CN113480543B - 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof - Google Patents
2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof Download PDFInfo
- Publication number
- CN113480543B CN113480543B CN202110765383.1A CN202110765383A CN113480543B CN 113480543 B CN113480543 B CN 113480543B CN 202110765383 A CN202110765383 A CN 202110765383A CN 113480543 B CN113480543 B CN 113480543B
- Authority
- CN
- China
- Prior art keywords
- mmol
- imidazo
- nmr
- pyrazine
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000005236 imidazo[1,2-a]pyrazines Chemical class 0.000 title abstract description 8
- 238000010189 synthetic method Methods 0.000 title 1
- 238000002360 preparation method Methods 0.000 claims abstract description 36
- 150000003839 salts Chemical class 0.000 claims abstract description 15
- 239000003814 drug Substances 0.000 claims abstract description 4
- 208000027866 inflammatory disease Diseases 0.000 claims abstract description 3
- 150000001875 compounds Chemical class 0.000 claims description 25
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 239000003937 drug carrier Substances 0.000 claims description 6
- 210000004881 tumor cell Anatomy 0.000 claims description 5
- 230000006907 apoptotic process Effects 0.000 claims description 3
- 239000002775 capsule Substances 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 230000012010 growth Effects 0.000 claims description 3
- 239000003112 inhibitor Substances 0.000 claims description 3
- 238000001764 infiltration Methods 0.000 claims description 2
- 230000008595 infiltration Effects 0.000 claims description 2
- 238000013508 migration Methods 0.000 claims description 2
- 230000005012 migration Effects 0.000 claims description 2
- 230000035755 proliferation Effects 0.000 claims description 2
- 239000012669 liquid formulation Substances 0.000 claims 1
- 230000001613 neoplastic effect Effects 0.000 claims 1
- 230000002265 prevention Effects 0.000 claims 1
- 206010028980 Neoplasm Diseases 0.000 abstract description 13
- 229940123371 Tyrosine kinase 2 inhibitor Drugs 0.000 abstract description 7
- 238000000034 method Methods 0.000 abstract description 6
- 230000002194 synthesizing effect Effects 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 51
- 238000006243 chemical reaction Methods 0.000 description 35
- -1 amino, substituted amino, hydroxyl Chemical group 0.000 description 28
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 25
- 239000000243 solution Substances 0.000 description 20
- 102000015774 TYK2 Kinase Human genes 0.000 description 19
- 108010010057 TYK2 Kinase Proteins 0.000 description 19
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 18
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 17
- 238000004440 column chromatography Methods 0.000 description 17
- 239000012074 organic phase Substances 0.000 description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 17
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 16
- 239000000741 silica gel Substances 0.000 description 16
- 229910002027 silica gel Inorganic materials 0.000 description 16
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 16
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 14
- 239000003960 organic solvent Substances 0.000 description 13
- 229910052786 argon Inorganic materials 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 7
- 101150003085 Pdcl gene Proteins 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- DCEVLHOTPRPEAA-UHFFFAOYSA-N 6-(3-aminophenyl)-N-(5-fluoropyrimidin-2-yl)imidazo[1,2-a]pyrazin-8-amine Chemical compound NC1=CC=CC(C(N=C2NC(N=C3)=NC=C3F)=CN3C2=NC=C3)=C1 DCEVLHOTPRPEAA-UHFFFAOYSA-N 0.000 description 6
- 125000001072 heteroaryl group Chemical group 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- WCLQGWQXRCNKHS-UHFFFAOYSA-N 6-(5-aminopyridin-3-yl)-n-(4-morpholin-4-ylphenyl)imidazo[1,2-a]pyrazin-8-amine Chemical compound NC1=CN=CC(C=2N=C(NC=3C=CC(=CC=3)N3CCOCC3)C3=NC=CN3C=2)=C1 WCLQGWQXRCNKHS-UHFFFAOYSA-N 0.000 description 5
- KOVKCUZEQFHAJL-UHFFFAOYSA-N N-[5-[8-(cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl]pyridin-3-yl]-3-fluorobenzamide Chemical compound O=C(C1=CC(F)=CC=C1)NC1=CC(C(N=C2NC3CC3)=CN3C2=NC=C3)=CN=C1 KOVKCUZEQFHAJL-UHFFFAOYSA-N 0.000 description 5
- BYOQRKUZWLLFDN-UHFFFAOYSA-N 6-bromo-n-(4-morpholin-4-ylphenyl)imidazo[1,2-a]pyrazin-8-amine Chemical compound N=1C(Br)=CN2C=CN=C2C=1NC(C=C1)=CC=C1N1CCOCC1 BYOQRKUZWLLFDN-UHFFFAOYSA-N 0.000 description 4
- NUFYZLBIPJYMKB-UHFFFAOYSA-N 6-bromo-n-cyclopropylimidazo[1,2-a]pyrazin-8-amine Chemical compound N=1C(Br)=CN2C=CN=C2C=1NC1CC1 NUFYZLBIPJYMKB-UHFFFAOYSA-N 0.000 description 4
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 4
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 201000009020 malignant peripheral nerve sheath tumor Diseases 0.000 description 4
- 208000029974 neurofibrosarcoma Diseases 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- IAJDSUYFELYZCS-UHFFFAOYSA-N 4-(4-nitrophenyl)morpholine Chemical compound C1=CC([N+](=O)[O-])=CC=C1N1CCOCC1 IAJDSUYFELYZCS-UHFFFAOYSA-N 0.000 description 3
- UQCZZGIPIMJBCL-UHFFFAOYSA-N 6,8-dibromoimidazo[1,2-a]pyrazine Chemical compound BrC1=NC(Br)=CN2C=CN=C21 UQCZZGIPIMJBCL-UHFFFAOYSA-N 0.000 description 3
- FIQWVYXKAVIOED-UHFFFAOYSA-N 6-(3-aminophenyl)-N-(4-morpholin-4-ylphenyl)imidazo[1,2-a]pyrazin-8-amine Chemical compound NC=1C=C(C=CC=1)C=1N=C(C=2N(C=1)C=CN=2)NC1=CC=C(C=C1)N1CCOCC1 FIQWVYXKAVIOED-UHFFFAOYSA-N 0.000 description 3
- WLYDMJJWNTZAMU-UHFFFAOYSA-N 6-(5-aminopyridin-3-yl)-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine Chemical compound NC1=CN=CC(C(N=C2NC3CC3)=CN3C2=NC=C3)=C1 WLYDMJJWNTZAMU-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- ONYDCYFHPPFRGX-UHFFFAOYSA-N ethyl 6,8-dibromoimidazo[1,2-a]pyrazine-2-carboxylate Chemical compound C1=C(Br)N=C(Br)C2=NC(C(=O)OCC)=CN21 ONYDCYFHPPFRGX-UHFFFAOYSA-N 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 125000003107 substituted aryl group Chemical group 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- GYLKKXHEIIFTJH-UHFFFAOYSA-N 3-cyanobenzoic acid Chemical compound OC(=O)C1=CC=CC(C#N)=C1 GYLKKXHEIIFTJH-UHFFFAOYSA-N 0.000 description 2
- PHNDZBFLOPIMSM-UHFFFAOYSA-N 4-morpholin-4-ylaniline Chemical compound C1=CC(N)=CC=C1N1CCOCC1 PHNDZBFLOPIMSM-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 102000042838 JAK family Human genes 0.000 description 2
- 108091082332 JAK family Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000001952 enzyme assay Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- RZLXZEJRGRNLQR-UHFFFAOYSA-N imidazo[1,2-a]pyrazin-8-amine Chemical compound NC1=NC=CN2C=CN=C12 RZLXZEJRGRNLQR-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- BMQDAIUNAGXSKR-UHFFFAOYSA-N (3-hydroxy-2,3-dimethylbutan-2-yl)oxyboronic acid Chemical compound CC(C)(O)C(C)(C)OB(O)O BMQDAIUNAGXSKR-UHFFFAOYSA-N 0.000 description 1
- WFQDTOYDVUWQMS-UHFFFAOYSA-N 1-fluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1 WFQDTOYDVUWQMS-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- LJVQHXICFCZRJN-UHFFFAOYSA-N 1h-1,2,4-triazole-5-carboxylic acid Chemical compound OC(=O)C1=NC=NN1 LJVQHXICFCZRJN-UHFFFAOYSA-N 0.000 description 1
- DTLBKXRFWUERQN-UHFFFAOYSA-N 3,5-dibromopyrazin-2-amine Chemical compound NC1=NC=C(Br)N=C1Br DTLBKXRFWUERQN-UHFFFAOYSA-N 0.000 description 1
- YMXIIVIQLHYKOT-UHFFFAOYSA-N 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC(N)=C1 YMXIIVIQLHYKOT-UHFFFAOYSA-N 0.000 description 1
- FIGQEPXOSAFKTA-UHFFFAOYSA-N 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC(C#N)=C1 FIGQEPXOSAFKTA-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- MXNBDFWNYRNIBH-UHFFFAOYSA-N 3-fluorobenzoic acid Chemical compound OC(=O)C1=CC=CC(F)=C1 MXNBDFWNYRNIBH-UHFFFAOYSA-N 0.000 description 1
- JGSIAOZAXBWRFO-UHFFFAOYSA-N 3-methylsulfanyl-1-phenyl-4,5-dihydrobenzo[g]indazole Chemical compound C1CC2=CC=CC=C2C2=C1C(SC)=NN2C1=CC=CC=C1 JGSIAOZAXBWRFO-UHFFFAOYSA-N 0.000 description 1
- FQXQBFUUVCDIRK-UHFFFAOYSA-N 3-trifluoromethylbenzoic acid Chemical compound OC(=O)C1=CC=CC(C(F)(F)F)=C1 FQXQBFUUVCDIRK-UHFFFAOYSA-N 0.000 description 1
- DAISWHFZWZZBBD-UHFFFAOYSA-N 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-amine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN=CC(N)=C1 DAISWHFZWZZBBD-UHFFFAOYSA-N 0.000 description 1
- YJTXQLYMECWULH-UHFFFAOYSA-N 5-fluoropyridin-2-amine Chemical compound NC1=CC=C(F)C=N1 YJTXQLYMECWULH-UHFFFAOYSA-N 0.000 description 1
- GXWNSJYVSIJRLS-UHFFFAOYSA-N 6-bromo-8-methylimidazo[1,2-a]pyrazine Chemical compound CC1=NC(Br)=CN2C=CN=C12 GXWNSJYVSIJRLS-UHFFFAOYSA-N 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 230000004163 JAK-STAT signaling pathway Effects 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 208000031839 Peripheral nerve sheath tumour malignant Diseases 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 1
- 108010087230 Sincalide Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 238000010609 cell counting kit-8 assay Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 238000003366 endpoint assay Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- VICYTAYPKBLQFB-UHFFFAOYSA-N ethyl 3-bromo-2-oxopropanoate Chemical compound CCOC(=O)C(=O)CBr VICYTAYPKBLQFB-UHFFFAOYSA-N 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 238000003674 kinase activity assay Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 238000010979 pH adjustment Methods 0.000 description 1
- 239000013610 patient sample Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明公开了一类如式(I)所述的2,6,8‑多取代咪唑并[1,2‑a]吡嗪或其药学上可接受的盐。本发明还公开了所述2,6,8‑多取代咪唑并[1,2‑a]吡嗪或其药学上可接受的盐作为TYK2抑制剂,在制备防治肿瘤或炎症类疾病的药物中的应用。本发明还提供了所述2,6,8‑多取代咪唑并[1,2‑a]吡嗪或其药学上可接受的盐的合成方法。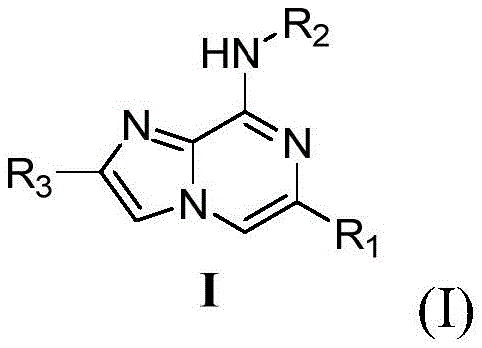
Description
技术领域technical field
本发明属于医药合成化工领域,涉及2,6,8-多取代咪唑并[1,2-a]吡嗪及其合成方法和应用,具体涉及一种作为TYK2抑制剂的2,6,8-多取代咪唑并 [1,2-a]吡嗪化合物及其制备方法和应用。The invention belongs to the field of pharmaceutical synthesis and chemical industry, relates to 2,6,8-polysubstituted imidazo[1,2-a]pyrazine, a synthesis method and application thereof, and in particular relates to a 2,6,8- Polysubstituted imidazo[1,2-a]pyrazine compound and preparation method and application thereof.
背景技术Background technique
酪氨酸激酶2(Tyrosine kinase 2,TYK2)是一类非受体蛋白酪氨酸激酶JAK 家族中的一员,是多种信号转导通路的交汇点,对正常肿瘤细胞增殖、分化和凋亡都发挥着重要的作用。下游蛋白STAT可被激活了的JAK招募,既而被JAK 磷酸化,形成同源或异源二聚体,进入细胞核,与下游基因的启动子结合,调控下游基因的转录。JAK-STAT信号通路与肿瘤的发生发展密切相关,在许多恶性肿瘤中呈现过度激活。Tyrosine kinase 2 (TYK2) is a member of the JAK family of non-receptor protein tyrosine kinases. death plays an important role. The downstream protein STAT can be recruited by the activated JAK, and then phosphorylated by JAK to form a homologous or heterodimer, enter the nucleus, bind to the promoter of the downstream gene, and regulate the transcription of the downstream gene. The JAK-STAT signaling pathway is closely related to the occurrence and development of tumors and is over-activated in many malignant tumors.
近年来,越来越多的研究表明TYK2与诸多肿瘤关系密切。在间变性大细胞淋巴瘤(ALCL)中,TYK2高表达,并通过TYK2-STAT1/3-Bcl2通路的异常活化促进ALCL细胞的生长,通过使用TYK2抑制剂可促使ALCL细胞凋亡。在恶性外周神经鞘瘤(MPNSTs)病人样本中发现TYK2普遍高表达,通过在鼠和人的MPNST细胞中较低TYK2可显著增加细胞死亡,同时在MPNST中也检测到 TYK2-STAT1/3-Bcl2通路的异常活化。在动物体内的研究中发现,分别在TYK2(-/-)鼠和TYK2(+/+)鼠体注射4T1乳腺癌细胞,观察其肿瘤生长情况, TYK2(-/-)鼠肿瘤生长迅速,转移能力明显强于TYK2(+/+)鼠。因此,基于TYK2 的生物学功能和在肿瘤中的广泛研究结果,可以断定TYK2在肿瘤的发生发展过程中具有重要的调控作用,TYK2是一个非常有潜力的肿瘤治疗的靶点。In recent years, more and more studies have shown that TYK2 is closely related to many tumors. In anaplastic large cell lymphoma (ALCL), TYK2 is highly expressed and promotes the growth of ALCL cells through the abnormal activation of the TYK2-STAT1/3-Bcl2 pathway, and the use of TYK2 inhibitors can promote the apoptosis of ALCL cells. TYK2 was found to be generally highly expressed in malignant peripheral nerve sheath tumor (MPNSTs) patient samples, and cell death was significantly increased by lowering TYK2 in murine and human MPNST cells, while TYK2-STAT1/3-Bcl2 was also detected in MPNST Abnormal activation of pathways. In animal studies, it was found that 4T1 breast cancer cells were injected into TYK2(-/-) mice and TYK2(+/+) mice, respectively, and the tumor growth was observed. The ability is obviously stronger than that of TYK2(+/+) mice. Therefore, based on the biological function of TYK2 and the extensive research results in tumors, it can be concluded that TYK2 has an important regulatory role in the occurrence and development of tumors, and TYK2 is a very potential tumor therapy target.
总之,在多种类型肿瘤病人样本中,TYK2的表达水平异常,这与肿瘤的发生密切相关。虽然有报道过的TYK2抑制剂已进入临床试验阶段,但并未有应用于肿瘤治疗的TYK2抑制剂。TYK2作为潜在的抗肿瘤治疗的药物靶标,其靶向抑制剂用于抗肿瘤药物的开发是非常有潜力的,迫切需要开发用于肿瘤治疗的TYK2抑制剂。In conclusion, abnormal expression levels of TYK2 are found in samples from patients with various types of tumors, which are closely related to tumorigenesis. Although the reported TYK2 inhibitors have entered the clinical trial stage, there is no TYK2 inhibitor used in tumor therapy. TYK2 is a potential drug target for anti-tumor therapy, and its targeted inhibitors have great potential for the development of anti-tumor drugs. There is an urgent need to develop TYK2 inhibitors for tumor therapy.
发明内容SUMMARY OF THE INVENTION
本发明提供的2,6,8-多取代咪唑并[1,2-a]吡嗪化合物是TYK2抑制剂,可用于治疗多种肿瘤,包括淋巴瘤、卵巢癌、胃癌、乳腺癌、肺癌等多种恶性肿瘤。The 2,6,8-polysubstituted imidazo[1,2-a]pyrazine compounds provided by the present invention are TYK2 inhibitors and can be used to treat various tumors, including lymphoma, ovarian cancer, gastric cancer, breast cancer, lung cancer, etc. various malignant tumors.
本发明所提供的2,6,8-多取代咪唑并[1,2-a]吡嗪及其在药学上可接受的盐,其结构通式如(I)表示:The 2,6,8-polysubstituted imidazo[1,2-a]pyrazine and its pharmaceutically acceptable salts provided by the present invention are represented by the general structural formula (I):

其中:in:
R1选自



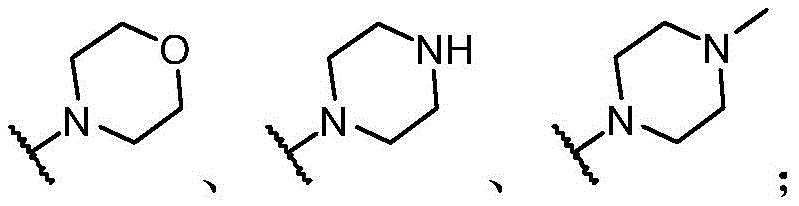
R2选自环烷基或含取代基的环烷基、杂原子环烷基或含有取代基的杂原子环烷基或
R3选自氢原子、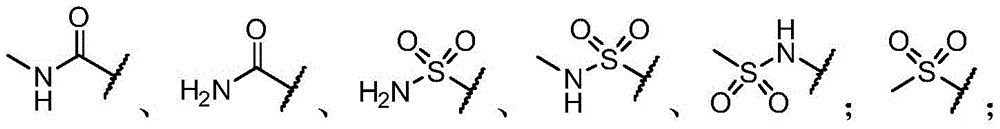

优选地,R1选自





进一步优选地,R1选自





本发明还提供了一种药物组合物,所述药物组合物包括式(I)所示的化合物或其药学上可接受的盐与一种或多种药用载体和/或稀释剂,制成适用于临床的药物制剂。其中,所述药物组合物被配制成片剂、胶囊剂或液体制剂等。本发明所述的化合物可以根据常规药物配制技术与药物载体或赋形剂(例如药学上可接受的载体和赋形剂)混合形成药物制剂。可以将所述2,6,8-多取代咪唑并[1,2-a]吡嗪化合物作为活性成分混合在任何常用的口服剂型中,所述口服剂型包括片剂、胶囊剂和液体制剂(例如酏剂和混悬剂),其中包含着色剂、矫味剂、稳定剂和掩盖味道的物质。对于混合口服剂型来说,所述2,6,8- 多取代咪唑并[1,2-a]吡嗪化合物作为活性成分可以与各种普通片剂材料(例如淀粉、碳酸钙、乳糖、蔗糖和磷酸二钙)混合以助于压片和装入胶囊,可以将所述2,6,8-多取代咪唑并[1,2-a]吡嗪化合物在药学上可接受的无菌液体载体例如无菌水、无菌有机溶剂或者两者的混合物中溶解或混悬。液体载体可以是适合注射剂的载体,比如生理盐水、丙二醇或者聚乙二醇水溶液。在其他情况下,还可以将微粉化的活性成分分散在淀粉或羧甲基纤维素钠的水溶液中或分散在适当的油(例如花生油)中来制得。液体药物制剂(指无菌溶液或混悬剂)可以用于静脉注射、肌肉注射、腹膜内注射或者皮下注射。所述药物组合物还可以包含一种或多种无机或有机、固体或液体的药学上可接受的载体或者赋形剂。术语“药学上可接受的”是指当给药至动物例如哺乳动物(例如人类)时生理学上可耐受且通常不会产生过敏或类似的不良反应(例如头晕等) 的添加剂或组合物。药物载体和赋形剂可以包括但不限于稀释剂,例如乳糖、葡萄糖、甘露糖和/或甘油;润滑剂;聚乙二醇;粘合剂,例如硅酸铝镁、淀粉、明胶、甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮;并且,如果需要的话,还包括崩解剂,例如淀粉、琼脂、海藻酸或其盐如海藻酸钠;和/或吸附剂、着色剂、防腐剂、稳定剂、矫味剂和甜味剂。The present invention also provides a pharmaceutical composition, which comprises the compound represented by formula (I) or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers and/or diluents to prepare Pharmaceutical preparations suitable for clinical use. Wherein, the pharmaceutical composition is formulated into tablets, capsules or liquid preparations and the like. The compounds described in the present invention can be mixed with pharmaceutical carriers or excipients (eg, pharmaceutically acceptable carriers and excipients) according to conventional pharmaceutical formulation techniques to form pharmaceutical formulations. The 2,6,8-polysubstituted imidazo[1,2-a]pyrazine compound can be mixed as an active ingredient in any commonly used oral dosage forms including tablets, capsules and liquids ( such as elixirs and suspensions), which contain coloring agents, flavoring agents, stabilizers and taste-masking substances. For mixed oral dosage forms, the 2,6,8-polysubstituted imidazo[1,2-a]pyrazine compound as an active ingredient can be combined with various common tablet materials (eg starch, calcium carbonate, lactose, sucrose) and dicalcium phosphate) to aid in tableting and encapsulation, the 2,6,8-polysubstituted imidazo[1,2-a]pyrazine compound may be mixed with a pharmaceutically acceptable sterile liquid carrier For example, dissolved or suspended in sterile water, sterile organic solvent, or a mixture of the two. The liquid carrier can be a carrier suitable for injection, such as physiological saline, propylene glycol or aqueous polyethylene glycol solution. In other cases, the micronized active ingredient can also be prepared by dispersing the micronized active ingredient in an aqueous solution of starch or sodium carboxymethyl cellulose or in a suitable oil such as peanut oil. Liquid pharmaceutical preparations (referring to sterile solutions or suspensions) can be administered intravenously, intramuscularly, intraperitoneally or subcutaneously. The pharmaceutical composition may also comprise one or more inorganic or organic, solid or liquid pharmaceutically acceptable carriers or excipients. The term "pharmaceutically acceptable" refers to an additive or composition that is physiologically tolerable and generally does not produce allergic or similar adverse reactions (eg, dizziness, etc.) when administered to an animal such as a mammal (eg, a human). Pharmaceutical carriers and excipients may include, but are not limited to, diluents such as lactose, dextrose, mannose and/or glycerol; lubricants; polyethylene glycols; binders such as magnesium aluminum silicate, starch, gelatin, methyl Cellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; and, if desired, disintegrating agents such as starch, agar, alginic acid or its salts such as sodium alginate; and/or adsorbents, colorants Agents, Preservatives, Stabilizers, Flavoring and Sweetening Agents.
本发明还提供了式(I)所示的化合物或其药学上可接受的盐、或所述的药物组合物在制备TYK2抑制剂中的应用。The present invention also provides the use of the compound represented by formula (I) or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition, in the preparation of a TYK2 inhibitor.
本发明还提供了式(I)所示的化合物或其药学上可接受的盐、或所述的药物组合物在制备防治肿瘤或炎症类疾病的药物中的应用。其中所述肿瘤包括淋巴瘤、卵巢癌、胃癌、乳腺癌、肺癌等多种恶性肿瘤。The present invention also provides the use of the compound represented by formula (I) or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition, in the preparation of a medicine for preventing and treating tumors or inflammatory diseases. The tumors include lymphoma, ovarian cancer, gastric cancer, breast cancer, lung cancer and other malignant tumors.
所述的化合物或其药学上可接受的盐、或所述的药物组合物用于抑制肿瘤细胞的增殖、生长、浸润和迁移,或促进肿瘤细胞的凋亡。The compound or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition is used for inhibiting the proliferation, growth, infiltration and migration of tumor cells, or promoting the apoptosis of tumor cells.
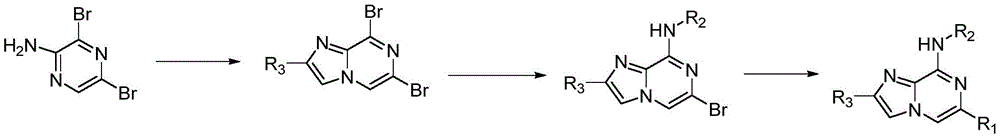
本发明还提供了式(I)所示的化合物或其药学上可接受的盐的制备方法:通过2-氨基-3,5-二溴吡嗪关环得到2-取代-6,8-二溴咪唑并[1,2-a]吡嗪,然后发生取代反应得到2,6-多取代-8-二溴咪唑并[1,2-a]吡嗪,最后经过偶练反应得到目标化合物2,6,8-多取代咪唑并[1,2-a]吡嗪:The present invention also provides a preparation method of the compound represented by formula (I) or a pharmaceutically acceptable salt thereof: obtaining 2-substituted-6,8-dibromopyrazine through ring closure of 2-amino-3,5-dibromopyrazine bromoimidazo[1,2-a]pyrazine, and then undergo a substitution reaction to obtain 2,6-polysubstituted-8-dibromoimidazo[1,2-a]pyrazine, and finally obtain the target compound 2 through coupling reaction ,6,8-Polysubstituted imidazo[1,2-a]pyrazine:
本发明的有益效果是,将本发明所述化合物进行细胞活性测定实验和TYK2 酶活性测定实验,实验结果表明本发明化合物对于防治癌症或炎症类相关疾病有良好的应用。The beneficial effect of the present invention is that the compounds of the present invention are subjected to cell activity assay experiments and TYK2 enzyme activity assay experiments, and the experimental results show that the present compounds have good applications in preventing and treating cancer or inflammation-related diseases.
具体实施方式Detailed ways
下面对本发明的较佳实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。The preferred embodiments of the present invention are described in detail below, so that the advantages and features of the present invention can be more easily understood by those skilled in the art, and the protection scope of the present invention can be more clearly defined.
下面对本发明的各个方面和特点作进一步的描述。Various aspects and features of the present invention are further described below.
本文所用的缩略语通常为本领域技术人员所熟知的,或者可以是根据基础知识易于理解的。Abbreviations used herein are generally known to those skilled in the art, or may be readily understood from basic knowledge.
在本发明化合物的制备中所采用的起始原料是已知的、能够根据已知方法制备的或者可商购获得的。The starting materials employed in the preparation of the compounds of the present invention are known, can be prepared according to known methods, or are commercially available.
本发明还涉及新的中间体和/或起始原料。特别优选与实施例中提到的那些相同或者相似的反应条件和新中间体。The present invention also relates to novel intermediates and/or starting materials. The same or similar reaction conditions and novel intermediates as those mentioned in the examples are particularly preferred.
中间体和终产物都可以根据常规方法进行后处理和/或纯化,所述常规方法包括调节pH、萃取、过滤、干燥、浓缩、色谱法、研磨、结晶等。Both intermediates and final products can be worked up and/or purified according to conventional methods including pH adjustment, extraction, filtration, drying, concentration, chromatography, trituration, crystallization, and the like.
另外,本发明化合物还可以通过本领域已知的各种方法或者本文所述方法的变通方法进行制备。Additionally, the compounds of the present invention can also be prepared by various methods known in the art or variations on the methods described herein.
下列实施例仅用于举例说明本发明,不以任何方式对本发明进行限制。The following examples are intended to illustrate the invention only and not to limit the invention in any way.
实施例1Example 1
3-氰基-N-(3-(8-((4-吗啡啉苯基)氨基)咪唑并[1,2-a]吡嗪-6-基)苯基)苯甲酰胺的制备Preparation of 3-cyano-N-(3-(8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazin-6-yl)phenyl)benzamide


步骤1.1:4-(4-硝基苯基)吗啡啉(1b)的制备Step 1.1: Preparation of 4-(4-nitrophenyl)morpholine (1b)

在100mL单口瓶中对氟硝基苯(35.44mmol)和过量的吗啡啉(205mmol) 在80℃下搅拌5小时。反应完毕,将反应混合物缓慢倒入至1000mL冰水中,有黄色固体析出,过滤,烘干,得到黄色粉末状固体,收率99%。1H NMR(400 MHz,CDCl3)δ8.14(d,J=9.4Hz,2H),6.84(d,J=9.4Hz,2H),3.90–3.83(m, 4H),3.37(dd,J=5.9,4.0Hz,4H).13C NMR(101MHz,CDCl3)δ155.0,138.9, 125.9,112.6,66.4,47.1.In a 100 mL one-neck flask, p-fluoronitrobenzene (35.44 mmol) and excess morpholine (205 mmol) were stirred at 80° C. for 5 hours. After the reaction was completed, the reaction mixture was slowly poured into 1000 mL of ice water, and a yellow solid was precipitated, which was filtered and dried to obtain a yellow powdery solid with a yield of 99%. 1 H NMR (400 MHz, CDCl 3 ) δ 8.14 (d, J=9.4 Hz, 2H), 6.84 (d, J=9.4 Hz, 2H), 3.90-3.83 (m, 4H), 3.37 (dd, J =5.9, 4.0Hz, 4H). 13 C NMR (101MHz, CDCl 3 ) δ 155.0, 138.9, 125.9, 112.6, 66.4, 47.1.
步骤1.2:4-吗啡啉苯胺(1c)的制备Step 1.2: Preparation of 4-morpholinoaniline (1c)

在氩气保护下,在两口瓶中将4-(4-硝基苯基)吗啡啉(19mmol)溶解于60 mL乙醇,依次加入10%钯碳和水合肼,加热到75℃反应8小时。反应结束后,用硅藻土过滤,减压蒸除有机溶剂,柱层析分离得到目标化合物,收率95%。1H NMR(400MHz,DMSO-d6)δ6.68(d,J=8.8Hz,2H),6.51(d,J=8.8Hz,2H),4.58 (s,2H),3.73–3.65(m,4H),2.88–2.85(m,4H);13CNMR(101MHz,DMSO-d6)δ 142.8,142.8,118.0,115.2,66.8,51.1.Under argon protection, 4-(4-nitrophenyl)morpholine (19 mmol) was dissolved in 60 mL of ethanol in a two-necked flask, 10% palladium on carbon and hydrazine hydrate were added successively, and the reaction was heated to 75° C. for 8 hours. After the reaction, filter through celite, evaporate the organic solvent under reduced pressure, and separate the target compound by column chromatography with a yield of 95%. 1 H NMR (400 MHz, DMSO-d 6 ) δ 6.68 (d, J=8.8 Hz, 2H), 6.51 (d, J=8.8 Hz, 2H), 4.58 (s, 2H), 3.73-3.65 (m, 4H), 2.88–2.85 (m, 4H); 13 CNMR (101 MHz, DMSO-d 6 ) δ 142.8, 142.8, 118.0, 115.2, 66.8, 51.1.
步骤1.3:6-溴-N-(4-吗啡啉苯基)咪唑[1,2-a]吡嗪-8-胺(1d)的制备Step 1.3: Preparation of 6-bromo-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine (1d)
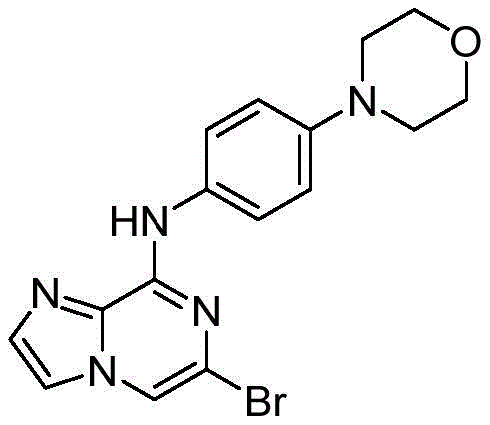
将4-吗啡啉苯胺(5.6mmol)溶解于60mL异丙醇溶液,依次加入DIPEA (8.4mmol)和6,8-二溴-咪唑[1,2-a]吡嗪(6.72mmol),加热到85℃反应8小时。反应结束后,冷却至室温,减压除去有机溶剂,残余物加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤一次,有机相用无水硫酸钠干燥,过滤,用柱层析硅胶分离得到白色固体,收率85%。1H NMR(400MHz,DMSO-d6)δ9.81(s, 1H),8.18(d,J=5.7Hz,1H),7.91(d,J=3.1Hz,1H),7.80(d,J=9.3Hz,2H),7.60 (d,J=2.4Hz,1H),6.92(d,J=8.3Hz,2H),3.73(d,J=3.1Hz,4H),3.06(dt,J=8.3,4.6Hz,4H);13C NMR(101MHz,DMSO-d6)δ147.58,144.9,132.9,132.1, 131.8,122.0,121.3,116.7,115.7,111.2,66.6,49.4.Dissolve 4-morpholine aniline (5.6 mmol) in 60 mL of isopropanol solution, add DIPEA (8.4 mmol) and 6,8-dibromo-imidazo[1,2-a]pyrazine (6.72 mmol) in turn, heat to The reaction was carried out at 85°C for 8 hours. After the reaction, it was cooled to room temperature, the organic solvent was removed under reduced pressure, the residue was dissolved in ethyl acetate, washed with water and saturated brine in turn, and the organic phase was dried with anhydrous sodium sulfate, filtered, and separated by column chromatography on silica gel to obtain White solid, 85% yield. 1 H NMR (400 MHz, DMSO-d 6 ) δ 9.81 (s, 1H), 8.18 (d, J=5.7 Hz, 1H), 7.91 (d, J=3.1 Hz, 1H), 7.80 (d, J= 9.3Hz, 2H), 7.60 (d, J=2.4Hz, 1H), 6.92 (d, J=8.3Hz, 2H), 3.73 (d, J=3.1Hz, 4H), 3.06 (dt, J=8.3, 4.6Hz, 4H); 13 C NMR (101MHz, DMSO-d 6 ) δ 147.58, 144.9, 132.9, 132.1, 131.8, 122.0, 121.3, 116.7, 115.7, 111.2, 66.6, 49.4.
步骤1.4:6-(3-氨基苯基)-N-(4-吗啡啉苯基)咪唑[1,2-a]吡嗪-8-胺(1e)的制备Step 1.4: Preparation of 6-(3-Aminophenyl)-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine (1e)

在氩气流下,向两口瓶中依次加入6-溴-N-(4-吗啡啉苯基)咪唑[1,2-a]吡嗪-8-胺(1mmol)、氨基硼酸酯(1.2mmol)、PdCl2(dppf)(0.1mmol)、1M碳酸钠水溶液(1.5mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率54%。1H NMR(400MHz,DMSO-d6)δ9.45(s,1H),8.39(s,1H),8.05(d,J=8.6Hz,2H),7.98(s,1H),7.61(s,1H),7.22(d,J=2.2Hz,1H),7.12(d,J=4.7Hz,2H),6.99(d, J=8.6Hz,2H),6.59(dt,J=6.1,2.7Hz,1H),5.42–5.13(m,2H),3.76–3.72(m, 4H),3.07(t,J=4.7Hz,4H);13C NMR(101MHz,DMSO-d6)δ149.4,146.8,145.1, 138.4,137.6,133.2,132.6,132.6,129.6,121.2,116.7,116.0,114.3,113.8,111.9, 108.3,66.6,49.6.Under the flow of argon, 6-bromo-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine (1 mmol), aminoboronic acid ester (1.2 mmol) were successively added to the two-necked flask. ), PdCl 2 (dppf) (0.1 mmol), 1M aqueous sodium carbonate solution (1.5 mmol), heated to 100° C. to react overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 54%. 1 H NMR (400MHz, DMSO-d 6 )δ9.45(s,1H),8.39(s,1H),8.05(d,J=8.6Hz,2H),7.98(s,1H),7.61(s, 1H), 7.22(d, J=2.2Hz, 1H), 7.12(d, J=4.7Hz, 2H), 6.99(d, J=8.6Hz, 2H), 6.59(dt, J=6.1, 2.7Hz, 1H), 5.42–5.13 (m, 2H), 3.76–3.72 (m, 4H), 3.07 (t, J=4.7Hz, 4H); 13 C NMR (101 MHz, DMSO-d 6 ) δ 149.4, 146.8, 145.1, 138.4, 137.6, 133.2, 132.6, 132.6, 129.6, 121.2, 116.7, 116.0, 114.3, 113.8, 111.9, 108.3, 66.6, 49.6.
步骤1.5:3-氰基-N-(3-(8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-6-基)苯基)苯甲酰胺(1)的制备Step 1.5: 3-Cyano-N-(3-(8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazin-6-yl)phenyl)benzamide (1 ) preparation

常温下,向单口瓶中依次加入化合物6-(3-氨基苯基)-N-(4-吗啡啉苯基) 咪唑[1,2-a]吡嗪-8-胺(0.5mmol)、3-氰基苯甲酸(0.7mmol)、以及EDCI(0.9 mmol)、HOBt(0.9mmol)、DIEA(1.2mmol),搅拌过夜。TLC监测反应结束后,用乙酸乙酯稀释反应液,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品收率53%。1H NMR(400MHz, DMSO-d6)δ10.61(s,1H),9.58(s,1H),8.70(s,1H),8.56(s,1H),8.49(s,1H), 8.34(d,J=8.0Hz,1H),8.10(dd,J=8.3,5.8Hz,3H),8.03(s,1H),7.81–7.72 (m,2H),7.67(d,J=9.9Hz,2H),7.48(t,J=7.9Hz,1H),7.03(d,J=8.6Hz, 2H),3.74(t,J=4.6Hz,4H),3.07(t,J=4.8Hz,4H).13C NMR(101MHz, DMSO-d6)δ164.04,146.79,145.13,139.69,138.12,136.70,136.31,135.51, 133.08,132.96,132.43,131.88,130.31,129.45,121.52,121.48,120.55,118.83, 118.73,117.02,116.07,112.04,66.60,49.62.Under normal temperature, compound 6-(3-aminophenyl)-N-(4-morpholine phenyl) imidazo[1,2-a]pyrazin-8-amine (0.5mmol), 3 -Cyanobenzoic acid (0.7 mmol), and EDCI (0.9 mmol), HOBt (0.9 mmol), DIEA (1.2 mmol), stirred overnight. After the completion of the reaction monitored by TLC, the reaction solution was diluted with ethyl acetate, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain a pure product with a yield of 53%. 1 H NMR (400MHz, DMSO-d 6 )δ10.61(s,1H), 9.58(s,1H), 8.70(s,1H), 8.56(s,1H), 8.49(s,1H), 8.34( d, J=8.0Hz, 1H), 8.10 (dd, J=8.3, 5.8Hz, 3H), 8.03 (s, 1H), 7.81–7.72 (m, 2H), 7.67 (d, J=9.9Hz, 2H) ), 7.48(t, J=7.9Hz, 1H), 7.03(d, J=8.6Hz, 2H), 3.74(t, J=4.6Hz, 4H), 3.07(t, J=4.8Hz, 4H). 13 C NMR(101MHz, DMSO-d 6 )δ164.04,146.79,145.13,139.69,138.12,136.70,136.31,135.51, 133.08,132.96,132.43,131.88,130.31,129.45,121.52,121.48,120.55,118.83, 118.73,117.02 ,116.07,112.04,66.60,49.62.
实施例2Example 2
6-(5-氨基吡啶-3-基)-N-(4-吗啡啉苯基)咪唑[1,2-a]吡嗪-8-胺(2)的制备Preparation of 6-(5-aminopyridin-3-yl)-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine (2)
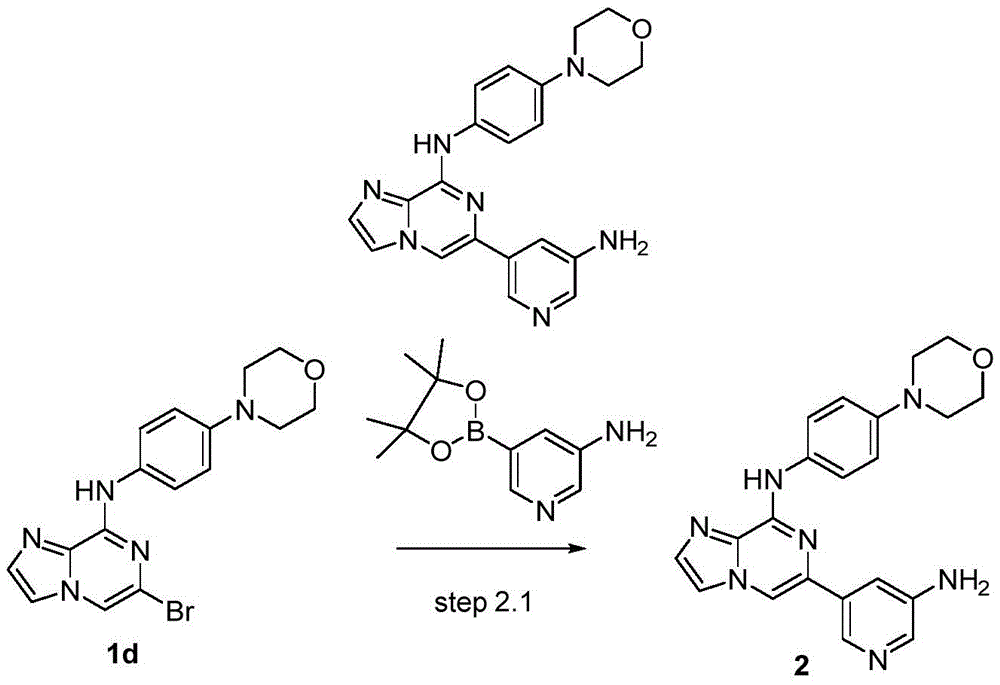
在氩气流下,向两口瓶中依次加入6-溴-N-(4-吗啡啉苯基)咪唑[1,2-a]吡嗪-8-胺(1.5mmol)、5-氨基吡啶-3-硼酸频哪醇酯(1.8mmol)、PdCl2(dppf) (0.15mmol)、1M碳酸钠水溶液(3mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,旋蒸除去有机溶剂,加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率73%。1H NMR(400MHz,DMSO-d6)δ9.51(s,1H),8.51(s,1H),8.35(s,1H),8.07–7.91(m,4H),7.63(s,1H),7.48(t,J=2.3Hz,1H),6.99(d, J=8.5Hz,2H),5.51(s,2H),3.75(t,J=4.8Hz,4H),3.08(t,J=4.8Hz,4H).;13C NMR(101MHz,DMSO-d6)δ146.51,145.00,135.80,134.65,134.45,133.85, 132.97,132.46,132.25,132.14,120.97,116.79,116.44,115.48,108.41,66.17,49.11.Under argon flow, 6-bromo-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine (1.5mmol) and 5-aminopyridine-3 were successively added to the two-necked flask. -Pinacol borate (1.8 mmol), PdCl 2 (dppf) (0.15 mmol), 1 M aqueous sodium carbonate solution (3 mmol), heated to 100° C. to react overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, the organic solvent was removed by rotary evaporation, ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried with anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 73%. 1 H NMR (400MHz, DMSO-d 6 )δ9.51(s,1H), 8.51(s,1H), 8.35(s,1H), 8.07–7.91(m,4H), 7.63(s,1H), 7.48(t, J=2.3Hz, 1H), 6.99(d, J=8.5Hz, 2H), 5.51(s, 2H), 3.75(t, J=4.8Hz, 4H), 3.08(t, J=4.8 Hz, 4H).; 13 C NMR (101MHz, DMSO-d 6 )δ146.51, 145.00, 135.80, 134.65, 134.45, 133.85, 132.97, 132.46, 132.25, 132.14, 120.97, 116.79, 116.44, 11, 6, 48, 10. .
实施例3Example 3
N-(5-(8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-6-基)-吡啶-3-基)-3-(三氟甲基)苯甲酰胺(3)的制备N-(5-(8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazin-6-yl)-pyridin-3-yl)-3-(trifluoromethyl) Preparation of benzamide (3)

常温下,向单口瓶中依次加入化合物6-(3-氨基苯基)-N-(4-吗啡啉苯基) 咪唑[1,2-a]吡嗪-8-胺(0.26mmol)、3-三氟甲基苯甲酸(0.28mmol)、EDCI(0.39 mmol)、HOBt(0.39mmol)和DIEA(0.52mmol),常温搅拌过夜。TLC监测反应结束后,用乙酸乙酯稀释反应液,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率66%。1H NMR(400 MHz,DMSO-d6)δ10.83(s,1H),9.64(s,1H),9.04(s,1H),8.95(s,1H),8.85(d, J=2.4Hz,1H),8.71(s,1H),8.44–8.32(m,2H),8.11–7.99(m,4H),7.83(t,J=7.8Hz,1H),7.66(s,1H),7.02(d,J=8.6Hz,2H),3.77–3.69(m,4H),3.11– 3.02(m,4H).13C NMR(101MHz,DMSO-d6)δ164.48,146.58,145.12,141.87, 141.08,135.71,135.23,133.55,132.88,132.47,132.41,132.21,131.93,129.89, 125.37,124.59,124.46,124.42,121.29,116.71,115.52,108.97,66.17,49.56, 49.08.Under normal temperature, compound 6-(3-aminophenyl)-N-(4-morpholinophenyl) imidazo[1,2-a]pyrazin-8-amine (0.26mmol), 3 - Trifluoromethylbenzoic acid (0.28 mmol), EDCI (0.39 mmol), HOBt (0.39 mmol) and DIEA (0.52 mmol), stirred at room temperature overnight. After the completion of the reaction monitored by TLC, the reaction solution was diluted with ethyl acetate, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 66%. 1 H NMR (400 MHz, DMSO-d 6 ) δ 10.83(s, 1H), 9.64(s, 1H), 9.04(s, 1H), 8.95(s, 1H), 8.85(d, J=2.4Hz ,1H),8.71(s,1H),8.44-8.32(m,2H),8.11-7.99(m,4H),7.83(t,J=7.8Hz,1H),7.66(s,1H),7.02( d, J=8.6Hz, 2H), 3.77–3.69 (m, 4H), 3.11– 3.02 (m, 4H). 13 C NMR (101 MHz, DMSO-d 6 ) δ 164.48, 146.58, 145.12, 141.87, 141.08, 135.71 ,135.23,133.55,132.88,132.47,132.41,132.21,131.93,129.89, 125.37,124.59,124.46,124.42,121.29,116.71,115.52,108.97,66.17,49.586
实施例4Example 4
N-(5-(8-(环丙基氨基)咪唑[1,2-a]吡嗪-6-基)-吡啶-3-基)-3-氟苯甲酰胺(4)的制备Preparation of N-(5-(8-(cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)-pyridin-3-yl)-3-fluorobenzamide (4)

步骤4.1:6-溴-N-环丙基咪唑[1,2-a]吡嗪-8-胺(4b)的制备Step 4.1: Preparation of 6-bromo-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine (4b)
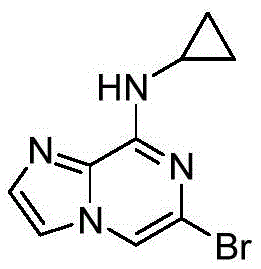
在100mL两口瓶中加入6,8-二溴-咪唑[1,2-a]吡嗪(9.1mmol)溶解于20mL 异丙醇中,依次加入环丙氨(13.6mmol)和DIPEA(13.6mmol),加热至80℃,反应3小时。反应结束后,冷却至室温,减压除去有机溶剂,残余物加入乙酸乙酯溶解后有黄色固体析出,过滤,得纯品,收率92%。1H NMR(400MHz, DMSO-d6)δ8.13(d,J=4.3Hz,1H),8.04(s,1H),7.83(s,1H),7.49(s,1H), 2.91(tt,J=8.1,3.8Hz,1H),0.75–0.63(m,4H).13C NMR(101MHz,DMSO-d6) δ148.47,132.03,131.47,122.16,115.81,109.40,23.85,6.16.In a 100mL two-necked flask, add 6,8-dibromo-imidazo[1,2-a]pyrazine (9.1mmol) and dissolve it in 20mL isopropanol, followed by adding cyclopropylamine (13.6mmol) and DIPEA (13.6mmol) , heated to 80°C, and reacted for 3 hours. After the reaction was completed, it was cooled to room temperature, and the organic solvent was removed under reduced pressure. After the residue was dissolved in ethyl acetate, a yellow solid was precipitated, which was filtered to obtain pure product with a yield of 92%. 1 H NMR (400MHz, DMSO-d 6 ) δ 8.13(d, J=4.3Hz, 1H), 8.04(s, 1H), 7.83(s, 1H), 7.49(s, 1H), 2.91(tt, J=8.1, 3.8Hz, 1H), 0.75–0.63 (m, 4H). 13 C NMR (101MHz, DMSO-d 6 ) δ148.47, 132.03, 131.47, 122.16, 115.81, 109.40, 23.85, 6.16.
步骤4.2:6-(5-氨基吡啶-3-基)-N-环丙基咪唑[1,2-a]吡嗪-8-胺(4c)的制备Step 4.2: Preparation of 6-(5-Aminopyridin-3-yl)-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine (4c)
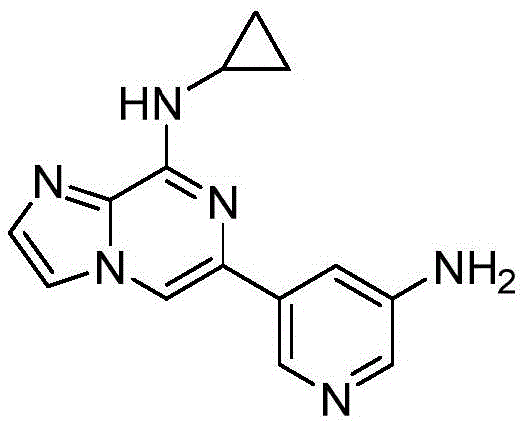
在氩气流下,向两口瓶中依次加入6-溴-N-环丙基咪唑[1,2-a]吡嗪-8-胺(2.1mmol)、5-氨基吡啶-3-硼酸频哪醇酯(2.5mmol)、PdCl2(dppf)(0.2mmol)、 1M碳酸钠水溶液(4.2mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率81%。1H NMR(400MHz,DMSO-d6)δ8.37(s,2H),8.03–7.84(m,2H),7.75 (d,J=4.2Hz,1H),7.56–7.49(m,2H),5.41(s,2H),3.07(tq,J=7.7,4.0Hz,1H),0.77(dh,J=8.1,3.9Hz,2H),0.69(hept,J=4.1Hz,2H).13C NMR(101MHz, DMSO-d6)δ148.72,135.72,135.03,134.62,131.87,116.99,115.87,113.79,111.51, 109.58,107.07,23.68,6.23.Under argon flow, 6-bromo-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine (2.1 mmol) and 5-aminopyridine-3-boronic acid pinacol were added to the two-necked flask in turn. Ester (2.5 mmol), PdCl 2 (dppf) (0.2 mmol), 1M aqueous sodium carbonate solution (4.2 mmol), heated to 100°C and reacted overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 81%. 1 H NMR (400MHz, DMSO-d 6 )δ8.37(s, 2H), 8.03-7.84(m, 2H), 7.75 (d, J=4.2Hz, 1H), 7.56-7.49(m, 2H), 5.41(s, 2H), 3.07(tq, J=7.7, 4.0Hz, 1H), 0.77(dh, J=8.1, 3.9Hz, 2H), 0.69(hept, J=4.1Hz, 2H). 13 C NMR (101MHz, DMSO-d 6 )δ148.72,135.72,135.03,134.62,131.87,116.99,115.87,113.79,111.51,109.58,107.07,23.68,6.23.
步骤4.3:N-(5-(8-(环丙基氨基)咪唑[1,2-a]吡嗪-6-基)-吡啶-3-基)-3-氟苯甲酰胺(4)的制备Step 4.3: N-(5-(8-(Cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)-pyridin-3-yl)-3-fluorobenzamide (4) preparation

常温下,向单口瓶中依次加入化合物6-(5-氨基吡啶-3-基)-N-环丙基咪唑 [1,2-a]吡嗪-8-胺(0.4mmol)、3-氟苯甲酸(0.4mmol)、以及EDCI(0.6mmol)、 HOBt(0.6mmol)、DIPEA(0.6mmol),常温下搅拌过夜。TLC监测反应结束后,用乙酸乙酯稀释反应液,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率28%。1H NMR(400MHz, DMSO-d6)δ10.63(s,1H),8.96(d,J=2.0Hz,1H),8.91(d,J=2.4Hz,1H),8.83(t,J=2.3Hz,1H),8.53(s,1H),7.94(s,1H),7.85(dd,J=16.8,7.9Hz, 3H),7.63(td,J=8.0,5.8Hz,1H),7.54(s,1H),7.49(td,J=8.6,2.7Hz,1H), 3.11(tq,J=7.6,4.0Hz,1H),0.80(dt,J=6.7,3.2Hz,2H),0.72(q,J=4.1Hz, 2H).13C NMR(101MHz,DMSO-d6)δ164.68,163.16,160.73,148.88,142.14, 141.21,135.55,134.05,133.09,132.10,132.08,130.76,130.68,124.47,124.05, 124.02,118.71,116.14,114.75,114.53,107.69,23.73,6.26.Under normal temperature, add compound 6-(5-aminopyridin-3-yl)-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine (0.4mmol), 3-fluoro Benzoic acid (0.4 mmol), EDCI (0.6 mmol), HOBt (0.6 mmol), and DIPEA (0.6 mmol) were stirred at room temperature overnight. After the completion of the reaction monitored by TLC, the reaction solution was diluted with ethyl acetate, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 28%. 1 H NMR (400MHz, DMSO-d 6 )δ10.63(s, 1H), 8.96(d, J=2.0Hz, 1H), 8.91(d, J=2.4Hz, 1H), 8.83(t, J= 2.3Hz, 1H), 8.53(s, 1H), 7.94(s, 1H), 7.85(dd, J=16.8, 7.9Hz, 3H), 7.63(td, J=8.0, 5.8Hz, 1H), 7.54( s, 1H), 7.49 (td, J=8.6, 2.7Hz, 1H), 3.11 (tq, J=7.6, 4.0Hz, 1H), 0.80 (dt, J=6.7, 3.2Hz, 2H), 0.72 (q , J=4.1Hz, 2H). 13 C NMR (101MHz, DMSO-d 6 )δ164.68, 163.16, 160.73, 148.88, 142.14, 141.21, 135.55, 134.05, 133.09, 132.10, 132.08, 130.76, 124.4.68, 124.47 124.02, 118.71, 116.14, 114.75, 114.53, 107.69, 23.73, 6.26.
实施例5Example 5
N-(3-(8-环丙基氨基)咪唑[1,2-a]吡嗪-6-基)苯基)-1H-1,2,4-三氮唑-3-甲酰胺(5)的制备N-(3-(8-Cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)phenyl)-1H-1,2,4-triazole-3-carboxamide (5 ) preparation

步骤5.1 6-(3-氨基苯基)-N-环丙基咪唑[1,2-a]吡嗪-8-胺(5a)的制备Step 5.1 Preparation of 6-(3-aminophenyl)-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine (5a)
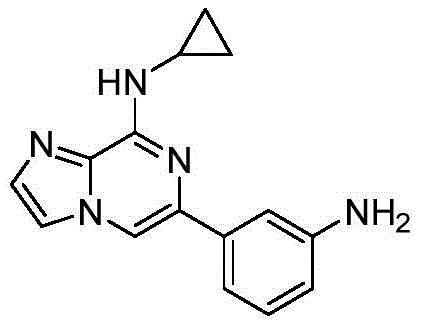
在氩气保护下,向两口瓶中依次加入6-溴-N-环丙基咪唑[1,2-a]吡嗪-8-胺(4.0mmol)、3-氨基苯硼酸频哪醇酯(4.8mmol)、PdCl2(dppf)(0.4mmol)、1M 碳酸钠水溶液(8.0mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率69%。1H NMR(400MHz,DMSO-d6)δ8.24(s,1H),7.88(s,1H),7.64(d,J=4.2Hz, 1H),7.49(s,1H),7.26(t,J=1.9Hz,1H),7.09(dt,J=15.3,7.7Hz,2H),6.56(dt,J =7.8,1.6Hz,1H),5.12(s,2H),3.08(tq,J=7.7,4.0Hz,1H),0.77(td,J=7.1,4.6Hz,2H),0.68(q,J=4.0Hz,2H).13C NMR(101MHz,DMSO)δ148.71,148.39, 138.04,137.42,132.03,131.71,128.88,115.69,113.66,113.43,111.54,106.46, 23.68,6.24.Under argon protection, 6-bromo-N-cyclopropylimidazo[1,2-a]pyrazin-8-amine (4.0 mmol), 3-aminophenylboronic acid pinacol ester ( 4.8 mmol), PdCl 2 (dppf) (0.4 mmol), 1 M aqueous sodium carbonate solution (8.0 mmol), heated to 100° C. to react overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 69%. 1 H NMR (400MHz, DMSO-d 6 )δ8.24(s,1H),7.88(s,1H),7.64(d,J=4.2Hz,1H),7.49(s,1H),7.26(t, J=1.9Hz, 1H), 7.09 (dt, J=15.3, 7.7Hz, 2H), 6.56 (dt, J=7.8, 1.6Hz, 1H), 5.12 (s, 2H), 3.08 (tq, J=7.7 , 4.0Hz, 1H), 0.77 (td, J=7.1, 4.6Hz, 2H), 0.68 (q, J=4.0Hz, 2H). 13 C NMR (101MHz, DMSO) δ 148.71, 148.39, 138.04, 137.42, 132.03 ,131.71,128.88,115.69,113.66,113.43,111.54,106.46,23.68,6.24.
步骤5.2 N-(3-(8-环丙基氨基)咪唑[1,2-a]吡嗪-6-基)苯基)-1H-1,2,4-三氮唑-3-甲酰胺(5)的制备Step 5.2 N-(3-(8-Cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)phenyl)-1H-1,2,4-triazole-3-carboxamide (5) Preparation

常温下,向单口瓶中依次加入化合物6-(3-氨基苯基)-N-环丙基咪唑[1,2-a] 吡嗪-8-胺(0.4mmol)、1H-1,2,4-三氮唑-3-羧酸(0.5mmol)、EDCI(0.7mmol)、 HOBt(0.7mmol)、DIPEA(1.02mmol),常温下搅拌过夜。TLC监测反应结束后,用乙酸乙酯稀释反应液,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率35%。1H NMR(400MHz, DMSO-d6)δ14.81(s,1H),10.56(s,1H),8.59(d,J=34.5Hz,2H),8.38(s,1H), 7.93(s,1H),7.82–7.72(m,3H),7.52(s,1H),7.43(t,J=7.9Hz,1H),3.13(tq, J=7.9,4.1Hz,1H),0.80(dq,J=6.9,4.3Hz,2H),0.70(p,J=4.4Hz,2H).13C NMR(101MHz,DMSO)δ148.57,146.87,138.48,138.01,136.56,132.05, 131.91,131.43,128.75,121.44,120.21,118.18,115.94,112.74,106.98,23.71, 6.26.Under normal temperature, add compound 6-(3-aminophenyl)-N-cyclopropylimidazo[1,2-a]pyrazine-8-amine (0.4mmol), 1H-1,2, 4-Triazole-3-carboxylic acid (0.5 mmol), EDCI (0.7 mmol), HOBt (0.7 mmol), DIPEA (1.02 mmol), stirred at room temperature overnight. After the completion of the reaction monitored by TLC, the reaction solution was diluted with ethyl acetate, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 35%. 1 H NMR (400MHz, DMSO-d 6 ) δ 14.81(s, 1H), 10.56(s, 1H), 8.59(d, J=34.5Hz, 2H), 8.38(s, 1H), 7.93(s, 1H), 7.82–7.72(m, 3H), 7.52(s, 1H), 7.43(t, J=7.9Hz, 1H), 3.13(tq, J=7.9, 4.1Hz, 1H), 0.80(dq, J =6.9, 4.3Hz, 2H), 0.70 (p, J=4.4Hz, 2H). 13 C NMR (101MHz, DMSO) δ 148.57, 146.87, 138.48, 138.01, 136.56, 132.05, 131.91, 131.43, 128.75, 121.44, 120.21 ,118.18,115.94,112.74,106.98,23.71,6.26.
实施例6Example 6
N-(3-(8-环丙基氨基)咪唑[1,2-a]吡嗪-6-基)苯基)-3-(甲硫基)苯甲酰胺(6)的制备Preparation of N-(3-(8-cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)phenyl)-3-(methylthio)benzamide (6)

常温下,向单口瓶中依次加入化合物6-(3-氨基苯基)-N-环丙基咪唑[1,2-a] 吡嗪-8-胺(0.6mmol)、3-(甲硫基)苯甲酸(0.9mmol)、EDCI(1.2mmol)、 HOBt(1.2mmol)和DIPEA(1.6mmol),常温下搅拌过夜。TLC监测反应结束后,用乙酸乙酯稀释反应液,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率72%。1H NMR(400MHz, DMSO-d6)δ10.41(s,1H),8.49–8.42(m,1H),8.38(s,1H),7.97–7.91(m,1H),7.84(s,1H),7.76(q,J=5.7,3.9Hz,4H),7.54–7.42(m,4H),3.13(tt,J= 7.5,3.7Hz,1H),2.56(s,3H),0.80(dh,J=7.8,3.5Hz,2H),0.71(q,J=3.9Hz, 2H).13C NMR(101MHz,DMSO)δ165.18,148.59,139.28,138.74,137.97, 136.63,135.67,132.06,131.89,128.99,128.72,124.71,124.19,121.24,120.23, 120.19,118.08,115.95,106.93,23.73,14.62,6.28.Under normal temperature, add compound 6-(3-aminophenyl)-N-cyclopropylimidazo[1,2-a]pyrazine-8-amine (0.6mmol), 3-(methylthio) successively to single-necked flask ) benzoic acid (0.9 mmol), EDCI (1.2 mmol), HOBt (1.2 mmol) and DIPEA (1.6 mmol), and stirred overnight at room temperature. After the completion of the reaction monitored by TLC, the reaction solution was diluted with ethyl acetate, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 72%. 1 H NMR (400MHz, DMSO-d 6 )δ10.41(s,1H), 8.49-8.42(m,1H), 8.38(s,1H), 7.97-7.91(m,1H), 7.84(s,1H) ), 7.76(q, J=5.7, 3.9Hz, 4H), 7.54–7.42(m, 4H), 3.13(tt, J=7.5, 3.7Hz, 1H), 2.56(s, 3H), 0.80(dh, J=7.8, 3.5Hz, 2H), 0.71 (q, J=3.9Hz, 2H). 13 C NMR (101MHz, DMSO) δ165.18, 148.59, 139.28, 138.74, 137.97, 136.63, 135.67, 132.06, 131.89, 128.99, 128.72, 124.71, 124.19, 121.24, 120.23, 120.19, 118.08, 115.95, 106.93, 23.73, 14.62, 6.28.
实施例7Example 7
N-(3-(8-环丙基氨基)咪唑[1,2-a]吡嗪-6-基)苯基)-3-(甲硫基)苯甲酰胺(7)的制备Preparation of N-(3-(8-cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)phenyl)-3-(methylthio)benzamide (7)

在0℃下,将N-(3-(8-环丙基氨基)咪唑[1,2-a]吡嗪-6-基)苯基)-3-(甲硫基) 苯甲酰胺(0.4mmol)溶于二氯甲烷,搅拌下加入过量间氯过氧苯甲酸(1.6 mmol)。反应结束后,加入亚硫酸钠饱和溶液,分液,有机相用无水硫酸钠干燥,柱分析分离,得白色固体,收率65%。1H NMR(400MHz,DMSO-d6)δ 10.64(s,1H),8.51(t,J=1.8Hz,1H),8.45(t,J=2.0Hz,1H),8.39(s,1H),8.33 (dt,J=7.8,1.4Hz,1H),8.15(dt,J=7.7,1.4Hz,1H),7.94(d,J=1.1Hz,1H), 7.85(t,J=7.8Hz,1H),7.80–7.74(m,3H),7.52(d,J=1.1Hz,1H),7.46(t,J =7.9Hz,1H),3.13(tt,J=7.2,3.9Hz,1H),0.80(td,J=7.1,4.7Hz,2H),0.73– 0.67(m,2H).13C NMR(101MHz,DMSO)δ164.09,148.61,141.13,139.01, 138.07,136.53,135.99,132.75,132.06,131.93,129.86,129.83,128.85,126.20, 121.48,120.35,118.23,115.99,107.00,43.50,23.74,6.29.At 0 °C, N-(3-(8-cyclopropylamino)imidazo[1,2-a]pyrazin-6-yl)phenyl)-3-(methylthio)benzamide (0.4 mmol) was dissolved in dichloromethane and excess m-chloroperoxybenzoic acid (1.6 mmol) was added with stirring. After the reaction, a saturated solution of sodium sulfite was added to separate the layers, the organic phase was dried with anhydrous sodium sulfate, and separated by column analysis to obtain a white solid with a yield of 65%. 1 H NMR (400MHz, DMSO-d 6 )δ 10.64(s, 1H), 8.51(t, J=1.8Hz, 1H), 8.45(t, J=2.0Hz, 1H), 8.39(s, 1H), 8.33(dt,J=7.8,1.4Hz,1H),8.15(dt,J=7.7,1.4Hz,1H),7.94(d,J=1.1Hz,1H), 7.85(t,J=7.8Hz,1H) ),7.80–7.74(m,3H),7.52(d,J=1.1Hz,1H),7.46(t,J=7.9Hz,1H),3.13(tt,J=7.2,3.9Hz,1H),0.80 (td, J=7.1, 4.7 Hz, 2H), 0.73– 0.67 (m, 2H). 13 C NMR (101 MHz, DMSO) δ 164.09, 148.61, 141.13, 139.01, 138.07, 136.53, 135.99, 132.75, 132.06, 131.93, 129.86,129.83,128.85,126.20, 121.48,120.35,118.23,115.99,107.00,43.50,23.74,6.29.
实施例8Example 8
6-(5-氨基吡啶-3-基)-N-(4-((1R,4R)-5-异丙基-2,5-二氮杂二环[2.2.1]庚烷-2-基)苯基)咪唑[1,2-a]吡嗪-8-胺(8)的制备6-(5-Aminopyridin-3-yl)-N-(4-((1R,4R)-5-isopropyl-2,5-diazabicyclo[2.2.1]heptane-2- Preparation of yl)phenyl)imidazo[1,2-a]pyrazin-8-amine (8)

在氩气流下,向两口瓶中依次加入6-溴-N-(4-((1R,4R)-5-异丙基-2,5-二氮杂二环[2.2.1]庚烷-2-基)苯基)咪唑[1,2-a]吡嗪-8-胺(1.0mmol)、5-氨基吡啶-3-硼酸频哪醇酯(1.2mmol)、PdCl2(dppf)(0.1mmol)、1M碳酸钠水溶液(2.0 mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率30%。1HNMR(400 MHz,DMSO-d6)δ9.43(s,1H),8.47(s,1H),8.34(s,1H),7.98–7.92(m,4H),7.62(s,1H),7.45(d,J=2.7Hz,1H),6.68(d,J=8.5Hz,2H),5.48(s,2H),4.40(s,1H), 3.21(s,5H),2.89–2.59(m,2H),1.97(s,1H),1.12–1.01(m,6H).13C NMR(101 MHz,DMSO)δ145.05,144.73,142.50,142.32,135.96,134.81,134.68,133.01, 132.21,132.17,121.61,116.71,116.45,112.85,108.16,59.81,58.55,57.98,56.40, 55.02,20.82,14.13.Under argon flow, 6-bromo-N-(4-((1R,4R)-5-isopropyl-2,5-diazabicyclo[2.2.1]heptane- 2-yl)phenyl)imidazo[1,2-a]pyrazin-8-amine (1.0 mmol), 5-aminopyridine-3-boronic acid pinacol ester (1.2 mmol), PdCl 2 (dppf) (0.1 mmol), 1M aqueous sodium carbonate solution (2.0 mmol), heated to 100 °C and reacted overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 30%. 1 HNMR (400 MHz, DMSO-d 6 )δ9.43(s,1H), 8.47(s,1H), 8.34(s,1H), 7.98–7.92(m,4H), 7.62(s,1H), 7.45(d, J=2.7Hz, 1H), 6.68(d, J=8.5Hz, 2H), 5.48(s, 2H), 4.40(s, 1H), 3.21(s, 5H), 2.89–2.59(m , 2H), 1.97(s, 1H), 1.12–1.01(m, 6H). 13 C NMR(101 MHz, DMSO) δ145.05, 144.73, 142.50, 142.32, 135.96, 134.81, 134.68, 133.01, 132.21, 132.17, 121.61 ,116.71,116.45,112.85,108.16,59.81,58.55,57.98,56.40,55.02,20.82,14.13.
实施例9Example 9
6-(3-氨基苯基)-N-(5-氟嘧啶-2-基)咪唑[1,2-a]吡嗪-8-胺(9)的制备Preparation of 6-(3-aminophenyl)-N-(5-fluoropyrimidin-2-yl)imidazo[1,2-a]pyrazin-8-amine (9)

步骤9.1 6-溴-N-(5-氟嘧啶-2-基)咪唑[1,2-a]吡嗪-8-胺(9b)的制备Step 9.1 Preparation of 6-bromo-N-(5-fluoropyrimidin-2-yl)imidazo[1,2-a]pyrazin-8-amine (9b)

在氩气流下,向两口瓶中依次加入6,8-二溴-咪唑[1,2-a]吡嗪(3.6mmol)、 2-氨基-5-氟-吡啶(5.4mmol)、Pd2(dba)3(0.36mmol)、Xantphos(0.72mmol)和碳酸铯(7.2mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率45%。1H NMR(400MHz,DMSO-d6)δ9.33(s,1H),8.41(s,1H),8.38(d,J=3.1Hz,1H), 8.33(dd,J=9.2,4.1Hz,1H),8.01(d,J=1.1Hz,1H),7.87(td,J=8.7,3.1Hz,1H),7.68(d,J=1.1Hz,1H).13C NMR(101MHz,DMSO)δ147.95,143.08,135.76, 135.51,133.42,131.35,125.58,125.38,119.71,116.84,114.32,113.32.Under argon flow, 6,8-dibromo-imidazo[1,2-a]pyrazine (3.6 mmol), 2-amino-5-fluoro-pyridine (5.4 mmol), Pd 2 ( dba) 3 (0.36 mmol), Xantphos (0.72 mmol) and cesium carbonate (7.2 mmol), heated to 100°C and reacted overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 45%. 1 H NMR (400MHz, DMSO-d 6 ) δ 9.33 (s, 1H), 8.41 (s, 1H), 8.38 (d, J=3.1 Hz, 1H), 8.33 (dd, J=9.2, 4.1 Hz, 1H), 8.01(d, J=1.1Hz, 1H), 7.87(td, J=8.7, 3.1Hz, 1H), 7.68(d, J=1.1Hz, 1H). 13 C NMR(101MHz, DMSO)δ147 .95,143.08,135.76, 135.51,133.42,131.35,125.58,125.38,119.71,116.84,114.32,113.32.
步骤9.2 6-(3-氨基苯基)-N-(5-氟嘧啶-2-基)咪唑[1,2-a]吡嗪-8-胺(9)的制备Step 9.2 Preparation of 6-(3-aminophenyl)-N-(5-fluoropyrimidin-2-yl)imidazo[1,2-a]pyrazin-8-amine (9)

在氩气流下,向两口瓶中依次加入6-溴-N-(5-氟嘧啶-2-基)咪唑[1,2-a]吡嗪-8-胺(2.0mmol)、3-氨基苯硼酸频哪醇酯(2.4mmol)、PdCl2(dppf)(0.2 mmol)、1M碳酸钠水溶液(4.0mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率45%。1H NMR(400MHz,DMSO-d6)δ8.96(s,1H),8.74(dd,J=9.2,4.0Hz,1H),8.58(s,1H),8.37(d,J=3.1Hz,1H),8.06(d,J=1.1Hz,1H),7.89(td, J=8.7,3.1Hz,1H),7.68(d,J=1.1Hz,1H),7.24(t,J=1.9Hz,1H),7.16–7.09(m, 2H),6.61(dt,J=7.5,1.9Hz,1H),5.27(s,2H).13C NMR(101MHz,DMSO)δ 156.42,153.97,149.05,148.66,143.08,137.27,136.70,135.63,135.38,133.07, 131.69,129.31,125.61,125.42,116.85,113.97,113.74,113.70,113.21,111.42, 109.89.Under argon flow, 6-bromo-N-(5-fluoropyrimidin-2-yl)imidazo[1,2-a]pyrazin-8-amine (2.0 mmol) and 3-aminobenzene were successively added to the two-necked flask. Pinacol borate (2.4 mmol), PdCl 2 (dppf) (0.2 mmol), and 1M aqueous sodium carbonate solution (4.0 mmol) were heated to 100° C. to react overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 45%. 1 H NMR (400MHz, DMSO-d 6 ) δ 8.96 (s, 1H), 8.74 (dd, J=9.2, 4.0 Hz, 1H), 8.58 (s, 1H), 8.37 (d, J=3.1 Hz, 1H), 8.06(d, J=1.1Hz, 1H), 7.89(td, J=8.7, 3.1Hz, 1H), 7.68(d, J=1.1Hz, 1H), 7.24(t, J=1.9Hz, 1H), 7.16–7.09(m, 2H), 6.61(dt, J=7.5, 1.9Hz, 1H), 5.27(s, 2H). 13 C NMR (101MHz, DMSO) δ 156.42, 153.97, 149.05, 148.66, 143.08, 137.27, 136.70, 135.63, 135.38, 133.07, 131.69, 129.31, 125.61, 125.42, 116.85, 113.97, 113.74, 113.70, 113.21, 111.42, 109.89.
实施例10Example 10
3-氰基-N-(3-(8-((5-氟嘧啶-2-基)氨基)咪唑[1,2-a]吡嗪-6-基)苯基)苯甲酰胺的制备Preparation of 3-cyano-N-(3-(8-((5-fluoropyrimidin-2-yl)amino)imidazo[1,2-a]pyrazin-6-yl)phenyl)benzamide

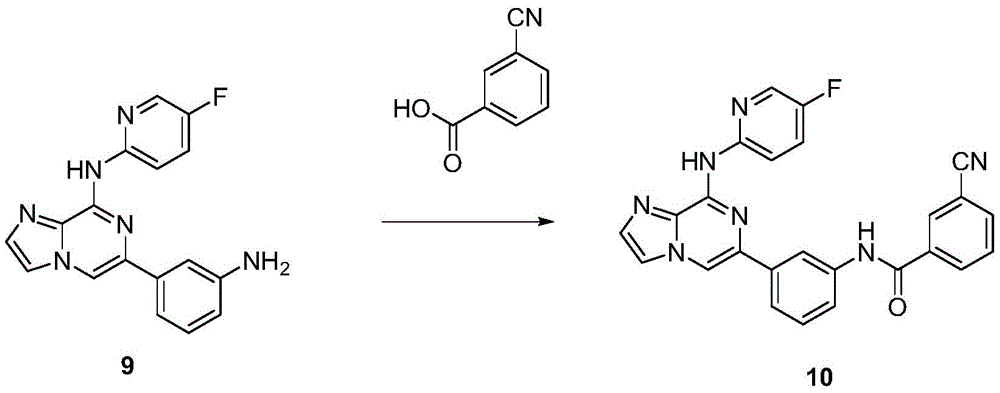
常温下,向单口瓶中依次加入化合物6-(3-氨基苯基)-N-(5-氟嘧啶-2-基) 咪唑[1,2-a]吡嗪-8-胺(0.5mmol)、3-氰基苯甲酸(0.7mmol)、以及EDCI(0.9 mmol)、HOBt(0.9mmol)、DIPEA(1.2mmol),常温下搅拌过夜。TLC监测反应结束后,用乙酸乙酯稀释反应液,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率77%。1H NMR(400 MHz,DMSO-d6)δ10.54(s,1H),8.96(s,1H),8.86(dd,J=9.2,4.1Hz,1H), 8.73(s,1H),8.68(s,1H),8.47(s,1H),8.33(dd,J=9.5,5.4Hz,2H),8.08(d,J=8.9Hz,2H),7.83(ddd,J=21.8,10.9,5.5Hz,2H),7.67(td,J=11.3,9.2,4.8 Hz,3H),7.46(t,J=7.9Hz,1H).13C NMR(101MHz,DMSO)δ163.78,156.46, 154.01,148.55,143.15,139.40,137.13,135.96,135.70,135.56,135.31,135.02, 133.14,132.58,131.70,131.35,129.87,129.05,125.54,125.35,120.91,119.96, 118.39,117.84,117.04,114.08,111.55,110.31.At room temperature, the compound 6-(3-aminophenyl)-N-(5-fluoropyrimidin-2-yl)imidazo[1,2-a]pyrazin-8-amine (0.5mmol) was added successively to the single-necked flask , 3-cyanobenzoic acid (0.7 mmol), and EDCI (0.9 mmol), HOBt (0.9 mmol), DIPEA (1.2 mmol), and stirred overnight at room temperature. After the completion of the reaction monitored by TLC, the reaction solution was diluted with ethyl acetate, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 77%. 1 H NMR (400 MHz, DMSO-d 6 ) δ 10.54 (s, 1H), 8.96 (s, 1H), 8.86 (dd, J=9.2, 4.1 Hz, 1H), 8.73 (s, 1H), 8.68 (s,1H),8.47(s,1H),8.33(dd,J=9.5,5.4Hz,2H),8.08(d,J=8.9Hz,2H),7.83(ddd,J=21.8,10.9,5.5 Hz, 2H), 7.67 (td, J=11.3, 9.2, 4.8 Hz, 3H), 7.46 (t, J=7.9 Hz, 1H). 13 C NMR (101 MHz, DMSO) δ 163.78, 156.46, 154.01, 148.55, 143.15 ,139.40,137.13,135.96,135.70,135.56,135.31,135.02, 133.14,132.58,131.70,131.35,129.87,129.05,125.54,125.35,120.91,119.96, 118.39,117.84,117.04,114.08,111.55,110.31.
实施例11Example 11
6-(3-氰基苯基)-N-甲基-8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-2-苯甲酰胺(11)的制备Preparation of 6-(3-cyanophenyl)-N-methyl-8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazine-2-benzamide (11)

步骤11.1 6,8-二溴咪唑[1,2-a]吡嗪-2-羧酸乙酯(11b)的制备Step 11.1 Preparation of ethyl 6,8-dibromoimidazo[1,2-a]pyrazine-2-carboxylate (11b)

在100mL单口瓶中,将2-氨基-3,5-二溴吡嗪(20mmol)和3-溴丙酮酸乙酯(24mmol)溶于40mL乙二醇二甲醚,在100℃下搅拌过夜。反应完毕,将反应混合物中加入乙醚,过滤,烘干,得棕色固体,收率47%。1H NMR(400MHz, CDCl3)δ8.31(dd,J=7.2,1.7Hz,2H),4.48(qd,J=7.1,1.6Hz,2H),1.43(td,J= 7.1,1.7Hz,3H).13C NMR(101MHz,CDCl3)δ161.81,139.73,138.31,135.93, 121.68,119.55,119.32,62.14,14.46.In a 100 mL single-necked flask, 2-amino-3,5-dibromopyrazine (20 mmol) and ethyl 3-bromopyruvate (24 mmol) were dissolved in 40 mL of ethylene glycol dimethyl ether, and stirred at 100° C. overnight. After the reaction was completed, ether was added to the reaction mixture, filtered and dried to obtain a brown solid with a yield of 47%. 1 H NMR (400 MHz, CDCl 3 ) δ 8.31 (dd, J=7.2, 1.7 Hz, 2H), 4.48 (qd, J=7.1, 1.6 Hz, 2H), 1.43 (td, J=7.1, 1.7 Hz, 3H). 13 C NMR (101MHz, CDCl 3 ) δ 161.81, 139.73, 138.31, 135.93, 121.68, 119.55, 119.32, 62.14, 14.46.
步骤11. 2:6-溴-8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-2-羧酸乙酯(11c)的制备Step 11.2: Preparation of ethyl 6-bromo-8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazine-2-carboxylate (11c)

将4-吗啡啉苯胺(7.2mmol)溶解于30mL乙腈溶液,依次加入DIPEA (16.2mmol)和6,8-二溴咪唑[1,2-a]吡嗪-2-羧酸乙酯(6.0mmol),加热到85℃反应过夜。反应结束后,冷却至室温,减压除去有机溶剂,残余物加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤一次,有机相用无水硫酸钠干燥,过滤,用柱层析硅胶分离得到黄色固体,收率39%。1H NMR(400MHz,CDCl3) δ8.13(s,1H),8.00(s,1H),7.70(d,J=8.6Hz,2H),7.60(s,1H),6.92(d,J= 8.5Hz,2H),4.44(q,J=7.1Hz,2H),3.85(t,J=4.5Hz,5H),3.12(t,J=4.6Hz, 4H),1.41(d,J=14.1Hz,3H).13C NMR(101MHz,CDCl3)δ162.31,148.02, 144.66,136.04,132.43,130.76,123.83,121.06,118.96,116.32,110.01,66.94, 61.57,49.68,14.47.4-Mormorpholinaniline (7.2 mmol) was dissolved in 30 mL of acetonitrile solution, DIPEA (16.2 mmol) and ethyl 6,8-dibromoimidazo[1,2-a]pyrazine-2-carboxylate (6.0 mmol) were added successively ), heated to 85°C and reacted overnight. After the reaction, it was cooled to room temperature, the organic solvent was removed under reduced pressure, the residue was dissolved in ethyl acetate, washed with water and saturated brine in turn, and the organic phase was dried with anhydrous sodium sulfate, filtered, and separated by column chromatography on silica gel to obtain Yellow solid, 39% yield. 1 H NMR (400MHz, CDCl 3 ) δ 8.13(s, 1H), 8.00(s, 1H), 7.70(d, J=8.6Hz, 2H), 7.60(s, 1H), 6.92(d, J= 8.5Hz, 2H), 4.44 (q, J=7.1Hz, 2H), 3.85 (t, J=4.5Hz, 5H), 3.12 (t, J=4.6Hz, 4H), 1.41 (d, J=14.1Hz) , 3H). 13 C NMR (101MHz, CDCl 3 )δ162.31, 148.02, 144.66, 136.04, 132.43, 130.76, 123.83, 121.06, 118.96, 116.32, 110.01, 66.94, 61.57, 49.68, 14.47
步骤11.3:6-溴-N-甲基-8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-2-甲酰胺(11d)的制备Step 11.3: Preparation of 6-bromo-N-methyl-8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazine-2-carboxamide (11d)

将6-溴-8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-2-羧酸乙酯(2.35mmol)溶解于10mL四氢呋喃溶液中,加入12mL 30%的甲胺水溶液,于室温下搅拌过夜。反应结束后,过滤,得到淡黄色固体,收率85%。1H NMR(400MHz, CDCl3)δ8.01(s,1H),7.79(s,1H),7.74(d,J=8.5Hz,2H),7.67(s,1H),7.23(d,J =5.6Hz,1H),7.00(s,2H),3.90(s,4H),3.17(t,J=4.8Hz,4H),3.05(d,J=5.0Hz, 3H).13C NMR(101MHz,CDCl3)δ162.84,149.54,144.59,137.06,133.61,130.59, 123.82,121.06,119.08,116.31,110.76,77.48,77.16,76.84,66.92,61.67,26.07.Dissolve ethyl 6-bromo-8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazine-2-carboxylate (2.35mmol) in 10mL of tetrahydrofuran solution, add 12mL of 30% The aqueous methylamine solution was stirred at room temperature overnight. After the reaction was completed, it was filtered to obtain a pale yellow solid with a yield of 85%. 1 H NMR (400MHz, CDCl 3 ) δ 8.01(s, 1H), 7.79(s, 1H), 7.74(d, J=8.5Hz, 2H), 7.67(s, 1H), 7.23(d, J= 5.6Hz, 1H), 7.00(s, 2H), 3.90(s, 4H), 3.17(t, J=4.8Hz, 4H), 3.05(d, J=5.0Hz, 3H). 13 C NMR(101MHz, CDCl3)δ162.84,149.54,144.59,137.06,133.61,130.59, 123.82,121.06,119.08,116.31,110.76,77.48,77.16,76.84,66.92,61.67,26.07.
步骤11.4:6-(3-氰基苯基)-N-甲基-8-((4-吗啡啉苯基)氨基)咪唑[1,2-a]吡嗪-2-苯甲酰胺(11)的制备Step 11.4: 6-(3-Cyanophenyl)-N-methyl-8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazine-2-benzamide (11 ) preparation

在氩气流下,向两口瓶中依次加入6-溴-N-甲基-8-((4-吗啡啉苯基)氨基) 咪唑[1,2-a]吡嗪-2-甲酰胺(0.23mmol)、3-氰基苯硼酸频哪醇酯(0.27mmol)、 PdCl2(dppf)(0.02mmol)、1M碳酸钠水溶液(0.46mmol),加热至100℃反应过夜。反应结束后,冷却至室温,硅藻土过滤,蒸除有机溶剂。加入乙酸乙酯溶解,依次用水洗、饱和食盐水洗涤,有机相用无水硫酸钠干燥。用柱层析硅胶分离,得到纯品,收率42%。1H NMR(400MHz,DMSO-d6)δ9.41(s, 1H),8.72(s,1H),8.35(d,J=1.8Hz,1H),8.32–8.28(m,2H),8.14(q,J=4.8Hz,1H),7.92–7.88(m,2H),7.85(dt,J=7.7,1.4Hz,1H),7.71(t,J=7.8Hz,1H),7.02 –6.97(m,2H),3.77–3.73(m,4H),3.12–3.07(m,4H),2.86(d,J=4.8Hz,3H). 13C NMR(101MHz,DMSO-d6)δ161.85,147.01,145.19,139.32,138.14,135.12, 131.70,131.62,131.55,130.22,130.13,128.96,121.36,118.83,117.14,115.38, 111.88,109.47,66.17,48.95,25.60.Under argon flow, 6-bromo-N-methyl-8-((4-morpholinophenyl)amino)imidazo[1,2-a]pyrazine-2-carboxamide (0.23 mmol), 3-cyanophenylboronic acid pinacol ester (0.27 mmol), PdCl 2 (dppf) (0.02 mmol), 1M aqueous sodium carbonate solution (0.46 mmol), heated to 100° C. to react overnight. After the reaction was completed, it was cooled to room temperature, filtered through celite, and the organic solvent was evaporated. Ethyl acetate was added to dissolve, washed with water and saturated brine successively, and the organic phase was dried over anhydrous sodium sulfate. It was separated by column chromatography on silica gel to obtain pure product with a yield of 42%. 1 H NMR (400MHz, DMSO-d 6 )δ9.41(s, 1H), 8.72(s, 1H), 8.35(d, J=1.8Hz, 1H), 8.32-8.28(m, 2H), 8.14( q,J=4.8Hz,1H),7.92-7.88(m,2H),7.85(dt,J=7.7,1.4Hz,1H),7.71(t,J=7.8Hz,1H),7.02-6.97(m , 2H), 3.77–3.73 (m, 4H), 3.12–3.07 (m, 4H), 2.86 (d, J=4.8Hz, 3H). 13 C NMR (101MHz, DMSO-d 6 )δ161.85, 147.01, 145.19 ,139.32,138.14,135.12, 131.70,131.62,131.55,130.22,130.13,128.96,121.36,118.83,117.14,115.38, 111.88,109.47,66.17,48.95,25.60
实施例12Example 12
生物学测定biological assay
TYK2酶活性测定TYK2 enzyme activity assay
384-孔微孔板TR-FRET(时间分辨-荧光能量转移)终点测定用于TYK2激酶活性测定。相同的测定用于小分子抑制剂的IC50测定。一般地,激酶反应是在 25μL体积包含如下成分的反应溶液中进行的:5μL化合物(在10%的DMSO 中)、20μL在测定缓冲液(25mM HEPES,pH 7.5,0.001%Brij-35,0.01%Triton,0.5 mM EGTA)中的TYK2、多肽和ATP的混合物。所有反应在室温下在384孔白色平底光密度板(Perkin Elmer)中进行60min,然后加入25μL终止反应液(100mM HEPES,pH 7.5,0.015%Brij-35,0.2%Coating Reagent#3,50mM EDTA)。在 Evision Multilabel读数器(Perkin Elmer,Envision 2102-0010)上读数。A 384-well microplate TR-FRET (time-resolved-fluorescence energy transfer) endpoint assay was used for TYK2 kinase activity assay. The same assay was used for IC50 determination of small molecule inhibitors. Typically, kinase reactions are performed in a 25 μL volume of reaction solution containing: 5 μL compound (in 10% DMSO), 20 μL in assay buffer (25 mM HEPES, pH 7.5, 0.001% Brij-35, 0.01% A mixture of TYK2, polypeptide and ATP in Triton, 0.5 mM EGTA). All reactions were performed in 384-well white flat-bottom optical density plates (Perkin Elmer) for 60 min at room temperature, followed by the addition of 25 μL of stop reaction (100 mM HEPES, pH 7.5, 0.015% Brij-35, 0.2% Coating Reagent #3, 50 mM EDTA) . Read on an Evision Multilabel reader (Perkin Elmer, Envision 2102-0010).
表1Table 1
细胞活性测定Cell viability assay
取对数生长期细胞按每孔3000个细胞/100μL密度接种于96孔板,待细胞贴壁后,加入100μL不同浓度的待测化合物,取6-8个浓度梯度。每组设五个平行孔,设置对照组。化合物和肿瘤细胞共孵育72小时后,每孔加10μL CCK-8溶液。在细胞培养箱中孵育1-2小时后,用酶联免疫检测仪测定每孔吸光度(OD 值),计算抑制率:抑制率(IR%)=(1-TOD/COD)x 100%,TOD:给药组OD均值; COD:溶剂对照组OD均值。The cells in the logarithmic growth phase were seeded in a 96-well plate at a density of 3000 cells/100 μL per well. After the cells adhered, 100 μL of different concentrations of the compounds to be tested were added to obtain 6-8 concentration gradients. Five parallel wells were set in each group, and a control group was set. After the compound and tumor cells were incubated for 72 hours, 10 μL of CCK-8 solution was added to each well. After 1-2 hours of incubation in the cell incubator, measure the absorbance (OD value) of each well with an enzyme-linked immunosorbent assay, and calculate the inhibition rate: inhibition rate (IR%) = (1-TOD/COD) x 100%, TOD : mean OD value of drug group; COD: mean OD value of solvent control group.
表2Table 2


以上实施方式只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人了解本发明的内容并加以实施,并不能以此限制本发明的保护范围,凡根据本发明精神实质所做的等效变化或修饰,都应涵盖在本发明的保护范围内。The above embodiments are only intended to illustrate the technical concept and characteristics of the present invention, and the purpose is to allow those who are familiar with the technology to understand the content of the present invention and implement it, and cannot limit the scope of protection of the present invention. Equivalent changes or modifications made should all be included within the protection scope of the present invention.
Claims (6)

Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110765383.1A CN113480543B (en) | 2021-07-07 | 2021-07-07 | 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof |
| PCT/CN2022/099407 WO2023279938A1 (en) | 2021-07-07 | 2022-06-17 | 2,6,8-polysubstituted imidazo[1,2-a]pyrazine, and synthesis method therefor and use thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110765383.1A CN113480543B (en) | 2021-07-07 | 2021-07-07 | 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN113480543A CN113480543A (en) | 2021-10-08 |
| CN113480543B true CN113480543B (en) | 2022-05-17 |
Family
ID=77941466
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110765383.1A Active CN113480543B (en) | 2021-07-07 | 2021-07-07 | 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN113480543B (en) |
| WO (1) | WO2023279938A1 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113480543B (en) * | 2021-07-07 | 2022-05-17 | 无锡市第二人民医院 | 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof |
| WO2023076161A1 (en) | 2021-10-25 | 2023-05-04 | Kymera Therapeutics, Inc. | Tyk2 degraders and uses thereof |
| CN116283831B (en) * | 2023-03-13 | 2024-07-23 | 中国医学科学院医药生物技术研究所 | P-nitrobenzene derivative and preparation method and application thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005014599A1 (en) * | 2003-06-04 | 2005-02-17 | Cellular Genomics, Inc. | Imidazo[1,2-a]pyrazin-8-ylamines and method of inhibition of bruton’s tyrosine kinase by such compounds |
| CN102056927A (en) * | 2008-05-13 | 2011-05-11 | Irm责任有限公司 | Fused nitrogen containing heterocycles and compositions thereof as kinase inhibitors |
| CN102307474A (en) * | 2008-12-08 | 2012-01-04 | 吉利德康涅狄格股份有限公司 | Imidazopyrazine SYK inhibitors |
| CN103443100A (en) * | 2010-12-17 | 2013-12-11 | 拜耳知识产权有限责任公司 | Substituted 6-midazopyrazines for use as mps-1 and TKK inhibitors in the treatment of hyperproliferative disorders |
| CN106459048A (en) * | 2014-05-14 | 2017-02-22 | 辉瑞公司 | Pyrazolopyridines and pyrazolopyrimidines |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI735853B (en) * | 2013-12-23 | 2021-08-11 | 美商克洛諾斯生技有限公司 | Syk inhibitors |
| WO2018195471A1 (en) * | 2017-04-21 | 2018-10-25 | Gilead Sciences, Inc. | Syk inhibitors in combination with hypomethylating agents |
| CN112939983A (en) * | 2021-02-01 | 2021-06-11 | 暨明医药科技(苏州)有限公司 | Synthesis method of SYK kinase inhibitor Lanraplenib |
| CN113480543B (en) * | 2021-07-07 | 2022-05-17 | 无锡市第二人民医院 | 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof |
-
2021
- 2021-07-07 CN CN202110765383.1A patent/CN113480543B/en active Active
-
2022
- 2022-06-17 WO PCT/CN2022/099407 patent/WO2023279938A1/en not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005014599A1 (en) * | 2003-06-04 | 2005-02-17 | Cellular Genomics, Inc. | Imidazo[1,2-a]pyrazin-8-ylamines and method of inhibition of bruton’s tyrosine kinase by such compounds |
| CN102056927A (en) * | 2008-05-13 | 2011-05-11 | Irm责任有限公司 | Fused nitrogen containing heterocycles and compositions thereof as kinase inhibitors |
| CN102307474A (en) * | 2008-12-08 | 2012-01-04 | 吉利德康涅狄格股份有限公司 | Imidazopyrazine SYK inhibitors |
| CN103443100A (en) * | 2010-12-17 | 2013-12-11 | 拜耳知识产权有限责任公司 | Substituted 6-midazopyrazines for use as mps-1 and TKK inhibitors in the treatment of hyperproliferative disorders |
| CN106459048A (en) * | 2014-05-14 | 2017-02-22 | 辉瑞公司 | Pyrazolopyridines and pyrazolopyrimidines |
Non-Patent Citations (2)
| Title |
|---|
| Cheng Wang等.Design and optimization of orally spleen tyrosine kinase (SYK) inhibitors for treatment of solid tumor.《Bioorganic Chemistry》.2019,第65卷. * |
| Design and optimization of orally spleen tyrosine kinase (SYK) inhibitors for treatment of solid tumor;Cheng Wang等;《Bioorganic Chemistry》;20191224;第1-13页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023279938A1 (en) | 2023-01-12 |
| CN113480543A (en) | 2021-10-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108349981B (en) | Novel pyrazolo [3, 4-d ] pyrimidine compound or salt thereof | |
| RU2671847C2 (en) | 2,3-dihydro-isoindol-1-one derivatives and methods for use thereof as bruton's tyrosine kinase inhibitors | |
| WO2021073439A1 (en) | Pyrazine derivative for inhibiting shp2 activity | |
| CN101952287B (en) | Protein kinase inhibitors and use thereof | |
| CN113480543B (en) | 2,6, 8-polysubstituted imidazo [1,2-a ] pyrazine and synthetic method and application thereof | |
| CN101143845B (en) | Substituted quinoline formyl guanidine derivative, its preparation method and medical use | |
| EA019723B1 (en) | cMET INHIBITORS | |
| TW201506028A (en) | 1,2-disubstituted heterocyclic compounds | |
| JP2014510037A (en) | Tyrosine kinase inhibitors containing diarylacetylene hydrazides | |
| WO2015188747A1 (en) | Phenyl-substituted triazine compound serving as egfr inhibitor and applications thereof | |
| CN107056755B (en) | Five-membered heterocyclic amide WNT pathway inhibitors | |
| KR101905295B1 (en) | Naphthyridinedione derivatives | |
| AU2019218187A1 (en) | Dioxinoquinoline compounds, preparation method and uses thereof | |
| CN112236430B (en) | A class of cyclic pyrazolone carboxamide compounds and their preparation method, pharmaceutical composition and use | |
| CN105153026B (en) | Sorafenib derivative of the amide structure containing biaryl and its preparation method and application | |
| CN106831812A (en) | Heterocycle containing biaryl amide structure and pyrimidine or pyrazine compounds and its application | |
| CN104211686A (en) | Quinoline derivatives and treatment use thereof | |
| CN107151233B (en) | Hydrazone-containing pyrimidine derivative and application thereof | |
| CN108601773B (en) | Condensed quinoline compounds as PI3K/MTOR inhibitors | |
| CN118976029A (en) | Pharmaceutical composition for preventing or treating Parkinson's disease or non-alcoholic steatohepatitis | |
| CN110054584B (en) | 1-Aryl-3-{4-[(pyridin-2-ylmethyl)thio]phenyl}urea compounds and their applications | |
| CN113461687B (en) | 2, 8-aza- [4,5] decaspirocyclic ketone derivative and preparation method and application thereof | |
| CN111440177B (en) | A kind of substituted pyrazolo[1,5-a]pyrimidine compound and its preparation method and application | |
| US11407760B2 (en) | Dioxinoquinoline compounds, preparation method and uses thereof | |
| CN110407839B (en) | Preparation and application of triazole heterocyclic compound containing heteroaryl amide structure |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |