CN112876503B - Borate compound for cancer boron neutron capture therapeutic drug and preparation thereof - Google Patents
Borate compound for cancer boron neutron capture therapeutic drug and preparation thereof Download PDFInfo
- Publication number
- CN112876503B CN112876503B CN202110289828.3A CN202110289828A CN112876503B CN 112876503 B CN112876503 B CN 112876503B CN 202110289828 A CN202110289828 A CN 202110289828A CN 112876503 B CN112876503 B CN 112876503B
- Authority
- CN
- China
- Prior art keywords
- reaction
- borate compound
- preparation
- sodium
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 Borate compound Chemical class 0.000 title claims abstract description 90
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 title claims abstract description 33
- 229910052796 boron Inorganic materials 0.000 title claims abstract description 31
- 238000002360 preparation method Methods 0.000 title claims abstract description 31
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 23
- 201000011510 cancer Diseases 0.000 title claims abstract description 17
- 229940126585 therapeutic drug Drugs 0.000 title claims abstract description 9
- 238000006243 chemical reaction Methods 0.000 claims description 97
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 45
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 42
- 239000007787 solid Substances 0.000 claims description 26
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 24
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 15
- 239000003153 chemical reaction reagent Substances 0.000 claims description 15
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 12
- 239000011734 sodium Substances 0.000 claims description 12
- 229910052708 sodium Inorganic materials 0.000 claims description 12
- AKQNYQDSIDKVJZ-UHFFFAOYSA-N triphenylsilane Chemical compound C1=CC=CC=C1[SiH](C=1C=CC=CC=1)C1=CC=CC=C1 AKQNYQDSIDKVJZ-UHFFFAOYSA-N 0.000 claims description 12
- 239000004327 boric acid Substances 0.000 claims description 11
- 230000000087 stabilizing effect Effects 0.000 claims description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 9
- 239000007810 chemical reaction solvent Substances 0.000 claims description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 6
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 claims description 6
- XBHPFCIWRHJDCP-UHFFFAOYSA-N (2-trimethylsilylphenyl) trifluoromethanesulfonate Chemical compound C[Si](C)(C)C1=CC=CC=C1OS(=O)(=O)C(F)(F)F XBHPFCIWRHJDCP-UHFFFAOYSA-N 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- WWLVEMOKDAVHDW-UHFFFAOYSA-N C(CO)O.[Na].[Na] Chemical compound C(CO)O.[Na].[Na] WWLVEMOKDAVHDW-UHFFFAOYSA-N 0.000 claims description 3
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 3
- UCXUKTLCVSGCNR-UHFFFAOYSA-N diethylsilane Chemical compound CC[SiH2]CC UCXUKTLCVSGCNR-UHFFFAOYSA-N 0.000 claims description 3
- VDCSGNNYCFPWFK-UHFFFAOYSA-N diphenylsilane Chemical compound C=1C=CC=CC=1[SiH2]C1=CC=CC=C1 VDCSGNNYCFPWFK-UHFFFAOYSA-N 0.000 claims description 3
- NEXSMEBSBIABKL-UHFFFAOYSA-N hexamethyldisilane Chemical compound C[Si](C)(C)[Si](C)(C)C NEXSMEBSBIABKL-UHFFFAOYSA-N 0.000 claims description 3
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 claims description 3
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- PARWUHTVGZSQPD-UHFFFAOYSA-N phenylsilane Chemical compound [SiH3]C1=CC=CC=C1 PARWUHTVGZSQPD-UHFFFAOYSA-N 0.000 claims description 3
- AMCPECLBZPXAPB-UHFFFAOYSA-N propane-1,2,3-triol;sodium Chemical compound [Na].OCC(O)CO AMCPECLBZPXAPB-UHFFFAOYSA-N 0.000 claims description 3
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 claims description 3
- 159000000000 sodium salts Chemical class 0.000 claims description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 3
- WVMSIBFANXCZKT-UHFFFAOYSA-N triethyl(hydroxy)silane Chemical compound CC[Si](O)(CC)CC WVMSIBFANXCZKT-UHFFFAOYSA-N 0.000 claims description 3
- AAPLIUHOKVUFCC-UHFFFAOYSA-N trimethylsilanol Chemical compound C[Si](C)(C)O AAPLIUHOKVUFCC-UHFFFAOYSA-N 0.000 claims description 3
- 239000008096 xylene Substances 0.000 claims description 3
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 claims description 2
- 229910000077 silane Inorganic materials 0.000 claims description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims 1
- 150000001408 amides Chemical class 0.000 claims 1
- ZMHATUZXFSOVSC-UHFFFAOYSA-N triphenyl(triphenylsilyl)silane Chemical compound C1=CC=CC=C1[Si]([Si](C=1C=CC=CC=1)(C=1C=CC=CC=1)C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 ZMHATUZXFSOVSC-UHFFFAOYSA-N 0.000 claims 1
- 125000003118 aryl group Chemical group 0.000 abstract description 8
- 201000010536 head and neck cancer Diseases 0.000 abstract description 5
- 208000014829 head and neck neoplasm Diseases 0.000 abstract description 5
- 239000002253 acid Substances 0.000 abstract description 4
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 abstract description 3
- 125000000217 alkyl group Chemical group 0.000 abstract description 3
- 229910052710 silicon Inorganic materials 0.000 abstract 1
- 239000010703 silicon Substances 0.000 abstract 1
- 239000000047 product Substances 0.000 description 36
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 24
- 238000000034 method Methods 0.000 description 24
- 210000004027 cell Anatomy 0.000 description 19
- 239000012298 atmosphere Substances 0.000 description 18
- 239000007795 chemical reaction product Substances 0.000 description 18
- 239000011521 glass Substances 0.000 description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 14
- 239000003814 drug Substances 0.000 description 13
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 12
- 229940079593 drug Drugs 0.000 description 11
- 150000001642 boronic acid derivatives Chemical class 0.000 description 10
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 9
- 230000000694 effects Effects 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 6
- NFIVJOSXJDORSP-QMMMGPOBSA-N (2s)-2-amino-3-(4-boronophenyl)propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(B(O)O)C=C1 NFIVJOSXJDORSP-QMMMGPOBSA-N 0.000 description 5
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 210000004881 tumor cell Anatomy 0.000 description 4
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- 229940126062 Compound A Drugs 0.000 description 3
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 108010059712 Pronase Proteins 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 150000001543 aryl boronic acids Chemical class 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 125000005620 boronic acid group Chemical class 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 125000006165 cyclic alkyl group Chemical group 0.000 description 2
- 125000006575 electron-withdrawing group Chemical group 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- 238000003760 magnetic stirring Methods 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical group 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- GOXICVKOZJFRMB-UHFFFAOYSA-N (3-phenylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(C=2C=CC=CC=2)=C1 GOXICVKOZJFRMB-UHFFFAOYSA-N 0.000 description 1
- UGZUUTHZEATQAM-UHFFFAOYSA-N (4-butylphenyl)boronic acid Chemical compound CCCCC1=CC=C(B(O)O)C=C1 UGZUUTHZEATQAM-UHFFFAOYSA-N 0.000 description 1
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 1
- SJGGDZCTGBKBCK-UHFFFAOYSA-N 3-acetylphenylboronic acid Chemical compound CC(=O)C1=CC=CC(B(O)O)=C1 SJGGDZCTGBKBCK-UHFFFAOYSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-BJUDXGSMSA-N Boron-10 Chemical compound [10B] ZOXJGFHDIHLPTG-BJUDXGSMSA-N 0.000 description 1
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical class [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 1
- 101710088172 HTH-type transcriptional regulator RipA Proteins 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- YAWNFGAHEUCLNJ-UHFFFAOYSA-N [B].OB(O)O Chemical compound [B].OB(O)O YAWNFGAHEUCLNJ-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- PKWBMOXZIMVOJT-UHFFFAOYSA-N anthracen-2-ylboronic acid Chemical compound C1=CC=CC2=CC3=CC(B(O)O)=CC=C3C=C21 PKWBMOXZIMVOJT-UHFFFAOYSA-N 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical class OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- URSLCTBXQMKCFE-UHFFFAOYSA-N dihydrogenborate Chemical compound OB(O)[O-] URSLCTBXQMKCFE-UHFFFAOYSA-N 0.000 description 1
- PZPGRFITIJYNEJ-UHFFFAOYSA-N disilane Chemical compound [SiH3][SiH3] PZPGRFITIJYNEJ-UHFFFAOYSA-N 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000004992 fission Effects 0.000 description 1
- 238000000799 fluorescence microscopy Methods 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 238000001095 inductively coupled plasma mass spectrometry Methods 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- KTMKRRPZPWUYKK-UHFFFAOYSA-N methylboronic acid Chemical compound CB(O)O KTMKRRPZPWUYKK-UHFFFAOYSA-N 0.000 description 1
- HUMMCEUVDBVXTQ-UHFFFAOYSA-N naphthalen-1-ylboronic acid Chemical compound C1=CC=C2C(B(O)O)=CC=CC2=C1 HUMMCEUVDBVXTQ-UHFFFAOYSA-N 0.000 description 1
- 238000009205 neutron capture therapy of cancer Methods 0.000 description 1
- RQJRLVCBMAKXFG-UHFFFAOYSA-N phenyl(silyl)silane Chemical compound [SiH3][SiH2]C1=CC=CC=C1 RQJRLVCBMAKXFG-UHFFFAOYSA-N 0.000 description 1
- 150000002993 phenylalanine derivatives Chemical class 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000010814 radioimmunoprecipitation assay Methods 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon or a metal, e.g. chelates or vitamin B12
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
Abstract

Description
技术领域technical field
本发明涉及药物化学和辐射医学领域,尤其涉及用于癌症硼中子俘获治疗药物的硼酸盐化合物及其制备。The present invention relates to the field of medicinal chemistry and radiation medicine, in particular to borate compounds used for cancer boron neutron capture therapeutic drugs and their preparation.
背景技术Background technique
头颈癌是一种发病率高的癌症,治疗难度大,死亡率高,对其进行根除是人类医学上面临的巨大挑战。近年来,随着国内外药物研发的进程不断推进,人们在抗癌药物开发方面已经取得了长足的进展,但仍亟需新的、更有效的癌症治疗方法。Head and neck cancer is a kind of cancer with high incidence, difficult to treat and high mortality. Its eradication is a huge challenge for human medicine. In recent years, with the continuous advancement of domestic and foreign drug research and development, people have made great progress in the development of anticancer drugs, but there is still an urgent need for new and more effective cancer treatment methods.
硼中子俘获疗法(BNCT)是一种靶向肿瘤细胞的新型放疗技术,BNCT是一种基于核俘获和裂变反应的二元方法,与传统的放射疗法和化学疗法相比具有明显的低副作用、高选择性、高效率的优势。当用低能的热/超热中子照射非放射性10B原子时,会产生高线性能量转移(LET)α粒子和7Li反冲核。这些高LET粒子行程为5~9 μm,与一个细胞直径相近,从而仅选择性与含有10B分子的肿瘤细胞发生强烈热作用,杀死癌细胞的同时不对周围的正常细胞造成过度损害。Boron neutron capture therapy (BNCT) is a novel radiotherapy technique targeting tumor cells. BNCT is a binary approach based on nuclear capture and fission reactions with significantly lower side effects compared to conventional radiotherapy and chemotherapy. , high selectivity and high efficiency. When non-radioactive 10 B atoms are irradiated with low-energy thermal/epithermal neutrons, highly linear energy transfer (LET) alpha particles and 7 Li recoil nuclei are produced. These high-LET particles have a length of 5–9 μm, which is similar to the diameter of a cell, and thus selectively generate strong thermal effects only with tumor cells containing 10 B molecules, killing cancer cells without causing excessive damage to surrounding normal cells.
目前,临床上已将巯基十二硼烷二钠盐(BSH)和(L)-4-二羟基硼基苯丙氨酸(BPA)用于BNCT治疗肿瘤的硼药[专利公布号CN 111971563 A和CN 111204736 A];其中,BPA于2020年3月在日本获批上市成为全球首个获批上市的BNCT硼药。但BSH与BPA在BNCT临床研究中,均发现存在肿瘤靶向性不足的问题。对硼酸及硼烷进行化学结构修饰是目前主要的研究策略,但是效率仍需要进一步提升。专利申请号201980041340.X提出一种聚集性硼10药剂可以用于硼中子俘获疗法中,并能够在短时间内选择性地或局部地靶向肿瘤组织,具有副作用小、侵袭性低且可以对肿瘤组织发挥局部的杀伤效果的优点。中国专利202011268512.8公布了碳硼烷基塞来昔布及其制备和在头颈癌硼中子俘获治疗药物中的应用,该化合物不仅载硼量高,肿瘤选择性强,并且在发挥放疗疗效的同时可以发挥抗炎效果,降低治疗毒副作用。中国专利202010020070.9公开了一种含硼碳量子点的制备及其在肿瘤诊断及硼中子俘获治疗药物中的应用,其中的含硼碳量子点的生物相容性好,具有优异的体内荧光成像效果。At present, mercaptododecaborane disodium salt (BSH) and (L)-4-dihydroxyboryl phenylalanine (BPA) have been clinically used as boron drugs for BNCT treatment of tumors [Patent Publication No. CN 111971563 A and CN 111204736 A]; among them, BPA was approved for marketing in Japan in March 2020, becoming the world's first BNCT boron drug approved for marketing. However, both BSH and BPA were found to have insufficient tumor targeting in BNCT clinical studies. The chemical structure modification of boronic acid and borane is the main research strategy at present, but the efficiency still needs to be further improved. Patent application No. 201980041340.X proposes an aggregated boron 10 agent that can be used in boron neutron capture therapy, and can selectively or locally target tumor tissue in a short time, with small side effects, low invasiveness and can It has the advantage of exerting local killing effect on tumor tissue. Chinese patent 202011268512.8 discloses carborane celecoxib and its preparation and application in boron neutron capture therapy for head and neck cancer. The compound not only has high boron loading, strong tumor selectivity, but also exerts the curative effect of radiotherapy at the same time It can exert anti-inflammatory effect and reduce the side effects of treatment. Chinese patent 202010020070.9 discloses the preparation of boron-containing carbon quantum dots and its application in tumor diagnosis and boron neutron capture therapy. The boron-containing carbon quantum dots have good biocompatibility and excellent in vivo fluorescence imaging. Effect.
肿瘤细胞微环境呈现弱酸性(pH = 6.5 ~ 6.8),而肿瘤细胞溶酶体的酸性更强(pH为5.0左右)。如果增加含硼分子的碱性,有望进一步提升硼药在癌细胞中的选择性。四配位硼酸类化合物具有一定的弱碱性,其是有机硼酸类分子化学反应中常见的中间体,由于具有较高的不稳定性,对这类化合物的研究极其有限。2020年,李跃辉等人报道了在有硅烷存在的情况下,芳基硼酸类化合物可以被有效地稳定化,并将芳基硼酸钠盐作为催化剂应用于非均相催化反应中[Green Chem., 2020, 22, 5317-5324]。因此,在提升硼酸盐稳定性的基础上,对硼酸类硼药分子进行改造应能得到效率更高的癌症硼中子俘获治疗的药物。The tumor cell microenvironment is weakly acidic (pH = 6.5 ~ 6.8), while the tumor cell lysosome is more acidic (pH around 5.0). If the basicity of boron-containing molecules is increased, it is expected to further improve the selectivity of boron drugs in cancer cells. Tetracoordinated boronic acids have certain weak basicity, and are common intermediates in the chemical reactions of organic boronic acids. Due to their high instability, the research on these compounds is extremely limited. In 2020, Li Yuehui et al. reported that arylboronic acid compounds could be effectively stabilized in the presence of silane, and arylboronic acid sodium salts were used as catalysts in heterogeneous catalytic reactions [ Green Chem. , 2020, 22, 5317-5324]. Therefore, on the basis of improving the stability of borates, the modification of boronic acid-based boron drug molecules should be able to obtain more efficient drugs for boron neutron capture therapy of cancer.
发明内容SUMMARY OF THE INVENTION
本发明所要解决的技术问题是提供一种具有较好的稳定性和耐酸性的用于癌症硼中子俘获治疗药物的硼酸盐化合物。The technical problem to be solved by the present invention is to provide a borate compound with better stability and acid resistance, which is used as a boron neutron capture therapy drug for cancer.
本发明所要解决的另一个技术问题是提供该用于癌症硼中子俘获治疗药物的硼酸盐化合物的制备。Another technical problem to be solved by the present invention is to provide the preparation of the borate compound for the treatment of cancer boron neutron capture.
为解决上述问题,本发明所述的用于癌症硼中子俘获治疗药物的硼酸盐化合物,其特征在于:该硼酸盐化合物的结构通式如下:In order to solve the above-mentioned problems, the borate compound for the treatment of cancer boron neutron capture therapy according to the present invention is characterized in that: the general structural formula of the borate compound is as follows:

如上所述的用于癌症硼中子俘获治疗药物的硼酸盐化合物的制备方法,其特征在于:在反应容器中加入硼酸化合物、强碱和反应溶剂,于25 ~ 180 ℃反应30 s ~ 48 h 后冷却至室温,得到反应液A;所述反应液A经过滤,得到固体A,该固体A经重结晶、分离即得硼酸盐化合物A;所述硼酸化合物与所述强碱的摩尔比为1:0.5~1:10。The above-mentioned preparation method of borate compound for cancer boron neutron capture therapeutic drug is characterized in that: adding boric acid compound, strong base and reaction solvent in reaction vessel, and reacting at 25~180 ℃ for 30 s~48 h after cooling to room temperature to obtain reaction solution A; the reaction solution A is filtered to obtain solid A, which is recrystallized and separated to obtain borate compound A; the molar ratio of the boric acid compound and the strong base The ratio is 1:0.5~1:10.
其化学反应式如下(式I):Its chemical reaction formula is as follows (Formula I):

如上所述的用于癌症硼中子俘获治疗药物的硼酸盐化合物的制备方法,其特征在于:在反应容器中加入硼酸化合物、强碱和反应溶剂,于25 ~ 180 ℃反应30 s ~ 48 h后加入稳定化试剂,反应结束冷却至室温,得到反应液B;所述反应液B经过滤,得到固体B,该固体B经重结晶、分离即得硼酸盐化合物B;所述硼酸化合物与所述强碱的摩尔比为1:0.5~1:10;所述硼酸化合物与所述稳定化试剂的摩尔比为1:0.5~5。The above-mentioned preparation method of borate compound for cancer boron neutron capture therapeutic drug is characterized in that: adding boric acid compound, strong base and reaction solvent in reaction vessel, and reacting at 25~180 ℃ for 30 s~48 After h, a stabilizing reagent was added, and the reaction was cooled to room temperature to obtain a reaction solution B; the reaction solution B was filtered to obtain a solid B, and the solid B was recrystallized and separated to obtain the borate compound B; the boric acid compound The molar ratio to the strong base is 1:0.5~1:10; the molar ratio of the boric acid compound to the stabilizing reagent is 1:0.5~5.
其化学反应式如下(式II):Its chemical reaction formula is as follows (Formula II):

所述硼酸化合物是指芳基硼酸或脂肪基硼酸。The boronic acid compound refers to aryl boronic acid or aliphatic boronic acid.
所述硼酸化合物是指苯环上连有B(OH)2基团的苯丙氨酸类化合物,其结构式为
所述硼酸化合物的结构式为下述之一:The structural formula of the boronic acid compound is one of the following:

所述强碱是指金属的氢氧化物或烷氧基化物。The strong base refers to metal hydroxides or alkoxylates.
所述强碱是指NaOH、KOH、LiOH、甲醇钠、乙醇钠、乙二醇二钠、丙三醇钠、糖的钠盐中的一种。The strong base refers to one of NaOH, KOH, LiOH, sodium methoxide, sodium ethoxide, disodium ethylene glycol, sodium glycerol, and sodium salt of sugar.
所述稳定化试剂是指任何带有吸电子基团的试剂。The stabilizing reagent refers to any reagent with an electron withdrawing group.
所述稳定化试剂是指五氟苯基钠、苯基硅烷、二苯基硅烷、三苯基硅烷、2-(三甲基硅)苯基三氟甲烷磺酸、六甲基二硅烷、六苯基二硅烷、叔丁基二甲基氯硅烷、二乙基硅烷、三乙基硅烷、三甲基硅醇、三乙基硅醇试剂中的一种。The stabilizing reagents refer to sodium pentafluorophenyl, phenylsilane, diphenylsilane, triphenylsilane, 2-(trimethylsilyl)phenyltrifluoromethanesulfonic acid, hexamethyldisilane, One of the reagents of phenyldisilane, tert-butyldimethylsilyl chloride, diethylsilane, triethylsilane, trimethylsilanol and triethylsilanol.
所述反应溶剂是指有机溶剂,包括但不限于二甲亚砜、N,N-二甲基甲酰胺、四氢呋喃、乙腈、甲醇、二氧六环、N-甲基吡咯烷酮、甲苯、二甲苯、均三甲苯中的一种或者多种。The reaction solvent refers to organic solvents, including but not limited to dimethyl sulfoxide, N,N-dimethylformamide, tetrahydrofuran, acetonitrile, methanol, dioxane, N-methylpyrrolidone, toluene, xylene, One or more of mesitylene.
反应温度优选60 ~ 100 ℃;反应时间优选1 h ~ 6 h。The reaction temperature is preferably 60 to 100 °C; the reaction time is preferably 1 h to 6 h.
本发明与现有技术相比具有以下优点:Compared with the prior art, the present invention has the following advantages:
1、相较于之前的研究对象局限于硼酸类化合物,本发明公开了一种基于稳定化策略的四配位硼酸盐的制备方法,所得硼酸盐具有较好的稳定性和耐酸性,打破了人们常规对硼酸盐不稳定、易分解的认识。1. Compared with the previous research objects limited to boric acid compounds, the present invention discloses a preparation method of a four-coordination borate based on a stabilization strategy, and the obtained borate has better stability and acid resistance, It breaks people's conventional understanding that borates are unstable and easy to decompose.
2、本发明通过利用具有一定Lewis酸性的金属、Si基、B基等基团分散硼酸盐阴离子的电荷,有效实现了硼酸盐稳定性的提升。2. The present invention effectively improves the stability of the borate by dispersing the charge of the borate anion by using groups such as metals, Si groups, and B groups with certain Lewis acidity.
3、本发明是对已知BNCT硼药范畴的全新提升,成功丰富、扩充了硼药的范围,填补了现有硼酸盐制备技术的缺陷,实现了多种不同结构硼酸盐的高效、低成本的生产。3. The present invention is a brand-new improvement to the category of known BNCT boron drugs, successfully enriches and expands the scope of boron drugs, fills up the defects of the existing borate preparation technology, and realizes the high-efficiency, high-efficiency, and high-efficiency of borate with different structures. low cost production.
4、本发明提供的硼酸盐的生物活性测定结果表明,BPA衍生的硼酸盐对头颈癌细胞(CAL27)具有较强的选择性,在硼中子俘获治疗中对头颈癌细胞具有明显的诱导凋亡作用,表现出了比BPA更好的效率,从而有望降低硼药的计量,相较于传统的硼酸类硼药表现出了更好的应用前景。4. The biological activity assay results of the borate provided by the present invention show that the BPA-derived borate has strong selectivity for head and neck cancer cells (CAL27), and has obvious effects on head and neck cancer cells in boron neutron capture therapy. Inducing apoptosis, it shows better efficiency than BPA, which is expected to reduce the dosage of boron drugs, and shows better application prospects than traditional boric acid boron drugs.
5、本发明工艺简单、合成效率高。5. The process of the invention is simple and the synthesis efficiency is high.
具体实施方式Detailed ways
用于癌症硼中子俘获治疗药物的硼酸盐化合物,该硼酸盐化合物的结构通式如下:The borate compound used for the treatment of cancer boron neutron capture, the general structural formula of the borate compound is as follows:

下述实施例中所用的材料,如无特殊说明,均可从商业途径得到。The materials used in the following examples can be obtained from commercial sources unless otherwise specified.
【一步法】【One-step method】
用于癌症硼中子俘获治疗药物的硼酸盐化合物的制备方法:Preparation method of borate compound for cancer boron neutron capture therapeutic drug:
在反应容器中加入硼酸化合物、强碱和反应溶剂,于25 ~ 180 ℃反应30 s ~ 48h 后冷却至室温,得到反应液A;反应液A经过滤,得到固体A,该固体A经重结晶、分离即得硼酸盐化合物A。Add boric acid compound, strong base and reaction solvent to the reaction vessel, react at 25-180 °C for 30 s-48 h, and then cool to room temperature to obtain reaction solution A; Reaction solution A is filtered to obtain solid A, which is recrystallized , and the borate compound A is obtained by separation.

【两步法】【Two-step method】
用于癌症硼中子俘获治疗药物的硼酸盐化合物的制备方法:Preparation method of borate compound for cancer boron neutron capture therapeutic drug:
在反应容器中加入硼酸化合物、强碱和反应溶剂,于25 ~ 180 ℃反应30 s ~ 48h后加入稳定化试剂,反应结束冷却至室温,得到反应液B;反应液B经过滤,得到固体B,该固体B经重结晶、分离即得硼酸盐化合物B。Add boric acid compound, strong base and reaction solvent to the reaction vessel, react at 25-180 ℃ for 30 s-48 h, add stabilizing reagent, and cool down to room temperature after the reaction to obtain reaction solution B; reaction solution B is filtered to obtain solid B , the solid B is recrystallized and separated to obtain borate compound B.

其中:硼酸化合物是指芳基硼酸或脂肪基硼酸。如:4-二羟基硼基苯丙氨酸(BPA),苯硼酸,1-萘硼酸,2-蒽硼酸,甲基硼酸,3-乙酰基苯硼酸,3-联苯硼酸,4-联苯硼酸,4-正丁基苯硼酸,4-乙酰苯硼酸等。Wherein: the boronic acid compound refers to aryl boronic acid or aliphatic boronic acid. Such as: 4-dihydroxyboronyl phenylalanine (BPA), phenylboronic acid, 1-naphthalene boronic acid, 2-anthracene boronic acid, methyl boronic acid, 3-acetylphenyl boronic acid, 3-biphenyl boronic acid, 4-biphenyl Boric acid, 4-n-butylbenzeneboronic acid, 4-acetophenylboronic acid, etc.
硼酸化合物是指苯环上连有B(OH)2基团的苯丙氨酸类化合物,其结构式为
硼酸化合物的结构式为下述之一:The structural formula of the boronic acid compound is one of the following:

强碱是指金属的氢氧化物或烷氧基化物。Strong bases refer to hydroxides or alkoxylates of metals.
强碱是指NaOH、KOH、LiOH、甲醇钠、乙醇钠、乙二醇二钠、丙三醇钠、糖的钠盐中的一种。Strong base refers to one of NaOH, KOH, LiOH, sodium methoxide, sodium ethoxide, disodium ethylene glycol, sodium glycerol, and sodium salt of sugar.
稳定化试剂是指任何带有吸电子基团的试剂。Stabilizing reagent refers to any reagent with electron withdrawing groups.
稳定化试剂是指五氟苯基钠、苯基硅烷、二苯基硅烷、三苯基硅烷、2-(三甲基硅)苯基三氟甲烷磺酸、六甲基二硅烷、六苯基二硅烷、叔丁基二甲基氯硅烷、二乙基硅烷、三乙基硅烷、三甲基硅醇、三乙基硅醇试剂中的一种。Stabilizing reagents refer to sodium pentafluorophenyl, phenylsilane, diphenylsilane, triphenylsilane, 2-(trimethylsilyl)phenyltrifluoromethanesulfonic acid, hexamethyldisilane, hexaphenylsilane One of the reagents of disilane, tert-butyldimethylsilyl chloride, diethylsilane, triethylsilane, trimethylsilanol and triethylsilanol.
反应溶剂是指有机溶剂,包括但不限于二甲亚砜、N,N-二甲基甲酰胺、四氢呋喃、乙腈、甲醇、二氧六环、N-甲基吡咯烷酮、甲苯、二甲苯、均三甲苯中的一种或者多种。Reaction solvent refers to organic solvents, including but not limited to dimethyl sulfoxide, N,N-dimethylformamide, tetrahydrofuran, acetonitrile, methanol, dioxane, N-methylpyrrolidone, toluene, xylene, mesitylene One or more of toluene.
反应温度优选60 ~ 100 ℃;反应时间优选1 h ~ 6 h。The reaction temperature is preferably 60 to 100 °C; the reaction time is preferably 1 h to 6 h.
强碱是预先制备好的纯品或者商业渠道直接购买;硼酸化合物可以提前制备出来并进行分离纯化后使用,也可以在反应过程中原位制备不做任何处理直接使用。Strong bases are pre-prepared pure products or purchased directly from commercial channels; boronic acid compounds can be prepared in advance and used after separation and purification, or they can be prepared in situ during the reaction process without any treatment and used directly.
硼酸盐化合物A和硼酸盐化合物B为四配位硼酸盐,其化学结构式如式I和II中所示,优选结构为:The borate compound A and the borate compound B are tetracoordinate borates, and their chemical structural formulas are shown in formulas I and II, and the preferred structures are:

实施例1:硼酸盐化合物(1)的制备(两步法)。Example 1: Preparation of borate compound (1) (two-step process).

在空气氛围下,将(L)-4-二羟基硼基苯丙氨酸(0.05 mmol, 10.45 mg),氢氧化钠(0.15 mmol, 6 mg ), 甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15 mmol, 39 mg),反应管在90 ℃反应6小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为78%。Under air atmosphere, (L)-4-dihydroxyboryl phenylalanine (0.05 mmol, 10.45 mg), sodium hydroxide (0.15 mmol, 6 mg), toluene (4 mL) and a magnetic stirrer were added to 35 mL glass pressure-resistant tube. Then triphenylsilane (0.15 mmol, 39 mg) was added, and the reaction tube was reacted at 90° C. for 6 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 78%.
1H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 8.71 (d, 2H), 7.75 (d, 2H),7.46 (m, 18H), 7.39-7.35 (m, 27H), 7.2(d, 2H), 4.18(s, 1H), 3.42(d, 2H). 1 H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 8.71 (d, 2H), 7.75 (d, 2H), 7.46 (m, 18H), 7.39-7.35 (m, 27H), 7.2(d , 2H), 4.18(s, 1H), 3.42(d, 2H).
13C NMR (100 MHz, DMSO) δ 174.7, 138.3, 136.6, 135.7, 133.3, 132.5,130.0, 129.5, 127.7, 56.7, 37.3. 13 C NMR (100 MHz, DMSO) δ 174.7, 138.3, 136.6, 135.7, 133.3, 132.5, 130.0, 129.5, 127.7, 56.7, 37.3.
实施例2: 硼酸盐化合物(2)的制备(一步法)Example 2: Preparation of borate compound (2) (one-step method)

在空气氛围下,将苯硼酸(0.05 mmol, 6.1 mg),三苯基硅醇钠(0.025 mmol,7.46 mg ),磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入甲苯(4 mL),反应管在90℃反应6小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为75%。Under air atmosphere, phenylboronic acid (0.05 mmol, 6.1 mg), sodium triphenylsiliconate (0.025 mmol, 7.46 mg), and a magnetic stirring bar were added to a 35 mL glass pressure-resistant tube. Then, toluene (4 mL) was added, and the reaction tube was reacted at 90°C for 6 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 75%.
1H NMR (300 MHz, DMSO) δ 7.75(d, 2H), 7.48 - 7.44 (m, 6H),7.37-7.39(m, 10m) 7.36(s, 1H), 7.35(s, 1H), 4.2 (d, 2H). 1 H NMR (300 MHz, DMSO) δ 7.75(d, 2H), 7.48 - 7.44 (m, 6H), 7.37-7.39(m, 10m) 7.36(s, 1H), 7.35(s, 1H), 4.2 ( d, 2H).
13C NMR (101 MHz, DMSO) δ 138.5, 138.3, 133.4, 132.5, 130.0, 129.5,128.7. 13 C NMR (101 MHz, DMSO) δ 138.5, 138.3, 133.4, 132.5, 130.0, 129.5, 128.7.
实施例3: 硼酸盐化合物(3)的制备(两步法)Example 3: Preparation of borate compound (3) (two-step method)
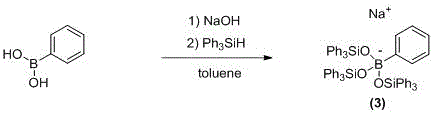
在空气氛围下,将苯硼酸(0.05 mmol, 6.1 mg),氢氧化钠(0.5 mmol, 20 mg),甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15 mmol,39 mg),反应管在90 ℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为75%。Under air atmosphere, phenylboronic acid (0.05 mmol, 6.1 mg), sodium hydroxide (0.5 mmol, 20 mg), toluene (4 mL) and a magnetic stir bar were added to a 35 mL glass pressure-resistant tube. Then triphenylsilane (0.15 mmol, 39 mg) was added, and the reaction tube was reacted at 90° C. for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 75%.
1H NMR (300 MHz, DMSO) δ 7.75 (d, 2H), 7.48- 7.44 (m, 18H), 7.34 -7.38 (m, 30H). 1 H NMR (300 MHz, DMSO) δ 7.75 (d, 2H), 7.48-7.44 (m, 18H), 7.34-7.38 (m, 30H).
13C NMR (101 MHz, DMSO) δ 138.3, 133.4, 132.5, 130.0, 129.5, 128.7. 13 C NMR (101 MHz, DMSO) δ 138.3, 133.4, 132.5, 130.0, 129.5, 128.7.
实施例4: 硼酸盐化合物(4)的制备(一步法)Example 4: Preparation of borate compound (4) (one-step method)

在空气氛围下,将F-BPA(0.05 mmol, 11.35 mg),三苯基硅醇钠(0.2 mmol,59.68 mg ),磁力搅拌子加入到35 mL的玻璃耐压管中。反应管在90 ℃反应6小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为85%。Under air atmosphere, F-BPA (0.05 mmol, 11.35 mg), sodium triphenylsiliconate (0.2 mmol, 59.68 mg), and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. The reaction tube was reacted at 90°C for 6 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 85%.
1H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 8.71 (d, 2H), 7.52 (s, 1H),7.46 (m, 6H), 7.35-7.39 (m, 9H), 7.18 (s, 1H), 7.04 (s, 1H), 4.2(d, 2H), 4.18(m, 1H), 3.42 (d, 2H). 1 H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 8.71 (d, 2H), 7.52 (s, 1H), 7.46 (m, 6H), 7.35-7.39 (m, 9H), 7.18 (s , 1H), 7.04 (s, 1H), 4.2(d, 2H), 4.18(m, 1H), 3.42 (d, 2H).
13C NMR (101 MHz, DMSO) δ 174.7, 160.2, 138.3, 137.3, 132.5, 130.0,129.5, 128.1, 115.6, 56.7, 32.4. 13 C NMR (101 MHz, DMSO) δ 174.7, 160.2, 138.3, 137.3, 132.5, 130.0, 129.5, 128.1, 115.6, 56.7, 32.4.
实施例5: 硼酸盐化合物(5)的制备(两步法)Example 5: Preparation of borate compound (5) (two-step method)

在空气氛围下,将BPA-Tyr(0.05 mmol, 18.61 mg),氢氧化钾(0.2 mmol, 11.2mg),甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.25mmol, 65.10 mg),反应管在100 ℃反应10小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为70%。Under air atmosphere, BPA-Tyr (0.05 mmol, 18.61 mg), potassium hydroxide (0.2 mmol, 11.2 mg), toluene (4 mL) and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Then triphenylsilane (0.25 mmol, 65.10 mg) was added, and the reaction tube was reacted at 100° C. for 10 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 70%.
1H NMR (300 MHz, DMSO) δ 12.8 9 (m, 1H), 9.06 (m, 1H), 8.86 (m, 2H),8.32 (m, 1H), 7.75 (d, 2H), 7.48-7.44 (m, 24H), 7.38-7.34 (m, 20), 7.2 (m,2H), 6.96 (m, 2H), 6.68 (m, 2H), 4.72 (m, 1H), 3.95 (d, 1H), 3.44 (m, 2H),3.12 (m, 2H). 1 H NMR (300 MHz, DMSO) δ 12.8 9 (m, 1H), 9.06 (m, 1H), 8.86 (m, 2H), 8.32 (m, 1H), 7.75 (d, 2H), 7.48-7.44 ( m, 24H), 7.38-7.34 (m, 20), 7.2 (m, 2H), 6.96 (m, 2H), 6.68 (m, 2H), 4.72 (m, 1H), 3.95 (d, 1H), 3.44 (m, 2H), 3.12 (m, 2H).
13C NMR (101 MHz, DMSO) δ 174.7, 171.7, 155.7, 138.3, 136.6, 135.7,132.5, 130.2, 130.0, 129.5, 129.2,127.7, 115.8, 59.2, 56.0, 38.7, 36.5. 13 C NMR (101 MHz, DMSO) δ 174.7, 171.7, 155.7, 138.3, 136.6, 135.7, 132.5, 130.2, 130.0, 129.5, 129.2, 127.7, 115.8, 59.2, 56.0, 38.7, 3
实施例6: 硼酸盐化合物(6)的制备(一步法)Example 6: Preparation of borate compound (6) (one-step method)

在空气氛围下,将BPA-Tyr (0.05 mmol, 18.61 mg),三苯基硅醇钠(0.05 mmol,14.92 mg),磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入甲苯(10 mL),反应管在90℃反应6小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为85%。Under air atmosphere, BPA-Tyr (0.05 mmol, 18.61 mg), sodium triphenylsiliconate (0.05 mmol, 14.92 mg), and a magnetic stirring bar were added to a 35 mL glass pressure-resistant tube. Then, toluene (10 mL) was added, and the reaction tube was reacted at 90° C. for 6 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 85%.
1H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 9.06 (s, 1H), 8.86 (m, 2H),8.32 (s, 1H), 7.75 (m, 2H) 7.46(m, 6H), 7.38-7.34 (m, 9H), 7.20 (m, 2H), 6.96(m, 2H), 6.68 (m, 2H), 4.72 (m, 1H), 4.20 (m, 2H), 3.95 (m, 1H), 3.44 (m,2H), 3.21 (m, 2H). 1 H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 9.06 (s, 1H), 8.86 (m, 2H), 8.32 (s, 1H), 7.75 (m, 2H) 7.46 (m, 6H) , 7.38-7.34 (m, 9H), 7.20 (m, 2H), 6.96(m, 2H), 6.68 (m, 2H), 4.72 (m, 1H), 4.20 (m, 2H), 3.95 (m, 1H) ), 3.44 (m, 2H), 3.21 (m, 2H).
13C NMR (101 MHz, DMSO) δ 174.7, 171.7, 155.7, 138.3,136.6, 135.7,133.3, 132.5, 130.2, 130.0, 129.5, 129.2, 127.7, 115.8, 59.2, 56.0, 38.7,36.5. 13 C NMR (101 MHz, DMSO) δ 174.7, 171.7, 155.7, 138.3, 136.6, 135.7, 133.3, 132.5, 130.2, 130.0, 129.5, 129.2, 127.7, 115.8, 59.2, 5.56.0,
实施例7: 硼酸盐化合物(7)的制备(两步法)Example 7: Preparation of borate compound (7) (two-step method)

在空气氛围下,将(L)-4-二羟基硼基苯丙氨酸(0.05 mmol, 10.45 mg),氢氧化钾(0.2 mmol, 11.2 mg),甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.025 mmol, 6.51 mg),反应管在100 ℃反应10小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为75%。Under air atmosphere, (L)-4-dihydroxyboryl phenylalanine (0.05 mmol, 10.45 mg), potassium hydroxide (0.2 mmol, 11.2 mg), toluene (4 mL) and a magnetic stir bar were added to 35 mL glass pressure-resistant tube. Triphenylsilane (0.025 mmol, 6.51 mg) was then added, and the reaction tube was reacted at 100° C. for 10 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 75%.
1H NMR (500 MHz, Chloroform-d) δ 7.64–7.57 (m, 7H), 7.61–7.53 (m,1H), 7.39–7.29 (m, 9H), 7.12 (dt, J = 9.5, 1.1 Hz, 1H), 3.05–2.91 (m, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64–7.57 (m, 7H), 7.61–7.53 (m, 1H), 7.39–7.29 (m, 9H), 7.12 (dt, J = 9.5, 1.1 Hz, 1H), 3.05–2.91 (m, 1H).
13C NMR (125 MHz, Chloroform-d) δ 174.88, 139.48, 135.07, 134.68,133.60, 133.39, 132.18, 129.80, 127.46, 56.92, 37.79. 13 C NMR (125 MHz, Chloroform-d) δ 174.88, 139.48, 135.07, 134.68, 133.60, 133.39, 132.18, 129.80, 127.46, 56.92, 37.79.
实施例8: 硼酸盐化合物(8)的制备(一步法)Example 8: Preparation of borate compound (8) (one-step method)

在空气氛围下,将BPA-Tyr(0.05 mmol, 18.61 mg),三苯基硅醇钠(0.2 mmol,59.68 mg),磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入甲苯(4 mL),反应管在80℃反应48小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为78%。Under air atmosphere, BPA-Tyr (0.05 mmol, 18.61 mg), sodium triphenylsiliconate (0.2 mmol, 59.68 mg), and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Then, toluene (4 mL) was added, and the reaction tube was reacted at 80°C for 48 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 78%.
1H NMR (500 MHz, Chloroform-d) δ 7.90 (d, J = 9.0 Hz, 1H), 7.64-7.57(m, 7H), 7.59-7.52 (m, 2H), 7.39-7.29 (m, 10H), 7.14 (dt, J = 9.1, 1.1 Hz,2H), 6.95 (dt, J = 8.6, 1.1 Hz, 2H), 6.75-6.69 (m, 2H), 6.14 (s, 1H), 5.92(s, 2H), 4.54 (dt, J = 9.0, 6.8 Hz, 1H), 3.88 (p, J = 5.9 Hz, 1H), 3.02 (m, J= 15.4, 5.7, 1.0 Hz, 1H), 2.95 (m, J = 6.8, 2.7, 1.0 Hz, 2H), 2.88 (m, J =15.2, 5.7, 1.0 Hz, 1H), 2.58 (dd, J = 8.0, 6.0 Hz, 1H), 2.42 (dd, J = 8.0,5.9 Hz, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.90 (d, J = 9.0 Hz, 1H), 7.64-7.57(m, 7H), 7.59-7.52 (m, 2H), 7.39-7.29 (m, 10H) , 7.14 (dt, J = 9.1, 1.1 Hz, 2H), 6.95 (dt, J = 8.6, 1.1 Hz, 2H), 6.75-6.69 (m, 2H), 6.14 (s, 1H), 5.92(s, 2H) ), 4.54 (dt, J = 9.0, 6.8 Hz, 1H), 3.88 (p, J = 5.9 Hz, 1H), 3.02 (m, J= 15.4, 5.7, 1.0 Hz, 1H), 2.95 (m, J = 6.8, 2.7, 1.0 Hz, 2H), 2.88 (m, J = 15.2, 5.7, 1.0 Hz, 1H), 2.58 (dd, J = 8.0, 6.0 Hz, 1H), 2.42 (dd, J = 8.0, 5.9 Hz , 1H).
13C NMR (125 MHz, Chloroform-d) δ 176.68, 173.61, 156.46, 136.89,135.07, 134.96, 134.52, 132.75, 130.65, 129.99, 129.80, 128.56, 127.46,115.72, 54.89, 54.55, 38.40, 37.66. 13 C NMR (125 MHz, Chloroform-d) δ 176.68, 173.61, 156.46, 136.89,135.07, 134.96, 134.52, 132.75, 130.65, 129.99, 129.80, 128.56, 127.46,115.72, 54.89, 54.55, 38.40, 37.66.
实施例9: 硼酸盐化合物(9)的制备(两步法)Example 9: Preparation of borate compound (9) (two-step method)

在空气氛围下,将Tyr-BPA(0.05 mmol, 18.61 mg),氢氧化钠(0.2 mmol, 8 mg),甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15mmol, 39 mg),反应管在90 ℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为90%。Under air atmosphere, Tyr-BPA (0.05 mmol, 18.61 mg), sodium hydroxide (0.2 mmol, 8 mg), toluene (4 mL) and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Then triphenylsilane (0.15 mmol, 39 mg) was added, and the reaction tube was reacted at 90° C. for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 90%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.57 (m, 7H), 7.61-7.53 (m,1H), 7.39-7.29 (m, 11H), 7.07 (m, J = 19.2, 8.6, 1.1 Hz, 2H), 6.75-6.69 (m,1H), 4.54 (dt, J = 9.0, 6.8 Hz, 0H), 3.06-2.96 (m, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.57 (m, 7H), 7.61-7.53 (m, 1H), 7.39-7.29 (m, 11H), 7.07 (m, J = 19.2, 8.6, 1.1 Hz, 2H), 6.75-6.69 (m, 1H), 4.54 (dt, J = 9.0, 6.8 Hz, 0H), 3.06-2.96 (m, 1H).
13C NMR (125 MHz, Chloroform-d) δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 133.11, 132.17, 130.70, 129.80, 127.46, 127.25,115.38, 54.88, 54.55, 38.39, 37.19. 13 C NMR (125 MHz, Chloroform-D) Δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 132.17, 130.70, 127.46, 127.25,115.38, 54.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.
实施例10: 硼酸盐化合物(10)的制备(一步法)Example 10: Preparation of borate compound (10) (one-step method)

在空气氛围下,将(L)-4-二羟基硼基苯丙氨酸(0.05 mmol, 10.45 mg),三苯基硅醇钠(0.05 mmol, 14.92 mg)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入DCE(4mL),反应管在80 ℃反应24小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为89%。Under air atmosphere, (L)-4-dihydroxyboryl phenylalanine (0.05 mmol, 10.45 mg), sodium triphenylsiliconate (0.05 mmol, 14.92 mg) and a magnetic stirrer were added to 35 mL of in glass pressure-resistant tubes. Then DCE (4 mL) was added, and the reaction tube was reacted at 80°C for 24 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 89%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.57 (m, 3H), 7.61-7.52 (m,1H), 7.39-7.29 (m, 4H), 7.13 (dt, J = 9.0, 1.0 Hz, 1H), 5.92 (s, 1H), 3.87(tt, J = 6.6, 5.1 Hz, 0H), 3.05-2.91 (m, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.57 (m, 3H), 7.61-7.52 (m, 1H), 7.39-7.29 (m, 4H), 7.13 (dt, J = 9.0, 1.0 Hz, 1H), 5.92 (s, 1H), 3.87(tt, J = 6.6, 5.1 Hz, 0H), 3.05-2.91 (m, 1H).
13C NMR (125 MHz, Chloroform-d) δ 174.88, 136.89, 135.07, 134.96,134.63, 132.84, 129.99, 129.80, 127.46, 56.92, 37.79. 13 C NMR (125 MHz, Chloroform-d) δ 174.88, 136.89, 135.07, 134.96, 134.63, 132.84, 129.99, 129.80, 127.46, 56.92, 37.79.
实施例11: 硼酸盐化合物(11)的制备(两步法)Example 11: Preparation of borate compound (11) (two-step method)

在空气氛围下,将(L)-4-二羟基硼基苯丙氨酸(0.05 mmol, 10.45 mg),氢氧化锂(0.15 mmol, 3.6 mg ),DMF(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15 mmol, 39 mg),反应管在90 ℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为72%。Under air atmosphere, (L)-4-dihydroxyboryl phenylalanine (0.05 mmol, 10.45 mg), lithium hydroxide (0.15 mmol, 3.6 mg), DMF (4 mL) and magnetic stirrer were added to 35 mL glass pressure-resistant tube. Then triphenylsilane (0.15 mmol, 39 mg) was added, and the reaction tube was reacted at 90° C. for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 72%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.57 (m, 7H), 7.61-7.53 (m,1H), 7.39-7.29 (m, 9H), 7.12 (dt, J = 9.5, 1.1 Hz, 1H), 3.05-2.91 (m, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.57 (m, 7H), 7.61-7.53 (m, 1H), 7.39-7.29 (m, 9H), 7.12 (dt, J = 9.5, 1.1 Hz, 1H), 3.05-2.91 (m, 1H).
13C NMR (125 MHz, Chloroform-d) δ 174.88, 139.48, 135.07, 134.68,133.60, 133.39, 132.18, 129.80, 127.46, 56.92, 37.79. 13 C NMR (125 MHz, Chloroform-d) δ 174.88, 139.48, 135.07, 134.68, 133.60, 133.39, 132.18, 129.80, 127.46, 56.92, 37.79.
实施例12: 硼酸盐化合物(12)的制备(一步法)Example 12: Preparation of borate compound (12) (one-step method)

在空气氛围下,将Tyr-BPA(0.05 mmol, 18.61 mg),三苯基硅醇钠(0.1 mmol,29.84 mg)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入甲苯(4 mL) ,反应管在90℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为90%。Under air atmosphere, Tyr-BPA (0.05 mmol, 18.61 mg), sodium triphenylsiliconate (0.1 mmol, 29.84 mg) and a magnetic stir bar were added to a 35 mL glass pressure-resistant tube. Then, toluene (4 mL) was added, and the reaction tube was reacted at 90°C for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 90%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.56 (m, 18H), 7.47-7.41 (m,2H), 7.37-7.27 (m, 27H), 7.10-7.01 (m, 4H), 6.73-6.67 (m, 2H), 4.38 (dt, J =9.1, 6.8 Hz, 1H), 3.84 (p, J = 5.9 Hz, 1H), 3.08-3.00 (m, 1H), 3.04-2.95 (m,2H), 2.84 (m, J = 15.4, 5.8, 1.1 Hz, 1H), 2.70 (dd, J = 7.9, 6.0 Hz, 1H),2.50 (dd, J = 8.0, 5.9 Hz, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.56 (m, 18H), 7.47-7.41 (m, 2H), 7.37-7.27 (m, 27H), 7.10-7.01 (m, 4H), 6.73- 6.67 (m, 2H), 4.38 (dt, J =9.1, 6.8 Hz, 1H), 3.84 (p, J = 5.9 Hz, 1H), 3.08-3.00 (m, 1H), 3.04-2.95 (m,2H) , 2.84 (m, J = 15.4, 5.8, 1.1 Hz, 1H), 2.70 (dd, J = 7.9, 6.0 Hz, 1H), 2.50 (dd, J = 8.0, 5.9 Hz, 1H).
13C NMR (125 MHz, Chloroform-d) δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 133.11, 132.17, 130.70, 129.80, 127.46, 127.25,115.38, 54.88, 54.55, 38.39, 37.19. 13 C NMR (125 MHz, Chloroform-D) Δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 132.17, 130.70, 127.46, 127.25,115.38, 54.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.
实施例13: 硼酸盐化合物(13)的制备(两步法)Example 13: Preparation of borate compound (13) (two-step method)

在空气氛围下,将F-BPA(0.05 mmol, 11.35 mg),氢氧化钠(0.2 mmol, 8 mg),甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15 mmol,39 mg),反应管在80 ℃反应24小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为55%,。Under air atmosphere, F-BPA (0.05 mmol, 11.35 mg), sodium hydroxide (0.2 mmol, 8 mg), toluene (4 mL) and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Then triphenylsilane (0.15 mmol, 39 mg) was added, and the reaction tube was reacted at 80° C. for 24 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 55%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.56 (m, 8H), 7.43 (dd, J =10.5, 2.0 Hz, 0H), 7.39-7.23 (m, 10H), 3.89 (tt, J = 7.5, 6.6 Hz, 0H), 3.18-3.07 (m, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.56 (m, 8H), 7.43 (dd, J =10.5, 2.0 Hz, 0H), 7.39-7.23 (m, 10H), 3.89 (tt, J = 7.5, 6.6 Hz, 0H), 3.18-3.07 (m, 1H).
13C NMR (125 MHz, Chloroform-d) δ 173.42, 135.07, 133.63, 129.80,127.46, 32.61, 32.57. 13 C NMR (125 MHz, Chloroform-d) δ 173.42, 135.07, 133.63, 129.80, 127.46, 32.61, 32.57.
实施例14: 硼酸盐化合物(14)的制备(一步法)Example 14: Preparation of borate compound (14) (one-step method)

在空气氛围下,Tyr-BPA(0.05 mmol, 18.61 mg),三苯基硅醇钠(0.2 mmol,59.68 mg)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入1,2-二氯乙烷(4 mL),反应管在90 ℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为85%。Under air atmosphere, Tyr-BPA (0.05 mmol, 18.61 mg), sodium triphenylsiliconate (0.2 mmol, 59.68 mg) and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Subsequently, 1,2-dichloroethane (4 mL) was added, and the reaction tube was reacted at 90° C. for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 85%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.55 (m, 18H), 7.59-7.53 (m,2H), 7.39-7.29 (m, 28H), 7.07 (m, J = 19.2, 8.6, 1.1 Hz, 4H), 6.75-6.69 (m,2H), 4.53 (dt, J = 9.0, 6.7 Hz, 1H), 3.88 (p, J = 5.9 Hz, 1H), 3.06-2.98 (m,1H), 3.00 (d, J = 1.1 Hz, 1H), 3.01-2.96 (m, 1H), 2.89 (m, J = 15.4, 5.7, 1.1Hz, 1H), 2.58 (dd, J = 8.0, 6.0 Hz, 1H), 2.42 (dd, J = 8.0, 5.9 Hz, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.55 (m, 18H), 7.59-7.53 (m, 2H), 7.39-7.29 (m, 28H), 7.07 (m, J = 19.2, 8.6, 1.1 Hz, 4H), 6.75-6.69 (m,2H), 4.53 (dt, J = 9.0, 6.7 Hz, 1H), 3.88 (p, J = 5.9 Hz, 1H), 3.06-2.98 (m,1H), 3.00 (d, J = 1.1 Hz, 1H), 3.01-2.96 (m, 1H), 2.89 (m, J = 15.4, 5.7, 1.1Hz, 1H), 2.58 (dd, J = 8.0, 6.0 Hz, 1H), 2.42 (dd, J = 8.0, 5.9 Hz, 1H).
13C NMR (125 MHz, Chloroform-d) δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 133.11, 132.17, 130.70, 129.80, 127.46, 127.25,115.38, 54.88, 54.55, 38.39, 37.19. 13 C NMR (125 MHz, Chloroform-D) Δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 132.17, 130.70, 127.46, 127.25,115.38, 54.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.
实施例15: 硼酸盐化合物(15)的制备(两步法)Example 15: Preparation of borate compound (15) (two-step method)

在空气氛围下,F-BPA衍生物(0.05 mmol, 11.35 mg),氢氧化钠(0.2 mmol, 8mg),甲苯(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15mmol, 39 mg),反应管在90 ℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为88%。Under air atmosphere, F-BPA derivative (0.05 mmol, 11.35 mg), sodium hydroxide (0.2 mmol, 8 mg), toluene (4 mL) and magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Then triphenylsilane (0.15 mmol, 39 mg) was added, and the reaction tube was reacted at 90° C. for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 88%.
1H NMR (500 MHz, Chloroform-d) δ 7.61 (dq, J = 8.4, 2.4, 1.9 Hz,26H), 7.39-7.27 (m, 34H), 4.59 (dt, J = 6.2, 3.7 Hz, 1H), 4.18-4.08 (m, 2H),4.03-3.91 (m, 2H), 3.93-3.84 (m, 1H), 3.18-3.07 (m, 2H). 1 H NMR (500 MHz, Chloroform-d) δ 7.61 (dq, J = 8.4, 2.4, 1.9 Hz, 26H), 7.39-7.27 (m, 34H), 4.59 (dt, J = 6.2, 3.7 Hz, 1H) , 4.18-4.08 (m, 2H), 4.03-3.91 (m, 2H), 3.93-3.84 (m, 1H), 3.18-3.07 (m, 2H).
13C NMR (125 MHz, Chloroform-d) δ 174.56, 161.90, 136.15, 136.08,135.54, 135.09, 133.91, 133.72, 133.38, 133.36, 132.29, 131.42, 129.88,128.87, 127.42, 127.40, 122.21, 120.65, 104.54, 77.48, 76.42, 72.57, 64.44,61.18, 54.64, 30.56. 13 C NMR (125 MHz, Chloroform-d) δ 174.56, 161.90, 136.15, 136.08,135.54, 135.09, 133.91, 133.72, 133.38, 133.36, 132.29, 131.42, 129.88,128.87, 127.42, 127.40, 122.21, 120.65, 104.54, 77.48, 76.42, 72.57, 64.44, 61.18, 54.64, 30.56.
实施例16: 硼酸盐化合物(16)的制备(一步法)Example 16: Preparation of borate compound (16) (one-step method)

在空气氛围下,Tyr-BPA(0.05 mmol, 18.61 mg),三苯基硅醇钠(0.2 mmol,59.68 mg)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入N,N-二甲基甲酰胺(4mL),反应管在100 ℃反应3小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为81%。Under air atmosphere, Tyr-BPA (0.05 mmol, 18.61 mg), sodium triphenylsiliconate (0.2 mmol, 59.68 mg) and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Subsequently, N,N-dimethylformamide (4 mL) was added, and the reaction tube was reacted at 100° C. for 3 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure yield of the target product was 81%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.55 (m, 18H), 7.59-7.53 (m,2H), 7.39-7.29 (m, 28H), 7.07 (m, J = 19.2, 8.6, 1.1 Hz, 4H), 6.75-6.69 (m,2H), 4.53 (dt, J = 9.0, 6.7 Hz, 1H), 3.88 (p, J = 5.9 Hz, 1H), 3.06-2.98 (m,1H), 3.00 (d, J = 1.1 Hz, 1H), 3.01-2.96 (m, 1H), 2.89 (m, J = 15.4, 5.7, 1.1Hz, 1H), 2.58 (dd, J = 8.0, 6.0 Hz, 1H), 2.42 (dd, J = 8.0, 5.9 Hz, 1H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.55 (m, 18H), 7.59-7.53 (m, 2H), 7.39-7.29 (m, 28H), 7.07 (m, J = 19.2, 8.6, 1.1 Hz, 4H), 6.75-6.69 (m,2H), 4.53 (dt, J = 9.0, 6.7 Hz, 1H), 3.88 (p, J = 5.9 Hz, 1H), 3.06-2.98 (m,1H), 3.00 (d, J = 1.1 Hz, 1H), 3.01-2.96 (m, 1H), 2.89 (m, J = 15.4, 5.7, 1.1Hz, 1H), 2.58 (dd, J = 8.0, 6.0 Hz, 1H), 2.42 (dd, J = 8.0, 5.9 Hz, 1H).
13C NMR (125 MHz, Chloroform-d) δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 133.11, 132.17, 130.70, 129.80, 127.46, 127.25,115.38, 54.88, 54.55, 38.39, 37.19. 13 C NMR (125 MHz, Chloroform-D) Δ 176.69, 173.61, 156.84, 139.48,135.16, 135.07, 133.60, 132.17, 130.70, 127.46, 127.25,115.38, 54.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.55, 38.
实施例17: 硼酸盐化合物(17)的制备(两步法)Example 17: Preparation of borate compound (17) (two-step method)

在空气氛围下,F-BPA衍生物(0.05 mmol, 11.35 mg),氢氧化锂(0.15 mmol, 3.6mg),N,N-二甲基甲酰胺(4 mL)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入三苯基硅烷(0.15 mmol, 39 mg),反应管在80 ℃反应24时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为80%。Under air atmosphere, F-BPA derivative (0.05 mmol, 11.35 mg), lithium hydroxide (0.15 mmol, 3.6 mg), N,N-dimethylformamide (4 mL) and magnetic stirrer were added to 35 mL in the glass pressure-resistant tube. Triphenylsilane (0.15 mmol, 39 mg) was then added, and the reaction tube was reacted at 80 °C for 24 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 80%.
1H NMR (500 MHz, Chloroform-d) δ 7.61 (m, J = 8.4, 2.4, 1.9 Hz, 26H),7.39-7.27 (m, 34H), 4.59 (dt, J = 6.2, 3.7 Hz, 1H), 4.18-4.08 (m, 2H), 4.03-3.91 (m, 2H), 3.93-3.84 (m, 1H), 3.18-3.07 (m, 2H). 1 H NMR (500 MHz, Chloroform-d) δ 7.61 (m, J = 8.4, 2.4, 1.9 Hz, 26H), 7.39-7.27 (m, 34H), 4.59 (dt, J = 6.2, 3.7 Hz, 1H) , 4.18-4.08 (m, 2H), 4.03-3.91 (m, 2H), 3.93-3.84 (m, 1H), 3.18-3.07 (m, 2H).
13C NMR (125 MHz, Chloroform-d) δ 174.56, 161.90, 136.15, 136.08,135.54, 135.09, 133.91, 133.72, 133.38, 133.36, 132.29, 131.42, 129.88,128.87, 127.42, 127.40, 122.21, 120.65, 104.54, 77.48, 76.42, 72.57, 64.44,61.18, 54.64, 30.56. 13 C NMR (125 MHz, Chloroform-d) δ 174.56, 161.90, 136.15, 136.08,135.54, 135.09, 133.91, 133.72, 133.38, 133.36, 132.29, 131.42, 129.88,128.87, 127.42, 127.40, 122.21, 120.65, 104.54, 77.48, 76.42, 72.57, 64.44, 61.18, 54.64, 30.56.
实施例18: 硼酸盐化合物(18)的制备(一步法)Example 18: Preparation of borate compound (18) (one-step method)

在空气氛围下,F-BPA衍生物(0.05 mmol, 11.35 mg),三苯基硅醇钠(0.2 mmol,59.68 mg)和磁力搅拌子加入到35 mL的玻璃耐压管中。随后加入1,2-二氯乙烷(4 mL),反应管在90 ℃反应12小时。待反应完毕,将反应体系冷却至室温,在室温下打开反应容器,将其进行过滤;所得固体重结晶后分离得到反应产物。目标产物纯品产率为78%。Under air atmosphere, F-BPA derivative (0.05 mmol, 11.35 mg), sodium triphenylsiliconate (0.2 mmol, 59.68 mg) and a magnetic stirrer were added to a 35 mL glass pressure-resistant tube. Subsequently, 1,2-dichloroethane (4 mL) was added, and the reaction tube was reacted at 90° C. for 12 hours. After the reaction is completed, the reaction system is cooled to room temperature, the reaction vessel is opened at room temperature, and filtered; the obtained solid is recrystallized and separated to obtain the reaction product. The pure product yield of the target product was 78%.
1H NMR (500 MHz, Chloroform-d) δ 7.64-7.56 (m, 6H), 7.44 (dd, J =10.6, 1.8 Hz, 1H), 7.39-7.31 (m, 5H), 7.34-7.27 (m, 4H), 7.26 (s, 1H), 7.11(m, J = 10.5, 5.0, 1.0 Hz, 1H), 4.59-4.52 (m, 2H), 4.49 (dd, J = 7.3, 6.6 Hz,1H), 4.27 (dd, J = 7.3, 6.6 Hz, 1H), 4.15 (t, J = 6.2 Hz, 1H), 4.06 (s, 1H),4.01-3.94 (m, 2H), 3.97-3.84 (m, 2H), 3.78-.68 (m, 2H), 3.46 (d, J = 5.1 Hz,1H), 3.20-3.06 (m, 2H). 1 H NMR (500 MHz, Chloroform-d) δ 7.64-7.56 (m, 6H), 7.44 (dd, J =10.6, 1.8 Hz, 1H), 7.39-7.31 (m, 5H), 7.34-7.27 (m, 4H), 7.26 (s, 1H), 7.11(m, J = 10.5, 5.0, 1.0 Hz, 1H), 4.59-4.52 (m, 2H), 4.49 (dd, J = 7.3, 6.6 Hz, 1H), 4.27 (dd, J = 7.3, 6.6 Hz, 1H), 4.15 (t, J = 6.2 Hz, 1H), 4.06 (s, 1H), 4.01-3.94 (m, 2H), 3.97-3.84 (m, 2H), 3.78-.68 (m, 2H), 3.46 (d, J = 5.1 Hz, 1H), 3.20-3.06 (m, 2H).
13C NMR (125 MHz, Chloroform-d) δ 173.42, 161.19, 159.18, 135.07,133.94, 133.88, 133.67, 131.79, 131.72, 129.80, 128.72, 128.70, 127.46,123.85, 123.69, 122.75, 122.59, 98.00, 78.45, 72.97, 72.74, 62.44, 60.93,55.32, 55.30, 32.74, 32.70. 13 C NMR (125 MHz, Chloroform-d) δ 173.42, 161.19, 159.18, 135.07,133.94, 133.88, 133.67, 131.79, 131.72, 129.80, 128.72, 128.70, 127.46,123.85, 123.69, 122.75, 122.59, 98.00, 78.45, 72.97, 72.74, 62.44, 60.93, 55.32, 55.30, 32.74, 32.70.
【硼酸盐在CAL27细胞内的摄取检测】【Detection of borate uptake in CAL27 cells】
将细胞含量为60-75%的处于对数生长期的CAL27细胞接种至6孔板(每孔1×106个细胞)。待细胞贴壁,用浓度分别为20或100 μM的对照组BPA或硼酸盐化合物分别作用于细胞48 h(每组设置3个平行实验,并设置空白对照组)。作用完毕,弃去培养液,用PBS洗涤细胞3次;随后每孔加入1mL浓度为2mg/mL的链霉蛋白酶溶液并于4 ℃下孵育4 h。弃去含链霉蛋白酶溶液,PBS洗涤细胞3次,用RIPA裂解液裂解细胞,收集细胞裂解液,离心收集上清液。通过ICP-MS检测上清液中的10B浓度。CAL27 cells in logarithmic growth phase at 60-75% cell content were seeded into 6 -well plates (1 x 106 cells per well). After the cells adhered, the control group BPA or borate compounds were used to act on the cells for 48 h respectively (3 parallel experiments were set for each group, and a blank control group was set). After the action was completed, the culture medium was discarded, and the cells were washed three times with PBS; then, 1 mL of pronase solution with a concentration of 2 mg/mL was added to each well and incubated at 4 °C for 4 h. The pronase-containing solution was discarded, the cells were washed 3 times with PBS, the cells were lysed with RIPA lysis buffer, the cell lysate was collected, and the supernatant was collected by centrifugation. The concentration of 10 B in the supernatant was detected by ICP-MS.
表1硼酸盐化合物在CAL27细胞内的摄取含量检测结果Table 1 Test results of uptake content of borate compounds in CAL27 cells
(应该是对比于正常细胞结果的)(should be compared to normal cell results)

如表1所示,本发明制备的硼酸盐化合物在CAL27细胞内的摄取与作用浓度正相关,同时硼酸盐化合物在CAL27细胞内的摄取高于同浓度下的阳性对照组BPA。As shown in Table 1, the uptake of borate compounds prepared by the present invention in CAL27 cells is positively correlated with the concentration, and the uptake of borate compounds in CAL27 cells is higher than that of the positive control group BPA at the same concentration.
Claims (2)


Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110289828.3A CN112876503B (en) | 2021-03-18 | 2021-03-18 | Borate compound for cancer boron neutron capture therapeutic drug and preparation thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110289828.3A CN112876503B (en) | 2021-03-18 | 2021-03-18 | Borate compound for cancer boron neutron capture therapeutic drug and preparation thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112876503A CN112876503A (en) | 2021-06-01 |
| CN112876503B true CN112876503B (en) | 2022-04-29 |
Family
ID=76042692
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110289828.3A Active CN112876503B (en) | 2021-03-18 | 2021-03-18 | Borate compound for cancer boron neutron capture therapeutic drug and preparation thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN112876503B (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116410216B (en) * | 2023-04-12 | 2024-05-07 | 东莞市人民医院 | Small molecule boron drug, preparation method thereof, pharmaceutical composition and application thereof |
| WO2025082423A1 (en) * | 2023-10-20 | 2025-04-24 | 江苏中硼医疗科技研究院有限公司 | Lipoic acid derivative, vesicle compound and preparation therefor and use thereof |
| CN120192337A (en) * | 2023-12-23 | 2025-06-24 | 江苏中硼医疗科技研究院有限公司 | A polyboron complex and its preparation and application |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2740451B1 (en) * | 1995-10-27 | 1998-01-16 | Seripharm | NOVEL INTERMEDIATES FOR THE HEMISYNTHESIS OF TAXANES, THEIR PREPARATION METHODS AND THEIR USE IN THE GENERAL SYNTHESIS OF TAXANES |
| UY29149A1 (en) * | 2004-10-07 | 2006-05-31 | Boehringer Ingelheim Int | TIAZOLIL-DIHIDRO-INDAZOLES |
| JOP20190230A1 (en) * | 2009-01-15 | 2017-06-16 | Incyte Corp | Methods for repairing JAK inhibitors and related intermediates |
| CN103848856B (en) * | 2012-11-30 | 2016-08-03 | 苏州汉德景曦新药研发有限公司 | There is the camptothecin derivative of anti-tumor activity |
-
2021
- 2021-03-18 CN CN202110289828.3A patent/CN112876503B/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN112876503A (en) | 2021-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112876503B (en) | Borate compound for cancer boron neutron capture therapeutic drug and preparation thereof | |
| JP6831834B2 (en) | Method of preparing L-BPA | |
| CN109970630A (en) | A two-photon fluorescent probe capable of targeting mitochondria and its preparation method and application | |
| JP7455429B2 (en) | Boron carrier integrated with tumor diagnosis and treatment, its preparation method and use | |
| WO2020035011A1 (en) | Method for preparing 18f-bpa and intermediate | |
| CN117105948A (en) | A kind of dideuterated camptothecin derivatives and preparation method | |
| CN115353527A (en) | Boron-containing compound and application thereof | |
| CN108218672A (en) | Application of Metal Compound/Palladium Compound Catalytic Reduction System in Deallylation and Deuteration | |
| CN116969848B (en) | A method for preparing a drug intermediate for the treatment of advanced breast cancer | |
| CN102329263A (en) | Preparation method of N-acetyl-5-methoxytryptamine | |
| CN110642878A (en) | Preparation method of fluorine-labeled BPA | |
| CN115028652A (en) | High-light-sensitivity fluorescent probe for ovarian cancer photodynamic diagnosis and treatment | |
| CN107789623B (en) | Piperazine Substituted Silicon Phthalocyanine and Its Application in Photothermal Therapy | |
| CN116606313B (en) | Aza-BODIPY-based high-brightness NIR-II region photothermal agent and preparation method and application thereof | |
| CN112358492B (en) | Carborane celecoxib and its preparation and application in boron neutron capture therapy for head and neck cancer | |
| CN102924362B (en) | Preparation method of hexahydro-2-cyclopentyl-pyrryl amine hydrochloride | |
| CN105348309B (en) | F-BPA nucleophilic synthesis method | |
| US8680265B2 (en) | Process for the preparation of a boron-substituted porphyrin | |
| CN117126184A (en) | Supermolecule photosensitizer and preparation method and application thereof | |
| CN108295882A (en) | The preparation of core-shell nano catalyst and for the application in Buddhist nun's class medicine preparation | |
| CN110642879A (en) | Preparation method of fluorine standard BPA | |
| CN107224580B (en) | Application of alpha-amino acid-like boron trifluoride in boron neutron capture therapy | |
| CN111747872A (en) | A kind of synthetic method of p-toluenesulfonylurea | |
| EP3677581A1 (en) | Deuterated indoleamine 2,3-dioxygenase inhibitor and application thereof | |
| CN109810042B (en) | A kind of preparation method of asymmetric bisindolylmethane and derivatives thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |