Petroleum resin hydrogenation catalyst and preparation method thereof
Technical Field
The invention relates to a petroleum resin hydrogenation catalyst and a preparation method thereof.
Background
C 5 Hydrogenated petroleum treeThe fat is obtained by reacting C with hydrogenation catalyst 5 The double bonds in the petroleum resin are saturated and the residual halogen elements in the polymerization process of the resin are removed to prepare the resin. The hydrogenation process improves the chroma and the photo-thermal stability of the resin and greatly widens the C 5 The application field of petroleum resin. C 5 Hydrogenation of petroleum resin will be China C 5 The petroleum resin realizes the important ways of serialization and commercialization, and is also an important means for improving economic benefit; thus, C is accelerated 5 Research and development of hydrogenated petroleum resins are essential.
CN 102935367B discloses a C 5 A petroleum resin hydrogenation catalyst and a preparation method thereof. The catalyst comprises an alumina-titania composite carrier and metallic palladium and metallic molybdenum or metallic tungsten loaded on the composite carrier. The catalyst is used in C 5 The petroleum resin hydrogenation process not only has low-temperature hydrogenation activity, but also has good impurity resistance and good stability. However, the catalyst adopts noble metal active components, the sulfur resistance and chlorine poisoning resistance of the catalyst are poor, the catalyst is easy to be poisoned and inactivated, and meanwhile, the catalyst adopts noble metals and is expensive.
CN 101700494B discloses the preparation and use of a hydrogenation catalyst. The catalyst adopts aluminum hydroxide, diatomite, a pore-expanding agent and rare earth element modified aluminum trioxide as carriers, bi-component noble metal loading is carried out on the carriers, and simultaneously, rare earth elements are used for carrying out catalytic activity blending to obtain a hydrogenation catalyst with high activity and long service life, so that hydrogenated petroleum resin with light hue and high softening point can be prepared. But the catalyst has poor capability of resisting sulfur and poisoning, is easy to be poisoned and deactivated, and simultaneously adopts noble metal, so the cost is high.
CN 107876049A discloses a petroleum resin hydrogenation catalyst with sulfur resistance, a preparation method and application thereof. The catalyst is prepared from alumina sol serving as a raw material by adopting a multi-time slurry dipping method 2 O 3 The membrane-coated active carbon carrier is then immersed in gamma-Al by the equal volume immersion method 2 O 3 Active component palladium and catalyst surface modifier CeO are loaded on membrane-coated active carbon carrier 2 Then drying and roasting to prepareObtaining the catalyst for petroleum resin hydrogenation. The catalyst of the invention is particularly suitable for use in C 5 The petroleum resin hydrogenation decoloring process has better activity stability and sulfur resistance effect. However, the catalyst adopts a multi-time slurry dipping method to prepare the carrier, so that the specific surface area of the catalyst is reduced, the number of active digits of the catalyst is reduced, and the activity of the catalyst is reduced.
Disclosure of Invention
Aiming at the defects of the prior art, the invention provides a petroleum resin hydrogenation catalyst and a preparation method thereof. The catalyst has higher hydrogenation activity and stability, is applied to the hydrogenation process of petroleum resin, and has high capability of resisting impurity poisoning such as sulfur, chlorine and the like.
The petroleum resin hydrogenation catalyst comprises hydrogenation active components and a hydrogenation catalyst carrier, wherein the hydrogenation active components are VIII group metal sulfides and VIB group metal oxides, the VIII group metal is preferably Co and/or Ni, and the VIB group metal is preferably Mo and/or W; based on the total weight of the catalyst, the content of the VIII group metal sulfide is 3wt% -20wt%, preferably 10wt% -20wt%, the content of the VIB group metal oxide is 2wt% -15wt%, preferably 3wt% -10wt%, and the content of the hydrogenation catalyst carrier is 65% -95%.
The catalyst is analyzed by XPS energy spectrum, and the molar proportion of the sulfurized state content of the VIII group metal with the +2 valence state to the total VIII group metal content is 80-100%.
After the catalyst is vulcanized, CO-FTIR infrared spectrum analysis is adopted, and the molar proportion of the content mol of the VIB group metal-VIII group metal-S species (such as Mo-Co-S, mo-Ni-S, W-Co-S, W-Ni-S and the like) in the total VIB group metal content is 60-100%. The vulcanization treatment conditions are as follows: adopting dry vulcanization or wet vulcanization, wherein the dry vulcanization agent is hydrogen sulfide, and the wet vulcanization agent is one or more of carbon disulfide, dimethyl disulfide, methyl sulfide and n-butyl sulfide; the vulcanization pressure is 3.2-6.4MPa, the vulcanization temperature is 250-400 ℃, and the vulcanization time is 4-12h.
The hydrogenation catalyst carrier component is one or more of porous inorganic refractory oxides selected from the oxides of elements in the II group, the III group, the IV group and the IVB group in the periodic table, more preferably one or more of silica, alumina, magnesia, zirconia, titania, silica-alumina, magnesia-silica and alumina-magnesia, and further preferably alumina. The hydrogenation catalyst carrier component can be modified according to the need, for example, modification elements such as B, P, F are adopted for modification, and the weight percentage of the modification elements is 0.5wt% -10wt% based on the weight of the modified hydrogenation catalyst carrier component.
The invention provides a preparation method of a petroleum resin hydrogenation catalyst, which comprises the following steps:
(l) Dipping a hydrogenation catalyst carrier by using dipping liquid containing VIII group metal, then drying, and vulcanizing the dried material to obtain a catalyst precursor;
(2) And (2) impregnating the catalyst precursor obtained in the step (1) with an impregnating solution containing a VIB group metal, and then drying and roasting the catalyst precursor in an inert atmosphere to obtain the petroleum resin hydrogenation catalyst.
In the method of the present invention, the second step described in step (1)
The impregnation of the group metals is well known to those skilled in the art and typically involves the use of nitrate, acetate, sulfate solutions and the like
The mass concentration of the group metal oxide is 0.1 g/mL-1.2 g/mL, an equal volume impregnation mode can be adopted, and the second step is
The group metals are preferably Ni and/or Co.
In the method of the invention, the drying conditions in the step (1) are as follows: the drying temperature is 90-300 ℃, and the drying time is 3-6 hours.
In the method of the present invention, the vulcanization treatment in step (1) is well known to those skilled in the art, and usually adopts dry vulcanization or wet vulcanization, wherein the dry vulcanization agent is hydrogen sulfide, and the wet vulcanization agent is one or two of carbon disulfide, dimethyl disulfide, methyl sulfide, and n-butyl sulfide; the vulcanization pressure is 2.5 to 6.4MPa, the vulcanization temperature is 250 to 400 ℃, and the vulcanization time is 4 to 12h.
In the method, the preparation method of the group VIB metal impregnation liquid in the step (2) is well known by those skilled in the art, for example, a phosphate or ammonium salt solution is generally adopted, and the mass concentration of the group VIB metal oxide in the impregnation liquid is 0.1 g/mL-1.0 g/mL. The group VIB metal is preferably Mo and/or W.
In the method of the invention, the inert atmosphere in the step (2) is N 2 And an inert gas; the drying temperature is 20 to 90 ℃, and the drying time is 4 to 16 hours; the roasting temperature is 200 to 500 ℃, and the roasting time is 2~5 hours.
The petroleum resin hydrogenation catalyst is applied to the hydrogenation process of petroleum resin, and is particularly suitable for C 5 Petroleum resin hydrogenation process.
The hydrogenation catalyst of the invention needs to be vulcanized before application, and the general vulcanization conditions are as follows: adopting dry vulcanization or wet vulcanization, wherein the dry vulcanization agent is hydrogen sulfide, and the wet vulcanization agent is one or more of carbon disulfide, dimethyl disulfide, methyl sulfide and n-butyl sulfide; the vulcanization pressure is 2.0 to 6.4MPa, the vulcanization temperature is 250 to 400 ℃, and the vulcanization time is 4 to 12h.
The invention provides the application of the petroleum resin hydrogenation catalyst obtained by the preparation method, when the petroleum resin hydrogenation catalyst is used in the hydrogenation reaction of petroleum resin, the reaction is carried out in a fixed bed reactor, and the reaction temperature is 200-220 ℃; the reaction pressure is 4.0-15.0 MPa, and the volume space velocity of the reaction raw material is 0.25-2.5 h -1 (ii) a The molar ratio of unsaturated hydrocarbon to hydrogen in the petroleum resin is preferably 1:3 to 1:4. during the hydrogenation reaction, the solvent is any one or more of cyclohexane, cyclopentane, toluene or xylene, preferably toluene or xylene, and the weight ratio of the raw material to the solvent is 1:1 to 1:2.
the VIII group metal is impregnated into the carrier, then vulcanized and then impregnated with the VIB group metal, the VIII group metal in the catalyst is firstly vulcanized to form more active edges, corners and edges, the VIB group metal is loaded on the active sites to promote the interaction of the VIB group metal and the VIII group metal to form active sites with impurity poisoning resistance, so that the activity and the impurity poisoning resistance of the catalyst are improved, meanwhile, the VIB group metal content in the catalyst is low, the cracking reaction in the hydrogenation reaction process of petroleum resin can be effectively weakened, and the reduction of the softening point of the petroleum resin is prevented.
Detailed Description
The following examples further illustrate the present invention and the effects thereof, but are not intended to limit the present invention. The catalyst composition provided by the invention can be characterized by combining inductively coupled plasma ICP and XPS energy spectrum, and the total content of VIB group metals and the total content of XPS group metals in the catalyst are firstly characterized by ICP
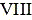
And (3) quantitatively characterizing the content of metal elements with different valence states in the catalyst by an XPS spectrometer. The molar content of the interacted species of the VIB group metal and the VIII group metal of the catalyst provided by the invention accounts for the total content of the VIB group metal, and is expressed by the content of Mo-Co-S, mo-Ni-S, W-Co-S, W-Ni-S. An infrared spectrum of CO adsorbed by the catalyst is measured by a Nicolet 6700 Fourier transform infrared spectrometer, then peak-splitting fitting is carried out on the spectrogram, and the content of Mo-Co-S, mo-Ni-S, W-Co-S, W-Ni-S is obtained by calculation according to the peak area.
Analysis and detection instrument and execution standard: color values: the United states Hunter LabColour Quest EX colorimetric analyzer, implements the standard ASTM E313. Bromine number: mettler TOLEDO model DL58, USA, implements standard ASTM D1159-93. Softening point: a domestic SYD-2806F softening point tester, which is used for executing the standard GB/T12007.6-1989. Chlorine content: a domestic RPA-200A microcoulometric titrator, performing standard large hospital-connected DIPP81.
Example 1
Dissolving nickel acetate into deionized water to prepare a solution, wherein the NiO concentration in the solution is 0.15g/mL, 100mL of the solution was impregnated into 120g of an alumina support, followed by drying at 120 ℃ and then by a dry mass of 2.0% H 2 Sulfurizing S hydrogen at 250 deg.C under 3.0MPa for 8 hr, and adding N 2 And cooling to room temperature in the atmosphere to obtain the catalyst precursor.
Dissolving ammonium metatungstate in deionized water to prepare a solution, wherein WO is contained in the solution 3 At a concentration of 0.26g/mL, 30mL of the solution was immersed in the catalyst precursor in equal volume, followed by N 2 Drying at 110 ℃ for 3h under the atmosphere, and roasting at 320 ℃ for 3h to obtain the catalyst C-1.
Example 2
Dissolving nickel acetate in deionized water to obtain a solution with NiO concentration of 0.15g/mL, soaking 100mL of the solution in 120g of silica-modified alumina support, drying at 120 deg.C, and subjecting to a solution containing 2.0% H 2 Sulfurizing S hydrogen at 300 deg.C under 3.5MPa for 8 hr, and adding N 2 And cooling to room temperature in the atmosphere to obtain the catalyst precursor.
Dissolving ammonium molybdate into deionized water to prepare solution, wherein MoO is contained in the solution 3 At a concentration of 0.3g/mL, 30mL of the solution was immersed in the catalyst precursor in equal volume, followed by N 2 Drying at 110 ℃ for 3h and roasting at 300 ℃ for 3h under the atmosphere to obtain the catalyst C-2.
Example 3
Dissolving cobalt nitrate in deionized water to give a solution having a CoO concentration of 0.4g/mL, impregnating 100mL of the solution into 150g of a P-modified alumina support, drying at 120 deg.C, and subjecting to a solution containing 2.0% H 2 Sulfurizing S hydrogen at 250 deg.C under 3.0MPa for 8 hr, and adding N 2 And cooling to room temperature in the atmosphere to obtain the catalyst precursor.
Dissolving ammonium metatungstate in deionized water to prepare solution, and adding WO in the solution 3 At a concentration of 0.26g/mL, 45mL of the solution was immersed in the catalyst precursor in equal volume, followed by N 2 Drying at 110 deg.C for 3h, and calcining at 320 deg.C for 3h under atmosphere to obtain the catalystAgent C-3.
Example 4
Dissolving nickel nitrate and cobalt nitrate in deionized water to prepare a solution having a NiO concentration of 0.1g/mL and a CoO concentration of 0.12g/mL, impregnating 100mL of the solution or the like into 120g of an alumina support, drying at 120 ℃, and then adjusting the H content by using a solution having a content of 2.0% 2 Sulfurizing S hydrogen at 350 deg.C under 3.0MPa for 8 hr, and adding N 2 And cooling to room temperature in the atmosphere to obtain the catalyst precursor.
Dissolving ammonium metatungstate in deionized water to prepare solution, and adding WO in the solution 3 At a concentration of 0.26g/mL, 30mL of the solution was immersed in the catalyst precursor in equal volume, followed by N 2 Drying at 110 ℃ for 3h and roasting at 320 ℃ for 3h under the atmosphere to obtain the catalyst C-4.
Example 5
Dissolving nickel acetate in deionized water to give a solution with NiO concentration of 0.25g/mL, soaking 100mL of the solution in an equal volume into 130g of an alumina support, drying at 120 deg.C, and subjecting to a solution containing 2.0% H 2 Sulfurizing S hydrogen at 250 deg.C under 3.0MPa for 8 hr, and adding N 2 And cooling to room temperature in the atmosphere to obtain the catalyst precursor.
Dissolving ammonium metatungstate and ammonium molybdate into deionized water to prepare solution, and adding WO into the solution 3 The concentration is 0.2g/mL, moO 3 At a concentration of 0.12g/mL, 30mL of the solution was immersed in the catalyst precursor in equal volume, followed by N 2 Drying at 110 ℃ for 3h and roasting at 320 ℃ for 3h under the atmosphere to obtain the catalyst C-5.
Comparative example 1
Dissolving nickel acetate into deionized water to prepare a solution, wherein the concentration of NiO in the solution is 0.15g/mL, soaking 100mL of the solution into 120g of an alumina carrier, and then drying at 120 ℃ to obtain a catalyst precursor.
Dissolving ammonium metatungstate in deionized water to prepare solution, and adding WO in the solution 3 30mL of the impregnation solution was immersed in the catalyst precursor at a concentration of 0.26g/mL, followed by N 2 Drying at 110 ℃ for 3h and roasting at 320 ℃ for 3h under the atmosphere to obtain the catalyst DC-1.
Comparative example 2
Dissolving nickel acetate and ammonium metatungstate into deionized water to prepare a solution, wherein the concentration of NiO in the solution is 0.15g/mL, and WO 3 The concentration was 0.078g/mL, and 100mL of the solution was impregnated into 120g of an alumina carrier, followed by drying at 120 ℃ and calcination at 320 ℃ for 3 hours to obtain catalyst DC-2.
Comparative example 3
120g of alumina carrier is put into 90ml of deionized water containing 0.006g/ml of palladium chloride, after the completion of impregnation, 120ml of hydrazine hydrate with the concentration of 40 percent is used for reduction for 2 hours, the obtained product is washed by the deionized water until no chloride ion exists, and then the obtained product is dried for 3 hours at the temperature of 120 ℃ and roasted for 3 hours at the temperature of 700 ℃ to obtain the catalyst DC-3. Wherein the palladium content is 0.26% of the total weight of the catalyst.
TABLE 1 catalyst active Metal composition
Example 6
This example demonstrates the performance of the catalyst provided by the present invention for hydrogenation reactions on petroleum resins.
The petroleum resin evaluated was a commercially available mixed carbon five petroleum resin, which was dissolved in a cyclohexane solvent to form a 40wt% raw material solution having a chlorine content of 986ppm and a sulfur content of 42.3ppm.
The hydrogenation performance of the catalysts C-1 to C-5 and the comparative examples DC-1 to DC-3 were evaluated by using a 200mL fixed bed petroleum resin hydrogenation apparatus.
Presulfiding conditions for catalysts C-1 to C-5, comparative examples DC-1 to DC-2 catalysts: using a catalyst containing 3wt% of CS 2 At the airspeed of 1.0h -1 Hydrogen to oil volume ratio 500, presulfiding the catalyst at an operating pressure of 5.0 MPa.
The prevulcanisation process is as follows: and (2) feeding pre-vulcanized oil at 120 ℃, after feeding the oil for 2 hours, vulcanizing at constant temperature for 2 hours, heating to 150 ℃ at a speed of 15 ℃/hour, vulcanizing at constant temperature for 4 hours, heating to 300 ℃ at a speed of 10 ℃/hour, vulcanizing at constant temperature for 6 hours, and naturally cooling to 150 ℃, thus finishing the pre-vulcanization.
Catalysts C-1 to C-5, comparative examples DC-1 to DC-3 the reaction conditions were evaluated as follows: the reaction temperature is 220 ℃, the operation pressure is 8.0Mpa, and the liquid raw material volume space velocity is 0.3h -1 Hydrogen volume space velocity of 200h -1 After the reaction is carried out for 200 hours, the reaction mixture is firstly subjected to alkali washing and water washing, and then the solvent and byproducts are removed by a falling film evaporator to obtain a solid resin product, and the solid resin product is analyzed and detected, wherein the evaluation result is shown in table 2.
TABLE 2 Properties and evaluation results of the catalyst after vulcanization
The evaluation results in table 2 can show that the petroleum resin hydrogenation catalyst of the present invention achieves higher hydrogenation activity, impurity poisoning resistance and stability when used in the hydrogenation reaction of petroleum resin.




