CN113727975A - 作为jak抑制剂的被取代的吡咯并吡啶 - Google Patents
作为jak抑制剂的被取代的吡咯并吡啶 Download PDFInfo
- Publication number
- CN113727975A CN113727975A CN202080023588.6A CN202080023588A CN113727975A CN 113727975 A CN113727975 A CN 113727975A CN 202080023588 A CN202080023588 A CN 202080023588A CN 113727975 A CN113727975 A CN 113727975A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- group
- cycloalkyl
- disease
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Steroid Compounds (AREA)
- Furan Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本发明涉及具有式(I)‑(IV)的结构的新型吡咯并吡啶化合物(其中R基团、A、B、C、D和n如详细说明中所定义)和组合物及其作为用于治疗疾病的药物的应用。
Description
概述
本文实施方案针对具有式(I)至式(IV)的结构的化合物或其衍生物,其中R基团、环标记和n值在本文中定义:

本文公开了新型吡咯并吡啶化合物和组合物及其作为用于治疗疾病的药物的应用。还提供在人受试者或动物受试者中抑制JAK激酶活性的方法,以用于治疗JAK介导的疾病。
詹纳斯激酶(Janus Kinase)(JAK)是非受体酪氨酸激酶的亚组,其对于转导来源于I型细胞因子受体和II型细胞因子受体的信号是必不可少的,并且其的酶活性对于细胞因子的生物学活性而言必不可少。JAK激酶家族由四个家族成员组成:JAK1、JAK2、JAK3和Tyk2,并且这些激酶对于免疫系统中以及更广泛的其他组织中的细胞因子信号传导的调节而言是重要的。JAK的激酶活性针对JAK本身、细胞因子受体的细胞内部分和几种其他底物(包含STAT转录因子家族的成员)。STAT(STAT1至STAT6)对多种类型的细胞(包含免疫细胞)中的基因转录具有特定且独特的作用,并且在过程(如细胞增殖和分化)中是关键的。由于这些激酶在免疫和炎症中具有广泛的作用,因此已经开发出许多小分子药物来干预其中JAK激酶信号传导致病的疾病。这些药物起初被开发用于全身性施用,以预防器官移植排斥。后来,它们已经被开发为用于血液恶性肿瘤、以及自身免疫性疾病和炎性疾病(举例来说,包含类风湿性关节炎、溃疡性结肠炎、炎性肠病、强硬性脊椎炎、银屑病、特应性皮炎、脱发紊乱和白癜风)的潜在疗法。最近,由于与JAK激酶的全身性抑制有关的血液学毒性、免疫抑制毒性和代谢毒性,已经描述了这些抑制剂作为局部药剂的局部递送。这些包含斑秃、特应性皮炎、白癜风、银屑病、炎性肠病、和干眼等。本文描述了具有出色的口服生物利用度和局部生物利用度并且对于全身性自身免疫疾病有用的化合物,以及被设计成具有有限的稳定性且从而限制全身性暴露、因此最适合于局限性(例如,局部)药物递送的化合物。
已经证明由细胞因子激活的细胞因子受体的信号转导通过与受体胞质域相关的JAK激酶发生。受体刺激造成JAK的激活和相关受体链的胞质域的随后磷酸化。这产生了SH2结合域,SH2结合域用于招募被称为STAT(信号转导子和转录激活子(Signal Transducerand Activator of Transcription))的潜在的胞质转录因子。当与磷酸化的细胞因子受体结合时,STAT自身在酪氨酸残基上被磷酸化,这导致SH2域介导的同二聚体和异二聚体形成并易位至细胞核。一旦到达细胞核,这些蛋白质就诱导与原始细胞因子受体的激活相关的基因的转录。这种事件(在几分钟内STAT蛋白磷酸化,并且在几小时内STAT诱导的基因转录)顺序适于表征化合物的细胞效能和告知结构-活性关系两者。
详细说明
在描述本发明的组合物和方法之前,应当理解的是,本发明不限于所描述的特定过程、制剂、组合物、或方法,因为这些可以变化。还应当理解的是,说明书中使用的术语仅出于描述特定型式或实施方案的目的,而非旨在限制本文实施方案的范围,本文实施方案的范围将仅由所附权利要求限定。除非另外定义,否则本文中使用的全部技术术语和科学术语具有与本领域普通技术人员通常理解的含义相同的含义。尽管与本文描述的那些方法和材料相似或等同的任何方法和材料都可以用于本文实施方案的实施方案的实践或测试中,但现在描述优选的方法、装置、和材料。本文提及的全部出版物都通过引用被整体并入。本文中的任何内容均不应当解释为承认本文的实施方案无权借助在先发明而早于这样的公开。
定义
还必须注意的是,除非上下文另外明确指出,否则如本文和所附权利要求中所使用的,单数形式“一(a)”、“一(an)”和“所述(the)”包含复数提及。因此,例如,对“JAK抑制剂”的提及是对本领域技术人员已知的一种或更多种JAK抑制剂及其等同物等的提及。
如本文所使用的,术语“约”旨在限定其修饰的数值,从而将这样的值表示为在误差范围内的变量。在没有列举特定的误差范围(如图表或数据表中给出的平均值的标准偏差)时,术语“约”应当被理解为与其一起使用的数字的数值的平均值加或减10%。因此,约50%意为在45%-55%的范围内。
与“包含”、“含有”或“特征在于”同义的过渡术语“包括”是包含性的或开放式的,并且不排除附加的未列举的要素或方法步骤。在术语“包括”用作过渡短语的实施方案或权利要求中,这样的实施方案也可以被预期为用术语“由……组成”或“基本上由……组成”代替术语“包括”。
如本文所使用的,术语“由……组成(consists of)”或“由……组成(consistingof)”意为药物组合物、组合物或方法仅包含在特定要求保护的实施方案或权利要求中具体列举的要素、步骤、或成分。
如本文所使用的,术语“基本上由……组成(consisting essentially of)”或“基本上由……组成(consists essentially of)”意为药物组合物或方法仅包含在特定要求保护的实施方案或权利要求中具体列举的要素、步骤或成分,并且可以可选地包含实质上不影响特定实施方案或权利要求的基本特性和新颖特性的附加要素、步骤或成分。例如,治疗特定病况(例如,营养耗竭)的组合物或方法中的一种或多种唯一的活性成分是特定实施方案或权利要求中具体列举的一种或多种治疗剂。
如本文所使用的,当一个实施方案被定义为与另一实施方案不同时,两个实施方案是“互斥的”。例如,其中两个基团结合形成环烷基的实施方案与其中一个基团为乙基而另一个基团为氢的实施方案互斥。类似地,其中一个基团为CH2的实施方案与其中相同基团为NH的实施方案互斥。
如本文所使用的,术语“其衍生物”指其盐、其药学上可接受的盐、其酯、其游离酸形式、其游离碱形式、其溶剂合物、其共晶、其氘化衍生物、其水合物、其N-氧化物、其笼合物、其前体药物、其多晶型物、其立体异构体、其几何异构体、其互变异构体、其互变异构体的混合物、其对映异构体、其非对映异构体、其外消旋体、其立体异构体的混合物、其同位素(例如,氚、氘)、或其组合。
如本文所使用的,术语“药学上可接受的盐”指由碱或酸制备的对于向患者(如哺乳动物)的施用是可接受的盐。术语“药学上可接受的盐”包含通常用于形成碱金属盐和形成游离酸或游离碱的加成盐的盐。盐的性质并非关键的,条件是其是药学上可接受的。这样的盐可以来源于药学上可接受的无机碱或有机碱以及来源于药学上可接受的无机酸或有机酸。
如本文所使用的,术语“肠限制性(gut-restricted)”或“肠限制性化合物”指优先在肠腔内起作用而在体循环中未达到足以引发显著药理学反应的暴露的化合物。不受理论的束缚,由于最小化药理学药剂或其代谢物对与疾病治疗无关的细胞、组织和器官/器官系统的全身性暴露,这造成分子的增强的安全性。
术语“改善”用于表示本文实施方案的化合物改变其被提供、施加或施用至的组织的外观、形式、特性和/或物理属性。
术语“抑制”意为限制、防止或阻断靶酶的作用或功能和/或防止、减轻或消除与疾病、病况或紊乱相关的一种或更多种症状的发作,或者预防、减轻或消除疾病、病况或紊乱。
当公开数值范围,并且使用标记法“从n1…至n2”或“在n1…与n2之间”(其中n1和n2是数字)时,则除非另有规定,否则该标记法旨在包含数字本身及其之间的范围。该范围在端值之间(包含端值)可以是整数的或者连续的。例如,范围“从2个至6个碳原子”旨在包含两个、三个、四个、五个、和六个碳原子,因为碳原子以整数单位出现。相对比的,例如,范围“从1μM(微摩尔)至3μM”旨在包含1μM、3μM、以及其之间的一切任何有效数字(例如,1.255μM、2.1μM、2.9999μM等)。
如本文所单独地或组合地使用的,术语“酰基”指与烯基、烷基、芳基、环烷基、杂芳基、杂环、或任何其他部分(其中与羰基连接的原子是碳)连接的羰基。“乙酰基”基团指-C(O)CH3基团。“烷基羰基”基团或“烷酰基”基团指通过羰基基团与母体分子部分连接的烷基基团。这样的基团的实例包含甲基羰基和乙基羰基。酰基基团的实例包含甲酰基、烷酰基和芳酰基。
如本文所单独地或组合地使用的,术语“烯基”指具有一个或更多个双键且含有从2个至20个碳原子的直链或支链烃基。在某些实施方案中,所述烯基将包括从2个至6个碳原子。术语“亚烯基”指在两个或更多个位置处连接的碳-碳双键系统,如亚乙烯基[(-CH=CH-)、(-C::C-)]。适合的烯基基团的实例包含乙烯基、丙烯基、2-甲基丙烯基、1,4-丁二烯基等。除非另有规定,否则术语“烯基”可以包含“亚烯基”基团。
如本文所单独地或组合地使用的,术语“烷氧基”指烷基醚基团,其中术语烷基如下文所定义。适合的烷基醚基团的实例包含甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基等。
如本文所单独地或组合地使用的,术语“烷基”指含有从1个至20个碳原子的直链或支链烷基基团。在某些实施方案中,所述烷基将包括从1个至10个碳原子。在另外的实施方案中,所述烷基将包括从1个至8个碳原子。烷基基团可以如本文所定义的被可选地取代。
烷基基团的实例包含甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、戊基、异戊基、己基、辛基、壬基等。如本文所单独地或组合地使用的,术语“亚烷基”指在两个或更多个位置处连接的来源于直链或支链饱和烃的饱和脂肪族基团,如亚甲基(-CH2-)。除非另有规定,否则术语“烷基”可以包含“亚烷基”基团。
如本文所单独地或组合地使用的,术语“烷基氨基”指通过氨基基团与母体分子部分连接的烷基基团。适合的烷基氨基基团可以是单烷基化的或二烷基化的形成的基团,如例如N-甲基氨基、N-乙基氨基、N,N-二甲基氨基、N,N-乙基甲基氨基等。
如本文所单独地或组合地使用的,术语“次烷基”指烯基基团,其中碳-碳双键的一个碳原子属于烯基基团所连接的部分。
如本文所单独地或组合地使用的,术语“烷基硫代”指烷基硫醚(R-S-)基团,其中术语烷基如上文所定义,并且其中硫可以是单氧化的或双氧化的。适合的烷基硫醚基团的实例包含甲基硫代、乙基硫代、正丙基硫代、异丙基硫代、正丁基硫代、异丁基硫代、仲丁基硫代、叔丁基硫代、甲烷亚磺酰基、乙烷亚磺酰基等。
如本文所单独地或组合地使用的,术语“炔基”指具有一个或更多个三键且含有从2个至20个碳原子的直链或支链烃基。在某些实施方案中,所述炔基包括从2个至6个碳原子。在另外的实施方案中,所述炔基包括从2个至4个碳原子。术语“亚炔基”指在两个位置处连接的碳-碳三键,如亚乙炔基(-C:::C-、-C≡C-)。
炔基基团的实例包含乙炔基、丙炔基、羟基丙炔基、丁炔-1-基、丁炔-2-基、戊炔-1-基、3-甲基丁炔-1-基、己炔-2-基等。除非另有规定,否则术语“炔基”可以包含“亚炔基”基团。
如本文所单独地或组合地使用的,术语“酰胺基”和“氨基甲酰基”指如下文所描述的通过羰基基团与母体分子部分连接的氨基基团,反之亦然。如本文所单独地或组合地使用的,术语“C-酰胺基”指-C(O)N(RR’)基团,其中R和R’如本文所定义或者如通过所指定的具体列举的“R”基团所定义。如本文所单独地或组合地使用的,术语“N-酰胺基”指RC(O)NH(R’)-基团,其中R和R’如本文所定义或者如通过所指定的具体列举的“R”基团所定义。如本文所单独地或组合地使用的,术语“酰氨基”包含通过氨基基团与母体部分连接的酰基基团。“酰氨基”基团的实例是乙酰氨基(CH3C(O)NH-)。
如本文所单独地或组合地使用的,术语“氨基”指-NRR’,其中R和R’独立地选自氢、烷基、酰基、杂烷基、芳基、环烷基、杂芳基、和杂环烷基,其中的任何一者可以本身可选地被取代。此外,R和R’可以结合以形成杂环烷基,其中的任一者可以可选地被取代。
如本文所单独地或组合地使用的,术语“芳基”意为含有一个、两个或三个环的碳环芳族系统,其中这样的多环环系被稠合在一起。术语“芳基”包含芳族基团,如苯基、萘基、蒽基、和菲基。
如本文所单独地或组合地使用的,术语“芳基烯基”或“芳烯基”指通过烯基基团与母体分子部分连接的芳基基团。
如本文所单独地或组合地使用的,术语“芳基烷氧基”或“芳烷氧基”指通过烷氧基基团与母体分子部分连接的芳基基团。
如本文所单独地或组合地使用的,术语“芳基烷基”或“芳烷基”指通过烷基基团与母体分子部分连接的芳基基团。
如本文所单独地或组合地使用的,术语“芳基炔基”或“芳炔基”指通过炔基基团与母体分子部分连接的芳基基团。
如本文所单独地或组合地使用的,术语“芳基烷酰基”或“芳烷酰基”或“芳酰基”指来源于芳基取代的烷烃羧酸的酰基基团(如苯甲酰基、萘甲酰基、苯基乙酰基、3-苯基丙酰基(氢化肉桂酰基)、4-苯基丁酰基、(2-萘基)乙酰基、4-氯氢化肉桂酰基等)。
如本文所单独地或组合地使用的,术语芳氧基指通过氧基与母体分子部分连接的芳基基团。
如本文所单独地或组合地使用的,术语“苯并(benzo)”和“苯并(benz)”指来源于苯的二价基C6H4=。实例包含苯并噻吩和苯并咪唑。
如本文所单独地或组合地使用的,术语“氨基甲酸酯”指氨基甲酸的酯(-NHCOO-),其可以从氮端或酸端与母体分子部分连接,并且其可以如本文所定义的被可选地取代。
如本文所单独地或组合地使用的,术语“O-氨甲酰基”指-OC(O)NRR’基团,其中R和R’如本文所定义。
如本文所单独地或组合地使用的,术语“N-氨甲酰基”指ROC(O)NR’-基团,其中R和R’如本文所定义。
如本文所使用的,当单独地使用时,术语“羰基”包含甲酰基[-C(O)H],并且当组合地使用时,术语“羰基”是-C(O)-基团。
如本文所使用的,术语“羧基(carboxyl)”或“羧基(carboxy)”指-C(O)OH或相应的“羧化物”阴离子(如在羧酸盐中)。“O-羧基”基团指RC(O)O-基团,其中R如本文所定义。“C-羧基”基团指-C(O)OR基团,其中R如本文所定义。
如本文所使用的,术语“化合物”意在包含所描述的结构的全部立体异构体、几何异构体、互变异构体和同位素(例如,氚、氘)。
如本文所单独地或组合地使用的,术语“氰基”指-CN。
如本文所单独地或组合地使用的,术语“环烷基”或可替代地“碳环”指饱和的或部分饱和的单环的、双环的或三环的烷基基团,其中每个环部分含有从3个至12个碳原子环成员,并且可以可选地为如本文所定义的可选地被取代的苯并稠合环系。在某些实施方案中,所述环烷基将包括从5个至7个碳原子。这样的环烷基基团的实例包含环丙基、环丁基、环戊基、环己基、环庚基、四氢萘基、茚满基、八氢萘基、2,3-二氢-1H-茚基、金刚烷基等。如本文所使用的,“双环的”和“三环的”旨在包含稠合环系,如十氢化萘、八氢化萘、以及多环的(多中心的)饱和类型或部分不饱和类型。通常通过双环[1,1,1]戊烷、樟脑、金刚烷和双环[3,2,1]辛烷来例示异构体的后者类型。
如本文所单独地或组合地使用的,术语“酯”指对在碳原子处连接的两个部分进行桥接的羧基基团。
如本文所单独地或组合地使用的,术语“醚”指对在碳原子处连接的两个部分进行桥接的氧基基团。
如本文所单独地或组合地使用的,术语“卤素”或“卤族元素”指氟、氯、溴、或碘。
如本文所单独地或组合地使用的,术语“卤代烷氧基”指通过氧原子与母体分子部分连接的卤代烷基基团。
如本文所单独地或组合地使用的,术语“卤代烷基”指具有如上文所定义的含义的烷基基团,其中一个或更多个氢被卤族元素代替。具体地包含单卤代烷基基团、双卤代烷基基团和多卤代烷基基团。例如,单卤代烷基基团可以在基团内具有碘原子、溴原子、氯原子或氟原子。双卤代烷基基团和多卤代烷基基团可以具有两个或更多个相同卤素原子、或者不同卤素基团的组合。卤代烷基基团的实例包含氟甲基、二氟甲基、三氟甲基、氯甲基、二氯甲基、三氯甲基、五氟乙基、七氟丙基、二氟氯甲基、二氯氟甲基、二氟乙基、二氟丙基、二氯乙基和二氯丙基。“卤代亚烷基”指在两个或更多个位置处连接的卤代烷基基团。实例包含氟亚甲基(-CFH-)、二氟亚甲基(-CF2-)、氯亚甲基(-CHCl-)等。
如本文所单独地或组合地使用的,术语“卤代环烷基”指具有如上文定义的含义的环烷基基团,其中一个或更多个氢被卤族元素代替。具体包含单卤代环烷基基团、双卤代环烷基基团和多卤代环烷基基团。例如,单卤代烷基基团可以在基团内具有碘原子、溴原子、氯原子或氟原子。双卤代烷基基团和多卤代烷基基团可以具有两个或更多个相同卤素原子、或者不同卤素基团的组合。卤代烷基基团的实例包含氟环丙基、二氟环丙基、氟环丁基、氯环丁基、和氯环戊基。
如本文所单独地或组合地使用的,术语“杂烷基”指稳定的直链或支链、或其组合,完全饱和的或含有从1个至3个不饱和度,由规定数量的碳原子和选自N、O、和S的一个至三个杂原子组成,并且其中N原子和S原子可以可选地被氧化,并且N杂原子可以可选地被季铵化。一个或多个杂原子可以位于杂烷基基团的任何内部位置处。多达两个杂原子可以是连续的(如例如,-CH2-NH-OCH3)。
如本文所单独地或组合地使用的,术语“杂芳基”指5元至15元不饱和的杂单环、或者稠合的单环环系、双环环系、或三环环系,其中稠环中的至少一个是芳族的,含有选自N、O、和S的至少一个原子。在某些实施方案中,所述杂芳基将包括从1个至4个杂原子作为环成员。在另外的实施方案中,所述杂芳基将包括从从1个至2个杂原子作为环成员。在某些实施方案中,所述杂芳基将包括从5个至7个原子。术语还包含稠合的多环基团,其中杂环与芳基环稠合,其中杂芳基环与其他杂芳基环稠合,其中杂芳基环与杂环烷基环稠合,或者其中杂芳基环与环烷基环稠合。杂芳基基团的实例包含吡咯基、吡咯啉基、咪唑基、吡唑基、吡啶基、嘧啶基、吡嗪基、哒嗪基、三唑基、吡喃基、呋喃基、噻吩基、噁唑基、异噁唑基、噁二唑基、噻唑基、噻二唑基、异噻唑基、吲哚基、异吲哚基、吲嗪基、苯并咪唑基、喹啉基、异喹啉基、喹喔啉基、喹唑啉基、吲唑基、苯并三唑基、苯并二氧戊环基、苯并吡喃基、苯并噁唑基、苯并噁二唑基、苯并噻唑基、苯并噻二唑基、苯并呋喃基、苯并噻吩基、色酮基(chromonyl)、香豆素基(coumarinyl)、苯并吡喃基、四氢喹啉基、四唑并哒嗪基、四氢异喹啉基、噻吩并吡啶基、呋喃并吡啶基、吡咯并吡啶基等。示例性三环杂环基团包含咔唑基、苯并吲哚基、菲咯啉基、二苯并呋喃基、吖啶基、菲啶基、呫吨基等。
如本文所单独地或组合地使用的,术语“杂环烷基”和可互换的“杂环”各自指含有至少一个杂原子作为环成员的饱和的、部分不饱和的、或完全不饱和的(但非芳族的)单环的、双环的、或三环的杂环基团,其中每个所述杂原子可以独立地选自氮、氧、和硫。在某些实施方案中,杂环烷基将包括从1个至4个杂原子作为环成员。在另外的实施方案中,所述杂环烷基将包括从1个至2个杂原子作为环成员。在某些实施方案中,所述杂环烷基在每个环中将包括从3个至8个环成员。在另外的实施方案中,所述杂环烷基在每个环中将包括从3个至7个环成员。在又另外的实施方案中,所述杂环烷基在每个环中将包括从5个至6个环成员。“杂环烷基”和“杂环”旨在包含砜、亚砜、具有叔氮环成员的N-氧化物、以及碳环稠合的环系和苯并稠合的环系;此外,两个术语都还包含杂环与如本文所定义的芳基基团或附加的杂环基团稠合的系统。杂环基团的实例包含吖丙啶基、氮杂环丁烷基、1,3-苯并二氧戊环基、二氢异吲哚基、二氢异喹啉基、二氢噌啉基、二氢苯并二噁英基、二氢[1,3]噁唑并[4,5-b]吡啶基、苯并噻唑基、二氢吲哚基、二氢吡啶基、1,3-二噁烷基、1,4-二噁烷基、1,3-二氧戊环基、异吲哚啉基、吗啉基、哌嗪基、吡咯烷基、四氢吡啶基、哌啶基、硫代吗啉基等。除非明确禁止,否则杂环基团可以可选地被取代。
如本文所单独地或组合地使用的,术语“肼基”指由单键连接的两个氨基基团(即,-N-N-)。
如本文所单独地或组合地使用的,术语“羟基”指-OH。
如本文所单独地或组合地使用的,术语“羟基烷基”指通过烷基基团与母体分子部分连接的羟基基团。
如本文所单独地或组合地使用的,术语“亚氨基”指=N-。
如本文所单独地或组合地使用的,术语“亚氨基羟基”指=N(OH)和=N-O-。
短语“在主链中”指起始于基团与具有本文公开的式中的任何一种的化合物的连接点处的碳原子的最长连续链或相邻链。
术语“异氰酸基”指-NCO基团。
术语“异硫氰基”指-NCS基团。
短语“原子的线性链”指独立地选自碳、氮、氧和硫的原子的最长直链。
如本文所单独地或组合地使用的,在没有另外明确定义的情况下,术语“低级烷基”意为含有从1个至6个碳原子(包含6个碳原子)(即,C1-C6烷基)。
如本文所单独地或组合地使用的,术语“低级芳基”意为苯基或萘基,其中任一者可以如所规定的可选地被取代。
如本文所单独地或组合地使用的,术语“低级杂芳基”意为:1)包括五个或六个环成员的单环杂芳基,所述环成员中的一个成员与四个成员之间可以是选自N、O、和S的杂原子;或者2)双环杂芳基,其中稠环中的每个包括五个或六个环成员,环成员在其之间包括选自N、O、和S的一个至四个杂原子。
如本文所单独地或组合地使用的,术语“低级环烷基”意为具有三个环成员与六个环成员之间的单环环烷基(即,C3-C6环烷基)。低级环烷基可以是不饱和的。低级环烷基的实例包含环丙基、环丁基、环戊基、和环己基。
如本文所单独地或组合地使用的,术语“低级杂环烷基”意为具有四个环成员与六个环成员之间的单环杂环烷基,环成员中的一个与四个之间可以是选自N、O、和S的杂原子(即,C3-C6杂环烷基)。低级杂环烷基的实例包含氧杂环丁烷、氮杂环丁烷、吡咯烷基、咪唑烷基、吡唑烷基、哌啶基、哌嗪基、和吗啉基。低级杂环烷基可以是不饱和的。
如本文所单独地或组合地使用的,术语“低级氨基”指-NRR’,其中R和R’独立地选自氢和低级烷基,其中的任一者可以可选地被取代。
如本文所单独地或组合地使用的,术语“巯基”指RS-基团,其中R如本文所定义。
如本文所单独地或组合地使用的,术语“硝基”指-NO2。
如本文所使用的,“N-氧化物”是使用方便的氧化剂由分子中存在的叔碱性胺或亚胺形成的。
如本文所单独地或组合地使用的,术语“氧基”或“氧杂”指-O-。
如本文所单独地或组合地使用的,术语“氧代”指=O。
术语“全卤代烷氧基”指烷氧基基团,其中全部氢原子都被卤族元素原子代替。
如本文所单独地或组合地使用的,术语“全卤代烷基”指烷基基团,其中全部氢原子都被卤族元素原子代替。
如本文所单独地或组合地使用的,术语“基本上不含”指在通过任何手段(包含核磁共振(NMR)、气相色谱/质谱(GC/MS)、或液相色谱/质谱(LC/MS))测量的检测限内不含全部其他化合物的化合物。
如本文所单独地或组合地使用的,术语“磺酸基(sulfonate)”、“磺酸”和“磺基(sulfonic)”指-SO3H基团,并且以盐形式使用其作为磺酸的阴离子。
如本文所单独地或组合地使用的,术语“硫烷基”指-S-。
如本文所单独地或组合地使用的,术语“亚磺酰基”指-S(O)-。
如本文所单独地或组合地使用的,术语“磺酰基”指-S(O)2-。
术语“N-磺酰胺基”指RS(=O)2NR’-基团,其中R和R’如本文所定义。
术语“S-磺酰胺基”指-S(=O)2NRR’基团,其中R和R’如本文所定义。
如本文所单独地或组合地使用的,术语“硫杂”和“硫代”指-S-基团或其中氧被硫代替的醚。硫代基团的被氧化的衍生物(即,亚磺酰基和磺酰基)包含在硫杂和硫代的定义中。
如本文所单独地或组合地使用的,术语“硫醇基”指-SH基团。
如本文所使用的,当单独地使用时,术语“硫代羰基”包含硫代甲酰基-C(S)H,并且当组合地使用时,术语“硫代羰基”是-C(S)-基团。
术语“N-硫代氨甲酰基”指ROC(S)NR’-基团,其中R和R’如本文所定义。
术语“O-硫代氨甲酰基”指-OC(S)NRR’基团,其中R和R’如本文所定义。
术语“氰硫基”指-CNS基团。
术语“三卤代甲烷磺酰胺基”指X3CS(O)2NR-基团,其中X是卤族元素,并且R如本文所定义。
术语“三卤代甲烷亚磺酰基”指X3CS(O)2-基团,其中X是卤族元素。
术语“三卤代甲氧基”指X3CO-基团,其中X是卤族元素。
如本文所单独地或组合地使用的,术语“三取代的甲硅烷基”指硅酮基团,硅酮基团在其三个自由价处被如本文在氨基的定义下所列出的基团取代。实例包含三甲基甲硅烷基、叔丁基二甲基甲硅烷基、三苯基甲硅烷基等。
本文的任何定义可以与任何其他定义组合地使用以描述复合结构基团。按照惯例,任何这样的定义的尾随要素是与母体部分连接的要素。例如,复合基团烷基酰胺基表示通过酰胺基基团与母体分子连接的烷基基团,并且术语烷氧基烷基表示通过烷基基团与母体分子连接的烷氧基基团。
当基团被定义为“无(null)”时,意为所述基团不存在。
术语“可选地被取代”意为在先的基团可以被取代或不被取代。当被取代时,“可选地被取代”基团的取代基可以包含(不限于)独立地选自下列基团或特别指定基团组的一个或更多个取代基(单独地或组合地):低级烷基、低级烯基、低级炔基、低级烷酰基、低级杂烷基、低级杂环烷基、低级卤代烷基、低级卤代烯基、低级卤代炔基、低级全卤代烷基、低级全卤代烷氧基、低级环烷基、苯基、芳基、芳氧基、低级烷氧基、低级卤代烷氧基、氧代、低级酰氧基、羰基、羧基、低级烷基羰基、低级羧酸酯、低级甲酰胺基、氰基、氢、卤族元素、羟基、氨基、低级烷基氨基、芳基氨基、酰胺基、硝基、硫醇、低级烷基硫代、低级卤代烷基硫代、低级全卤代烷基硫代、芳基硫代、磺酸基、磺酸、三取代的甲硅烷基、N3、SH、SCH3、C(O)CH3、CO2CH3、CO2H、吡啶基、噻吩、呋喃基、低级氨基甲酸酯、和低级脲。在结构上可行的情况下,可以将两个取代基连接在一起以形成稠合的五元、六元、或七元的由零个至三个杂原子组成的碳环或杂环,例如形成亚甲基二氧基或亚乙基二氧基。可选地被取代的基团可以未被取代(例如,-CH2CH3)、完全被取代(例如,-CF2CF3)、被单取代(例如,-CH2CH2F)、或者以完全被取代与单取代之间的任何水平被取代(例如,-CH2CF3)。在列举取代基而未定性为取代的情况下,包含取代形式和未取代形式两者。在取代基被定性为“取代的”情况下,特别地意指取代形式。此外,可以根据需要定义特定部分的不同组的可选取代基;在这些情况下,可选的取代将通常紧接在短语之后被定义为“可选地被...取代”。
除非另有定义,否则术语R或术语R’在独自出现且没有数字标号的情况下指选自氢、烷基、环烷基、杂烷基、芳基、杂芳基和杂环烷基(其中的任何一者可以可选地被取代)的部分。这样的R和R’基团应当被理解为如本文所定义的可选地被取代。无论R基团是否具有数字标号,都应当理解的是,每个R基团(包含R、R’和Rn(其中n=(1、2、3、…n))、每个取代基和每个术语就从组的选择而言彼此独立。如果任何变量、取代基、或术语(例如,芳基、杂环、R等)在式或族性结构中出现超过一次,则其在每次出现时的定义都独立于其他每次出现时的定义。本领域技术人员还将认识到,某些基团可以与母体分子连接、或者可以从如所编写的任一端占据元素的链中的位置。例如,不对称的基团(如-C(O)N(R)-)可以在碳或氮处与母体部分连接。
立体异构源中心存在于本文公开的化合物中。根据立体异构源中心周围的取代基的构型,这些中心由符号“R”或“S”指定。应当理解的是,本发明包含全部立体异构形式(包含非对映异构体形式、对映异构体形式和差向异构体形式,以及d-异构体和l-异构体,及其混合物)。化合物的单独立体异构体可以由可商业获得的含有确定立体化学构型的起始材料以合成方式制备,或者通过分离立体异构产物的混合物(通过转化成非对映异构体的混合物,随后分离或重结晶、色谱技术、通过手性色谱柱直接分离立体异构体)、或本领域中已知的任何其他适当的方法制备。特定构型的起始化合物是可商业获得的、或者可以通过本领域中已知的技术制造并拆分。此外,本文公开的化合物可以作为几何异构体存在。本发明包含全部的顺式异构体、反式异构体、同式异构体、逆式异构体、内型异构体、外型异构体、同侧(E)异构体、和异侧(Z)异构体、及其适当的混合物。此外,化合物可以作为互变异构体存在;本发明提供全部互变异构的异构体。此外,本文公开的化合物可以以非溶剂化形式、以及用药学上可接受的溶剂(如水、乙醇等)溶剂化的形式存在。通常,认为溶剂化形式等同于非溶剂化形式。
当通过键连接的原子被认为是较大子结构的部分时,术语“键”指在两个原子、或两个部分之间的共价键。除非另有规定,否则键可以是单键、双键、或三键。分子图中两个原子之间的虚线表示附加的键在该位置处可以存在或不存在。
如本文所使用的,术语“疾病”旨在与术语“紊乱”、“病征”、和“病况”(在医学状况下)大体上同义并且可互换地使用,因为全部都反映人体或动物体或其部分之一的异常状况,该异常状况损害正常功能,通常表现为有区别的病征和症状,并且使人或动物具有降低的寿命或生活品质。
术语“组合疗法”意为施用两种或更多种治疗剂以治疗本公开中描述的病况或紊乱。这样的施用包含以基本上同时的方式共同施用这些治疗剂,如在具有固定比例的活性成分的单个胶囊中、或者在每种活性成分的多个单独的胶囊中。此外,这样的施用还包含以连续方式使用每种类型的治疗剂。在任一情况下,治疗方案将在治疗本文中描述的病况或紊乱中提供药物组合的有益作用。
“JAK抑制剂”在本文中用于指展现出不超过约100μM并且更典型地不超过约50μM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50(如在本文大致描述的JAK1、JAK2、JAK3和Tyk-2酶测定中所测量的)的化合物。在一些实施方案中,化合物将展现出约1μM至约50μM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50。IC50是将酶(例如,JAK1、JAK2、JAK3和Tyk-2)的活性降低至半最大水平的抑制剂的浓度。已经发现本文公开的某些化合物展现出针对JAK1、JAK2、JAK3和Tyk-2的抑制。在一些实施方案中,化合物将展现出不超过约300nM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50。在一些实施方案中,化合物将展现出不超过约1nM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50。在某些实施方案中,化合物将展现出不超过约50μM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50;在进一步的实施方案中,化合物将展现出不超过约10μM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50;在又进一步的实施方案中,化合物将展现出不超过约5μM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50;在又进一步的实施方案中,化合物将展现出不超过约1μM的针对JAK1、JAK2、JAK3和Tyk-2活性的IC50(如在本文所描述的JAK1、JAK2、JAK3和Tyk-2测定中所测量的)。
短语“治疗有效的”旨在限定用于疾病或紊乱的治疗或者临床终点的影响的活性成分的量。
如本文所使用的,术语“治疗剂(therapeutic)”或“治疗剂(therapeutic agent)”或“药学活性剂”意为用于治疗、对抗、改良、预防或改善患者的不希望的病况或疾病的药剂。在某种程度上,本发明的实施方案针对JAK介导的疾病的治疗。
组合物的“治疗有效的量”或“有效的量”是达到期望的效果(例如,抑制、阻滞、逆转细胞的激活、迁移、或增殖)计算的预定量。本方法预期的活性包含(视情况而定)医学治疗性治疗和/或预防疾病的治疗两者。根据本发明施用以获得治疗性效果和/或预防性效果的化合物的具体剂量当然将通过围绕病例的特定情况来确定,包含例如所施用的化合物、施用途径和正在被治疗的病况。化合物在宽的剂量范围内是有效的,并且例如,每天的剂量通常将在0.001mg/kg至10mg/kg的范围内,更通常在0.01mg/kg至1mg/kg的范围内。然而,将理解的是,施用的有效的量将由医师根据相关情况(包含待治疗的病况、待施用的化合物的选择、以及所选择的施用途径)来确定,并且因此,以上剂量范围并非旨在以任何方式限制本发明的范围。本发明的化合物的治疗有效的量通常是使得当所述化合物在生理学上可耐受的赋形剂组合物中被施用时,足以在组织中达到有效系统性浓度或局部浓度的量。
如本文所使用的,对患者的“治疗”的提及旨在包含预防。治疗也可以在本质上是先发制人的,即,其可以包含疾病的预防。疾病的预防可以涉及完全免于疾病(例如,如在预防病原体感染的情况下),或者可以涉及疾病进展的预防。例如,疾病的预防可能不意味着在任何水平上与疾病有关的任何影响的完全消除,而是可能意味着将疾病的症状预防到临床上显著的或可检测的水平。疾病的预防也可以意味着预防疾病进展到疾病晚期。
如本文所使用的,术语“治疗(treat)”、“治疗(treated)”、“治疗(treating)”或“治疗(treatment)”指治疗性治疗和/或预防疾病的措施或预防性措施两者,其中目的是预防或减缓(减轻)不期望的生理病况、紊乱或疾病,或者是获得有益的或期望的临床结果。为了本发明的目的,有益的或期望的临床结果包含(但不限于)症状的缓和;病况、紊乱或疾病的程度的减弱;病况、紊乱或疾病的状态的稳定(即,不恶化);病况、紊乱或疾病的发作的延迟或病况、紊乱或疾病的进展的减缓;病况、紊乱或疾病状态的减轻;和病况、紊乱或疾病的缓解(无论是部分或全部的,无论是诱导或维持),无论是可检测的还是不可检测的,或病况、紊乱或疾病的改进(enhancement)或改善。治疗包含引起临床上显著的响应,而无过度水平的副作用。与未接受治疗的预期存活相比,治疗还包含延长存活。治疗也可以在本质上是先发制人的,即,其可以包含疾病的预防。疾病的预防可以涉及完全免于疾病(例如,如在预防病原体感染的情况下),或者可以涉及疾病进展的预防。例如,疾病的预防可能不意味着在任何水平上与疾病有关的任何影响的完全消除,而是可能意味着将疾病的症状预防到临床上显著的或可检测的水平。疾病的预防也可以意味着预防疾病进展到疾病晚期,并且与未接受治疗的无疾病存活相比延长无疾病存活,并且与未接受治疗的无疾病存活相比延长无疾病存活。
当与治疗手段结合使用时,“施用”意为将治疗剂直接施用到靶标组织内或靶标组织上、或者向患者施用治疗剂,由此治疗剂正向地影响其所靶向的组织。因此,如本文所使用的,当与本文实施方案的化合物结合使用时,术语“施用”可以包含(但不限于):将化合物提供到靶标组织内或靶标组织上;通过例如静脉内注射向患者全身性地提供化合物,由此使治疗剂到达靶标组织;向靶标组织提供编码其序列的形式的化合物(例如,通过所谓的基因疗法技术)。可以通过注射、局部、口服、或者通过这些方法中的任何一种与其他已知技术的组合来实现“施用”组合物。
术语“患者”与术语“受试者”大体上同义,并且包含全部哺乳动物(包含人)。患者的实例包含人、牲畜(如牛、山羊、绵羊、猪、和兔)、和伴侣动物(如狗、猫、兔、和马)。优选地,患者是人。
如本文所使用的,术语“赋形剂”和“药学上可接受的赋形剂”旨在与术语“载体”、“药学上可接受的载体”、“稀释剂”、“药学上可接受的稀释剂”大体上同义,并可互换地使用。
如本文所使用的,术语“治疗上可接受的盐”表示本文公开的化合物的盐或两性离子形式,其为水溶性的或油溶性的或可分散的并且如本文所定义的治疗上可接受的。盐可以在化合物的最终分离和纯化期间制备,或者通过使游离碱形式的适当化合物与适合的酸反应而单独地制备。代表性的酸加成盐包含乙酸盐、己二酸盐、藻酸盐、L-抗坏血酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐(benzenesulfonate)(苯磺酸盐(besylate))、硫酸氢盐、丁酸盐、樟脑酸盐、樟脑磺酸盐、柠檬酸盐、二葡萄糖酸盐、甲酸盐、延胡索酸盐、龙胆酸盐、戊二酸盐、甘油磷酸盐、乙醇酸盐、半硫酸盐、庚酸盐、己酸盐、马尿酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、2-羟乙基磺酸盐(依西酸盐)、乳酸盐、马来酸盐、丙二酸盐、DL-扁桃酸盐、均三甲苯磺酸盐、甲磺酸盐、萘磺酸盐、烟酸盐、2-萘磺酸盐、草酸盐、双羟萘酸盐、果胶酸盐、过硫酸盐、3-苯丙酸盐、膦酸盐、苦味酸盐、新戊酸盐、丙酸盐、焦谷氨酸盐、琥珀酸盐、磺酸盐、酒石酸盐、L-酒石酸盐、三氯乙酸盐、三氟乙酸盐、磷酸盐、谷氨酸盐、碳酸氢盐、对甲苯磺酸盐(para-toluenesulfonate)(对甲苯磺酸盐(p-tosylate))和十一烷酸盐。另外,可以用下列物质使本文公开的化合物中的碱性基团季铵化:甲基、乙基、丙基和丁基的氯化物、溴化物、和碘化物;二甲基、二乙基、二丁基和二戊基的硫化物;癸基、月桂基、肉豆蔻基和甾醇基的氯化物、溴化物、和碘化物;以及苄基和苯乙基的溴化物。可以用于形成治疗上可接受的加成盐的酸的实例包含无机酸(如盐酸、氢溴酸、硫酸、和磷酸)和有机酸(如草酸、马来酸、琥珀酸、和柠檬酸)。还可以通过化合物与碱金属或碱土离子的配位来形成盐。因此,本发明预期本文公开的化合物的钠盐、钾盐、镁盐、和钙盐等。
本发明的实施方案针对化合物和包括这样的化合物的药物组合物(已经被发现抑制JAK激酶)、以及合成和使用化合物的方法(包含(但不限于)用于通过施用化合物来在患者中治疗JAK介导的疾病的方法)。在一些实施方案中,化合物和药物组合物被局部施用。
本发明的化合物可以以各种方式在JAK同种型中具有选择性。例如,本文所描述的化合物对JAK1、JAK2、JAK3和/或Tyk-2的选择性可以超过其他同种型、可以针对全部同种型具有等同效能、或者可以仅对一种同种型具有选择性。在某些实施方案中,本发明的化合物对JAK1的选择性超过其他同种型。在一些实施方案中,本文公开的化合物对JAK1的选择性超过JAK2和Tyk-2。可以使用酶测定、细胞测定或两者来确定选择性。在一些实施方案中,与JAK2相关受体相比,本文公开的化合物对JAK1相关受体的效能高出至少约10×。在一些实施方案中,本文公开的化合物对JAK1相关受体的选择性超过Tyk-2相关受体至少约10×。
化合物
本发明的实施方案针对化合物和包括这样的化合物的药物组合物(已经被发现抑制JAK激酶)、以及合成和使用化合物的方法。一些实施方案包含用于通过施用本文实施方案的化合物来在患者中治疗疾病的方法。
本文公开的某些化合物可以具有有用的JAK抑制活性,并且可以用于治疗或预防其中JAK激酶起活性作用的疾病或病况。因此,实施方案还针对包括一种或更多种本文公开的化合物以及药学上可接受的载体的药物组合物、以及制造和使用化合物和组合物的方法。某些实施方案针对用于抑制JAK激酶的方法。其他实施方案针对用于在需要这样的治疗的患者中治疗JAK介导的紊乱的方法,方法包括向所述患者施用治疗有效量的根据本发明的化合物或组合物。还提供本文公开的某些化合物在制造用于治疗通过JAK激酶的抑制而改善的疾病或病况的药物中的应用。
在一些实施方案中,本文公开的化合物已经被设计成吸收不良以最小化全身性暴露。在一些实施方案中,本文公开的化合物已经被设计成快速代谢以最小化全身性暴露。在一些实施方案中,本文公开的化合物被设计成在期望的作用位点处(例如,在胃肠道(例如,结肠)中)发挥其作用,以减小显著全身性作用或不利全身性作用的风险。在一些实施方案中,本文公开的化合物是肠限制性化合物。
还提供了实施方案,其中除非另有说明和规定组合不是互斥的,否则本文的任何实施方案可以与其他实施方案中的任何一者或更多者组合。
还提供选自本文公开的实施例的化合物。本文实施方案的化合物也可以指其衍生物、或本文实施方案的化合物的前述的组合。
本文所描述的化合物可以含有一个或更多个立体异构源中心,并且因此可以作为立体异构体存在。本文的实施方案包含作为基本上纯的经拆分的立体异构体、其外消旋混合物、以及非对映异构体的混合物的全部这样的可能的立体异构体。在一些实施方案中,示出的式在某些位置处没有确定的立体化学。在其他实施方案中,将化合物分离成单一的立体异构体,但立体异构源中心的绝对构型是未知的,或者仅相对的立体化学构型(即,顺式异构或反式异构)是已知的。在这样的实施方案中,式被以临时分配的绝对指定示出,以表示它们是单一的立体异构体,并且同样描述了相对立体化学构型。本文的实施方案包含这样的式的全部立体异构体及其药学上可接受的盐。可以通过例如分级结晶来从适合的溶剂中分离立体异构体的非对映异构体对,并且可以通过常规手段(例如,通过使用旋光性酸或旋光性碱作为拆分剂或者在手性HPLC柱上)来将由此获得的对映异构体对分离成单独的立体异构体。此外,可以使用光学纯的或对映异构体富集的起始材料或者已知构型的试剂通过立体定向合成或立体选择性合成来获得通式化合物的任何立体异构体。本文所描述和所要求保护的实施方案的范围涵盖化合物的外消旋形式以及单独的对映异构体、非对映异构体、立体异构体和立体异构体富集的混合物。
用于单独对映异构体的制备/分离的常规技术包含从适合的对映异构体富集的或光学纯的前体进行手性合成或者使用例如手性高压液相色谱(HPLC)拆分外消旋体。可替代地,外消旋体(或外消旋前体)可以与适合的旋光化合物(例如,醇、或者酸或碱(如酒石酸或1-苯乙胺)(在化合物含有酸性部分或碱性部分的情况下))反应。可以通过色谱和/或分级结晶分离所得到的非对映异构体混合物,并且通过本领域技术人员公知的手段将非对映异构体中的一者或两者转化成相应的一种或多种纯的对映异构体。可以使用色谱(通常为HPLC)(其中流动相由烃(通常为庚烷或己烷)组成、含有从0至50%(通常从2至20%)的异丙醇和从0至5%的烷基胺(通常0.1%二乙胺))在不对称树脂上以对映异构体富集的形式获得本文实施方案的手性化合物(及其手性前体)。洗脱液的浓缩提供富集的混合物。可以通过本领域技术人员已知的常规技术分离立体异构体聚集物。参见,例如,“Stereochemistryof Organic Compounds”by Ernest L.Eliel(Wiley,New York,1994)。
阻转异构体是由于绕单键的受阻旋转而产生的立体异构体,其中旋转的立体应变屏障足够高以允许构象异构体的分离。Oki(Oki,M;Topics in Stereochemistry 1983,1)将阻转异构体定义为在给定温度以超过1000秒的半衰期互变的构象异构体。本文所描述和所要求保护的实施方案的范围涵盖化合物的外消旋形式以及单独阻转异构体(“基本上不含”其相应对映异构体的阻转异构体)和立体异构体富集的混合物(即,阻转异构体的混合物)。
可能通过手性拆分方法(如选择性结晶)进行阻转异构体的分离。在阻转异构-对映选择性合成或阻转异构选择性合成中,一种阻转异构体以另一种阻转异构体为代价而被形成。当相较于另一种阻转异构体,异构化反应有利于一种阻转异构体时,可以通过在火把莲酮(knipholone)的全合成中使用手性助剂(如科里-巴克什-柴田(Corey-Bakshi-Shibata)(CBS)催化剂(来源于脯氨酸的不对称催化剂))、或者通过基于热力学平衡的方法来进行阻转异构选择性合成。
可以由无机酸或有机酸制备本文实施方案的化合物的适合的药学上可接受的酸加成盐。可以通过常规手段由本文实施方案的相应化合物通过例如用适当的酸或碱处理化合物来制备全部这些盐。
药学上可接受的酸包含无机酸(例如,盐酸、氢溴酸、氢碘酸、硝酸、碳酸、硫酸、磷酸和焦磷酸)和有机酸(例如,甲酸、乙酸、三氟乙酸、丙酸、琥珀酸、乙醇酸、恩波酸(扑酸)、甲磺酸、乙磺酸、2-羟基乙磺酸、泛酸、苯磺酸、甲苯磺酸、磺胺酸、甲磺酸(mesylic acid)、环己基氨基磺酸、硬脂酸、藻酸(algenic acid)、β-羟丁酸、丙二酸、半乳酸(galacticacid)、半乳糖醛酸、柠檬酸、富马酸、葡萄糖酸、谷氨酸、乳酸(lactic acid)、马来酸、苹果酸、扁桃酸、粘酸、抗坏血酸、草酸、泛酸、琥珀酸、酒石酸、苯甲酸、乙酸、昔萘酸(xinafoicacid)(1-羟基-2-萘甲酸)、萘二磺酸(napadisilic acid)(1,5-萘二磺酸(1,5-naphthalenedisulfonic acid))等)两者。
来源于药学上可接受的无机碱的盐包含铝、铵、钙、铜、铁、亚铁、锂、镁、锰(manganic)、锰(manganous)、钾、钠、锌等。来源于药学上可接受的有机碱的盐包含伯胺、仲胺和叔胺的盐,伯胺、仲胺和叔胺包含烷基胺、芳烷基胺、杂环基胺、环胺、天然存在的胺等,如精氨酸、甜菜碱、咖啡因、胆碱、氯普鲁卡因、二乙醇胺、N-甲葡糖胺、N,N’-二苄基乙二胺、二乙胺、2-二乙基氨基乙醇、2-二甲基氨基乙醇、乙醇胺、乙二胺、N-乙基吗啉、N-乙基哌啶、葡糖胺、葡萄糖胺、组氨酸、哈胺(hydrabamine)、异丙胺、赖氨酸、甲葡糖胺、吗啉、哌嗪、哌啶、多胺树脂、普鲁卡因、嘌呤、可可碱、三乙胺、三甲胺、三丙胺、氨丁三醇等。
根据本文实施方案的其他优选的盐是季铵化合物,其中阴离子(X-)的当量与N原子的正电荷有关。X-可以是各种无机酸的阴离子(如,例如,氯离子、溴离子、碘离子、硫酸根、硝酸根、磷酸根)、或者有机酸的阴离子(如,例如,乙酸根、马来酸根、延胡索酸根、柠檬酸根、草酸根、琥珀酸根、酒石酸根、苹果酸根、扁桃酸根、三氟乙酸根、甲磺酸根和对甲苯磺酸根)。X-优选为选自氯离子、溴离子、碘离子、硫酸根、硝酸根、乙酸根、马来酸根、草酸根、琥珀酸根或三氟乙酸根的阴离子。更优选地,X-为氯离子、溴离子、三氟乙酸盐或甲磺酸根。
本文实施方案的化合物可以以未溶剂化形式和溶剂化形式存在。术语溶剂合物在本文中用于描述包括本文实施方案的化合物与一定量的一种或更多种药学上可接受的溶剂分子的分子复合物。当所述溶剂是水时,采用术语水合物。溶剂合物形式的实例包含(但不限于)与水、丙酮、二氯甲烷、2-丙醇、乙醇、甲醇、二甲亚砜(DMSO)、乙酸乙酯、乙酸、乙醇胺、或其混合物缔合的本文实施方案的化合物。特别预期的是,在本文实施方案中,一个溶剂分子可以与本文实施方案的化合物的一个分子缔合,如水合物。
此外,特别预期的是,在本文实施方案中,超过一个溶剂分子可以与本文实施方案的化合物的一个分子缔合,如二水合物。此外,特别预期的是,在本文实施方案中,少于一个溶剂分子可以与本文实施方案的化合物的一分子缔合,如半水合物。此外,本文实施方案的溶剂合物被预期为保留化合物的非溶剂合物形式的生物有效性的本文实施方案的化合物的溶剂合物。
本文的实施方案还包含本文实施方案的经同位素标记的化合物,其中一个或更多个原子被具有相同原子序数但原子质量或质量数不同于自然界中通常发现的原子质量或质量数的原子代替。适合于包含在本文实施方案的化合物中的同位素的实例包含氢的同位素(如2H和3H)、碳的同位素(如11C、13C和14C)、氯的同位素(如31Cl)、氟的同位素(如18F)、碘的同位素(如123I和125I)、氮的同位素(如13N和15N)、氧的同位素(如15O、17O和18O)、磷的同位素(如32P)、以及硫的同位素(如35S)。本文实施方案的某些经同位素标记的化合物(例如,并入放射性同位素的那些化合物)在药物和/或底物组织分布研究中是有用的。鉴于放射性同位素氚(3H)和碳-14(14C)的易于并入和现成的检测手段,其对于该目的特别有用。用较重的同位素(如氘2H)取代可以提供由较大的代谢稳定性造成的某些治疗优势(例如,增加的体内半衰期或降低的剂量要求),并且因此在一些情况下可以是优选的。用正电子发射同位素(如11C、18F、15O和13N)取代可以在用于检查底物受体占有率的正电子发射体层摄影(PET)研究中是有用的。
通常可以使用适当的经同位素标记的试剂代替以其他方式采用的未经标记的试剂通过本领域技术人员已知的常规技术或者通过与本文所描述的那些方法类似的方法来制备本文实施方案的经同位素标记的化合物。
优选的经同位素标记的化合物包含本文实施方案的化合物的氘化衍生物。如本文所使用的,术语氘化衍生物包含其中特定位置处的至少一个氢原子被氘代替的本文实施方案的化合物。氘(D或2H)是氢的稳定同位素,其以0.015摩尔%的天然丰度存在。
氢氘交换(氘并入)是其中共价键合的氢原子被氘原子代替的化学反应。所述交换(并入)反应可以是全部的或部分的。
通常,本文实施方案的化合物的氘化衍生物对于在化合物上被指定为潜在氘化位点的位点处存在的每个氘具有至少3500(52.5%氘并入)的同位素富集因子(同位素丰度与该同位素的天然丰度之间的比率,即,氘在分子中给定位置处代替氢的并入百分比)。
在一些实施方案中,同位素富集因子为至少5000(75%氘)。在一些实施方案中,同位素富集因子为至少6333.3(95%氘并入)。在一些实施方案中,同位素富集因子为至少6633.3(99.5%氘并入)。应当理解的是,在被指定为氘化位点的位点处存在的每个氘的同位素富集因子与其他氘化位点无关。
可以使用本领域普通技术人员已知的常规分析方法(包含质谱(MS)和核磁共振(NMR))来测定同位素富集因子。
术语“前体药物”指在体内变得更具活性的化合物。本文公开的某些化合物也可以作为前体药物存在,如在Hydrolysis in Drug and Prodrug Metabolism:Chemistry,Biochemistry,and Enzymology(Testa,Bernard and Mayer,Joachim M.Wiley-VHCA,Zurich,Switzerland2003)中所描述的。本文所描述的化合物的前体药物是在生理条件下容易经受化学改变而提供化合物的化合物的经结构上修饰的形式。此外,前体药物可以在离体环境下通过化学方法或生物化学方法转化成化合物。例如,当前体药物与适合的酶或化学试剂放置于经皮贴剂贮器中时,前体药物可以缓慢地转化成化合物。前体药物通常是有用的,因为在一些情况下,前体药物可以比化合物或母体药物更易于施用。例如,前体药物可以是通过口服施用生物可利用的,而母体药物则不然。与母体药物相比,前体药物还可以在药物组合物中具有改进的溶解度。多种前体药物衍生物是本领域中已知的,如依赖于前体药物的水解裂解或氧化激活的那些。前体药物的实例(不限于)是作为酯(“前体药物”)施用但然后被代谢水解成羧酸、活性实体的化合物。另外的实例包含化合物的肽基衍生物。
本文所描述的化合物的前体药物也在本文实施方案的范围内。因此,本文实施方案的化合物的某些衍生物(所述衍生物本身可能具有很少的药理活性或者不具有药理活性)在被施用至身体内或身体上时可以例如通过水解切割而被转化成具有期望活性的本文实施方案的化合物。这样的衍生物被称为‘前体药物’。可以在Pro-drugs as NovelDelivery Systems,Vol.14,ACS Symposium Series(T.Higuchi and W.Stella)和Bioreversible Carriers in Drug Design,Pergamon Press,1987(ed.E.B.Roche,American Pharmaceutical Association)中找到关于前体药物的应用的进一步信息。
可以例如通过用本领域技术人员已知作为‘前体部分(pro-moiety)’的某些部分代替本文实施方案的化合物中存在的适当官能团来制备根据本文实施方案的前体药物,如例如Design of Prodrugs by H.Bundgaard(Elsevier,1985)中所描述的。
在本文实施方案的化合物为固体的情况下,本领域技术人员应当理解的是,本发明的化合物和盐可以以不同的结晶形式或多晶型形式或者以无定形形式存在,其中的全部旨在在本文实施方案的范围内。
本文公开的化合物可以作为全部立体异构体、互变异构体、构象异构体、及其全部比例的混合物、以及同位素形式(如氘化化合物)存在,并且因此包含全部立体异构体、构象异构体、及其全部比例的混合物、以及同位素形式(如氘化化合物)。
本文公开的化合物可以作为治疗上可接受的盐存在。本发明包含以盐(包含酸加成盐)形式在上文列出的化合物。适合的盐包含用有机酸或无机酸形成的盐。这样的酸加成盐通常是药学上可接受的。然而,非药学上可接受的盐的盐可以在所讨论的化合物的制备和纯化中具有效用。碱加成盐也可以被形成,并且是药学上可接受的。对于盐的制备和选择的更完整的讨论,参考Pharmaceutical Salts:Properties,Selection,and Use(Stahl,P.Heinrich.Wiley-VCHA,Zurich,Switzerland,2002)。
通过使羧基基团与适合的碱(如金属阳离子的氢氧化物、碳酸盐、或碳酸氢盐)或者与氨或有机伯胺、仲胺、或叔胺反应,可以在化合物最终分离和纯化期间制备碱加成盐。治疗上可接受的盐的阳离子包含锂、钠、钾、钙、镁、和铝,以及无毒季胺阳离子(如铵、四甲铵、四乙铵、甲胺、二甲胺、三甲胺、三乙胺、二乙胺、乙胺、三丁胺、吡啶、N,N-二甲基苯胺、N-甲基哌啶、N-甲基吗啉、二环己胺、普鲁卡因、二苄胺、N,N-二苄基苯乙胺、1-二苯羟甲胺和N,N’-二苄基乙二胺)。对于碱加成盐的形成有用的其他代表性有机胺包含乙二胺、乙醇胺、二乙醇胺、哌啶和哌嗪。
在某些实施方案中,化合物具有结构式(I):

其中:
R1选自CN或杂芳基基团,并且可选地在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C0-C6烷基C3-C6环烷基、-(C0-C6烷基)C3-C6杂环、-OH、-SO2R9、-SOR9、-SR9、-NHSO2R9、-OSO2R9、-C0-C6烷基SO2R9、C0-C6烷基COR9、C0-C6烷基NR7C(O)NR7R8、C0-C6烷基OC(O)NR7R8、C0-C6烷基NR7SO2R9、-C0-C6烷基NR7COR9-OC1-C6烷基、-OC0-C6烷基C3-C6环烷基、-OC0-C6烷基C3-C6杂环、-OC0-C6烷基NR7C(O)NR7R8、-OC0-C6烷基OC(O)NR7R8、-OC0-C6烷基NR7SO2R9、-OC0-C6烷基NR7COR9、-NR7R8、-NR7C0-C6烷基C1-C6烷基、-NR7C0-C6烷基C3-C6环烷基、-NR7C0-C6烷基NR7C(O)NR7R8、-NR7C0-C6烷基OC(O)NR7R8、-NR7C0-C6烷基NR7SO2R9、-NR7C0-C6烷基NR7COR9、-NR7C0-C6烷基C3-C6杂环、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团、杂环基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、-C0-C6烷基NR7R8、-C0-C6烷基OH、-SO2R9、-SOR9、-NHSO2R9、-C0-C6烷基NR7R4、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R2选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环A在一个或更多个碳处被一个、两个、或三个R3取代基取代,其中每个R3取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
环A的相同碳原子或不同碳原子上的两个R3基团可以可选地连接以与环A形成螺环环系或双环环系;
R4选自-C(O)-R6、-CH2R6、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R5选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R6选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR7R8、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团可以可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R7和R8独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团可以可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R7和R8可以可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环,其中杂环可以可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代;并且
R9选自H、-C1-C5烷基、-OC1-C5烷基、-C3-C6环烷基、和NR7R8,其中烷基基团、杂环基团、或环烷基基团可以可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代。
在某些实施方案中,化合物具有结构式(II):

其中:
R10选自CN或杂芳基基团,并且可选地在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C0-C6烷基C3-C6环烷基、-(C0-C6烷基)C3-C6杂环、-OH、-SO2R90、-NHSO2R90、-OSO2R90、-C0-C6烷基SO2R90、C0-C6烷基COR90、C0-C6烷基NR70C(O)NR70R80、C0-C6烷基OC(O)NR70R80、C0-C6烷基NR70SO2R90、-C0-C6烷基NR70COR90-OC1-C6烷基、-OC0-C6烷基C3-C6环烷基、-OC0-C6烷基C3-C6杂环、-OC0-C6烷基NR70C(O)NR70R80、-OC0-C6烷基OC(O)NR70R80、-OC0-C6烷基NR70SO2R90、-OC0-C6烷基NR70COR90、-NR70R80、-NR70C0-C6烷基C1-C6烷基、-NR70C0-C6烷基C3-C6环烷基、-NR70C0-C6烷基NR7C(O)NR70R80、-NR70C0-C6烷基OC(O)NR70R80、-NR70C0-C6烷基NR70SO2R90、-NR70C0-C6烷基NR70COR90、-NR70C0-C6烷基C3-C6杂环、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团、杂环基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、-C0-C6烷基NR70R80、-C0-C6烷基OH、-SO2R90、-SOR90、-NHSO2R90、-C0-C6烷基NR70R40、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R20选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环B在一个或更多个碳处被一个、两个、或三个R30取代基取代,其中每个R30取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
环B的相同碳原子或不同碳原子上的两个R30基团可以可选地连接以与环B形成螺环环系或双环环系;
R40选自-C(O)-R60、-CH2R60、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R50选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R60选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR70R80、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团可以可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R70和R80独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团可以可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R70和R80可以可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环,其中杂环可以可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代;并且
R90选自H、-C1-C5烷基、-C3-C6环烷基、和NR70R80,其中烷基基团、杂环基团、或环烷基基团可以可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代。
在某些实施方案中,化合物具有结构式(III):

其中:
R100选自CN或杂芳基基团,并且在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C3-C6环烷基、-OH、-O-C1-C5烷基、-SO2R900、-NHSO2R900、-OSO2R900、-CH2SO2R900、(CH2)nCOR900、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、NH2、NH-C1-C5烷基、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R200选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环C在一个或更多个碳处被一个、两个、或三个R300取代基取代,其中每个R300取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
环C的相同碳原子或不同碳原子上的两个R300基团可以可选地连接以与环C形成螺环环系或双环环系;
R400选自-C(O)-R600、-CH2R600、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R500选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R600选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR700R800、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团可以可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R700和R800独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团可以可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R700和R800可以可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环;并且
R900选自H、-C1-C5烷基、-C3-C6环烷基、和NR700R800,其中烷基基团可以可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代。
在某些实施方案中,化合物具有结构式(IV):

其中:
R1000选自CN或杂芳基基团,并且在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C3-C6环烷基、-OH、-O-C1-C5烷基、-SO2R9000、-NHSO2R9000、-OSO2R9000、-CH2SO2R9000、(CH2)nCOR9000、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、NH2、NH-C1-C5烷基、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R2000选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环D在一个或更多个碳处被一个、两个、或三个R3000取代基取代,其中每个R3000取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
环D的相同碳原子或不同碳原子上的两个R3000基团可以可选地连接以与环D形成螺环环系或双环环系;
R4000选自-C(O)-R6000、-CH2R6000、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R5000选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团可以可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R6000选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR7000R8000、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团可以可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R7000和R8000独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团可以可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R7000和R8000可以可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环;并且
R9000选自H、-C1-C5烷基、-C3-C6环烷基、和NR7000R8000,其中烷基基团可以可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代。
通过式(I)的化合物的下列实施例进一步说明本发明。


















通过式(II)的化合物的下列实施例进一步说明本发明。


在一些实施方案中,本文公开的化合物是肠限制性化合物。
药物组合物
本文的一些实施方案针对药物组合物,药物组合物包括本文实施方案的化合物和药学上可接受的赋形剂。
还提供药物组合物,药物组合物包括本文公开的化合物和药学上可接受的赋形剂。
在一些实施方案中,本文公开的药物组合物是肠限制性化合物。
虽然本文所描述的化合物可能以原始材料的形式施用,但还可能的是将其呈现为药物组合物。因此,本文提供药物组合物,药物组合物包括本文公开的某些化合物中的一种或更多种、或其衍生物、以及一种或更多种其药学上可接受的赋形剂和可选的一种或更多种其他治疗成分。一种或多种赋形剂必须在与制剂的其他成分相容且对于其接受者无害的意义上是“可接受的”。药物组合物的适当制剂取决于所选择的施用途径。可以如本领域中适合且理解的那样使用公知的技术和赋形剂中的任何一种。可以以本领域中已知的任何方式制造本文公开的药物组合物,例如,通过常规的混合过程、溶解过程、造粒过程、糖衣丸制造过程、研磨过程、乳化过程、包封(encapsulating)过程、包埋(entrapping)过程或压缩过程。
在一些实施方案中,可以使用一种或更多种生理学上可接受的赋形剂以常规方式配制用于根据本文实施方案使用的药物组合物。
尽管最合适的途径可以取决于例如接受者的病况和紊乱,但组合物包含适合于口服施用、肠胃外(包含皮下、皮内、肌内、静脉内、关节内和髓内)施用、腹膜内施用、鞘内施用、硬膜内施用、经粘膜施用、经皮施用、直肠施用、鼻内施用、局部(包含例如皮肤、颊、舌下和眼内)施用、玻璃体内施用、或阴道内施用的那些组合物。组合物可以包含适合于通过储库注射或通过植入施用的那些组合物。组合物可以包含适合于通过吸入(如,例如,气体、蒸汽、或粉末)施用的那些组合物。组合物可以包含适合于例如经由喷雾器、增湿器、吸入器和汽化器等以气雾剂形式施用的那些组合物。组合物可以方便地以单位剂型呈现,并且可以通过药学领域中公知的任何方法制备。通常,这些方法包含使本文公开的化合物或其衍生物(“活性成分”)与构成一种或更多种辅助成分的载体结合的步骤。通常,通过使活性成分与液体载体或精细粉碎的固体载体或者液体载体和精细粉碎的固体载体两者均匀地且密切地结合,并且然后使产品成形为期望的组合物(如果需要的话)来制备组合物。
适合于口服施用的本文公开的化合物的组合物可以以下列形式呈现:离散单元,如各自含有预定量的活性成分的胶囊剂、扁囊剂或片剂;粉剂或颗粒剂;水性液体或非水性液体中的溶液或混悬剂;或者水包油液体乳剂或油包水液体乳剂。活性成分还可以以大丸剂、药糖剂或糊剂的形式呈现。
可以口服使用的药物制剂包含片剂、由明胶制成的推入配合式胶囊剂(push-fitcapsule)、以及由明胶和增塑剂(如甘油或山梨糖醇)制成的密封式软胶囊剂。可以通过可选地与一种或更多种辅助成分压制或模制来制造片剂。可以通过在适合的机器中压缩处于自由流动形式的活性成分(如粉末或颗粒)(可选地与粘合剂、惰性稀释剂、或润滑剂、表面活性剂或分散剂混合)来制备压制片剂。可以通过在适合的机器中模制用惰性液体稀释剂润湿的粉状化合物的混合物来制造模印片剂。片剂可以可选地被包衣或刻划,并且可以被配制以便提供其中的活性成分的缓慢释放或控制释放。用于口服施用的全部组合物应当处于适合于这样的施用的剂量。推入配合式胶囊剂可以含有与填充剂(如乳糖)、粘合剂(如淀粉)、和/或润滑剂(如滑石或硬脂酸镁)、以及可选的稳定剂混合的活性成分。在软胶囊剂中,活性化合物可以溶解于或悬浮于适合的液体(如脂肪油、液体石蜡、或液体聚乙二醇)中。此外,可以添加稳定剂。糖衣丸芯提供有适合的包衣。为此,可以使用浓缩的糖溶液,浓缩的糖溶液可以可选地含有阿拉伯树胶、滑石、聚乙烯吡咯烷酮、卡波姆凝胶、聚乙二醇、和/或二氧化钛、漆溶液、和适合的有机溶剂或溶剂混合物。可以将着色剂或色素添加到片剂或糖衣丸包衣中,以鉴别或表征活性化合物剂量的不同组合。
化合物可以被配制用于通过注射(例如,通过团注法(bolus injection)或持续输注(continuous infusion))进行肠胃外施用。用于注射的组合物可以与添加的防腐剂以单位剂型存在于例如安瓿或多剂量容器中。药物组合物可以采用这样的形式(如油性或水性溶媒(vehicle)中的混悬剂、溶液或乳剂),并且可以含有配制剂(formulatory agent)(如悬浮剂、稳定剂和/或分散剂)。组合物可以存在于单位剂量容器或多剂量容器(例如,密封式安瓿和小瓶)中,并且可以以粉剂形式储存或者储存于仅需要在使用之前添加无菌液体载体(例如,盐水或无菌无热原水)的冷冻干燥(冻干)条件下。可以由前述种类的无菌粉剂、颗粒剂和片剂制备临时的注射液和混悬剂。
用于肠胃外施用的药物组合物包含活性化合物的水性和非水性(油性)无菌注射液(其可以含有抗氧化剂、缓冲剂、抑菌剂和使组合物与预期接受者的血液等渗的溶质);以及水性和非水性无菌混悬剂(其可以包含悬浮剂和增稠剂)。适合的亲脂性溶剂或溶媒包含脂肪油(如芝麻油)、或合成的脂肪酸酯(如油酸乙酯或甘油三酯)、或脂质体。水性注射混悬剂可以含有增加混悬剂的粘度的物质(如羧甲基纤维素钠、山梨糖醇或葡聚糖)。可选地,混悬剂还可以含有适合的稳定剂或增加化合物的溶解度的剂,以允许制备高度浓缩的溶液。
除了先前描述的药物组合物之外,化合物还可以被配制成贮库制剂。可以通过植入(例如,皮下地或肌内地)或通过肌内注射施用这样的长效组合物。因此,例如,化合物可以与适合的聚合物材料或疏水性材料(例如,作为可接受的油中的乳剂)或离子交换树脂一起被配制,或者被配制成微溶性衍生物(例如,被配制成微溶性盐)。
对于颊施用或舌下施用,药物组合物可以采用以常规方式配制的片剂、锭剂、软锭剂、或凝胶剂的形式。这样的组合物可以在矫味的基料(flavored basis)(如蔗糖和阿拉伯胶或黄芪胶)中包括活性成分。
化合物还可以被配制成例如含有常规的栓剂基质(如可可脂、聚乙二醇、或其他甘油酯)的直肠用组合物(如栓剂或保留灌肠剂)。
可以局部地施用(即,通过非全身性施用)本文公开的某些化合物。这包含将本文公开的化合物外部地施加至表皮或颊腔、以及将这样的化合物滴入耳、眼和鼻内。相比之下,全身性施用指口服施用、静脉内施用、腹膜内施用和肌内施用。
在一些实施方案中,本文公开的化合物可以眼部施用。在一些实施方案中,本文公开的化合物可以以眼用组合物的形式施用。本文实施方案的化合物可以以例如液体制剂(包含用于局部施用的眼用洗剂、喷雾剂或眼用滴剂)的形式施用。在一些实施方案中,本文公开的化合物可以以半固体制剂(如乳膏剂、洗剂、凝胶剂、软膏剂或糊剂)的形式施用(例如,施加至眼睑)。在一些实施方案中,本文公开的化合物可以以固体剂型(如粉剂)的形式施用(例如,施加至眼表面)以产生改善的释放。在一些实施方案中,通过用于外科手术植入的装置、肠胃外产品(例如,角膜内产品或玻璃体内产品)、用于冲洗的液体等施用本文实施方案的化合物。在一些实施方案中,包括本文公开的化合物的药物组合物是无菌的,并且不含颗粒物质。在一些实施方案中,可以通过眼内注射、眶内注射、或玻璃体内注射施用本文公开的化合物。在一些实施方案中,眼内注射可以是针对眼的前房、眼的后房、或其组合。例如,可以向眼的后部眶内区域施用本文公开的化合物。
在一些实施方案中,适合于局部施用的药物组合物包含适合于渗透通过皮肤到达炎症位点的液体制剂或半液体制剂(如溶液、粉剂、液体乳剂、液体混悬剂、半固体、软膏剂、糊剂、乳膏剂、凝胶剂、胶状物、泡沫剂、搽剂、洗剂、和适合于向眼、耳或鼻施用的滴剂)。用于局部施用的活性成分可以占组合物的例如从0.001%w/w至10%w/w(按重量计)。在某些实施方案中,活性成分可以占多达10%w/w。在其他实施方案中,其可以占少于5%w/w。在某些实施方案中,活性成分可以占从2%w/w至5%w/w。在其他实施方案中,其可以占组合物的从0.1%w/w至1%w/w。
用于局部施用或经皮施用的凝胶剂通常可以包括挥发性溶剂、非挥发性溶剂、和水的混合物。在某些实施方案中,缓冲溶剂系统的挥发性溶剂组分可以包含低级(C1-C6)烷基醇、低级烷基二醇和低级二醇聚合物。在另外的实施方案中,挥发性溶剂是乙醇。挥发性溶剂组分被认为充当渗透促进剂,同时在蒸发时也对皮肤产生冷却作用。缓冲溶剂系统的非挥发性溶剂部分选自低级亚烷基二醇和低级二醇聚合物。在某些实施方案中,使用丙二醇。非挥发性溶剂减缓挥发性溶剂的蒸发并且降低缓冲溶剂系统的蒸汽压力。与挥发性溶剂一样,通过所使用的药物化合物或药物来确定这种非挥发性溶剂组分的量。当系统中的非挥发性溶剂太少时,药物化合物可能由于挥发性溶剂的蒸发而结晶,而过量可能由于从溶剂混合物中释放药物不良而造成生物利用度的缺乏。缓冲溶剂系统的缓冲剂组分可以选自本领域中通常使用的任何缓冲剂;在某些实施方案中,使用水。成分的常见比例为约20%的非挥发性溶剂、约40%的挥发性溶剂、和约40%的水。存在可以添加至局部组合物中的几种可选成分。这些包含(但不限于)螯合剂和胶凝剂。适当的胶凝剂可以包含(但不限于)半合成纤维素衍生物(如羟丙基甲基纤维素)和合成的聚合物以及美容剂。
洗剂包含适合于向皮肤或眼施加的那些洗剂。眼用洗剂可以包括可选地含有杀细菌剂的无菌水性溶液,并且可以通过与制备滴剂的那些方法类似的方法来制备。用于向皮肤施加的洗剂或搽剂还可以包含加速干燥和使皮肤冷却的剂(如醇或丙酮)、和/或保湿剂(如甘油)或油(如蓖麻油或花生油)。
乳膏剂、软膏剂或糊剂是用于外用的活性成分的半固体药物组合物。其可以通过将精细粉碎或粉状形式的活性成分(单独地或在水性或非水性流体中的溶液或混悬剂中)借助于适合的机械与油脂性基质或非油脂性基质混合来制造。基质可以包括烃类,如硬石蜡、软石蜡或液体石蜡、甘油、蜂蜡、金属皂;胶浆;天然来源的油,如杏仁油、玉米油、花生油、蓖麻油或橄榄油;羊毛脂或其衍生物或脂肪酸(如硬脂或油酸)和醇(如丙二醇)或大粒凝胶。药物组合物可以并入任何适合的表面活性剂,如阴离子型表面活性剂、阳离子型表面活性剂或非离子型表面活性剂(如失水山梨糖醇酯或其聚氧化乙烯衍生物)。还可以包含悬浮剂(如天然树胶)、纤维素衍生物或无机材料(如硅质二氧化硅)、以及其他成分(如羊毛脂)。
滴剂可以包括无菌的水性或油性溶液或混悬剂,并且可以通过将活性成分溶解在杀细菌剂和/或杀真菌剂和/或任何其他适合的防腐剂(并且在某些实施方案中包含表面活性剂)的适合的水溶液中来制备。然后,可以通过过滤澄清所得到的溶液,转移至适合的之后密封的容器中,并且通过高压灭菌或在98℃-100℃保持半小时来无菌化。可替代地,可以通过过滤使溶液无菌化,并且通过无菌技术转移到容器中。适合于包含在滴剂中的杀细菌剂和杀真菌剂的实例是硝酸苯汞或乙酸盐(0.002%)、苯扎氯铵(0.01%)和醋酸氯己定(0.01%)。用于制备油性溶液的适合的溶剂包含甘油、经稀释的醇和丙二醇。
用于在口中局部施用(例如颊施用或舌下施用)的药物组合物包含锭剂(在矫味的基料(如蔗糖和阿拉伯胶或黄芪胶)中包括活性成分)和软锭剂(在基料(如明胶和甘油或蔗糖和阿拉伯胶)中包括活性成分)。
对于通过吸入施用,可以从吹入器、喷雾器加压包装或其他递送气雾剂喷雾的方便的手段来方便地递送化合物。加压包装可以包括适合的喷射剂,如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其他适合的气体。在加压气雾剂的情况下,可以通过提供阀来递送经计量的量而确定剂量单位。可替代地,对于通过吸入或吹入施用,根据本发明的化合物可以采用干粉组合物的形式(例如,化合物与适合的粉末基质(如乳糖或淀粉)的粉末混合物)。粉末组合物可以以单位剂型存在于例如胶囊剂、灌流器(cartridge)、明胶包装或泡罩包装中,可以借助于吸入器或吹入器从其中施用粉末。
优选的单位剂量药物组合物是含有如本文中下文列举的有效剂量的活性成分或其适当部分的那些药物组合物。
应当理解的是,除了上文特别提及的成分之外,考虑到所讨论的药物组合物类型,上文所描述的药物组合物还可以包含其他本领域中常规的剂,例如适合于口服施用的那些制剂可以包含矫味剂。
可以以从每天0.1mg/kg至每天500mg/kg的剂量施用化合物。成人的剂量范围通常为从5mg/天至2g/天。离散单元中提供的片剂或其他呈现形式可以方便地含有在这样的剂量或作为相等剂量的多倍有效的量的一种或更多种化合物,例如含有5mg至500mg(通常大约10mg至200mg)的单元。
可以与载体材料组合以产生单剂型的活性成分的量将根据治疗的宿主和特定的施用方式而改变。
当用作药物时,可以以药物组合物的形式施用化合物。这些组合物可以以药学领域公知的方式制备,并且可以根据是否期望局部治疗或全身性治疗和待治疗的区域通过多种途径施用。公开的化合物或组合物的施用可以是口服施用、肠胃外(包含皮下、皮内、肌内、静脉内、关节内和髓内)施用、肺部(例如,通过粉剂或气雾剂的吸入或吹入(包含通过喷雾器);气管内或鼻内)施用、腹膜内施用、囊内施用、硬膜内施用、经粘膜施用、经皮施用、直肠施用、局部(包含皮肤、颊、舌下和眼内)施用、或阴道内施用。肠胃外施用包含静脉内施用、动脉内施用、皮下施用、腹膜内施用、肌内施用或者注射施用或输注施用;或颅内(例如,鞘内或脑室内)施用。肠胃外施用可以为单次剂量的形式,或者可以例如通过持续灌注泵进行。用于局部施用的药物组合物可以包含泡沫剂、经皮贴剂、软膏剂、洗剂、乳膏剂、凝胶剂、溶液剂、液体乳剂、液体混悬液、半固体、糊剂、滴剂、栓剂、喷雾剂、液体和粉剂。常规的药物载体、含水粉剂或油性基质、增稠剂等可以是必需的或期望的。经涂覆的避孕套、手套等也可以是有用的。在一些实施方案中,化合物可以与药学上可接受的稀释剂、填充剂、崩解剂、粘结剂、润滑剂、表面活性剂、疏水性溶媒、水溶性溶媒、乳化剂、缓冲剂、湿润剂、保湿剂、增溶剂、防腐剂等一起含有在这样的药物组合物中。技术人员可以参考各种药学参考文献作为指导。例如,可以查阅Modern Pharmaceutics,5th Edition,Banker&Rhodes,CRC Press(2009);和Goodman&Gilman’s The Pharmaceutical Basis of Therapeutics,13thEdition,McGraw Hill,New York(2018)。
在一些实施方案中,一种通过施用本文公开的实施方案的药物组合物来治疗JAK激酶介导的疾病的方法。在一些实施方案中,化合物为治疗有效的量。在一些实施方案中,治疗有效的量为本文公开的量。
本文公开的一些实施方案还包含药物组合物,药物组合物含有作为活性成分的本文公开的化合物中的一种或更多种与一种或更多种药学上可接受的载体(赋形剂)的组合。
在一些实施方案中,制造药物组合物的方法包括将活性成分与赋形剂混合,使用赋形剂稀释活性成分,或者将活性成分以例如胶囊、囊袋、纸或其他容器的形式封闭在载体内。当赋形剂充当稀释剂时,其可以是固体材料、半固体材料、或液体材料,其用作活性成分的溶媒(vehicle)、载体(carrier)或介质。因此,药物组合物可以是下列形式:片剂、丸剂、粉剂、锭剂、囊袋剂、扁囊剂、酏剂、混悬液、乳剂、溶液剂、糖浆、气雾剂(作为固体或者在液体介质中)、软膏剂(含有例如多达10重量%的活性化合物)、软明胶胶囊剂和硬明胶胶囊剂、栓剂、无菌注射液、以及无菌包装粉剂。
适合的赋形剂的一些实例包含含有低共熔溶剂、低共熔型离子液体、或离子液体的下列:乳糖、右旋糖、蔗糖、山梨糖醇、甘露糖醇、淀粉、阿拉伯胶、磷酸钙、藻酸盐、黄蓍胶、明胶、硅酸钙、微晶纤维素、聚乙烯吡咯烷酮、纤维素、水、糖浆和甲基纤维素。药物组合物可以附加地包含:润滑剂,如滑石、硬脂酸镁和矿物油;润湿剂;乳化剂和悬浮剂;防腐剂,如羟基苯甲酸甲酯和羟基苯甲酸丙酯;甜味剂;以及矫味剂。通过采用本领域已知的程序,药物组合物可以被配制成使得在施用至患者之后提供活性成分的快速释放、持续释放或延迟释放。
可以将药物组合物配制成单位剂型。术语“单位剂型”指适合作为用于人受试者和其他哺乳动物的单位剂量的物理上不相关连的单元,每个单元含有经计算产生期望的治疗效果的预定量的活性材料、联同适合的药物赋形剂。
活性化合物可以在宽剂量范围内有效,并且通常可以以治疗有效的量被施用。然而,将理解的是,将通常由医师根据相关情况(包含待治疗的病况,选择的施用途径,实际施用的化合物,个体患者的年龄、体重和反应,患者症状的严重程度等)决定实际施用的化合物的量。
在一些实施方案中,药物组合物可以包括约0.01%至约50%的本文公开的一种或更多种化合物。在一些实施方案中,一种或更多种化合物的含量为约0.01%至约50%、约0.01%至约45%、约0.01%至约40%、约0.01%至约30%、约0.01%至约20%、约0.01%至约10%、约0.01%至约5%、约0.05%至约50%、约0.05%至约45%、约0.05%至约40%、约0.05%至约30%、约0.05%至约20%、约0.05%至约10%、约0.1%至约50%、约0.1%至约45%、约0.1%至约40%、约0.1%至约30%、约0.1%至约20%、约0.1%至约10%、约0.1%至约5%、约0.5%至约50%、约0.5%至约45%、约0.5%至约40%、约0.5%至约30%、约0.5%至约20%、约0.5%至约10%、约0.5%至约5%、约1%至约50%、约1%至约45%、约1%至约40%、约1%至约35%、约1%至约30%、约1%至约25%、约1%至约20%、约1%至约15%、约1%至约10%、约1%至约5%、约5%至约45%、约5%至约40%、约5%至约35%、约5%至约30%、约5%至约25%、约5%至约20%、约5%至约15%、约5%至约10%、约10%至约45%、约10%至约40%、约10%至约35%、约10%至约30%、约10%至约25%、约10%至约20%、约10%至约15%、或者这些范围之一内的值。具体的实例可以包含约0.01%、约0.05%、约0.1%、约0.25%、约0.5%、约0.75%、约1%、约5%、约10%、约15%、约20%、约25%、约30%、约35%、约40%、约45%、约50%、约60%、约70%、约80%、约90%、或者这些值中任何两个之间的范围。前述全部代表药物组合物的重量百分比。在一些实施方案中,药物组合物适合于局部施用。在一些实施方案中,药物组合物适合于口服施用、肠胃外(包含皮下、皮内、肌内、静脉内、关节内和髓内)施用、腹膜内施用、鞘内施用、硬膜内施用、经粘膜施用、经皮施用、直肠施用、鼻内施用、局部(包含例如皮肤、颊、舌下和眼内)施用、玻璃体内施用、或阴道内施用。
在一些实施方案中,化合物为治疗有效的量。在一些实施方案中,治疗有效的量可以为约1mg至约1000mg、约1mg至约900mg、约1mg至约800mg、约1mg至约700mg、约1mg至约600mg、约1mg至约500mg、约1mg至约400mg、约1mg至约300mg、约1mg至约200mg、约1mg至约100mg、约10mg至约1000mg、约50mg至约1000mg、约100mg至约1000mg、约200mg至约1000mg、约300mg至约1000mg、约400mg至约1000mg、约500mg至约1000mg、约10mg至约500mg、约50mg至约500mg、约100mg至约500mg、约10mg至约300mg、约50mg至约300mg、从约100mg至约300mg、约10mg至约150mg、约50mg至约150mg、约60mg至约120mg、约50mg至约120mg、或者这些值中任何两个之间的范围。具体的实例包含例如约1000mg、约900mg、约800mg、约700mg、约750mg、约600mg、约500mg、约400mg、约450mg、约300mg、约250mg、约200mg、约175mg、约150mg、约125mg、约120mg、约110mg、约100mg、约90mg、约80mg、约70mg、约60mg、约50mg、约30mg、约20mg、或者上文公开的范围之间的任何值。
在一些实施方案中,治疗有效的量可以根据例如进行治疗的特定应用、化合物的施用方式、患者的健康和病况、以及处方医师的判断而变化。化合物在药物组合物中的比例或浓度可以根据许多因素(包含剂量、化学特性(例如,疏水性)、和施用途径)而变化。例如,可以在用于肠胃外施用的含有约0.1%w/v至约10%w/v的化合物的生理缓冲水溶液中提供化合物。化合物的一些典型剂量范围为从每天约1μg/kg体重至每天约1g/kg体重。在一些实施方案中,剂量范围为从每天约0.01mg/kg体重至每天约100mg/kg体重。剂量可能取决于变量,如疾病或紊乱的类型和进展程度、特定患者的整体健康状况、所选择的化合物的相对生物学功效、赋形剂的组成、及其施用途径。可以从来源于体外或动物模型测试系统的剂量响应曲线中推断有效剂量。
向患者施用的化合物或组合物的量将根据所施用的内容、施用目的(如预防或治疗)、患者的状态、施用方式等而变化。在治疗应用中,可以以足以治愈或至少部分地阻止疾病及其并发症的症状的量向已经患有疾病的患者施用组合物。
为了制备固体组合物(如片剂),可以将主要活性成分与药物赋形剂混合以形成含有本发明化合物的均匀混合物的固体预制剂组合物。当将这些预制剂组合物称为均匀的时,活性成分通常均匀地分散在整个药物组合物中,使得药物组合物可以容易地再平均分成治疗有效的单位剂型(如片剂、丸剂和胶囊剂)。然后,将该固体预制剂再分成含有例如从约0.1mg至约1000mg的活性成分的上述类型的单位剂型。
本发明的片剂或丸剂可以被包衣或以其他方式复合以提供给予延长作用的优势的剂型。例如,片剂或丸剂可以包括内部剂量组分和外部剂量组分,后者为前者上的包裹物的形式。这两种组分可以由肠溶层隔开,肠溶层用于抵抗在胃中的崩解,并且容许内部组分完整地进入十二指肠或者被延迟释放。各种材料可以用于这样的肠溶层或包衣,这样的材料包含许多聚合酸以及聚合酸与材料(如虫胶、鲸蜡醇和乙酸纤维素)的混合物。
用于口服施用或者通过注射施用的可以并入本发明的化合物和组合物的液体形式包含水溶液、经适当矫味的糖浆、水性混悬液或油性混悬液、以及具有可食用油(如棉籽油、芝麻油、椰子油或花生油)的经矫味的乳剂、以及酏剂和类似的药物溶媒。
用于吸入或吹入的组合物包含药学上可接受的水性溶剂或有机溶剂、或其混合物中的溶液和混悬液、以及粉末。液体组合物或固体组合物可以含有如上所述的适合的药学上可接受的赋形剂。在一些实施方案中,通过口服途径或鼻部呼吸途径施用药物组合物以产生局部作用或全身性作用。可以通过使用惰性气体雾化其中的组合物。可以直接从雾化装置中呼吸经雾化的溶液,或者可以将雾化装置与面罩帐(face mask tent)或间歇性正压呼吸机连接。可以从以适当方式递送组合物的装置口服施用或鼻施用溶液组合物、混悬液组合物或粉末组合物。
在一些实施方案中,向患者施用的药物组合物可以是上述药物组合物的形式。在一些实施方案中,这些组合物可以通过常规灭菌技术被灭菌,或者可以被无菌过滤。水性溶液可以被包装以原样使用或被冻干(经冻干的制剂在施用之前与无菌水性载体组合)。在一些实施方案中,化合物制剂的pH为约3至约11、约5至约9、约5.5至约6.5、或约5.5至约7.5。将理解的是,前述赋形剂、载体、或稳定剂中某些的使用将造成药物盐的形成。
使用方法
本发明涉及在受试者中调节JAK介导的功能的方法,方法包括治疗有效量的本文公开的化合物或含有该化合物的药物组合物的施用。
本发明还涉及通过本文所描述的化合物与至少一种JAK激酶同种型之间的相互作用来抑制JAK介导的疾病的方法。这种相互作用可能造成下列方面的变化:JAK激酶产生的生物化学输出、JAK激酶的表达、JAK激酶与正常结合伴侣(binding partner)的结合、细胞表型或细胞增殖。可以监测这些变化,以确定由本文所描述的化合物实现的调节程度。这样的方法可以是疾病的治疗方式、生物学测定、细胞测定、生物化学测定等。
本文还提供治疗JAK介导的疾病的方法,方法包括向有需要的患者施用治疗有效量的本文公开的化合物、本文公开的化合物的衍生物、或其组合。在某些实施方案中,治疗有效量的本文公开的化合物、本文公开的化合物的衍生物、或其组合可以是药物组合物的形式。在实施方案中,药物组合物可以包含药学上可接受的赋形剂。
在实施方案中,通过本发明的化合物治疗的与JAK激酶相关的疾病或紊乱包含自身免疫性紊乱、慢性炎性紊乱、急性炎性紊乱、自身炎性紊乱、纤维化紊乱、代谢紊乱、瘤形成、或心血管紊乱或脑血管紊乱。因此,在一些实施方案中,本发明提供用于在有需要的患者中治疗JAK介导的疾病或紊乱的方法,其中所述方法包括向所述患者施用治疗有效量的所提供的化合物或其组合物。这样的JAK介导的疾病或紊乱包含(但不限于)本文所描述的那些。
本文的一些实施方案针对在受试者中调节JAK介导的功能的方法,方法包括治疗有效量的如本文公开的肠限制性化合物的施用。在一些实施方案中,在受试者中抑制JAK的方法包括向受试者施用本文实施方案的肠限制性化合物。
本发明还涉及抑制至少一种JAK功能的方法,方法包括使JAK激酶与如本文所描述的肠限制性化合物接触的步骤。可以监测细胞表型、细胞增殖、JAK激酶的活性、活性JAK激酶产生的生物化学输出的变化、JAK激酶的表达、或者JAK激酶与天然结合伴侣的结合。这样的方法可以是疾病的治疗方式、生物学测定、细胞测定、生物化学测定等。
本文还提供治疗JAK介导的疾病的方法,方法包括向有需要的患者施用治疗有效量的本文公开的肠限制性化合物。在一些实施方案中,施用操作是肠限制性的。在某些实施方案中,治疗有效量的本文公开的肠限制性化合物可以是药物组合物的形式。在实施方案中,药物组合物还可以包含药学上可接受的赋形剂。
在一些实施方案中,所述JAK介导的疾病或紊乱选自皮肤紊乱、瘙痒、毛发缺损紊乱、癌症、赘生物、阿尔茨海默病、炎性病况、结缔组织病和自身免疫病况。
在某些实施方案中,所述JAK介导的疾病或紊乱是赘生物、恶性肿瘤、骨髓增生性紊乱、造血组织赘生物、脊髓赘生物、淋巴组织赘生物(包含骨髓纤维化、原发性骨髓纤维化、真性红细胞增多、原发性血小板增多、急性白血病和慢性白血病、淋巴瘤、皮肤淋巴瘤(包含蕈样肉芽肿病)、其他骨髓性恶性肿瘤和骨髓增生异常综合征)。
在某些实施方案中,所述JAK介导的疾病选自由下列组成的组:自身免疫紊乱或自身免疫应答、免疫应答的广泛激活、细菌感染、病毒感染、炎症、慢性和/或急性炎性紊乱或病况、和/或自身炎性紊乱、纤维化紊乱、代谢紊乱、赘生物、或心血管紊乱或脑血管紊乱、皮肤紊乱、瘙痒、毛发缺损紊乱、癌症或恶性肿瘤、自身免疫性结缔组织病和自身免疫病况;斯蒂尔病、成年起病型斯蒂尔病、Th17相关性炎症、多软骨炎(例如,复发性多软骨炎);肌炎、多肌炎、自身免疫性肌炎、皮肌炎、幼年型皮肌炎;重症肌无力;关节炎(例如,类风湿性关节炎、幼年型类风湿性关节炎、全身起病型幼年类风湿性关节炎、骨关节炎、感染性关节炎、炎症性关节炎、炎性肠病相关性关节炎、特发性关节炎、幼年型特发性关节炎、全身型幼年特发性关节炎、银屑病性关节炎)、脊椎炎/脊椎关节炎/脊椎关节病(强硬性脊椎炎)、痛风、硬皮病(全身性硬皮病、幼年型硬皮病)、赖特综合征/反应性关节炎、莱姆病、狼疮/系统性红斑狼疮(SLE)(红斑狼疮、儿童系统性红斑狼疮、皮肤狼疮(亚急性皮肤狼疮、慢性皮肤狼疮/盘状狼疮、冻疮样红斑狼疮))、风湿性多肌痛、起止点炎、混合型结缔组织病、起止点病、心脏炎、心肌炎、血管生成紊乱、骨髓增生异常综合征、动脉粥样硬化、再狭窄(动脉粥样硬化性冠状动脉再狭窄)、急性冠状动脉综合征、心肌梗死、心脏移植物血管病变、移植性动脉病;血管炎(大血管血管炎、小血管血管炎、巨细胞动脉炎、结节性多动脉炎、血管炎综合征,所述血管炎综合征包含大动脉炎、韦格纳肉芽肿、白塞病)、干扰素基因刺激蛋白(STING)相关的婴儿期起病型血管病变(SAVI);胃肠道紊乱、小肠结肠炎、结肠炎、炎性肠病(溃疡性结肠炎、克罗恩病)、肠易激综合征、肠炎综合征/痉挛性结肠、乳糜泻;急性胰腺炎和慢性胰腺炎;原发性胆汁性肝硬化、原发性硬化性胆管炎、黄疸、肝硬化(例如,原发性胆汁性肝硬化、或归因于脂肪性肝病(例如,酒精性脂肪变性和非酒精性脂肪变性)的肝硬化);食管炎、胃炎、胃溃疡和十二指肠溃疡、腹膜炎;肾病:免疫介导的肾小球性肾病、自身免疫性肾病、膜性肾小球病、慢性进行性肾病、糖尿病性肾脏疾病/糖尿病性肾病、肾纤维化、肾缺血/再灌注损伤、HIV相关性肾病、输尿管梗阻性肾病、肾小球硬化症、蛋白尿、肾病综合征、多囊性肾脏疾病、常染色体显性多囊性肾脏疾病、肾病是免疫介导的肾病、自身免疫性肾病、慢性进行性肾病、糖尿病性肾病、肾纤维化、缺血性/再灌注损伤相关性肾病、HIV相关性肾病、输尿管梗阻性肾病、肾小球肾炎、慢性肾脏疾病(例如,糖尿病性肾病)、高血压诱导的肾病、肾小球硬化症、蛋白尿、肾病综合征、多囊性肾脏疾病、常染色体显性多囊性肾脏疾病、糖尿病性肾脏疾病、狼疮性肾炎;间质性膀胱炎;牙周炎、龈炎;肺部炎症、窦炎、肺炎、支气管炎、哮喘、支气管哮喘、变应性哮喘、非变应性哮喘、变应性支气管肺真菌病、阿斯匹林诱导的哮喘、成年起病型哮喘、固定气流阻塞型哮喘、运动诱导的哮喘、咳嗽变异性哮喘、工作相关性哮喘、夜间哮喘、哮喘并发肥胖症、嗜酸细胞性哮喘、激素抵抗型哮喘(steroid-resistantasthma)/重度哮喘、外源性哮喘、内源性哮喘/隐源性哮喘(cryptogenic asthma)、丘-施综合征、细支气管炎、闭塞性细支气管炎、慢性阻塞性肺病(COPD)、间质性肺疾病(肺纤维化、特发性肺纤维化)、急性肺损伤、肺纤维化(例如,特发性肺纤维化或囊性纤维化)、慢性阻塞性肺病、成人呼吸窘迫综合征、急性肺损伤、药物诱导的肺损伤;梅尼埃病;眼部紊乱,所述眼部紊乱包含(例如)眼部炎症、葡萄膜炎、干眼/干燥性角膜结膜炎、巩膜炎、巩膜外层炎、角膜炎/角膜病、脉络膜炎、视网膜血管炎、视神经炎、视网膜病变(糖尿病性视网膜病变、免疫介导的视网膜病变、黄斑变性、湿性黄斑变性、干性(老年性)黄斑变性);肥大细胞增生病、缺铁性贫血、尿毒症、嗜酸细胞增多综合征(HES)、系统性肥大细胞病(SMCD)、骨髓增生异常综合征、特发性血小板减少性紫癜;骨吸收疾病;神经退行性紊乱、神经紊乱/神经肌肉紊乱(例如),多发性硬化、帕金森病、亨廷顿病、肌萎缩性侧索硬化(ALS)(家族性ALS、散发性ALS)、阿尔茨海默病、重症肌无力、兰-伊肌无力综合征(LEMS)、格林-巴利综合征、脑膜炎、脑炎、创伤性脑损伤;神经系统损害、寄生虫妄想症、神经元过程和感觉知觉的调节异常、中风/神经元缺血、脊髓损伤、外周神经病变、触幻觉、脊髓损伤、精神疾病;感觉异常性疼痛(急性疼痛、慢性疼痛、神经性疼痛、或纤维肌痛)、神经刺激、外周神经病变;瘙痒/痒病(特应性瘙痒、干燥性瘙痒、与银屑病相关的瘙痒/银屑病性痒病/银屑病相关性痒病)、急性瘙痒、慢性瘙痒、特发性瘙痒、慢性特发性痒病、胆汁性痒病、肝胆相关性痒病、肾相关性痒病/肾痒病、尿毒症性痒病、胆汁郁积、妊娠期肝内胆汁郁积、慢性单纯性苔癣相关性瘙痒、淋巴瘤相关性痒病、白血病相关性痒病、结节性痒疹、特应性皮炎相关性痒病、特应性痒病/特应性瘙痒、大疱性痒病、肱桡肌瘙痒、神经原性痒病、神经性痒病、感觉异常性背痛、HIV相关性瘙痒性丘疹、精神性痒病、游泳者痒病、瘙痒或尿毒症性痒病、荨麻疹性痒病;皮肤病(例如),皮肤病药物反应/药疹、干燥病/皮肤干燥、皮疹、皮肤致敏、皮肤刺激、晒斑、修面、体虱、头虱/虱病、阴虱、皮肤幼虫移行症、疥疮、寄生虫感染、昆虫感染、荨麻疹(urticarial)/荨麻疹(hives)、丘疹性荨麻疹、昆虫咬伤、昆虫刺伤、头皮屑、皮肤上的异物或装置、真菌感染、疱疹、水痘(varicella)/水痘(chicken pox)、嗜酸性毛囊炎、妊娠皮肤病/妊娠瘙痒性荨麻疹性丘疹及斑块病(PUPP)、炎性皮肤病、嗜中性皮肤病、组织细胞样嗜中性皮肤病、肠旁路综合征皮肤病、银屑病/寻常型银屑病、扁平苔藓、硬化性萎缩性苔藓、痤疮(寻常型痤疮、粉刺型痤疮、炎性痤疮、结节囊肿性痤疮、瘢痕性痤疮、颈项部疤痕疙瘩性痤疮)、特应症(变应性接触性致敏、变应性皮炎)、皮炎(特应性皮炎/湿疹、接触性皮炎、光照性皮炎、脂溢性皮炎、瘀滞性皮炎)、急性发热性嗜中性皮肤病(斯威特综合征)、慢性非典型中性粒细胞与脂肪代谢障碍性皮肤病和高温综合征(CANDLE综合征)、化脓性汗腺炎、荨麻疹、坏疽性脓皮病、脱发(眉毛脱落、鼻内毛发脱落、瘢痕性脱发(例如,疤痕性脱发、中央离心性疤痕性脱发、毛发扁平苔藓、前额纤维化性脱发、脱发性毛囊炎)、非瘢痕性脱发(斑秃(AA)(斑块型AA、全秃(AT)、普秃(AU)、蛇形斑秃、马蹄形斑秃))、雄激素源性/雄激素性脱发(AGA)/男性及女性型AGA)、静止期脱发、头癣、稀毛症(单纯性遗传性稀毛症)、毛发扁平苔藓(前额纤维化性脱发)、点状掌跖角皮病、持久隆起性红斑(EED)、嗜中性外分泌性汗腺炎、栅栏状嗜中性肉芽肿性皮炎、嗜中性荨麻疹皮肤病、白癜风,所述白癜风包含节段型白癜风(单节段型白癜风、双节段型白癜风、多节段型白癜风)、非节段型白癜风(肢端型白癜风、面部型白癜风、或肢端面部型白癜风、面部中央型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、泛发型白癜风、普发型白癜风)、混合型白癜风/非节段型白癜风伴发节段型白癜风、局限型白癜风、孤立性粘膜型白癜风、或者伴有或不伴有白发(身体毛发受累)的白癜风;大疱性疾病、免疫性大疱性疾病(大疱性类天疱疮、疤痕性类天疱疮、寻常型天疱疮、线性IgA疾病)、妊娠性类天疱疮、着色性干皮病;纤维化和瘢痕形成的紊乱:纤维瘤、肝脏纤维化、肺纤维化、特发性肺纤维化、诸如硬皮病、纤维化增加、疤痕疙瘩、术后瘢痕的低度瘢痕形成;伤口愈合、手术瘢痕形成、辐射诱导的纤维化(例如,头颈部、胃肠道或肺部)、中枢神经系统瘢痕形成、消化道或胃肠道纤维化、肾纤维化、肝脏纤维化或胆管纤维化、肝纤维化(例如,非酒精性脂肪性肝炎、丙型肝炎、或肝细胞癌)、心脏纤维化(例如,心内膜心肌纤维化或心房纤维化)、眼部瘢痕形成、纤维硬化、瘢痕生长、伤口或痂愈合、疤痕疙瘩、纵隔纤维化、骨髓纤维化、腹膜后纤维化/奥蒙德病、进行性大块纤维化、肾源性系统性纤维化;斯耶格伦综合征、结节病、家族性地中海热、冷吡啉相关周期性综合征(默-韦综合征、家族性寒冷性自身炎性综合征/家族性寒冷性荨麻疹/TNF受体相关周期性综合征、新生儿起病多系统炎性疾病)、氧过多诱导的炎症、再灌注损伤、术后创伤、组织损伤、高温综合征;糖尿病(diabetes)(I型糖尿病、II型糖尿病)/糖尿病(diabetes mellitus)、桥本甲状腺炎、格雷夫斯病、艾迪生病、卡斯尔门病、甲状旁腺功能亢进、绝经期、肥胖症、类固醇耐药性、葡萄糖耐受不良、代谢综合征、甲状腺疾病、垂体炎;系统性免疫衰老;自身免疫性萎缩性胃炎、恶性贫血的自身免疫性萎缩性胃炎、自身免疫性脑脊髓炎、自身免疫性睾丸炎、古德帕斯彻病、斯耶格伦综合征、自身免疫性血小板减少、交感性眼炎;自身免疫性疾病的继发血液学表现(例如,贫血)、自身免疫性溶血综合征(自身免疫性溶血性贫血)、自身免疫性肝炎和炎性肝炎、自身免疫性卵巢衰竭、自身免疫性血小板减少、硅酮植入相关的自身免疫性疾病、药物诱导的自身免疫、HIV相关的自身免疫性综合征、金属诱导的自身免疫、自身免疫性聋、自身免疫性甲状腺紊乱;过敏反应和变态反应,所述过敏反应和变态反应包含超敏反应,如I型超敏反应(例如,包含过敏症)、II型超敏反应(例如,古德帕斯彻病、自身免疫性溶血性贫血)、III型超敏反应疾病(例如,阿图斯反应、血清病)、和IV型超敏反应(例如,接触性皮炎、同种异体移植排斥);急性感染和慢性感染、脓毒症综合征(脓毒症、脓毒性休克、内毒素性休克、外毒素诱导的中毒性休克、革兰氏阴性脓毒症、革兰氏阳性脓毒症、真菌性脓毒症、中毒性休克综合征);急性感染和慢性感染、脓毒症综合征(脓毒症、脓毒性休克、内毒素性休克、外毒素诱导的中毒性休克、革兰氏阴性脓毒症、革兰氏阳性脓毒症、真菌性脓毒症、中毒性休克综合征);排斥反应:移植物抗宿主反应/移植物抗宿主病、同种异体移植排斥(例如,急性同种异体移植排斥或慢性同种异体移植排斥)、早期移植排斥;恶性肿瘤、癌症、淋巴瘤、白血病、多发性骨髓瘤、实体瘤、畸胎瘤、转移性紊乱和骨紊乱、内部癌症、骨癌、口腔癌/咽癌、食管癌、喉癌、胃癌、肠癌、结肠癌、直肠癌、肺癌(例如,非小细胞肺癌或小细胞肺癌)、肝癌(肝脏癌)、胰腺癌、神经癌、脑癌(例如,神经胶质瘤、多形性成胶质细胞瘤、星形细胞瘤、神经母细胞瘤、和神经鞘瘤)、头颈癌、咽喉癌、卵巢癌、子宫癌、前列腺癌、睾丸癌、膀胱癌、肾癌(肾脏癌)、乳腺癌、胆囊癌、宫颈癌、甲状腺癌、前列腺癌、眼癌(眼部恶性肿瘤)、和皮肤癌(黑素瘤、角化棘皮瘤);以及纤维化癌症、纤维瘤、纤维腺瘤、纤维肉瘤、骨髓增生性紊乱、赘生物(造血组织赘生物、脊髓赘生物、淋巴组织赘生物(骨髓纤维化、原发性骨髓纤维化、真性红细胞增多、原发性血小板增多))、白血病(急性淋巴细胞白血病、急性髓细胞白血病和慢性髓细胞白血病、慢性淋巴细胞白血病、急性淋巴细胞性白血病、慢性粒-单核细胞型白血病(CMML)、或前髓细胞白血病)、多发性骨髓瘤及其他骨髓性恶性肿瘤(骨髓纤维化伴髓样化生(MMM)、原发性骨髓纤维化(PMF)、特发性骨髓纤维化(IMF))、淋巴瘤(霍奇金病、皮肤淋巴瘤(皮肤T细胞淋巴瘤、蕈样肉芽肿病))、淋巴瘤(例如,B细胞淋巴瘤、T细胞淋巴瘤、套细胞淋巴瘤、毛细胞淋巴瘤、伯基特淋巴瘤、肥大细胞瘤、霍奇金病、或非霍奇金病);卡波西肉瘤、横纹肌肉瘤、精原细胞瘤、畸胎癌、骨肉瘤、甲状腺滤泡状癌;外源性阿片类物质或合成阿片类物质的增加的累积、感觉异常性背痛、强迫性障碍、与强迫性障碍相关的怀乡症、及其组合。
在一些实施方案中,附加的示例性紊乱包含(但不限于):来自器官移植(包含异种移植)的并发症,如移植物抗宿主反应(例如,移植物抗宿主病)、同种异体移植排斥(例如,急性同种异体移植排斥或慢性同种异体移植排斥)、早期移植、糖尿病、骨髓增生性紊乱、排斥反应(例如,急性同种异体移植排斥);骨吸收疾病、哮喘(例如,支气管哮喘)、特应性、自身免疫性甲状腺紊乱、慢性非典型中性粒细胞与脂肪代谢障碍性皮肤病和高温综合征(CANDLE综合征)、SAVI(干扰素基因刺激蛋白(STING)相关的婴儿期起病血管病变)、溃疡性结肠炎、炎性肠病、克罗恩病、乳糜泻、溃疡性结肠炎、白塞病、重症肌无力、肾病、和心肌炎、自身免疫性疾病的继发血液学表现(例如,贫血)、自身免疫性溶血综合征、自身免疫性肝炎和炎性肝炎、自身免疫性卵巢衰竭、自身免疫性睾丸炎、自身免疫性血小板减少、硅酮植入相关的自身免疫性疾病、药物诱导的自身免疫、HIV相关的自身免疫性综合征;急性感染和慢性感染、脓毒症综合征(例如,脓毒症、脓毒性休克、内毒素性休克、外毒素诱导的中毒性休克、革兰氏阴性脓毒症、革兰氏阳性脓毒症、真菌性脓毒症、中毒性休克综合征);氧过多诱导的炎症、再灌注损伤、术后创伤、组织损伤、疼痛(例如,急性疼痛、慢性疼痛、神经性疼痛、或纤维肌痛)。
在实施方案中,所述哮喘是变应性哮喘、非变应性哮喘、变应性支气管肺真菌病、阿斯匹林诱导的哮喘、成年起病型哮喘、固定气流阻塞型哮喘、运动诱导的哮喘、咳嗽变异性哮喘、工作相关性哮喘、夜间哮喘、哮喘并发肥胖症、嗜酸细胞性哮喘、激素抵抗型哮喘/重度哮喘、外源性哮喘、或内源性哮喘/隐源性哮喘。
在实施方案中,所述白癜风是节段型白癜风(包含单节段型白癜风、双节段型白癜风或多节段型白癜风)、非节段型白癜风(包含肢端型白癜风、面部型白癜风、或肢端面部型白癜风、面部中央型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、泛发型白癜风、普发型白癜风)、混合型白癜风(非节段型白癜风伴发节段型白癜风)、局限型白癜风、孤立性粘膜型白癜风、或者伴有或不伴有白发(身体毛发受累)的白癜风、或者下文表1中示出的任何类型的白癜风:
表1白癜风的分类

在实施方案中,所述皮肤紊乱是特应性皮炎、银屑病、寻常型银屑病、皮肤致敏、皮肤刺激、皮疹、接触性皮炎、变应性接触致敏、变应性皮炎、炎性皮肤病、或嗜中性皮肤病。
如本文所使用的,“瘙痒”与“痒病”是可互换的。在一些实施方案中,瘙痒包含慢性特发性瘙痒、以及其他瘙痒紊乱的瘙痒成分。在一些实施方案中,瘙痒可以是选自由下列组成的组的疾病或病况的症状:变态反应、节肢动物咬伤、脚癣、特应性皮炎(AD)、特应性痒病、特应性皮炎相关性痒病、自身免疫应答、自身免疫性结缔组织病、细菌感染、胆汁性痒病、免疫应答的广泛激活、体虱、大疱性疾病、肱桡肌瘙痒、脑肿瘤、慢性特发性瘙痒、接触性皮炎、胆汁郁积、皮肤幼虫移行症、皮肤T细胞淋巴瘤、神经系统损害、头皮屑、寄生虫妄想症、皮肌炎、妊娠皮肤病、糖尿病、药疹、神经元过程和感觉知觉的调节异常、湿疹、嗜酸性毛囊炎、皮肤上的异物或装置、真菌感染、妊娠性类天疱疮、头虱、疱疹、化脓性汗腺炎、荨麻疹、霍奇金病、甲状旁腺功能亢进、特发性慢性痒病、炎症、昆虫感染、昆虫咬伤、昆虫刺伤、妊娠期肝内胆汁郁积、缺铁性贫血、外源性阿片类物质或合成阿片类物质的增加的累积、内部癌症、黄疸、扁平苔癣、硬化性萎缩性苔藓、红斑狼疮、淋巴瘤、淋巴瘤相关性痒病、白血病相关性痒病、恶性肿瘤、肥大细胞增生病、绝经期、多发性硬化、赘生物、神经刺激、神经原性痒病、神经性痒病、感觉异常性背痛、感觉异常性背痛、强迫性障碍、寄生虫感染、丘疹性荨麻疹、虱病、外周神经病变、光照性皮炎、真性红细胞增多、精神疾病、精神性痒病、HIV相关性瘙痒性丘疹、妊娠瘙痒性荨麻疹性丘疹及斑块病(PUPPP)、银屑病、银屑病相关性痒病、银屑病性痒病、阴虱、点状掌跖角皮病、肾痒病、类风湿性关节炎、疥疮、瘢痕生长、修面、脂溢性皮炎、瘀滞性皮炎、晒斑、游泳者痒病、系统性免疫衰老、触幻觉、Th17相关性炎症、甲状腺疾病、尿毒症、瘙痒或尿毒症性痒病、荨麻疹、荨麻疹性痒病、水痘、病毒感染、伤口或痂愈合、以及干燥病。
在实施方案中,毛发缺损紊乱选自脱发、斑秃、斑块型斑秃、全秃、普秃、蛇形斑秃、马蹄形斑秃、雄激素源性脱发(男性及女性型毛发缺损)、静止期脱发、头癣、稀毛症、单纯性遗传性稀毛症、瘢痕性脱发、毛发扁平苔藓、中央离心性疤痕性脱发、脱发性毛囊炎、或前额纤维化性脱发。
在实施方案中,结缔组织病选自SLE(系统性红斑狼疮)、皮肤狼疮(例如,SCLE、盘状狼疮)、冻疮样红斑狼疮、肌炎、多肌炎、皮肌炎、硬皮病、斯耶格伦综合征、多软骨炎(复发性多软骨炎)、血管炎、或大血管血管炎。
在实施方案中,肾病选自免疫介导的肾病、自身免疫性肾病、慢性进行性肾病、糖尿病性肾病、肾纤维化、缺血/再灌注损伤相关性肾病、HIV相关性肾病、输尿管梗阻性肾病、肾小球硬化症、蛋白尿、肾病综合征、多囊性肾脏疾病、常染色体显性多囊性肾脏疾病或糖尿病性肾脏疾病。
在实施方案中,所述癌症是实体瘤。
在实施方案中,所述癌症是前列腺癌、肾癌、肝脏癌、乳腺癌、肺癌、甲状腺癌、卡波西肉瘤、卡斯尔门病、或胰腺癌。
在实施方案中,所述癌症是淋巴瘤、白血病、或多发性骨髓瘤。
在实施方案中,所述骨髓增生性紊乱(MPD)是真性红细胞增多(PV)、原发性血小板增多(ET)、骨髓纤维化伴髓样化生(MMM)、原发性骨髓纤维化(PMF)、慢性髓细胞白血病(CML)、慢性粒-单核细胞型白血病(CMML)、嗜酸细胞增多综合征(HES)、特发性骨髓纤维化(IMF)、或系统性肥大细胞病(SMCD)。
在实施方案中,所述骨髓增生性紊乱是骨髓纤维化。
在实施方案中,所述骨髓增生性紊乱是原发性骨髓纤维化(PMF)。
在一些实施方案中,JAK介导的疾病或紊乱是癌症、前列腺癌、肾癌、肝癌、乳腺癌、肺癌、甲状腺癌、卡波西肉瘤、卡斯尔门病、胰腺癌、淋巴瘤、白血病、多发性骨髓瘤、瘤形成、原发性恶性肿瘤、继发性恶性肿瘤或复发性恶性肿瘤、转移性恶性肿瘤、血管生成紊乱、急性淋巴细胞白血病、急性髓细胞白血病和慢性髓细胞白血病、慢性淋巴细胞白血病、急性淋巴细胞性白血病、前髓细胞白血病、B细胞淋巴瘤、T细胞淋巴瘤、套细胞淋巴瘤、毛细胞淋巴瘤、伯基特淋巴瘤、肥大细胞瘤、霍奇金病或非霍奇金病、骨髓增生异常综合征、肉瘤、纤维肉瘤、横纹肌肉瘤;星形细胞瘤、神经母细胞瘤、神经胶质瘤、神经鞘瘤、非黑素瘤皮肤癌、鳞状细胞癌、基底细胞癌、默克尔细胞癌、精原细胞瘤、畸胎癌、骨肉瘤、着色性干皮病、角化棘皮瘤、甲状腺滤泡状癌、黑素瘤、畸胎瘤、横纹肌肉瘤、转移性紊乱和骨紊乱、多形性成胶质细胞瘤、恶性肿瘤、骨髓增生性紊乱、造血组织赘生物、脊髓赘生物、淋巴赘生物、骨髓纤维化、原发性骨髓纤维化、真性红细胞增多、原发性血小板增多、急性白血病和慢性白血病、淋巴瘤、皮肤淋巴瘤、蕈样肉芽肿病、其他骨髓性恶性肿瘤、骨髓增生异常综合征、骨髓增生性紊乱、真性红细胞增多、原发性血小板增多、骨髓纤维化伴髓样化生、原发性骨髓纤维化、慢性髓细胞白血病(CML)、慢性粒单核细胞白血病、嗜酸细胞增多综合征、特发性骨髓纤维化(IMF)、系统性肥大细胞病、及其组合。
在实施方案中,所述骨吸收疾病是骨质疏松症、骨关节炎、与激素失衡相关的骨吸收、与激素疗法相关的骨吸收、与自身免疫性疾病相关的骨吸收、或与癌症相关的骨吸收。
在一些实施方案中,JAK介导的疾病或紊乱是纤维化紊乱。示例性纤维化紊乱包含系统性硬化/硬皮病、狼疮性肾炎、结缔组织病、伤口愈合、手术瘢痕形成、脊髓损伤、中枢神经系统瘢痕形成、急性肺损伤、肺纤维化(例如,特发性肺纤维化或囊性纤维化)、慢性阻塞性肺病、成人呼吸窘迫综合征、急性肺损伤、药物诱导的肺损伤、肾小球肾炎、慢性肾脏疾病(例如,糖尿病性肾病)、高血压诱导的肾病、消化道或胃肠道纤维化、肾纤维化、肝脏纤维化或胆管纤维化、肝纤维化(例如,非酒精性脂肪性肝炎、丙型肝炎、或肝细胞癌)、肝硬化(例如,原发性胆汁性肝硬化或归因于脂肪性肝病的肝硬化(例如,酒精性脂肪变性和非酒精性脂肪变性))、辐射诱导的纤维化(例如,头颈部、胃肠道或肺部)、原发性硬化性胆管炎、再狭窄、心脏纤维化(例如,心内膜心肌纤维化或心房纤维化)、眼部瘢痕形成、纤维硬化、纤维化癌症、纤维瘤(fibroids)、纤维瘤(fibroma)、纤维腺瘤、纤维肉瘤、移植性动脉病、疤痕疙瘩、纵隔纤维化、骨髓纤维化、腹膜后纤维化、进行性大块纤维化、和肾源性系统性纤维化。
在一些实施方案中,JAK介导的疾病或紊乱是代谢紊乱。示例性代谢紊乱包含肥胖症、类固醇耐药性、葡萄糖耐受不良和代谢综合征。在一些实施方案中,JAK介导的疾病或紊乱是瘤形成。示例性瘤形成包含癌症。在一些实施方案中,赘生物包含原发性恶性肿瘤、继发性恶性肿瘤或复发性恶性肿瘤、或转移性恶性肿瘤。在一些实施方案中,示例性瘤形成包含血管生成紊乱、多发性骨髓瘤、白血病(例如,急性淋巴细胞白血病、急性髓细胞白血病和慢性髓细胞白血病、慢性淋巴细胞白血病、急性淋巴细胞性白血病、或前髓细胞白血病)、淋巴瘤(例如,B细胞淋巴瘤、T细胞淋巴瘤、套细胞淋巴瘤、毛细胞淋巴瘤、伯基特淋巴瘤、肥大细胞瘤、霍奇金病或非霍奇金病)、骨髓增生异常综合征、肉瘤、纤维肉瘤、横纹肌肉瘤;星形细胞瘤、神经母细胞瘤、神经胶质瘤和神经鞘瘤;黑素瘤、非黑素瘤皮肤癌(例如,鳞状细胞癌、基底细胞癌、默克尔细胞癌)、精原细胞瘤、畸胎癌、骨肉瘤、着色性干皮病、角化棘皮瘤、甲状腺滤泡状癌、卡波西肉瘤、黑素瘤、畸胎瘤、横纹肌肉瘤、转移性紊乱和骨紊乱、以及骨癌、口腔癌/咽癌、食管癌、喉癌、胃癌、肠癌、结肠癌、直肠癌、肺癌(例如,非小细胞肺癌或小细胞肺癌)、肝癌、胰腺癌、神经癌、脑癌(例如,神经胶质瘤或多形性成胶质细胞瘤)、头颈癌、咽喉癌、卵巢癌、子宫癌、前列腺癌、睾丸癌、膀胱癌、肾癌、乳腺癌、胆囊癌、宫颈癌、甲状腺癌、前列腺癌、和皮肤癌。
在一些实施方案中,JAK介导的紊乱是心血管紊乱和脑血管紊乱。示例性心血管紊乱包含动脉粥样硬化、动脉粥样硬化性冠状动脉再狭窄、急性冠状动脉综合征、心肌梗死、心脏移植物血管病变和中风。示例性脑血管疾病包含具有炎性成分或细胞凋亡成分的中枢神经系统紊乱、阿尔茨海默病、帕金森病、亨廷顿病、肌萎缩性侧索硬化、脊髓损伤、神经元缺血和外周神经病变。
在一些实施方案中,JAK介导的紊乱是胃肠道紊乱,胃肠道紊乱包含(但不限于)炎性肠病、克罗恩病、溃疡性结肠炎(例如,直肠乙状结肠炎、全结肠炎、溃疡性直肠炎、左半结肠炎)、胶原性结肠炎、淋巴细胞结肠炎、免疫检查点抑制剂诱导的结肠炎、回肠炎、嗜酸细胞性食管炎、移植物抗宿主病、移植物抗宿主病相关性结肠炎、感染性结肠炎、不确定型结肠炎(indeterminant colitis)、非典型结肠炎(atypical colitis)、自身免疫性肠病、肠易激综合征、痉挛性结肠炎、急性胰腺炎和慢性胰腺炎、乳糜泻、白塞病、原发性胆汁性肝硬化、原发性硬化性胆管炎、牙周炎、龈炎、食管炎、胃炎、胃溃疡和十二指肠溃疡、腹膜炎、牙周炎、肠炎、结肠炎、口腔炎、以及口周坏疽性脓皮病/造口周围坏疽性脓皮病(stomal/peristomal pyoderma gangrenosum)。
在一些实施方案中,JAK介导的紊乱是炎性肠病。
本文还提供用作药物的本文公开的化合物。
本文还提供用作用于治疗JAK介导的疾病的药物的本文公开的化合物。
还提供本文公开的化合物作为药物的应用。
还提供本文公开的化合物作为用于治疗JAK介导的疾病的药物的应用。
还提供用于制造用于治疗JAK介导的疾病的药物的本文公开的化合物。
还提供本文公开的化合物用于治疗JAK介导的疾病的应用。
本文还提供JAK的抑制方法,方法包括使JAK酶与本文公开的化合物或本文公开的化合物的衍生物接触。
在一些实施方案中,化合物是肠限制性化合物。
本文还提供用于在患者中实现效果的方法,方法包括向患者施用治疗有效量的本文公开的化合物或本文公开的化合物的盐,其中效果选自认知增强。
在某些实施方案中,JAK介导的疾病选自瘙痒、脱发、斑秃、白癜风、男性型雄激素源性脱发、女性型雄激素源性脱发、特应性皮炎、类风湿性关节炎、银屑病性关节炎、和银屑病。
可以以各种方式施用化合物,例如,口服施用、肠胃外(包含皮下、皮内、肌内、静脉内、关节内、和髓内)施用、腹膜内施用、鞘内施用、硬膜内施用、经粘膜施用、经皮施用、直肠施用、鼻内施用、局部(包含例如皮肤、颊、舌下和眼内)施用、玻璃体内施用、或阴道内施用。任何特定患者的特定剂量水平将取决于多种因素,所述因素包含所采用的特定化合物的活性、年龄、体重、总体健康状况、性别、饮食、施用时间、施用途径、排泄率、药物组合、正在治疗的确切紊乱、以及正在治疗的适应症或病况的严重程度。另外,施用途径可以根据病况及其严重程度而变化。
在某些实施方案中,单独地用于斑秃(例如,斑块型斑秃、全秃、普秃)的治疗,或者与局部用或病灶内用皮质类固醇、局部用米诺地尔、口服用米诺地尔、局部用或全身性抗雄激素、口服用非那雄胺、口服用度他雄胺、局部用或口服用皮甾酮(cortexolone)17α-丙酸酯、酮康唑、螺内酯、前列腺素F2类似物(例如,比马前列素或拉坦前列素)、接触致敏疗法(如用方酸二丁酯、二硝基氯苯、二苯环丙烯酮)、局部用或口服用甲氧沙林(methoxalen)和紫外线A(PUVA)、局部用蒽林、毛发移植术、微针、低强度激光疗法、低强度非激光疗法、富血小板血浆(PRP)疗法、或者已知对病况具有有益效果的其他疗法组合地用于斑秃(例如,斑块型斑秃、全秃、普秃)的治疗的局部施用或口服施用的本文所描述的JAK抑制剂/拮抗剂。
在某些实施方案中,单独地用于男性型秃发或女性型秃发(雄激素源性脱发)的治疗,或者与局部用米诺地尔、口服用米诺地尔、局部用或全身性抗雄激素、口服用非那雄胺、口服用度他雄胺、局部用或口服用皮甾酮17α-丙酸酯、酮康唑、螺内酯、前列腺素F2类似物(例如,比马前列素或拉坦前列素)、接触致敏疗法(如用方酸二丁酯、二硝基氯苯、二苯环丙烯酮)、局部用或口服用甲氧沙林和紫外线A(PUVA)、局部用蒽林、毛发移植术、微针、低强度激光疗法、低强度非激光疗法、富血小板血浆(PRP)疗法、或者已知对病况具有有益效果的其他疗法组合地用于男性型秃发或女性型秃发(雄激素源性脱发)的治疗的局部施用或口服施用的本文公开的JAK抑制剂/拮抗剂。
在某些实施方案中,局部施用或口服施用的本文公开的JAK抑制剂/拮抗剂可以单独地用于瘢痕性脱发(例如,疤痕性脱发、中央离心性疤痕性脱发、毛发扁平苔藓、前额纤维化性脱发、脱发性毛囊炎)的治疗,或者与局部用米诺地尔、口服用米诺地尔、局部用或全身性抗雄激素、口服用非那雄胺、口服用度他雄胺、局部用或口服用皮甾酮17α-丙酸酯、酮康唑、螺内酯、前列腺素F2类似物(例如,比马前列素或拉坦前列素)、接触致敏疗法(如用方酸二丁酯、二硝基氯苯、二苯环丙烯酮)、局部用或口服用甲氧沙林和紫外线A(PUVA)、局部用蒽林、毛发移植术、微针、低强度激光疗法、低强度非激光疗法、富血小板血浆(PRP)疗法、或者已知对病况具有有益效果的其他疗法组合地用于瘢痕性脱发(例如,疤痕性脱发、中央离心性疤痕性脱发、毛发扁平苔藓、前额纤维化性脱发、脱发性毛囊炎)的治疗。
在某些实施方案中,化合物可以单独地用于白癜风(例如,局限型白癜风(localized vitiligo)、局限型白癜风(focal vitiligo)、泛发型白癜风、节段型白癜风、肢端型白癜风、面部型白癜风、肢端面部型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、混合型肢端面部型和寻常型白癜风、或普发型白癜风)的治疗,或者与局部用皮质类固醇、局部用他克莫司、局部用吡美莫司(pimecrolimus)、光线疗法(如用UVB、窄带UVB的紫外线疗法、口服用或局部用补骨脂素加上紫外线A(PUVA)、卡泊三醇(calcipotriene)或其他局部用维生素D类似物、准分子激光光线疗法)、全身性免疫抑制剂、手术治疗(如皮肤微型移植、自体表皮悬液的移植、遮盖剂(如用化妆品或二羟基丙酮等)、或者已知对病况具有有益效果的其他疗法组合地用于白癜风(例如,局限型白癜风(localized vitiligo)、局限型白癜风(focalvitiligo)、泛发型白癜风、节段型白癜风、肢端型白癜风、面部型白癜风、肢端面部型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、混合型肢端面部型和寻常型白癜风、或普发型白癜风)的治疗。
待通过本文公开的化合物、组合物和方法治疗的特定的JAK介导的疾病包含皮肤紊乱、瘙痒、癌症、阿尔茨海默病、炎性病况、和自身免疫病况。
在实施方案中,所述皮肤紊乱是瘙痒、特应性皮炎、银屑病、寻常型痤疮、粉刺型痤疮、炎性痤疮、结节囊肿性痤疮、瘢痕性痤疮、化脓性汗腺炎、坏疽性脓皮病、皮肤致敏、皮肤刺激、皮疹、接触性皮炎或变应性接触致敏。
除了对于人治疗有用之外,本文公开的某些化合物和组合物对于伴侣动物、外来动物和农场动物(包含哺乳动物、啮齿动物等)的兽医治疗也可以是有用的。更优选的动物包含马、狗、和猫。
组合疗法
本公开的化合物和药物组合物可以通过另外的药物药剂的顺序施用或共同施用用于预防或治疗JAK介导的紊乱。
本发明的化合物可以单独地或者与其他药物活性化合物组合地用于治疗病况(如上文先前所描述的那些病况)。本发明的一种或多种化合物和其他一种或多种药物活性化合物可以同时施用(以相同的剂型或者以单独的剂型)或者顺序施用。因此,在一个实施方案中,本发明包括通过向受试者施用治疗有效量的一种或更多种本发明的化合物和一种或更多种附加的药物活性化合物来治疗病况的方法。
在某些情况下,将本文所描述的化合物中的至少一种或所述化合物的衍生物与另外的药物药剂组合施用可以是适当的。仅作为示例,如果患者在接受本文化合物之一时经历的副作用之一是高血压,则将抗高血压药剂与初始药物药剂组合施用可以是适当的。或者,仅作为示例,可以通过佐剂的施用来增强本文所描述的化合物之一的治疗效果(即,佐剂本身仅可以具有最小的治疗益处,但在与另外的药物药剂组合时,增强了对患者的整体治疗益处)。或者,仅作为示例,可以通过将本文所描述的化合物之一与也具有治疗益处的另外的药物药剂(其也包含治疗方案)一起施用来增加患者所经历的益处。仅作为示例,在涉及本文所描述的化合物之一的施用的针对糖尿病的治疗中,也可以通过向患者提供针对糖尿病的另外的药物药剂来产生增加的治疗益处。在任何情况下,无论正在治疗的疾病、紊乱或病况,患者所经历的整体益处可以仅是两种药物药剂的累加,或者患者可以经历协同益处。
可能的组合疗法的具体非限制性实例包含本文实施方案的化合物与下列物质的使用:化疗治疗剂或抗增殖剂、抗炎药剂、免疫调节剂或免疫抑制剂、神经营养因子、用于治疗心血管疾病的药剂、用于治疗糖尿病的药剂、或者用于治疗免疫缺陷紊乱的药剂。
用于炎症的可能的组合疗法的具体非限制性实例包含本公开的某些化合物与下列物质的使用:(1)皮质类固醇,包含但不限于可的松、地塞米松、和甲泼尼龙;(2)非甾体类抗炎药(NSAID),包含但不限于布洛芬、萘普生、对乙酰氨基酚、阿司匹林、非诺洛芬(NALFONTM)、氟比洛芬(ANSAIDTM)、酮洛芬、奥沙普秦(DAYPROTM)、双氯芬酸钠(VOLTARENTM)、双氯芬酸钾(CATAFLAMTM)、依托度酸(etodolac)(LODINETM)、吲哚美辛(INDOCINTM)、酮咯酸(TORADOLTM)、舒林酸(CLINORILTM)、托美汀(TOLECTINTM)、甲氯芬那酸(MECLOMENTM)、甲芬那酸(PONSTELTM)、萘丁美酮(RELAFENTM)和吡罗昔康(FELDENETM);(3)免疫抑制剂,包含但不限于甲氨蝶呤(RHEUMATREXTM)、来氟米特(ARAVATM)、硫唑嘌呤(IMURANTM)、环孢菌素(NEORALTM、SANDIMMUNETM)、他克莫司和环磷酰胺(CYTOXANTM);(4)CD20阻滞剂,包含但不限于利妥昔单抗(RITUXANTM);(5)肿瘤坏死因子(TNF)阻滞剂,包含但不限于依那西普(ENBRELTM)、英利昔单抗(infliximab)(REMICADETM)和阿达木单抗(HUMIRATM);(6)白介素-1受体拮抗剂,包含但不限于阿那白滞素(KINERETTM);(7)白介素-6抑制剂,包含但不限于托珠单抗(ACTEMRATM);(8)白介素-17抑制剂,包含但不限于AIN457;(9)詹纳斯激酶抑制剂,包含但不限于他索西替尼(tasocitinib);以及(10)syk抑制剂,包含但不限于福坦替尼(fostamatinib)。
用于癌症的治疗的可能的组合疗法的具体非限制性实例包含本公开的某些化合物与下列物质的使用:(1)烷化剂,包含但不限于顺铂(PLATINTM)、卡铂(PARAPLATINTM)、奥沙利铂(ELOXATINTM)、链佐星(ZANOSARTM)、白消安(MYLERANTM)和环磷酰胺(ENDOXAN);(2)抗代谢物,包含但不限于巯基嘌呤(PURINETHOLTM)、硫鸟嘌呤、喷司他丁(NIPENTTM)、胞嘧啶阿拉伯糖苷(ARA-CTM)、吉西他滨(GEMZARTM)、氟尿嘧啶(CARACTM)、亚叶酸(FUSILEVTM)和甲氨蝶呤(RHEUMATREXTM);(3)植物类碱和萜类化合物,包含但不限于长春新碱(ONCOVINTM)、长春碱和紫杉酚(TAXOLTM);(4)拓扑异构酶抑制剂,包含但不限于伊立替康(CAMPTOSARTM)、拓扑替康(HYCAMTINTM)和依托泊苷(EPOSINTM);(5)细胞毒性抗生素,包含但不限于放线菌素D(COSMEGENTM)、阿霉素(ADRIAMYCINTM)、博来霉素(BLENOXANETM)和丝裂霉素(MITOSOLTM);(6)血管生成抑制剂,包含但不限于舒尼替尼(SUTENTTM)和贝伐单抗(AVASTINTM);(7)酪氨酸激酶抑制剂,包含但不限于伊马替尼(GLEEVECTM)、埃罗替尼(TARCEVATM)、拉帕替尼(lapatininb)(TYKERBTM)和阿昔替尼(INLYTATM);以及(8)免疫检查点抑制剂,包含但不限于阿特珠单抗(atezolizumab)(TECENTRIQTM)、阿维鲁单抗(avelumab)(BAVENCIOTM)、德瓦鲁单抗(durvalumab)(IMFINZITM)、伊匹单抗(ipilimumab)(YERVOYTM)、派姆单抗(pembrolizumab)(KEYTRUDATM)、纳武单抗(nivolumab)(OPDIVOTM)、和替西木单抗(tremelimumab)。
本公开的化合物和药物组合物可以用于通过顺序施用或共同施用另外的药物药剂来预防或治疗JAK介导的疾病。
在一些实施方案中,本文实施方案中公开的化合物也可以与多种其他药物药剂或治疗(例如,全身性(如口服或肠胃外)施用的药物药剂或治疗)共同施用(同时地或顺序地)。这样的全身性治疗的实例包含局部用或全身性皮质类固醇(如泼尼松)、抗生素(如红霉素、四环素和双氯西林)、抗真菌剂(如以商标名称DiflucanTM出售的酮康唑和氟康唑)、抗病毒剂(如以商标名称ValtrexTM出售的伐昔洛韦(valacyclovir)、阿昔洛韦(acyclovir)和以商标名称FamvirTM出售的泛昔洛韦(famciclovir))、皮质类固醇、免疫抑制剂(如以商标名称CytoxanTM出售的环磷酰胺、硫唑嘌呤、甲氨蝶呤、霉酚酸酯(mycophenolate))、生物制剂(如以商标名称RituxanTM出售的利妥昔单抗、以商标名称EnbrelTM出售的依那西普、以商标名称HumiraTM出售的阿达木单抗、以商标名称RemicadeTM出售的英利昔单抗、以商标名称StelaraTM出售的优特克单抗(ustenkinumab)、以及以商标名称AmeviveTM出售的阿法西普(alefacept))、和/或甲状腺激素代替物。
在一些实施方案中,可以与本文公开的化合物组合使用的其他疗法包含例如巯基嘌呤、局部用或全身性皮质类固醇(如泼尼松、甲泼尼龙和泼尼松龙)、烷化剂(如环磷酰胺)、钙调磷酸酶抑制剂(如环孢菌素、西罗莫司和他克莫司)、肌苷一磷酸脱氢酶(IMPDH)的抑制剂(如麦考酚酯、吗替麦考酚酯(mycophenolate mofetil)、硫唑嘌呤)、各种抗体(例如,抗淋巴细胞球蛋白(ALG)、抗胸腺细胞球蛋白(ATG)、单克隆抗T细胞抗体(OKT3))、和辐射。这些各种药剂可以根据其标准剂量或常用剂量(如附随可商业获得的药物表格的处方信息中所规定的(另外参见The Physician’s Desk Reference(2006版)中的处方信息))被使用。在一些实施方案中,当与本文实施方案的化合物组合使用时,可以降低这些药剂的标准剂量。在不限制本公开的范围的情况下,据信这样的组合可以产生具有更好的功效、更少的毒性、更长的作用持续时间、或更快的对疗法的响应的协同结果。在一些实施方案中,本文实施方案中的组合疗法可以以亚治疗量的本文实施方案的化合物、或附加的药物药剂、或本文实施方案的化合物与附加的药物药剂两者的形式施用。硫唑嘌呤目前可以品牌名称AzasanTM从Salix Pharmaceuticals,Inc.获得;巯基嘌呤目前可以品牌名称PurinetholTM从Gate Pharmaceuticals,Inc.获得;泼尼松和泼尼松龙目前可从Roxane Laboratories,Inc.获得;甲泼尼龙目前可从Pfizer获得;西罗莫司(雷帕霉素)目前可以品牌名称RapamuneTM从Wyeth-Ayerst获得;他克莫司目前可以品牌名称PrografTM从Fujisawa获得;环孢菌素目前可以品牌名称SandimmuneTM从Novartis获得以及以品牌GengrafTM从Abbott获得;IMPDH抑制剂(如吗替麦考酚酯和麦考酚酸)目前可以品牌名称CellceptTM从Roche获得以及以品牌名称MyforticTM从Novartis获得;硫唑嘌呤目前可以品牌名称ImuranTM从Glaxo Smith Kline获得;并且抗体目前可以品牌名称OrthocloneTM从Ortho Biotech获得,以品牌名称SimulectTM(巴利昔单抗(basiliximab))从Novartis获得,以及以品牌名称ZenapaxTM(达利珠单抗(daclizumab))从Roche获得。
在一些实施方案中,本文实施方案的化合物与以上药物药剂或疗法和/或与针对另外的疾病的药物药剂或疗法联合施用、附随施用或辅助施用。例如,本文实施方案的化合物可以与甲状腺激素替代疗法或与抗炎疗法或免疫调节疗法组合。
在一些实施方案中,本文实施方案中的组合疗法可以以亚治疗量的本文实施方案的化合物、或附加的药物药剂、或本文实施方案的化合物与附加的药物药剂两者的形式施用。
在任何情况下,可以以任何顺序或者甚至同时地施用多种药物药剂(其中至少一种为本文公开的化合物)。如果同时施用,则可以以单一统一的形式或者以多种形式(仅作为示例,作为单个丸剂或作为两个单独的丸剂)提供多种药物药剂。可以以多剂量给予药物药剂之一,或者可以以多剂量给予两种药物药剂。如果不同时施用,则多剂量之间的时间可以是从几分钟至八周的任何持续时间,或者在适合于维持期望的治疗功效的任何间隔。在一些实施方案中,多剂量之间的时间可以是一分钟、一小时、六小时、一天、两天、三天、四天、五天、六天、一周、两周、三周、四周、五周、六周、七周或八周。
因此,在另一方面,某些实施方案提供用于在需要这样的治疗的人受试者或动物受试者中治疗JAK介导的紊乱的方法,方法包括向所述受试者施用有效降低或预防受试者中的所述紊乱的一定量的本文公开的化合物与本领域已知的用于治疗所述紊乱的至少一种附加药剂的组合。在相关方面,某些实施方案提供治疗组合物,治疗组合物包括至少一种本文公开的化合物与用于治疗JAK介导的紊乱的一种或更多种附加药剂的组合。
在另外的实施方案中,提供药物组合物,该药物组合物包括一种或更多种本发明的化合物、一种或更多种附加的药物活性化合物、以及药学上可接受的载体。
在另外的实施方案中,一种或更多种附加的药物活性化合物选自由下列组成的组:抗炎药、抗动脉粥样硬化药、免疫抑制药、免疫调节药、细胞生长抑制药物、抗增殖剂、血管生成抑制剂、激酶抑制剂、细胞因子阻滞剂、以及细胞粘附分子的抑制剂。
在另外的实施方案中,药物组合物还可以包含一种或更多种附加的药物药剂(如化学治疗剂、类固醇、抗炎化合物、或免疫抑制剂)。
本文所描述的JAK抑制剂组合物还可选地与其他治疗试剂组合使用,根据对待治疗的病况的治疗价值来选择其他治疗试剂。通常,在采用组合疗法的实施方案中,本文所描述的药物组合物和其他药剂不必在同一药物组合物中施用,并且由于不同的物理特性和化学特性而可选地通过不同途径施用。通常根据已确立的方案进行初始施用,并且然后基于观察到的效果来改变剂量、施用模式和施用时间。在某些情况下,将本文所描述的JAK抑制剂组合物与另外的治疗剂组合施用是适当的。仅作为示例,如果患者在接受本文所描述的JAK抑制剂组合物时经历的副作用之一是皮疹,则将抗组胺药剂与初始治疗剂组合施用是适当的。或者,仅作为示例,通过也具有治疗益处的另外的药物药剂(其也包含治疗方案)的施用来增强JAK抑制剂的治疗效果。在任何情况下,无论正在治疗的疾病、紊乱或病况,患者所经历的整体益处仅是两种治疗剂的累加,或者患者经历协同益处。
当药物用于治疗组合时,治疗有效的剂量不同。用于用实验方法确定在组合治疗方案中使用的药物和其他药剂的治疗有效的剂量的方法是备有证明文件的方法学。组合治疗还包含在各种时间开始和停止的周期治疗,以协助患者的临床管理。在任何情况下,以任何顺序或者甚至同时地施用多种治疗剂(其中一种为本文所描述的JAK抑制剂)。如果同时施用,则以单一统一的形式或者以多种形式(仅作为示例,作为单个丸剂或作为两个单独的丸剂)可选地提供多种治疗剂。
在一些实施方案中,以多剂量给予治疗剂之一,或者以多剂量给予两种治疗剂。如果不同时施用,则多剂量之间的时间可选地从超过零周至小于十二周变化。
此外,组合方法和组合物不限于仅使用两种药剂,还预期多种治疗组合的使用。应当理解的是,根据多种因素可选地改变用于治疗、预防或改善寻求缓解的一种或多种病况的剂量方案。这些因素包含受试者患有的紊乱、以及受试者的年龄、体重、性别、饮食和身体状况。因此,在一些实施方案中,实际采用的剂量方案在很大范围内变化,并且因此与本文阐述的剂量方案不同。
组成本文公开的组合疗法的药物药剂可选地为旨在基本上同时施用的组合剂型或单独剂型。组成组合疗法的药物药剂也可选地被顺序施用,其中任一种药剂都通过需要两步施用方案被施用。两步施用方案可选地要求活性药剂的顺序施用或单独活性药剂的间隔施用。根据每种药物药剂的性质(如药物药剂的效能、溶解度、生物利用度、血浆半衰期和动力学特性),多个施用步骤之间的时间段的范围为从几分钟至几小时。目标分子浓度的昼夜节律变化可选地用于确定最优剂量间隔。
在另外的实施方案中,JAK抑制剂可选地与向患者提供附加益处或协同益处的程序结合使用。在疾病或病况的发生之前、在疾病或病况的发生期间或者在疾病或病况的发生之后,可选地施用JAK抑制剂和一种或多种附加的疗法,并且在一些实施方案中,施用含有JAK抑制剂的药物组合物的时间不同。因此,例如,将JAK抑制剂用作预防剂,并且连续地向具有发展病况或疾病倾向的受试者施用,以预防该疾病或病况的发生。在症状发作期间或者在症状发作之后立即可选地向受试者施用JAK抑制剂和组合物。虽然已经在本文中示出和描述了本发明的实施方案,但对于本领域技术人员将显而易见的是,这样的实施方案仅以示例的方式提供。在不脱离本发明的情况下,本领域技术人员继而将想到许多变化、改变和替换。应当理解的是,在本发明的一些实施方案中,在实践本发明中采用本文所描述的实施方案的各种替代方案。
JAK抑制剂可以与来自下列类别的药物组合施用:NSAID、免疫抑制药、免疫调节药、细胞生长抑制药物、抗增殖剂、血管生成抑制剂、生物制剂、类固醇、维生素D3类似物、类维生素A、其他激酶抑制剂、细胞因子阻滞剂、皮质类固醇和细胞粘附分子的抑制剂。在受试者患有动脉粥样硬化或与动脉粥样硬化相关的病况或者受试者处于患有动脉粥样硬化或与动脉粥样硬化相关的病况的风险的情况下,本文所描述的JAK抑制剂组合物可选地以任何组合与用于治疗动脉粥样硬化或与动脉粥样硬化相关的病况的一种或更多种药剂或方法一起使用。用于治疗动脉粥样硬化或与动脉粥样硬化相关的病况的治疗剂/治疗的实例包含(但不限于)下列中的任何一种:托彻普(torcetrapib)、阿斯匹林、烟酸、HMG CoA还原酶抑制剂(例如,阿托伐他汀(atorvastatin)、氟伐他汀(fluvastatin)、洛伐他汀(lovastatin)、普伐他汀(pravastatin)、罗苏伐他汀(rosuvastatin)和辛伐他汀(simvastatin))、考来维纶(colesevelam)、消胆胺、考来替泊(colestipol)、吉非贝齐(gemfibrozil)、普罗布考(probucol)和氯贝特(clofibrate)。
在受试者患有炎性病况或处于患有炎性病况的风险的情况下,本文所描述的JAK抑制剂组合物可选地以任何组合与用于治疗炎性病况的一种或更多种药剂或方法一起使用。
一种或更多种附加的药物药剂(如,例如,抗炎药剂、类固醇、免疫抑制剂、以及一种或更多种其他ITK激酶抑制剂和/或其他激酶抑制剂(如JAK3激酶抑制剂、JAK1激酶抑制剂、JAK1/2激酶抑制剂、或JAK2激酶抑制剂(如例如WO 99/65909、WO 00/00202和/或WO/2004/099205中所描述的那些))或其他药剂)可以与用于治疗JAK相关的疾病、紊乱或病况的本发明的化合物组合使用。
在某些实施方案中,附加的药物药剂选自紫杉烷类、bcr-abl的抑制剂、EGFR的抑制剂、DNA破坏剂、抗代谢物、紫杉酚、伊马替尼、达沙替尼、尼洛替尼、厄洛替尼、吉非替尼、顺铂、奥沙利铂、卡铂、蒽环类药物、AraC、5-FU、喜树碱、阿霉素、伊达比星、紫杉酚、多西他赛、长春新碱、MEK抑制剂、U0126、KSP抑制剂、伏立诺他、派姆单抗、纳武单抗、阿特珠单抗、阿维鲁单抗、替西木单抗、和德瓦鲁单抗。
在一些实施方案中,所述组合物还包括附加的药物药剂,附加的药物药剂选自化学治疗剂或抗增殖剂、抗病毒剂、抗生素、抗组胺剂、润滑剂、全身性光线疗法、补骨脂素光化学疗法、激光疗法、激素替代疗法、抗炎药剂、免疫调节剂或免疫抑制剂、神经营养因子、用于治疗心血管疾病的药剂、用于治疗糖尿病的药剂、以及用于治疗免疫缺陷紊乱的药剂。
在一些实施方案中,本文实施方案的一种或更多种化合物可以与ITK介导的紊乱的治疗中使用的一种或更多种其他治疗剂组合使用,并且与对单独的其他治疗剂的响应相比,可以改进治疗响应,而不会加剧其毒性作用。在一些实施方案中,本文实施方案的化合物可以与一种或更多种其他ITK抑制剂和/或JAK抑制剂组合使用用于ITK介导的紊乱的治疗。累加效应或协同效应是这样的组合的期望的结果。附加的药剂可以与本发明化合物以单一剂型或连续剂型组合,或者药剂可以作为单独的剂型同时施用或顺序施用。在一些实施方案中,一种或更多种附加的药剂可以与本文所描述的至少一种JAK抑制剂/拮抗剂组合地向患者施用,其中附加的药剂被间歇地(而非连续地)施用。
例如,在某些实施方案中,局部施用或口服施用的本文所描述的JAK抑制剂/拮抗剂可以单独地用于斑秃(例如,斑块型斑秃、全秃、普秃)的治疗,或者与局部用或病灶内用皮质类固醇、局部用米诺地尔、口服用非那雄胺、口服用度他雄胺、接触致敏疗法(如用方酸二丁酯、二硝基氯苯、二苯环丙烯酮)、局部用或口服用甲氧沙林和紫外线A(PUVA)、局部用蒽林、毛发移植术、或者已知对病况具有有益效果的其他疗法组合地用于斑秃(例如,斑块型斑秃、全秃、普秃)的治疗。
例如,在某些实施方案中,局部施用或口服施用的本文公开的JAK抑制剂/拮抗剂可以单独地用于男性型秃发或女性型秃发(雄激素源性脱发)的治疗,或者与局部用米诺地尔、口服用非那雄胺(在男性中)、口服用度他雄胺(在男性中)、局部用抗雄激素、毛发移植术、或者已知对病况具有有益效果的其他疗法组合地用于男性型秃发或女性型秃发(雄激素源性脱发)的治疗。
例如,在某些实施方案中,化合物可以单独地用于白癜风(例如,局限型白癜风(localized vitiligo)、局限型白癜风(focal vitiligo)、泛发型白癜风、节段型白癜风、肢端型白癜风、面部型白癜风、肢端面部型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、混合型肢端面部型和寻常型白癜风、或普发型白癜风)的治疗,或者与局部用皮质类固醇、局部用他克莫司、局部用吡美莫司(pimecrolimus)、光线疗法(如用UVB、窄带UVB的紫外线疗法、口服用或局部用补骨脂素加上紫外线A(PUVA)、卡泊三醇(calcipotriene)或其他局部用维生素D类似物、准分子激光光线疗法)、全身性免疫抑制剂、手术治疗(如皮肤微型移植、自体表皮悬液的移植、遮盖剂(如用化妆品或二羟基丙酮等)、或者已知对病况具有有益效果的其他疗法组合地用于白癜风(例如,局限型白癜风(localized vitiligo)、局限型白癜风(focalvitiligo)、泛发型白癜风、节段型白癜风、肢端型白癜风、面部型白癜风、肢端面部型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、混合型肢端面部型和寻常型白癜风、或普发型白癜风)的治疗。
在某些实施方案中,本公开的化合物可以与通过相同机制或通过不同机制起作用以实现胃肠道紊乱的治疗的一种或更多种药剂组合使用。可以顺序地或同时地(以单独的组合物或以同一组合物)施用不同的药剂。用于组合疗法的有用的药剂类别包含(但不限于)氨基水杨酸酯、类固醇、全身性免疫抑制剂、抗TNFα抗体、TNFα配体抑制剂、TNF结合剂、抗VLA-4抗体、抗整合素Cv37抗体、抗菌剂、糖皮质激素激动剂、核因子κB抑制剂、5-脂氧合酶抑制剂、整合素α-4/β-7拮抗剂、环氧合酶抑制剂、IL-23拮抗剂、白三烯BLT受体拮抗剂、IL-6拮抗剂、IL-8拮抗剂、整合素拮抗剂、烟碱型乙酰胆碱受体激动剂、PPARγ激动剂、鞘氨醇-1-磷酸受体-1调节剂、B淋巴细胞抗原CD20抑制剂、钙调磷酸酶抑制剂、CD3拮抗剂、细胞粘附分子抑制剂、嗜酸粒细胞过氧化物酶抑制剂、肝素激动剂、ICAM1基因抑制剂、IL-13拮抗剂、IL-2受体α亚基抑制剂、胰岛素增敏剂、干扰素β配体、干扰素γ受体拮抗剂、白介素-1β配体调节剂、MAdCAM抑制剂、PDE 4抑制剂、鞘氨醇-1-磷酸受体1激动剂、TLR-9激动剂、乙酰胆碱酯酶抑制剂、ACTH受体激动剂、激活素受体拮抗剂、CCR5趋化因子拮抗剂、CCR9趋化因子拮抗剂、以及止泻药。
可以与本公开的化合物组合使用的氨基水杨酸酯包含(但不限于)氨水杨酸、奥沙拉嗪(osalazine)和柳氮磺胺吡啶。类固醇的实例包含(但不限于)泼尼松、泼尼松龙、氢化可的松、布地奈德(budesonide)、倍氯米松(beclomethasone)、以及氟替卡松(fluticasone)。对于炎性紊乱的治疗有用的全身性免疫抑制剂包含(但不限于)环孢菌素、硫唑嘌呤、甲氨蝶呤、6-巯基嘌呤、以及他克莫司(tacrolimus)。此外,包含(但不限于)英夫利昔单抗(infliximab)、阿达木单抗(adalimumab)、戈利木单抗(golimumab)和赛妥珠单抗(certolizumab)的抗TNFα抗体可以用于组合疗法。通过其他机制起作用的有用的化合物包含抗VLA-4抗体(如那他珠单抗(natalizumab))、抗整合素α4β7抗体(如维多珠单抗(vedolizumab))、抗细菌剂(如利福昔明(rifaximin))和止泻药(如洛哌丁胺(loperamide))。(Mozaffari et al.Expert Opin.Biol.Ther.2014,14,583-600;Danese,Gut,2012,61,918-932;Lam et al.,Immunotherapy,2014,6,963-971)。
可以与本公开的化合物组合使用的其他化合物包含(但不限于)奥帕加尼(opaganib)、阿巴西普(abatacept)、蒙格森(mongersen)、非戈替尼(filgotinib)、LYC-30937、BI-655130、米吉珠单抗(mirikizumab)、阿达木单抗(adalimumab)、他克莫司、利妥昔单抗(rituximab)、GSK-2982772、安德西昔单抗(andecaliximab)、纳曲酮(naltrexone)、瑞莎珠单抗(risankizumab)、QBECO、阿里卡福森(alicaforsen)、依曲利珠单抗(etrolizumab)、福雷芦单抗(foralumab)、奥克雷珠单抗(crelizumab)、维多珠单抗(edolizumab)、阿米莫德(amiselimod)、奥扎莫德(ozanimod)、朵堪那替(olcanatide)、卡曲得考(catridecacog)、布地奈德(budesonide)、STNM-01、大麻二酚、特罗司他依替拉(telotristat etiprate)、SHP-647、甲基卡特格拉斯特(carotegrast methyl)、peg-伊洛白介素(ilodecakin)、TOP-1288、伊伯加斯特(iberogast)N、PF-06480605、培非替尼(peficitinib)、倍氯米松、重组干扰素β-1a、英夫利昔单抗、戈利木单抗、曲洛单抗(tralokinumab)、乌司奴单抗(ustekinumab)、塞妥珠单抗聚乙二醇(certolizumabpegol)、沙利度胺(thalidomide)、乌帕替尼(upadacitinib)、阿普斯特(apremilast)、那他珠单抗、干扰素β-1a、利福昔明、RBX-2660、艾曲莫德(etrasimod)、齐留通(zileuton)、芬戈莫德(fingolimod)、可比托莫德(cobitolimod)、罗哌卡因(ropivacaine)、ABX-464、PF-06700841、泼尼松龙、GLPG-0974、缬更昔洛韦(valganciclovir)、环孢素(ciclosporin)、VB-201、妥仑西普(tulinercept)、MDGN-002、PTG-100、地塞米松(dexamethasone)、GED-0507-34-Levo、柏替木单抗(bertilimumab)、布拉兹库单抗(brazikumab)、KHK-4083、罗格列酮(rosiglitazone)、莫拉维莫德(mocravimod)、司曲吐瑞(sotrastaurin)、KAG-308、PUR-0110、E-6007、巴柳氮(balsalazide)、巴利昔单抗(basiliximab)、LP-02、ASP-3291、猪鞭虫卵(Trichuris suis ova)、K(D)PT、米地玛(midismase)、DNVX-078、维特立珠单抗(vatelizumab)、艾奎尔(alequel)、低分子量肝素、甲基脑啡肽(metenkefalin)、曲卡克肽(tridecactide)、HMPL-004、SB-012、奥沙拉秦(olsalazine)、巴柳氮、丙酰-L-肉毒碱、丁酸梭菌(Clostridium butyricum)、倍氯米松和乙酰吗喃(acemannan)。
用于制备化合物的通用合成方法
可以使用下文详述的通用合成方案和实验程序中说明的方法来制备本发明的化合物。通用合成方案和实验程序是出于说明的目的而被呈现,而不旨在成为限制性的。用于制备本发明的化合物的起始材料是可商业获得的,或者可以使用本领域已知的常规方法制备。下文方案1-4中概述了用于制备本发明的化合物的代表性程序。可以在Sigma-Aldrich或Fisher Scientific购买合成制备未在下文描述的溶剂和试剂。
方案1描绘了式(I)的化合物(其中R1、R2、R3、和R4如上文所定义,n是0、1或2,并且X是溴或碘)的一般合成。PG1是吲哚保护基团,如苯磺酰基、甲苯磺酰基、均三甲苯磺酰基、氨基甲酸叔丁酯(Boc)、烯丙基、苄基、三异丙基甲硅烷基(TIPS)、2-(三甲基甲硅烷基)乙氧基甲基(SEM)、或对甲氧基苄基。PG2是仲胺保护基团,如氨基甲酸叔丁酯(Boc)、氨基甲酸苄酯(Cbz)、苄基(Bn)、对甲氧基苄基(PMB)、或N-乙酰基(Ac)。按照本领域技术人员已知的方法,通过用适当的吲哚保护基团保护1a来形成化合物1b。在热条件下在溶剂(如N-甲基-2-吡咯烷酮(NMP))中在存在三乙胺的情况下胺1c与1b的偶联提供1d。可替代地,布赫瓦尔德条件可以用于1b与1c的偶联。通过在铃木条件下使用钯催化剂在存在碱(如碳酸铯)的情况下在二噁烷/水溶剂混合物中用芳基硼酸酯或杂芳基硼酸酯处理1d来形成芳基1e。在适当保护基团(PG2)的标准条件下1e的脱保护提供1f。在存在胺碱的情况下使用肽偶联条件(如EDC和HOBT或卤素-R4)的1f与式HOR4的化合物的偶联产生化合物1g。最后,1g的吲哚保护基团PG1的去除提供1h。可替代地,可以去除1e的两个保护基团,并且将中间产物与式HOR4的化合物偶联以产生化合物1h。
方案1:式(I)的吡咯并吡啶杂环-胺类似物的一般合成

方案2描绘了式(I)的化合物(其中R1、R2、R3、和R4如上文所定义,n是0、1或2,并且X是溴或碘)的一般合成。通过用适当的吲哚保护基团保护2a来形成化合物2b。在热条件下在溶剂(如NMP)中在存在三乙胺的情况下胺2c与2b的偶联提供2d。可替代地,布赫瓦尔德条件可以用于1b与1c的偶联。通过在铃木条件下使用钯催化剂在存在碱(如碳酸铯)的情况下在二噁烷/水溶剂混合物中用芳基硼酸酯或杂芳基硼酸酯处理2d来形成芳基2e。在适当保护基团(PG1)的标准条件下2e的脱保护提供2f。在一些情况下,可以通过本领域技术人员已知的方法修饰2d、2e或2f上的R3取代基,以提供不同的R3基团。
方案2:式(II)的吡咯并吡啶环烷基胺类似物的一般合成

方案3描绘了式(I)的化合物(其中R1、R2、R3、和R4如上文所定义,n是0、1或2,并且X是溴或碘)的一般合成。通过在存在钯催化剂和配体(如X-Phos)的情况下使1d与双(频哪醇合)二硼(B-Pin)反应来形成硼酸酯3a。通过在铃木条件下使用钯催化剂在存在碱(如碳酸铯)的情况下在二噁烷/水溶剂混合物中用芳基溴化物或杂芳基溴化物处理3a来形成芳基3b。在适当保护基团(PG2)的标准条件下3b的脱保护提供3c。在存在胺碱的情况下使用肽偶联条件(如EDC和HOBT或卤素-R4)的3c与式HOR4的化合物的偶联产生化合物3d。3d的吲哚保护基团PG1的去除提供3e。可替代地,可以去除3b的两个保护基团,并且将中间产物与式HOR4的化合物偶联以产生化合物3e。
方案3.通过反向铃木偶联式(I)的吡咯并吡啶杂环-胺类似物的一般合成

方案4描绘了式(I)的化合物(其中R1、R2、R3、和R4如上文所定义,n是0、1或2,并且X是溴或碘)的一般合成。通过在存在钯催化剂和配体(如X-Phos)的情况下使1d与B-Pin反应来形成硼酸酯4a。通过在铃木条件下使用钯催化剂在存在碱(如碳酸铯)的情况下在二噁烷/水溶剂混合物中用芳基溴化物或杂芳基溴化物处理4a来形成芳基4b。4b的吲哚保护基团PG1的去除提供4e。
方案4.通过反向铃木偶联的式(II)的吡咯并吡啶环烷基胺类似物的一般合成

通过下列进一步说明在实施例部分中详述的某些化合物的示例性合成方法。
实施例1:(R)-3-氧代-3-(3-((5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

方案5:(R)-3-氧代-3-(3-((5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

步骤1:5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶的制备

用氢化钠(1.65g,41.47mmol,矿物油中的60%悬浮液)处理0℃的5-溴-4-氯-1H-吡咯并[2,3-b]吡啶(6g,25.92mmol)在干二甲基甲酰胺(60mL)中的溶液,并且将悬浮液搅拌30分钟。然后,添加2-(三甲基甲硅烷基)乙氧基甲基氯(9.56mL,53.91mmol),并且将混合物在环境温度搅拌1.5小时。用饱和氯化铵溶液淬灭反应混合物,并且用乙酸乙酯提取。用盐水洗涤合并的有机提取物,在无水硫酸钠上干燥,过滤溶液,并且在真空中浓缩。通过柱色谱(乙酸乙酯/己烷)纯化粗材料,以提供为黄色油状物的5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(8.6g,92%产率):MS(ES)m/z361.0(M+H)。
步骤2:(R)-N-(1-苄基哌啶-3-基)-5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(3g,8.29mmol)和(R)-1-苄基哌啶-3-胺(3.15g,16.58mmol)的混合物在密封管中加热至180℃。在7小时之后,将反应冷却至室温,并且通过柱色谱(乙酸乙酯/己烷)纯化粗材料,以提供为黄色油状物的(R)-N-(1-苄基哌啶-3-基)-5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-胺(3.7g,45%产率):MS(ES)m/z 517.1(M+2H)。
步骤3:(R)-(5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)(哌啶-3-基)氨基甲酸乙酯的制备

在室温向(R)-N-(1-苄基哌啶-3-基)-5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-胺(3.7g,7.17mmol)在干1,4-二噁烷(50mL)中的经搅拌的溶液添加氯甲酸乙酯(1.03mL,10.76mmol),并且将所得的混合物在100℃加热。在15小时之后,将反应冷却至室温,并且在真空中浓缩以去除挥发物。通过柱色谱(乙酸乙酯/己烷)纯化粗材料,以提供为黄色油状物的(R)-(5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)(哌啶-3-基)氨基甲酸乙酯(2.7g,76%产率):MS(ES)m/z 496.9(M+H)。
步骤4:(R)-5-溴-N-(哌啶-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

向(R)-(5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)(哌啶-3-基)氨基甲酸乙酯(0.5g,1.0mmol)在甲醇:水(12mL:4mL)中的经搅拌的溶液添加氢氧化钾(2.81g,50.25mmol),并且使所得的混合物在110℃经受微波辐照2小时。将反应冷却至室温,并且用二氯甲烷稀释,并且用水洗涤。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为黄色半固体的(R)-5-溴-N-(哌啶-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-胺(2g,94%产率):MS(ES)m/z424.9(M+H)。
步骤5:(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向0℃的(R)-5-溴-N-(哌啶-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-胺(2g,4.70mmol)在干二氯甲烷(30mL)中的经搅拌的溶液添加4-二甲基氨基吡啶(0.11g,0.94mmol)和三乙胺(1.32mL,9.4mmol),然后添加二碳酸二叔丁酯(1.23mL,5.64mmol)。将所得的混合物在环境温度搅拌15小时。用二氯甲烷稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过柱色谱(乙酸乙酯/己烷)纯化粗材料,以提供为黄色油状物的(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.9g,77%产率):MS(ES)m/z 524.9(M+H)。
步骤6:(R)-3-((5-(噻唑-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.5g,0.94mmol)、4-(三丁基甲锡烷基)噻唑(0.46g,1.23mmol)和双(三苯基膦)二氯化钯(II)(0.13g,0.18mmol)在N-甲基-2-吡咯烷酮(10mL)中的溶液在密封管中在氮气气氛下在130℃加热。在4小时之后,将反应冷却至环境温度,用乙酸乙酯稀释,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为粉色胶状固体的(R)-3-((5-(噻唑-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.22g,44%产率):MS(ES)m/z 529.9(M+H)。
步骤7:(R)-N-(哌啶-3-基)-5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(噻唑-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.21g,0.39mmol)在二氯甲烷:三氟乙酸(3.0mL:3.0mL)中的溶液在环境温度搅拌2小时。在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(3.0mL:3.0mL)中,并且然后在环境温度搅拌过夜。在真空中浓缩反应混合物,以提供为灰白色固体的(R)-N-(哌啶-3-基)-5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.08g,66%产率):MS(ES)m/z 300.0(M+H)。
步骤8:(R)-3-氧代-3-(3-((5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

将氰基乙酸(0.024g,0.28mmol)、N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.07g,0.34mmol)、1-羟基苯并三唑(0.04g,0.26mmol)和N,N-二异丙基乙胺(0.13mL,0.68mmol)在二氯甲烷(4mL)中的溶液在环境温度搅拌5分钟。然后,添加(R)-N-(哌啶-3-基)-5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.07g,0.23mmol),并且将所得的混合物在环境温度搅拌18小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-氧代-3-(3-((5-(噻唑-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.01g,12%产率):1H NMR(400MHz,DMSO-d6)δ11.37(brs,1H),9.20(br s,1H),8.30-8.57(m,1H),7.96(d,J=14.8Hz,1H),7.16(br s,1H),6.60(s,1H),3.92-4.48(m,3H),3.55-3.70(m,1H),3.05-3.27(m,3H),1.92-2.33(m,2H),1.46-1.75(m,3H);MS(ES)m/z 366.9(M+H)。
实施例2:(R)-3-(3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案6:(R)-3-(3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.4g,0.76mmol,如通过方案5、步骤-5中描述的方法制备)、1-(二氟甲基)-3-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡唑(0.24g,0.10mmol)、双(三苯基磷)二氯化钯(II)(0.11g,0.15mmol)和2M磷酸三钾(0.48g,2.28mmol)水溶液在1,4-二噁烷(20mL)中的混合物在氮气气氛下在80℃加热过夜。将反应混合物冷却至环境温度,用乙酸乙酯稀释,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(己烷中50%乙酸乙酯)纯化粗材料,以提供为橙色胶状固体的(R)-3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.34g,80%产率):MS(ES)m/z562.9(M+H)。
步骤2:(R)-5-(1-(二氟甲基)-1H-吡唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

通过实施例1、步骤7中描述的方法制备(R)-5-(1-(二氟甲基)-1H-吡唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺,并且分离为浅黄色胶状物(0.23g,98%):MS(ES)m/z 333.0(M+H)+。
步骤3:(R)-3-(3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例1、步骤8中描述的方法制备(R)-3-(3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生灰白色固体(0.05g,18%产率):1H NMR(400MHz,DMSO-d6)δ11.46(br s,1H),8.41-8.46(m,2H),8.28-8.31(m,1H),7.60-7.91(m,1H),7.10-7.20(m,2H),6.65(s,1H),4.40(s,1H),3.91-4.18(m,3H),3.43-3.68(m,2H),3.18-3.26(m,1H),2.06-2.07(m,1H),1.57-1.77(m,3H);MS(ES)m/z399.9(M+H)。
分析条件:
柱:Kinetex C18(100mm×4.6mm×2.6μm)
流动相(A):水中0.1%三氟乙酸
流动相(B):ACN
流速:0.75mL/min
实施例3:(R)-3-(3-((5-(2-甲基噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案7:(R)-3-(3-((5-(2-甲基噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(2-甲基噻唑-5-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

用碳酸钾(0.16g,0.76mmol)处理(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.2g,0.38mmol,如通过方案-1、步骤-5中描述的方法制备)在1,4-二噁烷:水(4.5mL:0.5mL)中的溶液,然后用2-甲基-5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)噻唑(0.17g,0.76mmol)和[1,1’-双(二苯基膦)二茂铁]二氯化钯(II)二氯甲烷络合物(0.03g,0.04mmol)处理。将所得的混合物在氮气气氛下在密封管中在100℃加热16小时。将反应冷却至环境温度,用乙酸乙酯和水稀释。分离有机层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为棕色液体的(R)-3-((5-(2-甲基噻唑-5-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.15g,36%产率):MS(ES)m/z 543.9(M+H)。
步骤2:(R)-5-(2-甲基噻唑-5-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

通过实施例-1、步骤-7中描述的方法制备(R)-5-(2-甲基噻唑-5-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺,其被分离为浅黄色胶状物(0.13g,94%产率):MS(ES)m/z 314.0(M+H)+。
步骤3:(R)-3-(3-((5-(2-甲基噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例1、步骤-8中描述的方法制备(R)-3-(3-((5-(2-甲基噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生灰白色固体(0.01g,5%产率):1H NMR(400MHz,DMSO-d6)δ11.43(s,1H),7.77-7.75(m,1H),7.59-7.57(m,1H),7.22(s,1H),6.61-6.57(m,1H),5.16-5.14(m,1H),4.16-4.13(m,1H),4.03(s,2H),3.89-3.77(m,3H),2.67-2.65(m,3H),1.96(s,2H),1.60-1.57(m,3H);MS(ES)m/z 381.3(M+H)。HPLC纯度:98.58%,
分析条件:
柱:X Bridge(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):甲醇
流速:1.0mL/min
实施例4:(R)-3-氧代-3-(3-((5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

方案8:(R)-3-氧代-3-(3-((5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

步骤1:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑的制备

将5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(1.5g,4.15mmol,通过方案1、步骤-1中描述的方法制备)、2-(三丁基甲锡烷基)噻唑(1.7g,4.57mmol)和双(三苯基膦)二氯化钯(II)(0.58g,0.83mmol)在N-甲基-2-吡咯烷酮中的溶液在氮气气氛下在密封管中在130℃加热。在4小时之后,将反应混合物冷却至环境温度,用乙酸乙酯稀释,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(己烷中10%乙酸乙酯)纯化粗材料,以提供为无色油状物的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑(0.75g,49%产率):MS(ES)m/z 365.8(M+H)。
步骤2:(R)-3-((5-(噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

使2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑(0.7g,1.92mmol)、(R)-3-氨基哌啶-1-羧酸叔丁酯(0.57g,2.87mmol)和三乙胺(0.27mL,1.92mmol)在N-甲基-2-吡咯烷酮(10mL)中的溶液在130℃经受微波辐照3小时。将反应冷却至环境温度,用乙酸乙酯稀释,用水、盐水洗涤,经无水硫酸钠干燥,过滤并且在真空中浓缩。通过使用快速色谱(己烷中50%乙酸乙酯)纯化粗材料,以提供为浅黄色固体的(R)-3-((5-(噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.24g,24%产率):MS(ES)m/z 529.9(M+H)。
步骤3:(R)-N-(哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

通过实施例1、步骤-7中描述的方法制备(R)-N-(哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺,其被分离为灰白色固体(0.11g,81%产率):MS(ES)m/z 300.0(M+H)+。
步骤4:(R)-3-氧代-3-(3-((5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

通过实施例1、步骤-8中描述的方法制备(R)-3-氧代-3-(3-((5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈,以产生黄色固体(0.05g,38%产率):1HNMR(400MHz,DMSO-d6)δ11.44(br s,1H),9.75(br s,1H),7.41(d,J=10.4Hz,1H),7.74-7.80(m,1H),7.59(s,1H),7.22(s,1H),6.69(s,1H),4.15-4.45(m,1H),3.90-4.10(m,2H),3.61-3.82(m,1H),3.25-3.55(m,2H),1.95-2.20(m,2H),1.45-1.85(m,3H);MS(ES)m/z366.9(M+H)。
实施例5:(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案9:(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑的制备

将5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(2g,5.54mmol,通过方案5、步骤-1中描述的方法制备)、叔丁醇钾(1.24g,11.08mmol)、双(三苯基膦)二氯化钯(II)(0.19g,0.27mmol)和噁唑(0.68g,9.97mmol)在1,4-二噁烷(20mL)中的混合物在氮气下在密封管中加热至110℃。在5小时之后,将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.43g,22%产率):MS(ES)m/z 349.9(M+H)。
步骤2:(R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

使2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.43g,1.28mmol)、三乙胺(0.03mL,将0.24mmol)和(R)-3-氨基哌啶-1-羧酸叔丁酯(0.464g,2.32mmol)在N-甲基吡咯烷酮(4mL)中的经搅拌的溶液在130℃经受微波辐照5小时。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.35g,53%):MS(ES)m/z 514.0(M+H)。
步骤3:(R)-5-(噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

通过实施例1、步骤-7中描述的方法制备(R)-5-(噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺:MS(ES)m/z 284.1(M+H)。
步骤4:(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例1、步骤-8中描述的方法制备(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生灰白色固体(0.05g,27%产率):1HNMR(400MHz,DMSO-d6)δ11.61(br s,J=8.8Hz,1H),9.11-9.05(m,1H),8.53(d,J=6.8Hz,1H),8.10(s,1H),7.33-7.22(m,2H),6.69(s,1H),4.37(br s,1H),4.17-3.58(m,6H),2.04(s,1H),1.73-1.54(m,3H);MS(ES)m/z 351.1(M+H)。
实施例6:(R)-3-(3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案10:(R)-3-(3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯的制备

使4-氯-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(0.2g,0.89mmol)、三乙胺(0.06mL,0.44mmol)和(R)-3-氨基哌啶-1-羧酸苄酯(0.31g,1.33mmol)在N-甲基吡咯烷酮(1mL)中的经搅拌的溶液在165℃经受微波辐照2小时。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以产生为浅黄色固的(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯:MS(ES)m/z 422.9[M+H]+。
步骤2:(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸的制备

向0℃的(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(1.5g,3.55mmol)在乙醇(20mL)中的经搅拌的溶液添加氢氧化钠(5N,10mL),并且将混合物在环境温度搅拌12小时。在真空中浓缩反应混合物,并且将剩余物溶解在水中,并且用二乙醚洗涤。在0℃用饱和硫酸氢钾溶液将水性层酸化至pH~2.0,并且然后用5%甲醇/二氯甲烷提取。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。用正戊烷研磨获得的粗产物,以提供为灰白色固体的(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(1.10g,粗品):MS(ES)m/z 394.9(M+H)。
步骤3:(R,E)-3-((5-((1-氨基亚乙基)氨基甲酰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

向(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.5g,1.26mmol)在N,N-二甲基甲酰胺(10mL)中的经搅拌的溶液添加1-双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.53g,1.39mmol)、N,N-二异丙基乙胺(1.2g,1.73mmol)和盐酸乙脒(0.178g,1.90mmol)。将混合物在环境温度搅拌18小时。过滤反应混合物,并且在真空中浓缩以提供为白色固体的(R,E)-3-((5-((1-氨基亚乙基)氨基甲酰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.4g,粗品):MS(ES)m/z 435.0(M+H)。
步骤4:(R)-3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将(R,E)-3-((5-((1-氨基亚乙基)氨基甲酰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.1g,0.23mmol)和盐酸羟胺(0.024g,0.345mmol)在乙酸(0.2mL)中的经搅拌的溶液加热至90℃。在3小时之后,将反应混合物冷却至环境温度,用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为白色固体的(R)-3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.01g,10%产率):MS(ES)m/z 433.3(M+H)。
步骤5:(R)-5-(3-甲基-1,2,4-噁二唑-5-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺盐酸盐的制备

将(R)-3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.34g,0.78mmol)和浓HCl(3mL)的经搅拌的溶液在密封管中在70℃加热。在3小时之后,将反应冷却至室温,并且在真空中浓缩,以提供为灰白色固体的((R)-5-(3-甲基-1,2,4-噁二唑-5-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.25g,粗品):MS(ES)m/z 299.0(M+H)。
步骤6:(R)-3-(3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向((R)-5-(3-甲基-1,2,4-噁二唑-5-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.25g,0.83mmol)在N,N-二甲基甲酰胺(0.5mL)中的经搅拌的溶液添加1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.34g,0.91mmol)、N,N-二异丙基乙胺(1.2g,1.73mmol)和氰基乙酸(0.118g,1.25mmol)。将混合物在环境温度搅拌18小时。在真空中浓缩反应混合物,并且通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(3-甲基-1,2,4-噁二唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.04g,13%产率):1H NMR(400MHz,DMSO-d6)δ11.82(br s,1H),8.71-8.76(m,1H),8.56-8.57(m,1H),7.28(s,1H),6.74(s,1H),4.50(br s,1H),4.28(br s,1H),4.03-4.14(m,1H),3.82-3.91(m,1H),3.57-3.60(m,1H),3.39(m,2H),3.29(m,1H),2.39-2.41(m,3H),2.04(m,1H),1.56-1.78(m,2H);MS(ES)m/z 365.9(M+H)。
实施例7:(R)-3-(3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案11:(R)-3-(3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(肼羰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(0.5g,1.18mmol,通过实施例6、步骤-1中描述的方法制备)和水合肼(32mL)在乙醇(8mL)中的经搅拌的溶液在110℃加热18小时。在冷却至室温之后,在真空中浓缩混合物以去除挥发物,并且用二乙醚中10%甲醇研磨剩余物以提供为黄色固体的(R)-3-((5-(肼羰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.3g,62%产率):MS(ES)m/z409.2(M+H)。
步骤2:(R)-3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将(R)-3-((5-(肼羰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.1g,0.24mmol)和原甲酸三乙酯(2mL,12.0mmol)在乙醇(8mL)中的经搅拌的溶液在密封管中加热至120℃。在18小时之后,将反应冷却至室温,并且在真空中浓缩混合物以去除挥发物。通过柱色谱(甲醇/二氯甲烷)纯化粗材料,以提供为浅黄色固体的(R)-3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.16g,40%产率):MS(ES)m/z 419.2(M+H)。
步骤3:(R)-5-(1,3,4-噁二唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

向(R)-3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.16g,0.38mmol)在四氢呋喃:乙醇(8mL:5mL)中的溶液添加钯碳(10%)(0.03g),并且将非均相混合物在氢气气氛下在环境温度搅拌32小时。通过硅藻土过滤反应混合物,并且用甲醇洗涤。在真空中浓缩滤液以提供粗材料,用二乙醚研磨粗材料以提供为黄色固体的(R)-5-(1,3,4-噁二唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.05g,52%产率):MS(ES)m/z 285.2(M+H)。
步骤4:(R)-3-(3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例1、步骤-8中描述的方法制备(R)-3-(3-((5-(1,3,4-噁二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生浅棕色固体(0.01g,14%产率):1H NMR(400MHz,DMSO-d6)δ11.77(s,1H),9.24(s,1H),8.40-8.47(m,2H),7.28(s,1H),6.75(s,1H),4.20(d,J=10.0Hz,1H),4.04(q,J1=18.8Hz,J2=18.4Hz,2H),3.73-3.84(m,2H),3.47(d,J=10.0Hz,1H),3.14(d,J=4.8Hz,1H),2.11(bs,1H),1.98(t,J=8.0Hz,1H),1.70-1.75(m,2H);MS(ES)m/z 352.0(M+H)。
分析条件:
柱:Inertsil ODS 3V(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):ACN
流速:1.0mL/min
B的组成:0/10、12/70、25/90、27/10、30/10
实施例8:(R)-3-(3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案12:(R)-3-(3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯的制备

向0℃的4-氯-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(5g,22.2mol)在干二甲基甲酰胺(20mL)中的经搅拌的溶液添加氢化钠(1.42g,35.6mol,矿物油中的60%悬浮液),并且将悬浮液搅拌30分钟。然后,添加2-(三甲基甲硅烷基)乙氧基甲基氯(7.88mL,44.5mol),并且将所得的混合物在环境温度搅拌1.5小时。用饱和氯化铵溶液淬灭反应混合物,并且用乙酸乙酯提取。用盐水洗涤合并的有机提取物,在无水硫酸钠上干燥,过滤溶液,并且在真空中浓缩。通过柱色谱(乙酸乙酯/己烷)纯化粗材料,以提供为黄色油状物的4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(5.5g,70%产率):MS(ES)m/z 354.9(M+H)。
步骤2:(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯的制备

在密封管中,向4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(2.0g,5.64mmol)在N-甲基-2-吡咯烷酮(20mL)中的溶液添加(R)-3-氨基哌啶-1-羧酸叔丁酯(1.7g,2.11mmol),然后添加三乙胺(0.8mL)。将混合物加热3小时至135℃。在冷却至环境温度之后,用水淬灭反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(17%乙酸乙酯/己烷)纯化粗材料,以提供为无色油状物的(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(1.0g,35%产率):MS(ES)m/z 519.0[M+H]+。
步骤3:(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸的制备

在环境温度向(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(1.0g,1.93mmol)在甲醇(3mL)、四氢呋喃:水(3mL:3mL)中的经搅拌的溶液添加氢氧化钠(0.15g,3.85mmol)。将混合物在60℃搅拌8小时。将反应冷却至环境温度,并且在真空中浓缩以去除挥发物。将剩余物溶解在水中,并且用1N HCl酸化,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。用正戊烷研磨粗材料以提供为灰白色固体的(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.8g,84%产率):MS(ES)m/z 490.9[M+H]+。
步骤4:(R)-3-((5-(2-甲酰肼-1-羰基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.4g,0.752mmol)在二甲基甲酰胺(5mL)中的溶液添加甲酸肼(0.07g,1.13mmol)、1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.15g,0.37mmol)和N,N-二异丙基乙胺(0.07mL,0.376mmol),并且将混合物在环境温度搅拌3小时。用水淬灭反应,并且用乙酸乙酯提取。用碳酸氢钠溶液、盐水洗涤有机层,在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩,以提供为浅黄色液体的(R)-3-((5-(2-甲酰肼-1-羰基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.4g,90%产率):MS(ES)m/z 533.0[M+H]+。
步骤5:(R)-3-((5-(1,3,4-噻二唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

在环境温度向(R)-3-((5-(2-甲酰肼-1-羰基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.37g,0.695mmol)在四氢呋喃(20mL)中的溶液添加劳森试剂(0.84g,2.08mmol),并且将混合物在70℃搅拌16小时。将反应冷却至环境温度,用10%柠檬酸淬灭,并且将溶液搅拌5分钟,然后添加碳酸氢钠溶液,并且搅拌另外的10分钟。用乙酸乙酯提取水性层,用饱和碳酸氢钠和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中干燥。通过快速色谱(35%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色液体的(R)-3-((5-(1,3,4-噻二唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.28g,78%产率):MS(ES)m/z 530.9[M+H]+。
步骤6:(R)-N-(哌啶-3-基)-5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

通过实施例1、步骤-7中描述的方法制备(R)-N-(哌啶-3-基)-5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺,以产生黄色液体(0.2g,%产率):MS(ES)m/z 301.2[M+H]+。
步骤7:(R)-3-(3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例1、步骤-8中描述的方法制备(R)-3-(3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生灰白色固体(0.05g,20%产率):1H NMR(400MHz,DMSO-d6)δ11.71(s,1H),9.4-9.5(m,2H),8.35(d,J=6.4Hz,1H),7.25(s,1H),6.78(s,1H),4.39(s,1H),3.60-4.21(m,6H),2.09(s,1H),1.65-1.74(m,3H);MS(ES)m/z351.3(M+H)。
实施例9和实施例10:3-((3R,5S)-3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈和3-((3S,5R)-3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过下述方法制备3-氨基-5-甲基哌啶-1-羧酸苄酯盐酸盐。

步骤1:(5-甲基哌啶-3-基)氨基甲酸叔丁酯的制备

在Parr振动器器皿中,在氮气气氛下向(5-甲基吡啶-3-基)氨基甲酸叔丁酯(0.8g)在乙酸(30mL)中的经搅拌的溶液添加二氧化铂(0.2g)和铑碳(0.2g)。将反应混合物在环境温度在氢气(100psi)下搅拌24小时。通过硅藻土过滤混合物,并且在真空中浓缩过滤,以提供为玻璃态液体的(5-甲基哌啶-3-基)氨基甲酸叔丁酯(0.74g,90%产率):MS(ES)m/z 215.1[M+H]+。
步骤2:3-((叔丁氧基羰基)氨基)-5-甲基哌啶-1-羧酸苄酯的制备

在0℃向(5-甲基哌啶-3-基)氨基甲酸叔丁酯(1.5g,6.99mmol)在四氢呋喃(12mL)和水(3mL)中的经搅拌的溶液添加氢氧化钠(1.4g,34.99mmol)和氯甲酸苄酯(2.4mL,13.99)。将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用盐水洗涤,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(23%乙酸乙酯/己烷)纯化粗材料,以提供为白色固体的3-((叔丁氧基羰基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.8g,33%产率):MS(ES)m/z 293.2[M-56]+。
步骤3:3-氨基-5-甲基哌啶-1-羧酸苄酯盐酸盐的制备

在0℃向3-((叔丁氧基羰基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.8g)在二氯甲烷(20mL)中的经搅拌的溶液添加1,4-二噁烷中的4M盐酸(8mL)。将反应混合物在环境温度搅拌3小时,然后在真空中浓缩,以提供为白色固体的3-氨基-5-甲基哌啶-1-羧酸苄酯盐酸盐(0.54g,95%产率):MS(ES)m/z 249.0[M+H]+。
方案13:3-((3S,5R)-3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈和3-((3S,5S)-3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:3-甲基-5-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

使2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.35g,1.00mmol,通过实施例5、步骤-1中描述的方法制备)、三乙胺(0.4mL,3.00mmol)、3-氨基-5-甲基哌啶-1-羧酸苄酯盐酸盐(0.43g,1.50mmol)在N-甲基吡咯烷酮(10mL)中的经搅拌的溶液在135℃经受微波辐照5小时。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(21%乙酸乙酯/己烷)纯化粗材料以提供为深棕色液体的3-甲基-5-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.13g,23%):MS(ES)m/z=561.9[M+H]+。
步骤2:N-(5-甲基哌啶-3-基)-5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将3-甲基-5-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.13g,0.23mmol)在三氟乙酸(3.0mL)中的溶液在100℃搅拌1小时。在冷却之后,在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(5.0mL:5.0mL)中,并且然后在环境温度搅拌过夜。在真空中浓缩反应混合物,以提供为浓稠液体的N-(5-甲基哌啶-3-基)-5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.08g,粗品):MS(ES)m/z 298.0[M+H]+。
步骤3:3-(3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

在0℃向N-(5-甲基哌啶-3-基)-5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.07g,0.235mmol)在二氯甲烷(10mL)中的溶液添加三乙胺(0.1mL,0.706mmol)、丙基膦酸酐(0.4mL,0.706mmol)和氰基乙酸(0.03g,0.353mmol)。将反应混合物在环境温度搅拌3小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(70%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的外消旋3-(3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.025g,31%产率):MS(ES)m/z 351.1[M+H]+。
通过手性制备型HPLC进一步纯化外消旋化合物,以分离对映异构体。
手性HPLC
分析条件:
柱:CHIRALPAC IC(10mm×4.6mm×3mic)
流动相(A):正己烷:具有0.1%DEA的乙醇(80:20)
流速:1.0mL/min
实施例9:3-((3R,5S)-3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(已经临时指定立体化学)

分离为白色固体(0.028g,11%产率):1H NMR(400MHz,CDCl3)δ11.84(s,1H),10.37-10.35(m,1H),8.56(m,2H),7.75(s,1H),6.975(m,2H),5.12(d,J=12.4Hz,1H),4.2(br s,1H),3.75-2.00(m,7H),1.49(s,1H),0.96(d,3H);MS(ES)m/z=365.2[M+H]+;HPLC纯度:99.45%。保留时间11.4min。
实施例10:3-((3S,5R)-3-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(已经临时指定立体化学)

分离为白色固体(0.003g,13%产率):1H NMR(400MHz,CDCl3)δ11.86(s,1H),10.37-10.35(m,1H),8.56(m,2H),7.75(s,1H),6.98(m,2H),5.12(d,J=12.4Hz,1H),4.18(br s,1H),3.8-2.20(m,7H),2.00(s,1H),0.9(d,3H);MS(ES)m/z=365.1[M+H]+;HPLC纯度:98.16%。保留时间16.22min。
实施例11:(R)-3-(3-((5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案14:(R)-3-(3-((5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-氨基甲酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

向(R)-4-((1-((苄氧基)羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.1g,0.25mmol,由实施例9、步骤-1中描述的方法制备)在N,N-二甲基甲酰胺(1mL)中的经搅拌的溶液添加1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.19g,0.508mmol)、三乙胺(0.08g,0.76mmol)和氯化铵(0.07g,1.27mmol)。将混合物在环境温度搅拌18小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为灰白色固体的(R)-3-((5-氨基甲酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.1g,粗品):MS(ES)m/z394.1(M+H)。
步骤2:(R,E)-3-((5-(((二甲基氨基)亚甲基)氨基甲酰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将(R)-3-((5-氨基甲酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.3g,0.76mmol)、N,N-二甲基甲酰胺二甲基缩醛(5mL)和N,N-二甲基甲酰胺(0.3mL)的经搅拌的溶液在90℃加热3小时。在真空中浓缩反应混合物,以提供为灰白色固体的(R,E)-3-((5-(((二甲基氨基)亚甲基)氨基甲酰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.25g,粗品):MS(ES)m/z 448.9(M+H)。
步骤3:(R)-3-((5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将(R,E)-3-((5-(((二甲基氨基)亚甲基)氨基甲酰基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.25g,0.55mmol)和水合肼(1.2mL)在乙酸(2.5mL)中的经搅拌的溶液在90℃搅拌。在3小时之后,将反应冷却至环境温度,用水淬灭,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-((5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.25g,粗品):MS(ES)m/z417.9(M+H)。
步骤4:(R)-N-(哌啶-3-基)-5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺盐酸盐的制备

通过实施例6、步骤5中描述的方法(提供灰白色固体)制备(R)-N-(哌啶-3-基)-5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺盐酸盐(0.15g,98%产率):MS(ES)m/z 284.0(M+H)+。
步骤5:(R)-3-(3-((5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例9、步骤5中描述的方法制备(R)-3-(3-((5-(4H-1,2,4-三唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生灰白色固体(0.09g,7.5%产率):1H NMR(400MHz,DMSO-d6)14.13(br s,1H),11.49(br s,1H),9.2(br s,1H),8.58-8.61(br s,2H),7.2(s,1H),6.66(s,1H),4.35(br s,1H),3.96-4.12(m,3H),3.32-3.69(m,2H),2.06(m,1H),1.53-1.76(m,3H),1.21(s,1H);MS(ES)m/z 351.2(M+H)。
实施例12:(1S,3R)-3-((5-(吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

通过下列方法制备(1S,3R)-3-氨基环戊烷-1-醇盐酸盐。

用1,4-二噁烷(2.5mL)中(4M)盐酸处理((1R,3S)-3-羟基环戊基)氨基甲酸叔丁酯(0.5g,2.48mmol)在1,4-二噁烷(1mL)中的溶液,并且在氮气气氛下在环境温度搅拌12小时。在真空中浓缩反应混合物,以提供为灰白色固体的(1S,3R)-3-氨基环戊烷-1-醇盐酸盐(0.25g,粗品):MS(ES)m/z 102.1(M+H)。
方案15:(1S,3R)-3-((5-(吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

步骤1:5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶的制备

向5-溴-4-氯-1H-吡咯并[2,3-b]吡啶(5g,21.7mmol)在二氯甲烷(40mL)中的溶液添加4-二甲基氨基吡啶(0.26g,2.17mmol)、对甲苯磺酰氯(5.3g,28.2mmol)和三乙胺(4.3g,43.4mmol),并且将混合物在室温搅拌12小时。用水淬灭反应,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为灰白色固体的5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(7g,96%产率):MS(ES)m/z 384.7(M+H)。
步骤2:(1S,3R)-3-((5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

向5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(0.1g,0.260mmol)在N-甲基-2-吡咯烷酮(0.6mL)中的经搅拌的溶液添加(1S,3R)-3-氨基环戊烷-1-醇盐酸盐(0.03g,0.33mmol)和三乙胺(0.1mL),并且使混合物在150℃经受微波辐照2.5小时。将反应冷却至室温,用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为无色油状物的(1S,3R)-3-((5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.15g,粗品):MS(ES)m/z 449.0(M+H)。
步骤3:(1S,3R)-3-((5-(吡啶-2-基)-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

用氮气使(1S,3R)-3-((5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.3g,0.66mmol)、2-(三丁基甲锡烷基)吡啶(0.32g,0.86mmol)、氯化锂(0.002g,0.06mmol)、碘化铜(0.01g,0.06mmol)和四(三苯基膦)铂(0)(0.03g,0.033mmol)在N,N-二甲基甲酰胺(0.5mL)中的混合物脱气5分钟。将反应混合物在密封管中在120℃加热2小时。将反应混合物冷却至环境温度,并且通过硅藻土过滤。用乙酸乙酯稀释滤液,并且用水洗涤。用盐水洗涤有机相,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗产物,以提供为灰白色固体的(1S,3R)-3-((5-(吡啶-2-基)-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.17g,56%产率):MS(ES)m/z449.3(M+H)。
步骤4:(1S,3R)-3-((5-(吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

向(1S,3R)-3-((5-(吡啶-2-基)-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.17g,0.38mmol)在N-甲基-2-吡咯烷酮(1.5mL)中的溶液添加叔丁醇钾(0.09g,0.83mmol),并且将混合物在室温搅拌3小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(1S,3R)-3-((5-(吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.02g,10%产率):1H NMR(400MHz,DMSO-d6)11.28(br s,1H),9.99(d,J=8.0Hz,1H),8.52-8.54(m,1H),8.31(m,1H),7.81-7.82(m,2H),7.20-7.22(m,1H),7.09(s,1H),6.60(s,1H),4.62(s,1H),4.59-4.61(m,1H),4.18-4.19(m,1H),2.24-2.31(m,1H),1.95-2.04(m,1H),1.62-1.83(m,2H),1.48-1.52(m,1H),1.22(s,1H);MS(ES)m/z 295.0(M+H)。
实施例13:(1S,3R)-3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

方案16:(1S,3R)-3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

步骤1:(1S,3R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

向2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.2g,0.573mmol,如通过实施例5、步骤-1中描述的方法制备)在N-甲基-2-吡咯烷酮(0.5mL)中的经搅拌的溶液添加(1S,3R)-3-氨基环戊烷-1-醇盐酸盐(0.09g,0.86mmol)和三乙胺(0.1mL),并且使混合物在140℃经受微波辐照6小时。将反应混合物冷却至室温,用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为无色油状物的(1S,3R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.19g,80%产率):MS(ES)m/z 415.0(M+H)。
步骤2:(1S,3R)-3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇的制备

将(1S,3R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.19g,0.46mmol)在二氯甲烷:三氟乙酸(2mL:2mL)中的溶液在环境温度搅拌3小时。在真空中去除挥发物,并且将剩余物溶解在二噁烷:氨水(5mL:5mL)中,并且将混合物在环境温度搅拌12小时。用水淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为灰白色固体的(1S,3R)-3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)环戊烷-1-醇(0.06g,50%产率):1H NMR(400MHz,DMSO-d6)11.52(br s,1H),9.13(d,J=8.0Hz,1H),8.50(s,1H),8.08(s,1H),7.36(s,1H),7.16(s,1H),6.65(s,1H),4.65-4.66(m,1H),4.52(m,1H),4.18-4.19(m,1H),2.31-2.48(m,1H),2.09-2.18(m,1H),1.66-1.81(m,3H),1.48-1.53(m,1H);MS(ES)m/z 285.3(M+H)。
实施例14:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

方案17:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

步骤1:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯的制备

在室温向(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.8g,1.522mmol,如通过方案1、步骤5中描述的方法制备)在1,4-二噁烷(8mL)中的经搅拌的溶液添加噁唑-4-羧酸乙酯(0.03g,1.83mmol)和碳酸铯(0.99g,3.044mmol),并且用氮气使溶液脱气15分钟。添加乙酸钯(0.017g,0.0761mmol)和CyJohnphos(0.053g,0.1522mmol),并且将所得的混合物在密封管中加热16小时至110℃。将反应冷却至环境温度,通过硅藻土过滤,并且在真空中浓缩滤液。通过柱色谱(乙酸乙酯/己烷)纯化粗品,以提供为黄色固体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.35g,39%产率):MS(ES)m/z 585.9(M+H)。
步骤2:(R)-3-((5-(4-氨基甲酰基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)-乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.35g,0.597mmol)和23%氨水(16mL)在甲醇(5mL)中的溶液在密封管中搅拌40小时。在真空中浓缩反应混合物,并且用二氯甲烷提取水性部分。用盐水洗涤有机提取物,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过柱色谱(乙酸乙酯/己烷)纯化粗品,以提供为黄色固体的(R)-3-((5-(4-氨基甲酰基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,36%产率):MS(ES)m/z 556.9(M+H)。
步骤3:(R)-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

通过实施例1、步骤-7中描述的方法制备(R)-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺,以产生标题化合物(0.1g粗品):MS(ES)m/z 327.0(M+H)。
步骤4:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

通过实施例9、步骤-5中描述的方法制备(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺,以产生浅棕色固体(0.01g,13%产率):1H NMR(400MHz,DMSO-d6)δ11.66(d,J=12.0Hz,1H),8.71(d,J=8.4Hz,1H),8.52-8.53(m,2H),7.56-7.67(m,2H),7.24(s,1H),6.72(s,1H),4.14-4.29(m,2H),4.07(s,1H),3.77-3.94(m,2H),3.52(d,J=14.4Hz,1H),3.00-3.25(m,1H),2.08(bs,1H),1.85(bs,2H),1.50-1.70(m,1H);MS(ES)m/z 394.1(M+H)。
分析条件:
柱:X Bridge C18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):ACN
流速:1.0mL/min
B的组成:0/10、12/60、25/90、27/10、30/10
实施例15:(R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案18:(R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,0.55mmol,通过方案23、步骤-4中描述的方法制备)在N,N-二甲基甲酰胺(5mL)中的溶液添加氯化锂(0.048g,1.1mmol)、4-甲基-2-(三丁基甲锡烷基)吡啶(0.25g,0.66mmol)、碘化铜(0.011g,0.05mmol)和四(三苯基膦)钯(0)(0.03g,0.02mmol)。将混合物在氮气气氛下在密封管中在120℃搅拌3小时。将反应冷却至环境温度,用乙酸乙酯和水稀释。分离有机层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.02g,8%产率):1H NMR(400MHz,DMSO-d6)δ11.42-11.37(m,1H),10.13-10.02(m,1H),8.42-8.32(m,2H),7.75-7.72(m,1H),7.15-7.08(m,2H),6.61(s,1H),4.38(m,1H),4.12-3.94(m,3H),3.73-3.55(m,3H),2.31(s,3H),1.97(s,1H),1.69-1.52(s,3H);MS(ES)m/z 375.1(M+H)。HPLC纯度:99.38%。
分析条件:
柱:Inertsil ODS 3V(150mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):ACN
流速:1.0mL/min
实施例16:(R)-3-氧代-3-(3-((5-(噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

方案19:(R)-3-氧代-3-(3-((5-(噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

将(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,0.55mmol,通过方案23、步骤4中描述的方法制备)、5-(三丁基甲锡烷基)噻唑(0.24g,0.66mmol)和双(三苯基膦)二氯化钯(II)(0.09g,0.027mmol)在乙腈(5mL)中的溶液在密封管中加热15小时至80℃。将反应混合物冷却至环境温度,并且用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-氧代-3-(3-((5-(噻唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.04g,22%产率):1H NMR(400MHz,DMSO-d6,在70℃)δ11.29(s,1H),9.10(s,1H),7.86(s,1H),7.79(s,1H),7.20(s,1H),6.58(s,1H),5.04(s,1H),4.13(br s,1H),3.82-3.99(m,3H),3.53-3.65(m,1H),3.35-3.44(m,1H),1.94(br s,1H),1.40-1.65(m,3H),1.29-1.30(m,1H);MS(ES)m/z 367.2(M+1)。
实施例17:(R)-3-(3-((5-(1-甲基-1H-咪唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案20:(R)-3-(3-((5-(1-甲基-1H-咪唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈

使(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.3g,0.82mmol,通过方案23、步骤4中描述的方法制备)、1-甲基-2-(三丁基甲锡烷基)-1H-咪唑(0.46g,1.24mmol)和四(三苯基膦)钯(0)(0.09g,0.08mmol)在1,4-二噁烷(4mL)中的溶液在140℃经受微波辐照2小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(1-甲基-1H-咪唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.04g,15%产率):1HNMR(400MHz,DMSO-d6,在70℃)δ11.41(s,1H),7.72-7.74(m,1H),7.63-7.64(m,1H),7.21(s,1H),6.88(s,1H),6.59-6.64(m,1H),4.85-4.99(m,2H),3.79-4.20(m,5H),2.68-3.07(m,3H),1.91(bs,2H),1.49-1.56(m,3H);MS(ES)m/z 364.1(M+1)。
实施例18:(R)-3-(3-((5-(1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案21:(R)-3-(3-((5-(1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

使(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,0.55mmol,通过方案23、步骤4中描述的方法制备)、5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡唑(0.16g,0.83mmol)、碳酸钠(0.17g,1.65mmol)和四(三苯基膦)钯(0)(0.032g,0.03mmol)在1,4-二噁烷(4mL)/水(1mL)中的混合物在100℃经受微波辐照3小时。将反应混合物冷却至环境温度,并且用乙酸乙酯稀释。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.045g,15%产率):1H NMR(400MHz,DMSO-d6,在70℃)δ12.67(s,1H),11.11(s,1H),8.63(s,1H),8.31(s,1H),7.75(s,1H),7.12(s,1H),6.70(s,1H),6.60(s,1H),4.41(br s,1H),3.81-4.09(m,3H),3.51(br s,1H),3.22(s,1H),2.99(s,1H),2.11(br s,1H),1.61-1.81(m,3H);MS(ES)m/z 350.3(M+1)。
实施例19:(R)-3-(3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案22:(R)-3-(3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-5-溴-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-1-羧酸叔丁酯的制备

向(R)-5-溴-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(粗品)(5.0g,17.0mmol,通过方案23、步骤3中描述的方法制备)在二氯甲烷(70mL)中的溶液添加二碳酸二叔丁酯(3.9mL,17.0mmol)、三乙胺(4.7mL,34mmol)和4-二甲基氨基吡啶(0.02g,0.17mmol),并且将混合物在环境温度搅拌16小时。用二氯甲烷稀释反应混合物,并且用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(R)-5-溴-4-((1-(叔丁氧基羰基)-哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-1-羧酸叔丁酯(1.2g,14%产率):MS(ES)m/z 496.8(M+2)。
步骤2:(R)-3-((5-氰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-5-溴-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-1-羧酸叔丁酯(1.5g,3.02mmol)、氰化锌(0.53g,4.54mmol)和四(三苯基膦)钯(0)(0.87g,0.75mmol)在二甲基乙酰胺(15mL)中的悬浮液在氩气气氛下在密封管中在130℃加热16小时。将反应混合物冷却至环境温度,并且用乙酸乙酯稀释。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(R)-3-((5-氰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.7g,粗品):MS(ES)m/z 342.2(M+H)。
步骤3:(R,Z)-3-((5-(N’-羟基甲脒基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-氰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.2g,0.58mmol)、盐酸羟胺(0.06g,0.88mmol)和三乙胺(0.16mL,1.17mmol)在乙醇(5mL)中的悬浮液在氩气气氛下在90℃加热15小时。将反应混合物冷却至环境温度,并且在真空中浓缩以去除挥发物。将剩余物溶解在二氯甲烷中,并且用水、盐水溶液洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为粘性固体的(R,Z)-3-((5-(N’-羟基甲脒基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.21g,95%产率):MS(ES)m/z375.2(M+1)。
步骤4:(R)-3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R,Z)-3-((5-(N’-羟基甲脒基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.1g,0.27mmol)在乙酸酐(2mL)中的溶液在140℃加热45分钟。将反应混合物冷却至环境温度,使用氨水中和,并且用二氯甲烷(30mL)提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。将粗物质溶解在甲醇(4mL)中,并且添加3M氢氧化钠水溶液(2mL),并且然后将溶液在环境温度搅拌2小时。在真空中浓缩反应混合物以去除挥发物;将剩余物溶解在二氯甲烷中,用水、盐水洗涤,在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩,以提供为灰白色固体的(R)-3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,粗品):MS(ES)m/z 399.0(M+H)。
步骤5:(R)-5-(5-甲基-1,2,4-噁二唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺制备

将(R)-3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,0.3mmol)在二氯甲烷:三氟乙酸(3mL:0.5mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物以去除挥发物,通过使用氨水中和,并且用二氯甲烷(30mL)提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为灰白色固体的(R)-5-(5-甲基-1,2,4-噁二唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.1g,粗品):MS(ES)m/z 299.0(M+1)。
步骤5:(R)-3-(3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

通过实施例1、步骤8中描述的方法制备(R)-3-(3-((5-(5-甲基-1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈,以产生灰白色固体(0.01g,9%产率):1H NMR(400MHz,DMSO-d6,在60℃)δ11.64(s,1H),8.67(s,1H),7.49-7.57(m,1H),7.25(s,1H),6.70(s,1H),4.21(br s,1H),4.03-4.12(m,2H),3.66-3.84(m,1H),3.42-3.53(m,2H),2.65(s,3H),2.06(br s,2H),1.65-1.73(m,3H);MS(ES)m/z 366.1(M+H)。
实施例20-25:5-芳基取代的吡咯并吡啶的制备

方案23:(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶的制备

在0℃向5-溴-4-氯-1H-吡咯并[2,3-b]吡啶(15g,65.0mmol)在二氯甲烷(200mL)中的溶液添加三乙胺(18.2mL,130mmol)、对甲苯磺酰氯(18.5g,97mmol)和4-二甲基氨基吡啶(0.79g,6.0mmol)。将溶液在环境温度搅拌16小时。用二氯甲烷稀释反应混合物,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中干燥。用正戊烷研磨粗产物,并且干燥以提供为灰白色固体的5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(23.0g,92%产率):MS(ES)m/z 386.7(M+2)。
步骤2:(R)-N-(1-苄基哌啶-3-基)-5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(10.0g,25.0mmol)和(R)-1-苄基哌啶-3-胺(9.88g,52mmol)的混合物在密封管中加热3小时至180℃。将反应混合物冷却至环境温度,并且通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色粘性固体的(R)-N-(1-苄基哌啶-3-基)-5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-胺(9.1g,66%产率):MS(ES)m/z 541.1(M+2)。
步骤3:(R)-5-溴-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

向(R)-N-(1-苄基哌啶-3-基)-5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-胺(10.0g,18.5mmol)在1,4-二噁烷(200mL)中的溶液添加氯甲酸乙酯(2.1mL,22.6mmol),并且将所得的混合物加热至100℃。在16小时之后,将反应混合物冷却至环境温度,并且在真空中浓缩。将粗中间产物溶解在甲醇:水(150mL:70mL)中,并且添加氢氧化钾(20.75g,370mmol),并且将混合物在90℃加热16小时。将反应混合物冷却至环境温度,并且在真空中浓缩以去除挥发物。将剩余物溶解在二氯甲烷中,并且用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为棕色油状物的(R)-5-溴-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(8.4g,粗品):MS(ES)m/z 297.0(M+2)。
步骤-4:(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向(R)-5-溴-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(8.4g,28.4mmol)在二氯甲烷(150mL)中的溶液添加氰基乙酸(4.84g,56.9mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(10.94g,56.9mmol)、羟基苯并三唑(7.68g,56.9mmol)和N,N-二异丙基乙胺(14.7mL,85.4mmol)。将反应混合物在环境温度搅拌16小时。用二氯甲烷(150mL)稀释反应混合物,并且用水、盐水溶液洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(2.8g,27%产率):1H NMR(400MHz,DMSO-d6,在60℃)δ11.37(b,1H),7.96(s,1H),7.20(s,1H),6.57(s,1H),5.19(d,J=8.8Hz,1H),4.20(br s,1H),3.80–4.10(m,4H),3.52-3.54(m,1H),3.01(br s,1H),2.02(br s,1H),1.51-1.75(m,3H);MS(ES)m/z 364.2(M+2)。
交叉偶联反应的一般程序

将(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(1.0当量)、芳基/杂芳基(三丁基或甲锡烷基)(1.2当量)、氯化锂(2.0当量)、碘化铜(0.1当量)和四(三苯基膦)钯(0)在N,N-二甲基甲酰胺(5mL)中的混合物在氩气气氛下加热3小时至120℃。将反应混合物冷却至环境温度,并且用乙酸乙酯稀释。用水、盐水洗涤乙酸乙酯层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供表2中所示的期望的产物。
表2通过上文描述的方法制备下列化合物:



实施例25:(R)-4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-腈的制备

方案24:(R)-4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-腈的制备

将(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,0.55mmol,如通过方案23、步骤4中描述的方法制备)、氰化锌(0.01mg,0.83mmol)和四(三苯基膦)钯(0)(0.16g,0.14mmol)在二甲基甲酰胺(5mL)中的悬浮液在氩气气氛下在密封管中在130℃加热16小时。将反应混合物冷却至室温,并且用乙酸乙酯稀释。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗产物,以提供为灰白色固体的(R)-4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-腈(0.04g,26%产率):1H NMR(400MHz,DMSO-d6在70℃)11.67(s,1H),8.07(s,1H),7.23(s,1H),6.71(s,1H),6.21-6.66(m,1H),4.05-4.29(m,2H),3.99(s,2H),3.53-3.91(m,1H),2.85-3.02(m,1H),2.03-2.10(m,2H),1.68-1.78(m,3H);MS(ES)m/z 309.2(M+H)。HPLC纯度:99.58%。
制备型HPLC条件:
柱:Kinetex C18(100mm×4.6mm×2.6μm)
流动相A:水中0.1%TFA
流动相B:ACN
流速:0.75mL/min
实施例26:(R)-3-(3-((5-(3-甲基-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案25:(R)-3-(3-((5-(3-甲基-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:3-((3R)-3-((5-(3-甲基-1-(四氢-2H-吡喃-2-基)-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.5g,1.38mmol,如通过方案23、步骤4中描述的方法制备)、3-甲基-1-(四氢-2H-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡唑(0.6g,2.07mmol)、碳酸铯(1.35g,4.14mmol)和四(三苯基膦)钯(0)(0.08g,0.07mmol)在1,4-二噁烷:水(4mL:1mL)中的混合物在氩气气氛下在密封管中在100℃加热5小时。将反应混合物冷却至环境温度,并且用乙酸乙酯和水稀释。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(80%乙酸乙酯/己烷)纯化粗材料,以提供为棕色固体的3-((3R)-3-((5-(3-甲基-1-(四氢-2H-吡喃-2-基)-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,32%产率):MS(ES)m/z 448(M+H)。
步骤2:(R)-3-(3-((5-(3-甲基-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将3-((3R)-3-((5-(3-甲基-1-(四氢-2H-吡喃-2-基)-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,0.44mmol)和对甲苯磺酸(0.15g,0.89mmol)在甲醇(5mL)中的经搅拌的溶液在60℃加热12小时。将反应混合物冷却至室温,并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(3-甲基-1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.01g,6%产率):1H NMR(400MHz,DMSO-d6)δ12.52(s,1H),11.35(s,1H),8.24-8.27(m,1H),7.44-7.46(m,1H),7.08-7.16(m,2H),6.47-6.50(m,1H),4.31(s,1H),3.89-4.11(m,2H),3.49-3.69(m,2H),3.20(s,3H),2.94(s,2H),2.27(m,3H),2.05-2.10(m,1H);MS(ES)m/z 364.1(M+)。
分析条件:流速:0.3mL/min
柱:BEH C18(100mm×2.1mm×1.7μm)
流动相(A):水中0.1%甲酸
流动相(B):ACN
实施例27:(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

方案26:(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

步骤1:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑的制备

将5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(2.00g,5.54mmol)、噁唑(0.68g,9.97mmol)、叔丁醇钾(1.24g,11.1mmol)和双(三苯基膦)二氯化钯(II)(0.19g,0.27mmol)在1,4-二噁烷(20mL)中的混合物在氮气下在密封管中在110℃加热。在5小时之后,将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(10%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.43g,22%产率):MS(ES)m/z 349.9(M+H)。
步骤2:(R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

使2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.43g,1.28mmol)、三乙胺(0.03mL,0.24mmol)和(R)-3-氨基哌啶-1-羧酸叔丁酯(0.46g,2.32mmol)在N-甲基吡咯烷酮(4mL)中的经搅拌的溶液在130℃经受微波辐照5小时。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.35g,53%产率):MS(ES)m/z 514.0(M+H)。
步骤3:(R)-5-(噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,0.23mmol)在二氯甲烷:三氟乙酸(3mL:3mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且将混合物在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为灰白色固体的(R)-5-(噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.05g,粗品):MS(ES)m/z 284.2(M+H)。
步骤4:(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

向(R)-5-(噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.05g,0.17mmol)在乙醇(3mL)中的经搅拌的溶液添加丙烯腈(0.09g,1.7mmol),然后添加三甲胺(0.05mL,0.34mmol),并且将溶液在80℃搅拌3小时。将反应冷却至环境温度,并且在真空中浓缩。使用快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.03g,45%产率):1H NMR(400MHz,DMSO-d6)δ11.53(br s,1H),9.17(br s,1H),8.50(s,1H),8.06(s,1H),7.36(s,1H),7.17(s,1H),6.56(s,1H),4.14-4.27(m,1H),2.85-2.98(m,1H),2.55-2.66(m,2H),2.83-2.97(m,2H),1.52-1.69(m,2H),1.42-1.60(m,3H),1.21(s,2H);MS(ES)m/z337.2(M+H)。
实施例28:3-((2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案27.3-((2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

向2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑(0.2g,0.57mmol)(从实施例27、步骤1获得)和(2S,5R)-5-氨基-2-甲基哌啶-1-羧酸苄酯(0.21g,0.85mmol)在1,4-二噁烷(5mL)中的经搅拌的溶液添加叔丁醇钾(0.16g,1.71mmol)、乙酸钯(II)(0.02g,0.08mmol)和(R)-(+)-2,2’-双(二苯基膦)-1,1’-联萘基(0.05g,0.08mmol),并且使所得的混合物在100℃经受微波辐照45分钟。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为无色胶状液体的(2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.12g,37%产率):MS(ES)m/z 561.9(M+H)。
步骤2:N-((3R,6S)-6-甲基哌啶-3-基)-5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(2.0g,0.35mmol)在三氟乙酸(2.0mL)中的溶液在100℃搅拌1小时。将反应冷却至环境温度,并且在真空中浓缩混合物,将剩余物溶解在1,4-二噁烷:氨水(2mL:2mL)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为浅黄色胶状液体的N-((3R,6S)-6-甲基哌啶-3-基)-5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.1g,粗品):MS(ES)m/z 298.0(M+H)。
步骤3:3-((2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向氰基乙酸(0.03g,0.4mmol)和1-羟基苯并三唑(0.055g,0.4mmol)在二氯甲烷(5mL)中的溶液添加N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.1g,0.51mmol),并且将混合物在环境温度搅拌5分钟。此时,添加N-((3R,6S)-6-甲基哌啶-3-基)-5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.1g,0.34mmol),然后添加N,N-二异丙基乙胺(0.18mL,1.02mmol),并且然后将所得的混合物在环境温度搅拌18小时。用水淬灭反应,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料并且通过反相色谱进一步纯化粗材料,以提供为灰白色固体的3-((2S,5R)-2-甲基-5-((5-(噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.04g,33%产率):1H NMR(400MHz,DMSO-d6,VT在70℃)δ11.43(brs,1H),8.92(d,J=7.6Hz,1H),8.55(s,1H),8.05(s,1H),7.36(s,1H),7.20(s,1H),6.68(s,1H),3.93-4.02(m,3H),3.56(s,2H),1.80-2.12(m,2H),1.60-1.79(m,2H),1.24(s,4H);MS(ES)m/z 365.1(M+H)。
分析条件:流速:0.3mL/min
柱:BEH C18(50mm×2.1mm×1.7μm)
流动相(A):水中0.1%甲酸
流动相(B):MeCN
实施例29:2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

方案28.2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

步骤1:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯的制备

向5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(0.8g,2.22mmol)和噁唑-4-羧酸乙酯(0.37g,2.66mmol)在1,4-二噁烷(30mL)中的经搅拌的溶液添加乙酸钯(0.024g,0.11mmol)、CyJohnphos(0.08g,0.22mmol)和碳酸铯(1.44g,4.44mmol),并且将混合物在密封管中在110℃加热16小时。将反应冷却至室温,通过硅藻土过滤,并且在真空中浓缩滤液。使用快速色谱(30%乙酸乙酯/己烷)纯化粗品,以提供为棕色液体的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.41g,44%产率):MS(ES)m/z 421.9(M+H)。
步骤2:2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯的制备

向2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.4g,0.95mmol)在N-甲基吡咯烷酮(5mL)中的经搅拌的溶液添加(1R,3S)-3-氨基环戊烷-1-醇盐酸盐(0.13g,0.95mmol),然后添加三甲胺(0.17mL,1.23mmol)。使所得的混合物在150℃经受微波辐照45分钟。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为无色浓稠液体的2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.31g,67%产率):MS(ES)m/z 486.9(M+H)。
步骤3:2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

将2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.30g,0.62mmol)、甲醇(5mL)和氨水(5mL,水中23%)的经搅拌的溶液在环境温度搅拌20小时。在真空中浓缩反应混合物,以提供为白色固体的2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.28g,99%产率):MS(ES)m/z 457.9(M+H)。
步骤4:2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

将2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.25g,0.55mmol)在二氯甲烷:三氟乙酸(3mL:3mL)中的溶液在环境温度搅拌3小时。在真空中浓缩混合物,将剩余物溶解在1,4-二噁烷:氨水(3mL:5mL,水中23%)中,并且将溶液在环境温度搅拌16小时。在真空中浓缩反应混合物,并且使用快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的2-(4-(((1S,3R)-3-羟基环戊基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.05g,35%产率):1H NMR(400MHz,DMSO-d6)δ11.56(br s,1H),9.35(d,J=8.4Hz,1H),8.48(d,J=8.4Hz,1H),7.58(s,1H),7.17(s,1H),6.71(s,1H),5.13(s,1H),4.63(s,1H),4.35(s,1H),2.05-2.19(m,2H),1.82-2.00(m,2H),1.75-1.80(m,1H),1.15-1.30(m,3H);MS(ES)m/z 328.1(M+H)。
中间产物

(1S,3R)-3-氨基环戊烷-1-醇盐酸盐的制备

向((1R,3S)-3-羟基环戊基)氨基甲酸叔丁酯(0.5g,2.48mmol)在1,4-二噁烷(1mL)中的溶液添加1,4-二噁烷中4N盐酸(2.5mL),并且在环境温度搅拌12小时。在真空中浓缩反应混合物,以提供为灰白色固体的(S)-4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯盐酸盐(0.25g,粗品):MS(ES)m/z 102.1(M+H)。
实施例30:2-(4-(((3R,6S)-1-(2-氰基乙酰基)-6-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

方案29.2-(4-(((3R,6S)-1-(2-氰基乙酰基)-6-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

步骤1:2-(4-(((3R,6S)-1-((苄氧基)羰基)-6-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯的制备

在10mL小瓶中,将2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酸乙酯(2.0g,4.74mmol)(从实施例29、步骤1获得)、(2S,5R)-5-氨基-2-甲基哌啶-1-羧酸苄酯(2.35g,9.48mmol)和二异丙基乙胺(1.29mL,14.2mmol)在N-甲基吡咯烷酮(15mL)中的溶液在130℃加热16h。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中干燥。使用快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色胶状液体的(2S,5R)-5-({5-[4-(乙氧基羰基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)-2-甲基哌啶-1-羧酸苄酯(0.71g,24%产率):MS(ES)m/z634.3(M+H)。
步骤2:2-(4-(((3R,6S)-1-((苄氧基)羰基)-6-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸的制备

将(2S,5R)-5-({5-[4-(乙氧基羰基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)-2-甲基哌啶-1-羧酸苄酯(0.60g,0.95mmol)、甲醇(20mL)和2M氢氧化钠(0.2g,4.73mmol)水溶液的溶液在环境温度搅拌5小时。在真空中去除挥发物,将剩余物溶解在水中,并且用1N盐酸酸化,并且调整pH~3。用二氯甲烷提取水性层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为无色浓稠液体的2-(4-{[(3R,6S)-1-[(苄氧基)羰基]-6-甲基哌啶-3-基]氨基}-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酸(0.51g,89%产率):MS(ES)m/z 606.3(M+H)。
步骤3:(2S,5R)-2-甲基-5-({5-[4-(甲基氨基甲酰基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)哌啶-1-羧酸苄酯的制备

向环境温度的2-(4-{[(3R,6S)-1-[(苄氧基)羰基]-6-甲基哌啶-3-基]氨基}-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酸(0.5g,0.82mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.48g,1.24mmol)在N,N-二甲基甲酰胺(8mL)中的经搅拌的溶液添加甲胺盐酸盐(0.13g,4.13mmol),然后添加三乙胺(0.6mL,4.13mmol),并且将混合物在环境温度搅拌10小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色粘性固体的(2S,5R)-2-甲基-5-({5-[4-(甲基氨基甲酰基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)哌啶-1-羧酸苄酯(0.32g,63%产率):MS(ES)m/z619.3(M+H)。
步骤4:N-甲基-2-(4-{[(3R,6S)-6-甲基哌啶-3-基]氨基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酰胺的制备

将(2S,5R)-2-甲基-5-({5-[4-(甲基氨基甲酰基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)哌啶-1-羧酸苄酯(0.32g,0.51mmol)在三氟乙酸(5.0mL)中的溶液在100℃搅拌1小时。将反应冷却至环境温度,并且在真空中浓缩混合物。将获得的剩余物溶解在1,4-二噁烷:氨水(5mL:5mL,水中23%)中,并且然后在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为浅黄色胶状液体的N-甲基-2-(4-{[(3R,6S)-6-甲基哌啶-3-基]氨基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酰胺(0.15g,粗品):MS(ES)m/z 355.2(M+H)。
步骤5:2-(4-(((3R,6S)-1-(2-氰基乙酰基)-6-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

将2-氰基乙酸(0.3g,0.27mmol)、1-羟基-1,2-二氢吡啶-2-酮(0.03g,0.27mmol)和2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基四氟硼酸铵(TBTU)(0.11g,0.34mmol)在N-甲基吡咯烷酮(3.0mL)中的溶液在环境温度搅拌2分钟。添加N-甲基-2-(4-{[(3R,6S)-6-甲基哌啶-3-基]氨基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酰胺(0.08g,0.23mmol),然后添加三乙胺(0.1mL,0.70mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相纯化纯化粗品,以获得为白色固体的2-(4-(((3R,6S)-1-(2-氰基乙酰基)-6-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺(0.03g,30%产率):1HNMR(400MHz,DMSO-d6,VT在80℃)δ11.46(br s,1H),8.46-8.53(m,3H),8.03(br s,1H),7.21(s,1H),6.68(s,1H),4.40-4.70(m,1H),3.92-4.01(m,5H),2.84(d,J=4.0Hz,3H),1.80-2.10(m,3H),1.20-1.35(m,4H);MS(ES)m/z 422.4(M+H)。
分析条件:
流动相A:H2O中0.1%NH4OH
流动相B:MeCN
柱:X-Bridge,C18 19*100*5微米
流速:20mL/min
实施例31:2-(4-(((3R,5S)-1-(2-氰基乙酰基)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

方案30.2-(4-(((3R,5S)-1-(2-氰基乙酰基)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

步骤1:2-(4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯的制备

将2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酸乙酯(7.0g,16.6mmol)(从实施例29、步骤1获得)、N-甲基吡咯烷酮(42mL)、(3S,5R)-3-氨基-5-甲基哌啶-1-羧酸苄酯(5.36g,21.6mmol)和三乙胺(11.7mL,82.9mmol)的经搅拌的溶液在密封管中加热32小时至150℃。将反应混合物冷却至环境温度,用水淬灭并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(乙酸乙酯/己烷)纯化粗品,以提供为浅黄色半固体的2-(4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(5.8g,55%产率):MS(ES)m/z634.3(M+H)。
步骤2:2-(4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸的制备

向(3R,5S)-3-({5-[4-(乙氧基羰基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)-5-甲基哌啶-1-羧酸苄酯(5.8g,9.15mmol)在甲醇:四氢呋喃(20mL:20mL)中的经搅拌的溶液添加2M单水氢氧化锂(0.8g,18.3mmol)水溶液,并且将混合物在环境温度搅拌5小时。在真空中去除挥发物,将剩余物溶解在水中,并且用10%NaHSO4溶液酸化,并且调整pH~2。用二氯甲烷提取水性层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为灰白色固体的2-(4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸(5.7g,粗品):MS(ES)m/z 606.3(M+H)。
步骤3:(3S,5R)-3-甲基-5-((5-(4-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将2-(4-{[(3R,5S)-1-[(苄氧基)羰基]-5-甲基哌啶-3-基]氨基}-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-1,3-噁唑-4-羧酸(5.7g,9.41mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.29g,0.74mmol)在N,N-二甲基甲酰胺(60mL)中的溶液在环境温度搅拌3分钟。添加甲胺盐酸盐(1.27g,18.8mmol),然后添加N,N-二异丙基乙胺(8.22mL,47.0mmol),并且将混合物在环境温度搅拌10小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色粘性固体的(3S,5R)-3-甲基-5-((5-(4-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(5.7g,97%产率):MS(ES)m/z 619.3(M+H)。
步骤4:N-甲基-2-(4-(((3R,5S)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

将(3S,5R)-3-甲基-5-((5-(4-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(5.7g,9.21mmol)在三氟乙酸(15mL)中的经搅拌的溶液在密封管中在100℃加热1小时。在冷却之后,将反应混合物浓缩至干,将剩余物溶解在1,4-二噁烷:氨水(10mL:20mL,水中23%)中,并且将反应混合物在环境温度搅拌15小时。将反应在真空中浓缩至干,以提供为灰白色固体的N-甲基-2-(4-(((3R,5S)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(9.2g,粗品):MS(ES)m/z 355.4(M+H)。
步骤5:2-(4-(((3R,5S)-1-(2-氰基乙酰基)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

将氰基乙酸(1.48g,17.4mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(11g,28.9mmol)在N,N-二甲基甲酰胺(45mL)中的溶液在环境温度搅拌10分钟。然后,添加N-甲基-2-(4-(((3R,5S)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(4.1g,11.6mmol),然后添加N,N-二异丙基乙胺(10mL,57.8mmol),并且将所得的混合物在环境温度搅拌12小时。用冰水淬灭反应,并且用乙酸乙酯提取,并且用水和盐水洗涤合并的有机部分,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩。使用反相色谱纯化粗材料,以产生白色固体(0.67g,13%产率):1HNMR(400MHz,DMSO-d6,VT在90℃)δ11.40(s,1H),8.54(s,1H),8.47-8.43(m,2H),7.85(brs,1H),7.21(s,1H),6.72(s,1H),4.80(br s,1H),4.15(s,1H),3.98(s,2H),3.75(br s,1H),2.85(s,3H),2.65(s,1H),2.30-2.27(m,2H),1.86(br s,1H),1.41(d,J=11.2Hz,1H),0.95(d,J=5.6Hz,3H);MS(ES)m/z 422.3(M+H)。
分析条件:
柱:X-BridgeC-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
实施例32:2-(4-(((3S,5R)-1-(2-氰基乙酰基)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

按照与实施例31相同的方法并且使用步骤1中的(3R,5S)-3-氨基-5-甲基哌啶-1-羧酸苄酯来制备2-(4-(((3S,5R)-1-(2-氰基乙酰基)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺,以产生白色固体(0.6g,11%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.45(s,1H),8.45-8.43(m,3H),7.91(br s,1H),7.21(s,1H),6.72(s,1H),4.80(br s,1H),4.15(s,1H),3.98(s,2H),3.75(br s,1H),2.85(s,3H),2.65(s,1H),2.30-2.27(m,2H),1.86(br s,1H),1.41(d,J=11.2Hz,1H),0.95(d,J=5.6Hz,3H);MS(ES)m/z 422.3(M+H)。
分析条件:
柱:X-BridgeC-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
实施例33:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

方案31.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

步骤1:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯的制备

向(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.2g,2.28mmol)和噁唑-4-羧酸乙酯(0.38g,2.74mmol)在1,4-二噁烷(18mL)中的经搅拌的溶液添加乙酸钯(0.03g,0.12mmol)、CyJohnphos(0.08g,0.23mmol)和碳酸铯(1.48g,4.56mmol),并且将所得的混合物在氮气气氛下在密封管中在110℃加热16小时。将反应混合物冷却至环境温度,通过硅藻土过滤,用乙酸乙酯洗涤,并且在真空中浓缩滤液。使用快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(1.1g,41%产率):MS(ES)m/z 585.9(M+H)。
步骤2:(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸的制备

向(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸乙酯(0.5g,0.85mmol)在乙醇(12mL)中的经搅拌的溶液添加2M氢氧化钠(0.1g,2.56mmol)水溶液,并且将所得的混合物在环境温度搅拌3小时。在真空中去除挥发物,将剩余物溶解在水中,并且用10%硫酸氢钾溶液酸化,并且调整pH~3。用二氯甲烷提取水性层,并且用盐水洗涤。在无水硫酸镁上干燥有机层,过滤并且在真空中浓缩,以提供为灰白色固体的(R)-4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.3g,64%产率):MS(ES)m/z 557.9(M+H)。
步骤3:(R)-3-((5-(4-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸(0.27g,0.48mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.24g,0.63mmol)在N,N-二甲基甲酰胺(10mL)中的溶液搅拌5分钟。添加甲胺盐酸盐(0.05g,0.726mmol),然后添加N,N-二异丙基乙胺(0.25mL,1.45mmol),并且将所得的混合物在环境温度搅拌3小时。用冰水淬灭反应,用乙酸乙酯提取,并且用水和盐水洗涤有机部分,并且然后在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩。使用快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为黄色油状物的(R)-3-((5-(4-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.27g,99%产率):MS(ES)m/z 571.3(M+H)。
步骤4:(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

将(R)-3-((5-(4-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.30g,0.52mmol)在二氯甲烷:三氟乙酸(5mL:4mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,并且将剩余物溶解在1,4-二噁烷:氨水(4mL:5mL,水中23%)中。将反应混合物在环境温度搅拌16小时。在真空中浓缩反应,以提供溶解在5%甲醇/二氯甲烷中的剩余物。用水、盐水洗涤有机层,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩,以提供为胶状液体的(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.12g,70%产率):MS(ES)m/z 341.2(M+H)。
步骤5:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺的制备

将氰基乙酸(0.05g,0.55mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.17g,0.45mmol)在N,N-二甲基甲酰胺(5mL)中的溶液搅拌10分钟。添加(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.12g,0.35mmol),然后添加N,N-二异丙基乙胺(0.18mL,1.05mmol),并且将所得的混合物在环境温度搅拌6小时。用冰水淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为白色固体的(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-4-羧酰胺(0.01g,8%产率):1H NMR(400MHz,DMSO-d6)δ11.68-11.64(brs,1H),8.63(d,J=7.2Hz,1H),8.55-8.52(m,2H),8.13(s,1H),7.24(s,1H),6.72(s,1H),4.38(br s,1H),4.22-4.19(m,1H),4.08(s,1H),3.96(s,1H),3.82-3.77(m,1H),3.48(s,1H),3.36(s,1H),2.83(d,J=3.6Hz,3H),2.07(br s,2H),1.85(br s,2H);MS(ES)m/z408.5(M+H)。
分析条件:
柱:X-Bridge C-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min。
实施例34:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N,N-二甲基-噁唑-4-羧酰胺的制备

方案32.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N,N-二甲基-噁唑-4-羧酰胺的制备

步骤1:(R)-3-((5-(4-(二甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酸(0.25g,0.44mmol)(从实施例32、步骤2获得)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.22g,0.58mmol)在N,N-二甲基甲酰胺(10mL)中的经搅拌的溶液添加二甲胺盐酸盐(0.05g,0.67mmol),然后添加N,N-二异丙基乙胺(0.23mL,1.34mmol),并且将混合物在环境温度搅拌30分钟。用水稀释反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过柱色谱(50%乙酸乙酯/己烷)纯化粗品,以提供为黄色油状物的标题产物(0.23g,88%产率):MS(ES)m/z 585.3(M+H)。
步骤2:(R)-N,N-二甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺的制备

将(R)-3-((5-(4-(二甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.23g,0.39mmol)在二氯甲烷:三氟乙酸(3mL:3mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供(R)-N,N-二甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.1g粗品):MS(ES)m/z 355.2(M+H)。
步骤3:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N,N-二甲基-噁唑-4-羧酰胺的制备

向氰基乙酸(0.03g,0.33mmol)、1-羟基-2-吡啶酮(0.03g,0.27mmol)和2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基四氟硼酸铵(0.09g,0.27mmol)在N-甲基吡咯烷酮(5mL)中的经搅拌的溶液添加(R)-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-4-羧酰胺(0.08g,0.22mmol),然后添加三乙胺(0.1mL,0.7mmol),并且将所得的混合物在环境温度搅拌2小时。用水稀释反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(10%甲醇/己烷)纯化粗品,以提供为灰白色固体的(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N,N-二甲基噁唑-4-羧酰胺(0.05g,52%产率):1H NMR(400MHz,DMSO-d6)δ11.4(brs,1H),8.73(br s,1H),8.55(s,1H),8.42(s,1H),7.22(s,1H),6.72(s,1H),4.20(m,2H),3.51-3.99(m,4H),3.09-3.29(m,6H),1.67-2.15(m,5H);MS(ES)m/z 422.1(M+H)。
实施例35:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-4-羧酰胺的制备

方案33.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-4-羧酰胺的制备

步骤1:2-溴-N-甲基噻唑-4-羧酰胺的制备

向2-溴噻唑-4-羧酸(1.0g,4.8mmol)和1-[双(二甲基氨基)-亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(4.5g,12.0mmol)在N,N-二甲基甲酰胺(20mL)中的经搅拌的溶液添加甲胺(3.6mL,7.2mmol,四氢呋喃中2M),然后添加二异丙基乙胺(4.2mL,24.0mmol),并且将所得的混合物在环境温度搅拌16小时。用冰水淬灭反应混合物,并且用乙酸乙酯提取两次。用盐水溶液洗涤有机层,在无水硫酸钠上干燥,并且在真空中浓缩。通过快速色谱(13%乙酸乙酯/己烷)纯化粗材料,以提供为白色固体的2-溴-N-甲基噻唑-4-羧酰胺(0.7g,70%产率):MS(ES)m/z 221.0(M+H)。
步骤2:(R)-3-((5-(4-(甲基氨基甲酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向2-溴-N-甲基噻唑-4-羧酰胺(0.7g,1.22mmol)、(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.4g,1.83mmol,中间产物7)在1,4-二噁烷(25mL)中的经搅拌的溶液添加四(三苯基膦)钯(0)(0.14g,0.12mmol)和2M碳酸钠(0.39g,3.66mmol)的水溶液,并且将反应混合物在100℃搅拌16小时。在冷却之后,用乙酸乙酯稀释反应混合物,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(35%乙酸乙酯/己烷)纯化粗材料,以提供为浅棕色液体的(R)-3-((5-(4-(甲基氨基甲酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.55g,79%产率):MS(ES)m/z 587.6(M+H)。
步骤3:(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-4-羧酰胺的制备

将(R)-3-((5-(4-(甲基氨基甲酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.55g,0.94mmol)在二氯甲烷:三氟乙酸(2.0mL:3.0mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(2.0mL:5.0mL,水中23%)中,并且然后在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为黄色液体的(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-4-羧酰胺(0.36g,粗品):MS(ES)m/z357.2(M+H)。
步骤4:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-4-羧酰胺的制备

将氰基乙酸(0.1g,1.2mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.9g,2.5mmol)在N,N-二甲基甲酰胺(10mL)中的溶液搅拌3分钟。此时,添加(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-4-羧酰胺(0.36g,1.0mmol),然后添加N,N-二异丙基乙胺(0.72mL,5.0mmol),并且将混合物在环境温度搅拌16小时。用冰水淬灭反应混合物,并且用乙酸乙酯提取。用盐水溶液洗涤有机层,在无水硫酸钠上干燥,并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-4-羧酰胺(0.01g,3%产率):1HNMR(400MHz,DMSO-d6)δ11.65(br s,1H),9.32(d,J=7.6Hz,1H),8.37(s,1H),8.30(s,1H),8.09(s,1H),7.23(s,1H),6.72(d,J=8.8Hz,1H),4.27(s,1H),4.11(s,1H),4.05(s,1H),3.89(s,1H),3.78(d,J=12.4Hz,1H),3.64(s,1H),3.51(s,1H),3.20-3.23(m,3H),3.00(s,1H),2.83(s,3H);MS(ES)m/z 424.2(M+H)。
分析条件:
柱:X Bridge C18(100mm×4.6mm×3.5μm)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:0.8mL/min。
实施例36:(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案34.(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(5-甲基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.30g,5.57mmol)、叔丁醇钾(0.13g,1.14mmol)、双(三苯基膦)二氯化钯(II)(0.02g,0.03mmol)和5-甲基噁唑(0.09g,1.14mmol)在1,4-二噁烷(5mL)中的混合物在氮气气氛下在密封管中在100℃加热。在6小时之后,将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为深黄色油状物的(R)-3-((5-(5-甲基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.2g,66%产率):MS(ES)m/z 528.0(M+H)。
步骤2:(R)-5-(5-甲基噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(5-甲基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.20g,0.37mmol)在二氯甲烷:三氟乙酸(2.0mL:2.0mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(3.0mL:3.0mL,水中23%)中,并且在环境温度搅拌16小时。在真空中浓缩反应,以提供为黄色固体的(R)-5-(5-甲基噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.17g,粗品):MS(ES)m/z 298.2(M+H)。
步骤3:(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.02g,0.24mmol)、N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.58g,0.30mmol)、1-羟基苯并三唑(0.03g,0.22mmol)和N,N-二异丙基乙胺(0.1mL,0.6mmol)在二氯甲烷(5mL)中的溶液在环境温度搅拌5分钟。然后,添加(R)-5-(5-甲基噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.06g,0.20mmol),并且将所得的混合物在环境温度搅拌18小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.01g,27%产率):1H NMR(400MHz,DMSO-d6,VT在100℃)δ11.38(br s,J=9.2Hz,1H),8.98(br s,1H),8.50(s,1H),7.19(s,1H),6.91(s,1H),6.66(s,1H),3.90-4.40(m,3H),3.52-3.91(m,1H),3.20-3.40(m,2H),2.31(s,3H),1.95-2.10(m,2H),1.50-1.90(m,3H);MS(ES)m/z 365.1(M+H)。
实施例37:(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

方案35.(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

将(R)-5-(5-甲基噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.08g,0.26mmol,实施例36、步骤2)和丙烯腈(0.04g,0.60mmol)、三乙胺(0.1mL,0.80mmol)在乙醇(10mL)中的溶液加热3小时至80℃。将反应冷却至环境温度,并且在真空中浓缩。通过快速色谱(10%甲醇/二氯甲烷)纯化粗品,以提供为白色固体的(R)-3-(3-((5-(5-甲基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.02g,30%产率):1H NMR(400MHz,DMSO-d6,)δ11.49(s,1H),9.10(d,J=8Hz,1H),8.46(s,1H),7.16(s,1H),6.96(s,1H),6.55(s,1H),4.18-4.20(m,1H),2.92-2.95(m,1H),2.56-2.64(m,5H),2.30-2.47(m,5H),1.91-1.20(m,1H),1.60-1.69(m,1H),1.49-1.53(m,2H);MS(ES)m/z351.1(M+H)。
实施例38:(R)-3-(3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案36.(R)-3-(3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-氰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

在密封管中,将4-氯-1H-吡咯并[2,3-b]吡啶-5-腈(1.0g,5.64mmol)、(R)-3-氨基哌啶-1-羧酸叔丁酯(1.69g,8.4mmol)和三乙胺(2.37mL,16.9mmol)在N-甲基-2-吡咯烷酮(15mL)中的经搅拌的溶液在130℃加热6小时。将反应混合物冷却至室温,用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以获得为灰白色固体的粗品(R)-3-((5-氰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(2.3g,粗品):MS(ES)m/z 342.2(M+H)。
步骤2:(R,Z)-3-((5-(N’-羟基甲脒基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-氰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(2.3g,6.74mmol)、盐酸羟胺(0.70g,10.1mmol)和三乙胺(1.89mL,13.4mmol)在乙醇(10mL)中的悬浮液在氩气气氛下在90℃加热15小时。将反应混合物冷却至环境温度,并且在真空中浓缩以去除挥发物。将剩余物溶解在二氯甲烷中,并且用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为半固体的粗(R,Z)-3-((5-(N’-羟基甲脒基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.2g,粗品):MS(ES)m/z 375.0(M+H)。
步骤3:(R)-3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R,Z)-3-((5-(N’-羟基甲脒基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.5g,1.3mmol)、三氟乙酸(0.05mL)和原甲酸三乙酯(0.5mL)的经搅拌的溶液在密封管中在60℃加热12小时。将反应冷却至环境温度,用二氯甲烷稀释,并且用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩,以提供为灰白色固体的(R)-3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,粗品):MS(ES)m/z 385.0(M+H)。
步骤4:(R)-5-(1,2,4-噁二唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,0.6mmol)在二氯甲烷:三氟乙酸(5mL:2mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物以去除挥发物,并且用氨水中和,并且用二氯甲烷(30mL)提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为无色液体的(R)-5-(1,2,4-噁二唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.11g,粗品):MS(ES)m/z 285.0(M+H)。
步骤5:(R)-3-(3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向氰基乙酸(0.044g,0.52mmol)、1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.17g,0.45mmol)在N,N-二甲基甲酰胺(15mL)中的经搅拌的溶液添加(R)-5-(1,2,4-噁二唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.1g,0.35mmol),然后添加N,N-二异丙基乙胺(0.19mL,1.05mmol),并且然后将所得的混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(1,2,4-噁二唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.01g,9%产率):1H NMR(400MHz,DMSO-d6 VT在70℃)δ11.51(br s,1H),9.56(s,1H),8.73(s,1H),8.34(s,1H),7.46(s,1H),7.23(s,1H),6.70(d,J=3.2Hz,1H),4.05-4.29(m,2H),3.89-4.02(m,2H),3.49-3.75(m,2H),3.40-3.51(m,2H),2.10(br s,1H),1.64-1.82(m,1H);MS(ES)m/z 352.2(M+H)。
分析条件:
柱:X-BridgeC-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
实施例39:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-5-羧酰胺的制备

方案37.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-5-羧酰胺的制备

步骤1:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸乙酯的制备

向(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(2.5g,4.36mmol)和2-溴噁唑-5-羧酸乙酯(1.05g,4.80mmol)在二噁烷(25mL)中的经搅拌的溶液添加双(三苯基膦)二氯化钯(II)和2M碳酸钾(1.8g,13.10mmol)水溶液,并且将所得的混合物在110℃加热16小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中干燥。使用快速色谱(30%乙酸乙酯/己烷)纯化粗品,以提供为粘性棕色液体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(tri甲基silyl)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸乙酯(0.35g,14%产率):MS(ES)m/z 586.3(M+H)。
步骤2:(R)-3-((5-(5-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸乙酯(0.5g,0.85mmol)和甲胺(5mL,四氢呋喃中2M)的溶液在环境温度搅拌48小时。在真空中浓缩反应混合物,以提供为粘性棕色液体的(R)-3-((5-(5-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.1g,粗品):MS(ES)m/z 571.3(M+H)。
步骤3:(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酰胺的制备

将(R)-3-((5-(5-(甲基氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.17g,0.29mmol)在二氯甲烷:三氟乙酸(2mL:2mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为胶状液体的(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酰胺(0.2g,粗品):MS(ES)m/z 341.1(M+H)。
步骤4:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-5-羧酰胺的制备

将氰基乙酸(0.07g,0.83mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.32g,0.83mmol)在N,N-二甲基甲酰胺(5mL)中的溶液搅拌5分钟。然后,添加(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酰胺(0.19g,0.56mmol),然后添加N,N-二异丙基乙胺(0.21mL,1.67mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应,并且用二氯甲烷提取。用水、盐水洗涤有机层,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩。通过使用快速色谱(6%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噁唑-5-羧酰胺(0.03g,12%产率):1HNMR(400MHz,DMSO-d6,80℃,VT在80℃)δ11.493(s,1H),8.90(br s,1H),8.80(s,1H),8.40(s,1H),7.72(s,1H)7.22(s,1H),6.69(s,1H),3.97(m,4H),3.45(m,5H),2.80-2.81(m,3H),2.07(m,1H),1.77(s,1H);MS(ES)m/z 408.4(M+H)。
实施例40:3-((3R,5S)-3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

方案38.3-((3R,5S)-3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

步骤1:4-溴-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶的制备

向冷却至0-5℃的氢化钠(10.55g,263mmol,矿物油中60%)在THF(400mL)中的悬浮液分批添加4-溴-1H-吡咯并[2,3-b]吡啶(40g,203mmol)达约20分钟。将反应在0-5℃搅拌30分钟。然后,在20分钟内逐滴添加三异丙基氯硅烷(64.7mL,304mmol),并且将反应加热3小时至70℃。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。分离层,并且用乙酸乙酯重新提取水性层。在无水硫酸钠上干燥合并的有机层,过滤并且在真空中浓缩。使用石油醚作为洗脱剂在230-400目硅胶上通过快速色谱纯化粗化合物,以产生为无色液体的4-溴-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶(70g,97%产率):MS(ES)m/z 355.2(M+H)。
步骤2:4-氟-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶的制备

将4-溴-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶(46g,133mmol)在THF(800mL)中的溶液冷却至-78℃。然后,使用注射器在约1小时内缓慢添加正丁基锂(己烷中1.2M,通过二苯乙酸法滴定)(167mL,200mmol)。将反应混合物在相同温度搅拌15分钟。然后,缓慢添加N-氟苯磺酰胺(84g,266mmol)在THF(235mL)中的溶液,并且将反应在相同温度搅拌1小时。用饱和NH4Cl水溶液淬灭反应,并且用MTBE(235mL)稀释。将反应缓慢升至环境温度,分离两层,并且用MTBE(235mL)提取水性层。在无水硫酸钠上干燥合并的有机层,过滤并且在真空中浓缩。使用230-400目硅胶和石油醚通过快速柱色谱纯化粗化合物,以产生为浅黄色液体的4-氟-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶(15.5g,40.6%产率):MS(ES)m/z 293.4(M+H)。
步骤3:5-溴-4-氟-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶的制备

将4-氟-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶(15g,51.31mmol)在THF(600mL)中的溶液冷却至-78℃。然后,在30分钟内缓慢添加仲丁基锂(环己烷中1.2M,64mL,76.97mmol),并且将混合物搅拌1小时。缓慢添加四溴化碳(30.57g,92.36mmol)在THF(45mL)中的溶液,并且将反应物质在-78℃搅拌1小时。通过缓慢添加饱和NH4Cl水溶液、然后添加MTBE(150mL,10vol)并且加温至环境温度来淬灭反应混合物。分离两层,并且用MTBE(150mL)提取水性层。在无水硫酸钠上干燥合并的有机层,过滤并且在真空中浓缩。使用230-400目硅胶和石油醚通过快速柱色谱纯化粗化合物,以产生为浅黄色液体的5-溴-4-氟-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶(15.6g,82%产率):MS(ES)m/z 371.3;373.3(1:1;M+H)。
步骤4:5-溴-4-氟-1H-吡咯并[2,3-b]吡啶的制备

使用冰浴将5-溴-4-氟-1-(三异丙基甲硅烷基)-1H-吡咯并[2,3-b]吡啶(26g,70.08mmol)在THF(260mL)中的溶液冷却至0-5℃。然后,在30分钟内缓慢添加氟化四丁基铵(THF中1.0M;84mL),并且将反应物质搅拌4小时。用水(260mL)稀释反应混合物,并且用乙酸乙酯(2×130mL)提取。在无水硫酸钠上干燥合并的有机层,过滤并且在真空中浓缩。使用230-400目硅胶和石油醚中15-20%乙酸乙酯通过快速柱纯化粗化合物,以产生为浅黄色固体的5-溴-4-氟-1H-吡咯并[2,3-b]吡啶(11g,73%产率):MS(ES)m/z 215.1;217.1(1:1;M+H)。
步骤5:5-溴-4-氟-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶的制备

使用冰盐混合物将氢化钠(2.94g,73.6mmol,矿物油中60%)在DMF(110mL)中的悬浮液冷却至0-5℃。向上述悬浮液分批添加5-溴-4-氟-1H-吡咯并[2,3-b]吡啶(11g,51.1mmol),并且将反应混合物在相同温度搅拌30分钟。然后,在30分钟内使用另外的漏斗逐滴添加2-(三甲基甲硅烷基)乙氧基甲基氯(13.83g,82mmol),并且将反应在环境温度搅拌4小时。将反应混合物倒入冷水中,并且用乙酸乙酯(2×)提取。在无水硫酸钠上干燥合并的有机层,过滤并且在真空中浓缩。使用石油醚作为洗脱剂使用100-200目硅胶通过快速色谱纯化粗化合物,以产生为浅黄色液体的5-溴-4-氟-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(16g,91%产率):MS(ES)m/z 345.1;347.1(1:1;M+H)。
步骤6:(3R,5S)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯的制备

在氮气气氛下向5-溴-4-氟-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(16g,46.5mmol)在二甲亚砜(96mL)中的经搅拌的溶液添加(3R,5S)-3-氨基-5-甲基哌啶-1-羧酸苄酯(13.85g,55.8mmol)。将反应物质在环境温度搅拌10min,然后添加N,N-二异丙基乙胺(20.7mL,116.2mmol)。将反应混合物缓慢加热至125-130℃,并且在氮气气氛下维持24小时。将反应混合物冷却至室温,并且倒入冷却水(240mL)中,搅拌15分钟,并且添加乙酸乙酯(160mL)。分离有机层,并且用乙酸乙酯(100mL)提取水性层。用水(2×100mL)洗涤合并的有机层,在无水硫酸钠上干燥,过滤并且在真空下浓缩,以提供为非常稠的浆状物质的粗产物。使用100-200目硅胶通过柱色谱纯化粗产物,以获得为浅黄色粘性物质的(3R,5S)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(19.5g,73.3%产率):MS(ES)m/z 573.3;575.4(1:1;M+H)。
步骤7:(3R,5S)-3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯的制备

向(3R,5S)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.6g,1.05mmol)和1-(二氟甲基)-3-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡唑(0.38g,1.57mmol)在1,4-二噁烷(35mL)中的经搅拌的溶液添加双-(三苯基膦)二氯化钯(II)(0.15g,0.21mmol)和2M磷酸三钾(0.90g,4.18mmol)水溶液,并且将混合物在氮气气氛下在80℃加热16小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为粘性浅黄色液体的(3R,5S)-3-({5-[1-(二氟甲基)-1H-吡唑-3-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)-5-甲基哌啶-1-羧酸苄酯(0.52g,81%产率):MS(ES)m/z 611.3(M+H)。
步骤8:5-(1-(二氟甲基)-1H-吡唑-3-基)-N-((3R,5S)-5-甲基哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3R,5S)-3-({5-[1-(二氟甲基)-1H-吡唑-3-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)-5-甲基哌啶-1-羧酸苄酯(0.56g,0.90mmol)在三氟乙酸(5mL)中的溶液在100℃搅拌1小时。将反应冷却至环境温度,并且在真空中浓缩混合物。将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为胶状物的5-(1-(二氟甲基)-1H-吡唑-3-基)-N-((3R,5S)-5-甲基哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.53g,粗品):MS(ES)m/z347.2(M+H。
步骤9:3-((3R,5S)-3-((5-(1-(二氟甲基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.11g,1.3mmol)、1-羟基苯并三唑(0.16g,1.04mmol)和N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.25g,1.30mmol)在二氯甲烷(10mL)中的溶液在环境温度搅拌5分钟。然后,添加5-(1-(二氟甲基)-1H-吡唑-3-基)-N-((3R,5S)-5-甲基哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.23g,0.86mmol),然后添加三乙胺(0.36mL,2.60mmol),并且然后将所得的混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用反相色谱纯化粗材料,以获得为白色固体的3-[(3R,5S)-3-({5-[1-(二氟甲基)-1H-吡唑-3-基]-1H-吡咯并[2,3-b]吡啶-4-基}氨基)-5-甲基哌啶-1-基]-3-氧代丙腈(0.04g,11%产率):1H NMR(400MHz,DMSO-d6,VT在90℃)δ11.17(br s,1H),8.14-8.37(m,3H),7.63-7.94(m,1H),7.16(s,1H),7.01(s,1H),6.67(s,1H),4.80-4.83(m,1H),3.60-4.36(m,3H),2.66-2.83(m,1H),2.18-2.39(m,2H),1.65-1.93(m,1H),1.11-1.26(m,2H),0.91(d,J=6.4Hz,3H);MS(ES)m/z 414.4(M+H)。
分析条件:
流速:20.0mL/min
柱:Sunfire C18 19*150*5微米
流动相(A):水中0.1%TFA
流动相(B):MeCN
实施例41:(R)-3-(3-((5-(异噻唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案39.(R)-3-(3-((5-(异噻唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(异噻唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.72g,1.25mmol)和3-溴异噻唑(0.3g,1.63mmol)在1,4-二噁烷(20mL)中的经搅拌的溶液添加四(三苯基膦)钯(0)(0.18g,0.25mmol),然后添加2M碳酸钠(0.4g,3.77mmol)水溶液,并且将反应混合物在100℃加热16小时。在冷却之后,用乙酸乙酯稀释反应混合物,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(25%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色液体的(R)-3-((5-(异噻唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.23g,35%产率):MS(ES)m/z 529.9(M+H)。
步骤2:(R)-5-(异噻唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(异噻唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.3g,0.56mmol)在二氯甲烷:三氟乙酸(3.0mL:3.0mL)中的经搅拌的溶液在环境温度搅拌5小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨(3.0mL:3.0mL,水中23%)中,并且在环境温度搅拌16小时。将反应混合物浓缩至干,以提供为粘性棕色液体的(R)-5-(异噻唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.22g,粗品):MS(ES)m/z 300.0(M+H)。
步骤3:(R)-3-(3-((5-(异噻唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.1g,1.00mmol)、1-羟基苯并三唑(0.15g,1.10mmol)和N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.21g,1.10mmol)在二氯甲烷(20mL)中的溶液在环境温度搅拌5分钟。然后,添加(R)-5-(异噻唑-3-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.22g,0.73mmol),然后添加N,N-二异丙基乙胺(0.4mL,2.20mmol),并且将所得的混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(3%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(异噻唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.04g,16%产率):1H NMR(400MHz,DMSO-d6)δ11.48(br s,1H),9.58(d,J=6.0Hz,1H),9.08(d,J=4.4Hz,1H),8.6(d,J=12.0Hz,1H),7.99(d,J=4.8Hz,1H),7.19(s,1H),6.67(s,1H),4.39(s,1H),4.02-3.95(m,6H),2.02(s,1H),1.72-1.60(m,3H);MS(ES)m/z 367.1(M+H)。
实施例42:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

方案40.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

步骤1:4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-硫代羧酰胺的制备

将4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(2.0g,6.1mmol)和劳森试剂(3.73g,9.20mmol)在四氢呋喃(20mL)中的悬浮液在环境温度搅拌30分钟。用水稀释反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱纯化粗品,以提供为黄色固体的4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-硫代羧酰胺(1g,50%产率):MS(ES)m/z 342.1(M+H)。
步骤2:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

向4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-硫代羧酰胺(0.9g,2.63mmol)和2-氯-3-氧代丙酸乙酯(0.39g,2.63mmol)在甲苯(10mL)中的经搅拌的溶液添加硫酸镁(0.317g,2.63mmol),并且将混合物在氮气气氛下加热1小时至110℃。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(20%乙酸乙酯/己烷)纯化粗材料,以提供为黄色固体的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.8g,70%产率):MS(ES)m/z 438.1(M+H)。
步骤3:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

在20mL微波小瓶中,将2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.60g,1.37mmol)、(R)-3-氨基哌啶-1-羧酸叔丁酯(0.411g,2.05mmol)、乙酸钯(II)(0.05g,0.20mmol)、(2,2’-双(二苯基膦)-1,1’-联萘基)(0.13g,0.20mmol)、叔丁醇钠(0.39g,4.11mmol)和1,4-二噁烷(7mL)合并,并且使所得的混合物在80℃经受微波辐照1小时。将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗品,以提供为粘性棕色液体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.23g,28%产率):MS(ES)m/z 602.3(M+H)。
步骤4:(R)-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

将2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.15g,0.24mmol)在二氯甲烷:三氟乙酸(3.0mL:3.0mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(5.0mL:4.0mL,水中23%)中,并且在环境温度搅拌16小时。在真空中浓缩反应,以提供为棕色固体的(R)-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.15g,粗品):MS(ES)m/z 372.1(M+H)。
步骤5:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

向氰基乙酸(0.05g,0.60mmol)、1-羟基-2-吡啶酮(0.05g,0.48mmol)和2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基四氟硼酸铵(0.15g,0.48mmol)在N-甲基吡咯烷酮(5mL)中的经搅拌的溶液添加2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.15g,0.40mmol),然后添加三乙胺(0.16mL,1.21mmol),并且在环境温度继续搅拌2小时。用水稀释反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(10%甲醇/二氯甲烷)纯化粗品,以提供为浅黄色固体的(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.02g,10%产率):1H NMR(400MHz,DMSO-d6)δ11.57(s,1H),9.71(m,1H),8.43-8.34(m,2H),7.20(s,1H),6.69(s,1H),4.30-4.35(q,J=6.4Hz,2H),4.02(m,1H),3.79-3.87(m,2H),3.56(m,1H),3.39(m,1H),2.03(m,2H),1.77(m,2H),1.62(m,2H),1.29-1.33(t,J=7.2Hz,3H);MS(ES)m/z 439.1(M+H)。
实施例43:(R)-3-(3-((5-(2-甲氧基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案41.(R)-3-(3-((5-(2-甲氧基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(2-甲氧基嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.36g,2.38mmol)和4-溴-2-甲氧基嘧啶(0.30g,1.58mmol)在1,4-二噁烷(15mL)中的经搅拌的溶液添加双(三苯基膦)二氯化钯(II)(0.22g,0.32mmol)和2M碳酸钾(0.87g,6.32mmol)水溶液,并且将混合物在105℃搅拌15小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为粘性浅黄色液体的(R)-3-((5-(2-甲氧基嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.62g,70%产率):MS(ES)m/z 555.5(M+H)。
步骤2:(R)-5-(2-甲氧基嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(2-甲氧基嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.3g,0.54mmol)在二氯甲烷:三氟乙酸(3mL:3mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且然后在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为胶状棕色液体的(R)-5-(2-甲氧基嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.18g,粗品):MS(ES)m/z 325.2(M+H)。
步骤3:(R)-3-(3-((5-(2-甲氧基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.03g,0.32mmol)、2-羟基吡啶-N-氧化物(0.03g,0.32mmol)和O-(苯并三唑-1-基)-N,N,N’,N’-四甲基脲四氟硼酸酯(0.11g,0.32mmol)在N-甲基吡咯烷酮(4mL)中的溶液在环境温度搅拌5分钟。然后,添加(R)-5-(2-甲氧基嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.08g,0.25mmol),然后添加三乙胺(0.1mL,0.74mmol)。将所得的混合物在环境温度搅拌16小时,用水淬灭并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(2-甲氧基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.04g,15%产率):1H NMR(400MHz,DMSO-d6,VT在100℃)δ11.27(br s,1H),9.89(br s,1H),8.44-8.59(m,2H),7.55(s,1H),7.15(s,1H),6.66(s,1H),4.16-4.27(m,2H),4.39(s,2H),3.83-3.87(m,2H),3.45-3.55(m,2H),2.05-2.10(m,2H),1.58-1.95(m,4H);MS(ES)m/z 392.2(M+H)。
实施例44:(R)-3-(3-((5-(6-氨基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案42.(R)-3-(3-((5-(6-氨基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(6-溴吡啶-2-基)氨基甲酸叔丁酯的制备

在-78℃向6-溴吡啶-2-胺(0.70g,4.09mmol)在四氢呋喃(15mL)中的溶液添加双(三甲基甲硅烷基)氨化钠(6.1mL,6.13mmol,THF中1.0M),并且将混合物搅拌0.5小时。在-78℃添加二碳酸二叔丁酯(0.98g,4.50mmol)溶解在四氢呋喃(5mL)中的溶液,并且然后将混合物缓慢加温至环境温度并且搅拌16小时。用饱和氯化铵淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(20%乙酸乙酯/己烷)纯化粗材料,以提供为无色半固体的(6-溴吡啶-2-基)氨基甲酸叔丁酯:1H NMR(400MHz,DMSO-d6)δ10.07(s,1H),7.76-7.78(m,1H),7.62-7.66(m,1H),7.20-7.22(m,1H),1.44(s,9H);MS(ES)m/z 174.2(M-Boc)。
步骤2:(R)-3-((5-(6-((叔丁氧基羰基)氨基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.2g,2.19mmol)和(6-溴吡啶-2-基)氨基甲酸叔丁酯(0.4g,1.58mmol)在1,4-二噁烷(40mL)中的经搅拌的溶液添加双(三苯基膦)二氯化钯(II)(0.2g,0.29mmol)和2M碳酸钾(0.81g,5.84mmol)水溶液,并且将混合物在100℃搅拌16小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为胶状浅色液体的(R)-3-((5-(6-((叔丁氧基羰基)氨基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.42g,45%产率):MS(ES)m/z 639.4(M+H)。
步骤3:(R)-5-(6-氨基吡啶-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(6-((叔丁氧基羰基)氨基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.42g,0.66mmol)在二氯甲烷:三氟乙酸(4mL:4mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL,水中23%)中,并且将溶液在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为灰白色固体的(R)-5-(6-氨基吡啶-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.45g,粗品):MS(ES)m/z 309.2(M+H)。
步骤4:(R)-3-(3-((5-(6-氨基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.07g,0.84mmol)、2-羟基吡啶-N-氧化物(0.08g,0.71mmol)和2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基四氟硼酸铵(0.27g,0.84mmol)在N-甲基吡咯烷酮(5mL)中的溶液在环境温度搅拌5分钟。然后,添加(R)-5-(6-氨基吡啶-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.08g,0.25mmol),然后添加三乙胺(0.27mL,1.95mmol),并且将所得的混合物在环境温度搅拌18小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(10%甲醇/二氯甲烷)纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(6-氨基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.11g,45%产率):1H NMR(400MHz,DMSO-d6,VT在70℃)δ11.11(br s,1H),9.58(br s,1H),8.19(s,1H),7.44(s,1H),7.10(s,1H),6.85(s,1H),6.57(s,1H),634-6.36(m,1H),5.79(s,2H),4.20-4.35(m,1H),3.95-4.07(m,1H),3.45-3.60(m,2H),2.10-2.15(m,2H),1.60-1.75(m,2H),1.15-1.30(m,3H);MS(ES)m/z 376.2(M+H)。
实施例45:3-((3R)-3-((5-(5-(甲基亚磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案43.3-((3R)-3-((5-(5-(甲基亚磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:5-(甲基硫代)噻唑-2-胺的制备

将5-溴噻唑-2-胺(4.5g,25.14mmol)和甲硫醇钠(3.52g,50.28mmol)在N,N-二甲基甲酰胺(30mL)中的溶液在50℃搅拌5小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(30%乙酸乙酯/己烷)纯化粗品,以提供为棕色固体的5-(甲基硫代)噻唑-2-胺(1.5g,41%产率):1H NMR(400MHz,CDCl3)δ7.08(s,1H),2.36(s,3H)。
步骤2:2-溴-5-(甲基硫代)噻唑的制备

在0℃向5-(甲基硫代)噻唑-2-胺(1.5g,10.27mmol)在乙腈(60mL)中的经搅拌的溶液添加亚硝酸叔丁酯(1.58g,15.40mmol),并且将混合物搅拌15分钟。添加溴化铜(II)(6.87g,30.81mmol),并且将混合物在0℃搅拌3小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(5%乙酸乙酯/己烷)纯化粗品,以提供为橙色油状物的2-溴-5-(甲基硫代)噻唑(0.75g,34%产率):MS(ES)m/z 209.8;211.0(1:1;M+H)。
步骤3:2-溴-5-甲烷亚磺酰基-1,3-噻唑的制备

将2-溴-5-(甲基硫代)-1,3-噻唑(0.59g,2.81mmol)和间氯过氧苯甲酸(0.48g,2.81mmol)在二氯甲烷(20mL)中的溶液在环境温度搅拌1小时。用饱和碳酸氢盐溶液淬灭反应混合物,并且搅拌30分钟。分离有机层,用水、盐水洗涤,在硫酸钠上干燥,过滤并且在真空中浓缩,以获得为粘性无色液体的2-溴-5-甲烷亚磺酰基-1,3-噻唑(0.35g粗品):MS(ES)m/z 226.0;228.0(1:1;M+H)。
步骤4:(3R)-3-((5-(5-(甲基亚磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(3R)-1-甲基-N-[5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(1.5g,2.62mmol)和2-溴-5-甲烷亚磺酰基-1,3-噻唑(0.39g,1.75mmol)在N,N-二甲基甲酰胺(4.00mL)中的经搅拌的溶液添加[1,1’-双(二苯基膦)二茂铁]二氯化钯(II)二氯甲烷络合物(0.28g,0.35mmol)和氟化铯(1.06g,6.99mmol),并且将所得的混合物在80℃加热5小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(50%乙酸乙酯/己烷)纯化粗品,以提供为棕色液体的(3R)-3-{[5-(5-甲烷亚磺酰基-1,3-噻唑-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸叔丁酯(0.12g,11%产率):MS(ES)m/z593.4(M+H)。
步骤5:5-(5-(甲基亚磺酰基)噻唑-2-基)-N-((R)-哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3R)-3-{[5-(5-甲烷亚磺酰基-1,3-噻唑-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸叔丁酯(0.12g,0.20mmol)在二氯甲烷:三氟乙酸(2mL:2mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且然后在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为胶状固体的(3R)-N-[5-(5-甲烷亚磺酰基-1,3-噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.2g,粗品):MS(ES)m/z 362.1(M+H)。
步骤6:3-((3R)-3-((5-(5-(甲基亚磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.04g,0.45mmol)和1-羟基苯并三唑(0.05g,0.30mmol)在二氯甲烷(5mL)中的溶液搅拌2分钟。然后,添加(3R)-N-[5-(5-甲烷亚磺酰基-1,3-噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.1g,0.3mmol)、N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.08g,0.41mmol)和三乙胺(0.11g,1.11mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为浅黄色固体的3-((3R)-3-((5-(5-(甲基亚磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(5mg,4%产率):1H NMR(400MHz,DMSO-d6)δ11.73(br s,1H),9.65(brs,1H),8.44(s,1H),8.17(s,1H),8.07(s,1H),7.24(s,1H),6.71(s,1H),5.91-6.11(m,1H),5.31(s,1H),4.12-4.50(m,1H),3.75-4.22(m,2H),3.42-3.72(m,2H),2.95-3.12(m,3H),1.95-2.10(m,1H),1.45-1.82(m,2H);MS(ES)m/z 429.2(M+H)。
分析条件:
流速:0.3mL/min
柱Ascentis Express C18(50mm×2.1mm×2.73μm)
流动相(A):水中0.1%甲酸
流动相(B):MeCN
实施例46:(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案44.(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:2-溴-5-(甲基磺酰基)噻唑的制备

将2-溴-5-(甲基硫代)-1,3-噻唑(0.59g,2.81mmol)和间氯过氧苯甲酸(1.2g,7.02mmol)在二氯甲烷(20mL)中的溶液在环境温度搅拌3小时。用饱和碳酸氢盐溶液淬灭反应混合物,并且搅拌30分钟。分离有机层,用水、盐水洗涤,在硫酸钠上干燥,过滤并且在真空中浓缩,以获得为灰白色固体的2-溴-5-甲烷磺酰基-1,3-噻唑(0.35g,粗品)(0.45g,70%产率):1H NMR(400MHz,DMSO-d6)δ8.30(s,1H),3.45(s,3H)。
步骤2:(R)-3-((5-(5-(甲基磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.7g,3.09mmol)、碳酸钾(1.14g,8.28mmol)、双(三苯基膦)二氯化钯(II)(0.30g,0.41mmol)和2-溴-5-(甲基磺酰基)噻唑(0.5g,2.07mmol)在1,4-二噁烷(50mL)中的溶液在氮气气氛下在密封管中在110℃加热。在16小时之后,将反应冷却至环境温度,用水稀释,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(乙酸乙酯/己烷)纯化粗材料,以提供为粘性棕色液体的(R)-3-((5-(5-(甲基磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.55g,30%产率):MS(ES)m/z 608.3(M+H)。
步骤3:(R)-5-(5-(甲基磺酰基)噻唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(5-(甲基磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.55g,0.91mmol)在二氯甲烷:三氟乙酸(5.0mL:5.0mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为胶状棕色固体的(R)-5-(5-(甲基磺酰基)噻唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.6g,粗品):MS(ES)m/z 378.1(M+H)。
步骤4:(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向氰基乙酸(0.03g,0.39mmol)、1-羟基-2-吡啶酮(0.03g,0.31mmol)和2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基四氟硼酸铵(0.10g,0.31mmol)在N-甲基吡咯烷酮(5mL)中的经搅拌的溶液添加(R)-5-(5-(甲基磺酰基)噻唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.1g,0.26mmol),然后添加三乙胺(0.11mL,0.79mmol),并且将所得的混合物在环境温度搅拌2小时。用水稀释反应混合物,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过柱色谱法纯化粗品,以提供为黄色固体的(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.01g,10%产率):1H NMR(400MHz,DMSO-d6,VT在100℃)δ11.58(br s,1H),9.65(br s,1H),8.42(s,1H),8.24(s,1H),7.21(s,1H),6.71(s,1H),3.54-4.44(m,6H),3.42(s,3H),1.21-2.04(m,5H);MS(ES)m/z 445.1(M+H)。
实施例47:(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

方案45.(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

向(R)-5-(5-(甲基磺酰基)噻唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.1g,0.26mmol,实施例46、步骤3)在乙醇(3mL)中的溶液添加丙烯腈(0.14g,2.65mmol)和三乙胺(0.06mL,0.79mmol),并且将溶液在80℃搅拌3小时。将反应冷却至环境温度,并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.03g,25%):1H NMR(400MHz,DMSO-d6)δ9.83(br s,1H),9.12(br s,1H),8.45(s,1H),8.24(s,1H),7.10(s,1H),6.69(s,1H),4.32(s,1H),3.24(s,3H),2.95-3.07(m,1H),2.65-2.85(m,2H),2.37-2.67(m,4H),1.80-2.04(m,2H),1.16-1.30(s,3H);MS(ES)m/z 431.2(M+H)。
分析条件:
流速:20.0mL/min
柱:Ascentis Express C18(50mm×2.1mm×2.7μm)
流动相(A):水中1%氨
流动相(B):MeCN
实施例48:(R)-3-(3-((5-(5-(甲基磺酰基)噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案46.(R)-3-(3-((5-(5-(甲基磺酰基)噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯的制备

在0℃向氢化钠(2.14g,89.28mmol,矿物油中60%)在N,N-二甲基甲酰胺(75mL)中的经搅拌的悬浮液添加4-氯-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(10g,44.64mmol)。在搅拌0.5小时之后,在0℃添加2-(三甲基甲硅烷基)乙氧基甲基氯(8.7mL,55.34mmol),并且将反应搅拌2小时。用冰水淬灭反应,并且用乙酸乙酯提取。在无水硫酸钠上干燥有机层,过滤并且浓缩。使用快速色谱(15%乙酸乙酯/己烷)纯化粗品,以提供为无色液体的4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(7.8g,49%产率):MS(ES)m/z 355.1(M+H)。
步骤2:4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸的制备

向4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(7.8,21.91mmol)在甲醇(80mL)中的溶液添加2M氢氧化钠(4.38g,109.55mmol)水溶液,并且将混合物在环境温度搅拌5小时。浓缩反应去除挥发物以获得剩余物,剩余物被溶解在水中并且用1N盐酸酸化以调整pH~3。用乙酸乙酯提取水性层,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为灰白色固体的4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(6.3g,88%产率):MS(ES)m/z:327.1(M+H)。
步骤3:4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺的制备

将4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(6.3g,19.32mmol)和羰基二咪唑(4.7g,28.98mmol)在二甲基甲酰胺(30mL)中的溶液在环境温度搅拌1小时。添加氨(10mL,水中23%),然后添加N,N-二异丙基乙胺(5.0mL,28.98mmol),并且将溶液搅拌3小时。用冰水淬灭反应,并且用乙酸乙酯提取。在无水硫酸钠上干燥有机层,过滤并且浓缩。使用快速色谱(30%乙酸乙酯/己烷)纯化粗品,以提供灰白色固体(4.56g,72%,产率):MS(ES)m/z:326.1(M+H)。
步骤4:4-氯-N-(2,2,2-三氯-1-羟基乙基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺的制备

将4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(3.0g,9.21mmol)和2,2,2-三氯乙醛(1.36g,9.21mmol)的固体混合物加热至90℃直至完全熔融。将反应混合物冷却至环境温度,并且使用快速色谱(30%乙酸乙酯/己烷)纯化粗产物,以提供为灰白色固体的4-氯-N-(2,2,2-三氯-1-羟基乙基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(2g,47%产率):MS(ES)m/z:474.2(M+H)。
步骤5:4-氯-N-(1,2,2,2-四氯乙基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺的制备

向4-氯-N-(2,2,2-三氯-1-羟基乙基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(1.30g,2.75mmol)在二氯甲烷(30mL)中的溶液添加亚硫酰氯(0.24mL,3.30mmol),并且在环境温度搅拌溶液。在3小时之后,在氮气气氛下在真空中浓缩反应,以提供为无色胶状物的4-氯-N-(1,2,2,2-四氯乙基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(1.35g,粗品)。将粗产物用于下一步骤而无需进一步纯化。
步骤6:(2,2-二氯-1-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺基)乙烯基)三苯基氯化膦的制备

向4-氯-N-(1,2,2,2-四氯乙基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(1.35g,2.75mmol)在甲苯(30mL)中的经搅拌的溶液添加三苯基膦(0.72g,2.75mmol),并且将溶液加热至80℃。在3小时之后,将反应冷却至环境温度,并且在真空中浓缩,以提供为无色胶状物的(2,2-二氯-1-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺基)乙烯基)三苯基氯化膦(2.07g,粗品):MS(ES)m/z:682.4(M-Cl)。
步骤7:2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-4-(三苯基-λ5-膦乙烯基)-4,5-二氢-1,3-噁唑-5-硫酮的制备

向(2,2-二氯-1-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-酰胺基)乙烯基)三苯基氯化膦(2.0g,2.78mmol)在甲醇(45mL)中的经搅拌的溶液添加硫氢化钠(0.5g,8.80mmol),并且在环境温度搅拌混合物。在3小时之后,用乙酸乙酯稀释反应混合物,并且用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且浓缩。使用快速色谱(50%乙酸乙酯/己烷)纯化粗品,以提供为浅黄色浓稠液体的2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-4-(三苯基-λ5-膦乙烯基)-4,5-二氢-1,3-噁唑-5-硫酮(1.23g,65%产率):MS(ES)m/z:642.2(M+H)。
步骤8:(2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)-5-(甲基硫代)噁唑-4-基)三苯基碘化膦的制备

向2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-4-(三苯基-λ5-膦乙烯基)-4,5-二氢-1,3-噁唑-5-硫酮(1.35g,2.10mmol)在甲醇(30mL)中的经搅拌的溶液添加碘甲烷(0.9g,6.31mmol),并且将混合物在环境温度搅拌16小时。将反应混合物在真空中浓缩至干,以提供为粘性浅黄色液体的[2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-5-(甲基硫烷基)-1,3-噁唑-4-基]三苯基碘化膦(1.50g,粗品):MS(ES)m/z:656.2(M-I)。
步骤9:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)-5-(甲基硫代)噁唑的制备

向2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-5-(甲基硫烷基)-1,3-噁唑-4-基]三苯基碘化膦(1.50g,2.28mL)在甲醇(2mL)中的经搅拌的溶液添加2M氢氧化钠(0.36g,9.13mmol)水溶液,并且将溶液在环境温度搅拌2小时。用乙酸乙酯稀释反应混合物,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(10%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色液体的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)-5-(甲基硫代)噁唑(0.22g,24%产率):MS(ES)m/z:396.1(M+H)。
步骤10:(R)-3-((5-(5-(甲基硫代)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

在10mL小瓶中,将2-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-5-(甲基硫烷基)-1,3-噁唑(0.4g,1.01mmol)、(3R)-3-氨基哌啶-1-羧酸叔丁酯(0.4g,2.02mmol)和N,N-二异丙基乙胺(0.39g,3.03mmol)在N-甲基-2-吡咯烷酮(10mL)中的溶液加热16小时至140℃。将反应混合物冷却至环境温度,用乙酸乙酯稀释,并且用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为粘性棕色液体的(3R)-3-({5-[5-(甲基硫烷基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)哌啶-1-羧酸叔丁酯(0.41g,72%产率):MS(ES)m/z:560.5(M+H)。
步骤11:(R)-3-((5-(5-(甲基磺酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(3R)-3-({5-[5-(甲基硫烷基)-1,3-噁唑-2-基]-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基}氨基)哌啶-1-羧酸叔丁酯(0.38g,0.68mmol)在四氢呋喃:水(10mL:3mL)中的经搅拌的溶液添加单过硫酸氢钾复合盐(1.68g,2.72mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗品,以提供为浅黄色胶状固体的(3R)-3-{[5-(5-甲烷磺酰基-1,3-噁唑-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸叔丁酯(0.32g,80%产率):MS(ES)m/z:592.5(M+H)。
步骤12:(R)-5-(5-(甲基磺酰基)噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3R)-3-{[5-(5-甲烷磺酰基-1,3-噁唑-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸叔丁酯(0.26g,439μmol)在二氯甲烷:三氟乙酸(5mL:5mL)中的溶液在环境温度搅拌3小时。在真空中浓缩混合物,将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL,水中23%)中,并且将溶液在环境温度搅拌16小时。将反应混合物在真空中浓缩至干,以提供为无色胶状固体的(3R)-N-[5-(5-甲烷磺酰基-1,3-噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.32g,80%产率):MS(ES)m/z:362.2(M+H)。
步骤13:(R)-3-(3-((5-(5-(甲基磺酰基)噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.04g,0.45mmol)和1-羟基苯并三唑(0.06g,0.4mmol)在二氯甲烷(10mL)中的溶液搅拌2分钟。然后,添加(3R)-N-[5-(5-甲烷磺酰基-1,3-噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.11g,0.30mmol)、N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.09g,0.46mmol)和三乙胺(0.18mL,1.12mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的3-[(3R)-3-{[5-(5-甲烷磺酰基-1,3-噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-基]-3-氧代丙腈(0.03g,60%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.58(br s,1H),8.74(d,J=5.6Hz,1H),8.62(s,1H),7.93(s,1H),7.23(s,1H),6.71(s,1H),4.24-4.43(m,1H),3.71-4.05(m,3H),3.41-3.55(m,6H),2.80-2.11(m,1H),1.55-1.85(m,3H);MS(ES)m/z 429.1(M+H)。
分析条件:
流速:0.3mL/min
柱:Ascentis Express C18(50mm×2.1mm×2.7μm)
流动相(A):水中0.1%甲酸
流动相(B):MeCN
实施例49:(R)-3-(3-((5-(2-(甲基硫代)嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案47.(R)-3-(3-((5-(2-(甲基硫代)嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(2-(甲基硫代)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向4-氯-2-(甲基硫代)嘧啶(0.30g,1.87mmol)和(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.07g,1.87mmol)在1,4-二噁烷(40mL)中的经搅拌的溶液添加[1,1’-双(二苯基膦)二茂铁]二氯化钯(II)二氯甲烷络合物(0.08g,0.09mmol)和碳酸钾(0.64g,4.68mmol),并且将混合物加热16小时至100℃。将反应冷却至环境温度,并且用乙酸乙酯和水稀释。分离有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗剩余物,以提供为黄色液体的(R)-3-((5-(2-(甲基硫代)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.5g,50%产率):MS(ES)m/z 571.4(M+H)。
步骤2:(R)-5-(2-(甲基硫代)嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(2-(甲基硫代)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.35g,0.61mmol)在二氯甲烷:三氟乙酸(3mL:6mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,并且将剩余物溶解在1,4-二噁烷:氨水(3mL:5mL,水中23%)中,并且将反应混合物在环境温度搅拌16小时。在真空中浓缩反应,以提供为黄色固体的(R)-5-(2-(甲基硫代)嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.3g,粗品):MS(ES)m/z 341.1(M+H)。
步骤3:(R)-3-(3-((5-(2-(甲基硫代)嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.11g,1.32mmol)和(1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.40g,1.05mmol)在N,N-二甲基甲酰胺(60mL)中的溶液搅拌3分钟。然后,添加(R)-5-(2-(甲基硫代)嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.30g,0.88mmol),然后添加N,N-二异丙基乙胺(0.48mL,2.64mmol)。将反应混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,并且用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相色谱纯化粗剩余物,以提供为浅黄色固体的(R)-3-(3-((5-(2-(甲基硫代)嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.03g,2%产率):1HNMR(400MHz,DMSO-d6,)δ11.61(s,1H),9.90-9.92(m,1H),8.50-8.54(m,2H),7.70(d,J=5.6Hz,1H),7.20-7.21(m,1H),6.70(s,1H),4.30-4.33(m,1H),4.05-4.11(m,1H),3.68-3.93(m,2H),3.48-3.52(m,1H),3.11-3.23(m,1H),2.98-3.03(m,1H),2.56(s,3H),2.15-2.20(m,1H),1.80-1.90(m,1H),1.61-1.70(m,2H);MS(ES)m/z 408.1(M+H)。
分析条件:
流速:0.3mL/min
柱:BEH C18(100mm×2.1mm×1.7μm)
流动相(A):水中0.1%甲酸
流动相(B):MeCN
实施例50:2-氰基-N-(1-(4-(4-(((R)-1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)嘧啶-2-基)吡咯烷-3-基)-N-甲基乙酰胺的制备

方案48.2-氰基-N-(1-(4-(4-(((R)-1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)嘧啶-2-基)吡咯烷-3-基)-N-甲基乙酰胺的制备

步骤1:(R)-3-((5-(2-(甲基磺酰基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-(2-(甲基硫代)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.6g,1.05mmol,实施例43、步骤1)在四氢呋喃:水(6mL:3mL)中的溶液添加单过硫酸氢钾复合盐(0.64g,4.21mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(60%乙酸乙酯/己烷)纯化粗品,以提供为棕色油状物的(R)-3-((5-(2-(甲基磺酰基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,39%产率):MS(ES)m/z 603.6(M+1)。
步骤2:(3R)-3-((5-(2-(3-((叔丁氧基羰基)(甲基)氨基)吡咯烷-1-基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-(2-(甲基磺酰基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸酯(0.1g,0.17mmol)、甲基(吡咯烷-3-基)氨基甲酸叔丁酯(0.04g,0.20mmol)和N,N-二异丙基乙胺(0.06mL,0.34mmol)在N,N-二甲基甲酰胺(2mL)中的溶液在60℃搅拌5小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(20%乙酸乙酯/己烷)纯化粗品,以提供为棕色胶状固体的(3R)-3-((5-(2-(3-((叔丁氧基羰基)(甲基)氨基)吡咯烷-1-基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.07g,58%产率):MS(ES)m/z 723.4(M+1)。
步骤3:5-(2-(3-(甲基氨基)吡咯烷-1-基)嘧啶-4-基)-N-((R)-哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3R)-3-((5-(2-(3-((叔丁氧基羰基)(甲基)氨基)吡咯烷-1-基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.15mg,0.21mmol)在二氯甲烷:三氟乙酸(2mL:2mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且然后在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为浅黄色浓稠液体的5-(2-(3-(甲基氨基)吡咯烷-1-基)嘧啶-4-基)-N-((R)-哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.07g,粗品):MS(ES)m/z 393.3(M+H)。
步骤4:2-氰基-N-(1-(4-(4-(((R)-1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)嘧啶-2-基)吡咯烷-3-基)-N-甲基乙酰胺的制备

将2-氰基乙酸(0.02g,0.23mmol)、1-羟基苯并三唑(0.03g,0.20mmol)和N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.08g,0.41mmol)在二氯甲烷(5mL)中的溶液在室温搅拌3分钟。然后,添加5-(2-(3-(甲基氨基)吡咯烷-1-基)嘧啶-4-基)-N-((R)-哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.07g,0.17mmol)和三乙胺(0.07mL,0.51mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为浅黄色固体的2-氰基-N-(1-(4-(4-(((R)-1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)嘧啶-2-基)吡咯烷-3-基)-N-甲基乙酰胺(0.02g,21%产率):1HNMR(400MHz,DMSO-d6,VT在100℃)δ11.23(br s,1H),9.49(br s,1H),8.38(s,1H),8.29(d,J=5.2Hz,1H),7.14(s,1H),7.04(d,J=5.2Hz,1H),6.65(s,1H),4.50-5.00(m,2H),3.72-4.20(m,7H),3.40-3.60(m,2H),2.88(s,3H),2.12-2.27(m,4H),1.45-1.80(m,5H);MS(ES)m/z 527.5(M+H)。
分析条件:
流速:0.8mL/min
柱:XB C18(100mm×4.6mm×3.5μm)
流动相(A):水中0.1%氨
流动相(B):MeCN
实施例51:(R)-3-(3-((5-(2-氨基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案49.(R)-3-(3-((5-(2-氨基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-5-(2-氨基嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(2-(甲基磺酰基)嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,0.41mmol,实施例50,步骤1)在二氯甲烷:三氟乙酸(3mL:3mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL,水中23%)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为粘性棕色液体的(R)-5-(2-氨基嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.3g,粗品):MS(ES)m/z 310.2(M+H)。
步骤2:(R)-3-(3-((5-(2-氨基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.04g,0.5mmol)、1-羟基苯并三唑(0.06g,0.45mmol)和N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.11g,0.58mmol)在二氯甲烷(5mL)中的溶液在室温搅拌3分钟。然后,添加(R)-5-(2-氨基嘧啶-4-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.12g,0.39mmol),然后添加三乙胺(0.27mL,1.95mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(2-氨基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.02g,13%产率):1H NMR(400MHz,DMSO-d6,VT在100℃)δ11.11(br s,1H),9.95(brs,1H),8.37(s,1H),8.17(s,1H),7.12(s,1H),7.00(s,1H),6.26(s,1H),6.22(s,2H),4.20-4.42(m,2H),3.20-3.80(m,5H),2.00-2.07(m,1H),1.45-1.80(m,3H);MS(ES)m/z377.4(M+H)。
分析条件:
流速:0.8mL/min
柱:X-Bridge C18(100mm×4.6mm×3.5μm)
流动相(A):水中0.1%氨
流动相(B):MeCN
实施例52:3-((3S,5R)-3-甲基-5-((5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案50.3-((3S,5R)-3-甲基-5-((5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:4-氯-N-甲氧基-N-甲基-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺的制备

将4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酸(2.00g,6.12mmol)和(1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(3.49g,9.18mmol)在N,N-二甲基甲酰胺(25.0mL)中的溶液在环境温度搅拌3分钟。然后,添加N,O-二甲基盐酸羟胺(1.49g,15.3mmol),然后添加三乙胺(4.28mL,30.6mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯和水稀释反应混合物。分离有机层,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色液体的4-氯-N-甲氧基-N-甲基-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(1.82g,80%产率):MS(ES)m/z 370.2(M+H)。
步骤2:1-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)丙-2-炔-1-酮的制备

在0℃向4-氯-N-甲氧基-N-甲基-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(2.20g,5.95mmol)在四氢呋喃(25mL)中的溶液添加溴(乙炔基)镁(18mL,8.92mmol,THF中3M溶液)。将反应混合物加温至环境温度,并且搅拌16小时。用饱和氯化铵水溶液淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色半固体的1-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)丙-2-炔-1-酮(1.25g,63%产率):MS(ES)m/z 335.1(M+H)。
步骤3:4-氯-5-(2-甲基嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶的制备

向1-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)丙-2-炔-1-酮(1.20g,3.58mmol)的经搅拌的悬浮液添加乙腈(10mL)中的N-氯乙脒(0.5mg,5.38mmol)和碳酸钠(1.14g,10.8mmol),并且使混合物在120℃经受微波辐照1小时。将反应冷却至环境温度,并且用乙酸乙酯和水稀释。分离有机层,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且浓缩以获得浓稠胶状物。使用快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为棕色液体的4-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-2-甲基嘧啶(0.72g,54%产率):MS(ES)m/z 375.1(M+H)。
步骤4:(3S,5R)-3-甲基-5-((5-(2-甲基嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

在20mL微波小瓶中,使4-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)-2-甲基嘧啶(0.6g,1.60mmol)、(3R,5S)-3-氨基-5-甲基哌啶-1-羧酸苄酯(0.6g,2.40mmol)和N,N-二异丙基乙胺(0.6mL,3.20mmol)在N-甲基吡咯烷-2-酮(7mL)中的溶液在190℃经受微波辐照6小时。将反应冷却至环境温度,用乙酸乙酯和水稀释。分离有机层,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为棕色浓稠液体的(3S,5R)-3-甲基-5-{[5-(2-甲基嘧啶-4-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸苄酯(0.34g,36%产率):MS(ES)m/z 587.6(M+H)。
步骤5:N-((3R,5S)-5-甲基哌啶-3-基)-5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3S,5R)-3-甲基-5-{[5-(2-甲基嘧啶-4-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸苄酯(0.33g,0.56mmol)在三氟乙酸(5mL)中的溶液在密封管中在100℃搅拌1小时。在1小时之后,将反应混合物冷却至环境温度,并且在真空中浓缩至干。将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL)中,并且在环境温度搅拌16小时。将反应混合物浓缩至干,以获得为无色胶状固体的(3R,5S)-5-甲基-N-[5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.35g,粗品):MS(ES)m/z323.2(M+H)。
步骤6:3-((3S,5R)-3-甲基-5-((5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.03g,0.32mol)、1-羟基-1,2-二氢吡啶-2-酮(0.31g,0.28mmol)和O-(苯并三唑-1-基)-N,N,N’,N’-四甲基脲四氟硼酸酯(0.1g,0.32mol)在N-甲基吡咯烷-2-酮(2mL)中的溶液在环境温度搅拌3分钟。然后,添加(3R,5S)-5-甲基-N-[5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.07g,0.22mmol),然后添加三乙胺(0.1mL,0.70mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用反相色谱纯化粗材料,以获得为白色固体的3-[(3S,5R)-3-甲基-5-{[5-(2-甲基嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-基]-3-氧代丙腈(0.02g,20%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.29(br s,1H),10.38(br s,1H),8.54-8.56(m,2H),7.75-7.76(m,1H),7.15(s,1H),6.70(s,1H),4.81-4.84(m,1H),3.93-4.33(m,5H),2.65(s,4H),2.27-2.30(m,1H),1.65-1.95(m,1H),1.17-1.30(m,1H),0.95(d,J=6.4Hz,3H);MS(ES)m/z 390.4(M+H)。
分析条件:
柱:X-Bridge,C18 19*100*5微米
流动相(A):H2O中0.1%氨
流动相(B):MeCN
流速:20.0mL/min
实施例53:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-5-羧酰胺的制备

方案51.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-5-羧酰胺的制备

步骤1:4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-硫代羧酰胺的制备

向4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(7.0g,21.5mmol,实施例48、步骤3)在四氢呋喃(30mL)中的溶液添加劳森试剂(8.70g,21.5mmol),并且将混合物在环境温度搅拌3小时。用冰冷的饱和碳酸氢钠溶液淬灭反应混合物,用乙酸乙酯提取,并且用盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩,以提供为浅黄色浓稠液体的4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-硫代羧酰胺(1.5g,47%产率):MS(ES)m/z 342.0(M+H)。
步骤2:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

向4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-硫代羧酰胺(6.0g,17.5mmol)在甲苯(60mL)中的溶液添加2-氯-3-氧代丙酸乙酯(7.93g,52.6mmol)和硫酸镁(4.22g,35.1mmol),并且将混合物加热12小时至90℃。将反应冷却至环境温度,用水淬灭,并且用乙酸乙酯提取,并且用盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩,以提供为粘性棕色液体的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.6g,7.81%产率):MS(ES)m/z438.1(M+H)。
步骤3:2-(4-氯-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

将2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.50g,0.22mmol)在二氯甲烷:三氟乙酸(5mL:5mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物。将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL,水中23%)中,并且将混合物在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为灰白色固体的2-(4-氯-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.52g,粗品):MS(ES)m/z 308.0(M+H)。
步骤4:2-(4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

向2-(4-氯-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.20g,0.65mmol)、4-二甲基氨基吡啶(0.02g,0.13mmol)和三乙胺(0.09mL,0.65mmol)在二氯甲烷(3mL)中的经搅拌的溶液添加4-甲基-苯磺酰氯(0.15g,0.78mmol),并且将反应混合物在环境温度搅拌3小时。用饱和碳酸氢钠溶液淬灭反应混合物,用二氯甲烷提取,并且用盐水洗涤。在硫酸钠上干燥有机层,过滤并且在真空中浓缩,以产生为棕色固体的2-(4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.2g,34%产率):MS(ES)m/z462.1(M+H)。
步骤5:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯的制备

向4-氯-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(0.2g,0.75mmol)在乙醇(0.5mL)中的溶液添加(R)-3-氨基哌啶-1-羧酸叔丁酯(0.35g,1.73mmol)和三乙胺(0.5mL,3.6mmol),并且使混合物在130℃经受微波辐照15分钟。将反应冷却至环境温度,用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(50%乙酸乙酯/己烷)纯化粗材料,以提供为粘性无色液体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.17g,62%产率):MS(ES)m/z 626.2(M+H)。
步骤6:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸的制备

向(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸乙酯(0.35g,0.55mmol)在乙醇(1mL)中的经搅拌的溶液添加2M氢氧化锂(0.07g,2.8mmol)水溶液,并且使混合物在90℃经受加热3小时。将反应冷却至环境温度,并且在真空中浓缩以去除挥发物。将剩余材料溶解在水中,并且用1N盐酸酸化以调整pH至~3。过滤沉淀的固体,用水洗涤,并且在高真空中干燥,以提供为灰白色固体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸(0.2g,83%产率):MS(ES)m/z 444.2(M+H)。
步骤7:(R)-3-((5-(5-(甲基氨基甲酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酸(0.25g,0.56mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓-3-氧化物六氟磷酸酯(0.43g,1.12mmol)在N,N-二甲基甲酰胺(5mL)中的溶液在环境温度搅拌5分钟。添加甲胺盐酸盐(0.07g,0.68mmol),然后添加三乙胺(0.4mL,2.77mmol),并且将混合物搅拌12小时。用水淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(80%乙酸乙酯/己烷)纯化粗材料,以提供为粘性无色液体的(R)-3-((5-(5-(甲基氨基甲酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,97%产率):MS(ES)m/z 457.2(M+H)。
步骤8:(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酰胺盐酸盐的制备

向4N盐酸和1,4-二噁烷(5mL)添加(R)-3-((5-(5-(甲基氨基甲酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸酯(0.25g,0.54mmol),并且将混合物在环境温度搅拌4小时。在真空中浓缩反应混合物并且干燥,以提供为灰白色固体的(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酰胺盐酸盐(0.16g,77%产率):MS(ES)m/z 357.3(M+H)+。
步骤9:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基噻唑-5-羧酰胺的制备

将氰基乙酸(0.04g,0.64mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.24g,0.63mmol)在N,N-二甲基甲酰胺(4mL)中的溶液在环境温度搅拌3分钟。然后,添加(R)-N-甲基-2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑-5-羧酰胺盐酸盐(0.15g,0.42mmol),然后添加N,N-二异丙基乙胺(0.5mL,1.42mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺(0.01g,6%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.45(s,1H),9.65(br s,1H),8.20-8.31(m,3H),7.19(s,1H),6.68(s,1H),3.89-4.50(m,4H),3.68(m,2H),3.43(m,2H),2.79(s,2H),2.07(s,1H),1.64-1.75(m,3H);MS(ES)m/z 424.2(M+H)。
分析条件:
柱:Ascentis Express C18(50mm×2.1mm×2.7μm)
流动相(A):水中0.1%%甲酸
流动相(B):MeCN
流速:1.0mL/min
实施例54:(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)N,N-二甲基硫酰胺的制备

方案52.(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)N,N-二甲基硫酰胺的制备

步骤1:[(2-溴吡啶-4-基)氨磺酰基]二甲胺的制备

向2-溴吡啶-4-胺(2.0g,11.5mmol)在吡啶(20mL)中的溶液添加二甲基氨磺酰氯(1.63g,11.5mmol),并且将混合物在氮气气氛下在密封管中在80℃搅拌12小时。将反应冷却至环境温度,用乙酸乙酯和水稀释。分离有机层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(20%乙酸乙酯/己烷)纯化粗剩余物,以提供为无色胶状液体的[(2-溴吡啶-4-基)氨磺酰基]二甲胺(1.2g,37%产率):MS(ES)m/z 280.0(M+)。
步骤2:(R)-3-((5-(4-((N,N-二甲基氨磺酰基)氨基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向[(2-溴吡啶-4-基)氨磺酰基]二甲胺(0.8g,2.85mmol)和(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.96g,3.42mmol)在二噁烷(9mL)中的溶液添加[1,1’-双(二苯基膦)二茂铁]二氯化钯(II)二氯甲烷络合物(0.1g,0.13mmol)和2M碳酸钾(1.18g,8.55mmol)水溶液,并且将混合物在氮气气氛下在100℃加热12小时。将反应冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤,并且在减压下蒸发以获得粗化合物。使用快速色谱(50%乙酸乙酯/己烷)纯化粗剩余物,以产生为胶状棕色固体的(R)-3-((5-(4-((N,N-二甲基氨磺酰基)氨基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,54%产率):MS(ES)m/z 645.9(M+H)。
步骤3:(3R)-N-(5-{4-[(二甲基氨磺酰基)氨基]吡啶-2-基}-1H-吡咯并[2,3-b]吡啶-4-基)哌啶-3-胺的制备

将(R)-3-((5-(4-((N,N-二甲基氨磺酰基)氨基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.25g,0.38mmol)在二氯甲烷:三氟乙酸(5mL:5mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,并且将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL,水中23%)中,并且在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩,以提供为胶状黄色固体的(R)-5-(5-甲基噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.15g,粗品):MS(ES)m/z 413.9(M-H)。
步骤4:(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)N,N-二甲基硫酰胺的制备

将氰基乙酸(0.036g,0.43mmol)和(1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.22g,0.43mmol)在N,N-二甲基甲酰胺(5mL)中的溶液搅拌5分钟。然后,添加(R)-5-(5-甲基噁唑-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.15g,0.36mmol),然后添加N,N-二异丙基乙胺(0.19mL,1.08mmol),并且将反应混合物在环境温度搅拌12小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的3-[(3R)-3-{[5-(4-{[(5-溴-3-甲酰基-1H-吲哚-7-基)(甲基)氨磺酰基]氨基}吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-基]-3-氧代丙腈(0.01g,6%产率):1HNMR(400MHz,DMSO-d6)δ12.37(s,1H),10.81(s,2H),8.45-8.46(m,1H),8.24-8.26(m,1H),7.41-7.48(m,2H)7.16-7.17(m,1H),6.91(s,1H),4.08(s,1H),3.60-3.76(m,4H),2.80(s,6H),2.04-21.30(m,6H);MS(ES)m/z 483.2(M+H)。
分析条件:
流速:0.3mL/min
柱:BEH C18(50mm×2.1mm×1.7μm)
流动相(A):水中0.1%TFA
流动相(B):MeCN
实施例55:(R)-3-(3-((5-(5-(甲基磺酰基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案53.(R)-3-(3-((5-(5-(甲基磺酰基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-((5-(5-(甲基磺酰基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(1.0g,1.75mmol)和2-溴-5-(甲基磺酰基)吡啶(0.62g,2.6mmol)在1,4-二噁烷(10mL)中的经搅拌的溶液添加双(三苯基膦)二氯化钯(II)(0.07g,0.09mmol),然后添加2M碳酸钾(0.72g,5.25mmol)水溶液,并且将混合物在密封管中在氮气下在90℃加热12小时。将反应冷却至环境温度,用乙酸乙酯和水稀释。分离有机层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为粘性浅黄色固体的(R)-3-((5-(5-(甲基磺酰基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.19g,18%产率):MS(ES)m/z 602.3(M+H)。
步骤2:(R)-5-(5-(甲基磺酰基)吡啶-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(R)-3-((5-(5-(甲基磺酰基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.19g,0.32mmol)在二氯甲烷:三氟乙酸(3mL:3mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将剩余物溶解在1,4-二噁烷:氨水(3mL:3mL,水中23%)中,并且将溶液在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为灰白色固体的(R)-5-(5-(甲基磺酰基)吡啶-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.15g,粗品):MS(ES)m/z 372.2(M+H)。
步骤3:(R)-3-(3-((5-(5-(甲基磺酰基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.051g,0.6mmol)、N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.085g,0.6mmol)、1-羟基苯并三唑(0.82g,0.6mmol)和N,N-二异丙基乙胺(0.2mL,1.2mmol)在二氯甲烷(15mL)中的溶液在环境温度搅拌5分钟。然后,添加(R)-5-(5-(甲基磺酰基)吡啶-2-基)-N-(哌啶-3-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.15g,0.4mmol),并且将所得的混合物在环境温度搅拌12小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(5-(甲基磺酰基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.03g,16%产率):1H NMR(400MHz,DMSO-d6VT在100℃)δ11.27(br s,1H),9.96(br s,1H),8.98(s,1H),8.51(s,1H),8.24(d,J=2.2Hz,1H),8.13(d,J=2.2Hz,1H),7.16(s,1H),6.66(s,1H),4.29(br s,1H),3.89(br s,2H),3.40-3.45(m,3H),3.27(s,3H),2.08(br s,1H),1.64-1.77(m,3H),1.25(s,1H);MS(ES)m/z439.1(M+H)。
分析条件:
柱:X bridge(250mm×4.6mm×5微米)
流动相(A):水中0.1%氨
流动相(B):甲醇
流速:1.0mL/min
实施例56:(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺的制备

方案54.(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺的制备

步骤1:N-(2-溴吡啶-4-基)甲烷磺酰胺的制备

在0℃向2-溴吡啶-4-胺(1.0g,5.81mmol)和吡啶(1mL)在二氯甲烷(5mL)中的经搅拌的溶液添加甲烷磺酰氯(0.79mL,6.97mmol),并且将溶液加温至环境温度并且搅拌12小时。用冰冷水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩。通过使用快速色谱(20%乙酸乙酯/己烷)纯化粗材料,以提供为棕色固体的N-(2-溴吡啶-4-基)甲烷磺酰胺(0.4g,28%产率):MS(ES)m/z252.8(M+H)。
步骤2:(R)-3-((5-(4-(甲基磺酰胺基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.80g,1.39mmol)和N-(2-溴吡啶-4-基)甲烷磺酰胺(0.28g,1.58mmol)在1,4-二噁烷(8mL)中的溶液添加双(三苯基膦)二氯化钯(II)(0.09g,0.02mmol),然后添加2M碳酸钠(0.45g,4.17mmol)水溶液,并且将混合物在氮气气氛下在100℃搅拌12小时。将反应混合物冷却至环境温度,用乙酸乙酯稀释,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为无色浓稠液体的(R)-3-((5-(4-(甲基磺酰胺基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,12%产率):MS(ES)m/z 616.9(M+H)。
步骤3:(R)-N-(2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺的制备

将(R)-3-((5-(4-(甲基磺酰胺基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸酯(0.140g,0.22mmol)在二氯甲烷:三氟乙酸(2mL:2mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(2mL:4mL,水中23%)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为灰白色固体的(R)-N-(2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺(0.11g,粗品):MS(ES)m/z 385.1(M-H)+。
步骤4:(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺的制备

向氰基乙酸(0.036g,0.426mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.140g,0.37mmol)在N,N-二甲基甲酰胺(5mL)中的经搅拌的溶液添加(R)-N-(2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺(0.11g,0.284mmol),然后添加N,N-二异丙基乙胺(0.45mL,1.42mmol),并且将混合物在环境温度搅拌16小时。用水淬灭反应,并且用乙酸乙酯提取。用水、盐水洗涤有机层,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-N-(2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-4-基)甲烷磺酰胺(0.02g,15%产率):H NMR(400MHz,DMSO-d6)δ11.37-11.41(m,1H),9.90(br s,1H),8.17-8.31(m,3H),7.40-7.42(s,1H),7.15(s,1H)6.94(s,1H),6.61(s,1H),4.04(m,1H),3.92-3.99(m,3H),3.55-3.67(m,2H),3.55(m,2H),3.11(s,2H),2.87(s,1H),1.97(s,2H),1.60-1.69(s,1H);MS(ES)m/z 454.1(M+H)。
分析条件:
柱:X-BridgeC-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
实施例57:3-((3R,5S)-3-甲基-5-((5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案55.3-((3R,5S)-3-甲基-5-((5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:4-氯-N-甲氧基-N-甲基-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酰胺的制备

将化合物4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酸(5.0g,15.3mmol)和[(二甲基氨基)-1H-1,2,3-三唑并-[4,5-b]吡啶-1-基亚甲基]-N-甲基甲铵六氟磷酸酯N-氧化物(11.6g,30.6mmol)在二氯甲烷(50mL)中的溶液搅拌5分钟。添加甲氧基(甲基)胺(1.40g,22.9mmol)和N,N-二异丙基乙胺(7.99mL,45.9mmol),并且在环境温度搅拌混合物。在5小时之后,用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且浓缩。通过快速色谱(20%乙酸乙酯/己烷)纯化粗材料,以提供为胶状棕色固体的4-氯-N-甲氧基-N-甲基-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(4g,71%产率):MS(ES)m/z 370.2(M+H)。
步骤2:1-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)乙烷-1-酮的制备

在0℃向4-氯-N-甲氧基-N-甲基-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-羧酰胺(10g,27mmol)在四氢呋喃(100mL)中的经搅拌的溶液添加溴(甲基)镁(27mL,81.1mmol),并且溶液被允许在环境温度搅拌2小时。用饱和氯化铵溶液淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且浓缩以提供为浅黄色浓稠液体的1-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)乙烷-1-酮(6.5g,68%产率):MS(ES)m/z 325.2(M+H)。
步骤3:4-氯-5-(嘧啶-4-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶的制备

使1-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)乙烷-1-酮(2.0g,6.16mmol)、甲酰胺(6mL)、对甲苯磺酸(0.30g,1.54mmol)和双(三甲基甲硅烷基)胺(1.61mL,7.70mmol)的混合物在215℃经受微波辐照40分钟。将反应混合物冷却至室温,并且用碎冰淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为棕色固体的4-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)嘧啶(0.4g,18%产率):MS(ES)m/z 361.1(M+H)。
步骤4:4-氯-5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶的制备

将4-(4-氯-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-5-基)嘧啶(0.80g,0.29mmol)在二氯甲烷:三氟乙酸(0.5mL:2mL)中的溶液在环境温度搅拌3小时。在真空中浓缩反应混合物,并且将剩余物溶解在1,4-二噁烷:氨水(0.2mL:2mL,水中23%)中,并且将混合物在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为棕色固体的4-氯-5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶(0.5g,粗品):MS(ES)m/z 231.1(M+H)。
步骤5:(3R,5S)-3-甲基-5-((5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将4-{4-氯-1H-吡咯并[2,3-b]吡啶-5-基}嘧啶(0.10g,0.43mmol)、1-甲基吡咯烷-2-酮(2mL)、(3S,5R)-3-氨基-5-甲基哌啶-1-羧酸苄酯(118mg,1.48mmol)和N,N-二异丙基乙胺(0.1mL,0.58mmol)的经搅拌的溶液在10mL密封管中在170℃加热32小时。将反应混合物冷却至环境温度,并且用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在硫酸钠上干燥,过滤并且在真空中浓缩,以提供为棕色液体的(3R,5S)-3-甲基-5-{[5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸苄酯(0.15g,44.57%产率):MS(ES)m/z 443.2(M+H)。
步骤6:N-((3S,5R)-5-甲基哌啶-3-基)-5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3R,5S)-3-甲基-5-{[5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸苄酯(0.25g,0.56mmol)在三氟乙酸(1mL)中的经搅拌的溶液在密封管中加热1小时至100℃。将反应混合物冷却至环境温度,并且在真空中浓缩,以提供为无色胶状固体的N-((3S,5R)-5-甲基哌啶-3-基)-5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.17g,99%产率):MS(ES)m/z 309.2(M+H)。
步骤7:3-((3R,5S)-3-甲基-5-((5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将氰基乙酸(0.06g,0.71mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.252g,1.07mmol)在N,N-二甲基甲酰胺(1mL)中的溶液在环境温度搅拌3分钟。然后,添加(3S,5R)-5-甲基-N-[5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.22g,0.71mmol),然后添加N,N-二异丙基乙胺(0.5mL,2.14mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的3-((3R,5S)-3-甲基-5-((5-(嘧啶-4-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.012g,4.5%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.36(s,1H),10.06(br s,1H),9.08(s,1H),8.65-8.66(s,1H),8.54(s,1H),7.96-7.97(m,1H),7.16(s,1H),6.71(s,1H),4.77(s,1H),3.98(m,4H),3.65(m,1H),2.70(m,1H),2.30(m,1H),1.85(m,1H),1.29(m,1H),0.93-0.94(m,3H);MS(ES)m/z 376.2(M+H)。
分析条件:
柱:BEH C18(50mm×2.1mm×1.7μm)
流动相(A):水中0.1%甲酸
流动相(B):MeCN
流速:0.3mL/min
实施例58:(R)-3-(3-((5-(1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案56.(R)-3-(3-((5-(1H-吡唑-5-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(2-氯乙基)(甲基)氨基甲酸叔丁酯的制备

向2-氯-N-甲基乙烷-1-胺(4.0g,42.9mmol)和三乙胺(17.99mL,128.7)在二氯甲烷(30mL)中的经搅拌的溶液添加二碳酸二叔丁酯(12.8mL,55.8mmol),并且将溶液在环境温度搅拌12小时。用水淬灭反应,并且用乙酸乙酯提取。用水、盐水洗涤有机层,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩,以提供为半固体的(2-氯乙基)(甲基)氨基甲酸叔丁酯(3g,粗品)。
步骤2:(2-((6-溴吡啶-2-基)氧基)乙基)(甲基)氨基甲酸叔丁酯的制备

向(2-氯乙基)(甲基)氨基甲酸叔丁酯(1.5g,8.6mmol)中添加6-溴吡啶-2-醇(2.0g,10.4mmol)和碳酸钾(2.3g,17.2mmol)在N,N-二甲基甲酰胺(15mL)中的悬浮液,并且将混合物加热12小时至80℃。将反应冷却至环境温度,用水淬灭,并且用乙酸乙酯提取。用水、盐水洗涤有机层,并且在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(10%乙酸乙酯/己烷)纯化粗材料,以提供为无色胶状固体的(2-((6-溴吡啶-2-基)氧基)乙基)(甲基)氨基甲酸叔丁酯(0.65g,26%产率):MS(ES)m/z 330.1;333.1(1:1;M+H)。
步骤3:(R)-(2-((6-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-2-基)氧基)乙基)(甲基)氨基甲酸叔丁酯的制备

向(R)-3-氧代-3-(3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.2g,0.49mmol,实施例61、步骤1)和(2-((6-溴吡啶-2-基)氧基)乙基)(甲基)氨基甲酸叔丁酯(0.16g,0.49mmol)在1,4-二噁烷(8mL)中的经搅拌的溶液添加氯(2-二环己基膦-2’,4’,6’-三异丙基-1,1’-联苯基)[2-(2’-氨基-1,1’-联苯基)]钯(II)(0.04g,0.05mmol),然后添加2M磷酸钾(0.31g,1.46mmol)水溶液,并且将混合物在氮气气氛下在110℃搅拌12小时。将反应混合物冷却至环境温度,过滤并且在真空中干燥。通过快速色谱(5%甲醇/氯甲烷)纯化粗材料,以提供为粘性棕色液体的(R)-(2-((6-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-2-基)氧基)乙基)(甲基)氨基甲酸叔丁酯(0.06g,23%产率):MS(ES)m/z 534.2(M+H)。
步骤4:(R)-3-(3-((5-(6-(2-(甲基氨基)乙氧基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈盐酸盐的制备

将(R)-(2-((6-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-2-基)氧基)乙基)(甲基)氨基甲酸叔丁酯(0.05g,0.09mmol)在二噁烷(1mL):二噁烷中4N盐酸盐(0.5mL)中的溶液在环境温度搅拌40分钟。在真空中浓缩反应混合物,并且通过反相色谱纯化,以提供为灰白色固体的(R)-3-(3-((5-(6-(2-(甲基氨基)乙氧基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈氯化氢(0.01g,23%产率):1H NMR(400MHz,DMSO-d6)δ12.14(br s,1H),8.70(br s,2H),8.25-8.27(m,1H),7.90-7.93(m,1H),7.37-7.43(m,2H),6.87-6.89(m,2H),4.49-4.52(m,3H),4.30(m,1H),3.99-4.10(m,3H),3.75-3.80(m,1H),3.60(m,2H),2.79-2.85(m,1H),2.64(s,3H),2.13(s,1H),1.68(br s,3H);MS(ES)m/z 434.6(M+H)。
分析条件:
Kinetex EVO C18(100mm×2.1mm×2.6μm)
流动相(A):水中0.1%三氟乙酸
流动相(B):MeCN
流速:0.75mL/min
实施例59:(R)-3-(3-((5-(3-(甲基磺酰基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案57.(R)-3-(3-((5-(3-(甲基磺酰基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

(R)-3-(3-((5-(3-(甲基磺酰基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.05g,0.14mmol)在1,4-二噁烷(4mL)中的经搅拌的溶液添加3-(甲基磺酰基)苯基硼酸(0.04g,0.21mmol)和2M碳酸铯(0.13g,0.41mmol)水溶液,并且用氩气使混合物脱气15分钟。添加四三苯基膦钯(0)(0.01g,0.07mmol),并且将所得的混合物在密封管中在100℃加热3小时。将反应混合物冷却至室温,通过硅藻土过滤,并且收集滤液并且在真空中浓缩。通过柱色谱(5%甲醇/二氯甲烷)纯化粗品,以提供为灰白色固体的(R)-3-(3-((5-(3-(甲基磺酰基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.05g,42%产率):1H NMR(400MHz,DMSO-d6,VT在70℃)δ11.23(s,1H),7.81-7.87(m,2H),7.73(d,J=6.8Hz,3H),7.22(s,1H),6.61(s,1H),4.90(br s,1H),4.11(br s,1H),3.84(br s,5H),3.42(br s,1H),3.21(s,3H),1.93(br s,1H),1.51-1.58(m,3H);MS(ES)m/z 437.9(M+H)。
实施例60:(R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案58.(R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

(R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.4g,1.1mmol)在N,N-二甲基甲酰胺(10mL)中的溶液添加氯化锂(0.095g,1.1mmol)、4-甲基-2-(三丁基甲锡烷基)嘧啶(0.50g,1.3mmol)、碘化铜(0.021g,0.05mmol)和四(三苯基膦)钯(0)(0.07g,0.05mmol),并且将所得的混合物在氮气气氛下在密封管中在120℃搅拌3小时。将反应冷却至环境温度,用乙酸乙酯和水稀释。分离有机层,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的((R)-3-(3-((5-(4-甲基吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.01g,7%产率):1H NMR(400MHz,DMSO-d6)δ12.09(br s,1H),8.43(br s,1H),8.21(s,1H),7.49(s,1H),7.35(s,1H),7.16(d,J=4.8Hz,1H),6.87(s,1H),4.30-4.55(m,1H),3.55-4.05(m,3H),3.30-3.45(m,2H),2.82(s,3H),1.95-2.10(m,2H),1.60-1.85(m,3H);MS(ES)m/z 376.1(M+H)。
分析条件:
柱:X-BridgeC-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
实施例61:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基异烟酰胺的制备

方案59.(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基异烟酰胺的制备

步骤1:(R)-3-氧代-3-(3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

向(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.20g,0.55mmol)在1,4-二噁烷(6mL)中的经搅拌的溶液添加4,4,5,5-四甲基-2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1,3,2-二噁硼烷(0.14g,0.57mmol)、磷酸钾(0.35g,1.66mmol)和氯(2-二环己基膦-2’,6’-二异丙氧基-1,1’-联苯基)[2-(2’-氨基-1,1’-联苯基)]钯(II)(0.09g,0.11mmol),并且将反应混合物在氮气气氛下在110℃搅拌2小时。将反应冷却至环境温度,用乙酸乙酯稀释,并且用水洗涤。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为粘性棕色固体的(R)-3-氧代-3-(3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.45g,粗品)。
步骤2:(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基异烟酰胺的制备

向(R)-3-氧代-3-(3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(0.20g,0.49mmol)在1,4-二噁烷:水(10mL)中的经搅拌的溶液添加2-溴-N-甲基异烟酰胺(0.13g,0.58mmol)、氯(2-二环己基膦基-2’,6’-二异丙氧基-1,1’-联苯基)[2-(2’-氨基-1,1’-联苯基)]钯(II)(0.04g,4.89mmol)和2M磷酸钾(0.31g,0.15mmol)水溶液,并且将反应混合物在氮气气氛下在110℃搅拌。在16小时之后,将反应混合物冷却至环境温度,用DCM稀释,并且通过硅藻土过滤。在真空中浓缩滤液,并且通过反相色谱纯化粗材料,以提供为浅黄色固体的(R)-2-(4-((1-(2-氰基乙酰基)哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)-N-甲基异烟酰胺(0.08g,7%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)11.23(br s,1H),9.83(br s,1H),8.51-8.62(m,3H),8.20(s,1H),7.56-7.57(m,1H),7.15(s,1H),6.63(s,1H),4.19-4.38(m,2H),3.94(m,2H),3.64(m,2H),3.40–3.45(m,2H),2.84-2.85(m,2H),2.02(br s,2H),1.60(m,2H);MS(ES)m/z 418.3(M+H)+。
分析条件:
柱:X-BRIDGE,C18(19mm×100mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:20.0mL/min
2-溴-N-甲基吡啶-4-羧酰胺的制备

将2-溴异烟酸(1.0g,4.95mmol)和(1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(2.45g,6.44mmol)在N,N-二甲基甲酰胺(10mL)中的溶液在环境温度搅拌3分钟。添加甲胺(12.4mL,24.8mmol),然后添加三乙胺(1.04mL,7.43mmol),并且在环境温度搅拌混合物。在5小时之后,用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为粘性无色液体的2-溴-N-甲基吡啶-4-羧酰胺(0.85g,3.95mmol):MS(ES)m/z 214.9(M+1)。
实施例62:(R)-3-(3-((5-(1-(2-羟基乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案60.(R)-3-(3-((5-(1-(2-羟基乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-3-氧代-3-(3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈的制备

向(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.35g,0.96mmol)、4,4,4’,4’,5,5,5’,5’-八甲基-2,2’-双(1,3,2-二噁硼烷)(0.36g,1.45mmol)、磷酸钾(0.60g,2.89mmol)在1,4-二噁烷(10mL)中的悬浮液添加氯(2-二环己基膦-2’,6’-二异丙氧基-1,1’-联苯基)[2-(2’-氨基-1,1’-联苯基)]钯(II)(0.07g,0.09mmol),并且将混合物在110℃加热1小时。将反应冷却至环境温度,并且将混合物用于下一步骤而无需后处理。
步骤2:(R)-3-(3-((5-(1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向(R)-3-氧代-3-(3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)丙腈(~0.35g)、3-溴-1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑(0.39g,1.283mmol)在二噁烷(10mL)中的经搅拌的混合物添加双(三苯基膦)二氯化钯(II)(0.03g,0.04mmol),然后添加2M磷酸钾(0.54g,2.56mmol)水溶液,并且将反应混合物在110℃加热12小时。将反应混合物冷却至环境温度,通过硅藻土过滤,并且在真空中浓缩滤液。使用快速色谱(5%甲醇/二氯甲烷)纯化粗材料,以提供为棕色半固体的2(R)-3-(3-((5-(1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.2g,46%产率):MS(ES)m/z 514.3(M+H)。
步骤3:(R)-3-(3-((5-(1-(2-羟基乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

在0℃向2(R)-3-(3-((5-(1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.18g,0.35mmol)在二氯甲烷(5mL)中的溶液添加三氟乙酸(0.5mL),并且将混合物在环境温度搅拌3小时。用二氯甲烷稀释反应,用饱和碳酸氢钠溶液和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(1-(2-羟基乙基)-1H-吡唑-3-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.01g,7%产率):1H NMR(400MHz,DMSO-d6)δ12.22(s,1H),10.1(s,1H),8.43(d,J=6.0Hz,1H),7.88(s,1H),7.40(s,1H),6.91(d,J=14.8Hz,2H),4.60(br s,1H),4.32(br s,1H),4.22-4.23(m,2H),3.97-4.07(m,2H),3.66-3.79(m,4H),2.11-2.30(m,2H),1.77(m,4H);MS(ES)m/z 394.4(M+H)。
分析条件:
柱:X-BridgeC-18(250mm×4.6mm×5mic)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
3-溴-1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑的制备

步骤1:2-(3-溴-1H-吡唑-1-基)乙烷-1-醇和2-(5-溴-1H-吡唑-1-基)乙烷-1-醇的制备

将3-溴-1H-吡唑(2.5g,17.123mmol)、2-溴乙烷-1-醇(3.95g,31.67mmol)、碘化钾(4.26g,25.68mmol)和氢氧化钾(1.91g,34.24mmol)在乙醇(30mL)中的混合物在100℃加热12小时。将反应冷却至环境温度,过滤以去除固体,并且在真空中浓缩滤液。将剩余物溶解在乙酸乙酯中,并且用水、盐水洗涤,在硫酸钠上干燥,过滤并且在真空中浓缩。通过柱色谱(正己烷:具有0.1%DEA的乙醇)纯化粗品,以提供为液体的2-(3-溴-1H-吡唑-1-基)乙烷-1-醇(1.2g,37%产率)和为无色胶状固体的2-(5-溴-1H-吡唑-1-基)乙烷-1-醇(0.8g,24%):MS(ES)m/z:191.0(M+H)。
步骤2:3-溴-1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑的制备

在0℃向2-(3-溴-1H-吡唑-1-基)乙烷-1-醇(1.2g,6.34mmol)在N,N-二甲基甲酰胺(10mL)中的经搅拌的溶液添加氢化钠(0.4g,9.53mmol,60%矿物油分散液)。在20分钟之后,在0℃添加1-(氯甲基)-4-甲氧基苯(1.48g,9.52mmol),并且将反应加温3小时至环境温度。用冰水淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色油状物的溴-1-(2-((4-甲氧基苄基)氧基)乙基)-1H-吡唑(1.45g,73%):MS(ES)m/z 313.3(M+H)。
实施例63:(R)-3-(3-((5-(3-(羟基甲基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案61.(R)-3-(3-((5-(3-(羟基甲基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

(R)-3-(3-((5-(3-(羟基甲基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向(R)-3-(3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.35g,0.96mmL)在二噁烷(8mL)中的经搅拌的溶液添加双(频哪醇合)二硼(0.37g,1.45mmol)和磷酸钾(0.61g,2.9mmol),然后添加XPhos-Pd-G2(0.08g,0.1mmol)。将所得的悬浮液加热2小时至120℃。将反应混合物冷却至环境温度,并且然后添加(6-溴吡啶-2-基)甲醇(0.28g,1.45mmol)、2M磷酸钾(0.41g,1.93mmol)水溶液和XPhos-Pd-G2(0.08g,0.1mmol),并且将混合物在100℃加热2小时。将反应物质冷却至环境温度,通过硅藻土过滤,并且用乙酸乙酯洗涤。用水和盐水洗涤滤液。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为灰白色固体的(R)-3-(3-((5-(3-(羟基甲基)苯基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(16mg,19%产率):1HNMR(400MHz,DMSO-d6在70℃)δ11.20(s,1H),9.93-10.08(m,1H),8.38(s,1H),7.82(m,1H),7.70(d,J=8.0Hz,1H),7.29(d,J=7.23Hz,1H),7.14(s,1H),6.63(s,1H),5.25(s,1H),4.63(d,J=6.0Hz,2H),4.28-4.49(m,1H),3.94-4.02(m,2H),3.68-3.75(m,1H),3.54-3.62(m,1H),2.80-2.96(m,1H),1.92-2.20(m,2H),1.52-1.86(m,3H);MS(ES)m/z:391.1(M+H)。
分析条件:
Zorbax-Eclipse XDB C18(150mm×4.6mm×5μm)
流动相(A):水中0.1%氨
流动相(B):MeCN
流速:1.0mL/min
实施例64:3-((3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

方案62.3-((3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

步骤1:(3S,5R)-3-甲基-5-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

向(3R,5S)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.70g,1.22mmol)在甲苯(30.0mL)中的经搅拌的溶液添加氯(2-二环己基膦-2’,4’,6’-三异丙基-1,1’-联苯基)[2-(2’-氨基-1,1’-联苯基)]钯(II)(0.19g,0.24mmol)、4,4,5,5-四甲基-2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1,3,2-二噁硼烷(0.68g,2.68mmol)和叔丁醇钾(0.34g,3.05mmol),并且将混合物在110℃在预热的油浴器上搅拌15分钟。将反应冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且浓缩,以获得为粘性棕色液体的(3S,5R)-3-甲基-5-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(1.24g,粗品):MS(ES)m/z621.4(M+H)。
步骤2:(3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯的制备

将(3S,5R)-3-甲基-5-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(1.20g,1.93mmol)、(6-溴吡啶-2-基)甲醇(0.24mg,1.29mmol)、氯(2-二环己基膦-2’,4’,6’-三异丙基-1,1’-联苯基)[2-(2’-氨基-1,1’-联苯基)]钯(II)(0.20g,0.26mmol)和2M磷酸三钾(1.09g,5.16mmol)水溶液在1,4-二噁烷(35mL)中的经搅拌的混合物在110℃加热16小时。将反应冷却至环境温度,并且用乙酸乙酯和水稀释。分离有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过用0-50%乙酸乙酯/己烷洗脱的combi-flash纯化粗品,以获得为粘性棕色液体的(3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.51g,66%产率):MS(ES)m/z 602.4(M+H)。
步骤3:(6-(4-(((3R,5S)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-2-基)甲醇的制备

将(3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.56g,0.85mmol)在三氟乙酸(5.0mL)中的经搅拌的溶液加热1小时至100℃。将反应混合物冷却至环境温度,并且然后在真空中浓缩,将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为粘性棕色液体的(6-(4-(((3R,5S)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-2-基)甲醇(0.52g,粗品):MS(ES)m/z 338.2(M+H)。
步骤4:3-((3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.14g,1.69mmol)、1-羟基苯并三唑(0.21mg,1.35mmol)和N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.32g,1.69mmol)在二氯甲烷(4.0mL)中的溶液在环境温度搅拌3分钟。然后,添加(6-(4-(((3R,5S)-5-甲基哌啶-3-基)氨基)-1H-吡咯并[2,3-b]吡啶-5-基)吡啶-2-基)甲醇(0.38g,1.13mmol),然后添加三乙胺(1.08mL,5.63mmol),并且将混合物在环境温度搅拌16小时。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用反相色谱纯化粗品,以提供为灰白色固体的3-((3R,5S)-3-((5-(6-(羟基甲基)吡啶-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈(0.03g,7%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.16(br s,1H),10.05(br s,1H),8.39(s,1H),7.79-7.83(m,1H),7.70-7.72(m,1H),7.28(d,J=7.6Hz,1H),7.12(s,1H),6.65(s,1H),4.15-4.30(m,1H),4.75-4.85(m,1H),4.60-4.67(m,1H),3.87-4.07(m,1H),3.60-3.67(m,1H),3.55(s,2H),2.80-2.90(m,1H),2.15-2.25(m,2H),1.65-1.80(m,2H),1.10-1.30(m,1H),0.92(d,J=6.0Hz,3H);MS(ES)m/z405.2(M+H)。
分析条件:
流速:0.3mL/min
柱:Ascentis Express C18(50mm×2.1mm×2.73μm)
流动相(A):水中0.1%甲酸
流动相(B):MeCN
实施例65:3-((3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案63.3-((3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(3S,5R)-3-甲基-5-{[5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸苄酯(1.0g,1.61mmol)和2-溴-5-甲烷磺酰基-1,3-噻唑(0.28g,1.18mmol)在1,4-二噁烷(30mL)中的经搅拌的溶液添加双(三苯基膦)二氯化钯(II)(0.15g,0.21mmol)和2M磷酸三钾(0.91g,4.30mmol)水溶液,并且将混合物加热16小时至110℃。将反应冷却至环境温度,并且用乙酸乙酯和水稀释。分离有机层,在无水硫酸钠上干燥,并且在真空中浓缩。通过快速色谱(20%乙酸乙酯/己烷)纯化粗品,以提供为粘性棕色液体的(3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.32,45%产率):MS(ES)m/z 622.8(M+H)。
步骤2:N-((3R,5S)-5-甲基哌啶-3-基)-5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.52g,1.00mmol)在三氟乙酸(5.0mL)中的经搅拌的溶液在密封管中在100℃加热1小时。将反应混合物冷却至环境温度,并且然后在真空中浓缩,将剩余物溶解在1,4-二噁烷:氨水(5mL:5mL)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为棕色胶状固体的N-((3R,5S)-5-甲基哌啶-3-基)-5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.45g,粗品):MS(ES)m/z 392.1(M+H)。
步骤3:3-((3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.13g,1.53mmol)、1-羟基苯并三唑(0.19mg,1.23mmol)和N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.29g,1.53mmol)在二氯甲烷(4.0mL)中的溶液在环境温度搅拌3分钟。然后,添加N-((3R,5S)-5-甲基哌啶-3-基)-5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.40g,1.02mmol),然后添加三乙胺(0.70mL,5.11mmol),并且将混合物在环境温度搅拌过夜。用乙酸乙酯稀释反应混合物,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。通过反相色谱纯化粗品,以提供为灰白色固体的3-((3S,5R)-3-甲基-5-((5-(5-(甲基磺酰基)噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(0.05g,11%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.56(br s,1H),9.33(br s,1H),8.41(s,1H),8.30(s,1H),7.21(s,1H),6.76(s,1H),4.79(br s,1H),3.84-4.44(m,3H),3.60-3.74(m,1H),3.56(s,1H),3.39(s,3H),2.60-2.80(m,1H),2.20-2.40(m,1H),1.75-1.85(m,1H),1.15-1.27(m,1H),0.94(d,J=6.4Hz,3H);MS(ES)m/z 459.1(M+H)。
分析条件:
流动相A:H2O中0.1%NH4OH
流动相B:MeCN
柱:X-Bridge,C18 19*100*5微米
流速:20mL/min
实施例66:(R)-3-(3-((5-(5-乙酰基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

方案64.(R)-3-(3-((5-(5-乙酰基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

步骤1:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸乙酯的制备

向(3R)-3-{[5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-{[2-(三甲基甲硅烷基)乙氧基]甲基}-1H-吡咯并[2,3-b]吡啶-4-基]氨基}哌啶-1-羧酸叔丁酯(1.50g,2.62mmol)和2-溴-1,3-噁唑-5-羧酸乙酯(0.38g,1.74mmol)在1,4-二噁烷(40mL)中的经搅拌的溶液添加双(三苯基膦)二氯化钯(II)(0.25g,0.35mol)和2M磷酸三钾(1.48g,6.97mmol)水溶液,并且将混合物在110℃搅拌16小时。将反应冷却至环境温度,用乙酸乙酯稀释,用水和盐水洗涤。在无水硫酸钠上干燥有机层,过滤并且在真空中浓缩。使用快速色谱(20%乙酸乙酯/己烷)纯化粗品,以提供为粘性棕色液体的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸乙酯(0.65g,63%产率):MS(ES)m/z 586.3(M+H)。
步骤2:(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸的制备

向(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸乙酯(0.50g,0.85mmol)在甲醇(20mL)中的经搅拌的溶液添加2M氢氧化钠(0.17g,4.27mmol)水溶液,并且将溶液在环境温度搅拌4小时。浓缩反应以去除挥发物,将获得的剩余物溶解在水中,并且用1N盐酸酸化,并且调整至pH~3。用乙酸乙酯提取水性层,在无水硫酸钠上干燥,过滤并且浓缩,以提供为无色胶状物的(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸(0.22g,46%产率):MS(ES)m/z558.3(M+H)。
步骤3:(R)-3-((5-(5-(甲氧基(甲基)氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-2-(4-((1-(叔丁氧基羰基)哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-羧酸(0.22g,0.40mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(HATU)(0.23g,0.60mmol)在N,N-二甲基甲酰胺(5mL)中的溶液在环境温度搅拌3分钟。然后,添加N,O-二甲基盐酸羟胺(0.2g,1.2mmol),然后添加三乙胺(0.38mL,2.76mmol),并且将混合物在环境温度搅拌5小时。用水淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为粘性液体的(R)-3-((5-(5-(甲氧基(甲基)氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.2g,84%产率):MS(ES)m/z 601.6(M+H)。
步骤4:(R)-3-((5-(5-乙酰基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向0℃的(R)-3-((5-(5-(甲氧基(甲基)氨基甲酰基)噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.20g,0.33mmol)在四氢呋喃(4.0mL)中的溶液添加甲基溴化镁(0.34mL,1.00mmol)。将反应混合物加温至环境温度,并且搅拌过夜。用饱和氯化铵溶液淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(40%乙酸乙酯/己烷)纯化粗材料,以提供为浅黄色半固体的(R)-3-((5-(5-乙酰基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,65%产率):MS(ES)m/z 556.3(M+H)。
步骤5:(R)-1-(2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-基)乙烷-1-酮的制备

在环境温度搅拌(R)-3-((5-(5-乙酰基噁唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(0.12g,0.22mmol)在二氯甲烷:三氟乙酸(2.0mL:2.0mL)中的溶液。在3小时之后,将反应浓缩至干,将剩余物溶解在1,4-二噁烷:氨水(2mL:2mL)中,并且在环境温度搅拌16小时。在真空中浓缩反应混合物,以提供为无色胶状物的(R)-1-(2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-基)乙烷-1-酮(0.12g,粗品):MS(ES)m/z 326.3(M+H)。
步骤6:(R)-3-(3-((5-(5-乙酰基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈的制备

向2-氰基乙酸(0.04g,0.42mmol)和1-羟基苯并三唑(0.51g,0.33mmol)在二氯甲烷(5mL)中的经搅拌的溶液添加N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐(0.8g,0.42mmol),并且将混合物在环境温度搅拌5分钟。然后,添加(R)-1-(2-(4-(哌啶-3-基氨基)-1H-吡咯并[2,3-b]吡啶-5-基)噁唑-5-基)乙烷-1-酮(0.09g,0.28mmol),然后添加三乙胺(0.2mL,0.38mmol),并且将所得的混合物在环境温度搅拌18小时。用水淬灭反应混合物,并且用二氯甲烷提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过反相色谱纯化粗材料,以提供为浅黄色固体的(R)-3-(3-((5-(5-乙酰基噁唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-基)-3-氧代丙腈(7.0mg,7%产率):1HNMR(400MHz,DMSO-d6,VT在80℃)δ11.53(br s,1H),9.45(br s,1H),8.61(s,1H),8.11(s,1H),7.21(s,1H),6.70(s,1H),4.20-4.40(m,1H),3.80-4.00(m,3H),3.50-3.65(m,1H),3.40-3.50(m,3H),2.00-2.15(m,2H),1.55-1.84(m,4H);MS(ES)m/z 393.1(M+H)。
分析条件:
流动相A:H2O中0.1%NH4OH
流动相B:MeCN
柱:Phenomenex,C18,21.2mm*250mm*5微米,
流速:20mL/min
实施例67:N-((3R,5S)-1-((2,2-二氟环丙基)甲基)-5-甲基哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

方案65.N-((3R,5S)-1-((2,2-二氟环丙基)甲基)-5-甲基哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

步骤1:2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑的制备

将5-溴-4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶(1.5g,4.15mmol)、2-(三丁基甲锡烷基)噻唑(1.7g,4.57mmol)和双(三苯基膦)二氯化钯(II)(0.58g,0.83mmol)在N-甲基-2-吡咯烷酮(15mL)中的经搅拌的溶液在氮气气氛下在密封管中在130℃加热。在5小时之后,将反应混合物冷却至环境温度,用乙酸乙酯稀释,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(己烷中10%乙酸乙酯)纯化粗材料,以提供为无色油状物的2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑(0.75g,49%产率):MS(ES)m/z 365.8(M+H)。
步骤2:(3S,5R)-3-甲基-5-((5-(噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯的制备

将2-(4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-基)噻唑(1.0g,2.73mmol)、(3R,5S)-3-氨基-5-甲基哌啶-1-羧酸苄酯(0.74g,3.0mmol)和N,N-二异丙基乙胺(2.0mL,13.6mmol)在N-甲基-2-吡咯烷酮(20mL)中的经搅拌的溶液在175℃加热20小时。将反应冷却至环境温度,用乙酸乙酯稀释,用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过使用快速色谱(己烷中20%乙酸乙酯)纯化粗材料,以提供为黄色液体的(3S,5R)-3-甲基-5-((5-(噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.5g,32%产率):MS(ES)m/z578.9(M+H)。
步骤3:N-((3R,5S)-5-甲基哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3S,5R)-3-甲基-5-((5-(噻唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸苄酯(0.5g,0.39mmol)和三氟乙酸(5.0mL)的溶液在密封管中在100℃搅拌1小时。在冷却之后,在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(5.0mL:5.0mL)中,并且在环境温度搅拌过夜。在真空中浓缩反应混合物,以提供为灰白色固体的N-((3R,5S)-5-甲基哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.2g,粗品):MS(ES)m/z 314.0(M+H)。
步骤4:N-((3R,5S)-1-((2,2-二氟环丙基)甲基)-5-甲基哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

向(3R,5S)-5-甲基-N-[5-(1,3-噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基]哌啶-3-胺(0.1g,0.32mmol)在N-甲基吡咯烷-2-酮(1.5mL)中的经搅拌的溶液添加2-(溴甲基)-1,1-二氟环丙烷(0.87g,0.51mmol),并且将溶液在密封管中在70℃加热12小时。在冷却之后,用冰水淬灭反应,用乙酸乙酯提取,用盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用反相色谱纯化粗材料,以提供为灰白色固体的N-((3R,5S)-1-((2,2-二氟环丙基)甲基)-5-甲基哌啶-3-基)-5-(噻唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.003g,3%产率):1H NMR(400MHz,DMSO-d6,VT在80℃)δ11.35(m,1H),9.53(m,1H),8.37(m,1H),7.80(s,1H),7.50(s,1H),7.15(m,1H),6.55(m,1H),4.21(m,1H),3.3(m,1H),2.80-2.92(m,1H),2.17-2.31(m,1H),1.73-1.92(m,3H),1.42-1.59(m,1H),1.35(m,1H),1.24(s,3H),0.90-1.11(m,4H);MS(ES)m/z 404.3(M+H)。
分析条件:
柱:Sunfire C18 19*150*5微米
流动相(A):H2O中0.1%TFA
流动相(B):MeCN
流速:20mL/min
实施例68:3-((3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

方案66.3-((3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

步骤1:4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯的制备

向4-氯-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(1.0g,2.82mmol)在N-甲基-2-吡咯烷酮(15mL)中的经搅拌的溶液添加(3R,5S)-3-氨基-5-甲基哌啶-1-羧酸苄酯(0.84g,3.38mmol),然后添加N,N-二异丙基乙胺(1.0mL),并且将混合物在密封管中加热18小时至180℃。将反应混合物冷却至环境温度,用水淬灭,并且用乙酸乙酯提取。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(20%乙酸乙酯/己烷)纯化粗材料,以提供为无色油状物的4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(0.85g,53%产率):MS(ES)m/z 567.3[M+H]+。
步骤2:4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸的制备

向4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸乙酯(0.85g,1.50mmol)在甲醇(10mL)中的经搅拌的溶液添加2M单水氢氧化锂(0.126g,3.0mmol)水溶液,并且将溶液在室温搅拌6小时、在50℃搅拌2小时。将反应冷却至环境温度,并且在真空中浓缩以去除挥发物。将剩余物溶解在水中,并且用1N盐酸酸化以调整pH~3。用乙酸乙酯提取水性层。用盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。用正戊烷研磨粗材料,以提供为粘性固体的4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.7g,粗品):MS(ES)m/z 539.3[M+H]+。
步骤3:(3R,5S)-3-((5-(2-甲酰肼-1-羰基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯的制备

将4-(((3R,5S)-1-((苄氧基)羰基)-5-甲基哌啶-3-基)氨基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-5-羧酸(0.6g,1.11mmol)和1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸酯(0.50g,1.34mmol)在二甲基甲酰胺(10mL)中的溶液在环境温度搅拌3分钟。添加甲酰肼(0.13g,2.23mmol),然后添加N,N-二异丙基乙胺(0.6mL,3.34mmol),并且将混合物在环境温度搅拌3小时。用水淬灭反应,并且用乙酸乙酯提取。用盐水洗涤有机层,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩,以提供为灰白色固体的(3R,5S)-3-((5-(2-甲酰肼-1-羰基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.45g,69%产率):MS(ES)m/z 581.3[M+H]+。
步骤4:(3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯的制备

在环境温度向(3R,5S)-3-((5-(2-甲酰肼-1-羰基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.45g,0.77mmol)在四氢呋喃(20mL)中的溶液添加劳森试剂(0.47g,1.16mmol),并且将混合物在70℃搅拌16小时。将反应冷却至环境温度,并且用饱和碳酸氢钠溶液淬灭。用乙酸乙酯提取产物,用水和盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。通过快速色谱(30%乙酸乙酯/己烷)纯化粗材料,以提供为灰白色固体的(3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.35g,78%产率):MS(ES)m/z 579.3[M+H]+。
步骤5:N-((3R,5S)-5-甲基哌啶-3-基)-5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺的制备

将(3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-羧酸苄酯(0.35g,1.02mmol)和三氟乙酸(5.0mL)的溶液在密封管中在100℃搅拌1小时。在冷却之后,在真空中浓缩反应混合物,将获得的剩余物溶解在1,4-二噁烷:氨水(5.0mL:5.0mL)中,并且在环境温度搅拌过夜。在真空中浓缩反应混合物,以提供为灰白色固体的N-((3R,5S)-5-甲基哌啶-3-基)-5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.3g,粗品):MS(ES)m/z 315.2(M+H)。
步骤6:3-((3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈的制备

将2-氰基乙酸(0.16g,1.91mmol)、1-羟基苯并三唑(0.22g,1.43mmol)和1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(0.27g,1.43mmol)在N,N-二甲基甲酰胺(6mL)中的溶液在环境温度搅拌3分钟,然后添加N-((3R,5S)-5-甲基哌啶-3-基)-5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-胺(0.3g,0.95mmol),然后添加N,N-二异丙基乙胺(0.5mL,2.86mmol),并且将所得的混合物在环境温度搅拌3小时。用冰水淬灭反应混合物,并且用乙酸乙酯提取产物。用碳酸氢钠溶液、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用反相色谱纯化粗材料,以提供为灰白色固体的3-((3R,5S)-3-((5-(1,3,4-噻二唑-2-基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)-5-甲基哌啶-1-基)-3-氧代丙腈(0.02g,5%产率):1H NMR(400MHz,DMSO-d6)δ11.71(br s,1H),9.36-9.46(m,2H),8.34(s,1H),7.25(s,1H),6.79(s,1H),4.82-4.84(m,1H),4.30(m,1H),3.93-4.09(m,3H),3.58-3.61(m,1H),2.64-2.74(m,1H),2.19-2.30(m,1H),1.87(m,1H),1.32-1.35(m,1H),0.91(brs,3H);MS(ES)m/z 382.4(M+H)。
分析条件:
柱:X-Bridge,C18 19*100*5微米
流动相(A):水中0.1%氢氧化铵
流动相(B):MeCN
流速:20mL/min
(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备
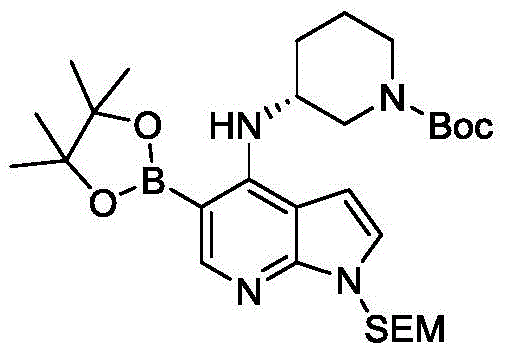
方案67.(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

步骤1:5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶的制备

在0℃向5-溴-4-氯-1H-吡咯并[2,3-b]吡啶(20g,87.3mmol)、三乙胺(36.4mL,262.0mmol)和4-二甲基氨基吡啶(1.06g,8.73mmol)在二氯甲烷(40mL)中的经搅拌的溶液添加对甲苯磺酰氯(24.9g,131.0mmol),并且将混合物在室温搅拌1小时。用水淬灭反应,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。用正己烷洗涤粗产物,以提供为灰白色固体的5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(32.5g,97%产率):MS(ES)m/z 387.0(M+2)。
步骤2:(R)-3-((5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

在密封管中,将5-溴-4-氯-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(5g,13.0mmol)、(R)-3-氨基哌啶-1-羧酸叔丁酯(3.9g,19.5mmol)和三乙胺(0.9mL,6.5mmol)在N-甲基吡咯烷酮(25mL)中的经搅拌的溶液在150℃加热16小时。将反应冷却至环境温度,并且用水淬灭反应,并且用乙酸乙酯提取。用水、盐水洗涤有机层,在无水硫酸钠上干燥,过滤并且在真空中浓缩。使用快速色谱(15%乙酸乙酯/己烷)纯化粗材料,以提供为棕色液体的(R)-3-((5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(4.0g,56%产率):MS(ES)m/z 549(M+)。
步骤3:(R)-3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

将(R)-3-((5-溴-1-甲苯磺酰基-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(18.4g,33.5mmol)和单水氢氧化锂(4.93g,117.5mmol)在THF:MeOH:水(25mL:100mL:25mL)中的经搅拌的溶液加热2小时至60℃。将反应冷却至环境温度,并且然后在真空中浓缩。将粗品溶解在乙酸乙酯中,并且用水、盐水洗涤,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为棕色液体的(R)-3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(12.4g,粗品):MS(ES)m/z 395.1(M+)。
步骤4:(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

在0℃向氢化钠(1.29g,32.4mmol,矿物油中60%悬浮液)在干二甲基甲酰胺(80mL)中的悬浮液缓慢添加(R)-3-((5-溴-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(8.0g,20.2mmol),并且将混合物搅拌30分钟。然后,在0℃添加2-(三甲基甲硅烷基)乙氧基甲基氯(6.45mL,36.4mmol),并且将混合物在环境温度搅拌1.5小时。用饱和氯化铵溶液淬灭反应混合物,并且用乙酸乙酯提取。用水、盐水洗涤有机提取物,并且在无水硫酸钠上干燥。过滤溶液,并且在真空中浓缩以提供粗材料,通过柱色谱(5%乙酸乙酯/己烷)纯化粗材料以提供为黄色油状物的(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(7.0g,66%产率):MS(ES)m/z525.0(M+)。
步骤5:(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯的制备

向(R)-3-((5-溴-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(2g,3.80mmol)在1,4-二噁烷:甲苯(4:1)中的溶液添加4,4,4’,4’,5,5,5’,5’-八甲基-2,2’-双(1,3,2-二噁硼烷)(1.25g,4.95mmol)和磷酸三钾(2.42g,11.4mmol)。将混合物用氮气吹扫15分钟。添加X-Phos Pd G2(0.60g,0.76mmol),并且将所得的混合物在110℃在预热的油浴器上搅拌15分钟。将反应冷却至环境温度,用乙酸乙酯稀释,并且通过硅藻土过滤。用水、盐水洗涤滤液,在无水硫酸钠上干燥,过滤并且在真空中浓缩,以提供为粗品的(R)-3-((5-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1H-吡咯并[2,3-b]吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(3g,粗品):MS(ES)m/z 573.0(M+H)+。
生物学活性测定
JAK1、JAK2、JAK3和Tyk-2酶活性测定
通过测量SRCtide(FAM-GEEPLYWSFPAKKK-NH2)的磷酸化来定量JAK3(a.a.781-1124,ThermoFisher)的活性。激酶反应在20mM HEPES(pH 7.5)、10mM MgCl2、0.01%BSA和0.0005%吐温20的缓冲液条件下在具有2%DMSO最终浓度的384孔Greiner板中进行。激酶反应组分为2.5nM JAK3、1μM SRCtide肽和1μM ATP。以2μM起始在剂量响应中测试实施例(11种浓度,3倍连续稀释,重复反应)。将反应在室温孵育40分钟,然后通过添加1:1体积的20mM HEPES(pH 7.5)中30mM EDTA(最终15mM EDTA)来终止反应。在反应终止后,分离磷酸化的肽和未磷酸化的肽,并且使用Caliper LC3000/EZ-Reader系统和HTS孔分析仪软件(Caliper,A PerkinElmer Company,Hopkinton,MA)定量。使用GraFit(ErithacusSoftware Ltd.,Horley,U.K.)通过将剂量响应数据拟合到4参数逻辑IC50方程来计算抑制剂效能。
在Thermo Fisher Scientific在其Selectscreen中使用Z-lyte测定完成JAK1、JAK2和Tyk-2的候选化合物的抑制效能。以下是每种酶的测定细节。
JAK1:在50mM HEPES(pH 6.5)、0.01%BRIJ-35、10mM MgCl2、1mM EGTA、0.02%NaN3中制备2×JAK-酶/Tyr 06混合物。最终10μL激酶反应由50mM HEPES(pH7.0)、0.01%BRIJ-35、10mM MgCl2、1mM EGTA、0.01%NaN3中的21.2-91.5ng JAK1和2μM Tyr 06组成。在一小时激酶反应孵育之后,添加5μL的1:128稀释度的显色剂。
JAK2:在50mM HEPES(pH 7.5)、0.01%BRIJ-35、10mM MgCl2、1mM EGTA中制备2×JAK2/Tyr 06混合物。最终10μL激酶反应由50mM HEPES(pH 7.5)、0.01%BRIJ-35、10mMMgCl2、1mM EGTA中的0.12-0.5ng JAK2和2μM Tyr 06组成。在一小时激酶反应孵育之后,添加5μL的1:128稀释度的显色剂A。
TYK2:在50mM HEPES(pH 6.5)、0.01%BRIJ-35、10mM MgCl2、1mM EGTA、0.02%NaN3中制备2×TYK2/Tyr 03混合物。最终10μL激酶反应由50mM HEPES(pH7.0)、0.01%BRIJ-35、10mM MgCl2、1mM EGTA、0.01%NaN3中的3.75-15ng TYK2和2μM Tyr 03组成。在一小时激酶反应孵育之后,添加5μL的1:4096稀释度的显色剂A。
JAK1、JAK2和TYK2的数据缩减相同(独立于酶运行)。总之,在不存在酶的情况下限定背景信号,并且在仅存在溶媒(2%DMSO)的情况下限定未被抑制的信号。以范围为20μM至0.34nM的11点剂量响应评估化合物。使用发射率的4参数逻辑拟合作为化合物浓度的函数来确定化合物的IC50值。表3中示出结果。
表3



JAK细胞靶标调节测定
靶标调节基于化合物抑制所选择底物的JAK同种型特异性磷酸化的能力。Tyr694上经IL-2刺激的STAT5磷酸化用于评估JAK1/3化合物选择性。Tyr694上经GM-CSF刺激的STAT5磷酸化用于评估JAK2化合物选择性。Tyr701上经IFNγ刺激的STAT1磷酸化用于评估JAK1/2化合物选择性。Tyr693上经IL-12刺激的STAT4磷酸化用于评估JAK2/TYK2化合物选择性。对于全部四种测定,将来自冷冻原料的人PBMC解冻、沉淀、重悬于完全培养基(90%RPMI、10%热灭活FBS、10mM HEPES、47μM 2-ME、青霉素/链霉素)中,并且以200000个/孔放置在120μl完全培养基、96孔V型底板的孔中。在具有1%DMSO的培养基(或者作为对照的具有1%DMSO的培养基)中添加为15μl/孔的10×工作储备溶液的化合物,并且放置在具有5%CO2的37℃孵育器中的板摇动器上(同时轻微摇动)(设置为3)1小时。刺激使用可溶性细胞因子的添加。对于JAK1/3磷酸化-STAT5测定,添加15μl的10×工作原料重组人IL-2至25ng/ml的最终浓度。对于JAK2磷酸化-STAT5测定,添加15μl的10×工作原料重组人GM-CSF至5ng/ml的最终浓度。对于JAK1/2磷酸化-STAT1测定,添加15μl重组人IFNγ的10×工作原料至10ng/ml的最终浓度。对于JAK2/TYK2磷酸化-STAT4测定,添加15μl重组人IL-12的10×工作原料至1.7ng/ml的最终浓度。然后,将板放置回孵育器中的板摇动器上分别达附加的5分钟、5分钟、10分钟和25分钟,在从孵育器中取出板时,用板密封件密封,并且以400×g使细胞沉淀5分钟。在沉淀之后,通过抽吸去除培养基,并且在测定特异性细胞裂解缓冲液中裂解细胞。使用来自Meso Scale Discovery的磷酸化(Tyr694)STAT5a,b全细胞裂解物试剂盒确定磷酸化-STAT5的水平。使用CST-PathScan磷酸化-STAT1(Tyr701)夹心ELISA(SandwichELISA)试剂盒确定磷酸化-STAT1的水平。使用来自Meso Scale Discovery的磷酸化-STAT4(Tyr693)全细胞裂解物试剂盒确定磷酸化-STAT4的水平。表4中示出结果。
表4


小鼠中的GI暴露PK
用~0.025%吐温20润湿测试化合物,并且在研钵和研杵中研磨,然后缓慢添加0.5%甲基纤维素以将最终体积补足至7.25mL。使BalbC测试动物经受用5%异氟烷和0.8-1.5L/min O2气流的混合物诱导的麻醉。然后,经由口服管饲法来将配制的测试化合物给药。在不同时间点(0.5h、1h、2h、4h、6h和8h)从门静脉(肝前)收集血液样品(~100μL),然后按照时间点心脏穿刺(循环)到含有2%w/v K2 EDTA水溶液的管中。在血液取样后,在同一时间点从小鼠收集胃肠(GI)组织。将GI组织解剖成为十二指肠、空肠、回肠和结肠的不同的部分。通过经由口服管饲针用PBS冲洗来洗涤分离的组织,以去除GI内容物。在收集后,将全部组织吸干、称重并且用PBS缓冲液(pH 7.4)均匀化。将血液样品立即放置在冰上,并且在30min内在4℃以8000rpm离心5min以获得血浆。通过移液器将血浆转移到预先标记的离心管中,并且立即转移到-80℃直至生物分析。将组织样品放置在预先标记的容器中,并且将全部样品保持在-80℃直至均匀化。将匀浆立即储存在-80℃直至进行生物分析。将研究样品解冻至室温,并且将50μL样品等分到预先标记的小瓶中。向其中添加400μL含有IS(100ng/mL;华法林、甲苯磺丁脲和洛哌丁胺)的100%乙腈。使小瓶充分混合,涡旋5min,然后在4℃以14000rpm离心5min。分离上清液,并且将其注射在LC-MS/MS上。使用文本矩阵中的标准曲线通过LC-MS分析确定测试化合物的血浆水平和GI组织水平。使用PhoenixWinNonlin(版本8.1)通过非房室分析(NCA)方法分析个体浓度-时间数据。表5中示出结果。
表5.在5mg/kg的口服剂量之后,雄性BalbC小鼠中实施例31的Cmax、Tmax和AUC计算

1.对于三个受试者中的两个受试者,低于第三个动物的定量极限(BLQ)。除了一个动物在
8小时处为13.5ng/mL之外,其他时间点为BLQ。
2.NRV(无报告值)-数据点不足以计算PK参数。
小鼠中急性DSS结肠炎模型
经由饮用水向小鼠口服施用硫酸化多糖葡聚糖硫酸钠(DSS)诱导严重的结肠炎,该结肠炎的特征在于体重减轻、血性腹泻、溃疡形成、上皮细胞损失以及嗜中性粒细胞、巨噬细胞和淋巴细胞的浸润(类似于人炎性肠病(IBD)的一些特征)。参见Okayasu,I.etal.Gastroenterology,98:694-702(1990)和Wirtz et al.Nature Protocols.12(7):1295(2017)。来自Charles River的9-12周龄雌性C57BL/6小鼠被给予无菌饮用水中4%DSS(结肠炎级DSS,分子量:36000-50000;MP Biomedicals目录号160110)达5天。在第6天,小鼠被转换成无菌饮用水,并且从第6天至第10天或第10天至第14天给药(口服,每日两次)。
每天监测体重,并且报告为初始体重的百分比。粪便稠度和血性腹泻也被评分(0-4)。在每个研究结束时,处死小鼠,并且去除整个结肠并且用PBS冲洗。测量结肠重量和长度,并且报告结肠重量与长度的比值。将末端结肠的0.5cm节段收集在微量离心管中,并且在干冰中冷冻。使用来自MSD的V-PLEX小鼠细胞因子29-plex试剂盒制备该组织的裂解物以用于结肠细胞因子确定。将末端结肠的3cm节段收集到盒中,并且固定在10%福尔马林中以用于组织学。制备载玻片,并且用苏木精和曙红(H&E)染色,并且由病理学家以盲法进行评分。H&E载玻片的评分包含炎症、腺体损失、糜烂、粘膜下水肿、粘膜厚度、嗜中性粒细胞评分和淋巴聚集体(lymphoid aggregate)计数和尺寸的类别。
本申请中引用的全部美国或外国的参考文献、专利或申请特此通过引用并入,如同整体写在本文中。在出现任何不一致的情况下,本文书面公开的材料进行约束。
由以上说明,本领域技术人员可以容易地确知本发明的本质特性,并且在不脱离本发明的精神和范围的情况下,可以对本发明作出各种变化和修改以使其适应各种用途和条件。
Claims (13)
1.一种式(I)的化合物、或所述式(I)的化合物的衍生物:

其中:
R1选自CN或杂芳基基团,并且可选地在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C0-C6烷基C3-C6环烷基、-(C0-C6烷基)C3-C6杂环、-OH、-SO2R9、-SOR9、-SR9、-NHSO2R9、-OSO2R9、-C0-C6烷基SO2R9、C0-C6烷基COR9、C0-C6烷基NR7C(O)NR7R8、C0-C6烷基OC(O)NR7R8、C0-C6烷基NR7SO2R9、-C0-C6烷基NR7COR9-OC1-C6烷基、-OC0-C6烷基C3-C6环烷基、-OC0-C6烷基C3-C6杂环、-OC0-C6烷基NR7C(O)NR7R8、-OC0-C6烷基OC(O)NR7R8、-OC0-C6烷基NR7SO2R9、-OC0-C6烷基NR7COR9、-NR7R8、-NR7C0-C6烷基C1-C6烷基、-NR7C0-C6烷基C3-C6环烷基、-NR7C0-C6烷基NR7C(O)NR7R8、-NR7C0-C6烷基OC(O)NR7R8、-NR7C0-C6烷基NR7SO2R9、-NR7C0-C6烷基NR7COR9、-NR7C0-C6烷基C3-C6杂环、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团、杂环基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、-C0-C6烷基NR7R8、-C0-C6烷基OH、-SO2R9、-SOR9、-NHSO2R9、-C0-C6烷基NR7R4、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R2选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环A在一个或更多个碳处被一个、两个、或三个R3取代基取代,其中每个R3取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
所述环A的相同碳原子或不同碳原子上的两个R3基团能够可选地连接以与环A形成螺环环系或双环环系;
R4选自-C(O)-R6、-CH2R6、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R5选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R6选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR7R8、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团能够可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R7和R8独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团能够可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R7和R8能够可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环,其中所述杂环能够可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代;并且
R9选自H、-C1-C5烷基、-OC1-C5烷基、-C3-C6环烷基、和NR7R8,其中烷基基团、杂环基团、或环烷基基团能够可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代。
2.一种式(II)的化合物、或所述式(II)的化合物的衍生物:

其中:
R10选自CN或杂芳基基团,并且可选地在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C0-C6烷基C3-C6环烷基、-(C0-C6烷基)C3-C6杂环、-OH、-SO2R90、-NHSO2R90、-OSO2R90、-C0-C6烷基SO2R90、C0-C6烷基COR90、C0-C6烷基NR70C(O)NR70R80、C0-C6烷基OC(O)NR70R80、C0-C6烷基NR70SO2R90、-C0-C6烷基NR70COR90-OC1-C6烷基、-OC0-C6烷基C3-C6环烷基、-OC0-C6烷基C3-C6杂环、-OC0-C6烷基NR70C(O)NR70R80、-OC0-C6烷基OC(O)NR70R80、-OC0-C6烷基NR70SO2R90、-OC0-C6烷基NR70COR90、-NR70R80、-NR70C0-C6烷基C1-C6烷基、-NR70C0-C6烷基C3-C6环烷基、-NR70C0-C6烷基NR7C(O)NR70R80、-NR70C0-C6烷基OC(O)NR70R80、-NR70C0-C6烷基NR70SO2R90、-NR70C0-C6烷基NR70COR90、-NR70C0-C6烷基C3-C6杂环、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团、杂环基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、-C0-C6烷基NR70R80、-C0-C6烷基OH、-SO2R90、-SOR90、-NHSO2R90、-C0-C6烷基NR70R40、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R20选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环B在一个或更多个碳处被一个、两个、或三个R30取代基取代,其中每个R30取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
所述环B的相同碳原子或不同碳原子上的两个R30基团能够可选地连接以与环B形成螺环环系或双环环系;
R40选自-C(O)-R60、-CH2R60、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R50选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R60选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR70R80、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团能够可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R70和R80独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团能够可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R70和R80能够可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环,其中所述杂环能够可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代;并且
R90选自H、-C1-C5烷基、-C3-C6环烷基、和NR70R80,其中烷基基团、杂环基团、或环烷基基团能够可选地被选自卤族元素、-OH、NH2、NHMe、NMe2、或-CN的一个或更多个基团取代。
3.一种式(III)的化合物、或所述式(III)的化合物的衍生物:

其中:
R100选自CN或杂芳基基团,并且在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C3-C6环烷基、-OH、-O-C1-C5烷基、-SO2R900、-NHSO2R900、-OSO2R900、-CH2SO2R900、(CH2)nCOR900、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、NH2、NH-C1-C5烷基、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R200选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环C在一个或更多个碳处被一个、两个、或三个R300取代基取代,其中每个R300取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
所述环C的相同碳原子或不同碳原子上的两个R300基团能够可选地连接以与环C形成螺环环系或双环环系;
R400选自-C(O)-R600、-CH2R600、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R500选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R600选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR700R800、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团能够可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R700和R800独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团能够可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R700和R800能够可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环;并且
R900选自H、-C1-C5烷基、-C3-C6环烷基、和NR700R800,其中烷基基团能够可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代。
4.一种式(IV)的化合物、或所述式(IV)的化合物的衍生物:

其中:
R1000选自CN或杂芳基基团,并且在一个或更多个可用的氮原子处被独立地选自H或C1-C5烷基的基团取代或者在一个或更多个可用的碳原子处被取代基取代,其中每个取代基独立地选自H、卤族元素、CN、-C1-C4烷基、-C3-C6环烷基、-OH、-O-C1-C5烷基、-SO2R9000、-NHSO2R9000、-OSO2R9000、-CH2SO2R9000、(CH2)nCOR9000、芳基和杂芳基,其中每个烷基基团、环烷基基团、芳基基团或杂芳基基团可选地被选自下列的一个或更多个基团取代:卤族元素、-OH、NH2、NH-C1-C5烷基、CN、-C1-C5烷基烷氧基、C1-C5烷氧基或-O-C1-C5烷基;
R2000选自H、-C1-C4烷基、-C3-C6环烷基、或-C1-C2烷基-C3-C6环烷基,其中烷基基团或环烷基基团可选地被选自卤族元素、-OH或-O-C1-C5烷基的一个或更多个基团取代;
n是0、1或2;
环D在一个或更多个碳处被一个、两个、或三个R3000取代基取代,其中每个R3000取代基独立地选自H、卤族元素、-C1-C4烷基、-C3-C6环烷基、-OH、或-O-C1-C5烷基,其中每个烷基基团或环烷基基团可选地被选自卤族元素、-OH、-C1-C5烷基烷氧基、或-O-C1-C5烷基的一个或更多个基团取代;
所述环D的相同碳原子或不同碳原子上的两个R3000基团能够可选地连接以与环D形成螺环环系或双环环系;
R4000选自-C(O)-R6000、-CH2R6000、-C(O)-C1-C5烷基、或-C(O)-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自-OH、卤族元素、炔烃、或-CN的一个或更多个基团取代;
R5000选自-C1-C5烷基、或-C3-C6环烷基,其中烷基基团或环烷基基团能够可选地被选自卤族元素、-OH、或-O-C1-C5烷基的一个或更多个基团取代;
R6000选自-C1-C5烷基、-C3-C6环烷基、-C1-C5烷基-C3-C6环烷基、-NR7000R8000、-O-芳基、-O-杂芳基、芳基、或杂芳基,其中烷基基团、环烷基基团、芳基基团或杂芳基基团能够可选地被选自卤族元素、-CN、炔烃、-OH、三氟甲基、-O-C1-C5烷基、或-O-C3-C6环烷基的一个或更多个基团取代;
R7000和R8000独立地选自H、-C1-C5烷基、-C1-C5烷氧基、或-C3-C5环烷基,其中烷基基团能够可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代;
R7000和R8000能够可选地连接成环以形成诸如哌啶、吡咯烷的杂环,或者与另外的杂原子连接以形成诸如吗啉的环;并且
R9000选自H、-C1-C5烷基、-C3-C6环烷基、和NR7000R8000,其中烷基基团能够可选地被选自卤族元素、-OH、或-CN的一个或更多个基团取代。
5.如权利要求1所述的化合物,其中所述化合物选自由下列组成的组:



















6.如权利要求2所述的化合物,其中所述化合物选自由下列组成的组:

7.一种药物组合物,所述药物组合物包括:权利要求1-6所述的化合物、所述化合物的衍生物、或其组合;以及药学上可接受的赋形剂。
8.一种在有需要的受试者中治疗JAK介导的疾病的方法,所述方法包括:以对于治疗所述JAK介导的疾病有效的量向患有所述JAK介导的疾病的受试者施用权利要求1-7所述的化合物、所述化合物的衍生物、或其组合。
9.如权利要求8所述的方法,所述方法还包括施用另外的治疗剂。
10.如权利要求8所述的方法,其中所述JAK介导的疾病选自由下列组成的组:自身免疫紊乱或自身免疫应答、免疫应答的广泛激活、细菌感染、病毒感染、炎症、慢性和/或急性炎性紊乱或病况、和/或自身炎性紊乱、纤维化紊乱、代谢紊乱、赘生物、或者心血管紊乱或脑血管紊乱、皮肤紊乱、瘙痒、毛发缺损紊乱、癌症或恶性肿瘤、自身免疫性结缔组织病和自身免疫病况;斯蒂尔病、成年起病型斯蒂尔病、Th17相关性炎症、多软骨炎(例如,复发性多软骨炎);肌炎、多肌炎、自身免疫性肌炎、皮肌炎、幼年型皮肌炎;重症肌无力;关节炎(例如,类风湿性关节炎、幼年型类风湿性关节炎、全身起病型幼年类风湿性关节炎、骨关节炎、感染性关节炎、炎症性关节炎、炎性肠病相关性关节炎、特发性关节炎、幼年型特发性关节炎、全身型幼年特发性关节炎、银屑病性关节炎)、脊椎炎/脊椎关节炎/脊椎关节病(强硬性脊椎炎)、痛风、硬皮病(全身性硬皮病、幼年型硬皮病)、赖特综合征/反应性关节炎、莱姆病、狼疮/系统性红斑狼疮(SLE)(红斑狼疮、儿童系统性红斑狼疮、皮肤狼疮(亚急性皮肤狼疮、慢性皮肤狼疮/盘状狼疮、冻疮样红斑狼疮))、风湿性多肌痛、起止点炎、混合型结缔组织病、起止点病、心脏炎、心肌炎、血管生成紊乱、骨髓增生异常综合征、动脉粥样硬化、再狭窄(动脉粥样硬化性冠状动脉再狭窄)、急性冠状动脉综合征、心肌梗死、心脏移植物血管病变、移植性动脉病;血管炎(大血管血管炎、小血管血管炎、巨细胞动脉炎、结节性多动脉炎、血管炎综合征,所述血管炎综合征包含大动脉炎、韦格纳肉芽肿、白塞病)、干扰素基因刺激蛋白(STING)相关的婴儿期起病型血管病变(SAVI);胃肠道紊乱、小肠结肠炎、结肠炎、炎性肠病(溃疡性结肠炎、克罗恩病)、肠易激综合征、肠炎综合征/痉挛性结肠、乳糜泻;急性胰腺炎和慢性胰腺炎;原发性胆汁性肝硬化、原发性硬化性胆管炎、黄疸、肝硬化(例如,原发性胆汁性肝硬化、或归因于脂肪性肝病(例如,酒精性脂肪变性和非酒精性脂肪变性)的肝硬化);食管炎、胃炎、胃溃疡和十二指肠溃疡、腹膜炎;肾病、免疫介导的肾小球性肾病、自身免疫性肾病、膜性肾小球病、慢性进行性肾病、糖尿病性肾脏疾病/糖尿病性肾病、肾纤维化、肾缺血/再灌注损伤、HIV相关性肾病、输尿管梗阻性肾病、肾小球硬化症、蛋白尿、肾病综合征、多囊性肾脏疾病、常染色体显性多囊性肾脏疾病、肾病是免疫介导的肾病、自身免疫性肾病、慢性进行性肾病、糖尿病性肾病、肾纤维化、缺血性/再灌注损伤相关性肾病、HIV相关性肾病、输尿管梗阻性肾病、肾小球肾炎、慢性肾脏疾病(例如,糖尿病性肾病)、高血压诱导的肾病、肾小球硬化症、蛋白尿、肾病综合征、多囊性肾脏疾病、常染色体显性多囊性肾脏疾病、糖尿病性肾脏疾病、狼疮性肾炎;间质性膀胱炎;牙周炎、龈炎;肺部炎症、窦炎、肺炎、支气管炎、哮喘、支气管哮喘、变应性哮喘、非变应性哮喘、变应性支气管肺真菌病、阿斯匹林诱导的哮喘、成年起病型哮喘、固定气流阻塞型哮喘、运动诱导的哮喘、咳嗽变异性哮喘、工作相关性哮喘、夜间哮喘、哮喘并发肥胖症、嗜酸细胞性哮喘、激素抵抗型哮喘/重度哮喘、外源性哮喘、内源性哮喘/隐源性哮喘、丘-施综合征、细支气管炎、闭塞性细支气管炎、慢性阻塞性肺病(COPD)、间质性肺疾病(肺纤维化、特发性肺纤维化)、急性肺损伤、肺纤维化(例如,特发性肺纤维化或囊性纤维化)、慢性阻塞性肺病、成人呼吸窘迫综合征、急性肺损伤、药物诱导的肺损伤;梅尼埃病;眼部紊乱,所述眼部紊乱包含(例如)眼部炎症、葡萄膜炎、干眼/干燥性角膜结膜炎、巩膜炎、巩膜外层炎、角膜炎/角膜病、脉络膜炎、视网膜血管炎、视神经炎、视网膜病变(糖尿病性视网膜病变、免疫介导的视网膜病变、黄斑变性、湿性黄斑变性、干性(老年性)黄斑变性);肥大细胞增生病、缺铁性贫血、尿毒症、嗜酸细胞增多综合征(HES)、系统性肥大细胞病(SMCD)、骨髓增生异常综合征、特发性血小板减少性紫癜;骨吸收疾病;神经退行性紊乱、神经紊乱/神经肌肉紊乱(例如),多发性硬化、帕金森病、亨廷顿病、肌萎缩性侧索硬化(ALS)(家族性ALS、散发性ALS)、阿尔茨海默病、重症肌无力、兰-伊肌无力综合征(LEMS)、格林-巴利综合征、脑膜炎、脑炎、创伤性脑损伤;神经系统损害、寄生虫妄想症、神经元过程和感觉知觉的调节异常、中风/神经元缺血、脊髓损伤、外周神经病变、触幻觉、脊髓损伤、精神疾病;感觉异常性疼痛(急性疼痛、慢性疼痛、神经性疼痛、或纤维肌痛)、神经刺激、外周神经病变;瘙痒/痒病(特应性瘙痒、干燥性瘙痒、与银屑病相关的瘙痒/银屑病性痒病/银屑病相关性痒病)、急性瘙痒、慢性瘙痒、特发性瘙痒、慢性特发性痒病、胆汁性痒病、肝胆相关性痒病、肾相关性痒病/肾痒病、尿毒症性痒病、胆汁郁积、妊娠期肝内胆汁郁积、慢性单纯性苔癣相关性瘙痒、淋巴瘤相关性痒病、白血病相关性痒病、结节性痒疹、特应性皮炎相关性痒病、特应性痒病/特应性瘙痒、大疱性痒病、肱桡肌瘙痒、神经原性痒病、神经性痒病、感觉异常性背痛、HIV相关性瘙痒性丘疹、精神性痒病、游泳者痒病、瘙痒或尿毒症性痒病、荨麻疹性痒病;皮肤病(例如),皮肤病药物反应/药疹、干燥病/皮肤干燥、皮疹、皮肤致敏、皮肤刺激、晒斑、修面、体虱、头虱/虱病、阴虱、皮肤幼虫移行症、疥疮、寄生虫感染、昆虫感染、荨麻疹/荨麻疹、丘疹性荨麻疹、昆虫咬伤、昆虫刺伤、头皮屑、皮肤上的异物或装置、真菌感染、疱疹、水痘/水痘、嗜酸性毛囊炎、妊娠皮肤病/妊娠瘙痒性荨麻疹性丘疹及斑块病(PUPP)、炎性皮肤病、嗜中性皮肤病、组织细胞样嗜中性皮肤病、肠旁路综合征皮肤病、银屑病/寻常型银屑病、扁平苔藓、硬化性萎缩性苔藓、痤疮(寻常型痤疮、粉刺型痤疮、炎性痤疮、结节囊肿性痤疮、瘢痕性痤疮、颈项部疤痕疙瘩性痤疮)、特应症(变应性接触性致敏、变应性皮炎)、皮炎(特应性皮炎/湿疹、接触性皮炎、光照性皮炎、脂溢性皮炎、瘀滞性皮炎)、急性发热性嗜中性皮肤病(斯威特综合征)、慢性非典型中性粒细胞与脂肪代谢障碍性皮肤病和高温综合征(CANDLE综合征)、化脓性汗腺炎、荨麻疹、坏疽性脓皮病、脱发(眉毛脱落、鼻内毛发脱落、瘢痕性脱发(例如,疤痕性脱发、中央离心性疤痕性脱发、毛发扁平苔藓、前额纤维化性脱发、脱发性毛囊炎)、非瘢痕性脱发(斑秃(AA)(斑块型AA、全秃(AT)、普秃(AU)、蛇形斑秃、马蹄形斑秃))、雄激素源性/雄激素性脱发(AGA)/男性及女性型AGA)、静止期脱发、头癣、稀毛症(单纯性遗传性稀毛症)、毛发扁平苔藓(前额纤维化性脱发)、点状掌跖角皮病、持久隆起性红斑(EED)、嗜中性外分泌性汗腺炎、栅栏状嗜中性肉芽肿性皮炎、嗜中性荨麻疹皮肤病、白癜风,所述白癜风包含节段型白癜风(单节段型白癜风、双节段型白癜风、多节段型白癜风)、非节段型白癜风(肢端型白癜风、面部型白癜风、或肢端面部型白癜风、面部中央型白癜风、粘膜型白癜风、碎纸样白癜风、三色白癜风、边缘性炎性白癜风、四色白癜风、蓝色白癜风、凯布内现象、寻常型白癜风、泛发型白癜风、普发型白癜风)、混合型白癜风/非节段型白癜风伴发节段型白癜风、局限型白癜风、孤立性粘膜型白癜风、或者伴有或不伴有白发(身体毛发受累)的白癜风;大疱性疾病、免疫性大疱性疾病(大疱性类天疱疮、疤痕性类天疱疮、寻常型天疱疮、线性IgA疾病)、妊娠性类天疱疮、着色性干皮病;纤维化和瘢痕形成的紊乱:纤维瘤、肝纤维化、肺纤维化、特发性肺纤维化、诸如硬皮病、纤维化增加、疤痕疙瘩、术后瘢痕的低度瘢痕形成;伤口愈合、手术瘢痕形成、辐射诱导的纤维化(例如,头颈部、胃肠道或肺部)、中枢神经系统瘢痕形成、消化道或胃肠道纤维化、肾纤维化、肝脏纤维化或胆管纤维化、肝纤维化(例如,非酒精性脂肪性肝炎、丙型肝炎、或肝细胞癌)、心脏纤维化(例如,心内膜心肌纤维化或心房纤维化)、眼部瘢痕形成、纤维硬化、瘢痕生长、伤口或痂愈合、疤痕疙瘩、纵隔纤维化、骨髓纤维化、腹膜后纤维化/奥蒙德病、进行性大块纤维化、肾源性系统性纤维化;斯耶格伦综合征、结节病、家族性地中海热、冷吡啉相关周期性综合征(默-韦综合征、家族性寒冷性自身炎性综合征/家族性寒冷性荨麻疹/TNF受体相关周期性综合征、新生儿起病多系统炎性疾病)、氧过多诱导的炎症、再灌注损伤、术后创伤、组织损伤、高温综合征;糖尿病(I型糖尿病、II型糖尿病)/糖尿病、桥本甲状腺炎、格雷夫斯病、艾迪生病、卡斯尔门病、甲状旁腺功能亢进、绝经期、肥胖症、类固醇耐药性、葡萄糖耐受不良、代谢综合征、甲状腺疾病、垂体炎;系统性免疫衰老;自身免疫性萎缩性胃炎、恶性贫血的自身免疫性萎缩性胃炎、自身免疫性脑脊髓炎、自身免疫性睾丸炎、古德帕斯彻病、斯耶格伦综合征、自身免疫性血小板减少、交感性眼炎;自身免疫性疾病的继发血液学表现(例如,贫血)、自身免疫性溶血综合征(自身免疫性溶血性贫血)、自身免疫性肝炎和炎性肝炎、自身免疫性卵巢衰竭、自身免疫性血小板减少、硅酮植入相关的自身免疫性疾病、药物诱导的自身免疫、HIV相关的自身免疫性综合征、金属诱导的自身免疫、自身免疫性聋、自身免疫性甲状腺紊乱;过敏反应和变态反应,所述过敏反应和变态反应包含超敏反应,如I型超敏反应(例如,包含过敏症)、II型超敏反应(例如,古德帕斯彻病、自身免疫性溶血性贫血)、III型超敏反应疾病(例如,阿图斯反应、血清病)和IV型超敏反应(例如,接触性皮炎、同种异体移植排斥);急性感染和慢性感染、脓毒症综合征(脓毒症、脓毒性休克、内毒素性休克、外毒素诱导的中毒性休克、革兰氏阴性脓毒症、革兰氏阳性脓毒症、真菌性脓毒症、中毒性休克综合征);急性感染和慢性感染、脓毒症综合征(脓毒症、脓毒性休克、内毒素性休克、外毒素诱导的中毒性休克、革兰氏阴性脓毒症、革兰氏阳性脓毒症、真菌性脓毒症、中毒性休克综合征);排斥反应:移植物抗宿主反应/移植物抗宿主病、同种异体移植排斥(例如,急性同种异体移植排斥或慢性同种异体移植排斥)、早期移植排斥;恶性肿瘤、癌症、淋巴瘤、白血病、多发性骨髓瘤、实体瘤、畸胎瘤、转移性紊乱和骨紊乱、内部癌症、骨癌、口腔癌/咽癌、食管癌、喉癌、胃癌、肠癌、结肠癌、直肠癌、肺癌(例如,非小细胞肺癌或小细胞肺癌)、肝癌(肝脏癌)、胰腺癌、神经癌、脑癌(例如,神经胶质瘤、多形性成胶质细胞瘤、星形细胞瘤、神经母细胞瘤、和神经鞘瘤)、头颈癌、咽喉癌、卵巢癌、子宫癌、前列腺癌、睾丸癌、膀胱癌、肾癌(肾脏癌)、乳腺癌、胆囊癌、宫颈癌、甲状腺癌、前列腺癌、眼癌(眼部恶性肿瘤)、和皮肤癌(黑素瘤、角化棘皮瘤);以及纤维化癌症、纤维瘤、纤维腺瘤、纤维肉瘤、骨髓增生性紊乱、赘生物(造血组织赘生物、脊髓赘生物、淋巴组织赘生物(骨髓纤维化、原发性骨髓纤维化、真性红细胞增多、原发性血小板增多))、白血病(急性淋巴细胞白血病、急性髓细胞白血病和慢性髓细胞白血病、慢性淋巴细胞白血病、急性淋巴细胞性白血病、慢性粒-单核细胞型白血病(CMML)、或前髓细胞白血病)、多发性骨髓瘤及其他骨髓性恶性肿瘤(骨髓纤维化伴髓样化生(MMM)、原发性骨髓纤维化(PMF)、特发性骨髓纤维化(IMF))、淋巴瘤(霍奇金病、皮肤淋巴瘤(皮肤T细胞淋巴瘤、蕈样肉芽肿病))、淋巴瘤(例如,B细胞淋巴瘤、T细胞淋巴瘤、套细胞淋巴瘤、毛细胞淋巴瘤、伯基特淋巴瘤、肥大细胞瘤、霍奇金病、或非霍奇金病);卡波西肉瘤、横纹肌肉瘤、精原细胞瘤、畸胎癌、骨肉瘤、甲状腺滤泡状癌;外源性阿片类物质或合成阿片类物质的增加的累积、感觉异常性背痛、强迫性障碍、与强迫性障碍相关的怀乡症、及其组合。
11.如权利要求9所述的方法,其中所述另外的治疗剂选自:化学治疗剂、抗增殖剂、抗炎药剂、免疫调节剂、免疫抑制剂、神经营养因子、用于治疗心血管疾病的药剂、用于治疗糖尿病的药剂、用于治疗免疫缺陷紊乱的药剂、及其组合。
12.如权利要求10所述的方法,其中所述JAK介导的疾病是炎性肠病。
13.如权利要求12所述的方法,其中在所述施用操作时,所述化合物在所述受试者中以肠限制性方式抑制JAK活性。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962842197P | 2019-05-02 | 2019-05-02 | |
| US62/842,197 | 2019-05-02 | ||
| PCT/US2020/031332 WO2020223728A1 (en) | 2019-05-02 | 2020-05-04 | Substituted pyrrolopyridines as jak inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113727975A true CN113727975A (zh) | 2021-11-30 |
Family
ID=73017367
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202080023588.6A Pending CN113727975A (zh) | 2019-05-02 | 2020-05-04 | 作为jak抑制剂的被取代的吡咯并吡啶 |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US11420966B2 (zh) |
| EP (1) | EP3962897A4 (zh) |
| JP (1) | JP2022531088A (zh) |
| KR (1) | KR20220004726A (zh) |
| CN (1) | CN113727975A (zh) |
| AU (1) | AU2020265828A1 (zh) |
| BR (1) | BR112021021477A2 (zh) |
| CA (1) | CA3138544A1 (zh) |
| IL (1) | IL287717A (zh) |
| MX (1) | MX2021013224A (zh) |
| PH (1) | PH12021552036A1 (zh) |
| SG (1) | SG11202108927UA (zh) |
| WO (1) | WO2020223728A1 (zh) |
| ZA (1) | ZA202105837B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116249533A (zh) * | 2020-07-17 | 2023-06-09 | 轶诺(浙江)药业有限公司 | 一类肠道裂解型共药及其制备和用途 |
| WO2024114814A1 (en) * | 2022-12-02 | 2024-06-06 | Onquality Pharmaceuticals China Ltd | Jak inhibitors, pharmaceutical compositions, and therapeutic applications |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10981906B2 (en) | 2017-11-03 | 2021-04-20 | Aclaris Therapeutics, Inc. | Substituted pyrrolopyridine JAK inhibitors and methods of making and using the same |
| BR112021002479A2 (pt) | 2018-08-10 | 2021-07-27 | Aclaris Therapeutics, Inc. | composto, composição farmacêutica, método para inibir a atividade de itk ou jak3 em uma população de células, método para tratar um distúrbio mediado por itk ou jak3 em um indivíduo necessitando do mesmo e uso de um composto |
| JP7248256B2 (ja) * | 2019-03-14 | 2023-03-29 | 上海華匯拓医薬科技有限公司 | Jakキナーゼ阻害剤及びその調製方法、並びにその医薬分野での使用 |
| AU2020358720A1 (en) | 2019-10-01 | 2022-04-21 | Empyrean Neuroscience, Inc. | Genetic engineering of fungi to modulate tryptamine expression |
| CN113200983B (zh) * | 2021-05-22 | 2023-01-03 | 中国药科大学 | 一种吡咯并吡啶结构的化合物、制备方法和医药用途 |
| WO2023130076A2 (en) | 2021-12-31 | 2023-07-06 | Empyrean Neuroscience, Inc. | Targets and pathways for the production of alkaloidal compounds |
| CN118459457A (zh) * | 2023-02-08 | 2024-08-09 | 南京羚诺生物医药技术研究院有限公司 | 一种吡咯并吡啶结构的化合物及其制备方法和用途 |
| CN118812508A (zh) * | 2023-04-07 | 2024-10-22 | 原研药港生命科学研究(辽宁)有限公司 | 1-异丙基-1h-吡唑类化合物及药物组合物和应用 |
| WO2025038910A1 (en) * | 2023-08-16 | 2025-02-20 | Aclaris Therapeutics, Inc. | Crystalline polymorph form a of a jak inhibitor and methods for its preparation |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1296481A (zh) * | 1998-04-10 | 2001-05-23 | 辉瑞产品公司 | (4-芳基磺酰氨基)-四氢吡喃-4-羧酸羟基酰胺 |
| CN101228161A (zh) * | 2005-05-20 | 2008-07-23 | 沃泰克斯药物股份有限公司 | 适用作蛋白激酶抑制剂的吡咯并吡啶类 |
| US20090264399A1 (en) * | 2005-07-14 | 2009-10-22 | Astellas Pharma Inc. | Heterocyclic janus kinase 3 inhibitors |
| US20100105661A1 (en) * | 2007-01-12 | 2010-04-29 | Astellas Pharma Inc. | Condensed pyridine compound |
| WO2012058645A1 (en) * | 2010-10-29 | 2012-05-03 | Biogen Idec Ma Inc. | Heterocyclic tyrosine kinase inhibitors |
| WO2015083028A1 (en) * | 2013-12-05 | 2015-06-11 | Pfizer Inc. | Pyrrolo[2,3-d]pyrimidinyl, pyrrolo[2,3-b]pyrazinyl and pyrrolo[2,3-d]pyridinyl acrylamides |
| US20170121352A1 (en) * | 2015-10-30 | 2017-05-04 | Calithera Biosciences, Inc. | Compositions and methods for inhibiting arginase activity |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK0924201T3 (da) | 1993-11-30 | 2002-05-21 | Searle & Co | Tricyclisk substitueret pyrazolylbenzensulfonamider og deres anvendelse som cyclooxygenase II-inhibitorer |
| PA8474101A1 (es) | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| AU4851599A (en) | 1998-06-30 | 2000-01-17 | Parker Hughes Institute | Method for inhibiting c-jun expression using jak-3 inhibitors |
| US7135575B2 (en) | 2003-03-03 | 2006-11-14 | Array Biopharma, Inc. | P38 inhibitors and methods of use thereof |
| SE0301372D0 (sv) | 2003-05-09 | 2003-05-09 | Astrazeneca Ab | Novel compounds |
| AR054416A1 (es) | 2004-12-22 | 2007-06-27 | Incyte Corp | Pirrolo [2,3-b]piridin-4-il-aminas y pirrolo [2,3-b]pirimidin-4-il-aminas como inhibidores de las quinasas janus. composiciones farmaceuticas. |
| CN102127078A (zh) | 2005-07-14 | 2011-07-20 | 安斯泰来制药株式会社 | Janus激酶3的杂环类抑制剂 |
| BRPI0619817B8 (pt) | 2005-12-13 | 2021-05-25 | Incyte Corp | composto 3-ciclopentil-3-[4-(7h-pirrol[2,3-d]pirimidin-4-il)-1h-pirazol-1-il]propanonitrila e sua composição |
| UA99467C2 (xx) | 2007-06-13 | 2012-08-27 | Инсайт Корпорейшин | СОЛІ ІНГІБІТОРА ЯНУС-КІНАЗИ (R)-3-(4-(7H-ПІРОЛО$2,3-d]ПІРИМІДИН-4-ІЛ)-1H-ПІРАЗОЛ-1-ІЛ)-3-ЦИКЛОПЕНТИЛПРОПАННІТРИЛУ$СОЛИ ИНГИБИТОРА ЯНУС-КИНАЗЫ (R)-3-(4-(7H-ПИРРОЛО$2,3-d]ПИРИМИДИН-4-ИЛ)-1H-ПИРАЗОЛ-1-ИЛ)-3-ЦИКЛОПЕНТИЛПРОПАННИТРИЛА |
| CL2009001884A1 (es) | 2008-10-02 | 2010-05-14 | Incyte Holdings Corp | Uso de 3-ciclopentil-3-[4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1-il)propanonitrilo, inhibidor de janus quinasa, y uso de una composición que lo comprende para el tratamiento del ojo seco. |
| US8716303B2 (en) | 2009-05-22 | 2014-05-06 | Incyte Corporation | N-(hetero)aryl-pyrrolidine derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines and pyrrol-3-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| PH12013501001A1 (en) | 2010-11-19 | 2019-09-02 | Incyte Holdings Corp | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as jak inhibitors |
| IN2014MN00228A (zh) | 2011-08-12 | 2015-09-25 | Nissan Chemical Ind Ltd | |
| EP2924026A1 (en) * | 2014-03-28 | 2015-09-30 | Novartis Tiergesundheit AG | Aminosulfonylmethylcyclohexanes as JAK inhibitors |
| ES2737696T3 (es) | 2014-05-14 | 2020-01-15 | Pfizer | Pirazolopiridinas y pirazolopirimidinas |
| EP3360878B9 (en) | 2015-12-11 | 2021-05-05 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Azetidine derivative, preparation method therefor, and use thereof |
| EP3504213A4 (en) | 2016-08-24 | 2020-01-15 | ArQule, Inc. | AMINO-PYRROLOPYRIMIDINONE COMPOUNDS AND METHODS OF USE |
| US10981906B2 (en) | 2017-11-03 | 2021-04-20 | Aclaris Therapeutics, Inc. | Substituted pyrrolopyridine JAK inhibitors and methods of making and using the same |
| US10800775B2 (en) | 2017-11-03 | 2020-10-13 | Aclaris Therapeutics, Inc. | Pyrazolyl pyrrolo[2,3-b]pyrmidine-5-carboxylate analogs and methods of making the same |
| BR112021002479A2 (pt) | 2018-08-10 | 2021-07-27 | Aclaris Therapeutics, Inc. | composto, composição farmacêutica, método para inibir a atividade de itk ou jak3 em uma população de células, método para tratar um distúrbio mediado por itk ou jak3 em um indivíduo necessitando do mesmo e uso de um composto |
-
2020
- 2020-05-04 US US16/866,182 patent/US11420966B2/en active Active
- 2020-05-04 CA CA3138544A patent/CA3138544A1/en active Pending
- 2020-05-04 PH PH1/2021/552036A patent/PH12021552036A1/en unknown
- 2020-05-04 AU AU2020265828A patent/AU2020265828A1/en not_active Abandoned
- 2020-05-04 BR BR112021021477A patent/BR112021021477A2/pt not_active Application Discontinuation
- 2020-05-04 WO PCT/US2020/031332 patent/WO2020223728A1/en unknown
- 2020-05-04 JP JP2021561745A patent/JP2022531088A/ja active Pending
- 2020-05-04 EP EP20798983.1A patent/EP3962897A4/en not_active Withdrawn
- 2020-05-04 SG SG11202108927UA patent/SG11202108927UA/en unknown
- 2020-05-04 CN CN202080023588.6A patent/CN113727975A/zh active Pending
- 2020-05-04 MX MX2021013224A patent/MX2021013224A/es unknown
- 2020-05-04 KR KR1020217039379A patent/KR20220004726A/ko not_active Withdrawn
-
2021
- 2021-08-16 ZA ZA2021/05837A patent/ZA202105837B/en unknown
- 2021-10-31 IL IL287717A patent/IL287717A/en unknown
-
2022
- 2022-06-30 US US17/809,984 patent/US20220372034A1/en not_active Abandoned
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1296481A (zh) * | 1998-04-10 | 2001-05-23 | 辉瑞产品公司 | (4-芳基磺酰氨基)-四氢吡喃-4-羧酸羟基酰胺 |
| CN101228161A (zh) * | 2005-05-20 | 2008-07-23 | 沃泰克斯药物股份有限公司 | 适用作蛋白激酶抑制剂的吡咯并吡啶类 |
| US20090264399A1 (en) * | 2005-07-14 | 2009-10-22 | Astellas Pharma Inc. | Heterocyclic janus kinase 3 inhibitors |
| US20100105661A1 (en) * | 2007-01-12 | 2010-04-29 | Astellas Pharma Inc. | Condensed pyridine compound |
| WO2012058645A1 (en) * | 2010-10-29 | 2012-05-03 | Biogen Idec Ma Inc. | Heterocyclic tyrosine kinase inhibitors |
| WO2015083028A1 (en) * | 2013-12-05 | 2015-06-11 | Pfizer Inc. | Pyrrolo[2,3-d]pyrimidinyl, pyrrolo[2,3-b]pyrazinyl and pyrrolo[2,3-d]pyridinyl acrylamides |
| US20170121352A1 (en) * | 2015-10-30 | 2017-05-04 | Calithera Biosciences, Inc. | Compositions and methods for inhibiting arginase activity |
Non-Patent Citations (2)
| Title |
|---|
| MARK ZAK,等: "Identification of C-2 Hydroxyethyl Imidazopyrrolopyridines as Potent JAK1 Inhibitors with Favorable Physicochemical Properties and High Selectivity over JAK2", 《JOURNAL OF MEDICINAL CHEMISTRY》, 9 May 2013 (2013-05-09), pages 4764 - 4785, XP055462006, DOI: 10.1021/jm4004895 * |
| 梁欢,等: "三氮唑骈吡啶类JAK抑制剂的合成及体外活性评价", 《中国药物化学杂志》, 30 April 2018 (2018-04-30), pages 97 - 104 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116249533A (zh) * | 2020-07-17 | 2023-06-09 | 轶诺(浙江)药业有限公司 | 一类肠道裂解型共药及其制备和用途 |
| WO2024114814A1 (en) * | 2022-12-02 | 2024-06-06 | Onquality Pharmaceuticals China Ltd | Jak inhibitors, pharmaceutical compositions, and therapeutic applications |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20220004726A (ko) | 2022-01-11 |
| SG11202108927UA (en) | 2021-09-29 |
| ZA202105837B (en) | 2022-07-27 |
| IL287717A (en) | 2021-12-01 |
| PH12021552036A1 (en) | 2022-05-23 |
| US20220372034A1 (en) | 2022-11-24 |
| MX2021013224A (es) | 2022-01-06 |
| EP3962897A4 (en) | 2022-12-14 |
| BR112021021477A2 (pt) | 2021-12-21 |
| CA3138544A1 (en) | 2020-11-05 |
| WO2020223728A1 (en) | 2020-11-05 |
| US20200347053A1 (en) | 2020-11-05 |
| EP3962897A1 (en) | 2022-03-09 |
| JP2022531088A (ja) | 2022-07-06 |
| AU2020265828A1 (en) | 2021-09-09 |
| US11420966B2 (en) | 2022-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11420966B2 (en) | Substituted pyrrolopyridines as JAK inhibitors | |
| CN112823005B (zh) | 吡咯并嘧啶itk抑制剂 | |
| CN111566095B (zh) | 被取代的吡咯并吡啶jak抑制剂及其制造方法和使用方法 | |
| US10800775B2 (en) | Pyrazolyl pyrrolo[2,3-b]pyrmidine-5-carboxylate analogs and methods of making the same | |
| US20220288070A1 (en) | Deuterated mk2 pathway inhibitors and methods of using the same | |
| US20190352300A1 (en) | Pyrrolopyrimidine itk inhibitors for treating inflammation and cancer | |
| US20220235043A1 (en) | Substituted sulfonamide pyrrolopyridines as jak inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20211130 |