CN113329750A - Compounds for the treatment of arenavirus infections - Google Patents
Compounds for the treatment of arenavirus infections Download PDFInfo
- Publication number
- CN113329750A CN113329750A CN201980080594.2A CN201980080594A CN113329750A CN 113329750 A CN113329750 A CN 113329750A CN 201980080594 A CN201980080594 A CN 201980080594A CN 113329750 A CN113329750 A CN 113329750A
- Authority
- CN
- China
- Prior art keywords
- compound
- independently selected
- compounds
- alkyl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4192—1,2,3-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/16—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms condensed with carbocyclic rings or ring systems
- C07D249/18—Benzotriazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
通过化合物A举例说明的化合物可用于治疗沙粒病毒感染和由沙粒病毒糖蛋白介导的病毒感染。 Compounds exemplified by Compound A are useful in the treatment of arenavirus infections and viral infections mediated by arenavirus glycoproteins. 
Description
Cross Reference to Related Applications
This patent application is a partial continuation of U.S. provisional patent application serial No. 62/776,390 filed 2018, 12, month 6 and claiming the benefit of priority, which is incorporated herein by reference in its entirety for all purposes.
Claims on inventions completed under federally sponsored research development
The invention was made with government support from R44 AI112097 awarded by the national institutes of health. The united states government has certain rights in the invention.
Accessories concerning "sequence lists", forms or computer program lists submitted on compact discs
Not applicable to
Technical Field
The present invention relates to the use of heterocyclic compounds to inhibit arenavirus infection in humans, other mammals, or in cell culture, methods of treating arenavirus infections such as Lassa (Lassa) hemorrhagic fever, bolivia (bolivia) hemorrhagic fever, Argentine (Argentine) hemorrhagic fever, Venezuelan (Venezuelan) hemorrhagic fever, basci (Brazilian) hemorrhagic fever, laparak (Chapare) hemorrhagic fever, and Lujo (Lujo) hemorrhagic fever, methods of inhibiting replication of arenavirus, methods of reducing the amount of arenavirus, and compositions that can be used in such methods.
Background
The Arenaviridae (Arenaviridae) family comprises a diverse family of 29 (and in enlargement) negative-strand enveloped RNA viruses. Arenaviruses were divided into two groups, the old world and the new world, based on serological, genetic and geographic data. The old world viruses are found predominantly throughout south and west africa and include the prototype lymphocytic choriomeningitis virus (LCMV), as well as lassa virus (LASV), luo virus (LUJV), mopenia (mopei) virus (MOPV), Ippy (Ippy) virus, and mobara (Mobala) virus (MOBV). Both LASV and LUJV can cause fatal Hemorrhagic Fever (HF), while LCMV infection is associated with aseptic meningitis. It is estimated that lassa virus (LASV) alone causes over 300,000 cases of disease annually in western africa, with 15% -20% of hospitalized patients dying and survivors often suffering from sequelae, including permanent bilateral hearing impairment. The larger new world complex group is located mainly in south america and is divided into 3 clades A, B and C, of which clade B is important because many viruses in this group can cause fatal HF. Clade B HF viruses include huinin (Junin) virus (JUNV), Machupo (Machupo) virus (MACV), guanaliito (Guanarito) virus (GTOV), Sabia (Sabia) virus (SABV) and laparv virus, as well as non-HF viruses such as Tacaribe (Tacaribe) virus (TCRV) and amapri (amapri) virus (AMPV). Human infections occur by contact with the excreta of infected rodents or by inhalation of fine particles (aerosol transmission) contaminated with rodent urine or saliva. There is also evidence that person-to-person transmission is primarily in a hospital environment (e.g., a hospital). The viral latency is 1-2 weeks, followed by fever, general malaise, weakness, sore throat, headache, cough, diarrhea and vomiting. These general symptoms make it difficult to differentially diagnose arenavirus infections. Worsening of symptoms to include pleural effusion, facial edema, neurological complications, and bleeding from mucosal surfaces indicate poor prognosis. Current arenavirus treatments are limited to the use of ribavirin (ribavirin), which is only partially effective when given early and is associated with significant side effects. Although vaccines against the huinin virus have been developed, their use is mainly limited to the highest risk group in argentina farm workers, and there is no approved vaccine against any other arenavirus. Although prophylactic vaccines are highly desirable, prophylactic vaccines may not always be an effective countermeasure against rapidly emerging, antigenically diverse new strains of virus, and existing vaccine development and production strategies do not adequately address the current or emerging diverse families of arenaviruses. Thus, new broad spectrum antiviral drugs may provide first line therapy and/or prophylaxis not only for areas of epidemic arenavirus infection, but also as a preventative measure against potential biowarfare agents.
Arenaviruses consist of a Nucleocapsid (NP) surrounded by an envelope, and the NP contains two ambisense RNA genome segments L and S that direct the synthesis of two polypeptides. The L segment encodes an RNA-dependent RNA polymerase (RdRp) and a small ring finger protein Z. The S segment encodes nucleoprotein and glycoprotein precursor GPC, which is cleaved by host proteases and undergoes post-translational modification into a mature complex consisting of glycoprotein GP1 (binding host proteins at the cell surface), GP2 (directing pH-dependent membrane fusion and release of genomic material in the cytoplasm), and a stable signal peptide (SSP 1). The mature glycoprotein complex (GP, otherwise known as glycoprotein) is formed in the viral envelope and is responsible for mediating viral entry. To enter/endocytose into cells, the old world arenavirus binds to the host d-dystrophic glycans, while the new world arenavirus binds to transferrin receptor 1. Upon binding to cell surface receptors, the virus is endocytosed and directed to an acidic late endosome, whereby GP2 mediates pH-dependent membrane fusion and release of genomic material into the cytoplasm for viral replication and transcription. Thus, viral entry inhibitors (e.g., small molecules) that target the viral GP complex or host factor are potential therapeutic/prophylactic methods of treating patients infected with arenavirus infection. Since the HF arenavirus species is classified as BSL-4, alternative methods are needed to identify viral entry inhibitors. To facilitate identification of arenavirus entry inhibitors, arenavirus GP complexes can be expressed in non-pathogenic BSL-2 enveloped viruses to produce a single round of infectious pseudoviruses whose viral entry function is determined by heterologous glycoproteins of interest. One viral expression system that may be utilized is the Vesicular Stomatitis Virus (VSV) system, whereby the envelope protein of VSV is replaced by an envelope glycoprotein from another virus (e.g., LASV) to mediate entry of the pseudovirion. The cellular entry and infection properties of GP pseudotype VSV virus have been shown against a variety of viruses including HIV, hepatitis B and C, Ebola virus, Lassa virus, Hantavirus, and the likeOgino,M.Et al Use of vascular storage viruses microorganisms associated with vascular or group viruses involved in proteins in a rapid and safe dilution test. Clin. Diagn. Lab. Immunol.(2003)10(1):154-60:Saha,M.N.Et al Formation of venous stock virus bearing surface proteins of hepatitis B virus.J.Virol (2005)79 (19): 12566-74; takada, A. et al A system for functional analysis of Ebola virus glycoprotein, Proc.Natl.Acad.Sci. (1997) 94: 14764-69;Garbutt,Met al Properties of reproduction-compatibility virus vectors of expressingglycoprotens of filovirus and arenaviruses.J. Virol (2004)78 (10): 5458-65]. The above documents are incorporated by reference herein in their entirety for all purposes. To monitor pseudoviral infection, a reporter gene, such as Green Fluorescent Protein (GFP) or luciferase, can be engineered into the pseudoviral genome, and viral infectivity in mammalian cell lines (e.g., Vero or Hek293) can be monitored using optical detection methods (e.g., plate readers) [ Cote, m.; misasi, j.; ren, T.; bruchez, a., Lee, k., Filone, c.m.; hensley, l.; li, Q.; ory, d.; chandran, k.; cunningham, j., Small molar apparatus innovative scientific Niemann-Pick C1 is scientific for Ebola virus infection, Nature (2011) 477: 344 through 348; elshabraway, h.a. et al Identification of a broad-specific aqueous all molecular acquisition of a segment serum respiratory syndrome, and Ebola, Hendra, and Nipah Viruses by using a novel high-through polypeptide screening assay.j.virol. (2014) 88: 4353-4365]. The above documents are incorporated by reference herein in their entirety for all purposes. Thus, "pseudoviruses" can be used to screen libraries of chemical compounds to identify inhibitors of arenavirus cell entry while avoiding the difficulties of working with highly pathogenic BSL-4 pathogens.
Introduction of deuterium (D) into a drug molecule is an attractive strategy that may help to improve the metabolism, pharmacokinetics and toxicity profile of the drug. Deuterium is a stable, non-toxic, non-radioactive isotope of hydrogen. Due to the larger atomic weight, deuterium forms a stronger bond with carbon than hydrogen, making the carbon-deuterium bond more difficult to break. In the case where the cleavage of carbon-hydrogen bonds is a partially or completely rate-limiting step in cytochrome P450-mediated drug metabolism, replacement of the hydrogen atom with deuterium may beSlowing the rate of metabolism, resulting in improved half-life, greater tolerability, improved efficacy and dosing regimen, lower side effects and reduced toxicity [ Foster, a.b. deuterium isotopopes efffects in students of drug metabolism. trends in pharmaceutical Sciences (1984) 5: 524-527; anderson, k.e.; stamler, d.; davis, M.D. et al Deuttrabenazine for treatment of innovative representations in properties with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 tertiary, Lancet Psychiary (2017) 4: 595-604; harbeson, s.; morgan, a.; liu, J. et al, alternating metabolic profiles of drugs by precision resolution 2: correlation of a determined analog of ivacai with a differentiated pharmacological determination J.Pharmacol.exp.ther, (2017) 362: 359-; T.;Feltmann,K.;Konradsson-Geuken,
T.;Feltmann,K.;Konradsson-Geuken, et al Deuterium-substasted l-DOPA display created behavial publication and dopamine output in an animal model of Parkinson's disease: composition with the effects produced by l-DOPA and an M4O-B inhibitor.J. neural.Transm. (Vienna) (2015) 122: 259-272; mutlib, a.e.; gerson, r.j.; meuner, P.C. et al The specifices-Dependent Metabolism of Effeavirenz products a neuropathoxic glutaminone Conjugate in rates. toxicol. appl. Pharmacol. (2000) 169: 102-113]. The above documents are incorporated by reference herein in their entirety for all purposes. However, in some cases, hydrogen-deuterium exchange may result in redirecting Metabolic sites ("Metabolic switching") [ Horning, m.g. et al Metabolic switching of drug pathways as d-con sequence of drug sub-protocol proceedings of the Second International Conference on Stable Isotopes (klein.e.r. and klein.p.d. editors) (1976) 41-54; miwa, g.t.; lu, A.Y.H.kinetic isotope effects and 'metabolic switching' in cytochrome P450-catalyzed reactions.Bioessays (1987) 7:215-219]. The above documents are incorporated by reference herein in their entirety for all purposes. At the same time, deuterium and hydrogen are essentially the same size, and in most cases, the deuteration of a drug will not be expected to affect the biochemical potency or selectivity of the deuteration drug for a biological target, as compared to its non-deuterated analogs. Even when deuterium atoms are incorporated at known metabolic sites, the effect of deuterium modification on drug metabolism and pharmacokinetic properties is unpredictable. The effect of deuterium incorporation on absorption, distribution, metabolism, excretion and/or toxicity (ADMET) properties can only be determined by preparing and testing actual deuterated compounds.
et al Deuterium-substasted l-DOPA display created behavial publication and dopamine output in an animal model of Parkinson's disease: composition with the effects produced by l-DOPA and an M4O-B inhibitor.J. neural.Transm. (Vienna) (2015) 122: 259-272; mutlib, a.e.; gerson, r.j.; meuner, P.C. et al The specifices-Dependent Metabolism of Effeavirenz products a neuropathoxic glutaminone Conjugate in rates. toxicol. appl. Pharmacol. (2000) 169: 102-113]. The above documents are incorporated by reference herein in their entirety for all purposes. However, in some cases, hydrogen-deuterium exchange may result in redirecting Metabolic sites ("Metabolic switching") [ Horning, m.g. et al Metabolic switching of drug pathways as d-con sequence of drug sub-protocol proceedings of the Second International Conference on Stable Isotopes (klein.e.r. and klein.p.d. editors) (1976) 41-54; miwa, g.t.; lu, A.Y.H.kinetic isotope effects and 'metabolic switching' in cytochrome P450-catalyzed reactions.Bioessays (1987) 7:215-219]. The above documents are incorporated by reference herein in their entirety for all purposes. At the same time, deuterium and hydrogen are essentially the same size, and in most cases, the deuteration of a drug will not be expected to affect the biochemical potency or selectivity of the deuteration drug for a biological target, as compared to its non-deuterated analogs. Even when deuterium atoms are incorporated at known metabolic sites, the effect of deuterium modification on drug metabolism and pharmacokinetic properties is unpredictable. The effect of deuterium incorporation on absorption, distribution, metabolism, excretion and/or toxicity (ADMET) properties can only be determined by preparing and testing actual deuterated compounds.
In the present invention, an arenavirus GP pseudovirus screen is used to identify the entry inhibitors and selected compounds are tested against the native non-HF virus TCRV to confirm activity against replicating arenaviruses. The selected superior compounds were then tested against native LASV to confirm activity against native highly pathogenic Human (HF) arenaviruses and to evaluate initial drug-like properties.
Disclosure of Invention
The present invention relates to the use of heterocyclic compounds to inhibit arenavirus infection in humans, other mammals, or in cell culture, methods of treating arenavirus infections such as lasa hemorrhagic fever, bolivia hemorrhagic fever, argentine hemorrhagic fever, venezuelan hemorrhagic fever, brazil hemorrhagic fever, copaene hemorrhagic fever, and lu hemorrhagic fever, methods of inhibiting replication of arenavirus, methods of reducing the amount of arenavirus, and compositions that can be used in such methods.
In one embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I

Or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein
A is independently selected from C and N;
g is independently selected from CH, CD and N;
e is independently selected from CH, CD and N;
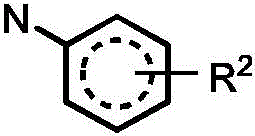
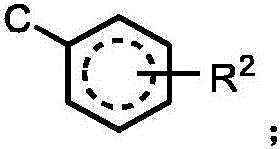
j is independently selected from
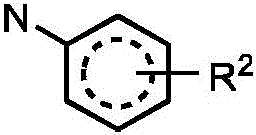
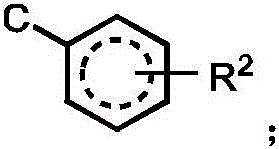
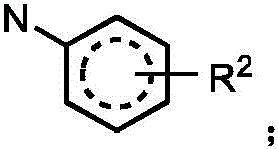
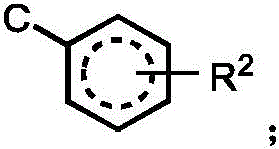
R2independently selected from H, D, -OR3、-R4、-NHR10、-CONHR10;
R3Independently selected from H, D, C1To C6Alkyl radical, C2To C6Alkenyl, (C)3To C10) Cycloalkyl group, (C)2To C9) Cycloheteroalkyl, -NHC (O) R4、-C(O)NHR10and-C (O) R10Wherein each C1To C6Alkyl is optionally substituted by D, halogen, -OH, -OR4、-NHR10Substitution;
R4independently selected from optionally substituted D, halogen, -OH, -OR10And NHR10Substituted C1To C6Alkyl and (C)2To C9) A cycloheteroalkyl group;
R5independently selected from H, D, C1To C6Alkyl radical, C2To C6Alkenyl radical, C2To C6Alkynyl, halogen, -OR3、-CO2R10、-NHC(O)R4、-C(O)NHR10、-NHR10、-CHNHR10、-CN、-CR4and-C (O) R10Wherein each C1To C6Alkyl optionally substituted with D;
R6independently selected from H, D, halogen, -OR3And R4;
R9Independently selected from H, D, halogen, C1To C6Alkyl and-OR10;
R10Independently of each otherSelected from H, D, -OH, C1To C6Alkyl and C2To C6An alkenyl group;
and when E is N, CH or CD, then A is C, G is CH or CD, and J is
And when A is N, then J is

With the proviso that the following compounds are excluded:
Detailed Description
In one embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I

Or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein
A is independently selected from C and N;
g is independently selected from CH, CD and N;
e is independently selected from CH, CD and N;
j is independently selected from
R2independently selected from H, D, -OR3、-R4、-NHR10、-CONHR10;
R3Independently selected from H, D, C1To C6Alkyl radical, C2To C6Alkenyl, (C)3To C10) Cycloalkyl group, (C)2To C9) Cycloheteroalkyl, -NHC (O) R4、-C(O)NHR10and-C (O) R10Wherein each C1To C6Alkyl is optionally substituted by D, halogen, -OH, -OR4、-NHR10Substitution;
R4independently selected from optionally substituted D, halogen, -OH, -OR10And NHR10Substituted C1To C6Alkyl and (C)2To C9) Cycloheteroalkyl group;
R5independently selected from H, D, C1To C6Alkyl radical, C2To C6Alkenyl radical, C2To C6Alkynyl, halogen, -OR3、-CO2R10、-NHC(O)R4、-C(O)NHR10、-NHR10、-CHNHR10、-CN、-CR4and-C (O) R10Wherein each C1To C6Alkyl optionally substituted with D;
R6independently selected from H, D, halogen, -OR3And R4;
R9Independently selected from H, D, halogen, -OR10And C1To C6An alkyl group;
R10independently selected from H, D, -OH, C1To C6Alkyl, and C2To C6An alkenyl group;
and when E is N, CH or CD, then A is C, G is CH or CD, and J is

And when A is N, then J is
With the proviso that the following compounds are excluded:


in another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
J is
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
J is
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, J, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
E is CH or CD.
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein E, G, J, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
A is C.
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein E, G, J, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
A is N.
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R9And R10As defined above and wherein
J is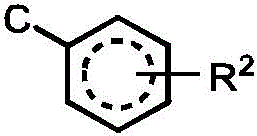 And R6 is
And R6 is Or
Or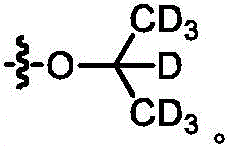
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample an effective amount of a compound represented by structural formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R9And R10As defined above and wherein
J is And R6 is
And R6 is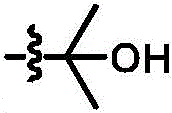 Or
Or
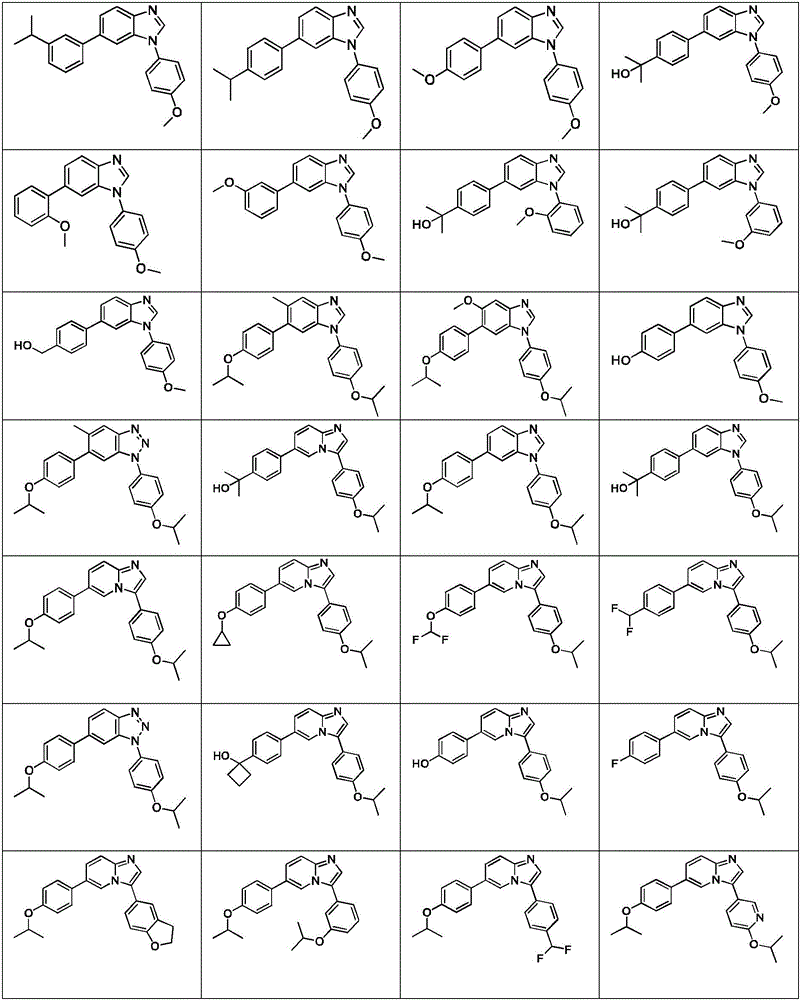
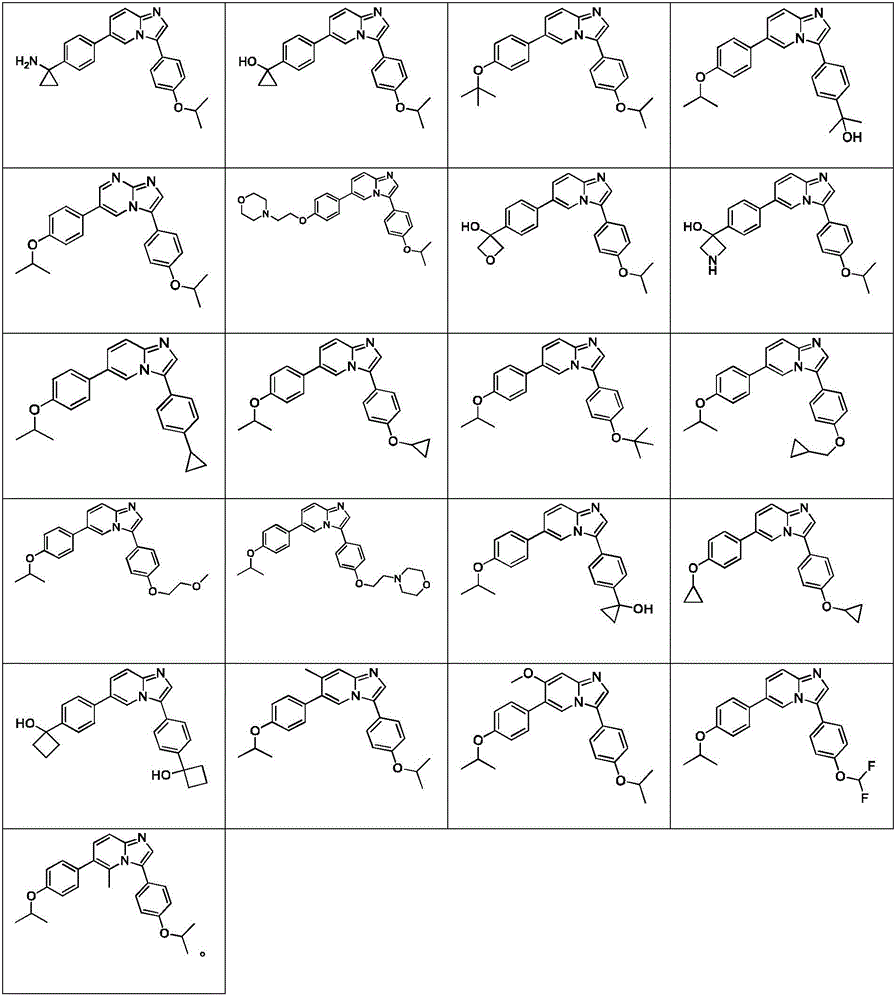
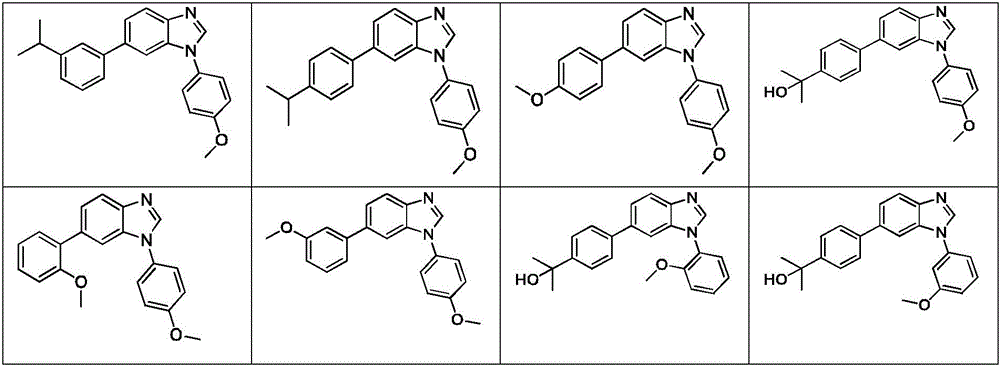
In another embodiment, the method comprises administering to a human, other mammal, cell culture or biological sample a pharmaceutically effective amount of a pharmaceutical composition comprising a compound selected from the compounds described in examples a1 to A3, B4 to B9, C10 to C26, D27 to D29 and E30, and a pharmaceutically acceptable carrier, diluent or vehicle.
In another embodiment, the method comprises administering a pharmaceutically effective amount of a pharmaceutical composition comprising a selected compound of structural formula I or a compound as shown above and a pharmaceutically acceptable carrier, diluent or vehicle and an additional therapeutically effective amount of a therapeutic agent selected from: ribavirin, polymerase inhibitors, Favipiravir (Favipiravir), texavirin (Triazavirin), small interfering rna (sirna), vaccines, monoclonal antibodies, immunomodulators and other arenavirus inhibitors.
In another embodiment, the present invention relates to compounds having structural formula I

Or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein
A is independently selected from C and N;
g is independently selected from CH, CD and N;
e is independently selected from CH, CD and N;
j is independently selected from
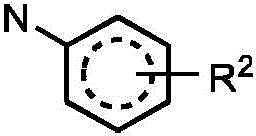

R2independently selected from H, D, -OR3、-R4、-NHR10、-CONHR10;
R3Independently selected from H, D, C1To C6Alkyl radical, C2To C6Alkenyl, (C)3To C10) Cycloalkyl group, (C)2To C9) Cycloheteroalkyl, -NHC (O) R4、-C(O)NHR10and-C (O) R10Wherein each C1To C6Alkyl is optionally substituted by D, halogen, -OH, -OR4、-NHR10Substitution;
R4independently selected from optionally substituted D, halogen, -OH, -OR10And NHR10Substituted C1To C6Alkyl and (C)2To C9) Cycloheteroalkyl group;
R5independently selected from H, D, C1To C6Alkyl radical, C2To C6Alkenyl radical, C2To C6Alkynyl, halogen, -OR3、-CO2R10、-NHC(O)R4、-C(O)NHR10、-NHR10、-CHNHR10、-CN、-CR4and-C (O) R10Wherein each C1To C6Alkyl optionally substituted with D;
R6independently selected from H, D, halogen, -OR3And R4;
R9Independently selected from H, D, halogen, -OR10And C1To C6An alkyl group;
R10independently selected from H, D, -OH, C1To C6Alkyl and C2To C6An alkenyl group;
and when E is N, CH or CD, then A is C, G is CH or CD, and J is
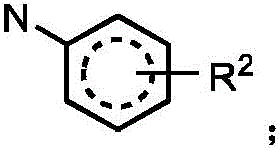
And when A is N, then J is

With the proviso that the following compounds are excluded:



in another embodiment, the invention relates toA compound or pharmaceutically acceptable salt of formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
J is
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt of structural formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
J is
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt of structural formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, J, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
E is CH or CD.
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt of structural formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein E, G, J, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
A is C.
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt of structural formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein E, G, J, R2、R3、R4、R5、R6、R9And R10As defined above and wherein
A is N.
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt of structural formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R9And R10As defined above and wherein
J is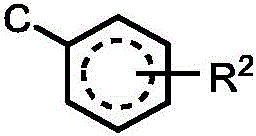 And R is6Is that
And R is6Is that Or
Or
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt of structural formula I, and a pharmaceutically acceptable carrier, diluent or vehicle thereof, wherein A, G, E, R2、R3、R4、R5、R9And R10As defined above and wherein
J is And R is6Is that
And R is6Is that Or
Or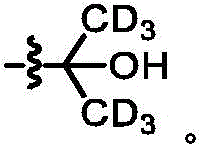
In another embodiment, the present invention relates to a compound or pharmaceutically acceptable salt selected from the compounds described in examples a1 to A3, B4 to B9, C10 to C26, D27 to D29 and E30, and a pharmaceutically acceptable carrier, diluent or vehicle thereof.
In another embodiment, the invention relates to a compound or pharmaceutically acceptable salt selected from the group consisting of:
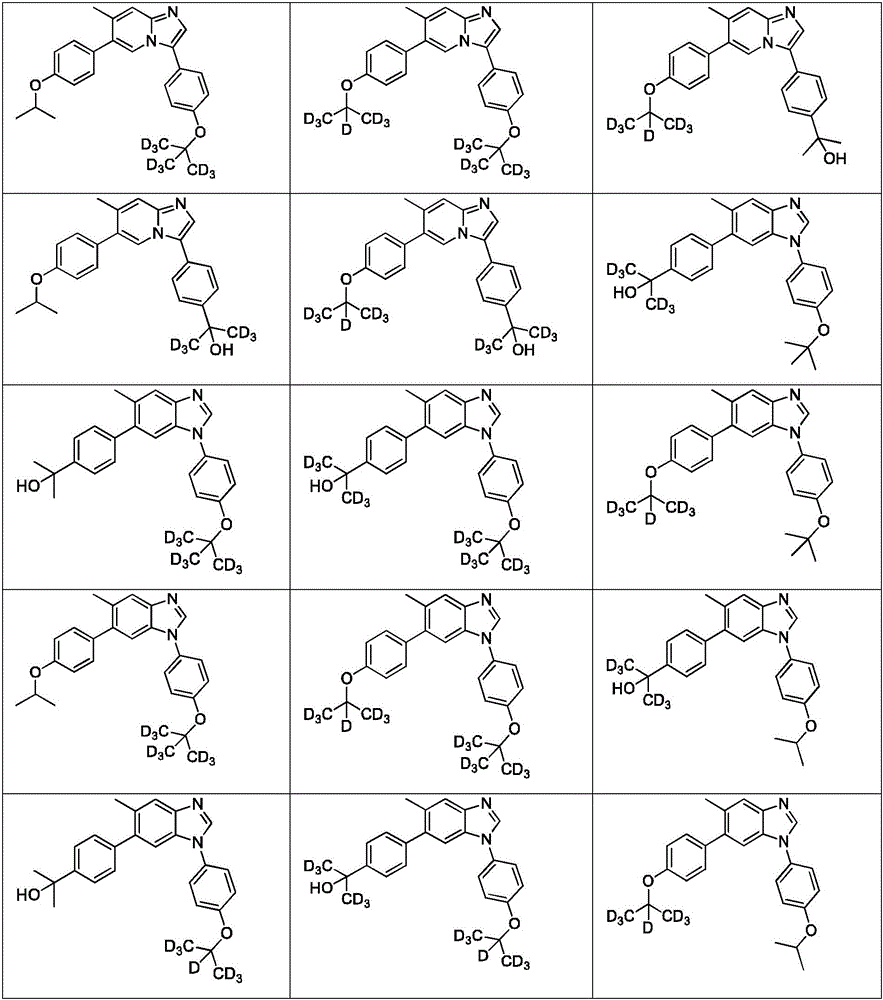

definition of
As used herein, the terms "comprises" and "comprising" are used in their open, non-limiting sense.
The term "halogen" and/or "halogen" refers to fluorine, chlorine, bromine or iodine.
Term "(C)1To C6) "alkyl" means a saturated aliphatic hydrocarbon group including straight and branched chain groups having 1 to 6 carbon atoms. (C)1To C6) Examples of alkyl groups include methyl, ethyl, propyl, 2-propyl, n-butyl, isobutyl, tert-butyl, pentyl, and the like. As used herein, the terms "Me" and "methyl" mean-CH3A group. As used herein, the terms "Et" and "ethyl" mean-C2H5A group.
As used herein, the term "(C)2To C8) Alkenyl "refers to an alkyl moiety having at least one carbon-carbon double bond comprising 2 to 8 carbons. The carbon-carbon double bond in this group can be anywhere along the 2 to 8 carbon chains to produce stable compounds. Such groups include the E and Z isomers of the alkenyl moiety. Examples of such groups include, but are not limited to, ethenyl, propenyl, butenyl, allyl, and pentenyl, and the like. As used herein, the term "allyl" means-CH2CH=CH2A group. As used herein, the term "c (R) ═ c (R)" denotes a carbon-carbon double bond in which each carbon atom is substituted with an R group, and includes E and Z isomers.
As used herein, the term "(C)2To C8) Alkynyl "means an alkyl moiety containing from 2 to 8 carbon atoms and having at least one carbon-carbon triple bond. The carbon-carbon triple bond in this group can be anywhere along the 2 to 8 carbon chains to produce stable compounds. Examples of such groups include, but are not limited to, acetylene, propyne, 1-butyne, 2-butyne, 1-pentyne, 2-pentyne, 1-hexyne, 2-hexyne, and 3-hexyne.
As used herein, the term "(C)1To C8) Alkoxy "means an O-alkyl group, wherein the alkyl group contains 1 to 8 carbon atoms and is straight, branched, or cyclic. Examples of such groups include, but are not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, t-butoxy, cyclopentyloxy, and cyclohexyloxy.
As used herein, the term "(C)6To C10) Aryl "means a group derived from an aromatic hydrocarbon containing from 6 to 10 carbon atoms. Examples of such groups include, but are not limited to, phenyl or naphthyl. As used herein, the terms "Ph" and "phenyl" mean-C6H5A group. As used herein, the term "benzyl" means-CH2C6H5A group.
As used herein, "(C)2To C9) Heteroaryl "means an aromatic heterocyclic group having a total of 5 to 10 atoms in its ring and containing 2 to 9 carbon atoms and 1 to 4 heteroatoms each independently selected from O, S and N, with the proviso that the ring of the group does not contain two adjacent O atoms or two adjacent S atoms. Heterocyclic groups include benzo-fused ring systems. Examples of aromatic heterocyclic groups are pyridyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolyl, isoquinolyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothienyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinoxalinyl, naphthyridinylAnd furopyridinyl. (C)2To C9) Heteroaryl groups may be C-linked or N-linked where possible. For example, a group derived from pyrrole may be pyrrol-1-yl (N-linked) or pyrrol-3-yl (C-linked). Furthermore, the groups derived from imidazole may be imidazol-1-yl (N-linked) or imidazol-3-yl (C-linked).
As used herein, "(C)2To C9) By cyclic heteroaryl "is meant a non-aromatic monocyclic, bicyclic, tricyclic, spirocyclic or tetracyclic group having a total of 4 to 13 atoms in its ring system and containing 5 to 9 carbon atoms and 1 to 4 heteroatoms each independently selected from O, S and N, with the proviso that the ring of said group does not contain two adjacent O atoms or two adjacent S atoms. Furthermore, such C2To C9The cycloheteroalkyl group may contain an oxo substituent on any available atom to produce a stable compound. For example, such groups may contain oxo atoms on available carbon or nitrogen atoms. Such groups may contain more than one oxo substituent, if chemically feasible. Furthermore, it is to be understood that when such C2 to C9 cycloheteroalkyl groups contain a sulfur atom, the sulfur atom may be oxidized by one or two oxygen atoms to give a sulfoxide or sulfone. An example of a 4-membered cycloheteroalkyl group is azetidinyl (derived from azetidine). An example of a 5-membered cycloheteroalkyl group is pyrrolidinyl. An example of a 6-membered cycloheteroalkyl group is piperidinyl. An example of a 9-membered cycloheteroalkyl group is indolinyl. An example of a 10-membered cycloheteroalkyl group is 4H-quinolizinyl. Additional examples of such C2 to C9 cycloheteroalkyl groups include, but are not limited to, tetrahydrofuryl, dihydrofuryl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl, oxetanyl (oxyethanyl), thietanyl, homopiperidinyl, oxepanyl (oxepanyl), thietanyl (thiepanyl), oxazepinyl (oxazepinyl), diazepinyl, thiazepinyl, 1, 2, 3, 6-tetrahydropyridinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1, 3-dioxolanyl, pyrazolinyl, dithianyl (dithianyl)) Dithiolanyl, dihydropyranyl, dihydrothienyl, dihydrofuryl, pyrazolidinyl, imidazolinyl, imidazolidinyl, 3-azabicyclo [3.1.0 ]]Hexyl, 3-azabicyclo [4.1.0]Heptyl, 3H-indolylquinolizinyl, 3-oxopiperazinyl, 4-methylpiperazinyl, 4-ethylpiperazinyl and 1-oxo-2, 8, diazaspiro [4.5]]Decan-8-yl.
Term "(C)3To C10) Cycloalkyl "means a saturated, monocyclic, fused, spiro or polycyclic ring structure having a total of 3 to 10 carbon ring atoms. Examples of such groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptyl, and adamantyl.
As used herein, the term "spiro" has its conventional meaning, i.e., any compound containing two or more rings, wherein the two rings have a common carbocyclic ring. As defined herein, the rings of a spiro compound independently have 3 to 20 ring atoms. Preferably, they have from 3 to 10 ring atoms. Non-limiting examples of spiro compounds include spiro [3.3] heptane, spiro [3.4] octane, and spiro [4.5] decane.
Term "(C)5To C8) Cycloalkenyl "means an unsaturated, monocyclic, fused, spiro ring structure having a total of 5 to 8 carbon ring atoms. Examples of such groups include, but are not limited to, cyclopentenyl, cyclohexenyl.
An "aldehyde" group refers to carbonyl-C (O) R, where R is hydrogen.
As defined herein, "alkoxy" refers to-O-alkyl and-O-cycloalkyl.
"alkoxycarbonyl" refers to-C (O) OR.
"alkylaminoalkyl" refers to-alkyl-NR-alkyl.
"alkylsulfonyl" means-SO2An alkyl group.
"amino" means-NH2Or a-NRR' group.
"aminoalkyl" refers to the group-alkyl-NRR'.
"aminocarbonyl" refers to-C (O) NRR'.
"arylalkyl" refers to-alkylaryl, wherein alkyl and aryl are defined herein.
As defined herein, "aryloxy" refers to-O-aryl and-O-heteroaryl.
"Aryloxycarbonyl" refers to-C (O) Oaryl.
"arylsulfonyl" means-SO2And (4) an aryl group.
"C-amido" refers to the group-C (O) NRR'.
"carbonyl" means-C (O) R.
"C-carboxy" refers to the group-C (O) OR.
A "carboxylic acid" group refers to a C-carboxyl group, wherein R is hydrogen.
"cyano" refers to the group-CN.
"Dialkylaminoalkyl" means- (alkyl) N (alkyl)2A group.
"halo" or "halogen" groups refer to fluoro, chloro, bromo, or iodo.
"haloalkyl" refers to an alkyl group substituted with one or more halogen atoms.
"heteroaryloxy" refers to a heteroaryl-O group having a heteroaryl group as defined herein.
"hydroxy" means an-OH group.
"N-amido" refers to the group-R' C (O) NR.
"N-carbamoyl" refers to the group-ROC (O) NR-.
"nitro" means-NO2A group.
"N-sulfonamido" means-NR-S (O)2And R group.
"N-thiocarbamoyl" refers to the ROC (S) NR' group.
"O-carbamoyl" refers to the group-OC (O) NRR'.
"O-carboxy" refers to an RC (O) O group.
"O-thiocarbamoyl" refers to the group-OC (S) NRR'.
An "oxo" group refers to a carbonyl moiety, such that an alkyl group substituted with oxo refers to a keto group.
"perfluoroalkyl" refers to an alkyl group in which all hydrogen atoms have been replaced with fluorine atoms.
"Phosphonyl" means-P (O) (OR)2A group.
"silyl" refers to-SiR3A group.
"S-sulfonamido" means-S (O)2An NR-group.
"sulfinyl" refers to the group-S (O) R.
"Sulfonyl" means-S (O)2And R group.
"thiocarbonyl" refers to the group-C (═ S) -R.
"trihalomethanecarbonyl" means Z3A CC (O) group, wherein Z is halogen.
"trihalomethanesulfonamido" means Z3CS(O)2NR-group, wherein Z is halogen.
"trihalomethanesulphonyl" means Z3CS(O)2A group wherein Z is halogen.
"Trihalomethyl" refers to the group-CZ3A group wherein Z is halogen.
"C-carboxy" refers to the group-C (O) OR.
The term "substituted" means that the specified group or moiety bears one or more substituents.
The term "unsubstituted" means that the specified group carries no substituents. The term "optionally substituted" means that the specified group is unsubstituted or substituted with one or more substituents. It will be understood that in the compounds of the present invention, when a group is said to be "unsubstituted" or "substituted" with a group having a lower valency than the valency at which all of the atoms in the compound are filled, the remaining valencies on the group are filled with hydrogen. For example, if C6Aryl (also referred to herein as "phenyl") is substituted with another substituent, as will be understood by those of ordinary skill in the art, this group having 4 residues left on C6Open positions on carbon atoms of the aryl ring (6 initial positions, minus one position bonded to the rest of the compound of the invention, minus another substituent, leaving 4). In such cases, the remaining 4 carbon atoms are eachBonded to a hydrogen atom to fill its valence. Similarly, if C is present in the compounds of the invention6Aryl is said to be "disubstituted," which, as will be understood by those of ordinary skill in the art, means C6Aryl has 3 unsubstituted remaining carbon atoms. Each of the three unsubstituted carbon atoms is bonded to a hydrogen atom to fill its valence.
The term "solvate" is used to describe a molecular complex between a compound of the invention and a solvent molecule. Examples of solvates include, but are not limited to, compounds of the present invention in combination with water, isopropanol, ethanol, methanol, dimethyl sulfoxide (DMSO), ethyl acetate, acetic acid, ethanolamine, or mixtures thereof. When the solvent is water, the term "hydrate" may be used. It is especially contemplated that in the present invention, one solvent molecule may be associated with one molecule of the present compound, such as a hydrate. Furthermore, it is especially contemplated that in the present invention, more than one solvent molecule may be associated with a molecule of the present compound, such as a dihydrate. Additionally, it is specifically contemplated that in the present invention, less than one solvent molecule may be associated with a molecule of the present compound, such as a hemihydrate. Furthermore, the solvates of the invention are contemplated to be solvates of the compounds of the invention that retain the biological effectiveness of the non-hydrated form of the compound.
As used herein, the term "pharmaceutically acceptable salt" means a biologically or otherwise desirable salt of a compound of the present invention that retains the biological effectiveness of the free acids and bases of the specified derivative.
As used herein, the term "pharmaceutically acceptable formulation" means a combination of a compound of the present invention or a salt or solvate thereof and a carrier, diluent and/or excipient that is compatible with the compound of the present invention and not deleterious to the recipient thereof. Pharmaceutical formulations may be prepared by procedures known to those of ordinary skill in the art. For example, the compounds of the present invention may be formulated with common excipients, diluents or carriers and formed into tablets, capsules and the like. Examples of excipients, diluents and carriers suitable for use in such formulations include the following: bulking and bulking agents such as starches, sugars, mannitol, and silicic acid derivatives; binders such as carboxymethyl cellulose and other cellulose derivatives, alginates, gelatin, and polyvinyl pyrrolidone; humectants, such as glycerol; disintegrants such as povidone, sodium starch glycolate, sodium carboxymethyl cellulose, agar, calcium carbonate and sodium bicarbonate; dissolution retarding agents, such as paraffin; resorption accelerators such as quaternary ammonium compounds; surfactants such as cetyl alcohol, glyceryl monostearate; adsorption carriers such as kaolin and bentonite; and lubricants such as talc, calcium stearate and magnesium stearate and solid polyethylene glycols. The final pharmaceutical form can be a pill, tablet, powder, lozenge, sachet, cachet, dragee, sterile packaged powder, or the like, depending on the type of excipient used. In addition, it is especially contemplated that the pharmaceutically acceptable formulations of the present invention may contain more than one active ingredient. For example, such formulations may contain more than one compound according to the invention. Alternatively, such formulations may contain one or more compounds of the invention and one or more additional agents that inhibit arenaviruses.
As used herein, the term "arenavirus GP inhibiting amount" refers to the amount of a compound of the invention, or a salt or solvate thereof, required to inhibit cellular entry in vivo, such as in a mammal, bird, or in vitro. The amount of such compounds required to cause such inhibition can be determined using the methods described herein and methods known to those of ordinary skill in the art without undue experimentation.
As used herein, the term "therapeutically effective amount" means an amount of a compound of the present invention or a salt thereof that is sufficient to effect treatment as defined herein when administered to a mammal in need of such treatment. Thus, a therapeutically effective amount of a compound of the invention, or a salt thereof, is an amount sufficient to modulate or inhibit the activity of an arenavirus GP protein such that cellular entry or replication of the arenavirus mediated by the activity of the arenavirus GP protein is reduced or mitigated.
In mammals, particularly humans, the term "treatment" with respect to arenavirus infection includes: (i) preventing the occurrence of a disease or disorder in a subject who may be predisposed to the disorder, such that treatment constitutes prophylactic treatment of the pathological disorder; (ii) modulating or inhibiting the disease or disorder, i.e., arresting its development; (iii) alleviating the disease or condition, i.e., causing regression of the disease or condition; or (iv) alleviating and/or alleviating the disease or disorder or symptoms resulting from the disease or disorder.
All references herein to compounds of the invention include references to salts, solvates and complexes thereof (including polymorphs, stereoisomers, tautomers and isotopically labeled forms thereof), unless otherwise indicated. For example, the compounds of the present invention may be pharmaceutically acceptable salts and/or pharmaceutically acceptable solvates.
The term "stereoisomers" refers to compounds having the same chemical composition, but differing with respect to the arrangement of their atoms or groups in space. In particular, the term "enantiomer" refers to two stereoisomers of a compound that are non-superimposable mirror images of each other. Pure enantiomers can be contaminated with up to about 10% of the opposite enantiomer.
As used herein, the term "racemic" or "racemic mixture" refers to a 1: 1 mixture of enantiomers of a particular compound. On the other hand, the term "diastereomer" refers to the relationship between a pair of stereoisomers that include two or more asymmetric centers and are not mirror images of each other. Symbols are used in the structural formulae herein to depict bonds, which are the points of attachment of a moiety or substituent to a core or backbone structure, according to conventions used in the art. According to another convention, in some formulae herein, carbon atoms and their bonded hydrogen atoms are not explicitly depicted, e.g. Represents a methyl group, and is represented by,
Represents a methyl group, and is represented by, represents an ethyl group, and represents a linear or branched alkyl group,
represents an ethyl group, and represents a linear or branched alkyl group, represents a cyclopentyl groupAnd the like.
represents a cyclopentyl groupAnd the like.
The compounds of the present invention may have asymmetric carbon atoms. Carbon-carbon bonds of the compounds of the invention may be represented herein by solid lines (__), solid wedge lines Or virtual wedge line
Or virtual wedge line Is shown. The use of a solid line to depict bonds to an asymmetric carbon atom is intended to indicate that all possible stereoisomers (e.g., specific enantiomers, racemic mixtures, etc.) are included on that carbon atom. The use of solid or dashed wedge lines to depict bonds to asymmetric carbon atoms is intended to indicate that only the stereoisomers shown are intended to be included. The compounds of the present invention may contain more than one asymmetric carbon atom. In these compounds, the use of a solid line to depict bonds to asymmetric carbon atoms is intended to indicate that all possible stereoisomers are meant to be included. For example, unless otherwise specified, it is contemplated that the compounds of the present invention may exist as enantiomers and diastereomers or as racemates and mixtures thereof. The use of solid lines to depict bonds to one or more asymmetric carbon atoms in compounds of the invention and the use of solid or dashed wedge lines to depict bonds to other asymmetric carbon atoms in the same compound is intended to indicate the presence of a mixture of diastereomers.
Is shown. The use of a solid line to depict bonds to an asymmetric carbon atom is intended to indicate that all possible stereoisomers (e.g., specific enantiomers, racemic mixtures, etc.) are included on that carbon atom. The use of solid or dashed wedge lines to depict bonds to asymmetric carbon atoms is intended to indicate that only the stereoisomers shown are intended to be included. The compounds of the present invention may contain more than one asymmetric carbon atom. In these compounds, the use of a solid line to depict bonds to asymmetric carbon atoms is intended to indicate that all possible stereoisomers are meant to be included. For example, unless otherwise specified, it is contemplated that the compounds of the present invention may exist as enantiomers and diastereomers or as racemates and mixtures thereof. The use of solid lines to depict bonds to one or more asymmetric carbon atoms in compounds of the invention and the use of solid or dashed wedge lines to depict bonds to other asymmetric carbon atoms in the same compound is intended to indicate the presence of a mixture of diastereomers.
Unless otherwise defined, a substituent "R" may be present on any atom of a ring system, provided that a stable structure is formed, provided that the hydrogen from one ring atom is replaced, either depicted, implied, or explicitly defined.
Conventional techniques for the preparation/separation of individual enantiomers include chiral synthesis from suitable optically pure precursors or resolution of the racemates using, for example, chiral High Pressure Liquid Chromatography (HPLC). Alternatively, the racemate (or racemic precursor) may be reacted with a suitable optically active compound (e.g. an alcohol), or, in the case where the compound contains an acidic or basic moiety, an acid or base (such as tartaric acid or 1-phenylethylamine). The resulting mixture of diastereomers may be separated by chromatography and/or fractional crystallization, and one or both of the diastereomers may be converted to the corresponding pure enantiomers by means well known to those skilled in the art. The chiral compounds of the invention (and chiral precursors thereof) can be obtained in enantiomerically enriched form using chromatography on asymmetric resins (typically HPLC) with a mobile phase consisting of a hydrocarbon (typically heptane or hexane) containing 0% to 50% isopropanol (typically 2% to 20%) and 0% to 5% alkylamine (typically 0.1% diethylamine). Concentration of the eluate yielded an enriched mixture. The collection of diastereomers may be separated by conventional techniques known to those skilled in the art. See, e.g., E L Eliel, "Stereochemistry of Organic Compounds" (Wiley, New York, 1994), the disclosure of which is incorporated by reference herein in its entirety.
When the compounds of the present invention contain an alkenyl or alkenylene group, geometric cis/trans (or Z/E) isomers are possible. In the case of compounds containing, for example, keto or oxime groups or aromatic moieties, tautomerism ("tautomerism") may occur. Examples of tautomerism include keto and enol tautomers. A single compound may exhibit more than one type of isomerism. All stereoisomers, geometric isomers and tautomeric forms of the compounds of the invention are included within the scope of the invention, including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. The cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, such as chromatography and fractional crystallization.
The compounds of the present invention may be administered as prodrugs. Thus, certain derivatives of the compounds of formula I (which may themselves have little or no pharmacological activity) may be converted to compounds of formula (I) having the desired activity when administered to a mammal, for example by hydrolytic cleavage. Such derivatives are referred to as "prodrugs". Prodrugs can be generated, for example, by replacing appropriate functional groups present in compounds having formula I with certain moieties known to those skilled in the art. See, e.g., "Pro-drugs as Novel Delivery Systems", volume 14, ACS Symposium Series (T Higuchi and W Stella) and "Bioreversible Carriers in Drug Design", Pergamon Press, 1987(E B Roche eds., American Pharmaceutical Association). Some examples of such prodrugs include: an ester moiety replacing the carboxylic acid functionality; an ether moiety or an amide moiety replacing the alcohol functional group; and amide moieties in place of primary or secondary amino functional groups. Further examples of displacing groups are known to the person skilled in the art. See, for example, H Bundgaard, "Design of Prodrugs" (Elsevier, 1985), the disclosure of which is incorporated herein by reference in its entirety. It is also possible that certain compounds having formula I may themselves act as prodrugs of other compounds having formula I.
The salts of the present invention may be prepared according to methods known to those skilled in the art. Examples of salts include, but are not limited to, acetate, acrylate, benzenesulfonate, benzoate (such as chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, and methoxybenzoate), bicarbonate, bisulfate, bisulfite, bitartrate, borate, bromide, butyne-1, 4-dioate, calcium edetate, camphorsulfonate, carbonate, chloride, hexanoate, octanoate, clavulanate, citrate, decanoate, dihydrochloride, dihydrogen phosphate, edetate, sulfonate, etonate (etolate), ethanesulfonate, ethylsuccinate, formate, fumarate, glucoheptonate, gluconate, glutamate, glycolate, glycollate, glycollylate, heptanoate, hexyne-1, 6-dioate, acetate, Hexylisophthalate (hexylresorcinate), hydrabamine (hydrabamine), hydrobromide, hydrochloride, gamma-hydroxybutyrate, iodide, isobutyrate, isothionate, lactate, lactobionate, laurate, malate, maleate, malonate, mandelate, methanesulfonate, metaphosphate, methanesulfonate, methylsulfate, monohydrogen phosphate, mucate, naphthalenesulfonate, naphthalene-1-sulfonate, naphthalene-2-sulfonate, nitrate, oleate, oxalate, pamoate (embonate), palmitate, pantothenate, phenylacetate, phenylbutyrate, phenylpropionate, phthalate, phosphate/diphosphate, polygalacturonate, propanesulfonate, propionate, propiolate, pyrosulfate, salicylate, stearate, subacetate, hydrabamine, salicylate, and mixtures thereof, Suberate, succinate, sulfate, sulfonate, sulfite, tannate, tartrate, 8-chlorotheophylline salt, tosylate, triethyliodide (triethiodode), and valerate.
The compounds of the present invention that are basic in nature are capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is generally necessary in practice to first isolate the compounds of the invention from the reaction mixture as a pharmaceutically unacceptable salt, then simply convert the latter back to the free base compound by treatment with a basic agent, and then convert the latter free base to a pharmaceutically acceptable acid addition salt. The acid addition salts of the base compounds of the present invention may be prepared by: the base compound is treated with a substantially equivalent amount of the selected mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent such as methanol or ethanol. After evaporation of the solvent, the desired solid salt was obtained. The desired acid salt may also be precipitated from a solution of the free base in an organic solvent by adding a suitable mineral or organic acid to the solution.
Those compounds of the present invention that are acidic in nature are capable of forming basic salts with various pharmaceutically acceptable cations. Examples of such salts include alkali metal or alkaline earth metal salts, and in particular sodium and potassium salts. These salts are prepared by conventional techniques. Chemical bases useful as reagents for preparing the pharmaceutically acceptable basic salts of the invention are those compounds that form non-toxic basic salts with the acidic compounds of the invention. Non-toxic basic salts at this point include those compounds derived from pharmaceutically acceptable cations such as sodium, potassium, calcium, and magnesium. These salts can be prepared by: the corresponding acidic compound is treated with an aqueous solution containing the desired pharmaceutically acceptable cation, and the resulting solution is then evaporated to dryness, preferably under reduced pressure. Alternatively, they can also be prepared by: the lower alcoholic solution of the acidic compound and the desired alkali metal alkoxide are mixed together and the resulting solution is then evaporated to dryness in the same manner as before. In either case, it is preferred to use stoichiometric amounts of the reagents to ensure completion of the reaction and maximum yield of the desired end product.
If the compound of the invention is a base, the desired salt may be prepared by any suitable method available in the art, for example, by treating the free base with an inorganic acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like) or with an organic acid such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, pyranosidyl acid (such as glucuronic acid or galacturonic acid), alpha-hydroxy acid (such as citric acid or tartaric acid), amino acid (such as aspartic acid or glutamic acid), aromatic acid (such as benzoic acid or cinnamic acid), sulfonic acid (such as p-toluenesulfonic acid or ethanesulfonic acid), and the like.
If the compound of the invention is an acid, the desired salt may be prepared by any suitable method, for example, with an inorganic or organic base such as an amine (primary, secondary or tertiary); alkali metal hydroxides or alkaline earth metal hydroxides. Illustrative examples of suitable salts include organic salts derived from amino acids (such as glycine and arginine), ammonia, primary, secondary and tertiary amines, and cyclic amines (such as piperidine, morpholine, and piperazine); and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum, and lithium.
In the case of pharmaceutical agents that are solids, those skilled in the art will appreciate that the compounds, agents, and salts of the present invention may exist in different crystalline or polymorphic forms, all of which are intended to be within the scope of the present invention and the specified formula.
The invention also includes isotopically-labeled compounds of the present invention, wherein one or more atoms are replaced by an atom having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes suitable for inclusion in compounds of the invention include isotopes of hydrogen, such as2H and3h; isotopes of carbon, such as11C、13C and14c; an isotope of chlorine,such as36Cl、35Cl and37cl; isotopes of fluorine, such as18F; isotopes of iodine, such as123I and125i; isotopes of nitrogen, such as13N and15n; isotopes of oxygen, such as15O、17O and18o; isotopes of phosphorus, such as32P; and isotopes of sulfur, such as35S。
Certain isotopically-labeled compounds of the present invention, for example those into which a radioactive isotope has been incorporated, are useful in drug and/or substrate tissue distribution studies. Radioisotope tritium3H and C-1414C is particularly useful for this purpose because of its ease of incorporation and the ease of detection means. Using heavier isotopes such as deuterium2Substitution of H may result in certain therapeutic advantages resulting from greater metabolic stability, e.g.35S has an increased in vivo half-life or reduced dosage requirements and is therefore preferred in some circumstances. With positron-emitting isotopes (such as11C、18F、15O and13n) substitution can be useful in Positron Emission Tomography (PET) studies to examine matrix receptor occupancy. Isotopically-labeled compounds of the present invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the unlabeled reagent employed.
The term "deuterated" refers to the replacement of one or more hydrogen atoms with a corresponding number of deuterium atoms. Unless otherwise specified, when a particular position in a compound of the invention is explicitly designated as "D", "deuterium", as "deuterated" or "having deuterium" (elemental deuterium is represented by the letter "D" in chemical structures and formulae and by the lower case letter "D" in chemical names), it is understood that the deuterium at that position is at an abundance that is at least 3000 times greater than the natural deuterium abundance (which is 0.015%) (i.e., the terms "D", "D" or "deuterium" indicate deuterium incorporation of at least 45%).
As used herein, the term "isotopic enrichment factor" means the ratio between the isotopic abundance and the natural abundance of a given isotope.
In some embodiments, the compounds of the invention have an isotopic enrichment factor for each deuterium atom present at a position designated as a potential site for deuterium on the compound of at least 3500 (52.5% deuterium incorporation), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
The compounds of the present invention may be formulated into pharmaceutical compositions as described below in any pharmaceutical form deemed appropriate by those skilled in the art. The pharmaceutical compositions of the present invention comprise a therapeutically effective amount of at least one compound of the present invention together with an inert pharmaceutically acceptable carrier or diluent.
For the treatment or prevention of diseases or conditions mediated in part or in whole by arenavirus infection or viruses expressing arenavirus glycoproteins, the pharmaceutical compositions of the invention are administered in suitable formulations prepared by combining a therapeutically effective amount (i.e., an arenavirus GP modulating, modulating or inhibiting amount effective to achieve therapeutic efficacy) of at least one compound of the invention (as active ingredient) with one or more pharmaceutically suitable carriers which may, for example, be selected from diluents, excipients and adjuvants which facilitate processing of the active compound into the final pharmaceutical formulation.
The pharmaceutical carrier employed may be a solid or a liquid. Exemplary solid carriers are lactose, sucrose, talc, gelatin, agar, pectin, gum arabic, magnesium stearate, stearic acid, and the like. Exemplary liquid carriers are syrup, peanut oil, olive oil, water, and the like. Similarly, the compositions of the present invention may include time delay or timed release materials known in the art, such as glyceryl monostearate or glyceryl distearate alone or with a wax, ethylcellulose, hydroxypropylmethylcellulose, methylmethacrylate or the like. Additional additives or excipients may be added to achieve desired formulation characteristics. For example, can addAdding bioavailability enhancing agents such as Labrasol, Gelucire, etc.; or a formulation, such as CMC (carboxymethylcellulose), PG (propylene glycol) or PEG (polyethylene glycol). For example, when preparing a capsule formulation, may be added A semi-solid excipient protects active ingredients from light, moisture and oxidation.
A semi-solid excipient protects active ingredients from light, moisture and oxidation.
If a solid carrier is used, the preparation may be tableted, placed in a hard gelatin capsule as a powder or pellet, or formed into dragees or lozenges. The amount of solid carrier can vary, but will generally be from about 25mg to about 1 g. If a liquid carrier is used, the preparation may be in the form of a syrup, emulsion, soft gelatin capsule, sterile injectable solution or suspension in an ampoule or vial, or non-aqueous liquid suspension. If a semi-solid carrier is used, the formulation may be in the form of hard and soft gelatin capsule formulations. The compositions of the present invention may be prepared in unit dosage form suitable for the mode of administration (e.g., parenteral or oral administration).
To obtain a stable water soluble dosage form, a salt of a compound of the present invention may be dissolved in an aqueous solution of an organic or inorganic acid, such as a 0.3M solution of succinic acid or citric acid. If a soluble salt form is not available, the agent may be dissolved in a suitable co-solvent or combination of co-solvents. Examples of suitable co-solvents include alcohols, propylene glycol, polyethylene glycol 300, polysorbate 80, glycerol, and the like, at concentrations ranging from 0% to 60% of the total volume. The compositions may also be in the form of a solution of the salt form of the active ingredient in a suitable aqueous vehicle such as water or isotonic saline or dextrose solution.
The appropriate formulation depends on the chosen route of administration. For injection, the pharmaceutical preparations of the compounds of the present invention may be formulated as aqueous solutions, preferably in physiologically compatible buffers such as hank's solution, ringer's solution or physiological saline buffer.
For transmucosal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
For oral administration, the compounds may be formulated by combining the active compound with pharmaceutically acceptable carriers known in the art. Such carriers enable the compounds of the invention to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject to be treated. Pharmaceutical formulations for oral use can be obtained by: the resulting mixture is optionally ground using a solid excipient mixed with the active ingredient (medicament) and, if necessary after addition of suitable auxiliaries, the mixture of granules is processed to obtain tablets or dragee cores. Suitable excipients include: fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; and cellulose preparations, such as corn starch, wheat starch, rice starch, potato starch, gelatin, gums, methyl cellulose, hydroxypropyl methyl cellulose, sodium carboxymethyl cellulose; or polyvinylpyrrolidone (PVP). If desired, disintegrating agents such as cross-linked polyvinylpyrrolidone, agar or alginic acid or a salt thereof such as sodium alginate may be added. Sugar-coated tablet cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain gum arabic, polyvinylpyrrolidone, carbomer gel, polyethylene glycol and/or titanium dioxide, lacquer solutions and suitable organic solvents or solvent mixtures. Dyes or pigments may be added to the tablets or dragee coatings for identifying or characterizing different combinations of active agents.
Pharmaceutical preparations which can be used orally include push-fit capsules made of gelatin, as well as soft-seal capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. Plug-in capsules may contain the active ingredient in admixture with fillers (such as lactose), binders (such as starches), and/or lubricants (such as talc or magnesium stearate) and, optionally, stabilizers. In soft capsules, the active agent may be dissolved or suspended in a suitable liquid, such as fatty oil, liquid paraffin, or liquid polyethylene glycol. In addition, stabilizers may be added. All formulations for oral administration should be in dosages suitable for such administration. For buccal administration, the compositions may take the form of tablets or lozenges formulated in conventional manner.
For intranasal or administration by inhalation, the compounds for use according to the invention may be conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable propellant (e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas). In the case of a pressurized aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount.
Capsules and cartridges of gelatin for use in an inhaler or insufflator or the like may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
The compounds may be formulated for parenteral administration by injection (e.g., by bolus injection or continuous infusion). Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. These compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
Pharmaceutical formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active agents can be prepared as appropriate oily injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils (such as sesame oil) or synthetic fatty acid esters (ethyl oleate or triglycerides) or liposomes. Aqueous injection suspensions may contain substances that increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents that increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
Alternatively, the active ingredient may be in powder form for constitution with a suitable vehicle (e.g., sterile, pyrogen-free water) before use.
In addition to the formulations described above, the compounds of the present invention may also be formulated as depot formulations. Such long-acting formulations may be administered by implantation (e.g., subcutaneously or intramuscularly) or by intramuscular injection. Thus, for example, the compounds may be formulated with suitable polymeric or hydrophobic materials (e.g., as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives (e.g., as a sparingly soluble salt). The pharmaceutical carrier for the hydrophobic compound is a co-solvent system comprising benzyl alcohol, a non-polar surfactant, a water-miscible organic polymer, and an aqueous phase. The co-solvent system may be a VPD co-solvent system. VPD was a solution of 3% w/v benzyl alcohol, 8% w/v non-polar surfactant polysorbate 80 and 65% w/v polyethylene glycol 300, made to volume in absolute ethanol. The VPD cosolvent system (VPD: 5W) contained VPD diluted 1: 1 with 5% dextrose in water. This co-solvent system dissolves hydrophobic compounds well and inherently produces less toxicity when administered systemically. The proportion of the co-solvent system can be varied appropriately without destroying its solubility and toxicity characteristics. Furthermore, the identity of the co-solvent component may vary: for example, other low toxicity non-polar surfactants may be used in place of polysorbate 80; the polyethylene glycol may vary in size; other biocompatible polymers, such as polyvinylpyrrolidone, may replace polyethylene glycol; other sugars or polysaccharides may replace dextrose.
Alternatively, other delivery systems for hydrophobic drug compounds may be employed. Liposomes and emulsions are known examples of delivery vehicles or carriers for hydrophobic drugs. Certain organic solvents, such as dimethyl sulfoxide, may also be employed, but typically at the expense of greater toxicity due to the toxic nature of DMSO. In addition, compounds can be delivered using sustained release systems, such as semipermeable matrices of solid hydrophobic polymers containing the therapeutic agent. Various sustained release materials have been established and are known to those skilled in the art. Depending on its chemical nature, a sustained release capsule can release the active ingredient over several weeks up to over 100 days depending on its chemical nature. Other protein stabilization strategies may be employed depending on the chemical nature and biological stability of the therapeutic agent.
The pharmaceutical compositions may also contain suitable solid or gel phase carriers or excipients. These carriers and excipients can provide significant improvements in the bioavailability of poorly soluble drugs. Examples of such carriers or excipients include calcium carbonate, calcium phosphate, sugars, starch, cellulose derivatives, gelatin, and polymers (such as polyethylene glycol). In addition, additives or excipients such as
 And the like.
And the like.
In addition, the pharmaceutical composition may be incorporated into a skin patch for direct delivery of the drug to the skin.
It will be understood that the actual dosage of the agents of the invention will vary depending upon the particular agent used, the particular composition formulated, the mode of administration and the particular site, host and disease being treated. Given the experimental data for a given compound, one skilled in the art can determine the optimal dosage for a given set of conditions using routine dosimetry assays. For oral administration, an exemplary daily dose generally employed will be from about 0.001 to about 1000mg/kg body weight, with the treatment cycle repeated at appropriate intervals.
Furthermore, the pharmaceutically acceptable formulation of the present invention may contain the compound of the present invention or a salt or solvate thereof in the following amounts: from about 10mg to about 2000mg, or from about 10mg to about 1500mg, or from about 10mg to about 1000mg, or from about 10mg to about 750mg, or from about 10mg to about 500mg, or from about 25mg to about 500mg, or from about 50 to about 500mg, or from about 100mg to about 500 mg.
In addition, the pharmaceutically acceptable formulation of the present invention may contain the compound of the present invention or a salt or solvate thereof in the following amounts: about 0.5 w/w% to about 95 w/w%, or about 1 w/w% to about 75 w/w%, or about 5 w/w% to about 75 w/w%, or about 10 w/w% to about 50 w/w%.
The compounds of the present invention or salts or solvates thereof may be administered to a mammal (such as a human) suffering from a condition or disease mediated by arenavirus or any virus expressing arenavirus glycoproteins, once a day, twice a day, three times a day, four times a day, or even more frequently, alone or in combination with one or more compounds selected from ribavirin, polymerase inhibitors, fapiravir, texavirine, small interfering rna (sirna), vaccines, monoclonal antibodies, immunomodulators and other arenavirus inhibitors as part of a pharmaceutically acceptable formulation.
A compound of the invention or a salt or solvate thereof may be administered to a mammal (such as a human) suffering from a condition or disease mediated by arenavirus, once a day, twice a day, three times a day, four times a day, or even more frequently, in combination with at least one other agent for treating arenavirus selected from the group consisting of: ribavirin (viral RNA-Dependent RNA polymerase inhibitors) (as shown by Ng KK, Arnold JJ and Cameron CE, Structure-Function Relationships RNA-Dependent RNA Polymerases, Curr Top Microbiol Immunol, 2008; 320: 137-, jaydeokar AV and Bandawane DD, Immunomodulators: a Pharmacological Review, Internat J Pharmacy and Pharmacological Sci, 2012; 4: 30-36, which are herein incorporated by reference in their entirety).
It will be understood by those of ordinary skill in the art that the particular pharmaceutical formulation, dosage and number of doses administered daily to a mammal in need of such treatment for the compounds of the present invention are all within the knowledge of those of ordinary skill in the art and can be determined without undue experimentation.
The compounds of the invention are useful for modulating or inhibiting arenavirus Glycoprotein (GP) in vitro and in vivo.
Thus, these compounds are useful for the prevention and/or treatment of disease states associated with arenavirus infection or the treatment of viruses expressing arenavirus glycoproteins.
The invention also relates to a method for treating an arenavirus infection in a mammal, including a human, comprising administering to the mammal an amount of a compound having formula I as defined above or a salt or solvate thereof effective to treat a disease state associated with an arenavirus infection or a virus expressing an arenavirus glycoprotein.
In the following preparations and examples, "Ac" means acetyl, "Me" means methyl, "Et" means ethyl, "Ph" means phenyl, "Py" means pyridine, "BOC", "Boc" or "BOC" means N-tert-butoxycarbonyl, "Ns" means 2-nitrobenzenesulfonyl, "DCM" (CH)2Cl2) Is intended to mean dichloromethane or methylene chloride, "dba" is intended to mean dibenzylideneacetone, "DCE" is intended to mean dichloroethane or ethylene chloride, "D" or "D" is intended to mean deuterium, "DIAD" is intended to mean diisopropylazadicarboxylate, "DIPEA" or "DIEA" is intended to mean diisopropylethylamine, "DMA" is intended to mean N, N-dimethylacetamide, "DMF" is intended to mean N-dimethylformamide, "DMSO" is intended to mean dimethylsulfoxide, "DPPP" is intended to mean 1, 3-bis (diphenylphosphino) propane, "HOAc" is intended to mean acetic acid, "IPA" is intended to mean isopropanol, "NMP" is intended to mean 1-methyl 2-pyrrolidone, "TEA" is intended to be triethylamine, "TFA" is intended to mean trifluoroacetic acid, "DCM" is intended to mean dichloromethane, "EtOAc" is intended to mean ethyl acetate, "MgSO4"means magnesium sulfate," Na2SO4"means sodium sulfate," MeOH "means methanol," Et2O "means diethyl ether," EtOH "means ethanol," H2O "means water," HCl "means hydrochloric acid," POCl3"means phosphorus oxychloride," SOCl2"means thionyl chloride," K2CO3"means potassium carbonate," THF "means tetrahydrofuran," DBU "means 1, 8-diazabicyclo [5.4.0]Undec-7-ene, "LiHMDS" or "LHMDS" means lithium hexamethyldisilazide, "TBME" or "MTBE" means t-butyl methyl ether, "LDA" means lithium diisopropylamide, "NBS" means N-bromosuccinimide, "NIS" means N-iodosuccinimide, "Xanthphos" means 4, 5-bis (diphenylphosphino) -9, 9-dimethylxanthene; "P (Ph)3) "means triphenylphosphine," N "means standard," M "means molar," mL "means milliliter," mmol "means millimole," μmol "means micromole," eq.
Preparation method
The compounds of the present invention can be prepared using the reaction routes and synthetic schemes described below, using readily available starting materials, using techniques available in the art. The preparation of certain embodiments of the present invention is described in detail in the following examples, but one of ordinary skill in the art will recognize that the preparation can be readily adjusted to prepare other embodiments of the present invention. For example, the synthesis of non-exemplified compounds according to the present invention may be carried out by modifications apparent to those skilled in the art, for example, by appropriately protecting interfering groups, by making other suitable reagents known in the art, or by making routine modifications to the reaction conditions. Alternatively, it will be appreciated that other reactions mentioned herein or known in the art have applicability for the preparation of other compounds of the invention.
Scheme 1 illustrates a method that can be used to synthesize compounds of structural formula I (where G is CH, J is N, E is CH and a is C). Can be in a solvent (such as THF or DMF), in a base (such as NaH or Cs)2CO3) Reacting compound 1-1(X ═ Cl, Br or I, and Y is F, Cl, Br or I) with an amine R in the presence of2NH2React to form compound 1-2. Using a reducing agent (such as Fe or SnCl) in a solvent (such as THF or methanol)2) Reduction of the nitro group affords the aniline 1-3, which can be reacted with formic acid HCO2H or ortho esters HC (OR)3React to form 1-4. In a solvent (such as dimethoxyethane) in a base (such as K)2CO3) In the presence of a catalyst (such as [1, 1' -bis (diphenylphosphino) ferrocene)]Palladium (II) dichloride) with boric acid or a borate R1B(OR)2Coupling may provide a compound having structural formula I.
Scheme 1

Scheme 2 depicts a method of synthesizing deuterated anilines 2-4, which deuterated anilines 2-4 can be used to prepare deuterated intermediates 1-2 for the synthesis of deuterated compounds of the invention as described in scheme 1 above. In a solvent (such as N, N-dimethylformamide), in a base (such as K)2CO3) Reaction of phenol 2-1 with deuterated alkyl halide 2-2 (X' ═ Br or I) in the presence of hydrogen can afford compound 2-3. Reduction of the nitro group with a reducing agent such as hydrogen in the presence of a catalyst such as palladium on carbon in a solvent such as methanol can provide the anilines 2-4.
Scheme 2

Scheme 3 depicts a method of synthesizing deuterated anilines 3-4, which deuterated anilines 3-4 can be used to prepare deuterated intermediates 1-2 to synthesize deuterated compounds of the invention as described in scheme 1 above. Arylation of alcohol 3-1 with diaryliodonium salt 3-2 in a solvent such as pentane in the presence of a base such as NaHMDS can provide compound 3-3[ Lindstedt, e.; stridfeldt, e.; olofsson, B.Mild synthesis of sequential conjugated alkyl aryl ethers.org.Lett. (2016) 18: 4234-4237]. The above documents are incorporated by reference herein in their entirety for all purposes. Reduction of the nitro group with a reducing agent such as hydrogen in the presence of a catalyst such as palladium on carbon in a solvent such as methanol can provide the aniline 3-4.
Scheme 3

Scheme 4 depicts a method that can be used to synthesize compounds of structural formula I (where a is N, E is CH and J is C). Can be in a solvent (such as dimethoxyethane) in a base (such as K)2CO3) In the presence of a catalyst (such as [1, 1' -bis (diphenylphosphino) ferrocene)]Palladium (II) dichloride) compound 4-1(X ═ Cl, Br) with boronic acid or boronic ester R1B(OR)2Coupling to form 4-2, 4-2 may be treated with a halogenating reagent, such as bromine or N-bromosuccinimide (NBS) or iodine or N-iodosuccinimide (NIS), to form compound 4-3(Y ═ Br, I)2CO3) With boric acid or borate R in the presence of a catalyst such as tetrakis (triphenylphosphine) palladium2B(OR)2Treatment 4-3 can provide a compound having structural formula I. Alternatively, it may be in a solvent (such as dioxane), in a base (such as K)2CO3) Reacting compound 4-4(X ═ Cl, Br) with boronic acid or boronic ester R in the presence of a catalyst such as tetrakis (triphenylphosphine) palladium2B(OR)2Reaction to provide compound 4-5, which may be in a solvent (such as dimethoxyethane), in a base (such as K)2CO3) In the presence of a catalyst (such as [1, 1' -bis (diphenylphosphino) ferrocene)]Palladium (II) dichloride) with a second diboronic acid or boronic ester R1B(OR)2Reacting to provide a compound having structural formula I.
Scheme 4

Scheme 5 illustrates a method that can be used to synthesize compounds of structural formula I (where G is CH, a is C, J is N, and E is N). Can be in a solvent (such as THF or DMF), in a base (such as NaH or K)2CO3) In the presence of a compound 5-1(X ═ Cl, Br or I, and Y is F, Cl, Br or I) with an amine R2NH2React to form compound 5-2. Using a reducing agent (such as Fe or SnCl) in a solvent (such as THF or methanol)2) Reduction of the nitro group can provide aniline 5-3, which can be reacted with nitrous acid to form 5-4. In a solvent (such as dimethoxyethane) in a base (such as K)2CO3) In the presence of a catalyst (such as [1, 1' -bis (diphenylphosphino) ferrocene)]Palladium (II) dichloride) with boric acid or a borate R1B(OR)2Coupling may provide a compound having structural formula I.
Scheme 5

Reaction schemes 6-8 illustrate methods of synthesizing borane reagents 6-4, 7-4, and 8-4, which borane reagents 6-4, 7-4, and 8-4 can be used to prepare deuterated intermediates and final compounds of the invention as described in schemes 1, 4, and 5 above to introduce R1And/or R2And (4) a substituent.
Scheme 6 depicts a method that can be used to synthesize deuterated boronic acids or boronic esters 6-4. In a solvent (such as N, N-2-methylformamide), in a base (such as K)2CO3) Reaction of phenol 6-1(X ═ Br or I) with deuterated alkyl halide 6-2(X ═ Br or I) in the presence of hydrogen can afford compound 6-3. Compound 6-3 can be converted to boronic acid or boronic ester 6-4 using standard boration reaction conditions well known to those skilled in the art. For example, compound 6-3 is metal-halogen exchanged with an organolithium reagent (such as n-butyllithium) followed by a trialkyl borate B (OR)3Treatment may provide a boronic ester 6-4, which boronic ester 6-4 may be hydrolysed to give the free boronic acid 6-4(R ═ H).
Scheme 6

Scheme 7 depicts a process that can be used to synthesize deuterated boronic acids or boronic acidsMethod of ester 7-4. In a solvent (such as toluene), in a base (such as NaHCO)3) Reaction of phenol 7-1(X ═ Br or I) with deuterated alkyl bromide 7-2 in the presence of a catalyst such as nickel (II) acetylacetonate can afford compound 7-3[ Hodous, b.l. U.S. patent application publication No. US2016/0031892, 2016/4/2/2016 @)]. The above patents are incorporated by reference herein in their entirety for all purposes. Compound 7-3 can be converted to boronic acid or boronic ester 7-4 using standard boration reaction conditions well known to those skilled in the art. For example, compound 7-3 is metal-halogen exchanged with an organolithium reagent (such as n-butyllithium) followed by a trialkyl borate B (OR)3Treatment may provide a boronic ester 7-4, which boronic ester 7-4 may be hydrolysed to give the free boronic acid 7-4(R ═ H).
Scheme 7
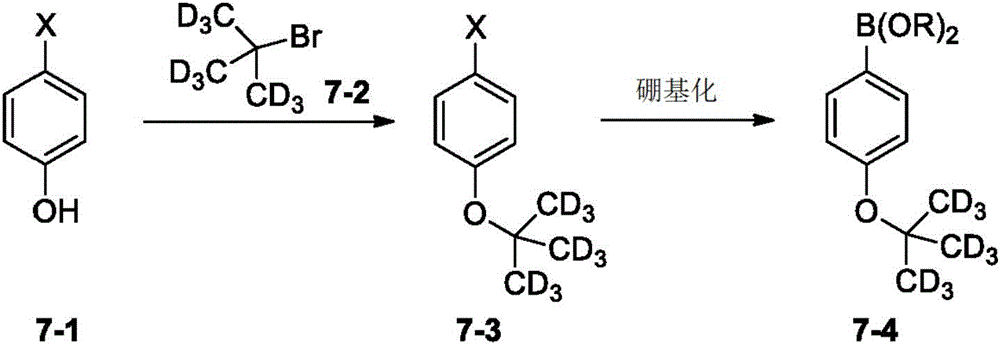
Scheme 8 depicts a method that can be used to synthesize deuterated boronic acids or boronic esters 8-4. Metal-halogen exchange of compound 8-1(X ═ Br or I) with an organolithium reagent such as n-butyllithium, followed by treatment with compound 8-2 in a solvent such as tetrahydrofuran, can afford compound 8-3. Compounds 8-3 can be converted to boronic acids or boronic esters 8-4 using standard boration reaction conditions well known to those skilled in the art. For example, coupling of compound 8-3 with a diboron-based reagent, such as bis (pinacolato) diboron, in a solvent, such as dioxane, in the presence of a base, such as potassium acetate, using a catalyst, such as [1, 1' -bis (diphenylphosphino) ferrocene ] palladium (II) dichloride, can provide boronic ester 8-4.
Scheme 8
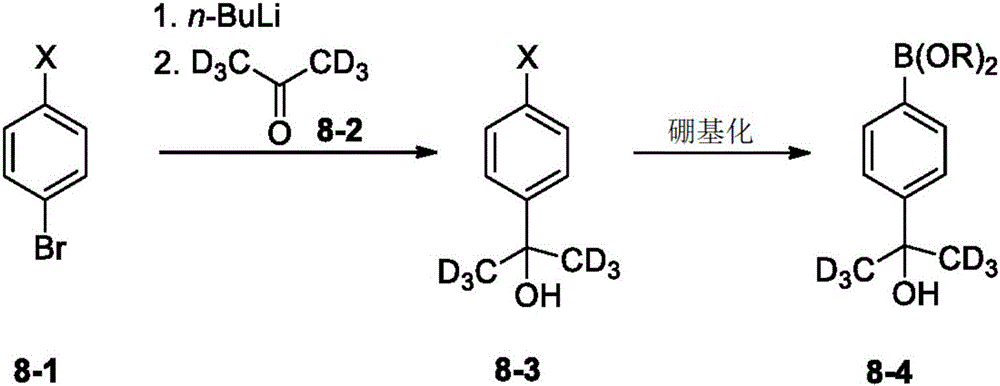
Examples
Preparation of the intermediates used in examples a1 to A3.
5-bromo-N1- (4-isopropoxyphenyl) -4-methylbenzene-1, 2-diamine
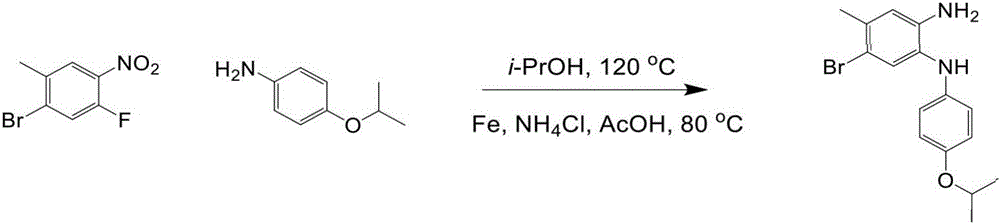
To a solution of 1-bromo-5-fluoro-2-methyl-4-nitrobenzene (200mg, 0.85mmol) in isopropanol (2mL) was added 4-isopropoxyaniline (129mg, 0.85 mmol). The resulting mixture was stirred at 120 ℃ for 30min under microwave irradiation. After cooling to room temperature, the reaction was concentrated under reduced pressure, and the residue was dissolved in ethanol (0.6mL), dioxane (0.6mL) and water (0.3 mL). To the solution were added iron (476mg, 8.5mmol) and NH4Cl (457mg, 8-5 mmol). The reaction was stirred at 80 ℃ for 2 hours. After cooling to room temperature, the reaction was filtered through a pad of celite. The filtrate was concentrated under reduced pressure, and the residue was poured into water and extracted with ethyl acetate. Passing the organic phase over Na2SO4Dried, filtered and concentrated in vacuo. Passing the residue through SiO2Purification by column chromatography (hexane/EtOAc ═ 3: 1) gave 198mg (69.2%) of the product as a white solid. LC/MS m/z: 335.13(79Br,M+H)+,337.19(81Br,M+H)+,376.28(79Br,M+H+CH3CN)+,378.25(81Br,M+H+CH3CN)+。
5-bromo-N1- (4- (tert-butoxy) phenyl) -4-methylbenzene-1, 2-diamine

With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described from 1-bromo-5-fluoro-2-methyl-4-nitrobenzene and 4- (tert-butoxy) aniline. LC/MS m/z: 351.24(79Br,M+H+CH3CN)+
6-bromo-5-methyl-1- [4- (prop-2-yloxy) phenyl ] -1H-1, 3-benzodiazole

To 5-bromo-N1To a solution of (4-isopropoxyphenyl) -4-methylbenzene-1, 2-diamine (50.1mg, 0.15mmol) in THF (1mL) was added trimethoxymethane (18.9mg, 0.18mmol), followed by addition of formic acid (100 uL). The resulting mixture was stirred at 80 ℃ for 2 hours. After cooling to room temperature, the reaction was poured into water and extracted with ethyl acetate. Passing the organic phase over Na2SO4Dried, filtered and concentrated in vacuo. Passing the residue through SiO2Purification by column chromatography (hexane/EtOAc 1: 1) gave 42.1mg (81.7%) of the product as a white solid. LC/MS m/z: 345.15(79Bt,M+H)+,347.21(81Br,M+H)+,386.28(79Br,M+H+CH3CN)+,388.20(81Br,M+H+CH3CN)+。
6-bromo-1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d ] imidazole

With a compound of formula (I) and (II) with a compound of formula (I) and (III) with a compound of formula (I) and (II) with a compound of formula (II) and (III)]-1H-1, 3-Benzodiazole the title compound was prepared from 5-bromo-N1- (4- (tert-butoxy) phenyl) -4-methylbenzene-1, 2-diamine in the same manner as described.1H NMR(500MHz,DMSO-d6)δ8.51(s,1H),7.77(s,1H),7.74(s,1H),7.57(d,2H),7.21(d,2H),2.47(s,3H),1.37(s,9H)。LC/MS m/z:359.16(79Br,M+H+CH3CN)+,361.17(81Br,M+H+CH3CN)+。
Example a 1: 2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d ] imidazol-6-yl) phenyl) propan-2-ol

To 6-bromo-1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]To a solution of imidazole (1g, 2.79mmo1) in 1, 4-dioxane (15mL) was added (4- (2-hydroxypropan-2-yl) phenyl) boronic acid (0.502g, 2.79mmol) and [1, 1' -bis (diphenylphosphino) ferrocene]Palladium (II) dichloride (230mg, 0.279mmol), potassium carbonate (1.15g, 8.4mmol) and water (5 mL). The resulting reaction mixture was degassed with nitrogen for 10min and then heated to 100 ℃ overnight. The reaction mixture was then diluted with ethyl acetate and washed with water. Passing the organic phase over Na2SO4Dried, filtered and concentrated in vacuo. Passing the residue through SiO2Purification by column chromatography (7: 3 to 1: 4 hexanes/EtOAc) provided 826mg of the product as a colorless oil.1H NMR(500MHz,DMSO-d6)δ8.48(s,1H),7.67(s,1H),7.58(d,2H),7.52(d,2H),7.31(s,1H),7.29(d,2H),7.17(d,2H),5.01(s,1H),2.32(s,3H),1.46(s,6H),1.34(d,9H)。LC/MS m/z:415.32(M+H)+
Examples a2 to A3 were prepared in the same manner as described above for 2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d ] imidazol-6-yl) phenyl) propan-2-ol (example a1) using the appropriate aryl halide described above and the appropriate commercially available boronic acid.


Preparation of the intermediates used in examples B4 to B9
6-bromoimidazo [1, 2-a ] pyridine-7-carbonitrile

To a solution of 2-amino-5-bromoisonicotinic carbonitrile (150mg, 0.76mmol) in i-PrOH (2mL) was added 0.6mL (1.5eq)2-chloro-1, 1-dimethoxyethane. The solution was capped and heated to 160 ℃ in a microwave reactor for 30 minutes. The mixture was cooled and evaporated in vacuo, the residue was dissolved in ethyl acetate and washed with saturated NaHCO3The aqueous solution is washed and evaporated in vacuo to give 0.47g of the title compound in a purity sufficient for further use. LC/MS m/z: 221.10(M + H)+
6-bromo-3-iodo-7-methylimidazo [1, 2-a ] pyridines
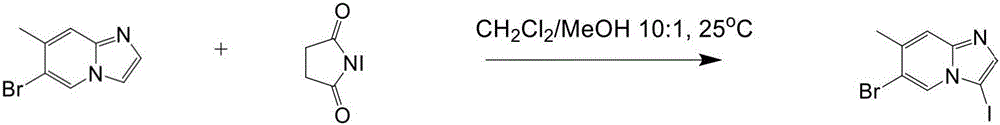
To 6-bromo-7-methylimidazo [1, 2-a ]]Pyridine (100mg, 0.47mmol) in CH2Cl2(1mL) to a solution was added 1-iodopyrrolidine-2, 5-dione (84mg, 0.47mmol) and MeOH (0.1 mL). The resulting mixture was stirred at room temperature for 2 h. The reaction was poured into water and extracted with ethyl acetate. Passing the organic phase over Na2SO4Dried, filtered and concentrated in vacuo. Passing the residue through SiO2Purification by column chromatography (hexane/EtOAc 1: 1) gave 122mg (77%) of the product as a white solid. LC/MS m/z: 337.00(M + H)+。
6-bromo-3-iodoimidazo [1, 2-a ] pyridine-7-carbonitrile

With a compound having a structure similar to that of 6-bromo-3-iodo-7-methylimidazo [1, 2-a ]]Pyridine is described in the same manner from 6-bromoimidazo [1, 2-a ]]Pyridine-7-carbonitrile and NIS the title compound was prepared. LC/MS m/z: 348.01(M + H)+。
6-bromo-3- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyridine
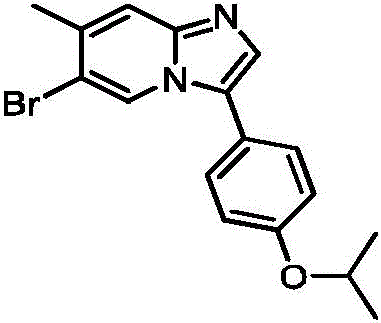
To react with 2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ 2 ], ]d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) was prepared in the same manner from 6-bromo-3-iodo-7-methylimidazo [1, 2-a ]]Pyridine and 4-Isopropoxybenzoic acid the title compound was prepared. LC/MSm/z: 345.20(M + H)+
6-bromo-3- (4- (tert-butoxy) phenyl) -7-methylimidazo [1, 2-a ] pyridine
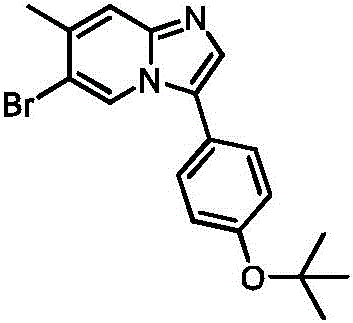
To react with p-2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) was prepared in the same manner from 6-bromo-3-iodo-7-methylimidazo [1, 2-a ]]Pyridine and (4- (tert-butoxy) phenyl) boronic acid the title compound was prepared. LC/MS m/z: 359.22(M + H)+
2- (4- (6-bromo-7-methylimidazo [1, 2-a ] pyridin-3-yl) phenyl) propan-2-ol
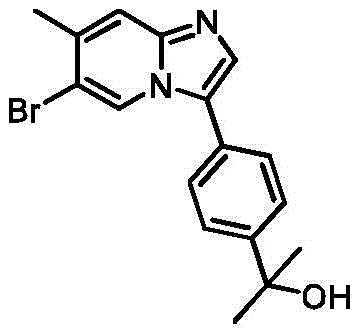
To react with p-2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) was prepared in the same manner from 6-bromo-3-iodo-7-methylimidazo [1, 2-a ]]Pyridine and (4- (2-hydroxyprop-2-yl) phenyl) boronic acid the title compound was prepared. LC/MS m/z: 345.10(M + H)+
Examples B4 to B9 were prepared in the same manner as described above for 2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d ] imidazol-6-yl) phenyl) propan-2-ol (example a1) using the appropriate aryl halide and a commercially available boronic acid. In the case of compounds having the same substituents of the aryl halide, 2 equivalents of boric acid are used.
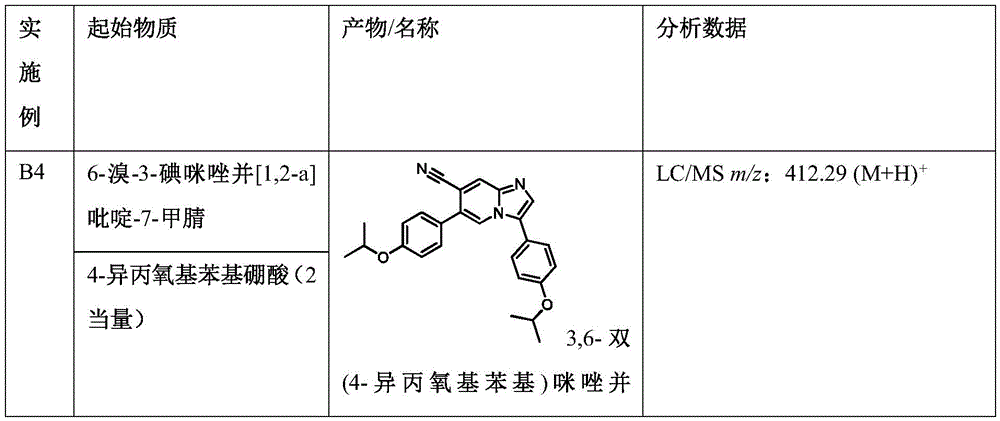


Preparation of intermediates used in examples C10 to C26
2-bromo-4- ((4-isopropoxyphenyl) amino) -5-nitrobenzoic acid methyl ester

With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described for 2-bromo-4-fluoro-5-nitrobenzoic acid methyl ester and 4-isopropoxyaniline. LC/MS m/z: 381.01(M + H)+,421.97(M+H+CH3CN)+
5-bromo-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine

With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described for 4-bromo-2-fluoro-1-nitrobenzene and 4-isopropoxyaniline. LCMS m/z: 321.20(79Br,M+H)+,323.19(81Br,M+H)+,362.20(79Br,M+H+CH3CN)+,364.24(81Br,M+H+CH3CN)+。
5-bromo-4-chloro-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine

With respect to 5-bromo-N1- (4-isopropoxyphenyl) -4-The title compound was prepared from 1-bromo-2-chloro-5-fluoro-4-nitrobenzene and 4-isopropoxyaniline in the same manner as described for methylbenzene-1, 2-diamine. LC/MS m/z: 355.05(79Br,M+H)+,357.12(81Br,M+H)+
5-bromo-6-fluoro-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine

With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described for 1-bromo-2, 3-difluoro-4-nitrobenzene and 4-isopropoxyaniline. LC/MSm/z: 339.13(79Br,M+H)+,341.27(81Br,M+H)+
5-bromo-4-fluoro-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine
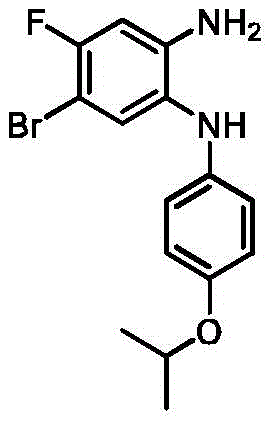
With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described for 1-bromo-2, 5-difluoro-4-nitrobenzene and 4-isopropoxyaniline. LC/MS m/z: 339.13(79Br,M+H)+,341.22(81Br,M+H)+
5-bromo-N1- (4-isopropoxyphenyl) -6-methylbenzene-1, 2-diamine
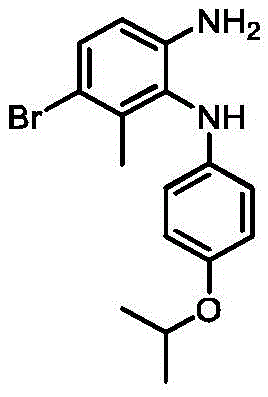
With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described from 1-bromo-3-fluoro-2-methyl-4-nitrobenzene and 4-isopropoxyaniline. LC/MSm/z: 335.19(M + H)+
5-bromo-N1- (4-isopropoxyphenyl) -4-methoxybenzene-1, 2-diamine
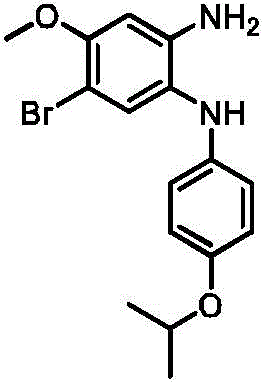
With respect to 5-bromo-N1- (4-Isopropoxyphenyl) -4-methylbenzene-1, 2-diamine the title compound was prepared in the same manner as described from 1-bromo-5-fluoro-2-methoxy-4-nitrobenzene and 4-isopropoxyaniline. LC/MSm/z: 418.30(M + H)+
6-bromo-1- [4- (prop-2-yloxy) phenyl ] -1H-1, 2, 3-benzotriazole

At 0 ℃ to 5-bromo-N1(4-Isopropoxyphenyl) benzene-1, 2-diamine (100mg, 0.3mmol) in acetic acid (3mL) was added PPh3(81.6mg, 0.3mmol) followed by the addition of nitrite (25.8mg, 0.36 mmol). The reaction was allowed to warm to room temperature and stirred for 1 h. The reaction was poured into water and extracted with ethyl acetate. Passing the organic phase over Na2SO4Dried, filtered and concentrated in vacuo. Passing the residue through SiO2Purification by column chromatography (hexane/EtOAc ═ 2: 1) gave 98mg (95%) of the product as a white solid. LC/MS m/z: 332.07(79Br,M+H)+,334.08(81Br,M+H)+,373.15(79Br,M+H+CH3CN)+,375.14(81Br,M+H+CH3CN)+。
6-bromo-1- (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole-5-carboxylic acid methyl ester

With a p-6-bromo-1- [4- (prop-2-yloxy) phenyl group]-1H-1, 2, 3-benzotriazole the title compound was prepared in the same manner as described for 2-bromo-4- ((4-isopropoxyphenyl) amino) -5-nitrobenzoic acid methyl ester, sodium nitrite and triphenylphosphine. LC/MS m/z: 390.17(79Br,M+H+CH3CN)+,392.16(81Br,M+H+CH3CN)+。
6-bromo-5-chloro-1- (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole
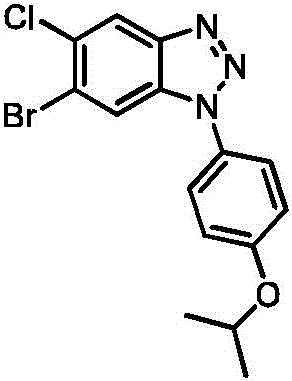
With a p-6-bromo-1- [4- (prop-2-yloxy) phenyl group]-1H-1, 2, 3-benzotriazole the title compound was prepared from 5-bromo-4-chloro-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine in the same manner as described. LC/MS m/z: 368.13(M + H)+,408.91(M+H+CH3CN)+
6-bromo-7-fluoro-1- (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole

With a p-6-bromo-1- [4- (prop-2-yloxy) phenyl group]-1H-1, 2, 3-benzotriazole the title compound was prepared from 5-bromo-6-fluoro-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine in the same manner as described.1H NMR(500MHz,DMSO-d6)δ7.98(d,1H),7.73-7.68(m,3H),7.16(d,2H),4.78-4.73(m,1H),1.34(d,6H)。LC/MS m/z:351.93(M+H)+
6-bromo-5-fluoro-1- (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole
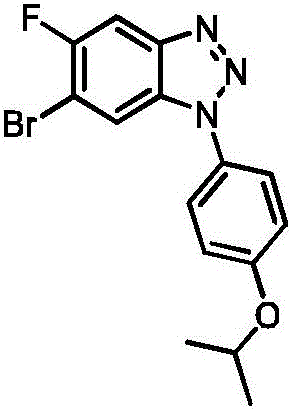
With a p-6-bromo-1- [4- (prop-2-yloxy) phenyl group]-1H-1, 2, 3-benzotriazole the title compound was prepared from 5-bromo-4-fluoro-N1- (4-isopropoxyphenyl) benzene-1, 2-diamine in the same manner as described. LC/MS m/z: 352.20(M + H)+,393.19(M+H+CH3CN)+
6-bromo-1- (4-isopropoxyphenyl) -7-methyl-1H-benzo [ d ] [1, 2, 3] triazole

With a p-6-bromo-1- [4- (prop-2-yloxy) phenyl group]-1H-1, 2, 3-benzotriazole the title compound was prepared from 5-bromo-N1- (4-isopropoxyphenyl) -6-methylbenzene-1, 2-diamine in the same manner as described. LC/MS m/z: 346.03(M + H)+
6-bromo-1- (4-isopropoxyphenyl) -5-methoxy-1H-benzo [ d ] [1, 2, 3] triazole
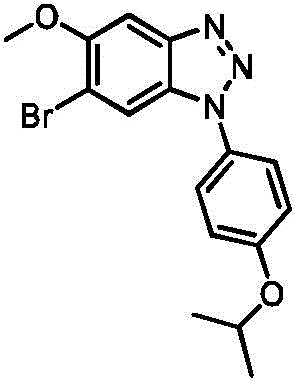
With a p-6-bromo-1- [4- (prop-2-yloxy) phenyl group]-1H-1, 2, 3-benzotriazole the title compound was prepared from 5-bromo-N1- (4-isopropoxyphenyl) -4-methoxybenzene-1, 2-diamine in the same manner as described. LC/MS m/z: 362.13(M + H)+
Examples C10 to C18 were prepared in the same manner as described above for 2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d ] imidazol-6-yl) phenyl) propan-2-ol (example a1) using the appropriate aryl halide described above and the appropriate commercially available boronic acid.

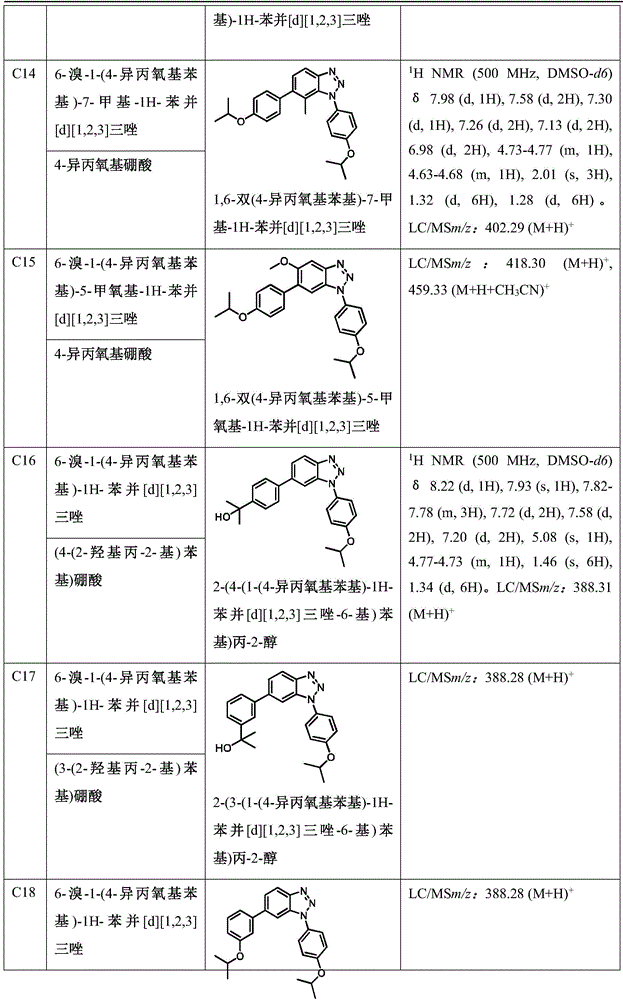

Example C19: (1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazol-5-yl) methanol

To 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ]][1,2,3]Triazole-5-carboxylic acid methyl ester (86mg, 1Eq) in THF (3mL) was added slowly to a 2.6M solution of lithium aluminum hydride in THF (80uL, 1 Eq). The mixture was stirred at room temperature for 3 hours and cooled saturated Na2SO4The solution was slowly quenched and filtered through a pad of celite. The filtrate was concentrated in vacuo and the residue was passed through SiO2Purification by column chromatography (hexane/EtOAc 7: 3 to 6: 4) gave 52mg of the title compound. LC/MS m/z: 418.31(M + H)+,835.60(2M+H)+
Example C20: 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole-5-carboxylic acid
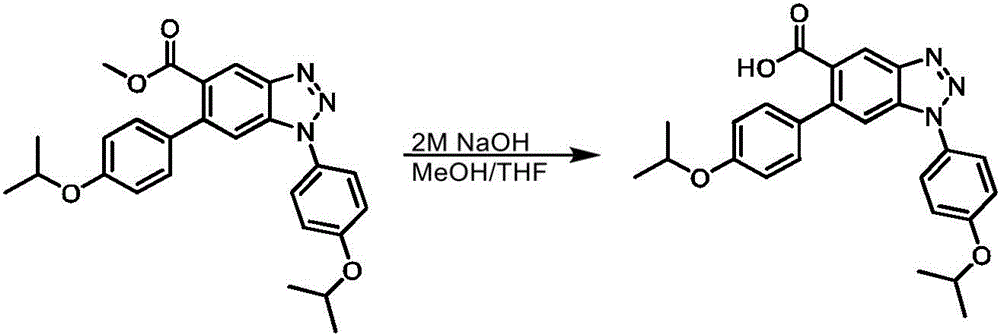
To 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ]][1,2,3]Triazole-5-carboxylic acid methyl ester (200mg, 1Eq) to a solution of 1: 1 MeOH/THF (8mL) was added 2M aqueous NaOH (4.25 mL). The mixture was stirred overnight, quenched by addition of 1M aqueous HCl, extracted with MTBE, and the organic phase evaporated in vacuo. Purification of the 10mg residue by preparative HPLC afforded 2.3mg of the title compound. LC/MS m/z: 432.35(M + H)+,473.25(M+H+CH3CN)+
Example C21: 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole-5-carboxamide

To 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ]][1,2,3]Triazole-5-carboxylic acid (9.5mg, 1Eq) to a solution in DMF (0.5mL) was added diisopropylethylamine (11uL, 3Eq) and HATU (12mg, 1.5 Eq). The mixture was stirred for 1 hour, at which time ammonium chloride (5mg, 4Eq) was added in one portion. The mixture was stirred overnightDiluted with ethyl acetate, washed twice with 1M aqueous HCl and evaporated in vacuo. The residue was purified by preparative HPLC to give 5mg of the title compound as a white solid. LC/MS m/z: 431.29(M + H)+,861.54(2M+H)+
Example C22: (1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazol-5-yl) methylamine
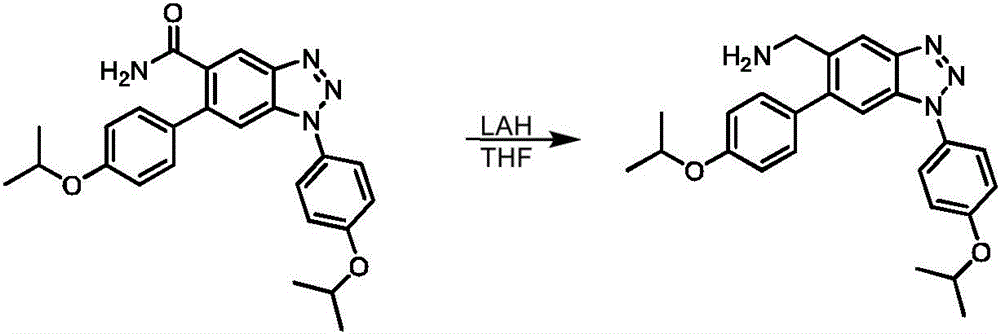
Stirring at reflux rather than room temperature to react with p- (1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d][1,2,3]Triazol-5-yl) methanol (example C19) in the same manner as described for 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ]][1,2,3]Triazole-5-carboxamide the title compound was prepared. LC/MS m/z: 417.41(M + H)+
Example C23: 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazol-5-amine
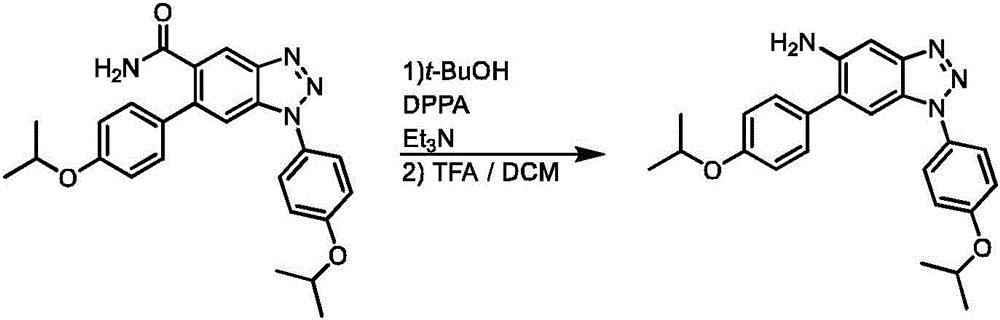
To 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ]][1,2,3]Triazole-5-carboxamide (51mg, 1Eq) to a solution of t-BuOH (0.5mL) was added triethylamine (33uL, 2Eq) and diphenylphosphoryl azide (25uL, 1 Eq). The mixture was heated to 85 ℃ and stirred for 6 hours, at which time it was diluted with ethyl acetate, saturated NH4Aqueous Cl and water were washed and evaporated in vacuo. The residue was extracted with DCM (1mL) and TFA (1mL) was added dropwise. The resulting solution was stirred overnight, then diluted with DCM and saturated NaHC03The aqueous solution was washed and evaporated in vacuo. Passing the residue through SiO2Purification by column chromatography (hexane/EtOAc ═ 7: 3 to 1: 2) gave 7mg of the title compound as a colourless oil. LC/MS m/z: 403.30(M + H)+,444.30(M+H+CH3CN)+
Example C24: 4, 4' - (5-methoxy-1H-benzo [ d ] [1, 2, 3] triazole-1, 6-diyl) biphenol
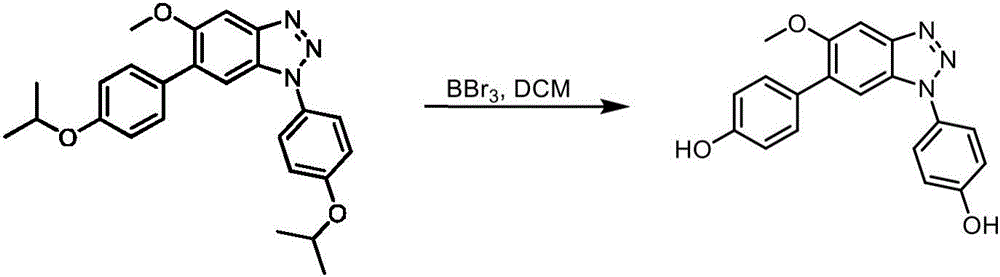
To 1, 6-bis (4-isopropoxyphenyl) -5-methoxy-1H-benzo [ d ] at 0 DEG C][1,2,3]Triazole (200mg, 0.5mmol) in DCM (4mL) was added BBr3(0.5mL, 0.5mmol) of a 1M solution. The mixture was allowed to warm to room temperature while stirring overnight, at which time it was quenched by pouring it onto ice, extracted twice with ethyl acetate, and evaporated in vacuo. The residue was purified by preparative HPLC to give 13mg of the title compound. LC/MS m/z: 334.27(M + H)+
Example C25: 1, 6-bis (4-isopropoxyphenyl) -5-vinyl-1H-benzo [ d ] [1, 2, 3] triazole
Step 1: 1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole-5-carbaldehyde

To (1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ]][1,2,3]To a solution of triazol-5-yl) methanol (30mg, 0.072mmol) was added DCM (0.5mL) and MnO2(12mg, 0.14 mmol). The mixture was stirred overnight, filtered through a pad of celite, and the filtrate was evaporated to give 24mg of the title compound without further purification. LC/MS m/z: 416.27(M + H)+
Step 2: 1, 6-bis (4-isopropoxyphenyl) -5-vinyl-1H-benzo [ d ] [1, 2, 3] triazole
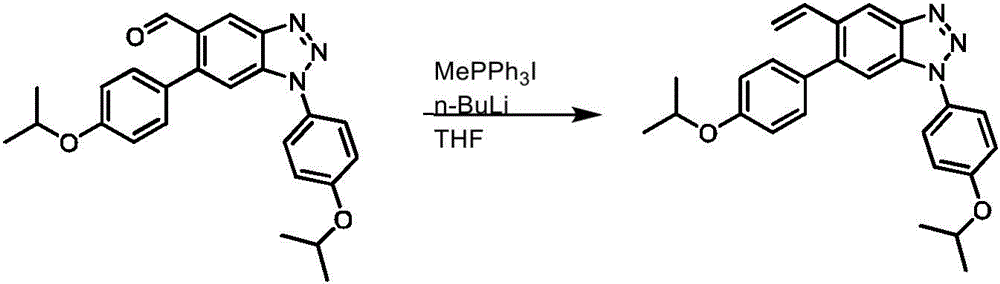
To a suspension of methyltriphenylphosphonium iodide (40mg, 0.1mmol) in THF (1mL) at 0 deg.C was added a 1.6M solution of n-butyllithium (0.06mL, 0.1 mmol). The mixture was stirred at 0 ℃ for 30 minutes, at which time 1, 6-bis (4-isopropoxyphenyl) -1 was addedH-benzo [ d ]][1,2,3]A solution of triazole-5-carbaldehyde (24mg, 0.057mmol) in THF (0.5mL) was stirred at room temperature for an additional 3 hours. The mixture is treated with NH4Aqueous Cl was quenched, extracted with ethyl acetate, and the organics evaporated in vacuo. The residue was purified by preparative HPLC to give 12.9mg of the title compound. LC/MS m/z: 414.29(M + H)+
Example C26: 5-Ethyl-1, 6-bis (4-isopropoxyphenyl) -1H-benzo [ d ] [1, 2, 3] triazole

Purge 1, 6-bis (4-isopropoxyphenyl) -5-vinyl-1H-benzo [ d ] by evacuation and backfilling with nitrogen gas twice][1,2,3]Air in a solution of triazole (11.5mg, 0.028mmol) in MeOH (0.5 mL). 10% palladium on carbon (5mg) was then added and the atmosphere replaced with hydrogen by pulling a vacuum and backfilling twice with a hydrogen balloon. The mixture was stirred overnight, diluted with ethyl acetate, filtered through a pad of celite, and the filtrate evaporated in vacuo to give 10mg of the title compound in sufficient purity to be used without further purification.1H NMR(500MHz,DMSO-d6)δ8.05(s,1H),7.74(d,2H),7.49(s,1H),7.29(d,2H),7.15(d,2H),6.98(d,2H),4.70-4.72(m,1H),4.65-4.67(m,1H),2.69-2.74(m,2H),1.31(d,6H),1.29(d,6H),1.08(t,3H)。LC/MS m/z:416.33(M+H)+
Example D27: 3, 6-bis (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidine
Step 1: 6-bromo-7-methylimidazo [1, 2-a ] pyrimidines

With respect to 6-bromoimidazo [1, 2-a ]]The title compound was prepared from 5-bromo-4-methylpyrimidin-2-amine in the same manner as described for pyridine-7-carbonitrile.1H NMR(500MHz,CDCl3)δ8.57(s,1H),7.85(s,1H),7.49(s,1H),2.79(s,3H)。LC/MS m/z:214.25(M+H)+
Step 2: 6- (4-Isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidine

To react with p-2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) in the same manner as described for 6-bromo-7-methylimidazo [1, 2-a]Pyrimidine and 4-isopropoxyphenylboronic acid the title compound was prepared. LC/MS m/z: 268.25(M + H)+
And step 3: 3-iodo-6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidine

The title compound was prepared from 6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidine and NIS in the same manner as described for 6-bromo-3-iodo-7-methylimidazo [1, 2-a ] pyridine. LC/MS m/z: 394.23
And 4, step 4: 3, 6-bis (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidine

To react with p-2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) was prepared in the same manner from 3-iodo-6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ]]Pyrimidine and 4-isopropoxyphenylboronic acid the title compound was prepared.1H NMR(500MHz,CDCl3)δ8.31(s,1H),7.74(s,1H),7.40(d,2H),7.22(d,2H),6.98(d,2H),6.96(d,2H),4.66-4.56(m,2H),2.55(s,3H),1.38(d,6H),1.36(d,6H)。LC/MS m/z:402.36(M+H)+
Example D28: 3- (4- (tert-butoxy) phenyl) -6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidine

To react with p-2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) was prepared in the same manner from 3-iodo-6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ]]Pyrimidine and 4-tert-butoxyphenylboronic acid the title compound was prepared. LC/MS m/z: 416.35(M + H)+
Example D29: 2- (4- (6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ] pyrimidin-3-yl) phenyl) propan-2-ol
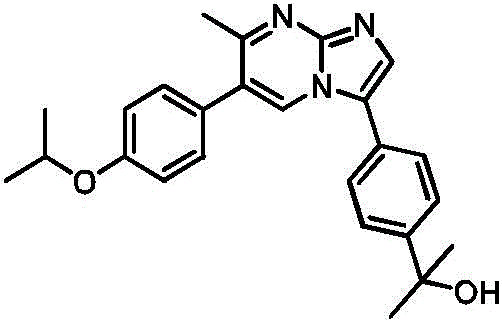
To react with p-2- (4- (1- (4- (tert-butoxy) phenyl) -5-methyl-1H-benzo [ d]Imidazol-6-yl) phenyl) propan-2-ol (example A1) was prepared in the same manner from 3-iodo-6- (4-isopropoxyphenyl) -7-methylimidazo [1, 2-a ]]Pyrimidine and (4- (2-hydroxyprop-2-yl) phenyl) boronic acid the title compound was prepared. LC/MS m/z: 402.39(M + H)+
Example E30: 3- (4- (tert-butoxy) phenyl) -6- (4- (isopropoxy-d)7) Phenyl) -7-methylimidazo [1, 2-a]Pyridine compound
Step 1: 4- (3- (4- (tert-butoxy) phenyl) -7-methylimidazo [1, 2-a ] pyridin-6-yl) phenol
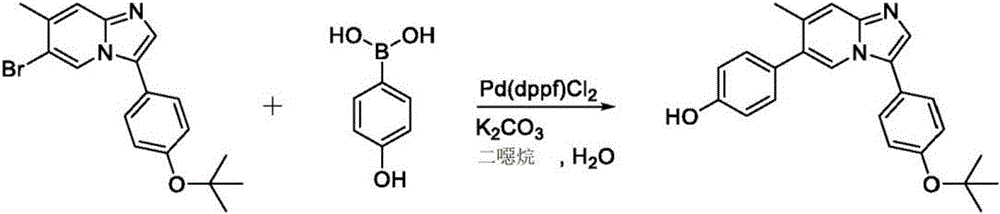
Addition of 6-bromo-3- (4- (tert-butoxy) phenyl) -7-methylimidazo [1, 2-a ] to a microwave reactor vial]Pyridine (25mg, 0.07mmol), 4-hydroxyphenylboronic acid (11mg,. 083mmol, 1.2Eq), Pd (dppf) Cl2(6mg, 10 mol%) and potassium carbonate (30mg,. 210mmol, 3 Eq). Then 1.5mL of 1, 4-dioxane and 0.5mL of water were added and the reaction was run throughThe mixture was degassed by bubbling nitrogen gas for 5 minutes. The vial was then capped and subjected to microwave radiation at 80 ℃ for 1 hour. The resulting mixture was diluted with ethyl acetate, and the aqueous layer was extracted 3 times with ethyl acetate, followed by Na2SO4Dried and evaporated to yield the crude product. The crude material was purified using silica gel flash chromatography with 100% ethyl acetate as eluent to give 20mg of the title compound as a light brown oil. LC/MS m/z: 373.35(M + H)+
Step 2: 3- (4- (tert-butoxy) phenyl) -6- (4- (isopropoxy-d)7) Phenyl) -7-methylimidazo [1, 2-a]Pyridine compound
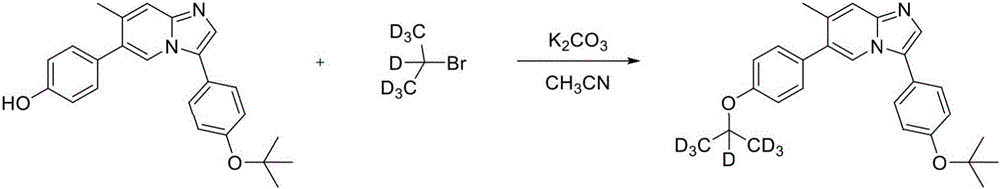
To 4- (3- (4- (tert-butoxy) phenyl) -7-methylimidazo [1, 2-a)]To a solution of pyridin-6-yl) phenol (142mg, 0.38mmol) in acetonitrile was added potassium carbonate (0.105g, 0.76mmol, 2 Eq). The mixture was stirred at reflux for 30 minutes at which time d 7-isopropyl bromide (57uL, 1.5Eq) was added in one portion. The mixture was stirred at reflux overnight and then evaporated to dryness. The residue was dissolved in ethyl acetate, washed twice with water and Na2SO4Drying and evaporation gave a crude solid which was purified using silica gel flash chromatography with 3: 7 hexane: ethyl acetate as eluent to give 42mg of the title compound. LC/MS m/z: 422.33(M + H)+
The following examples can be prepared according to schemes 2, 3, 6-8 and procedures above, using the appropriate starting materials.
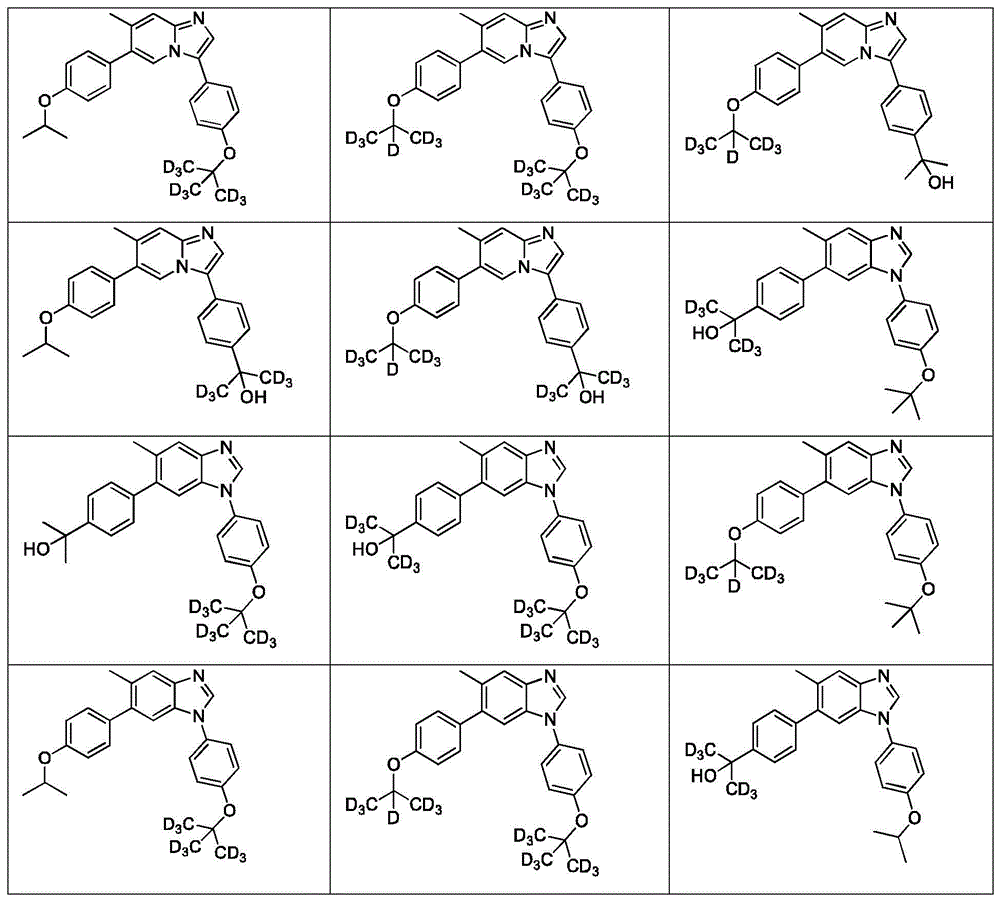

Arenavirus GP pseudotype assay.
The heterocyclic compounds were screened using the VSV pseudotype system expressing arenavirus glycoproteins (pseudotype viruses referred to herein as LASV-p, MACV-p, JUNV-p, GTOV-p, and TCRV-p) and Renilla luciferase reporter gene to identify individual compounds that inhibit the infectivity of the pseudotype virus but not the native VSV virus expressing the VSV glycoprotein. VSV viruses expressing or pseudotyped with LASV, MACV, JUNV, GTOV, and TCRV glycoproteins (LASV-p, MACV-p, JUNV-p, GTOV-p, and TCRV-p) were produced in cultured HEK-293T cells (ATCC CRL-3216) cultured in DMEM supplemented with 10% FBS, 1X penicillin-streptomycin, non-essential amino acids, and L-glutamine in 10cm dishes. When the cells reached approximately 80% confluence, they were transfected with a mixture of 15 μ g of pCAGGS plasmid encoding the desired glycoprotein and 45 μ l of PEI (polyethyleneimine) transfection reagent (PEI MAX, Polysciences inc., # 24765). Cells were incubated at 37 ℃ in 5% CO2Incubate with solution for 5 hours, then wash, and replace the mixture with supplemented DMEM and 5% CO at 37 ℃2The incubation was continued for about 16-18 hours. Subsequently, the cells were infected with approximately 50ul of VSV reporter virus, thereby replacing the VSV glycoprotein with the luciferase reporter gene. Cells were infected for 1 hour, then washed 1 time with PBS and incubated in supplemented media. 24 hours post infection, supernatants were collected, clarified by centrifugation and filtered through a 0.45um filter, aliquoted and stored at-80 ℃. Both VSV-luciferase and arenavirus glycoprotein pseudotypes were titrated for luminescent activity in Vero cells as described in the luciferase assay protocol (below). Vero cells (ATCC: CCL-81) were grown in supplemented DMEM medium in transparent 384-well plates (3000 cells/well). At 37 ℃ and 5% CO2After overnight incubation, cells were treated with the desired concentrations of compound and pseudotype virus in the assay medium. Assay medium consisted of 50% Opti-MEM, 50% DMEM, 1% FBS, penicillin-streptomycin, non-essential amino acids and L-glutamine. The resulting supernatants from each virus were diluted (from 1: 100 to 1: 2000) to yield similar luminescence signal/background values ≧ 200. Final DMSO concentration in compound test wells remained ≦ 1%, and control wells were incubated with assayNutrient and 1% DMSO treatment. Cells were incubated at 37 ℃ and 5% CO2Incubate for 24 hours. Compound-virus mixtures were aspirated from the cells 24 hours after infection and washed 1 time with PBS. Cells were then lysed using 20 μ l of lysis buffer from the luciferase kit diluted according to the manufacturer's instructions. After approximately 20 minutes of incubation, 5 μ l of cell lysate was transferred to an opaque white plate and mixed with 12.5ul coelenterazine diluted in buffer. This mixture was incubated at room temperature on a plate shaker for 10 minutes and the luminescence was then read using a plate reader (Beckman Coulter DTX 880 multimode detector with 535nm emission). Luminescence signals were obtained for compound-containing wells and control wells to determine the% activity (inhibition of luciferase signal) of each compound.
Cytotoxicity screening
The cytotoxicity of the active compounds in the pseudotype assay over a3 day period was also evaluated. Compounds were serially diluted and added to Vero cells (4000 cells/well) with the final DMSO concentration maintained at 1% in growth medium consisting of Minimum Essential Medium (MEM) and 1% FBS. The plates were incubated at 37 ℃ for 3 days, and then dead cells were removed by washing with Phosphate Buffered Saline (PBS). CPE was evaluated by: cells were stained with neutral red dye for 1 hour and then destained with 50% ethanol/1% acetic acid solution. The absorbance was read at 540nm and 690nm on a Spectramax Plus 384 spectrophotometer. Data were analyzed as (540nm-690nm) and then compared to untreated controls to obtain% cell viability.
Replicative LASV inhibitory Activity plaque assay
Confluent or near confluent cell culture monolayers were prepared in 12-well disposable cell culture plates. Cells were maintained in MEM or DMEM supplemented with 10% FBS. For antiviral assays, the same medium was used, but FBS was reduced to 2% or less and supplemented with 1% penicillin/streptomycin. Test compounds were prepared in 2 × MEM or 2 × DMEM at seven half-log 10 final concentrations (01-10 μ M). Test compound and positive control compound (Favipiravir or ribavirin)) Runs were performed in biological triplicate. The assay was initiated by first removing the growth medium from 12-well plates of cells, which were challenged with a given concentration of compound and 0.01 MOI of virus or approximately 50 to 100 plaque forming units (pfu). Cells were incubated for 60 min: 100 μ L inoculum/well at 37 ℃ 5% CO2Next, with constant gentle shaking. Virus inoculum was removed, cells were washed and overlaid with 1% agarose or 1% methylcellulose diluted 1: 1 with 2X MEM and supplemented with 2% FBS and 1% penicillin/streptomycin and corresponding drug concentrations. Cells were incubated at 37 ℃ and 5% CO2Incubate for 5 days. The cover was then removed and the plates were stained with 0.05% crystal violet in 10% buffered formalin for approximately 20 minutes at room temperature. Plates were washed, dried and plaque numbers were counted. Plaque numbers in each group of compound dilutions were converted to percentages relative to untreated viral controls. Then 50% Effective (EC) was calculated by linear regression analysis50Viral inhibitory) concentration. CC (challenge collapsar)50Divided by EC50The quotient of (a) gives the selectivity index (Si) value. Compounds exhibiting a SI value of > 10 are considered active.
Replication-competent LASV Virus production reduction assay
The VYR test is a direct measure of the concentration of test compound that inhibits viral replication. Compounds and virus were added to Vero cells for 3-4 days, at which time the supernatant was removed and tested for infectious particles. Supernatants were titrated with log10 dilutions of virus using 3 or 4 microwells per dilution on fresh monolayers of Vero cells in 96-well plates. After significant CPE was observed, wells were scored for the presence or absence of virus. Inhibitor concentrations were related to log of virus produced at each concentration10The plot allows calculation of 90% effective concentration by linear regression. In addition, compounds were run in parallel on Vero cells at different concentrations in the absence of virus to determine cytotoxic CC50The value is obtained. Selectivity Index (SI) calculated as CC50/EC90The ratio of.
Replication-competent Takaliba virus assay
Use ofSelected compounds were tested against native replication competent Tacarib (TCRV) virus (TRVL-11573, BEI source) based on ELISA assay. Vero cells (ATCC: CCL-81) were grown in 96-well format (5000 cells/well) in supplemented DMEM medium. After overnight incubation, cells were treated with TCRV and the desired concentration of compound in MEM medium with 1% FBS and supplements. The final DMSO concentration in the compound test wells remained ≦ 1%, and the control wells were treated with TCRV or media and 1% DMSO. At 5% CO2After 5 days of incubation at 37 ℃, cells were fixed with 2% paraformaldehyde for 45 minutes and then washed with PBS. Subsequently, the cells were permeabilized with 0.25% triton-X, and then assayed for TCRV using ELISA using the following protocol. Cells were stained with monoclonal anti-hunin virus antibody (BEI # NR 41860) cross-reactive with TCRV nucleoprotein. After washing, cells were treated with biotin-conjugated secondary antibodies and subsequently with streptavidin-conjugated horseradish peroxidase. TMB substrate was added to the wells and the reaction was stopped using 2M sulfuric acid. The absorbance was read using a plate reader (Beckman Coulter DTX 880 multimode detector with an emission of 450 nm). OD readings were obtained for compound-containing wells and control wells to determine% activity of each compound.
Microparticle assay
In addition to the ability of compounds to exhibit broad inhibitory activity against arenaviruses in vitro, compounds must also possess certain drug-like properties in order for them to be useful in inhibiting arenaviruses and to provide a method of treating arenavirus infections in mammals. Such compounds may exhibit drug-like Properties including, but not limited to, chemical stability against metabolic degradation by liver microsomal CYP p450 enzymes, cell permeability and oral bioavailability (if the drug is to be delivered orally), and no inhibition of the hERG ion channel (which is associated with cardiac safety) [ Kerns, e.h.li, d.drug-like Properties: concept, Structure Design and Methods from ADME to sensitivity Optimization, (2008) Academic Press, Burlington MA ]. The above publications are incorporated herein by reference for all purposes. To characterize the drug-like properties of the chemical series, the exemplified compounds were evaluated for metabolic stability (table 4) and inhibition of the hERG ion channel (table 5) in human, mouse, guinea pig, monkey, rat, mouse or dog liver microsomal assays. Compounds that exhibit > 60% parent residue indicate attractive chemical stability. Exhibiting good microsomal stability in human and non-human species facilitates the ability to test and optimize compounds in preclinical animal studies.
A reaction premix was set up containing 1uM of the compound of interest, 1mg/mL of liver microsomes of the desired species, 2.1mM MgCl2And 0.1M sodium phosphate buffer (pH 7.4). This pre-mix was incubated at 37 ℃ for 30 minutes with gentle stirring to dissolve the compound completely in the mixture. Then, a freshly prepared NADPH solution in 0.1M sodium phosphate buffer was added at a concentration of 2mM to start the reaction. Immediately after NADPH addition, a "time 0" sample (30uL) was taken and added to 140uL of cold acetonitrile containing 1uM of a predetermined internal standard. The remaining reaction mixture was incubated at 37 ℃ for the remaining period of time. The test compound was left in the reaction mixture for 60 minutes, and then the "time 60" sample was added to acetonitrile with an internal standard. Control compounds (Verapamil) for human, monkey and dog LM, Lidocaine (Lidocaine) for guinea pig LM and diphenhydramine (diphenhydramine) for rat and mouse LM) were incubated in the reaction mixture for 15 minutes, then "time 15" samples were collected and added to the cold acetonitrile containing the internal standard. The sample was then centrifuged at 4000rpm for 10 minutes in a centrifuge and the supernatant collected and mixed with an aliquot of distilled water. These were then analyzed on a Varian 500-MS.
hERG channel assay
Drugs belonging to different classes have been shown to be associated with QT prolongation and in some cases severe ventricular arrhythmias. The most common mechanism of these adverse events is the inhibition of one or more cardiac potassium channels, particularly hERG. This current is important for myocardial cell repolarization and is a common target for drugs that prolong the QT interval. Thus, the test preparations in this study were characterized to determine their ability to inhibit the hERG channel. Use the steady commentaries on classicsStained Chinese Hamster Ovary (CHO) cell lines expressing hERG mRNA measure ion channel activity. The pharmacology of this clonal channel expressed in the CHO cell line was very similar to that observed in native tissues. Cells were cultured in DMEM/F12 containing 10% FBS, 1% penicillin/streptomycin and 500. mu.g/ml G418. Prior to testing, cells were harvested using accumax (innovative Cell technologies). For electrophysiological recording, the following solutions were used: external solution: 2mM CaCl 2; 2mM MgCl 2; 4mM KCl; 150mM NaCl; 10mM glucose; 10mM HEPES; 305-315 mOsm; pH 7.4 (adjusted with 5M NaOH); internal solution: 140mM KCl; 10mM MgCl 2; 6mM EGTA; 5mM HEPES-Na; 5mM ATP-Mg; 295-305 mOsm; pH 7.25 (adjusted with 1M KOH). Sealchip with AVIVATMTechniques were performed for whole cell recordings using PX 7000a (axon instruments). Cells were clamped at a holding potential of-80 mV. Then, through the depolarization step hERG current activation to-50 mV, lasting 300 ms. This first step at-50 mV is used as a baseline to measure the peak amplitude of the tail current. Next, a voltage step of +20mV was applied for 5s to activate the channel. Finally, a step back to-50 mV for 5s removed the activation and the inactivated tail current was recorded. An external solution containing 0.1% DMSO (vehicle) was applied to the cells to establish a baseline. After allowing the current to stabilize for 3 to 10min, the test article was applied. The test article solution was added to the cells in 4 separate additions. The cells were kept in the test solution until the effect of the test article reached a steady state, up to 12 min. Next, 1 μ M cisapride (positive control) was added. Finally, washing off is carried out with an external solution until the recovery current reaches a steady state. Data analysis was performed using DataXpress (axon instruments), Clampfit (axon instruments), and origin (originLab corporation) software.
TABLE 1 pseudotyped viral activity. The example compounds and their observed inhibitory activity, shown as EC against LASV-p, MACV-p, JUNV-p, TCRV-p and GTOV-p50Values (VSV-p EC50 values all > 10uM) and CC for cytotoxicity50(ii) a nd: it is not determined.
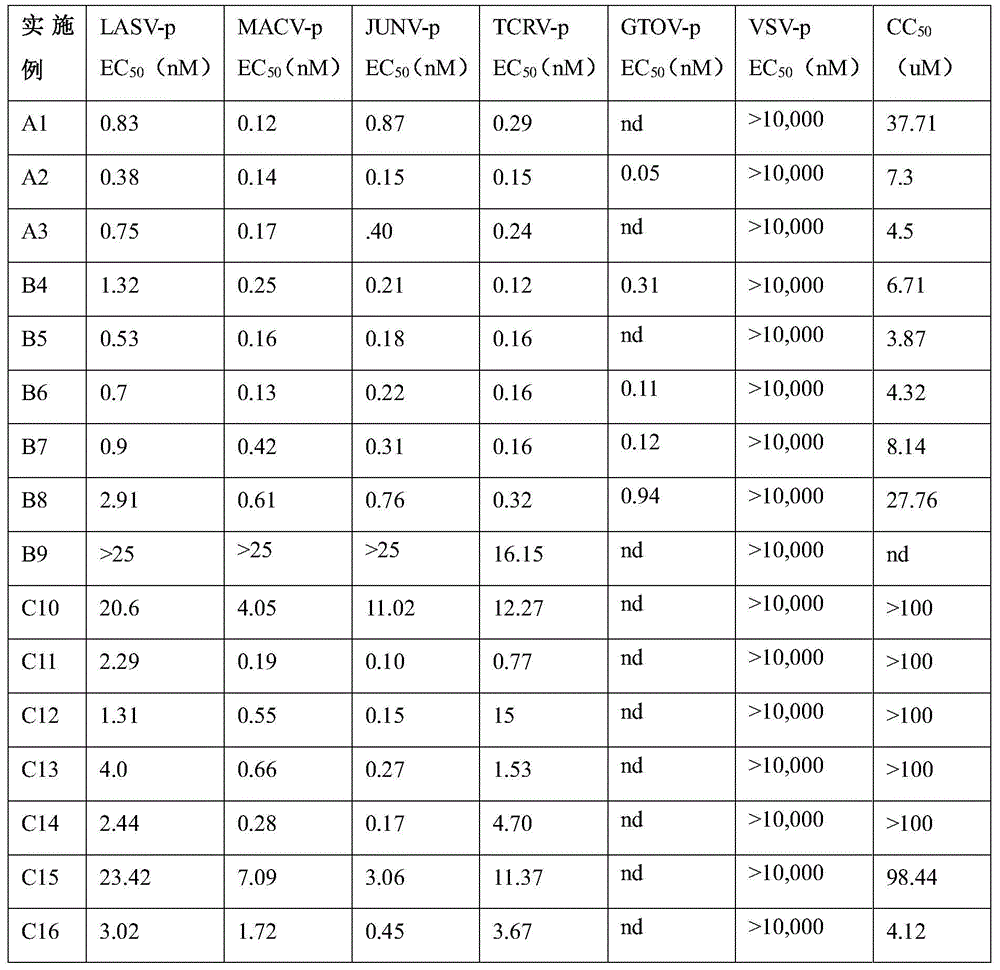

TABLE 2 comparison of inhibitory activity of pseudotyped versus replicative TCRV. The compounds of the examples and their observed inhibitory activity (EC) on pseudotyped or replicated TCRV50)。
| Examples | TCRV-p EC50(nM) | TCRV EC50(nM) |
| A1 | 0.24 | 0.89 |
| A2 | 0.12 | 0.33 |
| A3 | 0.21 | 0.87 |
| B4 | 0.16 | 0.74 |
| B5 | 0.10 | 0.88 |
| B6 | 0.10 | 0.95 |
| B7 | 0.20 | 0.74 |
| B8 | 0.32 | 2.16 |
| C11 | 0.61 | 0.84 |
| E30 | 0.2 | nd |
Surprisingly, a very close correlation between pseudotype and replicative virus inhibitory activity of the compounds of the present invention was found.
TABLE 3 inhibition of native Lassa virus. The compounds of the examples and their inhibitory activity and Selectivity Index (SI) observed in replicative LASV plaque and virus production reduction (VYR) assays.
| Examples | Plaque assay EC50(uM) | VYR determination of EC90(uM) | SI90 |
| B8 | <.003 | <0.003 | >9,000 |
| B7 | <.003 | <0.001 | >11,000 |
| A1 | <.003 | <0.001 | >33,000 |
| A2 | <.003 | <0.001 | >13,000 |
| E30 | nd | <0.014 | >1,510 |
Of all five compounds, compounds A1, A2, B7 and B8 exhibited very potent EC of less than 1-3nM in plaque and VYR assay formats50And EC90And compound E30 exhibited an EC of less than 14nM in the VYR assay format90。SI90Values (obtained from the VYR assay data) > 1510 clearly indicate that compound efficacy is due to antiviral activity and not cytotoxic effects. TABLE 1The results shown in-3 confirm the activity of the compounds against arenaviruses (including replicative LASV) and also strongly demonstrate the method of identifying true HF arenavirus inhibitors by using pseudotyped viral assays.
Table 4 multi-species microsomal stability. The parent compound remains% at 60min in liver microsomes
| Examples | Mouse | Monkey | Dog | Human being | Guinea pig | Rat |
| A1 | 82.7 | 8.6 | >95 | 68.3 | >95 | 93.8 |
| A2 | 93.9 | 53 | >95 | 95 | >95 | >95 |
| A3 | 25.4 | nd | nd | 89.9 | 67.6 | nd |
| B4 | 37.5 | 4.9 | 83.3 | 88.1 | 55.45 | 70.6 |
| B5 | 16.4 | nd | nd | 86.7 | 7.4 | nd |
| B6 | 81.7 | nd | nd | 81.5 | 61.5 | nd |
| B7 | 92.9 | 22.9 | >95 | >95 | >95 | >95 |
| B8 | 77.5 | 33.7 | 77.57 | 87.6 | 67.7 | 93 |
| C11 | >95 | nd | nd | >95 | >95 | nd |
| C12 | 90.3 | nd | nd | >95 | >95 | nd |
| C13 | nd | nd | nd | 79.5 | nd | nd |
| C14 | >95 | nd | nd | >95 | >95 | nd |
| C16 | 57.4 | nd | nd | 72.3 | 61.9 | nd |
| E30 | >95 | 74.4 | >95 | >95 | >95 | >95 |
The results of the multi-species microsomal stability study (table 4) show that deuterated compound E30 exhibits improved metabolic stability in the monkey liver microsomal assay compared to its non-deuterated analog B7, and thus good microsomal stability in both human and non-human species.
Table 5: hERG channel assay
| Examples | Inhibition at 3 uM% |
| A1 | <10 |
| B7 | <10 |
| B8 | <10 |
| C11 | <10 |
These data indicate no hERG channel inhibition, indicating good cardiac safety potential.
Table 6: mouse pharmacokinetic parameters

The compounds were administered in mice by intravenous (3mg/kg) and oral (30mg/kg) routes to determine pharmacokinetic parameters. IV time points included 0.083, 0.25, 0.5, 1, 2, 6 and 24h, and oral time points included 0.5, 1, 2, 4, 6, 8 and 24 h. Blood was drawn from 3 mice at each time point. Plasma was separated and measured by LC/MS/MS on a Varian 500-LC/MS. Both compounds exhibited low first-pass liver clearance, which is consistent with high levels of compound remaining after 1 hour in mouse liver microsomes (table 4). Both compounds exhibit reasonable oral bioavailability and long half-lives suitable for once-a-day administration. Finally, the distribution volume (Vd) values indicate that the compound is absorbed into the tissue, which further supports good oral biodistribution targeting arenavirus infection.
Mice are able to tolerate two compounds given orally daily for 3 days to at least 100mg/kg once daily (highest dose tested). There were no clinical signs of apparent toxicity as determined by daily monitoring of body weight, body temperature and behavior. On day 4 (24 hours after the last dose), plasma and liver samples were collected from the animals administered to measure compound levels. The liver was homogenized in 1: 1w/v phosphate buffered saline. Plasma and liver extracts were measured by LC/MS/MS on a Varian 500-MS (Table 7).
Table 7: compound concentration 24 hours after final application
| Examples | 24hr plasma concentration (ug/mL) | 24hr liver concentration (ug/g liver) |
| B7 | 9.1 | 63.4 |
| A1 | 8.8 | 240.3 |
In summary, the results show that the compounds of the invention exhibit potent broad-spectrum inhibition of HF arenavirus, as well as attractive drug-like properties for use as a treatment for viral infections mediated by arenavirus glycoproteins.
Claims (21)
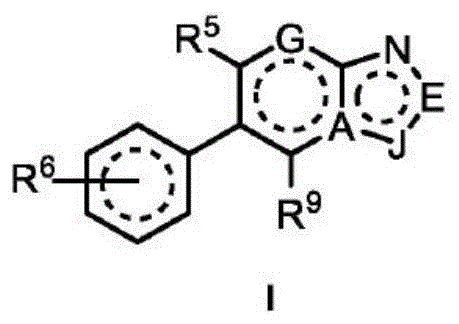
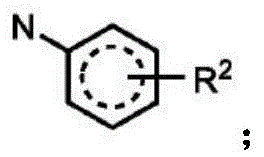
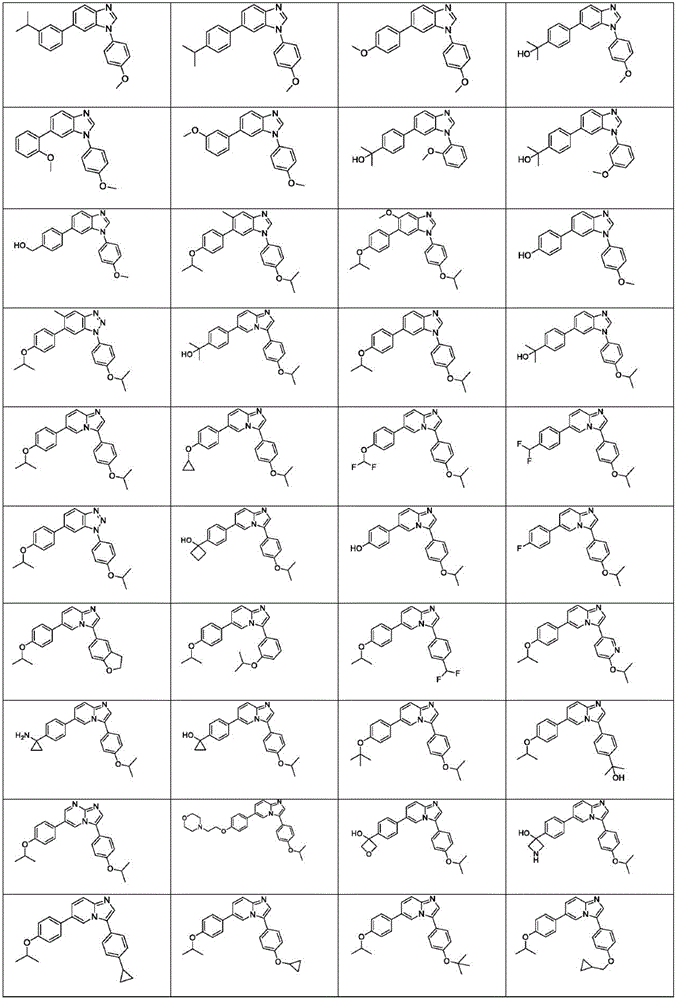



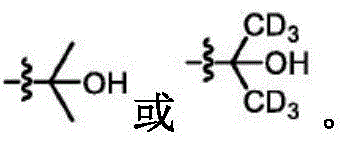

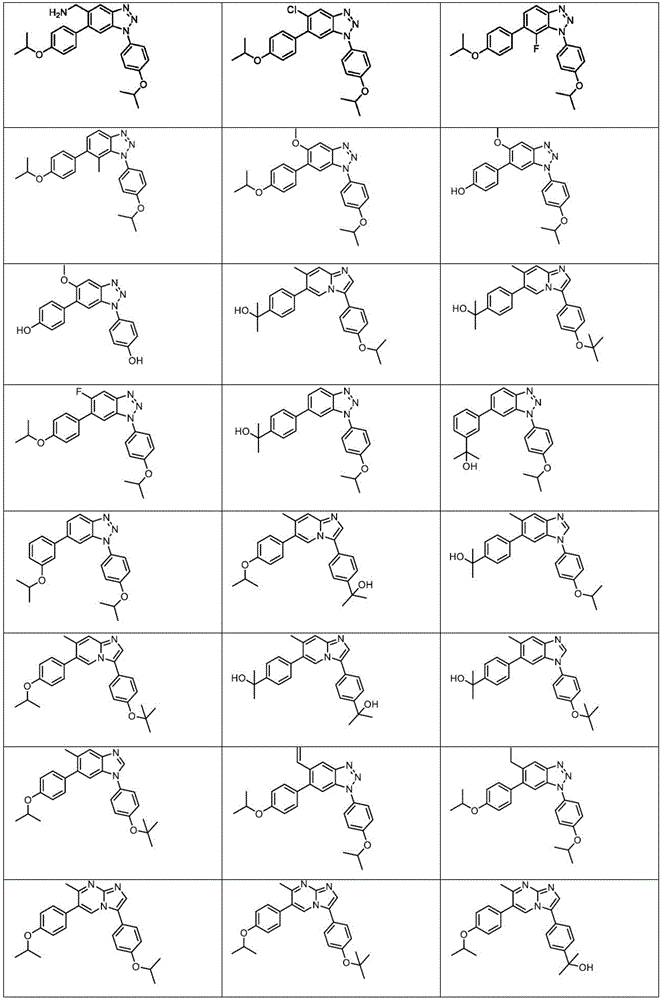
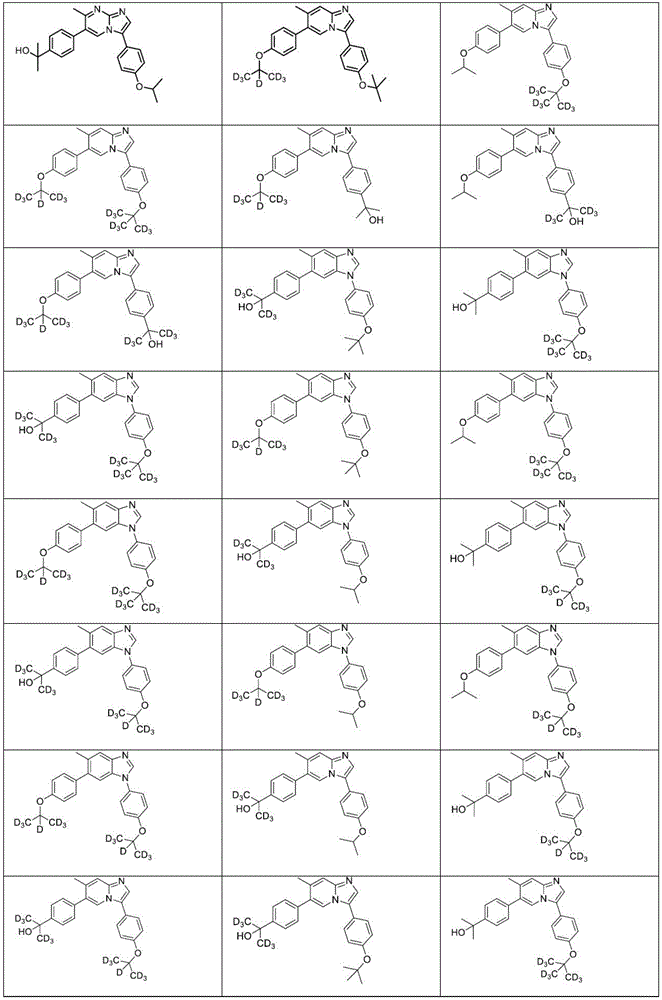
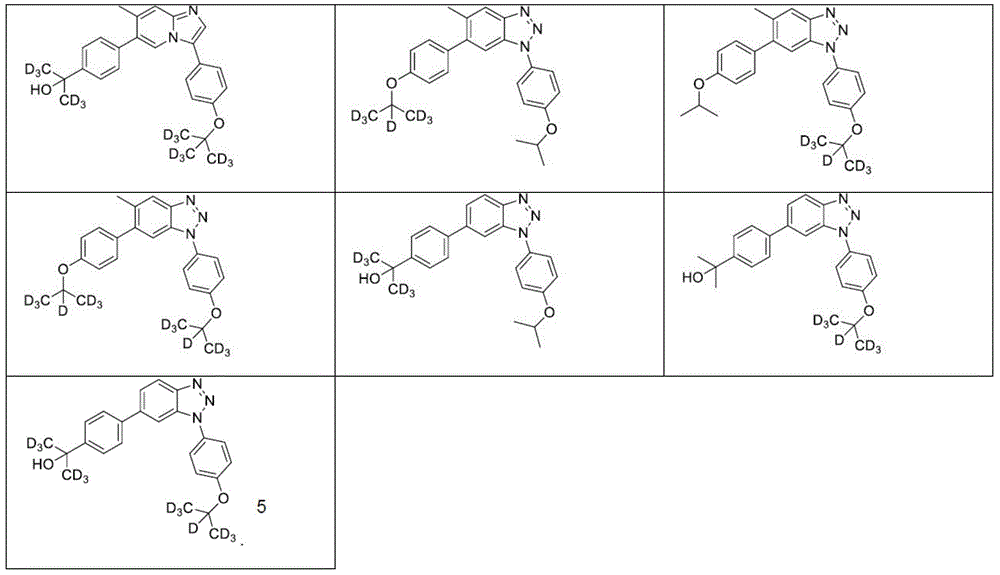
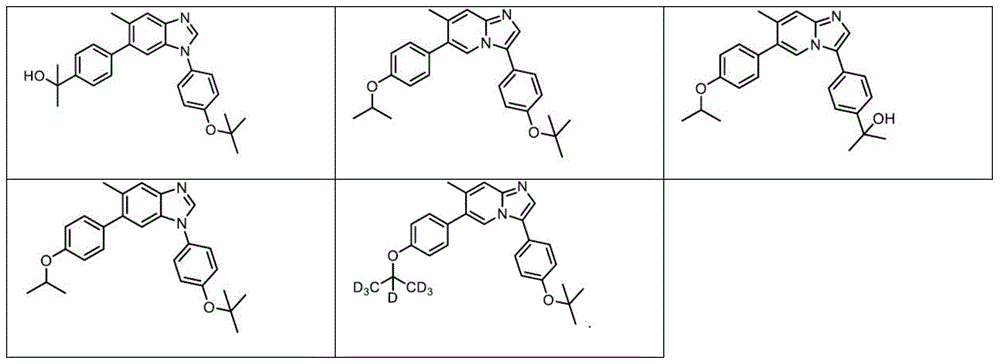


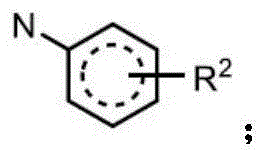


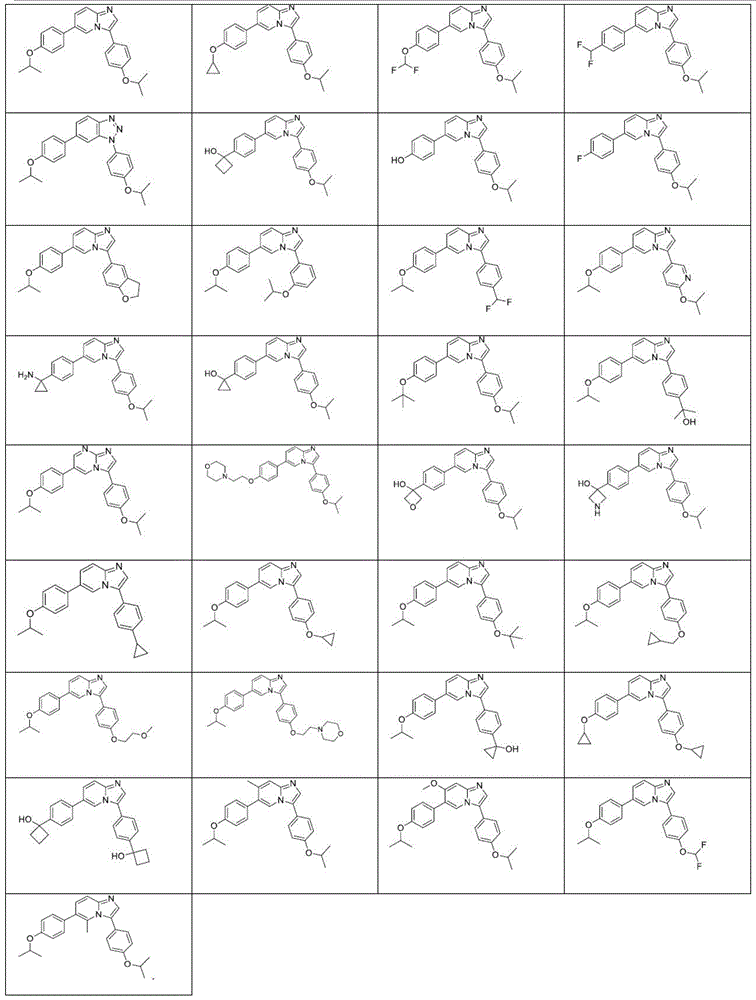
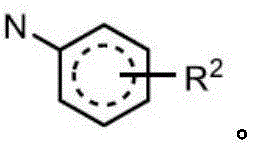

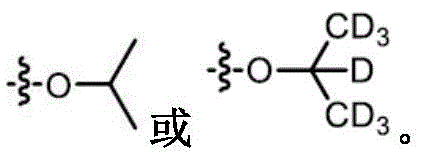


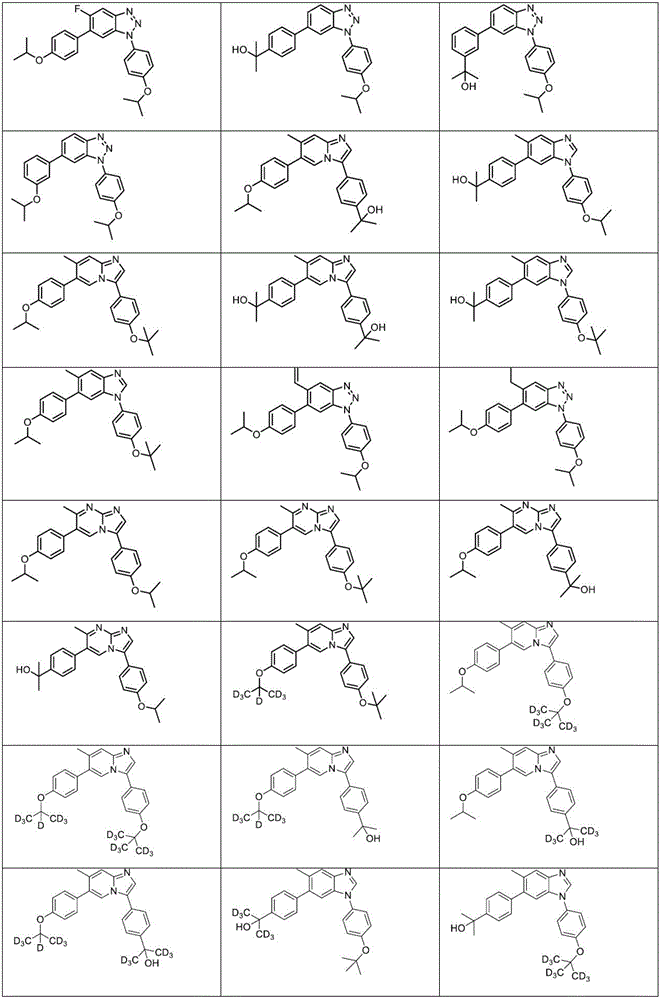

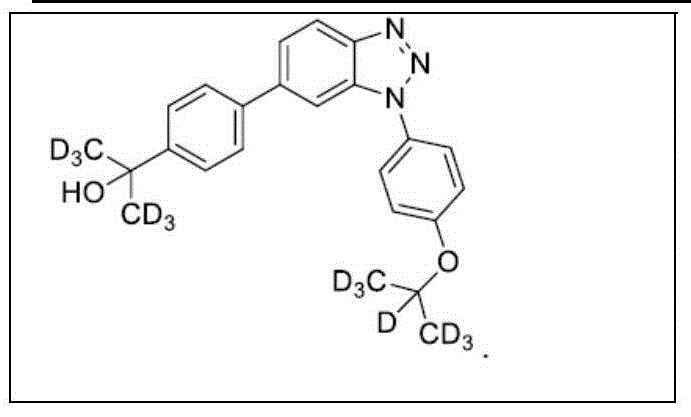

Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862776390P | 2018-12-06 | 2018-12-06 | |
| US62/776,390 | 2018-12-06 | ||
| PCT/US2019/064223 WO2020117794A1 (en) | 2018-12-06 | 2019-12-03 | Compounds for the treatment of arenavirus infection |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN113329750A true CN113329750A (en) | 2021-08-31 |
| CN113329750B CN113329750B (en) | 2025-10-17 |
Family
ID=70975517
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980080594.2A Active CN113329750B (en) | 2018-12-06 | 2019-12-03 | Compounds for the treatment of arenavirus infections |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP3890731A4 (en) |
| JP (1) | JP7675009B2 (en) |
| KR (1) | KR20210106997A (en) |
| CN (1) | CN113329750B (en) |
| AR (1) | AR117228A1 (en) |
| AU (1) | AU2019393784B2 (en) |
| BR (1) | BR112021010909A2 (en) |
| EA (1) | EA202191001A1 (en) |
| MX (2) | MX2021005234A (en) |
| WO (1) | WO2020117794A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116217490A (en) * | 2023-02-03 | 2023-06-06 | 中国人民解放军军事科学院军事医学研究院 | A kind of benzimidazole compound and its preparation method and application |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4434972A1 (en) * | 2023-03-22 | 2024-09-25 | Eberhard-Karls-Universität Tübingen | Atm kinase inhibitors |
| WO2025137345A1 (en) * | 2023-12-20 | 2025-06-26 | Arisan Therapeutics Inc. | Crystalline forms of an imidazopyridine derivative |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104507473A (en) * | 2012-02-17 | 2015-04-08 | 奇尼塔四有限责任公司 | Antiviral drugs used to treat arenavirus infection |
| WO2018013430A2 (en) * | 2016-07-12 | 2018-01-18 | Arisan Therapeutics Inc. | Heterocyclic compounds for the treatment of arenavirus infection |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE372966T1 (en) * | 1994-03-25 | 2007-09-15 | Isotechnika Inc | IMPROVED EFFECTIVENESS OF DRUGS THROUGH DEUTERATION |
| WO2012065297A1 (en) * | 2010-11-16 | 2012-05-24 | Impact Therapeutics, Inc. | 3-ARYL-6-ARYL-[1,2,4]TRIAZOLO[4,3-a]PYRIDINES AS INHIBITORS OF CELL PROLIFERATION AND THE USE THEREOF |
| US9688680B2 (en) | 2014-08-04 | 2017-06-27 | Blueprint Medicines Corporation | Compositions useful for treating disorders related to kit |
-
2019
- 2019-12-02 AR ARP190103517A patent/AR117228A1/en unknown
- 2019-12-03 EA EA202191001A patent/EA202191001A1/en unknown
- 2019-12-03 BR BR112021010909-9A patent/BR112021010909A2/en unknown
- 2019-12-03 KR KR1020217017726A patent/KR20210106997A/en not_active Ceased
- 2019-12-03 JP JP2021525159A patent/JP7675009B2/en active Active
- 2019-12-03 WO PCT/US2019/064223 patent/WO2020117794A1/en not_active Ceased
- 2019-12-03 EP EP19892030.8A patent/EP3890731A4/en active Pending
- 2019-12-03 AU AU2019393784A patent/AU2019393784B2/en active Active
- 2019-12-03 MX MX2021005234A patent/MX2021005234A/en unknown
- 2019-12-03 CN CN201980080594.2A patent/CN113329750B/en active Active
-
2021
- 2021-05-04 MX MX2024013042A patent/MX2024013042A/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104507473A (en) * | 2012-02-17 | 2015-04-08 | 奇尼塔四有限责任公司 | Antiviral drugs used to treat arenavirus infection |
| WO2018013430A2 (en) * | 2016-07-12 | 2018-01-18 | Arisan Therapeutics Inc. | Heterocyclic compounds for the treatment of arenavirus infection |
Non-Patent Citations (1)
| Title |
|---|
| PLEWE MB,等: "Discovery of a novel highly potent broad-spectrum heterocyclic chemical series of arenavirus cell entry inhibitors", BIOORG MED CHEM LETT, vol. 41, 5 May 2021 (2021-05-05), pages 127983, XP086566088, DOI: 10.1016/j.bmcl.2021.127983 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116217490A (en) * | 2023-02-03 | 2023-06-06 | 中国人民解放军军事科学院军事医学研究院 | A kind of benzimidazole compound and its preparation method and application |
| CN116217490B (en) * | 2023-02-03 | 2025-09-26 | 中国人民解放军军事科学院军事医学研究院 | A benzimidazole compound and its preparation method and application |
Also Published As
| Publication number | Publication date |
|---|---|
| AR117228A1 (en) | 2021-07-21 |
| EA202191001A1 (en) | 2021-08-16 |
| MX2021005234A (en) | 2021-08-11 |
| EP3890731A4 (en) | 2022-08-17 |
| KR20210106997A (en) | 2021-08-31 |
| JP7675009B2 (en) | 2025-05-12 |
| AU2019393784A1 (en) | 2021-05-27 |
| CA3118765A1 (en) | 2020-06-11 |
| AU2019393784B2 (en) | 2025-06-12 |
| EP3890731A1 (en) | 2021-10-13 |
| JP2022509763A (en) | 2022-01-24 |
| CN113329750B (en) | 2025-10-17 |
| BR112021010909A2 (en) | 2021-08-24 |
| WO2020117794A1 (en) | 2020-06-11 |
| MX2024013042A (en) | 2024-12-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11352328B2 (en) | Heterocyclic compounds for the treatment of arenavirus | |
| AU2018331172B2 (en) | Influenza virus replication inhibitor and use thereof | |
| CN112940010B (en) | Substituted polycyclic pyridone derivatives and prodrugs thereof | |
| CN113004304B (en) | Substituted polycyclic pyridone derivatives and prodrugs thereof | |
| CN102164924A (en) | 3H-imidaz0 [4, 5-B] pyridine- 6 -carboxamides as anti -inflammatory agents | |
| CN113329750A (en) | Compounds for the treatment of arenavirus infections | |
| TW202334123A (en) | Compounds, conjugates and methods thereof for preventing and treating coronavirus infection | |
| JP6859358B2 (en) | Tetrahydroindazole and its medical use | |
| JP2025503995A (en) | Compounds and their medical uses | |
| US12419865B2 (en) | Compounds for the treatment of arenavirus infection | |
| CN114466848A (en) | Antibacterial compounds | |
| KR102650441B1 (en) | Internally cyclic sulfiamidine amide-aryl amide compounds and their use for the treatment of hepatitis B | |
| JP2022550784A (en) | 4-quinolinone antibacterial compounds | |
| CA3118765C (en) | Compounds for the treatment of arenavirus infection | |
| EA044865B1 (en) | COMPOUNDS FOR TREATING ARENAVIRUS INFECTION | |
| KR20250116664A (en) | TYK2 inhibitor | |
| OA20266A (en) | ||
| WO2021194954A1 (en) | Substituted azole dione compounds with antiviral activity | |
| WO2021262426A9 (en) | Adamantane amides and thioamides for the treatment of ebolavirus infection | |
| ES2829254T3 (en) | Pyrrolopyrimidine nucleosides and analogs thereof useful as antiviral agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| TG01 | Patent term adjustment |