CN114433120A - Desulfurization catalyst and preparation method and application thereof - Google Patents
Desulfurization catalyst and preparation method and application thereof Download PDFInfo
- Publication number
- CN114433120A CN114433120A CN202210187860.5A CN202210187860A CN114433120A CN 114433120 A CN114433120 A CN 114433120A CN 202210187860 A CN202210187860 A CN 202210187860A CN 114433120 A CN114433120 A CN 114433120A
- Authority
- CN
- China
- Prior art keywords
- catalyst
- group
- solution
- metal
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 161
- 238000006477 desulfuration reaction Methods 0.000 title claims abstract description 20
- 230000023556 desulfurization Effects 0.000 title claims abstract description 20
- 238000002360 preparation method Methods 0.000 title abstract description 18
- 229910052751 metal Inorganic materials 0.000 claims abstract description 72
- 239000002184 metal Substances 0.000 claims abstract description 72
- 229910052755 nonmetal Inorganic materials 0.000 claims abstract description 32
- -1 VIB metals Chemical class 0.000 claims abstract description 15
- 238000006243 chemical reaction Methods 0.000 claims description 103
- 239000012018 catalyst precursor Substances 0.000 claims description 42
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 28
- 229910052717 sulfur Inorganic materials 0.000 claims description 27
- 239000011593 sulfur Substances 0.000 claims description 27
- 239000011148 porous material Substances 0.000 claims description 25
- 150000003839 salts Chemical class 0.000 claims description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 23
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 21
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 claims description 18
- 235000018660 ammonium molybdate Nutrition 0.000 claims description 18
- 239000011609 ammonium molybdate Substances 0.000 claims description 18
- 229940010552 ammonium molybdate Drugs 0.000 claims description 18
- 238000000034 method Methods 0.000 claims description 14
- 239000004115 Sodium Silicate Substances 0.000 claims description 12
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 12
- KBJMLQFLOWQJNF-UHFFFAOYSA-N nickel(ii) nitrate Chemical compound [Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O KBJMLQFLOWQJNF-UHFFFAOYSA-N 0.000 claims description 12
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 claims description 12
- 229910052911 sodium silicate Inorganic materials 0.000 claims description 12
- 239000000295 fuel oil Substances 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 239000001257 hydrogen Substances 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 229910052750 molybdenum Inorganic materials 0.000 claims description 8
- 150000002894 organic compounds Chemical class 0.000 claims description 8
- 238000002156 mixing Methods 0.000 claims description 7
- 239000002798 polar solvent Substances 0.000 claims description 7
- 229910021645 metal ion Inorganic materials 0.000 claims description 6
- 229910052721 tungsten Inorganic materials 0.000 claims description 6
- GVPFVAHMJGGAJG-UHFFFAOYSA-L cobalt dichloride Chemical compound [Cl-].[Cl-].[Co+2] GVPFVAHMJGGAJG-UHFFFAOYSA-L 0.000 claims description 5
- 150000001450 anions Chemical class 0.000 claims description 4
- 239000002904 solvent Substances 0.000 claims description 4
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 4
- KEZYHIPQRGTUDU-UHFFFAOYSA-N 2-[dithiocarboxy(methyl)amino]acetic acid Chemical compound SC(=S)N(C)CC(O)=O KEZYHIPQRGTUDU-UHFFFAOYSA-N 0.000 claims description 3
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 claims description 3
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 claims description 3
- 239000004111 Potassium silicate Substances 0.000 claims description 3
- MQRWBMAEBQOWAF-UHFFFAOYSA-N acetic acid;nickel Chemical compound [Ni].CC(O)=O.CC(O)=O MQRWBMAEBQOWAF-UHFFFAOYSA-N 0.000 claims description 3
- YVBOZGOAVJZITM-UHFFFAOYSA-P ammonium phosphomolybdate Chemical compound [NH4+].[NH4+].[NH4+].[NH4+].[O-]P([O-])=O.[O-][Mo]([O-])(=O)=O YVBOZGOAVJZITM-UHFFFAOYSA-P 0.000 claims description 3
- 229940011182 cobalt acetate Drugs 0.000 claims description 3
- UFMZWBIQTDUYBN-UHFFFAOYSA-N cobalt dinitrate Chemical compound [Co+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O UFMZWBIQTDUYBN-UHFFFAOYSA-N 0.000 claims description 3
- 229910001981 cobalt nitrate Inorganic materials 0.000 claims description 3
- 229910000361 cobalt sulfate Inorganic materials 0.000 claims description 3
- 229940044175 cobalt sulfate Drugs 0.000 claims description 3
- KTVIXTQDYHMGHF-UHFFFAOYSA-L cobalt(2+) sulfate Chemical compound [Co+2].[O-]S([O-])(=O)=O KTVIXTQDYHMGHF-UHFFFAOYSA-L 0.000 claims description 3
- QAHREYKOYSIQPH-UHFFFAOYSA-L cobalt(II) acetate Chemical compound [Co+2].CC([O-])=O.CC([O-])=O QAHREYKOYSIQPH-UHFFFAOYSA-L 0.000 claims description 3
- 150000002500 ions Chemical class 0.000 claims description 3
- PAZHGORSDKKUPI-UHFFFAOYSA-N lithium metasilicate Chemical compound [Li+].[Li+].[O-][Si]([O-])=O PAZHGORSDKKUPI-UHFFFAOYSA-N 0.000 claims description 3
- 229910052912 lithium silicate Inorganic materials 0.000 claims description 3
- 229910003455 mixed metal oxide Inorganic materials 0.000 claims description 3
- VLAPMBHFAWRUQP-UHFFFAOYSA-L molybdic acid Chemical compound O[Mo](O)(=O)=O VLAPMBHFAWRUQP-UHFFFAOYSA-L 0.000 claims description 3
- 229940078494 nickel acetate Drugs 0.000 claims description 3
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 claims description 3
- LGQLOGILCSXPEA-UHFFFAOYSA-L nickel sulfate Chemical compound [Ni+2].[O-]S([O-])(=O)=O LGQLOGILCSXPEA-UHFFFAOYSA-L 0.000 claims description 3
- 229910000363 nickel(II) sulfate Inorganic materials 0.000 claims description 3
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 claims description 3
- IYDGMDWEHDFVQI-UHFFFAOYSA-N phosphoric acid;trioxotungsten Chemical compound O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.OP(O)(O)=O IYDGMDWEHDFVQI-UHFFFAOYSA-N 0.000 claims description 3
- NNHHDJVEYQHLHG-UHFFFAOYSA-N potassium silicate Chemical compound [K+].[K+].[O-][Si]([O-])=O NNHHDJVEYQHLHG-UHFFFAOYSA-N 0.000 claims description 3
- 229910052913 potassium silicate Inorganic materials 0.000 claims description 3
- 235000019353 potassium silicate Nutrition 0.000 claims description 3
- 235000015393 sodium molybdate Nutrition 0.000 claims description 3
- 239000011684 sodium molybdate Substances 0.000 claims description 3
- TVXXNOYZHKPKGW-UHFFFAOYSA-N sodium molybdate (anhydrous) Chemical compound [Na+].[Na+].[O-][Mo]([O-])(=O)=O TVXXNOYZHKPKGW-UHFFFAOYSA-N 0.000 claims description 3
- XMVONEAAOPAGAO-UHFFFAOYSA-N sodium tungstate Chemical compound [Na+].[Na+].[O-][W]([O-])(=O)=O XMVONEAAOPAGAO-UHFFFAOYSA-N 0.000 claims description 3
- CMPGARWFYBADJI-UHFFFAOYSA-L tungstic acid Chemical compound O[W](O)(=O)=O CMPGARWFYBADJI-UHFFFAOYSA-L 0.000 claims description 3
- 239000005864 Sulphur Substances 0.000 claims description 2
- 229910052759 nickel Inorganic materials 0.000 claims description 2
- 239000000446 fuel Substances 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 20
- 239000002283 diesel fuel Substances 0.000 abstract description 10
- MYAQZIAVOLKEGW-UHFFFAOYSA-N 4,6-dimethyldibenzothiophene Chemical compound S1C2=C(C)C=CC=C2C2=C1C(C)=CC=C2 MYAQZIAVOLKEGW-UHFFFAOYSA-N 0.000 abstract description 5
- DGUACJDPTAAFMP-UHFFFAOYSA-N 1,9-dimethyldibenzo[2,1-b:1',2'-d]thiophene Natural products S1C2=CC=CC(C)=C2C2=C1C=CC=C2C DGUACJDPTAAFMP-UHFFFAOYSA-N 0.000 abstract description 3
- 150000002843 nonmetals Chemical class 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 60
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 38
- 229910052757 nitrogen Inorganic materials 0.000 description 19
- 238000001179 sorption measurement Methods 0.000 description 19
- 239000000843 powder Substances 0.000 description 17
- 239000002243 precursor Substances 0.000 description 17
- 238000010438 heat treatment Methods 0.000 description 16
- 239000002002 slurry Substances 0.000 description 16
- 239000002994 raw material Substances 0.000 description 14
- 239000012295 chemical reaction liquid Substances 0.000 description 12
- 238000010992 reflux Methods 0.000 description 12
- 239000004094 surface-active agent Substances 0.000 description 11
- 239000011259 mixed solution Substances 0.000 description 10
- 239000003960 organic solvent Substances 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 3
- 239000002071 nanotube Substances 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000005303 weighing Methods 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- WQOXQRCZOLPYPM-UHFFFAOYSA-N dimethyl disulfide Chemical compound CSSC WQOXQRCZOLPYPM-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- 239000011733 molybdenum Substances 0.000 description 2
- JKQOBWVOAYFWKG-UHFFFAOYSA-N molybdenum trioxide Chemical compound O=[Mo](=O)=O JKQOBWVOAYFWKG-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 229920001030 Polyethylene Glycol 4000 Polymers 0.000 description 1
- 229920002582 Polyethylene Glycol 600 Polymers 0.000 description 1
- 229920002593 Polyethylene Glycol 800 Polymers 0.000 description 1
- VBIIFPGSPJYLRR-UHFFFAOYSA-M Stearyltrimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)C VBIIFPGSPJYLRR-UHFFFAOYSA-M 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 238000003917 TEM image Methods 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 238000003916 acid precipitation Methods 0.000 description 1
- 238000003915 air pollution Methods 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- QGJOPFRUJISHPQ-NJFSPNSNSA-N carbon disulfide-14c Chemical compound S=[14C]=S QGJOPFRUJISHPQ-NJFSPNSNSA-N 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- WOWHHFRSBJGXCM-UHFFFAOYSA-M cetyltrimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+](C)(C)C WOWHHFRSBJGXCM-UHFFFAOYSA-M 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- UBEWDCMIDFGDOO-UHFFFAOYSA-N cobalt(II,III) oxide Inorganic materials [O-2].[O-2].[O-2].[O-2].[Co+2].[Co+3].[Co+3] UBEWDCMIDFGDOO-UHFFFAOYSA-N 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 238000010336 energy treatment Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 229910000008 nickel(II) carbonate Inorganic materials 0.000 description 1
- ZULUUIKRFGGGTL-UHFFFAOYSA-L nickel(ii) carbonate Chemical compound [Ni+2].[O-]C([O-])=O ZULUUIKRFGGGTL-UHFFFAOYSA-L 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000001741 organic sulfur group Chemical group 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000001878 scanning electron micrograph Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 150000004763 sulfides Chemical class 0.000 description 1
- 238000005987 sulfurization reaction Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- SZEMGTQCPRNXEG-UHFFFAOYSA-M trimethyl(octadecyl)azanium;bromide Chemical compound [Br-].CCCCCCCCCCCCCCCCCC[N+](C)(C)C SZEMGTQCPRNXEG-UHFFFAOYSA-M 0.000 description 1
- 150000003657 tungsten Chemical class 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images




Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/888—Tungsten
- B01J23/8885—Tungsten containing also molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/613—10-100 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/615—100-500 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/63—Pore volume
- B01J35/633—Pore volume less than 0.5 ml/g
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/02—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing
- C10G45/04—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used
- C10G45/06—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used containing nickel or cobalt metal, or compounds thereof
- C10G45/08—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used containing nickel or cobalt metal, or compounds thereof in combination with chromium, molybdenum, or tungsten metals, or compounds thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/201—Impurities
- C10G2300/202—Heteroatoms content, i.e. S, N, O, P
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2400/00—Products obtained by processes covered by groups C10G9/00 - C10G69/14
- C10G2400/04—Diesel oil
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Catalysts (AREA)
Abstract
Embodiments of the present disclosure relate to a desulfurization catalyst, and a preparation method and application thereof, wherein the catalyst comprises at least one group VIII metal, at least two group VIB metals, and a group IVA nonmetal. The invention also discloses a preparation method and application of the catalyst. The invention is used for hydrodesulfurization of diesel oil fractions containing 4, 6-dimethyldibenzothiophene, shows extremely high hydrodesulfurization activity, realizes ultra-deep desulfurization, and reduces the cost of a catalyst by introducing non-metals of IVA group.
Description
Technical Field
The disclosure relates to a desulfurization catalyst and a preparation method and application thereof, and belongs to the technical field of energy treatment.
Background
Because the petroleum reserves are reduced, the problems of petroleum heaviness and deterioration are more and more prominent, high-sulfur crude oil is increased year by year in the world, and the requirements of environmental legislation of various countries on the limitation of the sulfur content in fuel oil are increasingly strict, the development of the ultra-deep hydrodesulfurization catalyst with excellent performance not only becomes the core of the hydrodesulfurization field, but also makes the hydrotreating technology increasingly paid attention to in the petroleum processing industry. Sulfides in oil products are a main source of air pollution, SOx generated after organic sulfur-containing compounds in fuel oil are combusted can not only cause acid rain, but also can cause irreversible poisoning of a three-way catalyst of an automobile engine tail gas purification system, and can also participate in forming dust particles PM2.5 to cause gradually increased haze weather, so that the environment and human health are seriously harmed, and people are attracted to pay attention to the environment widely. Therefore, strict fuel oil sulfur content standards are promulgated by various countries, Europe has realized that the sulfur content of diesel oil is less than 10ppmw standard in 2005, China has executed the JingV clean diesel oil index with the sulfur content of less than 10ppmw in 6 and 1 days in 2012 first in Beijing, and has completely executed the diesel oil sulfur index equivalent to the Europe IV (<50ppmw) emission standard in 2015 nationwide, and has completely executed the clean diesel oil sulfur index equivalent to the Europe V (<10ppmw) emission standard in 2017 and 1 days.
With the increasing standards for sulfur content, the adjustment of process operating conditions and the use of new reactors require a significant investment cost, compared to the development of a new catalyst capable of ultra-deep hydrodesulfurization at existing plant operating conditions, which is a more economical and feasible process.
U.S. Pat. Nos. 09/518741, 08/900389, 09/869988 all report a NiMoW bulk catalyst. The synthesis method of said catalyst uses ammonia water as complexing agent and reacts with reaction raw material Ni2+Complexing, through slow heating process, the Ni-ammonia complex slowly decomposes to obtain Ni2+Reacting with molybdenum and tungsten in the solution to generate a NiMoW catalyst precursor, and then roasting and vulcanizing to form the NiMoWS sulfide catalyst. Chinese patent CN1339985A also discloses a method for synthesizing a NiMoW catalyst, which is mainly to react molybdenum and tungsten salts with basic nickel carbonate in an aqueous solution, at least ensure that part of the metal components exist in a solid state during the reaction, and finally obtain the catalyst by sulfurization.
From the reported work, it is not difficult to find that the existing synthesis method of the bulk catalyst has the following defects:
(1) the used raw materials are not friendly to the environment;
(2) the preparation cost of the catalyst is high;
(3) the specific surface area, pore volume and activity of the catalyst are yet to be further improved.
Disclosure of Invention
The purpose of the present disclosure is to provide a desulfurization catalyst, a preparation method and an application thereof.
To achieve the above objects, in one aspect of the present disclosure there is provided a desulfurization catalyst comprising a mixed metal oxide of at least one group VIII metal, at least two group VIB metals, and a group IVA nonmetal;
wherein, calculated by oxide, the catalyst contains 1-50 wt% of VIII group metal, 1-50 wt% of VIB group metal and 1-50 wt% of IVA group nonmetal.
Further, the catalyst comprises at least one group VIII metal, two group VIB metals and one group IVA nonmetal, and the molar ratio of the group VIII metal to the group VIB metal is 20:1-1: 20; the molar ratio of the group VIII metal to the group IVA nonmetal is 20:1 to 1: 1; the molar ratio of the two group VIB metals is 5:1-1: 5.
Further, the group VIII metal includes Ni or Co; the group VIB metal comprises Mo and W; the group IVA nonmetal comprises Si.
Further, the catalyst has a layered porous structure, and the specific surface area of the layered porous structure is 80-300m2The pore volume is 0.1-0.5 ml/g.
Another aspect of the present disclosure also provides a method of preparing a desulfurization catalyst, the method comprising:
adding at least one soluble salt of VIII group metal into a solvent to prepare a solution A, adding at least one soluble salt of IVA group nonmetal into water to prepare a solution B, and mixing and reacting the solution A and the solution B to obtain a catalyst precursor solution;
adding soluble salts of at least two VIB group metals into a polar solvent to prepare a solution C, and mixing and reacting the solution C with the catalyst precursor solution to obtain the catalyst.
Further, the concentration of the VIII group metal ions in the solution A is 0.01-0.3 mol/L, the concentration of the IVA group non-metal ions in the solution B is 0.01-0.3 mol/L, the concentration of the catalyst precursor solution is 0.01-0.9 mol/L, and the concentration of the VIB group metal ions in the solution C is 0.01-0.2 mol/L.
Further, the solution A and the solution B react for 4 to 50 hours at the temperature of 20 to 200 ℃, and the solution C and the catalyst precursor solution react for 4 to 50 hours at the temperature of 20 to 200 ℃.
Further, the soluble salt of the group IVA nonmetal comprises one or more of sodium silicate, potassium silicate, lithium silicate, ammonium silicate, sodium metasilicate pentahydrate, the soluble salt of the group VIII metal comprises one or more of nickel nitrate, nickel acetate, nickel sulfate or nickel chloride, cobalt nitrate, cobalt chloride, cobalt sulfate or cobalt acetate, and the soluble salt of the group VIB metal anion comprises one of ammonium molybdate, molybdic acid, ammonium phosphomolybdate, phosphomolybdic acid and sodium molybdate, and one of ammonium tungstate, phosphotungstic acid, ammonium metatungstate, tungstic acid or sodium tungstate.
In another aspect, the present disclosure also provides a use of the desulfurization catalyst in hydrodesulfurization reaction of sulfur-containing organic compound fuel oil.
Further, the hydrodesulfurization reaction conditions are as follows: the temperature is 250 ℃ and 450 ℃, the hydrogen pressure is 1-20MPa, and the volume ratio of the hydrogen to the sulfur-containing organic compound fuel oil is 50-1500Nm3/m3(ii) a The volume space velocity of the sulfur-containing organic compound fuel oil is 0.1-10h-1。
Compared with the prior art, the method has the advantages that:
1) in the method, the compound with the layered structure is used as a precursor of the synthetic catalyst, the multi-metal bulk catalyst with the layered structure is designed and synthesized, the synthetic process is easy to operate and environment-friendly, and the catalyst can be industrially produced.
2) The VIB group active metal is successfully introduced between the layers of the precursor, so that the distribution and dispersion of the active metal are more uniform, more active centers are formed, the catalyst is determined to have ultrahigh hydrodesulfurization activity, and under mild operation conditions, the sulfur in the 4, 6-dimethyldibenzothiophene in the diesel fraction can be removed from 521ppm to below 10ppm, so that ultra-deep desulfurization is realized.
3) By introducing the IVA group non-metal, the cost of the catalyst is reduced.
Drawings
FIG. 1 is a NiSiO solid prepared in example 1 of the present disclosure3XRD patterns of nanotubes and their catalyst precursors.
FIG. 2 shows a layered NiSiO crystal prepared according to the embodiments of the disclosure3TEM images of nanotubes.
Fig. 3 is an SEM image of a NiSiMoW catalyst prepared according to an example of the disclosure.
Fig. 4 is a graph showing the variation of hydrodesulfurization activity of the NiSiMoW catalysts prepared in examples 1 to 7 according to the present disclosure.
Detailed Description
The catalyst, the preparation method and the application thereof are disclosed and provided in detail in the following with reference to the accompanying drawings.
An embodiment of the present disclosure provides a desulfurization catalyst comprising a mixed metal oxide of at least one group VIII metal, at least two group VIB metals, and a group IVA nonmetal; wherein, calculated by oxide, the catalyst contains 1-50 wt% of VIII group metal, 1-50 wt% of VIB group metal and 1-50 wt% of IVA group nonmetal.
In other embodiments of the disclosure, the group VIII metal content may be 5 wt%, 10 wt%, 15 wt%, or 25 wt%, 30 wt%, calculated as oxide; the group VIB metal content may be 10 wt%, 15 wt%, 20 wt% or 30 wt%, 45 wt%; the content of the group IVA nonmetal may be 15 wt%, 20 wt%, 30 wt%, or 35 wt%, 45 wt%.
In one disclosed embodiment, the catalyst comprises at least one group VIII metal, two group VIB metals, and one group IVA nonmetal, wherein the group VIII metal and group VIB metal are in a molar ratio of 20:1 to 1: 20; the molar ratio of the VIII group metal to the IVA group nonmetal is 20:1 to 1: 1; the molar ratio of the two group VIB metals is 5:1-1: 5.
In other embodiments of the disclosure, the molar ratio of the group VIII metal and the VIB metal may be 10: 5. 10: 15. 15: 5 or 18: 15; the molar ratio of group VIII metal to group IVA nonmetal may be 5: 10. 10: 12. 15: 5. 15: 10 or 20: 5; the molar ratio of the two group VIB metals may be 1: 2. 2: 5 or 5: 2.
in one embodiment of the disclosure, the catalyst is NiSiMoW, i.e., the group VIII metal in the catalyst comprises Ni, the group VIB metal comprises Mo and W, and the group IVA nonmetal comprises Si.
In one embodiment of the disclosure, the group VIII metal comprises Co, the group VIB metal comprises Mo and W, and the group IVA nonmetal comprises Si.
In one embodiment of the present disclosure, the catalyst has a layered porous structure having a specific surface area of 80 to 300m2The pore volume is 0.1-0.5 ml/g. In other embodiments of the disclosure, the specific surface area of the catalyst may be 100-150m2/g、120-180m2/g、150-200m2(g or 200-)2The pore volume may be 0.1-0.3ml/g, 0.25-0.3ml/g or 0.3-0.35 ml/g.
In other embodiments of the present disclosure, a method of preparing a catalyst is provided, the method comprising:
s01: adding at least one soluble salt of VIII group metal into a solvent to prepare a solution A, adding at least one soluble salt of IVA group nonmetal into water to prepare a solution B, and mixing the solution A and the solution B for reaction to obtain a catalyst precursor solution;
s02: adding soluble salts of at least two VIB group metals into a polar solvent to prepare a solution C, and mixing the solution C and the catalyst precursor solution for reaction to obtain the catalyst.
In an embodiment of the present disclosure, S01 specifically includes: adding at least one soluble salt of VIII group metal into a mixed solution of water, an organic solvent and a surfactant to prepare a solution A of soluble salt, dissolving a soluble salt of IVA group nonmetal in water to prepare a solution B, and finally adding the solution B into the solution A containing the at least one soluble salt of VIII group metal to carry out coprecipitation reaction to obtain a catalyst precursor with a layered structure; s02 specifically includes: mixing the slurry of the catalyst precursor with a layered structure with a polar solvent containing soluble salts of at least two VIB group metal anions to carry out an ion exchange reaction, and after the reaction is finished, separating, washing, drying and roasting at 400-500 ℃ for 2-10 hours to obtain the multi-metal bulk catalyst with the layered structure, which contains at least one VIII group metal, one IVA group nonmetal and two VIB group metals.
In one embodiment of the present disclosure, the surfactant in S01 is one or more of cetyltrimethylammonium bromide, cetyltrimethylammonium chloride, octadecyltrimethylammonium bromide or octadecyltrimethylammonium chloride, PEG-400, PEG-600, PEG-800, PEG-4000, PVA, SA 20.
In one embodiment of the disclosure, the concentration of the group VIII metal ions in the solution A is 0.01-0.3 mol/L, the concentration of the group IVA non-metal ions in the solution B is 0.01-0.3 mol/L, the concentration of the catalyst precursor solution is 0.01-0.9 mol/L, and the concentration of the group VIB metal ions in the solution C is 0.01-0.2 mol/L.
In one embodiment of the disclosure, solution a is reacted with solution B at 20 ℃ to 200 ℃ for 4 to 50 hours, and solution C is reacted with the catalyst precursor solution at 20 ℃ to 200 ℃ for 4 to 50 hours.
In one embodiment of the disclosure, the soluble salt of a group IVA nonmetal comprises one or more of sodium silicate, potassium silicate, lithium silicate, ammonium silicate, sodium metasilicate pentahydrate, the soluble salt of a group VIII metal comprises one or more of nickel nitrate, nickel acetate, nickel sulfate or nickel chloride, cobalt nitrate, cobalt chloride, cobalt sulfate or cobalt acetate, and the soluble salt of a group VIB metal anion comprises one of ammonium molybdate, molybdic acid, ammonium phosphomolybdate, phosphomolybdic acid and sodium molybdate, and one of ammonium tungstate, phosphotungstic acid, ammonium metatungstate, tungstic acid or sodium tungstate.
In one embodiment of the present disclosure, the pH of the ion exchange reaction system is 1 to 11, which can be adjusted by an acid, such as nitric acid, or a base, such as aqueous ammonia.
To further illustrate the preparation of the catalysts of the present disclosure, the following examples are given.
Example 1
This example illustrates the preparation of a NiSiMoW catalyst in this disclosure:
a) separately weigh out nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol) were added, and they were dissolved in 200mlAdjusting the pH value to 12 in a mixed solution containing water, an organic solvent and a surfactant, heating to a reaction temperature to form a light green mixed reaction solution, and carrying out reflux reaction on the light green mixed reaction solution at a reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor A;
b) ammonium molybdate (5.4g, wherein Mo) was weighed out separately6+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol), dissolving the raw materials in a polar solvent (300ml) to form a solution, heating the solution to the reaction temperature, and continuously stirring to form a colorless transparent solution B; subsequently heating the slurry precursor a prepared in step a) to a reaction temperature; slowly adding the slurry into the colorless transparent solution B to form a light green reaction solution, and carrying out reflux reaction at the reaction temperature of 80 ℃ for 5 hours to obtain light green powder; the pale green powder obtained from the reaction was filtered, washed, and dried at 120 ℃ to obtain a NiSiMoW catalyst (16.0 g). The specific surface area of the catalyst, as determined by low temperature nitrogen adsorption, was 241.7m2The pore volume is 0.19 ml/g.
The catalyst is light green powder, and the molecular expression of the catalyst is NiO SiO measured by XRF2·MoO3·WO3. The catalyst synthesized in this example is represented by NiSiMoW-1. Its precursor NiSiO3And XRD characterization of the catalyst as shown in FIG. 1, it can be found in NiSiO3In the spectrum of (a), characteristic peaks of the layered structure were present at 10 °,20 °,26 °,36 ° and 62 °. The characteristic peaks of the catalyst, which are lamellar, remain after further reaction with the Mo and W salts, and some peaks shift from 20 ℃ to 17 ℃, which all well demonstrate the success of the synthesis of NiSiO with a lamellar structure3Mo and W active metals were also successfully introduced between the layers to form a bulk catalyst with highly dispersed active species. Precursor NiSiO3The TEM representation result is shown in FIG. 2, and NiSiO3 is seen to have a more obvious layered structure and a plurality of fragmented nanotube structures. SEM characterization of NiSiMoW-1 is shown in FIG. 3, where it can be seen that catalysis was after the introduction of Mo and WThe agent still retains the layered structure well.
Example 2
This example illustrates the preparation of a NiSiMoW bulk catalyst in the present disclosure:
a) separately weigh out nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form light green mixed reaction liquid, and carrying out reflux reaction on the light green mixed reaction liquid at the reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor;
b) ammonium molybdate (10.8g, Mo therein) was weighed out separately6+0.06mol) and ammonium metatungstate (14.4g, wherein W6+0.06mol) instead of ammonium molybdate (5.4g, in which Mo is present) as used in example 16+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol) in the same manner as in example 1 a multimetallic bulk catalyst (27.4g) was prepared, the synthesized multimetallic bulk catalyst being denoted by NiSiMoW-2, the material morphology of this catalyst being similar to that of the NiSiMoW-1 catalyst. Its ultrahigh hydrodesulfurization activity is shown in FIG. 4. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 206.8m2The pore volume was 0.19 ml/g.
Example 3
This example illustrates the preparation of a NiSiMoW bulk catalyst in the present disclosure:
a) separately weigh out nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form light green mixed reaction liquid, and carrying out reflux reaction on the light green mixed reaction liquid at the reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor;
b) ammonium molybdate (16.2g, Mo therein) was weighed out separately6+0.09mol) and ammonium metatungstate (21.6g, wherein W6+0.09mol) instead of ammonium molybdate (5.4g, in which Mo is present) as used in example 16+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol) in the same manner as in example 1 a multimetallic bulk catalyst (37.2g) was prepared, the synthesized multimetallic bulk catalyst being denoted by NiSiMoW-3, the material morphology of this catalyst being similar to that of the NiSiMoW-1 catalyst. Its ultrahigh hydrodesulfurization activity is shown in FIG. 4. The specific surface area of the catalyst was 183.5m as determined by low temperature nitrogen adsorption2The pore volume is 0.19 ml/g.
Example 4
This example illustrates the preparation of a NiSiMoW bulk catalyst in the present disclosure:
a) separately weighing nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form light green mixed reaction liquid, and carrying out reflux reaction on the light green mixed reaction liquid at the reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor;
b) ammonium molybdate (16.2g, Mo therein)6+0.09mol) and ammonium metatungstate (14.4g, wherein W6+0.06mol) instead of ammonium molybdate (5.4g, in which Mo is present) as used in example 16+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol) in the same manner as in example 1 a multimetallic bulk catalyst (29.1g) was prepared, the synthesized multimetallic bulk catalyst being denoted by NiSiMoW-4, the material morphology of this catalyst being similar to that of the NiSiMoW-1 catalyst. The hydrodesulfurization activity is shown in FIG. 4. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 174.3m2The pore volume is 0.19 ml/g.
Example 5
This example illustrates the preparation of a NiSiMoW bulk catalyst in the present disclosure:
a) separately weigh out nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form light green mixed reaction liquid, and carrying out reflux reaction on the light green mixed reaction liquid at the reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor;
b) ammonium molybdate (21.6g, Mo therein) was weighed separately6+0.12mol) and ammonium metatungstate (14.4g, wherein W6+0.06mol) instead of ammonium molybdate (5.4g, in which Mo is present) as used in example 16+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol) in the same manner as in example 1 a multimetallic bulk catalyst (37.8g) was prepared, the synthesized multimetallic bulk catalyst being denoted by NiSiMoW-5, the material morphology of this catalyst being similar to that of the NiSiMoW-1 catalyst. Its ultrahigh hydrodesulfurization activity is shown in FIG. 4. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 176.6m2The pore volume is 0.19 ml/g.
Example 6
This example illustrates the preparation of a NiSiMoW bulk catalyst in the present disclosure:
a) separately weigh out nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form light green mixed reaction liquid, and carrying out reflux reaction on the light green mixed reaction liquid at the reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); catalyzing thisAdding the agent precursor into 200ml of water to prepare a slurry precursor;
b) ammonium molybdate (7.2g, wherein Mo) was weighed out separately6+0.045mol) and ammonium metatungstate (10.8g, wherein W is6+0.045mol) instead of ammonium molybdate (5.4g, in which Mo is present) as used in example 16+0.03mol) and ammonium metatungstate (7.2g, wherein W6 +0.03mol) in the same manner as in example 1 a multimetallic bulk catalyst (20.1g) was prepared, the synthesized multimetallic bulk catalyst being denoted by NiSiMoW-2, the material morphology of this catalyst being similar to that of the NiSiMoW-2 catalyst. Its ultrahigh hydrodesulfurization activity is shown in fig. 4. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 210.4m2The pore volume is 0.19 ml/g.
Example 7
This example illustrates the preparation of a NiSiMoW bulk catalyst according to the invention:
a) separately weigh out nickel nitrate (29.08g, Ni therein)2+0.1mol), sodium silicate (28.42g, of which SiO is present3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form light green mixed reaction liquid, and carrying out reflux reaction on the light green mixed reaction liquid at the reaction temperature of 80 ℃ for 25 hours to obtain a light green product; the pale green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor;
b) ammonium molybdate (13.5g, wherein Mo) was weighed out separately6+0.075mol) and ammonium metatungstate (18.0g, where W is6+0.075mol) instead of ammonium molybdate (5.4g, in which Mo is present) as used in example 16+0.03mol) and ammonium metatungstate (7.2g, wherein W6 +0.03mol) in the same manner as in example 1 a multimetallic bulk catalyst (24.6g) was prepared, the synthesized multimetallic bulk catalyst being denoted by NiSiMoW-7, the material morphology of this catalyst being similar to that of the NiSiMoW-1 catalyst. Its ultrahigh hydrodesulfurization activity is shown in FIG. 4. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 182.1m2The pore volume is 0.19 ml/g.
Example 8
A multimetallic bulk catalyst (15.2g) was prepared in the same manner as described in example 1, except that in step a, a reaction at 100 ℃ for 10 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-a, which is a pale green powder. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 202.1m2The pore volume is 0.17 ml/g.
Example 9
A multimetallic bulk catalyst (15.8g) was prepared in the same manner as described in example 1, except that in step a reaction at 100 ℃ for 25 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-B, which is a pale green powder. The specific surface area of the catalyst, as determined by low temperature nitrogen adsorption, was 213.2m2The pore volume is 0.19 ml/g.
Example 10
A multimetallic bulk catalyst (15.5g) was prepared in the same manner as described in example 1, except that in step a, a reaction at 100 ℃ for 45 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-C, which is a pale green powder. The specific surface area of the catalyst, as determined by low temperature nitrogen adsorption, was 209.2m2The pore volume is 0.18 ml/g.
Example 11
A multimetallic bulk catalyst (16.1g) was prepared in the same manner as described in example 1, except that in step a, a reaction at 150 ℃ for 15 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-D, this Cat-D catalyst being a pale green powder. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 204.2m2The pore volume is 0.19 ml/g.
Example 12
Prepared in the same manner as described in example 1 except that the reaction at 150 ℃ for 25 hours was used in step a instead of the reaction at 80 ℃ for 25 hours used in example 1A multimetallic bulk catalyst (15.9g) was prepared and the synthesized multimetallic bulk catalyst was denoted Cat-E, which was a pale green powder. The specific surface area of the catalyst was 203.0m as measured by low temperature nitrogen adsorption2The pore volume is 0.19 ml/g.
Example 13
A multimetallic bulk catalyst (15.3g) was prepared in the same manner as described in example 1, except that in step a, a reaction at 150 ℃ for 45 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-F, which is a pale green powder. The specific surface area of the catalyst, as determined by low temperature nitrogen adsorption, was 205.7m2The pore volume is 0.19 ml/g.
Example 14
A multimetallic bulk catalyst (15.4G) was prepared in the same manner as described in example 1, except that in step b a reaction at 50 ℃ for 4 hours was used instead of the reaction at 80 ℃ for 5 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-G, which is a pale green powder. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 204.1m2The pore volume is 0.19 ml/g.
Example 15
A multimetallic bulk catalyst (15.2g) was prepared in the same manner as described in example 1, except that in step b, a reaction at 50 ℃ for 10 hours was used instead of the reaction at 80 ℃ for 5 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-H, this Cat-H catalyst being a pale green powder. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 204.5m2The pore volume is 0.18 ml/g.
Example 16
A multimetallic bulk catalyst (15.2g) was prepared in the same manner as described in example 1, except that in step b a reaction at 50 ℃ for 25 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-I, which is a pale green powder. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 202.3m2Per g, pore volume of 0.19ml/g。
Example 17
A multimetallic bulk catalyst (17.1g) was prepared in the same manner as described in example 1, except that in step b a reaction at 50 ℃ for 45 hours was used instead of the reaction at 80 ℃ for 25 hours used in example 1, the synthesized multimetallic bulk catalyst being denoted by Cat-J, which is a pale green powder. The specific surface area of the catalyst, determined by low-temperature nitrogen adsorption, was 210.7m2The pore volume is 0.19 ml/g.
Example 18
This example illustrates the preparation of a NiCoSiMoW bulk catalyst of the invention:
a) separately weighing nickel nitrate (23.264g, wherein Ni is present)2+0.08mol), cobalt chloride (4.758g, wherein Co2+0.02mol), sodium silicate (28.42g, SiO therein3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form a gray-green mixed reaction solution, and carrying out reflux reaction on the gray-green mixed reaction solution at the reaction temperature of 80 ℃ for 25 hours to obtain a gray-green product; the gray-green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiCoSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor A;
b) ammonium molybdate (5.4g, wherein Mo) was weighed out separately6+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol), dissolving the raw materials in a polar solvent (300ml) to form a solution, heating the solution to the reaction temperature, and continuously stirring to form a colorless transparent solution B; subsequently heating the slurry precursor a prepared in step a) to a reaction temperature; slowly adding the slurry into the colorless transparent solution B to form a gray-green reaction solution, and carrying out reflux reaction on the gray-green reaction solution at the reaction temperature of 80 ℃ for 5 hours to obtain gray-green powder; the grayish green powder obtained by the reaction was filtered, washed, and dried at 120 ℃ to obtain a NiCoSiMoW catalyst (15.8 g). The NiCoSiMoW catalyst synthesized in this example is represented by Cat-K. The specific surface area of the catalyst was 231.7m as determined by low temperature nitrogen adsorption2The pore volume is 0.18 ml/g.
Example 19
This example illustrates the preparation of a NiCoSiMoW bulk catalyst in this disclosure:
a) separately weighing nickel nitrate (17.448g, wherein Ni is present)2+0.06mol), cobalt chloride (9.516g, of which Co is present2+0.04mol), sodium silicate (28.42g, SiO therein3 2-0.1mol), dissolving the raw materials in 200ml of mixed solution containing water, organic solvent and surfactant, adjusting the pH value to 12, heating to the reaction temperature to form a gray-green mixed reaction solution, and carrying out reflux reaction on the gray-green mixed reaction solution at the reaction temperature of 80 ℃ for 25 hours to obtain a gray-green product; the gray-green product obtained by the reaction was filtered and washed to obtain a catalyst precursor (i.e., NiCoSiO having a layered structure)3A catalyst precursor); adding the catalyst precursor into 200ml of water to prepare a slurry precursor A;
b) ammonium molybdate (5.4g, wherein Mo) was weighed out separately6+0.03mol) and ammonium metatungstate (7.2g, wherein W6+0.03mol), dissolving the raw materials in a polar solvent (300ml) to form a solution, heating the solution to the reaction temperature, and continuously stirring to form a colorless transparent solution B; subsequently heating the slurry precursor a prepared in step a) to a reaction temperature; slowly adding the slurry into the colorless transparent solution B to form a gray-green reaction solution, and carrying out reflux reaction on the gray-green reaction solution at the reaction temperature of 80 ℃ for 5 hours to obtain gray-green powder; the grayish green powder obtained by the reaction was filtered, washed, and dried at 120 ℃ to obtain a NiCoSiMoW catalyst (15.8 g). The NiCoSiMoW catalyst synthesized in this example is represented by Cat-L. The specific surface area of the catalyst was 238.3m as determined by low temperature nitrogen adsorption2The pore volume is 0.18 ml/g.
In another embodiment of the present disclosure, there is provided the use of a catalyst as described above in the hydrodesulfurization of sulfur containing organic compound fuel oil.
In one embodiment of the present disclosure, the hydrodesulfurization reaction conditions of the catalyst prepared in the above embodiment are: the temperature is 250 ℃ and 450 ℃, the hydrogen pressure is 1-20MPa, and the volume ratio of the hydrogen to the sulfur-containing organic compound fuel oil is 50-1500Nm3/m3And sulfur-containing organic compoundsThe volume space velocity of the oil is 0.1-10h-1。
The catalyst is pretreated before the hydrodesulfurization reaction as follows: a) grinding, kneading and molding; and b) carrying out in-situ presulfurization on the mixed gas of the sulfur-containing compound and hydrogen at the temperature of 250-450 ℃ in a hydrodesulfurization fixed bed reactor; the prevulcanization time is 2-10 hours.
In one embodiment of the present disclosure, the sulfur-containing compound may be hydrogen sulfide, carbon disulfide, or dimethyl disulfide. Evaluation of catalyst Performance in hydrodesulfurization reactions
The performance of the catalyst in hydrodesulfurization reactions, carried out in a fixed bed reactor, was examined with a diesel fraction (4, 6-DMDBT dissolved in decalin solvent) having a sulphur content of 521 ppm. The reaction conditions are as follows: the catalyst mass was 0.5g, the reaction temperature was 300 deg.C, the hydrogen pressure was 3.0MPa, and the ratio of hydrogen to diesel oil fraction was 800Nm3Hydrogen/m3Diesel oil fraction, volume space velocity 9h-1. For the measurement of the sulfur content in the sample, an ANTEK sulfur determinator was used.
The hydrodesulfurization activity of the catalysts prepared in examples 1 to 7 is shown in fig. 4, and it can be seen that the desulfurization rate of these catalysts can be as high as 97% or more, and the sulfur in the diesel fraction, i.e., 4, 6-dimethyldibenzothiophene, can be removed from 521ppm to 10ppm or less, thereby achieving ultra-deep desulfurization.
The sulfur content of the diesel fractions after hydrodesulfurization over the catalysts prepared in examples 8-19 is given in Table 1.
In the hydrodesulfurization reaction of diesel oil fractions, the desulfurization result of the catalyst prepared by the embodiment of the disclosure shows that the desulfurization effect of all the catalysts is optimal, and the sulfur content in the raw material can be reduced from 521ppm to below 10 ppm.
TABLE 1 comparison of catalyst Performance in hydrodesulfurization reactions
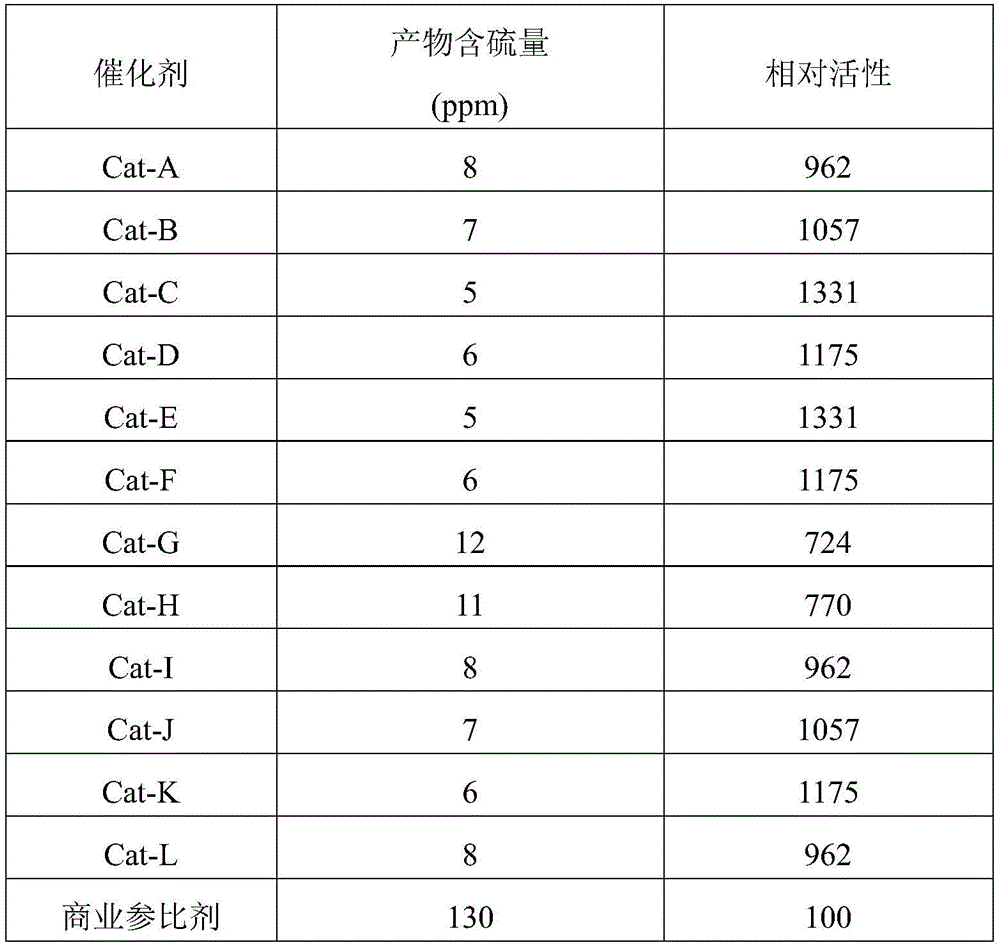
Commercial reference reagent of Table 1The national petrochemical company provides the composition of Co3O4·2.2NiO·5.9MoO3·2WO3。
The activity of the catalysts prepared in the examples of the present disclosure is expressed as relative activity, i.e., the activity of the reference agent at 200 hours of operation is 100, and the activity obtained by comparing the catalysts of the present invention with the reference agent represents the relative activity of the catalysts of the present invention. The relative desulfurization activity was calculated by the following formula:
relative desulfurization activity of 100 × [ (1/S)p)0.65-(1/Sf)0.65]/[(1/Spr)0.65-(1/Sfr)0.65]
In the formula Sfr、SprRespectively representing the sulfur concentration, S, of the diesel fraction used as a reference agent and the product obtained after hydrotreating the reference agentf、SpRespectively showing the sulfur concentration of the diesel oil fraction used by the catalyst and the product after the hydrogenation treatment by the catalyst.
In summary, the disclosed embodiments synthesize a multi-metal bulk catalyst with a layered structure, which has a higher specific surface area, a higher pore volume and an ultrahigh hydrodesulfurization activity than those of patents and literatures, the catalyst has a layered structure, and active metals are exchanged between layers, the dispersion degree of the active metals is higher, more active centers are displayed, and the catalyst can remove sulfur in a diesel oil fraction containing 4,6-DMDBT from 521ppmw to below 10ppmw through hydrodesulfurization reaction under mild operation conditions, so that the ultra-deep desulfurization is realized, and the catalyst shows a great industrial application potential.
Claims (10)
1. A desulfurization catalyst characterized by: mixed metal oxides comprising at least one group VIII metal, at least two group VIB metals, and a group IVA nonmetal;
wherein, calculated by oxide, the catalyst contains 1-50 wt% of VIII group metal, 1-50 wt% of VIB group metal and 1-50 wt% of IVA group nonmetal.
2. The catalyst of claim 1, wherein the catalyst comprises at least one group VIII metal, two group VIB metals, and one group IVA nonmetal, and the molar ratio of the group VIII metal to the group VIB metal is from 20:1 to 1: 20; the molar ratio of the group VIII metal to the group IVA nonmetal is 20:1 to 1: 1; the molar ratio of the two group VIB metals is 5:1-1: 5.
3. The catalyst of claim 1 wherein the group VIII metal comprises Ni or Co; the group VIB metal comprises Mo and W; the group IVA nonmetal comprises Si.
4. The catalyst according to claim 1, wherein the catalyst has a layered porous structure having a specific surface area of 80 to 300m2The pore volume is 0.1-0.5 ml/g.
5. A method for preparing a desulfurization catalyst, comprising:
adding at least one soluble salt of VIII group metal into a solvent to prepare a solution A, adding at least one soluble salt of IVA group nonmetal into water to prepare a solution B, and mixing and reacting the solution A and the solution B to obtain a catalyst precursor solution;
adding soluble salts of at least two VIB group metals into a polar solvent to prepare a solution C, and mixing and reacting the solution C with the catalyst precursor solution to obtain the catalyst.
6. The method according to claim 5, wherein the concentration of the group VIII metal ions in the solution A is 0.01-0.3 mol/L, the concentration of the group IVA non-metal ions in the solution B is 0.01-0.3 mol/L, the concentration of the catalyst precursor solution is 0.01-0.9 mol/L, and the concentration of the group VIB metal ions in the solution C is 0.01-0.2 mol/L.
7. The method of claim 5, wherein said solution A is reacted with said solution B at 20-200 ℃ for 4-50 hours, and said solution C is reacted with said catalyst precursor solution at 20-200 ℃ for 4-50 hours.
8. The method of claim 5, wherein the soluble salt of the group IVA nonmetal comprises one or more of sodium silicate, potassium silicate, lithium silicate, ammonium silicate, sodium metasilicate pentahydrate, the soluble salt of the group VIII metal comprises one or more of nickel nitrate, nickel acetate, nickel sulfate or nickel chloride, cobalt nitrate, cobalt chloride, cobalt sulfate or cobalt acetate, and the soluble salt of the group VIB metal anion comprises one of ammonium molybdate, molybdic acid, ammonium phosphomolybdate, phosphomolybdic acid and sodium molybdate, and one of ammonium tungstate, phosphotungstic acid, ammonium metatungstate, tungstic acid or sodium tungstate.
9. Use of a catalyst according to any one of claims 1 to 4 in the hydrodesulphurisation of sulphur containing organic compound fuels.
10. The use according to claim 9, characterized in that the hydrodesulfurization reaction conditions are: the temperature is 250 ℃ and 450 ℃, the hydrogen pressure is 1-20MPa, and the volume ratio of the hydrogen to the sulfur-containing organic compound fuel oil is 50-1500Nm3/m3(ii) a The volume space velocity of the sulfur-containing organic compound fuel oil is 0.1-10h-1。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210187860.5A CN114433120B (en) | 2022-02-28 | 2022-02-28 | Desulfurization catalyst and preparation method and application thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210187860.5A CN114433120B (en) | 2022-02-28 | 2022-02-28 | Desulfurization catalyst and preparation method and application thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN114433120A true CN114433120A (en) | 2022-05-06 |
| CN114433120B CN114433120B (en) | 2024-06-28 |
Family
ID=81373132
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210187860.5A Active CN114433120B (en) | 2022-02-28 | 2022-02-28 | Desulfurization catalyst and preparation method and application thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114433120B (en) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1359367A (en) * | 1970-08-31 | 1974-07-10 | Exxon Research Engineering Co | Layered complex metal silicate composition their preparation and use in hydrocarbon conversion reactions |
| JP2000325793A (en) * | 1999-05-18 | 2000-11-28 | Agency Of Ind Science & Technol | Catalyst for hydrogenating/desulfurizing thiophene and manufacture thereof |
| JP2002035589A (en) * | 2000-07-26 | 2002-02-05 | Agency Of Ind Science & Technol | Synthetic desulfurization catalyst and manufacturing method therefor |
| US20100133147A1 (en) * | 2006-07-31 | 2010-06-03 | Alexandra Chaumonnot | Catalyst based on an organic-inorganic hybrid support and its use in hydrorefining and hydroconversion |
| CN101733120A (en) * | 2009-12-23 | 2010-06-16 | 中国科学院大连化学物理研究所 | Multi-metal body catalyst with laminated structure, preparation method and application thereof |
-
2022
- 2022-02-28 CN CN202210187860.5A patent/CN114433120B/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1359367A (en) * | 1970-08-31 | 1974-07-10 | Exxon Research Engineering Co | Layered complex metal silicate composition their preparation and use in hydrocarbon conversion reactions |
| JP2000325793A (en) * | 1999-05-18 | 2000-11-28 | Agency Of Ind Science & Technol | Catalyst for hydrogenating/desulfurizing thiophene and manufacture thereof |
| JP2002035589A (en) * | 2000-07-26 | 2002-02-05 | Agency Of Ind Science & Technol | Synthetic desulfurization catalyst and manufacturing method therefor |
| US20100133147A1 (en) * | 2006-07-31 | 2010-06-03 | Alexandra Chaumonnot | Catalyst based on an organic-inorganic hybrid support and its use in hydrorefining and hydroconversion |
| CN101733120A (en) * | 2009-12-23 | 2010-06-16 | 中国科学院大连化学物理研究所 | Multi-metal body catalyst with laminated structure, preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114433120B (en) | 2024-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101544904B (en) | Composite metal oxide catalyst, preparation and application thereof | |
| CN101733120B (en) | Multi-metal body catalyst with laminated structure, preparation method and application thereof | |
| US20150190789A1 (en) | Preparation and application of ultra-deep hydrodesulfurization multi-metal bulk catalyst of layered structure | |
| CN109692693A (en) | A kind of Hydrobon catalyst and its preparation method | |
| CN111215094A (en) | A kind of multi-metal unsupported hydrofining catalyst and its preparation method and application | |
| RU2402380C1 (en) | Catalyst for hydrofining hydrocarbon material, method of preparing said catalyst and hydrofining process | |
| CN106268976A (en) | A kind of catalyst for selective hydrodesulfurizationof of gasoline and preparation thereof and application | |
| CN107812525A (en) | A kind of hydrogenating catalyst composition and the method for hydrotreating | |
| US5565091A (en) | Catalyst composition manufacturing method and sulfur-containing hydrocarbon hydrodesulfurization method using the same catalyst composition | |
| CN106179414B (en) | A kind of sulfurized hydrogenation catalyst for refining and preparation method thereof | |
| CN102451705B (en) | Preparation method of hydrotreatment catalyst composition | |
| CN101153228A (en) | Multi-metal bulk catalyst for diesel hydrodesulfurization and its preparation method and application | |
| CN106512984B (en) | Preparation method of a high-activity diesel hydrodesulfurization catalyst | |
| CN109821557A (en) | A kind of preparation method and application of MoS2/LDHS Hydrobon catalyst | |
| CN105312060A (en) | Multi-metal body catalyst with layered structure, preparation and applications thereof | |
| Han et al. | A novel and facile strategy for the preparation of highly reactive NiMo/γ-Al2O3 hydrodesulfurization catalyst | |
| CN100348700C (en) | Sulfide catalyst for hydrogenation desulfurization and denitrogenation and its preparation process and use | |
| CN114308000B (en) | Alumina carrier and preparation method thereof, hydrorefining catalyst and preparation method thereof | |
| EP0638361B1 (en) | Catalyst composition manufacturing method and sulfur-containing hydrocarbon hydrodesulfurization method using the same catalyst composition | |
| CN114433120A (en) | Desulfurization catalyst and preparation method and application thereof | |
| CN101468309B (en) | Method for preparing non-supported hydrogenation catalyst | |
| CN120286014A (en) | A method for preparing a desulfurization catalyst, a desulfurization catalyst and its application | |
| CN110038584A (en) | The method for preparing Hydrobon catalyst | |
| CN119425744B (en) | Diesel hydrorefining catalyst and preparation method thereof | |
| CN112742402B (en) | Hydrodesulfurization catalyst and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |