CN114621068A - Preparation method of 3-hydroxy-1-adamantane methyl ketone and method for synthesizing saxagliptin - Google Patents
Preparation method of 3-hydroxy-1-adamantane methyl ketone and method for synthesizing saxagliptin Download PDFInfo
- Publication number
- CN114621068A CN114621068A CN202210336222.5A CN202210336222A CN114621068A CN 114621068 A CN114621068 A CN 114621068A CN 202210336222 A CN202210336222 A CN 202210336222A CN 114621068 A CN114621068 A CN 114621068A
- Authority
- CN
- China
- Prior art keywords
- hydroxy
- reaction
- adamantane
- saxagliptin
- ketone
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 title claims abstract description 42
- 229960004937 saxagliptin Drugs 0.000 title claims abstract description 34
- QGJUIPDUBHWZPV-SGTAVMJGSA-N saxagliptin Chemical compound C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QGJUIPDUBHWZPV-SGTAVMJGSA-N 0.000 title claims abstract description 34
- 108010033693 saxagliptin Proteins 0.000 title claims abstract description 34
- 238000000034 method Methods 0.000 title claims abstract description 26
- 230000002194 synthesizing effect Effects 0.000 title claims abstract description 15
- 238000002360 preparation method Methods 0.000 title claims abstract description 14
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims abstract description 102
- 238000006243 chemical reaction Methods 0.000 claims abstract description 99
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 81
- 150000002576 ketones Chemical class 0.000 claims abstract description 39
- 238000010992 reflux Methods 0.000 claims abstract description 25
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims abstract description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims abstract description 16
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 claims abstract description 12
- JIMXXGFJRDUSRO-UHFFFAOYSA-N adamantane-1-carboxylic acid Chemical compound C1C(C2)CC3CC2CC1(C(=O)O)C3 JIMXXGFJRDUSRO-UHFFFAOYSA-N 0.000 claims abstract description 12
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims abstract description 11
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 9
- 238000003786 synthesis reaction Methods 0.000 claims abstract description 9
- 229960000583 acetic acid Drugs 0.000 claims abstract description 8
- 238000005660 chlorination reaction Methods 0.000 claims abstract description 8
- 239000012362 glacial acetic acid Substances 0.000 claims abstract description 8
- 238000004519 manufacturing process Methods 0.000 claims abstract description 8
- 239000002994 raw material Substances 0.000 claims abstract description 8
- 230000008569 process Effects 0.000 claims abstract description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 64
- -1 3-chloro-1-adamantyl formyl diethyl malonate Chemical compound 0.000 claims description 55
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 46
- 238000001035 drying Methods 0.000 claims description 46
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 38
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 36
- 238000001914 filtration Methods 0.000 claims description 34
- 239000012074 organic phase Substances 0.000 claims description 33
- 239000000706 filtrate Substances 0.000 claims description 32
- 238000005406 washing Methods 0.000 claims description 30
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 claims description 28
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 claims description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 27
- 239000003208 petroleum Substances 0.000 claims description 22
- 239000002904 solvent Substances 0.000 claims description 22
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 21
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 21
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 claims description 20
- 239000004471 Glycine Substances 0.000 claims description 19
- CGJIQTOWYDOCCS-UHFFFAOYSA-N 3-chloroadamantane-1-carbonyl chloride Chemical compound C1C(C2)CC3CC2(Cl)CC1(C(=O)Cl)C3 CGJIQTOWYDOCCS-UHFFFAOYSA-N 0.000 claims description 18
- 239000013078 crystal Substances 0.000 claims description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 15
- 159000000000 sodium salts Chemical class 0.000 claims description 15
- 239000007864 aqueous solution Substances 0.000 claims description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 14
- 239000012266 salt solution Substances 0.000 claims description 14
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 claims description 14
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 claims description 14
- 239000012286 potassium permanganate Substances 0.000 claims description 12
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 9
- 229910052708 sodium Inorganic materials 0.000 claims description 9
- 239000011734 sodium Substances 0.000 claims description 9
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 claims description 9
- 102000006746 NADH Dehydrogenase Human genes 0.000 claims description 7
- 108010086428 NADH Dehydrogenase Proteins 0.000 claims description 7
- 238000005859 coupling reaction Methods 0.000 claims description 7
- 238000002425 crystallisation Methods 0.000 claims description 7
- 230000008025 crystallization Effects 0.000 claims description 7
- 238000002156 mixing Methods 0.000 claims description 7
- 108010078226 phenylalanine oxidase Proteins 0.000 claims description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 7
- 239000012454 non-polar solvent Substances 0.000 claims description 6
- JBEPFZCYCKRHTN-UHFFFAOYSA-N C(=O)O.C12CC3CC(CC(C1)C3)C2 Chemical compound C(=O)O.C12CC3CC(CC(C1)C3)C2 JBEPFZCYCKRHTN-UHFFFAOYSA-N 0.000 claims description 5
- 230000009471 action Effects 0.000 claims description 5
- 239000003999 initiator Substances 0.000 claims description 4
- WPIYZQBALIBMAA-SHLRHQAISA-N (1s,3s,5s)-2-azabicyclo[3.1.0]hexane-3-carboxamide;methanesulfonic acid Chemical compound CS(O)(=O)=O.N1[C@H](C(=O)N)C[C@@H]2C[C@@H]21 WPIYZQBALIBMAA-SHLRHQAISA-N 0.000 claims description 3
- 238000001953 recrystallisation Methods 0.000 claims description 3
- 101710088194 Dehydrogenase Proteins 0.000 claims description 2
- 238000006482 condensation reaction Methods 0.000 claims description 2
- AWUCVROLDVIAJX-UHFFFAOYSA-N glycerol 1-phosphate Chemical compound OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 claims description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 2
- 238000005191 phase separation Methods 0.000 claims description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 2
- 239000000376 reactant Substances 0.000 claims description 2
- AVTLBBWTUPQRAY-UHFFFAOYSA-N 2-(2-cyanobutan-2-yldiazenyl)-2-methylbutanenitrile Chemical compound CCC(C)(C#N)N=NC(C)(CC)C#N AVTLBBWTUPQRAY-UHFFFAOYSA-N 0.000 abstract description 7
- 102100040409 Ameloblastin Human genes 0.000 abstract description 7
- 101000891247 Homo sapiens Ameloblastin Proteins 0.000 abstract description 7
- 239000003054 catalyst Substances 0.000 abstract description 4
- 229940079593 drug Drugs 0.000 abstract description 2
- 239000003814 drug Substances 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 105
- 238000003756 stirring Methods 0.000 description 71
- 239000000243 solution Substances 0.000 description 36
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 30
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- 238000004809 thin layer chromatography Methods 0.000 description 20
- 238000010438 heat treatment Methods 0.000 description 19
- 238000001816 cooling Methods 0.000 description 18
- 239000000203 mixture Substances 0.000 description 16
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 239000007788 liquid Substances 0.000 description 15
- 238000012544 monitoring process Methods 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 10
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 10
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 238000007792 addition Methods 0.000 description 10
- 238000001704 evaporation Methods 0.000 description 10
- 239000005457 ice water Substances 0.000 description 10
- 239000010410 layer Substances 0.000 description 10
- 239000012044 organic layer Substances 0.000 description 10
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 10
- 238000000605 extraction Methods 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- AFNTWHMDBNQQPX-NHKADLRUSA-N saxagliptin hydrate Chemical compound O.C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AFNTWHMDBNQQPX-NHKADLRUSA-N 0.000 description 7
- MTLWTRLYHAQCAM-UHFFFAOYSA-N 2-[(1-cyano-2-methylpropyl)diazenyl]-3-methylbutanenitrile Chemical compound CC(C)C(C#N)N=NC(C#N)C(C)C MTLWTRLYHAQCAM-UHFFFAOYSA-N 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 5
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 5
- 229910006124 SOCl2 Inorganic materials 0.000 description 5
- 239000012153 distilled water Substances 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 5
- 239000012046 mixed solvent Substances 0.000 description 5
- 230000007935 neutral effect Effects 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 239000011736 potassium bicarbonate Substances 0.000 description 5
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 235000010265 sodium sulphite Nutrition 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- 238000005303 weighing Methods 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 238000009738 saturating Methods 0.000 description 4
- 235000002639 sodium chloride Nutrition 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 239000000872 buffer Substances 0.000 description 3
- ZHNUHDYFZUAESO-UHFFFAOYSA-N formamide Substances NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 238000000967 suction filtration Methods 0.000 description 3
- GMOBHRQSNDDZAD-UHFFFAOYSA-N 3-chloroadamantane-1-carboxylic acid Chemical compound C1C(C2)CC3CC2(Cl)CC1(C(=O)O)C3 GMOBHRQSNDDZAD-UHFFFAOYSA-N 0.000 description 2
- CJJMAWPEZKYJAP-UHFFFAOYSA-N 3-hydroxyadamantane-1-carboxylic acid Chemical compound C1C(C2)CC3CC2(O)CC1(C(=O)O)C3 CJJMAWPEZKYJAP-UHFFFAOYSA-N 0.000 description 2
- QSJXEFYPDANLFS-UHFFFAOYSA-N Diacetyl Chemical compound CC(=O)C(C)=O QSJXEFYPDANLFS-UHFFFAOYSA-N 0.000 description 2
- 108010004460 Gastric Inhibitory Polypeptide Proteins 0.000 description 2
- 102100039994 Gastric inhibitory polypeptide Human genes 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 230000000911 decarboxylating effect Effects 0.000 description 2
- 230000000640 hydroxylating effect Effects 0.000 description 2
- 238000009776 industrial production Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 239000012345 acetylating agent Substances 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229940090124 dipeptidyl peptidase 4 (dpp-4) inhibitors for blood glucose lowering Drugs 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- GFAUNYMRSKVDJL-UHFFFAOYSA-N formyl chloride Chemical compound ClC=O GFAUNYMRSKVDJL-UHFFFAOYSA-N 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000006257 total synthesis reaction Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/64—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by introduction of functional groups containing oxygen only in singly bound form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/673—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton
- C07C45/676—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton by elimination of carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/58—Preparation of carboxylic acid halides
- C07C51/60—Preparation of carboxylic acid halides by conversion of carboxylic acids or their anhydrides or esters, lactones, salts into halides with the same carboxylic acid part
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/24—Preparation of oxygen-containing organic compounds containing a carbonyl group
- C12P7/26—Ketones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/56—Ring systems containing bridged rings
- C07C2603/58—Ring systems containing bridged rings containing three rings
- C07C2603/70—Ring systems containing bridged rings containing three rings containing only six-membered rings
- C07C2603/74—Adamantanes
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Zoology (AREA)
- Health & Medical Sciences (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention discloses a preparation method of 3-hydroxy-1-adamantanemethyl ketone and a method for synthesizing saxagliptin, relating to the technical field of drug synthesis, wherein the preparation method comprises the steps of adding adamantanecarboxylic acid and thionyl chloride into dichloromethane, adding NCS, ABVN and AMBN, performing chlorination reaction, reacting with diethyl malonate, adding glacial acetic acid, water and sulfuric acid, performing reflux reaction, and performing reflux reaction on the obtained 3-chloro-1-adamantanemethyl ketone and strong base in water to obtain the 3-hydroxy-1-adamantanemethyl ketone; the method for synthesizing saxagliptin takes 3-hydroxy-1-adamantane methyl ketone as a raw material for synthesis. The invention effectively reduces the using amount of thionyl chloride in the synthesis process and reduces the production cost by selecting a proper catalyst.
Description
Technical Field
The invention relates to the technical field of drug synthesis, in particular to a preparation method of 3-hydroxy-1-adamantane methyl ketone and a method for synthesizing saxagliptin.
Background
Saxagliptin, which has the chemical name { (1S,3S,5S) -2- [ (2S) -2-amino-2- (3-hydroxytricyclo [3.3.1.13, 7] decan-1-yl) acetyl ] -2-azabicyclo [3.1.0] -hexane-3-carbonitrile }, is a DPP-4 inhibitor. Saxagliptin can strongly and selectively act on DPP-4, inhibit the activity of the DPP-4, increase the in vivo concentration of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), prolong the action time and further reduce the blood sugar.
For the synthesis of saxagliptin, 3-hydroxy-1-adamantanemethyl ketone is an important intermediate in the synthetic process of saxagliptin. Through the literature reference of relevant synthetic routes, the synthetic routes are mainly five: the route is as follows: LI, Qun et al, 2016, showed that 3-hydroxy-1-adamantanecarboxylic acid was reacted with methyllithium and chlorotrimethylsilane in sequence as starting material to give 3-hydroxy-1-adamantanemethyl ketone with a yield of 34%; route two: KishiA uses adamantane as raw material and butanedione as acetylating agent in Co (OAc)2Reacting under catalysis to obtain 3-hydroxy-1-adamantane methyl ketone with the yield of 6 percent; route III: khusnutdinovor takes 1-adamantanemethyl ketone as a starting material, and Mo (CO)6Is a catalyst, with H2O/CBrCl3Reacting to obtain a compound 3-hydroxy-1-adamantane methyl ketone with the yield of 98 percent; route iv: takes adamantane formic acid as the initial raw material, firstly, the mixed acid (H) is added2SO4/HNO3) Hydroxylating at the 3-carbon to obtain 3-hydroxy-1-adamantanecarboxylic acid, and reacting with acetyl chlorideSynthesizing acid anhydride, then substituting sodium salt of malonic acid di, and finally decarboxylating to obtain 3-hydroxy-1-adamantane methyl ketone, wherein the yield is about 70%; a fifth route: takes adamantane formic acid as a starting material, firstly, mixed acid (H) is added2SO4/HNO3) Hydroxylating on the 3-carbon to obtain 3-hydroxy-1-adamantane formic acid, reacting with thionyl chloride to obtain 3-chloro-1-adamantane formyl chloride under a reflux condition, then adding prepared diethyl malonate to obtain 3-chloro-1-adamantane formyl diethyl malonate, refluxing and decarboxylating under an acidic condition to obtain 3-chloro-1-adamantane methyl ketone, and finally refluxing under an alkaline condition to obtain the target compound 3-hydroxy-1-adamantane methyl ketone, wherein the total yield is over 70 percent.
Among the methods, firstly, the method has low yield and cannot be used for industrial production, thirdly, the method has high yield but expensive metal catalyst and low industrial application value, and fourthly, a large amount of acetyl chloride or thionyl chloride is used in the method, so that the production cost is high.
Disclosure of Invention
The invention aims to provide a preparation method of 3-hydroxy-1-adamantanemethyl ketone and a method for synthesizing saxagliptin, so as to solve the problems in the background technology.
In order to achieve the purpose, the invention provides the following technical scheme:
a method for preparing 3-hydroxy-1-adamantanemethyl ketone, comprising the following steps carried out in sequence:
11) adding adamantane formic acid and thionyl chloride into dichloromethane, adding N-chlorosuccinimide (NCS), adding Azobisisoheptonitrile (ABVN) and Azobisisovaleronitrile (AMBN) as an initiator, and refluxing for chlorination reaction to obtain 3-chloro-1-adamantyl formyl chloride;
12) taking sodium and diethyl malonate to react in a nonpolar solvent for mixing reaction to obtain a sodium salt solution;
dissolving 3-chloro-1-adamantyl formyl chloride in a nonpolar solvent, slowly dripping sodium salt solution, reacting at room temperature after dripping, and obtaining 3-chloro-1-adamantyl formyl diethyl malonate after the reaction is finished;
13) taking 3-chloro-1-adamantane formyl diethyl malonate, adding glacial acetic acid, water and sulfuric acid, and performing reflux reaction to obtain 3-chloro-1-adamantane methyl ketone;
14) taking 3-chloro-1-adamantane methyl ketone to carry out reflux reaction with strong base in water to obtain the 3-hydroxy-1-adamantane methyl ketone, wherein the specific chemical reaction formula is as follows:

further, in the step 11), the molar ratio of the adamantane carboxylic acid to the dichloromethane to the N-chlorosuccinimide is 1: 2.5-3.0: 0.8 to 0.9;
the weight ratio of the adamantane carboxylic acid to the initiator is 1: 0.1 to 0.2;
the weight ratio of the azodiisoheptanonitrile to the azodiisovaleronitrile is 3-4: 1.
further, in the step 11), after the reaction is finished, concentrating to remove residual reactants and solvent, adding carbon tetrachloride, dissolving, filtering, and concentrating the filtrate to obtain the 3-chloro-1-adamantyl formyl chloride.
Further, in the step 11), the weight volume ratio of the adamantane carboxylic acid to the dichloromethane is 1 g: 2-2.5 mL.
Further, in the step 12), the molar ratio of the 3-chloro-1-adamantyl formyl chloride to the sodium to the diethyl malonate is 1: 2-2.1: 2.5 to 2.7.
Further, in step 12), the nonpolar solvent is petroleum ether.
A method for synthesizing saxagliptin by using the 3-hydroxy-1-adamantanemethyl ketone prepared by the preparation method as a raw material.
Further, the method for synthesizing saxagliptin comprises the following steps which are sequentially carried out:
21) the 3-hydroxy-1-adamantane methyl ketone is used as a raw material, and methyl is oxidized by potassium permanganate under an alkaline condition to obtain the 3-hydroxy-1-adamantane glyoxylic acid, wherein the specific chemical reaction formula is as follows:

22) the 3-hydroxy-1-adamantane glyoxylic acid and glycerol are subjected to coupling reaction under the action of NADH dehydrogenase, 3-phosphoglycerol dehydrogenase (GDH) and phenylalanine dehydrogenase, and then subjected to Boc amino protection reaction with Boc anhydride to obtain (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine, wherein the specific chemical reaction formula is as follows:

23) taking (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine and (1S,3S,5S) -2-azabicyclo [3.1.0] hexane-3-formamide mesylate to carry out condensation reaction to obtain tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, wherein the specific chemical reaction formula is as follows:

24) tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dehydrated by trifluoroacetic anhydride to give tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the specific chemical reaction formula is as follows:

25) taking ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid to remove Boc under the action of hydrochloric acid, and carrying out post-treatment to obtain the saxagliptin (namely, saxagliptin monohydrate), wherein the specific chemical reaction formula is as follows:
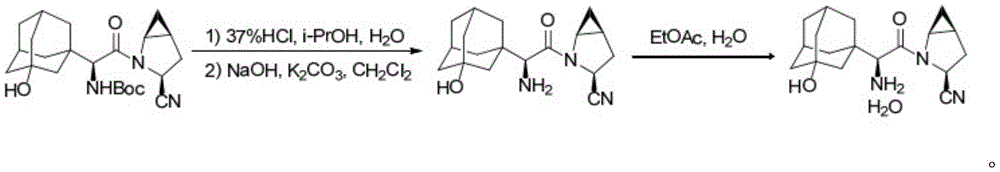
further, in the step 5), after removing Boc, adding water and dichloromethane in sequence, adding a sodium hydroxide aqueous solution and a potassium carbonate aqueous solution to adjust the pH value to be alkaline, carrying out phase separation, washing, drying and concentrating an organic phase, and recrystallizing an obtained residue to obtain the saxagliptin.
Further, in the step 5), the recrystallization is to dissolve the residue in methanol/ethanol, maintain 24 ℃, dropwise add water until white crystals are separated out, maintain 24 ℃ for crystallization, filter and dry to obtain the saxagliptin.
Compared with the prior art, the invention has the beneficial effects that:
by selecting a proper catalyst, the invention effectively reduces the using amount of thionyl chloride in the synthesis process and reduces the production cost;
the invention adopts adamantane formic acid as the starting material to complete the total synthesis of saxagliptin, the reaction process is simple, the intermediate is easy to purify, the raw materials used in the reaction are easy to obtain, and the reaction process is milder and safer than the prior art and is more suitable for industrial production;
according to the invention, a suitable recrystallization reagent is adopted to prepare the saxagliptin monohydrate with higher purity.
Detailed Description
The technical solutions in the embodiments of the present invention will be clearly and completely described below with reference to the embodiments of the present invention, and it is obvious that the described embodiments are only a part of the embodiments of the present invention, and not all of the embodiments. All other embodiments, which can be obtained by a person skilled in the art without making any creative effort based on the embodiments in the present invention, belong to the protection scope of the present invention.
The first embodiment is as follows:
the synthetic method of saxagliptin comprises the following steps:
mono) 3-hydroxy-1-adamantanemethyl ketone
11) 90.12g (0.5mol) of adamantanecarboxylic acid was added to 200mL of dichloromethane, and 119g (1.0mol) of SOCl was added2Refluxing with stirring for 4h, cooling to room temperature, 56.75g (0.425mol) NCS, 10.51g Azobisisoheptonitrile (ABVN), 3.00g Azobisisovaleronitrile (AMBN) and 47.6g (0.4mol) SOCl were added2Heating to reflux again for chlorination reaction for 4h, and rotary evaporating to remove residual SOCl2And methylene chloride, 500mL of CCl was added4Stirring for 10min to dissolve, filtering to remove insoluble substances, concentrating the filtrate, and evaporating to remove CCl4110.37g of 3-chloro-1-adamantyl formyl chloride is obtained as yellow oily liquid, and the crude yield is 94.69%;
12) adding 22.32g (0.97mol) of sodium into 2L of petroleum ether, then dripping 197.15g (1.23mol) of diethyl malonate, and reacting at room temperature for 8h under stirring to obtain a sodium salt solution after dripping;
110.37g (0.47mol) of the obtained 3-chloro-1-adamantyl formyl chloride is added into 500mL of petroleum ether and stirred for dissolution, sodium salt solution is dropwise added at room temperature, and the mixture is stirred and reacted for 12 hours at room temperature after the dropwise addition, so as to obtain yellow emulsion, 1L of water is added, and the mixture is stirred for 10 minutes; separating, collecting the upper petroleum ether layer, extracting the water layer with petroleum ether (500mL × 3), combining the organic layers, and concentrating to obtain 165.12 yellow oily 3-chloro-1-adamantyl diethyl malonate with crude yield of 97.74%;
13) 165.12g of the diethyl 3-chloro-1-adamantanecarbonyl malonate obtained above was added with 470mL of glacial acetic acid, 140mL of water and 47mL of concentrated sulfuric acid, and subjected to a reflux reaction for 8 hours, after the reaction was completed, the mixture was poured into ice water (3L), and extracted with dichloromethane (500 mL. times.3), the organic solvents were combined, the solvent was evaporated, and the residue was recrystallized from n-hexane to obtain 86.57g of white crystal 3-chloro-1-adamantanemethyl ketone with a yield of 87.95%.
14) To the 3-chloro-1-adamantanemethyl ketone obtained above were added 2.6L of water and 262g (4.67mol) of KOH, and the mixture was refluxed for 8 hours with stirring, poured into ice water (2.5L), extracted with ethyl acetate (500mL × 3), the organic layers were combined, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated, and the residue was recrystallized with a mixed solvent (cyclohexane/ethyl acetate ═ 10/1, V/V) to obtain 72.48g of white crystalline 3-hydroxy-1-adamantanemethyl ketone with a yield of 91.68%.
The specific chemical reaction formula of the preparation method of the 3-hydroxy-1-adamantane methyl ketone is as follows:
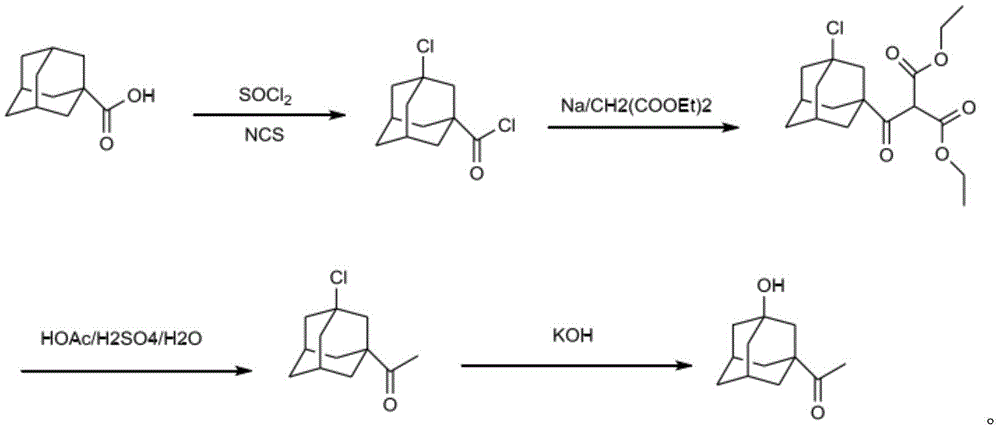
wherein, the total yield of the 3-hydroxy-1-adamantane methyl ketone is 74.62 percent.
II) method for synthesizing saxagliptin
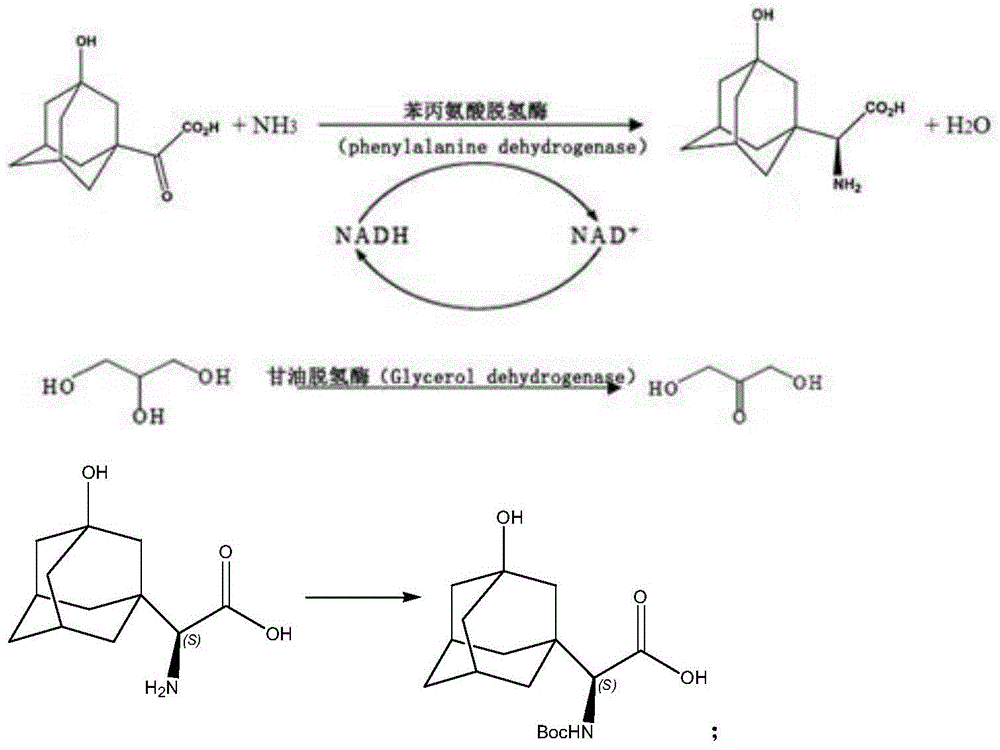
21) Weighing 68g (0.35mol) of 3-hydroxy-1-adamantane methyl ketone, adding the 3-hydroxy-1-adamantane methyl ketone into 350mL of t-BuOH for dissolving, then adding 1.7L of NaOH solution with the mass concentration of 2%, heating to 30 ℃, slowly adding 110.6g (0.7mol) of potassium permanganate in batches, reacting for 4 hours at 40 ℃, adding 680mL of sodium sulfite solution with the mass concentration of 10% after the reaction is finished to treat redundant potassium permanganate, filtering, adjusting the pH of the filtrate to 1 by hydrochloric acid, separating out white solids, filtering again, and drying to obtain 70.25g of white crystal 3-hydroxy-1-adamantane glyoxylic acid, wherein the yield is 89.50%, and the specific chemical reaction formula is as follows:

22) 31.2L of NH with pH 8.5 and a concentration of 1.0mol/L were taken4Cl-NH4Adding 70g (0.312mol) of 3-hydroxy-1-adamantane glyoxylic acid and 143.66g (1.56mol) of glycerol into an OH buffer aqueous solution, then adding 0.36g of NADH dehydrogenase, 31.2mg of GDH and 31.2mg of phenylalanine dehydrogenase, stirring at room temperature to perform coupling reaction, after the reaction is finished, adjusting the pH to 2, then adding ethyl acetate (5L multiplied by 3) for extraction, combining organic phases, drying by anhydrous magnesium sulfate, and concentrating to obtain 66.81g of white solid with (S) configuration, wherein the yield is 95.05%;
taking 66g (0.29mol) of (S) -configuration white solid, adding 300mL of distilled water for dissolving, adjusting the pH to 10 by concentrated hydrochloric acid, adding 300mL of tetrahydrofuran solution dissolved with 76.39g (0.35mol) of Boc anhydride, stirring at room temperature for reaction overnight, performing suction filtration, adjusting the pH of filtrate to 2 by concentrated hydrochloric acid, extracting by ethyl acetate (300mL multiplied by 3), combining extract liquor, washing by saturated common salt water, drying by anhydrous sodium sulfate, removing the solvent by spin drying, and recrystallizing the residual oily matter to obtain 79.13g of (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine, wherein the yield is 83.00%, and the specific chemical reaction formula is as follows:
23) taking 61g (0.473mol) DIPEA, 60mL acetonitrile and 50mL ethyl acetate, mixing uniformly, and keeping a diisopropylethylamine solution for later use;
69.8g (0.215mol) of (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine were successively added 50g (0.225mol) of (1S,3S,5S) -2-azabicyclo [3.1.0]Hexane-3-formamide methanesulfonate, 45.2g (0.236mol) EDCI, 29g (0.215mol) HOBt and 240mL acetonitrile, stirring, dropwise adding diisopropylethylamine solution at room temperature, heating to 40 ℃ for reaction for 1.5h after dropwise adding, monitoring by TLC that (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine reaction is completed, cooling to 10 ℃, adjusting the pH of the solution to 3 with 1mol/L diluted hydrochloric acid, adding 500mL ethyl acetate, stirring for 10min, standing, separating, washing an organic phase with 1mol/L diluted hydrochloric acid (200mL multiplied by 2), washing with water (200mL multiplied by 1), and saturating KHCO3Washing (200 mL. times.2), drying over anhydrous magnesium sulfate, filtering, and concentrating the filtrate to obtain 84.41g of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0]]Hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, yield 90.98%, specific chemical reaction formula:

24) 80.0g (0.185mol) of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dissolved in 400mL of ethyl acetate, the temperature is reduced to-5 ℃, 82.2g (0.812mol) of triethylamine is added dropwise, 85.4g (0.407mol) of trifluoroacetic anhydride is added dropwise while maintaining the temperature at-5 ℃, the reaction is maintained at-5 ℃ for 1h, 120mL of water is added after the TLC monitoring reaction is completed, liquid separation is carried out, the organic phase is washed with 80mL of 1mol/L dilute hydrochloric acid, potassium carbonate (76.6 g) with the mass concentration of 40% is added, 0.554mol) and 120mL of methanol, stirring, heating to 40 ℃ for reaction for 2h, monitoring by TLC (thin-layer chromatography) that the reaction is complete, cooling to room temperature, separating, adjusting the pH of the organic phase to 3 by using 2mol/L dilute hydrochloric acid, separating, washing the organic phase with saturated common salt water (100mL multiplied by 1), drying anhydrous magnesium sulfate, concentrating the filtrate to remove the solvent, dissolving the residue in 80mL of acetone, cooling to 15 ℃, dropwise adding 240mL of isopropyl ether, continuing stirring for 3h after dropwise addition, filtering, and drying to obtain 49.21g of white solid tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the yield is 64.36%, and the specific chemical reaction formula is as follows:

25) 46g (0.11mol) ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is added with 46mL isopropanol and 46mL water, the temperature is increased to 65 ℃, 37% concentrated hydrochloric acid (0.154mol) is added dropwise, the temperature is maintained for reaction for 3h, after TLC monitors that the reaction is completed, 90mL water is added, the temperature is reduced to room temperature, 400mL dichloromethane is added, stirring is carried out, ice bath is carried out to 10 ℃, 20mL of aqueous solution containing 4.4g of NaOH (2.2g) is added dropwise, the pH is adjusted to be neutral, then solution with the mass concentration of 50% is added, the pH is adjusted to be 10, stirring is carried out for 15min, separating, extracting the water phase with dichloromethane (50mL multiplied by 1), combining the organic phases, washing with saturated brine (100mL multiplied by 1), drying with anhydrous magnesium sulfate, concentrating under reduced pressure to evaporate the solvent, dissolving the residue in 80mL ethanol, maintaining the temperature at 24 ℃, slowly adding pure water dropwise until white crystals are separated out, maintaining the temperature at 24 ℃ for crystallization for 5 hours, filtering, and drying to obtain 27.83g of saxagliptin monohydrate, wherein the yield is 75.88%, the purity is 99.96%, and the specific chemical reaction formula is as follows:

example two:
the synthetic method of saxagliptin comprises the following steps:
mono) 3-hydroxy-1-adamantanemethyl ketone
11) 90.12g (0.5mol) of adamantanecarboxylic acid was added to 195mL of dichloromethane, and 119g (1.0mol) of SOCl was added2Refluxing with stirring for 4h, cooling to room temperature, 60.09g (0.45mol) NCS, 7.21g Azobisisoheptonitrile (ABVN), 1.8g Azobisisovaleronitrile (AMBN) and 35.7g (0.3mol) SOCl were added2Heating to reflux again for chlorination reaction for 4h, and rotary evaporating to remove residual SOCl2And methylene chloride, 500mL of CCl was added4Stirring for 10min to dissolve, filtering to remove insoluble substances, concentrating the filtrate, and evaporating to remove CCl4108.79g of 3-chloro-1-adamantyl formyl chloride is obtained as yellow oily liquid, and the crude yield is 93.33%;
12) adding 22.54g (0.98mol) of sodium into 2L of petroleum ether, then dripping 186.86g (1.17mol) of diethyl malonate, and reacting at room temperature for 8h under stirring to obtain a sodium salt solution after dripping;
adding the obtained 3-chloro-1-adamantyl formyl chloride into 500mL of petroleum ether, stirring and dissolving, dropwise adding a sodium salt solution at room temperature, continuing stirring and reacting at room temperature for 12 hours after dropwise adding is finished, obtaining yellow emulsion, adding 1L of water, and stirring for 10 min; separating, collecting the upper petroleum ether layer, extracting the water layer with petroleum ether (500mL × 3), combining the organic layers, and concentrating to obtain 164.39 yellow oily 3-chloro-1-adamantyl diethyl malonate with crude yield of 98.72%;
13) 470mL of glacial acetic acid, 140mL of water and 47mL of concentrated sulfuric acid are added into the obtained 3-chloro-1-adamantanecarbonyl diethyl malonate, reflux reaction is carried out for 8 hours, after the reaction is finished, the mixture is poured into ice water (3L), dichloromethane (500mL multiplied by 3) is used for extraction, organic solvents are combined, the solvents are evaporated, and the residue is recrystallized by normal hexane, so that 86.1g of white crystal 3-chloro-1-adamantanemethyl ketone is obtained, wherein the yield is 87.86%.
14) To the 3-chloro-1-adamantanemethyl ketone obtained above were added 2.6L of water and 261g (4.65mol) of KOH, and the mixture was refluxed for 8 hours with stirring, poured into ice water (2.8L), extracted with ethyl acetate (500mL × 3), the organic layers were combined, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated, and the residue was recrystallized with a mixed solvent (cyclohexane/ethyl acetate ═ 10/1, V/V) to obtain 72.07g of white crystalline 3-hydroxy-1-adamantanemethyl ketone with a yield of 91.65%.
Wherein the total yield of the 3-hydroxy-1-adamantanemethyl ketone is 74.20 percent.
II) method for synthesizing saxagliptin
21) Weighing 68g (0.35mol) of 3-hydroxy-1-adamantane methyl ketone, adding the mixture into 350mL of t-BuOH for dissolution, adding 1.7L of NaOH solution with the mass concentration of 2%, heating to 30 ℃, slowly adding 110.6g (0.7mol) of potassium permanganate in batches, reacting for 4 hours at 40 ℃, adding 680mL of sodium sulfite solution with the mass concentration of 10% after the reaction is finished to treat redundant potassium permanganate, filtering, adjusting the pH value of the filtrate to 1 by hydrochloric acid, separating out white solids, filtering again, and drying to obtain 70.18g of white crystal 3-hydroxy-1-adamantane glyoxylic acid with the yield of 89.42%;
22) 31.2L of NH with pH 8.5 and concentration of 1.0mol/L is taken4Cl-NH4Adding 70g (0.312mol) of 3-hydroxy-1-adamantane glyoxylic acid and 143.66g (1.56mol) of glycerol into an OH buffer aqueous solution, then adding 0.36g of NADH dehydrogenase, 31.2mg of GDH and 31.2mg of phenylalanine dehydrogenase, stirring at room temperature to perform coupling reaction, after the reaction is finished, adjusting the pH to 2, then adding ethyl acetate (5L multiplied by 3) for extraction, combining organic phases, drying by anhydrous magnesium sulfate, and concentrating to obtain 66.79g of white solid with (S) configuration, wherein the yield is 95.02%;
taking 66g (0.29mol) of (S) -configuration white solid, adding 300mL of distilled water for dissolving, adjusting the pH to 10 by concentrated hydrochloric acid, adding 300mL of tetrahydrofuran solution dissolved with 76.39g (0.35mol) of Boc anhydride, stirring at room temperature for reaction overnight, performing suction filtration, adjusting the pH of filtrate to 2 by concentrated hydrochloric acid, extracting by ethyl acetate (300mL multiplied by 3), combining extract liquor, washing by saturated salt water, drying by anhydrous sodium sulfate, removing the solvent by spin drying, and recrystallizing the residual oily matter to obtain 79.08g of (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine with the yield of 82.95%;
23) taking 61g (0.473mol) DIPEA, 60mL acetonitrile and 50mL ethyl acetate, mixing uniformly, and keeping a diisopropylethylamine solution for later use;
69.8g (0.215mol) of (S) -N-t-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine were added to 50g (0.225mol) of (1S,3S,5S) -2-azabicyclo [ 3.1.0)]Hexane-3-formamide mesylate, 45.2g (0.236mol) EDCI, 29g (0.215mol) HOBt and 240mL acetonitrile, uniformly stirring, dropwise adding diisopropylethylamine solution at room temperature, heating to 40 ℃ for reacting for 1.5h after completing dropwise addition, monitoring by TLC (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine reaction, cooling to 10 ℃, adjusting the pH of the solution to 3 by using 1mol/L dilute hydrochloric acid, adding 500mL ethyl acetate, stirring for 10min, standing, separating, washing an organic phase by using 1mol/L dilute hydrochloric acid (200mL multiplied by 2), washing by water (200mL multiplied by 1), and saturating KHCO3Washing (200 mL. times.2), drying over anhydrous magnesium sulfate, filtering, and concentrating the filtrate to yield 84.37g of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0]]Hex-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, yield 90.94%;
24) 80.0g (0.185mol) of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dissolved in 400mL of ethyl acetate, cooled to-5 ℃, 82.2g (0.812mol) of triethylamine is added dropwise, 85.4g (0.407mol) of trifluoroacetic anhydride is added dropwise while maintaining at-5 ℃, reaction is maintained for 1h while maintaining-5 ℃ until TLC monitoring reaction is completed, 120mL of water is added for quenching reaction, liquid separation is carried out, the organic phase is washed with 80mL of diluted hydrochloric acid with the concentration of 1mol/L, potassium carbonate with the mass concentration of 40% (76.6 g) is added, 0.554mol) of aqueous solution and 120mL of methanol, stirring, heating to 40 ℃ for 2h, monitoring by TLC (thin layer chromatography), cooling to room temperature after the reaction is completed, separating liquid, adjusting the pH of the organic phase to 3 by using 2mol/L diluted hydrochloric acid, separating liquid, washing the organic phase with saturated common salt water (100mL multiplied by 1), drying anhydrous magnesium sulfate, concentrating the filtrate to remove the solvent, dissolving the residue in 80mL of acetone, cooling to 15 ℃, dropwise adding 240mL of isopropyl ether, continuing stirring for 3h after dropwise addition, filtering, and drying to obtain 49.19g of white solid tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the yield is 64.34 percent;
25) 46g (0.11mol) ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is added with 46mL isopropanol and 46mL water, the temperature is increased to 65 ℃, 37% concentrated hydrochloric acid (0.154mol) is added dropwise, the temperature is maintained for reaction for 3h, after TLC monitors that the reaction is completed, 90mL water is added, the temperature is reduced to room temperature, 400mL dichloromethane is added, stirring is carried out, ice bath is carried out to 10 ℃, 20mL of aqueous solution containing 4.4g of NaOH (2.2g) is added dropwise, the pH is adjusted to be neutral, then solution with the mass concentration of 50% is added, the pH is adjusted to be 10, stirring is carried out for 15min, separating, extracting the water phase with dichloromethane (50mL multiplied by 1), combining the organic phases, washing with saturated brine (100mL multiplied by 1), drying with anhydrous magnesium sulfate, concentrating under reduced pressure to evaporate the solvent, dissolving the residue in 80mL of methanol, maintaining the temperature at 24 ℃, slowly adding pure water dropwise until white crystals are separated out, maintaining the temperature at 24 ℃ for crystallization for 5h, filtering, and drying to obtain 26.92g of saxagliptin monohydrate, wherein the yield is 73.40%, and the purity is 99.95%.
Example three:
the preparation method of the saxagliptin comprises the following steps:
mono) 3-hydroxy-1-adamantanemethyl ketone
11) 90.12g (0.5mol) of adamantanecarboxylic acid were added to 225.3mL of dichloromethane, and 119g (1.0mol) of SOCl were added2Refluxing with stirring for 4h, cooling to room temperature, 53.41g (0.4mol) NCS, 13.52g Azobisisoheptonitrile (ABVN), 4.51g Azobisisovaleronitrile (AMBN) and 29.75g (0.25mol) SOCl were added2Heating to reflux again for chlorination reaction for 4h, and rotary evaporating to remove residual SOCl2And methylene chloride, 500mL of CCl was added4Stirring for 10min to dissolve, filtering to remove insoluble substances, concentrating the filtrate, and evaporating to remove CCl4109.41g of 3-chloro-1-adamantyl formyl chloride is obtained as yellow oily liquid, and the crude yield is 93.86%;
12) adding 21.59g (0.94mol) of sodium into 2L of petroleum ether, then dripping 195.44g (1.22mol) of diethyl malonate, and reacting at room temperature for 8h under stirring to obtain a sodium salt solution after dripping;
adding the obtained 3-chloro-1-adamantyl formyl chloride into 500mL of petroleum ether, stirring and dissolving, dropwise adding a sodium salt solution at room temperature, continuing stirring and reacting at room temperature for 12 hours after dropwise adding is finished, obtaining yellow emulsion, adding 1L of water, and stirring for 10 min; separating, collecting the upper petroleum ether layer, extracting the water layer with petroleum ether (500mL × 3), combining the organic layers, and concentrating to obtain 163.78 yellow oily 3-chloro-1-adamantyl diethyl malonate with crude yield of 97.80%;
13) 470mL of glacial acetic acid, 140mL of water and 47mL of concentrated sulfuric acid are added into the obtained 3-chloro-1-adamantanecarboxylate diethyl ester, reflux reaction is carried out for 8 hours, after the reaction is finished, the mixture is poured into ice water (3L), dichloromethane (500mL multiplied by 3) is used for extraction, organic solvents are combined, the solvents are evaporated, and the residue is recrystallized by normal hexane to obtain 85.41g of white crystal 3-chloro-1-adamantanemethyl ketone with the yield of 87.48%.
14) To the 3-chloro-1-adamantanemethyl ketone obtained above were added 2.6L of water and 261g (4.65mol) of KOH, and the mixture was refluxed for 8 hours with stirring, poured into ice water (2.8L), extracted with ethyl acetate (500mL × 3), the organic layers were combined, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated, and the residue was recrystallized with a mixed solvent (cyclohexane/ethyl acetate ═ 10/1, V/V) to obtain 71.52g of white crystalline 3-hydroxy-1-adamantanemethyl ketone with a yield of 91.69%.
Wherein the total yield of the 3-hydroxy-1-adamantanemethyl ketone is 73.63 percent.
II) method for synthesizing saxagliptin
21) Weighing 68g (0.35mol) of 3-hydroxy-1-adamantane methyl ketone, adding the 3-hydroxy-1-adamantane methyl ketone into 350mL of t-BuOH for dissolving, then adding 1.7L of NaOH solution with the mass concentration of 2%, heating to 30 ℃, slowly adding 110.6g (0.7mol) of potassium permanganate in batches, reacting for 4 hours at 40 ℃, adding 680mL of sodium sulfite solution with the mass concentration of 10% after the reaction is finished to treat redundant potassium permanganate, filtering, adjusting the pH value of the filtrate to 1 by hydrochloric acid, separating out white solids, filtering again, and drying to obtain 70.31g of white crystal 3-hydroxy-1-adamantane glyoxylic acid with the yield of 89.58%;
22) 31.2L of NH with pH 8.5 and concentration of 1.0mol/L is taken4Cl-NH4An aqueous OH buffer solution to which 70g (0.312mol) of 3-hydroxy-1-adamantane acetaldehyde was addedAcid and 143.66g (1.56mol) of glycerol are added, then 0.36g of NADH dehydrogenase, 31.2mg of GDH and 31.2mg of phenylalanine dehydrogenase are added, the coupling reaction is carried out by stirring at room temperature, after the reaction is finished, the pH value is adjusted to 2, ethyl acetate (5L multiplied by 3) is added for extraction, organic phases are combined, the organic phases are dried by anhydrous magnesium sulfate and concentrated, and 66.83g of white solid with (S) configuration is obtained, and the yield is 95.08%;
taking 66g (0.29mol) of (S) -configuration white solid, adding 300mL of distilled water for dissolving, adjusting the pH to 10 by concentrated hydrochloric acid, adding 300mL of tetrahydrofuran solution dissolved with 76.39g (0.35mol) of Boc anhydride, stirring at room temperature for reacting overnight, filtering, adjusting the pH of filtrate to 2 by concentrated hydrochloric acid, extracting by ethyl acetate (300mL multiplied by 3), combining extracts, washing by saturated salt water, drying by anhydrous sodium sulfate, spin-drying to remove the solvent, recrystallizing the residual oily matter to obtain 79.13g of (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine, wherein the yield is 83.00%;
23) taking 61g (0.473mol) DIPEA, 60mL acetonitrile and 50mL ethyl acetate, mixing uniformly, and keeping a diisopropylethylamine solution for later use;
69.8g (0.215mol) of (S) -N-t-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine were added to 50g (0.225mol) of (1S,3S,5S) -2-azabicyclo [ 3.1.0)]Hexane-3-formamide methanesulfonate, 45.2g (0.236mol) EDCI, 29g (0.215mol) HOBt and 240mL acetonitrile, stirring, dropwise adding diisopropylethylamine solution at room temperature, heating to 40 ℃ for reaction for 1.5h after dropwise adding, monitoring by TLC that (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine reaction is completed, cooling to 10 ℃, adjusting the pH of the solution to 3 with 1mol/L diluted hydrochloric acid, adding 500mL ethyl acetate, stirring for 10min, standing, separating, washing an organic phase with 1mol/L diluted hydrochloric acid (200mL multiplied by 2), washing with water (200mL multiplied by 1), and saturating KHCO3Washing (200 mL. times.2), drying over anhydrous magnesium sulfate, filtering, and concentrating the filtrate to obtain 84.34g of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0]]Hex-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, yield 90.91%;
24) 80.0g (0.185mol) of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dissolved in 400mL of ethyl acetate, cooled to-5 ℃, 82.2g (0.812mol) of triethylamine is added dropwise, 85.4g (0.407mol) of trifluoroacetic anhydride is added dropwise while maintaining at-5 ℃, reaction is maintained for 1h while maintaining-5 ℃ until TLC monitoring reaction is completed, 120mL of water is added for quenching reaction, liquid separation is carried out, the organic phase is washed with 80mL of diluted hydrochloric acid with the concentration of 1mol/L, potassium carbonate with the mass concentration of 40% (76.6 g) is added, 0.554mol) of aqueous solution and 120mL of methanol, stirring, heating to 40 ℃ for 2h, monitoring by TLC (thin layer chromatography), cooling to room temperature after the reaction is completed, separating liquid, adjusting the pH of the organic phase to 3 by using 2mol/L diluted hydrochloric acid, separating liquid, washing the organic phase with saturated common salt water (100mL multiplied by 1), drying anhydrous magnesium sulfate, concentrating the filtrate to remove the solvent, dissolving the residue in 80mL of acetone, cooling to 15 ℃, dropwise adding 240mL of isopropyl ether, continuing stirring for 3h after dropwise addition, filtering, and drying to obtain 49.15g of white solid tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the yield is 64.28%;
25) 46g (0.11mol) ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is added with 46mL isopropanol and 46mL water, the temperature is increased to 65 ℃, 37% concentrated hydrochloric acid (0.154mol) is added dropwise, the temperature is maintained for reaction for 3h, after TLC monitors that the reaction is completed, 90mL water is added, the temperature is reduced to room temperature, 400mL dichloromethane is added, stirring is carried out, ice bath is carried out to 10 ℃, 20mL of aqueous solution containing 4.4g of NaOH (2.2g) is added dropwise, the pH is adjusted to be neutral, then solution with the mass concentration of 50% is added, the pH is adjusted to be 10, stirring is carried out for 15min, separating, extracting the water phase with dichloromethane (50mL multiplied by 1), combining the organic phases, washing with saturated brine (100mL multiplied by 1), drying with anhydrous magnesium sulfate, concentrating under reduced pressure to evaporate the solvent, dissolving the residue in 80mL methanol, maintaining the temperature at 24 ℃, slowly adding pure water dropwise until white crystals are separated out, maintaining the temperature at 24 ℃ for crystallization for 5h, filtering, and drying to obtain 27.14g of saxagliptin monohydrate, wherein the yield is 74.00%, and the purity is 99.96%.
Example four:
the preparation method of the saxagliptin comprises the following steps:
mono) 3-hydroxy-1-adamantanemethyl ketone
11) 90.12g (0.5mol) of adamantanecarboxylic acid was added to 180.2mL of dichloromethane, and 119g (1.0mol) of SOCl was added2Refluxed for 4h with stirring, cooled to room temperature, and 57.42g (0.43mol) NCS, 12.84g Azobisisoheptonitrile (ABVN), 3.38g Azobisisovaleronitrile (AMBN) and 41.65g (0.35mol) SOCl were added2Heating to reflux again for chlorination reaction for 4 hr, and rotary evaporating to remove residual SOCl2And methylene chloride, 500mL of CCl was added4Stirring for 10min to dissolve, filtering to remove insoluble substances, concentrating the filtrate, and evaporating to remove CCl4109.17g of 3-chloro-1-adamantyl formyl chloride is obtained as yellow oily liquid, and the crude yield is 93.66%;
12) 21.51g (0.94mol) of sodium is taken and added into 2L of petroleum ether, 202.51g (1.26mol) of diethyl malonate is added dropwise, and after the addition, the sodium salt solution is obtained after the reaction at room temperature for 8 hours under stirring;
adding the obtained 3-chloro-1-adamantyl formyl chloride into 500mL of petroleum ether, stirring and dissolving, dropwise adding a sodium salt solution at room temperature, continuing stirring and reacting at room temperature for 12 hours after dropwise adding is finished, obtaining yellow emulsion, adding 1L of water, and stirring for 10 min; separating, collecting the upper petroleum ether layer, extracting the water layer with petroleum ether (500mL × 3), combining the organic layers, and concentrating to obtain 164.47 yellow oily 3-chloro-1-adamantyl diethyl malonate with crude yield of 98.43%;
13) 470mL of glacial acetic acid, 140mL of water and 47mL of concentrated sulfuric acid are added into the obtained 3-chloro-1-adamantanecarboxylate diethyl ester, reflux reaction is carried out for 8 hours, after the reaction is finished, the mixture is poured into ice water (3L), dichloromethane (500mL multiplied by 3) is used for extraction, organic solvents are combined, the solvents are evaporated, and the residue is recrystallized by normal hexane to obtain 85.95g of white crystal 3-chloro-1-adamantanemethyl ketone with the yield of 87.66%.
14) To the 3-chloro-1-adamantanemethyl ketone obtained above were added 2.6L of water and 261g (4.65mol) of KOH, and the mixture was refluxed for 8 hours with stirring, poured into ice water (2.8L), extracted with ethyl acetate (500mL × 3), the organic layers were combined, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated, and the residue was recrystallized with a mixed solvent (cyclohexane/ethyl acetate ═ 10/1, V/V) to obtain 71.95g of white crystalline 3-hydroxy-1-adamantanemethyl ketone with a yield of 91.66%.
Wherein the total yield of the 3-hydroxy-1-adamantanemethyl ketone is 74.07 percent.
II) method for synthesizing saxagliptin
21) Weighing 68g (0.35mol) of 3-hydroxy-1-adamantane methyl ketone, adding the mixture into 350mL of t-BuOH for dissolution, adding 1.7L of NaOH solution with the mass concentration of 2%, heating to 30 ℃, slowly adding 110.6g (0.7mol) of potassium permanganate in batches, reacting for 4 hours at 40 ℃, adding 680mL of sodium sulfite solution with the mass concentration of 10% after the reaction is finished to treat redundant potassium permanganate, filtering, adjusting the pH value of the filtrate to 1 by hydrochloric acid, separating out white solids, filtering again, and drying to obtain 70.27g of white crystal 3-hydroxy-1-adamantane glyoxylic acid with the yield of 89.53%;
22) 31.2L of NH with pH 8.5 and concentration of 1.0mol/L is taken4Cl-NH4Adding 70g (0.312mol) of 3-hydroxy-1-adamantane glyoxylic acid and 143.66g (1.56mol) of glycerol into OH buffer aqueous solution, then adding 0.36g of NADH dehydrogenase, 31.2mg of GDH and 31.2mg of phenylalanine dehydrogenase, stirring at room temperature for coupling reaction, adjusting the pH to 2 after the reaction is finished, then adding ethyl acetate (5L multiplied by 3) for extraction, combining organic phases, drying by anhydrous magnesium sulfate and concentrating to obtain 66.74g of white solid with the (S) configuration, wherein the yield is 94.95%;
taking 66g (0.29mol) of (S) -configuration white solid, adding 300mL of distilled water for dissolving, adjusting the pH to 10 by concentrated hydrochloric acid, adding 300mL of tetrahydrofuran solution dissolved with 76.39g (0.35mol) of Boc anhydride, stirring at room temperature for reacting overnight, filtering, adjusting the pH of filtrate to 2 by concentrated hydrochloric acid, extracting by ethyl acetate (300mL multiplied by 3), combining extracts, washing by saturated salt water, drying by anhydrous sodium sulfate, spin-drying to remove the solvent, recrystallizing the residual oily matter to obtain 79.06g of (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine with the yield of 82.93%;
23) taking 61g (0.473mol) DIPEA, 60mL acetonitrile and 50mL ethyl acetate, mixing uniformly, and keeping a diisopropylethylamine solution for later use;
69.8g (0.215mol) of (S) -N-t-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine were added to 50g (0.225mol) of (1S,3S,5S) -2-azabicyclo [ 3.1.0)]Hexane-3-carboxylic acidAmine methanesulfonate, 45.2g (0.236mol) EDCI, 29g (0.215mol) HOBt and 240mL acetonitrile are stirred and mixed uniformly, diisopropylethylamine solution is added dropwise at room temperature after dropwise addition, the temperature is raised to 40 ℃ for reaction for 1.5h until TLC monitoring (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine reaction is completed, the temperature is reduced to 10 ℃, diluted hydrochloric acid with the concentration of 1mol/L is used for adjusting the pH of the solution to 3, 500mL ethyl acetate is added, stirring is carried out for 10min, standing and liquid separation are carried out, and an organic phase is washed by diluted hydrochloric acid with the concentration of 1mol/L (200mL multiplied by 2), washed by water (200mL multiplied by 1) and saturated KHCO3Washing (200 mL. times.2), drying over anhydrous magnesium sulfate, filtering, and concentrating the filtrate to obtain 84.42g of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0]]Hex-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, yield 90.99%;
24) 80.0g (0.185mol) of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dissolved in 400mL of ethyl acetate, the temperature is reduced to-5 ℃, 82.2g (0.812mol) of triethylamine is added dropwise, 85.4g (0.407mol) of trifluoroacetic anhydride is added dropwise while maintaining the temperature at-5 ℃, the reaction is maintained at-5 ℃ for 1h, 120mL of water is added after the TLC monitoring reaction is completed, liquid separation is carried out, the organic phase is washed with 80mL of 1mol/L dilute hydrochloric acid, potassium carbonate (76.6 g) with the mass concentration of 40% is added, 0.554mol) and 120mL of methanol, stirring, heating to 40 ℃ for reaction for 2h, monitoring by TLC (thin-layer chromatography) that the reaction is complete, cooling to room temperature, separating, adjusting the pH of the organic phase to 3 by using 2mol/L dilute hydrochloric acid, separating, washing the organic phase with saturated common salt water (100mL multiplied by 1), drying anhydrous magnesium sulfate, concentrating the filtrate to remove the solvent, dissolving the residue in 80mL of acetone, cooling to 15 ℃, dropwise adding 240mL of isopropyl ether, continuing stirring for 3h after dropwise addition, filtering, and drying to obtain 49.28g of white solid tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the yield is 64.45 percent;
25) 46g (0.11mol) ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is added with 46mL isopropanol and 46mL water, the temperature is increased to 65 ℃, 37% concentrated hydrochloric acid (0.154mol) is added dropwise, the temperature is maintained for reaction for 3h, after TLC monitors that the reaction is completed, 90mL water is added, the temperature is reduced to room temperature, 400mL dichloromethane is added, stirring is carried out, ice bath is carried out to 10 ℃, 20mL of aqueous solution containing 4.4g of NaOH (2.2g) is added dropwise, the pH is adjusted to be neutral, then solution with the mass concentration of 50% is added, the pH is adjusted to be 10, stirring is carried out for 15min, separating, extracting the water phase with dichloromethane (50mL multiplied by 1), combining the organic phases, washing with saturated saline (100mL multiplied by 1), drying with anhydrous magnesium sulfate, concentrating under reduced pressure to evaporate the solvent, dissolving the residue in 80mL ethanol, maintaining the temperature at 24 ℃, slowly adding pure water dropwise until white crystals are separated out, maintaining the temperature at 24 ℃ for crystallization for 5h, filtering, and drying to obtain 27.91g of saxagliptin monohydrate, wherein the yield is 76.09%, and the purity is 99.97%.
Example five:
the preparation method of the saxagliptin comprises the following steps:
mono) 3-hydroxy-1-adamantanemethyl ketone
11) 90.12g (0.5mol) of adamantanecarboxylic acid was added to 210mL of dichloromethane, and 119g (1.0mol) of SOCl was added2Refluxing with stirring for 4h, cooling to room temperature, 56.08g (0.42mol) NCS, 8.24g Azobisisoheptonitrile (ABVN), 2.57g Azobisisovaleronitrile (AMBN) and 59.5g (0.5mol) SOCl were added2Heating to reflux again for chlorination reaction for 4h, and rotary evaporating to remove residual SOCl2And methylene chloride, 500mL of CCl was added4Stirring for 10min to dissolve, filtering to remove insoluble substances, concentrating the filtrate, and evaporating to remove CCl4110.08g of 3-chloro-1-adamantyl formyl chloride is obtained as yellow oily liquid, and the crude yield is 94.44%;
12) 22.81g (0.99mol) of sodium is taken and added into 2L of petroleum ether, 196.64g (1.23mol) of diethyl malonate is added dropwise, and after the addition, the mixture reacts at room temperature for 8 hours under stirring to obtain sodium salt solution;
adding the obtained 3-chloro-1-adamantyl formyl chloride into 500mL of petroleum ether, stirring and dissolving, dropwise adding a sodium salt solution at room temperature, continuing stirring and reacting at room temperature for 12 hours after dropwise adding is finished, obtaining yellow emulsion, adding 1L of water, and stirring for 10 min; separating, collecting the upper petroleum ether layer, extracting the water layer with petroleum ether (500mL × 3), combining the organic layers, and concentrating to obtain 164.21 yellow oily 3-chloro-1-adamantyl diethyl malonate with crude yield of 97.46%;
13) 470mL of glacial acetic acid, 140mL of water and 47mL of concentrated sulfuric acid are added into the obtained 3-chloro-1-adamantanecarboxyl diethyl malonate, reflux reaction is carried out for 8 hours, after the reaction is finished, the mixture is poured into ice water (3L), dichloromethane (500mL multiplied by 3) is used for extraction, organic solvents are combined, the solvents are evaporated, and the residue is recrystallized by normal hexane to obtain 85.72g of white crystal 3-chloro-1-adamantanemethyl ketone with the yield of 87.57%.
14) To the 3-chloro-1-adamantanemethyl ketone obtained above were added 2.6L of water and 261g (4.65mol) of KOH, and the mixture was refluxed for 8 hours with stirring, poured into ice water (2.8L), extracted with ethyl acetate (500mL × 3), the organic layers were combined, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated, and the residue was recrystallized with a mixed solvent (cyclohexane/ethyl acetate ═ 10/1, V/V) to obtain 71.77g of 3-hydroxy-1-adamantanemethyl ketone as white crystals, with a yield of 91.68%.
Wherein the total yield of the 3-hydroxy-1-adamantanemethyl ketone is 73.89 percent.
II) method for synthesizing saxagliptin
21) Weighing 68g (0.35mol) of 3-hydroxy-1-adamantane methyl ketone, adding the mixture into 350mL of t-BuOH for dissolution, adding 1.7L of NaOH solution with the mass concentration of 2%, heating to 30 ℃, slowly adding 110.6g (0.7mol) of potassium permanganate in batches, reacting for 4 hours at 40 ℃, adding 680mL of sodium sulfite solution with the mass concentration of 10% after the reaction is finished to treat redundant potassium permanganate, filtering, adjusting the pH value of the filtrate to 1 by hydrochloric acid, separating out white solids, filtering again, and drying to obtain 70.29g of white crystal 3-hydroxy-1-adamantane glyoxylic acid with the yield of 89.56%;
22) 31.2L of NH with pH 8.5 and concentration of 1.0mol/L is taken4Cl-NH4Adding 70g (0.312mol) of 3-hydroxy-1-adamantane glyoxylic acid and 143.66g (1.56mol) of glycerol into OH buffer solution, adding 0.36g of NADH dehydrogenase, 31.2mg of GDH and 31.2mg of phenylalanine dehydrogenase, stirring at room temperature for coupling reaction, adjusting pH to 2 after the reaction is finished, adding ethyl acetate (5L multiplied by 3) for extraction, combining organic phases, drying by anhydrous magnesium sulfate, and concentrating to obtain 66 percent of solution76g of a white solid of (S) configuration, yield 94.98%;
taking 66g (0.29mol) of (S) -configuration white solid, adding 300mL of distilled water for dissolving, adjusting the pH to 10 by concentrated hydrochloric acid, adding 300mL of tetrahydrofuran solution dissolved with 76.39g (0.35mol) of Boc anhydride, stirring at room temperature for reaction overnight, performing suction filtration, adjusting the pH of filtrate to 2 by concentrated hydrochloric acid, extracting by ethyl acetate (300mL multiplied by 3), combining extract liquor, washing by saturated salt water, drying by anhydrous sodium sulfate, removing the solvent by spin drying, and recrystallizing the residual oily matter to obtain 79.16g of (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine with the yield of 83.04%;
23) taking 61g (0.473mol) DIPEA, 60mL acetonitrile and 50mL ethyl acetate, mixing uniformly, and keeping a diisopropylethylamine solution for later use;
69.8g (0.215mol) of (S) -N-t-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine were added to 50g (0.225mol) of (1S,3S,5S) -2-azabicyclo [ 3.1.0)]Hexane-3-formamide methanesulfonate, 45.2g (0.236mol) EDCI, 29g (0.215mol) HOBt and 240mL acetonitrile, stirring, dropwise adding diisopropylethylamine solution at room temperature, heating to 40 ℃ for reaction for 1.5h after dropwise adding, monitoring by TLC that (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine reaction is completed, cooling to 10 ℃, adjusting the pH of the solution to 3 with 1mol/L diluted hydrochloric acid, adding 500mL ethyl acetate, stirring for 10min, standing, separating, washing an organic phase with 1mol/L diluted hydrochloric acid (200mL multiplied by 2), washing with water (200mL multiplied by 1), and saturating KHCO3Washing (200 mL. times.2), drying over anhydrous magnesium sulfate, filtering, and concentrating the filtrate to obtain 84.38g of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0]]Hex-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, yield 90.95%;
24) 80.0g (0.185mol) of tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dissolved in 400mL of ethyl acetate, cooled to-5 ℃, 82.2g (0.812mol) of triethylamine is added dropwise, 85.4g (0.407mol) of trifluoroacetic anhydride is added dropwise while maintaining at-5 ℃, reaction is maintained for 1h while maintaining-5 ℃ until TLC monitoring reaction is completed, 120mL of water is added for quenching reaction, liquid separation is carried out, the organic phase is washed with 80mL of diluted hydrochloric acid with the concentration of 1mol/L, potassium carbonate with the mass concentration of 40% (76.6 g) is added, 0.554mol) and 120mL of methanol, stirring, heating to 40 ℃ for reaction for 2h, monitoring by TLC (thin-layer chromatography) that the reaction is completed, cooling to room temperature, separating, adjusting the pH of the organic phase to 3 by using 2mol/L diluted hydrochloric acid, separating, washing the organic phase with saturated common salt water (100mL multiplied by 1), drying anhydrous magnesium sulfate, concentrating the filtrate to remove the solvent, dissolving the residue in 80mL of acetone, cooling to 15 ℃, dropwise adding 240mL of isopropyl ether, continuing stirring for 3h after dropwise addition, filtering, and drying to obtain 49.23g of white solid tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the yield is 64.39%;
25) 46g (0.11mol) ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is added with 46mL isopropanol and 46mL water, the temperature is increased to 65 ℃, 37% concentrated hydrochloric acid (0.154mol) is added dropwise, the temperature is maintained for reaction for 3h, after TLC monitors that the reaction is completed, 90mL water is added, the temperature is reduced to room temperature, 400mL dichloromethane is added, stirring is carried out, ice bath is carried out to 10 ℃, 20mL of aqueous solution containing 4.4g of NaOH (2.2g) is added dropwise, the pH is adjusted to be neutral, then solution with the mass concentration of 50% is added, the pH is adjusted to be 10, stirring is carried out for 15min, separating, extracting the water phase with dichloromethane (50mL multiplied by 1), combining the organic phases, washing with saturated saline (100mL multiplied by 1), drying with anhydrous magnesium sulfate, concentrating under reduced pressure to evaporate the solvent, dissolving the residue in 80mL ethanol, maintaining the temperature at 24 ℃, slowly adding pure water dropwise until white crystals are separated out, maintaining the temperature at 24 ℃ for crystallization for 5h, filtering, and drying to obtain 27.82g saxagliptin monohydrate, wherein the yield is 75.85%, and the purity is 99.95%.
Although embodiments of the present invention have been shown and described, it will be appreciated by those skilled in the art that various changes, modifications, substitutions and alterations can be made in these embodiments without departing from the principles and spirit of the invention, the scope of which is defined in the appended claims and their equivalents.
Claims (10)
1. A preparation method of 3-hydroxy-1-adamantane methyl ketone is characterized by comprising the following steps: the preparation method comprises the following steps of:
11) adding adamantane formic acid and thionyl chloride into dichloromethane, adding N-chlorosuccinimide, adding azodiisoheptanonitrile and azodiisovaleronitrile as initiators, and refluxing for chlorination reaction to obtain 3-chloro-1-adamantyl formyl chloride;
12) taking sodium and diethyl malonate to react in a nonpolar solvent for mixing reaction to obtain a sodium salt solution;
dissolving 3-chloro-1-adamantyl formyl chloride in a nonpolar solvent, slowly dripping sodium salt solution, reacting at room temperature after dripping, and obtaining 3-chloro-1-adamantyl formyl diethyl malonate after the reaction is finished;
13) taking 3-chloro-1-adamantane formyl diethyl malonate, adding glacial acetic acid, water and sulfuric acid, and performing reflux reaction to obtain 3-chloro-1-adamantane methyl ketone;
14) taking 3-chloro-1-adamantane methyl ketone to perform reflux reaction with strong base in water to obtain the 3-hydroxy-1-adamantane methyl ketone, wherein the specific chemical reaction formula is as follows:

2. the method for producing 3-hydroxy-1-adamantanemethyl ketone according to claim 1, characterized in that: in the step 11), the molar ratio of the adamantane carboxylic acid to the dichloromethane to the N-chlorosuccinimide is 1: 2.5-3.0: 0.8 to 0.9;
the weight ratio of the adamantane carboxylic acid to the initiator is 1: 0.1 to 0.2;
the weight ratio of the azodiisoheptanonitrile to the azodiisovaleronitrile is 3-4: 1.
3. the method for producing 3-hydroxy-1-adamantanemethyl ketone according to claim 1 or 2, characterized in that: in the step 11), after the reaction is finished, concentrating to remove residual reactants and solvent, adding carbon tetrachloride, dissolving, filtering, and concentrating filtrate to obtain the 3-chloro-1-adamantyl formyl chloride.
4. The method for producing 3-hydroxy-1-adamantanemethyl ketone according to claim 1 or 2, characterized in that: in the step 11), the weight volume ratio of the adamantane carboxylic acid to the dichloromethane is 1 g: 2-2.5 mL.
5. The method for producing 3-hydroxy-1-adamantanemethyl ketone according to claim 1 or 2, characterized in that: in the step 12), the molar ratio of 3-chloro-1-adamantyl formyl chloride, sodium and diethyl malonate is 1: 2-2.1: 2.5 to 2.7.
6. The method for producing 3-hydroxy-1-adamantanemethyl ketone according to claim 1 or 2, characterized in that: in step 12), the nonpolar solvent is petroleum ether.
7. A method for synthesizing saxagliptin by using the 3-hydroxy-1-adamantanemethyl ketone prepared by the preparation method of any one of claims 1 to 6 as a raw material.
8. The process for the synthesis of saxagliptin according to claim 7, characterized in that: the method for synthesizing saxagliptin comprises the following steps of:
21) the 3-hydroxy-1-adamantane methyl ketone is used as a raw material, and methyl is oxidized by potassium permanganate under an alkaline condition to obtain the 3-hydroxy-1-adamantane glyoxylic acid, wherein the specific chemical reaction formula is as follows:

22) the 3-hydroxy-1-adamantane glyoxylic acid and glycerol are subjected to coupling reaction under the action of NADH dehydrogenase, 3-phosphoglycerol dehydrogenase and phenylalanine dehydrogenase, and then subjected to Boc amino protection reaction with Boc anhydride to obtain (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine, wherein the specific chemical reaction formula is as follows:

23) taking (S) -N-tert-butoxycarbonyl- (3-hydroxyadamantan-1-yl) glycine and (1S,3S,5S) -2-azabicyclo [3.1.0] hexane-3-formamide mesylate to carry out condensation reaction to obtain tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, wherein the specific chemical reaction formula is as follows:

24) tert-butyl ((S) -2- ((1S,3S,5S) -3-carbamoyl-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid is dehydrated by trifluoroacetic anhydride to give tert-butyl ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexan-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid, the specific chemical reaction formula is as follows:

25) taking ((S) -2- ((1S,3S,5S) -3-cyano-2-azabicyclo [3.1.0] hexane-2-yl) -1- ((1R,3R,5R,7S) -3-hydroxyadamantan-1-yl) -2-oxoethyl) carbamic acid to remove Boc under the action of hydrochloric acid, and carrying out post-treatment to obtain the saxagliptin, wherein the specific chemical reaction formula is as follows:

9. the process for the synthesis of saxagliptin according to claim 8, characterized in that: in the step 5), after removing Boc, sequentially adding water and dichloromethane, adding a sodium hydroxide aqueous solution and a potassium carbonate aqueous solution to adjust the pH value to be alkaline, carrying out phase separation, washing, drying and concentrating an organic phase, and recrystallizing the obtained residue to obtain the saxagliptin.
10. The process for the synthesis of saxagliptin according to claim 9, characterized in that: in the step 5), the recrystallization is to dissolve the residue in methanol/ethanol, maintain the temperature at 24 ℃, dropwise add water until white crystals are separated out, maintain the temperature at 24 ℃ for crystallization, filter and dry to obtain the saxagliptin.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210336222.5A CN114621068A (en) | 2022-03-31 | 2022-03-31 | Preparation method of 3-hydroxy-1-adamantane methyl ketone and method for synthesizing saxagliptin |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210336222.5A CN114621068A (en) | 2022-03-31 | 2022-03-31 | Preparation method of 3-hydroxy-1-adamantane methyl ketone and method for synthesizing saxagliptin |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN114621068A true CN114621068A (en) | 2022-06-14 |
Family
ID=81905197
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210336222.5A Pending CN114621068A (en) | 2022-03-31 | 2022-03-31 | Preparation method of 3-hydroxy-1-adamantane methyl ketone and method for synthesizing saxagliptin |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114621068A (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115417794A (en) * | 2022-09-21 | 2022-12-02 | 扬州市三药制药有限公司 | Preparation method of saxagliptin intermediate |
| CN115894207A (en) * | 2022-12-08 | 2023-04-04 | 南京康立瑞生物科技有限公司 | Synthesis method of 2- (3-hydroxy-1-adamantane) -2-oxoacetic acid |
| CN116023263A (en) * | 2022-11-17 | 2023-04-28 | 苏州汉德创宏生化科技有限公司 | Synthesis method of intermediate 3- (3, 5-dichlorophenyl) -3-oxo-ethyl propionate |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102070451A (en) * | 2002-12-09 | 2011-05-25 | 布里斯托尔-迈尔斯斯奎布公司 | Methods and compounds producing dipeptidyl peptidase IV inhibitors and intermediates thereof |
| CN104098505A (en) * | 2014-07-31 | 2014-10-15 | 天津民祥生物医药科技有限公司 | Preparation method for saxagliptin |
| CN104817476A (en) * | 2015-03-25 | 2015-08-05 | 重庆医科大学 | Preparation method for unnatural amino acids |
| CN105503698A (en) * | 2014-09-26 | 2016-04-20 | 深圳翰宇药业股份有限公司 | Method for synthesizing Saxagliptin and intermediate |
| CN109320425A (en) * | 2018-09-03 | 2019-02-12 | 赵旭萌 | A kind of synthetic method of rimantadine hydrochloride |
-
2022
- 2022-03-31 CN CN202210336222.5A patent/CN114621068A/en active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102070451A (en) * | 2002-12-09 | 2011-05-25 | 布里斯托尔-迈尔斯斯奎布公司 | Methods and compounds producing dipeptidyl peptidase IV inhibitors and intermediates thereof |
| CN104098505A (en) * | 2014-07-31 | 2014-10-15 | 天津民祥生物医药科技有限公司 | Preparation method for saxagliptin |
| CN105503698A (en) * | 2014-09-26 | 2016-04-20 | 深圳翰宇药业股份有限公司 | Method for synthesizing Saxagliptin and intermediate |
| CN104817476A (en) * | 2015-03-25 | 2015-08-05 | 重庆医科大学 | Preparation method for unnatural amino acids |
| CN109320425A (en) * | 2018-09-03 | 2019-02-12 | 赵旭萌 | A kind of synthetic method of rimantadine hydrochloride |
Non-Patent Citations (3)
| Title |
|---|
| A. G. YURCHENKO ET AL.: ""On The Mechanism of Liquid Phass Halogenation of Adamantane Derivatives"", 《TETRAHEDRON LETTERS》 * |
| A. L. FRIDMAN ET AL.: ""SYNTHESIS AND BIOLOGICAL ACTIVITY OF ADAMANTANE DERIVATIVES"" * |
| VLADIMIR BURMISTROV ET AL.: ""Effects of adamantane alterations on soluble epoxide hydrolase inhibition potency, physical properties and metabolic stability"", 《BIOORGANIC CHEMISTRY》 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115417794A (en) * | 2022-09-21 | 2022-12-02 | 扬州市三药制药有限公司 | Preparation method of saxagliptin intermediate |
| CN115417794B (en) * | 2022-09-21 | 2024-03-01 | 扬州市三药制药有限公司 | Preparation method of saxagliptin intermediate |
| CN116023263A (en) * | 2022-11-17 | 2023-04-28 | 苏州汉德创宏生化科技有限公司 | Synthesis method of intermediate 3- (3, 5-dichlorophenyl) -3-oxo-ethyl propionate |
| CN115894207A (en) * | 2022-12-08 | 2023-04-04 | 南京康立瑞生物科技有限公司 | Synthesis method of 2- (3-hydroxy-1-adamantane) -2-oxoacetic acid |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111423452B (en) | Intermediates of Relugoli and their preparation methods and applications | |
| CN114621068A (en) | Preparation method of 3-hydroxy-1-adamantane methyl ketone and method for synthesizing saxagliptin | |
| CN106748950A (en) | A kind of preparation method of Bu Waxitan and its intermediate | |
| CN106966947B (en) | A kind of preparation method of vildagliptin | |
| CN108191829B (en) | Method for preparing Vonoprazan fumarate by using Vonoprazan fumarate intermediate IV | |
| CN109206317B (en) | A kind of preparation technology of amantadine nitrate derivatives | |
| WO2019019795A1 (en) | Method for preparing sacubitril intermediate | |
| WO2014020555A2 (en) | An improved process for the preparation of dabigatran etexilate mesylate | |
| CN113717176B (en) | Method for preparing remazolam | |
| CN114213283A (en) | One-pot method for the preparation of [2-[1-(Fmoc-amino)ethoxy]ethoxy]acetic acid | |
| CN114478690A (en) | Preparation method of 6, 6-dimethyl-3-azabicyclo [3.1.0] hexane derivative | |
| WO2021238965A1 (en) | Method for preparing methyl (s)-2-amino-3-(4-(2,3-dimethylpyridin-4-yl)phenylpropionate and salt thereof | |
| WO2012167413A1 (en) | Method for preparing optically pure (-)-clausenamide compound | |
| CN116535384A (en) | The preparation method of ticagrelor intermediate | |
| CN108440409B (en) | Green and efficient preparation method of rebamipide | |
| CN116836150A (en) | Preparation method of lebo Lei Sheng and intermediate compound thereof | |
| CN116354855B (en) | A kind of preparation method of silvelestat sodium | |
| CN106608853A (en) | Preparation method of dipeptidyl peptidase IV inhibitor | |
| CN111205216A (en) | Method for preparing saxagliptin | |
| CN106117104B (en) | A kind of preparation method of vildagliptin | |
| CN117186018A (en) | A new method for synthesizing key intermediates of Haibo Maibu | |
| CN105622460B (en) | (R) synthetic method of N tertbutyloxycarbonyls biphenyl Propanolamine | |
| WO2024092892A1 (en) | Edoxaban intermediate and preparation method therefor | |
| CN108314688A (en) | A kind of synthetic method of sitagliptin | |
| CN111072543B (en) | Preparation method and application of (3R,4S) -4-ethylpyrrolidine-3-carboxylic acid compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20220614 |
|
| RJ01 | Rejection of invention patent application after publication |