gene: chr5:92914797:92915496; EGR1 gene: chr5:137802399:137803098; LARP1 gene: chr5:154133955:154134654; RARS gene: chr 5:167837780:167838499; TTBK1 gene: chr6:43215063:43215762; FAM20C gene: chr7:193512:194211; CREB5 gene: chr7:28449041:28449740; LIMK1 gene: chr7:73508743:73509442; PRKAG2 gene: chr7:151424814:151425513; SLC39a14 gene: chr8:22236914:22237613; EGR3 gene: chr8:22547976:22549090; DUSP26 gene: chr8:34104888:34105587; AGPAT2 gene: chr9:139581855:139582554; NRARP gene: chr9:140205734:140206433; EGR2 gene: chr10:64578269:64578968; PPIF gene: chr10:81001706:81002405; CHID1 gene: chr11:911289:911988; ADM gene: chr11:10328946:10329645; NAV2 gene: chr11:19734801:19736359; EHBP1L1 gene: chr11:65343387:65344086; PHLDB1 gene: chr11:118479144:118479843; PARP11 gene: chr12:4139935:4140634; ANO6 gene: chr12:45610331:45611030; PLXNC1 gene: chr12:94544076:94544775; ZNF219 gene: chr14:21559748:21560447; FOXA1 gene: chr14:3806876:38065575; PAPLN gene: chr14:73704629:73705328; UACA gene: chr15:70766881:70767580; PGPEP1L gene: chr15:99466242:99466941; ITPRIPL2 gene: chr16:19125694:19126393; TNK1 gene: chr17:7286958:7287657; RPL19 gene: chr17:37366033:37366732; ICAM2 gene: chr 17:6207008:6207677; TMC6 gene: chr 17:7613226:7624091; CEP295NL gene: chr17:7689761:768880460; the BAIAP2 gene: chr17:79060865:79061564; TBCD gene: chr17:80744791:80745490; METRNL gene: chr17:81083812:81084511; MED16 gene: chr19:883793:884492; SBNO2 gene: chr19:1177275:1177974; CIRBP gene: chr19:1265690:1266389; KLF16 gene: chr19:1860343:1861042; c19orf77 gene: chr19:34666:3435687; SNAPC2 gene: chr 19:7985709:7986108; ICAM1 gene: chr19:10381317:10382016; ICAM5 gene: chr19:10404832:10405531; IER2 gene: chr19:13266647:13267346; ASF1B gene: chr 19:14248133:14248172; CRTC1 gene: chr19:18770961:18771660; ZNF536 gene: chr19:31039247:31039946; LTBP4 gene: chr19:41105706:41106405; NOL4L-DT gene: chr20:31162101:31162800; KCNK15 gene: chr20:43374048:43374747; UCKL1 gene: chr20:62588113:62588812; RTN4R gene: chr22:20226373:20227274; BCR gene: chr22:23624092:23624791; TEF gene: chr22:41771229:41771928.
In some embodiments, the EGR3 gene, NAV2 gene, TMC6 gene, C19orf77 gene, and RTN4R gene may include the following two Hg coordinate regions:
EGR3 gene: chr8:22547976:22548675; chr8:22548391:22549090;
NAV2 gene: chr11:19734801:19735500; chr11:19735660:19736359;
TMC6 gene: chr 17:7613226:76101925; chr 17:7623399:7624091;
c19orf77 gene: chr19:34666:3435365; chr19:34988:3435687;
RTN4R gene: chr22:20226373:20227072; chr22:20226575:20227274.
In a further preferred embodiment, the Hg coordinate regions of one or more target markers described herein are respectively: PRDM16 gene: chr1:3155311:3155510; CAMK2N1 gene: chr1:20813453:20813652; TACSTD2 Gene: chr 1:59041685:59042064; CRABP2 Gene: chr1:15667684:156676723; IER5 gene: chr1:181074789:181074988; ITPKB gene: chr1:226924950:226925149; ITGB1BP1 gene: chr2:9527054:9527253; MTHFD2 gene: chr2:74454089:74454288; BIN1 gene: chr2, 127822446, 127822645; DNASE1L3 gene: chr3:58153461:58153660; LSG1 gene: chr3:194408777:194408976; SH3BP2 gene: chr 4:279282:2795581; SLC12A7 gene: chr5:1117911:1118110; NR2F1 gene: chr5:92915047:92915246; EGR1 gene: chr5:137802649:137802848; LARP1 gene: chr5:154134205:154134404; RARS gene: chr 5:167838020:167838129; TTBK1 gene: chr6:43215313:43215512; FAM20C gene: chr7:193762:193961; CREB5 gene: chr7:28449291:28449490; LIMK1 gene: chr7:73508993:73509192; PRKAG2 gene: chr7:151425064:151425263; SLC39a14 gene: chr8:22237164:22237363; EGR3 gene: chr8:22548226:22548425; EGR3 gene: chr8:22548641:22548840; DUSP26 gene: chr 8:3405138:34105337; AGPAT2 gene: chr9:139582105:13958234; NRARP gene: chr9:140205984:140206183; EGR2 gene: chr 10:64578519:6457878; PPIF gene: chr10:81001956:81002155; CHID1 gene: chr11:911539:911738; ADM gene: chr 11:1032996:10329395; NAV2 gene: chr11:19735051:19735250; NAV2 gene: chr11:19735910:19736109; EHBP1L1 gene: chr11:65343637:65343836; PHLDB1 gene: chr11:118479394:118479593; PARP11 gene: chr12, 4140185:4140384; ANO6 gene: chr12:45610581:45610780; PLXNC1 gene: chr12:94544326:94544525; ZNF219 gene: chr 14:21559998:2156097; FOXA1 gene: chr14:380665126:380565325; PAPLN gene: chr14:73704879:73705078; UACA gene: chr15:70767131:70767330; PGPEP1L gene: chr 15:99466492:466691; ITPRIPL2 gene: chr16:19125944:19126143; TNK1 gene: chr17:7287208:7287407; RPL19 gene: chr17:37366283:37366482; ICAM2 gene: chr 17:62076858:62057657; TMC6 gene: chr 17:7613476:7613675; TMC6 gene: chr 17:7623642:7623841; CEP295NL gene: chr 17:768880011:768880210; the BAIAP2 gene: chr17:79061115:79061314; TBCD gene: chr17, 80745041, 80745240; METRNL gene: chr17:81084062:81084261; MED16 gene: chr19:884043:884242; SBNO2 gene: chr19:1177525:1177724; CIRBP gene: chr19:1265940:1266139; KLF16 gene: chr19:1860593:1860792; c19orf77 gene: chr19:34916:3435115; c19orf77 gene: chr19:3435238:3435437; SNAPC2 gene: chr19:7985959:7986158; ICAM1 gene: chr19:10381567:10381766; ICAM5 gene: chr19:10405082:10405281; IER2 gene: chr19:13266897:13267096; ASF1B gene: chr19:14248383:14248582; CRTC1 gene: chr19:18771211:18771410; ZNF536 gene: chr19:31039497:31039696; LTBP4 gene: chr19:41105956:4106155; NOL4L-DT gene: chr20:31162351:31162550; KCNK15 gene: chr20:43374298:43374497; UCKL1 gene: chr20:62588863:6258562; RTN4R gene: chr22:20226623:20226822; RTN4R gene: chr22:20226825:20227024; BCR gene: chr22:23624342:23624541; TEF gene: chr22:41771479:41771678.
The target marker of the present invention also includes 5kb upstream of each start site and 5kb downstream of each end site of each region described above. The specific nucleotide sequences of the above Hg19 coordinates, as well as 5kb upstream of the respective start sites and 5kb downstream of the respective end sites of each region, can be obtained in a common database (e.g., UCSC Genome Browser, ensemble, and NCBI website).
The target markers of the invention (e.g., sequences of a gene and its genome, or fragments of each gene containing at least one CpG dinucleotide sequence, or sequences comprising a gene spacer) also include non-enzymatically transformed (e.g., the corresponding region after bisulfite conversion), and the corresponding region obtained after enzymatic transformation (e.g., MSRE conversion).
In some embodiments, the subject markers also include variants of each of the genes described above. Variants include nucleic acid sequences from the same region that have at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% sequence identity (i.e., have one or more deletions, insertions, substitutions, inverted sequences, etc.) to a gene or region described herein. Thus, the present disclosure should be understood to extend to such variants that achieve the same result, despite the fact that the actual nucleic acid sequences between individuals have minor genetic variations.
As used herein, the term "percent (%) sequence identity" refers to the same percentage of amino acid (or nucleic acid) residues of a candidate sequence and amino acid (or nucleic acid) residues of a reference sequence after sequence alignment, where a gap (if necessary) may be introduced to maximize the number of identical amino acids (or nucleic acids). In other words, the percent (%) sequence identity of an amino acid sequence (or nucleic acid sequence) can be calculated by dividing the number of amino acid residues (or bases) that are identical to a reference sequence by the total number of amino acid residues (or bases) in the candidate sequence or reference sequence, whichever is shorter. Conservative substitutions of amino acid residues may or may not be considered as identical residues. The percentage of amino acid (or Nucleic acid) sequence identity may be determined, for example, using published tools such as BLASTN, BLASTp (available on the National Center for Biotechnology Information (NCBI) website, see also Altschul S.F.et al, J.mol.biol.,215:403-410 (1990), stephen F.et al, nucleic Acids Res.,25:3389-3402 (1997)), clustalW2 (available on the European bioinformatics institute website), and Higgins D.G.et al., methods in Enzymology,266:383-402 (1996); larkin M.A. et al, bioinformation (Oxford, england), 23 (21): 2947-8 (2007)) and ALIGN or Megalign (DNASTAR) software. The default parameters provided by the tool may be used by those skilled in the art, or parameters suitable for alignment may be customized (e.g., by selecting an appropriate algorithm).
The target markers of the present invention also include the corresponding regions of the above genes 5kb upstream of the start site and 5kb downstream of the end site after non-enzymatic conversion (e.g., bisulfite conversion) or after enzymatic treatment (e.g., methylation sensitive restriction enzyme treatment).
II, source and preparation of target markers
Herein, the target marker may be from a biological sample of any individual of interest. The term "individual" as used herein includes both human and non-human animals. Non-human animals include all vertebrates, such as mammals and non-mammals. "individual" may also be livestock, such as cattle, pigs, sheep, poultry and horses; or rodents, e.g., rats, mice; non-human primates, e.g., apes, monkeys, rhesus monkeys; or a domestic animal such as a dog or cat. In some embodiments, the subject is a human or a non-human primate. In some embodiments, the subject is a human. In this application, "individual," "subject," and "subject" are used interchangeably.
It will be appreciated that the sequences given in section I above are human sequences. When referring to the sequence of a non-human animal, the corresponding position and corresponding sequence of the above-described genes in the genome of the non-human animal can be readily determined using prior art techniques.
The term "biological sample" as used herein refers to a biological composition obtained or derived from an individual, comprising cells and/or other molecular entities (e.g. DNA) to be characterized or identified based on physical, biochemical, chemical and/or physiological characteristics. Biological samples include, but are not limited to, cells, tissues, organs and/or biological fluids of an individual obtained by any method known to those of skill in the art. In some embodiments, the biological sample is selected from the group consisting of: histological sections, tissue biopsies, paraffin-embedded tissues, body fluids, surgical resection samples, isolated blood cells, cells isolated from blood, and any combination thereof. In some embodiments, the bodily fluid is selected from the group consisting of: whole blood, serum, plasma, and any combination thereof. The choice of the most suitable sample will depend on the nature of the context. In some embodiments, the biological sample is whole blood of an individual. In some embodiments, the biological sample is plasma of an individual. Those skilled in the art are aware of various methods for preparing plasma from whole blood. For example, in some embodiments, plasma is obtained by centrifuging whole blood from an individual one, two, three, four, five or more times. In some embodiments, the biological sample is a thyroid nodule biopsy, preferably a fine needle penetration biopsy.
The DNA to be detected may be isolated from the biological sample. The DNA to be detected may be isolated and purified from a biological sample by using various methods known in the art. Commercial kits can be used for isolation and purification. For example, DNA is isolated from cells and tissues by: the raw materials are cleaved under highly denaturing and reducing conditions, the protein degrading enzymes are used in part, the nucleic acid components obtained by the phenol/chloroform extraction process are purified, and the nucleic acids are recovered from the aqueous phase by dialysis or ethanol precipitation (see e.g. Sambrook, j., fritsch, e.f.in t.maniatis, CS H, molecular Cloning, 1989). For another example, there are many reagent systems now particularly suitable for purifying DNA fragments from agarose gels, isolating plasmid DNA from bacterial lysates, and isolating longer-chain nucleic acids (genomic DNA, total cellular RNA) from blood, tissue or cell cultures. Many of these commercially available purification systems are based on the rather well known principle of binding nucleic acids to mineral carriers in the presence of solutions of different chaotropic salts. In these systems, suspensions of finely ground glass powder, diatomaceous earth or silica gel are used as support materials. Some other methods of isolating and purifying DNA from biological samples are described, for example, in US7888006B2 and EP1626085 A1. The choice between methods will be affected by several factors, including time, cost and the amount of DNA required.
In some embodiments, the DNA contained in the biological sample comprises genomic DNA. The term "genomic DNA" as used herein refers to DNA comprising the complete genome of a cell or organism, as well as fragments or portions thereof. Genomic DNA is a large piece of DNA (e.g., longer than about 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, or 300 kb) derived from an individual and may have natural modifications, such as DNA methylation.
In some embodiments, the DNA contained in the biological sample comprises cellular DNA. The term "cellular DNA" as used herein refers to DNA that is present within a cell, or that is obtained from a cell in vivo and isolated in vitro, or otherwise manipulated in vitro, as long as the DNA is not removed from the cell in vivo.
In some embodiments, the DNA contained in the biological sample comprises extracellular free DNA. The term "extracellular free DNA" as used herein refers to a DNA fragment that exists extracellularly in vivo. The term may also be used to refer to DNA fragments obtained from an extracellular source in vivo and isolated, or manipulated, in vitro. The DNA fragment in the extracellular free DNA generally has a length of about 100 to 200bp, presumably related to the length of the DNA fragment encapsulated in nucleosomes. Extracellular free DNA (cfDNA) includes, for example, extracellular free fetal DNA and circulating tumor DNA. Extracellular free fetal DNA circulates in the body of a pregnant woman (e.g., blood), representing the fetal genome, while circulating tumor DNA circulates in the body of a cancer patient (e.g., blood). In some embodiments, the extracellular free DNA may be substantially free of cellular DNA of the individual. For example, the extracellular free DNA may comprise less than about 1,000ng/mL, less than about 100ng/mL, less than about 10ng/mL, less than about 1ng/mL of cellular DNA.
Extracellular free DNA can be prepared by using conventional techniques known in the art. For example, the extracellular free DNA of the blood sample may be obtained by centrifuging the blood sample at a speed of about 200-20,000g, about 200-10,000g, about 200-5,000g, about 300-4000g, etc., for about 3-30 minutes, about 3-15 minutes, about 3-10 minutes, about 3-5 minutes. For example, in some embodiments, extracellular free DNA of a blood sample may be obtained by centrifuging the plasma or serum of an individual one, two, three, four, five or more times. In some embodiments, the biological sample may be obtained by microfiltration in order to isolate cells and fragments thereof from cell-free components comprising soluble DNA. Typically, microfiltration may be performed using filters, for example, 0.1 micron to 0.45 micron membrane filters, such as 0.22 micron membrane filters.
In some embodiments, extracellular free DNA is extracted from whole blood, serum, or plasma for analysis using commercially available DNA extraction products. This extraction method is said to have high recovery (> 50%) of circulating DNA, and some products (e.g., QIAamp Circulating Nucleic Acid Kit by Qiagen) are said to extract DNA fragments of small size. Typical amounts of sample used are 1-5mL serum or plasma.
In some embodiments, the extracellular free DNA comprises circulating tumor DNA. Circulating tumor DNA ("ctDNA") is fragmented DNA of tumor origin in cell-independent body fluids (e.g., blood, urine, saliva, sputum, stool, pleural fluid, cerebrospinal fluid, etc.). Typically, ctDNA is highly fragmented, with an average length of about 150 base pairs. ctDNA generally comprises a very small fraction of extracellular free DNA in body fluids (e.g., plasma), e.g., ctDNA may constitute less than about 10% of plasma DNA. Typically, the percentage is less than about 1%, such as less than about 0.5% or less than about 0.01%. In addition, the total amount of plasma DNA is typically very low, e.g., about 10ng/mL plasma. The amount of ctDNA varies from person to person and depends on the type, location of the tumor, and for cancerous tumors, on the stage of the cancer. However, ctDNA is generally very rare in body fluids and can only be detected by extremely sensitive and specific techniques. Detection of ctDNA may be useful in detecting and diagnosing tumors, directing tumor-specific therapies, monitoring treatments, and monitoring remission of cancer.
III base conversion
Herein, DNA methylation is a biological process of adding a methyl group (e.g., by the action of a DNA methyltransferase) to a DNA molecule (e.g., to one or more cytosine bases of a DNA molecule). In mammals, DNA methylation occurs at the 5' position of a cytosine-phosphate-guanine (CpG) dinucleotide (i.e., a "CpG site"), which when present in the promoter or 5' -CpG-3' dinucleotide in the first exon of a gene, results in epigenetic inactivation of the gene. DNA methylation has been well documented to play an important role in regulating gene expression, tumorigenesis, and other genetic and epigenetic diseases.
As used herein, the term "methylated cytosine residue" refers to a derivative of a cytosine residue wherein a methyl group is attached to a carbon atom (e.g., C5) of the cytosine ring. The term "unmethylated cytosine residue" refers to an underivatized cytosine residue in which, in contrast to a "methylated cytosine residue", there is no methyl linkage on a carbon atom (e.g., C5) of the cytosine ring. CpG sites in which cytosine residues are methylated CpG sites, while CpG sites in which cytosine residues are unmethylated CpG sites.
As described herein, conversion can occur between bases of DNA or RNA. As used herein, "transformation", "cytosine transformation" or "CT transformation" is a process whereby a non-enzymatic or enzymatic process is used to treat DNA to convert an unmodified cytosine base (C) to a base that does not bind to guanine (G) (e.g., uracil base (U)). Some agents are capable of distinguishing between unmethylated and methylated CpG sites in DNA, thereby obtaining treated DNA. The agent can act selectively on unmethylated cytosine residues but not significantly on methylated cytosine residues. Alternatively, the agent may act selectively on methylated cytosine residues, but not significantly on unmethylated cytosine residues. For example, some reagents may selectively convert an unmethylated cytosine residue to uracil, thymine, or another base that is hybridized to cytosine, while the methylated cytosine residue remains in an unconverted state; for another example, some reagents may selectively cleave methylated residues, or selectively cleave unmethylated residues. Thus, the original DNA is converted into the treated DNA in a manner depending on whether it is methylated or not, so that the treated DNA can be distinguished from the original DNA by its hybridization behavior.
As used herein, "treated DNA," "treated sequence," "treated fragment" refers to DNA, nucleic acid sequence, gene fragment that has been treated with an agent capable of distinguishing between unmethylated and methylated CpG sites in the DNA, nucleic acid sequence, gene fragment.
More specifically, cytosine conversion can be performed using non-enzymatic or enzymatic methods. Illustratively, the non-enzymatic method includes a bisulfite or bisulfate treatment. In some embodiments, the reagent used in the non-enzymatic method comprises a bisulphite reagent. As used herein, the term "bisulfite reagent" refers to a reagent including bisulfite, bisulfite ions, or any combination thereof, such as disclosed herein, that can be used to distinguish between methylated and unmethylated CpG dinucleotide sequences. In this application, treatment of DNA with a bisulfite reagent is also described as a "bisulfite reaction" or "bisulfite treatment" and refers to a reaction that converts unmethylated cytosine residues, particularly in the presence of bisulfite ions, to uracil bases, thymine bases, or other bases that differ from cytosine in hybridization behavior, in nucleic acids, where methylated cytosine residues are not significantly converted. In other words, bisulfite treatment can be used to distinguish between methylated CpG dinucleotides and unmethylated CpG dinucleotides. The bisulfite reaction for detecting methylated cytosine residues is described in detail in Frommer, M., et al, proc Natl Acad Sci USA 89 (1992) 1827-31 and Grigg, G, clark, S., bioessays 16 (1994) 431-6. The bisulfite reaction includes a deamination step and a desulfonation step (see Grigg and Clark, supra). The statement that "methylated cytosine residues are not significantly converted" does not exclude that a very small percentage (e.g., less than 0.1%, less than 0.2%, less than 0.3%, less than 0.4%, less than 0.5%, less than 0.6%, less than 0.7%, less than 0.8%, less than 0.9%, less than 1%, less than 2%, less than 3%, less than 4%, less than 5%, less than 6%, less than 7%, less than 8%, less than 9%, less than 10%, less than 11%, less than 12%, less than 13%, less than 14%, less than 15%, less than 16%, less than 17%, less than 18%, less than 19%, less than 20%) of the methylated cytosine residues are converted to uracil, thymine, or other bases that differ in hybridization behavior, although it is intended to convert only unmethylated cytosine residues.
In the case of, for example, reference to from m., et al (supra) or Grigg and Clark (supra), which disclose basic parameters of the bisulfite treatment, the person skilled in the art knows how to carry out the bisulfite treatment, in particular the deamination step and the desulfonation step. The effect of incubation time and temperature on deamination efficiency, and parameters affecting DNA degradation, are disclosed.
In some embodiments, the bisulphite reagent is selected from the group consisting of: ammonium bisulfate, sodium bisulfate, potassium bisulfate, calcium bisulfate, magnesium bisulfate, aluminum bisulfate, bisulfite ions, and any combination thereof. In some embodiments, the bisulphite reagent is sodium bisulphite. In some embodiments, the bisulphite reagent is commercially available, e.g., methyl code TM Bisulfite Conversion Kit、EpiMark TM Bisulfite Conversion Kit、EpiJET TM Bisulfite Conversion Kit、EZDNAMethylation-Gold TM Kit, etc. In some embodiments, the bisulfite reaction is performed according to the instructions of the kit.
Exemplary enzymatic methods include deaminase treatment, and selective cleavage of unmethylated residues but not methylated residues or selective cleavage of methylated residues but not unmethylated residues using a reagent. Preferably, the agent is a Methylation Sensitive Restriction Enzyme (MSRE).
The term "methylation sensitive restriction enzyme" refers to an enzyme that selectively digests nucleic acid according to the methylation state of its recognition site. For restriction enzymes that cleave specifically only when the recognition site is unmethylated or hemimethylated, cleavage does not occur, or with significantly reduced efficiency, when the recognition site is methylated. For restriction enzymes that cleave specifically when the recognition site is methylated, cleavage does not occur, or with significantly reduced efficiency, when the recognition site is unmethylated. In some embodiments, the recognition sequence of the methylation sensitive restriction enzyme contains a CG dinucleotide (e.g., cgcg or cccggg). In some embodiments, the methylation sensitive restriction enzyme does not cleave when a cytosine in the CG dinucleotide is methylated at the C5 carbon atom.
Exemplary MSREs are selected from the group consisting of: hpaII enzyme, salI enzyme, Enzymes, scrFI enzymes, bbeI enzymes, notI enzymes, smaI enzymes, xmaI enzymes, mboI enzymes, bstBI enzymes, claI enzymes, mluI enzymes, naeI enzymes, narI enzymes, pvuI enzymes, sacII enzymes, hhaI enzymes, and any combination thereof.
Enzymes, scrFI enzymes, bbeI enzymes, notI enzymes, smaI enzymes, xmaI enzymes, mboI enzymes, bstBI enzymes, claI enzymes, mluI enzymes, naeI enzymes, narI enzymes, pvuI enzymes, sacII enzymes, hhaI enzymes, and any combination thereof.
Methylation is determined using methods known in the art using methylation sensitive restriction enzymes or a series of restriction enzyme reagents comprising methylation sensitive restriction enzymes, such as, but not limited to, differential methylation hybridization ("DMH"), that are capable of distinguishing between methylated and unmethylated CpG dinucleotides within a target region.
In some embodiments, DNA in the biological sample may be cleaved prior to treatment with the methylation sensitive restriction enzyme. Such methods are known in the art and may include both physical and enzymatic means. It is particularly preferred to use one or more restriction enzymes which are insensitive to methylation and whose recognition sites are AT-rich and do not contain CG dinucleotides. The use of such enzymes allows the preservation of CpG sites and CpG-enriched regions within the DNA fragment. In some embodiments, such restriction enzymes are selected from the group consisting of MseI enzymes, bfaI enzymes, csp6I15 enzymes, tru1I enzymes, tru9I enzymes, maeI enzymes, xspI enzymes, and any combination thereof.
The transformed DNA is optionally purified. DNA purification methods suitable for use herein are well known in the art.
Quantitative analysis
The methylation state or methylation level of at least one CpG dinucleotide in any 1, any 2, any 3, any 4, any 5, any 6, any 7, any 8, any 9, any 10, any 11, any 12, any 13, any 14, any 15, any 16, any 17, any 18, any 19, any 20 or more of the markers of interest described herein can be detected for identifying a benign or malignant thyroid nodule. The detection reagent and the diagnostic kit can be used for detecting the methylation state or the methylation level.
Herein, the terms "benign" and "malignant" refer to the nature of thyroid nodules. In general, benign manifestations are slow growth of nodules, uniform texture, good mobility, smooth surface, cystic changes, no lymphadenomegaly, no calcification, etc. Malignancy manifests as uncontrolled malignant cell growth, spread and tissue infiltration. Ultrasound signs that suggest a thyroid nodule as malignant include: the height of the nodule is larger than the width, the areola is lacked, the micro calcification is carried out, the boundary is irregular, the echo is reduced, the solid nodule is provided, the blood flow in the nodule is rich, etc. In some embodiments, the malignant thyroid nodule includes thyroid cancer.
Herein, "methylation state" refers to the presence or absence of one or more methylated nucleotide bases in a nucleic acid molecule. For example, a nucleic acid molecule containing a methylated cytosine is considered methylated (e.g., the methylation state of the nucleic acid molecule is methylated). Nucleic acid molecules that do not contain any methylated nucleotides are considered unmethylated. In some embodiments, a nucleic acid may be characterized as "unmethylated" if it is not methylated at a particular locus (e.g., a locus of a particular single CpG dinucleotide) or a particular combination of loci, even if it is methylated at other loci of the same gene or molecule.
Thus, methylation status describes the status of methylation of a nucleic acid (e.g., a genomic sequence or a marker of interest described herein). In addition, methylation state refers to a characteristic of a nucleic acid segment at a particular genomic locus that is associated with methylation. Such features include, but are not limited to, whether any cytosine (C) residues within this DNA sequence are methylated, the position of one or more methylated C residues, the frequency or percentage of methylated C throughout any particular region of the nucleic acid, and methylation allele differences due to, for example, differences in allele origins. "methylation state" refers to the relative concentration, absolute concentration, or pattern of methylated C or unmethylated C throughout any particular region of a nucleic acid in a biological sample. For example, one or more cytosine (C) residues within a nucleic acid sequence may be referred to as "hypermethylated" or have "increased methylation" if they are methylated, and one or more cytosine (C) residues within a DNA sequence may be referred to as "demethylated" or have "decreased methylation" if they are unmethylated. Likewise, if one or more cytosine (C) residues within a nucleic acid sequence are methylated compared to another nucleic acid sequence (e.g., from a different region or from a different individual, etc.), the sequence is considered hypermethylated or has increased methylation compared to the other nucleic acid sequence. Alternatively, if one or more cytosine (C) residues within a DNA sequence are unmethylated compared to another nucleic acid sequence (e.g., from a different region or from a different individual, etc.), then the sequence is considered to be demethylated or to have reduced methylation compared to the other nucleic acid sequence.
Herein, methylation level represents the proportion (or percentage, fraction, ratio, degree) of one or more sites in methylation state. The methylation level of a region (or group of sites) is the average of the methyl levels of all sites in the region (or all sites in the group). Thus, an increase or decrease in the methylation level of a region does not indicate an increase or decrease in the methylation level of all methylation sites in the region. The process of converting the results obtained by methods for detecting DNA methylation (e.g., simplified methylation sequencing) to methylation levels is known in the art. Methylation levels can be determined, for example, by quantitative analysis of the amount of intact DNA present after restriction digestion with methylation-sensitive restriction enzymes. In this example, if quantitative PCR is used to quantitatively analyze a particular sequence in DNA, an amount of template DNA approximately equal to the control of the mock-treatment indicates that the sequence is not highly methylated, and an amount of template significantly less than the amount of template in the mock-treated sample indicates that methylated DNA is present in the sequence. Thus, the methylation level as in the above examples can be used as a quantitative indicator of methylation status. This is particularly useful when it is desired to compare the methylation level of sequences in a sample to a threshold level.
In one or more embodiments, the methylation level (e.g., ct value) of the target marker is increased or decreased when compared to a reference level. When the methylation marker level (e.g., ct value) meets a certain threshold, then the thyroid nodule is identified as malignant. Alternatively, a mathematical analysis of the methylation level of the target marker may be performed to obtain a score. For the detected sample, when the score is greater than or less than the threshold, the result is judged to be positive, namely the thyroid nodule is malignant. Conventional mathematical analysis methods and processes for determining thresholds are known in the art, with an exemplary method being a Support Vector Machine (SVM) mathematical model. For example, for differential methylation markers, a Support Vector Machine (SVM) is constructed on training set samples, and test set sample prediction scores are counted using accuracy, sensitivity and specificity of model statistical test results and area under predictor feature curve (ROC) (AUC).
The methylation level/status of one or more CpG dinucleotide sequences within a DNA sequence (e.g. a marker of interest) can be determined by various analytical methods known in the art, preferably quantitative analytical methods. An exemplary analysis method includes: polymerase chain reactions, including real-time polymerase chain reactions, digital polymerase chain reactions, and bisulfite conversion-based PCR (e.g., methylation-specific PCR (MSP)); sequencing nucleic acid; whole genome methylation sequencing (RRBS); simplified methylation sequencing; mass-based separations (e.g., electrophoresis, mass spectrometry); target capture (e.g., hybridization, microarray); methylation sensitive restriction enzyme assays; methylation-sensitive high-resolution melting curve method; chip-based methylation profile analysis; mass spectrometry; and fluorescence quantification. Herein, detecting includes detecting any strand at a gene or site.
In some embodiments, the quantitative analysis is performed by real-time PCR. Non-limiting examples of real-time PCR include the HeavyMethyll described by Cottrell et al, nucl. Acids Res.32:e10, 2003 TM PCR; eads et al, cancer Res.59:2302-2306, 1999 described MethyLightTMPCR; rand et al, nucleic acids res.33:e127, 2005 describes Headloop PCR.
As used herein, the term "HeavyMethyl TM PCR "refers to a real-time PCR technique recognized in the art wherein one or more are non-extendableThe nucleic acid (e.g., oligonucleotide) is attached to the bisulfite treated nucleic acid in a methylation specific manner (i.e., the nucleic acid binds specifically to unmutated DNA under medium to high stringency conditions). The amplification reaction is performed using one or more primers, which may optionally be methylation specific, but flanked by one or more closures. In the presence of unmethylated nucleic acid (i.e., mutated DNA), the closure binds and no PCR products are produced. TaqMan is used as described essentially in Holland et al, proc.Natl. Acad.Sci.USA,88:7276-7280,1991, for example TM The methylation level of the nucleic acid in the sample is determined by the assay method.
As used herein, the term "MethyLightTMPCR" refers to a fluorescence-based real-time PCR technique based on the recognition in the art, wherein a technique called TaqMan is employed TM The probe's double-labeled fluorescent oligonucleotide probe is designed to hybridize to CpG-rich sequences located between forward and reverse amplification primers. The TaqMan probes comprise a fluorescent "reporter moiety" and a "quencher moiety" covalently bound to a linker moiety (e.g., phosphoramidite) that is attached to a nucleotide of the TaqMan oligonucleotide. During PCR amplification, the TaqMan probes hybridized to CpG-rich sequences are cleaved by the 5' nuclease activity of Taq polymerase, thereby generating a signal that is detected in real-time during the PCR reaction. In this approach, molecular beacons can be used as detectable probes, and the system is independent of the 5'-3' exonuclease activity of the DNA polymerase used (see Mhlanga and Malmberg, methods 25:463-471, 2001).
As used herein, the term "Headloop PCR" refers to a real-time PCR recognized in the art that selectively amplifies a target nucleic acid, but inhibits amplification of non-amplified target variants by extending the 3' stem loop to form a hairpin structure that does not provide further amplification template.
In some embodiments, the real-time PCR is multiplex real-time PCR. As used herein, the term "multiplex" may refer to an assay or other analytical method in which the presence and/or amount of multiple markers (e.g., multiple nucleic acid sequences) may be determined simultaneously by using more than one marker, each marker having at least one different detection characteristic, such as a fluorescent characteristic (e.g., excitation wavelength, emission intensity, FWHM (full width at half maximum) or fluorescent lifetime) or a unique nucleic acid or protein sequence characteristic.
In some embodiments, the quantitative analysis is performed by nucleic acid sequencing. Exemplary methods of nucleic acid sequencing are known in the art, see, e.g., from mer et al, proc.Natl. Acad.Sci.USA 89:1827-1831,1992; clark et al, nucleic acids Res.22:2990-2997,1994. For example, identification of methylated cytosines in a DNA sequence is facilitated by comparing the sequence obtained from a sample that has not been treated with bisulfite, or a known nucleotide sequence of a region of interest, to the sequence obtained from a sample that has been treated with bisulfite. In contrast to untreated samples, thymine residues detected at any cytosine position in the bisulfite treated sample can be considered as mutations resulting from bisulfite treatment, i.e., methylated cytosines are present at that position.
Methods for sequencing DNA are known in the art and include, for example, the dideoxy chain termination method or the Maxam-Gilbert method (see Sambrook et al, molecular Cloning, A Laboratory Manual (2) nd Ed., CSHP, new York 1989)), pyrosequencing (see Uhlmann et al, electrophoresis,23:4072-4079,2002), solid phase pyrosequencing (see Landegren et al, genome Res.,8 (8): 769-776, 1998), solid phase micro-sequencing (see, e.g., southern et al, genomics,13:1008-1017,1992), micro-sequencing using FRET (see, e.g., chen and Kwok, nucleic Acids Res.25:347-353, 1997), ligation sequencing or ultra-deep sequencing (see Margulles et al, nature 437 (7057): 376-80 (2005)).
In some embodiments, the quantitative analysis is performed by mass-based separation (e.g., electrophoresis, mass spectrometry). For example, the presence of methylated cytosine residues can be detected by a combination bisulfite restriction assay (COBRA), substantially as described by Xiong and Laird, nucleic acids Res.,25:2532-2534,2001. This method exploits the difference in restriction enzyme recognition sites between methylated and unmethylated nucleic acids after treatment with a compound that can selectively mutate unmethylated cytosine residues (e.g., bisulfite). For example, the restriction endonuclease Taq1 cleaves the sequence TCGA, which will be TTGA after bisulfite treatment of unmethylated nucleic acid, and will therefore not be cleaved. The digested and/or undigested nucleic acids are then detected using detection means known in the art, such as electrophoresis and/or mass spectrometry. As another example, nucleic acid differences in amplified products are detected using different techniques, such as methylation specific single strand conformation analysis (MS-SSCA) (Bianco et al, hum. Mutat, 14:289-293, 1999), methylation specific gradient gel electrophoresis (MS-DGGE) (Abrams and Stanton, methods enzymes, 212:71-74,1992) and methylation specific high performance liquid chromatography (MS-DHPLC) (Deng et al, chin. J. Cancer Res.,12:171-191,2000), based on differences in nucleotide sequence and/or secondary structure after treatment with compounds that selectively mutate unmethylated cytosine residues.
In some embodiments, the quantitative analysis is performed by target capture (e.g., hybridization, microarray). Suitable detection methods by hybridization are known in the art, for example Southern, dot blot, slot blot or other means of nucleic acid hybridization (Kawai et al, mol. Cell. Biol.14:7421-7427,1994;Gonzalgo et al, cancer Res.57:594-599, 1997). In some embodiments, the probe used in the hybridization assay is detectably labeled. In some embodiments, the nucleic acid-based probes used in hybridization assays are unlabeled. Such unlabeled probes may be immobilized on a solid support such as a microarray, and may hybridize to the detectably labeled target nucleic acid molecules. One example of a microarray is a methylation specific microarray that can be used to distinguish between sequences with converted cytosine residues and sequences with unconverted cytosine residues (see Adorjan et al, nucleic acids res, 30:e21, 2002). Hybridization-based assays can also be used for nucleic acids after treatment with methylation-sensitive restriction enzymes. For another example, the methylation status of CpG dinucleotide sequences within a DNA sequence can be determined by an oligonucleotide probe that hybridizes to the bisulfite treated DNA simultaneously with the PCR amplification primer (wherein the primer can be a methylation specific primer or a standard primer).
In some embodiments, the quantitative analysis is performed in the presence of a detection reagent. As used herein, the term "detection reagent" is a reagent used to detect the presence, absence, or amount of nucleic acid in a quantitative analysis step. Various detection reagents known in the art may be used in the present application. In some embodiments, the detection reagent is selected from the group consisting of: fluorescent probes, intercalating dyes, chromophore-labeled probes, radioisotope-labeled probes, and biotin-labeled probes.
In some embodiments, the quantitative analysis comprises amplifying the treated DNA using a quantitative primer pair and a DNA polymerase. As used herein, the term "quantitative primer pair" refers to one or more primer pairs used in a quantitative analysis step. Preferably, the quantitative primer pair is capable of hybridizing to at least 9 consecutive nucleotides of the treated DNA under stringent, moderately stringent or highly stringent conditions.
In some embodiments, the quantitative analysis comprises determining the methylation level of one or more markers of interest based on the presence or level of a plurality of CpG dinucleotides, tpG dinucleotides, or CpA dinucleotides in the treated DNA. In some embodiments, the quantitative analysis comprises determining the methylation level of cytosine residues based on the presence or level of one or more CpG dinucleotides in the treated DNA. In some embodiments, the quantitative analysis comprises determining the methylation level of cytosine residues based on the presence or level of one or more TpG dinucleotides in the treated DNA. In some embodiments, the quantitative analysis comprises determining the methylation level of cytosine residues based on the presence of CpA dinucleotides in the treated DNA.
In some embodiments, the step of quantifying is performed by separating the treated DNA product into a plurality of components. In some embodiments, a plurality of different quantitative analytical tests are performed on a plurality of components, wherein different combinations of the treated DNA products (if present in the components) are quantitatively analyzed in one of the plurality of components. In some embodiments, the control markers in each component are quantitatively analyzed.
In some embodiments, the methylation level of each target marker is separately quantitatively analyzed using MSP (see Herman, supra) based on pre-amplified DNA. For example, by using one or more primers that specifically hybridize to untransformed sequences under medium and/or high stringency conditions, amplification products are only produced when the template comprises methylated cytosines at the CpG sites.
In some embodiments, the quantitative primer pair is designed to amplify at least a portion of the treated DNA product, i.e., quantitative analysis is designed as nested PCR. Nested PCR is an improvement of PCR and aims at improving sensitivity and specificity. Nested PCR involves the use of two primer sets and two consecutive PCR reactions. A first round of amplification is performed to generate a first amplicon and a second round of amplification is performed using one primer pair, wherein one or both primers anneal to sites within the region defined by the initial primer pair, i.e., the second primer pair is considered "nested" within the first primer pair. In this way, background amplification products from the first PCR reaction that do not contain the correct internal sequences are not amplified any further in the second PCR reaction.
Typically, the reaction solution for PCR comprises Taq DNA polymerase, PCR buffer, primer, probe, dNTPs, mg 2+ . Preferably, the Taq DNA polymerase is a hot start Taq DNA polymerase. Illustratively, mg 2+ The final concentration is 1.0-20.0mM; the concentration of each primer is 100-500nM; each probe was at a concentration of 100-500nM. Exemplary PCR reaction conditions are, pre-denaturation at 95℃for 5min; denaturation at 95℃for 15s, annealing at 60℃for 60s,50 cycles.
In some embodiments, the methods of the invention comprise a pre-amplification step. One of the purposes of pre-amplifying the target markers is to increase the number of target markers in the treated DNA. As used herein, the term "amplification" refers generally to any process that can result in an increase in the copy number of a molecule or group of related molecules. When "amplification" is used with respect to a polynucleotide molecule, it is meant that multiple copies of the polynucleotide molecule or portions of the polynucleotide molecule are typically produced starting with a small amount of the polynucleotide, wherein the substance being amplified (amplicon, PCR amplicon) is typically detectable. Amplification of polynucleotides encompasses a number of chemical and enzymatic processes. Amplified forms include the generation of multiple copies of DNA from one or several copies of a template RNA or DNA molecule by polymerase chain reaction (reverse transcription PCR, PCR), strand Displacement Amplification (SDA) reaction, transcription Mediated Amplification (TMA) reaction, nucleic Acid Sequence Based Amplification (NASBA) reaction, or Ligase Chain Reaction (LCR).
The target marker in the treated DNA may be pre-amplified with a pre-amplification primer. As used herein, the term "primer" refers to a single stranded oligonucleotide that is capable of acting as a point of initiation of template-directed DNA synthesis in the presence of four different nucleoside triphosphates and reagents for polymerization (e.g., DNA polymerase) under suitable conditions (e.g., buffer and temperature). In any given case, the length of the primer depends on, for example, the intended use of the primer, and is typically in the range of 15 to 30 nucleotides. Short primer molecules typically require lower temperatures to form sufficiently stable hybridization complexes with the template. The primer need not reflect the exact sequence of the template, but must be sufficiently complementary to hybridize to the template. The primer site is the region of the template to which the primer hybridizes. The primer pair is a set of primers comprising a 5 'forward primer that hybridizes to the 5' end of the sequence to be amplified and a 3 'reverse primer that hybridizes to the complementary strand of the 3' end of the sequence to be amplified. The skilled person can design primers based on the markers to be amplified based on common knowledge in the art (see, e.g., PCR Primer: A Laboratory Manual, cold Spring Harbor Laboratories, NY, 1995). In addition, some software packages for designing optimal probes and/or primers for use in a wide variety of assays are disclosed, such as Primer 3 available from genomic research center (the Center for Genome Research, cambridge, mass., USA) of Cambridge, massachusetts. Obviously, the potential use of probes or primers should also be considered in designing them. For example, a primer designed for the purposes of the present invention may comprise at least one CpG site, or an amplification product obtained from the primer may comprise at least one CpG site. Tools for designing primers for detecting the methylation state of DNA are also known in the art, for example MethPrimer (Li LC and Dahiya R.MethPrimer: designing primers for methylation PCRs.Bioinformatics.2002Nov;18 (11): 1427-31). In this application, by using pre-amplification primers as primer pools, any target marker (each at least a portion of the target marker or one sub-region of the target marker) in the treated DNA can be pre-amplified.
As used herein, the term "complementary" refers to hybridization or base pairing between nucleotides or nucleic acids, e.g., between two strands of a double-stranded DNA molecule, or between a primer binding site and an oligonucleotide primer on a single-stranded nucleic acid to be sequenced or amplified. The complementary nucleotides are typically A and T (or A and U), or C and G. When nucleotides of one strand are optimally aligned, compared, and have appropriate nucleotide insertions or deletions, the two single stranded RNA or DNA molecules are said to be complementary, paired with at least about 80% (typically at least about 90% to 95%, more preferably about 98% to 100%) of the nucleotides of the other strand. Alternatively, complementarity exists when an RNA strand or DNA strand hybridizes to its complement under selective hybridization conditions. Typically, selective hybridization will occur when there is at least about 65% (preferably at least about 75%, more preferably at least about 90%) complementarity over a stretch of at least 14 to 25 nucleotides. See M. Kanehisa, nucleic Acids Res.12:203 (1984), incorporated herein by reference.
In some embodiments, the pre-amplification primer pool comprises at least one methylation specific primer pair. In some embodiments, the pre-amplification primer pool comprises a plurality of methylation specific primer pairs. In some embodiments, the pre-amplification step is performed by methylation specific PCR ("MSP"), which is PCR using methylation specific primers. Herman et al, methyl-specific PCR a novelPCRassay for Methylation status ofCpGislands, proc Natl Acad Sci USA 1996September 3;93 (18) this technique (i.e., MSP) has been described in 9821-6 and United States Patent No.6,265,171.
As used herein, the term "methylation specific primer pair" refers to a primer pair specifically designed to recognize CpG sites to exploit differences in methylation to amplify a particular target marker in treated DNA. The primer acts only on molecules with or without a specific methylation state. For example, the primer may be an oligonucleotide that hybridizes specifically to a specific CpG site with methylation, but not to a specific CpG site without methylation, under stringent, moderately stringent, or highly stringent conditions. Thus, the primers will specifically amplify a target marker that has methylation at a particular CpG site. For another example, the primer may be an oligonucleotide that hybridizes specifically to an unmethylated specific CpG site, but not to a methylated specific CpG site, under stringent, moderately stringent, or highly stringent conditions. Thus, the primers will specifically amplify target markers that are not methylated at a particular CpG site. Thus, in the present application, methylation specific primers are used in the pre-amplification of at least one target marker within the treated DNA, allowing to distinguish between methylated and unmethylated CpG sites. The methylation specific primer pairs of the present application comprise at least one primer that hybridizes to a bisulfite treated CpG dinucleotide. Thus, the sequence of the primer specific for methylated DNA comprises at least one CpG dinucleotide and the sequence of the primer specific for unmethylated DNA comprises a "T" at the C position of CpG and/or a "at the G position in CpG.
Methylation specific primer pairs typically comprise a forward primer and a reverse primer, each comprising an oligonucleotide sequence that hybridizes to at least 9 consecutive nucleotides of one of the markers (or a subregion of the marker) of interest under stringent conditions, moderately stringent conditions, or highly stringent conditions, wherein at least 9 consecutive nucleotides of one of the markers (or a subregion of the marker) of interest comprise at least one (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, or more) CpG site.
As used herein, the term "hybridization" may refer to a process in which two single-stranded polynucleotides are non-covalently bound to form a stable double-stranded polynucleotide. In one aspect, the resulting double-stranded polynucleotide may be a "hybrid" or "double-stranded". The salt concentration in the "hybridization conditions" is typically less than about 1M, often less than about 500mM and may be less than about 200mM. "hybridization buffer" includes buffered saline solutions, such as 5% sspe, or other such buffers known in the art. Hybridization temperatures can be as low as 5 ℃, but are typically above 22 ℃, and more typically above about 30 ℃, and typically above 37 ℃. The hybridization is usually performed under stringent conditions, i.e., conditions under which a sequence will hybridize to its target sequence but not to other non-complementary sequences. Stringent conditions will be sequence dependent and will be different in different circumstances. For example, longer fragments may require higher hybridization temperatures than shorter fragments to specifically hybridize. Parameter combinations are more important than absolute measurements using either parameter alone, as other factors may affect the stringency of hybridization, including base composition and length of the complementary strand, presence of organic solvents, and degree of base mismatch. Typically stringent conditions are selected to be about 5℃lower than the melting temperature (Tm) for a specific sequence at a specific ionic strength and pH. The Tm may be the temperature at which half of the population of double-stranded nucleic acid molecules are separated into single strands. Several equations for calculating the Tm of a nucleic acid are well known in the art. As shown in the standard reference, a simple estimated Tm value can be calculated by the formula tm=81.5+0.41 (% g+c) when the nucleic acid is in 1M aqueous NaCl solution (see, e.g., anderson and Young, quantitative Filter Hybridization, in Nucleic Acid Hybridization (1985)). Other references (e.g., allawi and SantaLucia, jr., biochemistry,36:10581-94 (1997)) include alternative calculation methods that take structural and environmental as well as sequence characteristics into account when calculating Tm.
Generally, the stability of a hybrid is a function of ion concentration and temperature. Typically, the hybridization reaction is performed under lower stringency conditions and then washed in wash solutions having different but higher stringency. Exemplary stringent conditions include those having a pH of about 7.0 to about 8.3, a temperature of at least 25℃and a sodium ion (or other salt) concentration of at least 0.01M and not more than 1M. For example, a temperature of 5 XSSPE (750 mM NaCl,50mM sodium phosphate, 5mM EDTA, pH 7.4) and about 30℃is suitable for allele-specific hybridization, although the suitable temperature depends on the length and/or GC content of the hybridization region. In one aspect, the "hybridization stringency" to determine the percentage of mismatches can be as follows: 1) High stringency: 0.1x SSPE,0.1%SDS,65 ℃; 2) Moderate stringency (also referred to as moderate stringency): 0.2x SSPE,0.1%SDS,50 ℃; 3) Low stringency: 1.0x SSPE,0.1%SDS,50 ℃. It will be appreciated that the same stringency can be achieved using alternative buffers, salts and temperatures. For example, moderately stringent hybridization may refer to conditions that allow a nucleic acid molecule (e.g., a probe) to bind to a complementary nucleic acid molecule. The hybridized nucleic acid molecules typically have at least 60% identity, including, for example, at least 70%, 75%, 80%, 85%, 90% or 95% identity. The moderately stringent conditions may be conditions that achieve the same effect as the following conditions: hybridization was performed at 42℃with 50% formamide, 5 XDenhardt's solution, 5 XSSPE, 0.2% SDS, and then washed with 42℃ 0.2x SSPE,0.2%SDS. High stringency conditions can be provided by, for example, hybridization with 42℃in 50% formamide, 5 XDenhardt's solution, 5 XSSPE, 0.2% SDS, followed by washing at 65℃in 0.1 XSSPE and 0.1% SDS. The low stringency hybridization may be performed under conditions equivalent to those described below: hybridization was performed at 22℃with 10% formamide, 5 XDenhardt's solution, 6 XSSPE, 0.2% SDS, followed by washing in 1 XSSPE, 0.2% SDS at 37 ℃. The Denhardt solution contained 1% polysucrose, 1% polyvinylpyrrolidone and 1% Bovine Serum Albumin (BSA). 20 XSSPE (sodium chloride, sodium phosphate, EDTA) comprises 3M sodium chloride, 0.2M sodium phosphate, and 0.025M EDTA. Other suitable moderately stringent and highly stringent hybridization buffers and conditions are well known to those skilled in the art and are described, for example, in Sambrook et al, molecular Cloning: A Laboratory Manual,2nd ed., cold Spring Harbor Press, planview, n.y. (1989) and Ausubel et al, short Protocols in Molecular Biology,4th ed., john Wiley & Sons (1999).
In some embodiments, the pre-amplification primer pool further comprises a control primer pair for amplifying a control marker. Typically, a control marker is a nucleic acid having a known characteristic (e.g., a known sequence, a known copy number per cell) for comparison to an experimental target (e.g., a nucleic acid of unknown concentration). The control may be an endogenous, preferably a constant gene against which the test nucleic acid or target nucleic acid under analysis may be normalized. Such controls, normalized for sample-to-sample variability, may occur, for example, in sample processing, analytical efficiency, etc., and allow for accurate sample-to-sample data comparisons, quantitative analysis of amplification efficiency and bias.
In some embodiments, the invention uses RRBS technology to detect the methylation level of CpG sites of a target marker of interest, and then calculates the average methylation ratio (average methylation fraction, AMF) of the marker as the DNA methylation level of the marker. The calculation of AMF may be performed as described in the embodiments of the present application.
V. identification of benign and malignant thyroid nodule
The present invention finds that the methylation level of one or more target markers described herein can be used to determine the benign malignancy of a thyroid nodule in an individual. In one or more embodiments, the methylation level of a CpG site in a target marker described herein can be detected, and then the average methylation ratio (AMF) of the target marker can be calculated as the DNA methylation level of the marker. Herein, the AMF can be calculated from the following formula:
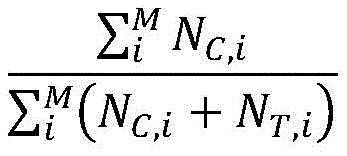
Wherein M is the total number of CpG sites in the marker, i is one of the CpG sites, N C,i Sequencing reads number, N, for methylation of the CpG sites T,i Sequencing reads that are unmethylated for the CpG sites.
And then calculating the malignancy prediction probability of the sample through the constructed mathematical model. The malignancy prediction probability is calculated using a logistic regression (Logistic Regression) model. First, the input z of the Sigmoid function is calculated, which is given by the following formula:
z=Σw*x+w0
then, the Sigmoid function is calculated as follows:
σ(z)=1/(1+e -z )
w is the regression model coefficient for each marker, w0 is the intercept, and x is the calculated DNA methylation level (i.e., AMF) for that marker. The sigma value is the malignancy prediction probability.
The training set is constructed according to the DNA methylation level of each marker in the training set sample, the threshold defined by the Johnson index of the training set is used as a malignancy prediction threshold, the malignancy prediction threshold of each marker is obtained, and the malignancy prediction threshold of each marker is shown in the table 6.
In some embodiments, the probability of malignancy prediction for each sample is calculated according to the above formula based on the methylation level of a single target marker described herein, and if the value is above the threshold for the target marker shown in table 6, then it is determined to be malignant, and vice versa. In preferred embodiments, the target marker is the PRDM16 sequence of the PRDM16 gene or genome, the BIN1 sequence of the BIN1 gene or genome, the LIMK1 sequence of the LIMK1 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the PPIF sequence of the PPIF gene or genome, the ZNF219 sequence of the ZNF219 gene or genome, the UACA sequence of the UACA gene or genome, the TNK1 sequence of the TNK1 gene or genome, the CEP295NL sequence of the CEP295NL gene or genome, the SBNO2 sequence of the SBNO2 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome, the ICAM5 sequence of the ICAM5 gene or genome, the CRTC1 sequence of the CRTC1 gene or genome, the RTN4R sequence of the CAMK2N1 gene or genome, the DNASE1 sequence of the DNASE1L3 gene or genome, the SP26 sequence of the DUK 1 gene or genome, the ASP 26 sequence of the ASE 2 gene or genome, the CSTP 2 sequence of the ASTP 2 gene or genome, the CSTP 2 sequence of the ASTE 2 gene or genome, the BAND 2 gene or genome, the CSTP 2 sequence of the ASTE 2 gene or genome, the ASTP 2 or the ASTP 2 gene or genome, the CSTP 2 sequence of the BAND 2 gene or genome.
In other embodiments, any 2, any 3, any 4, any 5, any 6, any 7, any 8, any 9, any 10, any 11, any 12, any 13, any 14, any 15, any 16, any 17, any 18, any 19, any 20 or more combinations of the markers of interest described herein may be determined using the methods described herein as a threshold for malignancy prediction when assessed, and the combination of the markers of interest is used as a marker for diagnosing benign malignancy of thyroid nodules, determining the probability of malignancy prediction of the combination of the markers of interest in an individual sample (preferably thyroid nodule tissue, such as a puncture) by comparing it to the threshold, indicating malignancy above the threshold, and conversely, indicating benign.
In some embodiments, the one or more target markers include at least one or more of the following target markers: the EGR3 sequence of the EGR3 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, the TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the BCR sequence of the BCR gene or genome.
In some embodiments, the one or more target markers comprise: the PRDM16 gene or genome PRDM16 sequence, BIN1 sequence of a BIN1 gene or genome, LIMK1 sequence of a LIMK1 gene or genome, EGR3 sequence of an EGR3 gene or genome, PPIF sequence of a PPIF gene or genome, ZNF219 sequence of a ZNF219 gene or genome, UACA sequence of a UACA gene or genome, TNK1 sequence of a TNK1 gene or genome, CEP295NL sequence of a CEP295NL gene or genome, SBNO2 sequence of a SBNO2 gene or genome, C19orf77 sequence of a C19orf77 gene or genome, ICAM5 sequence of an ICAM5 gene or genome, CRTC1 sequence of a CRTC1 gene or genome, and RTN4R sequence of an RTN4R gene or genome; and the threshold is 0.49.
In some embodiments, the one or more target markers comprise: the CAMK2N1 gene or genome, the DNASE1L3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the ICAM2 sequence of the ICAM2 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome and the NOL4L-DT sequence of the NOL4L-DT gene or genome; and the threshold is 0.58.
In some embodiments, the one or more target markers comprise: the TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the LSG1 sequence of the LSG1 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, and the BCR sequence of the BCR gene or genome; and the threshold is 0.52.
In some embodiments, the one or more target markers comprise: the TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, and the BCR sequence of the BCR gene or genome; and the threshold is 0.52.
In some embodiments, the one or more target markers comprise: the TACSTD2 sequence of the TACSTD2 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome and the BCR sequence of the BCR gene or genome; and the threshold is 0.52.
Particularly preferably, the Hg coordinates of each of the target markers are described herein, particularly as shown in table 6.
In addition to the above comparisons, one of skill in the art can determine whether an individual's thyroid nodule is malignant or at risk of malignancy based on various factors, such as age, gender, medical history, family history, symptoms, and the like.
VI compositions and kits
The present invention provides a methylation detection or diagnostic kit and diagnostic reagent or diagnostic composition for the identification of benign and malignant thyroid nodules comprising reagents for detecting the methylation status or level of at least one CpG dinucleotide of one or more markers of interest described herein. Depending on the target marker to be detected, the kit and composition may contain primer and/or probe molecules. Preferably, the primer comprises a primer pair capable of hybridizing to said target marker or target region thereof to be detected under stringent, moderately stringent or highly stringent conditions. Primers may also include primers that detect internal references such as ACTB.
In some embodiments, the primers are packaged in a single container or in separate containers. In some embodiments, the kit further comprises one or more blocking oligonucleotides.
In some embodiments, the kits and compositions further comprise a detection reagent. In some embodiments, the detection reagent is selected from the group consisting of: fluorescent probes, intercalating dyes, chromophore-labeled probes, radioisotope-labeled probes, and biotin-labeled probes.
In some embodiments, the kit may further comprise a DNA polymerase and/or a container suitable for storing a biological sample obtained from an individual. In some embodiments, the kit further comprises instructions for use and/or interpretation of the detection results of the kit.
In some embodiments, the kits and compositions may further comprise reagents for enzymatic or non-enzymatic conversion. In a preferred embodiment, the kit further comprises a bisulphite reagent or a methylation sensitive restriction enzymeMSRE). In some embodiments, the bisulphite reagent is selected from the group consisting of: ammonium bisulfate, sodium bisulfate, potassium bisulfate, calcium bisulfate, magnesium bisulfate, aluminum bisulfate, bisulfite ions, and any combination thereof. In some embodiments, the bisulphite reagent is sodium bisulphite. In some embodiments, the MSRE is selected from the group consisting of: hpaII enzyme, salI enzyme,  Enzymes, scrFI enzymes, bbeI enzymes, notI enzymes, smaI enzymes, xmaI enzymes, mboI enzymes, bstBI enzymes, claI enzymes, mluI enzymes, naeI enzymes, narI enzymes, pvuI enzymes, sacII enzymes, hhaI enzymes, and any combination thereof.
Enzymes, scrFI enzymes, bbeI enzymes, notI enzymes, smaI enzymes, xmaI enzymes, mboI enzymes, bstBI enzymes, claI enzymes, mluI enzymes, naeI enzymes, narI enzymes, pvuI enzymes, sacII enzymes, hhaI enzymes, and any combination thereof.
The kits and compositions can also include a positive standard for conversion, wherein unmethylated cytosines are converted to bases that do not bind guanine. The positive standard may be fully methylated.
The kits and compositions may also include PCR reaction reagents. Preferably, the PCR reagent comprises Taq DNA polymerase, PCR buffer, dNTPs, mg 2+ 。
In some embodiments, the kits and compositions further comprise standard reagents useful for performing CpG site-specific methylation assays, wherein the assays include one or more of the following techniques: MS-SNuPE, MSP, methyLightTM, heavyMethyl TM COBRA and nucleic acid sequencing.
In some embodiments, the kits and compositions may comprise additional reagents selected from the group consisting of: buffers (e.g., restriction enzymes, PCR, preservation or washing buffers), DNA recovery reagents or kits (e.g., precipitation, ultrafiltration, affinity columns), DNA recovery components, and the like.
The kit of the present application may further comprise one or several of the following components known in the DNA enrichment art: a protein component that selectively binds methylated DNA; a triplex forming nucleic acid component, one or more linkers, optionally in a suitable solution; substances or solutions for performing ligation, e.g. ligase, buffers; a substance or solution for performing column chromatography; a substance or solution for performing an immunological-based enrichment (e.g., immunoprecipitation); substances or solutions for performing nucleic acid amplification, such as PCR; a dye or dyes, if applicable to a coupling agent, if applicable in solution; substances or solutions for performing hybridization; and/or a substance or solution used to perform the washing step.
In other embodiments, the compositions of the invention contain an isolated nucleic acid molecule selected from one or more of the following: PRDM16 gene: chr1:3155051:3155760; CAMK2N1 gene: chr1:20813203:20813902; TACSTD2 Gene: chr 1:5904615:59042314; CRABP2 Gene: chr1:15667274:156676773; IER5 gene: chr1:181074539:181075238; ITPKB gene: chr1:226924700:226925399; ITGB1BP1 gene: chr2:9526804:9527503; MTHFD2 gene: chr2:74453839:74454538; BIN1 gene: chr2, 127822196, 127822895; DNASE1L3 gene: chr3:58153211:58153910; LSG1 gene: chr3:194408527:194409226; SH3BP2 gene: chr 4:2795932:2795331; SLC12A7 gene: chr5:1117661:1118360; NR2F1 gene: chr5:92914797:92915496; EGR1 gene: chr5:137802399:137803098; LARP1 gene: chr5:154133955:154134654; RARS gene: chr 5:167837780:167838499; TTBK1 gene: chr6:43215063:43215762; FAM20C gene: chr7:193512:194211; CREB5 gene: chr7:28449041:28449740; LIMK1 gene: chr7:73508743:73509442; PRKAG2 gene: chr7:151424814:151425513; SLC39a14 gene: chr8:22236914:22237613; EGR3 gene: chr8:22547976:22549090; DUSP26 gene: chr8:34104888:34105587; AGPAT2 gene: chr9:139581855:139582554; NRARP gene: chr9:140205734:140206433; EGR2 gene: chr10:64578269:64578968; PPIF gene: chr10:81001706:81002405; CHID1 gene: chr11:911289:911988; ADM gene: chr11:10328946:10329645; NAV2 gene: chr11:19734801:19736359; EHBP1L1 gene: chr11:65343387:65344086; PHLDB1 gene: chr11:118479144:118479843; PARP11 gene: chr12:4139935:4140634; ANO6 gene: chr12:45610331:45611030; PLXNC1 gene: chr12:94544076:94544775; ZNF219 gene: chr14:21559748:21560447; FOXA1 gene: chr14:3806876:38065575; PAPLN gene: chr14:73704629:73705328; UACA gene: chr15:70766881:70767580; PGPEP1L gene: chr15:99466242:99466941; ITPRIPL2 gene: chr16:19125694:19126393; TNK1 gene: chr17:7286958:7287657; RPL19 gene: chr17:37366033:37366732; ICAM2 gene: chr 17:6207008:6207677; TMC6 gene: chr 17:7613226:7624091; CEP295NL gene: chr17:7689761:768880460; the BAIAP2 gene: chr17:79060865:79061564; TBCD gene: chr17:80744791:80745490; METRNL gene: chr17:81083812:81084511; MED16 gene: chr19:883793:884492; SBNO2 gene: chr19:1177275:1177974; CIRBP gene: chr19:1265690:1266389; KLF16 gene: chr19:1860343:1861042; c19orf77 gene: chr19:34666:3435687; SNAPC2 gene: chr 19:7985709:7986108; ICAM1 gene: chr19:10381317:10382016; ICAM5 gene: chr19:10404832:10405531; IER2 gene: chr19:13266647:13267346; ASF1B gene: chr 19:14248133:14248172; CRTC1 gene: chr19:18770961:18771660; ZNF536 gene: chr19:31039247:31039946; LTBP4 gene: chr19:41105706:41106405; NOL4L-DT gene: chr20:31162101:31162800; KCNK15 gene: chr20:43374048:43374747; UCKL1 gene: chr20:62588113:62588812; RTN4R gene: chr22:20226373:20227274; BCR gene: chr22:23624092:23624791; TEF gene: chr22:41771229:41771928.
In other embodiments, the compositions of the invention contain an isolated nucleic acid molecule selected from one or more of the following: PRDM16 gene: chr1:3155311:3155510; CAMK2N1 gene: chr1:20813453:20813652; TACSTD2 Gene: chr 1:59041685:59042064; CRABP2 Gene: chr1:15667684:156676723; IER5 gene: chr1:181074789:181074988; ITPKB gene: chr1:226924950:226925149; ITGB1BP1 gene: chr2:9527054:9527253; MTHFD2 gene: chr2:74454089:74454288; BIN1 gene: chr2, 127822446, 127822645; DNASE1L3 gene: chr3:58153461:58153660; LSG1 gene: chr3:194408777:194408976; SH3BP2 gene: chr 4:279282:2795581; SLC12A7 gene: chr5:1117911:1118110; NR2F1 gene: chr5:92915047:92915246; EGR1 gene: chr5:137802649:137802848; LARP1 gene: chr5:154134205:154134404; RARS gene: chr 5:167838020:167838129; TTBK1 gene: chr6:43215313:43215512; FAM20C gene: chr7:193762:193961; CREB5 gene: chr7:28449291:28449490; LIMK1 gene: chr7:73508993:73509192; PRKAG2 gene: chr7:151425064:151425263; SLC39a14 gene: chr8:22237164:22237363; EGR3 gene: chr8:22548226:22548425; EGR3 gene: chr8:22548641:22548840; DUSP26 gene: chr 8:3405138:34105337; AGPAT2 gene: chr9:139582105:13958234; NRARP gene: chr9:140205984:140206183; EGR2 gene: chr 10:64578519:6457878; PPIF gene: chr10:81001956:81002155; CHID1 gene: chr11:911539:911738; ADM gene: chr 11:1032996:10329395; NAV2 gene: chr11:19735051:19735250; NAV2 gene: chr11:19735910:19736109; EHBP1L1 gene: chr11:65343637:65343836; PHLDB1 gene: chr11:118479394:118479593; PARP11 gene: chr12, 4140185:4140384; ANO6 gene: chr12:45610581:45610780; PLXNC1 gene: chr12:94544326:94544525; ZNF219 gene: chr 14:21559998:2156097; FOXA1 gene: chr14:380665126:380565325; PAPLN gene: chr14:73704879:73705078; UACA gene: chr15:70767131:70767330; PGPEP1L gene: chr 15:99466492:466691; ITPRIPL2 gene: chr16:19125944:19126143; TNK1 gene: chr17:7287208:7287407; RPL19 gene: chr17:37366283:37366482; ICAM2 gene: chr 17:62076858:62057657; TMC6 gene: chr 17:7613476:7613675; TMC6 gene: chr 17:7623642:7623841; CEP295NL gene: chr 17:768880011:768880210; the BAIAP2 gene: chr17:79061115:79061314; TBCD gene: chr17, 80745041, 80745240; METRNL gene: chr17:81084062:81084261; MED16 gene: chr19:884043:884242; SBNO2 gene: chr19:1177525:1177724; CIRBP gene: chr19:1265940:1266139; KLF16 gene: chr19:1860593:1860792; c19orf77 gene: chr19:34916:3435115; c19orf77 gene: chr19:3435238:3435437; SNAPC2 gene: chr19:7985959:7986158; ICAM1 gene: chr19:10381567:10381766; ICAM5 gene: chr19:10405082:10405281; IER2 gene: chr19:13266897:13267096; ASF1B gene: chr19:14248383:14248582; CRTC1 gene: chr19:18771211:18771410; ZNF536 gene: chr19:31039497:31039696; LTBP4 gene: chr19:41105956:4106155; NOL4L-DT gene: chr20:31162351:31162550; KCNK15 gene: chr20:43374298:43374497; UCKL1 gene: chr20:62588863:6258562; RTN4R gene: chr22:20226623:20226822; RTN4R gene: chr22:20226825:20227024; BCR gene: chr22:23624342:23624541; TEF gene: chr22:41771479:41771678.
The present application also includes a medium bearing the sequence of the isolated nucleic acid molecules described herein and optionally methylation information thereof for comparison with gene methylation sequencing data to determine the presence, amount, and/or methylation level of the nucleic acid molecules. Preferably, the medium is a card, such as paper, plastic, metal, glass card, printed with the sequence and optionally its methylation information. Preferably, the medium is a computer readable medium storing the sequence and optionally methylation information thereof and a computer program which, when executed by a processor, performs the steps of: comparing methylation sequencing data of a sample to said sequence, thereby obtaining the presence, amount and/or methylation level of a nucleic acid molecule comprising said sequence in said sample.
The present application also includes an apparatus for identifying a benign or malignant thyroid nodule, the apparatus comprising a memory, a processor, and a computer program stored on the memory and executable on the processor, the processor implementing the steps of when executing the program: (1) Obtaining in the sample a methylation level of a target marker selected from one or more of the following or a target region thereof described herein, (2) interpreting the benign and malignant thyroid nodule according to the methylation level of (1). Preferably, the step of obtaining is performed using any of the methods described in section IV of the present application; preferably, the interpretation is performed by any of the methods described in section V of the present application.
VII use of
The application also provides application of the isolated nucleic acid molecules as described in the application as detection targets in diagnosis of benign and malignant thyroid nodule.
The methylation marker of the invention identifies that the sensitivity of thyroid cancer reaches 100%; more importantly, the sensitivity of the thyroid nodule with undefined cytological classification is 100% identified by the invention. Compared with the existing technology for molecular diagnosis of benign and malignant thyroid nodule, the methylation marker and the technical scheme provided by the invention effectively solve the problem of low sensitivity of the existing diagnosis technology, and are beneficial to early diagnosis and early treatment of thyroid cancer so as to improve the cure rate.
The invention will be further illustrated with reference to specific examples. It is to be understood that these examples are illustrative of the present invention and are not intended to limit the scope of the present invention. The experimental methods, in which specific conditions are not noted in the following examples, are generally conducted under conventional conditions or under conditions recommended by the manufacturer. Percentages and parts are by weight unless otherwise indicated.
Examples
The invention will be illustrated by way of specific examples. It should be understood that these examples are illustrative only and are not intended to limit the scope of the invention. The method of the invention comprises the following steps:
1. The level of CpG site methylation in the above markers in the sample was detected using genome-simplified methylation sequencing (RRBS) techniques, and then the average methylation ratio (average methylation fraction, AMF) of the markers was calculated as the DNA methylation level of the markers. AMF is derived from the following equation:
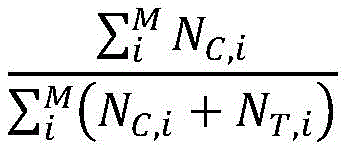
m is the total CpG site number in the marker, i is one of the CpG sites, N C,i Sequencing reads number, N, for methylation of the CpG sites T,i Sequencing reads that are unmethylated for the CpG sites.
2. And calculating the malignancy prediction probability of the sample through the constructed mathematical model. The malignancy prediction probability is calculated using a logistic regression (Logistic Regression) model. First, the input z of the Sigmoid function is calculated, which is given by the following formula:
z=Σw*x+w0
then, the Sigmoid function is calculated as follows:
σ(z)=1/(1+e -z )
w is the regression model coefficient for each marker, w0 is the intercept, and x is the sample DNA methylation level for that marker.
3. And identifying the benign and malignant of the thyroid nodule according to the sample malignant prediction probability. And (3) judging that the sample malignancy prediction probability threshold value calculated based on the data model constructed by the methylation marker combination is malignant if the sample malignancy prediction probability is larger than the threshold value, otherwise judging that the sample malignancy prediction probability is benign.
Using published data of published scientific papers, namely genome simplified methylation sequencing (RRBS) raw sequencing data [ Guerra a, carrano M, angrisani E et al, detection of RAS mutation by pyrosequencing in thyroid cytology samples, int J Surg,12suppl 1: s91-4, 2014 ], analyzing all samples of benign and malignant thyroid nodules, i.e., 145 surgical samples (65 benign nodules, 80 malignant nodules), to obtain methylation levels of CpG sites above 10x in sequencing depth; AMF is then calculated as the marker DNA methylation level based on the CpG sites detected in each methylation marker. For comparison with the published article [ Valderrabano P, khazai L, len ME et al, evaluation of ThyroSeq v performance in thyroid nodules with indeterminate cytology, endocr Relat Cancer,24:127-136,2017 ] identification of benign and malignant thyroid nodules, the present invention was consistent with the article sample grouping, with Developing cohort as the training set (28 benign nodules, 39 malignant nodules) and Testing cobort as the validation set 1 (37 benign nodules, 41 malignant nodules). In addition, 74 Chinese thyroid surgery samples are collected as verification set 2 (37 benign nodules and 37 malignant nodules), each sample obtains CpG sites detected in each methylation marker by using the RRBS technology and the analysis flow, and AMF is calculated and used as the DNA methylation level of the marker. In the following examples, two sets of validation set samples predict the area AUC (Area Under Curve) under the subject's operating characteristics curve (receiver operating characteristic curve, ROC) using a mathematical model constructed of training set samples.
Example 1
Methylation markers chr1:31550561:3155760, chr2:127822196:127822895, chr7:73508743:73509442, chr8:22547976:22548675, chr8:22548391:225090, chr10:81001706:81002405, chr14:21559748:21560447, chr15:70766881:70767580, chr17:7286958:7287657, chr17:768879761:768880460, chr19:1177275:1177974, chr19:34666:3435365, chr19:10404832:10405531, chr19:18770961:181660, chr22:20226373:27072 are models constructed in combination (methylation marker combination 1) to test AUC in both sets of validation samples. The logistic regression model coefficient w for each marker is shown in Table 1-1, with a logistic regression model intercept w0 of 0.305.
Table 1-1: logistic regression model coefficients for each marker
| Methylation markers | Gene name | Logistic regression model coefficients |
| chr1:3155061:3155760 | PRDM16 | 0.273 |
| chr2:127822196:127822895 | BIN1 | -0.347 |
| chr7:73508743:73509442 | LIMK1 | -0.258 |
| chr8:22547976:22548675 | EGR3 | 0.373 |
| chr8:22548391:22549090 | EGR3 | 0.239 |
| chr10:81001706:81002405 | PPIF | -0.228 |
| chr14:21559748:21560447 | ZNF219 | 0.413 |
| chr15:70766881:70767580 | UACA | -0.172 |
| chr17:7286958:7287657 | TNK1 | -0.143 |
| chr17:76879761:76880460 | CEP295NL | -0.170 |
| chr19:1177275:1177974 | SBNO2 | -0.230 |
| chr19:3434666:3435365 | C19orf77 | -0.184 |
| chr19:10404832:10405531 | ICAM5 | 0.423 |
| chr19:18770961:18771660 | CRTC1 | -0.251 |
| chr22:20226373:20227072 | RTN4R | -0.180 |
The results are shown in FIG. 1. The results show that the area under the ROC curve of the verification set 1 is 0.98, and the 95% CI is 0.97-0.99; the area under the ROC curve of validation set 2 was 0.95 and 95% CI was 0.93-0.97. When the training set specificity is 86% and the sensitivity is 92%, the malignancy prediction threshold is 0.49, namely the malignancy prediction probability is greater than 0.49, and the training set is judged to be malignant, otherwise, the training set is judged to be benign; the sensitivity of the threshold value to the diagnosis of the thyroid malignant nodule in the verification set 1 reaches 100%, the specificity reaches 76%, the PPV reaches 82%, and the NPV (negative predict value) reaches 100%; the sensitivity of the kit for diagnosing the thyroid malignant nodule in the validation set 2 reaches 87%, the specificity reaches 84%, the PPV reaches 84%, and the NPV reaches 86%. The results of the two sets of validation set samples predicted using the methylation marker combination 1 are shown in tables 1-2 and tables 1-3, respectively.
Table 1-2: results of the validation set 1 samples predicted with methylation marker combination 1

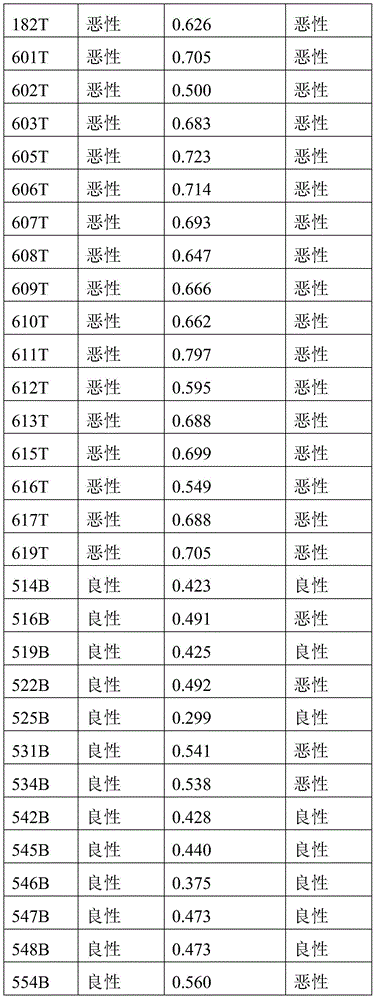
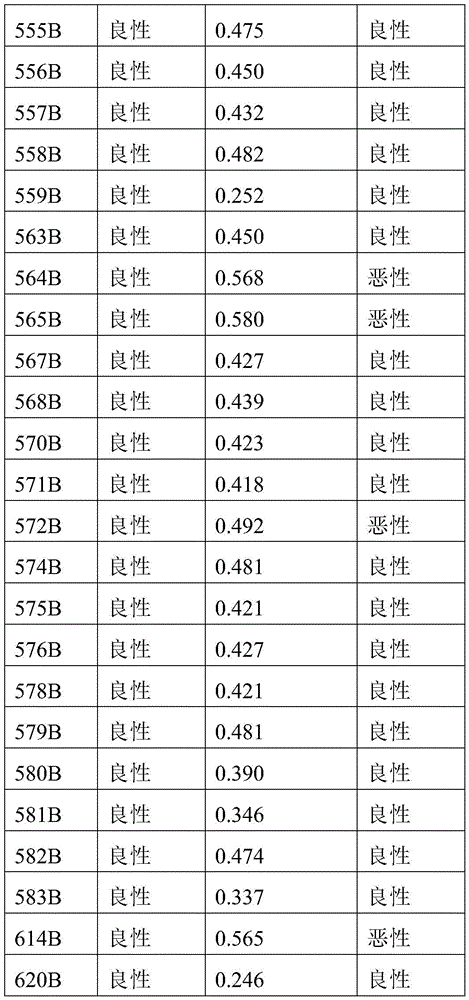
Tables 1-3: validation set 2 results of samples predicted with methylation marker combination 1
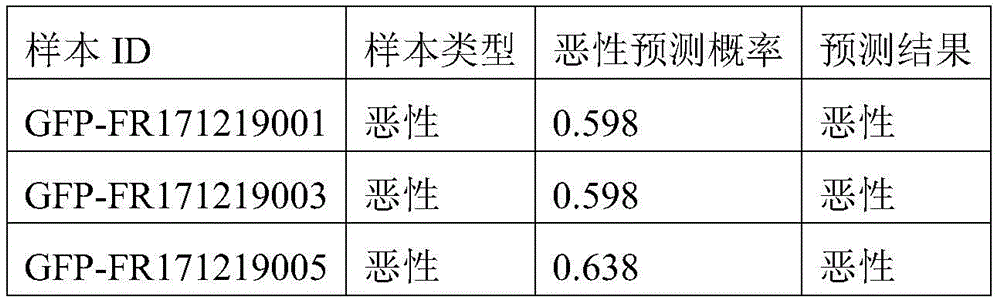
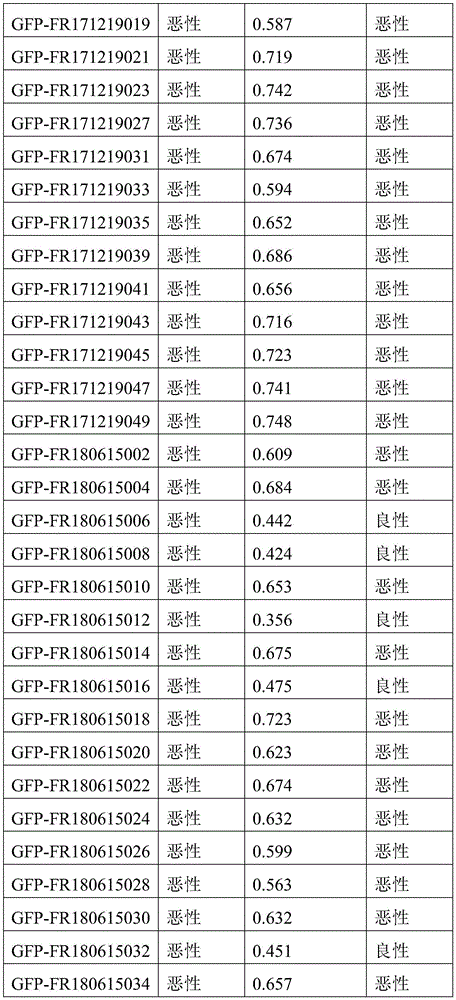
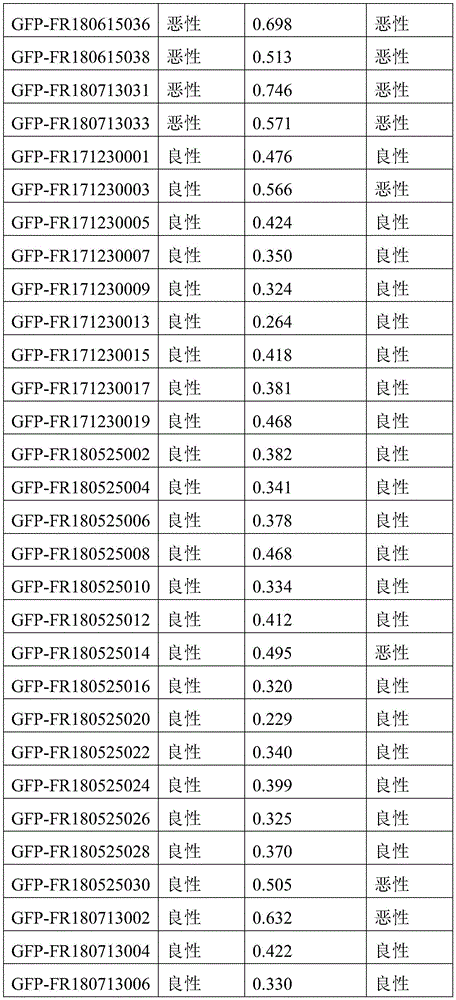
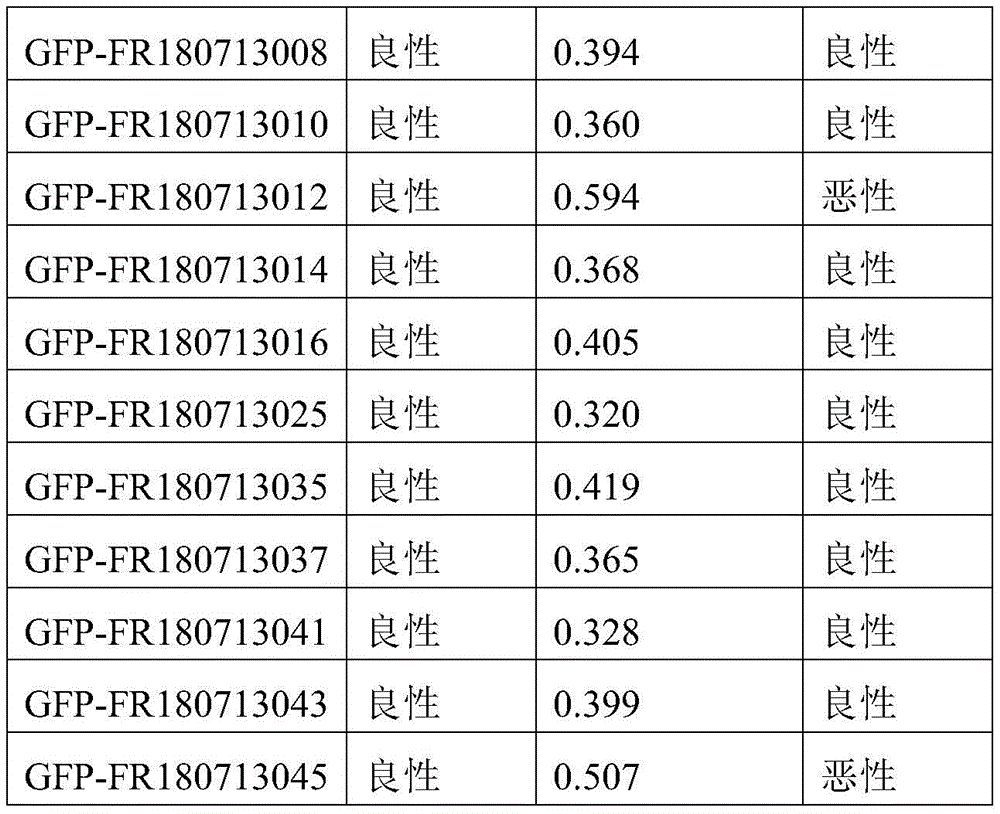
Example 2
The model constructed with the methylation markers chr1:20813203:20813902, chr3:58153211:58153910, chr8:22547976:22548675, chr8:34104888:34105587, chr17:6207008:62076775, chr17:79060865:79061564, chr19:883793:884492, chr19:3434988:3435687, chr20:31162101:31162800 in combination of methylation markers (methylation marker combination 2) was tested for AUC in both validation set samples. The logistic regression model coefficients for each marker are shown in table 2-1 with a logistic regression model intercept of 1.212.
Table 2-1: logistic regression model coefficients for each marker
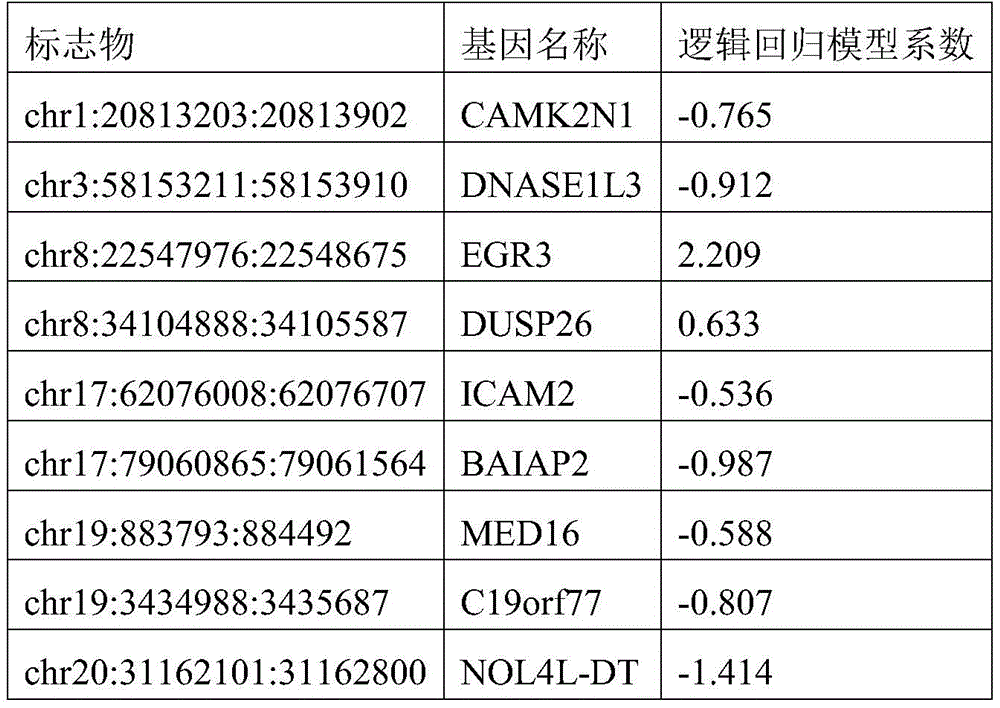
The results are shown in FIG. 2. The results show that the area under the ROC curve of the verification set 1 is 1.00, and the 95% CI is 1.00-1.00; the area under the ROC curve of validation set 2 was 0.96 and 95% CI was 0.95-0.98. When the training set specificity is 96% and the sensitivity is 85%, the malignancy prediction threshold is 0.58, namely the malignancy prediction probability is greater than 0.58, and the training set is judged to be malignant, otherwise, the training set is judged to be benign; the sensitivity of the threshold value to diagnosis of the thyroid malignant nodule in the verification set 1 reaches 88%, the specificity reaches 100%, the PPV reaches 100%, and the NPV reaches 88%; the sensitivity of the kit for diagnosing the thyroid malignant nodule in the validation set 2 reaches 76%, the specificity reaches 95%, the PPV reaches 93%, and the NPV reaches 80%. The prediction results of the methylation marker combination 2 for the two groups of verification set samples are shown in tables 2-2 and tables 2-3 respectively.
Table 2-2: validation of results predicted for set 1 samples with methylation marker combination 2
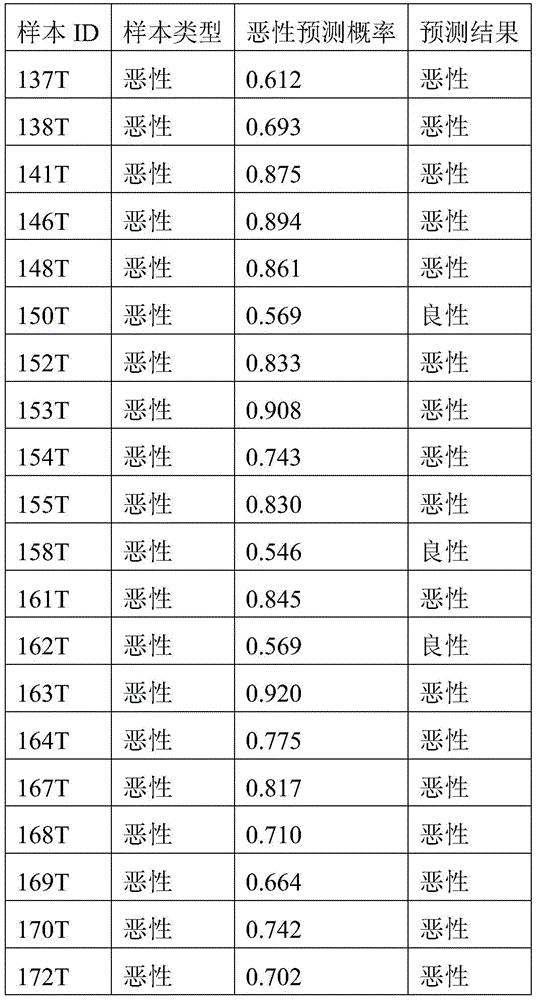

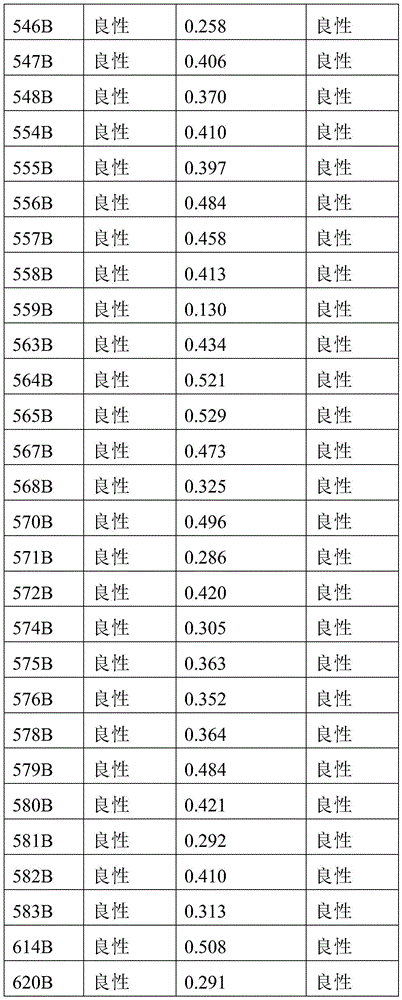
Table 2-3: validation set 2 results of samples predicted with methylation marker combination 2
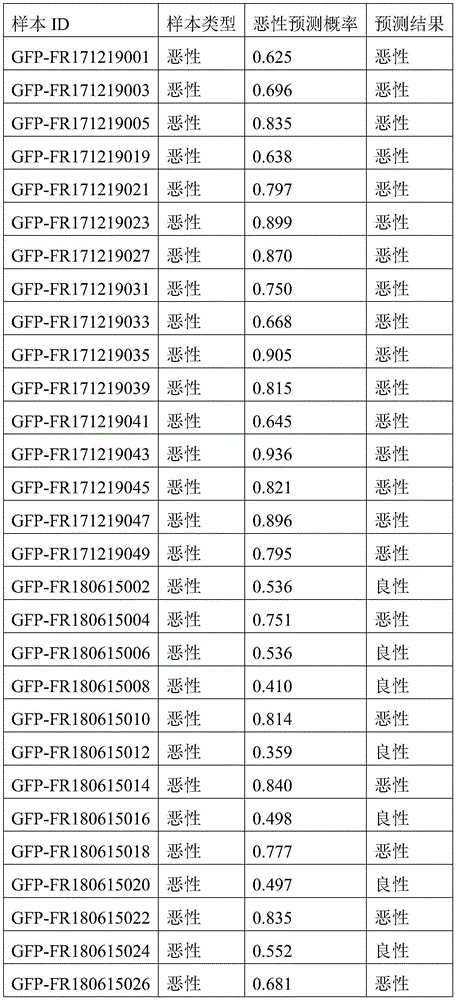
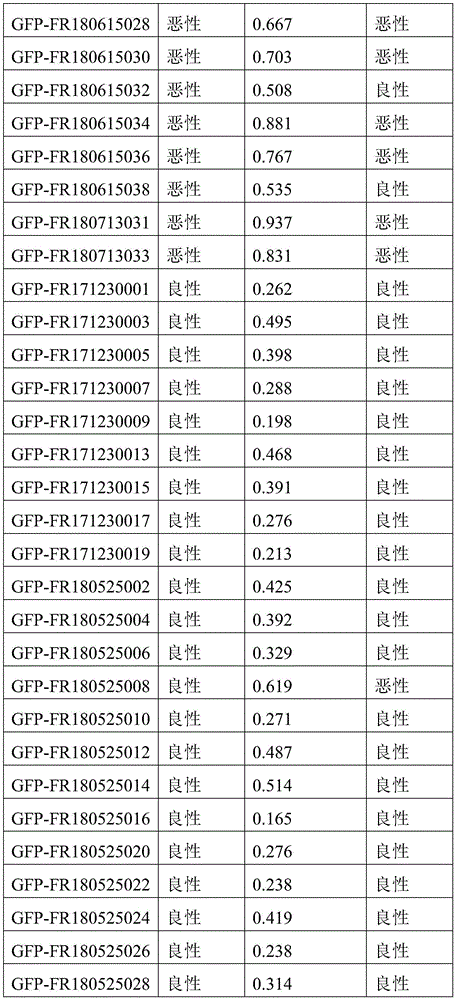

Example 3
The model constructed with the methylation markers chr1:5904615:59042314, chr1:15667274:1566767973, chr3:58153211:58153910, chr3:194408527:194409226, chr8:22547976:22548675, chr17:7286958:7287657, chr17:79060865:79061564, chr20:31162101:31162800, chr22:23624092:23624791 combinations (methylation marker combination 3) was tested for AUC in two sets of validation set samples. The logistic regression model coefficients for each marker are shown in table 3-1. The intercept of the logistic regression model was 1.681.
Table 3-1: logistic regression model coefficients for each marker
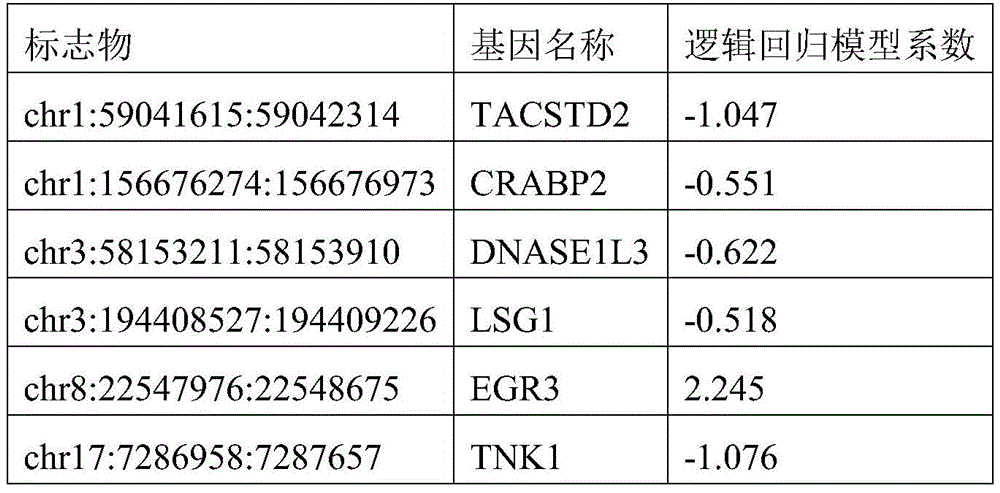

The results are shown in FIG. 3. The results show that the area under the ROC curve of the verification set 1 is 1.00, and the 95% CI is 0.99-1.00; the area under the ROC curve of validation set 2 was 0.97 and 95% CI was 0.95-0.98. When the training set specificity is 93% and the sensitivity is 95%, the malignancy prediction threshold is 0.52, namely the malignancy prediction probability is greater than 0.52, and the training set is judged to be malignant, otherwise, the training set is judged to be benign; the sensitivity of the threshold value to diagnosis of the thyroid malignant nodule in the verification set 1 reaches 98%, the specificity reaches 100%, the PPV reaches 100%, and the NPV reaches 97%; the sensitivity of the kit for diagnosing the thyroid malignant nodule in the validation set 2 reaches 92%, the specificity reaches 87%, the PPV reaches 87%, and the NPV reaches 91%. The prediction results of the methylation marker combination 3 for the two groups of validation set samples are shown in Table 3-2 and Table 3-3 respectively.
Table 3-2: validation of results predicted for set 1 samples with methylation marker combination 3




Table 3-3 verifies the results of the prediction of set 2 samples with methylation marker combination 3

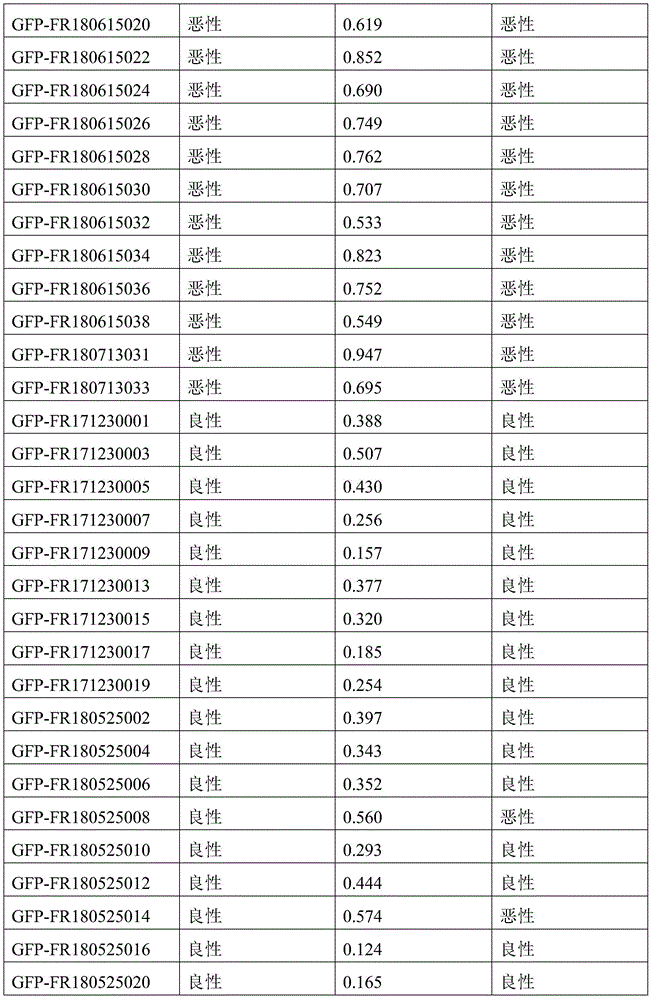

Example 4
The model constructed with the methylation markers chr1:5904615:59042314, chr1:15667274:1566767973, chr3:58153211:58153910, chr8:22547976:22548675, chr8:34104888:34105587, chr17:7286958:7287657, chr17:79060865:79061564, chr20:31162101:31162800, chr22:23624092:23624791 in combination of methylation markers 4 was tested for AUC in two sets of validation set samples. The logistic regression model coefficients for each marker are shown in table 4-1. The intercept of the logistic regression model is 1.358.
Table 4-1: logistic regression model coefficients for each marker


The results are shown in FIG. 4. The results show that the area under the ROC curve of the verification set 1 is 1.00, and the 95% CI is 0.99-1.00; the area under the ROC curve of validation set 2 was 0.97 and 95% ci was 0.95-0.98 (fig. 4). When the training set specificity is 93% and the sensitivity is 95%, the malignancy prediction threshold is 0.52, namely the malignancy prediction probability is greater than 0.52, and the training set is judged to be malignant, otherwise, the training set is judged to be benign; the sensitivity of the threshold value to diagnosis of the thyroid malignant nodule in the verification set 1 reaches 95%, the specificity reaches 97%, the PPV reaches 98%, and the NPV reaches 95%; the sensitivity of the kit for diagnosing the thyroid malignant nodule in the validation set 2 reaches 92%, the specificity reaches 87%, the PPV reaches 87%, and the NPV reaches 91%. The prediction results of the methylation marker combination 4 for the two groups of validation set samples are shown in Table 4-2 and Table 4-3 respectively.
Table 4-2: validation of results predicted for set 1 samples with methylation marker combination 4
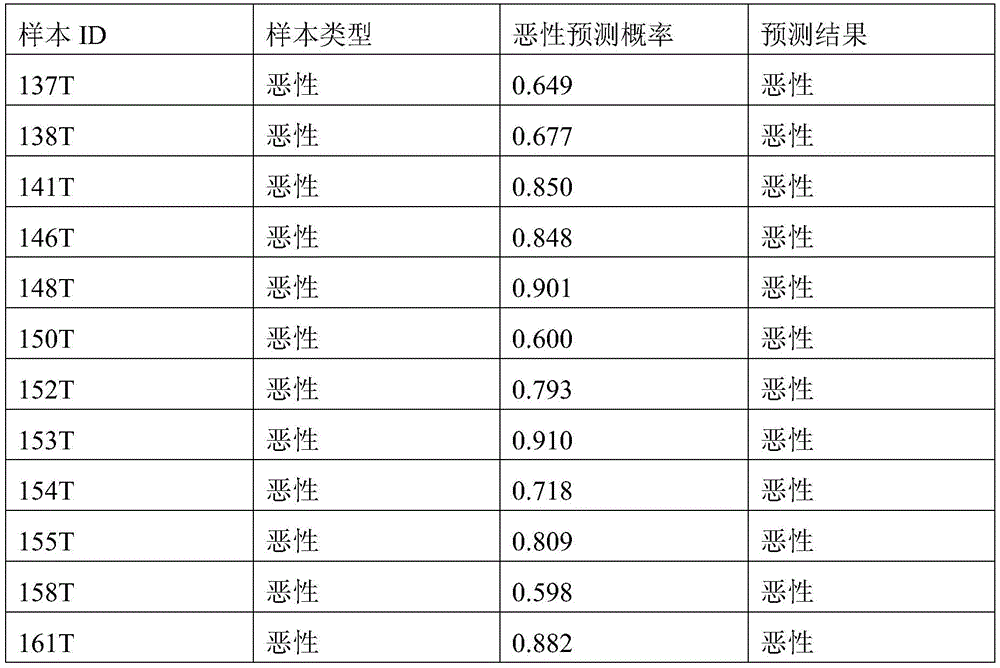
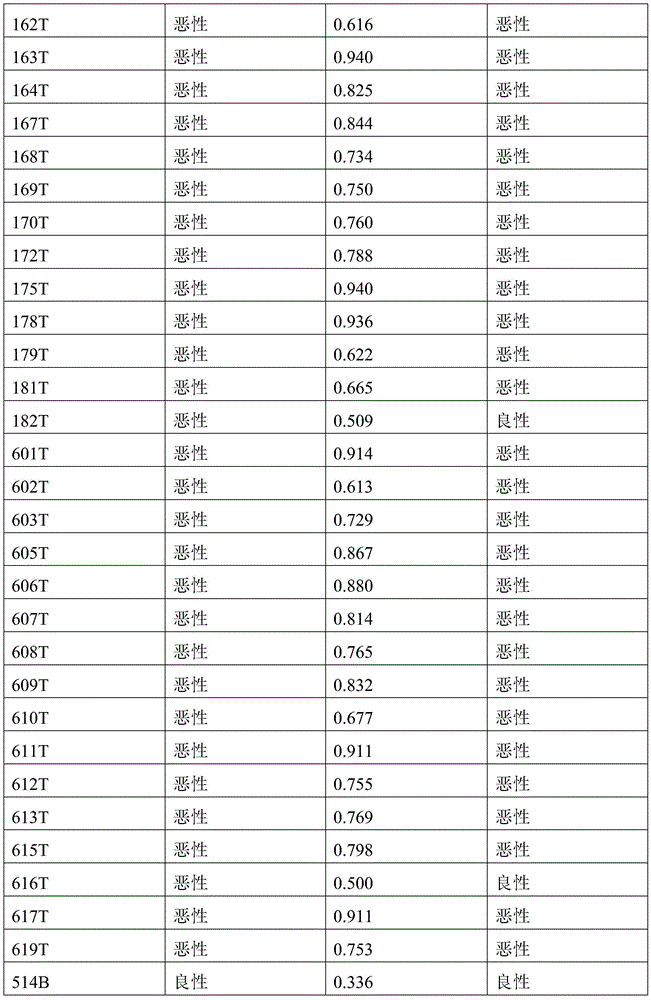
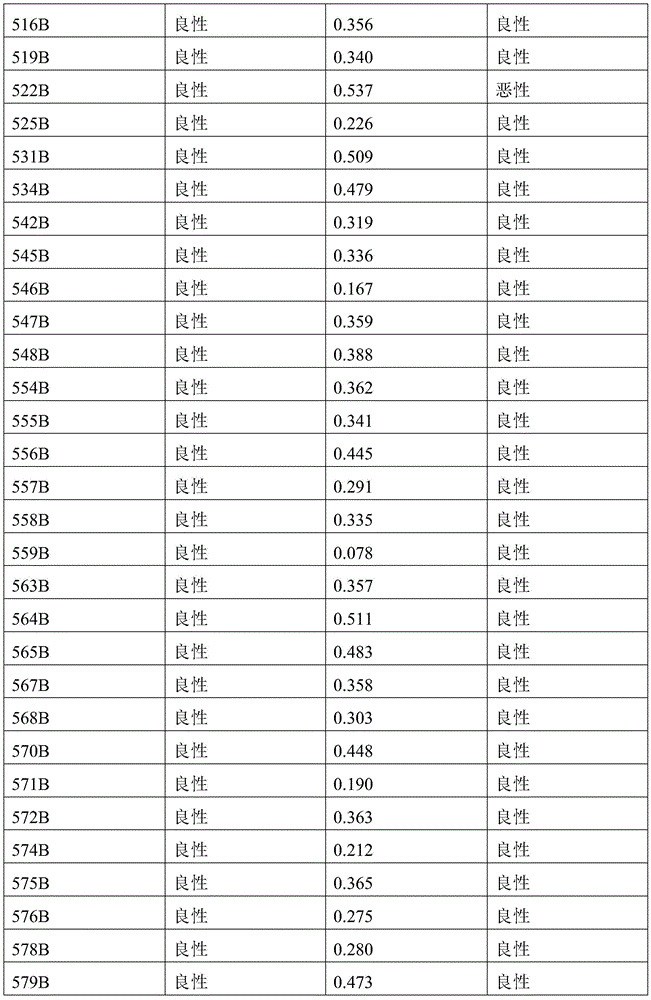

Table 4-3: validation set 2 samples predicted results with methylation marker combination 4
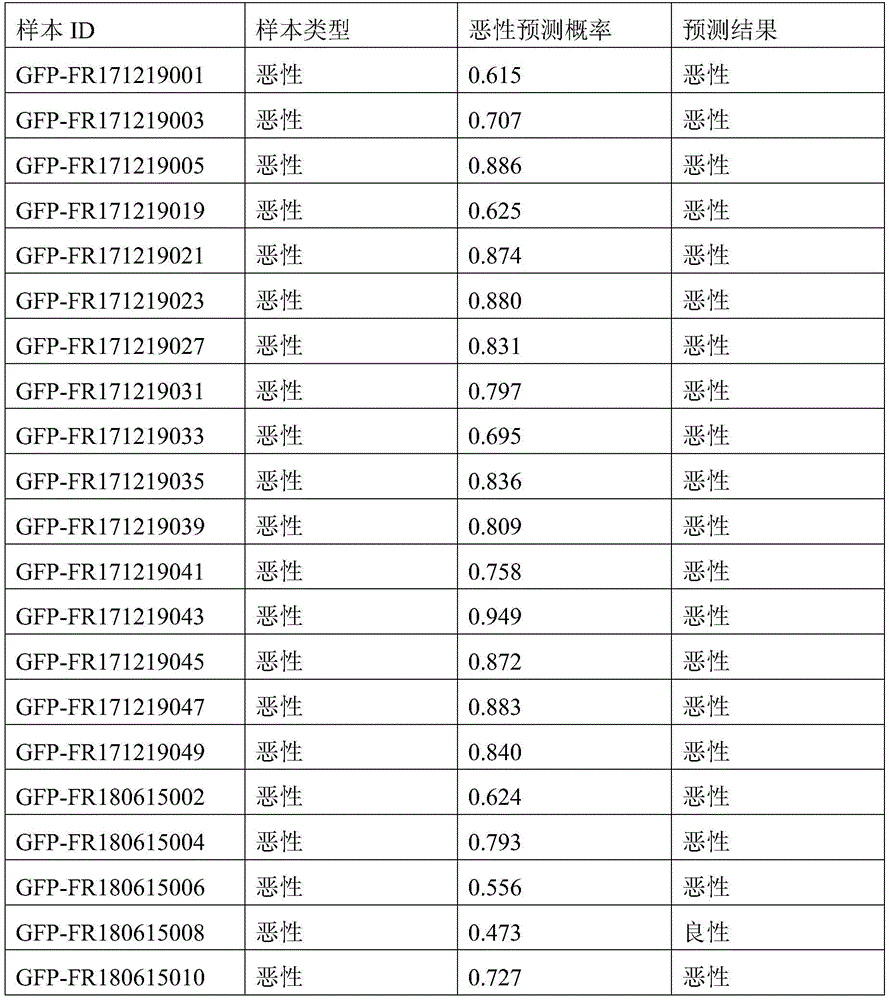
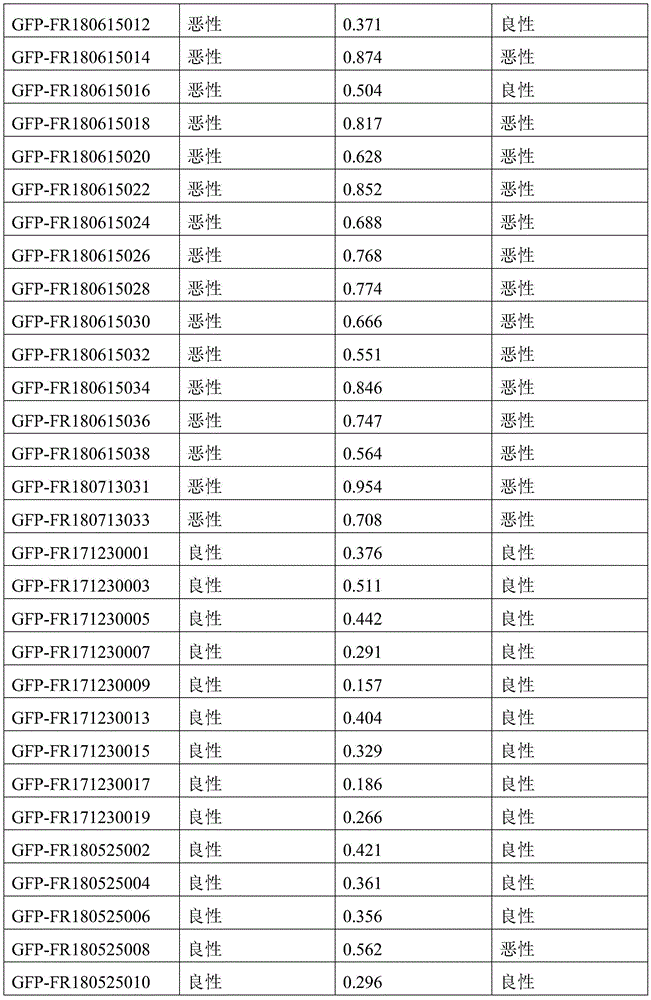
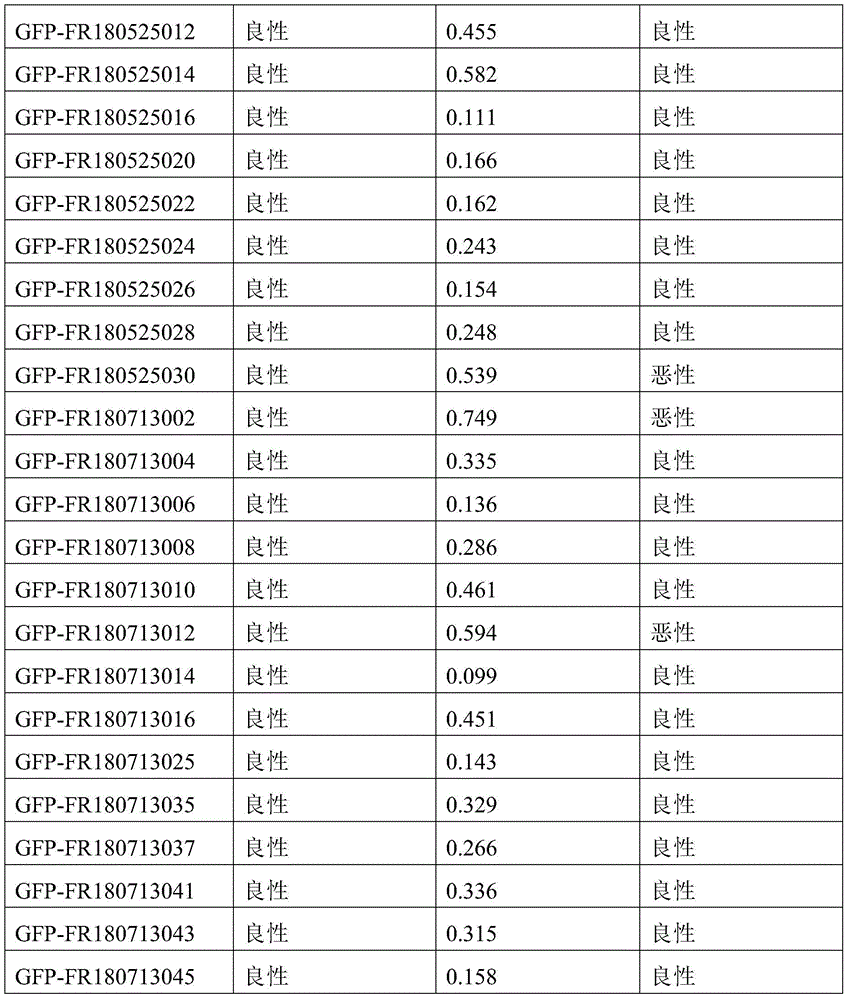
Example 5
The model constructed with the methylation markers chr1:5904615:59042314, chr3:58153211:58153910, chr8:22547976:22548675, chr8:34104888:34105587, chr17:7286958:7287657, chr17:79060865:79061564, chr19:883793:884492, chr20:31162101:31162800, chr22:23624092:23624791 in combination of methylation markers (methylation marker combination 5) was tested for AUC in both validation set samples. The logistic regression model coefficients for each marker are shown in table 5-1. The intercept of the logistic regression model was 1.447.
Table 5-1: logistic regression model coefficients for each marker
| Marker(s) | Gene name | Logistic regression model coefficients |
| chr1:59041615:59042314 | TACSTD2 | -1.122 |
| chr3:58153211:58153910 | DNASE1L3 | -0.724 |
| chr8:22547976:22548675 | EGR3 | 2.143 |
| chr8:34104888:34105587 | DUSP26 | 0.893 |
| chr17:7286958:7287657 | TNK1 | -1.212 |
| chr17:79060865:79061564 | BAIAP2 | -0.717 |
| chr19:883793:884492 | MED16 | -0.411 |
| chr20:31162101:31162800 | NOL4L-DT | -1.258 |
| chr22:23624092:23624791 | BCR | -0.682 |
The results are shown in FIG. 5. The results show that the area under the ROC curve of the verification set 1 is 1.00, and the 95% CI is 0.99-1.00; the area under the ROC curve of validation set 2 was 0.97 and 95% ci was 0.96-0.99 (fig. 4). When the training set specificity is 93% and the sensitivity is 95%, the malignancy prediction threshold is 0.52, namely the malignancy prediction probability is greater than 0.52, and the training set is judged to be malignant, otherwise, the training set is judged to be benign; the sensitivity of the threshold value to diagnosis of the thyroid malignant nodule in the verification set 1 reaches 95%, the specificity reaches 97%, the PPV reaches 98%, and the NPV reaches 95%; the sensitivity of the kit for diagnosing the thyroid malignant nodule in the validation set 2 reaches 92%, the specificity reaches 87%, the PPV reaches 87%, and the NPV reaches 91%. The prediction results of the methylation marker combination 5 for the two groups of validation set samples are shown in tables 5-2 and tables 5-3 respectively.
Table 5-2: validation of results predicted for set 1 samples with methylation marker combination 5
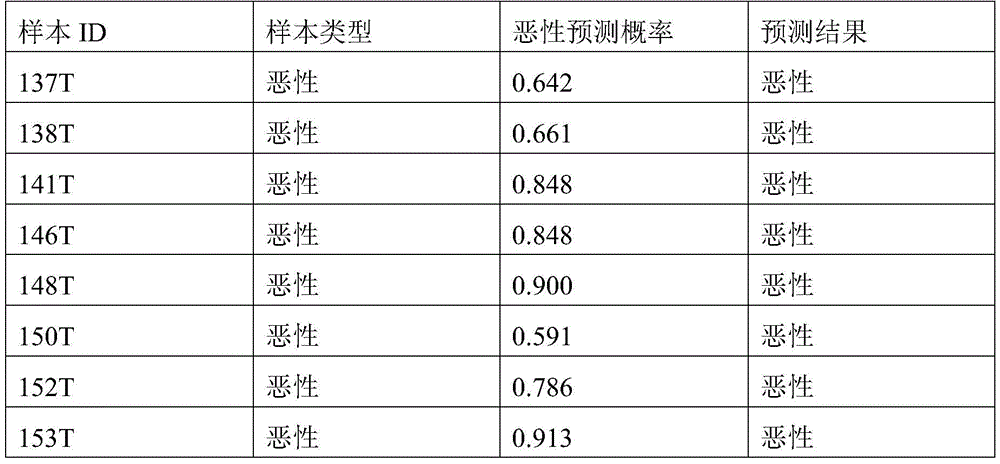

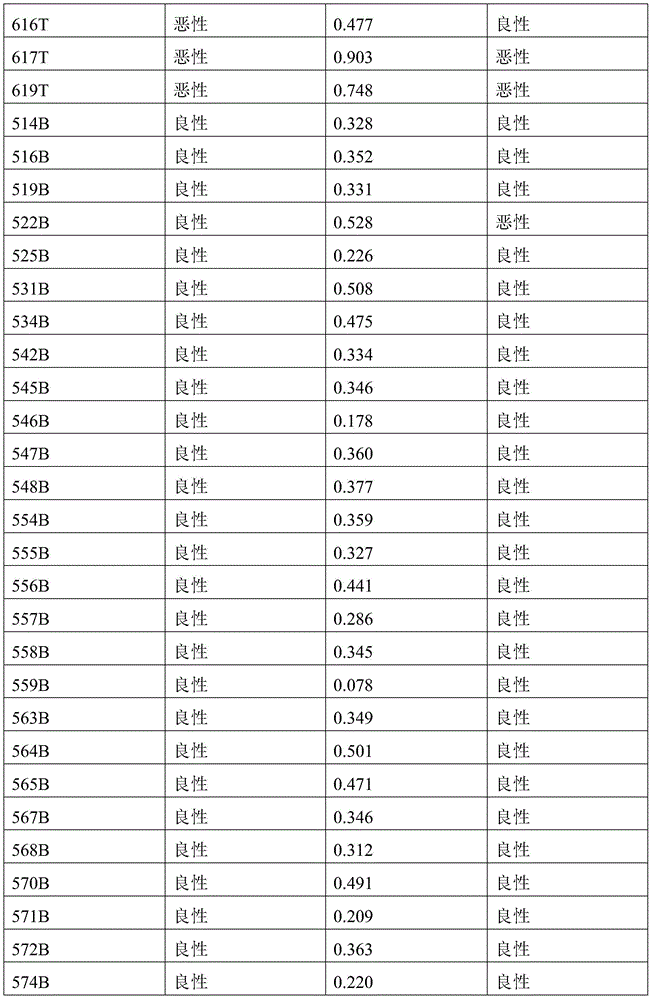
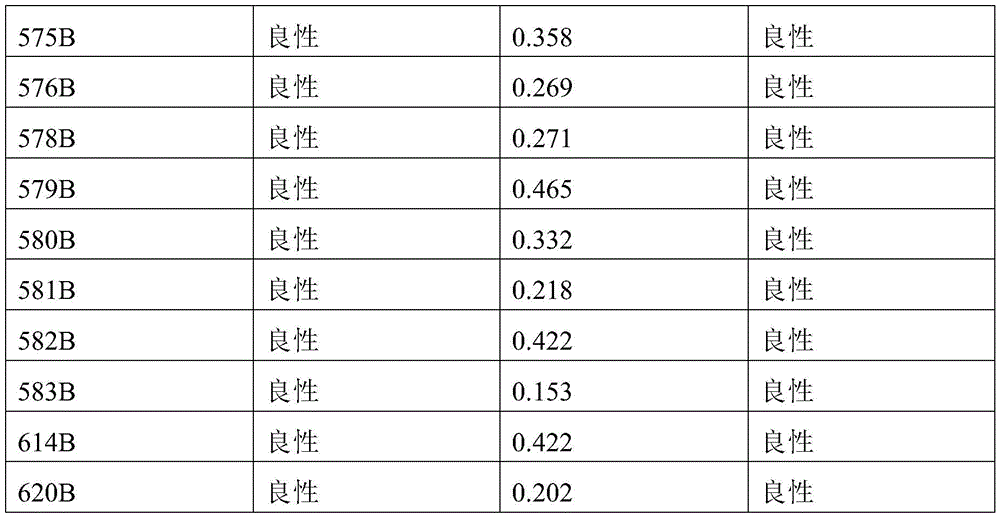
Table 5-3: validation set 2 samples predicted outcome with methylation marker combination 5
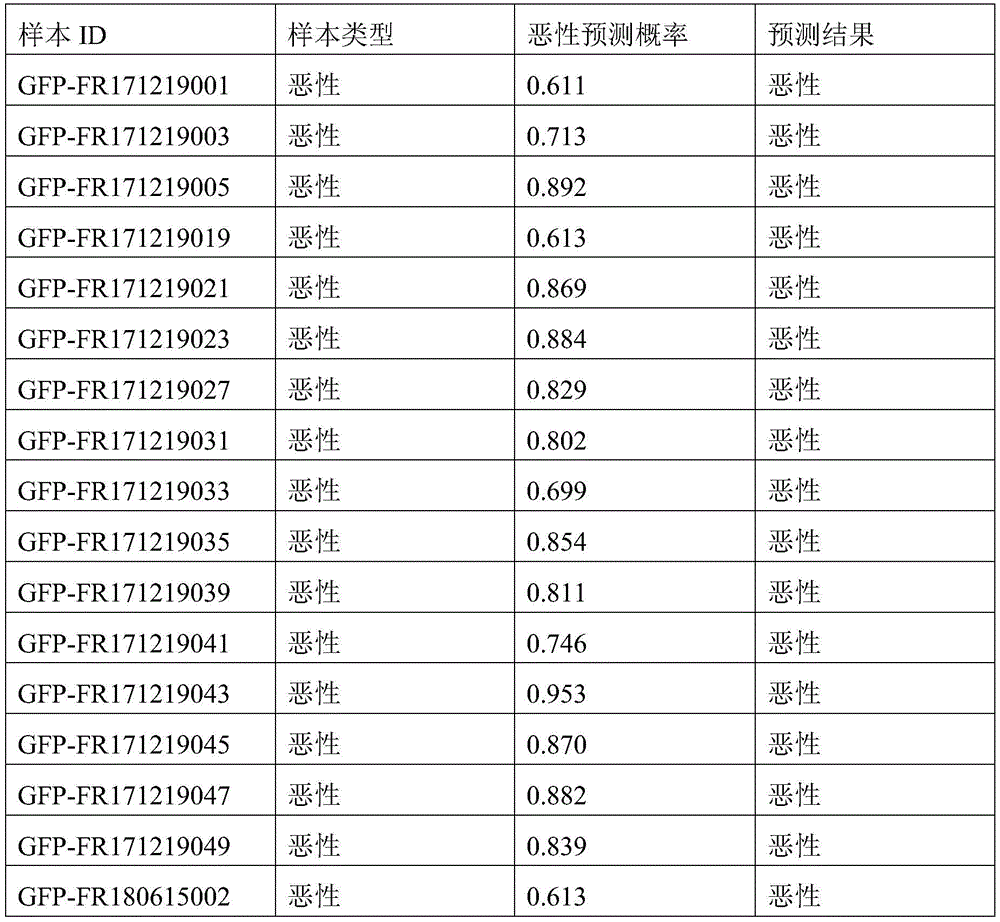

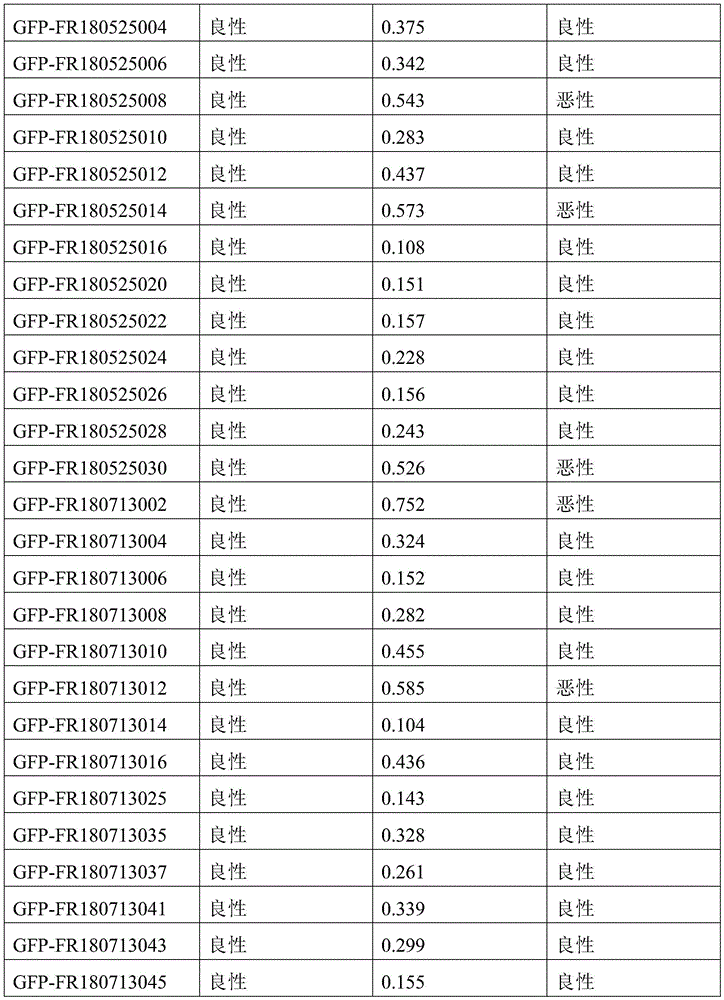
Example 6
AUC was tested in two sets of validation set samples using the model constructed with each methylation marker separately. A threshold defined by the Johnson index of the training set is used as a malignancy prediction threshold, and a person above the threshold is judged to be malignant, otherwise, the person is judged to be benign. The predictive performance of each methylation marker on two sets of validation set samples is shown in table 6.
Table 6: predicting performance of each methylation marker on test set samples
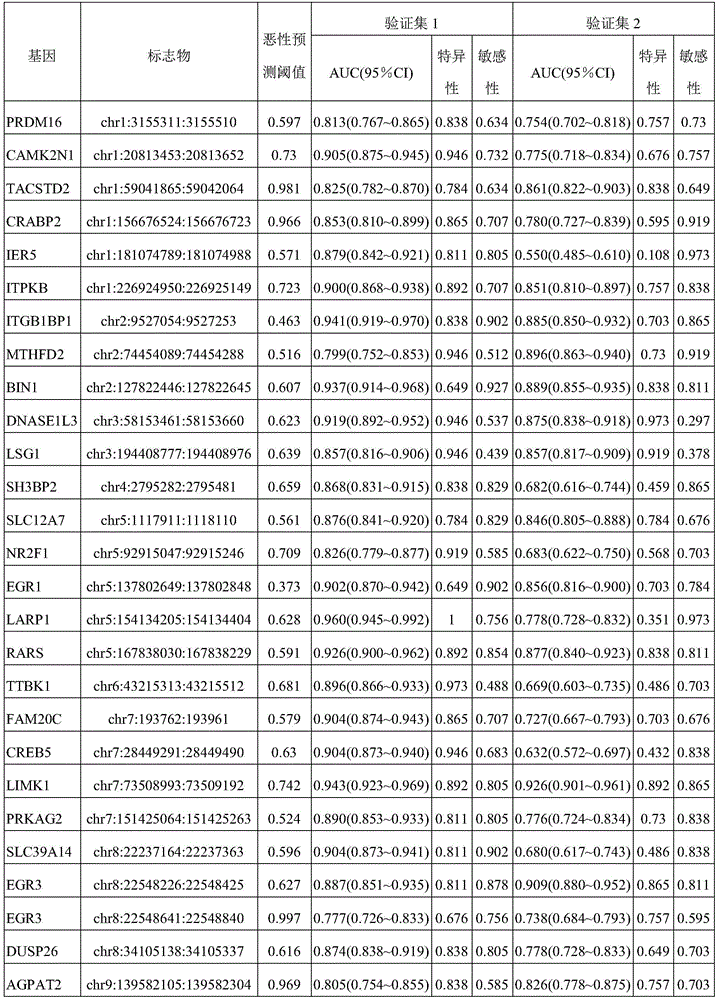
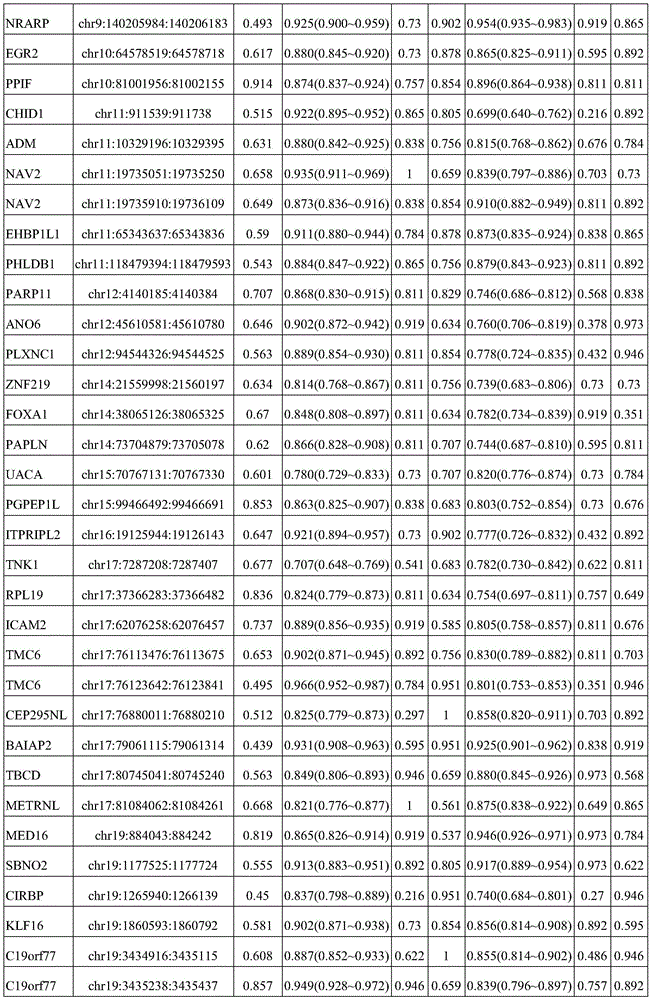
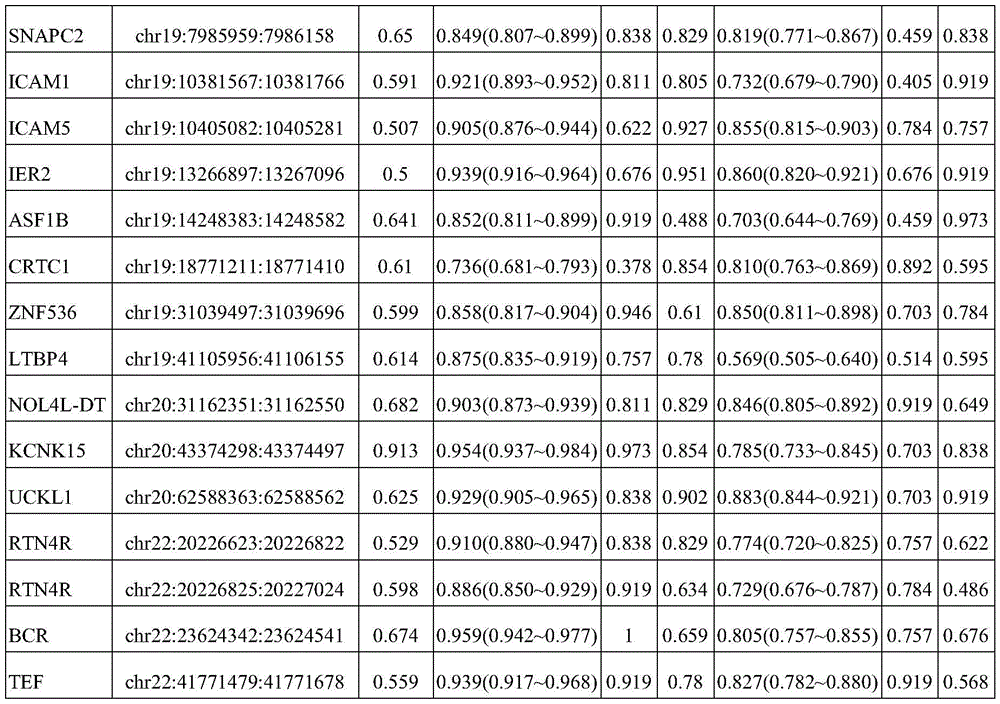
Example 7
Up to 30% of thyroid fine needle puncture samples are difficult to diagnose accurately by cytologic characterization. According to the Bethesda thyroid cytopathology classification criteria, 25 samples in validation set 1 were of ambiguous cytological classification, with a predictive accuracy of 84%, 100% sensitivity and 73% specificity using the methylation marker combination 1 of example 1. Validation set 1 samples with ambiguous cytological classification the predictive results of methylation marker combination 1 of example 1 are shown in table 7-1.
Table 7-1: validation set 1 sample with ambiguous cytological classification using methylation marker combination 1 predicted results
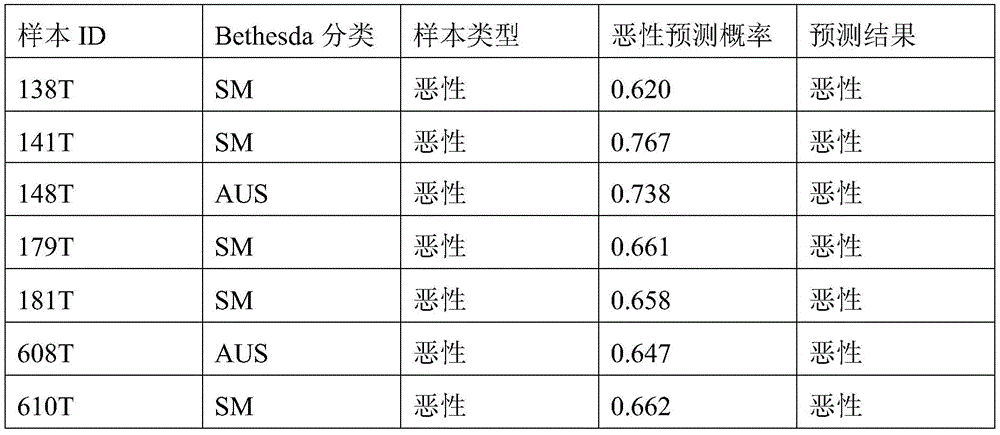
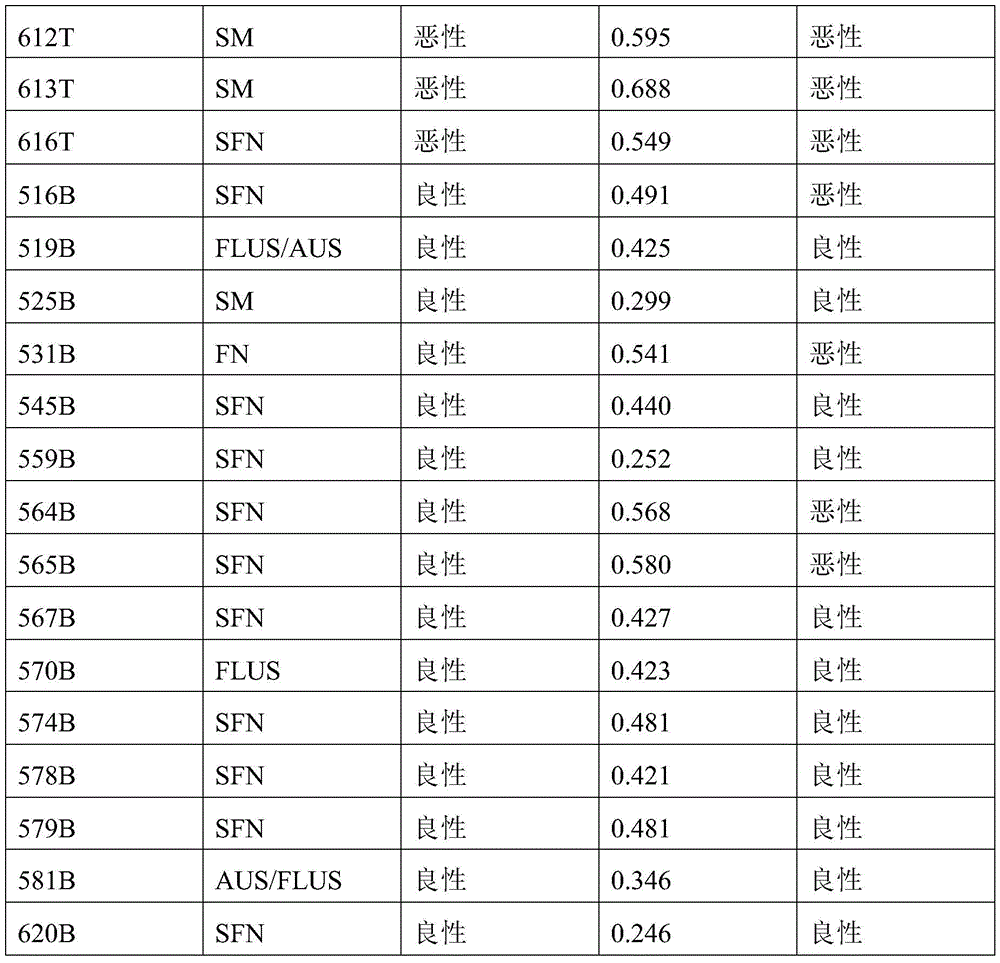
25 samples of the validation set 1 with ambiguous cytological classifications were predicted to have 96% accuracy, 90% sensitivity, and 100% specificity using the methylation marker combination 2 of example 2. The prediction of the cytological classification ambiguous validation set 1 samples using the methylation marker combinations of example 2 is shown in Table 7-2.
Table 7-2: validation set 1 sample with ambiguous cytological classification using methylation marker combination 2 predicted results
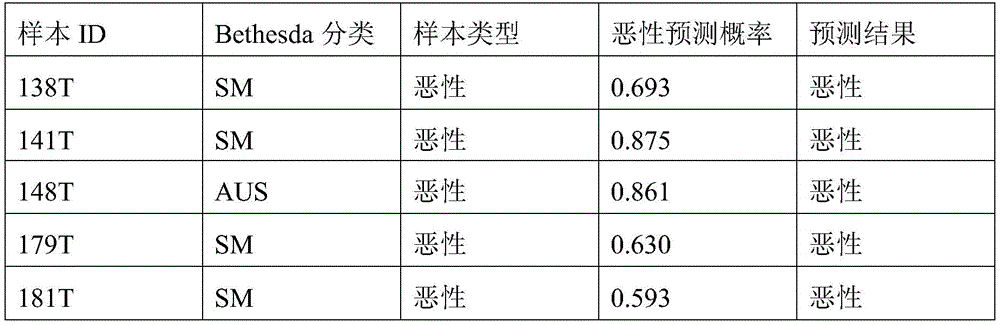
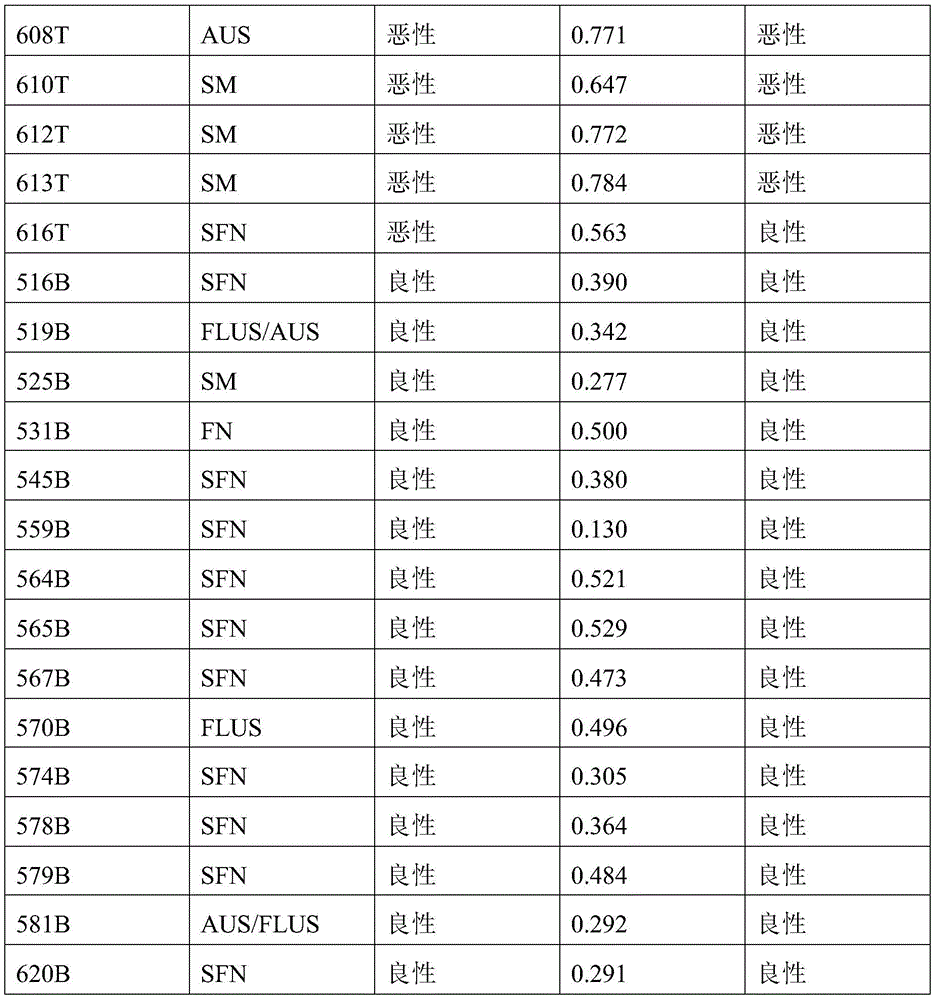
25 samples of validation set 1 with ambiguous cytological classifications were predicted to have 96% accuracy, 90% sensitivity, and 100% specificity using the methylation marker combination 3 of example 3. Validation set 1 samples with ambiguous cytological classification are shown in tables 7-3 using the methylation signature combination 3 prediction results of example 3.
Table 7-3: validation set 1 sample with ambiguous cytological classification using methylation marker combination 3 predicted results
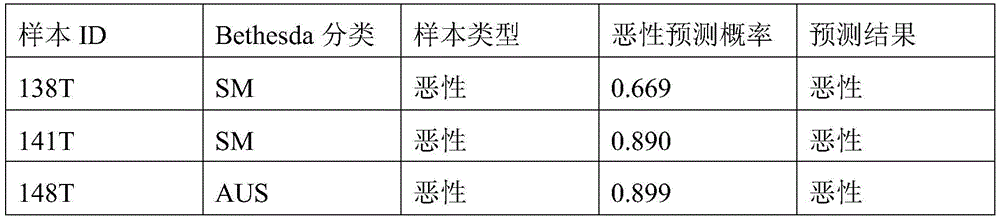
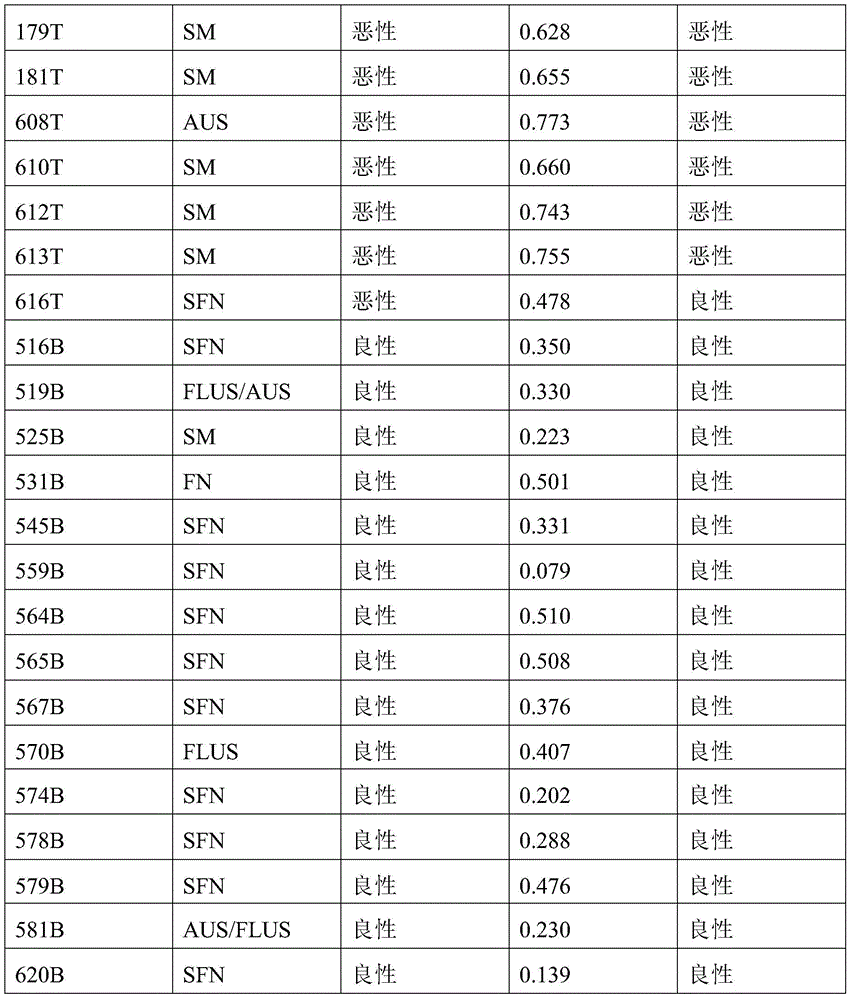
25 samples of the validation set 1 with ambiguous cytological classifications were predicted to have 96% accuracy, 90% sensitivity, and 100% specificity using the methylation marker combination 4 of example 4. Validation set 1 samples with ambiguous cytological classification are shown in tables 7-4 using the methylation signature combination 4 prediction results of example 4.
Table 7-4: validation set 1 sample with ambiguous cytological classification predicted outcome with methylation marker combination 4

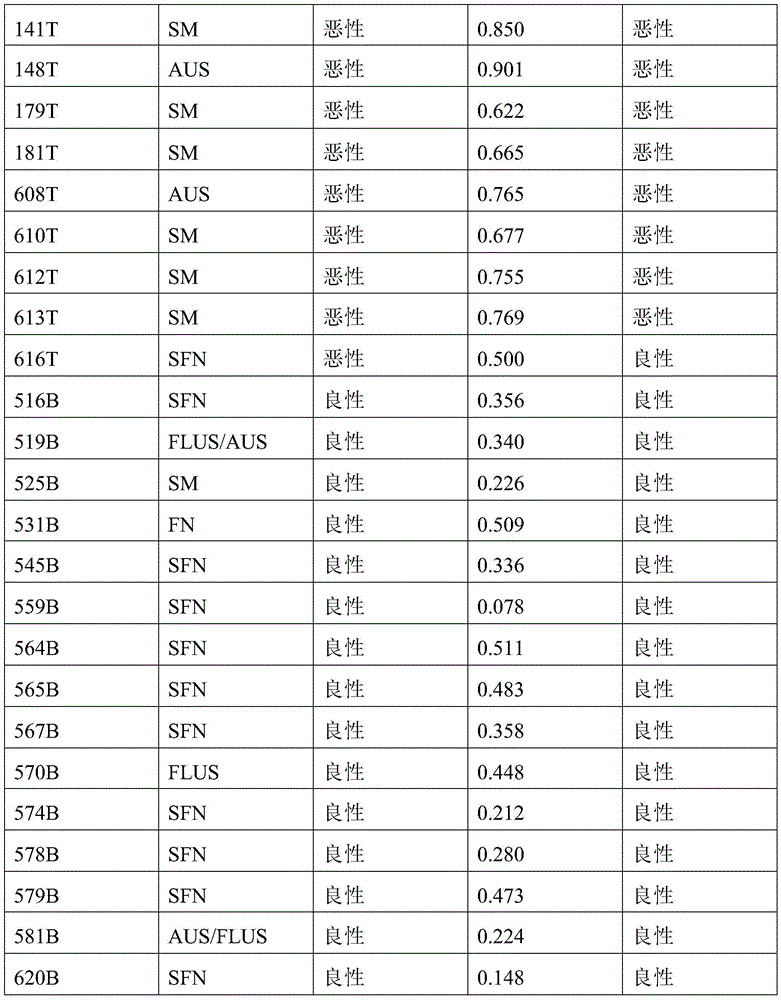
25 samples of the validation set 1 with ambiguous cytological classifications were predicted to have 96% accuracy, 90% sensitivity, and 100% specificity using the methylation marker combination 5 of example 5. Validation set 1 samples with ambiguous cytological classification are shown in tables 7-5 using the methylation signature combination 5 prediction results of example 5.
Table 7-5: validation set 1 sample with ambiguous cytological classification predicted outcome with methylation marker combination 5
| Sample ID | Bethesda classification | Sample type | Probability of malignancy prediction | Prediction result |
| 138T | SM | Malignant malignancy of | 0.661 | Malignant malignancy of |
| 141T | SM | Malignant malignancy of | 0.848 | Malignant malignancy of |
| 148T | AUS | Malignant malignancy of | 0.900 | Malignant malignancy of |
| 179T | SM | Malignant malignancy of | 0.624 | Malignant malignancy of |
| 181T | SM | Malignant malignancy of | 0.653 | Malignant malignancy of |
| 608T | AUS | Malignant malignancy of | 0.758 | Malignant malignancy of |
| 610T | SM | Malignant malignancy of | 0.668 | Malignant malignancy of |
| 612T | SM | Malignant malignancy of | 0.751 | Malignant malignancy of |
| 613T | SM | Malignant malignancy of | 0.769 | Malignant malignancy of |
| 616T | SFN | Malignant malignancy of | 0.477 | Benign |
| 516B | SFN | Benign | 0.352 | Benign |
| 519B | FLUS/AUS | Benign | 0.331 | Benign |
| 525B | SM | Benign | 0.226 | Benign |
| 531B | FN | Benign | 0.508 | Benign |
| 545B | SFN | Benign | 0.346 | Benign |
| 559B | SFN | Benign | 0.078 | Benign |
| 564B | SFN | Benign | 0.501 | Benign |
| 565B | SFN | Benign | 0.471 | Benign |
| 567B | SFN | Benign | 0.346 | Benign |
| 570B | FLUS | Benign | 0.491 | Benign |
| 574B | SFN | Benign | 0.220 | Benign |
| 578B | SFN | Benign | 0.271 | Benign |
| 579B | SFN | Benign | 0.465 | Benign |
| 581B | AUS/FLUS | Benign | 0.218 | Benign |
| 620B | SFN | Benign | 0.202 | Benign |
Bethesda classification description:
AUS: cannot be diagnosed explicitly (Atypia of Undetermined Significance);
FLUS: unknown follicular lesions (Follicular Lesion of Undetermined Significance);
FN: a follicular new growth (follicular neoplasms);
SFN: suspicious follicular tumors (suspicious for follicular neoplasm);
SM: suspected malignant tumor (suspicious for malignancy).
Claims (22)
1. Use of a reagent for detecting the methylation status or level of at least one CpG dinucleotide of one or more markers of interest for the manufacture of a detection reagent or a diagnostic kit for diagnosing benign and malignant thyroid nodules in an individual, and use of a device for determining the methylation status or level of at least one CpG dinucleotide of one or more markers of interest for the manufacture of a diagnostic kit for diagnosing benign and malignant thyroid nodules in an individual, wherein the one or more markers of interest are selected from the group consisting of: PRDM16 gene or genome PRDM16 sequence, CAMK2N1 gene or genome CAMK2N1 sequence, TACSTD2 gene or genome TACSTD2 sequence, CRABP2 gene or genome CRABP2 sequence, IER5 gene or genome IER5 sequence, ITPKB gene or genome ITPKB sequence, ITGB1BP1 gene or genome ITGB1BP1 sequence, MTHFD2 gene or genome MTHFD2 sequence, BIN1 gene or genome BIN1 sequence, DNASE1L3 gene or genome DNASE1L3 sequence, DNASE1L3 sequence LSG1 sequence of LSG1 gene or genome, SH3BP2 sequence of SH3BP2 gene or genome, SLC12A7 sequence of SLC12A7 gene or genome, NR2F1 sequence of NR2F1 gene or genome, EGR1 sequence of EGR1 gene or genome, LARP1 sequence of LARP1 gene or genome, RARS sequence of RARS gene or genome, TTBK1 sequence of TTBK1 gene or genome, FAM20C sequence of FAM20C gene or genome, CREB5 sequence of CREB5 gene or genome, LIMK1 sequence of LIMK1 gene or genome PRKAG2 sequence of PRKAG2 gene or genome, SLC39A14 sequence of SLC39A14 gene or genome, EGR3 sequence of EGR3 gene or genome, DUSP26 sequence of DUSP26 gene or genome, AGPAT2 sequence of AGPAT2 gene or genome, AGPAT2 sequence of gene or genome, gene, or genome, or sequence, or gene, or NRARP sequence of NRARP gene or genome, EGR2 sequence of EGR2 gene or genome, PPIF sequence of PPIF gene or genome, CHID1 sequence of CHID1 gene or genome, ADM sequence of ADM gene or genome, NAV2 sequence of NAV2 gene or genome, and/or its/their/EHBP 1L1 sequence of the EHBP1L1 gene or genome, PHLDB1 sequence of the PHLDB1 gene or genome, PARP11 sequence of the PARP11 gene or genome, ANO6 sequence of the ANO6 gene or genome, PLXNC1 sequence of the PLXNC1 gene or genome, ZNF219 sequence of the ZNF219 gene or genome, FOXA1 sequence of the FOXA1 gene or genome, PAPLN sequence of the PAPLN gene or genome, UACA sequence of the UACA gene or genome, PGPEP1L sequence of the PGPEP1L gene or genome, ITPRIPL2 sequence of the PLXNC1 gene or genome, TNK1 gene or genome's TNK1 sequence, RPL19 gene or genome's RPL19 sequence, ICAM2 gene or genome's ICAM2 sequence, TMC6 gene or genome's TMC6 sequence, CEP295NL gene or genome's CEP295NL sequence, BAIAP2 gene or genome's BAIAP2 sequence, TBCD gene or genome's TBCD sequence, METRNL gene or genome's METRL sequence, MED16 gene or genome's MED16 sequence, SBNO2 gene or genome's SBNO2 sequence, CIRBP gene or genome's CIRBP sequence, KLF16 gene or genome's KLF16 sequence, C19orf77 gene or genome's C19orf77 sequence, SNNK 2 gene or genome's SNN 2 sequence, ICAM1 gene or genome's ICAM5 sequence, IER2 gene or genome's IER2 sequence, ASF1B gene or genome's MED16 sequence, ASF1 gene or genome's CIRBP sequence, KLF16 gene or genome's KLF16 sequence, C19orf77 sequence, SNNK 2 gene or genome's SNNK 2 sequence, ICAM1 gene or genome's ICAM5 sequence, ICAM5 gene or genome's TC 2 gene's IER2 gene or genome's IEF 1, ASF1 gene or ASF1 gene's ASF1 or genome's ASF1, ASF 4 or gene's BCF 4 gene's 4 sequence, or its BCF 4 gene or genome's 4 sequence, and its KCR 4 gene or its 4 sequence.
2. The use of claim 1, wherein the one or more target markers are selected from the group consisting of: the PRDM16 gene or genome PRDM16 sequence, BIN1 sequence of a BIN1 gene or genome, LIMK1 sequence of a LIMK1 gene or genome, EGR3 sequence of a CRTC1 gene or genome, PPIF sequence of a PPIF gene or genome, ZNF219 sequence of a ZNF219 gene or genome, UACA sequence of a UACA gene or genome, TNK1 sequence of a TNK1 gene or genome, CEP295NL sequence of a CEP295NL gene or genome, SBNO2 sequence of a SBNO2 gene or genome, C19orf77 sequence of a C19orf77 gene or genome, ICAM5 sequence of a ICAM5 gene or genome, CRTC1 sequence of a CRTC1 gene or genome, RTN4R sequence of a RTN4 gene or genome, CAMK2N1 sequence of a CAMK 1 gene or genome, DNASE1L3 sequence of a DNASE1 gene or genome, DUSP26 sequence of a DUSP26 gene or genome, a cstr 2 sequence of a csag 2 gene or genome, a cstr 2 sequence of a cstr 2 gene or genome, a cstr 2 or a cstr 2 gene or genome.
3. The use of claim 1, wherein the one or more target markers comprise at least one or more of the following target markers: the EGR3 sequence of the EGR3 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, the TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the BCR sequence of the BCR gene or genome.
4. The use of claim 1, wherein the one or more target markers comprise:
the PRDM16 gene or genome PRDM16 sequence, BIN1 sequence of a BIN1 gene or genome, LIMK1 sequence of a LIMK1 gene or genome, EGR3 sequence of an EGR3 gene or genome, PPIF sequence of a PPIF gene or genome, ZNF219 sequence of a ZNF219 gene or genome, UACA sequence of a UACA gene or genome, TNK1 sequence of a TNK1 gene or genome, CEP295NL sequence of a CEP295NL gene or genome, SBNO2 sequence of a SBNO2 gene or genome, C19orf77 sequence of a C19orf77 gene or genome, ICAM5 sequence of an ICAM5 gene or genome, CRTC1 sequence of a CRTC1 gene or genome, and RTN4R sequence of an RTN4R gene or genome; or (b)
The CAMK2N1 gene or genome, the DNASE1L3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the ICAM2 sequence of the ICAM2 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the C19orf77 sequence of the C19orf77 gene or genome and the NOL4L-DT sequence of the NOL4L-DT gene or genome; or (b)
The TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the LSG1 sequence of the LSG1 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, and the BCR sequence of the BCR gene or genome; or (b)
The TACSTD2 sequence of the TACSTD2 gene or genome, the CRABP2 sequence of the CRABP2 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome, and the BCR sequence of the BCR gene or genome; or (b)
The TACSTD2 sequence of the TACSTD2 gene or genome, the DNASE1L3 sequence of the DNASE1L3 gene or genome, the EGR3 sequence of the EGR3 gene or genome, the DUSP26 sequence of the DUSP26 gene or genome, the TNK1 sequence of the TNK1 gene or genome, the BAIAP2 sequence of the BAIAP2 gene or genome, the MED16 sequence of the MED16 gene or genome, the NOL4L-DT sequence of the NOL4L-DT gene or genome and the BCR sequence of the BCR gene or genome.
5. The use according to any one of claims 1-4, wherein the Hg19 coordinates of the one or more target markers are as follows:
PRDM16 gene: chr1:3155051:3155760; CAMK2N1 gene: chr1:20813203:20813902; TACSTD2 Gene: chr 1:5904615:59042314; CRABP2 Gene: chr1:15667274:156676773; IER5 gene: chr1:181074539:181075238; ITPKB gene: chr1:226924700:226925399; ITGB1BP1 gene: chr2:9526804:9527503; MTHFD2 gene: chr2:74453839:74454538; BIN1 gene: chr2, 127822196, 127822895; DNASE1L3 gene: chr3:58153211:58153910; LSG1 gene: chr3:194408527:194409226; SH3BP2 gene: chr 4:2795932:2795331; SLC12A7 gene: chr5:1117661:1118360; NR2F1 gene: chr5:92914797:92915496; EGR1 gene: chr5:137802399:137803098; LARP1 gene: chr5:154133955:154134654; RARS gene: chr 5:167837780:167838499; TTBK1 gene: chr6:43215063:43215762; FAM20C gene: chr7:193512:194211; CREB5 gene: chr7:28449041:28449740; LIMK1 gene: chr7:73508743:73509442; PRKAG2 gene: chr7:151424814:151425513; SLC39a14 gene: chr8:22236914:22237613; EGR3 gene: chr8:22547976:22549090; DUSP26 gene: chr8:34104888:34105587; AGPAT2 gene: chr9:139581855:139582554; NRARP gene: chr9:140205734:140206433; EGR2 gene: chr10:64578269:64578968; PPIF gene: chr10:81001706:81002405; CHID1 gene: chr11:911289:911988; ADM gene: chr11:10328946:10329645; NAV2 gene: chr11:19734801:19736359; EHBP1L1 gene: chr11:65343387:65344086; PHLDB1 gene: chr11:118479144:118479843; PARP11 gene: chr12:4139935:4140634; ANO6 gene: chr12:45610331:45611030; PLXNC1 gene: chr12:94544076:94544775; ZNF219 gene: chr14:21559748:21560447; FOXA1 gene: chr14:3806876:38065575; PAPLN gene: chr14:73704629:73705328; UACA gene: chr15:70766881:70767580; PGPEP1L gene: chr15:99466242:99466941; ITPRIPL2 gene: chr16:19125694:19126393; TNK1 gene: chr17:7286958:7287657; RPL19 gene: chr17:37366033:37366732; ICAM2 gene: chr 17:6207008:6207677; TMC6 gene: chr 17:7613226:7624091; CEP295NL gene: chr17:7689761:768880460; the BAIAP2 gene: chr17:79060865:79061564; TBCD gene: chr17:80744791:80745490; METRNL gene: chr17:81083812:81084511; MED16 gene: chr19:883793:884492; SBNO2 gene: chr19:1177275:1177974; CIRBP gene: chr19:1265690:1266389; KLF16 gene: chr19:1860343:1861042; c19orf77 gene: chr19:34666:3435687; SNAPC2 gene: chr 19:7985709:7986108; ICAM1 gene: chr19:10381317:10382016; ICAM5 gene: chr19:10404832:10405531; IER2 gene: chr19:13266647:13267346; ASF1B gene: chr 19:14248133:14248172; CRTC1 gene: chr19:18770961:18771660; ZNF536 gene: chr19:31039247:31039946; LTBP4 gene: chr19:41105706:41106405; NOL4L-DT gene: chr20:31162101:31162800; KCNK15 gene: chr20:43374048:43374747; UCKL1 gene: chr20:62588113:62588812; RTN4R gene: chr22:20226373:20227274; BCR gene: chr22:23624092:23624791; TEF gene: chr22:41771229:41771928.
6. The use according to any one of claims 1-5, wherein the Hg19 coordinates of the one or more target markers are as follows:
PRDM16 gene: chr1:3155311:3155510; CAMK2N1 gene: chr1:20813453:20813652; TACSTD2 Gene: chr 1:59041685:59042064; CRABP2 Gene: chr1:15667684:156676723; IER5 gene: chr1:181074789:181074988; ITPKB gene: chr1:226924950:226925149; ITGB1BP1 gene: chr2:9527054:9527253; MTHFD2 gene: chr2:74454089:74454288; BIN1 gene: chr2, 127822446, 127822645; DNASE1L3 gene: chr3:58153461:58153660; LSG1 gene: chr3:194408777:194408976; SH3BP2 gene: chr 4:279282:2795581; SLC12A7 gene: chr5:1117911:1118110; NR2F1 gene: chr5:92915047:92915246; EGR1 gene: chr5:137802649:137802848; LARP1 gene: chr5:154134205:154134404; RARS gene: chr 5:167838020:167838129; TTBK1 gene: chr6:43215313:43215512; FAM20C gene: chr7:193762:193961; CREB5 gene: chr7:28449291:28449490; LIMK1 gene: chr7:73508993:73509192; PRKAG2 gene: chr7:151425064:151425263; SLC39a14 gene: chr8:22237164:22237363; EGR3 gene: chr8:22548226:22548425; EGR3 gene: chr8:22548641:22548840; DUSP26 gene: chr 8:3405138:34105337; AGPAT2 gene: chr9:139582105:13958234; NRARP gene: chr9:140205984:140206183; EGR2 gene: chr 10:64578519:6457878; PPIF gene: chr10:81001956:81002155; CHID1 gene: chr11:911539:911738; ADM gene: chr 11:1032996:10329395; NAV2 gene: chr11:19735051:19735250; NAV2 gene: chr11:19735910:19736109; EHBP1L1 gene: chr11:65343637:65343836; PHLDB1 gene: chr11:118479394:118479593; PARP11 gene: chr12, 4140185:4140384; ANO6 gene: chr12:45610581:45610780; PLXNC1 gene: chr12:94544326:94544525; ZNF219 gene: chr 14:21559998:2156097; FOXA1 gene: chr14:380665126:380565325; PAPLN gene: chr14:73704879:73705078; UACA gene: chr15:70767131:70767330; PGPEP1L gene: chr 15:99466492:466691; ITPRIPL2 gene: chr16:19125944:19126143; TNK1 gene: chr17:7287208:7287407; RPL19 gene: chr17:37366283:37366482; ICAM2 gene: chr 17:62076858:62057657; TMC6 gene: chr 17:7613476:7613675; TMC6 gene: chr 17:7623642:7623841; CEP295NL gene: chr 17:768880011:768880210; the BAIAP2 gene: chr17:79061115:79061314; TBCD gene: chr17, 80745041, 80745240; METRNL gene: chr17:81084062:81084261; MED16 gene: chr19:884043:884242; SBNO2 gene: chr19:1177525:1177724; CIRBP gene: chr19:1265940:1266139; KLF16 gene: chr19:1860593:1860792; c19orf77 gene: chr19:34916:3435115; c19orf77 gene: chr19:3435238:3435437; SNAPC2 gene: chr19:7985959:7986158; ICAM1 gene: chr19:10381567:10381766; ICAM5 gene: chr19:10405082:10405281; IER2 gene: chr19:13266897:13267096; ASF1B gene: chr19:14248383:14248582; CRTC1 gene: chr19:18771211:18771410; ZNF536 gene: chr19:31039497:31039696; LTBP4 gene: chr19:41105956:4106155; NOL4L-DT gene: chr20:31162351:31162550; KCNK15 gene: chr20:43374298:43374497; UCKL1 gene: chr20:62588863:6258562; RTN4R gene: chr22:20226623:20226822; RTN4R gene: chr22:20226825:20227024; BCR gene: chr22:23624342:23624541; TEF gene: chr22:41771479:41771678.
7. The use according to any one of claims 1 to 6, wherein the reagent comprises a primer and/or a probe molecule;
preferably, the primer molecule is identical, complementary or hybridizes under stringent conditions to the one or more target markers and comprises at least 9 consecutive nucleotides, and the probe molecule hybridizes under stringent conditions to the amplification product of the one or more target markers.
8. The use according to any one of claims 1 to 6, wherein the reagent is a reagent required to perform a genome-simplified methylation sequencing technique.
9. A diagnostic reagent or diagnostic kit for detecting the methylation state or methylation level of at least one CpG dinucleotide of one or more markers of interest to diagnose a benign or malignant thyroid nodule, comprising a reagent for detecting the methylation state or level of at least one CpG dinucleotide of one or more markers of interest; wherein the one or more markers of interest are as defined in any one of claims 1-6.
10. The diagnostic reagent or diagnostic kit according to claim 9, wherein the diagnostic reagent or diagnostic kit comprises a primer and/or a probe molecule; preferably, the primer molecule is identical, complementary or hybridizes under stringent conditions to the one or more target markers and comprises at least 9 consecutive nucleotides, and the probe molecule hybridizes under stringent conditions to an amplification product of the one or more target markers;
Optionally, the diagnostic reagent or diagnostic kit further comprises a primer molecule and/or a probe molecule for detecting the reference gene ACTB.
11. The diagnostic reagent or diagnostic kit of claim 9, further comprising one or more substances selected from the group consisting of: PCR buffer, polymerase, dNTP, restriction endonuclease, digestion buffer, fluorescent dye, fluorescence quencher, fluorescent reporter, exonuclease, alkaline phosphatase, internal standard, control, KCl, mgCl 2 And (NH) 4 ) 2 SO 4 。
12. The diagnostic reagent or diagnostic kit of claim 9, wherein the reagent further comprises reagents used in one or more of the following methods: bisulfite conversion-based PCR, DNA sequencing, methylation-sensitive restriction enzyme analysis, fluorescent quantitation, methylation-sensitive high resolution melting curve, chip-based methylation profile analysis, and mass spectrometry.
13. The diagnostic reagent or diagnostic kit of claim 12, wherein the reagent is selected from one or more of the following: bisulfite and derivatives thereof, fluorescent dyes, fluorescence quenchers, fluorescence reporters, internal standards and controls.
14. Use of at least one reagent or set of reagents that distinguish between methylated and unmethylated CpG dinucleotides within at least one target region of genomic DNA, wherein the method comprises contacting genomic DNA isolated from a biological sample of the individual with the at least one reagent or set of reagents, wherein the target region is identical or complementary to a sequence of at least 16 consecutive nucleotides of one or more target markers, wherein the consecutive nucleotides comprise at least one CpG dinucleotide sequence, thereby at least partially providing for detection and/or classification of benign and malignant thyroid nodules in an individual, wherein the one or more target markers are as defined in any one of claims 1-6.
15. Use of one or more reagents for converting an unmethylated cytosine base at position 5 to uracil or other bases detectably different from cytosine in terms of hybridization properties, an amplification enzyme, and at least one primer comprising at least 9 consecutive nucleotides, in the preparation of a kit for use in a method of detecting and/or classifying a benign malignancy of a thyroid nodule in an individual, wherein the method comprises:
a) Isolating genomic DNA from the individual biological sample;
b) Treating the genomic DNA of a) or fragment thereof with the one or more reagents;
c) Contacting the treated genomic DNA or a treated fragment thereof with the amplification enzyme and the at least one primer that is identical, complementary or hybridizes under stringent conditions to one or more target markers, wherein the treated genomic DNA or fragment thereof is amplified to produce at least one amplification product or is not amplified; and
d) Determining the methylation status or level of at least one CpG dinucleotide of the one or more markers of interest, or a mean or value reflecting the mean methylation status or level of a plurality of CpG dinucleotides of the one or more markers of interest, based on the presence or the nature of the amplificate, thereby at least partially detecting and/or classifying a benign malignancy of a thyroid nodule in an individual;
wherein the one or more markers of interest are as defined in any one of claims 1-6.
16. The use of claim 15, wherein in step b) the genomic DNA or fragment thereof is treated with an agent selected from the group consisting of bisulfites, acid sulfites, metabisulfites and combinations thereof.
17. The use according to claim 16, wherein in c) the contacting or amplification of the nucleic acid molecule is performed by using a thermostable DNA polymerase as the amplification enzyme, using a polymerase lacking 5'-3' exonuclease activity, using a polymerase chain reaction and/or producing an amplification product with a detectable label.
18. The use of claim 15, wherein the contacting or amplifying in c) comprises the use of methylation specific primers.
19. Use of one or more methylation sensitive restriction and amplification enzymes and at least one primer comprising at least 9 consecutive nucleotides, wherein the primer is identical, complementary or hybridizes under stringent conditions to one or more target markers, for the preparation of a kit for use in a method of detecting and/or classifying a benign and malignant thyroid nodule in an individual; the method comprises the following steps:
a) Isolating genomic DNA from the individual biological sample;
b) Digesting the genomic DNA of a) or a fragment thereof with the one or more methylation sensitive restriction enzymes, and contacting the resulting digestion product with the amplification enzyme and the at least one primer; and
c) Determining the methylation status or level of at least one CpG dinucleotide of the one or more markers of interest based on the presence or the nature of the amplificate, thereby at least partially detecting and/or classifying a benign malignancy of a thyroid nodule in an individual;
Wherein the one or more markers of interest are as defined in any one of claims 1-6.
20. The use of claim 19, wherein the presence or absence of amplification products is determined by hybridization of at least one nucleic acid or peptide nucleic acid that is identical or complementary to at least a 16 base long fragment of a sequence selected from the one or more markers of interest.
21. Use of a treated nucleic acid derived from one or more markers of interest, wherein the treatment is suitable for converting at least one unmethylated cytosine base of the one or more markers of interest to uracil or other bases detectably different from cytosine on hybridization, in the preparation of a kit for diagnosing a benign or malignant thyroid nodule.
22. An apparatus for detecting and diagnosing benign and malignant thyroid nodules in an individual, the apparatus comprising a memory, a processor, and a computer program stored on the memory and executable on the processor, the processor implementing the following steps when executing the program: (1) Obtaining the methylation level or methylation status of at least one CpG dinucleotide of one or more markers of interest in the sample, and (2) interpreting the benign and malignant thyroid nodule according to the methylation level or methylation status of (1);
Wherein the one or more markers of interest are as defined in any one of claims 1-6.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111496935.XA CN116287222A (en) | 2021-12-09 | 2021-12-09 | Methylation marker for diagnosis of benign and malignant thyroid cancer nodules and application thereof |
| PCT/CN2022/137459 WO2023104136A1 (en) | 2021-12-09 | 2022-12-08 | Methylation marker in diagnosis of benign and malignant nodules of thyroid cancer and applications thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111496935.XA CN116287222A (en) | 2021-12-09 | 2021-12-09 | Methylation marker for diagnosis of benign and malignant thyroid cancer nodules and application thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN116287222A true CN116287222A (en) | 2023-06-23 |
Family
ID=86729670
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111496935.XA Pending CN116287222A (en) | 2021-12-09 | 2021-12-09 | Methylation marker for diagnosis of benign and malignant thyroid cancer nodules and application thereof |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN116287222A (en) |
| WO (1) | WO2023104136A1 (en) |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090170087A1 (en) * | 2007-02-02 | 2009-07-02 | Orion Genomics Llc | Gene Methylation in Cervical Cancer Diagnosis |
| WO2016020551A1 (en) * | 2014-08-08 | 2016-02-11 | Ait Austrian Institute Of Technology Gmbh | Thyroid cancer diagnosis by dna methylation analysis |
| CN111197087B (en) * | 2020-01-14 | 2020-11-10 | 中山大学附属第一医院 | Thyroid cancer differential marker |
| CN113122634A (en) * | 2020-01-14 | 2021-07-16 | 上海鹍远生物技术有限公司 | Reagent for detecting DNA methylation and application |
| CN113186278B (en) * | 2021-07-01 | 2021-10-12 | 上海鹍远生物技术有限公司 | Thyroid nodule benign and malignant related marker and application thereof |
-
2021
- 2021-12-09 CN CN202111496935.XA patent/CN116287222A/en active Pending
-
2022
- 2022-12-08 WO PCT/CN2022/137459 patent/WO2023104136A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023104136A1 (en) | 2023-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113186278B (en) | Thyroid nodule benign and malignant related marker and application thereof | |
| CN119421958A (en) | Identification of methylation markers for cancer and their applications | |
| JP7801235B2 (en) | Methods and kits for screening for colorectal neoplasia | |
| AU2020445677B2 (en) | Tumor detection reagent and kit | |
| JP2022552400A (en) | COMPOSITION FOR DIAGNOSING LIVER CANCER USING CPG METHYLATION CHANGE IN SPECIFIC GENE AND USE THEREOF | |
| CN116219020B (en) | Methylation reference gene and application thereof | |
| CN118086498A (en) | Methods used to diagnose lung cancer | |
| CN113122637B (en) | Reagent for detecting DNA methylation and application thereof | |
| CN111197087A (en) | Thyroid Cancer Differential Markers | |
| WO2022170984A1 (en) | Screening, risk assessment, and prognosis method and kit for advanced colorectal adenomas | |
| US20250002987A1 (en) | Detection of methylation status of a dna sample | |
| CN113493835A (en) | Method and kit for screening large intestine tumor by detecting methylation state of BCAN gene region | |
| CN117778568A (en) | Marker for identifying gastric cancer and application thereof | |
| CN111100866A (en) | Gene segment for identifying benign and malignant thyroid nodules and application thereof | |
| CN115094139B (en) | Application of reagent for detecting methylation level in preparation of bladder cancer diagnosis product and bladder cancer diagnosis kit | |
| CN116287222A (en) | Methylation marker for diagnosis of benign and malignant thyroid cancer nodules and application thereof | |
| WO2025002157A1 (en) | Marker for detecting esophageal cancer and detection method | |
| CN116064820B (en) | Biomarkers, kits and methods of use for detecting early liver cancer | |
| CN117660622B (en) | Methylation molecular markers for detecting pulmonary nodules and their applications | |
| WO2023274350A1 (en) | Benign and malignant thyroid nodule related marker and use thereof | |
| US20100021902A1 (en) | Method for methylation-selective amplification | |
| CN120648795A (en) | Composition for detecting ovarian lesions and application thereof | |
| HK40071692A (en) | Methods and kits for screening colorectal neoplasm | |
| CN115961045A (en) | Tumor marker for judging early hepatocellular carcinoma, kit and use method thereof | |
| US20250066836A1 (en) | Methods for evaluating the methylation status of a polynucleotide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |