Disclosure of Invention
The invention aims to overcome the defects of the prior art and provide a carbon-11 labeled Larotrectinib compound and a preparation method thereof.
The invention provides that Larotrectinib can be marked by radionuclide carbon-11 and prepared into a [ alpha ], [ beta ] -cyclodextrin11C]The compound is used as a novel TRK positron emission tomography tracer, is used for PET online in-vivo imaging, has TRK specificity, is used for detecting the expression of TRK fusion protein in tumor tissues by means of the distribution change of PET on TRK kinase, thereby providing reliable diagnosis and molecular phenotype data for clinical examination, and can also be used for in-vitro cell receptor and drug molecule screening and evaluation. The research result of the present invention preliminarily shows that11C]the-Larotrectini molecule as a TRK kinase [ 2 ]11C]Potential imaging of positron emission tomography tracers.
In order to realize the purpose of the invention, the method can be realized by the following technical scheme:
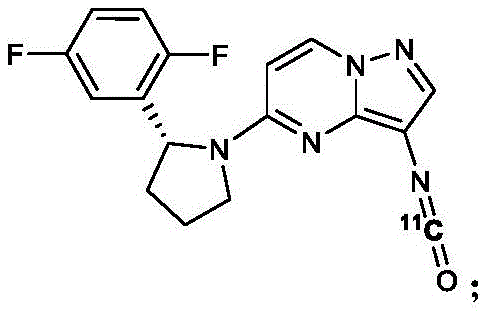
a carbon-11 labeled Larotrectinib compound has the following chemical structural formula:
the invention provides a method for preparing the carbon-11 labeled Larotrectinib compound, which comprises the following steps:
(1) mixing and reacting the compound shown in the formula 1, the compound shown in the formula 2 and carbon-11 marked fluorophosphone to obtain a compound shown in the formula 3,
wherein R is hydrogen or a hydroxyl protecting group;
alternatively, (2) when R is a hydroxyl protecting group, removing the hydroxyl protecting group of the compound shown in the formula 3.
Preferably, the hydroxyl protecting group is selected from trimethylsilyl, t-butyldimethylsilyl, triethylsilyl, t-butyldiphenylsilyl, methyl, benzyl, trityl, p-methoxytrityl, dimethoxytrityl, t-butyl, methoxymethyl, 2-methoxyethoxymethyl, methylthiomethyl, benzyloxymethyl, p-methoxybenzyl, p-methoxybenzyloxymethyl, 3, 4-dimethoxybenzyl, tetrahydropyranyl, methoxycarbonyl, ethoxycarbonyl, t-butoxycarbonyl, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl, trifluoroacetyl, chloroacetyl, dichloroacetyl, phenylacetyl, pivaloyl, methylsulfonyl, benzylsulfonyl, allylsulfonyl, allyloxycarbonyl, C1-C16 alkanoyl, C3-C16 alkenoyl, C3-C6 alkynoyl, methyl, benzyl, N-methyl, N-ethyl, N-propyl, N-butyl, N-methyl, N-butyl-methyl, N-butyl-methyl, N-methyl, N-N, N-, C4-C10 cycloalkyl, C7-C16 aroyl, C4-C10 heterocycloalkanoyl.
Preferably, the mixing reaction in step (1) comprises: a solution containing the compound represented by formula 1 and the compound represented by formula 2 is passed through carbon-11 labeled fluorophosphone.
Preferably, the concentration of the compound represented by formula 1 in the solution is 0.01-1000 mmol/L, more preferably 0.1-200 mmol/L, and most preferably 1-20 mmol/L.
Preferably, the molar ratio of the compound shown in the formula 1 to the compound shown in the formula 2 is 1: 0.1-10, more preferably, the molar ratio is 1: 0.5-5, and most preferably, the molar ratio is 1: 1-3.
Preferably, the carbon-11 labeled fluorophosphogas is introduced for 1 to 10min, more preferably for 2 to 7min, and most preferably for 4 to 6 min.
Preferably, the reaction solvent of the mixing reaction is selected from acetonitrile.
Preferably, when R is a hydroxyl protecting group, the compound shown in the formula 3 reacts at 60-180 ℃ under the action of acid, potassium carbonate or magnesium bromide to remove the hydroxyl protecting group, so that the carbon-11 labeled Larotrectinib compound is obtained.
Preferably, when R is a hydroxyl protecting group, the step (1) and the step (2) are completed by adopting a one-pot method.
The invention provides another method for preparing the carbon-11 labeled Larotrectinib compound, which comprises the following steps:
(1) introducing carbon-11-labeled carbon dioxide into a solution containing BEMP and the compound shown in the formula 1, and adding POCl after the reaction is finished3Continuing the reaction to obtain the compound shown in the formula 4,
(2) mixing the compound shown in the formula 4 and the compound shown in the formula 2 for reaction to obtain the compound shown in the formula 3,
wherein R is hydrogen or a hydroxyl protecting group;
alternatively, (3) when R is a hydroxyl protecting group, the hydroxyl protecting group of the compound represented by formula 3 is removed.
Preferably, the hydroxyl protecting group is selected from trimethylsilyl, t-butyldimethylsilyl, triethylsilyl, t-butyldiphenylsilyl, methyl, benzyl, trityl, p-methoxytrityl, dimethoxytrityl, t-butyl, methoxymethyl, 2-methoxyethoxymethyl, methylthiomethyl, benzyloxymethyl, p-methoxybenzyl, p-methoxybenzyloxymethyl, 3, 4-dimethoxybenzyl, tetrahydropyranyl, methoxycarbonyl, ethoxycarbonyl, t-butoxycarbonyl, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl, trifluoroacetyl, chloroacetyl, dichloroacetyl, phenylacetyl, pivaloyl, methylsulfonyl, benzylsulfonyl, allylsulfonyl, allyloxycarbonyl, C1-C16 alkanoyl, C3-C16 alkenoyl, C3-C6 alkynoyl, methyl, benzyl, N-methyl, N-ethyl, N-propyl, N-butyl, N-methyl, N-butyl-methyl, N-butyl-methyl, N-methyl, N-N, N-, C4-C10 cycloalkyl, C7-C16 aroyl, C4-C10 heterocycloalkanoyl.
Preferably, the concentration of the compound represented by formula 1 in the solution is 0.01-1000 mmol/L, more preferably 0.1-200 mmol/L, and most preferably 1-20 mmol/L.
Preferably, the volume percent concentration of BEMP in the solution is 1-30%, preferably 1-10%, more preferably 3-6%.
Preferably, in the step (1), the introduction of the carbon-11 labeled carbon dioxide is stopped after the radioactivity trapped in the solution reaches a peak, more preferably, the reaction is performed for 0.5 to 5 minutes, and most preferably, for 1 to 2 minutes after the introduction of the carbon-11 labeled carbon dioxide is stopped.
Preferably, POCl3The molar use ratio of the compound shown in the formula 1 to the compound shown in the formula 1 is 1-50: 1, more preferably 2-20: 1, and most preferably 3-10: 1.
Preferably, in step (1), POCl is added3And then reacting for 0.1-3 minutes, preferably 0.2-2 minutes, more preferably 0.5-1 minute.
Preferably, the molar use ratio of the compound shown in the formula 4 to the compound shown in the formula 2 is 1: 0.1-100, and preferably, the molar use ratio is 1: 1-100.
Preferably, the reaction solvent of step (1) is selected from acetonitrile.
Preferably, when R is a hydroxyl protecting group, the compound shown in the formula 3 reacts at 60-180 ℃ under the action of acid, potassium carbonate or magnesium bromide to remove the hydroxyl protecting group, so that the carbon-11 labeled Larotrectinib compound is obtained.
Preferably, step (1), step (2) and/or step (3) is accomplished using a one-pot process.
The invention also provides an intermediate for synthesizing the carbon-11 labeled Larotrectinib compound, wherein the intermediate has the following chemical structural formula:
the invention also provides an intermediate for synthesizing the carbon-11 labeled Larotrectinib compound, wherein the intermediate has the following chemical structural formula:
wherein R is selected from trimethylsilyl, tert-butyldimethylsilyl, triethylsilyl, tert-butyldiphenylsilyl, methyl, benzyl, trityl, p-methoxytrityl, dimethoxytrityl, tert-butyl, methoxymethyl, 2-methoxyethoxymethyl, methylthiomethyl, benzyloxymethyl, p-methoxybenzyl, p-methoxybenzyloxymethyl, 3, 4-dimethoxybenzyl, tetrahydropyranyl, methoxycarbonyl, ethoxycarbonyl, tert-butoxycarbonyl, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl, trifluoroacetyl, chloroacetyl, dichloroacetyl, phenylacetyl, pivaloyl, methylsulfonyl, benzylsulfonyl, allylsulfonyl, allyloxycarbonyl, C1-C16 alkanoyl, C3-C16 alkenoyl, C3-C6 alkynoyl, C4-C10 cycloalkyl, benzyl, N-methyl-ethyl, N-methyl, N-, C7-C16 aroyl, C4-C10 heterocycloalkanoyl.
Advantageous effects
Compared with the prior art, the invention has the following beneficial technical effects:
(1) the present invention successfully prepares carbon-11 labeled Larotrectinib compounds that have been desired in the art but have not been available.
(2) Compared with the prior art, the preparation method can obviously shorten the reaction time of the carbon-11 label, and enables the rapid synthesis of the radioactive tracer corresponding to the short half-life period of the carbon-11 in a short time, thereby successfully realizing the synthesis of the carbon-11 labeled Larotrectinib with the short half-life period.
(3) The preparation process provided by the invention has the advantages of few types of reaction reagents, small usage amount of reaction precursors, simple and feasible operation steps, simple post-treatment, capability of preparing the carrier-free radioactive labeled compound and high radiochemical purity. Is suitable for the clinical requirement of the carbon-11 marker Larotrectinib with short half-life.
(4) The present invention provides11C]The Larotrcetinib has the characteristic of emitting positrons, visually displays the distribution condition of Larotrcetiib compounds in vivo and in tumors by means of a PET-CT positron emission tomography technology, and provides a new imaging agent for early diagnosis of tumors.
Detailed Description
The present invention is described in further detail below with reference to specific examples and with reference to the data. It will be understood that this example is intended to illustrate the invention and not to limit the scope of the invention in any way.
Terms used in the present invention generally have meanings commonly understood by those of ordinary skill in the art, unless otherwise specified.
In the following examples, various procedures and methods not described in detail are conventional methods well known in the art. Materials, reagents, devices, instruments, apparatuses and the like used in the following examples are commercially available unless otherwise specified.
A carbon-11 labeled Larotrectinib compound has the following chemical structural formula:
in some embodiments of the present invention, the above-mentioned carbon-11 labeled Larotrectinib compound is prepared by a method comprising the steps of:
(1) a compound represented by the formula 1, a compound represented by the formula 2 and carbon-11 labeled fluorophosphone (, [ 2 ]11C]-COF2) Mixing and reacting to obtain the compound shown in the formula 3,
wherein R is hydrogen or a hydroxyl protecting group;
alternatively, (2) when R is a hydroxyl protecting group, removing the hydroxyl protecting group of the compound shown in the formula 3.
In some embodiments of the invention, the hydroxyl protecting group is selected from Trimethylsilyl (TMS), t-butyldimethylsilyl (TBDMS), Triethylsilyl (TES), t-butyldiphenylsilyl (TBDPS), methyl (Me), benzyl (Bn), trityl (Tr), p-methoxytrityl (MMT), Dimethoxytrityl (DMT), t-butyl (tBu), methoxymethyl (MOM), 2-methoxyethoxymethyl (MEM), methylthiomethyl (MTM), Benzyloxymethyl (BOM), p-methoxybenzyl (PMB), p-methoxybenzyloxymethyl (PMBOM), 3, 4-Dimethoxybenzyl (DMB), Tetrahydropyranyl (THP), methoxycarbonyl (Moc), ethoxycarbonyl (Eoc), t-butoxycarbonyl (Boc), benzyloxycarbonyl (Cbz), 9-fluorenylmethoxycarbonyl (Fmoc), trifluoroacetyl (TfAc), Chloroacetyl (CAc), dichloroacetyl (DAc), phenylacetyl (Bz), pivaloyl (Pv), methanesulfonyl (Ms), benzylsulfonyl (Bs), allylsulfonyl (Bs), allyloxycarbonyl (Als), C1-C16 alkanoyl, C3-C16 alkenoyl, C3-C6 alkynoyl, C4-C10 cycloalkyl, C7-C16 aroyl, C4-C10 heterocycloalkanoyl.
In some embodiments of the invention, the C1-C16 alkanoyl is selected from formyl, acetyl, propionyl, butyryl, pentanoyl, hexanoyl, heptanoyl, octanoyl, nonanoyl, decanoyl, undecanoyl, dodecanoyl, tridecanoyl, tetradecanoyl, pentadecanoyl, hexadecanoyl.
In some embodiments of the invention, the C3-C16 alkenoyl is selected from acryloyl, 2-butenoyl, 4-pentenoyl, 5-hexenoyl, 2-ethylacryloyl, 3-dimethyl-2-methylbutanoyl, 6-heptenoyl, 7-octenoyl, 3-methyl-hept-6-alkenoyl, 5-methyl-hept-6-alkenoyl, 8-nonenoyl.
In some embodiments of the invention, the C3-C6 alkynoyl is selected from propinyl, 2-butynoyl, 4-pentynoyl, 5-hexynoyl, 6-heptynoyl, 7-octynoyl, 8-nonynoyl.
In some embodiments of the invention, the C4-C10 cycloalkyl is selected from cyclopropylformyl, cyclobutylformyl, cyclopentylcarbonyl, cyclohexylformyl, 2-methylcyclohexylcarbonyl, 2, 6-dimethylcyclohexylformyl.
In some embodiments of the invention, the C7-C16 aroyl is selected from the group consisting of benzoyl, p-methylbenzoyl, m-methylbenzoyl, o-methylbenzoyl, 4-bibenzoyl, 1-naphthoyl, 2-naphthoyl, 1-methyl-2-naphthoyl, 6-methyl-2-naphthoyl, 3-quinolinecarbonyl, 8-quinolinecarbonyl, 9-anthraceneyl, 9-acridinecarbonyl.
In some embodiments of the present invention, the mixing reaction in step (1) comprises: a solution containing the compound represented by formula 1 and the compound represented by formula 2 is passed through carbon-11 labeled fluorophosphone.
The concentration of the compound of formula 1 in solution at the time of synthesis can be determined by one skilled in the art based on the amount required to perform online in vivo imaging. In some embodiments of the invention, the concentration of the compound of formula 1 in the solution is from 0.01 to 1000mmol/L, more preferably from 0.1 to 200mmol/L, and most preferably from 1 to 20 mmol/L.
In some embodiments of the invention, the molar ratio of the compound of formula 1 to the compound of formula 2 is 1:0.1 to 10, specifically 1:0.1, 1:0.2, 1:0.3, 1:0.4, 1:0.5, 1:0.6, 1:0.7, 1:0.8, 1:0.9, 1:1, 1:1.1, 1:1.2, 1:1.3, 1:1.4, 1:1.5, 1:1.6, 1:1.7, 1:1.8, 1:2, 1:2.2, 1:2.4, 1:2.6, 1:2.8, 1:3, 1:3.5, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1: 10. More preferably, the molar use ratio is 1: 0.5-5, and most preferably, the molar use ratio is 1: 1-3.
The carbon-11 labeled fluorophosphates were bubbled into a solution containing the compound represented by formula 1 and the compound represented by formula 2, and the compound represented by formula 1 or formula 2 was followed by Thin Layer Chromatography (TLC) until the reactants substantially disappeared, and the introduction of the fluorophosphates was stopped. The tail gas is absorbed by sodium hydroxide solution and the pipeline is purged with an inert gas such as helium.
In some embodiments of the invention, the carbon-11 labeled fluorophosphogas is introduced for a period of time ranging from 1 to 10 minutes, more preferably for a period of time ranging from 2 to 7 minutes, and most preferably for a period of time ranging from 4 to 6 minutes.
In some embodiments of the invention, the reaction solvent of the mixing reaction is selected from acetonitrile.
In some embodiments of the present invention, when R is a hydroxyl protecting group, the compound represented by formula 3 generated in step (1) is directly used in the treatment of removing the hydroxyl protecting group in step (2) without separation and purification, i.e., step (1) and step (2) are completed by a one-pot method.
The one-pot method of the invention means that the intermediate product obtained in the previous step is directly used for the reaction in the next step without separation and purification.
The hydroxyl protecting group (R) in the compound represented by formula 3 may be removed in the above step (2) by a method conventional in the art.
In some embodiments of the present invention, the hydroxyl protecting group on the compound represented by formula 3 is removed using the following method:
and heating the compound shown in the formula 3 in an acid solution to 60-100 ℃, reacting for 1-5 minutes, cooling, neutralizing to be neutral, separating and purifying by using preparative HPLC, and concentrating collected eluent to be dry to obtain the carbon-11 labeled Larotrectinib compound.
In some embodiments of the invention, the acid solution may be selected from hydrochloric acid having a concentration of 6 mol/L.
In some embodiments of the present invention, the hydroxyl protecting group on the compound represented by formula 3 is removed using the following method:
and (3) mixing the compound shown in the formula 3 with magnesium bromide, heating to 120-180 ℃, reacting for 2-7 minutes, cooling, neutralizing to be neutral, separating and purifying by using preparative HPLC, and concentrating collected eluent to be dry to obtain the carbon-11 labeled Larotrectinib compound.
In some embodiments of the present invention, the hydroxyl protecting group on the compound represented by formula 3 is removed using the following method:
and (3) mixing the compound shown in the formula 3 with potassium carbonate, heating to 80-120 ℃, reacting for 5-15 minutes, cooling, neutralizing to be neutral, separating and purifying by using preparative HPLC, and concentrating collected eluent to be dry to obtain the carbon-11 labeled Larotrectinib compound.
In some embodiments of the invention, the concentrating to dryness may be vacuum concentrating to dryness, nitrogen blow drying or evaporation to dryness.
In some embodiments of the invention, the preparative HPLC separation purification is performed using Waters C-18Sep-Pak, and the eluent is ethanol.
In some embodiments of the invention, upon completion using a one-pot method, the reaction of step (1) may be quenched with HPLC eluent/water (1/1, v/v) before the hydroxyl protecting group removal treatment of step (2) is performed.
In some embodiments of the present invention, the above-mentioned carbon-11 labeled Larotrectinib compound is prepared by a method comprising the steps of:
(1) introducing carbon-11-labeled carbon dioxide into a solution containing BEMP and the compound shown in the formula 1, and adding POCl after the reaction is finished3Continuing the reaction to obtain the compound shown in the formula 4,
(2) mixing the compound shown in the formula 4 and the compound shown in the formula 2 for reaction to obtain the compound shown in the formula 3,
wherein R is hydrogen or a hydroxyl protecting group;
alternatively, (3) when R is a hydroxyl protecting group, the hydroxyl protecting group of the compound represented by formula 3 is removed.
In some preferred embodiments of the invention, the hydroxyl protecting group is selected from trimethylsilyl, t-butyldimethylsilyl, triethylsilyl, t-butyldiphenylsilyl, methyl, benzyl, trityl, p-methoxytrityl, dimethoxytrityl, t-butyl, methoxymethyl, 2-methoxyethoxymethyl, methylthiomethyl, benzyloxymethyl, p-methoxybenzyl, p-methoxybenzyloxymethyl, 3, 4-dimethoxybenzyl, tetrahydropyranyl, methoxycarbonyl, ethoxycarbonyl, t-butoxycarbonyl, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl, trifluoroacetyl, chloroacetyl, dichloroacetyl, phenylacetyl, pivaloyl, methanesulfonyl, benzylsulfonyl, allyloxycarbonyl, C1-C16 alkanoyl, C3-C16 alkenoyl, vinylbenzyl, or a combination thereof, C3-C6 alkynoyl, C4-C10 cycloalkyl, C7-C16 aroyl, C4-C10 heterocycloalkanoyl.
In some embodiments of the invention, the C1-C16 alkanoyl is selected from formyl, acetyl, propionyl, butyryl, pentanoyl, hexanoyl, heptanoyl, octanoyl, nonanoyl, decanoyl, undecanoyl, dodecanoyl, tridecanoyl, tetradecanoyl, pentadecanoyl, hexadecanoyl.
In some embodiments of the invention, the C3-C16 alkenoyl is selected from acryloyl, 2-butenoyl, 4-pentenoyl, 5-hexenoyl, 2-ethylacryloyl, 3-dimethyl-2-methylbutanoyl, 6-heptenoyl, 7-octenoyl, 3-methyl-hept-6-alkenoyl, 5-methyl-hept-6-alkenoyl, 8-nonenoyl.
In some embodiments of the invention, the C3-C6 alkynoyl is selected from propinyl, 2-butynoyl, 4-pentynoyl, 5-hexynoyl, 6-heptynoyl, 7-octynoyl, 8-nonynoyl.
In some embodiments of the invention, the C4-C10 cycloalkyl is selected from cyclopropylformyl, cyclobutylformyl, cyclopentylcarbonyl, cyclohexylformyl, 2-methylcyclohexylcarbonyl, 2, 6-dimethylcyclohexylformyl.
In some embodiments of the invention, the C7-C16 aroyl is selected from the group consisting of benzoyl, p-methylbenzoyl, m-methylbenzoyl, o-methylbenzoyl, 4-bibenzoyl, 1-naphthoyl, 2-naphthoyl, 1-methyl-2-naphthoyl, 6-methyl-2-naphthoyl, 3-quinolinecarbonyl, 8-quinolinecarbonyl, 9-anthraceneyl, 9-acridinecarbonyl.
The concentration of the compound of formula 1 in solution at the time of synthesis can be determined by one skilled in the art based on the amount required to perform online in vivo imaging. In some embodiments of the invention, the concentration of the compound of formula 1 in the solution is from 0.01 to 1000mmol/L, more preferably from 0.1 to 200mmol/L, and most preferably from 1 to 20 mmol/L.
In some embodiments of the invention, the solution has a BEMP concentration of 1 to 30% by volume, specifically 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, 10.5%, 11%, 11.5%, 12%, 14%, 16%, 18%, 20%, 24%, 28%, 30% by volume. Preferably, the volume percentage concentration is 1-10%, and more preferably, the volume percentage concentration is 3-6%.
In some embodiments of the present invention, in step (1), the introduction of the carbon-11 labeled carbon dioxide is stopped after the radioactivity trapped in the solution reaches a peak, and more preferably, the reaction is stopped after the introduction of the carbon-11 labeled carbon dioxide is stopped, and the reaction is performed for 0.5 to 5 minutes, specifically, for 0.5 minute, 0.6 minute, 0.8 minute, 1 minute, 1.1 minute, 1.2 minute, 1.3 minute, 1.4 minute, 1.5 minute, 1.6 minute, 1.7 minute, 1.8 minute, 1.9 minute, 2 minutes, 2.5 minutes, 3 minutes, 3.5 minutes, 4 minutes, 4.5 minutes, or 5 minutes. Most preferably, the reaction is carried out for 1 to 2 minutes.
In some embodiments of the invention, the POCl3The molar use ratio of the compound to the compound shown as the formula 1 is 1-50: 1, specifically POCl3The molar ratio of the compound of formula 1 to the compound of formula 1 is 1:1, 1.5:1, 2:1, 2.5:1, 3:1, 3.5:1, 4:1, 4.5:1, 5:1, 5.5:1, 6:1, 6.5:1, 7:1, 7.5:1, 8:1, 8.5:1, 9:1, 9.5:1, 10:1, 12:1, 14:1, 16:1, 18:1 or 20: 1. More preferably, the molar use ratio is 2-20: 1, and most preferably, the molar use ratio is 3-10: 1.
In some embodiments of the invention, in step (1), POCl is added3Then, the reaction is carried out for 0.1 to 3 minutes, specifically, for 0.1 minute, 0.2 minute, 0.3 minute, 0.4 minute, 0.5 minute, 0.6 minute, 0.7 minute, 0.8 minute, 0.9 minute, 1 minute, 1.5 minute, 2 minutes, 2.5 minutes, and 3 minutes. Preferably, the reaction is carried out for 0.2 to 2 minutes, and more preferably, for 0.5 to 1 minute.
In some embodiments of the invention, the molar ratio of the compound of formula 4 to the compound of formula 2 is 1:0.1 to 100, more preferably, in order to make full use of the compound of formula 4, an excess of the compound of formula 2 may be added, such that the molar ratio of the compound of formula 4 to the compound of formula 2 is 1:1 to 100, such as 1:1, 1:10, 1:20, 1:30, 1:40, 1:50, 1:60, 1:70, 1:80, 1:90, 1:100, or intervals therebetween.
In some embodiments of the invention, the reaction solvent of step (1) is selected from acetonitrile.
In some embodiments of the present invention, the intermediates generated in the reactions of the respective steps are directly used for the next step without isolation and purification, i.e., step (1), step (2) and/or step (3) are completed by a one-pot method.
The one-pot method of the invention means that the intermediate product obtained in the previous step is directly used for the reaction in the next step without separation and purification.
The hydroxyl protecting group (R) in the compound represented by formula 3 may be removed in the above step (3) by a method conventional in the art.
In some embodiments of the present invention, the hydroxyl protecting group on the compound represented by formula 3 is removed using the following method:
and heating the compound shown in the formula 3 in an acid solution to 60-100 ℃, reacting for 1-5 minutes, cooling, neutralizing to be neutral, separating and purifying by using preparative HPLC, and concentrating collected eluent to be dry to obtain the carbon-11 labeled Larotrectinib compound.
In some embodiments of the invention, the acid solution may be selected from hydrochloric acid having a concentration of 6 mol/L.
In some embodiments of the present invention, the hydroxyl protecting group on the compound represented by formula 3 is removed using the following method:
and (3) mixing the compound shown in the formula 3 with magnesium bromide, heating to 120-180 ℃, reacting for 2-7 minutes, cooling, neutralizing to be neutral, separating and purifying by using preparative HPLC, and concentrating collected eluent to be dry to obtain the carbon-11 labeled Larotrectinib compound.
In some embodiments of the present invention, the hydroxyl protecting group on the compound represented by formula 3 is removed using the following method:
and (3) mixing the compound shown in the formula 3 with potassium carbonate, heating to 80-120 ℃, reacting for 5-15 minutes, cooling, neutralizing to be neutral, separating and purifying by using preparative HPLC, and concentrating collected eluent to be dry to obtain the carbon-11 labeled Larotrectinib compound.
In some embodiments of the invention, the concentrating to dryness may be vacuum concentrating to dryness, nitrogen blow drying or evaporation to dryness.
In some embodiments of the invention, the preparative HPLC separation purification is performed using Waters C-18Sep-Pak, and the eluent is ethanol.
In some embodiments of the invention, when a one-pot method is used, the reaction of step (2) may be quenched with HPLC eluent/water (1/1, v/v) before the dehydroxylation protecting group treatment of step (3) is performed.
The chemical structural formula of the BEMP is as follows:
CAS:98015-45-3。
the invention also provides an intermediate for synthesizing the carbon-11 labeled Larotrectinib compound, wherein the intermediate has the following chemical structural formula:
the invention also provides an intermediate for synthesizing the carbon-11 labeled Larotrectinib compound, wherein the intermediate has the following chemical structural formula:
wherein R is selected from trimethylsilyl, tert-butyldimethylsilyl, triethylsilyl, tert-butyldiphenylsilyl, methyl, benzyl, trityl, p-methoxytrityl, dimethoxytrityl, tert-butyl, methoxymethyl, 2-methoxyethoxymethyl, methylthiomethyl, benzyloxymethyl, p-methoxybenzyl, p-methoxybenzyloxymethyl, 3, 4-dimethoxybenzyl, tetrahydropyranyl, methoxycarbonyl, ethoxycarbonyl, tert-butoxycarbonyl, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl, trifluoroacetyl, chloroacetyl, dichloroacetyl, phenylacetyl, pivaloyl, methylsulfonyl, benzylsulfonyl, allylsulfonyl, allyloxycarbonyl, C1-C16 alkanoyl, C3-C16 alkenoyl, C3-C6 alkynoyl, C4-C10 cycloalkyl, benzyl, N-methyl-ethyl, N-methyl, N-, C7-C16 aroyl, C4-C10 heterocycloalkanoyl.
In some embodiments of the invention, the C1-C16 alkanoyl is selected from formyl, acetyl, propionyl, butyryl, pentanoyl, hexanoyl, heptanoyl, octanoyl, nonanoyl, decanoyl, undecanoyl, dodecanoyl, tridecanoyl, tetradecanoyl, pentadecanoyl, hexadecanoyl.
In some embodiments of the invention, the C3-C16 alkenoyl is selected from acryloyl, 2-butenoyl, 4-pentenoyl, 5-hexenoyl, 2-ethylacryloyl, 3-dimethyl-2-methylbutanoyl, 6-heptenoyl, 7-octenoyl, 3-methyl-hept-6-alkenoyl, 5-methyl-hept-6-alkenoyl, 8-nonenoyl.
In some embodiments of the invention, the C3-C6 alkynoyl is selected from propinyl, 2-butynoyl, 4-pentynoyl, 5-hexynoyl, 6-heptynoyl, 7-octynoyl, 8-nonynoyl.
In some embodiments of the invention, the C4-C10 cycloalkyl is selected from cyclopropylformyl, cyclobutylformyl, cyclopentylcarbonyl, cyclohexylformyl, 2-methylcyclohexylcarbonyl, 2, 6-dimethylcyclohexylformyl.
In some embodiments of the invention, the C7-C16 aroyl is selected from the group consisting of benzoyl, p-methylbenzoyl, m-methylbenzoyl, o-methylbenzoyl, 4-bibenzoyl, 1-naphthoyl, 2-naphthoyl, 1-methyl-2-naphthoyl, 6-methyl-2-naphthoyl, 3-quinolinecarbonyl, 8-quinolinecarbonyl, 9-anthraceneyl, 9-acridinecarbonyl.
Examples 1 to 5:
the method comprises the following steps: [11C]-3-(isocyanato)pyrazolo[1,5-a]Synthesis of pyrimidine analog intermediate (Compound 4)
1ml of V-type reaction flask, purged with nitrogen gas (10ml/min) for 5 minutes, then added with 100. mu.L of acetonitrile, 5. mu.L of BEMP, 100. mu.g of Compound 1, prepared with a cyclotron11CO2]CO2The above acetonitrile solution was passed under a nitrogen flow (10mL/min) and the radioactivity trapped in the solution was measured by a near-end small radiation detector until it peaked. After 1 minute of reaction, 100. mu.l of freshly prepared POCl was added3(0.2% v/v) acetonitrile, reacting for 30 seconds, sampling, analyzing and detecting to obtain a compound 4, and directly using the compound in the next reaction without purification.
Step two, 211C]Preparation of (E) -3-R-Larotrectinib compound
Adding 100 μ L of acetonitrile solution of Compound 2(0.2mg) to the reaction mixture of step one, reacting for 1 minute, sampling, analyzing and detecting, TLC tracing and labeling precursor compound 4 substantially disappears, and radioactive TLC tracing reaction mixture sampling (1-2 μ L) detected11C]the-3-R-Larotrectinib labeled product, namely the compound 3, is directly used for the next reaction without purification.
Step three, 211C]Deprotection of the (E) -3-R-Larotrectinib to give (E) (, [ 2 ]11C]-Larotrectinib
The reaction of step two was quenched with 750. mu.L of HPLC eluent/water (1/1, v/v). 6N hydrochloric acid (0.25mL) was then added to the reaction mixture, heated to 80 deg.C for 3 minutes, the reaction was cooled in an ice bath at 0 deg.C, and 10% NaHCO was added3The aqueous solution is neutralized to neutrality. The target product was isolated and purified by preparative HPLC: the neutralized mixture was buffered (60:40 CH)3CN:H2O +0.1N ammonium formate, 2mL) and activated by washing with ethanol (1mL) and water (5mL) in sequence. The Sep-Pak was washed with water (2mL) and the desired product was eluted with ethanol (1 mL). After separation and purification by preparative HPLC, the target product eluent is collected and concentrated to dryness in vacuum at 70 ℃. Dissolving again by using 10mL of normal saline containing 0.25% of Tween 80 and 5% of ethanol, filtering by using a sterile filter membrane when the container is closed, and collecting the filtrate into a sterile and pyrogen-free V-shaped bottle to obtain a sterile and pyrogen-free radiotracer preparation for PET imaging research of small animals.
The product identification, the radiochemical purity and the specific activity can be evaluated by analytical HPLC (high performance liquid chromatography) by adopting an internal standard method under different elution conditions. 810mCi (29GBq) [ solution ] produced by a cyclotron11C]CO2In the method, 48-98mCi (2.1-3.5GBq) participates in the tracer, and the preparation time is 25-30 min. The radiochemical purity of the product is more than 97.2 percent and the chemical purity is more than 98.4 percent. [11C]Uncorrected radiochemical yield of Larotrectinib was 12.5% with respect to the V-flask and a specific activity of 2200-.
Example 6:
the method comprises the following steps: [11C]-3-(isocyanato)pyrazolo[1,5-a]Synthesis of pyrimidine analog intermediate (Compound 4)
Same as example 1 step one.
Step two, 211C]Preparation of-Larotrectinib compound
To the reaction mixture in the first step, 100. mu.l of an acetonitrile solution of (S) -3-pyrrolidinol (0.15mg) was added, and after 1 minute of reaction, sampling analysis was conducted and detection was made, TLC tracing label precursor compound 4 was substantially disappeared, and radioactive TLC tracing reaction mixture sample (1-2. mu.L) was detected as being [, ]11C]Larotrectinib-labeled products. The reaction was quenched with 750. mu.l of ethanol/water (1/1, v/v), and the mixture was purified by preparative HPLC (the procedure was as in example 1), and the desired product was collectedConcentrating the eluate at 70 deg.C under vacuum to dryness to obtain the final product11C]-Larotrectinib compounds.
Examples 7 to 12:
step one, [ 2 ]11C]COF2Synthesizing;
nuclear reaction of cyclotron14N(p,α)11C produced [ alpha ], [ alpha ] and11C]CO2(3-4GBq), successively passed through a hot molybdenum target (875 deg.C), a sodium hydroxide absorption bottle, a liquid nitrogen cooled stainless steel conical bottle filled with 0.4g AgF2The whole process takes 11-12 minutes to complete11C]COF2Radiochemical yield (RCY)71 +/-2%. 0.4g of AgF at a flow rate of 5mL/min helium2The product can be used almost quantitatively11C]Conversion of CO into [ 2 ]11C]COF2. No detection was made in the NaOH system11C]COF2Or [ alpha ], [ alpha11C]CO2,AgF2Can also be recycled.
Reference, preparation [ 2 ]11C]Fluorophosphone, Jimmy E.Jakobsson et al, (2020) [11C ]]Carbonyl difluoride-a new and highly efficient[11C]carbonyl group transfer agent.Angew.Chem.Int.Ed.,DOI: 10.1002/anie.201915414
Step two, 211C]Preparation of (E) -3-R-Larotrectinib compound
The term "prepared in step one11C]COF2Bubbling through 0.5mL acetonitrile solution containing 1.0. mu. mol of Compound 1 and 1.2. mu. mol of Compound 2 for a flow time of about 5.5min, and passing the tail gas through sodium hydroxide solution. Pipeline connectorPurging with helium as inert gas. The sample was analyzed and examined, the TLC tracing-labeled precursor compound 1 was substantially disappeared, and the radioactive TLC tracing-reacted mixture sample (1-2uL) was examined11C]the-3-R-Larotrectinib labeled product is directly used for the next reaction without purification.
Step three, 211C]Deprotection of (E) -3-R-Larotrectinib to prepare (2)11C]-Larotrectinib
The reaction mixture from step two was quenched with 750ul of HPLC eluent/water (1/1, v/v). Magnesium bromide (0.6mg) was added to the reaction mixture, heated to 155 ℃ for 5 minutes, the reaction was cooled in an ice bath at 0 ℃ and 10% NaHCO was added3The aqueous solution is neutralized to neutrality. After separation and purification of the mixture by preparative HPLC (the procedure is the same as in example 1), the eluate of the desired product is collected and concentrated to dryness in vacuo at 70 ℃. Re-dissolving with 10mL of normal saline containing 0.25% Tween 80 and 5% ethanol, filtering with sterile filter membrane, and collecting into sterile pyrogen-free V-shaped bottle to obtain sterile pyrogen-free radiotracer preparation.
The product identification, the radiochemical purity and the specific activity can be evaluated by analytical HPLC (high performance liquid chromatography) by adopting an internal standard method under different elution conditions. 85mCi (3-4GBq), [ alpha ] produced by a cyclotron11C]CO2In the method, 5.5-7.5mCi (0.2-0.3GBq) participates in the tracer, and the preparation time is 18-19 min. The radiochemical purity of the product is more than 98.2 percent and the chemical purity is more than 98.4 percent. [11C]Uncorrected radiochemical yield of Larotrectinib was 10% in relation to the V-shaped reaction flask and a specific activity of 22.2 Ci/. mu.mol was obtained in the final formulation.
Example 13:
[11C]deprotection of (E) -3-R-Larotrectinib to prepare (2)11C]-Larotrectinib
The reaction mixture from step two, example 7 was quenched with 750 μ L of HPLC eluent/water (1/1, v/v). Adding anhydrous K to the reaction mixture2CO3(0.86mg), heated to 100 ℃ for 10 minutes, cooled in an ice bath at 0 ℃ and then neutralized by the addition of 10% aqueous HCl (15. mu.L). The reaction was further buffered (60:40 CH)3CN:H2O +0.1N ammonium formate, 2mL) and activated by washing with ethanol (1mL) and water (5mL) in sequence. Washing Sep-Pak with water (2mL), eluting the desired product with ethanol (1mL), rinsing into a sterile vacuum bottle, blow-drying with nitrogen at 60 ℃ for 20 minutes, re-compounding with saline, wherein the aqueous solution contains 100ul of 25% vitamin C, 100 muL of 20% Tween 80 in ethanol, and obtaining the final product11C]Larotrectinib marker injection.
The radiochemical conversion (RCC) was determined by analysis of the samples by radio TLC (silica gel plates, 100% ethyl acetate extension) by means of radioactive HPLC (60:40 CH)3CN:H2O +0.1N ammonium formate, Phenomenex Luna C-18 column) and radioactive TLC (silica gel plate, 100% ethyl acetate extension) for product identity and purity. Radiochemical purity of the product>93% and chemical purity>95 percent. And determining the consistency identification labeling product of the peak position by using a radioactive and non-radioactive reference substance co-injection method and a radioactive detector and a non-radioactive ultraviolet detector. The radiochemical yield is determined when the amino precursor (Compound 1) is diluted by adding to acetonitrile11C]CO2Or [ alpha ], [ alpha11C]COF2Solution, percentage of radioactivity separated as end product from the amount of activity in the V-vial, and no decay correction. [11C]Uncorrected radiochemical yield of Larotrectinib relative to that in a type V reaction vial (,)11C]CO2Or [ alpha ], [ alpha11C]COF2Typically 1.85mCi) of 28.56% and a specific activity (19.28 Ci/. mu.mol) was obtained in the final formulation. The removal rate of the hydroxyl protecting group R was 97%.
Example 14
Step one, [ 2 ]11C]COF2Synthesis of (2)
The same procedure as in step one of example 3.
Step two, 211C]Preparation of-Larotrectinib compound
The term "prepared in step one11C]COF2A0.5 mL acetonitrile solution containing Compound 1(2.5mg) and (S) -3-pyrrolidinol (0.9mg) was bubbled through, the flow through time was approximately 5.5min, and the tail gas was passed through sodium hydroxide solution. The tubing is purged with an inert gas, helium. Sampling, analyzing and detecting, wherein the TLC tracing marked precursor 2 is basically disappeared, and the radioactive TLC tracing reaction mixture sample (1-2 μ L) is detected to have11C]Larotrectinib-labeled products. After the target product was separated and purified by preparative HPLC (the specific procedure was the same as in example 1), the target product eluate was collected and concentrated to dryness in vacuo at 70 ℃.
The invention develops a novel TRK PET probe, which has TRK specificity and is used for detecting the expression of TRK fusion protein in tumor tissues, thereby providing reliable diagnosis and molecular phenotype data for clinical examination and being also used for screening and evaluating in vitro cell receptors and drug molecules.
Trk plays an important role in the physiology, development and function of the central and peripheral nervous systems. After being combined with a combined extramembranous ligand, a normal Trk receptor forms a dimer and causes phosphorylation and activation of an intramembranous kinase region, so that activation of a downstream signal path is triggered, and proliferation and differentiation of cells are promoted. NTRK gene rearrangement and fusion are common phenomena in various cancers, are widely existed in various cancers (such as rectal cancer, non-small cell lung cancer, breast cancer, glioma, astrocytoma, infantile fibrosarcoma, thyroid cancer, gastrointestinal stromal tumor and the like), and encode various prokinase Trk fusion proteins. These fusion proteins are capable of activating downstream pathways and promoting tumor formation and cell proliferation in the absence of a ligand. Therefore, Trk fusion protein also becomes a broad spectrum drug target and diagnostic marker in the cancer field.
So far, the clinical technical means for detecting NTRK fusion mainly include NGS sequencing, FISH and IHC. The invention modifies the existing small molecular compound aiming at the Trk fusion protein, thereby further forming a molecular probe capable of specifically diagnosing Trk fusion protein expression, thereby providing reliable molecular typing data for clinically performing molecular diagnosis on characteristics of tumors and guiding the clinical medication of related Trk fusion protein.
Example 15[ alpha ], [ alpha11C]-radioactivity distribution of Larotrectinib in normal mice:
experimental protocol
Male mice were completely randomly divided into 5 groups of 5 mice each, and the tail vein was injected with [11C ] -Larotrectinib imaging agent 37MBq, killed by decapitation after 5, 15, 30, 45, 60min, and then the brain, lung, heart, liver, spleen, stomach, intestine, kidney, muscle and other organs were removed, washed with physiological saline, weighed, and the radioactivity count (cpm) thereof was measured using a gamma counter, and after blood and urine were collected, weighed and the radioactivity count (cpm) thereof was measured using a gamma counter. The percent injection dose rate per gram of tissue (% ID/g wet tissue) was calculated after time decay correction.
Results of the experiment
The data in Table 1 show the radioactive biodistribution of [11C ] -Larotrectinib in normal mice.
[11C] Larotrectinib is rapidly absorbed in vivo, and is distributed most in lung, heart, liver, kidney and brain. After intravenous injection of [11C ] -Larotrectinib for 5min, the radioactivity distribution can reach a peak, and after 15min, the radioactivity distribution in each tissue is rapidly reduced; the radiation dose in each organ is obviously reduced 30min after the administration.
The intracerebral radioactivity was distributed moderately, 0.988 mer 0.056ID/g at 5min, 0.268 mer 0.043% ID/g at 15min, and 0.123. + -. 0.088% ID/g at 30min, indicating that [11C ] -Larotrectinib cleared more rapidly in the brain. The radioactive uptake is high in lung, heart, liver, kidney, blood and other organs, and the radioactivity is distributed more. The radioactive uptake in the spleen, stomach, intestine, muscle, bone and other organs is low, and the radioactivity distribution is less. The radioactivity distribution of liver and kidney tissues at 5min is 2.654 + -0.420% ID/g,1.373 + -0.312% ID/g, 0.231 + -0.039% ID/g and 0.153 + -0.165% ID/g, respectively. Blood clearance was rapid, with only 0.001% of the injected dose of 0.010 gauss remaining 30min post-injection. At 45min post injection, 88% of the dose was cleared from the urethra. The tracer has high liver uptake rate which is the highest in 5min and is 2.654 +/-0.420% ID/g (the uptake rate is 0.345 +/-0.003% ID/g in 45 min), and the liver is rapidly cleared, so that the liver tissue is small in absorption. The kidney tissue is the main excretory organ of [11C ] -Larotrectinib, and shows that [11C ] -Larotrectinib is metabolized through the liver and gall system and excreted through the kidney-urinary system.
Table 1 in vivo radioactivity distribution (% ID/g ± SD, n ═ 5) of the tracer [11C ] -larotrefectonib 1. hours after normal mouse injection.
Example 16[11C ] -Larotrectinib PET/CT imaging in tumor-bearing mice
Experimental protocol
Male tumor bearing (gene fusion human lung adenocarcinoma cell H2228 cell line) mice were completely randomly divided into 5 groups, each group had 5 mice, and PET/CT imaging was performed 5min, 15min, 30min, 45min and 60min after tail vein injection of 37MBq [11C ] -Lavitrectinib.
Results of the experiment
Table 2 summarizes the radiobiodistribution of the tracer [11C ] -Larotrectinib in tumor-bearing mice. The radioactive substances in the blood can be effectively removed, and only 0.010 +/-0.001% of dose remains in the whole blood. Its major route of excretion is very similar to that of normal mice. Uptake of this tracer by renal tissue at 5, 15, 30 and 60min post-injection was 1.899 deg.C 0.655, 0.689 + -0.004, 0.352 + -0.142 and 0.118 + -0.112% ID/g, respectively. Higher uptake of radioactivity was observed in the liver compared to normal mouse biodistribution data, with results of 3.864 ± 0.564% ID/g at 5min post-injection and 0.167 ± 0.012% ID/g at 60min post-injection. The uptake concentration of the transplanted H2228 tumor reaches the maximum value (4.545 +/-1.688% ID/g) at 15min after injection, the tumor concentration of the tracer still keeps a higher value at 30 and 45min after injection, which exceeds that of all normal tissues, 2.888 +/-0.456 and 2.166 +/-1.234% ID/g are respectively reserved, and the tumor is cleaned. It can be seen that the radioactive clearance of tumor tissue is much slower than that of other positively expressed organs such as heart, liver and kidney.
Table 2 tracer [11C ] -larotrytinib radiobiodistribution (% ID/g ± SD, n ═ 5) in vivo within 1 hour after injection in tumor-bearing (genetically fused human lung adenocarcinoma cell H2228 cell line) mice.
The data show that the [11C ] -Larotrectinib is rapidly absorbed in vivo, and is distributed in lung, liver, spleen, kidney, heart and brain more at 5min, and the urinary bladder and visible radioactive aggregates are accumulated; the radioactivity is mainly distributed in lung, heart, liver and kidney at 15min, and more radioactivity appears in bladder; after 30min, the liver shadow, the kidney shadow and the like become light obviously, and the radioactive accumulation of the bladder also declines; after 45min, the liver and kidneys were virtually undisvisualised and the bladder radio-aggregations resolved.
Finally, the ratio of tumor to background is high due to the rapid clearance of radioactive material in the body. For example, tumor to blood and tumor to muscle ratios were 33.6 and 32.0 times, respectively, from 15min after injection, and remained 36.1 and 28.3 times 30min after injection.
Micro PET/micro CT imaging of tumor-bearing (H2228) mice using [11C ] -Larotrectinib is shown in FIG. 1. The H2228 xenograft tumor is still clearly visible at 45min, a high-quality and high-contrast micro PET/micro CT image is generated, and the tumor-to-background ratio is good.
Research results show that the [11C ] -Larotrectinib imaging agent is feasible for tumor evaluation by combining PET/CT imaging, the distribution and the metabolic characteristics of the [11C ] -Larotrectinib imaging agent can be visually and dynamically observed, and quantitative analysis can be carried out.
The embodiments described above are described to facilitate an understanding and use of the invention by those skilled in the art. It will be readily apparent to those skilled in the art that various modifications to these embodiments may be made, and the generic principles described herein may be applied to other embodiments without the use of the inventive faculty. Therefore, the present invention is not limited to the above embodiments, and those skilled in the art should make improvements and modifications within the scope of the present invention based on the disclosure of the present invention.