CN112707943B - Combining 5'-terminal conjugate and neutral/cationic mixed lipid-encapsulated small interfering RNA and its modification method - Google Patents
Combining 5'-terminal conjugate and neutral/cationic mixed lipid-encapsulated small interfering RNA and its modification method Download PDFInfo
- Publication number
- CN112707943B CN112707943B CN202011644259.1A CN202011644259A CN112707943B CN 112707943 B CN112707943 B CN 112707943B CN 202011644259 A CN202011644259 A CN 202011644259A CN 112707943 B CN112707943 B CN 112707943B
- Authority
- CN
- China
- Prior art keywords
- chemical formula
- sirna
- antisense strand
- simb3
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/543—Lipids, e.g. triglycerides; Polyamines, e.g. spermine or spermidine
- A61K47/544—Phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5146—Organic macromolecular compounds; Dendrimers obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyamines, polyanhydrides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Optics & Photonics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Physics & Mathematics (AREA)
- Biomedical Technology (AREA)
- Nanotechnology (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biophysics (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本发明公开了结合5′‑末端缀合物和中性/阳离子混合脂材制剂包载修饰的小干扰RNA及其修饰方法。所述的修饰方法包括在siRNA的反义链或/和正义链的5′‑末端缀合一个或多个链接基团作为缀合基团,并利用中性脂材和阳离子混合脂材进行包载,还包括在链接基团的末端活泼酯上通过缩合反应链接靶向基团。通过本发明提供的化学修饰策略所获得的siRNA脂质复合物具有更好生物活性,细胞水平表现出优异的跨膜转运能力和靶mRNA的沉默活性,动物水平表现出高效的靶器官分布能力和抑制肿瘤生长的能力,而且在细胞水平和动物水平应用的安全性好,无明显毒性,为siRNA技术在临床的广泛应用奠定了基础。The invention discloses a modified small interfering RNA combined with a 5'-terminal conjugate and a neutral/cationic mixed lipid preparation and a modification method thereof. The modification method includes conjugating one or more linking groups at the 5′-end of the antisense strand or/and the sense strand of the siRNA as a conjugating group, and using neutral lipid material and cationic mixed lipid material for encapsulation. It also includes linking the targeting group through a condensation reaction on the terminal active ester of the linking group. The siRNA lipid complex obtained by the chemical modification strategy provided by the present invention has better biological activity, exhibits excellent transmembrane transport ability and silencing activity of target mRNA at the cellular level, and exhibits efficient target organ distribution at the animal level. The ability to inhibit tumor growth, and the safety of application at the cell level and animal level, without obvious toxicity, has laid a foundation for the wide application of siRNA technology in clinical practice.
Description
技术领域technical field
本发明涉及一种结合5′-末端缀合物和中性脂材/阳离子混合脂材包载修饰的小干扰RNA(siRNA)及其化学修饰方法和载体递送策略。本发明实现了对siRNA反义链的5′-末端缀合,缀合物的存在不影响siRNA的基因沉默活性。经过本发明方法缀合修饰并包载后的siRNA,具有稳定性好、递送效率高效、体内靶器官分布高、入胞能力强、毒性低且生物活性好等有点,能够广泛应用于抗肿瘤、抗病毒及代谢类疾病的药物研究中。本发明属于生物医药技术领域。The present invention relates to a modified small interfering RNA (siRNA) combined with a 5'-terminal conjugate and a neutral lipid/cationic mixed lipid material, as well as a chemical modification method and a carrier delivery strategy. The present invention realizes conjugation to the 5'-end of the antisense strand of siRNA, and the presence of the conjugate does not affect the gene silencing activity of siRNA. The siRNA conjugated, modified and encapsulated by the method of the present invention has the advantages of good stability, high delivery efficiency, high target organ distribution in vivo, strong cell entry ability, low toxicity and good biological activity, and can be widely used in anti-tumor, Antiviral and metabolic diseases drug research. The invention belongs to the technical field of biomedicine.
背景技术Background technique
小干扰RNA(siRNA)是一类具有巨大临床前景的药物候选分子,它的靶标是细胞内的mRNA,可以在基因水平上直接沉默靶标基因的表达,从根本上预防疾病的发生和发展。天然的siRNA由dsRNA在胞内被Dicer剪切至21-25mer,通常会有5′-磷酸基和3′-羟基。合成的siRNA的5′-羟基会在胞内被Clp1细胞激酶迅速磷酸化,这也是siRNA发挥活性的必要条件,因为siRNA只有在5′-磷酸化后,才可以进入RISC发挥其基因沉默活性。与传统的小分子药物相比,siRNA具有更高的特异性,更低的毒性和副作用,易于制备等优点。但是由于其血清稳定性差、跨膜困难、易脱靶、激发免疫应答和其他缺陷,使临床应用受到限制。Small interfering RNA (siRNA) is a class of drug candidate molecules with great clinical prospects. Its target is intracellular mRNA, which can directly silence the expression of target genes at the gene level and fundamentally prevent the occurrence and development of diseases. Native siRNA is cleaved intracellularly by Dicer from dsRNA to 21-25mer, usually with 5'-phosphate and 3'-hydroxyl groups. The 5′-hydroxyl group of the synthesized siRNA will be rapidly phosphorylated by the Clp1 cell kinase in the cell, which is also a necessary condition for the siRNA to exert its activity, because the siRNA can enter RISC to exert its gene silencing activity only after 5′-phosphorylation. Compared with traditional small molecule drugs, siRNA has the advantages of higher specificity, lower toxicity and side effects, and easy preparation. However, its clinical application is limited due to its poor serum stability, difficulty in transmembrane transmembrane, easy off-target, stimulation of immune response and other defects.
2018年,FDA(美国食品药品监督管理局)批准了第一例siRNA药物

阳离子脂材依靠其与siRNA的正负电荷间的库仑力作用结合并包载siRNA。但阳离子脂质体的毒性较强,且易于在生理条件下带负电的血清蛋白结合,产生免疫原性、肝脏毒性并使siRNA易在靶器官外释放。这些缺陷限制了阳离子脂质体在包载siRNA成药的进一步应用。Cationic lipids bind and encapsulate siRNA by the Coulomb force between the positive and negative charges of siRNA. However, cationic liposomes are highly toxic and easily bind to negatively charged serum proteins under physiological conditions, resulting in immunogenicity, liver toxicity, and easy release of siRNA outside target organs. These defects limit the further application of cationic liposomes to encapsulate siRNA into drugs.
本发明人前期设计合成了碱基乙酰胺甘油醚分子DNCA(CN108059619A),因其具有碱基性质的头部,可以通过氢键作用和π-π堆积作用结合并包载单链核酸药物和质粒(CN1084478807A)。结合DNCA及发明人前期设计合成的以赖氨酸作为头部的阳离子脂材CLD共同包载3′,3″-双肽缀siRNA已在细胞水平成功应用(Mol Pharm,2019,16,4920)。在此,我们对包载方法进行优化,重新制定并探索了最优的DNCA/CLD转染处方,并加入DSPE-PEG,以优化混合制剂体内应用的性质。结合使用磷酸二酯键作为siRNA链末端与缀合基团的链接的缀合策略和化学修饰方法,获得了高效低毒的缀合物/中性/阳离子混合脂材siRNA药物包载递送系统。The inventor designed and synthesized the base acetamide glyceryl ether molecule DNCA (CN108059619A) in the early stage. Because of its base head, it can be combined by hydrogen bonding and π-π stacking and encapsulate single-stranded nucleic acid drugs and plasmids (CN1084478807A). Combining DNCA and the cationic lipid CLD with lysine as the head designed and synthesized by the inventors to co-encapsulate 3′,3″-dipeptide-conjugated siRNA has been successfully applied at the cellular level (Mol Pharm, 2019, 16, 4920) Here, we optimized the encapsulation method, reformulated and explored the optimal DNCA/CLD transfection recipe, and added DSPE-PEG to optimize the properties of the mixed formulation for in vivo applications. Combining the use of phosphodiester bonds as siRNA The conjugation strategy and chemical modification method of linking the chain end with the conjugating group have obtained a conjugate/neutral/cationic mixed lipid siRNA drug-encapsulated delivery system with high efficiency and low toxicity.
发明内容SUMMARY OF THE INVENTION
为了提高小干扰RNA(siRNA)体内运送过程中靶器官递送的特异性,入胞效率及胞内释放的有效性,本发明提供了一种结合5′-末端缀合物和中性脂材/阳离子脂材混合载体包载的siRNA的化学修饰方法和载体递送策略。本发明通过将缀合基团制备为亚磷酰胺单体,使用固相合成仪实现缀合物的快速合成。本发明利用中性脂材和阳离子混合脂材包载siRNA缀合物,以实现更有效且安全无毒的体内siRNA递送,从而提高siRNA的成药性。In order to improve the specificity of target organ delivery, cell entry efficiency and intracellular release effectiveness of small interfering RNA (siRNA) in vivo delivery, the present invention provides a combination of 5'-terminal conjugates and neutral lipids/ Chemical modification method and carrier delivery strategy of siRNA encapsulated in cationic lipid mixed carrier. In the present invention, the conjugated group is prepared as a phosphoramidite monomer, and a solid-phase synthesizer is used to realize the rapid synthesis of the conjugate. The present invention utilizes neutral lipid material and cationic mixed lipid material to encapsulate the siRNA conjugate to achieve more effective, safe and non-toxic delivery of siRNA in vivo, thereby improving the druggability of siRNA.
为了达到上述目的,本发明采用的技术方案如下:In order to achieve the above object, the technical scheme adopted in the present invention is as follows:
本发明的一种对小干扰RNA(siRNA)的化学修饰方法,包括在siRNA的正义链或/和反义链的5′-末端缀合一个或多个链接基团作为缀合基团,并利用中性脂材和阳离子混合脂材进行包载,其中所述链接基团的结构式如化学式I或化学式II所示,其中,化学式I所示的结构为一个六碳单元的链接基团,化学式II所示的结构为一个三碳单元的链接基团;A chemical modification method for small interfering RNA (siRNA) of the present invention, comprising conjugating one or more linking groups as a conjugating group at the 5'-end of the sense strand or/and antisense strand of the siRNA, and Neutral lipid material and cationic mixed lipid material are used for encapsulation, wherein the structural formula of the linking group is shown in chemical formula I or chemical formula II, wherein the structure shown in chemical formula I is a six-carbon unit linking group, chemical formula The structure shown in II is a linking group of three carbon units;
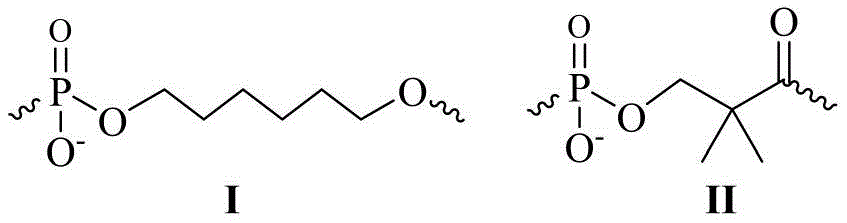
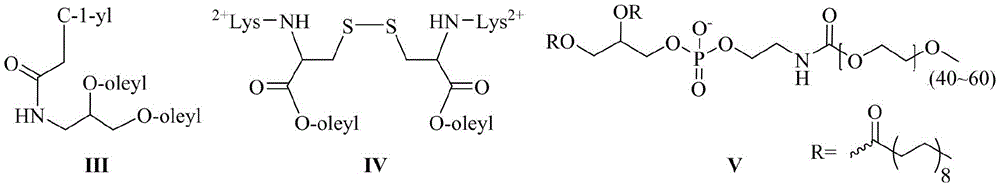
其中,所述的混合脂材由化学式III所示的中性胞苷脂材、化学式IV所示的阳离子脂材组成,或由化学式III所示的中性胞苷脂材、化学式IV所示的阳离子脂材、化学式V所示的PEG2000-DSPE组成:Wherein, the mixed resin is composed of neutral cytidine resin represented by chemical formula III, cationic resin represented by chemical formula IV, or neutral cytidine resin represented by chemical formula III, chemical formula IV The composition of cationic lipid material and PEG2000-DSPE shown in chemical formula V:

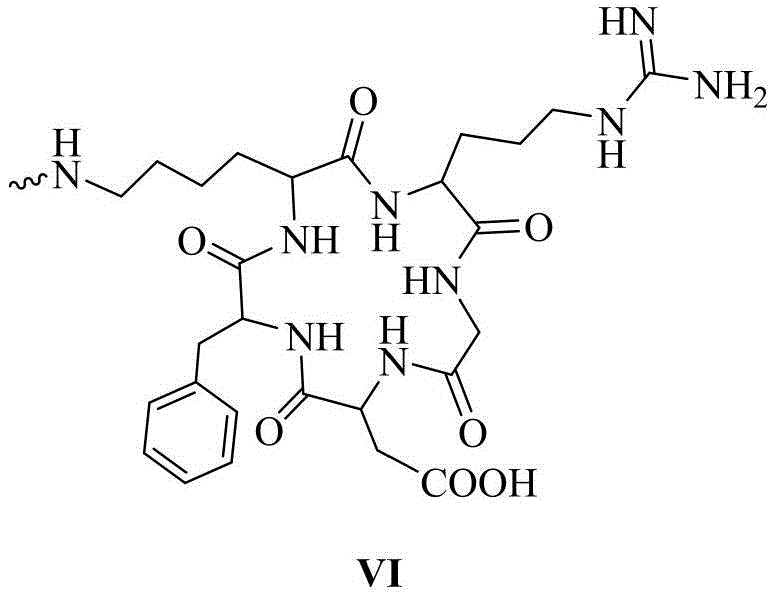
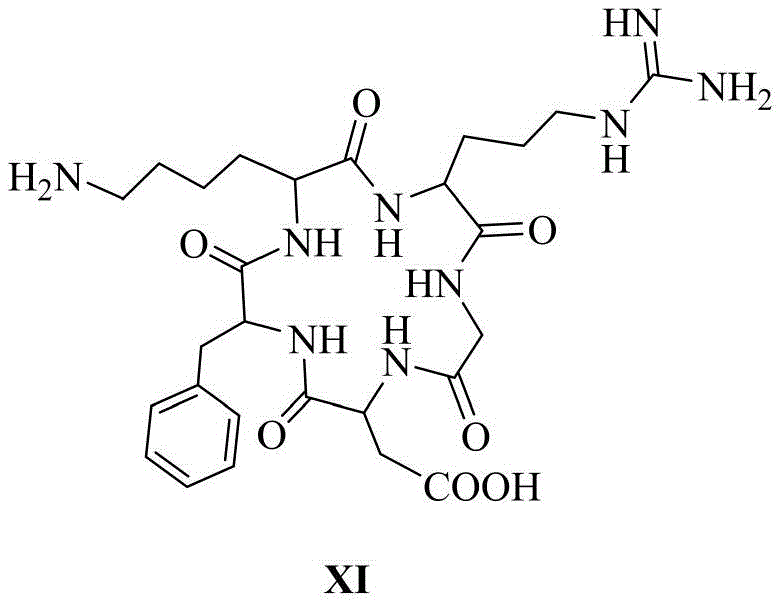
其中,优选的,在siRNA的反义链或/和正义链5′-末端缀合一个或多个链接基团的同时,还包括在链接基团的末端活泼酯上通过缩合反应链接靶向基团,所述的靶向基团结构式如化学式VI所示:Among them, preferably, when the antisense strand or/and the 5′-end of the sense strand of the siRNA are conjugated with one or more linking groups, it also includes linking the targeting group through a condensation reaction on the terminal active ester of the linking group group, the structural formula of the targeting group is shown in chemical formula VI:
其中,优选的,在对siRNA进行缀合前,将化学式VII、化学式VIII所示的链接基团分别制备成化学式IX、化学式X所示的亚磷酰胺单体,然后通过磷酸二酯键与siRNA进行缀合;随后在化学式IX或化学式X所示的亚磷酰胺单体的活泼酯上通过缩合反应连接化学式VI所示的靶向基团;Among them, preferably, before conjugating the siRNA, the linking groups shown in chemical formula VII and chemical formula VIII are prepared into phosphoramidite monomers shown in chemical formula IX and chemical formula X, respectively, and then the siRNA is linked to the siRNA through a phosphodiester bond. Carry out conjugation; then connect the targeting group shown in chemical formula VI through a condensation reaction on the active ester of the phosphoramidite monomer shown in chemical formula IX or chemical formula X;

其中,优选的,使用固相合成技术,采用亚磷酰胺法合成天然的及缀合链接基团的siRNA正义链及反义链序列,在RNA合成仪上,序列从3′-端至5′-端进行合成,每偶联一个核苷为一个循环,在天然核苷亚磷酰胺单体偶联21次后,将一个或几个所述的亚磷酰胺单体IX或X在序列的5′-末端相应位置进行偶联,每个循环包括四个反应:脱DMT、偶联、封闭、氧化。Among them, it is preferred to use solid-phase synthesis technology to synthesize the natural and conjugated linking group siRNA sense strand and antisense strand sequence by phosphoramidite method. On RNA synthesizer, the sequence is from 3'-end to 5' -Synthesis is carried out at the end, each coupling of a nucleoside is a cycle, after 21 couplings of the natural nucleoside phosphoramidite monomer, one or several of the phosphoramidite monomers IX or X in the
其中,优选的,每个循环亚磷酰胺单体进样后的偶联时间为600秒/次,偶联3次。Among them, preferably, the coupling time after the injection of the phosphoramidite monomer in each cycle is 600 seconds/time, and the coupling is performed 3 times.
其中,优选的,按照摩尔百分比计算,所述的混合脂材由72.7%化学式III所示的中性胞苷脂材、27.3%化学式IV所示的阳离子脂材组成,其中,优选的,总脂质与siRNA的摩尔比为10:1。或所述的混合脂材由39.7%的化学式III所示的中性胞苷脂材、59.6%化学式IV所示的阳离子脂材以及0.7%化学式V所示的PEG2000-DSPE组成,其中,优选的,总脂质与siRNA的摩尔比为5.3:1。Wherein, preferably, calculated according to the mole percentage, the mixed resin is composed of 72.7% of the neutral cytidine resin shown by the chemical formula III and 27.3% of the cationic resin shown by the chemical formula IV, wherein, preferably, the total lipid The molar ratio of mass to siRNA was 10:1. Or the mixed resin material is composed of 39.7% neutral cytidine resin shown in chemical formula III, 59.6% cationic resin material shown in chemical formula IV and 0.7% PEG2000-DSPE shown in chemical formula V, wherein, the preferred , the molar ratio of total lipid to siRNA was 5.3:1.
其中,优选的,利用中性脂材和阳离子脂材的混合脂材进行包载的方法为:将所述的混合脂材溶于乙醇溶液中,将反义链或/和正义链的5′-末端通过磷酸二酯键缀合一个或多个链接基团的siRNA溶于DEPC水中,使用未缓冲溶液或缓冲溶液混合,超声处理,使用0.22μm滤膜过滤,滤液中包含修饰后的siRNA,其中,优选的,所述的未缓冲溶液为生理盐水、水或任何转染优化溶液,所述的缓冲溶液包含乙酸盐、柠檬酸盐、碳酸盐、磷酸盐或其任何组合。Among them, preferably, the method for encapsulation by using the mixed lipid material of neutral lipid material and cationic lipid material is: dissolving the mixed lipid material in an ethanol solution, and 5' of the antisense strand or/and the sense strand - siRNA whose ends are conjugated with one or more linking groups through a phosphodiester bond is dissolved in DEPC water, mixed with unbuffered solution or buffered solution, sonicated, filtered through a 0.22 μm filter, and the filtrate contains the modified siRNA, Wherein, preferably, the unbuffered solution is physiological saline, water or any transfection optimized solution, and the buffered solution comprises acetate, citrate, carbonate, phosphate or any combination thereof.
其中,所述的化学修饰策略还包括与其它化学修饰策略的共同使用,包括siRNA的2′-O-甲基(2′-OMe)、2′-氟代(2′-F)、2′-O-甲氧乙基(2′-O-MOE)、锁核苷酸(LNA),磷硫骨架修饰及其他末端缀合方式等。Wherein, the chemical modification strategy also includes the co-use with other chemical modification strategies, including 2'-O-methyl (2'-OMe), 2'-fluoro (2'-F), 2'- siRNA of siRNA -O-methoxyethyl (2'-O-MOE), locked nucleotide (LNA), modification of phosphorus-sulfur backbone and other terminal conjugation methods, etc.
进一步的,本发明还提出了按照以上任一项所述的化学修饰方法合成的结合5′-末端缀合物和中性/阳离子混合脂材载体包载修饰的小干扰RNA。Further, the present invention also proposes a 5'-terminal conjugate synthesized according to any one of the chemical modification methods described above and a neutral/cationic mixed lipid material carrier to encapsulate the modified small interfering RNA.
本发明所述的5′-末端缀合方式与3′-末端缀合相比具有以下特点:Compared with the 3'-end conjugation, the 5'-end conjugation method of the present invention has the following characteristics:
a.将缀合基团制备成相应的亚磷酰胺单体,使缀合部分可以直接作为单体使用固相合成仪进行合成,合成简单快速;a. The conjugated group is prepared into the corresponding phosphoramidite monomer, so that the conjugated part can be directly synthesized as a monomer using a solid-phase synthesizer, and the synthesis is simple and fast;
b.链接基团X的末端保留活泼酯,作为活性反应位点,可利用缩合反应高效链接多种官能团,快速扩展缀合物规模;b. The end of the linking group X retains an active ester as an active reaction site, which can efficiently link a variety of functional groups by condensation reaction and rapidly expand the scale of the conjugate;
c.链接基团间以磷酸二酯键进行连接,可由细胞内的酶进行水解剪切,释放出带有磷酸末端的未缀合siRNA单链,最终达到更易载入RISC复合物、提高siRNA基因沉默活性的能力。c. The linking groups are connected by phosphodiester bonds, which can be hydrolyzed and cleaved by enzymes in the cell to release unconjugated siRNA single-strands with phosphate ends, which are ultimately easier to load into RISC complexes and improve siRNA genes. The ability to silence activity.
在本发明的具体实施例中,5-末端缀合物修饰后的序列选自以下序列所组成的群组:In a specific embodiment of the present invention, the modified sequence of the 5-terminal conjugate is selected from the group consisting of the following sequences:
(1)在小干扰RNA序列的正义链或反义链的5′-末端缀合一个化学式II所示的链接基团而得到的反义链或正义链;(1) An antisense strand or a sense strand obtained by conjugating a linking group shown in chemical formula II to the 5'-end of the sense strand or antisense strand of the small interfering RNA sequence;
(2)在小干扰RNA序列的正义链或反义链的5′-末端依次延3′-5′方向缀合一个或几个化学式I和一个化学式II所示的链接基团而得到的反义链或正义链;(2) The antisense obtained by conjugating one or more linking groups shown in chemical formula I and one chemical formula II to the 5′-end of the sense strand or antisense strand of the small interfering RNA sequence in turn in the 3′-5′ direction sense chain or sense chain;
(3)在小干扰RNA序列的正义链或反义链的5′-末端缀合一个化学式II所示的链接基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式X,随后在化学式X所示的基团的活泼酯上通过缩合反应连接化学式VI所示的靶向基团,而得到的正义链或反义链;(3) Conjugate a linking group shown in chemical formula II to the 5′-end of the sense strand or antisense strand of the small interfering RNA sequence, and the conjugation is completed by a solid-phase synthesizer, and the phosphoramidite monomer used is chemical formula X, then on the active ester of the group shown in chemical formula X, the targeting group shown in chemical formula VI is connected by condensation reaction, and the obtained sense strand or antisense strand;
(4)在小干扰RNA序列的反义链的正义链或5′-末端依次延3′-5′方向缀合一个或几个化学式I和一个化学式II所示的链接基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式IX和X,随后在化学式X所示的基团的活泼酯上通过缩合反应连接化学式VI所示的靶向基团,而得到的反义链或正义链缀合物;(4) Conjugate one or more linking groups shown in chemical formula I and one chemical formula II to the sense strand or 5'-end of the antisense strand of the small interfering RNA sequence in turn in the 3'-5' direction, and the conjugation uses The solid-phase synthesizer is completed, the phosphoramidite monomers used are chemical formula IX and X, and then the target group shown in chemical formula VI is connected by condensation reaction on the active ester of the group shown in chemical formula X, and the obtained reaction a sense strand or a sense strand conjugate;
上述(1)-(4)中的反义链和正义链为自由组合的形式。The antisense strand and the sense strand in the above (1)-(4) are in the form of free combination.
本发明所述的siRNA及其缀合物是按照需求,将缀合或未缀合修饰的反义链与正义链经退火后形成的RNA双链复合物。The siRNA and its conjugates of the present invention are RNA double-stranded complexes formed by annealing a conjugated or unconjugated modified antisense strand and a sense strand according to requirements.
以RL1As/RL0S-siRNA为例,在siRNA的反义链(As表示)5′-末端依次(3′-5′方向)通过固相合成缀合一个化学式I(数字表示化学式I所示的链接基团的数量)和一个化学式II(L表示)所示的链接基团,其中缀合基团间通过磷酸二酯键链接,随后通过缩合反应缀合一个化学式VI所示的靶向基团cRGD(R表示),即RL1As。在正义链(S表示)5′-末端通过固相合成缀合一个化学式II(L表示),随后通过缩合反应缀合一个化学式VI所示的靶向基团cRGD(R表示),即RL0S,随后将合成纯化完毕的反义链与正义链按照1:1.1的摩尔比进行退火,形成RL1As/RL0S-siRNA,其余缀合的siRNA以此类推。Taking RL1As/RLOS-siRNA as an example, at the 5'-end (3'-5' direction) of the antisense strand (represented by As) of the siRNA, a chemical formula I (the number represents the link shown in chemical formula I) is conjugated by solid-phase synthesis. number of groups) and a linking group shown in chemical formula II (represented by L), wherein the conjugating groups are linked by phosphodiester bonds, and then a targeting group cRGD shown in chemical formula VI is conjugated through a condensation reaction (R represents), namely RL1As. A chemical formula II (represented by L) is conjugated to the 5'-end of the sense strand (represented by S) by solid-phase synthesis, and then a targeting group cRGD (represented by R) represented by chemical formula VI is conjugated by a condensation reaction, namely RLOS, Then, the synthesized and purified antisense strand and the sense strand are annealed at a molar ratio of 1:1.1 to form RL1As/RLOS-siRNA, and the rest of the conjugated siRNA is deduced by analogy.
在本发明的一个具体实施例中,待修饰的siRNA为靶向ERK通路中BRAFV600E突变的mRNA的siMB3序列。siMB3修饰前的序列如下:In a specific embodiment of the present invention, the siRNA to be modified is the siMB3 sequence targeting the BRAF V600E mutated mRNA in the ERK pathway. The sequence of siMB3 before modification is as follows:
siMB3:正义链:5′-GCUACAGAGAAAUCUCGAUdtdt-3′siMB3: sense strand: 5′-GCUACAGAGAAAUCUCGAUdtdt-3′
反义链:5′-AUCGAGAUUUCUCUGUAGCdtdt-3′Antisense strand: 5'-AUCGAGAUUUCUCUGUAGCdtdt-3'
在本发明的一个具体实施例中,在以上所述siMB3序列由化学式III所示的中性脂材、化学式IV所示的化合物阳离子脂材以及化学式V所示的化合物组成的混合脂材包载时,能够以球形囊泡形态均匀分布,siRNA脂质复合物粒径均一,在100nm左右,表面电性中性偏负。In a specific embodiment of the present invention, the above-mentioned siMB3 sequence is encapsulated by a mixed lipid material consisting of a neutral lipid material represented by chemical formula III, a compound cationic lipid material represented by chemical formula IV, and a compound represented by chemical formula V. When , the siRNA lipoplexes can be uniformly distributed in the form of spherical vesicles, and the particle size of the siRNA lipoplexes is uniform.
在本发明的一个具体实施例中,siMB3及其反义链末端缀合一个化学式I和一个化学式VI所示的缀合基团,并使用商用阳离子转染试剂Lipofectamine2000或由化学式III所示的中性脂材、化学式IV所示的化合物阳离子脂材以及化学式V所示的化合物组成的混合脂材包载时,混合脂材具有更优的入胞效率和基因沉默活性。In a specific embodiment of the present invention, siMB3 and its antisense chain end are conjugated with a conjugation group shown in chemical formula I and a chemical formula VI, and use commercial cationic transfection reagent Lipofectamine2000 or by chemical formula III. When encapsulated by the mixed lipid material composed of the cationic lipid material, the compound of the chemical formula IV, and the compound of the chemical formula V, the mixed lipid material has better cell entry efficiency and gene silencing activity.
在本发明的一个具体实施例中,在以上所述的siMB3序列的反义链5-末端缀合依次缀合三个化学式I所示的缀合基团、一个化学式II所示的缀合基团,并通过活性酯反应缀合一个化学式VI所示的靶向基团cRGD,并由化学式III所示的中性脂材、化学式IV所示的化合物阳离子脂材以及化学式V所示的化合物组成的混合脂材包载时,具有最优的细胞摄取能力。In a specific embodiment of the present invention, three conjugating groups shown in chemical formula I and one conjugating group shown in chemical formula II are sequentially conjugated to the 5-terminal of the antisense strand of the siMB3 sequence described above. group, and a targeting group cRGD shown in chemical formula VI is conjugated through active ester reaction, and is composed of neutral lipid material shown in chemical formula III, compound cationic lipid material shown in chemical formula IV and compound shown in chemical formula V. When encapsulated in the mixed lipid material, it has the optimal cellular uptake capacity.
在本发明的一个具体实施例中,在以上所述的siMB3序列的反义链5-末端缀合依次缀合三个化学式I所示的缀合基团、一个化学式II所示的缀合基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式IX和X,其中缀合基团间通过磷酸二酯键链接,随后在化学式X所示的基团的活泼酯上通过缩合反应连接一个化学式VI所示的靶向基团cRGD,并由化学式III所示的中性脂材、化学式IV所示的化合物阳离子脂材以及化学式V所示的化合物组成的混合脂材包载时,具有最优的细胞水平基因沉默活性。In a specific embodiment of the present invention, three conjugating groups shown in chemical formula I and one conjugating group shown in chemical formula II are sequentially conjugated to the 5-terminal of the antisense strand of the siMB3 sequence described above. The conjugation was completed using a solid-phase synthesizer, and the phosphoramidite monomers used were chemical formulas IX and X, wherein the conjugation groups were linked by phosphodiester bonds, and then on the active ester of the group shown in chemical formula X A targeting group cRGD represented by chemical formula VI is connected through a condensation reaction, and a mixed lipid material package composed of a neutral lipid material represented by chemical formula III, a compound cationic lipid material represented by chemical formula IV and a compound represented by chemical formula V When loaded, it has the best cell-level gene silencing activity.
在本发明的一个具体实施例中,在以上所述的siMB3序列的反义链5-末端缀合依次缀合三个化学式I所示的缀合基团、一个化学式II所示的缀合基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式IX和X,其中缀合基团间通过磷酸二酯键链接,随后在化学式X所示的基团的活泼酯上通过缩合反应连接一个化学式VI所示的靶向基团cRGD,并由化学式III所示的中性脂材、化学式IV所示的化合物阳离子脂材以及化学式V所示的化合物组成的混合脂材包载时,具有最优的动物体内抗肿瘤活性。In a specific embodiment of the present invention, three conjugating groups shown in chemical formula I and one conjugating group shown in chemical formula II are sequentially conjugated to the 5-terminal of the antisense strand of the siMB3 sequence described above. The conjugation was completed using a solid-phase synthesizer, and the phosphoramidite monomers used were chemical formulas IX and X, wherein the conjugation groups were linked by phosphodiester bonds, and then on the active ester of the group shown in chemical formula X A targeting group cRGD represented by chemical formula VI is connected through a condensation reaction, and a mixed lipid material package composed of a neutral lipid material represented by chemical formula III, a compound cationic lipid material represented by chemical formula IV and a compound represented by chemical formula V When loaded, it has the best anti-tumor activity in animals.
在本发明的一个具体实施例中,在以上所述的siMB3序列的反义链5-末端缀合一个化学式II所示的缀合基团。缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式X,随后在化学式X所示的基团的活泼酯上通过缩合反应连接一个化学式VI所示的靶向基团cRGD,并由化学式III所示的中性脂材、化学式IV所示的化合物阳离子脂材以及化学式V所示的化合物组成的混合脂材包载时,具有最优的动物体内肿瘤组织分布。In a specific embodiment of the present invention, the 5-terminus of the antisense strand of the above-mentioned siMB3 sequence is conjugated with a conjugating group shown in chemical formula II. The conjugation is completed using a solid-phase synthesizer, and the phosphoramidite monomer used is chemical formula X, and then a targeting group cRGD shown in chemical formula VI is connected by condensation reaction on the active ester of the group shown in chemical formula X, and When encapsulated by the mixed lipid material consisting of the neutral lipid material represented by the chemical formula III, the compound cationic lipid material represented by the chemical formula IV, and the compound represented by the chemical formula V, it has the optimal tumor tissue distribution in animals.
相较于现有技术,本发明的优点在于:Compared with the prior art, the advantages of the present invention are:
1.本发明提供的结合靶向基团缀合及中性/阳离子混合载体包载的化学修饰策略,所获得的siRNA脂质复合物具有更好生物活性,细胞水平表现出优异的跨膜转运能力和靶mRNA的沉默活性,动物水平表现出高效的靶器官分布能力和抑制肿瘤生长的能力,而且在细胞水平和动物水平应用的安全性好,无明显毒性,为siRNA技术在临床的广泛应用奠定了基础;1. The chemical modification strategy of combining targeting group conjugation and neutral/cationic mixed carrier encapsulation provided by the present invention, the obtained siRNA lipid complex has better biological activity, and exhibits excellent transmembrane transport at the cellular level The ability and silencing activity of target mRNA, animal level shows efficient target organ distribution ability and tumor growth inhibition ability, and it has good safety and no obvious toxicity in cell level and animal level, which is widely used in clinical application of siRNA technology Foundation;
2.中性核苷脂材具有碱基头部,可以与siRNA通过氢键作用和π-π堆积作用结合,并且与阳离子脂材和siRNA间的电荷作用相比,在体内应用时更为稳定,在循环过程中不易吸附带电的颗粒或蛋白,避免了脂质复合物在靶器官外的解体与siRNA的释放。并且基于非电性作用的结合减少了阳离子脂材在制剂处方中的用量,阳离子脂材与siRNA的氮磷比仅为3/1,远低于其余报道中的阳离子脂材用量。2. The neutral nucleoside lipid material has a base head, which can be combined with siRNA through hydrogen bonding and π-π stacking, and is more stable in in vivo application than the charge interaction between cationic lipid material and siRNA , it is not easy to adsorb charged particles or proteins during the circulation process, avoiding the disassembly of lipid complexes outside the target organ and the release of siRNA. And the combination based on non-electric effect reduces the dosage of cationic resin in the formulation. The nitrogen-phosphorus ratio of cationic resin and siRNA is only 3/1, which is far lower than the dosage of cationic resin in other reports.
3.5′-缀合具有合成简单快速,可作为底物高效扩展缀合物组合,并且在胞内降解代谢时可释放出5′-末端为磷酸的siRNA反义链,更易进入RISC复合物发挥基因沉默活性。本发明证明了反义链5′-末端缀合不影响siRNA的基因沉默活性,扩展了siRNA缀合位点,丰富了siRNA的修饰策略。3. 5'-conjugation is simple and fast to synthesize, can be used as a substrate to efficiently expand the conjugate combination, and can release the siRNA antisense strand with 5'-terminal phosphoric acid during intracellular degradation and metabolism, making it easier to enter the RISC complex to play genes Silencing activity. The present invention proves that the 5'-end conjugation of the antisense strand does not affect the gene silencing activity of siRNA, expands the siRNA conjugation site, and enriches the modification strategy of siRNA.
附图说明Description of drawings
图1为siRNA/DNCA/CLD/PEG-DSPE脂质复合物和空载体的透射电镜(TEM)照片;Figure 1 is a transmission electron microscope (TEM) photograph of siRNA/DNCA/CLD/PEG-DSPE lipoplex and empty carrier;
图2为反义链5′-末端单缀合的siRNA中性/阳离子脂质复合物的细胞摄取(10nM,4h);Figure 2 is the cellular uptake (10 nM, 4h) of siRNA neutral/cationic lipid complexes monoconjugated at the 5'-end of the antisense strand;
图3为反义链5′-末端单缀合的siRNA中性/阳离子脂质复合物的胞内分布情况(50nM,4h),其中蓝色表示细胞核,红色表示Cy3标记的siRNA,绿色表示溶酶体,图中标尺为10μm;Figure 3 shows the intracellular distribution of siRNA neutral/cationic lipid complexes mono-conjugated at the 5′-end of the antisense strand (50nM, 4h), in which blue represents the nucleus, red represents Cy3-labeled siRNA, and green represents soluble siRNA Enzyme, the scale bar in the figure is 10 μm;
图4为5′-末端单缀合和5′,5″-双缀合的siRNA中性/阳离子脂质复合物的靶基因沉默活性(50nM,48h);Figure 4 is the target gene silencing activity of 5'-terminal mono-conjugated and 5',5"-diconjugated siRNA neutral/cationic lipid complexes (50nM, 48h);
图5为5′-末端单缀合和5′,5″-双缀合的siRNA中性/阳离子脂质复合物的抗肿瘤活性和载体细胞毒性(100nM,72h);Figure 5 shows the antitumor activity and carrier cytotoxicity of 5'-terminal monoconjugated and 5',5"-diconjugated siRNA neutral/cationic lipid complexes (100 nM, 72h);
图6为反义链5′-末端单缀合的siRNA中性/阳离子脂质复合物的动物体内(a)以及离体后(b)的分布情况,使用小动物活体成像系统拍摄,激发光为745nm,发射光为800nm;Figure 6 shows the distribution of neutral/cationic lipid complexes of siRNA mono-conjugated at the 5'-end of the antisense strand in vivo (a) and in vitro (b), photographed using a small animal in vivo imaging system, with excitation light is 745nm, and the emission light is 800nm;
图7为反义链5′-末端单缀合的siRNA(RL0As/S)的动物体内抗肿瘤活性(Balb/c-裸鼠,腋下接种A375);Figure 7 is the anti-tumor activity of 5'-terminal mono-conjugated siRNA (RLOAs/S) of the antisense strand in animals (Balb/c-nude mice, inoculated with A375 in the armpit);
a.当肿瘤体积达到约50mm3时为第0天,制剂在第1、3、5、8天通过静脉注射给药,每天测量并计算肿瘤体积;b.整个治疗过程中小鼠的体重变化曲线;c.最后一次给药后48h安乐死小鼠,收集肿瘤并对肿瘤称重记录;d.取出的肿瘤内BRAFV600EmRNA表达;e.免疫组化考察取出的肿瘤组织内BRAFV600E表达,黄褐色斑点代表BRAFV600E蛋白表达量,蓝色代表细胞核,黑色标尺为50μm。数据使用mean±SD表示,n=5,***p<0.001;a. When the tumor volume reached about 50mm3, it was the 0th day, the preparation was administered by intravenous injection on the 1st, 3rd , 5th, and 8th days, and the tumor volume was measured and calculated every day; b. The weight change curve of the mice during the whole treatment process ; c. Euthanize the mice 48h after the last administration, collect the tumor and record the weight of the tumor; d. BRAF V600E mRNA expression in the tumor taken out; e. BRAF V600E expression in the tumor tissue taken out by immunohistochemical investigation, yellowish brown Dots represent BRAF V600E protein expression, blue represents nuclei, and the black bar is 50 μm. Data are represented by mean±SD, n=5, ***p<0.001;
图8为反义链5′-末端单缀合的siRNA(RL2As/S)的动物体内抗肿瘤活性(Balb/c-裸鼠,腋下接种A375);Figure 8 is the anti-tumor activity of antisense strand 5'-terminal monoconjugated siRNA (RL2As/S) in animals (Balb/c-nude mice, inoculated with A375 in the armpit);
a.当肿瘤体积达到约50mm3时为第0天,制剂在第1、3、5、7、9天通过静脉注射给药,每天测量并计算肿瘤体积,每只小鼠以第0天的肿瘤体积为1进行归一化处理;b.最后一次给药后48h安乐死小鼠,收集肿瘤并对肿瘤称重记录;c.对取出的肿瘤拍照观察;d.取出的肿瘤内BRAFV600EmRNA表达;e.免疫组化考察取出的肿瘤组织内BRAFV600E表达,黄褐色斑点代表BRAFV600E蛋白表达量,蓝色代表细胞核,黑色标尺为50μm;f.整个治疗过程中小鼠的体重变化曲线。数据使用mean±SD表示,n=5,*p<0.05,**p<0.01,***p<0.001,****p<0.0001;a. When the tumor volume reached about 50mm3, it was the 0th day. The preparation was administered by intravenous injection on the 1st, 3rd , 5th, 7th, and 9th days. The tumor volume was measured and calculated every day. The tumor volume was normalized to 1; b. The mice were euthanized 48 hours after the last administration, and the tumors were collected and weighed for recording; c. The removed tumors were photographed and observed; d. BRAF V600E mRNA expression in the removed tumors ; e. The expression of BRAF V600E in the removed tumor tissue was investigated by immunohistochemistry, the yellow-brown spots represent the protein expression of BRAF V600E , the blue represents the nucleus, and the black scale is 50 μm; f. The weight change curve of the mice during the whole treatment process. Data are expressed as mean±SD, n=5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001;
图9为反义链5′-末端单缀合的siRNA(RL0As/S)在动物体内应用时的肝肾生化指标;Fig. 9 is the biochemical indexes of liver and kidney when the 5'-end single-conjugated siRNA of antisense strand (RLOAs/S) is applied in animals;
图10为反义链5′-末端单缀合的siRNA(RL2As/S)单次给药48h后瘤内的靶基因沉默活性,siRNA给药剂量为1.5mg/kg,每只小鼠2nmol siRNA,数据使用mean±SD表示,n=4,**p<0.01,***p<0.001。Figure 10 shows the target gene silencing activity in the tumor after a single administration of siRNA (RL2As/S) at the 5'-end of the antisense strand for 48 hours. The dose of siRNA was 1.5 mg/kg, and each mouse was 2 nmol siRNA , data are expressed as mean±SD, n=4, **p<0.01, ***p<0.001.
具体实施方式Detailed ways
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随具体实施例的描述而更为清楚。但实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。The present invention will be further described below with reference to specific embodiments, and the advantages and characteristics of the present invention will become more apparent with the description of the specific embodiments. However, the embodiments are only exemplary and do not constitute any limitation to the scope of the present invention. It should be understood by those skilled in the art that the details and forms of the technical solutions of the present invention can be modified or replaced without departing from the spirit and scope of the present invention, but these modifications and replacements all fall within the protection scope of the present invention.
实施例1 5-末端缀合的siRNA的单链固相合成Example 1 Single-stranded solid-phase synthesis of 5-terminally conjugated siRNA
RNA的合成采用Appllied Biosystems model 394RNA固相合成仪。RNA synthesis was performed using an Applied Biosystems model 394 RNA solid-phase synthesizer.
正常的核苷酸亚磷酰化单体(dT,rGibu,rABz,rCAc,rU)从安徽芜湖华仁科技有限公司购买;CPG(CPG-dT),CAP-A和CAP-B,氧化I2液,Cl3CCOOH从北京奥科生物科技公司购买;0.25M的5-乙巯基1H-四氮唑溶液从上海智研科技有限公司(上海)购买。Normal nucleotide phosphorylated monomers (dT, rGibu, rABz, rCAc, rU) were purchased from Anhui Wuhu Huaren Technology Co., Ltd.; CPG (CPG-dT), CAP-A and CAP - B, oxidized I solution, Cl 3 CCOOH was purchased from Beijing Aoke Biotechnology Company; 0.25M 5-ethylmercapto 1H-tetrazolium solution was purchased from Shanghai Zhiyan Technology Co., Ltd. (Shanghai).
按照文献(Biomacromolecules 2018,19,7,2526-2534)的方法,将化学式I/化学式II所示的链接基团分别制备成化学式IX、化学式X所示的亚磷酰胺单体,即:According to the method of the literature (
化学式VII所示的化合物(8.4g,4.2mmol)和三乙胺(0.43g,4.2mmol)溶解在10mL无水DCM中。然后将溶解在DCM中的DMTrCl(1.41g,4.2mmol)缓慢加入上述混合物中。将反应混合物在室温下搅拌4小时。减压除去溶剂,通过柱色谱(PE:EA=5:1)纯化,得产物(1.45g,41%)。在氩气保护下,将双(二异丙基氨基)(2-氰基乙氧基)膦(0.64g,2.1mmol)加入到产物(0.45g,1.1mmol)和四氮唑(0.18g,2.6mmol)的混合物中,用无水DCM(5mL)溶解。将反应混合物在室温下搅拌2小时。通过柱色谱(PE:EA=10:1)纯化,得化学式IX所示的化合物(0.40g,60.6%)。The compound of formula VII (8.4 g, 4.2 mmol) and triethylamine (0.43 g, 4.2 mmol) were dissolved in 10 mL of anhydrous DCM. Then DMTrCl (1.41 g, 4.2 mmol) dissolved in DCM was slowly added to the above mixture. The reaction mixture was stirred at room temperature for 4 hours. The solvent was removed under reduced pressure and purified by column chromatography (PE:EA=5:1) to give the product (1.45 g, 41%). Under argon, bis(diisopropylamino)(2-cyanoethoxy)phosphine (0.64 g, 2.1 mmol) was added to the product (0.45 g, 1.1 mmol) and tetrazolium (0.18 g, 2.6 mmol) was dissolved in dry DCM (5 mL). The reaction mixture was stirred at room temperature for 2 hours. Purified by column chromatography (PE:EA=10:1) to obtain the compound of formula IX (0.40 g, 60.6%).
化学式VIII所示的化合物(1.5g,12.7mmol)和1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC HCl)(2.7g,15.1mmol)溶解在10mL无水DCM(CH2Cl2)中。然后加入N-羟基琥珀酰亚胺(NHS)(1.6g,14.3mmol)并将混合物搅拌过夜。减压除去溶剂,通过柱色谱(PE:EA=1:1)纯化,得产物(1.9g,69.6%)。氩气保护下,将双(二异丙基氨基)(2-氰基乙氧基)膦(0.8g,2.6mmol加入到产物(0.3g,1.4mmol)和四氮唑(0.21g,3mmol)的混合物中用无水DCM(5mL)溶解。将反应混合物在室温下搅拌2小时。通过柱色谱(PE:EA=10:1)纯化,得到化学式X所示的化合物(0.38g,66%)。Compound of formula VIII (1.5 g, 12.7 mmol) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC HCl) (2.7 g, 15.1 mmol) were dissolved in 10 mL of dry DCM ( CH2Cl2 ) . Then N-hydroxysuccinimide (NHS) (1.6 g, 14.3 mmol) was added and the mixture was stirred overnight. The solvent was removed under reduced pressure and purified by column chromatography (PE:EA=1:1) to give the product (1.9 g, 69.6%). Under argon, bis(diisopropylamino)(2-cyanoethoxy)phosphine (0.8 g, 2.6 mmol) was added to the product (0.3 g, 1.4 mmol) and tetrazolium (0.21 g, 3 mmol) The mixture was dissolved in anhydrous DCM (5 mL). The reaction mixture was stirred at room temperature for 2 hours. Purification by column chromatography (PE:EA=10:1) gave the compound of formula X (0.38 g, 66%) .

合成规模:~1.0μmolSynthesis scale: ~1.0 μmol
核苷亚磷酰化单体溶液的配制:氩气保护下称量,加无水乙腈,1g单体对应20mL乙腈;Preparation of nucleoside phosphorylated monomer solution: weigh under argon protection, add anhydrous acetonitrile, 1g monomer corresponds to 20mL acetonitrile;
连接臂亚磷酰化单体溶液的配制:氩气保护下称量化学式IX、X所示的化合物,加无水乙腈,1g单体对应20mL乙腈。Preparation of linker arm phosphorylated monomer solution: weigh the compounds represented by chemical formulas IX and X under argon protection, add anhydrous acetonitrile, and 1 g of monomer corresponds to 20 mL of acetonitrile.
采用固相合成的方法合成了天然的及缀合的siRNA反义链及正义链序列。在RNA合成仪上,序列从3′-端至5′-端进行合成,每偶联一个核苷为一个循环,在天然核苷亚磷酰胺单体偶联21次后,将一个或几个所述的链接臂亚磷酰胺单体在序列的5′-末端相应位置进行偶联,每个循环包括四个反应:脱DMT、偶联、封闭、氧化。Native and conjugated siRNA antisense and sense strand sequences were synthesized by solid-phase synthesis. On an RNA synthesizer, the sequence is synthesized from the 3'-end to the 5'-end, and each coupling of one nucleoside is a cycle. After 21 couplings of the natural nucleoside phosphoramidite monomer, one or several The linking arm phosphoramidite monomer is coupled at the corresponding position at the 5'-end of the sequence, and each cycle includes four reactions: de-DMT, coupling, blocking, and oxidation.
合成序列:Synthetic sequence:
在本实施例中使用待修饰的siRNA为靶向ERK通路中BRAFV600E mRNA的siMB3,修饰前的序列如下:In this example, the siRNA to be modified is used as siMB3 targeting BRAF V600E mRNA in the ERK pathway, and the sequence before modification is as follows:
siMB3:正义链:5′-GCUACAGAGAAAUCU CGAUdtdt-3′siMB3: sense strand: 5′-GCUACAGAGAAAAUCU CGAUdtdt-3′
反义链:5′-AUC GAGAUU UCU CUG UAG C dtdt-3′Antisense strand: 5'-AUC GAGAUU UCU CUG UAG C dtdt-3'
修饰策略选择以下任意一种:Choose any of the following for the grooming strategy:
(1)在以上所述的siMB3序列反义链或正义链的5′-末端缀合化学式II的链接基团;(1) Conjugating a linking group of chemical formula II at the 5'-end of the antisense strand or the sense strand of the siMB3 sequence described above;

(2)在以上所述的siMB3序列反义链或正义链的5′-末端依次(3′-5′方向)缀合一个或多个化学式I和一个化学式II的链接基团,其中缀合基团间通过磷酸二酯键链接;(2) Conjugate one or more linking groups of chemical formula I and one chemical formula II in sequence (3'-5' direction) at the 5'-end of the antisense strand or sense strand of the siMB3 sequence described above, wherein the conjugation The groups are linked by phosphodiester bonds;
(3)在以上所述的siMB3序列反义链或正义链的5′-末端缀合一个化学式II所示的链接基团,缀合使用固相合成仪完成,使用的亚磷单体为化学式X,在化学式X所示的链接基团的活泼酯上通过缩合反应连接化学式VI所示的靶向基团cRGD;(3) Conjugate a linking group shown in chemical formula II to the 5'-end of the antisense strand or sense strand of the siMB3 sequence described above, and the conjugation is completed by a solid-phase synthesizer, and the phosphorous monomer used is chemical formula X, the targeting group cRGD shown in chemical formula VI is connected by condensation reaction on the active ester of the linking group shown in chemical formula X;
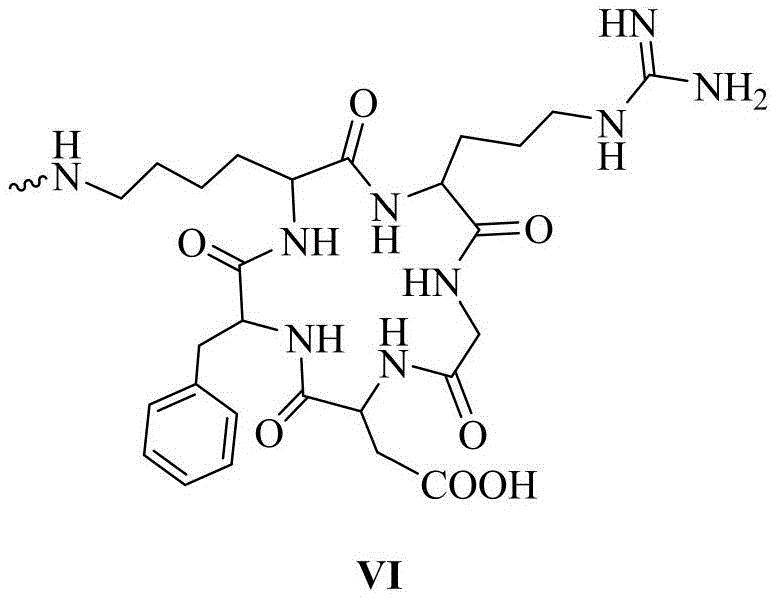
(4)在以上所述的siMB3序列反义链或正义链的5′-末端依次(3′-5′方向)缀合一个或几个化学式I和一个化学式II所示的链接基团,缀合使用固相合成仪完成,使用的亚磷单体为化学式IX和X,其中缀合基团间通过磷酸二酯键链接,随后在化学式X的活泼酯上通过缩合反应连接化学式VI所示的靶向基团cRGD。(4) Conjugate one or more linking groups shown in chemical formula I and one chemical formula II to the 5'-end of the antisense or sense strand of the siMB3 sequence described above in turn (3'-5' direction), The combination is completed by a solid-phase synthesizer, and the phosphorous monomers used are chemical formulas IX and X, wherein the conjugated groups are linked by phosphodiester bonds, and then the active ester of chemical formula X is connected by condensation reaction shown in chemical formula VI. Targeting group cRGD.
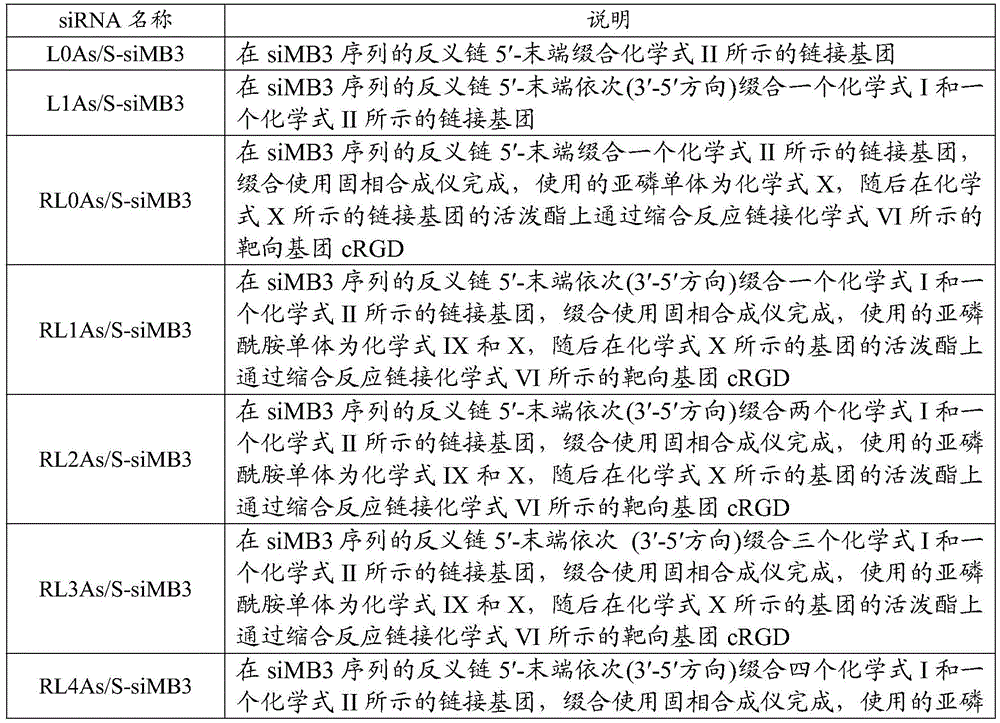
经过上述修饰策略后获得的siRNA如下表1所示:The siRNA obtained after the above modification strategy is shown in Table 1 below:
表1Table 1

合成步骤:每次称量约33mg CPG-dT装入合成柱中,按照ABI394核酸合成仪的RNA标准步骤,采用DMT-ON的合成策略。以合成RL2As为例,在合成仪序列面板上设定好合成序列。其中,将化学式IX和X所示的连接臂也视为亚磷单体编入序列中,即在As链5′-末端再编入2个化学式IX和1个化学式X所示的连接臂。cRGD官能团在合成完毕后手动单独反应,合成完毕后,将合成柱中的粉末转移到1.5mL玻璃反应瓶中,加入1mg化学式XI所示的化合物、100μl DIPEA、1mL DMF室温搅拌反应24小时,后弃去上清液,使用1mL乙醇洗涤两次,1mL乙醚洗涤一次,后晾干。Synthesis steps: About 33 mg of CPG-dT was weighed each time and loaded into the synthesis column, and the synthesis strategy of DMT-ON was adopted according to the RNA standard procedure of ABI394 nucleic acid synthesizer. Taking the synthesis of RL2As as an example, set the synthesis sequence on the synthesizer sequence panel. Wherein, the linking arms shown by chemical formulas IX and X are also regarded as phosphorous monomers programmed into the sequence, that is, two linking arms represented by chemical formula IX and one chemical formula X are added to the 5'-end of the As chain. After the synthesis, the cRGD functional group was manually reacted separately. After the synthesis, the powder in the synthesis column was transferred to a 1.5 mL glass reaction flask, and 1 mg of the compound represented by chemical formula XI, 100 μl of DIPEA, and 1 mL of DMF were added to stir and react at room temperature for 24 hours. The supernatant was discarded, washed twice with 1 mL of ethanol and once with 1 mL of ether, and then air-dried.
RNA的切割、脱保护:向晾干的粉末中加入750μl甲胺醇和750μl甲氨水切割,置于摇床上,60℃,80rpm,震摇90min。之后将上清液取出,用DEPC水洗涤三次,均匀分装至两个1.5mL EP管中,最后用冻干机冻干。在每管中加入100μl DMSO和125μl三乙胺三氢氟酸,封口膜缠住,80rpm,65℃,震摇90min。冷却后,加入100μl 3M CH3COONa溶液,涡旋,加入1mL正丁醇溶液,涡旋后放入-80℃静置30min。后取出12500rpm离心10min,弃去上清液,留下白色沉淀,加入0.75mL无水乙醇涡旋,放入-80℃静置30min,12500rpm离心10min,弃去上清,再重复一次该操作,后冻干。RNA cleavage and deprotection: add 750 μl methylamino alcohol and 750 μl methylamine water to the dried powder for cleavage, place on a shaker, 60° C., 80 rpm, and shake for 90 min. After that, the supernatant was taken out, washed three times with DEPC water, evenly distributed into two 1.5mL EP tubes, and finally lyophilized by a freeze dryer. 100 μl of DMSO and 125 μl of triethylamine trihydrofluoric acid were added to each tube, wrapped with parafilm, 80 rpm, 65° C., and shaken for 90 min. After cooling, 100 μl of 3M CH 3 COONa solution was added, vortexed, 1 mL of n-butanol solution was added, and after vortexing, it was placed at -80° C. for 30 min. Then take out and centrifuge at 12500rpm for 10min, discard the supernatant, leaving a white precipitate, add 0.75mL absolute ethanol, vortex, put it at -80°C for 30min, centrifuge at 12500rpm for 10min, discard the supernatant, repeat this operation again, freeze-dried.
RNA的分离纯化:将样品用400μl DEPC水稀释溶解,用0.22μm滤头过滤,在用200μlDEPC水洗涤两次,最终总体积大约为800μl。用进样针吸取,每针体积120μl进样。使用XBridgeTM Oligonucleotide BEH C18 OBDTM Prep Column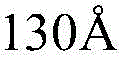
实施例2中性/阳离子混合制材包载siRNA形成脂质复合物(siRNA/DNCA/CLD/PEG2000-DSPE)Example 2 Neutral/cationic mixed material encapsulating siRNA to form lipid complex (siRNA/DNCA/CLD/PEG2000-DSPE)
将siRNA与混合制材混合,混合制材由39.7%的化学式III所示的化合物(DNCA,中性型)、59.6%的化学式IV所示的化合物(CLD,阳离子型)、0.7%的化学式V所示的化合物(PEG2000-DSPE)组成。这些比例指的是所有脂质总量的摩尔%。脂质与siRNA的摩尔比为5.3:1。按文献(CN108059619A)的方法合成DNCA,按文献(New JChem,2014,38(10),4952-4962)的方法合成CLD,PEG2000-DSPE购自源叶公司。The siRNA was mixed with a mixed material consisting of 39.7% compound of formula III (DNCA, neutral), 59.6% compound of formula IV (CLD, cationic), 0.7% compound of formula V Composition of the compound shown (PEG2000-DSPE). These ratios refer to mole % of the total amount of all lipids. The molar ratio of lipid to siRNA was 5.3:1. DNCA was synthesized according to the method in the literature (CN108059619A), CLD was synthesized according to the method in the literature (New JChem, 2014, 38(10), 4952-4962), and PEG2000-DSPE was purchased from Yuanye Company.
简要来说,将化学式III所示的化合物(DNCA,中性型)、化学式IV所示的化合物(CLD,阳离子型)以及化学式V所示的化合物溶于乙醇溶液中,将siRNA溶于DEPC水中,使用GenOpti(北京迈晨)混合,然后于70℃超声10min,超声频率150W,40kHz。超声结束恢复至室温后,使用0.22μm滤膜过滤,滤液中包含脂质复合物。Briefly, compounds of formula III (DNCA, neutral), compounds of formula IV (CLD, cationic), and compounds of formula V were dissolved in ethanol solution, and siRNA was dissolved in DEPC water , mixed with GenOpti (Beijing Maichen), and then ultrasonicated at 70°C for 10min, ultrasonic frequency 150W, 40kHz. After the ultrasound was completed and returned to room temperature, it was filtered through a 0.22 μm filter, and the filtrate contained lipid complexes.
例如,在一个具体的方法中,在乙醇中配置新鲜的脂质储液:称取156.5mg DNCA、315mg CLD、10.5mg PEG2000-DSPE,并在20mL乙醇中溶解以形成脂质储备液,将其在37℃超声4分钟以形成均匀的脂质混合物。随后,将20μl上述储备液加入480μl GenOpti中形成500μl工作脂质储液。将这个用量的脂质用于形成具有10nmol的siRNA脂质复合物。在siRNA干粉溶解在DEPC水中形成200μM的siRNA储备液。将50μl上述储备液加入450μl Genopti中形成500μl工作siRNA储液,将500μl的工作脂质储液与500μl的工作siRNA储液混合,短暂低速离心后,于70℃超声10min形成脂质复合物。体内和体外实验所用制剂使用0.22μm滤膜过滤(默克密理博,merck millipore),使用前使用PBS溶液稀释至1×siRNA脂质复合物溶液。For example, in one specific method, a fresh lipid stock solution is prepared in ethanol: 156.5 mg DNCA, 315 mg CLD, 10.5 mg PEG2000-DSPE are weighed and dissolved in 20 mL ethanol to form a lipid stock solution, which is Sonicate for 4 min at 37°C to form a homogeneous lipid mixture. Subsequently, 20 [mu]l of the above stock solution was added to 480 [mu]l GenOpti to form a 500 [mu]l working lipid stock. This amount of lipid was used to form siRNA lipoplexes with 10 nmol. Dissolve the siRNA dry powder in DEPC water to form a 200 μM siRNA stock solution. 50 μl of the above stock solution was added to 450 μl of Genopti to form 500 μl of working siRNA stock solution, 500 μl of working lipid stock solution was mixed with 500 μl of working siRNA stock solution, briefly centrifuged at low speed, and sonicated at 70°C for 10 min to form lipoplexes. The formulations used in the in vivo and in vitro experiments were filtered through a 0.22 μm filter (merck millipore) and diluted to a 1× siRNA lipoplex solution with a PBS solution before use.
图1显示通过这些方法制备的siRNA的脂质复合物和不含siRNA的空载体的示例性电性显微镜照片(TEM)。这些脂质体包含的siRNA为未缀合的靶向ERK通路中BRAFV600EmRNA的siMB3。不加入siRNA的空载体的颗粒较为松散,且颗粒表面皱缩。加入siRNA后的脂质复合物可以形成颗粒均一的球型纳米颗粒,而且边缘光滑形态较好。Figure 1 shows exemplary electron micrographs (TEM) of lipoplexes of siRNA prepared by these methods and an empty carrier without siRNA. The siRNA contained in these liposomes was unconjugated siMB3 targeting BRAF V600E mRNA in the ERK pathway. The particles of the empty carrier without siRNA were looser, and the particle surface shrunken. The lipoplex after adding siRNA can form spherical nanoparticles with uniform particles, and the edge is smooth and the shape is better.
动态光散射显示包载有siRNA的脂质复合物的平均直径是138.7±6.6(根据密度),多分散系数是0.188±0.051,其表面电性是-16.9±0.4mV;空载体的平均直径是363.7±20.1(根据密度),多分散系数是0.635±0.045,其表面电性是24.3±2.8mV。由于TEM拍摄前需对样品脱水干燥处理,颗粒会因水分的流失产生皱缩,而DLS测定的是纳米颗粒的水和粒径,因此TEM法所测得的粒径小于DLS法。Dynamic light scattering showed that the mean diameter of the siRNA-loaded lipoplex was 138.7±6.6 (according to density), the polydispersity coefficient was 0.188±0.051, and its surface electrical property was -16.9±0.4mV; the mean diameter of the empty vector was 363.7 ± 20.1 (based on density), the polydispersity coefficient is 0.635 ± 0.045, and its surface electrical properties are 24.3 ± 2.8 mV. Since the sample needs to be dehydrated and dried before TEM shooting, the particles will shrink due to the loss of water, and DLS measures the water and particle size of the nanoparticles, so the particle size measured by the TEM method is smaller than that by the DLS method.
实施例3利用流式细胞术考察反义链5-末端单缀合的siRNA中性/阳离子脂质复合物的细胞摄取Example 3 Cellular uptake of antisense 5-terminal monoconjugated siRNA neutral/cationic lipoplexes by flow cytometry
1.样品:1. Sample:
As/Cy3-S-siMB3、RL0As/Cy3-S-siMB3、RL1As/Cy3-S-siMB3、RL2As/Cy3-S-siMB3、RL3As/Cy3-S-siMB3、RL4As/Cy3-S-siMB3。siMB3的正义链5-末端使用荧光染料Cy3标记。反义链缀合物的合成参见实施例1,siRNA脂质复合物siRNA/DNCA/CLD的制备参见实施例2,区别在于混合制材由72.7%化学式III所示的中性胞苷脂材、27.3%化学式IV所示的阳离子脂材组成。这些比例指的是所有脂质总量的摩尔%。总脂质与siRNA的摩尔比为10:1。As/Cy3-S-siMB3, RL0As/Cy3-S-siMB3, RL1As/Cy3-S-siMB3, RL2As/Cy3-S-siMB3, RL3As/Cy3-S-siMB3, RL4As/Cy3-S-siMB3. The 5-terminus of the sense strand of siMB3 was labeled with the fluorescent dye Cy3. The synthesis of the antisense strand conjugate is shown in Example 1, and the preparation of the siRNA lipoplex siRNA/DNCA/CLD is shown in Example 2. The difference is that the mixed material is composed of 72.7% neutral cytidine lipid material represented by chemical formula III, 27.3% of the cationic resin composition of formula IV. These ratios refer to mole % of the total amount of all lipids. The molar ratio of total lipid to siRNA was 10:1.
2.方法:2. Method:
将A375以6万/孔铺24孔板,培养18h后进行转染。siMB3及其缀合物的给药浓度为10nM,转染使用的Lipofectamine2000用量为0.5ul,按照其使用说明书与siRNA混合制配置转染复合物。将复合物加入至培养板中,培养4h后,使用DMEM润洗细胞表面,胰酶消化收集细胞,流式细胞仪PE通道测定细胞内荧光强度,考察siMB3及其缀合物的细胞摄取能力,实验结果见图2。A375 was plated in a 24-well plate at 60,000/well, and transfected after culturing for 18 h. The administration concentration of siMB3 and its conjugates was 10 nM, and the amount of Lipofectamine 2000 used for transfection was 0.5 ul, and the transfection complex was prepared by mixing with siRNA according to its instruction manual. The complex was added to the culture plate, and after 4 h of culture, the cell surface was rinsed with DMEM, the cells were collected by trypsin digestion, and the intracellular fluorescence intensity was measured by the PE channel of the flow cytometer to investigate the cellular uptake ability of siMB3 and its conjugates. The experimental results are shown in Figure 2.
3.结果:3. Results:
与Lipofectamine2000相比,混合载体DNCA/CLD具有更高效的递送siRNA入胞的能力,说明血清蛋白的存在对DNCA/CLD/siMB3纳米复合物的摄取影响不大,即使血清中含有潜在的转染竞争者,例如玻连蛋白和纤连蛋白。Compared with Lipofectamine2000, the mixed vector DNCA/CLD has a more efficient ability to deliver siRNA into cells, indicating that the presence of serum protein has little effect on the uptake of DNCA/CLD/siMB3 nanocomplexes, even if the serum contains potential transfection competition , such as vitronectin and fibronectin.
对比不同长度链接臂连接的cRGD缀合物的摄取情况发现,随链接臂延长(RL0As/S至RL3As/S),摄取逐渐增加。细胞摄取RL4As/S的能力较低,提示继续延长链接臂长度已经不能增强siMB3缀合物的细胞摄取能力,这有可能是由于增加链接臂长度的同时会引入更多的带负电荷的磷酸二酯键,其可能影响siMB3缀合物/DNCA/CLD脂质复合物的形成。Comparing the uptake of cRGD conjugates linked by linking arms of different lengths, it was found that the uptake gradually increased as the linking arm lengthened (RL0As/S to RL3As/S). The ability of cells to uptake RL4As/S was low, suggesting that continuing to extend the link arm length could not enhance the cellular uptake ability of siMB3 conjugates, which may be due to the introduction of more negatively charged phosphodiesteric bisphosphonates while increasing the link arm length. Ester bond, which may affect the formation of the siMB3 conjugate/DNCA/CLD lipoplex.
实施例4利用共聚焦激光显微镜考察反义链5-末端单缀合的siRNA中性/阳离子脂质复合物的细胞内分布Example 4 Intracellular distribution of antisense 5-terminal monoconjugated siRNA neutral/cationic lipoplexes using confocal laser microscopy
1.样品:1. Sample:
As/Cy3-S-siMB3、RL0As/Cy3-S-siMB3、RL1As/Cy3-S-siMB3。siMB3的正义链5-末端使用荧光染料Cy3标记。反义链缀合物的合成参见实施例1,siRNA脂质复合物siRNA/DNCA/CLD的制备参见实施例3。As/Cy3-S-siMB3, RL0As/Cy3-S-siMB3, RL1As/Cy3-S-siMB3. The 5-terminus of the sense strand of siMB3 was labeled with the fluorescent dye Cy3. See Example 1 for the synthesis of the antisense strand conjugate and Example 3 for the preparation of the siRNA lipoplex siRNA/DNCA/CLD.
2.方法:2. Method:
将A375以6万/孔铺于共聚焦皿中,培养18h后进行转染。siMB3及其缀合物的给药浓度为50nM,转染使用的Lipofectamine2000用量为1μl,按照其使用说明书与siRNA混合制配置转染复合物。将复合物加入至培养板中,培养4h后,使用Hoechst、Lysobrite NIR分别对细胞核和溶酶体进行染色。加入染料后孵育30分钟。使用DMEM润洗细胞表面去除游离的染料,使用共聚焦显微镜拍照。考察siMB3及其缀合物的细胞内分布情况,实验结果见图3。A375 was plated in confocal dishes at 60,000/well, and transfected after culturing for 18 hours. The administration concentration of siMB3 and its conjugates was 50 nM, and the amount of Lipofectamine 2000 used for transfection was 1 μl, and the transfection complex was prepared by mixing it with siRNA according to its instruction manual. The complex was added to the culture plate, and after 4 hours of culture, the nucleus and lysosome were stained with Hoechst and Lysobrite NIR, respectively. Incubate for 30 minutes after adding dye. Cell surfaces were rinsed with DMEM to remove free dye and photographed using a confocal microscope. The intracellular distribution of siMB3 and its conjugates was investigated, and the experimental results are shown in Figure 3.
3.结果:3. Results:
共聚焦结果与流式细胞术结果相吻合,Lipofectamine2000递送siRNA入胞的效率远低于DNCA/CLD(Mix)。cRGD缀合于反义链5′-末端后,细胞摄取有明显增加,这表明部分缀合的cRGD可能不在DNCA/CLD/siMB3纳米复合物内部,从而通过靶向αvβ3整合素进入细胞内,增加αvβ3阳性细胞A375摄取脂质复合物的能力。Cy3标记的siRNA(红色)在细胞膜上及细胞内呈点状聚集,可能分布于细胞膜转运囊泡和内体中,从而进行胞内转运。siRNA经DNCA/CLD混合制剂包载后几乎不与溶酶体(绿色)共定位,提示这一脂质复合物可有效避免进入溶酶体或可快速从溶酶体中逃逸,从而最大程度减少siRNA在溶酶体内的酸化降解。Confocal results were consistent with flow cytometry results, and the efficiency of Lipofectamine2000 in delivering siRNA into cells was much lower than that of DNCA/CLD(Mix). After cRGD was conjugated to the 5′-terminus of the antisense strand, cellular uptake was significantly increased, suggesting that the partially conjugated cRGD may not be inside the DNCA/CLD/siMB3 nanocomplex, thereby increasing intracellular entry by targeting αvβ3 integrins. Capacity of αvβ3-positive cells A375 to uptake lipid complexes. Cy3-labeled siRNA (red) was punctately aggregated on the cell membrane and in the cell, and may be distributed in the cell membrane transport vesicles and endosomes for intracellular transport. The siRNA hardly co-localized with lysosomes (green) after being encapsulated in the DNCA/CLD mixture, suggesting that this lipid complex can effectively avoid entering or rapidly escape from lysosomes, thereby minimizing the Acidification and degradation of siRNA in lysosomes.
实施例5利用RT-qPCR技术考察5-末端单/双缀合的siRNA中性/阳离子脂质复合物的基因沉默活性Example 5 Investigating the gene silencing activity of 5-terminal single/double conjugated siRNA neutral/cationic lipoplexes by RT-qPCR technology
1.样品:1. Sample:
As/S-siMB3、L0As/S-siMB3、L1As/S-siMB3、RL0As/S-siMB3、RL1As/S-siMB3、RL2As/S-siMB3、RL3As/S-siMB3、RL4As/S-siMB3、L1As/L0S-siMB3、As/RL0S-siMB3、RL0As/RL0S-siMB3、RL1As/RL0S-siMB3、L0As/RL0S-siMB3、L1As/RL0S-siMB3。以上5-末端单双缀合物的合成参见实施例1。As/S-siMB3, L0As/S-siMB3, L1As/S-siMB3, RL0As/S-siMB3, RL1As/S-siMB3, RL2As/S-siMB3, RL3As/S-siMB3, RL4As/S-siMB3, L1As/ L0S-siMB3, As/RL0S-siMB3, RL0As/RL0S-siMB3, RL1As/RL0S-siMB3, L0As/RL0S-siMB3, L1As/RL0S-siMB3. See Example 1 for the synthesis of the above 5-terminal monobiconjugates.
2.RT-qPCR实验:2. RT-qPCR experiment:
将A375以8万/孔铺12孔板,培养18h后进行转染。siMB3及其缀合物的给药浓度为50nM,转染使用的Lipofectamine2000用量为1ul,按照其使用说明书与siRNA混合制配置转染复合物。脂质复合物siRNA/DNCA/CLD按照实施例3的方法制备。将复合物加入至培养板中,培养48h后,每孔加入0.5ml TRizol试剂提取总RNA,使用逆转录试剂盒将RNA逆转录为cDNA,进行实时荧光定量PCR,考察siMB3及其缀合物的靶mRNA沉默能力,实验结果见图4。A375 was plated in 12-well plates at 80,000/well, and transfected after culturing for 18 hours. The administration concentration of siMB3 and its conjugates was 50 nM, and the amount of Lipofectamine 2000 used for transfection was 1 ul, and the transfection complex was prepared by mixing with siRNA according to its instruction manual. Lipoplex siRNA/DNCA/CLD was prepared according to the method of Example 3. The complex was added to the culture plate. After culturing for 48 hours, 0.5 ml of TRizol reagent was added to each well to extract total RNA. The RNA was reverse transcribed into cDNA using a reverse transcription kit, and real-time fluorescent quantitative PCR was performed to investigate the activity of siMB3 and its conjugates. Target mRNA silencing ability, the experimental results are shown in Figure 4.
3.结果3. Results
首先比较使用商用转染试剂和DNCA/CLD混合制剂转染siRNA的效率,发现混合脂材DNCA/CLD包载的siRNA基因沉默活性优于使用商用转染试剂Lipofectamine2000包载(lipo组),且具有显著性差异(p<0.0001)。Firstly, the efficiency of siRNA transfection using commercial transfection reagent and DNCA/CLD mixed preparation was compared, and it was found that the gene silencing activity of siRNA encapsulated by mixed lipid material DNCA/CLD was better than that encapsulated using commercial transfection reagent Lipofectamine 2000 (lipo group), and had Significant difference (p<0.0001).
对反义链5′-单缀合物基因沉默活性考察发现,末端缀合短的连接臂(L0As/S和L1As/S)与未缀合的siMB3(As/S)的基因沉默能力没有显著性差异(p>0.05)。继续考察了RL0-RL4一这系列末端缀合了较大体积基团cRGD的缀合物的基因沉默活性,结果表明基因沉默活性存在一定差异且与链接臂长度相关,具有更长链接臂(RL2As/S,RL3As/S和RL4As/S)的5′-单缀合物的活性优于较短链接臂(RL1As/S和RL0As/S)连接的缀合物,这一结果可能是由于在反义链5′-末端和cRGD之间插入更长的链接臂会使缀合部分的结构更加松散,减少了siRNA缀合物载入RISC时的空间阻碍。RL2As/S-siMB3与siMB3的活性差异较小,可能是因其可以采用适当的构象避免与RISC的空间冲突,因此与未缀合物具有相似的基因沉默活性。The gene silencing activity of 5′-single conjugates of antisense strands was investigated, and it was found that the gene silencing ability of end-conjugated short linkers (LOAs/S and L1As/S) and unconjugated siMB3 (As/S) had no significant gene silencing ability Sex difference (p>0.05). Continued to investigate the gene silencing activity of R0-RL4, a series of conjugates with a larger bulk group cRGD at the end, the results showed that the gene silencing activity was different and related to the length of the linking arm, with a longer linking arm (RL2As /S, RL3As/S and RL4As/S) 5′-monoconjugates were more active than conjugates linked by shorter linking arms (RL1As/S and RL0As/S), this result may be due to The insertion of a longer linker arm between the 5'-end of the sense strand and cRGD will loosen the structure of the conjugated moiety and reduce the steric hindrance when loading the siRNA conjugate into RISC. RL2As/S-siMB3 showed less difference in activity from siMB3, probably because it could adopt an appropriate conformation to avoid steric conflict with RISC and thus have similar gene silencing activity to the unconjugated conjugate.
正义链5′-末端单缀合cRGD对基因沉默活性无明显影响(As/RL0S-siMB3),即使是在链接臂最短的情况下。而双缀合物与未缀合的siMB3相比,基因沉默活性略有下降,但在此浓度条件下活性差异不明显,当在正义链的5′-末端通过较短连接基团缀合cRGD后,反义链5′-缀合大基团的活性优于缀合较小基团。Single-conjugated cRGD at the 5'-end of the sense strand had no apparent effect on gene silencing activity (As/RLOS-siMB3), even with the shortest linker arm. Compared with unconjugated siMB3, the gene silencing activity of the biconjugate decreased slightly, but the difference in activity was not obvious at this concentration. When cRGD was conjugated to the 5′-end of the sense strand through a shorter linking group Afterwards, the antisense strand 5'-conjugated large groups were more active than conjugated smaller groups.
实施例6考察5-末端单/双缀合的siRNA中性/阳离子脂质复合物的抗肿瘤活性及空载体细胞毒性Example 6 Investigate the antitumor activity and empty vector cytotoxicity of 5-terminal single/double conjugated siRNA neutral/cationic lipid complexes
1.样品:1. Sample:
As/S-siMB3、L0As/S-siMB3、L1As/S-siMB3、RL0As/S-siMB3、RL1As/S-siMB3、L1As/L0S-siMB3、RL0As/RL0S-siMB3、RL1As/RL0S-siMB3、As/RL0S-siMB3、L0As/RL0S-siMB3、L1As/RL0S-siMB3。以上5-末端单双缀合物的制备参见实施例1,siRNA脂质复合物siRNA/DNCA/CLD的制备参见实施例3。As/S-siMB3, L0As/S-siMB3, L1As/S-siMB3, RL0As/S-siMB3, RL1As/S-siMB3, L1As/L0S-siMB3, RL0As/RL0S-siMB3, RL1As/RL0S-siMB3, As/ RL0S-siMB3, L0As/RL0S-siMB3, L1As/RL0S-siMB3. See Example 1 for the preparation of the above 5-terminal single-double conjugate, and see Example 3 for the preparation of siRNA lipoplex siRNA/DNCA/CLD.
2.cck-8实验2. cck-8 experiment
将A375以1万/孔铺96孔板,培养18h后进行转染。siMB3及其缀合物的给药浓度为100nM,将siRNA脂质复合物加入至培养板中,培养72h后,弃去培养基,加入10%cck-8试剂,37℃孵育2h,使用酶标仪测定450nm的吸光度。结果以control组的吸光度为1进行均一化处理。考察siMB3及其缀合物的抗肿瘤活性和空载体的细胞毒性,实验结果见图5。A375 was plated in a 96-well plate at 10,000/well, and transfected after culturing for 18 hours. The administration concentration of siMB3 and its conjugates was 100nM, and the siRNA lipid complex was added to the culture plate. After culturing for 72h, the medium was discarded, 10% cck-8 reagent was added, and the siRNA was incubated at 37°C for 2h. The absorbance at 450 nm was measured by the instrument. The results were normalized by taking the absorbance of the control group as 1. The antitumor activity of siMB3 and its conjugates and the cytotoxicity of the empty carrier were investigated. The experimental results are shown in Figure 5.
3.结果3. Results
对单缀合siMB3/DNCA/CLD脂质复合物的细胞水平抗肿瘤活性进行评价发现,对反义链5′-末端进行缀合会使其体外抗肿瘤活性降低,且与缀合基团的大小无关。正义链5′-末端缀合后,抗肿瘤活性也随之下降,双缀合物中,仅双缀合物L1As/L0S的基因沉默活性与非缀合物As/S相似,L0As/RL0S同样具有较好的抗肿瘤活性。DNCA/CLD空载体(mix组)的细胞毒性较低,提示体外应用脂质复合物安全低毒。The cell-level antitumor activity of single-conjugated siMB3/DNCA/CLD lipoplexes was evaluated, and it was found that conjugation to the 5′-terminus of the antisense strand reduced its antitumor activity in vitro, and was associated with the conjugation group. Size doesn't matter. After the 5′-end of the sense strand was conjugated, the antitumor activity also decreased. Among the biconjugates, only the gene silencing activity of the biconjugate L1As/LOS was similar to that of the non-conjugate As/S, and the same was true for L0As/RLOS Has good antitumor activity. The cytotoxicity of DNCA/CLD empty vector (mix group) was low, suggesting that the application of lipid complexes in vitro is safe and low in toxicity.
实施例7考察5-末端单缀合的siRNA中性/阳离子脂质复合物的动物体内分布Example 7 Investigating the distribution of 5-terminal monoconjugated siRNA neutral/cationic lipid complexes in animals
1.样品:1. Sample:
As/Cy7-S-siMB3、RL0As/Cy7-S-siMB3,siMB3的正义链5′-末端使用荧光染料Cy7标记。反义链缀合物的合成参见实施例1,siRNA脂质复合物siRNA/DNCA/CLD/PEG2000-DSPE的制备参见实施例2。As/Cy7-S-siMB3, RL0As/Cy7-S-siMB3, the 5'-end of the sense strand of siMB3 was labeled with the fluorescent dye Cy7. See Example 1 for the synthesis of the antisense strand conjugate, and Example 2 for the preparation of the siRNA lipoplex siRNA/DNCA/CLD/PEG2000-DSPE.
2.方法:2. Method:
在6周龄的裸鼠腋下接种A375细胞,在10天后肿瘤体积达到约500mm3。随后,通过尾静脉注射DNCA/CLD/PEG2000-DSPE包载的Cy7标记的siMB3(As/S)或cRGD缀合物(RL0As/S),siRNA给药剂量1.5mg/kg。给药后1.5h至36h通过体内成像系统测量siRNA的荧光信号。给药后36h,安乐死动物,取出肿瘤组织和器官并拍照。实验结果见图6。A375 cells were inoculated in the armpit of 6-week-old nude mice, and the tumor volume reached approximately 500 mm 3 after 10 days. Subsequently, DNCA/CLD/PEG2000-DSPE-encapsulated Cy7-labeled siMB3 (As/S) or cRGD conjugate (RL0As/S) was injected via tail vein, and the dose of siRNA was 1.5 mg/kg. The fluorescence signal of siRNA was measured by in vivo imaging system from 1.5h to 36h after administration. 36h after administration, animals were euthanized, and tumor tissues and organs were removed and photographed. The experimental results are shown in Figure 6.
3.结果:3. Results:
整个拍照过程中可以明显观察到RL0As/S在瘤内蓄积,但未缀合cRGD的As/S仅有微弱的荧光信号(图6a)。对瘤内荧光信号进行定量,结果显示cRGD-缀合的siMB3的肿瘤蓄积比未缀合的siMB3高约90%(p<0.05),混合载体可有效递送siRNA至肿瘤,As/S组的瘤内荧光信号显著高于BLANK组(p<0.01)。然而,由于胃肠道中食物的自发荧光,难以观察到肝脏和肾脏中的siRNA信号,但其与肿瘤内的荧光信号无关。The intratumoral accumulation of RL0As/S could be clearly observed during the whole photographing process, but the As/S without cRGD had only a weak fluorescence signal (Fig. 6a). The intratumoral fluorescence signal was quantified, and the results showed that the tumor accumulation of cRGD-conjugated siMB3 was about 90% higher than that of unconjugated siMB3 (p<0.05). The internal fluorescence signal was significantly higher than that of the BLANK group (p<0.01). However, due to autofluorescence of food in the gastrointestinal tract, it was difficult to observe the siRNA signal in liver and kidney, but it was not related to the fluorescence signal in the tumor.
离体结果可以发现(图6b),Cy7标记的siMB3主要聚集在肿瘤和肝脏中,肿瘤中RL0As/S的荧光信号比As/S强,离体肿瘤内荧光定量结果与体内分布的肿瘤定量结果相对应,cRGD缀合的siMB3(RL0As/S)瘤内蓄积能力强于未缀合siMB3(p<0.05)。以上结果表明,DNCA/CLD/PEG2000-DSPE混合载体可高效递送siMB3及其缀合物至肿瘤组织,并且给药后36小时仍有明显的瘤内和肝脏分布,这种组合的缀合和递送策略为全身递送siRNA体内治疗肿瘤提供了平台。In vitro results (Figure 6b) can be found that Cy7-labeled siMB3 mainly accumulates in tumors and livers, and the fluorescence signal of RL0As/S in tumors is stronger than that of As/S. Correspondingly, the intratumoral accumulation ability of cRGD-conjugated siMB3 (RLOAs/S) was stronger than that of unconjugated siMB3 (p<0.05). The above results show that the DNCA/CLD/PEG2000-DSPE mixed carrier can efficiently deliver siMB3 and its conjugates to tumor tissues, and there is still obvious intratumor and liver distribution 36 hours after administration. The conjugation and delivery of this combination The strategy provides a platform for systemic delivery of siRNA to treat tumors in vivo.
实施例8考察5-末端单缀合的siRNA中性/阳离子脂质复合物的动物体内抗肿瘤活性及其机制Example 8 Investigate the antitumor activity of 5-terminal monoconjugated siRNA neutral/cationic lipid complex in animals and its mechanism
1.样品:1. Sample:
As/S-siMB3、RL0As/S-siMB3、RL2As/S-siMB3。siRNA及缀合物的合成参见实施例1,siRNA脂质复合物siRNA/DNCA/CLD/PEG2000-DSPE的制备参见实施例2。As/S-siMB3, RL0As/S-siMB3, RL2As/S-siMB3. See Example 1 for the synthesis of siRNA and conjugate, and Example 2 for the preparation of siRNA lipoplex siRNA/DNCA/CLD/PEG2000-DSPE.
2.方法:2. Method:
将160w黑色素瘤细胞A375接种至3周龄裸鼠腋下,待7天后肿瘤成型后将裸鼠分为5组,此时平均肿瘤体积约为50mm3(第0天),每组5只。尾静脉注射siMB3及缀合物混合制剂进行治疗,空白对照为BLANK(尾静脉注射制剂溶剂Genopti),阴性对照为siMB3(未经包载的siMB3)、mix(不含有siRNA的DNCA/CLD/PEG2000-DSPE空载体)、mix-NC(经mix包载的靶向firefly萤光素酶mRNA的siFL)。在第1、3、5、8天(RL0As/S)或第1、3、5、7、9天(RL2As/S)通过尾静脉注射给药,siRNA的给药剂量为2mg/kg(RL0As/S)或1.48mg/kg(RL2As/S)。给药治疗期间每日或每两日量取肿瘤大小及小鼠体重,并按照公式肿瘤体积V=长×宽2×0.5计算肿瘤体积。最后一次给药48h后,取1ml血液,随后安乐死小鼠,分离肿瘤组织称重并拍照记录。取部分瘤组织(50-100mg)加入TRizol后匀浆提取总RNA,使用逆转录试剂盒将RNA逆转录为cDNA,进行实时荧光定量PCR,考察siMB3及其缀合物的瘤内mRNA沉默能力。另取部分瘤组织(50-100mg)于多聚甲醛中固定过夜,随后使用石蜡包埋,并制成切片,对细胞核和BRAFV600E蛋白标记染色处理。小鼠血液4℃静止一夜,低速离心15分钟分离出血清,随后对血清中的肝肾生化指标进行检测。实验结果见图7、图8、图9、图10。160w melanoma cells A375 were inoculated into the armpits of 3-week-old nude mice. After 7 days of tumor formation, the nude mice were divided into 5 groups. At this time, the average tumor volume was about 50 mm 3 (day 0), with 5 mice in each group. Tail vein injection of siMB3 and conjugate mixture for treatment, blank control is BLANK (tail vein injection preparation solvent Genopti), negative control is siMB3 (unencapsulated siMB3), mix (DNCA/CLD/PEG2000 without siRNA) -DSPE empty vector), mix-NC (mix-encapsulated siFL targeting firefly luciferase mRNA). The siRNA was administered at a dose of 2 mg/kg (RL0As) by tail vein injection on
3.结果:3. Results:
首先考察较短链接臂链接的RL0As/S-siMB3的体内抗肿瘤活性(图7)。结果表明,siMB3及其缀合物包载后在整个治疗过程中可明显抑制肿瘤生长(图7a,p<0.001),但缀合物RL0As/S抑制肿瘤生长的能力的弱于非缀合物As/S,尽管p值为0.25,无显著性差异。整个给药治疗过程中,小鼠体重稳定增长,各组间无差异,说明缀合不影响siRNA脂质复合物的安全性(图7b)。最后一次给药后48h安乐死小鼠并取出肿瘤称重(图7c),mix-As/S肿瘤体积最小,mix-RL0As/S的肿瘤体积相比空白和阴性对照也有一定降低,但整组内的治愈失败率为2/5,五只小鼠中有两只肿瘤体积与对照组相近。RT-qPCR测定瘤内BRAFV600EmRNA水平发现,As/S相较于RL0As/S可以更有效的沉默靶标mRNA(图7d)。免疫组化结果与qPCR结果一致(图7e),在小鼠的肿瘤石蜡切片中,siMB3及缀合物治疗组均明显降低了BRAFV600E蛋白的表达(褐色斑点)。The in vivo antitumor activity of RLOAs/S-siMB3 linked by the shorter linker arm was first examined (Figure 7). The results showed that siMB3 and its conjugates could significantly inhibit tumor growth during the entire treatment process after encapsulation (Fig. 7a, p<0.001), but the ability of conjugate RLOAs/S to inhibit tumor growth was weaker than that of non-conjugate As/S, despite the p-value of 0.25, was not significantly different. During the whole administration and treatment process, the body weight of the mice increased steadily, and there was no difference between the groups, indicating that the conjugation did not affect the safety of the siRNA lipoplex (Fig. 7b). The mice were euthanized 48 hours after the last administration and the tumors were taken out and weighed (Fig. 7c). The tumor volume of mix-As/S was the smallest, and the tumor volume of mix-RL0As/S was also reduced to a certain extent compared with the blank and negative controls. The cure failure rate was 2/5, and two of the five mice had tumors similar in size to the control group. RT-qPCR assay of intratumoral BRAF V600E mRNA levels found that As/S could silence target mRNA more effectively than RLOAs/S (Figure 7d). The immunohistochemical results were consistent with the qPCR results (Fig. 7e), in tumor paraffin sections of mice, both siMB3 and conjugate-treated groups significantly reduced the expression of BRAF V600E protein (brown spots).
体内抗肿瘤实验说明,尽管具有cRGD靶向基团的RL0As/S具有更优的肿瘤的蓄积能力,但由于其缀合基团与反义链间的空间距离过近,极大的影响了基因沉默活性,因此RL0As/S缀合物的治疗效果弱于未缀合的siMB3。结合细胞实验,对RL2As/S体内抗肿瘤活性进行考察(图8)。In vivo anti-tumor experiments show that although RLOAs/S with cRGD targeting groups have better tumor accumulation ability, the spatial distance between their conjugating groups and antisense strands is too close, which greatly affects gene expression. silencing activity, so the therapeutic effect of the RLOAs/S conjugate was weaker than that of the unconjugated siMB3. Combined with cell experiments, the in vivo antitumor activity of RL2As/S was investigated (Fig. 8).
经过5次尾静脉给药后,mix-siMB3及其缀合物mix-RL2As/S的肿瘤生长速度显着低于BLANK(p<0.0001),也低于未包载的siRNA(p<0.001)及mix-NC组(p<0.001)(图8a)。空白及阴性对照组的的肿瘤体积是第0天的25倍以上,mix-As治疗组增加了12倍,而mix-RL2As/S组的肿瘤生长速度比mix-As/S组慢得多(p<0.05),mix-RL2As/S治疗组的肿瘤体积仅增加了约6倍。反义链5′-末端经较长链接臂连接cRGD缀合基团的siMB3脂质复合物相比未缀合的siMB3脂质复合物可更有效的治疗肿瘤。最后一次给药48h后,安乐死小鼠,分离肿瘤组织称重(图8b)并拍照记录(图8c)。如图8b所示,mix-As/S组肿瘤重量明显小于空白及阴性对照组(p<0.05),缀合物mix-RL2As/S组的肿瘤重量与空白及阴性对照组相比也显著减小(p<0.01)。缀合物与未缀合的肿瘤肿瘤没有显著性差异(p>0.5),但mix-RL2As/S组的肿瘤重量均值小于mix-As/S组。各组离体肿瘤体积的大小与肿瘤生长曲线一致(图8c)。RT-qPCR测定瘤内BRAFV600E mRNA水平发现,As/S与RL2As/S可以更有效的沉默约40%靶标mRNA表达(图8d),且与空白及阴性对照组有显著性差异(p<0.001)。通过免疫组化实验同步评估了瘤内BRAFV600E的蛋白水平,黄色斑点代表BRAFV600E蛋白的表达(图8e)。与NC相比,siMB3及RL2As/S均下调了BRAFV600E,而RL2As/S与As/S的沉默效率相似。RL2As/S出色的抗肿瘤能力既与靶向基团的缀合,使肿瘤组织siMB3的蓄积量增加有关,也与缀合基团与反义链5′-末端的链接臂延长,使siRNA的基因沉默活性恢复有关。因为在体内实验结束时,经RL0As/S治疗的肿瘤生长速度大于未缀合的siMB3(图7a),尽管二者的肿瘤体积没有显著性差异(p=0.25)。这些结果表明,肿瘤靶向缀合基团cRGD增加了siMB3缀合物的纳米颗粒在肿瘤组织中的积累,并且cRGD与5′-末端之间的延长的空间距离(L2与L0相比)是反义链5′-末端缀合物优异抗肿瘤效率的关键点。此外,稳定变化的体重和治疗期间无小鼠死亡均表明所有的缀合物在体内应用是足够安全的(图8f)。After 5 tail vein administrations, the tumor growth rate of mix-siMB3 and its conjugate mix-RL2As/S was significantly lower than that of BLANK (p<0.0001) and lower than that of unencapsulated siRNA (p<0.001) and mix-NC group (p<0.001) (Fig. 8a). The tumor volume of the blank and negative control groups was more than 25 times that of
为验证缀合物及制剂的肝肾毒性,最后一次给药后48小时,收集全血,测量小鼠血液生化指标(图9)。各组的代表肾功能的CREA-J、UA、UREA和肝功能的ALT、AST、ALP、TP、ALB血生化指标与未给药组无明显差别,说明缀合和DNCA/CLD/PEG2000-DSPE混合脂材制剂的安全性佳,几乎不存在肝肾毒性,siMB3/DNCA/CLD/PEG2000-DSPE脂质复合物可作为安全有效的小鼠肿瘤疗法。In order to verify the liver and kidney toxicity of the conjugates and preparations, 48 hours after the last administration, whole blood was collected, and the blood biochemical indexes of the mice were measured (Fig. 9). The blood biochemical indexes of CREA-J, UA, UREA representing renal function and ALT, AST, ALP, TP, and ALB of liver function in each group were not significantly different from those of the non-administered group, indicating that the conjugation and DNCA/CLD/PEG2000-DSPE The mixed lipid preparation has good safety and almost no liver and kidney toxicity. The siMB3/DNCA/CLD/PEG2000-DSPE lipid complex can be used as a safe and effective tumor therapy in mice.
为验证siMB3缀合物治疗肿瘤的机制,测定单次给药后瘤内BRAFV600EmRNA的表达水平。使用Balb/c-nude 4周龄鼠,右侧腋下接种A375细胞160w,接种后20日,肿瘤体积约为500-800mm3时随机分成四组,分别经尾静脉注射给药。给药后48h,安乐死动物,取肿瘤组织50-100mg,定量mRNA的表达量。如图10所示,一次给药后,siMB3可沉默瘤内BRAFV600EmRNA的表达。未缀合的As/S可沉默14%靶mRNA,反义链5′-末端缀合cRGD的RL2As/S沉默22%的靶mRNA表达,且与mix-NC组相比有显著性差异(NC vs.As/S,p<0.05;NC vs.RL2As/S,p<0.001)。siRNA瘤内靶基因抑制水平略低可能是因为给药时肿瘤体积较大,siRNA不能完全转染进入肿瘤组织中的所有细胞,多次给药后的基因沉默活性更为明显(图8d)。RL2As/S在动物水平抑制肿瘤生长与BRAFV600EmRNA的沉默有关。To verify the mechanism of siMB3 conjugates in treating tumors, the expression level of intratumoral BRAF V600E mRNA was determined after a single dose. Balb/c-nude 4-week-old mice were used to inoculate 160w of A375 cells in the right armpit. 20 days after inoculation, when the tumor volume was about 500-800mm 3 , the mice were randomly divided into four groups and administered by tail vein injection respectively. 48h after administration, the animals were euthanized, and 50-100 mg of tumor tissue was taken to quantify the mRNA expression. As shown in Figure 10, siMB3 silenced intratumoral BRAF V600E mRNA expression after a single administration. Unconjugated As/S silenced 14% of the target mRNA, and RL2As/S with cRGD conjugated to the 5′-end of the antisense strand silenced 22% of the target mRNA expression, which was significantly different from the mix-NC group (NC). vs. As/S, p<0.05; NC vs. RL2As/S, p<0.001). The slightly lower level of intratumoral target gene inhibition by siRNA may be due to the large tumor volume at the time of administration, and the siRNA could not be fully transfected into all cells in the tumor tissue, and the gene silencing activity was more pronounced after multiple administrations (Figure 8d). The inhibition of tumor growth by RL2As/S at the animal level is associated with silencing of BRAF V600E mRNA.
本文显示并详细描述的信息足以实现本发明的上述目的,因此本发明的优选实施方案代表本发明的主题,该主题为本发明所广泛涵盖。本发明的范围完全涵盖其它对本领域技术人员来说显而易见的实施方案,因此,本发明的范围不被除所附权利要求之外的任何内容所限制,其中除了明确说明外,所用元素的单数形式并不是指“一个和唯一”,而是指“一个或更多”。对本领域一般技术人员来说,所有公知的上述优选的实施方案和附加实施方案部分的结构、组成和功能上的等价物因此引入本文作参考,而且试图被本发明的权利要求所涵盖。The information shown and described in detail herein is sufficient to accomplish the foregoing objects of the invention, and thus preferred embodiments of the invention represent the subject matter of the invention, which is broadly encompassed by the invention. The scope of the present invention fully encompasses other embodiments apparent to those skilled in the art, therefore, the scope of the present invention is not to be limited by anything other than the appended claims, wherein the singular form of elements used is used unless explicitly stated otherwise. It does not mean "one and only", but "one or more". All known structural, compositional and functional equivalents of the above-described preferred embodiments and parts of the additional embodiments to those of ordinary skill in the art are hereby incorporated by reference and are intended to be encompassed by the present claims.
此外,不需要某种设备或方法来表达本发明所解决的每个问题,因为它们都已包括在本发明的权利要求之内。另外,无论本发明公开事实中的所有部分、成分,或者方法步骤是否在权利要求中被明确叙述,它们都没有贡献给公众。但是,对本领域普通技术人员来说,很明显在不背离如所附权利要求中所阐明的本发明的实质和范围的前提下,可以在形式、试剂和合成细节上做出各种改变和修饰。Furthermore, no particular apparatus or method is required to express each and every problem solved by the present invention, for they are intended to be encompassed within the claims of the present invention. Furthermore, no matter whether or not all parts, elements, or method steps of the present disclosure are expressly recited in the claims, they are not dedicated to the public. However, it will be apparent to those of ordinary skill in the art that various changes and modifications in form, reagents and details of synthesis can be made without departing from the spirit and scope of the invention as set forth in the appended claims .
Claims (9)

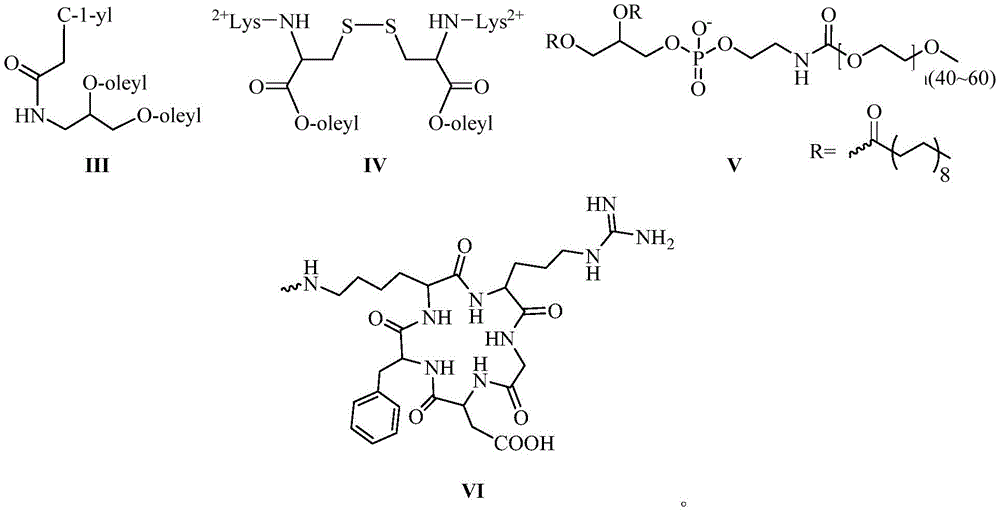
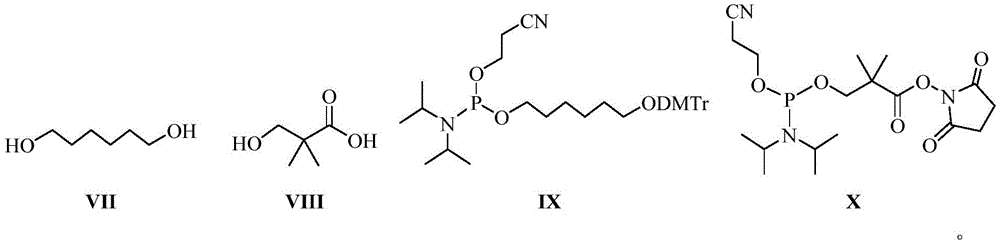
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011644259.1A CN112707943B (en) | 2020-12-31 | 2020-12-31 | Combining 5'-terminal conjugate and neutral/cationic mixed lipid-encapsulated small interfering RNA and its modification method |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011644259.1A CN112707943B (en) | 2020-12-31 | 2020-12-31 | Combining 5'-terminal conjugate and neutral/cationic mixed lipid-encapsulated small interfering RNA and its modification method |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112707943A CN112707943A (en) | 2021-04-27 |
| CN112707943B true CN112707943B (en) | 2022-09-09 |
Family
ID=75548063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202011644259.1A Active CN112707943B (en) | 2020-12-31 | 2020-12-31 | Combining 5'-terminal conjugate and neutral/cationic mixed lipid-encapsulated small interfering RNA and its modification method |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN112707943B (en) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113797327A (en) * | 2021-09-24 | 2021-12-17 | 北京大学 | A kind of nucleic acid drug delivery carrier for carrying mRNA and preparation method and application thereof |
| CN114246829B (en) * | 2021-12-31 | 2023-03-24 | 北京大学 | Method for encapsulating antisense nucleic acid medicament, medicinal preparation prepared by method and application of medicinal preparation in liver cancer treatment |
| CN118615256A (en) * | 2023-03-08 | 2024-09-10 | 北京大码方诚生物科技有限公司 | A mixed nanolipid delivery system for mRNA and its preparation method and use |
| CN116570615B (en) * | 2023-05-09 | 2026-01-27 | 北京大学 | SiRNA pharmaceutical preparation for treating pancreatic cancer and preparation method and application thereof |
| CN119639745B (en) * | 2024-12-12 | 2025-11-25 | 制能(北京)生物科技有限公司 | An antisense nucleic acid targeting the KRAS gene, its drug formulation and application |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101346393B (en) * | 2005-11-02 | 2015-07-22 | 普洛体维生物治疗公司 | Modified siRNA molecules and uses thereof |
| CN102660543A (en) * | 2012-01-21 | 2012-09-12 | 北京大学 | Method, product and application of chemical modification of siRNA sequence using isonucleoside |
| CN110540992B (en) * | 2019-08-21 | 2021-02-09 | 武汉泽智生物医药有限公司 | siRNA molecule for enhancing silencing efficiency of target gene and application thereof |
-
2020
- 2020-12-31 CN CN202011644259.1A patent/CN112707943B/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN112707943A (en) | 2021-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112707943B (en) | Combining 5'-terminal conjugate and neutral/cationic mixed lipid-encapsulated small interfering RNA and its modification method | |
| EP2549986B1 (en) | Multi-compartmental macrophage delivery | |
| CN104244928B (en) | Magnetic nanoparticle-SAMiRNA complex and preparation method thereof | |
| RU2599449C1 (en) | New oligonucleotides conjugates and use thereof | |
| JP2011503070A (en) | Self-assembled micelle-like nanoparticles for systemic gene delivery | |
| BR112014016562B1 (en) | DOUBLE HELIX OLIGO-RNA STRUCTURE, NANOPARTICLE AND PREPARATION METHOD | |
| JP2021525508A (en) | Compositions and Methods of Adjustable Co-Coupling Polypeptide Nanoparticle Delivery Systems for Nucleic Acid Therapeutics | |
| JP2009504179A (en) | Conjugate between siRNA and hydrophilic polymer for intracellular transmission of siRNA, and method for producing the same | |
| WO2008058457A1 (en) | A biodegradable crosslinked polyethyleneimine and its uses | |
| CN106177975A (en) | Reversible cross-linked biodegradable polymer vesicles with asymmetric membrane structure and its preparation method and application in nucleic acid medicine | |
| CN116425973B (en) | PH-responsive liver-targeted drug delivery carrier and preparation method and application thereof | |
| CN111481679A (en) | siRNA nanocapsule and its preparation method and application | |
| CN116077670B (en) | Targeting drug-carrying system based on DNA tetrahedral nanoclusters and construction method and application thereof | |
| US12201644B2 (en) | Conjugation of lipophilic albumin-binding moiety to RNA for improved carrier-free in vivo pharmacokinetics and gene silencing | |
| JP2022550901A (en) | Tumor-targeted polypeptide nanoparticle delivery system for nucleic acid therapeutics | |
| CN113633613B (en) | siRNA micelle, preparation method, composition and application thereof | |
| CN117919202A (en) | A lipid nanoparticle, anionic polymer synergistic lipid nanoparticle nucleic acid delivery system and its preparation and application | |
| CN108904811A (en) | It is a kind of based on aptamers modification hollow mesoporous silicon oxide be total to charge material grain multifunctional nano drug preparation and its application | |
| CN111803517A (en) | Based on nucleic acid material loaded platinum drug conjugate, platinum drug delivery system and preparation method and application thereof | |
| CN120249289B (en) | T-cell-targeting nanoparticles based on nucleic acid aptamers and their preparation and application | |
| CN114432262A (en) | Liver targeting small nucleic acid drug sustained-release delivery system and application thereof | |
| JP7578988B2 (en) | Temperature-responsive drug-polymer conjugates and their application to drug delivery | |
| CN118994011A (en) | SiRNA nano micelle and preparation method and application thereof | |
| CN121081678A (en) | A mRNA-LNP vector that can regulate muscle and lymph node organ and cell targeting | |
| KR101722948B1 (en) | Double stranded oligo RNA molecule with a targeting ligand and method of preparing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |