CN114524767A - Synthetic method of sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride - Google Patents
Synthetic method of sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride Download PDFInfo
- Publication number
- CN114524767A CN114524767A CN202210254550.0A CN202210254550A CN114524767A CN 114524767 A CN114524767 A CN 114524767A CN 202210254550 A CN202210254550 A CN 202210254550A CN 114524767 A CN114524767 A CN 114524767A
- Authority
- CN
- China
- Prior art keywords
- compound
- reaction
- stage
- dichloro
- synthesis method
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- OQLXYGUEFSFZIX-UHFFFAOYSA-N 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline;hydrochloride Chemical compound Cl.C1CNCC2=CC(Cl)=CC(Cl)=C21 OQLXYGUEFSFZIX-UHFFFAOYSA-N 0.000 title claims abstract description 22
- 238000010189 synthetic method Methods 0.000 title description 3
- 238000006243 chemical reaction Methods 0.000 claims abstract description 44
- 150000001875 compounds Chemical class 0.000 claims abstract description 32
- 229940125782 compound 2 Drugs 0.000 claims abstract description 24
- 239000003054 catalyst Substances 0.000 claims abstract description 19
- 238000001308 synthesis method Methods 0.000 claims abstract description 19
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims abstract description 18
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 18
- 229940126214 compound 3 Drugs 0.000 claims abstract description 16
- 229940125898 compound 5 Drugs 0.000 claims abstract description 16
- 239000003513 alkali Substances 0.000 claims abstract description 10
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 10
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims abstract description 9
- 229940125904 compound 1 Drugs 0.000 claims abstract description 9
- 239000011630 iodine Substances 0.000 claims abstract description 9
- 229910052740 iodine Inorganic materials 0.000 claims abstract description 9
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 claims abstract description 8
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 claims abstract description 8
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims abstract description 6
- 239000002535 acidifier Substances 0.000 claims abstract description 6
- 235000011114 ammonium hydroxide Nutrition 0.000 claims abstract description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 4
- 239000001257 hydrogen Substances 0.000 claims abstract description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 39
- 239000007810 chemical reaction solvent Substances 0.000 claims description 27
- 239000003153 chemical reaction reagent Substances 0.000 claims description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical group Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 12
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical group CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 10
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 claims description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 9
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical group CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 8
- 229910052802 copper Inorganic materials 0.000 claims description 8
- 239000010949 copper Substances 0.000 claims description 8
- 238000006722 reduction reaction Methods 0.000 claims description 8
- 239000002841 Lewis acid Substances 0.000 claims description 7
- 150000007517 lewis acids Chemical class 0.000 claims description 7
- WJPYOCIWVYDFDT-UHFFFAOYSA-N ethyl 3-oxo-4-(2,4,5-trifluorophenyl)butanoate Chemical compound CCOC(=O)CC(=O)CC1=CC(F)=C(F)C=C1F WJPYOCIWVYDFDT-UHFFFAOYSA-N 0.000 claims description 6
- 150000007530 organic bases Chemical class 0.000 claims description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical group C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 6
- 229910021595 Copper(I) iodide Inorganic materials 0.000 claims description 5
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical group I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 claims description 5
- 229910052763 palladium Inorganic materials 0.000 claims description 5
- 229910000073 phosphorus hydride Inorganic materials 0.000 claims description 5
- LAIZPRYFQUWUBN-UHFFFAOYSA-L nickel chloride hexahydrate Chemical compound O.O.O.O.O.O.[Cl-].[Cl-].[Ni+2] LAIZPRYFQUWUBN-UHFFFAOYSA-L 0.000 claims description 4
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 claims description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 claims description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 3
- 238000007112 amidation reaction Methods 0.000 claims description 3
- 238000005859 coupling reaction Methods 0.000 claims description 3
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical group Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 claims description 3
- 238000006192 iodination reaction Methods 0.000 claims description 3
- 229910052700 potassium Inorganic materials 0.000 claims description 3
- 239000011591 potassium Substances 0.000 claims description 3
- 239000012279 sodium borohydride Substances 0.000 claims description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 3
- 239000002585 base Substances 0.000 claims description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 claims description 2
- 239000002994 raw material Substances 0.000 abstract description 9
- 238000009776 industrial production Methods 0.000 abstract description 4
- 230000015572 biosynthetic process Effects 0.000 abstract description 2
- 229940079593 drug Drugs 0.000 abstract description 2
- 239000003814 drug Substances 0.000 abstract description 2
- 238000003786 synthesis reaction Methods 0.000 abstract description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- 239000012074 organic phase Substances 0.000 description 16
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 8
- 206010013774 Dry eye Diseases 0.000 description 8
- 238000000034 method Methods 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- QCSLOZFEMQTPJA-UHFFFAOYSA-N 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline Chemical compound C1CNCC2=CC(Cl)=CC(Cl)=C21 QCSLOZFEMQTPJA-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- CASRSOJWLARCRX-UHFFFAOYSA-N 3,5-dichlorobenzaldehyde Chemical compound ClC1=CC(Cl)=CC(C=O)=C1 CASRSOJWLARCRX-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- JFOZKMSJYSPYLN-QHCPKHFHSA-N lifitegrast Chemical compound CS(=O)(=O)C1=CC=CC(C[C@H](NC(=O)C=2C(=C3CCN(CC3=CC=2Cl)C(=O)C=2C=C3OC=CC3=CC=2)Cl)C(O)=O)=C1 JFOZKMSJYSPYLN-QHCPKHFHSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000013341 scale-up Methods 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- QKWWDTYDYOFRJL-UHFFFAOYSA-N 2,2-dimethoxyethanamine Chemical compound COC(CN)OC QKWWDTYDYOFRJL-UHFFFAOYSA-N 0.000 description 1
- VHJKDOLGYMULOP-UHFFFAOYSA-N 2-(2,4-dichlorophenyl)ethanamine Chemical compound NCCC1=CC=C(Cl)C=C1Cl VHJKDOLGYMULOP-UHFFFAOYSA-N 0.000 description 1
- ONRREFWJTRBDRA-UHFFFAOYSA-N 2-chloroethanamine;hydron;chloride Chemical compound [Cl-].[NH3+]CCCl ONRREFWJTRBDRA-UHFFFAOYSA-N 0.000 description 1
- PWKJMPFEQOHBAC-UHFFFAOYSA-N 4-Ethyloctanoic acid Chemical compound CCCCC(CC)CCC(O)=O PWKJMPFEQOHBAC-UHFFFAOYSA-N 0.000 description 1
- LRTAHCUSBZEOBO-UHFFFAOYSA-N 5,7-dichloroisoquinoline Chemical compound C1=CN=CC2=CC(Cl)=CC(Cl)=C21 LRTAHCUSBZEOBO-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000009319 Keratoconjunctivitis Sicca Diseases 0.000 description 1
- 206010052143 Ocular discomfort Diseases 0.000 description 1
- 208000023715 Ocular surface disease Diseases 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 239000002262 Schiff base Substances 0.000 description 1
- 150000004753 Schiff bases Chemical class 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 description 1
- 206010047571 Visual impairment Diseases 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 229960005381 lifitegrast Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 229960004034 sitagliptin Drugs 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 208000029257 vision disease Diseases 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- 229940023106 xiidra Drugs 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
The invention relates to the technical field of drug synthesis, and discloses a synthesis method of a sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride, which comprises the following steps: after hydrogen is extracted from the compound 1 by n-butyl lithium, the compound reacts with iodine to obtain a compound 2; condensing the compound 2 with a condensing agent to obtain an intermediate state; condensing with methyl methoxy amine hydrochloride in the presence of organic alkali to obtain a compound 3; reducing the compound 3 with a reducing agent to obtain a compound 4; in the first stage, a compound 4 is catalyzed by a first catalyst in the presence of alkali and is coupled with trimethylsilyl acetylene; in the second stage, the second catalyst is used for catalyzing, and the second catalyst reacts with ammonia water to close a ring to obtain a compound 5; and reducing the compound 5 by a reducing agent, and acidifying by an acidifying agent to obtain a compound 6. The synthesis method has the advantages of low price and easy obtainment of used raw materials, mild reaction conditions, no use of high-risk reaction, simple requirement on used equipment, high yield and lower total cost, and is suitable for large-scale industrial production.
Description
Technical Field
The invention relates to the technical field of drug synthesis, in particular to a synthesis method of a sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride.
Background
Dry eye, also known as keratoconjunctivitis sicca, is a multifactorial tear and ocular surface disease that can lead to symptoms such as ocular discomfort, visual impairment, and tear film instability, with concomitant increases in tear film permeability and ocular surface inflammation. Dry eye is a chronic, and often progressive, disorder. Depending on the cause and severity, dry eye sometimes does not heal completely. In most cases, however, disease management of dry eye can be successful, and by administering appropriate treatment, eye comfort can be significantly improved, dry eye symptoms can be alleviated, and sometimes even clearer vision can be achieved.
Ritatest (Lifitegrast) was a former research by Shire pharmaceutical company, uk, for the treatment of signs and symptoms of dry eye, with trade names: xiidra. The eye drops of Lifitestast approved by Shire corporation in 2016, 7, 11 days in the United states by FDA are the first new drug approved by FDA for treating dry eye, and are used 2 times a day, about 12 hours apart.
The sitagliptin consists of three fragments, wherein one key fragment is 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride, the structural formula of which is shown as the following,

the literature reports methods for synthesizing 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride to date mainly include the following methods:
the first method is to take 3, 5-dichlorobenzaldehyde as a raw material, condense the raw material with 2-chloroethylamine hydrochloride, reduce the raw material by sodium cyanoborohydride, and then close a ring in the presence of aluminum trichloride to obtain 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline (the technical route is shown as follows). Although the reaction process is short, the reaction temperature of the second step is as high as 185 ℃, general equipment cannot be used, only special high-temperature kettles can be used, and the process is not suitable for scale-up production (ACS Medicinal Chemistry Letters 2012,3(3), 203-.

In the second method, 2, 4-dichlorobenzyl cyanide is used as a raw material, nickel is used for catalytic hydrogenation to obtain 2, 4-dichlorophenethylamine, acetic anhydride is used for acetylation, 5, 7-dichlorotetrahydroisoquinoline protected by acetyl is obtained by cyclization with paraformaldehyde in the presence of sulfuric acid and acetic acid, and finally, the 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline is obtained by hydrochloric acid hydrolysis (the technical route is shown as follows). The scheme has a problem that the first step hydrogenation reduction cyano-group reaction belongs to high-risk reaction, is easy to cause safety accidents, and has higher safety risk in production (the patent publication number is CN 111057003A).

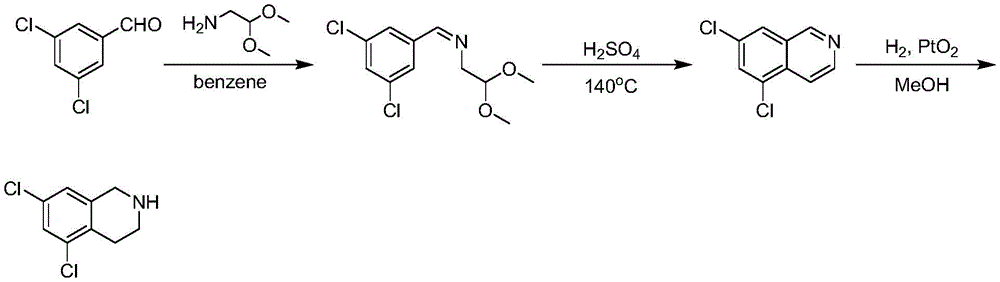
The third method is to take 3, 5-dichlorobenzaldehyde as raw material, condense with 2-aminoacetaldehyde dimethyl acetal to obtain Schiff base, close the ring in sulfuric acid to obtain 5, 7-dichloroisoquinoline, then hydrogenate with platinum dioxide to obtain 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline (the technical route is shown as follows). The problem with this route is that the second step reaction temperature is 140 ℃, the temperature is relatively high, scale-up production is not required, and the reaction solvent is concentrated sulfuric acid, which is very dangerous at high temperatures. The third step is that the hydrogenation reaction belongs to high-risk reaction and has larger safety hazard (Journal of Medicinal Chemistry 1980,23(5), 506-.
In summary, the existing method for synthesizing 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride has certain limitations, and the safety risk of mass production is higher.
Disclosure of Invention
< problems to be solved by the present invention >
The prior synthesis method of 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride is not suitable for large-scale production and has the problem of safety risk in production.
< technical solution adopted in the present invention >
Aiming at the technical problems, the invention aims to provide a synthesis method of a sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride, which has the advantages of easily obtained raw materials, no high-risk reaction, mild conditions, high yield and suitability for industrial production.
The specific contents are as follows:
the synthesis method of the sitagliptin intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride comprises the following steps:
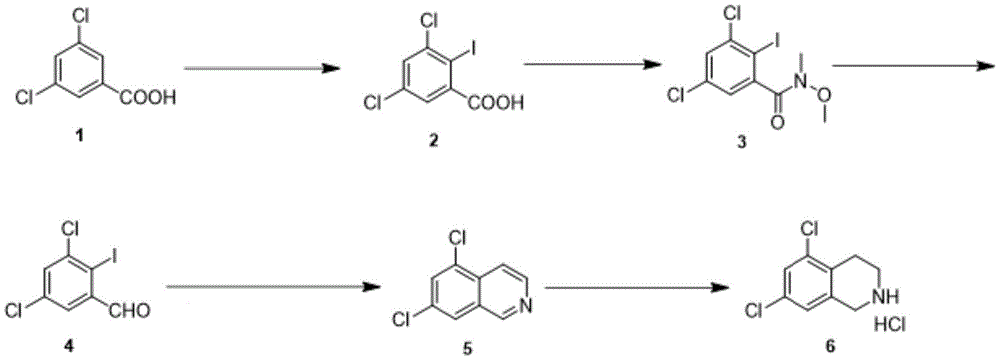
(1) iodination reaction: after hydrogen is extracted from the compound 1 by n-butyl lithium, the compound reacts with iodine to obtain a compound 2;
(2) amidation reaction: condensing the compound 2 with a condensing agent to obtain an intermediate state; then condensing with methyl methoxy amine hydrochloride in the presence of organic alkali to obtain a compound 3;
(3) reduction reaction: reducing the compound 3 to obtain a compound 4;
(4) coupling reaction: in the first stage, a compound 4 is catalyzed by a first catalyst in the presence of alkali and is coupled with trimethylsilyl acetylene; in the second stage, the second catalyst is used for catalyzing, and the second catalyst reacts with ammonia water to close the ring to obtain a compound 5;
(5) reduction reaction: the compound 5 is reduced by a reducing agent in the presence of Lewis acid, and is acidified by an acidifying agent to obtain a compound 6.
< technical mechanism adopted in the present invention >
By adopting a Sonogashira reaction, alkynyl is introduced on a benzene ring under the action of a first catalyst, then the ring is closed to form isoquinoline under the action of a second catalyst, and finally the tetrahydroisoquinoline structure is obtained by reduction through a reducing agent.
< advantageous effects of the present invention >
The synthesis method has the advantages of low price and easy obtainment of used raw materials, mild reaction conditions, no use of high-risk reaction, simple requirement on used equipment, high yield, lower total cost, suitability for large-scale industrial production and good application prospect.
Drawings
FIG. 1 is a spectrum of Compound 6 of example 8.
Detailed Description
In order to make the objects, technical solutions and advantages of the embodiments of the present invention clearer, the technical solutions in the embodiments of the present invention will be clearly and completely described below. The examples, in which specific conditions are not specified, were conducted under conventional conditions or conditions recommended by the manufacturer. The reagents or instruments used are not indicated by the manufacturer, and are all conventional products available commercially.
The synthesis method of the sitagliptin intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride comprises the following steps:
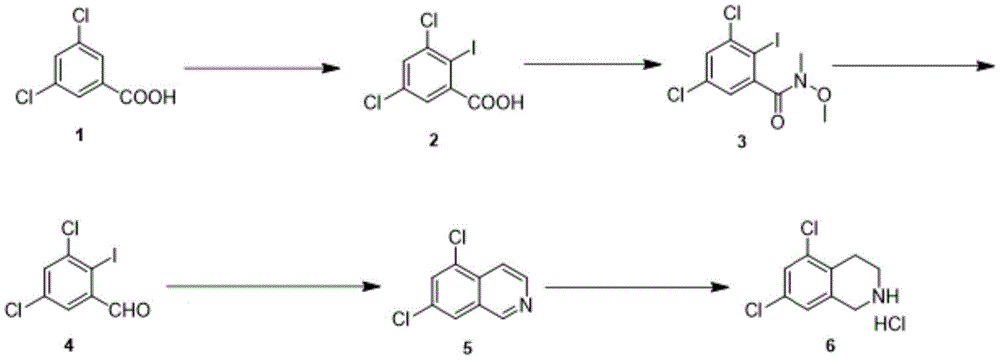
(1) iodination reaction: after hydrogen is extracted from the compound 1 by n-butyl lithium, the compound reacts with iodine to obtain a compound 2;
(2) amidation reaction: condensing the compound 2 with a condensing agent to obtain an intermediate state; then condensing with methyl methoxy amine hydrochloride in the presence of organic alkali to obtain a compound 3;
(3) reduction reaction: reducing the compound 3 to obtain a compound 4;
(4) coupling reaction: in the first stage, a compound 4 is catalyzed by a first catalyst in the presence of alkali and is coupled with trimethylsilyl acetylene; in the second stage, the second catalyst is used for catalyzing, and the second catalyst reacts with ammonia water to close the ring to obtain a compound 5;
(5) reduction reaction: the compound 5 is reduced by a reducing agent in the presence of Lewis acid, and is acidified by an acidifying agent to obtain a compound 6.
The condensing agent used in the step (2) is CDI or EDCI; the organic base is diisopropylethylamine or triethylamine;
and/or the reducing agent used in the step (3) is diisobutylaluminum hydride;
and/or, in the step (4), in the first stage, the used base is triethylamine or diisopropylethylamine, and the first catalyst is a mixed reagent comprising a palladium reagent, a copper reagent and a phosphine reagent; in the second stage, the second catalyst is a copper reagent;
and/or, in the step (5), the reducing agent is sodium borohydride or potassium borohydride, and the Lewis acid is nickel chloride hexahydrate; the acidifying agent is hydrochloric acid.
In the invention, in the step (1), the molar ratio of the compound 1, n-butyllithium and iodine is 1.0: 2.0-3.0: 1.0-1.5.
And/or in the step (1), the volume ratio of the compound 1 to the reaction solvent is 1.0: 8.0.
In the invention, in the step (2), the molar ratio of the compound 2, the condensing agent, the methyl methoxylamine hydrochloride and the organic base is 1.0: 1.0-1.3: 1.2-2.5;
and/or in the step (2), the volume ratio of the compound 2 to the reaction solvent is 1.0: 8.0.
In the present invention, in the step (3), the molar ratio of the compound 3 to the reducing agent is 1.0:1.0 to 1.5.
And/or in the step (3), the volume ratio of the compound 2 to the reaction solvent is 1.0: 8.0.
In the invention, in the step (4), in the first stage, the molar ratio of the compound 4, the alkali, the palladium reagent, the copper reagent, the phosphine reagent and the trimethylsilyl acetylene is 1.0: 2.0-2.5: 0.02-0.05: 0.1-0.15: 0.04-0.1: 1.2-1.5; in the second stage, the molar ratio of the compound 4 to the second catalyst to the ammonia water is 1.0: 0.05-0.1: 2.0-3.0;
and/or, in the step (4), in the first stage, the volume ratio of the compound 2 to the reaction solvent is 1.0: 8.0; in the second stage, the volume ratio of compound 2 to the reaction solvent was 1.0: 7.0.
In the invention, in the step (5), the molar ratio of the compound 5, the reducing agent and the Lewis acid is 1.0: 2.5-3.0: 1.0-1.5.
And/or in the step (5), the volume ratio of the compound 2 to the reaction solvent is 1: 10.0.
In the invention, the reaction solvent used in the step (1) is tetrahydrofuran;
and/or the reaction solvent used in the step (2) is tetrahydrofuran or dichloromethane;
and/or the reaction solvent used in the step (3) is dichloromethane, toluene or tetrahydrofuran;
and/or, in the step (4), the reaction solvent used in the first stage is dichloromethane or dichloroethane; the reaction solvent used in the second stage is ethanol;
and/or the reaction solvent used in step (5) is methanol.
In the invention, in the step (1), the reaction temperature of the dropwise adding n-butyllithium is-80 to-70 ℃, and the reaction temperature of the reaction solvent of the dropwise adding iodine is-80 to-70 ℃;
and/or in the step (2), the reaction temperature of adding the condensing agent is 20-25 ℃, and the reaction temperature of dropwise adding the organic base is 20-25 ℃;
and/or, in the step (3), the reaction temperature of dropwise adding the reducing agent is-80 to-70 ℃;
and/or in the step (4), in the first stage, the reaction temperature is 30-40 ℃, and in the second stage, the reaction temperature is 65-70 ℃;
and/or in the step (5), the reaction temperature of adding the reducing agent is 0-10 ℃.
< example >
The reaction route of the synthetic method adopted by the invention is as follows:

EXAMPLE 1 preparation of Compound 2
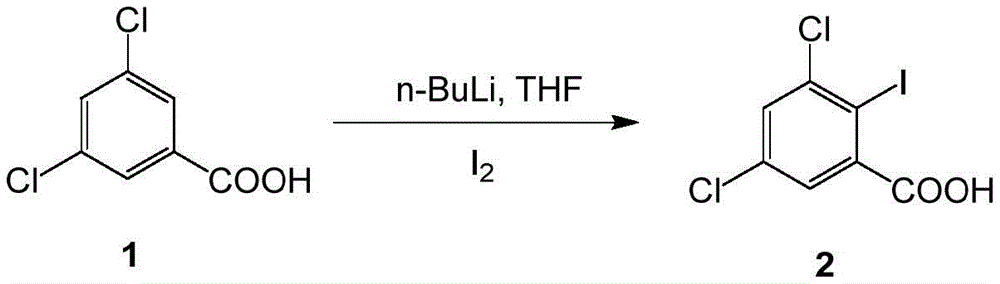
To compound 1(100.0g,0.524mol,1.0eq) was added tetrahydrofuran (800 mL). Under the protection of nitrogen, the temperature is reduced to-80 to-70 ℃, 2.5M butyl lithium (0.524mL,1.31mol,2.5eq) is added dropwise, and the temperature is controlled to-80 to-70 ℃. After the dripping is finished, the reaction is kept for 1 h. Iodine (146.2g,0.576mol,1.1eq) was dissolved in tetrahydrofuran (500mL) and added dropwise, controlling the temperature between-80 ℃ and-70 ℃. After the dripping is finished, the temperature is kept for 1h, and then the temperature is slowly raised to-30 ℃. The reaction was slowly quenched into 20% citric acid (800mL) and extracted with ethyl acetate (600mL × 2). The organic phases were combined and washed with 5% sodium bisulfite (500mL) and saturated brine (500 mL). Concentration of the organic phase afforded compound 2(122.9g,0.388mol) in 74% yield.
EXAMPLE 2 preparation of Compound 3

To a solution of compound 2(110g,0.347mol,1.0eq) in tetrahydrofuran (880mL) was added CDI (61.9g,0.382mol,1.1eq) in portions under nitrogen and stirred at 20-25 ℃ for 2 h. Adding N-methylmethoxyamine hydrochloride (37.1g,0.382mol,1.1eq), dropwise adding diisopropylethylamine (58.2g,0.451mol,1.3eq), controlling the temperature to be 20-25 ℃, and stirring for 12 hours after dropwise adding. Water (880mL) was added, the pH was adjusted to 4-5 with concentrated hydrochloric acid, and extraction was performed with ethyl acetate (550mLx 2). The organic phases were combined and washed with saturated brine (550 mL). Concentration of the organic phase afforded compound 3(113.4g,0.316mol) in 91% yield.
EXAMPLE 3 preparation of Compound 3 II
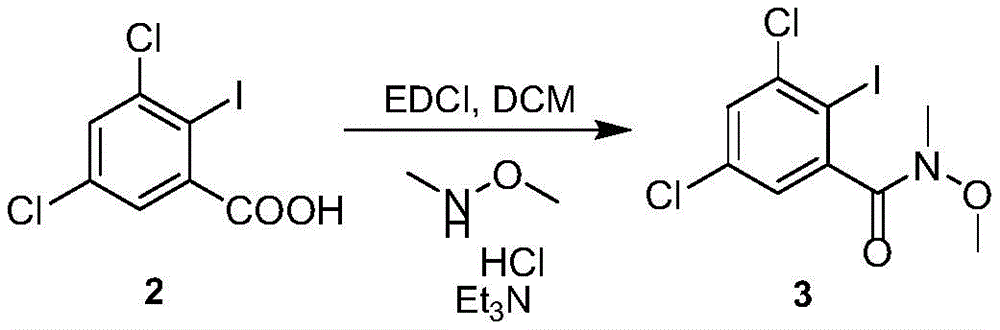
To a solution of compound 2(110g,0.347mol,1.0eq) in dichloromethane (880mL) was added EDCI (86.5g,0.451mol,1.3eq) in portions under nitrogen, N-methylmethoxyamine hydrochloride (44.0g,0.451mol,1.3eq) in portions at 20-25 deg.C, triethylamine (87.6g,0.868mol,2.5eq) was added dropwise, the temperature was controlled at 20-25 deg.C, and stirring was carried out for 12h after dropping. Water (880mL) was added, the pH was adjusted to 4-5 with concentrated hydrochloric acid, the layers were separated, and the aqueous layer was extracted with dichloromethane (550mLx 2). The organic phases were combined and washed with saturated brine (550 mL). Concentration of the organic phase afforded compound 3(107.3g,0.298mol) in 86% yield.
EXAMPLE 4 preparation of Compound 4
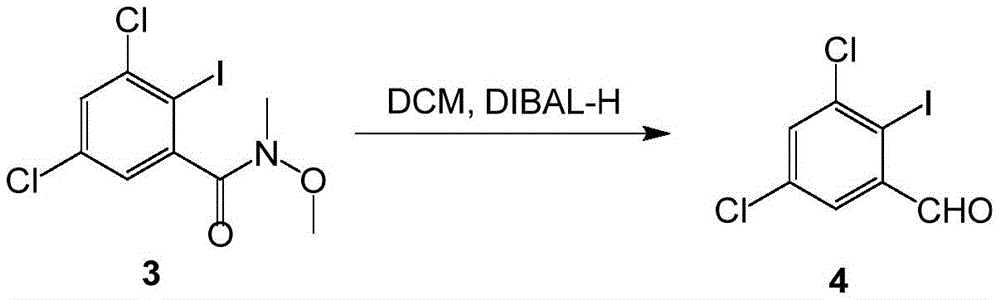
Dissolving the compound 3(100g,0.278mol,1.0eq) in dichloromethane (800mL), and cooling to-80-70 ℃ under the protection of nitrogen. 1.0M DIBAL-H (361mL,0.361mol,1.3eq) is added dropwise, the temperature is controlled between minus 80 ℃ and minus 70 ℃, and the temperature is kept for 2 hours after the dropwise addition. The reaction was slowly quenched into saturated ammonium chloride (800mL) and the temperature controlled at less than 5 ℃. Filtering with diatomite, and separating. The aqueous phase was extracted with dichloromethane (500mL x2), and the organic phases were combined and washed with 10% citric acid (800mL) and saturated brine (500mL), respectively. Concentration of the organic phase afforded compound 4(69.5g,0.231mol) in 83% yield.
EXAMPLE 5 preparation of Compound 4

Dissolving compound 3(100g,0.278mol,1.0eq) in toluene (800mL), and cooling to-80-70 ℃ under the protection of nitrogen. 1.0M DIBAL-H (361mL,0.361mol,1.3eq) is added dropwise, the temperature is controlled between minus 80 ℃ and minus 70 ℃, and the temperature is kept for 2 hours after the dropwise addition. The reaction was slowly quenched into saturated ammonium chloride (800mL) and the temperature controlled at less than 5 ℃. Filtering with diatomite, and separating. The aqueous phase was extracted with toluene (500mL × 2), and the organic phases were combined and washed with 10% citric acid (800mL) and saturated brine (500mL), respectively. Concentration of the organic phase afforded compound 4(62.7g,0.209mol) in 75% yield.
EXAMPLE 6 preparation of Compound 5

Compound 4(60g,0.199mol,1.0eq), triethylamine (40.2g,0.398mol,2.0eq), cuprous iodide (3.8g,0.02mol,0.1eq), triphenylphosphine (2.1g,0.008mol,0.04eq) and dichloromethane (600mL) were added to the reaction flask, respectively. After nitrogen substitution, bis (triphenylphosphine) palladium dichloride (2.8g,0.004mol,0.02eq) was added, trimethylsilylacetylene (27.5g,0.28mol,1.4eq) was added dropwise, and the temperature was controlled at 30-40 ℃. After dropping, the reaction is kept for 12 h. The temperature was reduced to 20-25 deg.C, water (240mL) was added, the pH was adjusted to 6-7 with 1N HCl, and the layers were separated. The organic phase was concentrated to dryness. Ethanol (420mL), cuprous iodide (1.9g,0.01mol,0.05eq), 28% ammonia (50.0g,0.4mol,2.0eq) were added. Heating to 65-70 ℃ and reacting for 8 hours. The reaction mixture was concentrated, ethyl acetate (120mL) and N-heptane (480mL) were added, and the mixture was washed with 1N HCl (200mL) and saturated brine (300mL), respectively, and the mixture was separated. The organic phase was filtered through a pad of silica gel and the filtrate was concentrated to give compound 5(28.1g,0.142mol) in 71% yield.
EXAMPLE 7 preparation of Compound 5

Compound 4(60g,0.199mol,1.0eq), diisopropylethylamine (51.3g,0.398mol,2.0eq), cuprous iodide (3.8g,0.02mol,0.1eq), triphenylphosphine (2.1g,0.008mol,0.04eq) and dichloroethane (600mL) were added to the reaction flask, respectively. After nitrogen substitution, bis (triphenylphosphine) palladium dichloride (2.8g,0.004mol,0.02eq) was added, trimethylsilylacetylene (27.5g,0.28mol,1.4eq) was added dropwise, and the temperature was controlled at 30-40 ℃. After dropping, the reaction is kept for 12 h. The temperature was reduced to 20-25 deg.C, water (240mL) was added, the pH was adjusted to 6-7 with 1N HCl, and the layers were separated. The organic phase was concentrated to dryness. Ethanol (420mL), cuprous iodide (1.9g,0.01mol,0.05eq), 28% ammonia (50.0g,0.4mol,2.0eq) were added. Heating to 65-70 ℃ and reacting for 8 hours. The reaction mixture was concentrated, ethyl acetate (120mL) and N-heptane (480mL) were added, and the mixture was washed with 1N HCl (200mL) and saturated brine (300mL), respectively, and the mixture was separated. The organic phase was filtered through a pad of silica gel and the filtrate was concentrated to give compound 5(27.2g,0.137mol) in 69% yield.
EXAMPLE 8 preparation of Compound 6

The reaction flask was charged with compound 5(20g,0.10mol,1.0eq), nickel chloride hexahydrate (23.7g,0.10mol,1.0eq), methanol (200 mL). Under the protection of nitrogen, sodium borohydride (9.5g,0.25mol,2.5eq) is added in batches, the temperature is controlled to be 0-10 ℃, after the addition is finished, the temperature is raised to 20-30 ℃, and the reaction is kept for 8 hours. The reaction was quenched slowly into water (200 mL). Concentrated under reduced pressure, and the residue was extracted with 10% sodium hydroxide (250mL) and dichloromethane (100mL × 3). The organic phases were combined, filtered through celite, the filtrate was concentrated to the remaining 100mL, concentrated HCl (20mL) was added dropwise, the temperature was controlled at 0-10 deg.C and stirred for 2 h. Filtration and drying afforded compound 6(20.3g,0.085mol), 85% yield. HNMR (400MHz, DMSO-d6):9.81(s,2H),7.62(s,1H),7.43(s,1H),4.27(s,2H),3.41(s,2H),2.95(s,2H).
EXAMPLE 9 preparation of Compound 6

The reaction flask was charged with compound 5(20g,0.10mol,1.0eq), nickel chloride hexahydrate (23.7g,0.10mol,1.0eq), methanol (200 mL). Under the protection of nitrogen, potassium borohydride (13.5g,0.25mol,2.5eq) is added in batches, the temperature is controlled to be 0-10 ℃, after the addition is finished, the temperature is raised to 20-30 ℃, and the reaction is kept for 8 hours. The reaction was slowly quenched into water (200 mL). Concentrated under reduced pressure, and the residue was extracted with 10% sodium hydroxide (250mL) and dichloromethane (100mL × 3). The organic phases were combined, filtered through celite, the filtrate was concentrated to the remaining 100mL, concentrated HCl (20mL) was added dropwise, the temperature was controlled at 0-10 deg.C and stirred for 2 h. Filtration and drying gave compound 6(18.8g,0.079mol) in 79% yield. HNMR (400MHz, DMSO-d6):9.81(s,2H),7.62(s,1H),7.43(s,1H),4.27(s,2H),3.41(s,2H),2.95(s,2H).
The spectrum of compound 6 is shown in FIG. 1.
In conclusion, the synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride disclosed by the invention has the advantages of low price and easiness in obtaining of used raw materials, mild reaction conditions, no use of high-risk reaction, simple requirement on used equipment, high yield, lower total cost, suitability for large-scale industrial production and good application prospect.
The above description is only a preferred embodiment of the present invention and is not intended to limit the present invention, and various modifications and changes may be made by those skilled in the art. Any modification, equivalent replacement, or improvement made within the spirit and principle of the present invention should be included in the protection scope of the present invention.
Claims (10)
1. The synthesis method of the sitagliptin intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride is characterized by comprising the following steps:

(1) iodination reaction: after hydrogen is extracted from the compound 1 by n-butyl lithium, the compound reacts with iodine to obtain a compound 2;
(2) amidation reaction: condensing the compound 2 with a condensing agent to obtain an intermediate state; then condensing with methyl methoxy amine hydrochloride in the presence of organic alkali to obtain a compound 3;
(3) reduction reaction: reducing the compound 3 to obtain a compound 4;
(4) coupling reaction: in the first stage, a compound 4 is catalyzed by a first catalyst in the presence of alkali and is coupled with trimethylsilyl acetylene; in the second stage, the second catalyst is used for catalyzing, and the second catalyst reacts with ammonia water to close the ring to obtain a compound 5;
(5) reduction reaction: the compound 5 is reduced by a reducing agent in the presence of Lewis acid, and is acidified by an acidifying agent to obtain a compound 6.
2. The synthesis method of the sitagliptin intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to claim 1, characterized in that,
the condensing agent used in the step (2) is CDI or EDCI; the organic base is diisopropylethylamine or triethylamine;
and/or the reducing agent used in the step (3) is diisobutylaluminum hydride;
and/or, in the step (4), in the first stage, the used base is triethylamine or diisopropylethylamine, and the first catalyst is a mixed reagent comprising a palladium reagent, a copper reagent and a phosphine reagent; in the second stage, the second catalyst is a copper reagent;
and/or, in the step (5), the reducing agent is sodium borohydride or potassium borohydride, and the Lewis acid is nickel chloride hexahydrate; the acidifying agent is hydrochloric acid.
3. The synthesis method of the sitagliptin intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to claim 2, characterized in that in the step (4), the palladium reagent is bis (triphenylphosphine) palladium dichloride, the copper reagent is cuprous iodide, and the phosphine reagent is triphenylphosphine.
4. The synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to any one of claims 1 to 3,
in the step (1), the molar ratio of the compound 1, n-butyllithium and iodine is 1.0: 2.0-3.0: 1.0-1.5;
and/or in the step (1), the volume ratio of the compound 1 to the reaction solvent is 1.0: 8.0.
5. The synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to any one of claims 1 to 3,
in the step (2), the molar ratio of the compound 2, the condensing agent, the methyl methoxylamine hydrochloride and the organic base is 1.0: 1.0-1.3: 1.2-2.5;
and/or in the step (2), the volume ratio of the compound 2 to the reaction solvent is 1.0: 8.0.
6. The synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to any one of claims 1 to 3,
in the step (3), the molar ratio of the compound 3 to the reducing agent is 1.0: 1.0-1.5;
and/or in the step (3), the volume ratio of the compound 2 to the reaction solvent is 1.0: 8.0.
7. The synthesis method of the sitagliptin intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to claim 2, characterized in that,
in the step (4), in the first stage, the molar ratio of the compound 4, alkali, a palladium reagent, a copper reagent, a phosphine reagent and trimethylsilyl acetylene is 1.0: 2.0-2.5: 0.02-0.05: 0.1-0.15: 0.04-0.1: 1.2-1.5; in the second stage, the molar ratio of the compound 4, the copper reagent and the ammonia water is 1.0: 0.05-0.1: 2.0-3.0;
and/or, in the step (4), in the first stage, the volume ratio of the compound 2 to the reaction solvent is 1.0: 8.0; in the second stage, the volume ratio of compound 2 to the reaction solvent was 1.0: 7.0.
8. The synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to any one of claims 1 to 3,
in the step (5), the molar ratio of the compound 5, the reducing agent and the Lewis acid is 1.0: 2.5-3.0: 1.0-1.5;
and/or in the step (5), the volume ratio of the compound 2 to the reaction solvent is 1: 10.0.
9. The synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to any one of claims 1 to 3,
the reaction solvent used in the step (1) is tetrahydrofuran;
and/or the reaction solvent used in the step (2) is tetrahydrofuran or dichloromethane;
and/or the reaction solvent used in the step (3) is dichloromethane, toluene or tetrahydrofuran;
and/or, in the step (4), the reaction solvent used in the first stage is dichloromethane or dichloroethane; the reaction solvent used in the second stage is ethanol;
and/or the reaction solvent used in step (5) is methanol.
10. The synthesis method of the sitaxel intermediate 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride according to any one of claims 1 to 3,
in the step (1), the reaction temperature is-80 to-70 ℃ when the n-butyllithium is dripped, and the reaction temperature of the reaction solvent is-80 to-70 ℃ when the iodine is dripped;
and/or in the step (2), the reaction temperature of adding the condensing agent is 20-25 ℃, and the reaction temperature of dropwise adding the organic base is 20-25 ℃;
and/or, in the step (3), the reaction temperature of dropwise adding the reducing agent is-80 to-70 ℃;
and/or, in the step (4), in the first stage, the reaction temperature is 30-40 ℃, and in the second stage, the reaction temperature is 65-70 ℃;
and/or in the step (5), the reaction temperature of adding the reducing agent is 0-10 ℃.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210254550.0A CN114524767B (en) | 2022-03-11 | 2022-03-11 | Synthesis method of sitaxel intermediate 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline hydrochloride |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210254550.0A CN114524767B (en) | 2022-03-11 | 2022-03-11 | Synthesis method of sitaxel intermediate 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline hydrochloride |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN114524767A true CN114524767A (en) | 2022-05-24 |
| CN114524767B CN114524767B (en) | 2022-10-18 |
Family
ID=81626548
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210254550.0A Active CN114524767B (en) | 2022-03-11 | 2022-03-11 | Synthesis method of sitaxel intermediate 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline hydrochloride |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114524767B (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104797574A (en) * | 2012-07-25 | 2015-07-22 | 原生质生物科学股份有限公司 | LFA-1 inhibitors and polymorphs thereof |
| CN112321506A (en) * | 2020-11-26 | 2021-02-05 | 江西天戌药业有限公司 | Preparation method of 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline |
| CN112500343A (en) * | 2020-12-26 | 2021-03-16 | 山东金城柯瑞化学有限公司 | Synthetic method of 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride |
-
2022
- 2022-03-11 CN CN202210254550.0A patent/CN114524767B/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104797574A (en) * | 2012-07-25 | 2015-07-22 | 原生质生物科学股份有限公司 | LFA-1 inhibitors and polymorphs thereof |
| CN112321506A (en) * | 2020-11-26 | 2021-02-05 | 江西天戌药业有限公司 | Preparation method of 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline |
| CN112500343A (en) * | 2020-12-26 | 2021-03-16 | 山东金城柯瑞化学有限公司 | Synthetic method of 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline hydrochloride |
Non-Patent Citations (2)
| Title |
|---|
| WANBIN XU等: "Scalable Process for Making 5,7-Dichlorotetrahydroisoquinoline-6-carboxylic Acid Using Methylene as the Protecting Group", 《ORGANIC PROCESS RESEARCH & DEVELOPMENT》 * |
| WILLIAM E. BONDINELL等: "Inhibitors of Phenylethanolamine JV-Methyltransferase and Epinephrine Biosynthesis. 1. Chloro-Substituted 1,2,3,4-Tetrahydroisoquinolines", 《JOURNAL OF MEDICINAL CHEMISTRY》 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114524767B (en) | 2022-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7500102B2 (en) | Intermediate for synthesizing camptothecin derivatives, production method thereof and use thereof | |
| CN108203404A (en) | (R) synthetic method of -3- Phenylpiperidines or/and the chiral intermediate of (S) -3- Phenylpiperidines and Ni Lapani | |
| EP3080086B1 (en) | Process of making adamantanamides | |
| CN102229613A (en) | New process for synthesis of asenapine | |
| US20040106818A1 (en) | Process for the preparation of cyclohexanol derivatives | |
| JPH0368022B2 (en) | ||
| CN114394884B (en) | Preparation method of allylphenol compound | |
| CN104370746B (en) | A kind of cost-effective preparation method to nitrobenzyl alcohol | |
| CN113248432B (en) | Novel method for preparing Luo Shasi other intermediates in high yield | |
| CN114524767B (en) | Synthesis method of sitaxel intermediate 5,7-dichloro-1,2,3,4-tetrahydroisoquinoline hydrochloride | |
| CN106831441A (en) | A kind of preparation method of cinacalcet hydrochloride | |
| WO2025256525A1 (en) | Method for preparing elacestrant and intermediate thereof | |
| CN109824516B (en) | Preparation method of trans-4-hydroxycyclohexanecarboxylic acid tert-butyl ester | |
| CN111100042B (en) | Preparation method of 2-methoxy-5-sulfonamide benzoic acid | |
| CN103030533B (en) | Process for synthesizing bis(4-hydroxy-1-naphthyl)benzyl alcohol | |
| CN105732445B (en) | Dapoxetine hydrochloride intermediate and preparation method thereof | |
| CN111825688B (en) | Preparation method of 6-tert-butyl-3 ', 3', 3-trimethylpyrano [3,2-a ] carbazole | |
| EP1730102B1 (en) | A novel catalytic process for the production of 3,3 , 4,4'-tetraminobiphenyl | |
| CN111689993B (en) | A new preparation method of chiral α-amino boronate, a key intermediate of boron-containing Zomib drugs | |
| CN101279986B (en) | Synthetic method of axis-unsymmetric chiral diphosphine ligand | |
| CN107445879B (en) | Preparation method of Latricinib intermediate | |
| CN114394933B (en) | Synthesis method of 11, 12-dihydro-gamma-oxo-dibenzo [ F ] azo-5- (6H) -butyric acid | |
| KR101479986B1 (en) | New process for the synthesis of ivabradine and addition salts thereof with a pharmaceutically acceptable acid | |
| CN105566150A (en) | Preparation method of aliskiren | |
| CN110724098A (en) | Synthetic method of 5, 7-dichloro-1, 2,3, 4-tetrahydroisoquinoline-6-carboxylic acid hydrochloride |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |