CN115925969A - An adenine base editor capable of precise deletion of small DNA fragments and its construction and application - Google Patents
An adenine base editor capable of precise deletion of small DNA fragments and its construction and application Download PDFInfo
- Publication number
- CN115925969A CN115925969A CN202210810655.XA CN202210810655A CN115925969A CN 115925969 A CN115925969 A CN 115925969A CN 202210810655 A CN202210810655 A CN 202210810655A CN 115925969 A CN115925969 A CN 115925969A
- Authority
- CN
- China
- Prior art keywords
- endov
- adenine
- abe8e
- deletion
- base
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images





Landscapes
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
The invention discloses an adenine base editor capable of realizing accurate deletion of small DNA fragments and construction and application thereof. The adenine single-base editor comprises a fusion protein ABE8e-EndoV, wherein the fusion protein sequentially comprises adenine deaminase, a nickase variant of Cas nuclease and inosine I base deletion repair endonuclease from an N end to a C end. The editor for accurately deleting the adenine base of the small DNA segment has the characteristics of efficient A-G base replacement and capability of generating small segment deletion of 9-13bp from a deaminated A base to a Cas nuclease protein cutting site interval. The adenine base editor is particularly suitable for the excavation and research of cis-acting elements in animal and plant genomes, and has wide application prospects in the aspects of single base substitution, saturation mutation screening and the like of target genes.
Description
Technical Field
The invention belongs to the field of biotechnology. More particularly, the invention relates to an adenine single base editor pYL-ABE8e-EndoV which can realize the efficient replacement of A-G bases in a target spot and can realize the accurate deletion of a small DNA fragment of 9-13bp in the target spot, and the construction and the application thereof.
Background
Spatio-temporal specific expression of eukaryotic genes is often regulated by careful interaction of more complex cis-acting elements, typically found in the promoter region, 5'UTR and 3' UTR regions, miRNA coding sequences and intron regions of genes of interest, and trans-acting factors. The TATA box is mainly studied in the promoter region, is usually positioned in a region from-25 to-30 upstream of a transcription starting point, controls the accuracy and frequency of transcription, and also comprises a GC box, a CAAT box and the like. Many studies have also found that at the 5' and 3' ends of introns, a branch point is present, which is an adenylate base, typically 20-30bp upstream of the 3' splice junction, and that deletion or mutation of this base often results in reduced base efficiency or in the inability of the precursor RNA to splice (Simpson et al, 1996). These cis-acting elements are usually composed of AT-rich base sequences, and it is therefore of great importance to develop a precise and predictable set of research tools suitable for AT-rich cis-acting elements.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated proteins (CRISPR/Cas) are acquired immune systems formed in evolutionary processes of bacteria and archaea, and can rapidly and accurately identify and degrade invasion of exogenous DNA. According to the diversity of the number and functions of Cas proteins in a CRISPR/Cas system, the CRISPR/Cas system can be divided into 2 major classes (Class 1 and Class 2) comprising 5 minor classes (Type I-Type V) (Makarova et al, 2017), and Class 1 comprises three types of Type I, type III and Type IV, which need a plurality of Cas proteins to participate in forming a complex simultaneously to play a role, so that great difficulty exists in designing and constructing a carrier; class 2 contains two types, type ii and Type v, which require only one Cas protein and guide RNA (gRNA) to form a complex to effect cleavage of a target site in a genome, and thus CRISPR/Cas systems from different families of bacteria and archaea have been explored and used (Mohanraju et al, 2016). Type II is represented by the Cas9 (Streptococcus pyogenes Cas9, spCas 9) system from Streptococcus pyogenes (Mali et al, 2013), and Type V is mainly represented by the CRISPR/Cas12a (also known as Cpf 1) system from Francisella noviviida (Zetsche et al, 2015).
Although CRISPR/Cas 9-mediated genome editing can cause double-strand break (DSB) at a target site to efficiently knock out a target gene by means of non-homologous end repair (NHEJ), CRISPR/Cas 9-mediated genome editing mainly causes random deletion of 1-3bp or sometimes large fragment deletion, and accurate study on the function of a target element is difficult. Furthermore, the CRISPR/Cas12a, although it may result in deletion of larger fragments, the deleted fragments are also random. In addition, although the high Cabernet task group is based on cytosine deamination and Base Excision Repair (BER) principles, combining wild-type SpCas9 with cytosine deaminase APOBEC, uracil glycosylase (UDG), and an apurinic pyrimidine site lyase (AP lyase) creates a set of predictable polynucleotide targeted Deletion Systems (AFIDs). The cytosine deaminase APOBEC used in the AFIDs system has great advantages on CG-rich sequences, but has great limitation on AT-rich cis-acting elements, and the system uses SpCas9 for recognizing NGG-PAM, thereby limiting the application range of the AFIDs.
In the Adenine Base Editor (ABE), the A base is deaminated to inosine I, which at the DNA level is recognized as a G base, and base substitutions of A-to-G are achieved through DNA repair and replication. Since the enzymatic activity of inosine base deletion repair is not high in eukaryotic cells, the substitution of the ABE base editing system A-to-G contained almost no byproducts of A-Y or indels. However, it has been found that mismatches in I: T trigger the AER repair pathway due to the deamination of a bases to I bases as a result of natural hydrolytic and nitrosative stress on cells following exposure to endogenous and/or exogenous agents (Kuraoka et al, 2015). In the AER pathway, endonuclease V (EndoV) hydrolyzes the second phosphodiester bond located 3' to deoxyinosine in the DNA strand. In the E.coli model, an EndoV-dependent AER pathway for the removal of deoxyinosine from DNA has been identified. However, the AER pathway in mammalian cells has not been elucidated. According to biochemical studies of mammalian endo V, it has a considerably high deoxyinosine 3' endonuclease activity in single-stranded DNA containing deoxyinosine, and then its activity in double-stranded DNA is very low. To date, AER pathways based on I: T mismatches in the ABE system are still poorly studied.
In summary, the development of adenine base editors suitable for cis-acting elements, miRNA coding sequences, conserved elements of intron regions, wide targeting, and precise deletion of small fragments of new DNA that can adapt to the high AT polynucleotide sequence environment is an important direction for gene editing technology optimization.
Disclosure of Invention
The invention aims to solve the technical problem of overcoming the defect that small fragment random deletion of 1-3bp is mainly caused by CRISPR/Cas9 system-mediated gene editing in the prior art, and limiting the accurate research on the functions of a cis-acting element, an miRNA coding sequence and a conservative element of an intron region, so that an adenine single base editor capable of accurately realizing DNA deletion of about 10bp needs to be invented.
The first purpose of the invention is to provide an adenine base editor fusion protein ABE8e-EndoV which can realize accurate deletion of small DNA fragments.
The second purpose of the invention is to provide a nucleotide sequence for encoding the fusion protein ABE8e-EndoV.
The third object of the present invention is to provide a plasmid vector containing the nucleotide sequence.
The fourth purpose of the invention is to provide the fusion protein ABE8e-EndoV or the nucleotide sequence or the application of the plasmid vector in preparing an adenine base editor.
The fifth object of the present invention is to provide an adenine base editor that can achieve accurate deletion of a small DNA fragment.
The sixth object of the present invention is to provide a method for constructing the adenine base editor capable of deleting a small DNA fragment accurately.
The seventh object of the present invention is to provide the use of the adenine base editor capable of realizing precise deletion of a small DNA fragment in gene editing in an organism.
The above object of the present invention is achieved by the following technical solutions:
an adenine base editor fusion protein ABE8e-EndoV capable of realizing accurate deletion of small DNA fragments sequentially comprises adenine deaminase, nickase variant protein of Cas nuclease and inosine I base deletion repair endonuclease from an N end to a C end;
in the fusion protein ABE8e-EndoV, adenine deaminase is used for base editing of A-to-G, a nickase variant protein of Cas nuclease is used for cutting a target sequence, and an inosine I base deletion repair endonuclease plays a key role in an inosine selective excision repair (AER) pathway. When the fusion protein ABE8e-EndoV is used for gene editing, based on the base mismatch repair (AER) pathway of I: T in ABE8e-EndoV, firstly, A is deaminated and hydrolyzed by adenine deaminase to form inosine I, so that the base mismatch of I: T in a DNA double strand is caused, a second phosphodiester bond at the 3' end of the inosine is hydrolyzed by an inosine I base deletion repair endonuclease, and the base substitution of A-to-G is realized through the repair and the replication of DNA; in addition, the mutation of A-to-Deletions is realized through non-homologous end connection repair, and 9-13bp small fragments with 5' -deaminated A bases to endonuclease cleavage sites are generated for accurate deletion. In theory, all adenine deaminases or mutants thereof, nickase variant proteins of Cas nuclease variants and endonucleases with inosine I base deletion repair can achieve the objectives of the present invention and are within the scope of the present invention.
Preferably, the adenine deaminase is an adenine deaminase mutant.
Further preferably, the adenine deaminase mutant is TadA8e, and the amino acid sequence thereof is shown in SEQ ID No. 1. The adenine deaminase TadA8e has high activity and high base editing efficiency of A-to-G.
Preferably, the nickase variant of a Cas nuclease is a nickase variant of a Cas9 protein.
Further preferably, the nicking enzyme variant of the Cas9 protein is a SpCas9 nicking enzyme variant SpGn, and the amino acid sequence of the nicking enzyme variant is shown in SEQ ID NO.2, so that the compact NG-PAM can be recognized.
Preferably, the inosine I base deletion repair endonuclease is EndoV, and the amino acid sequence of the EndoV base deletion repair endonuclease is shown in SEQ ID NO.3, or the amino acid sequence with 80% of sequence homology with the sequence shown in SEQ ID NO. 3.
Preferably, the kit further comprises two nuclear localization signals fused to the N-terminal and the C-terminal of the ABE8e-EndoV, respectively.
Further preferably, the amino acid sequence of the nuclear localization signal is shown as SEQ ID NO. 4.
Preferably, the polypeptide also comprises a protein linker sequence for respectively connecting adenine deaminase TadA8e and a SpCas9 variant SpGn, and connecting the SpCas9 variant SpGn and an inosine I base deletion repair key enzyme EndoV; preferably, the protein linker sequence is a flexible linker sequence.
Preferably, the adenine base editor fusion protein ABE8e-EndoV capable of realizing precise deletion of small DNA fragments sequentially comprises a nuclear localization signal bpNLS, adenine deaminase TadA8e, a flexible connecting sequence, a SpCas9 variant SpGn, a flexible connecting sequence, an inosine I base deletion repair key enzyme EndoV and a nuclear localization signal bpNLS from the N end to the C end, and the whole amino acid sequence of the fusion protein is shown as SEQ ID No. 5.
The invention also provides a nucleotide for coding any one of the adenine base editor fusion protein ABE8e-EndoV capable of realizing accurate deletion of the small DNA fragments.
Preferably, the nucleotide sequence is shown as SEQ ID NO. 6.
The invention also provides a plasmid vector containing any one of the nucleotide sequences.
The invention also provides an application of any one of the adenine base editor fusion protein ABE8e-EndoV capable of realizing accurate deletion of the small DNA fragment or any one of the nucleotides or the plasmid vector in preparation of the DNA small fragment accurate deletion adenine base editor.
The invention also provides an adenine base editor capable of realizing the precise deletion of the small DNA fragments, which comprises a nucleotide sequence for coding any one of the DNA small fragment precise deletion adenine base editor fusion protein ABE8e-EndoV, and is obtained by building the nucleotide sequence for coding any one of the DNA small fragment precise deletion adenine base editor fusion protein ABE8e-EndoV on a transformation carrier.
Preferably, the transformation vector is a plant transformation vector; including but not limited to pCAMBIA1300 and a vector modified on the basis of the pCAMBIA1300, such as pYRCISPR/Cas 9Pubi-H.
More preferably, the plant transformation vector is a binary vector pYLRISPR/Cas 9Pubi-H.
The invention also provides a construction method of the DNA small segment precise deletion adenine base editor, which comprises the steps of firstly preparing a complete fusion DNA sequence of the encoded DNA small segment precise deletion adenine base editor fusion protein ABE8e-EndoV, then inserting the complete fusion DNA sequence into an expression vector, transforming host bacteria, extracting positive plasmids, sequencing and obtaining the stable DNA small segment precise deletion adenine base editor;
preferably, the adenine base editor pYL-ABE8e-EndoV is obtained for insertion between Pst I and BamH I of the binary vector pYRCISPR/Cas 9Pubi-H.
As a preferable embodiment, the method for constructing the adenine base editor capable of realizing the precise deletion of the small DNA fragment comprises the following steps:
s1, respectively synthesizing a gene fragment 1 for coding bpNLS-TadA8e-linker 1 and a gene fragment 2 for coding linker2-EndoV-bpNLS, adding one enzyme cutting site at the 5 'end of the gene fragment 1 through PCR reaction, and adding the other enzyme cutting site at the 3' end of the gene fragment 2; synthesizing a gene fragment 3 encoding SpGn;
s2, connecting the C end of the gene fragment 1 with the enzyme cutting site with the N end of the gene fragment 3 by an overlapped PCR technology to obtain a gene fragment 4 of a fusion protein bpNLS-TadA8e-linker 1-SpGn; then connecting the C end of the gene fragment 4 with the N end of the gene fragment 2 with the enzyme cutting site to obtain a gene fragment 5 of a fusion protein bpNLS-TadA8e-linker 1-spGn-linker 2-EndoV-bpNLS;
s3, inserting the gene fragment 5 between two corresponding enzyme cutting sites of a plant vector pYLCRISPR/Cas9Pubi-H, transforming host bacteria, extracting positive plasmids, sequencing, and obtaining the stable and efficient ABE base editor for accurately deleting small DNA fragments.
Preferably, the insertion of the gene fragment 5 into the vector in step S3 is achieved by Gibson assembly technology.
Preferably, the host bacterium described in step S3 is Escherichia coli Top10F'.
The invention also provides application of the DNA small fragment precise adenine base deletion editor in genome editing in organisms. The adenine base editor for precisely deleting the small DNA fragment is particularly suitable for functional research of cis-acting elements, miRNA and key elements of intron splicing, and can simultaneously compare functional differences caused by target base substitution and fragment deletion. Therefore, the invention also provides the mining and research of the cis-acting element in the genome of the adenine base editor animal and plant and the application of the accurate deletion of small segments of organism genome DNA.
Preferably, the specific operations for mining and studying cis-acting elements in the genome are as follows:
(1) Determining a cis-acting element to be researched, and designing and synthesizing an sgRNA expression cassette element according to a target site;
(2) Integrating the sgRNA expression cassette element to an adenine base editor for accurately deleting the small DNA fragment to obtain a vector which can realize accurate deletion of the small DNA fragment and can realize adenine base replacement in the small DNA fragment;
(3) And transforming the adenine base editing vector for accurately deleting the small segment of the target gene DNA into host cells, and screening to obtain cells for accurately deleting the corresponding small segment of the target DNA or replacing adenine bases in the small segment of the target DNA.
Compared with the prior art, the invention has the following beneficial effects:
(1) When the fusion protein ABE8e-EndoV is used for gene editing, based on the base mismatch repair (AER) pathway of I: T in ABE8e-EndoV, A is deaminated and hydrolyzed into inosine I by adenine deaminase to cause the base mismatch of I: T in a DNA double strand, a second phosphodiester bond at the 3' end of the inosine is hydrolyzed by an inosine I base deletion repair endonuclease, and the base substitution of A-to-G is realized through the repair and the replication of DNA; in addition, the mutation of A-to-Deletions is realized through non-homologous end connection repair, and 9-13bp small fragments with 5' -deaminated A bases to endonuclease cleavage sites are generated for accurate deletion. The editor for accurately deleting the adenine base in the small DNA segment is particularly suitable for functional research of cis-acting elements, miRNA, intron splicing and other key elements, and is also suitable for substitution of key amino acids of target genes, large-scale saturation mutation and the like.
(2) The DNA small fragment accurate deletion adenine base editor pYL-ABE8e-EndoV tool constructed based on the DNA small fragment accurate deletion adenine base editor fusion protein ABE8e-EndoV can identify concise NG-PAM, has high A-to-G replacement efficiency, and has a base replacement activity window mainly concentrated between A3 and A8. In addition, the target site also has efficient DNA small fragment accurate deletion activity, the DNA small fragment is deleted from deaminated A base to 9-13bp of a Cas nuclease cutting site, and compared with other existing editing systems, the ABE8e-EndoV editing is more advantageous.
Drawings
FIG. 1 is a schematic diagram of the structure of the DNA small fragment precise deletion adenine base editor pYL-ABE8e-EndoV vector of the present invention. The figure shows pCAMBIA1300 carrier skeleton; the polynucleotide sequence of the ABE8e-EndoV fusion protein is expressed by a Pubi constitutive promoter; tadA8e, an adenine deaminase, having a base editing efficiency of high efficiency a-to-G; spGn, spCas9 variant, recognizing NG-PAM; endoV, endonuclease V, plays a key role in the inosine selective excision repair (AER) pathway.
FIG. 2 is a graph showing the editing efficiency and editing window results for pYL-ABE8e-EndoV. Wherein, (a) the editing efficiency of ABE8e-EndoV in 5 targets, and the total editing efficiency is that the number of mutant plants at the target is larger than that of all T0 generation transformants; the editing efficiency of the A-to-G is that the number of the plants subjected to A-to-G base replacement at the target point is larger than that of all transformants in the T0 generation; the Indels editing efficiency is for plants with small DNA fragment deletion at the target site of interest (including plants with deletion mutation and base substitution), and PAM in the target site is highlighted by underlining and bolding. (b) A-to-G average edit window and average edit efficiency for ABE8e-EndoV.
FIG. 3 shows that ABE8e-EndoV exhibits highly efficient predictable DNA fragment deletion. (a) ABE8e-EndoV produces precise deletions from the target 5' -deaminated a base to the SpGn nick site and efficiencies, PAM and precise editing efficiencies highlighted in light black, black bars representing the missing nucleotide fragments, spGn nick site, (b) Sanger sequencing chromatograms of some typical mutation types of ABE8e-EndoV. Het, heterozygosity mutation; del, deletion mutation.
FIG. 4 is an off-target analysis of ABE8e-EndoV. Candidate off-target sites are analyzed and selected for candidate off-target sites and targeting sequences with 1 to 3 base variations by the CRISPR-GE off-target subroutine, PAM of the target is highlighted by underlining and bolding, and base differences between candidate off-target sites and target are highlighted by bolding. The analysis result shows that the ABE8e-EndoV has lower off-target efficiency.
FIG. 5 is a schematic diagram of ABE8e-EndoV mediated repair pathway for precise adenine base deletion in small DNA fragments. In the ABE8e-EndoV system, the base mismatch repair pathway of I: T is that A is firstly deaminated and hydrolyzed into inosine I by adenine deaminase TadA8e, so that the base mismatch of I: T in a DNA double strand is caused, a second phosphodiester bond of inosine 3' is hydrolyzed by EndoV, and the mutation of A-to-deletion and the base substitution of A-to-G are realized through the repair and replication of DNA.
Detailed Description
The invention is further described with reference to the drawings and specific examples, which are not intended to limit the invention in any way. The reagents, methods and apparatus employed in the present invention are conventional in the art, except as otherwise indicated.
Unless otherwise indicated, reagents and materials used in the following examples are commercially available.
Example 1 construction of DNA Small fragment accurate deletion adenine base editor pYL-ABE8e-EndoV
The protein sequences and polynucleotide sequences of the N-terminal nuclear localization signals bpNLS1, deaminase TadA8e, 32 amino acid linker1 and the variant SpGn of SpCas9 used in the present invention have been reported and published earlier in the subject (Tan et al, 2020, plant Biotechnology journal,201 to 1584aa of SEQ ID NO.5) The polynucleotide sequence of bpNLS1-TadA8e-linker-SpGn1 is shown as ( Bases 4 to 4752 of SEQ ID NO.6) As shown. In addition, the main function of EndoV, a key enzyme for inosine deletion repair, was mainly referred to (Kuraoka et al, 2015, biomodules, 5SEQ ID NO.5 Amino acids 1585 to 1860Polynucleotides of linker2-EndoV-bpNLS2 such asNo.6 of SEQ ID NO.6 Base numbers 4753 to 5583). The optimally synthesized linker2-EndoV-bpNLS2 is respectively connected to the C end of bpNLS1-TadA8e-linker1-SpGn through Overlaping PCR to form completely fused bpNLS1-TadA8e-linker1-SpGn-linker2-EndoV-bpNLS2, and the completely fused bpNLS1-TadA8e-linker1-SpGn-linker2-EndoV-bpNLS2 is cloned between Pst I and BamH I of binary vector pYLCRISPR/Cas9Pubi-H (Ma et al, 2015, molecular plant, 8. As a control for comparison of editing efficiency, the ABE8e-SpG adenine base editor (Tan et al, 2020, plant Biotechnology journal, 20.
Amino acid sequence of adenine deaminase TadA8 e: (SEQ ID NO. 1)
SEVEFSHEYWMRHALTLAKRARDEREVPVGAVLVLNNRVIGEGWNRAIGLHDPTAHAEIMALRQGGLVMQNYRLIDATLYVTFEPCVMCAGAMIHSRIGRVVFGVRNSKRGAAGSLMNVLNYPGMNHRVEITEGILADECAALLCDFYRMPRQVFNAQKKAQSSIN
Amino acid sequence of SpCas9 variant SpGn: (SEQ ID NO. 2)
DKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKBPNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFLWPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAKQLQKGNELALPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENIIHLFTLTNLGAPAAFKYFDTTIDRKQYRSTKEVLDATLIHQSITGLYETRIDLSQLGGD
Amino acid sequence of inosine base deletion repair key enzyme EndoV (SEQ ID NO. 3)
DLASLRAQQIELASSVIREDRLDKDPPDLIAGADVGFEQGGEVTRAAMVLLKYPSLELVEYKVARIATTMPYIPGFLSFREYPALLAAWEMLSQKPDLVFVDGHGISHPRRLGVASHFGLLVDVPTIGVAKKRLCGKFEPLSSEPGALAPLMDKGEQLAWVWRSKARCNPLFIATGHRVSVDSALAWVQRCMKGYRLPEPTRWADAVASERPAFVRYTANQP
Amino acid sequence of nuclear localization signal (SEQ ID NO. 4)
KRTADGSEFESPKKKRKV
Amino acid complete sequence of fusion protein (SEQ ID NO. 5)
MKRTADGSEFESPKKKRKVSEVEFSHEYWMRHALTLAKRARDEREVPVGAVLVLNNRVIGEGWNRAIGLHDPTAHAEIMALRQGGLVMQNYRLIDATLYVTFEPCVMCAGAMIHSRIGRVVFGVRNSKRGAAGSLMNVLNYPGMNHRVEITEGILADECAALLCDFYRMPRQVFNAQKKAQSSINSGGSSGGSSGSETPGTSESATPESSGGSSGGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKBPNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVLTLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFLWPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAKQLQKGNELALPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENIIHLFTLTNLGAPAAFKYFDTTIDRKQYRSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSSGGSSGSETPGTSESATPESSGGSSGGSDLASLRAQQIELASSVIREDRLDKDPPDLIAGADVGFEQGGEVTRAAMVLLKYPSLELVEYKVARIATTMPYIPGFLSFREYPALLAAWEMLSQKPDLVFVDGHGISHPRRLGVASHFGLLVDVPTIGVAKKRLCGKFEPLSSEPGALAPLMDKGEQLAWVWRSKARCNPLFIATGHRVSVDSALAWVQRCMKGYRLPEPTRWADAVASERPAFVRYTANQPSGGSKRTADGSEFESPKKKRKV*
Nucleotide complete sequence encoding fusion protein (SEQ ID NO. 6)



The specific procedure is as follows:
the primers used in the construction are shown in table 1:
TABLE 1 primers for basic vector engineering for pYL-ABE8e-EndoV

(1) A fragment with a PstI enzyme cutting site is obtained by amplification by using F-ABE8e-EndoV-1/R-ABE8e-EndoV-1 (SEQ ID NO.7 and SEQ ID NO. 8) primers and ABE8e-SpG which is developed in the earlier stage of the laboratory as a template, and is named as a fragment D1.
PCR System (15. Mu.l): 2 × Phanta Max Buffer 7.5 μ L,10mmol/L dNTPs Mix 0.35 μ L, phanta Max Polymerase 0.35 μ L, ABE8e-SpG 10ng,10 μmol/L F-ABE8e-EndoV-10.35 μ L,10 μmol/L R-ABE8 e-EndoV-1.35 μ L, ddH 2 O make up to 15. Mu.l.
PCR procedure: pre-denaturation at 95 ℃ for 2min,28 PCR cycles (95 ℃ for 10s,56 ℃ for 15s,72 ℃ for 4 min), and extension at 72 ℃ for 5min.
(2) Using F-ABE8e-EndoV-2/R-ABE8e-EndoV-2 (SEQ ID NO.9 and SEQ ID NO. 10) primers and chemically synthesized linker2-EndoV-bpNLS2 target plasmids as templates, and amplifying to obtain a linker2-EndoV-bpNLS2 fragment named as fragment D2.
PCR System (15. Mu.l): 2X Phanta Max Buffer 7.5. Mu.l, 10mmol/L dNTPs Mix 0.35. Mu.l, phanta Max Polymerase 0.35. Mu.l, linker2-EndoV-bpNLS2 10ng,10μmol/L F-ABE8e-EndoV-2 0.35μl,10μmol/L R-ABE8e-EndoV-2 0.35μl,ddH 2 O make up to 15. Mu.l.
PCR procedure: pre-denaturation at 95 ℃ for 2min,28 PCR cycles (95 ℃ for 10s,56 ℃ for 15s,72 ℃ for 30 s), and extension at 72 ℃ for 5min.
(3) A fusion fragment of bpNLS1-TadA8e-linker1-SpGn-linker2-EndoV-bpNLS2 (TadA 8e-SpGn-EndoV fusion fragment for short) was amplified using F-ABE8e-EndoV-1/R-ABE8e-EndoV-2 (SEQ ID NO.7 and SEQ ID NO. 10) primers and the fragment D1 and the fragment D2 amplified in the first round as templates.
PCR System (50. Mu.l): 2 x Phanta Max Buffer 25. Mu.l, 10mmol/L dNTPs Mix 1.0. Mu.l, phanta Max Polymerase 1.0. Mu.l, first round amplified bpNLS1-TadA8e-linker1-SpGn fragment D1 and first round amplified linker2-EndoV-bpNLS2 fragment D2 0.5. Mu.l, 10. Mu.mol/L F-ABE8 e-EndoV-1.0. Mu.l, 10. Mu.mol/L R-ABE8 e-EndoV-2.0. Mu.l, ddH 2 O make up to 50. Mu.l.
PCR procedure: pre-denaturation at 95 ℃ for 2min,28 PCR cycles (95 ℃ for 10s,56 ℃ for 15s,72 ℃ for 4.5 min), and extension at 72 ℃ for 10min.
(4) And purifying the PCR product of the amplified TadA8e-SpGn-EndoV fusion DNA fragment by using a Genstar purification kit. pYRCISPR/Cas 9Pubi-H (Ma et al, 2015, molecular plant,8: 10 XFaster digest buffer, pst I0.5. Mu.l, bamH I0.5. Mu.l, pYRCISPR/Cas 9 Pubi-H300ng, ddH 2 O to 10. Mu.l, reaction at 37 ℃ for 1h, gel recovery of the support backbone for Gibson assembly (NEB # E5510S): 2 xMix 5. Mu.l, eCBE-Cas9n-NG-2 xUGI fusion fragment 60NG, gel recovery vector backbone 90ng, ddH 2 Make up to 10. Mu.l of O, and react at 50 ℃ for 50min. 1.5. Mu.l of the ligation product of Gibson was taken, E.coli Top10F' was electrically transformed, and transformed single clones were selected on kanamycin-resistant (Kana) LB plates. And the positive clones were sequenced to obtain pYL-ABE8e-EndoV basic vector plasmid (FIG. 1).
Example 2pYL-ABE8e-EndoV has both functions of precise deletion of DNA small fragment and adenine base substitution
Referring to earlier published literature by the present inventors (Ma et al, 2015, molecular plant, 8-1284, ma and Liu,2016, current Protocols in Molecular biology,115, 31.6.1-31.6.21; zaozhang et al, 2018, china science: life sciences, 48. The specific operation is as follows:
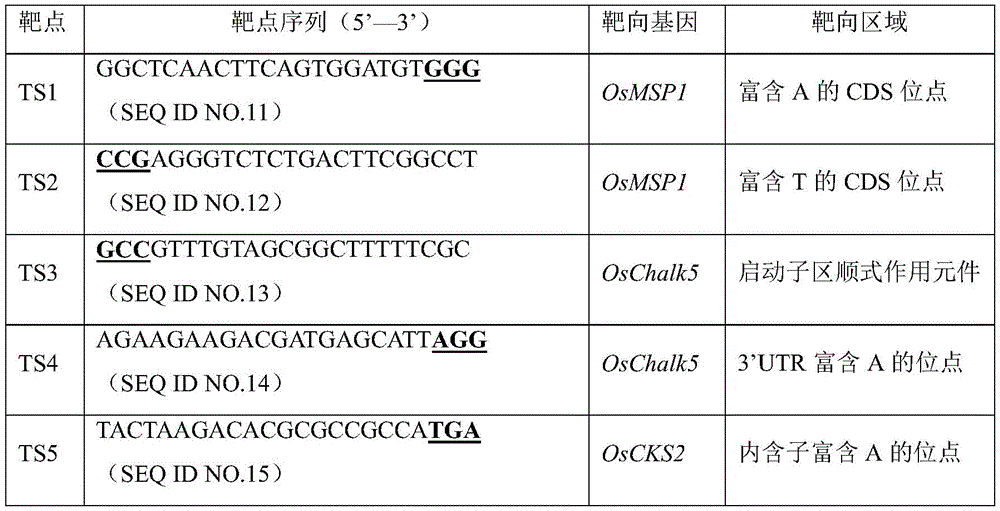
1.TS1 to TS5 target design
OsMSP1 gene (Os 01g 0917500), osChalk5 gene (Os 05g 0156900) and OsCKS2 gene (Os 01g 0197700) are selected from rice genome as target genes. 2 targets (SEQ ID NO.11 and 12) are respectively designed in a CDS region of OsMSP1, 1 target (SEQ ID NO.13 and 14) is respectively designed in a promoter region and a 3' UTR of OsChalk5, and 1 target (SEQ ID NO. 15) is designed in an intron region of OsCKS 2. The 5 target targets are all sequences rich in A or T bases and are used for testing the editing performance of pYL-ABE8e-EndoV.
TABLE 25 targets for testing pYL-ABE8e-EndoV editing Performance
2.design of sgRNA expression cassette primers from TS1 to TS5
The CRISPR-GE web page (http:// skl. Scau. Edu. Cn /) (Xie et al, 2018, molecular plant, 11.
Overlapping PCR splicing of sgRNA expression cassettes from T1 to T5
Two rounds of PCR were performed to obtain sgRNA expression cassettes driven by a small RNA promoter flanked by Bsa I cleavage sites according to our earlier published literature (Ma et al, 2015, molecular plant, 8.
TABLE 3 first round PCR target primer gR-T # and U # -T # sequences


In the first round of PCR, a designed U # -T #/gR-T # primer (SEQ ID NO. 16-SEQ ID NO.25, table 3) is used for introducing a target sequence into the downstream of an OsU6/OsU3 promoter and the upstream of an sgRNA sequence. In a PCR system, a U-F primer (SEQ ID NO.26, table 3) is paired with a gR-T # primer, and a promoter sequence containing a target spot is obtained through PCR amplification; and pairing a gR-R primer (SEQ ID NO.27, table 3) with a U # -T # primer, and carrying out PCR amplification to obtain a sgRNA sequence containing a target spot. PCR System (20. Mu.l): 2 × Phanta Max Buffer 10.0 μ L,10mmol/L dNTPs Mix 0.4 μ L, phanta Max Polymerase 0.3 μ L, pYLgRNA-OsU6/3 (containing promoter and sgRNA plasmid) (Ma et al 2015, molecular plant, 8. PCR procedure: pre-denaturation at 95 ℃ for 1min,28 PCR cycles (95 ℃ for 10s,58 ℃ for 15s,72 ℃ for 20 s), and extension at 72 ℃ for 1min.
Table 4 construction of universal primers for multiple sgRNA expression cassettes


Note 1: bsa I cleavage ends were designed as non-palindromic sequences, which resulted in efficient ligation (Golden Gate ligation).
Note 2: if more than 8 sgRNA expression cassettes are ligated, it is necessary to design by itself more sets of Pgs and Pps primers, each set containing complementary nonreciprocal Bsa I cleavage ends.
Table 5 second round PCR primer combinations to assemble different numbers of sgRNA expression cassettes

Second round PCR, splicing small RNA promoter driven sgRNA expression cassettes by using second round PCR primers Pps and Pgs (SEQ ID NO. 28-SEQ ID NO.42, table 4 and Table 5), and adding Bsa I enzyme cutting sites on both sides of PCR products. And constructing sgRNA expression cassette vectors of T1-T5. PCR System (50. Mu.l): 2 x Phanta Max Buffer 25.0. Mu.l, 10mmol/L dNTPs Mix 1.0. Mu.l, phanta Max Polymerase 1.0. Mu.l, 10 x Dilute the previous round of PCR product 1.0. Mu.l, primers for 5 targets respectively, 10. Mu. Mol/L Pps-L/Pgs-2 (T1), pps-2/Pgs-3 (T2), pps-3/Pgs-4 (T3), pps-4/Pgs-5 (T4), pps-5/Pgs-R (T5) each 1.0. Mu.l, ddH 2 O to 50. Mu.l, and the PCR procedure was the same as the first round of PCR described above. The second round PCR product was purified using the gentar purification kit.
4. Construction of DNA small fragment deletion adenine base editor vector containing sgRNA expression cassettes of different targets
A 'gold gate' cloning method based on Bsa I enzyme digestion and connection is used, and TS1-TS5 are assembled respectively in a 'side-cutting and side-connecting' mode (Ma and Liu,2016, current Protocols in Molecular biology,115, 31.6.1-31.6.21; zangchang et al, 2018, china science: life sciences, 48, 783-794); two groups of sgRNA expression cassettes driven by small nuclear RNA promoters are respectively cloned to a binary vector pYL-ABE8e-EndoV. 15 μ l reaction: 10 XCutSmart Buffer 1.5. Mu.l, 10mmol/L ATP 1.5. Mu.l, pYL-ABE8e-EndoV plasmid 80-100 ng, purified sgRNA expression cassette 10-15ng, bsa I-HF 10U, T4 DNA ligase 35U, ddH 2 O make up to 15. Mu.l. Carrying out enzyme digestion and ligation reaction by using a PCR instrument in a variable temperature cycle manner: 10min at 37 ℃ followed by 10-12 cycles (5 min at 37 ℃, 3min at 10 ℃, 5min at 20 ℃); finally 3min at 37 ℃. After dialysis of the ligation product, electro-stimulation is converted intoDH10B cells were selected on Carla-resistant (Kan) LB plates, and with the primer pair SP-L1/SP-R (SEQ ID NO.53 and SEQ ID NO.54, table 3), colony PCR was performed according to literature (Ma and Liu,2016, current Protocols in Molecular biology,115, 31.6.1-31.6.21; zengmarchand et al, 2018, china science: life sciences, 48 783-794), screening positive clones, and finally determined by sequencing with the primer SP-L1.
pYL-ABE8e-EndoV has higher editing efficiency
The agrobacterium-mediated rice (japonica rice middle flower 11) callus transformation is utilized, the 5-target vector containing sgRNA driven by different small RNA promoters of TS1-TS5 is transformed into rice callus, and the T0 generation rice plant is subjected to genetic transformation process of plants or T0 generation rice plant 0 Leaf DNA of the transformed plants was used as a template, DNA fragments of target editing target sites were amplified using amplification (amp) and Sequencing (SEQ) primers (SEQ ID No.45 to SEQ ID No.59, table 6), sanger sequencing was performed directly, and the editing efficiency of pYL-ABE8e-EndoV was counted by aligning the sequencing results with the reference sequence. The results of the T0 generation showed that stable editing occurred in ABE8e-EndoV-TS 1-TS5, and the total average editing efficiency of ABE8e-EndoV was 62.7%. Furthermore, the average efficiency of A-to-G substitutions for ABE8e-EndoV was essentially the same as for ABE8e-SpG (59.7%), indicating that increasing the structure of the inosine base deletion repair enzyme EndoV did not affect the deamination efficiency of ABE8e-SpG (FIG. 2 a). The result that ABE8e-EndoV produced an average of 17.3% of single-allele insertions or deletions in addition to the detection of base editing by A-to-G indicates that EndoV does play a critical role in inosine mismatch repair. In terms of editing or window, the base editing window of A-to-G of ABE8e-EndoV is mainly focused on A3-A8, and ABE8e-EndoV has a narrower editing window compared to ABE8e-SpGn (A4-A8, which can be expanded to A4-A11) (FIG. 2 b).
pYL-ABE8e-EndoV enables predictable accurate deletion of small fragments of DNA
The CRISPR/Cas9 has two nuclease structural domains HNH and RuvC to cut a target chain and a non-target chain respectively, so that double-strand DNA break is caused, and random insertion and deletion mutation are generated. In the genome of animals and plants, the presence of a large number of cis-acting elements consisting of small segments also plays a critical role. The property of CRISPR/Cas9 that requires recognition of NGG-PAM and random insertions and deletions presents challenges for application in specific small fragment functional studies. ABE8e-EndoV in the present invention detected on average 17.3% of insertion or deletion mutations in the target. We further analyzed these indels and found that ABE8e-EndoV gave a small precise deletion of the 5' -deaminated A base to the SpGn cleavage site in TS1 (33.3%), TS3 (33.3%), TS4 (55.5%) and TS5 (60%) (FIG. 3 a). The deletion window for ABE8e-EndoV was focused mainly on the A5-A8 to SpGn cleavage site, resulting in a deletion of 9-13bp (FIG. 3 b). These results indicate that ABE8e-EndoV has the function of predicting the precise deletion of small fragments of target DNA in addition to efficient A-to-G base substitution. The ABE8e-EndoV can be applied to the function research of key elements rich in A/T, and in addition, the ABE8e-EndoV uses CRISPR/Cas9 variant SpGn (recognizing NG-PAM) with wider target range, thereby being more beneficial to realizing accurate base substitution and deletion of target targets.
TABLE 6 T0 generation transformed plant target amplification and sequencing primer

Lower off-target efficiency of pYL-ABE8e-EndoV
A CRISPR-GE webpage (http:// skl.scau.edu.cn /) (Xie et al, 2018, molecular plant, 11. And (3) carrying out PCR amplification on potential off-target sites by using the resistant callus genome DNA as a template for high-throughput sequencing analysis. The results showed that no off-target was detected at any of the sites except for the mutation detected by ABE8e-EndoV in one transformant at TS2-off1 (FIG. 4). The results of the ABE8e-EndoV off-target assay also indicate that increasing the EndoV domain did not show a high frequency of off-target.
pYL-ABE8e-EndoV mediated DNA small fragment precision deletion and adenine base editing pathway
In the present invention, ABE8e-EndoV plays an important role in the base mismatch repair pathway of I: T. In ABE8e-EndoV editing, A is first deaminated and hydrolyzed by adenine deaminase TadA8e to inosine I, the second phosphodiester bond at the 3' end of inosine is hydrolyzed by EndoV, and A-to-G (56.7%) base substitution is achieved through DNA repair and replication, and A-to-Deletions (17.3%) mutation is also achieved through nonhomologous end-joining repair (FIG. 5).
In conclusion, compared with the ABE editing system, the novel DNA small fragment accurate deletion adenine base editor pYL-ABE8e-EndoV has more advantages, firstly, the pYL-ABE8e-EndoV has wide targeting (recognition of NG-PAM), in addition, efficient A-to-G base replacement editing can be realized, and the most prominent advantage is that the pYL-ABE8e-EndoV can realize 9-13bp efficient DNA small fragment accurate deletion at a target point. Therefore, pYL-ABE8e-EndoV is more widely applicable to cis-acting elements, miRNA coding sequences, conserved elements of intron regions and high-AT polynucleotide sequence environments, and is also applicable to operations such as gene function screening, large-scale saturation mutation, alternative splicing and the like.
The above embodiments are preferred embodiments of the present invention, but the present invention is not limited to the above embodiments, and any other changes, modifications, substitutions, combinations, and simplifications which do not depart from the spirit and principle of the present invention should be construed as equivalents thereof, and all such changes, modifications, substitutions, combinations, and simplifications are intended to be included in the scope of the present invention.
Claims (10)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210810655.XA CN115925969B (en) | 2022-07-11 | 2022-07-11 | An adenine base editor capable of precisely deleting small DNA fragments and its construction and application |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210810655.XA CN115925969B (en) | 2022-07-11 | 2022-07-11 | An adenine base editor capable of precisely deleting small DNA fragments and its construction and application |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN115925969A true CN115925969A (en) | 2023-04-07 |
| CN115925969B CN115925969B (en) | 2025-10-21 |
Family
ID=86554548
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210810655.XA Active CN115925969B (en) | 2022-07-11 | 2022-07-11 | An adenine base editor capable of precisely deleting small DNA fragments and its construction and application |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN115925969B (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117659210A (en) * | 2023-11-30 | 2024-03-08 | 华南农业大学 | A recombinant fusion protein used as a plant double base editor and its application |
| WO2024230760A1 (en) * | 2023-05-09 | 2024-11-14 | 北京齐禾生科生物科技有限公司 | Adenosine deaminase capable of acting on dna and use thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180073012A1 (en) * | 2016-08-03 | 2018-03-15 | President And Fellows Of Harvard College | Adenosine nucleobase editors and uses thereof |
| WO2020063520A1 (en) * | 2018-09-30 | 2020-04-02 | 中山大学 | Method for detecting off-target effect of adenine base editor system based on whole-genome sequencing and use thereof in gene editing |
| CN114524879A (en) * | 2021-12-24 | 2022-05-24 | 华南农业大学 | Efficient plant wide-target adenine single base editor and construction and application thereof |
-
2022
- 2022-07-11 CN CN202210810655.XA patent/CN115925969B/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180073012A1 (en) * | 2016-08-03 | 2018-03-15 | President And Fellows Of Harvard College | Adenosine nucleobase editors and uses thereof |
| WO2020063520A1 (en) * | 2018-09-30 | 2020-04-02 | 中山大学 | Method for detecting off-target effect of adenine base editor system based on whole-genome sequencing and use thereof in gene editing |
| CN114524879A (en) * | 2021-12-24 | 2022-05-24 | 华南农业大学 | Efficient plant wide-target adenine single base editor and construction and application thereof |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024230760A1 (en) * | 2023-05-09 | 2024-11-14 | 北京齐禾生科生物科技有限公司 | Adenosine deaminase capable of acting on dna and use thereof |
| CN117659210A (en) * | 2023-11-30 | 2024-03-08 | 华南农业大学 | A recombinant fusion protein used as a plant double base editor and its application |
Also Published As
| Publication number | Publication date |
|---|---|
| CN115925969B (en) | 2025-10-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112266420B (en) | Plant efficient cytosine single-base editor and construction and application thereof | |
| US20240336905A1 (en) | Class ii, type v crispr systems | |
| WO2019041296A1 (en) | Base editing system and method | |
| CN102864498B (en) | Establishment method of long mate pair library | |
| US12435323B2 (en) | Enzymes with RUVC domains | |
| CN115925969B (en) | An adenine base editor capable of precisely deleting small DNA fragments and its construction and application | |
| CN114524879B (en) | Efficient plant wide-targeting adenine single-base editor and construction and application thereof | |
| CN113234701A (en) | Cpf1 protein and gene editing system | |
| CN105567718A (en) | Building method of carrier for expressing multiple sgRNAs simultaneously | |
| CA3036443A1 (en) | Compositions and methods for polynucleotide assembly | |
| WO2022159742A1 (en) | Novel engineered and chimeric nucleases | |
| CN117659210A (en) | A recombinant fusion protein used as a plant double base editor and its application | |
| US20250002881A1 (en) | Class ii, type v crispr systems | |
| CN114901820A (en) | Method for constructing gene mutation library | |
| CN104109685A (en) | A biomedical molecular cloning method | |
| CN103088015A (en) | DNA (Deoxyribonucleic Acid) multi-site directed mutation method | |
| CN111876472A (en) | Method for detecting trace nucleic acid in multiple mixed nucleic acids | |
| CN111454367A (en) | Base editing molecule and application thereof | |
| CN115161316B (en) | Guided editing tool, fusion RNA and application thereof | |
| WO2023076952A1 (en) | Enzymes with hepn domains | |
| CN119913131B (en) | Accurate pichia pastoris base editor based on adenosine deaminase mutant | |
| CN120173141B (en) | A protein for guiding editing and its application | |
| CN119931999B (en) | An efficient Pichia pastoris base editor based on an adenosine deaminase mutant | |
| CN115851784B (en) | Plant cytosine base editing system constructed by Lbcpf1 variant and application thereof | |
| CN117904069B (en) | VirEN protein-mediated DNA splicing and gene editing method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |