CN115536823A - Catalyst for preparing polyester by ring-opening polymerization and method for preparing polyester - Google Patents
Catalyst for preparing polyester by ring-opening polymerization and method for preparing polyester Download PDFInfo
- Publication number
- CN115536823A CN115536823A CN202211191148.9A CN202211191148A CN115536823A CN 115536823 A CN115536823 A CN 115536823A CN 202211191148 A CN202211191148 A CN 202211191148A CN 115536823 A CN115536823 A CN 115536823A
- Authority
- CN
- China
- Prior art keywords
- reaction
- phenyl
- thio
- carbon atoms
- urea
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/87—Non-metals or inter-compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
- C08G63/08—Lactones or lactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/68—Polyesters containing atoms other than carbon, hydrogen and oxygen
- C08G63/682—Polyesters containing atoms other than carbon, hydrogen and oxygen containing halogens
- C08G63/6822—Polyesters containing atoms other than carbon, hydrogen and oxygen containing halogens derived from hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/81—Preparation processes using solvents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/823—Preparation processes characterised by the catalyst used for the preparation of polylactones or polylactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/02—Aliphatic polycarbonates
- C08G64/0208—Aliphatic polycarbonates saturated
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/02—Aliphatic polycarbonates
- C08G64/0208—Aliphatic polycarbonates saturated
- C08G64/0225—Aliphatic polycarbonates saturated containing atoms other than carbon, hydrogen or oxygen
- C08G64/0233—Aliphatic polycarbonates saturated containing atoms other than carbon, hydrogen or oxygen containing halogens
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/20—General preparatory processes
- C08G64/26—General preparatory processes using halocarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/20—General preparatory processes
- C08G64/30—General preparatory processes using carbonates
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyesters Or Polycarbonates (AREA)
Abstract
本发明公开了一种用于开环聚合制备聚酯的催化剂及其制备聚酯的方法,属于有机氢键催化高分子材料技术领域。本发明的步骤为(1)二苯基膦基取代的(硫)脲和丙烯酸酯原位合成两性(硫)脲离子对催化剂。(2)在反应条件下,(硫)脲离子对催化剂对环状单体和引发剂进行双重活化。(3)在醇类引发剂的存在下,(硫)脲离子对催化环状单体开环聚合得到聚酯。该方法高效、工艺简便、成本低廉、适用范围广、反应活性高,无转酯反应、催化剂无金属残留、而且制备的聚酯具有精确分子量和分子量分布低的优点。The invention discloses a catalyst for preparing polyester by ring-opening polymerization and a method for preparing polyester, belonging to the technical field of organic hydrogen bond catalyzed polymer materials. The steps of the invention are (1) in-situ synthesis of amphoteric (thio)urea ion-pair catalysts by diphenylphosphino-substituted (thio)urea and acrylate. (2) Under the reaction conditions, (thio)urea ions double activate the catalyst to the cyclic monomer and the initiator. (3) In the presence of alcohol initiators, (thio)urea ion pairs catalyze the ring-opening polymerization of cyclic monomers to obtain polyesters. The method has the advantages of high efficiency, simple process, low cost, wide application range, high reactivity, no transesterification reaction, no metal residue in the catalyst, and the prepared polyester has the advantages of precise molecular weight and low molecular weight distribution.
Description
技术领域technical field
本发明属于有机催化和高分子材料技术领域,具体涉及有机催化环酯开环聚合制备聚酯的方法。The invention belongs to the technical field of organic catalysis and macromolecular materials, and in particular relates to a method for preparing polyester by ring-opening polymerization of an organic catalyzed cyclic ester.
背景技术Background technique
石油化工催化技术和聚合方法的创新促进了石油基材料的发展,这是二十世纪最重要的成就之一。尽管石油基材料具有许多优点,但却无法再生和降解。而随着全球化石能源的不断消耗以及大量塑料污染的产生,人们迫切地寻求一种可再生和可降解的替代材料。脂肪族聚酯是一类具有生物相容性、生物可再生性和生物降解性的材料,可以较好地替代石油基材料。合成脂肪族聚酯的最有效方法之一是环酯的开环聚合(ROP)。与传统的高分子合成方法相比,开环聚合制备脂肪族聚酯是一种能源消耗小、原料可再生的环境友好型的方法,能够在温和的反应条件下得到结构明确的聚酯。Innovations in petrochemical catalysis and polymerization methods have facilitated the development of petroleum-based materials, one of the most important achievements of the twentieth century. Despite their many advantages, petroleum-based materials cannot be regenerated and degrade. With the continuous consumption of global fossil energy and the generation of a large amount of plastic pollution, people are urgently seeking a renewable and degradable alternative material. Aliphatic polyesters are a class of biocompatible, biorenewable, and biodegradable materials that can be a good alternative to petroleum-based materials. One of the most efficient methods for the synthesis of aliphatic polyesters is the ring-opening polymerization (ROP) of cyclic esters. Compared with traditional polymer synthesis methods, the preparation of aliphatic polyesters by ring-opening polymerization is an environmentally friendly method with low energy consumption and renewable raw materials, and can obtain polyesters with well-defined structures under mild reaction conditions.
在开环聚合的催化方法中,有机催化开环聚合具有反应条件温和、单体转化率高、产物分子量高和反应时间短等优点,而且其制备的聚合物中没有金属残留,在生物医药、食品包装和微电子材料等领域的应用不受限制。因为这些优点,在过去的二十年中,有机催化开环聚合迅猛发展,涌现出了大量有机开环聚合催化剂。其中,氢键催化剂催化的开环聚合反应温和、高效且无转酯反应,得到的聚酯结构明确且分子量分布低,是最具研究潜力的有机催化剂之一。In the catalytic method of ring-opening polymerization, organocatalytic ring-opening polymerization has the advantages of mild reaction conditions, high monomer conversion rate, high product molecular weight and short reaction time, and there is no metal residue in the prepared polymer. It is used in biomedicine, Applications in fields such as food packaging and microelectronic materials are not limited. Because of these advantages, organocatalytic ring-opening polymerization has developed rapidly in the past two decades, and a large number of organic ring-opening polymerization catalysts have emerged. Among them, the ring-opening polymerization catalyzed by hydrogen bond catalysts is mild, efficient and free of transesterification, and the obtained polyester has a clear structure and low molecular weight distribution. It is one of the organic catalysts with the most research potential.
传统的氢键催化剂往往需要碱作为共/助催化剂来实现环酯的开环聚合(Macromolecules 2006,39,7863-7871,Nature Chem.2016,8,1047-1053,:Macromolecules 2018,51,3203-3211),而碱的加入可能会带来一些其他的问题。例如,使用金属碱作为共/助催化剂时,可能会向聚合物中引入金属残留,影响聚合物在生物医药等金属敏感领域的应用;有机碱虽然不会引入金属残留,但有机碱一般价格昂贵,会增加聚合物的生产成本。此外,碱共/助催化剂的加入往往会增加开环聚合过程中转酯反应等副反应发生的概率,因此碱的加入量需要被严格控制。Traditional hydrogen bond catalysts often require bases as co/co-catalysts to achieve ring-opening polymerization of cyclic esters (Macromolecules 2006, 39, 7863-7871, Nature Chem. 2016, 8, 1047-1053,: Macromolecules 2018, 51, 3203- 3211), and the addition of alkali may bring some other problems. For example, when metal bases are used as co-/co-catalysts, metal residues may be introduced into the polymer, which affects the application of polymers in metal-sensitive fields such as biomedicine; although organic bases do not introduce metal residues, organic bases are generally expensive , will increase the production cost of the polymer. In addition, the addition of base co-catalysts often increases the probability of side reactions such as transesterification during ring-opening polymerization, so the amount of base addition needs to be strictly controlled.
发明内容Contents of the invention
本发明提供了一种用于开环聚合制备聚酯的催化剂,本发明能够更加简单、温和、高效地合成具有精确分子量的聚合物,基于有机氢键作用合成多种结构精确的生物可降解的高分子聚合物,同时避免碱类试剂参与开环聚合带来的不利影响。The invention provides a catalyst for preparing polyester by ring-opening polymerization. The invention can synthesize polymers with precise molecular weight more simply, gently and efficiently, and synthesize various biodegradable polymers with precise structures based on organic hydrogen bonding. High molecular polymers, while avoiding the adverse effects of alkaline reagents participating in ring-opening polymerization.
本发明的另一目的在于提供一种基于(硫)脲的单分子双官能催化体系催化环酯的开环聚合制备聚酯的方法。该方法工艺简便、成本低廉、反应活性高,反应速率高效、过程可控,而且制备的聚酯具有不含金属残留物、精确分子量和分子量分布低的优点。Another object of the present invention is to provide a method for preparing polyester by ring-opening polymerization of cyclic esters catalyzed by a (thio)urea-based monomolecular bifunctional catalytic system. The method has the advantages of simple process, low cost, high reactivity, high reaction rate and controllable process, and the prepared polyester has the advantages of no metal residue, precise molecular weight and low molecular weight distribution.
为了解决上述技术问题,本发明采用的技术方案如下:In order to solve the problems of the technologies described above, the technical scheme adopted in the present invention is as follows:
以本发明提供的二苯基膦基取代的(硫)脲为基础,其与丙烯酸酯反应生成(硫)脲离子对催化剂,得到的(硫)脲离子对催化剂可以催化环酯开环聚合制备聚酯。利用环酯作为反应单体,(硫)脲离子对催化剂作为催化剂,醇类化合物作为引发剂,在室温溶液环境下或高温本体条件下进行开环聚合反应,并经分离纯化得到聚酯。Based on the diphenylphosphino-substituted (thio)urea provided by the present invention, it reacts with acrylate to form a (thio)urea ion-pair catalyst, and the obtained (thio)urea ion-pair catalyst can be prepared by catalyzing ring-opening polymerization of cyclic esters polyester. Using cyclic esters as reaction monomers, (thio)urea ion pair catalysts as catalysts, and alcohol compounds as initiators, ring-opening polymerization is carried out under room temperature solution environment or high temperature bulk conditions, and polyester is obtained through separation and purification.
一种用于开环聚合制备聚酯的催化剂,所述的催化剂如式I所示的两性(硫) 脲离子结构,A kind of catalyst that is used for ring-opening polymerization to prepare polyester, described catalyst is as the amphoteric (thio) urea ion structure shown in formula I,

其中in
X选自O或S;X is selected from O or S;
R1选自具有1~6个碳原子的直链或支链烷基,环己基、苯基、多取代或单取代的苯基,所述的单取代或多取代的苯基中的取代基选自甲基、氟基或三氟甲基;R 1 is selected from linear or branched alkyl groups with 1 to 6 carbon atoms, cyclohexyl, phenyl, polysubstituted or monosubstituted phenyl, the substituents in the monosubstituted or polysubstituted phenyl selected from methyl, fluoro or trifluoromethyl;
R2选自H,苯基,苄基,异丙基或叔丁基;R is selected from H, phenyl, benzyl, isopropyl or tert - butyl;
R3选自H,具有1~4个碳原子的直链或支链烷基,苯基或苄基; R3 is selected from H, a straight or branched chain alkyl group with 1 to 4 carbon atoms, phenyl or benzyl;
R4选自具有1~10个碳原子的直链或支链烷基,乙烯基,烯丙基,苯基或苄基。R 4 is selected from straight or branched chain alkyl, vinyl, allyl, phenyl or benzyl having 1 to 10 carbon atoms.
优选X选自O,R1选自乙基、异丙基、叔丁基、环己基、3,5-双(三氟甲基)苯基、4-甲基苯基、4-三氟甲基苯基或4-氟苯基,R2选自H、苄基或异丙基, R3选自甲基、异丙基、苯基或苄基,R4选自甲基、叔丁基、癸基、乙烯基,烯丙基,苯基或苄基。Preferably X is selected from O, R is selected from ethyl, isopropyl, tert - butyl, cyclohexyl, 3,5-bis(trifluoromethyl)phenyl, 4-methylphenyl, 4-trifluoromethyl phenyl or 4 -fluorophenyl, R2 is selected from H, benzyl or isopropyl, R3 is selected from methyl, isopropyl, phenyl or benzyl, R4 is selected from methyl, tert - butyl , decyl, vinyl, allyl, phenyl or benzyl.
在如式I所示的催化剂中,(硫)脲结构是众所周知的氢键供体,可以有效地活化单体,氧负离子部分可以用来活化引发剂/增长链末端,进而促进环酯的开环聚合。首先,该方案提供了一种单分子双官能催化体系,保持有机氢键催化剂高控制性的同时提高了催化活性,所得聚合物结构明确且分子量分布窄。其次,该方案所选择的催化剂不需要碱作为共/助催化剂,这是传统氢键催化开环聚合中不常见的,避免了强Lewis碱对反应的影响,进一步确保了聚合的控制性。最后,该方案不需要碱作为共/助催化,避免了传统氢键催化过程中金属碱的金属残留问题和有机碱的高成本问题,具有更广泛的应用前景。In the catalyst shown in formula I, the (thio)urea structure is a well-known hydrogen bond donor, which can effectively activate the monomer, and the oxyanion moiety can be used to activate the initiator/extended chain end, thereby promoting the opening of the cyclic ester. ring polymerization. First of all, this scheme provides a monomolecular bifunctional catalytic system, which improves the catalytic activity while maintaining the high controllability of the organic hydrogen bond catalyst, and the resulting polymer has a clear structure and a narrow molecular weight distribution. Secondly, the catalyst selected in this scheme does not require a base as a co-/co-catalyst, which is uncommon in traditional hydrogen bond-catalyzed ring-opening polymerization, avoiding the influence of strong Lewis bases on the reaction, and further ensuring the controllability of polymerization. Finally, this scheme does not require bases as co/promoters, which avoids the metal residue problem of metal bases and the high cost of organic bases in the traditional hydrogen bond catalysis process, and has broader application prospects.
优选所述的如式I所示的两性(硫)脲离子结构如下:Preferably described amphoteric (thio) urea ion structure as shown in formula I is as follows:

一种开环聚合制备聚酯的方法,在引发剂存在下,采用如式II所示的二苯基膦基取代的(硫)脲与式III所示的丙烯酸酯原位合成如式I所示的两性(硫) 脲离子对催化环状单体开环聚合,得到聚酯类化合物,所述的如式II所示的二苯基膦基取代的(硫)脲与如式III所示的丙烯酸酯的反应过程如下:A method for preparing polyester by ring-opening polymerization, in the presence of an initiator, using a diphenylphosphino-substituted (thio)urea shown in formula II and an acrylate shown in formula III to synthesize in situ as shown in formula I The shown amphoteric (thio) urea ion pair catalyzes the ring-opening polymerization of cyclic monomers to obtain polyester compounds, and the described diphenylphosphino-substituted (thio) urea as shown in formula II and The reaction process of the acrylate is as follows:

其中in
X选自O或S;X is selected from O or S;
R1选自具有1~6个碳原子的直链或支链烷基,环己基、苯基、多取代或单取代的苯基,所述的单取代或多取代的苯基中的取代基选自甲基、氟基或三氟甲基;R 1 is selected from linear or branched alkyl groups with 1 to 6 carbon atoms, cyclohexyl, phenyl, polysubstituted or monosubstituted phenyl, the substituents in the monosubstituted or polysubstituted phenyl selected from methyl, fluoro or trifluoromethyl;
R2选自H,苯基,苄基,异丙基或叔丁基;R is selected from H, phenyl, benzyl, isopropyl or tert - butyl;
R3选自H,具有1~4个碳原子的直链或支链烷基,苯基或苄基; R3 is selected from H, a straight or branched chain alkyl group with 1 to 4 carbon atoms, phenyl or benzyl;
R4选自具有1~10个碳原子的直链或支链烷基,乙烯基,烯丙基,苯基或苄基。R 4 is selected from straight or branched chain alkyl, vinyl, allyl, phenyl or benzyl having 1 to 10 carbon atoms.
优选如式II所示的二苯基膦基取代的(硫)脲可有如编号1~9所示结构:Preferably, the diphenylphosphino-substituted (thio)ureas shown in formula II can have the structures shown in numbers 1-9:


同样代表性地,如式III所示的可有如编号10~19所示的代表的丙烯酸酯结构:Also representatively, as shown in formula III, there may be representative acrylate structures as shown in numbers 10-19:

以上制备方法所采用的环状单体选自于以下的一种或几种:The cyclic monomers used in the above preparation method are selected from one or more of the following:
(1)式IV所示的内酯单体:(1) lactone monomer shown in formula IV:

其中,A为[—(CR1R2)—]n,n为2~10的整数;R1、R2选自H,具有1~5个碳原子的烷基和具有1~5个碳原子并被卤原子或羟基取代的烷基中的相同或不同基团,如β-丙内酯、γ-丁内酯、δ-戊内酯、ε-己内酯、大环十内酯、氯代己内酯;Among them, A is [—(CR 1 R 2 )—] n , n is an integer of 2 to 10; R 1 and R 2 are selected from H, alkyl groups with 1 to 5 carbon atoms and alkyl groups with 1 to 5 carbon atoms Atoms and the same or different groups in the alkyl group substituted by a halogen atom or a hydroxyl group, such as β-propiolactone, γ-butyrolactone, δ-valerolactone, ε-caprolactone, macrocyclic decalactone, Chlorocaprolactone;
(2)或式V所示的交酯单体:(2) or the lactide monomer shown in formula V:

其中,A、B为[—(CR1R2)—]n,n为0~10的整数,A和B相同或不同;R1、 R2选自H,具有1~5个碳原子并被卤原子或羟基取代的烷基中的相同或不同基团,如乙交酯、丙交酯、溴代乙交酯、丁交酯、癸交酯、大环十二交酯、O-羧酸内酸酐。Wherein, A and B are [—(CR 1 R 2 )—] n , n is an integer from 0 to 10, A and B are the same or different; R 1 and R 2 are selected from H, have 1 to 5 carbon atoms and The same or different groups in the alkyl group substituted by a halogen atom or a hydroxyl group, such as glycolide, lactide, bromoglycolide, butyrolactide, decanide, macrocyclic dodecide, O-carboxylate Acid internal anhydride.
(3)或式VI所示的碳酸酯单体:(3) or the carbonate monomer shown in formula VI:

其中,R1、R2选自H,具有1~5个碳原子并被卤原子或羟基取代的烷基中的相同或不同基团,如三亚甲基碳酸酯、羟基三亚甲基碳酸酯、氯代三亚甲基碳酸酯。Among them, R 1 and R 2 are selected from H, the same or different groups in alkyl groups having 1 to 5 carbon atoms and substituted by halogen atoms or hydroxyl groups, such as trimethylene carbonate, hydroxytrimethylene carbonate, Chlorothrimethylene carbonate.
以上制备方法所述的方法使用的引发剂为醇类化合物,包括甲醇、乙醇、正丙醇、异丙醇、正丁醇、叔丁醇、苯甲醇、苯乙醇、苯丙醇、乙二醇、缩乙二醇或季戊四醇。The initiator used in the method described in the above preparation method is an alcohol compound, including methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol, benzyl alcohol, phenylethyl alcohol, phenylpropanol, ethylene glycol , ethylene glycol or pentaerythritol.
以上制备方法所述的溶液环境是指在室温下,向反应体系中添加合适的聚合溶剂,这些溶剂会使反应物分布均匀,避免局部反应。本发明主要使用了四氢呋喃和二氯甲烷作为溶剂,所述方法的反应为溶液环境聚合时,反应温度为 20~35℃;反应投料比主要选择为环状单体:引发剂为25~400:1。The solution environment described in the above preparation method refers to adding a suitable polymerization solvent to the reaction system at room temperature, and these solvents will make the reactants evenly distributed and avoid local reactions. The present invention mainly uses tetrahydrofuran and dichloromethane as solvents, and when the reaction of the method is solution environment polymerization, the reaction temperature is 20-35°C; the reaction feed ratio is mainly selected as cyclic monomer: the initiator is 25-400: 1.
以上制备方法所述的本体条件是指不使用反应溶剂,反应温度较高,保证反应体系处于一种熔融的状态,本发明的本体反应温度主要选择为60-150℃,反应投料比主要选择为环状单体:引发剂为25~400:1。The bulk conditions described in the above preparation method means that no reaction solvent is used, the reaction temperature is relatively high, and the reaction system is guaranteed to be in a molten state. The bulk reaction temperature of the present invention is mainly selected as 60-150 ° C, and the reaction feed ratio is mainly selected as Cyclic monomer: initiator is 25-400:1.
以上制备方法所述的分离纯化是指将反应产物用良溶剂溶解后再用沉出溶剂沉淀析出,所用良溶剂为二氯甲烷、三氯甲烷、甲苯、苯、丙酮或四氢呋喃,优选二氯甲烷、三氯甲烷或四氢呋喃,所用沉出溶剂为甲醇、乙醇或水。The separation and purification described in the above preparation method refers to dissolving the reaction product in a good solvent and then precipitating it with a settling solvent. The good solvent used is dichloromethane, chloroform, toluene, benzene, acetone or tetrahydrofuran, preferably dichloromethane , chloroform or tetrahydrofuran, and the precipitation solvent used is methanol, ethanol or water.
本发明中,(硫)脲部分的非膦基取代的取代基的不同,丙烯酸酯的取代基不同,均会影响催化效率。开环聚合反应需根据聚合产物的性状要求以及反应装置的工艺条件来确定适宜温度及温度变化范围,保证聚合反应在一定温度范围内有效的进行。聚酯末端结构和分子量的可控分布,例如窄分子量分布,可以通过向开环聚合体系中加入含活泼氢的化合物(R-O-H)作为引发剂来实现,其引发后的单体链端结构分别为R-O-和-OH,而环酯单体与引发剂的投料比决定了所得聚酯的目标分子量。In the present invention, the difference in the non-phosphine-substituted substituents of the (thio)urea part and the difference in the acrylate substituents will affect the catalytic efficiency. The ring-opening polymerization reaction needs to determine the appropriate temperature and temperature variation range according to the property requirements of the polymerization product and the process conditions of the reaction device, so as to ensure that the polymerization reaction can be carried out effectively within a certain temperature range. The controllable distribution of polyester terminal structure and molecular weight, such as narrow molecular weight distribution, can be achieved by adding active hydrogen-containing compounds (R-O-H) as initiators to the ring-opening polymerization system. The monomer chain end structures after initiation are R-O- and -OH, and the feed ratio of the cyclic ester monomer to the initiator determines the target molecular weight of the resulting polyester.
有益效果Beneficial effect
本发明利用基于(硫)脲的单分子双官能催化体系来催化环酯开环聚合制备聚酯,而且本发明中参与开环聚合反应的催化剂为(硫)脲离子对,是一种热稳定性的有机催化剂。所以,该方法既可以采用溶液聚合的方法,具有极快快的反应速率,得到的聚合物不含金属残留物、分子量和末端结构可控、分子量分布窄;The present invention uses (thio)urea-based monomolecular bifunctional catalytic system to catalyze the ring-opening polymerization of cyclic esters to prepare polyester, and the catalyst participating in the ring-opening polymerization reaction in the present invention is (thio)urea ion pair, which is a thermally stable active organic catalysts. Therefore, this method can adopt the method of solution polymerization, has an extremely fast reaction rate, and the obtained polymer does not contain metal residues, the molecular weight and terminal structure are controllable, and the molecular weight distribution is narrow;
也可以采用本体聚合的方法,无需在反应体系中引入额外的反应溶剂,有利于工业化生产,而且在本体聚合体系中,一般反应温度较高,使得反应体系对空气和水的敏感度大大降低,方便工业操作。The method of bulk polymerization can also be adopted without introducing additional reaction solvent in the reaction system, which is beneficial to industrial production, and in the bulk polymerization system, the general reaction temperature is relatively high, which greatly reduces the sensitivity of the reaction system to air and water. Convenient for industrial operation.
反应还可以根据需求,受控地合成具有高目标分子量的产品聚酯,产品产率高,无单体残留。而且所采用的催化体系均为中性物质,避免了有机酸碱直接参与开环聚合反应,避免了转酯反应的的发生,进一步确保了反应的控制性。The reaction can also be controlled to synthesize the product polyester with high target molecular weight according to the demand, the product yield is high, and there is no monomer residue. Moreover, the catalytic system adopted is all neutral substances, which avoids the direct participation of organic acids and bases in the ring-opening polymerization reaction, avoids the occurrence of transesterification reaction, and further ensures the controllability of the reaction.
本发明采用的制备方法,有益效果如下The preparation method that the present invention adopts, beneficial effect is as follows
(1)利用丙烯酸酯和二苯基膦基取代的(硫)脲反应,生成包含氧负离子的(硫)脲离子对催化剂。(1) Utilize the reaction of acrylate and diphenylphosphino-substituted (thio)urea to generate (thio)urea ion-pair catalysts containing oxyanions.
(2)在反应条件下,(硫)脲离子对催化剂中(硫)脲部分作为氢键供体活化环酯,同时其氧负离子部分作为氢键受体活化引发剂/增长链末端。(2) Under the reaction conditions, the (thio)urea part in the catalyst acts as a hydrogen bond donor to activate the cyclic ester, while its oxyanion part acts as a hydrogen bond acceptor to activate the initiator/end of the extended chain.
(3)在醇类引发剂的存在下,步骤(1)得到的(硫)脲离子对催化剂催化环酯开环聚合得到聚酯。(3) In the presence of an alcohol initiator, the (thio)urea ion pair catalyst obtained in step (1) catalyzes ring-opening polymerization of cyclic esters to obtain polyester.
综上所述,该方法高效、工艺简便、成本低廉、适用范围广、反应活性高,无转酯反应、催化剂无金属残留、而且制备的聚酯具有精确分子量和分子量分布低的优点。In summary, the method is efficient, simple in process, low in cost, wide in application range, high in reactivity, free of transesterification reaction, free of metal residue in the catalyst, and the prepared polyester has the advantages of precise molecular weight and low molecular weight distribution.
附图说明Description of drawings
结合附图来详细说明本发明的实施例,其中Embodiments of the present invention are described in detail in conjunction with the accompanying drawings, wherein
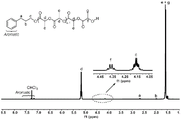
图1.用(硫)脲离子对作催化剂制备得到的聚丙交酯的1H NMR谱图;Fig. 1. uses (thio) urea ion pair as the 1 H NMR spectrogram of the polylactide that catalyst prepares;
图2.用(硫)脲离子对作催化剂制备得到的聚丙交酯在体积排阻色谱分析中的谱图;Fig. 2. use (thio) urea ion pair as the spectrogram of the polylactide that catalyzer prepares in the size exclusion chromatographic analysis;
图3.用(硫)脲离子对作催化剂制备得到的聚三亚甲基碳酸酯的1H NMR 谱图;Fig. 3. use (thio) urea ion pair as the 1 H NMR spectrogram of the polytrimethylene carbonate that catalyst prepares;
图4.用(硫)脲离子对作催化剂制备得到的聚三亚甲基碳酸酯体积排阻色谱分析中的谱图;Fig. 4. use (thio) urea ion pair as the spectrogram in the polytrimethylene carbonate size exclusion chromatographic analysis that catalyst prepares;
图5.用(硫)脲离子对作催化剂制备得到的聚戊内酯的1H NMR谱图;Fig. 5. uses (thio) urea ion pair as the 1 H NMR spectrogram of the polyvalerolactone that catalyst prepares;
图6.用(硫)脲离子对作催化剂制备得到的聚戊内酯体积排阻色谱分析中的谱图。Fig. 6. The spectrogram in the size exclusion chromatography analysis of polyvalerolactone prepared by using (thio)urea ion pair as a catalyst.
图7.用(硫)脲离子对作催化剂制备得到的聚己内酯的1H NMR谱图;Fig. 7. uses (thio) urea ion pair as the 1 H NMR spectrogram of the polycaprolactone that catalyst prepares;
图8.用(硫)脲离子对作催化剂制备得到的聚己内酯体积排阻色谱分析中的谱图。Fig. 8. The spectrogram in the size exclusion chromatography analysis of polycaprolactone prepared by using (thio)urea ion pair as a catalyst.
具体实施方式detailed description
通过下列实施例可以进一步说明本发明,实施例是为了说明而非限制本发明的。本领域的任何普通技术人员都能够理解这些实施例不以任何方式限制本发明,可以对其做适当的修改和数据变换而不违背本发明的实质和偏离本发明的范围。The present invention can be further illustrated by the following examples, which are intended to illustrate and not limit the invention. Any person skilled in the art can understand that these embodiments do not limit the present invention in any way, and can make appropriate modifications and data transformations without departing from the essence of the present invention and the scope of the present invention.
实施例中所用的二苯基膦基取代的(硫)脲具有如下结构:The diphenylphosphino-substituted (thio)urea used in the examples has the following structure:


实施例中所用的丙烯酸酯具有如下结构:The acrylates used in the examples have the following structure:

溶液聚合solution polymerization
实施例1Example 1
在10mL的聚合管中,加入化合物(3)(0.018g,0.05mmol)和化合物(10) (4.5μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌4h,停止反应,把反应液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物0.34g,转化率为96%,聚L-丙交酯的数均分子量Mn为7340g/mol,分子量分布PDI为1.08。In a 10mL polymerization tube, add compound (3) (0.018g, 0.05mmol) and compound (10) (4.5μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 4 hours, stop the reaction, slowly drop the reaction solution into cold methanol, the polymer precipitates, centrifuge and vacuum dry to obtain 0.34g of the product, the conversion rate is 96%, the poly-L-lactide The number average molecular weight M n was 7340 g/mol, the molecular weight distribution PDI was 1.08.
实施例2Example 2
在10mL的聚合管中,加入化合物(5)(0.021g,0.05mmol)和化合物(10) (4.5μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌1h,停止反应,把反应液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物0.31g,转化率为97%,聚L-丙交酯的数均分子量Mn为6334g/mol,分子量分布PDI为1.09。In a 10mL polymerization tube, add compound (5) (0.021g, 0.05mmol) and compound (10) (4.5μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 1 h, stop the reaction, slowly drop the reaction solution into cold methanol, and polymers are precipitated. After centrifugation and vacuum drying, 0.31 g of the product is obtained, and the conversion rate is 97%. Poly-L-lactide The number average molecular weight M n was 6334 g/mol, the molecular weight distribution PDI was 1.09.
实施例3Example 3
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(10) (4.5μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌5min,停止反应,把反应液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物0.30g,转化率为93%,聚L-丙交酯的数均分子量 Mn为7991g/mol,分子量分布PDI为1.11。(附图1、2)In a 10mL polymerization tube, add compound (6) (0.024g, 0.05mmol) and compound (10) (4.5μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 5 minutes, stop the reaction, slowly drop the reaction solution into cold methanol, and the polymer is precipitated, centrifuged and vacuum-dried to obtain 0.30 g of the product, the conversion rate is 93%, the poly-L-lactide The number average molecular weight M n was 7991 g/mol, the molecular weight distribution PDI was 1.11. (
实施例4Example 4
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(11) (5.3μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌24h,停止反应,把反应液缓慢滴入冷甲醇,有少量聚合物析出,经离心、真空干燥得到产物0.11g,转化率为30%,聚L-丙交酯的数均分子量Mn为2472g/mol,分子量分布PDI为1.16。In a 10mL polymerization tube, add compound (6) (0.024g, 0.05mmol) and compound (11) (5.3μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 24 hours, stop the reaction, slowly drop the reaction solution into cold methanol, a small amount of polymer precipitates, centrifuge and vacuum dry to obtain the product 0.11g, the conversion rate is 30%, poly L-lactide The number-average molecular weight M n of 2472 g/mol, the molecular weight distribution PDI is 1.16.
实施例5Example 5
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(13) (5.4μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌15min,停止反应,把反应液缓慢滴入冷甲醇,有少量聚合物析出,经离心、真空干燥得到产物0.34g,转化率为98%,聚L-丙交酯的数均分子量Mn为7567g/mol,分子量分布PDI为1.12。In a 10mL polymerization tube, add compound (6) (0.024g, 0.05mmol) and compound (13) (5.4μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 15 minutes, stop the reaction, slowly drop the reaction solution into cold methanol, a small amount of polymer precipitates, centrifuge and vacuum dry to obtain the product 0.34g, the conversion rate is 98%, poly L-lactide The number-average molecular weight M n of the compound is 7567 g/mol, and the molecular weight distribution PDI is 1.12.
实施例6Example 6
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(14) (7.3μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌2h,停止反应,把反应液缓慢滴入冷甲醇,有少量聚合物析出,经离心、真空干燥得到产物0.32g,转化率为98%,聚L-丙交酯的数均分子量 Mn为7643g/mol,分子量分布PDI为1.12。In a 10mL polymerization tube, add compound (6) (0.024g, 0.05mmol) and compound (14) (7.3μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 2 hours, stop the reaction, slowly drop the reaction solution into cold methanol, a small amount of polymer precipitates, centrifuge and vacuum dry to obtain the product 0.32g, the conversion rate is 98%, poly L-lactide The number-average molecular weight M n of the product is 7643 g/mol, and the molecular weight distribution PDI is 1.12.
实施例7Example 7
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(15) (7.2μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌15min,停止反应,把反应液缓慢滴入冷甲醇,有少量聚合物析出,经离心、真空干燥得到产物0.30g,转化率为90%,聚L-丙交酯的数均分子量Mn为6974g/mol,分子量分布PDI为1.14。In a 10mL polymerization tube, add compound (6) (0.024g, 0.05mmol) and compound (15) (7.2μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 15 minutes, stop the reaction, slowly drop the reaction solution into cold methanol, a small amount of polymer precipitates, centrifuge and vacuum dry to obtain 0.30 g of the product, the conversion rate is 90%, poly L-lactide The number-average molecular weight M n of the product is 6974 g/mol, and the molecular weight distribution PDI is 1.14.
实施例8Example 8
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(19) (7.5μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(0.360g,2.5mmol)和苯丙醇(6.8μL,0.05mmol)。在氩气保护下,室温搅拌15min,停止反应,把反应液缓慢滴入冷甲醇,有少量聚合物析出,经离心、真空干燥得到产物0.33g,转化率为96%,聚L-丙交酯的数均分子量Mn为6889g/mol,分子量分布PDI为1.13。In a 10mL polymerization tube, add compound (6) (0.024g, 0.05mmol) and compound (19) (7.5μL, 0.05mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (0.360 g, 2.5 mmol) and phenylpropanol (6.8 μL, 0.05 mmol). Under the protection of argon, stir at room temperature for 15 minutes, stop the reaction, slowly drop the reaction solution into cold methanol, a small amount of polymer precipitates, centrifuge and vacuum dry to obtain the product 0.33g, the conversion rate is 96%, poly L-lactide The number-average molecular weight M n of the product is 6889 g/mol, and the molecular weight distribution PDI is 1.13.
实施例9Example 9
在10mL的聚合管中,加入化合物(6)(0.015g,0.03mmol)和化合物(10) (2.7μL,0.03mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入L-丙交酯(1.730g,12mmol)和苯丙醇(4.08μL,0.03mmol)。在氩气保护下,室温搅拌4h,停止反应,把反应液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物1.52g,转化率为95%,聚L-丙交酯的数均分子量Mn为51350g/mol,分子量分布PDI为1.10。In a 10mL polymerization tube, add compound (6) (0.015g, 0.03mmol) and compound (10) (2.7μL, 0.03mmol), dissolve with 2.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add L - Lactide (1.730 g, 12 mmol) and phenylpropanol (4.08 μL, 0.03 mmol). Under the protection of argon, stir at room temperature for 4 hours, stop the reaction, slowly drop the reaction solution into cold methanol, the polymer precipitates, centrifuge and vacuum dry to obtain 1.52g of the product, the conversion rate is 95%, the poly-L-lactide The number average molecular weight M n was 51350 g/mol, the molecular weight distribution PDI was 1.10.
实施例10Example 10
在10mL的聚合管中,加入化合物(6)(0.015g,0.03mmol)和化合物(10) (2.7μL,0.03mmol),用0.5ml二氯甲烷溶解,室温搅拌10分钟,再向其中加入三亚甲基碳酸酯(0.153g,1.5mmol)和苯丙醇(4.08μL,0.03mmol)。在氩气保护下,室温搅拌4h,停止反应,把反应液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物0.14g,转化率为98%,聚三亚甲基碳酸酯的数均分子量Mn为6306g/mol,分子量分布PDI为1.12。(附图3、4)In a 10mL polymerization tube, add compound (6) (0.015g, 0.03mmol) and compound (10) (2.7μL, 0.03mmol), dissolve with 0.5ml of dichloromethane, stir at room temperature for 10 minutes, and then add Sanya Methyl carbonate (0.153 g, 1.5 mmol) and phenylpropanol (4.08 μL, 0.03 mmol). Under the protection of argon, stir at room temperature for 4 hours, stop the reaction, slowly drop the reaction solution into cold methanol, the polymer precipitates, centrifuge and vacuum dry to obtain 0.14g of the product, the conversion rate is 98%, the polytrimethylene carbonate The number average molecular weight M n is 6306 g/mol, the molecular weight distribution PDI is 1.12. (Attachments 3 and 4)
本体聚合Ontology aggregation
实施例1Example 1
在10mL的聚合管中,加入化合物(6)(0.024g,0.05mmol)和化合物(10) (4.5μL,0.05mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟。在水泵上小心抽除溶剂,氩气保护下向其中加入D-丙交酯(0.360g,2.5mmol)和季戊四醇(4.9μL,0.05mmol)。140℃条件下磁力搅拌2h,停止反应,向所得混合物中滴加少量二氯甲烷溶解,再把所得混合液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物0.27g,转化率为96%,聚D-丙交酯的数均分子量Mn为6978g/mol,分子量分布PDI为1.20。Compound (6) (0.024 g, 0.05 mmol) and compound (10) (4.5 μL, 0.05 mmol) were added to a 10 mL polymerization tube, dissolved in 2.5 ml of dichloromethane, and stirred at room temperature for 10 minutes. The solvent was carefully pumped off on a water pump, and D-lactide (0.360 g, 2.5 mmol) and pentaerythritol (4.9 μL, 0.05 mmol) were added thereto under the protection of argon. Stir magnetically at 140°C for 2 hours, stop the reaction, add a small amount of dichloromethane dropwise to the resulting mixture to dissolve, then slowly drop the resulting mixture into cold methanol, a polymer precipitates, centrifuge and vacuum dry to obtain 0.27g of the product, transform The ratio was 96%, the number average molecular weight Mn of poly-D-lactide was 6978 g/mol, and the molecular weight distribution PDI was 1.20.
实施例2Example 2
在10mL的聚合管中,加入化合物(2)(0.035g,0.1mmol)和化合物(10) (9.0μL,0.1mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟。在水泵上小心抽除溶剂,氩气保护下向其中加入乙交酯(0.348g,3mmol)和苯甲醇(10.0μL, 0.1mmol)。130℃条件下磁力搅拌22h,停止反应,向所得混合物中滴加少量四氢呋喃溶解,再把所得混合液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物0.28g,转化率为92%,聚乙交酯的数均分子量Mn为3770g/mol,分子量分布PDI为1.20。Compound (2) (0.035 g, 0.1 mmol) and compound (10) (9.0 μL, 0.1 mmol) were added to a 10 mL polymerization tube, dissolved in 2.5 ml of dichloromethane, and stirred at room temperature for 10 minutes. The solvent was carefully pumped off on a water pump, and glycolide (0.348 g, 3 mmol) and benzyl alcohol (10.0 μL, 0.1 mmol) were added thereto under argon protection. Stir magnetically at 130°C for 22 hours, stop the reaction, add a small amount of tetrahydrofuran dropwise to the obtained mixture to dissolve, and then slowly drop the obtained mixture into cold methanol, and a polymer is precipitated. After centrifugation and vacuum drying, 0.28 g of the product is obtained. The conversion rate is 92%, the number average molecular weight M n of polyglycolide is 3770 g/mol, and the molecular weight distribution PDI is 1.20.
实施例3Example 3
在10mL的聚合管中,加入化合物(6)(0.048g,0.1mmol)和化合物(10) (9.0μL,0.1mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟。在水泵上小心抽除溶剂,氩气保护下向其中加入L-丁交酯(1.512g,9mmol)和苯甲醇(10.0 μL,0.1mmol)。150℃条件下磁力搅拌12h,停止反应,向所得混合物中滴加少量四氢呋喃溶解,再把所得混合液缓慢滴入冷甲醇,有聚合物析出,经离心、真空干燥得到产物1.2g,转化率为98%,聚L-丁交酯的数均分子量Mn为14730 g/mol,分子量分布PDI为1.23。Compound (6) (0.048 g, 0.1 mmol) and compound (10) (9.0 μL, 0.1 mmol) were added to a 10 mL polymerization tube, dissolved in 2.5 ml of dichloromethane, and stirred at room temperature for 10 minutes. The solvent was carefully pumped off on a water pump, and L-butyrolactide (1.512 g, 9 mmol) and benzyl alcohol (10.0 μL, 0.1 mmol) were added thereto under argon protection. Stir magnetically at 150°C for 12 hours, stop the reaction, add a small amount of tetrahydrofuran dropwise to the resulting mixture to dissolve, and then slowly drop the resulting mixture into cold methanol, and a polymer is precipitated. After centrifugation and vacuum drying, 1.2 g of the product is obtained. The conversion rate is 98%, the number average molecular weight M n of poly-L-butyrolactide is 14730 g/mol, and the molecular weight distribution PDI is 1.23.
实施例4Example 4
在10mL的聚合管中,加入化合物(6)(0.048g,0.1mmol)和化合物(10) (9.0μL,0.1mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟。在水泵上小心抽除溶剂,氩气保护下向其中加入三亚甲基碳酸酯(0.306g,3mmol)和苯甲醇(10.0μL,0.1mmol)。60℃条件下磁力搅拌8h,停止反应,向所得混合物中滴加少量三氯甲烷溶解,再把所得混合液缓慢滴入冷乙醇,有聚合物析出,经离心、真空干燥得到产物0.25g,转化率为88%,聚三亚甲基碳酸酯的数均分子量Mn为2972g/mol,分子量分布PDI为1.13。Compound (6) (0.048 g, 0.1 mmol) and compound (10) (9.0 μL, 0.1 mmol) were added to a 10 mL polymerization tube, dissolved in 2.5 ml of dichloromethane, and stirred at room temperature for 10 minutes. The solvent was carefully pumped off on a water pump, and trimethylene carbonate (0.306 g, 3 mmol) and benzyl alcohol (10.0 μL, 0.1 mmol) were added thereto under argon protection. Stir magnetically at 60°C for 8 hours, stop the reaction, add a small amount of chloroform to the obtained mixture to dissolve, and then slowly drop the obtained mixture into cold ethanol, and a polymer is precipitated. After centrifugation and vacuum drying, 0.25 g of the product is obtained. The ratio was 88%, the number average molecular weight M n of polytrimethylene carbonate was 2972 g/mol, and the molecular weight distribution PDI was 1.13.
实施例5Example 5
在10mL的聚合管中,加入化合物(6)(0.048g,0.1mmol)和化合物(10) (9.0μL,0.1mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟。在水泵上小心抽除溶剂,氩气保护下向其中加入羟基三亚甲基碳酸酯(0.714g,6mmol) 和异丙醇(7.6μL,0.1mmol)。60℃下条件下磁力搅拌5h,停止反应,向所得混合物中滴加少量三氯甲烷溶解,再把混合液缓慢滴入冷乙醇,有聚合物析出,经离心、真空干燥得到产物0.64g,转化率为95%,聚羟基三亚甲基碳酸酯的数均分子量Mn为6267g/mol,分子量分布PDI为1.22。Compound (6) (0.048 g, 0.1 mmol) and compound (10) (9.0 μL, 0.1 mmol) were added to a 10 mL polymerization tube, dissolved in 2.5 ml of dichloromethane, and stirred at room temperature for 10 minutes. The solvent was carefully pumped off on a water pump, and hydroxytrimethylene carbonate (0.714 g, 6 mmol) and isopropanol (7.6 μL, 0.1 mmol) were added thereto under argon protection. Stir magnetically at 60°C for 5 hours, stop the reaction, add a small amount of chloroform to the obtained mixture to dissolve, then slowly drop the mixed solution into cold ethanol, a polymer precipitates, centrifuge and vacuum dry to obtain 0.64g of the product, transform The ratio was 95%, the number average molecular weight M n of polyhydroxytrimethylene carbonate was 6267 g/mol, and the molecular weight distribution PDI was 1.22.
实施例6Example 6
在10mL的聚合管中,加入化合物(6)(0.048g,0.1mmol)和化合物(10) (9.0μL,0.1mmol),用2.5ml二氯甲烷溶解,室温搅拌10分钟。在水泵上小心抽除溶剂,氩气保护下向其中加入氯代三亚甲基碳酸酯(0.825g,6mmol) 和正丁醇(9.1μL,0.1mmol)。60℃下条件下磁力搅拌18h,停止反应,向所得混合物中滴加少量三氯甲烷溶解,再把混合液缓慢滴入冷乙醇,有聚合物析出,经离心、真空干燥得到产物0.63g,转化率为96%,聚氯代三亚甲基碳酸酯的数均分子量Mn为6754g/mol,分子量分布PDI为1.15。Compound (6) (0.048 g, 0.1 mmol) and compound (10) (9.0 μL, 0.1 mmol) were added to a 10 mL polymerization tube, dissolved in 2.5 ml of dichloromethane, and stirred at room temperature for 10 minutes. The solvent was carefully pumped off on a water pump, and chlorotrimethylene carbonate (0.825 g, 6 mmol) and n-butanol (9.1 μL, 0.1 mmol) were added thereto under argon protection. Stir magnetically at 60°C for 18 hours, stop the reaction, add a small amount of chloroform to the obtained mixture to dissolve, then slowly drop the mixed solution into cold ethanol, a polymer precipitates, centrifuge and vacuum dry to obtain 0.63g of the product, transform The ratio was 96%, the number average molecular weight Mn of polychlorotrimethylene carbonate was 6754 g/mol, and the molecular weight distribution PDI was 1.15.
实施例7Example 7
在10mL的聚合管中,加入δ-戊内酯(0.45mL,5mmol)、化合物(6)(0.048 g,0.1mmol)和化合物(10)(9.0μL,0.1mmol),室温搅拌10分钟后向其中加入苯丙醇(13.6μL,0.1mmol)。90℃下条件下磁力搅拌2.5h,停止反应,向所得混合物中滴加少量二氯甲烷溶解,过滤除去不溶物,再把所得混合液缓慢滴入冷乙醇,有聚合物析出,经离心、真空干燥得到产物0.33g,转化率为89%,聚戊内酯的数均分子量Mn为5660g/mol,分子量分布PDI为1.14。(附图5、6)In a 10mL polymerization tube, add δ-valerolactone (0.45mL, 5mmol), compound (6) (0.048 g, 0.1mmol) and compound (10) (9.0μL, 0.1mmol), stir at room temperature for 10 minutes and pour To this was added phenylpropanol (13.6 μL, 0.1 mmol). Stir magnetically at 90°C for 2.5 hours, stop the reaction, add a small amount of dichloromethane dropwise to the obtained mixture to dissolve, filter to remove insoluble matter, and then slowly drop the obtained mixture into cold ethanol, and polymers are precipitated. After drying, 0.33 g of the product was obtained, the conversion rate was 89%, the number average molecular weight M n of polyvalerolactone was 5660 g/mol, and the molecular weight distribution PDI was 1.14. (Figures 5 and 6)
实施例8Example 8
在10mL的聚合管中,加入γ-氯-δ-戊内酯(7.60mL,40mmol)、化合物(6) (0.048g,0.1mmol)和化合物(10)(9.0μL,0.1mmol),室温搅拌10分钟后向其中加入苯丙醇(13.6μL,0.1mmol)。90℃下条件下磁力搅拌28h,停止反应,向所得混合物中滴加少量二氯甲烷溶解,过滤除去不溶物,再把所得混合液缓慢滴入冷乙醇,有聚合物析出,经离心、真空干燥得到产物5.4g,转化率为84%,所得聚合物数均分子量Mn为45740g/mol,分子量分布PDI为1.25。In a 10mL polymerization tube, add γ-chloro-δ-valerolactone (7.60mL, 40mmol), compound (6) (0.048g, 0.1mmol) and compound (10) (9.0μL, 0.1mmol), and stir at room temperature After 10 minutes, phenylpropanol (13.6 μL, 0.1 mmol) was added thereto. Stir magnetically at 90°C for 28 hours, stop the reaction, add a small amount of dichloromethane dropwise to the resulting mixture to dissolve, filter to remove insoluble matter, then slowly drop the resulting mixture into cold ethanol, and polymers are precipitated, centrifuged and vacuum-dried 5.4 g of the product was obtained, the conversion rate was 84%, the number average molecular weight M n of the obtained polymer was 45740 g/mol, and the molecular weight distribution PDI was 1.25.
实施例9Example 9
在10mL的聚合管中,加入ε-己内酯(0.55mL,5mmol)、化合物(6)(0.048 g,0.1mmol)和化合物(10)(9.0μL,0.1mmol),室温搅拌10分钟后向其中加入苯丙醇(13.6μL,0.1mmol)。90℃下条件下磁力搅拌36h,停止反应,向所得混合物中滴加少量二氯甲烷溶解,再把所得混合液缓慢滴入冷乙醇,有聚合物析出,经离心、真空干燥得到产物0.44g,转化率为99%,聚己内酯的数均分子量Mn为5138g/mol,分子量分布PDI为1.10。(附图7、8)。In a 10mL polymerization tube, add ε-caprolactone (0.55mL, 5mmol), compound (6) (0.048 g, 0.1mmol) and compound (10) (9.0μL, 0.1mmol), stir at room temperature for 10 minutes To this was added phenylpropanol (13.6 μL, 0.1 mmol). Stir magnetically at 90°C for 36 hours, stop the reaction, add a small amount of dichloromethane dropwise to the resulting mixture to dissolve, then slowly drop the resulting mixture into cold ethanol, a polymer is precipitated, centrifuged and vacuum-dried to obtain 0.44g of the product, The conversion rate was 99%, the number average molecular weight M n of polycaprolactone was 5138 g/mol, and the molecular weight distribution PDI was 1.10. (accompanying drawing 7,8).
Claims (10)








Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211191148.9A CN115536823B (en) | 2022-09-28 | 2022-09-28 | Catalyst for preparing polyester by ring-opening polymerization and method for preparing polyester by using catalyst |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211191148.9A CN115536823B (en) | 2022-09-28 | 2022-09-28 | Catalyst for preparing polyester by ring-opening polymerization and method for preparing polyester by using catalyst |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN115536823A true CN115536823A (en) | 2022-12-30 |
| CN115536823B CN115536823B (en) | 2023-10-31 |
Family
ID=84730373
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211191148.9A Active CN115536823B (en) | 2022-09-28 | 2022-09-28 | Catalyst for preparing polyester by ring-opening polymerization and method for preparing polyester by using catalyst |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN115536823B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116217901A (en) * | 2023-03-24 | 2023-06-06 | 吉林大学 | A kind of preparation method of high isotactic polylactide |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002057335A1 (en) * | 2001-01-18 | 2002-07-25 | Toyo Boseki Kabushiki Kaisha | Polymerization catalyst for polyester, polyester, and process for producing the same |
| TWI276646B (en) * | 2001-01-18 | 2007-03-21 | Toyo Boseki | Catalyst for polymerization of polyester, polyester and process for preparing polyester |
| CN103497314A (en) * | 2013-10-10 | 2014-01-08 | 大连理工大学 | Linear comb polycaprolactones and preparation method thereof |
| CN107022070A (en) * | 2017-04-28 | 2017-08-08 | 南京工业大学 | Method for preparing polyester by ring-opening polymerization |
| CN110092892A (en) * | 2019-04-25 | 2019-08-06 | 南京工业大学 | Preparation method of polyester |
-
2022
- 2022-09-28 CN CN202211191148.9A patent/CN115536823B/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002057335A1 (en) * | 2001-01-18 | 2002-07-25 | Toyo Boseki Kabushiki Kaisha | Polymerization catalyst for polyester, polyester, and process for producing the same |
| TWI276646B (en) * | 2001-01-18 | 2007-03-21 | Toyo Boseki | Catalyst for polymerization of polyester, polyester and process for preparing polyester |
| CN103497314A (en) * | 2013-10-10 | 2014-01-08 | 大连理工大学 | Linear comb polycaprolactones and preparation method thereof |
| CN107022070A (en) * | 2017-04-28 | 2017-08-08 | 南京工业大学 | Method for preparing polyester by ring-opening polymerization |
| CN110092892A (en) * | 2019-04-25 | 2019-08-06 | 南京工业大学 | Preparation method of polyester |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116217901A (en) * | 2023-03-24 | 2023-06-06 | 吉林大学 | A kind of preparation method of high isotactic polylactide |
Also Published As
| Publication number | Publication date |
|---|---|
| CN115536823B (en) | 2023-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Lecomte et al. | Recent developments in ring-opening polymerization of lactones | |
| CN110092900B (en) | Preparation method of carbon dioxide-based block copolymer | |
| CN109880073B (en) | Preparation method of polylactone | |
| CN109679077B (en) | Method for preparing polyester by (thio) urea/organic base catalytic ring-opening copolymerization of epoxide and cyclic anhydride | |
| CN112079999B (en) | A kind of method for zinc catalyst catalyzed ring-opening polymerization of cyclic ester | |
| Naves et al. | Enzymatic syntheses of unsaturated polyesters based on isosorbide and isomannide | |
| CN109851764B (en) | A kind of preparation method of polylactone | |
| CN110092892B (en) | Preparation method of polyester | |
| CN105153408B (en) | Preparation method of polyester-polycarbonate-polyester multi-block copolymer | |
| WO2022041326A1 (en) | Zinc catalyst for catalyzing ring-opening polymerization of cyclic esters and controlled depolymerization of polyester materials and catalytic method therefor | |
| CN102060986A (en) | Aromatic-aliphatic block copolyester and preparation method thereof | |
| Liu et al. | Squaramide and amine binary H-bond organocatalysis in polymerizations of cyclic carbonates, lactones, and lactides | |
| CN107022070B (en) | A kind of method that ring-opening polymerization prepares polyester | |
| CN106947067B (en) | Preparation method of polyester | |
| CN106083907A (en) | A kind of Schiff's base aluminum complex and its preparation method and application | |
| Chen et al. | Controlled/living ring-opening polymerization of ɛ-caprolactone catalyzed by phosphoric acid | |
| CN115536823A (en) | Catalyst for preparing polyester by ring-opening polymerization and method for preparing polyester | |
| CN110483753A (en) | The method of Lewis Acid-Base System controlled catalytic O- carboxylic acid anhydride monomer's ring-opening polymerisation based on metal salt and organic base | |
| CN114685764A (en) | Polyhydroxyalkanoate and preparation method thereof | |
| CN105367763A (en) | Method for preparing polyester by ring-opening polymerization | |
| Omar et al. | Star-shaped Poly (hydroxybutyrate) s from bio-based polyol cores via zinc catalyzed ring-opening polymerization of β-Butyrolactone | |
| CN110591064A (en) | Furanyl copolyester and its preparation method | |
| Mili et al. | Designing of poly (l-lactide)–nicotine conjugates: mechanistic and kinetic studies and thermal release behavior of nicotine | |
| CN115947934B (en) | A catalyst for preparing polyester by ring-opening polymerization and a method for preparing polyester | |
| Liu et al. | Protecting-group-free synthesis of thiol-functionalized degradable polyesters |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |