CN113827562A - Stable pharmaceutical compositions of clopidogrel free base for oral and parenteral administration - Google Patents
Stable pharmaceutical compositions of clopidogrel free base for oral and parenteral administration Download PDFInfo
- Publication number
- CN113827562A CN113827562A CN202111171945.6A CN202111171945A CN113827562A CN 113827562 A CN113827562 A CN 113827562A CN 202111171945 A CN202111171945 A CN 202111171945A CN 113827562 A CN113827562 A CN 113827562A
- Authority
- CN
- China
- Prior art keywords
- oil
- clopidogrel
- free base
- emulsion
- composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 title claims abstract description 225
- 229960003009 clopidogrel Drugs 0.000 title claims abstract description 195
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 title claims abstract description 194
- 239000012458 free base Substances 0.000 title claims abstract description 86
- 238000007911 parenteral administration Methods 0.000 title claims description 13
- 239000008194 pharmaceutical composition Substances 0.000 title description 11
- 239000000839 emulsion Substances 0.000 claims abstract description 139
- 239000000203 mixture Substances 0.000 claims abstract description 104
- 239000003921 oil Substances 0.000 claims abstract description 89
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 58
- 229910001868 water Inorganic materials 0.000 claims abstract description 57
- 239000004094 surface-active agent Substances 0.000 claims abstract description 42
- -1 pH adjusters Substances 0.000 claims abstract description 14
- 239000003963 antioxidant agent Substances 0.000 claims abstract description 7
- 239000003755 preservative agent Substances 0.000 claims abstract description 4
- 239000000375 suspending agent Substances 0.000 claims abstract description 4
- 239000002357 osmotic agent Substances 0.000 claims abstract 2
- 235000019198 oils Nutrition 0.000 claims description 83
- 239000012071 phase Substances 0.000 claims description 68
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 66
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 55
- 239000008346 aqueous phase Substances 0.000 claims description 42
- 238000000034 method Methods 0.000 claims description 35
- 239000012535 impurity Substances 0.000 claims description 34
- 239000003549 soybean oil Substances 0.000 claims description 29
- 235000012424 soybean oil Nutrition 0.000 claims description 29
- JQWAHKMIYCERGA-UHFFFAOYSA-N (2-nonanoyloxy-3-octadeca-9,12-dienoyloxypropoxy)-[2-(trimethylazaniumyl)ethyl]phosphinate Chemical group CCCCCCCCC(=O)OC(COP([O-])(=O)CC[N+](C)(C)C)COC(=O)CCCCCCCC=CCC=CCCCCC JQWAHKMIYCERGA-UHFFFAOYSA-N 0.000 claims description 20
- 235000011187 glycerol Nutrition 0.000 claims description 19
- 229950010477 clopidogrel hydrogen sulphate Drugs 0.000 claims description 16
- FDEODCTUSIWGLK-UHFFFAOYSA-N hydrogen sulfate;hydron;methyl 2-(2-chlorophenyl)-2-(6,7-dihydro-4h-thieno[3,2-c]pyridin-5-yl)acetate Chemical compound OS(O)(=O)=O.C1CC=2SC=CC=2CN1C(C(=O)OC)C1=CC=CC=C1Cl FDEODCTUSIWGLK-UHFFFAOYSA-N 0.000 claims description 16
- 239000004615 ingredient Substances 0.000 claims description 13
- 239000003513 alkali Substances 0.000 claims description 12
- 239000003002 pH adjusting agent Substances 0.000 claims description 8
- 239000002245 particle Substances 0.000 claims description 8
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 claims description 7
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 claims description 7
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 claims description 7
- 239000005642 Oleic acid Substances 0.000 claims description 7
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 claims description 7
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 claims description 7
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 claims description 7
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid group Chemical group C(CCCCCCC\C=C/CCCCCCCC)(=O)O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 claims description 7
- 238000000265 homogenisation Methods 0.000 claims description 6
- 229940057917 medium chain triglycerides Drugs 0.000 claims description 6
- 239000002105 nanoparticle Substances 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 6
- 150000003839 salts Chemical class 0.000 claims description 6
- 229960003964 deoxycholic acid Drugs 0.000 claims description 5
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 claims description 5
- 235000021323 fish oil Nutrition 0.000 claims description 5
- 239000007788 liquid Substances 0.000 claims description 5
- 239000004006 olive oil Substances 0.000 claims description 5
- 235000008390 olive oil Nutrition 0.000 claims description 5
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 claims description 4
- 239000004380 Cholic acid Substances 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 4
- 229960002471 cholic acid Drugs 0.000 claims description 4
- 235000019416 cholic acid Nutrition 0.000 claims description 4
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 claims description 4
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 4
- 229930195729 fatty acid Natural products 0.000 claims description 4
- 239000000194 fatty acid Substances 0.000 claims description 4
- 102000002322 Egg Proteins Human genes 0.000 claims description 3
- 108010000912 Egg Proteins Proteins 0.000 claims description 3
- 235000010469 Glycine max Nutrition 0.000 claims description 3
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 claims description 3
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 claims description 3
- 239000002738 chelating agent Substances 0.000 claims description 3
- 235000013345 egg yolk Nutrition 0.000 claims description 3
- 210000002969 egg yolk Anatomy 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 235000019483 Peanut oil Nutrition 0.000 claims description 2
- 229920001214 Polysorbate 60 Polymers 0.000 claims description 2
- 239000002199 base oil Substances 0.000 claims description 2
- 239000006172 buffering agent Substances 0.000 claims description 2
- 239000004359 castor oil Substances 0.000 claims description 2
- 235000019438 castor oil Nutrition 0.000 claims description 2
- 235000005687 corn oil Nutrition 0.000 claims description 2
- 239000002285 corn oil Substances 0.000 claims description 2
- 238000001914 filtration Methods 0.000 claims description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 claims description 2
- 230000036512 infertility Effects 0.000 claims description 2
- 239000002563 ionic surfactant Substances 0.000 claims description 2
- 239000002736 nonionic surfactant Substances 0.000 claims description 2
- 239000000312 peanut oil Substances 0.000 claims description 2
- 239000008180 pharmaceutical surfactant Substances 0.000 claims description 2
- 229920001983 poloxamer Polymers 0.000 claims description 2
- 229920001993 poloxamer 188 Polymers 0.000 claims description 2
- 229940044519 poloxamer 188 Drugs 0.000 claims description 2
- 229920001992 poloxamer 407 Polymers 0.000 claims description 2
- 229940044476 poloxamer 407 Drugs 0.000 claims description 2
- 239000003813 safflower oil Substances 0.000 claims description 2
- 239000008159 sesame oil Substances 0.000 claims description 2
- 235000011803 sesame oil Nutrition 0.000 claims description 2
- NRHMKIHPTBHXPF-TUJRSCDTSA-M sodium cholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 NRHMKIHPTBHXPF-TUJRSCDTSA-M 0.000 claims description 2
- 230000001954 sterilising effect Effects 0.000 claims description 2
- 238000004659 sterilization and disinfection Methods 0.000 claims description 2
- 239000008348 synthetic phosphatidyl choline Substances 0.000 claims description 2
- 150000003626 triacylglycerols Chemical class 0.000 claims description 2
- 150000003904 phospholipids Chemical class 0.000 claims 2
- 239000008349 purified phosphatidyl choline Substances 0.000 claims 1
- 235000020238 sunflower seed Nutrition 0.000 claims 1
- 238000002360 preparation method Methods 0.000 abstract description 38
- 239000003814 drug Substances 0.000 abstract description 30
- 229940079593 drug Drugs 0.000 abstract description 26
- 229920000858 Cyclodextrin Polymers 0.000 abstract description 13
- 238000006243 chemical reaction Methods 0.000 abstract description 11
- 239000008186 active pharmaceutical agent Substances 0.000 abstract description 10
- 238000006731 degradation reaction Methods 0.000 abstract description 10
- 230000007062 hydrolysis Effects 0.000 abstract description 10
- 238000006460 hydrolysis reaction Methods 0.000 abstract description 10
- 239000011159 matrix material Substances 0.000 abstract description 10
- 230000015556 catabolic process Effects 0.000 abstract description 9
- 229940088679 drug related substance Drugs 0.000 abstract description 9
- 238000005057 refrigeration Methods 0.000 abstract description 6
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 abstract description 6
- 239000002994 raw material Substances 0.000 abstract description 5
- 229940127218 antiplatelet drug Drugs 0.000 abstract description 4
- 235000003599 food sweetener Nutrition 0.000 abstract description 3
- 239000003765 sweetening agent Substances 0.000 abstract description 3
- 239000000546 pharmaceutical excipient Substances 0.000 abstract description 2
- 238000009472 formulation Methods 0.000 description 39
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 30
- 238000002156 mixing Methods 0.000 description 27
- 239000000047 product Substances 0.000 description 24
- 239000007764 o/w emulsion Substances 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- 239000000126 substance Substances 0.000 description 15
- 238000010438 heat treatment Methods 0.000 description 14
- 238000003756 stirring Methods 0.000 description 14
- 238000003860 storage Methods 0.000 description 13
- 239000012467 final product Substances 0.000 description 11
- 230000000694 effects Effects 0.000 description 9
- OMOVVBIIQSXZSZ-UHFFFAOYSA-N [6-(4-acetyloxy-5,9a-dimethyl-2,7-dioxo-4,5a,6,9-tetrahydro-3h-pyrano[3,4-b]oxepin-5-yl)-5-formyloxy-3-(furan-3-yl)-3a-methyl-7-methylidene-1a,2,3,4,5,6-hexahydroindeno[1,7a-b]oxiren-4-yl] 2-hydroxy-3-methylpentanoate Chemical compound CC12C(OC(=O)C(O)C(C)CC)C(OC=O)C(C3(C)C(CC(=O)OC4(C)COC(=O)CC43)OC(C)=O)C(=C)C32OC3CC1C=1C=COC=1 OMOVVBIIQSXZSZ-UHFFFAOYSA-N 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 238000011049 filling Methods 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- 229960003958 clopidogrel bisulfate Drugs 0.000 description 6
- 238000009826 distribution Methods 0.000 description 6
- 238000001990 intravenous administration Methods 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 235000006708 antioxidants Nutrition 0.000 description 5
- 239000000787 lecithin Substances 0.000 description 5
- 235000010445 lecithin Nutrition 0.000 description 5
- 239000012669 liquid formulation Substances 0.000 description 5
- 125000004492 methyl ester group Chemical group 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 4
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 239000004064 cosurfactant Substances 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 229940067606 lecithin Drugs 0.000 description 4
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 4
- 229940097346 sulfobutylether-beta-cyclodextrin Drugs 0.000 description 4
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 3
- XTWYTFMLZFPYCI-KQYNXXCUSA-N 5'-adenylphosphoric acid Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XTWYTFMLZFPYCI-KQYNXXCUSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 3
- XTWYTFMLZFPYCI-UHFFFAOYSA-N Adenosine diphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(O)=O)C(O)C1O XTWYTFMLZFPYCI-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 230000003078 antioxidant effect Effects 0.000 description 3
- 239000012736 aqueous medium Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 230000000740 bleeding effect Effects 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 239000007935 oral tablet Substances 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 235000019640 taste Nutrition 0.000 description 3
- 238000009210 therapy by ultrasound Methods 0.000 description 3
- 102000013392 Carboxylesterase Human genes 0.000 description 2
- 108010051152 Carboxylesterase Proteins 0.000 description 2
- 244000068988 Glycine max Species 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- 229930003427 Vitamin E Natural products 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- JAZBEHYOTPTENJ-JLNKQSITSA-N all-cis-5,8,11,14,17-icosapentaenoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O JAZBEHYOTPTENJ-JLNKQSITSA-N 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 238000012865 aseptic processing Methods 0.000 description 2
- 230000003143 atherosclerotic effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- 229940097362 cyclodextrins Drugs 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 235000020673 eicosapentaenoic acid Nutrition 0.000 description 2
- 229960005135 eicosapentaenoic acid Drugs 0.000 description 2
- JAZBEHYOTPTENJ-UHFFFAOYSA-N eicosapentaenoic acid Natural products CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O JAZBEHYOTPTENJ-UHFFFAOYSA-N 0.000 description 2
- 150000002148 esters Chemical group 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 2
- 239000008350 hydrogenated phosphatidyl choline Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 230000000873 masking effect Effects 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 2
- 235000019799 monosodium phosphate Nutrition 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- 239000006070 nanosuspension Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 238000013146 percutaneous coronary intervention Methods 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- 208000001297 phlebitis Diseases 0.000 description 2
- 150000008105 phosphatidylcholines Chemical class 0.000 description 2
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000006340 racemization Effects 0.000 description 2
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 238000010257 thawing Methods 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- 235000019165 vitamin E Nutrition 0.000 description 2
- 229940046009 vitamin E Drugs 0.000 description 2
- 239000011709 vitamin E Substances 0.000 description 2
- DVSZKTAMJJTWFG-SKCDLICFSA-N (2e,4e,6e,8e,10e,12e)-docosa-2,4,6,8,10,12-hexaenoic acid Chemical compound CCCCCCCCC\C=C\C=C\C=C\C=C\C=C\C=C\C(O)=O DVSZKTAMJJTWFG-SKCDLICFSA-N 0.000 description 1
- XUKUURHRXDUEBC-SXOMAYOGSA-N (3s,5r)-7-[2-(4-fluorophenyl)-3-phenyl-4-(phenylcarbamoyl)-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-SXOMAYOGSA-N 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- KMZHZAAOEWVPSE-UHFFFAOYSA-N 2,3-dihydroxypropyl acetate Chemical compound CC(=O)OCC(O)CO KMZHZAAOEWVPSE-UHFFFAOYSA-N 0.000 description 1
- VSWICNJIUPRZIK-UHFFFAOYSA-N 2-piperideine Chemical compound C1CNC=CC1 VSWICNJIUPRZIK-UHFFFAOYSA-N 0.000 description 1
- GZJLLYHBALOKEX-UHFFFAOYSA-N 6-Ketone, O18-Me-Ussuriedine Natural products CC=CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O GZJLLYHBALOKEX-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- CZGKDNMEVCHYQW-UHFFFAOYSA-N CC1=C(SCC1)C1=C(C=CC=C1)Cl Chemical compound CC1=C(SCC1)C1=C(C=CC=C1)Cl CZGKDNMEVCHYQW-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- CTKXFMQHOOWWEB-UHFFFAOYSA-N Ethylene oxide/propylene oxide copolymer Chemical compound CCCOC(C)COCCO CTKXFMQHOOWWEB-UHFFFAOYSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 206010029155 Nephropathy toxic Diseases 0.000 description 1
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 1
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 1
- 206010050661 Platelet aggregation inhibition Diseases 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 230000002744 anti-aggregatory effect Effects 0.000 description 1
- 230000000702 anti-platelet effect Effects 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 230000010100 anticoagulation Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical class CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 235000019658 bitter taste Nutrition 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000036471 bradycardia Effects 0.000 description 1
- 208000006218 bradycardia Diseases 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000036461 convulsion Effects 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000007857 degradation product Substances 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 235000020669 docosahexaenoic acid Nutrition 0.000 description 1
- KAUVQQXNCKESLC-UHFFFAOYSA-N docosahexaenoic acid (DHA) Natural products COC(=O)C(C)NOCC1=CC=CC=C1 KAUVQQXNCKESLC-UHFFFAOYSA-N 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 230000002949 hemolytic effect Effects 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 239000000413 hydrolysate Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 230000014508 negative regulation of coagulation Effects 0.000 description 1
- 230000007694 nephrotoxicity Effects 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 235000020660 omega-3 fatty acid Nutrition 0.000 description 1
- 229940012843 omega-3 fatty acid Drugs 0.000 description 1
- 239000006014 omega-3 oil Substances 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 229940096978 oral tablet Drugs 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- 208000030613 peripheral artery disease Diseases 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000008137 solubility enhancer Substances 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
Images






Landscapes
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides a ready-to-use oil/water emulsion composition having an average oil droplet size (average intensity, nm) of between 100 and 500nm, wherein the oil phase comprises clopidogrel free base dispersed in a pharmaceutically acceptable oil. The present emulsions use clopidogrel free base or a pre-mixture of clopidogrel free base in oil as the drug substance and may also contain one or more excipients, such as: surfactants and/or co-surfactants, osmotic agents, pH adjusters, antioxidants, preservatives, sweeteners, and/or suspending agents, and the like. Compared with other aqueous matrix formulas (for example, a cyclodextrin matrix formula and an emulsion which use a clopidogrel salt as a raw material drug), the formula and the preparation method of the emulsion obviously improve the stability of clopidogrel in the aspects of chiral conversion, hydrolysis and thermal degradation. The ready-to-use emulsion composition is administered parenterally or orally, with a single dose of up to 300mg of clopidogrel; it can be stored at ambient conditions for at least 19 weeks after preparation, or at refrigeration temperatures for at least 1 year. The composition can be used as an antiplatelet agent to play a rapid treatment role for patients who need emergency treatment, intensive care or can not swallow tablets.
Description
The present application is a divisional application of chinese patent application 201480007796.1 entitled "stable clopidogrel free base pharmaceutical composition for oral and parenteral administration", filed 2/6/2014.
Priority
This application claims priority to U.S. provisional patent application No. 61/761,234, filed 2013, 2, 6, d.c., the content of which is incorporated herein by reference.
Technical Field
The invention relates to a clopidogrel oil/water emulsion composition, which obviously improves the stability of clopidogrel against chiral conversion, hydrolysis and thermal degradation by dispersing clopidogrel free alkali in an oil phase; methods of preparing and using clopidogrel compositions are described herein when a mammalian, and particularly a human subject, is in need of treatment with clopidogrel.
Background
Clopidogrel, its chemical name is: methyl (+) - (S) -alpha- (0-chlorophenyl) -6, 7-dihydrothieno [3,2-C ]) pyridine-5 (4H) -acetate, clopidogrel is an anticoagulant that inhibits platelet aggregation by selectively inhibiting the binding of Adenosine Diphosphate (ADP) to platelet receptors for ADP. It is widely used for preventing atherosclerotic events such as myocardial infarction, stroke, peripheral artery disease, acute coronary syndrome, cardiovascular death. The S-form enantiomer structure of clopidogrel is as follows:

clopidogrel (for example: and other pharmaceuticals) has been marketed as a tablet formulation containing 75mg of clopidogrel free base, clopidogrel being present in the formulation as a sulfate. In the marketed products, no parenteral or oral liquid preparations have been presented.
and other pharmaceuticals) has been marketed as a tablet formulation containing 75mg of clopidogrel free base, clopidogrel being present in the formulation as a sulfate. In the marketed products, no parenteral or oral liquid preparations have been presented. Is an anti-platelet aggregation drug approved by the Food and Drug Administration (FDA) to reduce thrombotic events and acute coronary syndrome. For the average dose (300 mg)
Is an anti-platelet aggregation drug approved by the Food and Drug Administration (FDA) to reduce thrombotic events and acute coronary syndrome. For the average dose (300 mg) Or clopidogrel) to achieve the desired therapeutic effect (e.g.: inhibiting platelet aggregation) requires between 2-5h, which can be attributed to delayed absorption, delayed availability of the system, poor bioavailability. If this treatment is required immediately (e.g. Percutaneous Coronary Intervention (PCI) is required in less than 2 to 3 hours), it is generally necessary to administer to the patient a greater than average dose of clopidogrel, in order to achieve a rapid onset of action, but this may cause potentially fatal side effects such as: bleeding and prolonged bleeding.
Or clopidogrel) to achieve the desired therapeutic effect (e.g.: inhibiting platelet aggregation) requires between 2-5h, which can be attributed to delayed absorption, delayed availability of the system, poor bioavailability. If this treatment is required immediately (e.g. Percutaneous Coronary Intervention (PCI) is required in less than 2 to 3 hours), it is generally necessary to administer to the patient a greater than average dose of clopidogrel, in order to achieve a rapid onset of action, but this may cause potentially fatal side effects such as: bleeding and prolonged bleeding.
Therefore, there is an urgent need for a clopidogrel liquid preparation which can be administered parenterally or orally to achieve rapid onset and gradual controlled administration dosage. The key to the use of clopidogrel for the preparation of a intravenous parenteral or oral liquid formulation is the ability to place clopidogrel in a biocompatible solvent with low side effects and a suitable pharmacodynamic profile and to prepare it into a prescription; clopidogrel formulations present a serious challenge due to its lipid solubility, pH dependence and extremely low solubility at physiological pH, and extreme chemical instability at alkaline pH.
Clopidogrel is a weak base with pKa 4.5, which is almost insoluble in water at neutral pH, but is very soluble in solution at pH1, is very soluble in methanol, partially soluble in dichloromethane, and almost insoluble in diethyl ether. Its specific rotation is about +56 °. Clopidogrel free base is a semi-solid high viscosity oily substance which causes various problems during storage or handling thereof. In addition, clopidogrel free base is reported to be unsuitable for the preparation of pharmaceutical formulations because clopidogrel free base is unstable under an environment of increased humidity and temperature. Because of the proton instability of clopidogrel free base at the chiral center and the methyl ester group, clopidogrel free base is susceptible to racemization, oxidation, and hydrolysis of the methyl ester group. Antioxidants are reported to be unable to inhibit their degradation, and higher pH values exacerbate their instability. Literature reports so far indicate that clopidogrel can ensure the stability only by combining with acid to form a salt before being prepared into a preparation.
Clopidogrel hydrogen sulfate, currently applied to marketed oral tablet products (SanofiAventis), which is an example of the clopidogrel salt form used in oral formulations, provides clopidogrel free base in a tablet dosage form having a specification of 75 mg. Like clopidogrel free base, clopidogrel bisulfate is relatively unstable under conditions of increased humidity, temperature, alkaline pH media due to its sensitivity to racemization, oxidation, hydrolysis of methyl ester groups. Clopidogrel is a chiral molecule with R, S enantiomers; the S enantiomer has biological activity, and the R enantiomer (impurity C) does not have any anticoagulation activity and has poor tolerance; animals can induce convulsions at high doses. Most of the clopidogrel entering the general circulation after administration is converted into an inactive carboxylic acid derivative, which is formed by the hydrolysis of ester functions catalyzed by a carboxylesterase. Carboxylic acid derivative (+) -S- (o-chlorophenyl) -6, 7-dihydrothiophene [3,2-c]Pyridine-5 (4H) -acetic acid (clopidogrel acid, impurity A)Obtainable by hydrolysis of an ester function; the carboxylic acid derivatives as hydrolysis products, whether catalytically produced by increasing humidity, pH, temperature in vitro or by carboxylesterase catalysis in vivo, are major degradation products with no pharmacological activity. This means that the clopidogrel drug substance or formulation product must have a fine control of the content of the R-enantiomer as well as the inactive carboxylic acid derivative. The main impurities of clopidogrel are recorded in the 32 th edition of the United states pharmacopoeia and the structure diagram of the European pharmacopoeia as follows:
(SanofiAventis), which is an example of the clopidogrel salt form used in oral formulations, provides clopidogrel free base in a tablet dosage form having a specification of 75 mg. Like clopidogrel free base, clopidogrel bisulfate is relatively unstable under conditions of increased humidity, temperature, alkaline pH media due to its sensitivity to racemization, oxidation, hydrolysis of methyl ester groups. Clopidogrel is a chiral molecule with R, S enantiomers; the S enantiomer has biological activity, and the R enantiomer (impurity C) does not have any anticoagulation activity and has poor tolerance; animals can induce convulsions at high doses. Most of the clopidogrel entering the general circulation after administration is converted into an inactive carboxylic acid derivative, which is formed by the hydrolysis of ester functions catalyzed by a carboxylesterase. Carboxylic acid derivative (+) -S- (o-chlorophenyl) -6, 7-dihydrothiophene [3,2-c]Pyridine-5 (4H) -acetic acid (clopidogrel acid, impurity A)Obtainable by hydrolysis of an ester function; the carboxylic acid derivatives as hydrolysis products, whether catalytically produced by increasing humidity, pH, temperature in vitro or by carboxylesterase catalysis in vivo, are major degradation products with no pharmacological activity. This means that the clopidogrel drug substance or formulation product must have a fine control of the content of the R-enantiomer as well as the inactive carboxylic acid derivative. The main impurities of clopidogrel are recorded in the 32 th edition of the United states pharmacopoeia and the structure diagram of the European pharmacopoeia as follows:
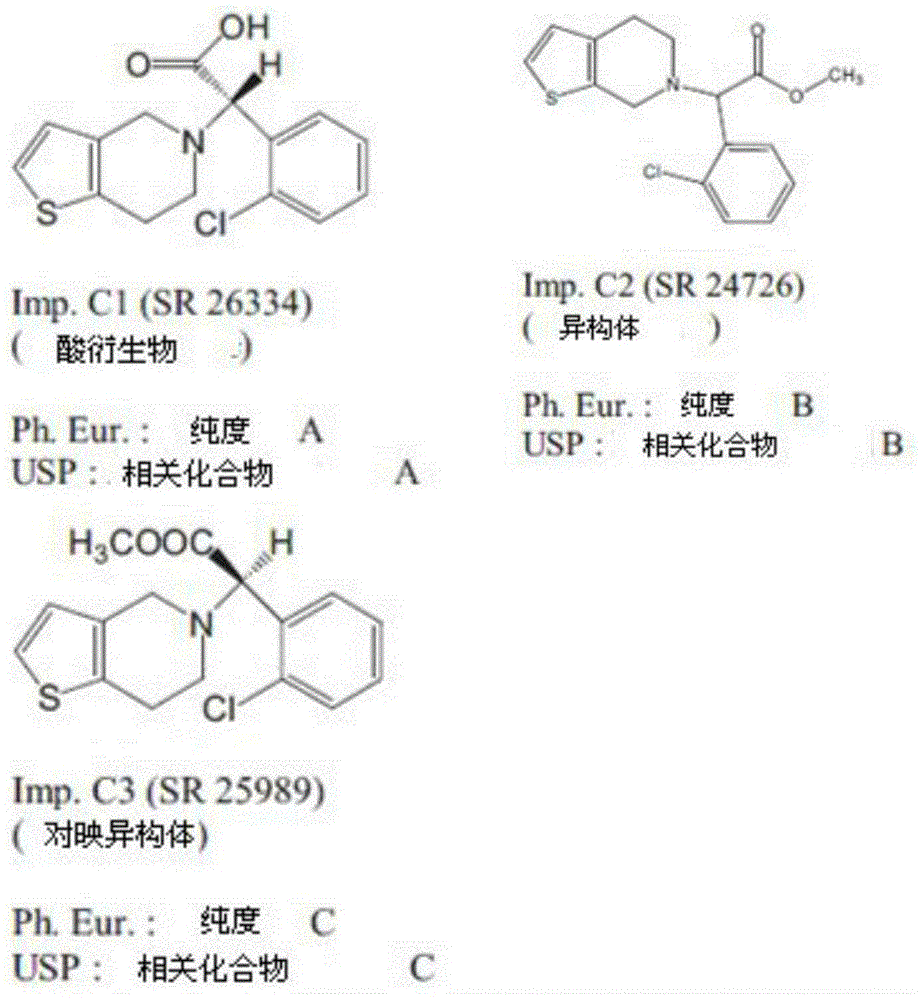
clopidogrel has low solubility in an aqueous solution of neutral pH, making it difficult to develop a bioavailable and physico-chemically stable formulation product, particularly when patients require intravenous administration or oral solutions. The solubility of clopidogrel is highly pH-dependent, which makes it a great challenge to develop an aqueous parenteral preparation that is suitable for use, does not precipitate when it comes into contact with body fluids, and does not cause pain in the parenteral region, phlebitis, or vascular embolism. In addition, the chemical instability of clopidogrel, which is manifested by the instability of clopidogrel in wet, hot and alkaline pH environments, eliminates the use of aqueous solvents in the formulation, limits the product formulation to liquid formulations or freeze-dried solid formulations using organic solvents as a matrix, and strictly limits the storage conditions to lower storage temperatures, such as refrigeration or freezing.
A number of processes are available for preparing intravenous parenteral and oral liquid compositions of poorly soluble or poorly soluble drugs. These techniques mainly include: micellar solubilization or formation of nanoparticle suspensions of the drug by surfactants; forming a composition with cyclodextrin and its derivatives (hydroxypropyl β -cyclodextrin (HPBCD) and sulfobutyl ether- β -cyclodextrin (SBECD)); the use of multiple co-solvent systems; salt formation with strong acids in low pH solutions. However, for micellar systems, the surfactants used therein have a close correlation with adverse effects, such as: such as hemolytic and histamine reactions and severe allergic reactions; for nanosuspension systems, since nanoparticle size stability of the drug particles requires a certain amount of polymer and surfactant, catalytic degradation of the drug due to greater contact area of the drug with the aqueous medium and the peripherally surrounding surfactant has been reported; taste masking and parenteral pain are still a great problem faced by micellar/nanosuspension systems due to the higher concentration of free drug in the aqueous medium. It is well known that cosolvent systems cause precipitation, parenteral pain and phlebitis. Cyclodextrins and their derivatives can cause potential nephrotoxicity and bradycardia and blood pressure reduction; concerns have been raised about the association of cyclodextrins with co-administered lipophilic drugs. Formation of weakly basic salts such as clopidogrel hydrogen sulfate with strong acids in a low pH solution will cause problems in stability of the drug-excipient and the drug product and will cause problems in taste (clopidogrel has a bitter taste), and the drug, when present as a free base, will precipitate upon contact with blood at neutral pH, causing irritation and pain at the parenteral site. In summary, the above methods have their limitations, and it is difficult to prepare clopidogrel as an intravenous parenteral or oral solution with a suitable biocompatible solvent, minimal side effects, and a proper pharmacodynamic profile.
WO2008/060934 discloses an emulsion composition that stabilizes oil droplets of a prepared pure tetrahydropyridine antiplatelet drug by micronization and addition of a surfactant. The preparation does not use vegetable oil, and only uses pure antiplatelet drugs and surfactants. Although the patent mentions that the matrix can be applied to clopidogrel, the patent only discloses an emulsion composition using clopidogrel bisulfate as a raw material drug, and the preparation of an emulsion composition using clopidogrel free base as a raw material drug is not disclosed in the patent; the effect of the formulation on the product stability of clopidogrel free base or various salts of clopidogrel, particularly the effect of the composition on the distribution of impurities related to clopidogrel, is not disclosed in this patent.
CN102697724 discloses a method for preparing oil-in-water emulsion by using clopidogrel hydrogen sulfate and amino acid salt thereof as raw material medicines. However, this patent does not embrace a process for preparing an emulsion composition using clopidogrel free base as the drug substance, and clopidogrel is too low in concentration (< 0.15% w/v) in the formulation that ultimately forms the emulsion composition, which may require large amounts of emulsion to achieve a dosage requirement of 300 mg; and the influence of the dosage form on the stability of products caused by various salt forms of clopidogrel, particularly the influence of the combination on the related impurity distribution of clopidogrel is not disclosed in the patent.
Summary of The Invention
In order to solve the drawbacks and problems of the prior art, there is a need in the art to develop a clopidogrel liquid dosage form, particularly a parenteral formulation that is stable in quality, ready to use, can provide a single intravenous parenteral 300mg clopidogrel dose, is rapid in onset of action, and can be formulated and stored at room temperature or refrigerated temperature. The present patent provides compositions that can provide antiplatelet drugs to patients in emergency and intensive care situations, or in situations where oral administration is not available to the patient. The stability and purity of the product both meet the preparation requirements of the United states Food and Drug Administration (FDA) and GMP for pharmaceutical preparations.
In view of the above problems, the present invention provides a clopidogrel free base-containing oil/water parenteral or oral emulsion composition having an average particle size of 100-. Wherein the oil phase comprises clopidogrel free base dispersed in an oil containing a surfactant and/or co-surfactant at acceptable concentration limits, unexpectedly resulting in a product having good stability and an excellent profile of impurities when the oil phase is subjected to a pH of about 9 to 10, or to an autoclave with water or steam; the product still has excellent quality after long-term storage.
The present invention describes a method for controlling clopidogrel impurity content within a range conforming to the impurity content regulation of clopidogrel tablets formulated in the United States Pharmacopeia (USP)32 edition when clopidogrel is placed as an active substance in a pharmaceutical composition. Compared with other aqueous matrix formulas (such as cyclodextrin matrix formulas) and formulas using clopidogrel salt (such as clopidogrel hydrogen sulfate), the emulsion formula has the advantages that the stability of clopidogrel in chiral conversion, hydrolysis and thermal degradation is remarkably improved.
1. One aspect of the present invention is that the aqueous matrix emulsions prepared significantly reduce hydrolysis of the methyl ester groups (impurity a); although clopidogrel itself is very unstable in an alkaline pH environment, the emulsion formulation of the present invention is prepared in a pH9-10 environment while minimizing hydrolysis of clopidogrel during its shelf life, whereas clopidogrel not formulated is completely degraded in the same solution as the above conditions, which is formulated with NaOH, in only 5 minutes.
2. Another aspect of the present invention is that the prepared emulsion effectively inhibits thermal degradation of clopidogrel. It is reported in the literature that clopidogrel is easily oxidized due to unstable proton of the chiral center thereof, and degradation thereof cannot be inhibited even by adding an antioxidant. Contrary to literature conclusions, the emulsion formulation of the present invention effectively inhibits thermal degradation of clopidogrel without using an antioxidant.
3. Another aspect of the invention is that the emulsions prepared according to the invention effectively inhibit the chiral conversion of clopidogrel from the S-enantiomer (biologically active) to the R-enantiomer (in the absence of any anti-aggregation activity and poorly tolerated) (impurity C < 1.5%). In contrast, cyclodextrin inclusion compound solutions or emulsions using clopidogrel salt forms (e.g., clopidogrel bisulfate) as the drug substance form appreciable amounts of the R-enantiomer during preparation or storage.
4. Another aspect of the present invention is that pharmaceutical compositions prepared using the processes described herein minimize or reduce the levels of drug-related impurities, either during manufacture or during storage.
a) This is particularly true. The pharmaceutical composition described in the present invention uses clopidogrel as an effective active substance, and the content of impurities conforms to the impurities regulation for clopidogrel tablets in the United States Pharmacopeia (USP)32 edition.
b) More specifically, the pharmaceutical composition described herein uses clopidogrel as an active substance, and the pharmaceutical composition contains only clopidogrel-related substance a of not more than 1.2%, clopidogrel-related substance C of not more than 1.5%, none of the other single-drug-related impurities exceeds 0.2% (excluding clopidogrel-related substance B), and total impurities does not exceed 2.5% (excluding clopidogrel-related substance B).
5. In another aspect, the present invention is directed to a method of treating or alleviating a disease state in a subject in need of the formulation, comprising providing to the subject a pharmaceutical composition having clopidogrel free base as an effective active substance and containing an effective amount of less impurities or minimized impurities, the impurities not exceeding 2.5% of the total drug content (except for clopidogrel-related substance B). Any disease or condition such as those described herein can be treated with a medicament comprising clopidogrel, thereby preventing atherosclerotic events such as myocardial infarction, stroke, peripheral arterial disease, acute coronary syndrome, cardiovascular and cerebrovascular death and other cardiovascular diseases.
It is an object of the present invention to provide a stable, nano-sized oil droplet containing, oil/water emulsion composition for parenteral or oral use, which emulsion is composed of clopidogrel free base, surfactant, optionally co-surfactant, aqueous phase substantially free of clopidogrel, pH adjusting agent dispersed in oil phase.
It is another object of the present invention to provide a method for preparing a stable, parenteral or oral oil-in-water emulsion composition comprising nano-scale oil droplets, the method essentially comprising the steps of: a) dispersing clopidogrel free alkali in an oily carrier to prepare an oil phase; b) preparing an aqueous phase comprising water and a pH adjusting agent; c) adding a surfactant or optionally a co-surfactant to the aqueous or oil phase; d) dispersing the oil phase in the aqueous phase to form a crude emulsion, and if necessary adjusting the pH to-9; e) c, carrying out ultrasonic treatment or high-pressure homogenization treatment on the emulsion obtained in the step d to form a final emulsion, and adjusting the pH to be 5.5-10; f) filtering the final emulsion; g) the bioburden or sterility of the product is controlled by aseptic processing or terminal sterilization.
It is another object of the present invention to provide a method of treatment for a patient in need of a single high dose administration of clopidogrel, comprising: a) providing a liquid formulation of an oil/water emulsion composition by dispersing clopidogrel free base in an oily carrier; preparing water phase from water and a pH regulator; dispersing the oil phase into the water phase through ultrasonic treatment or high-pressure homogenization treatment to form oil drops with nanometer particle size, and finally preparing the oil drop; b) a single oral or parenteral dose of the composition comprising up to 300mg of clopidogrel free base.
Drawings
FIG. 1 illustrates the average droplet size (average intensity, nm) of emulsions prepared from clopidogrel free base (example 10) or clopidogrel bisulfate (example 8) after autoclaving, or freeze-thaw conditions, or exposure to refrigeration (-5 ℃) conditions for 1 year
FIG. 2 illustrates an HPLC chromatogram of a clopidogrel free base emulsion (example 10) after being subjected to refrigeration conditions (-5 ℃) for 1 year.
FIG. 3 compares the percentage of chiral conversion of clopidogrel from the S-enantiomer to the R-enantiomer after storage of the clopidogrel emulsion of the present invention (example 10) with clopidogrel-HPBCD and SBECD compositions at about pH8, all at 40 ℃.
FIG. 4 compares the percentage of chiral conversion of clopidogrel from the S-enantiomer to the R-enantiomer of clopidogrel (example 10) with clopidogrel-HPBCD and SBECD compositions at about pH8, all stored at 25 ℃.
FIG. 5 compares the percentage of chiral conversion of clopidogrel from the S-enantiomer to the R-enantiomer when both the clopidogrel emulsion of the present invention (example 10) and the clopidogrel salt type emulsion (example 8) are stored at 40 ℃.
FIG. 6 is a graph showing the droplet size distribution of a newly formulated emulsion sample, an emulsion sample stored at room temperature for 19 weeks, and an emulsion sample stored at 40 ℃ for 19 weeks, all superimposed on the emulsion described in example 6.
Detailed description of the invention
"clopidogrel drug substance" or "clopidogrel free base" is defined as: methyl (+) - (S) -alpha- (0-chlorophenyl) -6, 7-dihydrothieno [3,2-C ]) pyridine-5 (4H) -acetate.
"clopidogrel-related substance a" or "impurity a" is defined as: (+) -S- (o-chlorophenyl) -6, 7-dihydrothiophene [3,2-c ] pyridine-5 (4H) -acetic acid.
"clopidogrel-related substance B" or "impurity B" is defined as: methyl (±) - (o-chlorophenyl) -4, 5-dihydrothiophene [2,3-c ] pyridine-6 (7H) -acetate.
"clopidogrel-related substance C" or "impurity C" is defined as: methyl (-) - (R) - (o-chlorophenyl) -6, 7-dihydrothiophene [3,2-c ] pyridine-5 (4H) -acetate.
The invention provides an aqueous base oil/water emulsion formula composition with the average particle size of 100-500nm, which comprises clopidogrel free alkali, a surfactant and/or a cosurfactant and an aqueous liquid carrier in oil. The prescription of the invention comprises the following components:
a) clopidogrel free base;
b) an oil phase;
c) a surfactant and/or co-surfactant;
d) water and a pH regulator.
Optional ingredients for the emulsion formulation further include: chelating agent, antioxidant, penetrant, suspending agent, preservative and buffering agent.
In some embodiments, the formulation further comprises a solubilizing agent, a flavoring agent, a sweetener, a viscosity increasing agent, an electrolyte, another therapeutic agent, or a combination thereof.
Different combinations of clopidogrel with other ingredients within the allowed dosage ranges, as described in the claims of the present invention, can be used to provide different embodiments of the present invention. The invention also provides a method for administration of clopidogrel for immediate use, which comprises dissolving clopidogrel free base in an oil phase, and encapsulating the oil phase in an aqueous phase.
The emulsion of the invention has better stability and less side effect compared with other organic matrix or aqueous matrix preparations (such as cyclodextrin matrix preparation). The oil/water emulsion also inhibits the adhesion of lipophilic clopidogrel to the walls of plastic parenteral devices. The invention provides the clopidogrel emulsion which can be prepared at room temperature or under refrigeration condition and can keep stable, and the administration mode does not need to be diluted. In addition, the emulsion has the advantages of rapid release and rapid onset of action compared with other oral tablets.
In some embodiments, the formulation is administered to a patient without dilution. In other embodiments, the liquid formulation may be diluted and ensure that clopidogrel precipitation does not occur. The formulations of the present invention may be administered in a single dose or in multiple doses.
Modes of administration of the liquid formulation of some embodiments of the present invention include parenteral administration, oral administration, or enteral administration. The present invention provides a method of treatment which prevents or reduces the development of complications of platelet aggregation, or which is effective in treating conditions requiring clopidogrel therapy, which method includes administration to a subject as required by the present invention. The invention also provides a method for reducing the time to onset of therapeutic action or reducing the time to achieve a target therapeutic effect by administering the clopidogrel formulation of the invention to a subject in a parenteral, oral, enteral mode as desired. The formulations of the present invention may reduce the onset of therapeutic action or reduce the time required to achieve a target therapeutic effect compared to oral tablets. The formulation of the present invention allows administration of a low dose of clopidogrel to achieve a target therapeutic effect, for example, with respect to bleeding time or a platelet aggregation inhibition target, as compared to a solid oral formulation, administration of a low dose of clopidogrel using the formulation of the present invention to achieve the same target therapeutic effect.
According to one embodiment of the invention, the emulsion comprises
a) 0.01-10% w/w clopidogrel free base;
b) 1-30% w/w of an oil phase;
c) 0.5-5.4% w/w surfactant;
d) 0-0.5% of any co-surfactant; and
d) 60-99% w/w water with a pH adjusting agent, e.g. sodium hydroxide to adjust the pH
Clopidogrel free base uses its S-enantiomer as the bulk drug. The S-enantiomer is biologically active, the R enantiomer (impurity C) is free of any anticoagulant activity and poorly tolerated.
Clopidogrel bisulfate, an active pharmaceutical ingredient used in the marketed tablet product, is mainly the S-enantiomer according to the 32 th edition of the United States Pharmacopeia (USP), and contains not more than 0.2% of a clopidogrel-related substance a, not more than 0.3% of a first enantiomer of a clopidogrel-related substance B, not more than 1.0% of a clopidogrel-related substance C, not more than 0.1% of any other found drug-related impurities, and a total drug-related impurity content of not more than 1.5%.
According to the United States Pharmacopeia (USP)32 edition, as a tablet product on the market, the content of the clopidogrel-related substance a is not more than 1.2%, the content of the clopidogrel-related substance C is not more than 1.5%, the content of any single impurity is not more than 0.2% (except for the clopidogrel-related substance B), and the content of total impurities of the drug is not more than 2.5% (except for the clopidogrel-related substance B) after the preparation and during the storage.
Because the free base of the clopidogrel is unstable under the conditions of increased temperature and humidity, and the methyl ester group of the free base is easy to racemize, oxidize and hydrolyze; the free base of the clopidogrel can be stabilized only by being acidified into a salt before being prepared into a pharmaceutical preparation; it is therefore generally accepted that clopidogrel free base is not suitable for use in the preparation of pharmaceutical formulations. The present invention surprisingly found a new phenomenon, which, unlike the previous general opinion, shows better stability after emulsion made with clopidogrel free base than after emulsion made with clopidogrel salt. Because of its low solubility in the oils available for formulation, clopidogrel salt has a low partition ratio between the oil phase and the aqueous phase. Therefore, the encapsulation efficiency of the clopidogrel salt in the oil phase is low. Unless very high doses of surfactant or solvent are used, it is clearly not suitable to use clopidogrel salt as the drug substance to prepare emulsions and achieve higher drug loadings (> 0.15% clopidogrel free base loading) in the range of doses that can be tolerated by a single dose administered (e.g. 300mg in 100ml solvent or less by volume). Otherwise, the clopidogrel may be separated out from the oil phase, dissolved or precipitated in the water phase; this will cause problems of homogeneity of the product (there are two forms of dispersion, one in which the clopidogrel is dissolved in the oil phase and the other in which the free clopidogrel is suspended in the aqueous medium); as a result of direct exposure of clopidogrel to water, ionizing catalytic degradation reactions, clopidogrel will present stability problems; problems with parenteral irritation/pain, taste masking of oral formulations, physical stability (sedimentation) of intravenous parenteral administration, etc. will also occur. In one embodiment of the invention, clopidogrel free base is used as a drug substance, which is dissolved in oil that can be used in formulation formulations to make compositions; in another embodiment, the clopidogrel free base is obtained using a technique in which a clopidogrel salt is converted to the free base, and the counter ion is separated from the free base prior to the preparation of the formulation.
The preferred range for clopidogrel free base in the formulation is 0.15 to 10%, most preferably 0.2 to 3%.
The oil phase in the emulsion is a pharmaceutical grade oil, preferably triglycerides, such as, but not limited to, soybean oil, safflower oil, olive oil, cottonseed oil, sunflower oil, fish oil (including omega-3 fatty acids, eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA)), castor oil, sesame oil, peanut oil, corn oil, medium chain triglycerides (e.g., Miglyol812or 810). The oil phase may contain surfactants and/or co-surfactants such as egg yolk lecithin, soy lecithin, other lecithins, propylene glycol diesters, oleic acid, or monoglycerides (e.g., acetyl monoglyceride). The oil phase may also be a mixed component.
Preferred oil phases are soybean oil, Medium Chain Triglycerides (MCT), olive oil, fish oil, either as individual ingredients or as a mixture with other ingredients.
The most preferred oil phase is soybean oil. The preferred range of oil carrier is 5-30%, the most preferred range of oil carrier is 10-20%.
The surfactant is any pharmaceutically acceptable surfactant, preferably phospholipid compounds extracted from egg yolk or soybean, synthetic phosphatidylcholine, and phosphatidylcholine purified from plants. It is also possible to use hydrogenated derivatives, such as: hydrogenated phosphatidylcholine (egg yolk) and hydrogenated phosphatidylcholine (soybean). The surfactant may also be a non-ionic surfactant, for example: poloxamers (e.g., poloxamer 188, poloxamer 407), losamide, polyoxyethylene stearate, polyoxyethylene sorbitan fatty acid esters or sorbitan fatty acid esters. Ionic surfactants may also be used, for example: cholic acid,
The preferred surfactant is egg yolk lecithin. A preferred range is 0.6-2.4%, and a most preferred range is 1.2-1.8%.
The cosurfactant is selected from oleic acid, sodium oleate, cholic acid, sodium cholate, deoxycholic acid, sodium deoxycholate, or mixture thereof; wherein the co-surfactant according to the invention is present in the composition in the range of 0-0.5 w/v%.
The preferred buffer has water in the range of 70-90%.
The emulsion may also contain co-solvents or other solubility enhancers, chelating agents, preservatives, antioxidants, stabilizers, pH adjusting agents or tonicity adjusting agents, such as: glycerin, polymers as suspending agents, sweeteners, and the like.
The ideal emulsion is a stable system with a mean droplet size of 100-1000nm, white to off-white. The preferred average droplet size is 100-500 nm; the most preferred mean droplet size is 100-300 nm.
Preferred emulsions have a pH range of 5.5 and above 5.5 after preparation and during storage. In one embodiment, the pH of the emulsion is controlled to be in the range of 5.5-7; in another embodiment, the pH of the emulsion is controlled to be in the range of 7 to 10. The pH range of the preferred emulsion is 6.5-9. The pH adjusting agent may be a buffer or sodium hydroxide or other pH adjusting agent, or a mixture of the above.
The emulsion of the present invention can be prepared by the following method: for the aqueous phase, the pharmaceutical grade water is dispersed in a vessel and heated to 40-80 deg.C, egg yolk lecithin and glycerol are added to the aqueous phase, and the pH is finally adjusted to 9-10. For the oil phase, the soybean oil should be dispersed in another container and heated to 40-80 deg.C; adding clopidogrel and any cosurfactant into soybean oil, and heating to 40-80 ℃; in addition, egg yolk lecithin may also be added to the oil phase. The aqueous phase was mixed with the oil phase using a high shear mixer to form a coarse emulsion. The coarse emulsion is prepared into emulsion with ideal microdroplet particle size by ultrasonic treatment, high-pressure homogenization treatment or microfluidization technology under the conditions of 5000-. The pH is adjusted to 5.5-10 by means of a pH adjusting agent, for example 1N sodium hydroxide solution. In one embodiment, the pH is adjusted to 9-10; in another embodiment, the pH is adjusted to 7-10; in yet another embodiment, the pH is adjusted to 8-10. The sample was filtered and dispersed in a clean grade bottle, the external environment was typically nitrogen blanketed, plugged with a siliconized rubber stopper, and finally sealed with an aluminum cap. The product should be prepared by aseptic processing or terminal sterilized. The preferred unit dose is a sterile and stable emulsion obtained by autoclaving techniques. In one embodiment, the emulsion should be autoclaved at 121 ℃ for 15-20 minutes. In another embodiment, the emulsion should be prepared in a sterile environment without the use of autoclaving techniques.
The invention will now be described by way of non-limiting examples. The invention consists of embodiments of the examples and detailed descriptions of various aspects of the invention. Accordingly, the invention includes single elements of the embodiments of the examples as well as different combinations of aspects of the invention as described herein. Furthermore, the present invention includes other features, advantages and embodiments that will be derived by those skilled in the art from the technical disclosure and related examples of the following sections. The disclosure herein includes all possible variations and modifications of the factors and methods. Furthermore, the embodiments are defined and illustrated for exemplary purposes, and are not intended to be exclusive or limited to what is described. The invention includes variations and modifications that would be recognized by those skilled in the art without departing from the spirit of the invention.
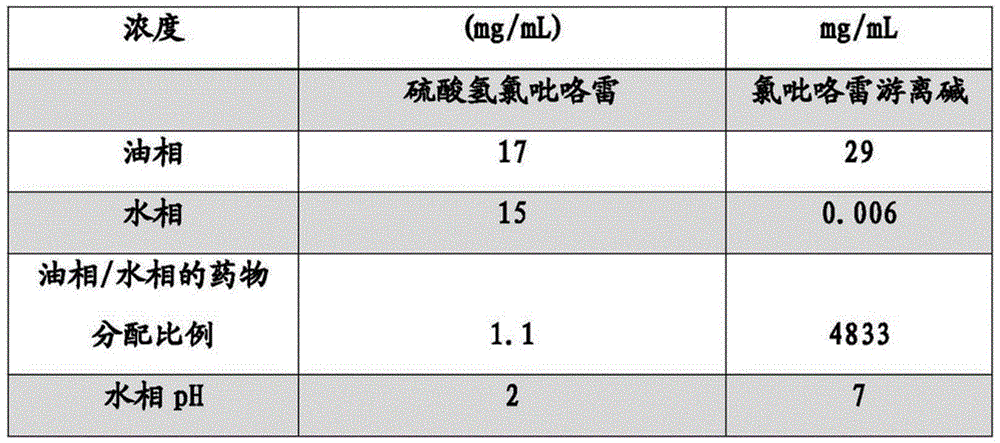
Example 1 comparison of drug partitioning ratio of clopidogrel hydrogen sulfate and clopidogrel free base between oil and aqueous phases
In order to determine the proportion of the clopidogrel in the oil phase and the water phase, the invention respectively determines the proportion of the clopidogrel hydrogen sulfate and the clopidogrel free alkali in the oil phase and the water phase, and 600mg of the drug is weighed and added into a beaker containing 20g of each of soybean oil and deionized water. The mixture was stirred at room temperature for 24 hours. Thereafter, the samples were separated from the two phases and tested using the HPLC chromatographic conditions described in example 14. And the pH of the aqueous phase was measured.
Table 1 shows that about 50% of clopidogrel will enter the aqueous phase when experiments are carried out with clopidogrel hydrogen sulfate. In contrast, when the experiment was carried out using clopidogrel free base, only a very small amount of clopidogrel entered the aqueous phase. This indicates that clopidogrel free base is more suitable than clopidogrel hydrogen sulfate as a drug substance in an emulsion if we wish to minimize the amount of clopidogrel entering the aqueous phase. Otherwise, the formulation will have problems of homogenization and stability.
TABLE 1 partitioning of drugs between oil and aqueous phases
Example 2 preparation of an emulsion Using Clopyrrolidine free base obtained from a supplier
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 0.20 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.2 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
All preparation processes were carried out under nitrogen protection.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the aqueous phase was prepared using glycerol and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
2. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
3. Meanwhile, the soybean oil filtered by a 0.22 micron filter, the clopidogrel free alkali and the egg yolk lecithin are put into a mixing container to prepare an oil phase. Mixing with stirring was continued and heating to 60 ℃ was continued until all ingredients had dissolved.
4. And adding the mixed oil phase into the water phase.
5. The mixture was stirred using a high shear mixer (Polytron PT3100) at 10,000rpm for 5 minutes to give a crude emulsion. Adjusting the pH of the emulsion to 9-10.
6. The crude emulsion was sonicated for 30 minutes using a sonicator (Fisher Scientific Sonic Dismembator, Model 500). And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; if necessary, adjusting the pH to 9-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers, for example. Finally autoclaving at 121 ℃ for 20 minutes.
9. The final product had a pH of 8.
Example 3 preparation of an emulsion Using Clopyrrolidine free base obtained from the supplier
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 0.6 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.8 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
All preparation processes were carried out under nitrogen protection.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the aqueous phase was prepared using egg yolk lecithin, glycerol and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
2. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
3. Meanwhile, the soybean oil filtered by a 0.22 micron filter and the clopidogrel free alkali are placed in a mixing container to prepare an oil phase. Mixing with stirring was continued and heating to 60 ℃ was continued until all ingredients had dissolved.
4. And adding the mixed oil phase into the water phase.
5. The mixture was stirred using a high shear mixer (polytron pt3100) at 10,000rpm for 5 minutes to give a crude emulsion. Adjusting the pH of the emulsion to 9-10.
6. The crude emulsion was homogenized using a high pressure homogenizer (APV2000) at a pressure of 10,000psi for 10 cycles. And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; if necessary, adjusting the pH to 9-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers, for example. Finally autoclaving at 121 ℃ for 20 minutes.
9. The final product had a pH of 8.
Example 4 preparation of an emulsion Using Clopyrrolidine free base obtained from the supplier
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 3.0 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.2 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
All preparation processes were carried out under nitrogen protection.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the aqueous phase was prepared using glycerol and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
2. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
3. Meanwhile, the soybean oil filtered by a 0.22 micron filter, lecithin and clopidogrel free alkali are placed in a mixing container to prepare an oil phase. Mixing with stirring was continued and heating to 60 ℃ was continued until all ingredients had dissolved.
4. And adding the mixed oil phase into the water phase which is continuously stirred at high speed.
5. The mixture was stirred using a high shear mixer (polytron pt3100) at 10,000rpm for 5 minutes to give a crude emulsion. Adjusting the pH of the emulsion to 9-10.
6. The crude emulsion was homogenized using a high pressure homogenizer (APV2000) at a pressure of 10,000psi for 10 cycles. And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; if necessary, adjusting the pH to 9-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers, for example. Finally autoclaving at 121 ℃ for 20 minutes.
9. The final product had a pH of 8.
Example 5 preparation of an emulsion by aseptic technique Using clopidogrel free base obtained from the supplier
Preparation of emulsions Using Clopyrrolidine free base obtained from the supplier as drug substance
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 3.0 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.8 |
| Glycerol | 2.25 |
| Vitamin E | 0.06 |
| Sodium hydroxide | Adjusting pH to 8-10 |
| Water for parenteral use | To 100g |
All preparation processes were carried out under nitrogen protection.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the aqueous phase was prepared using glycerol and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
2. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
3. Meanwhile, the soybean oil filtered by a 0.22 micron filter, lecithin and clopidogrel free alkali are placed in a mixing container to prepare an oil phase. Mixing with stirring was continued and heating to 60 ℃ was continued until all ingredients had dissolved.
4. And adding the mixed oil phase into the water phase.
5. The mixture was stirred using a high shear mixer (polytron pt3100) at 10,000rpm for 5 minutes to give a crude emulsion. Adjusting the pH of the emulsion to 9-10.
6. The crude emulsion was homogenized using a high pressure homogenizer (APV2000) at a pressure of 10,000psi for 10 cycles. And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; if necessary, adjusting the pH value to 8-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers, for example.
9. The final product had a pH of 8.
Example 6 preparation of an emulsion by aseptic Process Using Clopyrrolidine free base obtained from the supplier
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 3.0 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.2 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 8-10 |
| Water for parenteral use | To 100g |
All preparation processes were carried out under nitrogen protection.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the aqueous phase was prepared using glycerol and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
2. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
3. Meanwhile, the soybean oil filtered by a 0.22 micron filter, lecithin and clopidogrel free alkali are placed in a mixing container to prepare an oil phase. Mixing with stirring was continued and heating to 60 ℃ was continued until all ingredients had dissolved.
4. And adding the mixed oil phase into the water phase which is continuously stirred at high speed.
5. The mixture was stirred using a high shear mixer (polytron pt3100) at 6,000rpm for 5 minutes to give a crude emulsion. Adjusting the pH of the emulsion to 9-10.
6. The crude emulsion was homogenized using a high pressure homogenizer (APV2000) at a pressure of 10,000psi for 10 cycles. And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; if necessary, adjusting the pH value to 8-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers, for example.
9. The pH of the final product was about 8.
Example 7 preparation of an emulsion Using clopidogrel free base obtained from a supplier
Emulsion is prepared by using clopidogrel free alkali as a raw material medicine, lecithin as a surfactant and oleic acid as a cosurfactant
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 0.2 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.2 |
| Oleic acid | 0.03 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
All preparation processes were carried out under nitrogen protection.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the aqueous phase was prepared using glycerol, egg yolk lecithin and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
2. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
3. Meanwhile, the soybean oil filtered by a 0.22 micron filter, oleic acid and clopidogrel free alkali are placed in a mixing container to prepare an oil phase. Mixing with stirring was continued and heating to 60 ℃ was continued until all ingredients had dissolved.
4. And adding the mixed oil phase into the water phase which is continuously stirred at high speed.
5. The mixture was stirred using a high shear mixer (polytron pt3100) at 10,000rpm for 5 minutes to give a crude emulsion. Adjusting the pH of the emulsion to 9-10.
6. The crude emulsion was sonicated for 30 minutes using a sonicator (FisherScientific sonic Dismembator, Model 500). And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; adjusting the pH value to 9-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers, for example. Finally autoclaving at 121 ℃ for 20 minutes.
9. The pH of the final product was about 8.
EXAMPLE 8 preparation of an emulsion using clopidogrel hydrogen sulfate (reference preparation)

This example is prepared by the same procedure as described in example 2 without autoclaving. The final product pH was 7.4.
Example 9 preparation of an emulsion Using clopidogrel free base
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 2.0 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.2 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
The procedure for this example was as described in example 2. The final product had a pH of 8.
Example 10 an emulsion was prepared using clopidogrel free base premixed in soybean oil;
the clopidogrel hydrogen sulfate is converted into clopidogrel free base before the emulsion is prepared, and sulfate ions are separated from the clopidogrel free base.

All preparation processes were carried out under nitrogen protection.
A preparation method of premixing free base of clopidogrel in soybean oil.
Dissolving clopidogrel bisulfate in sufficient water. While stirring continuously, the desired soybean oil is dispersed in the aqueous phase, and a 1N sodium hydroxide solution is gradually dropped, for example, into the aqueous phase mixture until the pH of the aqueous phase reaches 6.5 or more. Separating an oil phase containing clopidogrel free base from an aqueous phase containing sulfate ions and sodium ions; if necessary, the oil phase is washed with water.
The sterile aqueous oil-in-water emulsion for parenteral administration is prepared as follows:
1. the drug-oil premix was stirred at 60 ℃, the oil phase filtered through a 0.22 micron filter, and charged to a mixing vessel.
2. The aqueous phase was prepared using glycerol, egg yolk lecithin and parenteral water. The pH of the aqueous phase was adjusted to 9-10 using 1N sodium hydroxide solution. Mixing was continued with stirring and heating to 60 ℃.
3. The aqueous phase was filtered using a 0.22 micron filter and charged to a mixing vessel.
4. And adding the mixed oil phase into the water phase which is continuously stirred at high speed.
5. The mixture was stirred using a high shear mixer (polytron pt3100) at 10,000rpm for 5 minutes to give a crude emulsion.
6. The crude emulsion was homogenized using a high pressure homogenizer at a pressure of 10,000psi for 5 cycles. And the product temperature was controlled at 45 ℃.
7. Cooling the resulting oil-in-water emulsion; adjusting the pH value to 9-10; finally the emulsion was transferred to a filling container.
8. The emulsion was filtered under nitrogen using a 0.45 micron filter and filled into containers. Finally autoclaving at 121 ℃ for 20 minutes.
9. The final product had a pH of 8.
Example 11 preparation of an emulsion Using clopidogrel free base
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 0.20 |
| Soybean oil | 10.0 |
| Egg yolk lecithin | 1.8 |
| Glycerol | 2.25 |
| Vitamin E | 0.06 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
The procedure for this example was as described in example 3. The final product had a pH of 8.
Example 12 preparation of an emulsion Using clopidogrel free base
| Formulation of | Quantity: |
| keke (Chinese character of 'Keke') | |
| Clopidogrel free base | 2.8 |
| Soybean oil | 0.17 |
| Egg yolk lecithin | 0.54 |
| Glycerol | 2.25 |
| Sodium hydroxide | Adjusting pH to 9-10 |
| Water for parenteral use | To 100g |
The procedure for this example was as described in example 3. The final product had a pH of 8.
Example 13 comparison of the differences in droplet size distribution characteristics of emulsions prepared using clopidogrel free base and clopidogrel hydrogen sulfate by a NanoZetasizer
Particle size distribution testing was performed using the emulsions prepared in examples 8 and 10. The emulsion was tested for droplet size distribution using a Malvern Zetasizer Nano-ZSSEN 3600. FIG. 1 shows the average droplet size (average intensity, nm) of emulsions prepared using clopidogrel free base (example 10) and clopidogrel hydrogen sulfate (example 8), respectively. Emulsions prepared using clopidogrel free base are very stable after storage for 1 year under autoclaving, freeze-thawing and refrigeration conditions, whereas emulsions prepared using clopidogrel hydrogen sulfate show drastic changes in particle size after autoclaving, freeze-thawing.
Example 14 comparison of the chemical stability of compositions Using emulsions of the invention with SB-E-CD and HP-B-CD
HPLC chromatographic conditions
Chiral liquid chromatography was used to detect impurity and enantiomer contents. The stationary phase used ULTRONES-OVM column, 5um (4.6 mm. times.150 mm. i.d.). The composition of the mobile phase was: phase A, adding 1.36g sodium dihydrogen phosphate (NaH2PO4.H2O) into 1L pure water; phase B, acetonitrile; isoconcentrate elution was performed at a constant flow rate of 1.0mL/min for 18 min run time; the sample injection volume is 5-10 ul; the UV detection wavelength was 220 nm.
The main degradation impurity of the clopidogrel emulsion prepared by the invention is impurity C (R-enantiomer); according to observations, hydrolysate impurity a (< 1.2%) in the emulsion product appeared less and did not change significantly. Thus, the indicator monitored for chemical stability of the emulsion of the invention is impurity C (fig. 2). The emulsions prepared using clopidogrel free base had better stability data than the cyclodextrin based clopidogrel solutions (fig. 3, fig. 4).
FIG. 3 shows the percentage conversion of the chiral structure of clopidogrel from the S enantiomer to the R enantiomer after storage of the clopidogrel emulsion prepared according to the invention (example 10) with a composition of clopidogrel-HPBCD, clopidogrel-SBECD (US20100292268) having a pH of about 8 at 40 ℃.
FIG. 4 shows the percentage conversion of the chiral structure of clopidogrel from the S enantiomer to the R enantiomer after storage of the clopidogrel emulsion prepared according to the invention (example 10) with a composition of clopidogrel-HPBCD, clopidogrel-SBECD (US20100292268) having a pH of about 8 at 25 ℃.
Example 15 comparison of the differences in chemical stability of emulsions prepared using clopidogrel free base (example 10) and clopidogrel sulfate (example 8)
The HPLC chromatographic conditions were the same as described in example 14.
The emulsion prepared using clopidogrel free base had better stability data than the emulsion prepared using clopidogrel hydrogen sulfate (figure 5).
FIG. 5 shows the percentage conversion of the chiral structure of clopidogrel from the S enantiomer to the R enantiomer after storage at 40 ℃ of an emulsion prepared with clopidogrel free base (example 10) and an emulsion prepared with clopidogrel hydrogen sulfate (example 8).
Example 16 summary of emulsion stability data for example 10
Table 2 shows stability data for the emulsions of example 10 stored under refrigerated conditions for at least 52 weeks
TABLE 2 stability data of the emulsions of example 10 stored under refrigerated conditions

Example 17 summary of emulsion stability data for example 6
Table 3, Table 4 show stability data for the emulsions of example 6 stored under refrigerated (-5 ℃) and room temperature (-25 ℃) conditions for at least 19 weeks
TABLE 3 stability of example 6 (25 ℃ C.)

TABLE 4 stability of example 6 (5 degree C)

Claims (10)
1. A stable pharmaceutical oil/water emulsion composition containing nano-sized oil droplets for parenteral or oral administration comprising the following ingredients:
clopidogrel free base dispersed in the oil phase;
a surfactant and optionally a co-surfactant;
an aqueous phase substantially free of clopidogrel; and
a pH regulator.
Preferably, wherein the composition further comprises one or more ingredients selected from the group consisting of chelating agents, antioxidants, osmotic agents, preservatives, suspending agents and buffering agents.
Preferably, wherein the average oil droplet size (average intensity, nm) is 100-500 nm.
More preferably, wherein the average oil droplet particle diameter (average intensity, nm) is 100-300 nm.
2. The composition of claim 1, wherein the composition comprises no more than 1.2% impurity a, no more than 1.5% impurity C, and wherein the pH of the composition is above 5.5.
Preferably wherein the pH of the composition is from 5.5 to 10.
Preferably wherein the oil phase is present in the composition in a proportion of about 5 to 30% (w/v).
Preferably, wherein the oily phase is selected from the group of pharmaceutically acceptable oils comprising: triglycerides, such as: soybean oil, safflower seed oil, olive oil, sunflower seed oil, fish oil, castor oil, sesame oil, peanut oil and corn oil; medium chain triglycerides; or a mixture of the above ingredients.
More preferably wherein the oil is selected from soybean oil, fish oil, medium chain triglycerides, olive oil, or mixtures thereof.
Still more preferably, wherein the oil is soybean oil.
Preferably wherein the composition comprises 0.1-10% (w/v) clopidogrel free base.
3. The composition of claim 1 or 2, wherein the composition comprises 0.15-3% (w/v) clopidogrel free base.
Preferably wherein the total weight of the surfactant and optional co-surfactant is 0.5-6% (w/v) of the composition.
Preferably, the composition comprises 0.5-5.5% (w/v) surfactant with 0-0.5% (w/v) optional co-surfactant:
wherein said surfactant is selected from the group consisting of pharmaceutically acceptable surfactants including phospholipids extracted from egg yolk or soy, synthetic phosphatidylcholines or plant-purified phosphatidylcholines, hydrogenated phospholipid derivatives; nonionic surfactants include poloxamers (e.g., poloxamer 188, poloxamer 407), losamide, polyoxyethylene stearate, polyoxyethylene sorbitan fatty acid esters or sorbitan fatty acid esters; the ionic surfactant comprises cholic acid, deoxycholic acid or surface active derivatives and salts thereof; the optional co-surfactant is selected from oleic acid, sodium oleate, cholic acid, sodium cholate, deoxycholic acid, sodium deoxycholate, or a mixture thereof.
Preferably, the composition comprises 0.6-2.4% (w/v) surfactant and 0.0-0.5% (w/v) optional co-surfactant; wherein the surfactant is egg yolk lecithin and the co-surfactant is oleic acid or sodium oleate.
4. The composition of any one of claims 1-3, wherein the composition comprises 0.15-3% (w/v) clopidogrel free base, 10-20% (w/v) soybean oil, 1.2-1.8% (w/v) egg yolk lecithin, 2.25% (w/v) glycerin, sodium hydroxide, and water.
5. A process for preparing a stable pharmaceutical oil/water emulsion composition containing nano-sized oil droplets for parenteral or oral administration, said process comprising the steps of:
a) preparing an oil phase by dispersing clopidogrel free base in an oil carrier;
b) preparing an aqueous phase comprising water and a pH adjusting agent;
c) adding a surfactant or optionally a co-surfactant to the oil or water phase;
d) dispersing the oil phase in the aqueous phase to form a coarse emulsion and adjusting the pH to 9-10;
e) preparing the crude emulsion prepared in step d into a final emulsion by ultrasonic or high-pressure homogenization, and adjusting the pH to 5.5-10;
f) filtering the final emulsion; and
g) the bioburden or sterility of the product is controlled by the aseptic process or terminal sterilization.
Preferably, wherein the clopidogrel free base is obtained by converting clopidogrel hydrogen sulfate into clopidogrel free base and separating sulfate ions from the clopidogrel free base.
Preferably, wherein the clopidogrel free base is provided as a free base-oil carrier premix.
6. The method of claim 5, wherein the oil carrier is soybean oil, fish oil, medium chain triglycerides, olive oil, or mixtures thereof, and the surfactant is egg yolk lecithin.
7. The process of claim 5 or 6, wherein the product of step e is adjusted to a pH of 7-10.
8. A method of treatment for administering to a patient in need of a single high dose of clopidogrel, the method comprising:
a) providing a pharmaceutical oil/water emulsion composition in liquid form; the composition is prepared by the following method: dispersing clopidogrel free alkali in an oil carrier to prepare an oil phase, preparing a water phase by using water and a pH regulator, dispersing the oil phase in the water phase, and finally forming oil drops with nano particle sizes by using ultrasonic or high-pressure homogenization treatment; and
b) by oral or parenteral administration containing a single dose of the composition, the single dose should contain up to 300mg of clopidogrel free base.
9. The method according to claim 8, wherein a single dose of the composition should contain no more than 1.2% of impurity a and no more than 1.5% of impurity C.
10. The method of claim 8, wherein the composition is adjusted to a pH of 7-10 in step a.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361761234P | 2013-02-06 | 2013-02-06 | |
| US201314174351A | 2013-02-06 | 2013-02-06 | |
| US61/761,234 | 2013-02-06 | ||
| US14/174,351 | 2014-02-06 | ||
| CN201480007796.1A CN105188671A (en) | 2013-02-06 | 2014-02-06 | Stable clopidogrel free base pharmaceutical composition for oral and parenteral administration |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201480007796.1A Division CN105188671A (en) | 2013-02-06 | 2014-02-06 | Stable clopidogrel free base pharmaceutical composition for oral and parenteral administration |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113827562A true CN113827562A (en) | 2021-12-24 |
Family
ID=54910104
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111171945.6A Pending CN113827562A (en) | 2013-02-06 | 2014-02-06 | Stable pharmaceutical compositions of clopidogrel free base for oral and parenteral administration |
| CN201480007796.1A Pending CN105188671A (en) | 2013-02-06 | 2014-02-06 | Stable clopidogrel free base pharmaceutical composition for oral and parenteral administration |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201480007796.1A Pending CN105188671A (en) | 2013-02-06 | 2014-02-06 | Stable clopidogrel free base pharmaceutical composition for oral and parenteral administration |
Country Status (1)
| Country | Link |
|---|---|
| CN (2) | CN113827562A (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101212954A (en) * | 2005-05-10 | 2008-07-02 | 伊兰制药国际有限公司 | Nanoparticle Clopidogrel Formulation |
| US20100062066A1 (en) * | 2006-11-14 | 2010-03-11 | Acusphere, Inc | Formulations of Tetrahydropyridine Antiplatelet Agents for Parenteral or Oral Administration |
| CN102697724A (en) * | 2012-06-07 | 2012-10-03 | 沈阳药科大学 | Clopidogrel and salt submicron emulsion injection thereof as well as preparation method of same |
-
2014
- 2014-02-06 CN CN202111171945.6A patent/CN113827562A/en active Pending
- 2014-02-06 CN CN201480007796.1A patent/CN105188671A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101212954A (en) * | 2005-05-10 | 2008-07-02 | 伊兰制药国际有限公司 | Nanoparticle Clopidogrel Formulation |
| US20100062066A1 (en) * | 2006-11-14 | 2010-03-11 | Acusphere, Inc | Formulations of Tetrahydropyridine Antiplatelet Agents for Parenteral or Oral Administration |
| CN102697724A (en) * | 2012-06-07 | 2012-10-03 | 沈阳药科大学 | Clopidogrel and salt submicron emulsion injection thereof as well as preparation method of same |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105188671A (en) | 2015-12-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6577548B2 (en) | Depot formulation of hydrophobic active ingredient and method for preparing the same | |
| RU2642234C2 (en) | Antagonists neurokinin-1 compositions for intravenous introduction | |
| US20180125782A1 (en) | Stable pharmaceutical composition of clopidogrel free base for oral and parenteral delivery | |
| BR112012027279B1 (en) | low oil pharmaceutical emulsion compositions comprising progestogen | |
| CN104940939B (en) | Heavy dose of glycerol application in can tolerate freeze thawing lipomul | |
| CN102802624A (en) | Nanodispersion Of A Drug And Process For Its Preparation | |
| CN101653414A (en) | Long-circulating solid lipid docetaxel nanoparticles and preparation method thereof | |
| ES2782106T3 (en) | Improved formulations of levosimendan for intravenous administration as an infusion or injection and as an infusion concentrate | |
| WO2022160970A1 (en) | Concentrated solution of insoluble drug not containing ethanol, and micellar solution prepared therefrom | |
| WO2022160971A1 (en) | Concentrate containing poorly soluble drug, and emulsion prepared therefrom | |
| US20100130619A1 (en) | Pharmaceutical composition for parenteral administration of idebenone | |
| CN113827562A (en) | Stable pharmaceutical compositions of clopidogrel free base for oral and parenteral administration | |
| HK1218081B (en) | Stable pharmaceutical composition of clopidogrel free base for oral and parenteral delivery | |
| CN107184587B (en) | 2-methoxyestradiol oral pharmaceutical composition, preparation method thereof and 2-methoxyestradiol soft capsule | |
| JPWO2018182039A1 (en) | Non-aqueous composition holding drug and method for producing same | |
| CN108888594B (en) | Nano fat emulsion for intravenous injection | |
| KR20230166337A (en) | Sustained release drug delivery system and method for manufacturing the same | |
| CN102805732A (en) | Tanshinone IIA lipid microsphere preparation and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |