CN112501169A - Method for constructing MSH2 gene knockout cell line - Google Patents
Method for constructing MSH2 gene knockout cell line Download PDFInfo
- Publication number
- CN112501169A CN112501169A CN202011373830.0A CN202011373830A CN112501169A CN 112501169 A CN112501169 A CN 112501169A CN 202011373830 A CN202011373830 A CN 202011373830A CN 112501169 A CN112501169 A CN 112501169A
- Authority
- CN
- China
- Prior art keywords
- msh2
- px459m
- sgrna
- sgrna1
- gene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/66—General methods for inserting a gene into a vector to form a recombinant vector using cleavage and ligation; Use of non-functional linkers or adaptors, e.g. linkers containing the sequence for a restriction endonuclease
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
Landscapes
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
The invention relates to a method for constructing an MSH2 gene knockout cell line, and relates to the technical field of genetic engineering. The method is characterized in that a CRISPR/Cas9 system is adopted to prepare an MSH2 gene knockout cell line, firstly, two sgRNA sequences are designed aiming at an MSH2 gene, and a recombinant plasmid is constructed by utilizing a molecular cloning technology; and then transfecting the recombinant plasmid to a HeLa cell, verifying the activity of sgRNA by PCR, performing puromycin drug screening and monoclonality treatment, extracting the genomic DNA and total protein of a monoclonal cell strain, and performing MSH2 gene level sequencing identification and protein level expression detection to obtain an MSH2 gene knockout HeLa cell line. The HeLa cell line knocked out by the MSH2 gene constructed by the invention is a cell line with stable genetic genes. The method provided by the invention can be used for directional knockout of the MSH2 gene to inactivate the function of the MSH2 gene, has the characteristics of simplicity, convenience, high efficiency, rapidness, low cost and the like, and has important significance for research on the function of the MSH2 gene and related pathways.
Description
[ technical field ] A method for producing a semiconductor device
The invention relates to the technical field of genetic engineering, in particular to a method for constructing an MSH2 gene knockout cell line.
[ background of the invention ]
Human MutS homologous protein 2(Human MutS homologue 2, hMSH2) is an important member of the DNA mismatch repair protein family, usually localized to the nucleus, forming heterodimers with MSH3 or MSH6, and involved in the repair of base mismatches in DNA synthesis. The hMSH2 is expressed ectopically, is commonly found in various tumor cells, and can be infected by EBV virus, H2O2Induced by stimulation. The ectopically expressed hMSH2 can be used as a ligand molecule of a gamma delta T cell receptor (TCR gamma delta), is doubly recognized by TCR gamma delta and NK cell receptor member 2D (Natural killer group 2member D, NKG2D), and mediates the killing effect of the gamma delta T cells on tumor cells.
In DNA mismatch repair, hMSH2 protein first forms a dimeric complex with hMSH6, called hMutS α dimer, and binds to DNA base mismatch regions, and then binds to the dimeric hMutL α formed by hMLH1 and the hPSM2 genes, where the heterodimeric hMutS α functions to recognize the nicked nascent DNA strand. Binding of hmutL α and hmutS α to the DNA mismatch region activates the mutH-related GATC endonuclease, which cleaves the unmodified methylated GATC sequence. Depending on the synergistic effect of MutS, MutL and DNA helicase (MutU), the entire repair reaction is initiated following excision of the mismatch region of the nascent strand.
The MSH2 protein is almost present in all colon normal mucosa, and the positive rate reaches 94%. The positive products are located in the nucleus, and cytoplasmic staining occurs simultaneously in part of the cells. The positive cells are distributed throughout the mucosa, more densely arranged under the fossa of the gland 1/3-2/3.
Expression of MSH2 protein was occasionally seen in goblet cells. The MSH2 protein is also expressed on mesenchymal fibroblasts, vascular endothelial cells and lymphocytes, in karyotype or karyotype, but with less expression in plasma cells.
Mutations in the MSH2 gene are associated with microsatellite instability and some cancers. The protein positive expression rate of the MSH2 gene is found to be gradually reduced from normal tissues, adenoma atypical hyperplasia, adenoma carcinoma to adenocarcinoma, which indicates that the MMR gene has mutation or dysfunction in the adenoma stage, and the mutation frequency of the MMR gene is gradually increased along with the increase of the malignancy degree. Studies have shown that mutations in the MMR gene, and in particular in MSH2, may be one of the early events in colorectal cancer development. Hereditary nonpolyposis colorectal cancer (HNPCC), sometimes referred to as Lynch syndrome, is inherited in an autosomal dominant fashion, where inheritance of only one copy of a mutated mismatch repair gene is sufficient to cause the disease phenotype, accounting for approximately 15% to 18% of the total number of colorectal cancers, accounting for 50% of familial colorectal cancers. Mutations in the MSH2 gene account for 40% of the disease-associated gene alterations, and HNPCC-associated mutations are widely distributed in all domains of MSH2 and are the major cause of MLH1 mutations. In addition, mutations in MSH2 and other mismatch repair genes can also result in unrepaired DNA damage, resulting in increased mutation frequencies.
The researchers have proved that the expression level of the mismatch repair protein MSH2 in most visceral organs such as brain, lung, colon, heart, muscle, spleen, stomach and prostate gradually decreases with age, which indicates that it is closely related to cell aging.
CRISPR/Cas9 is a gene editing technology developed based on the natural immune mechanisms of bacteria. The system is an immune system used by prokaryotes to combat invasion by foreign genetic material. The simplified engineered CRISPR/Cas9 system is a third generation precise genome editing technology that appears following zinc finger endonucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). The Double-stranded DNA chip consists of two parts, namely endonuclease Cas9 and gRNA (Guide RNA, gRNA), wherein the Cas9 can recognize and combine an original spacer adjacent motif (PAM) on a genome and form DNA Double-stranded unwinding, and if the crRNA part of the gRNA can be successfully and complementarily paired with an upstream sequence of the PAM at the moment, the Cas9 activates the endonuclease activity of the gRNA and forms Double-stranded DNA Break (DBS) at a specific position upstream of the PAM. DBS can activate cellular DNA damage repair mechanisms, Non-homologous End Joining (NHEJ) or homologous recombination-mediated repair (HDR). NHEJ, an error-prone repair, causes random insertions or deletions at the repair site that result in a frameshift mutation such that the gene is no longer expressed, thereby creating a gene knockout. HDR, i.e., precision repair, can mediate gene replacement or insertion by means of exogenously introduced single-or double-stranded DNA as a template, which allows for precise insertion of a DNA sequence into a specific genomic site, thereby completing knock-in or replacement.
At present, no report about the MSH2 gene knockout by using a CRISPR/Cas9 system exists, so that a method for constructing an MSH2 gene knockout cell line needs to be researched on the basis of the CRISPR/Cas9 system.
[ summary of the invention ]
The invention aims to provide a method for constructing an MSH2 gene knockout cell line, which can knock out an MSH2 gene in a cell efficiently, quickly and conveniently, construct an MSH2 gene knockout cell line, and can be used for researching the function, related pathways and drug development of the MSH2 gene.
In view of this, the present invention provides an sgRNA for knocking out MSH2 gene, including the sgRNA shown in SEQ ID NO: 1 and sgRNA1 as set forth in SEQ ID NO: 2, sgRNA 2.
Another objective of the invention is to provide a recombinant plasmid PX459M-MSH2-sgRNAs of the sgRNA.
The invention also aims to provide a method for constructing the recombinant plasmid PX459M-MSH2-sgRNAs of the sgRNAs, which comprises the following steps:
1. synthesizing a sgRNA DNA sequence with an enzyme cutting site and a complementary sequence thereof based on the sgRNA1 and the sgRNA2, and respectively annealing to obtain two double-stranded DNA fragments of MSH2-sgRNA1 and MSH2-sgRNA 2;
2. respectively carrying out enzyme digestion and recycling on PX459M and EZ-GuideXH two carriers;
3. the two double-stranded DNA fragments are respectively connected with a PX459M and an EZ-guide XH vector which are recovered by enzyme digestion, and product transformation and sequencing identification are carried out to obtain PX459M-MSH2-sgRNA1 and EZ-guide XH-MSH2-sgRNA 2;
4. PX459M-MSH2-sgRNA1 and EZ-guide XH-MSH2-sgRNA2 are subjected to enzyme digestion, the former recovers a vector skeleton containing MSH2-sgRNA1, the latter recovers a DNA fragment containing MSH2-sgRNA2, and then product connection is carried out to construct recombinant plasmid PX459M-MSH 2-sgRNAs.
Further, in the step 1: the sgRNA DNA sequence of the sgRNA1 with the enzyme cutting site is SEQ ID NO: 3, the complementary sequence is SEQ ID NO: 4; the sgRNA DNA sequence of the sgRNA2 with the enzyme cutting site is SEQ ID NO: 5, the complementary sequence is SEQ ID NO: 6.
further, the method for transforming and identifying the DNA ligation product in the step 3 comprises the following steps: adding a connection product of the enzyme-cut vector skeleton and the double-stranded DNA fragment into competent bacteria, supplementing a culture medium, culturing for half an hour, coating the mixture on a resistant plate, culturing overnight, selecting a single colony, carrying out amplification culture, harvesting a bacterial liquid, extracting a recombinant plasmid, carrying out sequencing identification, and storing correctly identified recombinant plasmids PX459M-MSH2-sgRNA1 and EZ-GuideXH-MSH2-sgRNA 2.
Further, the step 4 is: recovering a vector framework containing MSH2-sgRNA1 for PX459M-MSH2-sgRNA1, recovering a DNA fragment containing MSH2-sgRNA2 for EZ-guide XH-MSH2-sgRNA2, and connecting the recovered enzyme digestion product by using DNA ligase; then, the transformation and identification of the ligation products are carried out in the same step 3, and the recombinant plasmid with correct sequencing identification is named as PX459M-MSH 2-sgRNAs.
The invention also aims to provide application of the recombinant plasmid PX459M-MSH2-sgRNAs in preparation of a HeLa cell line with a knocked-out MSH2 gene.
Further, the method for constructing the MSH2 gene knockout HeLa cell line by the recombinant plasmid PX459M-MSH2-sgRNA of the sgRNA comprises the following steps:
1) transfecting HeLa cells with recombinant plasmid PX459M-MSH 2-sgRNAs;
2) extracting transfected cell genome, performing PCR amplification identification aiming at MSH2 gene, and detecting the activity of sgRNA;
3) screening puromycin drugs for transfected cells, and then performing cell monoclonality treatment;
4) extracting the genomic DNA of the monoclonal cell strain, and performing MSH2 gene sequencing identification to obtain a HeLa cell line knocked out by the MSH2 gene.
Further, the method for detecting the activity of the sgRNA in the step 2) comprises the following steps: after recombinant plasmid PX459M-MSH2-sgRNAs transfects HeLa cells, a rat tail genomic DNA detection kit is adopted to detect the genomic DNA of the extracted cells as a template, and the DNA sequence shown in SEQ ID NO: 7 and SEQ ID NO: 8, carrying out PCR amplification on the primers shown in the specification, carrying out agarose gel electrophoresis detection on the amplification products, and judging the activity of the sgRNA by observing and comparing the sizes of the DNA fragments of the control group and the experimental group.
11. Further, the traditional Chinese medicine screening method in the step 3) comprises the following steps: after the recombinant plasmid PX459M-MSH2-sgRNAs transfects HeLa cells, complete medium containing puromycin is added to culture the cells.
Compared with the prior art, the invention has the beneficial effects that:
1. the invention successfully constructs the HeLa cell line knocked out by the MSH2 gene, and the MSH2 gene fragment is deleted to cause gene inactivation, so that the MSH2 protein can not be correctly expressed.
2. According to the invention, a HeLa cell line knocked out by MSH2 gene constructed by using a CRISPR-Cas9 system is determined to successfully construct a HeLa cell line 1 knocked out by MSH2 gene through gene sequencing identification and Western blot verification.
3. The MSH2 gene knockout HeLa cell line constructed by the invention is large-fragment gene deletion caused on the genome level, so that the MSH2 gene knockout HeLa cell line can be stably inherited to daughter cells along with division and proliferation of the cells.
4. The PX459M-MSH2-sgRNAs plasmid constructed by the invention has puromycin resistance, and puromycin is added to screen cells, so that the positive rate of obtaining the MSH2 gene knockout monoclonal cell strain is improved.
5. The method can be used for directional knockout of the MSH2 gene to inactivate the function of the MSH2 gene, has the characteristics of simplicity, convenience, high efficiency, rapidness, low cost and the like, and has important significance for researching the function of the MSH2 gene and related channels.
[ description of the drawings ]
In order to more clearly illustrate the technical solutions of the embodiments of the present invention, the drawings needed to be used in the embodiments will be briefly described below, it should be understood that the following drawings only illustrate some embodiments of the present invention and therefore should not be considered as limiting the scope, and for those skilled in the art, other related drawings can be obtained according to the drawings without inventive efforts.
FIG. 1 shows the PCR identification result of sgRNA activity detection after PX459M-MSH2-sgRNA plasmid of the invention is transfected into HeLa cells.
FIG. 2 shows the PCR identification result of the monoclonal cell strain MSH2 gene after PX459M-MSH2-sgRNAs plasmid of the present invention is transfected into HeLa cells.
FIG. 3 shows the sequencing comparison result of the monoclonal cell strain MSH2 gene after PX459M-MSH2-sgRNAs plasmid of the present invention is transfected into HeLa cells.
FIG. 4 shows the Western-Blotting detection result of the expression of the monoclonal cell strain MSH2 protein after PX459M-MSH2-sgRNAs plasmid of the invention is transfected into HeLa cells.
[ detailed description ] embodiments
The following examples are intended to illustrate the invention without limiting its scope. It is intended that all modifications or alterations to the methods, procedures or conditions of the present invention be made without departing from the spirit and substance of the invention.
Example 1: vector construction
1) sgRNA design of MSH2 Gene
Aiming at an MSH2 gene (the gene name: MSH2, the gene ID number: 4436, the detailed information of the gene is https:// www.ncbi.nlm.nih.gov/gene/:
AAGCTGATTGGGTGTGGTCGCCGTGGCCGGACGCCGCTCGGGGGACGTGGGAGGGGAGGCGGGAAACAGCTTAGTGGGTGTGGGGTCGCGCATTTTCTTCAACCAGGAGGTGAGGAGGTTTCGACATGGCGGTGCAGCCG, respectively; in the mRNA introduction, the Ensemble website is linked to, and the exon sequences are found and labeled.
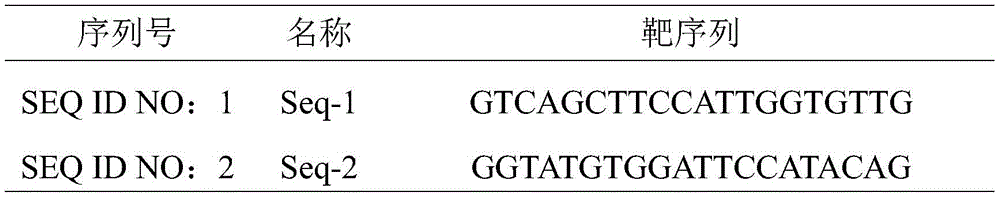
Designing sgRNA by using online software (http:// crispor.tefor. net /), inputting the exon sequences, setting and searching to obtain a plurality of sgRNA sequences, and respectively selecting 1 optimal upstream sgRNA sequence from the sgRNA sequences by analyzing the position of the sgRNA on the gene sequence and off-target (off-target) information of the sgRNA, as shown in Seq-1; 1 optimal downstream sgRNA sequence as shown in Seq-2, in particular as shown in table 1:
table 1: target sequence
2) Synthesis of sgRNA fragments and annealing treatment
The sgRNA optimized for the above design was intended to be cloned into PX459M and EZ-guideeXH vectors, respectively. The sgrna dna sequence was added to the cohesive end of the Bbs1 restriction enzyme and sent to the obendaceae biosynthetic single nucleotide chain.
Adding a joint to the Seq-1 sequence to synthesize an insert MSH2-sgRNA 1:
Seq-1F:5’-CACCGTCAGCTTCCATTGGTGTTG(SEQ ID NO:3)
Seq-1R:5’-AAACCAACACCAATGGAAGCTGAC(SEQ ID NO:4);
adding an adaptor to the Seq-2 sequence to obtain an insert MSH2-sgRNA 2:
Seq-2F:5’-CACCGGTATGTGGATTCCATACAG(SEQ ID NO:5)
Seq-2R:5’-AAACCTGTATGGAATCCACATACC(SEQ ID NO:6);
and (2) mixing each group of Seq-F and Seq-R in equal volume respectively to carry out annealing reaction, wherein an annealing reaction system comprises 4.5 mu L of each primer shown by the Seq-F and the Seq-R with the concentration of 100 mu M and 21 mu L of 10 XNEB Buffer, placing the primers in a PCR instrument for reaction, and the reaction procedure is that the primers react for 5min at 95 ℃, and then the temperature is reduced to 25 ℃ at the speed of 5 ℃/30s to obtain a double-stranded DNA fragment of the sgRNA, wherein the double-stranded DNA fragment can be connected with an enzyme digestion carrier at the moment or stored at the temperature of-20 ℃ for later use.
3) Digestion and recovery of the vector
Taking 2 mu g of PX459M vector and 4 mu g of EZ-guideXH vector, and respectively carrying out enzyme digestion by Bbs1, wherein the enzyme digestion system is as follows: PX459M 2 μ G, Bbs11 μ L, 10 XBuffer G2 μ L, plus ddH2O complement to 20. mu.L, and EZ-GuideXH 4. mu.g, Bbs 11. mu.L, 10 XBuffer G2. mu.L, plus ddH2O make up to 20. mu.L. Respectively and uniformly mixed, and then reacted in water bath at 37 ℃ for 2-4 hours. The agarose DNA recovery kit is used for the aboveThe enzyme digestion product is recovered and purified, and the specific experimental steps are shown in the specification of the agarose DNA recovery kit.
4) Two MSH2-sgRNA fragments are respectively connected with DNAs of PX459M and EZ-guideXH vectors
The following reagents were added to sterile 1.5mL EP tubes, respectively: digested PX459M 50ng, 2 Xquick ligation buffer 10. mu. L, MSH2-sgRNA137.5ng, DNADNA ligase 1. mu.L, and dd H2O complement to 20. mu.L, EZ-guide XH 50ng, 2 XQuick Ligationbuffer 10. mu. L, MSH2-sgRNA237.5ng, DNA ligase 1. mu.L, plus dd H2And O is added to the system to 20 mu L, mixed evenly and reacted in water bath at 25 ℃ for 5 min.
5) Ligation product transformation and colony PCR identification
Adding 5 μ L of the ligation product into 50uL of competent bacteria, and standing on ice for 30 min; heating in 42 deg.C water bath for 90s, and standing on ice for 5 min; adding 1mL of antibiotic-free culture medium, and performing shaking culture at 37 ℃ for half an hour; coating the bacterial liquid on a resistant plate, and placing the resistant plate in a constant-temperature incubator at 37 ℃ for inverted culture overnight; and selecting a single colony for amplification culture, harvesting a bacterial liquid to extract recombinant plasmids, then performing sequencing identification, and storing and identifying correct recombinant plasmids PX459M-MSH2-sgRNA1 and EZ-guideXH-MSH2-sgRNA 2.
6) Construction of recombinant plasmid PX459M-MSH2-sgRNAs
Recombinant plasmids PX459M-MSH2-sgRNA1 and EZ-guideXH-MSH2-sgRNA2 were double digested with Spe1 and Kpn1, respectively. The enzyme cutting system is as follows: PX459M-MSH2-sgRNA 12. mu.g, Spe 11. mu. L, Kpn 11. mu.L, 10 XBufferTango 2. mu.L, plus dd H2O complement to 20. mu.L, and EZ-GuideXH-MSH2-sgRNA 25. mu.g, Spe 12. mu. L, Kpn 12. mu.L, 10 XBufferTango 2. mu.L, plus dd H2And (3) complementing the system to 20 mu L, reacting in water bath at 37 ℃ for 2-4 hours, running 1% agarose gel, cutting the agarose gel, and recovering the enzyme digestion vector. For PX459M-MSH2-sgRNA1 plasmid, cutting gel and recovering a vector framework containing a sgRNA1 double-stranded DNA fragment; for EZ-GuideXH-MSH2-sgRNA2 plasmid, the sgRNA2 double-stranded DNA fragment was recovered by gel cutting. Then using a DNA ligase kit to connect the enzyme digestion product after glue recovery, wherein a connection reaction system comprises PX459M-MSH2-sgRNA150ng after enzyme digestion, EZ-guideXH-MSH2-sgRNA237.5ng after enzyme digestion,2 Xquick ligation buffer 10. mu. L, DNA ligase 1. mu.L, plus dd H2And O is added to the system to 20 mu L, mixed evenly and reacted in water bath at 25 ℃ for 5 min. Then, the step 5) is carried out to transform and identify the ligation product, and the recombinant plasmid PX459M-MSH2-sgRNAs with correct sequencing identification are preserved and identified.
Example 2 preparation of MSH2 Gene knockout HeLa cell line
PX459M-MSH2-sgRNAs transfected HeLa cells
1) Cells were seeded on 12-well plates, and HeLa cells (from wuhan kino life technologies ltd., cat #: CL-0101) suspension at 2.5X 105Inoculating the cell number per well, uniformly spreading the cell number per well into 2 wells of a 12-well plate, respectively supplementing 1mL of complete culture medium (containing 10% fetal calf serum and 1% double antibody), and placing the cell culture box for culture for 12-16 hours;
2) preparing a transfection compound, taking out 2 sterile EP tubes, respectively adding 50 mu L of serum-free antibiotic-free DMEM basic culture medium, adding 3 mu g of PX459M-MSH2-sgRNAs recombinant plasmids (experimental group) into the 1 st sterile EP tube, adding 3 mu g of PX459M-GFP plasmids (control group) into the 2 nd sterile EP tube, and uniformly mixing to obtain solution A; taking out 1 sterile EP tube, adding 100 μ L serum-free and antibiotic-free DMEM culture medium, adding 12 μ L HighGene transfection reagent, and mixing to obtain solution B;
3) respectively taking out 50 μ LB solution, slightly dropping into solution A, mixing well (blowing with gun for 3-5 times) with total volume of 100 μ L, standing at room temperature for 15-20 min;
4) the A/B mixed solution is gently dripped into the corresponding 12-hole plate, the cell culture plate is gently shaken back and forth, and then the 12-hole plate is put back into the cell culture box for continuous culture.
5) After 4-6 hours of cell transfection, half of the fresh complete medium was replaced and culture was continued for 24 hours.
2. Puromycin drug screening
1) After the cell transfection is carried out for 24 hours, observing whether a control group has green fluorescence under a fluorescence microscope, and if so, indicating that the cell transfection is successful;
2) adding fresh complete culture medium containing puromycin medicine with final concentration of 1.6 mu g/mL into cells of an experimental group and cells of a control group respectively;
3) after the puromycin is screened for 24 hours, replacing a fresh complete culture medium containing puromycin with the same concentration as the puromycin to continue cell culture;
4) after 48 hours of puromycin screening, massive cell death floating of a control group is observed under a microscope, 40% -50% of death floating of an experimental group is observed, and puromycin screening is stopped.
Detection of sgRNA Activity
1) Using pancreatin to digest the cells of the control group and the experimental group, and collecting 30-50% cell suspension;
2) extracting genomes of a control group and experimental cells by using a rat tail genome detection kit;
3) PCR amplification was performed using the extracted genomic DNA of the cells as a template with the following primers:
seq-F:5’-TGGTGGTGTGCGCCTGTATT(SEQ ID NO:7)
seq-R:5’-TTCCCCATGTCTCCAGCAGTC(SEQ ID NO:8);
the PCR reaction system included 2 Xmix 10. mu.L of each of the primers Seq-F and Seq-R at a concentration of 10. mu.M, 0.5. mu.L of each of the primers Seq-F and Seq-R, 1. mu.L of the template DNA, and ddH2O8 mu L; the PCR reaction program is pre-denaturation reaction at 95 ℃ for 5 min; denaturation reaction at 95 ℃ for 10s, annealing reaction at 60 ℃ for 10s, extension reaction at 72 ℃ for 20s, and 30 cycles; further extension at 72 deg.C for 5 min;
4) the amplified product was detected by agarose gel electrophoresis, and the sgRNA activity was determined by observing and comparing the band sizes of the amplified products of the control and experimental groups, as shown in FIG. 1, lane 1 is DL2000Marker, lane 2 is normal HeLa cell (control group), and lane 3 is HeLa cell transfected with PX459M-MSH2-sgRNA plasmid.
4. Cell monoclonality culture
1) Discarding a cell culture medium of a 12-well plate experimental group, rinsing cells by PBS, adding 0.5mL of pancreatin digested cells, then adding 1mL of complete culture medium to terminate digestion, and re-suspending the cells;
2) after the cells are evenly resuspended, 200 mu L of cell suspension is taken out to be put into an EP tube, and a blood counting chamber is used for counting;
3) taking 500 cells out of the cell suspension, adding the cells into 20mL of complete culture medium, simultaneously supplementing 1mL of clone-easy growth factor solution, and uniformly mixing the cells;
4) spreading the cell suspension into 2 96-well culture plates by using a discharging gun, wherein each well is 100 mu L;
5) after 10 days of cell culture, the growth of the cells was observed under a microscope to mark the monoclonal cell colonies.
5. Gene level verification of monoclonal cell strain
1) When the monoclonal cell density grows to 50-60% of the area of the bottom of the hole, using pancreatin to digest the cells, taking out 50% of the cells to an EP tube, extracting cell genomes to carry out gene sequencing verification, and continuously culturing and amplifying the remaining 50% of the cells in a 96-well plate;
2) PCR amplification was performed using the extracted genomic DNA of the cells as a template with the following primers:
seq-F:5’-TGGTGGTGTGCGCCTGTATT(SEQ ID NO:7)
seq-R:5’-TTCCCCATGTCTCCAGCAGTC(SEQ ID NO:8);
the PCR reaction system comprises 2 xMix 10 muL, 0.5 muL of each of the primers shown by Seq-F and Seq-R with the concentration of 10 muM, 1 muL of template DNA and 8 muL of ddH2O 8; the PCR reaction program is: performing pre-denaturation reaction at 95 ℃ for 5 min; denaturation reaction at 95 ℃ for 10s, annealing reaction at 60 ℃ for 10s, extension reaction at 72 ℃ for 20s, and 30 cycles; further extension at 72 deg.C for 5 min;
4) carrying out agarose gel electrophoresis detection on the amplification product, and comparing the size of the band of the amplification product with that of a control group to judge whether the monoclonal cell strain is targeted; as shown in FIG. 2, lane 1 is DL2000Marker, lane 2 is normal HeLa cells (control group), and lane 3 is HeLa-MSH2-KO monoclonal cells in which MSH2 gene was knocked out.
5) And cutting the gel, recovering the targeted PCR amplification product, sequencing, identifying positive cells through sequencing comparison, carrying out amplification culture on the positive cells, freezing and preserving seeds, and naming the cells as HeLa-MSH2-KO, wherein the comparison chart of the sequencing result is shown in figure 3.
Western Blot for detecting expression condition of monoclonal cell strain MSH2 protein
In order to verify the knockout effect of the MSH2 gene, a Western Blot experiment is adopted to detect the expression condition of the MSH2 protein in a HeLa cell line knocked out by the MSH2 gene.
1) Cell collection: digesting the HeLa-MSH2-KO monoclonal cells and normal HeLa cells by pancreatin, collecting the cells into a 1.5mL centrifuge tube, centrifuging, removing supernatant, and reserving cell precipitates;
2) cell lysis: adding RIPA lysis buffer containing protease inhibitor, performing lysis on ice for 30min, performing centrifugation for 15min at 12000r/min, taking supernatant, transferring to a new 1.5mL centrifuge tube, and determining protein concentration;
3) preparing a cell sample: mixing the cell total protein supernatant with appropriate amount of 5 xSDS protein sample buffer solution to make the final concentration of cell total protein 2mg/mL, boiling at 95 deg.C for 5 min;
4) electrophoresis: preparing 10% SDS-PAGE gel, and performing electrophoretic separation of protein, wherein the voltage is 80V and the time is 30 minutes when the gel is concentrated, and the voltage is 120V and the time is 1 hour when the gel is separated;
5) film transfer: adopting a wet transfer method, performing constant current of 240mA, and transferring the film for 2 hours;
6) and (3) sealing: the membrane was incubated for 2 hours at room temperature using TBST solution of 5% skimmed milk powder at final concentration;
7) primary antibody incubation: primary anti-MSH 2 mAb (ABclonal: a8740) was diluted 1:1000 in TBST solution of 5% skimmed milk powder final concentration and incubated overnight at 4 ℃;
8) and (3) secondary antibody incubation: prior to incubation of the secondary antibody, the membrane was washed 3 times for 10 minutes each using TBST; a secondary antibody Peroxidase affinity Goat Anti-Rabbit IgG (H + L) (Jackson: 111-;
9) exposure imaging: membranes were washed 3 times for 10 minutes each using TBST. And uniformly mixing equal amounts of ECL developing solution A and solution B, and imaging by using a gel imager.
The results show that: no MSH2 protein signal is detected in a HeLa-MSH2-KO monoclonal cell, and the MSH2 gene knockout is realized on the protein level of the monoclonal cell. In particular, as shown in FIG. 4, lane 1 shows normal HeLa cells, and MSH2 protein signal was detected; lane 2 shows HeLa-MSH2-KO monoclonal cells with MSH2 gene knockout, and no MSH2 protein signal was detected.
The invention utilizes the MSH2 gene knockout HeLa cell line constructed by the CRICPR/Cas9 system to edit the MSH2 gene at the DNA level, completely silence the expression of the MSH2 protein and provide a practical tool for the research of the MSH2 gene function.
Sequence listing
<110> Wuhan Ebola Biotech Co., Ltd
<120> a method for constructing MSH2 gene knockout cell line
<141> 2020-11-25
<160> 8
<170> SIPOSequenceListing 1.0
<210> 1
<211> 20
<212> DNA
<213> Artificial Sequence
<400> 1
gtcagcttcc attggtgttg 20
<210> 2
<211> 20
<212> DNA
<213> Artificial Sequence
<400> 2
ggtatgtgga ttccatacag 20
<210> 3
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 3
caccgtcagc ttccattggt gttg 24
<210> 4
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 4
aaaccaacac caatggaagc tgac 24
<210> 5
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 5
caccggtatg tggattccat acag 24
<210> 6
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 6
aaacctgtat ggaatccaca tacc 24
<210> 7
<211> 20
<212> DNA
<213> Artificial Sequence
<400> 7
tggtggtgtg cgcctgtatt 20
<210> 8
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 8
ttccccatgt ctccagcagt c 21
Claims (10)
1. An sgRNA knockout of MSH2 gene comprising the amino acid sequence set forth in SEQ ID NO: 1 and sgRNA1 as set forth in SEQ ID NO: 2, sgRNA 2.
2. The recombinant plasmid PX459M-MSH2-sgRNAs of the sgRNA of claim 1.
3. The method of claim 2 for constructing recombinant plasmid PX459M-MSH2-sgRNAs, comprising the steps of:
s1, synthesizing a sgRNA DNA sequence with an enzyme cutting site and a complementary sequence thereof based on the sgRNA1 and the sgRNA2, and respectively annealing to obtain two double-stranded DNA fragments of MSH2-sgRNA1 and MSH2-sgRNA 2;
s2, respectively carrying out enzyme digestion and recycling on PX459M and EZ-guideXH two carriers;
s3, connecting the two double-stranded DNA fragments with a PX459M and an EZ-guide XH vector which are recovered by enzyme digestion respectively, and performing product transformation and sequencing identification to obtain PX459M-MSH2-sgRNA1 and EZ-guide XH-MSH2-sgRNA 2;
s4, carrying out enzyme digestion on PX459M-MSH2-sgRNA1 and EZ-guide XH-MSH2-sgRNA2, recovering a vector skeleton containing MSH2-sgRNA1 from the former and a DNA fragment containing MSH2-sgRNA2 from the latter, and then carrying out product connection to construct a recombinant plasmid PX459M-MSH 2-sgRNAs.
4. The method for constructing recombinant plasmids of sgRNA1, sgRNA2 according to claim 3, characterized in that in step S1: the sgRNA sequence of the sgRNA1 with the enzyme cutting site is SEQ ID NO: 3, the complementary sequence is SEQ ID NO: 4; the sgRNA sequence of the sgRNA2 with the enzyme cutting site is SEQ ID NO: 5, the complementary sequence is SEQ ID NO: 6.
5. the method for constructing recombinant plasmids of sgRNA1, sgRNA2 of claim 3, wherein the method for transforming and identifying the product in step S3 is: adding a connection product of the enzyme-cut vector skeleton and the double-stranded DNA fragment into competent bacteria, supplementing a culture medium, culturing for half an hour, coating the mixture on a resistant plate, culturing overnight, selecting a single colony, carrying out amplification culture, harvesting a bacterial liquid, extracting a recombinant plasmid, carrying out sequencing identification, and storing the correctly identified recombinant plasmids PX459M-MSH2-sgRNA1 and EZ-Guide XH-MSH2-sgRNA 2.
6. The method for constructing recombinant plasmid PX459M-MSH2-sgRNAs of sgRNA of claim 3, wherein said step S4 is: recovering a vector framework containing MSH2-sgRNA1 for PX459M-MSH2-sgRNA1, recovering a DNA fragment containing MSH2-sgRNA2 for EZ-guide XH-MSH2-sgRNA2, and connecting the recovered enzyme digestion product by using DNA ligase; then the transformation and identification of the ligation products are carried out in the same step S3, and the recombinant plasmid with correct sequencing identification is named as PX459M-MSH 2-sgRNAs.
7. The use of the recombinant plasmid PX459M-MSH2-sgRNAs of claim 2 in the preparation of MSH2 gene knockout HeLa cell line.
8. The method for constructing a HeLa knockout cell line of the MSH2 gene by the recombinant plasmid PX459M-MSH2-sgRNAs of claim 2, which comprises the following steps:
t1, transfecting HeLa cells by using recombinant plasmids PX459M-MSH 2-sgRNAs;
t2, extracting a transfected cell genome, performing PCR amplification identification aiming at the MSH2 gene, and detecting the activity of sgRNA;
t3, screening puromycin drugs for transfected cells, and then performing cell monoclonality treatment;
and T4, extracting the genomic DNA of the monoclonal cell strain, and performing MSH2 gene sequencing identification to obtain a HeLa cell line knocked out by the MSH2 gene.
9. The method according to claim 8, wherein the sgRNA activity in step T2 is detected by: after recombinant plasmid PX459M-MSH2-sgRNAs transfects HeLa cells, extracting genomic DNA of the cells by using a rat tail genomic DNA detection kit as a template, and performing PCR amplification by using a DNA sequence shown in SEQ ID NO: 7 and SEQ ID NO: 8, carrying out PCR amplification on the primers shown in the specification, carrying out agarose gel electrophoresis detection on the amplification products, and judging the activity of the sgRNA by observing and comparing the sizes of the DNA fragments of the control group and the experimental group.
10. The method of claim 8, wherein the step T3 is performed by adding puromycin-containing complete medium to the recombinant plasmid PX459M-MSH2-sgRNA transfected HeLa cells.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011373830.0A CN112501169A (en) | 2020-11-30 | 2020-11-30 | Method for constructing MSH2 gene knockout cell line |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011373830.0A CN112501169A (en) | 2020-11-30 | 2020-11-30 | Method for constructing MSH2 gene knockout cell line |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112501169A true CN112501169A (en) | 2021-03-16 |
Family
ID=74968072
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202011373830.0A Pending CN112501169A (en) | 2020-11-30 | 2020-11-30 | Method for constructing MSH2 gene knockout cell line |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN112501169A (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117645975A (en) * | 2024-01-29 | 2024-03-05 | 天津科技大学 | Cell line for high-throughput screening of TRPV1 receptor inhibitor, construction method and application thereof |
| WO2024051871A3 (en) * | 2023-07-24 | 2024-06-13 | 华北理工大学 | Crispr/cas9-based wsb2 gene knockout method and use thereof |
| CN118995818A (en) * | 2024-10-10 | 2024-11-22 | 远见生物科技(上海)有限公司 | Simple substance granule double sgRNA gene knockout kit and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110016481A (en) * | 2019-05-24 | 2019-07-16 | 青岛农业大学 | A kind of pX335-xCas9n vector and its construction method and application |
| CN111349654A (en) * | 2018-12-20 | 2020-06-30 | 北京大学 | Compositions and methods for efficient genetic screening using tagged guide RNA constructs |
-
2020
- 2020-11-30 CN CN202011373830.0A patent/CN112501169A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111349654A (en) * | 2018-12-20 | 2020-06-30 | 北京大学 | Compositions and methods for efficient genetic screening using tagged guide RNA constructs |
| CN110016481A (en) * | 2019-05-24 | 2019-07-16 | 青岛农业大学 | A kind of pX335-xCas9n vector and its construction method and application |
Non-Patent Citations (3)
| Title |
|---|
| CHAO SUI等: "CRISPR-Cas9 Mediated RNase L Knockout Regulates Cellular Function of PK-15 Cells and Increases PRV Replication", 《BIOMED RESEARCH INTERNATIONAL》 * |
| GENBANK等: "Homo sapiens mutS homolog 2 (MSH2), RefSeqGene (LRG_218) on chromosome 2", 《GENBANK》 * |
| GENKI HAYASHIDA等: "Differential genomic destabilisation in human cells with pathogenic MSH2 mutations introduced by genome editing", 《EXPERIMENTAL CELL RESEARCH》 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024051871A3 (en) * | 2023-07-24 | 2024-06-13 | 华北理工大学 | Crispr/cas9-based wsb2 gene knockout method and use thereof |
| CN117645975A (en) * | 2024-01-29 | 2024-03-05 | 天津科技大学 | Cell line for high-throughput screening of TRPV1 receptor inhibitor, construction method and application thereof |
| CN117645975B (en) * | 2024-01-29 | 2024-06-07 | 天津科技大学 | Cell line for high-throughput screening of TRPV1 receptor inhibitor, construction method and application thereof |
| CN118995818A (en) * | 2024-10-10 | 2024-11-22 | 远见生物科技(上海)有限公司 | Simple substance granule double sgRNA gene knockout kit and application thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108410911B (en) | LMNA knockout cell line based on CRISPR/Cas9 technology | |
| CN112501170A (en) | Method for constructing MLH1 gene knockout cell line | |
| CN108410907B (en) | Method for realizing HMGCR gene knockout based on CRISPR/Cas9 technology | |
| CN107760652A (en) | The cell models of caco 2 and its method that CRISPR/CAS9 mediate drugs transporter target knocks out | |
| CN112501169A (en) | Method for constructing MSH2 gene knockout cell line | |
| Maraia | The subset of mouse B1 (Alu-equivalent) sequences expressed as small processed cytoplasmic transcripts | |
| CN111607594A (en) | A cell line knocking out porcine IRF8 gene based on CRISPR-Cas9 editing technology and its construction method | |
| HK1039962A1 (en) | Cell-free chimeraplasty and eukaryotic use of heteroduplex mutational vectors | |
| EP2474614A2 (en) | Dna fragment for improving translation efficiency, and recombinant vector containing same | |
| WO2019086007A1 (en) | Sgrna for targeting and guiding cas9 protein to efficiently cleave tcr and b2m gene loci | |
| CN110305908B (en) | An efficient and precise targeting gene integration system and its application | |
| CN109266651A (en) | SgRNA based on CRISPR/Cas9 technical editor's HBB-41/42 deletion mutation site | |
| CN111254164A (en) | A method and cell line for rapidly establishing CRISPR gene editing liver cancer cell line | |
| CN116286931B (en) | Double-plasmid system for rapid gene editing of Ralstonia eutropha and application thereof | |
| CN117821462B (en) | Gene editing repair of Alzheimer's disease-related PSEN1 site mutation | |
| CN119242630A (en) | MDA-MB-231 cell line expressing moesin-EGFP and construction method thereof | |
| CN108467864A (en) | A kind of ICAM-1 gene knockouts tumor cell line and its application | |
| CN107841509B (en) | Kit for knocking out glioblastoma DHC2 gene | |
| CN111793653A (en) | A construction method of dpy19l1l gene deletion zebrafish | |
| CN109943589A (en) | A single base mutation method and system used | |
| CN117625621A (en) | A plasmid for targeted knockout of WNT5A gene and its preparation method and application | |
| CN118755721A (en) | A sgRNA for knocking out human Profilin gene, a recombinant expression vector and a construction method | |
| LU102620B1 (en) | A gRNA and Related Kit and Application | |
| CN113088497A (en) | HEK293 cell line stably knocking out abhd16a gene and construction method thereof | |
| CN112921040A (en) | Construction method of pig TFF1 gene knockout cell line based on CRISPR-Cas9 gene editing technology |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |