CN111868032B - Kynurenine pathway inhibitors - Google Patents
Kynurenine pathway inhibitors Download PDFInfo
- Publication number
- CN111868032B CN111868032B CN201980012783.6A CN201980012783A CN111868032B CN 111868032 B CN111868032 B CN 111868032B CN 201980012783 A CN201980012783 A CN 201980012783A CN 111868032 B CN111868032 B CN 111868032B
- Authority
- CN
- China
- Prior art keywords
- compound
- group
- added
- solution
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明公开了新的作为犬尿氨酸通路抑制剂的化合物,具体公开了式(I)所示化合物及其药学上可接受的盐。
Description
本申请主张如下优先权:This application claims the following priority:
CN201810143151.0,申请日2018年2月11日。CN201810143151.0, filed on February 11, 2018.
技术领域technical field
本发明涉及新的作为犬尿氨酸通路抑制剂的化合物,具体公开了式(I)所示化合物及其药学上可接受的盐。本发明还涉及式(I)所示化合物及其药学上可接受的盐在制备治疗肿瘤药物中的应用。The present invention relates to novel compounds as kynurenine pathway inhibitors, and specifically discloses compounds represented by formula (I) and pharmaceutically acceptable salts thereof. The present invention also relates to the application of the compound represented by formula (I) and its pharmaceutically acceptable salts in the preparation of medicaments for treating tumors.
背景技术Background technique
恶性肿瘤是现如今危害人类生命安全的主要疾病之一。近百年来,人类为了对抗恶性肿瘤,发展出了多种包括最常用的化疗,手术,放疗以及靶向疗法在内的诊疗手段与治疗方法。这些疗法在一定程度上延缓的肿瘤的发展,延长了患者生命。但是由于恶性肿瘤的无限制生长,浸润与转移的特性,以上疗法依旧无法达到理想的抑制效果。同时,以上的疗法的毒副作用也是限制其应用的关键因素。Malignant tumors are one of the major diseases that endanger human life today. In the past 100 years, in order to combat malignant tumors, human beings have developed a variety of diagnosis and treatment methods, including the most commonly used chemotherapy, surgery, radiotherapy and targeted therapy. These therapies delay the development of tumors to a certain extent, prolonging the lives of patients. However, due to the unrestricted growth, infiltration and metastasis of malignant tumors, the above treatments still cannot achieve the desired inhibitory effect. At the same time, the toxic and side effects of the above treatments are also a key factor limiting their application.
近年来,免疫疗法作为恶性肿瘤治疗的新兴疗法被发展起来,其特点是通过调动宿主的天然防御机制来抵抗肿瘤的生长。其主要途径主要通过激活宿主原有的免疫系统,增强宿主集体对肿瘤细胞的免疫反应,利用免疫系统能力达到精准有效的杀伤肿瘤,遏制其发展。In recent years, immunotherapy has been developed as an emerging therapy for the treatment of malignant tumors, which is characterized by mobilizing the host's natural defense mechanisms to resist tumor growth. The main way is to activate the host's original immune system, enhance the host's collective immune response to tumor cells, and use the ability of the immune system to kill tumors accurately and effectively, and curb their development.
肿瘤内部的犬尿酸的产生主要受吲哚胺2,3-双加氧酶(Indoleamine-2,3-dioxygenase,IDO)及色氨酸-2,3双加氧酶(Tryptophan2,3-dioxygenase,TDO)的调控,其功能在于催化了色氨酸向犬尿胺酸代谢的决速步骤。以上酶可以迅速消耗肿瘤微环境内色氨酸,使得浸润至肿瘤内部的T细胞由于缺乏色氨酸而停滞于G1期中期,从而抑制了T细胞增殖并使T细胞信号传导受阻,功能丧失。同时犬尿氨酸的浓度上升会导致肿瘤形成免疫抑制的微环境。因此,抑制肿瘤内犬尿氨酸的生成有助于激活集体免疫系统,抵制肿瘤生长。The production of kynuric acid inside the tumor is mainly regulated by indoleamine-2,3-dioxygenase (IDO) and tryptophan2,3-dioxygenase (Tryptophan2,3-dioxygenase, TDO), its function is to catalyze the rate-determining step in the metabolism of tryptophan to kynurenine. The above enzymes can quickly deplete tryptophan in the tumor microenvironment, so that T cells infiltrating into the tumor are arrested in the mid-G1 phase due to the lack of tryptophan, thereby inhibiting T cell proliferation and blocking T cell signaling and loss of function. At the same time, the increased concentration of kynurenine can lead to tumor formation of an immunosuppressive microenvironment. Therefore, inhibiting the production of kynurenine in tumors helps to activate the collective immune system and resist tumor growth.
犬尿氨酸通路抑制剂作为药物在医药行业具有良好的应用前景,但是目前尚无犬尿氨酸通路抑制剂上市。处于临床的犬尿氨酸通路抑制剂只有NewLink Genetics公司开发的(D)-1-甲基色胺酸,NLG-0919与Incyte公司的INCB-24360及百时美施贵宝的BMS-985205。但是,现有临床犬尿氨酸通路抑制剂存在诸如CYP抑制(NLG 0919)、给药剂量大、半衰期短、给药次数多(INCB-24360),溶解性差(BMS-986205)等问题。因此,无上述缺点的犬尿氨酸通路抑制剂依旧是未被满足的医疗需求。As a drug, kynurenine pathway inhibitors have good application prospects in the pharmaceutical industry, but no kynurenine pathway inhibitors are currently on the market. The only clinical kynurenine pathway inhibitors are (D)-1-methyltryptophan developed by NewLink Genetics, NLG-0919, INCB-24360 from Incyte and BMS-985205 from Bristol-Myers Squibb. However, the existing clinical kynurenine pathway inhibitors have problems such as CYP inhibition (NLG 0919), large dosage, short half-life, many times of administration (INCB-24360), and poor solubility (BMS-986205). Therefore, kynurenine pathway inhibitors without the above-mentioned disadvantages remain an unmet medical need.
NLG-0919,INCB-24360及BMS-986205的具体结构如下:The specific structures of NLG-0919, INCB-24360 and BMS-986205 are as follows:

发明内容SUMMARY OF THE INVENTION
本发明提供式(I)所示化合物或其药学上可接受的盐,The present invention provides a compound represented by formula (I) or a pharmaceutically acceptable salt thereof,

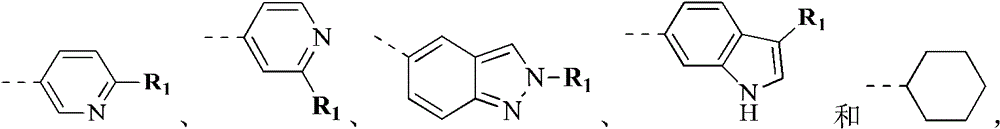
其中,in,
环A选自苯基、吡啶基、环戊基1H-吲唑基、2H-吲唑基、1H-吲哚基和环己基;Ring A is selected from phenyl, pyridyl, cyclopentyl 1H-indazolyl, 2H-indazolyl, 1H-indolyl and cyclohexyl;
环B选自苯基、吡啶基、喹啉基、异喹啉基、1H-吲唑基和2H-吲唑基;Ring B is selected from phenyl, pyridyl, quinolyl, isoquinolyl, 1H-indazolyl and 2H-indazolyl;
L选自单键、-O-、-CH2-、-CH2CH2-和-CH2CH2CH2-;L is selected from single bond , -O- , -CH2- , -CH2CH2- and -CH2CH2CH2- ;
X选自CH和N;X is selected from CH and N;
L’选自单键和-O-;L' is selected from single bond and -O-;
R1分别独立地选自卤素、NH2、CN、C1-3烷基、CF3、CHF2、CH2F、C1-3杂烷基;R 1 is each independently selected from halogen, NH 2 , CN, C 1-3 alkyl, CF 3 , CHF 2 , CH 2 F, C 1-3 heteroalkyl;
R2分别独立地选自卤素、CN、C1-3烷基、CF3、CHF2、CH2F、C1-3杂烷基、3-6元环烷基、3-6元杂环烷基、3-6元环烷基-O-、3-6元杂环烷基-O-、3-6元环烷基-NH-、3-6元杂环烷基-NH-;R 2 is independently selected from halogen, CN, C 1-3 alkyl, CF 3 , CHF 2 , CH 2 F, C 1-3 heteroalkyl, 3-6 membered cycloalkyl, 3-6 membered heterocycle Alkyl, 3-6-membered cycloalkyl-O-, 3-6-membered heterocycloalkyl-O-, 3-6-membered cycloalkyl-NH-, 3-6-membered heterocycloalkyl-NH-;
m和n分别独立地选自0、1、2和3;m and n are independently selected from 0, 1, 2 and 3;
“杂”表示杂原子或杂原子团,所述C1-3杂烷基、3-6元杂环烷基、3-6元杂环烷基-O-和3-6元杂环烷基-NH-之“杂”选自N、-NH-、-O-、-S-、-C(=O)-、-C(=S)-、-C(=O)O-、-S(=O)-和-S(=O)2-;"Hetero" means a heteroatom or heteroatom group, the C 1-3 heteroalkyl, 3-6 membered heterocycloalkyl, 3-6 membered heterocycloalkyl-O- and 3-6 membered heterocycloalkyl- The "hetero" of NH- is selected from N, -NH-, -O-, -S-, -C(=O)-, -C(=S)-, -C(=O)O-, -S( =O)- and -S(=O) 2 -;
以上任何一种情况下,杂原子或杂原子团的数目分别独立地选自1、2和3。In any of the above cases, the number of heteroatoms or heteroatomic groups is independently selected from 1, 2 and 3, respectively.
在本发明的一些方案中,上述R1分别独立地选自F、Cl、Br、I、NH2、CN、CH3、CF3、CH3CH2、CH3O和CH3CH2O。In some aspects of the present invention, the above R 1 are each independently selected from F, Cl, Br, I, NH 2 , CN, CH 3 , CF 3 , CH 3 CH 2 , CH 3 O and CH 3 CH 2 O.
在本发明的一些方案中,上述R2分别独立地选自F、Cl、Br、I、CN、CH3、CF3、CHF2、CH2F、CH3CH2、CH3O-、CH3CH2O-、
在本发明的一些方案中,上述结构单元
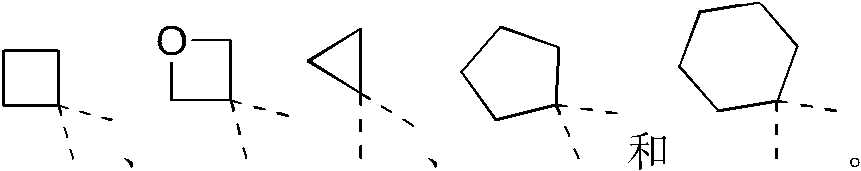
在本发明的一些方案中,上述结构单元


在本发明的一些方案中,上述结构单元
在本发明的一些方案中,上述结构单元



在本发明的一些方案中,上述结构单元


在本发明的一些方案中,上述R1分别独立地选自F、Cl、Br、I、NH2、CN、CH3、CF3、CH3CH2、CH3O和CH3CH2O,其他变量如上述所定义。In some aspects of the present invention, the above R 1 are each independently selected from F, Cl, Br, I, NH 2 , CN, CH 3 , CF 3 , CH 3 CH 2 , CH 3 O and CH 3 CH 2 O, Other variables are as defined above.
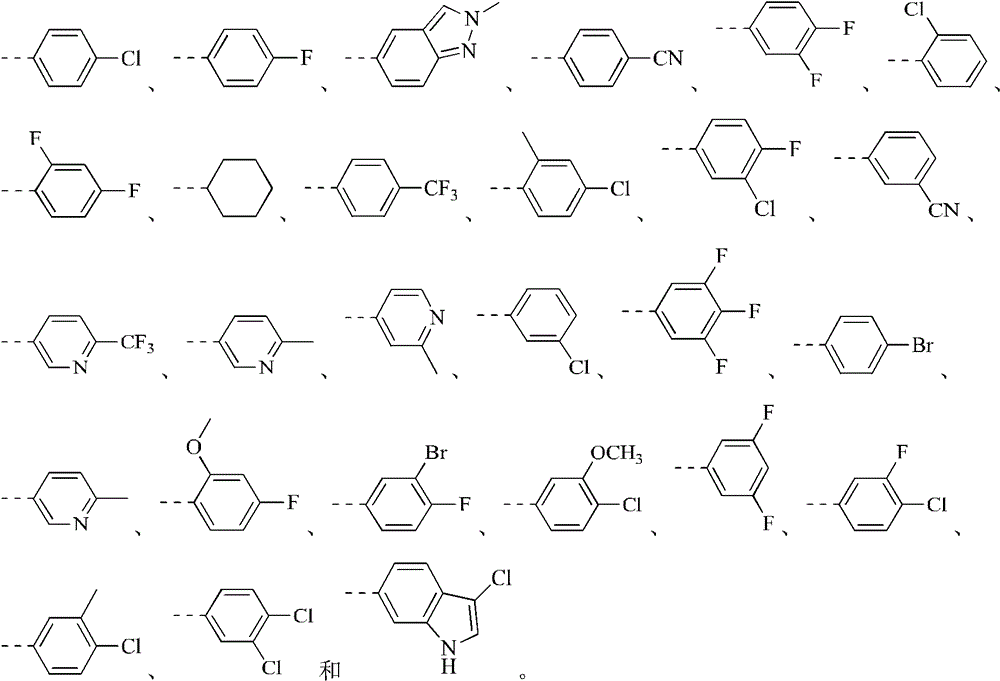
在本发明的一些方案中,上述R2分别独立地选自F、Cl、Br、I、CN、CH3、CF3、CHF2、CH2F、CH3CH2、CH3O-、CH3CH2O-、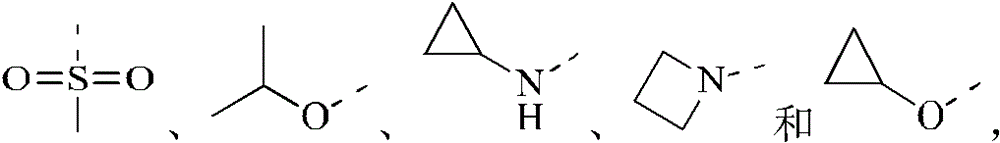
在本发明的一些方案中,上述结构单元

在本发明的一些方案中,上述结构单元



在本发明的一些方案中,上述结构单元


在本发明的一些方案中,上述结构单元




在本发明的一些方案中,上述结构单元



在本发明的一些方案中,上述化合物或其药学上可接受的盐,选自In some aspects of the present invention, the above-mentioned compound, or a pharmaceutically acceptable salt thereof, is selected from

其中,环A、X、m、n、L、L’、R1和R2如上述所定义。wherein Rings A, X, m, n, L, L', R 1 and R 2 are as defined above.
在本发明的一些方案中,上述化合物或其药学上可接受的盐,选自In some aspects of the present invention, the above-mentioned compound, or a pharmaceutically acceptable salt thereof, is selected from

其中,L、R1和R2如上述所定义。wherein L, R 1 and R 2 are as defined above.
在本发明的一些方案中,上述R1分别独立地选自F、Cl、Br、I、NH2、CN、CH3、CF3、CH3CH2、CH3O和CH3CH2O,其他变量如本发明所定义。In some aspects of the present invention, the above R 1 are each independently selected from F, Cl, Br, I, NH 2 , CN, CH 3 , CF 3 , CH 3 CH 2 , CH 3 O and CH 3 CH 2 O, Other variables are as defined in the present invention.
在本发明的一些方案中,上述R2分别独立地选自F、Cl、Br、I、CN、CH3、CF3、CHF2、CH2F、CH3CH2、CH3O-、CH3CH2O-、
在本发明的一些方案中,上述结构单元

在本发明的一些方案中,上述结构单元

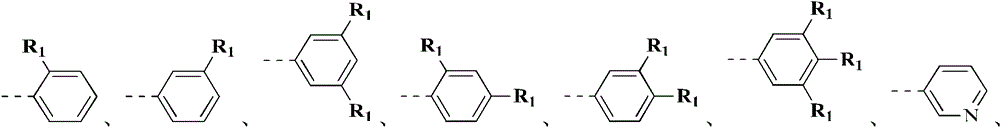
在本发明的一些方案中,上述结构单元


在本发明的一些方案中,上述结构单元



在本发明的一些方案中,上述结构单元



在本发明的一些方案中,上述化合物或其药学上可接受的盐,选自In some aspects of the present invention, the above-mentioned compound, or a pharmaceutically acceptable salt thereof, is selected from

其中,环A、m、n、X、L、L’、R1和R2如本发明所定义。wherein, rings A, m, n, X, L, L', R 1 and R 2 are as defined in the present invention.
10.根据权利要求9所述的化合物或其药学上可接受的盐,选自10. The compound of

其中,L、R1和R2如本发明所定义。Wherein, L, R 1 and R 2 are as defined in the present invention.
本发明还提供化合物或其药学上可接受的盐,选自The present invention also provides a compound or a pharmaceutically acceptable salt thereof selected from the group consisting of



本发明提供一种药物组合物,其含有治疗有效量的上述的化合物或其药学上可接受的盐和药学上可接受的载体。The present invention provides a pharmaceutical composition comprising a therapeutically effective amount of the above-mentioned compound or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
本发明还提供上述的化合物或其药学上可接受的盐或上述的药物组合物在制备治疗癌症药物中的应用。The present invention also provides the use of the above-mentioned compound or a pharmaceutically acceptable salt thereof or the above-mentioned pharmaceutical composition in the preparation of a medicament for treating cancer.
本发明还有一些方案是由上述各变量任意组合而来。There are still some solutions of the present invention which are obtained by any combination of the above variables.
技术效果technical effect
本发明提供一种新型结构的犬尿氨酸通路抑制剂,具有优异的体外细胞活性;结构中酰胺邻位的环状结构可以增强该系列化合物的体内代谢稳定性。相同剂量下,该发明中的结构相比BMS-986205具有更高的AUC,同时半衰期更长。同时该系列的化合物相比BMS-986205溶解度更好,口服生物利用度更高,小鼠体内组织分布更优,综合药代动力学性质更优。本发明提供的新型结构很可能是一个选择性的IDO抑制剂。The invention provides a kynurenine pathway inhibitor with a novel structure, which has excellent in vitro cell activity; the cyclic structure of the amide ortho position in the structure can enhance the in vivo metabolic stability of the series of compounds. At the same dose, the structure in this invention has higher AUC and longer half-life than BMS-986205. At the same time, compared with BMS-986205, this series of compounds has better solubility, higher oral bioavailability, better tissue distribution in mice, and better comprehensive pharmacokinetic properties. The novel structure provided by the present invention is likely to be a selective IDO inhibitor.
定义和说明Definition and Explanation
除非另有说明,本文所用的下列术语和短语旨在具有下列含义。一个特定的术语或短语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文中出现商品名时,意在指代其对应的商品或其活性成分。这里所采用的术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。Unless otherwise specified, the following terms and phrases used herein are intended to have the following meanings. A particular term or phrase should not be considered indeterminate or unclear without specific definitions, but should be understood in its ordinary meaning. When a trade name appears herein, it is intended to refer to its corresponding commercial product or its active ingredient. As used herein, the term "pharmaceutically acceptable" refers to those compounds, materials, compositions and/or dosage forms that, within the scope of sound medical judgment, are suitable for use in contact with human and animal tissue , without excessive toxicity, irritation, allergic reactions or other problems or complications, commensurate with a reasonable benefit/risk ratio.
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定取代基的化合物与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物的中性形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机胺或镁盐或类似的盐。当本发明的化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物的中性形式接触的方式获得酸加成盐。药学上可接受的酸加成盐的实例包括无机酸盐,所述无机酸包括例如盐酸、氢溴酸、硝酸、碳酸,碳酸氢根,磷酸、磷酸一氢根、磷酸二氢根、硫酸、硫酸氢根、氢碘酸、亚磷酸等;以及有机酸盐,所述有机酸包括如乙酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸和甲磺酸等类似的酸;还包括氨基酸(如精氨酸等)的盐,以及如葡糖醛酸等有机酸的盐。本发明的某些特定的化合物含有碱性和酸性的官能团,从而可以被转换成任一碱或酸加成盐。The term "pharmaceutically acceptable salts" refers to salts of the compounds of the present invention, prepared from compounds with specific substituents discovered by the present invention and relatively non-toxic acids or bases. When the compounds of the present invention contain relatively acidic functional groups, base addition salts can be obtained by contacting the neutral forms of such compounds with a sufficient amount of base in neat solution or in a suitable inert solvent. Pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amine or magnesium salts or similar salts. When the compounds of the present invention contain relatively basic functional groups, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of acid in neat solution or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include inorganic acid salts including, for example, hydrochloric acid, hydrobromic acid, nitric acid, carbonic acid, bicarbonate, phosphoric acid, monohydrogen phosphate, dihydrogen phosphate, sulfuric acid, Hydrogen sulfate, hydroiodic acid, phosphorous acid, etc.; and organic acid salts including, for example, acetic acid, propionic acid, isobutyric acid, maleic acid, malonic acid, benzoic acid, succinic acid, suberic acid, Similar acids such as fumaric, lactic, mandelic, phthalic, benzenesulfonic, p-toluenesulfonic, citric, tartaric, and methanesulfonic acids; also include salts of amino acids such as arginine, etc. , and salts of organic acids such as glucuronic acid. Certain specific compounds of the present invention contain both basic and acidic functional groups and thus can be converted into either base or acid addition salts.
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的这些化合物与化学计量的适当的碱或酸反应来制备。The pharmaceutically acceptable salts of the present invention can be synthesized from the acid or base containing parent compound by conventional chemical methods. Generally, such salts are prepared by reacting the free acid or base form of these compounds with a stoichiometric amount of the appropriate base or acid in water or an organic solvent or a mixture of the two.
除了盐的形式,本发明所提供的化合物还存在前药形式。本文所描述的化合物的前药容易地在生理条件下发生化学变化从而转化成本发明的化合物。此外,前体药物可以在体内环境中通过化学或生化方法被转换到本发明的化合物。In addition to salt forms, the compounds provided herein also exist in prodrug forms. Prodrugs of the compounds described herein are readily chemically altered under physiological conditions to convert to the compounds of the present invention. Furthermore, prodrugs can be converted to the compounds of the present invention by chemical or biochemical methods in an in vivo environment.
本发明的某些化合物可以以非溶剂化形式或者溶剂化形式存在,包括水合物形式。一般而言,溶剂化形式与非溶剂化的形式相当,都包含在本发明的范围之内。Certain compounds of the present invention may exist in unsolvated as well as solvated forms, including hydrated forms. In general, solvated and unsolvated forms are equivalent and are intended to be included within the scope of the present invention.
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对对映体、(R)-和(S)-对映体、非对映异构体、(D)-异构体、(L)-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。烷基等取代基中可存在另外的不对称碳原子。所有这些异构体以及它们的混合物,均包括在本发明的范围之内。The compounds of the present invention may exist in specific geometric or stereoisomeric forms. The present invention contemplates all such compounds, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers isomers, (D)-isomers, (L)-isomers, and racemic and other mixtures thereof, such as enantiomerically or diastereomerically enriched mixtures, all of which belong to within the scope of the present invention. Additional asymmetric carbon atoms may be present in substituents such as alkyl. All such isomers, as well as mixtures thereof, are included within the scope of the present invention.
除非另有说明,术语“对映异构体”或者“旋光异构体”是指互为镜像关系的立体异构体。Unless otherwise indicated, the terms "enantiomers" or "optical isomers" refer to stereoisomers that are mirror images of each other.
除非另有说明,术语“顺反异构体”或者“几何异构体”系由因双键或者成环碳原子单键不能自由旋转而引起。Unless otherwise specified, the terms "cis-trans isomer" or "geometric isomer" result from the inability to rotate freely due to double bonds or single bonds to ring carbon atoms.
除非另有说明,术语“非对映异构体”是指分子具有两个或多个手性中心,并且分子间为非镜像的关系的立体异构体。Unless otherwise indicated, the term "diastereomer" refers to a stereoisomer in which the molecule has two or more chiral centers and the molecules are in a non-mirror-image relationship.
除非另有说明,“(D)”或者“(+)”表示右旋,“(L)”或者“(-)”表示左旋,“(DL)”或者“(±)”表示外消旋。Unless otherwise specified, "(D)" or "(+)" means dextrorotatory, "(L)" or "(-)" means levorotatory, and "(DL)" or "(±)" means racemic.
除非另有说明,用楔形实线键








本发明的化合物可以存在特定的。除非另有说明,术语“互变异构体”或“互变异构体形式”是指在室温下,不同官能团异构体处于动态平衡,并能很快的相互转化。若互变异构体是可能的(如在溶液中),则可以达到互变异构体的化学平衡。例如,质子互变异构体(proton tautomer)(也称质子转移互变异构体(prototropic tautomer))包括通过质子迁移来进行的互相转化,如酮-烯醇异构化和亚胺-烯胺异构化。价键异构体(valencetautomer)包括一些成键电子的重组来进行的相互转化。其中酮-烯醇互变异构化的具体实例是戊烷-2,4-二酮与4-羟基戊-3-烯-2-酮两个互变异构体之间的互变。The compounds of the present invention may exist in particular. Unless otherwise specified, the term "tautomer" or "tautomeric form" refers to isomers of different functional groups that are in dynamic equilibrium and are rapidly interconverted at room temperature. A chemical equilibrium of tautomers can be achieved if tautomers are possible (eg, in solution). For example, proton tautomers (also called prototropic tautomers) include interconversions by migration of protons, such as keto-enol isomerization and imine-ene Amine isomerization. Valence isomers (valencetautomer) include some interconversions by the recombination of bonding electrons. A specific example of keto-enol tautomerization is the interconversion between two tautomers, pentane-2,4-dione and 4-hydroxypent-3-en-2-one.
除非另有说明,术语“富含一种异构体”、“异构体富集”、“富含一种对映体”或者“对映体富集”指其中一种异构体或对映体的含量小于100%,并且,该异构体或对映体的含量大于等于60%,或者大于等于70%,或者大于等于80%,或者大于等于90%,或者大于等于95%,或者大于等于96%,或者大于等于97%,或者大于等于98%,或者大于等于99%,或者大于等于99.5%,或者大于等于99.6%,或者大于等于99.7%,或者大于等于99.8%,或者大于等于99.9%。Unless otherwise indicated, the terms "enriched in one isomer", "enriched in isomers", "enriched in one enantiomer" or "enriched in one enantiomer" refer to one of the isomers or pairs The enantiomer content is less than 100%, and the isomer or enantiomer content is greater than or equal to 60%, or greater than or equal to 70%, or greater than or equal to 80%, or greater than or equal to 90%, or greater than or equal to 95%, or Greater than or equal to 96%, or greater than or equal to 97%, or greater than or equal to 98%, or greater than or equal to 99%, or greater than or equal to 99.5%, or greater than or equal to 99.6%, or greater than or equal to 99.7%, or greater than or equal to 99.8%, or greater than or equal to 99.9%.
除非另有说明,术语“异构体过量”或“对映体过量”指两种异构体或两种对映体相对百分数之间的差值。例如,其中一种异构体或对映体的含量为90%,另一种异构体或对映体的含量为10%,则异构体或对映体过量(ee值)为80%。Unless otherwise indicated, the terms "isomeric excess" or "enantiomeric excess" refer to the difference between two isomers or relative percentages of two enantiomers. For example, if the content of one isomer or enantiomer is 90% and the content of the other isomer or enantiomer is 10%, the isomer or enantiomeric excess (ee value) is 80% .
可以通过的手性合成或手性试剂或者其他常规技术制备光学活性的(R)-和(S)-异构体以及D和L异构体。如果想得到本发明某化合物的一种对映体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对映体混合物分离,并且辅助基团裂开以提供纯的所需对映异构体。或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的常规方法进行非对映异构体拆分,然后回收得到纯的对映体。此外,对映异构体和非对映异构体的分离通常是通过使用色谱法完成的,所述色谱法采用手性固定相,并任选地与化学衍生法相结合(例如由胺生成氨基甲酸盐)。本发明的化合物可以在一个或多个构成该化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素标记化合物,比如氚(3H),碘-125(125I)或C-14(14C)。又例如,可用重氢取代氢形成氘代药物,氘与碳构成的键比普通氢与碳构成的键更坚固,相比于未氘化药物,氘代药物有降低毒副作用、增加药物稳定性、增强疗效、延长药物生物半衰期等优势。本发明的化合物的所有同位素组成的变换,无论放射性与否,都包括在本发明的范围之内。“任选”或“任选地”指的是随后描述的事件或状况可能但不是必需出现的,并且该描述包括其中所述事件或状况发生的情况以及所述事件或状况不发生的情况。Optically active (R)- and (S)-isomers, as well as D and L isomers, can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If one enantiomer of a compound of the present invention is desired, it can be prepared by asymmetric synthesis or derivatization with a chiral auxiliary, wherein the resulting mixture of diastereomers is separated and the auxiliary group is cleaved to provide pure desired enantiomer. Alternatively, when the molecule contains a basic functional group (such as an amino group) or an acidic functional group (such as a carboxyl group), a diastereomeric salt is formed with an appropriate optically active acid or base, followed by conventional methods known in the art The diastereoisomers were resolved and the pure enantiomers recovered. In addition, separation of enantiomers and diastereomers is usually accomplished by the use of chromatography employing a chiral stationary phase, optionally in combination with chemical derivatization (eg, from amines to amino groups) formate). The compounds of the present invention may contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute the compound. For example, compounds can be labeled with radioisotopes, such as tritium ( 3 H), iodine-125 ( 125 I) or C-14 ( 14 C). For another example, deuterated drugs can be formed by replacing hydrogen with deuterium, and the bonds formed by deuterium and carbon are stronger than those formed by ordinary hydrogen and carbon. Compared with non-deuterated drugs, deuterated drugs can reduce toxic side effects and increase drug stability. , enhance the efficacy, prolong the biological half-life of drugs and other advantages. All transformations of the isotopic composition of the compounds of the present invention, whether radioactive or not, are included within the scope of the present invention. "Optional" or "optionally" means that the subsequently described event or circumstance may, but need not, occur, and that the description includes instances where said event or circumstance occurs and instances in which it does not.
术语“被取代的”是指特定原子上的任意一个或多个氢原子被取代基取代,可以包括重氢和氢的变体,只要特定原子的价态是正常的并且取代后的化合物是稳定的。当取代基为氧(即=O)时,意味着两个氢原子被取代。氧取代不会发生在芳香基上。术语“任选被取代的”是指可以被取代,也可以不被取代,除非另有规定,取代基的种类和数目在化学上可以实现的基础上可以是任意的。The term "substituted" means that any one or more hydrogen atoms on a specified atom are replaced by a substituent, which may include deuterium and hydrogen variants, as long as the valence of the specified atom is normal and the substituted compound is stable of. When the substituent is oxygen (ie =O), it means that two hydrogen atoms are substituted. Oxygen substitution does not occur on aromatic groups. The term "optionally substituted" means that it may or may not be substituted, and unless otherwise specified, the type and number of substituents may be arbitrary on a chemically achievable basis.
当任何变量(例如R)在化合物的组成或结构中出现一次以上时,其在每一种情况下的定义都是独立的。因此,例如,如果一个基团被0-2个R所取代,则所述基团可以任选地至多被两个R所取代,并且每种情况下的R都有独立的选项。此外,取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。When any variable (eg, R) occurs more than once in the composition or structure of a compound, its definition in each case is independent. Thus, for example, if a group is substituted with 0-2 Rs, the group may optionally be substituted with up to two Rs, with independent options for R in each case. Furthermore, combinations of substituents and/or variants thereof are permissible only if such combinations result in stable compounds.
当一个连接基团的数量为0时,比如-(CRR)0-,表示该连接基团为单键。When the number of a linking group is 0, such as -(CRR) 0 -, it means that the linking group is a single bond.
当其中一个变量选自单键时,表示其连接的两个基团直接相连,比如A-L-Z中L代表单键时表示该结构实际上是A-Z。When one of the variables is selected from a single bond, it means that the two groups connected to it are directly connected, for example, when L in A-L-Z represents a single bond, it means that the structure is actually A-Z.
当一个取代基为空缺时,表示该取代基是不存在的,比如A-X中X为空缺时表示该结构实际上是A。当所列举的取代基中没有指明其通过哪一个原子连接到被取代的基团上时,这种取代基可以通过其任何原子相键合,例如,吡啶基作为取代基可以通过吡啶环上任意一个碳原子连接到被取代的基团上。When a substituent is vacant, it means that the substituent does not exist. For example, when X in A-X is vacant, it means that the structure is actually A. When the listed substituents do not indicate through which atom it is attached to the substituted group, such substituents may be bonded through any of its atoms, for example, pyridyl as a substituent may be through any one of the pyridine ring The carbon atom is attached to the substituted group.
当所列举的连接基团没有指明其连接方向,其连接方向是任意的,例如,


除非另有规定,术语“杂”表示杂原子或杂原子团(即含有杂原子的原子团),包括碳(C)和氢(H)以外的原子以及含有这些杂原子的原子团,例如包括氧(O)、氮(N)、硫(S)、硅(Si)、锗(Ge)、铝(Al)、硼(B)、-O-、-S-、、-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)2-,以及任选被取代的-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)2N(H)-或-S(=O)N(H)-。Unless otherwise specified, the term "hetero" means a heteroatom or heteroatom group (ie, a group containing a heteroatom), including atoms other than carbon (C) and hydrogen (H) and groups containing such heteroatoms, including, for example, oxygen (O ), nitrogen (N), sulfur (S), silicon (Si), germanium (Ge), aluminum (Al), boron (B), -O-, -S-,, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O) 2 -, and optionally substituted -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H) - or -S(=O)N(H)-.
除非另有规定,“环”表示被取代或未被取代的环烷基、杂环烷基、环烯基、杂环烯基、环炔基、杂环炔基、芳基或杂芳基。所述的环包括单环,也包括螺环、并环和桥环等双环或多环体系。环上原子的数目通常被定义为环的元数,例如,“5~7元环”是指环绕排列5~7个原子。除非另有规定,该环任选地包含1~3个杂原子。因此,“5~7元环”包括例如苯基、吡啶基和哌啶基;另一方面,术语“5~7元杂环烷基”包括吡啶基和哌啶基,但不包括苯基。术语“环”还包括含有至少一个环的环系,其中的每一个“环”均独立地符合上述定义。Unless otherwise specified, "ring" means substituted or unsubstituted cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, heterocycloalkynyl, aryl, or heteroaryl. Said ring includes a single ring, and also includes a bicyclic or polycyclic ring system such as a spiro ring, a paracyclic ring and a bridged ring. The number of atoms in a ring is generally defined as the number of ring members, eg, "5-7 membered ring" means 5-7 atoms arranged around it. Unless otherwise specified, the ring optionally contains 1 to 3 heteroatoms. Thus, "5-7 membered ring" includes, for example, phenyl, pyridyl, and piperidinyl; on the other hand, the term "5-7 membered heterocycloalkyl" includes pyridyl and piperidinyl, but does not include phenyl. The term "ring" also includes ring systems containing at least one ring, wherein each "ring" independently meets the above definition.
除非另有规定,术语“烷基”用于表示直链或支链的饱和的碳氢基团,在一些实施方案中,所述烷基为C1-12烷基;在另一些实施方案中,所述烷基为C1-6烷基;在另一些实施方案中,所述烷基为C1-3烷基。其可以是单取代(如-CH2F)或多取代的(如-CF3),可以是一价(如甲基)、二价(如亚甲基)或者多价(如次甲基)。烷基的实例包括但不限于甲基(Me),乙基(Et),丙基(包括n-丙基和异丙基),丁基(包括n-丁基,异丁基,s-丁基和t-丁基),戊基(包括n-戊基,异戊基和新戊基)、己基等。Unless otherwise specified, the term "alkyl" is used to refer to a straight or branched chain saturated hydrocarbon group, which in some embodiments is a C 1-12 alkyl; in other embodiments , the alkyl group is a C 1-6 alkyl group; in other embodiments, the alkyl group is a C 1-3 alkyl group. It may be monosubstituted (eg -CH2F ) or polysubstituted (eg -CF3 ), monovalent (eg methyl), divalent (eg methylene) or multivalent (eg methine) . Examples of alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n-propyl and isopropyl), butyl (including n-butyl, isobutyl, s-butyl) and t-butyl), pentyl (including n-pentyl, isopentyl and neopentyl), hexyl and the like.
除非另有规定,“烯基”用于表示直链或支链的包含一个或多个碳-碳双键的碳氢基团,碳-碳双键可以位于该基团的任何位置上。在一些实施方案中,所述烯基为C2-8烯基;在另一些实施方案中,所述烯基为C2-6烯基;在另一些实施方案中,所述烯基为C2-4烯基。其可以是单取代或多取代的,可以是一价、二价或者多价。烯基的实例包括但不限于乙烯基,丙烯基,丁烯基,戊烯基,己烯基,丁间二烯基,戊间二烯基,己间二烯基等。Unless otherwise specified, "alkenyl" is used to denote a straight or branched chain hydrocarbon group containing one or more carbon-carbon double bonds, which may be located anywhere in the group. In some embodiments, the alkenyl group is a C 2-8 alkenyl group; in other embodiments, the alkenyl group is a C 2-6 alkenyl group; in other embodiments, the alkenyl group is C 2-4 alkenyl. It may be mono- or polysubstituted, and may be monovalent, divalent or polyvalent. Examples of alkenyl groups include, but are not limited to, vinyl, propenyl, butenyl, pentenyl, hexenyl, butadienyl, piperylene, hexamadienyl, and the like.
除非另有规定,“炔基”用于表示直链或支链的包含一个或多个碳-碳三键的碳氢基团,碳-碳三键可以位于该基团的任何位置上。在一些实施方案中,所述炔基为C2-8炔基;在另一些实施方案中,所述炔基为C2-6炔基;在另一些实施方案中,所述炔基为C2-4炔基。其可以是单取代或多取代的,可以是一价、二价或者多价。炔基的实例包括但不限于乙炔基,丙炔基,丁炔基,戊炔基等。Unless otherwise specified, "alkynyl" is used to denote a straight or branched chain hydrocarbon group containing one or more carbon-carbon triple bonds, which may be located anywhere in the group. In some embodiments, the alkynyl group is a C 2-8 alkynyl group; in other embodiments, the alkynyl group is a C 2-6 alkynyl group; in other embodiments, the alkynyl group is C 2-4 alkynyl. It may be mono- or polysubstituted, and may be monovalent, divalent or polyvalent. Examples of alkynyl groups include, but are not limited to, ethynyl, propynyl, butynyl, pentynyl, and the like.
除非另有规定,术语“杂烷基”本身或者与另一术语联合,表示由一定数目碳原子和至少一个杂原子或杂原子团组成的,稳定的直链或支链的烷基原子团或其组合物。在一些实施方案中,杂原子选自B、O、N和S,其中氮和硫原子任选地被氧化,氮杂原子任选地被季铵化。在另一些实施方案中,杂原子团选自-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)2-、-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)2N(H)-和-S(=O)N(H)-。在一些实施方案中,所述杂烷基为C1-6杂烷基;在另一些实施方案中,所述杂烷基为C1-3杂烷基。杂原子或杂原子团可以位于杂烷基的任何内部位置,包括该烷基与分子其余部分的连接位置,但术语“烷氧基”、“烷氨基”和“烷硫基”(或硫代烷氧基)属于惯用表达,是指分别通过一个氧原子、氨基或硫原子连接到分子的其余部分的那些烷基基团。杂烷基的实例包括但不限于-OCH3、-OCH2CH3、-OCH2CH2CH3、-OCH2(CH3)2、-CH2-CH2-O-CH3、-NHCH3、-N(CH3)2、-NHCH2CH3、-N(CH3)(CH2CH3)、-CH2-CH2-NH-CH3、-CH2-CH2-N(CH3)-CH3、-SCH3、-SCH2CH3、-SCH2CH2CH3、-SCH2(CH3)2、-CH2-S-CH2-CH3、-CH2-CH2、-S(=O)-CH3、-CH2-CH2-S(=O)2-CH3、-CH=CH-O-CH3、-CH2-CH=N-OCH3和-CH=CH-N(CH3)-CH3。至多两个杂原子可以是连续的,例如-CH2-NH-OCH3。Unless otherwise specified, the term "heteroalkyl" by itself or in combination with another term means a stable straight or branched chain alkyl radical or a combination thereof consisting of a certain number of carbon atoms and at least one heteroatom or heteroatom thing. In some embodiments, the heteroatoms are selected from the group consisting of B, O, N, and S, wherein nitrogen and sulfur atoms are optionally oxidized, and nitrogen heteroatoms are optionally quaternized. In other embodiments, the heteroatomic group is selected from the group consisting of -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O) 2 -, -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O) 2 N(H)- and -S(=O)N( H)-. In some embodiments, the heteroalkyl group is a C 1-6 heteroalkyl group; in other embodiments, the heteroalkyl group is a C 1-3 heteroalkyl group. A heteroatom or group of heteroatoms can be located at any internal position within a heteroalkyl group, including where the alkyl group is attached to the rest of the molecule, except that the terms "alkoxy,""alkylamino," and "alkylthio" (or thioalkyl) Oxygenyl) is a customary expression referring to those alkyl groups attached to the rest of the molecule through an oxygen, amino or sulfur atom, respectively. Examples of heteroalkyl include, but are not limited to, -OCH3 , -OCH2CH3, -OCH2CH2CH3, -OCH2 (CH3)2 , -CH2 - CH2 - O - CH3 , -NHCH 3 , -N(CH 3 ) 2 , -NHCH 2 CH 3 , -N(CH 3 )(CH 2 CH 3 ), -CH 2 -CH 2 -NH-CH 3 , -CH 2 -CH 2 -N( CH3 ) -CH3 , -SCH3 ,-SCH2CH3,-SCH2CH2CH3, -SCH2 ( CH3 ) 2 , -CH2 - S - CH2 - CH3 , -CH2- CH 2 , -S(=O)-CH 3 , -CH 2 -CH 2 -S(=O) 2 -CH 3 , -CH=CH-O-CH 3 , -CH 2 -CH=N-OCH 3 and -CH=CH-N( CH3 ) -CH3 . Up to two heteroatoms may be consecutive, eg -CH2 -NH- OCH3 .
除非另有规定,术语“杂烯基”本身或者与另一术语联合,表示由一定数目碳原子和至少一个杂原子或杂原子团组成的,稳定的直链或支链的烯基原子团或其组合物。在一些实施方案中,杂原子选自B、O、N和S,其中氮和硫原子任选地被氧化,氮杂原子任选地被季铵化。在另一些实施方案中,杂原子团选自-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)2-、-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)2N(H)-和-S(=O)N(H)-。在一些实施方案中,所述杂烯基为C2-6杂烯基;在另一些实施方案中,所述杂烷基为C2-4杂烯基。杂原子或杂原子团可以位于杂烯基的任何内部位置,包括该烯基与分子其余部分的连接位置,但术语“烯基氧基”、“烯基氨基”和“烯基硫基”属于惯用表达,是指分别通过一个氧原子、氨基或硫原子连接到分子的其余部分的那些烯基基团。杂烯基的实例包括但不限于-O-CH=CH2、-O-CH=CHCH3、-O-CH=C(CH3)2、-CH=CH-O-CH3、-O-CH=CHCH2CH3、-CH2-CH=CH-OCH3、-NH-CH=CH2、-N(CH=CH2)-CH3、-CH=CH-NH-CH3、-CH=CH-N(CH3)2、-S-CH=CH2、-S-CH=CHCH3、-S-CH=C(CH3)2、-CH2-S-CH=CH2、-S(=O)-CH=CH2和-CH=CH-S(=O)2-CH3。至多两个杂原子可以是连续的,例如-CH=CH-NH-OCH3。Unless otherwise specified, the term "heteroalkenyl" by itself or in combination with another term means a stable straight or branched chain alkenyl radical or a combination thereof consisting of a certain number of carbon atoms and at least one heteroatom or heteroatom thing. In some embodiments, the heteroatoms are selected from the group consisting of B, O, N, and S, wherein nitrogen and sulfur atoms are optionally oxidized, and nitrogen heteroatoms are optionally quaternized. In other embodiments, the heteroatomic group is selected from the group consisting of -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O) 2 -, -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O) 2 N(H)- and -S(=O)N( H)-. In some embodiments, the heteroalkenyl is a C2-6 heteroalkenyl; in other embodiments, the heteroalkyl is a C2-4 heteroalkenyl. A heteroatom or group of heteroatoms may be located at any internal position within a heteroalkenyl group, including where the alkenyl group is attached to the rest of the molecule, but the terms "alkenyloxy", "alkenylamino" and "alkenylthio" are customary Expressed, refers to those alkenyl groups that are attached to the rest of the molecule through an oxygen, amino or sulfur atom, respectively. Examples of heteroalkenyl groups include, but are not limited to, -O-CH=CH 2 , -O-CH=CHCH 3 , -O-CH=C(CH 3 ) 2 , -CH=CH-O-CH 3 , -O- CH=CHCH 2 CH 3 , -CH 2 -CH=CH-OCH 3 , -NH-CH=CH 2 , -N(CH=CH 2 )-CH 3 , -CH=CH-NH-CH 3 , -CH =CH-N(CH 3 ) 2 , -S-CH=CH 2 , -S-CH=CHCH 3 , -S-CH=C(CH 3 ) 2 , -CH 2 -S-CH=CH 2 , - S(=O)-CH= CH2 and -CH=CH-S(=O) 2 - CH3 . Up to two heteroatoms may be consecutive, eg -CH=CH-NH- OCH3 .
除非另有规定,术语“杂炔基”本身或者与另一术语联合,表示由一定数目碳原子和至少一个杂原子或杂原子团组成的,稳定的直链或支链的炔基原子团或其组合物。在一些实施方案中,杂原子选自B、O、N和S,其中氮和硫原子任选地被氧化,氮杂原子任选地被季铵化。在另一些实施方案中,杂原子团选自-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)2-、-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)2N(H)-和-S(=O)N(H)-。在一些实施方案中,所述杂炔基为C2-6杂炔基;在另一些实施方案中,所述杂烷基为C2-4杂炔基。杂原子或杂原子团可以位于杂炔基的任何内部位置,包括该炔基与分子其余部分的连接位置,但术语“炔基氧基”、“炔基氨基”和“炔基硫基”属于惯用表达,是指分别通过一个氧原子、氨基或硫原子连接到分子的其余部分的那些炔基基团。杂炔基的实例包括但不限于



除非另有规定,“环烷基”包括任何稳定的环状烷基,其包括单环、双环或者三环体系,其中双环和三环体系包括螺环、并环和桥环。在一些实施方案中,所述环烷基为C3-8环烷基;在另一些实施方案中,所述环烷基为C3-6环烷基;在另一些实施方案中,所述环烷基为C5-6环烷基。其可以是单取代或多取代的,可以是一价、二价或者多价。这些环烷基的实例包括,但不限于,环丙基、环丁基、环戊基、环己基、环庚基、降冰片烷基、[2.2.2]二环辛烷、[4.4.0]二环癸烷等。Unless otherwise specified, "cycloalkyl" includes any stable cyclic alkyl group, including monocyclic, bicyclic, or tricyclic ring systems, wherein bicyclic and tricyclic rings include spiro, paracyclic, and bridged rings. In some embodiments, the cycloalkyl is C 3-8 cycloalkyl; in other embodiments, the cycloalkyl is C 3-6 cycloalkyl; in other embodiments, the cycloalkyl Cycloalkyl is C 5-6 cycloalkyl. It may be mono- or polysubstituted, and may be monovalent, divalent or polyvalent. Examples of such cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, [2.2.2]bicyclooctane, [4.4.0 ] Bicyclodecane, etc.
除非另有规定,“环烯基”包括任何稳定的环状烯基,在该基团的任何位点含有一个或多个不饱和的碳-碳双键,其包括单环、双环或者三环体系,其中双环和三环体系包括螺环、并环和桥环,但是此体系的任意环都是非芳香性的。在一些实施方案中,所述环烯基为C3-8环烯基;在另一些实施方案中,所述环烯基为C3-6环烯基;在另一些实施方案中,所述环烯基为C5-6环烯基。其可以是单取代或多取代的,可以是一价、二价或者多价。这些环烯基的实例包括,但不限于,环戊烯基、环己烯基等。Unless otherwise specified, "cycloalkenyl" includes any stable cyclic alkenyl group containing one or more unsaturated carbon-carbon double bonds at any position in the group, including monocyclic, bicyclic, or tricyclic systems in which bicyclic and tricyclic systems include spiro, paracyclic, and bridged rings, but any ring of this system is non-aromatic. In some embodiments, the cycloalkenyl is a C 3-8 cycloalkenyl; in other embodiments, the cycloalkenyl is a C 3-6 cycloalkenyl; in other embodiments, the cycloalkenyl Cycloalkenyl is C 5-6 cycloalkenyl. It may be mono- or polysubstituted, and may be monovalent, divalent or polyvalent. Examples of such cycloalkenyl groups include, but are not limited to, cyclopentenyl, cyclohexenyl, and the like.
除非另有规定,“环炔基”包括任何稳定的环状炔基,在该基团的任何位点含有一个或多个碳-碳三键,其包含单环、双环或者三环体系,其中双环和三环体系包括螺环、并环和桥环。其可以是单取代或多取代的,可以是一价、二价或者多价。Unless otherwise specified, "cycloalkynyl" includes any stable cyclic alkynyl group containing one or more carbon-carbon triple bonds at any position in the group, comprising a monocyclic, bicyclic or tricyclic ring system, wherein Bicyclic and tricyclic systems include spiro, paracyclic, and bridged rings. It may be mono- or polysubstituted, and may be monovalent, divalent or polyvalent.
除非另有规定,术语“杂环烷基”本身或者与其他术语联合分别表示环化的“杂烷基”,其包括单环、双环和三环体系,其中双环和三环体系包括螺环、并环和桥环。此外,就该“杂环烷基”而言,杂原子可以占据杂环烷基与分子其余部分的连接位置。在一些实施方案中,所述杂环烷基为4~6元杂环烷基;在另一些实施方案中,所述杂环烷基为5~6元杂环烷基。杂环烷基的实例包括但不限于氮杂环丁基、氧杂环丁基、硫杂环丁基、吡咯烷基、吡唑烷基、咪唑烷基、四氢噻吩基(包括四氢噻吩-2-基和四氢噻吩-3-基等)、四氢呋喃基(包括四氢呋喃-2-基等)、四氢吡喃基、哌啶基(包括1-哌啶基、2-哌啶基和3-哌啶基等)、哌嗪基(包括1-哌嗪基和2-哌嗪基等)、吗啉基(包括3-吗啉基和4-吗啉基等)、二噁烷基、二噻烷基、异噁唑烷基、异噻唑烷基、1,2-噁嗪基、1,2-噻嗪基、六氢哒嗪基、高哌嗪基、高哌啶基或氧杂环庚烷基。Unless otherwise specified, the term "heterocycloalkyl" by itself or in combination with other terms means cyclized "heteroalkyl", respectively, which includes monocyclic, bicyclic, and tricyclic rings, wherein bicyclic and tricyclic systems include spiro, Union ring and bridge ring. Furthermore, with respect to the "heterocycloalkyl", a heteroatom may occupy the position of attachment of the heterocycloalkyl to the remainder of the molecule. In some embodiments, the heterocycloalkyl is a 4-6 membered heterocycloalkyl; in other embodiments, the heterocycloalkyl is a 5-6 membered heterocycloalkyl. Examples of heterocycloalkyl include, but are not limited to, azetidinyl, oxetanyl, thietanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, tetrahydrothienyl (including tetrahydrothiophene) -2-yl and tetrahydrothiophen-3-yl, etc.), tetrahydrofuranyl (including tetrahydrofuran-2-yl, etc.), tetrahydropyranyl, piperidinyl (including 1-piperidinyl, 2-piperidinyl and 3-piperidyl, etc.), piperazinyl (including 1-piperazinyl and 2-piperazinyl, etc.), morpholinyl (including 3-morpholinyl and 4-morpholinyl, etc.), dioxanyl , dithianyl, isoxazolidinyl, isothiazolidinyl, 1,2-oxazinyl, 1,2-thiazinyl, hexahydropyridazinyl, homopiperazinyl, homopiperidinyl or oxygen Heteroheptyl.
除非另有规定,术语“杂环烯基”本身或者与其他术语联合分别表示环化的“杂烯基”,其包括单环、双环和三环体系,其中双环和三环体系包括螺环、并环和桥环,但是此体系的任意环都是非芳香性的。此外,就该“杂环烯基”而言,杂原子可以占据杂环烯基与分子其余部分的连接位置。在一些实施方案中,所述杂环烯基为4~6元杂环烯基;在另一些实施方案中,所述杂环烯基为5~6元杂环烯基。杂环烯基的实例包括但不限于

除非另有规定,术语“杂环炔基”本身或者与其他术语联合分别表示环化的“杂炔基”,其包括单环、双环和三环体系,其中双环和三环体系包括螺环、并环和桥环。此外,就该“杂环炔基”而言,杂原子可以占据杂环炔基与分子其余部分的连接位置。在一些实施方案中,所述杂环炔基为4~6元杂环炔基;在另一些实施方案中,所述杂环炔基为5~6元杂环炔基。除非另有规定,术语“卤代素”或“卤素”本身或作为另一取代基的一部分表示氟、氯、溴或碘原子。此外,术语“卤代烷基”意在包括单卤代烷基和多卤代烷基。例如,术语“卤代(C1-C4)烷基”意在包括但不仅限于三氟甲基、2,2,2-三氟乙基、4-氯丁基和3-溴丙基等等。除非另有规定,卤代烷基的实例包括但不仅限于:三氟甲基、三氯甲基、五氟乙基,和五氯乙基。Unless otherwise specified, the term "heterocycloalkynyl" by itself or in combination with other terms means cyclized "heteroalkynyl", respectively, which includes monocyclic, bicyclic, and tricyclic systems, wherein bicyclic and tricyclic systems include spiro, Union ring and bridge ring. Furthermore, with respect to the "heterocycloalkynyl", a heteroatom may occupy the position of attachment of the heterocycloalkynyl to the rest of the molecule. In some embodiments, the heterocyclic alkynyl group is a 4-6 membered heterocyclic alkynyl group; in other embodiments, the heterocyclic alkynyl group is a 5-6 membered heterocyclic alkynyl group. Unless otherwise specified, the term "halogen" or "halogen" by itself or as part of another substituent means a fluorine, chlorine, bromine or iodine atom. Furthermore, the term "haloalkyl" is intended to include monohaloalkyl and polyhaloalkyl. For example, the term "halo( C1 - C4 )alkyl" is intended to include, but is not limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like Wait. Unless otherwise specified, examples of haloalkyl include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.
“烷氧基”代表通过氧桥连接的具有特定数目碳原子的上述烷基,除非另有规定,C1-6烷氧基包括C1、C2、C3、C4、C5和C6的烷氧基。在一些实施方案中,所述烷氧基为C1-3烷氧基。烷氧基的实例包括但不限于:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基和S-戊氧基。"Alkoxy" represents the above-mentioned alkyl groups having the specified number of carbon atoms attached through an oxygen bridge, and unless otherwise specified, C 1-6 alkoxy includes C 1 , C 2 , C 3 , C 4 , C 5 and C 6 of the alkoxy. In some embodiments, the alkoxy group is a C 1-3 alkoxy group. Examples of alkoxy groups include, but are not limited to: methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, and S- pentoxy.
除非另有规定,本发明术语“芳环”和“芳基”可以互换使用,术语“芳环”或“芳基”表示多不饱和的碳环体系,它可以是单环、双环或多环体系,其中至少一个环是芳香性的,所述双环和多环体系中的各个环稠合在一起。其可以是单取代或多取代的,可以是一价、二价或者多价,在一些实施方案中,所述芳基为C6-12芳基;在另一些实施方案中,所述芳基为C6-10芳基。芳基的实例包括但不限于苯基、萘基(包括1-萘基和2-萘基等)。上述任意一个芳基环系的取代基选自本发明所述的可接受的取代基。Unless otherwise specified, the terms "aromatic ring" and "aryl" are used interchangeably herein, and the term "aromatic ring" or "aryl" refers to a polyunsaturated carbocyclic ring system, which may be monocyclic, bicyclic or polycyclic Ring systems wherein at least one ring is aromatic and the individual rings in the bicyclic and polycyclic systems are fused together. It can be mono- or poly-substituted, and can be monovalent, divalent or multivalent, in some embodiments, the aryl group is a C 6-12 aryl group; in other embodiments, the aryl group For C 6-10 aryl. Examples of aryl groups include, but are not limited to, phenyl, naphthyl (including 1-naphthyl and 2-naphthyl, and the like). Substituents for any of the foregoing aryl ring systems are selected from the acceptable substituents described herein.
除非另有规定,本发明术语“杂芳环”和“杂芳基”可以互换使用,术语“杂芳基”是指含有1、2、3或4个独立选自B、N、O和S的杂原子的芳基(或芳环),其可以是单环、双环或三环体系,其中氮原子可以是被取代的或未取代的(即N或NR,其中R是H或本文已经定义过的其他取代基),且任选地被季铵化,氮和硫杂原子可任选被氧化(即NO和S(O)p,p是1或2)。杂芳基可通过杂原子连接到分子的其余部分。在一些实施方案中,所述杂芳基为5-10元杂芳基;在另一些实施方案中,所述杂芳基为5-6元杂芳基。所述杂芳基的实例包括但不限于吡咯基(包括N-吡咯基、2-吡咯基和3-吡咯基等)、吡唑基(包括2-吡唑基和3-吡唑基等)、咪唑基(包括N-咪唑基、2-咪唑基、4-咪唑基和5-咪唑基等)、噁唑基(包括2-噁唑基、4-噁唑基和5-噁唑基等)、三唑基(1H-1,2,3-三唑基、2H-1,2,3-三唑基、1H-1,2,4-三唑基和4H-1,2,4-三唑基等)、四唑基、异噁唑基(3-异噁唑基、4-异噁唑基和5-异噁唑基等)、噻唑基(包括2-噻唑基、4-噻唑基和5-噻唑基等)、呋喃基(包括2-呋喃基和3-呋喃基等)、噻吩基(包括2-噻吩基和3-噻吩基等)、吡啶基(包括2-吡啶基、3-吡啶基和4-吡啶基等)、吡嗪基、嘧啶基(包括2-嘧啶基和4-嘧啶基等)、苯并噻唑基(包括5-苯并噻唑基等)、嘌呤基、苯并咪唑基(包括2-苯并咪唑基等)、吲哚基(包括5-吲哚基等)、异喹啉基(包括1-异喹啉基和5-异喹啉基等)、喹喔啉基(包括2-喹喔啉基和5-喹喔啉基等)、喹啉基(包括3-喹啉基和6-喹啉基等)、吡嗪基、嘌呤基、苯基并噁唑基、吲唑基(包括1H-吲唑基和2H-吲唑基)。上述任意一个杂芳基环系的取代基选自本发明所述的可接受的取代基。Unless otherwise specified, the terms "heteroaromatic ring" and "heteroaryl" are used interchangeably herein, and the term "heteroaryl" refers to a group containing 1, 2, 3 or 4 atoms independently selected from B, N, O and The aryl group (or aromatic ring) of the heteroatom of S, which may be a monocyclic, bicyclic, or tricyclic ring system, in which the nitrogen atom may be substituted or unsubstituted (ie, N or NR, where R is H or has been described herein other substituents as defined), and optionally quaternized, nitrogen and sulfur heteroatoms may be optionally oxidized (ie, NO and S(O) p , p is 1 or 2). A heteroaryl group can be attached to the remainder of the molecule through a heteroatom. In some embodiments, the heteroaryl group is a 5-10 membered heteroaryl group; in other embodiments, the heteroaryl group is a 5-6 membered heteroaryl group. Examples of such heteroaryl groups include, but are not limited to, pyrrolyl (including N-pyrrolyl, 2-pyrrolyl, and 3-pyrrolyl, etc.), pyrazolyl (including 2-pyrazolyl, 3-pyrazolyl, and the like) , imidazolyl (including N-imidazolyl, 2-imidazolyl, 4-imidazolyl and 5-imidazolyl, etc.), oxazolyl (including 2-oxazolyl, 4-oxazolyl and 5-oxazolyl, etc. ), triazolyl (1H-1,2,3-triazolyl, 2H-1,2,3-triazolyl, 1H-1,2,4-triazolyl and 4H-1,2,4-triazolyl Triazolyl, etc.), tetrazolyl, isoxazolyl (3-isoxazolyl, 4-isoxazolyl and 5-isoxazolyl, etc.), thiazolyl (including 2-thiazolyl, 4-thiazole base and 5-thiazolyl, etc.), furyl (including 2-furyl and 3-furyl, etc.), thienyl (including 2-thienyl and 3-thienyl, etc.), pyridyl (including 2-pyridyl, 3-pyridyl and 4-pyridyl, etc.), pyrazinyl, pyrimidinyl (including 2-pyrimidinyl and 4-pyrimidinyl, etc.), benzothiazolyl (including 5-benzothiazolyl, etc.), purinyl, Benzimidazolyl (including 2-benzimidazolyl, etc.), indolyl (including 5-indolyl, etc.), isoquinolinyl (including 1-isoquinolinyl and 5-isoquinolinyl, etc.), Quinoxalinyl (including 2-quinoxalinyl and 5-quinoxalinyl, etc.), quinolinyl (including 3-quinolinyl and 6-quinolinyl, etc.), pyrazinyl, purinyl, phenyl oxazolyl, indazolyl (including 1H-indazolyl and 2H-indazolyl). Substituents for any of the above heteroaryl ring systems are selected from the acceptable substituents described herein.
除非另有规定,术语“芳烷基”意在包括芳基附着于烷基的那些基团,在一些实施方案中,所述芳烷基为C6-10芳基-C1-4烷基;在另一些实施方案中,所述芳烷基为C6-10芳基-C1-2烷基。芳烷基的实例包括但不限于苄基、苯乙基、萘甲基等。“芳氧基”和“芳硫基”分别表示芳烷基中的碳原子(如甲基)已经被氧或硫原子代替的那些基团,在一些实施方案中,所述芳氧基为C6-10芳基-O-C1-2烷基;在另一些实施方案中,芳氧基为C6-10芳基-C1-2烷基-O-。在一些实施方案中,所述芳硫基为C6-10芳基-S-C1-2烷基;在另一些实施方案中,芳硫基为C6-10芳基-C1-2烷基-S-。芳氧基和芳硫基的实例包括但不限于苯氧基甲基、3-(1-萘氧基)丙基,苯硫基甲基等。Unless otherwise specified, the term "aralkyl" is intended to include those groups in which an aryl group is attached to an alkyl group, which, in some embodiments, is a C6-10 aryl- C1-4 alkyl group ; In other embodiments, the aralkyl group is a C 6-10 aryl-C 1-2 alkyl group. Examples of aralkyl groups include, but are not limited to, benzyl, phenethyl, naphthylmethyl, and the like. "Aryloxy" and "arylthio" respectively refer to those groups in which a carbon atom (eg, methyl) in an aralkyl group has been replaced by an oxygen or sulfur atom, and in some embodiments, the aryloxy group is C 6-10 aryl-OC 1-2 alkyl; in other embodiments, aryloxy is C 6-10 aryl-C 1-2 alkyl-O-. In some embodiments, the arylthio group is C 6-10 aryl-SC 1-2 alkyl; in other embodiments, the arylthio group is C 6-10 aryl-C 1-2 alkyl -S-. Examples of aryloxy and arylthio groups include, but are not limited to, phenoxymethyl, 3-(1-naphthyloxy)propyl, phenylthiomethyl, and the like.
除非另有规定,术语“杂芳烷基”意在包括杂芳基附着于烷基的那些基团,在一些实施方案中,所述杂芳烷基为5-8元杂芳基-C1-4烷基;在另一些实施方案中,所述杂芳烷基为5-6元杂芳基-C1-2烷基。杂芳烷基的实例包括但不限于吡咯基甲基、吡唑基甲基、吡啶基甲基、嘧啶基甲基等。“杂芳氧基”和“杂芳硫基”分别表示杂芳烷基中的碳原子(如甲基)已经被氧或硫原子代替的那些基团,在一些实施方案中,所述杂芳氧基为5-8元杂芳基-O-C1-2烷基;在另一些实施方案中,杂芳氧基为5-6元杂芳基-C1-2烷基-O-。在一些实施方案中,所述杂芳硫基为5-8元杂芳基-S-C1-2烷基;在另一些实施方案中,杂芳硫基为5-6元杂芳基-C1-2烷基-S-。杂芳氧基和杂芳硫基的实例包括但不限于吡咯氧甲基、吡唑氧甲基、2-吡啶氧甲基、吡咯硫甲基、吡唑硫甲基、2-吡啶硫甲基等。Unless otherwise specified, the term "heteroaralkyl" is intended to include those groups in which a heteroaryl group is attached to an alkyl group, which in some embodiments is a 5-8 membered heteroaryl-C 1 -4 alkyl; in other embodiments, the heteroaralkyl is a 5-6 membered heteroaryl-C 1-2 alkyl. Examples of heteroaralkyl groups include, but are not limited to, pyrrolylmethyl, pyrazolylmethyl, pyridylmethyl, pyrimidinylmethyl, and the like. "Heteroaryloxy" and "heteroarylthio" respectively refer to those groups in which a carbon atom (eg, methyl) in a heteroaralkyl group has been replaced by an oxygen or sulfur atom. In some embodiments, the heteroaryl group Oxygen is 5-8 membered heteroaryl-OC 1-2 alkyl; in other embodiments, heteroaryloxy is 5-6 membered heteroaryl-C 1-2 alkyl-O-. In some embodiments, the heteroarylthio group is 5-8 membered heteroaryl-SC 1-2 alkyl; in other embodiments, the heteroarylthio group is 5-6 membered heteroaryl-C 1 -2 alkyl-S-. Examples of heteroaryloxy and heteroarylthio include, but are not limited to, pyrroleoxymethyl, pyrazolyloxymethyl, 2-pyridyloxymethyl, pyrrolethiomethyl, pyrazolethiomethyl, 2-pyridylthiomethyl Wait.
除非另有规定,Cn-n+m或Cn-Cn+m包括n至n+m个碳的任何一种具体情况,例如C1-12包括C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、和C12,也包括n至n+m中的任何一个范围,例如C1-12包括C1-3、C1-6、C1-9、C3-6、C3-9、C3-12、C6-9、C6-12、和C9-12等;同理,n元至n+m元表示环上原子数为n至n+m个,例如3-12元环包括3元环、4元环、5元环、6元环、7元环、8元环、9元环、10元环、11元环、和12元环,也包括n至n+m中的任何一个范围,例如3-12元环包括3-6元环、3-9元环、5-6元环、5-7元环、6-7元环、6-8元环、和6-10元环等。Unless otherwise specified, Cn-n+m or Cn - Cn+m includes any particular instance of n to n+ m carbons, eg C1-12 includes C1 , C2 , C3, C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 , and C 12 , also including any one range from n to n+m, eg C 1-12 includes C 1-3 , C 1-6 , C 1-9 , C 3-6 , C 3-9 , C 3-12 , C 6-9 , C 6-12 , and C 9-12 , etc.; in the same way, n yuan to n +m-membered means that the number of atoms in the ring is from n to n+m, for example, 3-12-membered ring includes 3-membered ring, 4-membered ring, 5-membered ring, 6-membered ring, 7-membered ring, 8-membered ring, 9-membered ring , 10-membered ring, 11-membered ring, and 12-membered ring, also including any one range from n to n+m, for example, 3-12-membered ring includes 3-6 membered ring, 3-9 membered ring, 5-6 membered ring ring, 5-7 membered ring, 6-7 membered ring, 6-8 membered ring, and 6-10 membered ring, etc.
术语“离去基团”是指可以被另一种官能团或原子通过取代反应(例如亲和取代反应)所取代的官能团或原子。例如,代表性的离去基团包括三氟甲磺酸酯;氯、溴、碘;磺酸酯基,如甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯、对甲苯磺酸酯等;酰氧基,如乙酰氧基、三氟乙酰氧基等等。The term "leaving group" refers to a functional group or atom that can be replaced by another functional group or atom through a substitution reaction (eg, affinity substitution reaction). For example, representative leaving groups include triflate; chlorine, bromine, iodine; sulfonate groups such as mesylate, tosylate, p-bromobenzenesulfonate, p-toluenesulfonic acid Esters, etc.; acyloxy, such as acetoxy, trifluoroacetoxy, and the like.
术语“保护基”包括但不限于“氨基保护基”、“羟基保护基”或“巯基保护基”。术语“氨基保护基”是指适合用于阻止氨基氮位上副反应的保护基团。代表性的氨基保护基包括但不限于:甲酰基;酰基,例如链烷酰基(如乙酰基、三氯乙酰基或三氟乙酰基);烷氧基羰基,如叔丁氧基羰基(Boc);芳基甲氧羰基,如苄氧羰基(Cbz)和9-芴甲氧羰基(Fmoc);芳基甲基,如苄基(Bn)、三苯甲基(Tr)、1,1-二-(4′-甲氧基苯基)甲基;甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。术语“羟基保护基”是指适合用于阻止羟基副反应的保护基。代表性羟基保护基包括但不限于:烷基,如甲基、乙基和叔丁基;酰基,例如链烷酰基(如乙酰基);芳基甲基,如苄基(Bn),对甲氧基苄基(PMB)、9-芴基甲基(Fm)和二苯基甲基(二苯甲基,DPM);甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。The term "protecting group" includes, but is not limited to, "amino protecting group", "hydroxy protecting group" or "thiol protecting group". The term "amino protecting group" refers to a protecting group suitable for preventing side reactions at the amino nitrogen position. Representative amino protecting groups include, but are not limited to: formyl; acyl groups, such as alkanoyl groups (eg, acetyl, trichloroacetyl, or trifluoroacetyl); alkoxycarbonyl groups, such as tert-butoxycarbonyl (Boc) ; Arylmethoxycarbonyl, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); Arylmethyl, such as benzyl (Bn), trityl (Tr), 1,1-di -(4'-Methoxyphenyl)methyl; silyl groups such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS) and the like. The term "hydroxy protecting group" refers to a protecting group suitable for preventing hydroxyl side reactions. Representative hydroxy protecting groups include, but are not limited to: alkyl groups such as methyl, ethyl and tert-butyl; acyl groups such as alkanoyl (eg acetyl); arylmethyl groups such as benzyl (Bn), p-methyl Oxybenzyl (PMB), 9-fluorenylmethyl (Fm) and diphenylmethyl (diphenylmethyl, DPM); silyl groups such as trimethylsilyl (TMS) and tert-butyl Dimethylsilyl (TBS) and the like.
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。The compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments enumerated below, embodiments formed in combination with other chemical synthesis methods, and those well known to those skilled in the art Equivalent to alternatives, preferred embodiments include, but are not limited to, the embodiments of the present invention.
本发明采用下述缩略词:DCM代表二氯甲烷;DMF代表N,N-二甲基甲酰胺;DMSO代表二甲亚砜;EtOAc和EA代表乙酸乙酯;MeOH代表甲醇;BOC代表叔丁氧羰基是一种胺保护基团;THF代表四氢呋喃;Boc2O代表二-叔丁基二碳酸酯;Na2SO4代表硫酸钠;TEA代表三乙胺;Pd(dppf)Cl2代表[1,1′-双(二苯基膦基)二茂铁]二氯化钯;NaOH代表氢氧化钠;NaHMDS代表双(三甲基硅基)氨基钠;HATU代表2-(7-偶氮苯并三氮唑)-N,N,N,N-四甲基脲六氟磷酸酯;DIEA代表二异丙基乙基胺;HCl代表氯化氢;CDI代表N,N′-羰基二咪唑;MTBE代表甲基叔丁基醚;Pd2(dba)3代表三(二亚苄基丙酮)二钯;Xanphos代表4,5-双二苯基膦-9,9-二甲基氧杂蒽;PGE代表聚乙二醇。The following abbreviations are used in the present invention: DCM for dichloromethane; DMF for N,N-dimethylformamide; DMSO for dimethyl sulfoxide; EtOAc and EA for ethyl acetate; MeOH for methanol; BOC for tert-butyl Oxycarbonyl is an amine protecting group; THF stands for tetrahydrofuran; Boc 2 O stands for di-tert-butyl dicarbonate; Na 2 SO 4 stands for sodium sulfate; TEA stands for triethylamine; Pd(dppf)Cl 2 stands for [1 , 1'-bis(diphenylphosphino)ferrocene]palladium dichloride; NaOH stands for sodium hydroxide; NaHMDS stands for bis(trimethylsilyl)sodium amide; HATU stands for 2-(7-azobenzene triazole)-N,N,N,N-tetramethylurea hexafluorophosphate; DIEA for diisopropylethylamine; HCl for hydrogen chloride; CDI for N,N'-carbonyldiimidazole; MTBE for Methyl tert-butyl ether; Pd 2 (dba) 3 for tris(dibenzylideneacetone)dipalladium; Xanphos for 4,5-bisdiphenylphosphine-9,9-dimethylxanthene; PGE for polyethylene glycol.
化合物经手工或者
附图说明Description of drawings
图1为CT26小鼠同系移植瘤模型中的药效结果。Figure 1 shows the efficacy results in the CT26 mouse syngeneic xenograft model.
图2为CT26小鼠同系移植瘤模型中小鼠体重变化。Figure 2 shows the changes of mouse body weight in the CT26 mouse syngeneic xenograft model.
具体实施方式Detailed ways
下面通过实施例对本发明进行详细描述,但并不意味着对本发明任何不利限制。本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。对本领域的技术人员而言,在不脱离本发明精神和范围的情况下针对本发明具体实施方式进行各种变化和改进将是显而易见的。The present invention will be described in detail by the following examples, but it does not mean any unfavorable limitation of the present invention. The compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments enumerated below, embodiments formed in combination with other chemical synthesis methods, and those well known to those skilled in the art Equivalent to alternatives, preferred embodiments include, but are not limited to, the embodiments of the present invention. It will be apparent to those skilled in the art that various changes and modifications can be made to the specific embodiments of the present invention without departing from the spirit and scope of the invention.
实施例1Example 1


化合物1-b的合成:Synthesis of compound 1-b:
0℃下,向化合物1-a(98.5g)的丙酮溶液(1500mL)中加入碘甲烷(185.33g),在25℃反应1小时后,过滤,滤饼旋干得到化合物1-b。At 0°C, iodomethane (185.33g) was added to the acetone solution (1500mL) of compound 1-a (98.5g), reacted at 25°C for 1 hour, filtered, and the filter cake was spin-dried to obtain compound 1-b.
化合物1-d的合成:Synthesis of compound 1-d:
0℃下,向将二氯亚砜(96.92g,814.69mmol)慢慢滴加到无水乙醇溶液(1000mL)中,然后加入化合物1-c(95g,盐酸盐),在80度下反应12小时后,减压浓缩得到化合物1-d。At 0 °C, thionyl chloride (96.92 g, 814.69 mmol) was slowly added dropwise to anhydrous ethanol solution (1000 mL), then compound 1-c (95 g, hydrochloride) was added, and the reaction was carried out at 80 °C After 12 hours, it was concentrated under reduced pressure to give compound 1-d.
化合物1-e的合成:Synthesis of compound 1-e:
90℃下,向化合物1-d(98g,盐酸盐)和N,N-二异丙基乙胺(141.01g,1.09mol)的二氧六环(1400mL)和水(350mL)的溶液中分批加入化合物1-b(208.74g,818.28mmol),在9008下反应12小时后,减压浓缩移除溶剂二氧六环,加入1000毫升乙酸乙酯,溶液用饱和氯化钠溶液洗涤3次,每次1000毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到化合物1-e。To a solution of compound 1-d (98 g, hydrochloride) and N,N-diisopropylethylamine (141.01 g, 1.09 mol) in dioxane (1400 mL) and water (350 mL) at 90 °C Compound 1-b (208.74 g, 818.28 mmol) was added in batches, reacted at 9008 for 12 hours, concentrated under reduced pressure to remove the solvent dioxane, added 1000 ml of ethyl acetate, and the solution was washed with saturated
化合物1-f的合成:Synthesis of compound 1-f:
-70℃下,向化合物1-e(76g)和N,N-二(三氟甲烷磺酰基)苯胺(126.55g)的四氢呋喃(1500mL)溶液中滴加入NaHMDS(1M,371.09mL),保持温度不高于-60℃,滴加完后在25℃反应12小时,加入1500mL盐酸(0.2M)溶液淬灭反应,减压浓缩移除溶剂四氢呋喃,加入1500毫升甲基叔丁基醚,溶液依次用盐酸(0.2M)溶液洗涤2次,每次1500毫升,用5%NaOH溶液洗涤3次,每次1500毫升,用饱和氯化钠溶液洗涤1次,每次1500毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到棕色胶状化合物1-f。To a solution of compound 1-e (76 g) and N,N-bis(trifluoromethanesulfonyl)aniline (126.55 g) in tetrahydrofuran (1500 mL) was added dropwise NaHMDS (1 M, 371.09 mL) at -70°C, maintaining the temperature Not higher than -60°C, react at 25°C for 12 hours after the dropwise addition, add 1500 mL of hydrochloric acid (0.2M) solution to quench the reaction, concentrate under reduced pressure to remove the solvent tetrahydrofuran, add 1500 mL of methyl tert-butyl ether, and the solutions are sequentially Wash 2 times with hydrochloric acid (0.2M) solution, 1500 mL each time, wash 3 times with 5% NaOH solution, 1500 mL each time, wash 1 time with saturated sodium chloride solution, 1500 mL each time, the organic phase is washed with anhydrous Dry over sodium sulfate, filter, and concentrate to give compound 1-f as a brown gum.
化合物1-h的合成:Synthesis of compound 1-h:
向化合物1-f(93.5g,261.65mmol)和双频哪醇硼酯(73.09g,287.82mmol)的二氧六环(1000mL)溶液中加入溴化钠(10.77g,104.66mmol,3.37mL)、醋酸钾(77.04g)和Pd(dppf)Cl2(9.57g),在氮气保护和80℃反应12小时后,,加入2000毫升乙酸乙酯,过滤,滤液用饱和氯化钠溶液洗涤2次,每次1500毫升,然后加入1000毫升盐酸溶液(0.2M),分离得水相,有机相用盐酸溶液(0.2M)萃取两次,每次1000毫升,联合的水相用乙酸乙酯洗涤2次,每次2000毫升,然后用饱和碳酸钠溶液调节pH到9,用乙酸乙酯萃取两次,每次1500毫升,两次萃取联合的有机相用饱和氯化钠溶液洗涤2次,每次2000毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到化合物1-h。To a solution of compound 1-f (93.5 g, 261.65 mmol) and bispinacol boronate (73.09 g, 287.82 mmol) in dioxane (1000 mL) was added sodium bromide (10.77 g, 104.66 mmol, 3.37 mL) , potassium acetate (77.04g) and Pd(dppf)Cl 2 (9.57g), after 12 hours of reaction at 80°C under nitrogen protection, 2000 ml of ethyl acetate was added, filtered, and the filtrate was washed twice with saturated sodium chloride solution , 1500 ml each time, then 1000 ml hydrochloric acid solution (0.2M) was added, the aqueous phase was separated, the organic phase was extracted twice with 1000 ml hydrochloric acid solution (0.2M) each time, and the combined aqueous phase was washed with ethyl acetate 2 times, 2000 ml each time, then adjusted to
化合物1-j的合成:Synthesis of compound 1-j:
向化合物1-h(29.85g)和4-氯-6-氟喹啉(14.7g)的二氧六环(240mL)和水(60mL)溶液中加入三乙胺(24.57g)和Pd(dppf)Cl2(2.96g),在氮气保护和100℃反应12小时后,加入1000毫升乙酸乙酯,过滤,滤液用饱和氯化钠溶液洗涤2次,每次500毫升,然后用盐酸溶液(1M)调节pH到1,分离得水相,有机相用盐酸溶液(0.2M)萃取两次,每次500毫升,联合的水相用乙酸乙酯洗涤2次,每次1000毫升,然后用饱和碳酸钠溶液调节pH到9,用乙酸乙酯萃取两次,每次750毫升,两次萃取联合的有机相用饱和氯化钠溶液洗涤2次,每次1000毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到30g粗产品,粗产品加入到150毫升乙腈中,在25℃下拌1小时,过滤,滤饼旋干得到化合物1-j。To a solution of compound 1-h (29.85 g) and 4-chloro-6-fluoroquinoline (14.7 g) in dioxane (240 mL) and water (60 mL) was added triethylamine (24.57 g) and Pd (dppf )Cl2 (2.96g), after 12 hours of reaction at 100 °C under nitrogen protection, 1000 ml of ethyl acetate was added, filtered, the filtrate was washed twice with saturated sodium chloride solution, 500 ml each time, and then with hydrochloric acid solution (1M) The pH was adjusted to 1, the aqueous phase was separated, the organic phase was extracted twice with 500 ml of hydrochloric acid solution (0.2M), and the combined aqueous phase was washed with ethyl acetate twice, 1000 ml each, and then with saturated sodium carbonate The pH of the solution was adjusted to 9, extracted with ethyl acetate twice, 750 mL each time, the combined organic phase of the two extractions was washed twice with 1000 mL saturated sodium chloride solution, and the organic phase was dried with anhydrous sodium sulfate, Filtration and concentration to obtain 30 g of crude product, which was added to 150 ml of acetonitrile, stirred at 25° C. for 1 hour, filtered, and the filter cake was spin-dried to obtain compound 1-j.
化合物1-k的合成:Synthesis of compound 1-k:
向化合物1-j(21g)和吡啶(7.03g)的异丙醇溶液(400mL)中加入钯碳(4g,10%纯度),在50℃和氢气氛围(15psi)下反应1小时后,用硅藻土过滤,浓缩得到化合物1-k。Palladium carbon (4 g, 10% purity) was added to a solution of compound 1-j (21 g) and pyridine (7.03 g) in isopropanol (400 mL), and the reaction was carried out at 50° C. under a hydrogen atmosphere (15 psi) for 1 hour. Filter through celite and concentrate to give compound 1-k.
化合物1-l的合成:Synthesis of compound 1-1:
向化合物1-k(20g)的甲醇(100mL),四氢呋喃(100mL)和水(50mL)溶液中加入氢氧化钠(6.73g),在70℃下反应12小时后,减压浓缩移除溶剂甲醇和四氢呋喃,然后用盐酸(1M)将pH调到6-7,大量固体析出,过滤,滤饼减压旋干得到化合物1-l。To a solution of compound 1-k (20 g) in methanol (100 mL), tetrahydrofuran (100 mL) and water (50 mL) was added sodium hydroxide (6.73 g), reacted at 70°C for 12 hours, concentrated under reduced pressure to remove the solvent methanol and tetrahydrofuran, then the pH was adjusted to 6-7 with hydrochloric acid (1M), a large amount of solid was precipitated, filtered, and the filter cake was spin-dried under reduced pressure to obtain compound 1-1.
化合物1的合成:Synthesis of compound 1:
向化合物1-l(5g)和对氯苯氨(2.33g)的DMF溶液(100mL)中加入N,N-二异丙基乙胺(5.90g)和HATU(8.11g),在氮气保护和25℃下反应12小时后,加入400毫升乙酸乙酯稀释,溶液用饱和氯化钠溶液洗涤3次,每次500毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到9g粗品,粗产品加入到45毫升乙腈中,在25乙腈搅拌12小时,过滤,滤饼旋干得到纯化后的粗品,粗品通过制备级高效液相色谱(甲酸体系)制备纯化得到化合物1。To a DMF solution (100 mL) of compound 1-1 (5 g) and p-chloroaniline (2.33 g) was added N,N-diisopropylethylamine (5.90 g) and HATU (8.11 g), under nitrogen protection and After reacting at 25°C for 12 hours, 400 ml of ethyl acetate was added to dilute the solution. The solution was washed three times with saturated sodium chloride solution, 500 ml each time. The organic phase was dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 9 g of crude product. It was added to 45 ml of acetonitrile, stirred at 25 acetonitrile for 12 hours, filtered, and the filter cake was spin-dried to obtain the purified crude product.
1H NMR(400MHz,DMSO-d6)δ=9.58(s,1H),8.82(d,J=4.5Hz,1H),8.08(dd,J=6.0,9.2Hz,1H),8.02(dd,J=2.8,11.2Hz,1H),,7.82-7.75(m,2H),7.65(dt,J=2.8,8.7Hz,1H),7.48(d,J=4.6Hz,1H),7.41-7.34(m,2H),3.39-3.34(m,1H),2.99(d,J=11.2Hz,2H),2.45-2.23(m,6H),1.91-1.68(m,6H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.58 (s, 1H), 8.82 (d, J=4.5 Hz, 1H), 8.08 (dd, J=6.0, 9.2 Hz, 1H), 8.02 (dd, J=2.8, 11.2Hz, 1H), 7.82-7.75(m, 2H), 7.65(dt, J=2.8, 8.7Hz, 1H), 7.48(d, J=4.6Hz, 1H), 7.41-7.34( m, 2H), 3.39-3.34 (m, 1H), 2.99 (d, J=11.2Hz, 2H), 2.45-2.23 (m, 6H), 1.91-1.68 (m, 6H).
实施例2Example 2

化合物2的合成Synthesis of Compound 2
将化合物1-l(50mg)溶于DMF(5mL)中,并在25℃下,往溶液中加入DIEA(98.39mg)和HATU(86.84mg).所得混合物在25℃下搅拌30分钟.然后向反应液中加2-a(23.59mg),所得混合物在25℃下搅拌12小时.向反应液中加入水(10mL)淬灭反应并用乙酸乙酯(30mL*3)萃取.合并有机相用Na2SO4干燥,过滤后减压浓缩得到粗品.粗品通过制备色谱纯化得到化合物2.Compound 1-1 (50 mg) was dissolved in DMF (5 mL), and DIEA (98.39 mg) and HATU (86.84 mg) were added to the solution at 25°C. The resulting mixture was stirred at 25°C for 30 minutes. 2-a (23.59 mg) was added to the reaction solution, and the resulting mixture was stirred at 25°C for 12 hours. Water (10 mL) was added to the reaction solution to quench the reaction and extracted with ethyl acetate (30 mL*3). The organic phases were combined with Na 2 SO 4 was dried, filtered and concentrated under reduced pressure to obtain the crude product. The crude product was purified by preparative chromatography to obtain compound 2.
1H NMR(400MHz,CCl3D)δppm 1.69-1.91(m,3H)2.00-2.10(m,3H)2.33-2.56(m,6H)3.00(br d,J=11.74Hz,2H)3.18(br t,J=11.80Hz,1H)6.77-6.88(m,2H)7.31(d,J=4.65Hz,1H)7.39-7.48(m,1H)7.61(dd,J=10.33,2.63Hz,1H)8.03-8.15(m,1H)8.28(td,J=9.23,5.99Hz,1H)8.80(d,J=4.65Hz,1H)9.42(br s,1H). 1 H NMR (400MHz, CCl 3 D) δppm 1.69-1.91 (m, 3H) 2.00-2.10 (m, 3H) 2.33-2.56 (m, 6H) 3.00 (br d, J=11.74Hz, 2H) 3.18 (br d t, J=11.80Hz, 1H) 6.77-6.88 (m, 2H) 7.31 (d, J=4.65Hz, 1H) 7.39-7.48 (m, 1H) 7.61 (dd, J=10.33, 2.63Hz, 1H) 8.03 -8.15(m, 1H) 8.28(td, J=9.23, 5.99Hz, 1H) 8.80(d, J=4.65Hz, 1H) 9.42(br s, 1H).
实施例3Example 3

化合物3的合成Synthesis of
将化合物1-l(50mg)溶于DMF(3mL)中,并在25℃下,往溶液中加入DIEA(98.39mg)和HATU(86.84mg).所得混合物在25℃下搅拌30分钟.然后向反应液中加3-a(23.31mg),所得混合物在25℃下搅拌12小时.向反应液中加入水(10mL)淬灭反应并用乙酸乙酯(20mL*3)萃取.合并有机相用Na2SO4干燥,过滤后减压浓缩得到粗品.粗品通过制备色谱纯化得到化合物3.Compound 1-1 (50 mg) was dissolved in DMF (3 mL), and DIEA (98.39 mg) and HATU (86.84 mg) were added to the solution at 25°C. The resulting mixture was stirred at 25°C for 30 minutes. 3-a (23.31 mg) was added to the reaction solution, and the resulting mixture was stirred at 25°C for 12 hours. Water (10 mL) was added to the reaction solution to quench the reaction and extracted with ethyl acetate (20 mL*3). The organic phases were combined with Na 2 SO 4 was dried, filtered and concentrated under reduced pressure to obtain the crude product. The crude product was purified by preparative chromatography to obtain
1H NMR(400MHz,CCl3D)δppm 1.68-1.92(m,3H)1.96-2.10(m,3H)2.25-2.58(m,6H)3.02(br d,J=11.25Hz,2H)3.18(br t,J=12.10Hz,1H)7.01(dd,J=8.01,1.04Hz,1H)7.17(s,1H)7.21(s,1H)7.37-7.48(m,2H)7.60(dd,J=10.33,2.63Hz,1H)7.66(s,1H)8.07-8.14(m,1H)8.80(d,J=4.65Hz,1H)9.11(br s,1H). 1 H NMR (400MHz, CCl 3 D) δppm 1.68-1.92 (m, 3H) 1.96-2.10 (m, 3H) 2.25-2.58 (m, 6H) 3.02 (br d, J=11.25Hz, 2H) 3.18 (br d t, J=12.10Hz, 1H) 7.01 (dd, J=8.01, 1.04Hz, 1H) 7.17 (s, 1H) 7.21 (s, 1H) 7.37-7.48 (m, 2H) 7.60 (dd, J=10.33, 2.63Hz, 1H) 7.66(s, 1H) 8.07-8.14(m, 1H) 8.80(d, J=4.65Hz, 1H) 9.11(br s, 1H).
实施例4Example 4

化合物4的合成Synthesis of compound 4
将化合物1-l(100mg)溶解在DMF(3mL)中,然后再加入DIEA(118.07mg)和HATU(231.58mg,),再将4-a(58.26mg)加入到混合物中,该反应在25℃N2保护下反应12小时,反应液为棕色溶液。将反应液倾倒入水50mL,随后用EA 120mL(40mL*3)萃取,合并有机相用饱和食盐水洗60mL(30mL*2),Na2SO4干燥,过滤浓缩后得到粗品.将粗品通过制备色谱纯化后得到4。Compound 1-1 (100 mg) was dissolved in DMF (3 mL), then DIEA (118.07 mg) and HATU (231.58 mg, ) were added, and 4-a (58.26 mg) was added to the mixture, and the reaction was carried out at 25 The reaction was carried out under the protection of N 2 for 12 hours, and the reaction solution was a brown solution. The reaction solution was poured into 50 mL of water, then extracted with EA 120 mL (40 mL*3), the combined organic phases were washed with saturated brine 60 mL (30 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified by preparative chromatography After getting 4.
1H NMR(400MHz,DMSO-d6)δppm 1.73-1.94(m,6H)2.24-2.45(m,7H)3.02(br d,J=11.2Hz,2H)4.14(s,3H)7.42(dd,J=9.2,1.6Hz,1H)7.49-7.57(m,2H)7.66(td,J=8.8,2.6Hz,1H)7.99-8.12(m,2H)8.26(s,1H)8.83(d,J=4.4Hz,1H)9.40(s,1H) 1 H NMR (400 MHz, DMSO-d 6 ) δppm 1.73-1.94 (m, 6H) 2.24-2.45 (m, 7H) 3.02 (br d, J=11.2 Hz, 2H) 4.14 (s, 3H) 7.42 (dd, J=9.2, 1.6Hz, 1H) 7.49-7.57(m, 2H) 7.66(td, J=8.8, 2.6Hz, 1H) 7.99-8.12(m, 2H) 8.26(s, 1H) 8.83(d, J= 4.4Hz, 1H) 9.40(s, 1H)
实施例5Example 5

化合物5的合成Synthesis of
将化合物1-l(100mg)溶解于DMF(5mL)中,然后加入DIEA(196.79mg),HATU(173.68mg),5-a(43.25mg).氮气置换三次,所得混合物在25℃下搅拌12小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物5。Compound 1-1 (100 mg) was dissolved in DMF (5 mL), then DIEA (196.79 mg), HATU (173.68 mg), 5-a (43.25 mg). Nitrogen was replaced three times, and the resulting mixture was stirred at 25 °C for 12 h. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain a crude product, which was purified by preparative chromatography to obtain
1H NMR(400MHz,DMSO-d6)δ=9.67(s,1H),8.83(d,J=4.4Hz,1H),8.20(s,1H),8.11-8.01(m,2H),7.96(br dd,J=2.4,7.6Hz,1H),7.92(br dd,J=2.4,7.5Hz,1H),7.66(dt,J=2.8,8.7Hz,1H),7.58-7.48(m,1H),7.44-7.36(m,1H),3.56-3.28(m,1H),3.17(brs,2H),3.00(s,2H),2.97(br s,4H),2.54-2.53(m,4H),2.47-2.22(m,2H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.67 (s, 1H), 8.83 (d, J=4.4 Hz, 1H), 8.20 (s, 1H), 8.11-8.01 (m, 2H), 7.96 ( br dd, J=2.4, 7.6Hz, 1H), 7.92 (br dd, J=2.4, 7.5Hz, 1H), 7.66 (dt, J=2.8, 8.7Hz, 1H), 7.58-7.48 (m, 1H) , 7.44-7.36(m, 1H), 3.56-3.28(m, 1H), 3.17(brs, 2H), 3.00(s, 2H), 2.97(brs, 4H), 2.54-2.53(m, 4H), 2.47-2.22(m, 2H).
实施例6Example 6

化合物6的合成Synthesis of compound 6
将化合物1-l(100mg)溶解于DMF(5mL)中,然后加入DIEA(196.79mg),HATU(173.68mg),6-a(33.22mg)。氮气置换三次,所得混合物在25℃下搅拌12小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物6。Compound 1-1 (100 mg) was dissolved in DMF (5 mL), then DIEA (196.79 mg), HATU (173.68 mg), 6-a (33.22 mg) were added. Nitrogen was replaced three times, and the resulting mixture was stirred at 25°C for 12 hours. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product , the crude product was purified by preparative chromatography to obtain compound 6.
1H NMR(400MHz,DMSO-d6)δ=8.82(br d,J=3.8Hz,1H),8.19(s,1H),8.14-8.04(m,1H),8.01(s,1H),7.99(br s,1H),7.66(br t,J=7.6Hz,1H),7.47(br d,J=3.8Hz,1H),3.64(br s,1H),3.35-3.25(m,1H),2.90(br d,J=10.4Hz,2H),2.43-2.27(m,2H),2.16(br dd,J=8.8,19.4Hz,4H),1.92-1.80(m,2H),1.79-1.63(m,8H),1.58(br d,J=12.6Hz,1H),1.36-1.18(m,4H),1.14(br s,1H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=8.82 (br d, J=3.8 Hz, 1H), 8.19 (s, 1H), 8.14-8.04 (m, 1H), 8.01 (s, 1H), 7.99 (br s, 1H), 7.66 (br t, J=7.6Hz, 1H), 7.47 (br d, J=3.8Hz, 1H), 3.64 (br s, 1H), 3.35-3.25 (m, 1H), 2.90(br d, J=10.4Hz, 2H), 2.43-2.27(m, 2H), 2.16(br dd, J=8.8, 19.4Hz, 4H), 1.92-1.80(m, 2H), 1.79-1.63( m, 8H), 1.58 (br d, J=12.6Hz, 1H), 1.36-1.18 (m, 4H), 1.14 (br s, 1H).
实施例7Example 7

化合物7的合成Synthesis of
将化合物1-l(50mg)溶解于DMF(5mL)中,然后加入DIEA(98.39mg),HATU(86.84mg),7-a(28.81mg).氮气置换三次,所得混合物在25℃下搅拌12小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物7.Compound 1-1 (50 mg) was dissolved in DMF (5 mL), then DIEA (98.39 mg), HATU (86.84 mg), 7-a (28.81 mg). Nitrogen replacement three times, the resulting mixture was stirred at 25°C for 12 h. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product, which was purified by preparative chromatography to obtain
1H NMR(400MHz,DMSO-d6)δ=9.59(s,1H),8.83(d,J=4.4Hz,1H),8.31(br s,1H),8.12-8.07(m,2H),8.04(d,J=10.4Hz,2H),7.76-7.64(m,1H),7.54-7.47(m,3H),3.63(brs,1H),3.35(br t,J=11.2Hz,2H),2.99(br d,J=11.0Hz,2H),2.47-2.24(m,4H),1.92-1.68(m,6H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.59 (s, 1H), 8.83 (d, J=4.4 Hz, 1H), 8.31 (br s, 1H), 8.12-8.07 (m, 2H), 8.04 (d, J=10.4Hz, 2H), 7.76-7.64 (m, 1H), 7.54-7.47 (m, 3H), 3.63 (brs, 1H), 3.35 (br t, J=11.2Hz, 2H), 2.99 (br d, J=11.0Hz, 2H), 2.47-2.24 (m, 4H), 1.92-1.68 (m, 6H).
实施例8Example 8

化合物8的合成Synthesis of Compound 8
将化合物1-l(50mg)溶解于DMF(5mL)中,然后加入DIEA(98.39mg),HATU(86.84mg),8-a(18.61mg).氮气置换三次,所得混合物在25℃下搅拌12小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物8。Compound 1-1 (50 mg) was dissolved in DMF (5 mL), then DIEA (98.39 mg), HATU (86.84 mg), 8-a (18.61 mg). Nitrogen replacement three times, the resulting mixture was stirred at 25°C for 12 h. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product, which was purified by preparative chromatography to obtain compound 8.
1H NMR(400MHz,DMSO-d6)δ=9.52(s,1H),8.83(d,J=4.6Hz,1H),8.25(s,1H),8.12-8.01(m,2H),7.79-7.73(m,2H),7.67(dt,J=2.8,8.7Hz,1H),7.50(d,J=4.4Hz,1H),7.17(t,J=8.2Hz,2H),3.36(br t,J=12Hz,1H),3.26(br s,2H),3.00(br d,J=10Hz,2H),2.46-2.22(m,4H),1.93-1.68(m,6H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.52 (s, 1H), 8.83 (d, J=4.6 Hz, 1H), 8.25 (s, 1H), 8.12-8.01 (m, 2H), 7.79- 7.73(m, 2H), 7.67(dt, J=2.8, 8.7Hz, 1H), 7.50(d, J=4.4Hz, 1H), 7.17(t, J=8.2Hz, 2H), 3.36(br t, J=12Hz, 1H), 3.26 (br s, 2H), 3.00 (br d, J=10Hz, 2H), 2.46-2.22 (m, 4H), 1.93-1.68 (m, 6H).
实施例9Example 9

化合物9的合成Synthesis of
将化合物1-l(50mg)溶解于DMF(5mL)中,然后加入DIEA(98.39mg),HATU(86.84mg),9-a(18.11mg)。氮气置换三次,所得混合物在25℃下搅拌12小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物9。Compound 1-1 (50 mg) was dissolved in DMF (5 mL), then DIEA (98.39 mg), HATU (86.84 mg), 9-a (18.11 mg) were added. Nitrogen was replaced three times, and the resulting mixture was stirred at 25°C for 12 hours. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product , the crude product was purified by preparative chromatography to obtain
1H NMR(400MHz,DMSO-d6)δ=9.56(s,1H),8.83(d,J=4.4Hz,1H),8.76(d,J=2.2Hz,1H),8.22(s,1H),8.13-7.97(m,3H),7.66(dt,J=2.8,8.7Hz,1H),7.50(d,J=4.4Hz,1H),7.22(d,J=8.4Hz,1H),3.63(br s,1H),3.35(br t,J=11.2Hz,2H),3.10(brd,J=11.2Hz,4H),3.00(br d,J=11.2Hz,4H),2.46-2.24(m,2H),1.93-1.68(m,4H),1.65-1.42(m,2H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.56 (s, 1H), 8.83 (d, J=4.4 Hz, 1H), 8.76 (d, J=2.2 Hz, 1H), 8.22 (s, 1H) , 8.13-7.97 (m, 3H), 7.66 (dt, J=2.8, 8.7Hz, 1H), 7.50 (d, J=4.4Hz, 1H), 7.22 (d, J=8.4Hz, 1H), 3.63 ( br s, 1H), 3.35 (br t, J=11.2Hz, 2H), 3.10 (brd, J=11.2Hz, 4H), 3.00 (br d, J=11.2Hz, 4H), 2.46-2.24 (m, 2H), 1.93-1.68 (m, 4H), 1.65-1.42 (m, 2H).
实施例10Example 10
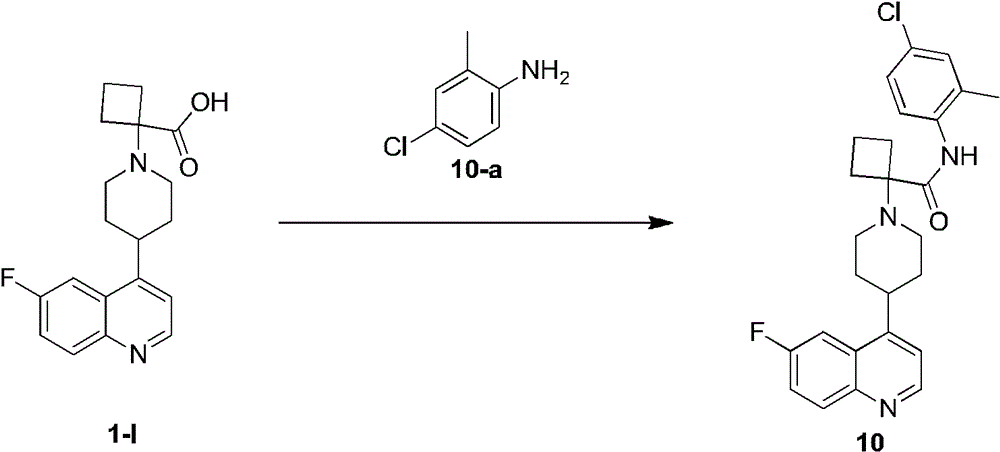
化合物10的合成Synthesis of
将化合物1-l(62.68mg)溶解于DMF(2mL)中,之后添加HATU(108.86mg)和DIEA(74.00mg)在氮气氛围.将混合液在25℃搅拌30分钟。之后添加10-a(32.43mg)。将混合液在25℃搅拌11.5小时。混合液保持淡黄色。将反应液稀释使用水(30mL)并萃取使用EA(50mL*2).将结合的有机相清洗使用饱和食盐水(30mL*4),用Na2SO4干燥,过滤,浓缩得到粗品。将粗品纯化使用制备色谱纯化得到化合物10。Compound 1-1 (62.68 mg) was dissolved in DMF (2 mL), followed by addition of HATU (108.86 mg) and DIEA (74.00 mg) under nitrogen atmosphere. The mixture was stirred at 25°C for 30 minutes. 10-a (32.43 mg) was then added. The mixture was stirred at 25°C for 11.5 hours. The mixture remained pale yellow. The reaction solution was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phase was washed with saturated brine (30 mL*4), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified using preparative chromatography to give
1H NMR(400MHz,DMSO-d6)δ=10.13(br s,1H),8.92(d,J=4.4Hz,1H),8.23(dd,J=2.8,10.8Hz,1H),8.15(dd,J=5.6,9.2Hz,1H),7.75(dt,J=2.8,8.8Hz,1H),7.50-7.41(m,3H),7.39-7.33(m,1H),3.86-3.31(m,4H),2.79-2.65(m,4H),2.44-2.31(m,1H),2.28(s,3H),2.18-2.08(m,4H),2.04-1.84(m,2H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=10.13 (br s, 1H), 8.92 (d, J=4.4 Hz, 1H), 8.23 (dd, J=2.8, 10.8 Hz, 1H), 8.15 (dd , J=5.6, 9.2Hz, 1H), 7.75 (dt, J=2.8, 8.8Hz, 1H), 7.50-7.41 (m, 3H), 7.39-7.33 (m, 1H), 3.86-3.31 (m, 4H) ), 2.79-2.65 (m, 4H), 2.44-2.31 (m, 1H), 2.28 (s, 3H), 2.18-2.08 (m, 4H), 2.04-1.84 (m, 2H).
实施例11Example 11

化合物11的合成Synthesis of Compound 11
将化合物1-l(54mg)溶解于DMF(1mL)中,之后添加HATU(93.79mg)和DIEA(63.76mg,)在氮气氛围。将混合液在25℃搅拌30分钟。之后添加11-a(28.72mg)。将混合液在25℃搅拌11.5小时。将反应液稀释使用水(30mL)并萃取使用EA(50mL*2).将结合的有机相清洗使用饱和食盐水(30mL*4),用Na2SO4干燥,过滤,浓缩得到粗品。将粗品纯化使用制备色谱纯化得到化合物11。Compound 1-1 (54 mg) was dissolved in DMF (1 mL), after which HATU (93.79 mg) and DIEA (63.76 mg, ) were added under nitrogen atmosphere. The mixture was stirred at 25°C for 30 minutes. Then 11-a (28.72 mg) was added. The mixture was stirred at 25°C for 11.5 hours. The reaction solution was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phase was washed with saturated brine (30 mL*4), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified using preparative chromatography to give compound 11.
1H NMR(400MHz,DMSO-d6)δ=10.41(br s,1H),8.94(d,J=4.4Hz,1H),8.24-8.12(m,2H),8.07(dd,J=2.4,6.8Hz,1H),7.79-7.70(m,2H),7.54-7.46(m,2H),3.81(br s,1H),3.62(br d,J=11.2Hz,2H),3.28(br s,2H),2.84-2.61(m,4H),2.18-2.03(m,4H),2.00-1.77(m,2H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=10.41 (br s, 1H), 8.94 (d, J=4.4Hz, 1H), 8.24-8.12 (m, 2H), 8.07 (dd, J=2.4, 6.8Hz, 1H), 7.79-7.70 (m, 2H), 7.54-7.46 (m, 2H), 3.81 (br s, 1H), 3.62 (br d, J=11.2Hz, 2H), 3.28 (br s, 2H), 2.84-2.61 (m, 4H), 2.18-2.03 (m, 4H), 2.00-1.77 (m, 2H).
实施例12Example 12

化合物12的合成Synthesis of Compound 12
将化合物1-l(62.68mg)溶解于DMF(1mL)中,之后添加HATU(108.86mg)和DIEA(74.00mg)在氮气氛围。将混合液在25℃搅拌30分钟。之后化合物12-a(24.77mg)加入反应液中。将混合液在25℃搅拌11.5小时。将混合液稀释使用水(30mL)并萃取使用EA(50mL*2)。将结合的有机相清洗使用饱和食盐水(30mL*4),干燥使用Na2SO4,过滤并在真空中浓缩得粗品。将获得的粗品通过制备色谱纯化得到化合物12。Compound 1-1 (62.68 mg) was dissolved in DMF (1 mL), after which HATU (108.86 mg) and DIEA (74.00 mg) were added under nitrogen atmosphere. The mixture was stirred at 25°C for 30 minutes. Then compound 12-a (24.77 mg) was added to the reaction solution. The mixture was stirred at 25°C for 11.5 hours. The mixture was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phases were washed with saturated brine (30 mL*4), dried with Na2SO4, filtered and concentrated in vacuo to give crude product. The obtained crude product was purified by preparative chromatography to give compound 12.
1H NMR(400MHz,DMSO-d6)δ=9.69(s,1H),8.83(d,J=4.8Hz,1H),8.30(d,J=5.6Hz,1H),8.08(dd,J=6.0,9.2Hz,1H),8.03(dd,J=2.8,11.2Hz,1H),7.69-7.61(m,2H),7.59(dd,J=1.6,5.6Hz,1H),7.48(d,J=4.4Hz,1H),3.43-3.21(m,1H),2.98(br d,J=11.2Hz,2H),2.42(s,3H),2.41-2.35(m,2H),2.32(br d,J=8.4Hz,4H),1.91-1.68(m,6H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.69 (s, 1H), 8.83 (d, J=4.8 Hz, 1H), 8.30 (d, J=5.6 Hz, 1H), 8.08 (dd, J= 6.0, 9.2Hz, 1H), 8.03 (dd, J=2.8, 11.2Hz, 1H), 7.69-7.61 (m, 2H), 7.59 (dd, J=1.6, 5.6Hz, 1H), 7.48 (d, J =4.4Hz, 1H), 3.43-3.21(m, 1H), 2.98(br d, J=11.2Hz, 2H), 2.42(s, 3H), 2.41-2.35(m, 2H), 2.32(br d, J=8.4Hz, 4H), 1.91-1.68 (m, 6H).
实施例13Example 13

化合物13-b的合成:Synthesis of compound 13-b:
向二氧六环(8mL)和水(2mL)中依次加入反应物1-h(784.29mg,2.34mmol),反应物13-a(500mg,2.13mmol),钯催化剂(77.81mg,106.34μmol),三乙基胺(645.62mg,6.38mmol,888.07uL),反应液在氮气保护下加热到90℃搅拌12小时,将反应液过滤,滤液加入水稀释,用乙酸乙酯萃取三次(50mL*3),有机相合并用饱和食盐水洗涤三次(50mL*3),用无水硫酸钠干燥,过滤,将滤液浓缩得到粗产品,柱层析纯化得到化合物13-b。To dioxane (8 mL) and water (2 mL) were sequentially added reactant 1-h (784.29 mg, 2.34 mmol), reactant 13-a (500 mg, 2.13 mmol), palladium catalyst (77.81 mg, 106.34 μmol) , triethylamine (645.62mg, 6.38mmol, 888.07uL), the reaction solution was heated to 90°C and stirred for 12 hours under nitrogen protection, the reaction solution was filtered, the filtrate was diluted with water, and extracted three times with ethyl acetate (50mL*3 ), the organic phases were combined and washed three times with saturated brine (50 mL*3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated to obtain a crude product, which was purified by column chromatography to obtain compound 13-b.
化合物13-c的合成:Synthesis of compound 13-c:
向乙酸乙酯(10mL)中依次加入反应物13-b(750mg,2.06mmol),湿钯碳(70mg,10%纯度),所得混合物用氢气置换三次。在氢气氛围下,常压下加热到50℃,搅拌2小时,将反应液过滤,滤液浓缩得到化合物13-c。To ethyl acetate (10 mL) were added reactant 13-b (750 mg, 2.06 mmol), wet palladium on carbon (70 mg, 10% purity), and the resulting mixture was replaced with hydrogen three times. Under a hydrogen atmosphere, the mixture was heated to 50° C. under normal pressure, stirred for 2 hours, the reaction solution was filtered, and the filtrate was concentrated to obtain compound 13-c.
化合物13-d的合成:Synthesis of compound 13-d:
向乙醇(8mL)和水(2mL)中依次加入反应物13-c(700mg,1.92mmol),氢氧化钠(229.81mg,5.75mmol),加热到70℃搅拌2小时,将反应液浓缩,加入水稀释,用乙酸乙酯萃取三次(30mL*3),水相加入稀盐酸(1M)调节pH至3,用乙酸乙酯萃取三次(30mL*3),所得有机相合并用饱和食盐水洗涤三次(50mL*3),用无水硫酸钠干燥,过滤,将滤液浓缩得到化合物13-d。To ethanol (8 mL) and water (2 mL) were sequentially added reactant 13-c (700 mg, 1.92 mmol), sodium hydroxide (229.81 mg, 5.75 mmol), heated to 70 ° C and stirred for 2 hours, the reaction solution was concentrated, added Diluted with water, extracted three times with ethyl acetate (30mL*3), added dilute hydrochloric acid (1M) to the aqueous phase to adjust the pH to 3, extracted three times with ethyl acetate (30mL*3), the obtained organic phases were combined and washed three times with saturated brine (50 mL*3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated to obtain compound 13-d.
化合物13的合成:Synthesis of compound 13:
向DMF(10mL)中依次加入反应物13-d(500mg,1.48mmol),反应物1-n(226.84mg,1.78mmol),缩合剂HATU(845.12mg,2.22mmol),N,N-二异丙基乙基胺(574.51mg,4.45mmol,774.28uL),反应液在室温下搅拌12小时,将反应液加入乙酸乙酯(30mL)稀释,用饱和氯化铵溶液洗涤三次(30mL*3),再用饱和食盐水洗涤三次(30mL*3),有机相用无水硫酸钠干燥,过滤,将滤液浓缩得到粗产品,反相色谱纯化得到化合物13。To DMF (10 mL) were sequentially added reactant 13-d (500 mg, 1.48 mmol), reactant 1-n (226.84 mg, 1.78 mmol), condensing agent HATU (845.12 mg, 2.22 mmol), N,N-diiso Propylethylamine (574.51mg, 4.45mmol, 774.28uL), the reaction solution was stirred at room temperature for 12 hours, the reaction solution was diluted with ethyl acetate (30mL), washed three times with saturated ammonium chloride solution (30mL*3) , and then washed three times with saturated brine (30 mL*3), the organic phase was dried with anhydrous sodium sulfate, filtered, the filtrate was concentrated to obtain the crude product, and the
1H NMR(400MHz,MeOD-d4)δppm 1.85-2.02(m,6H)2.40-2.55(m,6H)2.73-2.82(m,1H)3.12(s,3H)3.16(br d,J=11.49Hz,2H)7.36(d,J=7.78Hz,2H)7.57(d,J=8.31Hz,2H)7.65(d,J=8.46Hz,2H)7.91(d,J=8.44Hz,2H)8.13(s,1H)。 1 H NMR (400 MHz, MeOD-d 4 ) δppm 1.85-2.02 (m, 6H) 2.40-2.55 (m, 6H) 2.73-2.82 (m, 1H) 3.12 (s, 3H) 3.16 (br d, J=11.49 Hz, 2H) 7.36(d, J=7.78Hz, 2H) 7.57(d, J=8.31Hz, 2H) 7.65(d, J=8.46Hz, 2H) 7.91(d, J=8.44Hz, 2H) 8.13( s, 1H).
实施例14Example 14

化合物14-a的合成Synthesis of compound 14-a
向化合物1-l(1g)的DCM(30mL)溶液中加入CDI(740.68mg),在25℃下反应12小时后,加入100mLDCM稀释,然后用饱和食盐水洗(100mL*3),Na2SO4干燥,过滤浓缩后得到化合物14-a。CDI (740.68 mg) was added to a solution of compound 1-1 (1 g) in DCM (30 mL), and after reacting at 25°C for 12 hours, 100 mL of DCM was added to dilute, and then washed with saturated brine (100 mL*3), Na 2 SO 4 After drying, filtration and concentration, compound 14-a was obtained.
化合物14的合成Synthesis of compound 14
将化合物14-b(20.47mg)溶解于THF(5mL)中,然后在0℃下逐滴加入NaHMDS(1M,158.54uL),氮气置换三次,然后让其在0℃下反应半小时。然后在0℃下加入化合物14-a(50mg)。所得混合物在0℃下搅拌1小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物14。Compound 14-b (20.47 mg) was dissolved in THF (5 mL), then NaHMDS (1 M, 158.54 uL) was added dropwise at 0 °C, nitrogen purged three times, and then allowed to react at 0 °C for half an hour. Compound 14-a (50 mg) was then added at 0°C. The resulting mixture was stirred at 0 °C for 1 hour. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain a crude product, which was prepared by Chromatographic purification afforded compound 14.
1H NMR(400MHz,DMSO-d6)δ=9.80(s,1H),8.83(d,J=4.4Hz,1H),8.45(s,1H),8.09(t,J=7.0Hz,2H),8.04(d,J=10.4Hz,4H),7.69-7.55(m,1H),2.98(br d,J=11.0Hz,2H),2.81-2.60(m,1H),2.57-2.53(m,6H),2.44-2.37(m,4H),2.37-2.22(m,2H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.80 (s, 1H), 8.83 (d, J=4.4 Hz, 1H), 8.45 (s, 1H), 8.09 (t, J=7.0 Hz, 2H) , 8.04(d, J=10.4Hz, 4H), 7.69-7.55(m, 1H), 2.98(br d, J=11.0Hz, 2H), 2.81-2.60(m, 1H), 2.57-2.53(m, 6H), 2.44-2.37 (m, 4H), 2.37-2.22 (m, 2H).
实施例15Example 15

化合物15的合成Synthesis of
将化合物15-a(30.84mg)溶解于THF(2mL)中,之后逐滴添加NaHMDS(1M,190.25uL)在-75℃在10mins内在氮气氛围。将混合液在-75℃搅拌20min。之后添加14-a(60mg,158.54μmol)。将混合液在25℃搅拌9.5小时。将反应液稀释使用水(30mL)并萃取使用EA(50mL*2)。将结合的有机相清洗使用饱和食盐水(30mL*4),用Na2SO4干燥,过滤,浓缩得到粗品。将粗品使用制备色谱纯纯化得到化合物15。Compound 15-a (30.84 mg) was dissolved in THF (2 mL), followed by the dropwise addition of NaHMDS (1 M, 190.25 uL) at -75°C for 10 mins under nitrogen atmosphere. The mixture was stirred at -75 °C for 20 min. Then 14-a (60 mg, 158.54 μmol) was added. The mixture was stirred at 25°C for 9.5 hours. The reaction solution was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phase was washed with saturated brine (30 mL*4), dried over Na 2 SO 4 , filtered and concentrated to obtain crude product. The crude product was purified using preparative chromatography to give
1H NMR(400MHz,DMSO-d6)δ=10.05(s,1H),9.10(d,J=2.4Hz,1H),8.82(d,J=4.4Hz,1H),8.48(dd,J=2.0,8.4Hz,1H),8.08(dd,J=6.0,9.2Hz,1H),8.02(dd,J=2.8,11.2Hz,1H),7.89(d,J=8.8Hz,1H),7.65(dt,J=2.8,8.8Hz,1H),7.48(d,J=4.4Hz,1H),3.01(br d,J=11.2Hz,2H),2.67(s,1H),2.47-2.29(m,6H),1.92-1.67(m,6H)。 1 H NMR (400MHz, DMSO-d 6 ) δ=10.05 (s, 1H), 9.10 (d, J=2.4Hz, 1H), 8.82 (d, J=4.4Hz, 1H), 8.48 (dd, J= 2.0, 8.4Hz, 1H), 8.08 (dd, J=6.0, 9.2Hz, 1H), 8.02 (dd, J=2.8, 11.2Hz, 1H), 7.89 (d, J=8.8Hz, 1H), 7.65 ( dt, J=2.8, 8.8Hz, 1H), 7.48 (d, J=4.4Hz, 1H), 3.01 (br d, J=11.2Hz, 2H), 2.67 (s, 1H), 2.47-2.29 (m, 6H), 1.92-1.67 (m, 6H).
实施例16Example 16

化合物16的合成Synthesis of compound 16
将化合物16-a(30.82mg)溶解于THF(2mL)中,之后逐滴添加NaHMDS(1M,190.25uL)在0℃在10分钟内在氮气氛围。将混合液在25℃搅拌20分钟。之后添加14-a(60mg)。将混合液在25℃搅拌2小时。将反应液稀释使用水(30mL)并萃取使用EA(50mL*2)。将结合的有机相使用饱和食盐水(30mL*4)洗涤,Na2SO4干燥,过滤,浓缩得到粗品。将粗品纯化使用制备色谱纯化得到化合物16。Compound 16-a (30.82 mg) was dissolved in THF (2 mL) followed by the dropwise addition of NaHMDS (1 M, 190.25 uL) at 0°C for 10 min under nitrogen atmosphere. The mixture was stirred at 25°C for 20 minutes. Then 14-a (60 mg) was added. The mixture was stirred at 25°C for 2 hours. The reaction solution was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phases were washed with saturated brine (30 mL*4), dried over Na 2 SO 4 , filtered, and concentrated to obtain crude product. The crude product was purified using preparative chromatography to give compound 16.
1H NMR(400MHz,DMSO-d6)δ=9.72(s,1H),8.83(d,J=4.4Hz,1H),8.31(s,1H),8.15(d,J=2.4Hz,1H),8.08(dd,J=5.6,9.2Hz,1H),8.03(dd,J=2.8,11.2Hz,1H),7.76(dd,J=2.4,8.8Hz,1H),7.65(dt,J=2.8,8.8Hz,1H),7.58(d,J=8.8Hz,1H),7.48(d,J=4.8Hz,1H),2.98(br d,J=11.2Hz,2H),2.67(br d,J=1.6Hz,1H),2.41(br t,J=10.4Hz,2H),2.35-2.25(m,4H),1.93-1.79(m,4H),1.79-1.67(m,2H)。 1 H NMR (400MHz, DMSO-d6) δ=9.72(s, 1H), 8.83(d, J=4.4Hz, 1H), 8.31(s, 1H), 8.15(d, J=2.4Hz, 1H), 8.08 (dd, J=5.6, 9.2Hz, 1H), 8.03 (dd, J=2.8, 11.2Hz, 1H), 7.76 (dd, J=2.4, 8.8Hz, 1H), 7.65 (dt, J=2.8, 8.8Hz, 1H), 7.58 (d, J=8.8Hz, 1H), 7.48 (d, J=4.8Hz, 1H), 2.98 (br d, J=11.2Hz, 2H), 2.67 (br d, J= 1.6Hz, 1H), 2.41 (br t, J=10.4Hz, 2H), 2.35-2.25 (m, 4H), 1.93-1.79 (m, 4H), 1.79-1.67 (m, 2H).
实施例17Example 17

化合物17的合成Synthesis of
将化合物17-a(20.23mg)溶解于THF(5mL)中,然后在0℃下逐滴加入NaHMDS(1M,158.54uL).氮气置换三次,然后让其在0℃下反应半小时,然后在0℃下加入化合物14-a(50mg)。所得混合物在0℃下搅拌1小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物17。Compound 17-a (20.23 mg) was dissolved in THF (5 mL), then NaHMDS (1 M, 158.54 uL) was added dropwise at 0 °C. Nitrogen was replaced three times, and then it was allowed to react at 0 °C for half an hour, and then at 0 °C. Compound 14-a (50 mg) was added at 0°C. The resulting mixture was stirred at 0 °C for 1 hour. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain a crude product, which was prepared by Chromatographic purification afforded
1H NMR(400MHz,DMSO-d6)δ=9.94(s,1H),8.85(d,J=4.4Hz,1H),8.26(dd,J=1.4,8.2Hz,1H),8.11(t,J=7.0Hz,2H),8.05(d,J=10.4Hz,1H),7.68(dt,J=2.8,8.7Hz,1H),7.53(d,J=7.6Hz,1H),7.47(d,J=4.8Hz,1H),7.37(t,J=7.8Hz,1H),2.96(br d,J=11.4Hz,2H),2.69-2.53(m,1H),2.48-2.39(m,4H),2.39-2.31(m,2H),2.30-2.18(m,2H),2.01-1.72(m,4H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.94 (s, 1H), 8.85 (d, J=4.4 Hz, 1H), 8.26 (dd, J=1.4, 8.2 Hz, 1H), 8.11 (t, J=7.0Hz, 2H), 8.05 (d, J=10.4Hz, 1H), 7.68 (dt, J=2.8, 8.7Hz, 1H), 7.53 (d, J=7.6Hz, 1H), 7.47 (d, J=4.8Hz, 1H), 7.37 (t, J=7.8Hz, 1H), 2.96 (br d, J=11.4Hz, 2H), 2.69-2.53 (m, 1H), 2.48-2.39 (m, 4H) , 2.39-2.31 (m, 2H), 2.30-2.18 (m, 2H), 2.01-1.72 (m, 4H).
实施例18Example 18

化合物18的合成Synthesis of compound 18
将化合物18-a(25.55mg)溶解于THF(5mL)中,然后在-78℃下逐滴加入NaHMDS(1M,158.54μL).氮气置换三次,然后让其在-78℃下反应半小时。然后在-78℃下加入化合物14-a(50mg)。所得混合物在25℃下搅拌11.5小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物18。Compound 18-a (25.55 mg) was dissolved in THF (5 mL), then NaHMDS (1 M, 158.54 μL) was added dropwise at -78 °C. Nitrogen was replaced three times, and then it was allowed to react at -78 °C for half an hour. Compound 14-a (50 mg) was then added at -78°C. The resulting mixture was stirred at 25°C for 11.5 hours. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product, which was prepared by Chromatographic purification afforded compound 18.
1H NMR(400MHz,DMSO-d6)δ=9.81(s,1H),8.83(d,J=4.5Hz,1H),8.12-7.93(m,4H),7.72-7.63(m,3H),7.49(d,J=4.5Hz,1H),3.62(br s,1H),3.35(br t,J=11.2Hz,2H),3.01(br d,J=11.1Hz,2H),2.47-2.25(m,4H),1.93-1.70(m,4H),1.68-1.47(m,2H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.81 (s, 1H), 8.83 (d, J=4.5 Hz, 1H), 8.12-7.93 (m, 4H), 7.72-7.63 (m, 3H), 7.49(d, J=4.5Hz, 1H), 3.62(br s, 1H), 3.35(br t, J=11.2Hz, 2H), 3.01(br d, J=11.1Hz, 2H), 2.47-2.25( m, 4H), 1.93-1.70 (m, 4H), 1.68-1.47 (m, 2H).
实施例19Example 19

化合物19的合成Synthesis of
将化合物19-a(18.73mg)溶解于THF(5mL)中,然后在-78℃下逐滴加入NaHMDS(1M,158.54μL)。氮气置换三次,然后让其在-78℃下反应半小时。然后在-78℃下加入化合物14-a(50mg)。所得混合物在25℃下搅拌11.5小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗涤(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物19。Compound 19-a (18.73 mg) was dissolved in THF (5 mL), then NaHMDS (1 M, 158.54 μL) was added dropwise at -78°C. The nitrogen was replaced three times and then allowed to react at -78°C for half an hour. Then compound 14-a (50 mg) was added at -78°C. The resulting mixture was stirred at 25°C for 11.5 hours. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product, which was passed through Purification by preparative chromatography gave
1H NMR(400MHz,DMSO-d6)δ=9.79(s,1H),8.84(d,J=4.4Hz,1H),8.28(s,2H),8.25(br s,3H),8.12-8.01(m,1H),7.66(dt,J=2.8,8.7Hz,3H),3.48-3.21(m,1H),3.00(br d,J=11.4Hz,2H),2.83-2.62(m,2H),2.57-2.53(m,4H),2.47-2.23(m,4H),1.93-1.69(m,2H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.79 (s, 1H), 8.84 (d, J=4.4 Hz, 1H), 8.28 (s, 2H), 8.25 (br s, 3H), 8.12-8.01 (m, 1H), 7.66 (dt, J=2.8, 8.7Hz, 3H), 3.48-3.21 (m, 1H), 3.00 (br d, J=11.4Hz, 2H), 2.83-2.62 (m, 2H) , 2.57-2.53(m, 4H), 2.47-2.23(m, 4H), 1.93-1.69(m, 2H).
实施例20Example 20

化合物20的合成Synthesis of
将化合物20-a(20.29mg)溶解在THF(2mL)中,将NaHMDS(1M,158.54μL)在0℃滴加,混合物在0℃搅拌1小时,再将14-a(50mg)溶于THF(1mL)加入到该混合物中,反应在0℃N2保护下反应1小时。将水(15mL)加入至反应液,随后用EA 45mL(15mL*3)萃取,合并有机相用饱和食盐水洗40mL(20mL*2),Na2SO4干燥,过滤浓缩后得到粗品。粗品通过制备色谱纯化得到产物20。Compound 20-a (20.29 mg) was dissolved in THF (2 mL), NaHMDS (1 M, 158.54 μL) was added dropwise at 0 °C, the mixture was stirred at 0 °C for 1 hour, and 14-a (50 mg) was dissolved in THF (1 mL) was added to the mixture, and the reaction was allowed to react under N2 protection at 0°C for 1 hour. Water (15 mL) was added to the reaction solution, followed by extraction with EA 45 mL (15 mL*3), the combined organic phases were washed with saturated brine 40 mL (20 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified by preparative chromatography to give
1H NMR(400MHz,DMSO-d6)δppm 1.72-1.91(m,6H)2.29-2.35(m,4H)2.43(br d,J=11.00Hz,2H)2.99(br d,J=10.88Hz,2H)3.35(br t,J=11.25Hz,1H)7.48(d,J=4.52Hz,1H)7.65(td,J=8.71,2.63Hz,1H)7.79(d,J=8.68Hz,2H)7.96-8.11(m,4H)8.82(d,J=4.52Hz,1H)9.87(s,1H) 1 H NMR (400MHz, DMSO-d 6 ) δppm 1.72-1.91 (m, 6H) 2.29-2.35 (m, 4H) 2.43 (br d, J=11.00Hz, 2H) 2.99 (br d, J=10.88Hz, 2H) 3.35(br t, J=11.25Hz, 1H) 7.48(d, J=4.52Hz, 1H) 7.65(td, J=8.71, 2.63Hz, 1H) 7.79(d, J=8.68Hz, 2H) 7.96 -8.11(m, 4H) 8.82(d, J=4.52Hz, 1H) 9.87(s, 1H)
实施例21Example 21
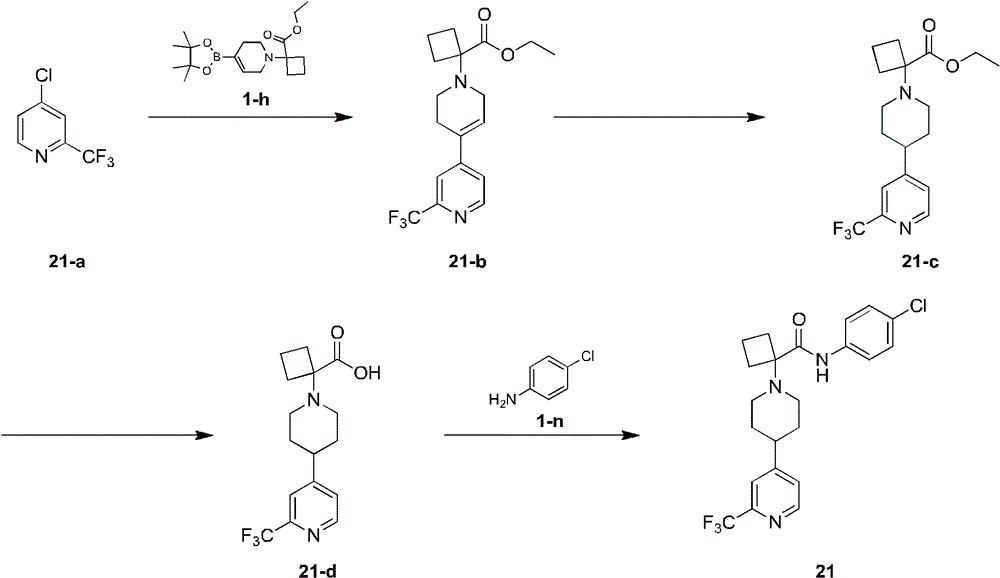
化合物21-b的合成Synthesis of compound 21-b
将混合物21-a(500mg)和1-h(1.02g)溶解在二氧六环(12mL)和水(3mL)中,然后加入TEA(836.08mg),之后Pd(dppf)Cl2(201.52mg)在N2保护下加入至该混合物中,所得混合物用N2置换数次,该反应在85℃下搅拌12小时得黑色溶液,将反应液过滤后,加入水50mL,随后用EA(50mL*3)萃取,合并有机相用饱和食盐水洗50mL(25mL*2),Na2SO4干燥,过滤浓缩后得到粗品。粗品通过柱层析纯化得到化合物21-b。The mixture 21-a (500 mg) and 1-h (1.02 g) were dissolved in dioxane (12 mL) and water (3 mL), then TEA (836.08 mg) was added followed by Pd(dppf)Cl2 (201.52 mg) It was added to the mixture under the protection of N, and the resulting mixture was replaced with N several times. The reaction was stirred at 85 °C for 12 hours to obtain a black solution. After filtering the reaction solution, 50 mL of water was added, followed by EA (50 mL*3 ) extraction, the combined organic phases were washed with saturated brine with 50 mL (25 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified by column chromatography to obtain compound 21-b.
化合物21-c的合成Synthesis of compound 21-c
将化合物21-b(590mg)溶解于EtOAc(10mL)中,然后在N2保护下加入Pd/C(60mg,10%纯度),所得混合物不断通入H2并置换数次,反应在25℃、H2(15psi)下搅拌12小时得黑色悬浊液。将反应液过滤浓缩后得21-c。Compound 21-b (590 mg) was dissolved in EtOAc (10 mL), then Pd/C (60 mg, 10% purity) was added under N2 protection, the resulting mixture was continuously passed into H2 and replaced several times, the reaction was carried out at 25 °C, Stir under H2 (15 psi) for 12 hours to give a black suspension. The reaction solution was filtered and concentrated to obtain 21-c.
化合物21-d的合成Synthesis of compound 21-d
将化合物21-c(510mg)溶于MeOH(8mL)中,并加入NaOH(228.95mg)的水(2mL)溶液,所得混合物在75℃下搅拌12小时得黄色溶液。将反应液浓缩,然后加入水6mL,该溶液用3MHCl调节pH至7左右,将溶液在0℃搅拌30min,大量白色沉淀形成,过滤、然后将滤饼浓缩得21-d。Compound 21-c (510 mg) was dissolved in MeOH (8 mL), and a solution of NaOH (228.95 mg) in water (2 mL) was added, and the resulting mixture was stirred at 75° C. for 12 hours to obtain a yellow solution. The reaction solution was concentrated, then 6 mL of water was added, the pH of the solution was adjusted to about 7 with 3M HCl, the solution was stirred at 0°C for 30 min, a large amount of white precipitate formed, filtered, and then the filter cake was concentrated to obtain 21-d.
化合物21的合成Synthesis of
将化合物21-d(160mg)溶解在DMF(4mL)中,然后再加入DIEA(188.95mg)和HATU(240.88mg),将该混合物在25℃下搅拌1小时,再将1-n(80.82mg)加入到混合物中,该反应在50℃ N2保护下反应11小时,反应液为黄色溶液。然后将水40mL加入至反应液,随后用EA60mL(20mL*3)萃取,合并有机相用饱和食盐水洗40mL(20mL*2),Na2SO4干燥,过滤浓缩后得到粗品。粗品通过制备色谱纯化得到化合物21。Compound 21-d (160 mg) was dissolved in DMF (4 mL), then DIEA (188.95 mg) and HATU (240.88 mg) were added, the mixture was stirred at 25°C for 1 hour, and 1-n (80.82 mg) was added. ) was added to the mixture, the reaction was carried out under the protection of N2 at 50° C. for 11 hours, and the reaction solution was a yellow solution. Then 40 mL of water was added to the reaction solution, followed by extraction with 60 mL of EA (20 mL*3), the combined organic phases were washed with 40 mL of saturated brine (20 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified by preparative chromatography to give
1H NMR(400MHz,DMSO-d6)δppm 1.83-2.01(m,1H)1.83-2.01(m,2H)2.16(br s,4H)2.77(br s,4H)2.99-3.25(m,3H)3.61(br s,2H)7.53(br d,J=7.6Hz,2H)7.65(br s,1H)7.77-7.86(m,3H)8.79(br d,J=3.6Hz,1H)10.36(br s,1H) 1 H NMR (400 MHz, DMSO-d 6 ) δppm 1.83-2.01 (m, 1H) 1.83-2.01 (m, 2H) 2.16 (br s, 4H) 2.77 (br s, 4H) 2.99-3.25 (m, 3H) 3.61(br s, 2H) 7.53(br d, J=7.6Hz, 2H) 7.65(br s, 1H) 7.77-7.86(m, 3H) 8.79(br d, J=3.6Hz, 1H) 10.36(br s , 1H)
实施例22Example 22

化合物22-b的合成Synthesis of compound 22-b
将混合物22-a(500mg)和1-h(980.66mg)溶解在二氧六环(12mL)和水(3mL)中,然后加入TEA(807.27mg),之后Pd(dppf)Cl2(194.58mg)在N2保护下加入至该混合物中,所得混合物用N2置换数次,该反应在85℃下搅拌12小时得棕色溶液,将反应液过滤后,加入水50mL,随后用EA 150(50mL*3)萃取,合并有机相用饱和食盐水洗50mL(25mL*2),Na2SO4干燥,过滤浓缩后得到粗品。粗品通过柱层析纯化,得到化合物22-b。The mixture 22-a (500 mg) and 1-h (980.66 mg) were dissolved in dioxane (12 mL) and water (3 mL), then TEA (807.27 mg) was added followed by Pd(dppf)Cl2 (194.58 mg) It was added to the mixture under the protection of N2 , the resulting mixture was replaced with N2 several times, the reaction was stirred at 85 °C for 12 hours to obtain a brown solution, after filtering the reaction solution, 50 mL of water was added, followed by EA 150 (50 mL* 3) Extraction, the combined organic phases were washed with saturated brine with 50 mL (25 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified by column chromatography to give compound 22-b.
化合物22-c的合成Synthesis of compound 22-c
将化合物22-b(500mg)溶解于EtOAc(10mL)中,然后在N2保护下加入Pd/C(50mg,10%purity),所得混合物不断通入H2并置换数次,反应在50℃、H2(15psi)下搅拌3小时得黑色悬浊液。将反应液过滤浓缩后得22-c。Compound 22-b (500 mg) was dissolved in EtOAc (10 mL), then Pd/C (50 mg, 10% purity) was added under the protection of N2, the resulting mixture was continuously passed into H2 and replaced several times, the reaction was carried out at 50 °C under H2 Stir at 2 (15 psi) for 3 hours to obtain a black suspension. The reaction solution was filtered and concentrated to obtain 22-c.
化合物22-d的合成Synthesis of compound 22-d
将化合物22-c(520mg)(粗品)溶于MeOH(10mL)中,并加入NaOH(228.62mg)的水(2mL)溶液,所得混合物在70℃下搅拌12小时得黄色溶液。反应液浓缩,然后加入水15mL,该溶液用2M HCl调节pH至6左右,然后将混合液浓缩得粗品。将MeOH 15mL加入至粗品中,搅拌30min后过滤,将滤液浓缩得22-d。Compound 22-c (520 mg) (crude) was dissolved in MeOH (10 mL), and a solution of NaOH (228.62 mg) in water (2 mL) was added, and the resulting mixture was stirred at 70° C. for 12 hours to obtain a yellow solution. The reaction solution was concentrated, then 15 mL of water was added, the pH of the solution was adjusted to about 6 with 2M HCl, and the mixture was concentrated to obtain a crude product. 15 mL of MeOH was added to the crude product, stirred for 30 min, filtered, and the filtrate was concentrated to obtain 22-d.
化合物22的合成Synthesis of Compound 22
将化合物22-d(200mg)溶解在DMF(5mL)中,然后再加入DIEA(267.07mg)和HATU(340.48mg),再将1-n(114.23mg)加入到混合物中,该反应在25℃N2保护下反应12小时,反应液为黄色溶液。将水40mL加入至反应液,随后用EA 60(20mL*3)萃取,合并有机相用饱和食盐水洗40mL(20mL*2),Na2SO4干燥,过滤浓缩后得到粗品。将粗品通过制备色谱纯化,得化合物22。Compound 22-d (200 mg) was dissolved in DMF (5 mL), then DIEA (267.07 mg) and HATU (340.48 mg) were added, and 1-n (114.23 mg) was added to the mixture, and the reaction was carried out at 25°C The reaction was carried out under the protection of N 2 for 12 hours, and the reaction solution was a yellow solution. 40 mL of water was added to the reaction solution, followed by extraction with EA 60 (20 mL*3), the combined organic phases were washed with saturated brine 40 mL (20 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The crude product was purified by preparative chromatography to give compound 22.
1H NMR(400MHz,DMSO-d6)δppm 1.67-1.83(m,6H)2.13-2.35(m,7H)2.94(br d,J=11.2Hz,2H)3.82(s,3H)6.67(s,1H)6.90(d,J=5.2Hz,1H)7.37(d,J=8.80Hz,2H)7.73-7.78(m,2H)8.06(d,J=5.26Hz,1H)9.54(s,1H)。 1 H NMR (400MHz, DMSO-d 6 ) δppm 1.67-1.83(m, 6H) 2.13-2.35(m, 7H) 2.94(br d, J=11.2Hz, 2H) 3.82(s, 3H) 6.67(s, 1H) 6.90 (d, J=5.2 Hz, 1H) 7.37 (d, J=8.80 Hz, 2H) 7.73-7.78 (m, 2H) 8.06 (d, J=5.26 Hz, 1H) 9.54 (s, 1H).
实施例23Example 23

化合物23-b的合成Synthesis of compound 23-b
将化合物23-a(3.2g)溶解于EA(30mL)中,之后添加Me3OBF4(4.52g)在氮气氛围。将混合液在25℃搅拌6小时。将所得悬浊液过滤并将滤饼真空干燥得粗品23-b。Compound 23-a (3.2 g) was dissolved in EA (30 mL), after which Me3OBF4 ( 4.52 g) was added under nitrogen atmosphere. The mixture was stirred at 25°C for 6 hours. The obtained suspension was filtered and the filter cake was vacuum dried to obtain crude product 23-b.
合物23-c的合成Synthesis of Compound 23-c
将化合物23-b(1.63g,10.86mmol)溶解于DCM(17mL)中,之后添加吡啶(1.29g)和二(三氟乙酸)碘苯(5.60g)在氮气氛围。将所得混合液在30℃搅拌30分钟,之后添加I2(3.31g)。将所得混合液在30℃搅拌15.5小时。将所得悬浊液过滤并将滤饼真空干燥得粗品23-c。Compound 23-b (1.63 g, 10.86 mmol) was dissolved in DCM (17 mL), followed by addition of pyridine (1.29 g) and bis(trifluoroacetic acid)iodobenzene (5.60 g) under nitrogen atmosphere. The resulting mixture was stirred at 30°C for 30 minutes, after which I2 (3.31 g) was added. The resulting mixture was stirred at 30°C for 15.5 hours. The obtained suspension was filtered and the filter cake was vacuum dried to obtain crude product 23-c.
化合物23-d的合成Synthesis of compound 23-d
将化合物1-h(2.40g,7.17mmol,1eq)和23-c(1.98g,7.17mmol,1eq)溶解于二氧六环(20mL)和水(5mL)中,之后添加Pd(dppf)Cl2(262.41mg,358.63μmol,0.05eq)和Et3N(2.18g,21.52mmol,3.00mL,3eq)在氮气氛围。将混合液使用氮气置换三次。将混合液在100℃搅拌8小时。将混合液过滤并清洗滤饼使用EA(80mL),之后添加水(40mL),分液。将水相萃取使用EA(70mL*2)。并将合并的有机相清洗使用饱和食盐水(50mL*2),Na2SO4干燥,过滤浓缩后得到粗品。将所得粗品使用柱层析纯化得到化合物23-d。Compounds 1-h (2.40 g, 7.17 mmol, 1 eq) and 23-c (1.98 g, 7.17 mmol, 1 eq) were dissolved in dioxane (20 mL) and water (5 mL), followed by the addition of Pd(dppf)Cl 2 (262.41 mg, 358.63 μmol, 0.05 eq) and Et3N (2.18 g, 21.52 mmol, 3.00 mL, 3 eq) under nitrogen. The mixture was replaced three times with nitrogen. The mixture was stirred at 100°C for 8 hours. The mixed solution was filtered, and the filter cake was washed with EA (80 mL), and then water (40 mL) was added for liquid separation. The aqueous phase was extracted using EA (70 mL*2). The combined organic phase was washed with saturated brine (50 mL*2), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product. The obtained crude product was purified using column chromatography to obtain compound 23-d.
化合物23-e的合成Synthesis of Compound 23-e
将化合物23-d(800mg,2.24mmol)溶解于MeOH(10mL)中,之后添加Pd/C(80mg,10%纯度)在H2氛围(35psi)。将混合液置换气体三次使用H2。将混合液在H2(15psi)氛围在50℃搅拌20小时。将混合液过滤并将所得溶液浓缩得到化合物23-e。Compound 23-d (800 mg, 2.24 mmol) was dissolved in MeOH (10 mL) followed by the addition of Pd/C (80 mg, 10% purity) under H2 atmosphere (35 psi). The mixed liquid was replaced with gas three times using H 2 . The mixture was stirred at 50°C for 20 hours under an atmosphere of H2 (15 psi). The mixture was filtered and the resulting solution was concentrated to give compound 23-e.
化合物23-f的合成Synthesis of compound 23-f
将化合物23-e(803mg,2.23mmol)溶解于MeOH(10mL)和水(5mL)中,之后添加NaOH(268.09mg,6.70mmol)在氮气氛围。将所得混合液在70℃搅拌12小时。使用3M HCl调节溶液pH约为pH=8,此时有固体析出,过滤并用水(10mL)清洗滤饼,将滤饼真空干燥得粗品23-f。Compound 23-e (803 mg, 2.23 mmol) was dissolved in MeOH (10 mL) and water (5 mL), followed by the addition of NaOH (268.09 mg, 6.70 mmol) under nitrogen atmosphere. The resulting mixture was stirred at 70°C for 12 hours. Use 3M HCl to adjust the pH of the solution to about pH=8, at this time a solid precipitates out, filter and wash the filter cake with water (10 mL), and vacuum dry the filter cake to obtain the crude product 23-f.
化合物23的合成Synthesis of compound 23
将化合物23-f(100mg,301.76μmol)溶解于DMF(2mL)之后添加DIEA(117.00mg,905.29μmol,157.68uL)和HATU(172.11mg,452.65μmol)在氮气氛围。将所得混合液在25℃搅拌30min。之后添加1-n(46.20mg,362.12μmol)。将混合液在25℃搅拌11.5小时。将反应液稀释使用水(30mL)并萃取使用EA(50mL*2)。将结合的有机相使用饱和食盐水(30mL*4)洗涤,用Na2SO4干燥,过滤,浓缩得到粗品。粗品通过制备色谱纯化得到化合物23。Compound 23-f (100 mg, 301.76 μmol) was dissolved in DMF (2 mL) followed by the addition of DIEA (117.00 mg, 905.29 μmol, 157.68 uL) and HATU (172.11 mg, 452.65 μmol) under nitrogen atmosphere. The resulting mixture was stirred at 25 °C for 30 min. 1-n (46.20 mg, 362.12 μmol) was then added. The mixture was stirred at 25°C for 11.5 hours. The reaction solution was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phases were washed with saturated brine (30 mL*4), dried over Na 2 SO 4 , filtered, and concentrated to obtain crude product. The crude product was purified by preparative chromatography to give compound 23.
1H NMR(400MHz,DMSO-d6)δ=9.66(s,1H),7.79(d,J=8.8Hz,2H),7.65-7.53(m,2H),7.38(d,J=8.8Hz,2H),7.09(dt,J=2.4,9.2Hz,1H),4.09(s,3H),2.99(br d,J=11.2Hz,2H),2.67(s,1H),2.37-2.22(m,6H),2.19-2.04(m,2H),1.92-1.83(m,2H),1.79-1.69(m,2H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.66 (s, 1H), 7.79 (d, J=8.8 Hz, 2H), 7.65-7.53 (m, 2H), 7.38 (d, J=8.8 Hz, 2H), 7.09(dt, J=2.4, 9.2Hz, 1H), 4.09(s, 3H), 2.99(br d, J=11.2Hz, 2H), 2.67(s, 1H), 2.37-2.22(m, 6H), 2.19-2.04 (m, 2H), 1.92-1.83 (m, 2H), 1.79-1.69 (m, 2H).
实施例24Example 24

化合物24的合成Synthesis of compound 24
将化合物14-a(23.32mg)溶于THF(3mL)中,并在-78℃下,往溶液中滴加NaHMDS(1M,158.54μL).所得混合物在-78℃下搅拌30分钟.然后在该温度下向反应液中滴加溶于THF(3mL)的24-a(50mg,132.12μmol),所得混合物在25℃下搅拌12小时。向反应液中加入水(10mL)淬灭反应并用乙酸乙酯(20mL*3)萃取.合并有机相用Na2SO4干燥,过滤后减压浓缩得到粗品.粗品通过制备色谱纯化得到化合物24.Compound 14-a (23.32 mg) was dissolved in THF (3 mL), and NaHMDS (1 M, 158.54 μL) was added dropwise to the solution at -78 °C. The resulting mixture was stirred at -78 °C for 30 min. 24-a (50 mg, 132.12 μmol) dissolved in THF (3 mL) was added dropwise to the reaction solution at this temperature, and the resulting mixture was stirred at 25° C. for 12 hours. Water (10 mL) was added to the reaction solution to quench the reaction and extracted with ethyl acetate (20 mL*3). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure to obtain the crude product. The crude product was purified by preparative chromatography to obtain compound 24.
1H NMR(400MHz,DMSO-d6)δppm 1.67-1.97(m,6H)2.24-2.37(m,5H)2.42(br t,J=10.45Hz,2H)2.97(br d,J=11.37Hz,2H)7.49(d,J=4.52Hz,1H)7.67(td,J=8.71,2.63Hz,1H)7.78(dd,J=10.58,6.54Hz,2H)7.98-8.14(m,2H)8.84(d,J=4.52Hz,1H)9.80(s,1H). 1 H NMR (400MHz, DMSO-d 6 ) δppm 1.67-1.97 (m, 6H) 2.24-2.37 (m, 5H) 2.42 (br t, J=10.45Hz, 2H) 2.97 (br d, J=11.37Hz, 2H) 7.49(d, J=4.52Hz, 1H) 7.67(td, J=8.71, 2.63Hz, 1H) 7.78(dd, J=10.58, 6.54Hz, 2H) 7.98-8.14(m, 2H) 8.84(d , J=4.52Hz, 1H) 9.80(s, 1H).
实施例25Example 25

化合物25的合成Synthesis of compound 25
将化合物25-a(22.38mg)溶于THF(3mL)中,并在-78℃下,往溶液中滴加NaHMDS(1M,158.54μL).所得混合物在-78℃下搅拌30分钟.然后在该温度下向反应液中滴加溶于THF(3mL)的14-a(50mg),所得混合物在20℃下搅拌12小时.向反应液中加入水(10mL)淬灭反应并用乙酸乙酯(20mL*3)萃取.合并有机相用Na2SO4干燥,过滤后减压浓缩得到粗品.相品通过制备色谱纯化得到化合物25.Compound 25-a (22.38 mg) was dissolved in THF (3 mL), and NaHMDS (1 M, 158.54 μL) was added dropwise to the solution at -78 °C. The resulting mixture was stirred at -78 °C for 30 min. 14-a (50 mg) dissolved in THF (3 mL) was added dropwise to the reaction at this temperature, and the resulting mixture was stirred at 20°C for 12 hours. Water (10 mL) was added to the reaction to quench the reaction and ethyl acetate ( 20mL*3) extraction. The combined organic phases were dried with Na 2 SO 4 , filtered and concentrated under reduced pressure to obtain the crude product. The phase product was purified by preparative chromatography to obtain compound 25.
1H NMR(400MHz,DMSO-d6)δppm 1.74-2.03(m,6H)2.21(br t,J=8.86Hz,2H)2.31-2.36(m,1H)2.34(br s,1H)2.33-2.46(m,1H)2.38-2.46(m,1H)2.38-2.44(m,1H)2.93(brd,J=11.00Hz,2H)3.92(s,3H)6.78(td,J=8.74,2.69Hz,1H)7.01(dd,J=10.76,2.69Hz,1H)7.50(d,J=4.52Hz,1H)7.63-7.74(m,1H)8.00-8.22(m,3H)8.88(d,J=4.52Hz,1H)9.71(s,1H)。 1 H NMR (400MHz, DMSO-d 6 ) δppm 1.74-2.03(m, 6H) 2.21(br t, J=8.86Hz, 2H) 2.31-2.36(m, 1H) 2.34(br s, 1H) 2.33-2.46 (m, 1H) 2.38-2.46 (m, 1H) 2.38-2.44 (m, 1H) 2.93 (brd, J=11.00Hz, 2H) 3.92 (s, 3H) 6.78 (td, J=8.74, 2.69Hz, 1H) ) 7.01 (dd, J=10.76, 2.69Hz, 1H) 7.50 (d, J=4.52Hz, 1H) 7.63-7.74 (m, 1H) 8.00-8.22 (m, 3H) 8.88 (d, J=4.52Hz, 1H) 9.71 (s, 1H).
实施例26Example 26

化合物26-a的合成Synthesis of compound 26-a
向化合物1-e(2g)的乙醇溶液(30mL)中加入硼氢化钠(503.80mg),在25℃下反应12小时后,加入100毫升乙酸乙酯稀释,溶液用饱和氯化钠溶液洗涤三次,每次100毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到化合物26-a。Sodium borohydride (503.80 mg) was added to an ethanol solution (30 mL) of compound 1-e (2 g), and after reacting at 25°C for 12 hours, 100 mL of ethyl acetate was added to dilute, and the solution was washed three times with saturated sodium chloride solution , each 100 mL, the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated to obtain compound 26-a.
化合物26-b的合成Synthesis of compound 26-b
向化合物26-a(50mg)和4-氯-6-氟喹啉(39.95mg)的DMF溶液(5mL)中加入叔丁醇钾(74.05mg),在90℃下反应12小时,加入30毫升乙酸乙酯稀释,溶液用饱和氯化钠溶液洗涤3次,每次30毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到黄色胶状化合26-b(300mg),直接用于下一步反应。To a DMF solution (5 mL) of compound 26-a (50 mg) and 4-chloro-6-fluoroquinoline (39.95 mg), potassium tert-butoxide (74.05 mg) was added, and the reaction was carried out at 90° C. for 12 hours, and 30 mL of Diluted with ethyl acetate, the solution was washed three times with saturated sodium chloride solution, 30 mL each time, the organic phase was dried with anhydrous sodium sulfate, filtered, and concentrated to give yellow gum compound 26-b (300 mg), which was used directly in the next step reaction.
化合物26-c的合成Synthesis of compound 26-c
向化合物26-b(300mg)的甲醇(5mL)和水(5mL)溶液中加入氢氧化钠(161.09mg),在65℃下反应12小时后,用盐酸(1M)将pH调到6-7,减压浓缩得到粗品,粗品通过反相快速柱层系纯化得到化合物26-c。Sodium hydroxide (161.09 mg) was added to a solution of compound 26-b (300 mg) in methanol (5 mL) and water (5 mL), and after reacting at 65°C for 12 hours, the pH was adjusted to 6-7 with hydrochloric acid (1 M). , concentrated under reduced pressure to obtain the crude product, and the crude product was purified by reverse-phase flash column chromatography to obtain compound 26-c.
化合物26的合成Synthesis of compound 26
向化合物26-c(40mg)和1-n(16.30mg)的DMF溶液(5mL)中加入N,N-二异丙基乙胺((75.06mg)和HATU(66.25mg),在氮气保护和25℃下反应12小时后,加入30毫升乙酸乙酯稀释,溶液用饱和氯化钠溶液洗涤3次,每次30毫升,有机相用无水硫酸钠干燥,过滤,浓缩得到粗品,粗品通过制备色谱(甲酸体系)纯化得到26。To a solution of compound 26-c (40 mg) and 1-n (16.30 mg) in DMF (5 mL) was added N,N-diisopropylethylamine (75.06 mg) and HATU (66.25 mg), under nitrogen protection and After 12 hours of reaction at 25°C, 30 ml of ethyl acetate was added to dilute the solution. The solution was washed three times with saturated sodium chloride solution, 30 ml each time. The organic phase was dried with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was prepared by Purification by chromatography (formic acid system) afforded 26.
1H NMR(400MHz,DMSO-d6)δ=9.56(s,1H),8.67(d,J=5.1Hz,1H),8.00(dd,J=5.4,9.2Hz,1H),7.77-7.60(m,4H),7.36(d,J=8.9Hz,2H),7.14(d,J=5.1Hz,1H),4.81(brs,1H),2.80-2.70(m,2H),2.50-2.40(m,2H),2.32-2.17(m,4H),2.15-2.05(m,2H),1.95-1.80(m,2H),1.80-1.57(m,2H). 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.56 (s, 1H), 8.67 (d, J=5.1 Hz, 1H), 8.00 (dd, J=5.4, 9.2 Hz, 1H), 7.77-7.60 ( m, 4H), 7.36 (d, J=8.9Hz, 2H), 7.14 (d, J=5.1Hz, 1H), 4.81 (brs, 1H), 2.80-2.70 (m, 2H), 2.50-2.40 (m , 2H), 2.32-2.17(m, 4H), 2.15-2.05(m, 2H), 1.95-1.80(m, 2H), 1.80-1.57(m, 2H).
实施例27Example 27

化合物27-b的合成Synthesis of compound 27-b
将化合物26-a(50mg),27-a(39.93mg)溶解在DMF(5mL)中,然后加入t-BuOK(74.05mg)氮气置换三次,所得混合物在氮气保护下,90℃下搅拌12小时。将反应物用EtOAc(15mL*3)萃取.合并有机相用饱和食盐水洗(15mL*3),Na2SO4干燥,过滤浓缩后得到27-b。Compounds 26-a (50 mg) and 27-a (39.93 mg) were dissolved in DMF (5 mL), and then t-BuOK (74.05 mg) was added for nitrogen replacement three times, and the resulting mixture was stirred at 90 °C for 12 hours under nitrogen protection . The reactant was extracted with EtOAc (15 mL*3). The combined organic phases were washed with saturated brine (15 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain 27-b.
化合物27-c的合成Synthesis of compound 27-c
将化合物27-b(460mg)溶解于MeOH(5mL)和水(5mL)中,加入NaOH(148.22mg,3.71mmol),所得混合物在60℃下氮气保护下搅拌12小时.将甲醇拉干后,在0℃下用1M HCl调节PH为7,水相直接旋干。粗品通过制备色谱纯化得到化合物27-c。Compound 27-b (460 mg) was dissolved in MeOH (5 mL) and water (5 mL), NaOH (148.22 mg, 3.71 mmol) was added, and the resulting mixture was stirred at 60° C. under nitrogen protection for 12 hours. After methanol was dried, The pH was adjusted to 7 with 1M HCl at 0°C, and the aqueous phase was directly spun dry. The crude product was purified by preparative chromatography to give compound 27-c.
化合物27的合成Synthesis of compound 27
将化合物27-c(50mg)溶解于DMF(5mL)中,然后加入DIEA(93.84mg,726.05μmol,126.47uL),HATU(82.82mg),对氯苯胺(20.38mg).氮气置换三次,所得混合物在25℃下搅拌12小时.将反应液用EtOAc(10mL*3)萃取,合并有机相用饱和食盐水洗(10mL*3),Na2SO4干燥,过滤浓缩后得到粗品,粗品通过制备色谱纯化得到化合物27。Compound 27-c (50 mg) was dissolved in DMF (5 mL), then DIEA (93.84 mg, 726.05 μmol, 126.47 uL), HATU (82.82 mg), p-chloroaniline (20.38 mg) were added. The resulting mixture was replaced with nitrogen three times. Stir at 25°C for 12 hours. The reaction solution was extracted with EtOAc (10 mL*3), the combined organic phases were washed with saturated brine (10 mL*3), dried over Na 2 SO 4 , filtered and concentrated to obtain the crude product, which was purified by preparative chromatography Compound 27 was obtained.
1H NMR(400MHz,DMSO-d6)δ=9.55(s,1H),8.53(d,J=5.6Hz,1H),7.75(d,J=8.0Hz,2H),7.44-7.34(m,1H),7.31-7.25(m,2H),7.29(dd,J=2.4,5.7Hz,1H),4.72(td,J=4.4,8.3Hz,1H),3.43(br s,2H),2.81-2.71(m,4H),2.44-2.17(m,2H),2.12-1.94(m,2H),1.78-1.63(m,4H)。 1 H NMR (400 MHz, DMSO-d 6 ) δ=9.55 (s, 1H), 8.53 (d, J=5.6 Hz, 1H), 7.75 (d, J=8.0 Hz, 2H), 7.44-7.34 (m, 1H), 7.31-7.25 (m, 2H), 7.29 (dd, J=2.4, 5.7Hz, 1H), 4.72 (td, J=4.4, 8.3Hz, 1H), 3.43 (br s, 2H), 2.81- 2.71 (m, 4H), 2.44-2.17 (m, 2H), 2.12-1.94 (m, 2H), 1.78-1.63 (m, 4H).
实施例28Example 28

化合物28-b的合成Synthesis of compound 28-b
将化合物28-a(1g,6.04mmol,HCl)溶解于二氧六环(8mL)和水(2mL)中,之后添加DIEA(1.56g)。之后添加1-b(2.31g,9.06mmol)到混合液中在90℃。将所得混合液在90℃搅拌12小时。将混合液萃取使用EtOAc(10mL*3),将结合的有机相清洗使用饱和食盐水(10mL*3),干燥使用Na2SO4,过滤,过滤浓缩后得到28-b。Compound 28-a (1 g, 6.04 mmol, HCl) was dissolved in dioxane (8 mL) and water (2 mL), followed by the addition of DIEA (1.56 g). Then 1-b (2.31 g, 9.06 mmol) was added to the mixture at 90°C. The resulting mixture was stirred at 90°C for 12 hours. The mixture was extracted with EtOAc (10 mL*3), the combined organic phase was washed with saturated brine (10 mL*3), dried with Na 2 SO 4 , filtered, filtered and concentrated to obtain 28-b.
化合物28-c的合成Synthesis of compound 28-c
将化合物28-b(670mg,3.17mmol)溶解于THF(10mL)中,之后添加N,N-二(三氟甲烷磺酰基)苯胺(1.25g,3.49mmol)。将混合液使用氮气置换三次。之后将NaHMDS(1M,4.12mL)逐滴添加到混合液中在-78℃。将所得混合液在-78℃搅拌1小时。之后将混合液在25℃搅拌11小时。将混合液淬灭通过添加0.2M HCl(10mL),之后萃取使用MTBE(10mL*3),将结合的有机相使用0.2M HCl(10mL*3),0.5%NaOH(10mL*3)和饱和食盐水(10mL*3)洗涤,Na2SO4干燥,过滤,浓缩得化合物28-c。Compound 28-b (670 mg, 3.17 mmol) was dissolved in THF (10 mL), followed by the addition of N,N-bis(trifluoromethanesulfonyl)aniline (1.25 g, 3.49 mmol). The mixture was replaced three times with nitrogen. NaHMDS (1 M, 4.12 mL) was then added dropwise to the mixture at -78°C. The resulting mixture was stirred at -78°C for 1 hour. The mixture was then stirred at 25°C for 11 hours. The mixture was quenched by adding 0.2M HCl (10 mL), followed by extraction using MTBE (10 mL*3), the combined organic phase was 0.2M HCl (10 mL*3), 0.5% NaOH (10 mL*3) and saturated common salt Washed with water (10 mL*3), dried over Na2SO4, filtered and concentrated to obtain compound 28-c.
化合物28-d的合成Synthesis of compound 28-d
将化合物28-c(200mg,582.55μmol),双联频哪醇硼酸酯(192.31mg,757.31μmol),KOAc(171.51mg,1.75mmol),NaBr(23.98mg,233.02μmol,7.49uL)溶解于二氧六环(2mL)中,之后在氮气氛围下添加Pd(dppf)Cl2(21.31mg,29.13μmol)。将混合液使用氮气置换三次。将混合液在100℃搅拌12小时。将反应液过滤并且清洗使用EA(80mL),之后加入水(40mL)。将获得有机相清洗使用饱和食盐水(30mL*2)。调节溶液pH为pH=3使用1M HCl。将获得水相使用EA(30mL*2)洗涤。使用饱和碳酸钠溶液调节水相pH为pH=10,使用EA(40mL*2)萃取。将获得的有机相使用饱和食盐水(20mL*2)洗涤,干燥使用Na2SO4,过滤并浓缩得粗产品28-d。Compound 28-c (200 mg, 582.55 μmol), double pinacol boronate (192.31 mg, 757.31 μmol), KOAc (171.51 mg, 1.75 mmol), NaBr (23.98 mg, 233.02 μmol, 7.49 uL) were dissolved in into dioxane ( 2 mL), followed by the addition of Pd(dppf)Cl2 (21.31 mg, 29.13 μmol) under nitrogen. The mixture was replaced three times with nitrogen. The mixture was stirred at 100°C for 12 hours. The reaction was filtered and washed with EA (80 mL), after which water (40 mL) was added. The obtained organic phase was washed with saturated brine (30 mL*2). The pH of the solution was adjusted to pH=3 using 1M HCl. The obtained aqueous phase was washed with EA (30 mL*2). The pH of the aqueous phase was adjusted to pH=10 with saturated sodium carbonate solution and extracted with EA (40 mL*2). The obtained organic phase was washed with saturated brine (20 mL*2), dried with Na 2 SO 4 , filtered and concentrated to obtain crude product 28-d.
化合物28-e的合成Synthesis of Compound 28-e
将化合物28-d(210mg,653.76μmol)和1-i(98.93mg,544.80μmol)溶解于二氧六环(4mL)和水(1mL)中,之后添加Et3N(165.39mg,1.63mmol,227.49μL)和Pd(dppf)Cl2(19.93mg,27.24μmol)在氮气氛围。将混合液使用氮气置换三次。将混合液在110℃搅拌8小时。将反应液过滤使用EA(40mL),之后清洗使用水(20mL)。将有机相清洗使用饱和食盐水(15mL*2)。调节溶液pH为pH=3使用1M HCl。之后清洗水相使用EA(15mL*2)。调节溶液pH为pH=10使用饱和碳酸钠溶液,萃取使用EA(20mL*2)。将获得的有机相清洗使用饱和食盐水(20mL*2),干燥使用Na2SO4,过滤并浓缩得28-e。Compounds 28-d (210 mg, 653.76 μmol) and 1-i (98.93 mg, 544.80 μmol) were dissolved in dioxane (4 mL) and water (1 mL), followed by the addition of Et3N (165.39 mg, 1.63 mmol, 227.49 μL) and Pd(dppf)Cl2 (19.93 mg , 27.24 μmol) under nitrogen. The mixture was replaced three times with nitrogen. The mixture was stirred at 110°C for 8 hours. The reaction solution was filtered using EA (40 mL), and then washed with water (20 mL). The organic phase was washed with saturated brine (15 mL*2). The pH of the solution was adjusted to pH=3 using 1M HCl. After that, EA (15mL*2) was used to wash the aqueous phase. The pH of the solution was adjusted to pH=10 using saturated sodium carbonate solution, and EA (20 mL*2) was used for extraction. The obtained organic phase was washed with saturated brine (20 mL*2), dried with Na 2 SO 4 , filtered and concentrated to obtain 28-e.
化合物28-f的合成Synthesis of compound 28-f
将化合物28-e(156mg,458.30μmol)和吡啶(54.38mg,687.44μmol,55.49uL)溶解于IPA(2mL)中,之后添加Pd/C(32mg,10%纯度)在H2(15psi)氛围。将混合液使用氢气置换三次。将混合液在H2(15psi)氛围在50℃搅拌8小时。将混合液过滤,浓缩滤液得到化合物28-f。Compound 28-e (156 mg, 458.30 μmol) and pyridine (54.38 mg, 687.44 μmol, 55.49 uL) were dissolved in IPA (2 mL) followed by the addition of Pd/C (32 mg, 10% purity) under H 2 (15 psi) atmosphere . The mixture was replaced three times with hydrogen. The mixture was stirred at 50°C for 8 hours under an atmosphere of H2 (15 psi). The mixture was filtered, and the filtrate was concentrated to obtain compound 28-f.
化合物28-g的合成Synthesis of compound 28-g
将化合物28-f(152mg,443.92μmol)溶解于MeOH(2mL)和水(1mL)中,之后添加NaOH(71.03mg,1.78mmol)在氮气氛围。将混合液在70℃搅拌12小时。将混合液浓缩得粗品,通过制备色谱纯化得到28-g。Compound 28-f (152 mg, 443.92 μmol) was dissolved in MeOH (2 mL) and water (1 mL) before adding NaOH (71.03 mg, 1.78 mmol) under nitrogen atmosphere. The mixture was stirred at 70°C for 12 hours. The mixture was concentrated to give crude product, which was purified by preparative chromatography to give 28-g.
化合物28的合成Synthesis of Compound 28
化合物28-g(26mg,82.71μmol)溶解于DMF(1mL)中,之后添加HATU(47.17mg,124.06μmol)和DIEA(32.07mg,248.13μmol,43.22uL)在氮气氛围。将混合液在25℃搅拌30min。之后加入1-n(12.66mg,99.25μmol)。将混合液在25℃搅拌11.5小时。将混合液稀释使用水(30mL)并使用EA(50mL*2)萃取。将结合的有机相使用饱和食盐水(30mL*4)洗涤,使用Na2SO4干燥,过滤并在真空中浓缩得粗品。粗品通过制备色谱纯化得到化合物28。Compound 28-g (26 mg, 82.71 μmol) was dissolved in DMF (1 mL), followed by the addition of HATU (47.17 mg, 124.06 μmol) and DIEA (32.07 mg, 248.13 μmol, 43.22 uL) under nitrogen atmosphere. The mixture was stirred at 25 °C for 30 min. 1-n (12.66 mg, 99.25 μmol) was then added. The mixture was stirred at 25°C for 11.5 hours. The mixture was diluted with water (30 mL) and extracted with EA (50 mL*2). The combined organic phases were washed with saturated brine (30 mL*4), dried over Na2SO4 , filtered and concentrated in vacuo to give crude product. The crude product was purified by preparative chromatography to give compound 28.
1H NMR(400MHz,DMSO-d6)δ=10.11(s,1H),8.86(d,J=4.4Hz,1H),8.18-8.05(m,2H),7.98(dd,J=2.8,10.8Hz,1H),7.77(d,J=8.8Hz,2H),7.68(dt,J=2.8,8.8Hz,1H),7.58(d,J=4.4Hz,1H),7.39(d,J=8.8Hz,2H),3.29-3.25(m,1H),2.88(br d,J=11.1Hz,2H),2.54(br s,2H),2.09-2.01(m,2H),1.86(br d,J=12.0Hz,2H),1.15(br d,J=15.6Hz,4H). 1 H NMR (400MHz, DMSO-d6) δ=10.11 (s, 1H), 8.86 (d, J=4.4Hz, 1H), 8.18-8.05 (m, 2H), 7.98 (dd, J=2.8, 10.8Hz , 1H), 7.77 (d, J=8.8Hz, 2H), 7.68 (dt, J=2.8, 8.8Hz, 1H), 7.58 (d, J=4.4Hz, 1H), 7.39 (d, J=8.8Hz) , 2H), 3.29-3.25(m, 1H), 2.88(br d, J=11.1Hz, 2H), 2.54(br s, 2H), 2.09-2.01(m, 2H), 1.86(br d, J= 12.0Hz, 2H), 1.15 (br d, J=15.6Hz, 4H).
实施例29Example 29

化合物29的合成Synthesis of compound 29
向DMF(5mL)中依次加入反应物1-l(50mg),反应物29-a(34.72mg),HATU(86.84mg),二异丙基乙基胺(59.04mg),反应液在室温下搅拌12小时,向反应体系中加入乙酸乙酯(30mL),然后用饱和氯化铵溶液洗涤三次(30mL*3),再用饱和食盐水洗涤三次(30mL*3),用无水硫酸钠干燥,过滤,将滤液浓缩得到粗产品,粗产品用常规反相色谱柱进行纯化得到化合物29。To DMF (5 mL) were sequentially added reactant 1-1 (50mg), reactant 29-a (34.72mg), HATU (86.84mg), diisopropylethylamine (59.04mg), the reaction solution was at room temperature Stir for 12 hours, add ethyl acetate (30 mL) to the reaction system, then wash with saturated ammonium chloride solution three times (30 mL*3), then wash with saturated brine three times (30 mL*3), and dry with anhydrous sodium sulfate , filtered, and the filtrate was concentrated to obtain the crude product, which was purified by conventional reverse-phase chromatography to obtain compound 29.
1H NMR(400MHz,MeOD-d4)δppm 1.83-1.93(m,2H)1.94-2.01(m,2H)2.04(br s,2H)2.36-2.44(m,2H)2.45-2.52(m,2H)2.54-2.61(m,2H)3.10-3.15(m,2H)3.36-3.39(m,1H)7.21(t,J=8.68Hz,1H)7.58(d,J=4.65Hz,1H)7.61(d,J=2.69Hz,1H)7.63(d,J=2.81Hz,1H)7.94(dd,J=10.70,2.63Hz,1H)8.07-8.08(m,1H)8.08-8.12(m,1H)8.79(d,J=4.65Hz,1H)。 1 H NMR (400 MHz, MeOD-d 4 ) δppm 1.83-1.93 (m, 2H) 1.94-2.01 (m, 2H) 2.04 (br s, 2H) 2.36-2.44 (m, 2H) 2.45-2.52 (m, 2H) ) 2.54-2.61(m, 2H) 3.10-3.15(m, 2H) 3.36-3.39(m, 1H) 7.21(t, J=8.68Hz, 1H) 7.58(d, J=4.65Hz, 1H) 7.61(d , J=2.69Hz, 1H) 7.63(d, J=2.81Hz, 1H) 7.94(dd, J=10.70, 2.63Hz, 1H) 8.07-8.08(m, 1H) 8.08-8.12(m, 1H) 8.79( d, J=4.65 Hz, 1H).
实施例30Example 30

化合物30的合成Synthesis of
向DMF(5mL)中依次加入反应物1-l(50mg),反应物30-a(28.80mg),HATU(86.84mg),二异丙基乙基胺(59.04mg),反应液在室温下搅拌12小时,向反应体系中加入乙酸乙酯(30mL),然后用饱和氯化铵溶液洗涤三次(30mL*3),再用饱和食盐水洗涤三次(30mL*3),用无水硫酸钠干燥,过滤,将滤液浓缩得到粗产品,粗产品用常规反相色谱柱进行纯化,得到化合物30。To DMF (5 mL) were sequentially added reactant 1-1 (50mg), reactant 30-a (28.80mg), HATU (86.84mg), diisopropylethylamine (59.04mg), the reaction solution was at room temperature Stir for 12 hours, add ethyl acetate (30 mL) to the reaction system, then wash with saturated ammonium chloride solution three times (30 mL*3), then wash with saturated brine three times (30 mL*3), and dry with anhydrous sodium sulfate , filtered, and the filtrate was concentrated to obtain a crude product, which was purified by conventional reverse-phase chromatography to obtain
1H NMR(400MHz,METHANOL-d4)δppm 1.92-2.00(m,2H)2.01-2.09(m,2H)2.09-2.16(m,2H)2.48-2.63(m,4H)2.71-2.83(m,2H)3.23-3.29(m,2H)3.48-3.53(m,1H)3.93(s,3H)7.21(dd,J=8.56,2.20Hz,1H)7.33(d,J=8.56Hz,1H)7.57(d,J=4.65Hz,1H)7.61(d,J=2.08Hz,1H)7.64(dd,J=8.74,2.14Hz,1H)7.96(dd,J=10.51,2.69Hz,1H)8.12(d,J=3.55Hz,1H)8.81(d,J=4.65Hz,1H)。 1 H NMR (400 MHz, METHANOL-d 4 ) δppm 1.92-2.00 (m, 2H) 2.01-2.09 (m, 2H) 2.09-2.16 (m, 2H) 2.48-2.63 (m, 4H) 2.71-2.83 (m, 2H) 3.23-3.29(m, 2H) 3.48-3.53(m, 1H) 3.93(s, 3H) 7.21(dd, J=8.56, 2.20Hz, 1H) 7.33(d, J=8.56Hz, 1H) 7.57( d, J=4.65Hz, 1H) 7.61 (d, J=2.08Hz, 1H) 7.64 (dd, J=8.74, 2.14Hz, 1H) 7.96 (dd, J=10.51, 2.69Hz, 1H) 8.12 (d, J=3.55Hz, 1H) 8.81 (d, J=4.65Hz, 1H).
实施例31Example 31

化合物31的合成Synthesis of compound 31
向四氢呋喃(5mL)中加入反应物14-a(23.08mg),在-78℃下缓慢滴加六甲基二硅基氨基钠(1M,264.24uL),反应液在该温度下搅拌半小时,然后加入反应物31-a(50mg),反应液在室温下搅拌1小时,向反应体系中加入乙酸乙酯(30mL),然后用饱和氯化铵溶液洗涤三次(30mL*3),再用饱和食盐水洗涤三次(30mL*3),用无水硫酸钠干燥,过滤,将滤液浓缩得到粗产品,粗产品用常规反相色谱柱进行纯化,得到化合物31。The reactant 14-a (23.08 mg) was added to tetrahydrofuran (5 mL), sodium hexamethyldisilazide (1 M, 264.24 uL) was slowly added dropwise at -78 °C, and the reaction solution was stirred at this temperature for half an hour, Then reactant 31-a (50 mg) was added, the reaction solution was stirred at room temperature for 1 hour, ethyl acetate (30 mL) was added to the reaction system, then washed three times with saturated ammonium chloride solution (30 mL*3), and then saturated Washed with brine three times (30 mL*3), dried with anhydrous sodium sulfate, filtered, and the filtrate was concentrated to obtain a crude product. The crude product was purified by a conventional reverse-phase chromatography column to obtain compound 31.
1H NMR(400MHz,MeOD-d4)δppm 1.84-1.95(m,2H)1.98(br d,J=12.72Hz,2H)2.04(br d,J=5.50Hz,2H)2.35-2.43(m,2H)2.46-2.60(m,4H)3.11(br d,J=10.88Hz,2H)3.37-3.47(m,1H)7.43(br s,2H)7.56-7.65(m,2H)7.81(br d,J=11.86Hz,1H)7.93(br d,J=10.76Hz,1H)8.08-8.13(m,1H)8.77-8.81(m,1H)。 1 H NMR (400MHz, MeOD-d 4 ) δppm 1.84-1.95 (m, 2H) 1.98 (br d, J=12.72Hz, 2H) 2.04 (br d, J=5.50Hz, 2H) 2.35-2.43 (m, 2H) 2.46-2.60(m, 4H) 3.11(br d, J=10.88Hz, 2H) 3.37-3.47(m, 1H) 7.43(br s, 2H) 7.56-7.65(m, 2H) 7.81(br d, J=11.86Hz, 1H) 7.93 (br d, J=10.76Hz, 1H) 8.08-8.13 (m, 1H) 8.77-8.81 (m, 1H).
实施例32Example 32

化合物32的合成Synthesis of compound 32
向DMF(5mL)中依次加入反应物1-l(50mg),反应物32-a(25.87mg),HATU(86.84mg),二异丙基乙基胺(59.04mg),反应液在室温下搅拌12小时,向反应体系中加入乙酸乙酯(30mL),然后用饱和氯化铵溶液洗涤三次(30mL*3),再用饱和食盐水洗涤三次(30mL*3),用无水硫酸钠干燥,过滤,将滤液浓缩得到粗产品,粗产品用常规反相色谱柱进行纯化,得到化合物32。To DMF (5 mL) were sequentially added reactant 1-1 (50mg), reactant 32-a (25.87mg), HATU (86.84mg), diisopropylethylamine (59.04mg), the reaction solution was at room temperature Stir for 12 hours, add ethyl acetate (30 mL) to the reaction system, then wash with saturated ammonium chloride solution three times (30 mL*3), then wash with saturated brine three times (30 mL*3), and dry with anhydrous sodium sulfate , filtered, and the filtrate was concentrated to obtain a crude product, which was purified by a conventional reverse-phase chromatography column to obtain compound 32.
1H NMR(400MHz,DMSO-d6)δppm 1.81-1.99(m,2H)2.06-2.14(m,3H)2.28-2.42(m,5H)2.57-2.84(m,5H)2.86-2.93(m,2H)3.19-3.41(m,1H)7.42-7.50(m,2H)7.63(dd,J=8.50,2.26Hz,1H)7.69-7.77(m,2H)8.11-8.20(m,2H)8.91(d,J=4.52Hz,1H)10.15-10.28(m,1H)。 1 H NMR (400 MHz, DMSO-d 6 ) δppm 1.81-1.99 (m, 2H) 2.06-2.14 (m, 3H) 2.28-2.42 (m, 5H) 2.57-2.84 (m, 5H) 2.86-2.93 (m, 2H) 3.19-3.41(m, 1H) 7.42-7.50(m, 2H) 7.63(dd, J=8.50, 2.26Hz, 1H) 7.69-7.77(m, 2H) 8.11-8.20(m, 2H) 8.91(d , J=4.52Hz, 1H) 10.15-10.28 (m, 1H).
实施例33Example 33

化合物33-b的合成Synthesis of compound 33-b
将化合物33-a(2g,27.75mmol)溶于MTBE(30mL)后加入碘化锌(88.59mg,277.54μmol)和TMSCN(2.75g,27.75mmol,3.47mL).所得混合物在25℃下搅拌12小时.将反应液浓缩得到粗品化合物33-b.Compound 33-a (2 g, 27.75 mmol) was dissolved in MTBE (30 mL) and zinc iodide (88.59 mg, 277.54 μmol) and TMSCN (2.75 g, 27.75 mmol, 3.47 mL) were added. The resulting mixture was stirred at 25°C for 12 h. The reaction solution was concentrated to obtain crude compound 33-b.
化合物33-c的合成Synthesis of compound 33-c
将化合物33-b(3g,17.52mmol,1eq)和化合物33-h(3.26g,17.52mmol,1eq)溶于MTBE(50mL)后在25℃下搅拌12小时.将反应液浓缩得到粗品化合物33-c.Compound 33-b (3g, 17.52mmol, 1eq) and compound 33-h (3.26g, 17.52mmol, 1eq) were dissolved in MTBE (50mL) and stirred at 25°C for 12 hours. The reaction solution was concentrated to obtain crude compound 33 -c.
化合物33-d的合成Synthesis of compound 33-d
将33-c(2g,7.48mmol)溶于MeOH(20mL)和水(4mL)中后加入NaOH(897.72mg,22.44mmol).所得混合物在60℃下搅拌4小时.将反应液浓缩后用水(50mL)稀释EA(60mL*2)萃取.水相用HCl(1M,20mL)调节pH到5,EA(100mL*2)萃取.所得有机相用饱和食盐水(100mL*2)洗涤,Na2SO4干燥,过滤浓缩得到化合物33-d.33-c (2 g, 7.48 mmol) was dissolved in MeOH (20 mL) and water (4 mL) and NaOH (897.72 mg, 22.44 mmol) was added. The resulting mixture was stirred at 60 °C for 4 hours. The reaction solution was concentrated and water ( 50mL) diluted EA (60mL*2) for extraction. The aqueous phase was adjusted to
化合物33-f的合成Synthesis of compound 33-f
将33-d(780mg,2.73mmol),33-e(782.20mg,3.28mmol),Pd2(dba)3(250.32mg,273.36μmol),Xantphos(316.34mg,546.72μmol)和碳酸铯(1.78g,5.47mmol)溶于1,4二氧六环(20mL)。所得混合液用N2置换三次后在80℃下搅拌12小时.将反应液过滤,滤液用水(50mL)稀释,EA(80mL*3)萃取.所得混合有机相用饱和食盐水(80mL*2)洗涤,Na2SO4干燥,过滤浓缩得到粗品产物.粗品通过制备硅胶板纯化得到化合物33-f.Combine 33-d (780 mg, 2.73 mmol), 33-e (782.20 mg, 3.28 mmol), Pd 2 (dba) 3 (250.32 mg, 273.36 μmol), Xantphos (316.34 mg, 546.72 μmol) and cesium carbonate (1.78 g , 5.47 mmol) was dissolved in 1,4 dioxane (20 mL). The obtained mixed solution was replaced with N 3 three times and then stirred at 80°C for 12 hours. The reaction solution was filtered, the filtrate was diluted with water (50 mL), and extracted with EA (80 mL*3). The obtained mixed organic phase was mixed with saturated brine (80 mL*2) Washed, dried over Na 2 SO 4 , filtered and concentrated to give the crude product. The crude product was purified by preparative silica gel plate to give compound 33-f.
化合物33-g的合成Synthesis of compound 33-g
将化合物33-f(640mg,1.62mmol)溶于MeOH(10mL)后加入HCl二氧六环溶液(4M,4.04mL).所得混合物在25℃下搅拌2小时.将反应液浓缩得到化合物33-g.Compound 33-f (640 mg, 1.62 mmol) was dissolved in MeOH (10 mL) and HCl dioxane solution (4 M, 4.04 mL) was added. The resulting mixture was stirred at 25°C for 2 hours. The reaction solution was concentrated to obtain compound 33- g.
化合物33的合成Synthesis of compound 33
将化合物33-g(200mg,676.22μmol),1-i(61.40mg,338.11μmol),Pd2(dba)3(61.92mg,67.62μmol),Xantphos(78.25mg,135.24μmol)和碳酸铯(440.65mg,1.35mmol)溶于1,4二氧六环(10mL)后用N2置换三次。所得混合液在80℃下搅拌12小时.将反应液过滤后用水(20mL)稀释,乙酸乙酯(30mL*3)萃取.所得有机相用饱和食盐水(30mL*2)洗涤,Na2SO4干燥,过滤浓缩后通过制备色谱纯化得到化合物33.Compound 33-g (200 mg, 676.22 μmol), 1-i (61.40 mg, 338.11 μmol), Pd2(dba )3 ( 61.92 mg, 67.62 μmol), Xantphos (78.25 mg, 135.24 μmol) and cesium carbonate (440.65 mg, 1.35 mmol) was dissolved in 1,4 dioxane (10 mL) and replaced three times with N 2 . The resulting mixture was stirred at 80°C for 12 hours. The reaction solution was filtered, diluted with water (20 mL), and extracted with ethyl acetate (30 mL*3). The resulting organic phase was washed with saturated brine (30 mL*2), and Na 2 SO 4 After drying, filtration and concentration, purification by preparative chromatography gave compound 33.
1H NMR(400MHz,MeOD-d4)δppm 2.95-3.04(m,4H)3.88-4.08(m,4H)4.86(br s,4H)7.29(d,J=6.85Hz,1H)7.34-7.38(m,2H)7.64-7.70(m,2H)7.79-7.97(m,2H)8.05(dd,J=9.29,4.89Hz,1H)8.58(d,J=6.85Hz,1H). 1 H NMR (400 MHz, MeOD-d 4 ) δppm 2.95-3.04 (m, 4H) 3.88-4.08 (m, 4H) 4.86 (br s, 4H) 7.29 (d, J=6.85Hz, 1H) 7.34-7.38 ( m, 2H) 7.64-7.70 (m, 2H) 7.79-7.97 (m, 2H) 8.05 (dd, J=9.29, 4.89Hz, 1H) 8.58 (d, J=6.85Hz, 1H).
实施例34Example 34

化合物34-a的合成Synthesis of compound 34-a
将化合物23-f(100mg,301.76umol)溶于DCM(5mL)中,往溶液中加入CDI(73.40mg,452.65umol).所得混合物在25℃下搅拌反应12小时.向反应液中加入水(20mL)淬灭反应并用乙酸乙酯(20mL*3)萃取.合并有机相用饱和氯化钠溶液(20mL*3)洗涤,Na2SO4干燥,过滤后减压浓缩得到34-a.Compound 23-f (100 mg, 301.76 umol) was dissolved in DCM (5 mL), and CDI (73.40 mg, 452.65 umol) was added to the solution. The resulting mixture was stirred at 25 °C for 12 hours. To the reaction solution was added water ( 20mL) quenched the reaction and extracted with ethyl acetate (20mL*3). The combined organic phases were washed with saturated sodium chloride solution (20mL*3), dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to give 34-a.
化合物34的合成Synthesis of compound 34
将化合物20-a(29.73mg,251.67umol)溶于THF(5mL)中,并在-78℃下,往溶液中滴加NaHMDS(1M,251.67uL).所得混合物在-78℃下搅拌30分钟.然后在该温度下向反应液中滴加溶于THF(2mL)中34-a(80mg,209.73umol),所得混合物在20℃下搅拌12小时.向反应液中加入水(10mL)淬灭反应并用乙酸乙酯(20mL*3)萃取,合并有机相用Na2SO4干燥,过滤后减压浓缩得到粗品.粗品通过制备色谱纯化得到化合物34.Compound 20-a (29.73 mg, 251.67 umol) was dissolved in THF (5 mL), and NaHMDS (1 M, 251.67 uL) was added dropwise to the solution at -78 °C. The resulting mixture was stirred at -78 °C for 30 min 34-a (80 mg, 209.73 umol) dissolved in THF (2 mL) was then added dropwise to the reaction solution at this temperature, and the resulting mixture was stirred at 20°C for 12 hours. Water (10 mL) was added to the reaction solution to quench The reaction was followed by extraction with ethyl acetate (20 mL*3), the combined organic phases were dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to obtain the crude product. The crude product was purified by preparative chromatography to obtain compound 34.
1H NMR(400MHz,DMSO-d6)δppm 1.70-1.94(m,4H)2.03-2.21(m,2H)2.22-2.39(m,6H)2.99(brd,J=11.13Hz,2H)3.14(ddd,J=12.10,8.68,3.67Hz,1H)4.09(s,3H)7.09(td,J=9.26,2.38Hz,1H)7.52-7.65(m,2H)7.79(d,J=8.68Hz,2H)8.00(d,J=8.68Hz,2H)9.94(s,1H). 1 H NMR (400 MHz, DMSO-d 6 ) δppm 1.70-1.94 (m, 4H) 2.03-2.21 (m, 2H) 2.22-2.39 (m, 6H) 2.99 (brd, J=11.13 Hz, 2H) 3.14 (ddd , J=12.10, 8.68, 3.67Hz, 1H) 4.09 (s, 3H) 7.09 (td, J=9.26, 2.38Hz, 1H) 7.52-7.65 (m, 2H) 7.79 (d, J=8.68Hz, 2H) 8.00(d, J=8.68Hz, 2H) 9.94(s, 1H).
实验例1:化合物对Hela细胞犬尿氨酸通路活性抑制的评价Experimental Example 1: Evaluation of Compounds for Inhibition of Kynurenine Pathway Activity in Hela Cells
实验目的:Purpose:
检测本发明化合物对Hela细胞犬尿氨酸通路活性的抑制效应。The inhibitory effect of the compounds of the present invention on the activity of the kynurenine pathway in Hela cells was detected.
实验方法:experimental method:
以二甲亚砜将化合物溶解到3mM,取15μl化合物加入到96孔板。以3倍连续稀释化合物(10μl+20μl)。加入192μl检测培养基到一个96孔板中,向化合物板中加入78μl DMEM培养基(Dulbecco′s modified Eagle′s medium)并充分混匀,转移8μl DMEM培养基稀释过的化合物到已经加入检测培养基的96孔板。取出细胞培养板并弃去其中的培养基,转移100μl化合物到细胞培养板中,将细胞培养板转入培养箱中继续培养48小时。Compounds were dissolved to 3 mM in dimethyl sulfoxide, and 15 [mu]l of compounds were added to a 96-well plate. Compounds were serially diluted 3-fold (10 μl + 20 μl). Add 192 μl assay medium to a 96-well plate, add 78 μl DMEM medium (Dulbecco's modified Eagle's medium) to the compound plate and mix well, transfer 8 μl of compound diluted in DMEM medium to the assay culture that has been added based 96-well plate. Remove the cell culture plate and discard the medium in it, transfer 100 μl of compound to the cell culture plate, transfer the cell culture plate to the incubator and continue culturing for 48 hours.
取出细胞培养板,并转出50μl上清到一新的96孔板,加入200μl乙腈到上述板中并充分混匀,4度离心转移40μl上清到另一96孔板。加入360μl含有2μg/ml内参的去离子水,充分混匀对样品进行检测。Take out the cell culture plate, transfer 50 μl of supernatant to a new 96-well plate, add 200 μl of acetonitrile to the above plate and mix well, and transfer 40 μl of supernatant to another 96-well plate by centrifugation at 4 degrees. Add 360 μl of deionized water containing 2 μg/ml of internal reference, mix well to test the sample.
实验结果:见表1。Experimental results: see Table 1.
表1 体外筛选试验结果Table 1 In vitro screening test results
结论:本发明的化合物对Hela细胞内犬尿氨酸的产生具有明显的抑制作用。Conclusion: The compound of the present invention has obvious inhibitory effect on the production of kynurenine in Hela cells.
实验例2:化合物药代动力学的评价Experimental Example 2: Evaluation of Compound Pharmacokinetics
试验动物test animals
本研究使用的健康的成年雌性Balb/c小鼠,均购自上海灵畅生物科技有限公司。Healthy adult female Balb/c mice used in this study were purchased from Shanghai Lingchang Biotechnology Co., Ltd.
药物的配制formulation of drugs
静脉注射组给药溶液的配制Preparation of dosing solution for intravenous injection group
精确称量适量化合物,加入一定量的PEG400超声2分钟后得到澄清溶液,然后加入适量的纯水。得到终浓度为1mg/mL的澄清溶液,给药溶媒为60%PEG400+40%纯水。静脉注射组溶液在给药前用2um的滤膜进行过滤。Accurately weigh an appropriate amount of compound, add a certain amount of PEG400 and ultrasonicate for 2 minutes to obtain a clear solution, and then add an appropriate amount of pure water. A clear solution with a final concentration of 1 mg/mL was obtained, and the administration vehicle was 60% PEG400+40% pure water. The solution of intravenous injection group was filtered with 2um filter membrane before administration.
口服给组给药溶液的配制Preparation of Oral Dosing Group Dosing Solution
精确称量适量化合物,加入一定量的PEG400超声2分钟后得到澄清溶液,然后加入适量的纯水。得到终浓度为10mg/mL的混悬溶液,给药溶媒为60%PEG400+40%纯水。Accurately weigh an appropriate amount of compound, add a certain amount of PEG400 and ultrasonicate for 2 minutes to obtain a clear solution, and then add an appropriate amount of pure water. A suspension solution with a final concentration of 10 mg/mL was obtained, and the administration vehicle was 60% PEG400+40% pure water.
给药dosing
24只雌性Balb/c小鼠,按照每组6只,分为4组。第一组,静脉注射给予2mg/kg的BMS-986205;第二组,灌胃给予50mg/kg BMS-986205;第三组,静脉注射给予2mg/kg化合物1;第四组,灌胃给予50mg/kg化合物1。24 female Balb/c mice were divided into 4 groups according to 6 mice in each group. The first group was given BMS-986205 at 2 mg/kg by intravenous injection; the second group was given 50 mg/kg BMS-986205 by intragastric administration; the third group was given 2 mg/kg of
样品采集Sample Collection
采用交叉采血方式,每个时间点采集三只动物的血液。分别于给药前及给药后0.0833(仅静注组)、0.25、0.5、1、2、4、8、24小时采集40uL全血。将全血至于抗凝管中,在4℃条件先,3000g离心15分钟,制备血浆并保存于-80℃。应用LC/MS-MS测定血浆中的药物浓度。Blood was collected from three animals at each time point using a cross-bleeding method. 40uL whole blood was collected before administration and at 0.0833 (intravenous injection group only), 0.25, 0.5, 1, 2, 4, 8, and 24 hours after administration, respectively. The whole blood was placed in an anticoagulation tube, centrifuged at 3000g for 15 minutes at 4°C, and plasma was prepared and stored at -80°C. Drug concentrations in plasma were determined by LC/MS-MS.
表2 药代动力学参数Table 2 Pharmacokinetic parameters

“-”表示尚未得到数据"-" means no data yet
结论:本发明的实施例1相比临床分子BMS-986205具有更好的组织分布,相同剂量下具有更高的AUC,同时半衰期更长,口服生物利用度更高,综合药代动力学性质更优。Conclusion: Compared with the clinical molecule BMS-986205, Example 1 of the present invention has better tissue distribution, higher AUC at the same dose, longer half-life, higher oral bioavailability, and better comprehensive pharmacokinetic properties. excellent.
实验例3:化合物以及与PD-1单抗组合用药在CT26小鼠同系移植瘤模型中的药效评估Experimental Example 3: Pharmacodynamic evaluation of the compound and its combination with PD-1 monoclonal antibody in CT26 mouse syngeneic xenograft model
试验动物:Test animals:
BALB/c小鼠,雌性,18~22g,上海斯莱克。BALB/c mice, female, 18-22 g, Shanghai Slack.
供试药品:Tested drugs:
实施例1及对照化合物BMS-986205,PD-1单抗。Example 1 and control compound BMS-986205, PD-1 monoclonal antibody.
细胞及接种:Cells and seeding:
1.细胞系:CT26细胞(小鼠结肠癌细胞)1. Cell line: CT26 cells (mouse colon cancer cells)
2.接种量:3×105/0.1mL2. Inoculum volume: 3×10 5 /0.1mL
3.接种途径:皮下注射3. Vaccination route: subcutaneous injection
分组及给药方案:Grouping and dosing schedule:


实验结果:见图1、图2和表3。Experimental results: see Figure 1, Figure 2 and Table 3.
表3 CT26小鼠同系移植瘤模型中小鼠肿瘤体积数据Table 3 Mouse tumor volume data in CT26 mouse syngeneic xenograft model

a.相对肿瘤增殖率T/C(%)=TRTV/CRTV×100%(TRTV:治疗组平均RTV;CRTV:阴性对照组平均RTV)。a. Relative tumor proliferation rate T/C (%)=TRTV/CRTV×100% (TRTV: the average RTV of the treatment group; CRTV: the average RTV of the negative control group).
b.肿瘤生长抑制率TGI(%)=1-(治疗组测量当天平均肿瘤体积-治疗组分组当天平均肿瘤体积)/(溶媒组测量当天平均肿瘤体积-溶媒组分组当天平均肿瘤体积)×100%b. Tumor growth inhibition rate TGI(%)=1-(average tumor volume of the treatment group on the day of measurement-average tumor volume of the treatment group on the day)/(average tumor volume of the vehicle group on the day of measurement-average tumor volume of the vehicle group on the day)×100 %
c.P值:第18天肿瘤体积指与溶媒对照相比c. P value: Tumor volume on day 18 refers to vehicle control
结论:本发明化合物表现出优异的体内抗肿瘤活性,且能对PD-1单抗起到增效作用。相比临床分子BMS-986205,在相同剂量下,具有更优的单药及联用药效。Conclusion: The compound of the present invention exhibits excellent antitumor activity in vivo, and can synergize PD-1 monoclonal antibody. Compared with the clinical molecule BMS-986205, at the same dose, it has better single drug and combined drug efficacy.
Claims (12)


















Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2018101431510 | 2018-02-11 | ||
| CN201810143151 | 2018-02-11 | ||
| PCT/CN2019/074642 WO2019154380A1 (en) | 2018-02-11 | 2019-02-02 | Kynurenine pathway inhibitor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111868032A CN111868032A (en) | 2020-10-30 |
| CN111868032B true CN111868032B (en) | 2021-09-03 |
Family
ID=67548779
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980012783.6A Active CN111868032B (en) | 2018-02-11 | 2019-02-02 | Kynurenine pathway inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN111868032B (en) |
| WO (1) | WO2019154380A1 (en) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106999450A (en) * | 2014-11-05 | 2017-08-01 | 弗莱塞斯生物科学公司 | immunomodulator |
| CN107205970A (en) * | 2014-11-05 | 2017-09-26 | 弗莱塞斯生物科学公司 | Immunomodulator |
| WO2017192813A1 (en) * | 2016-05-04 | 2017-11-09 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| CN107427499A (en) * | 2014-11-05 | 2017-12-01 | 弗莱塞斯生物科学公司 | Immunomodulator |
-
2019
- 2019-02-02 CN CN201980012783.6A patent/CN111868032B/en active Active
- 2019-02-02 WO PCT/CN2019/074642 patent/WO2019154380A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106999450A (en) * | 2014-11-05 | 2017-08-01 | 弗莱塞斯生物科学公司 | immunomodulator |
| CN107205970A (en) * | 2014-11-05 | 2017-09-26 | 弗莱塞斯生物科学公司 | Immunomodulator |
| CN107427499A (en) * | 2014-11-05 | 2017-12-01 | 弗莱塞斯生物科学公司 | Immunomodulator |
| WO2017192813A1 (en) * | 2016-05-04 | 2017-11-09 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
Non-Patent Citations (1)
| Title |
|---|
| 442896-85-7;Registry;《Registry》;20020807;442896-85-7 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019154380A1 (en) | 2019-08-15 |
| CN111868032A (en) | 2020-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111836798B (en) | Prodrugs of ketamine, compositions and uses thereof | |
| JP6704535B2 (en) | Lanosterol prodrug compound, its production method and application | |
| CN108025004B (en) | muscarinic agonists | |
| CN114685488A (en) | Compounds as SOS1 inhibitors and uses thereof | |
| US9688654B2 (en) | Compounds inhibiting leucine-rich repeat kinase enzyme activity | |
| CA3111649A1 (en) | Tricyclic compounds acting on crbn proteins | |
| EP3390374A1 (en) | Hydroxyalkylamine- and hydroxycycloalkylamine-substituted diamine-arylsulfonamide compounds with selective activity in voltage-gated sodium channels | |
| BR112020020008A2 (en) | 6-AMINOISOQUINOLIN MONO ACID SALTS AND THEIR USES | |
| BRPI0712730A2 (en) | Compound of Formula I; METHOD FOR TREATMENT OF A CENTRAL NERVOUS SYSTEM DISORDER RELATED TO OR AFFECTED BY THE 5-HT6 RECEIVER IN A PATIENT NEEDING THIS; PHARMACEUTICAL COMPOSITION; PROCESS FOR PREPARING A COMPOUND OF FORMULA IA; AND USE OF A COMPOUND FORMULA I | |
| KR20170012444A (en) | Alkyl derivatives of 1-oxa-4,9-diazaspiro undecane compounds having multimodal activity against pain | |
| JP2017516817A (en) | Amide derivatives of 1-oxa-4,9-diazaspiroundecane compounds having various activities against pain | |
| US20240400547A1 (en) | 2,6-piperidinedione compound and application thereof | |
| JP6884974B2 (en) | Oxa-azaspiro compounds for the treatment of substance abuse and drug addiction | |
| CN115093428A (en) | 2, 6-dioxaspiro [4,5] decane derivative, preparation method and medical application thereof | |
| CN105339369B (en) | A kind of simultaneously [1,2 a] benzazolyl compounds and preparation method thereof and the application in medicine of pyrazine | |
| WO2019149244A1 (en) | Jak inhibitor and use thereof | |
| CN111868032B (en) | Kynurenine pathway inhibitors | |
| JP2019516736A (en) | Tetrahydropyran and thiopyran derivatives having various activities against pain | |
| WO2020135454A1 (en) | Class of steroid compounds and use thereof | |
| CN114105977B (en) | Estrogen receptor modulator compounds and uses thereof | |
| JP6884973B2 (en) | Oxadiazaspiro compounds that are active against pain | |
| JP2018531263A6 (en) | Oxadiazaspiro compounds with activity against pain | |
| EP3150598B1 (en) | Substituted tropane derivatives | |
| RU2779618C2 (en) | Oxadiazaspiro-compounds for treatment of drug abuse and addiction | |
| WO2024059017A2 (en) | Phenalkylamines and methods of making and using the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |