CN111393367A - N-para-sulfonium salt substituted pyrazoline derivative, photocuring composition and preparation method - Google Patents
N-para-sulfonium salt substituted pyrazoline derivative, photocuring composition and preparation method Download PDFInfo
- Publication number
- CN111393367A CN111393367A CN202010354947.8A CN202010354947A CN111393367A CN 111393367 A CN111393367 A CN 111393367A CN 202010354947 A CN202010354947 A CN 202010354947A CN 111393367 A CN111393367 A CN 111393367A
- Authority
- CN
- China
- Prior art keywords
- substituted
- formula
- unsubstituted
- sulfonium salt
- pyrazoline derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 150000003219 pyrazolines Chemical class 0.000 title claims abstract description 60
- 239000000203 mixture Substances 0.000 title claims abstract description 23
- 238000002360 preparation method Methods 0.000 title claims abstract description 21
- 238000000016 photochemical curing Methods 0.000 title description 11
- 238000000034 method Methods 0.000 claims abstract description 39
- 230000008569 process Effects 0.000 claims abstract description 18
- 150000001875 compounds Chemical class 0.000 claims description 81
- -1 aromatic heterocyclic radical Chemical class 0.000 claims description 71
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 55
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 45
- 238000006243 chemical reaction Methods 0.000 claims description 42
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 39
- 239000003054 catalyst Substances 0.000 claims description 29
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 28
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical class O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 claims description 27
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 23
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 22
- 150000001450 anions Chemical class 0.000 claims description 22
- HKOOXMFOFWEVGF-UHFFFAOYSA-N phenylhydrazine Chemical compound NNC1=CC=CC=C1 HKOOXMFOFWEVGF-UHFFFAOYSA-N 0.000 claims description 18
- 229940067157 phenylhydrazine Drugs 0.000 claims description 18
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 claims description 18
- 229910001868 water Inorganic materials 0.000 claims description 18
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 17
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 claims description 17
- 125000000623 heterocyclic group Chemical group 0.000 claims description 16
- 150000003254 radicals Chemical class 0.000 claims description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 15
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 15
- 229910052751 metal Inorganic materials 0.000 claims description 15
- 239000002184 metal Substances 0.000 claims description 15
- 239000002253 acid Substances 0.000 claims description 14
- 239000002585 base Substances 0.000 claims description 14
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 14
- 229910052717 sulfur Inorganic materials 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 125000003118 aryl group Chemical group 0.000 claims description 13
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 claims description 12
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 12
- 239000003513 alkali Substances 0.000 claims description 11
- 235000019256 formaldehyde Nutrition 0.000 claims description 11
- 125000003700 epoxy group Chemical group 0.000 claims description 10
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 9
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims description 9
- 239000007864 aqueous solution Substances 0.000 claims description 8
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 8
- 125000001424 substituent group Chemical group 0.000 claims description 8
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 7
- 229920000642 polymer Polymers 0.000 claims description 7
- 239000002244 precipitate Substances 0.000 claims description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 7
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 claims description 6
- 239000000178 monomer Substances 0.000 claims description 6
- 229910052757 nitrogen Inorganic materials 0.000 claims description 6
- 239000003960 organic solvent Substances 0.000 claims description 6
- 229910052763 palladium Inorganic materials 0.000 claims description 6
- 238000010992 reflux Methods 0.000 claims description 6
- 150000003462 sulfoxides Chemical class 0.000 claims description 6
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 5
- 239000002250 absorbent Substances 0.000 claims description 5
- 230000002745 absorbent Effects 0.000 claims description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 5
- 229910052794 bromium Inorganic materials 0.000 claims description 5
- 125000005842 heteroatom Chemical group 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical class CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 4
- 239000003377 acid catalyst Substances 0.000 claims description 4
- 230000009471 action Effects 0.000 claims description 4
- 125000001624 naphthyl group Chemical group 0.000 claims description 4
- 229910052760 oxygen Inorganic materials 0.000 claims description 4
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical class OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 claims description 4
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 claims description 4
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 3
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 3
- KAIPKTYOBMEXRR-UHFFFAOYSA-N 1-butyl-3-methyl-2h-imidazole Chemical compound CCCCN1CN(C)C=C1 KAIPKTYOBMEXRR-UHFFFAOYSA-N 0.000 claims description 3
- BXOAIZOIDUQOFA-UHFFFAOYSA-M 1-butyl-3-methylimidazol-3-ium;hydroxide Chemical compound [OH-].CCCC[N+]=1C=CN(C)C=1 BXOAIZOIDUQOFA-UHFFFAOYSA-M 0.000 claims description 3
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 claims description 3
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 3
- 229930194542 Keto Natural products 0.000 claims description 3
- 125000002837 carbocyclic group Chemical group 0.000 claims description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- FHBSGPWHCCIQPG-UHFFFAOYSA-N hydroxy-methyl-oxo-sulfanylidene-$l^{6}-sulfane Chemical class CS(S)(=O)=O FHBSGPWHCCIQPG-UHFFFAOYSA-N 0.000 claims description 3
- 239000002608 ionic liquid Substances 0.000 claims description 3
- 125000000468 ketone group Chemical group 0.000 claims description 3
- 239000000126 substance Substances 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 2
- 125000001318 4-trifluoromethylbenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])*)C(F)(F)F 0.000 claims description 2
- 229910019142 PO4 Inorganic materials 0.000 claims description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 claims description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 claims description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 claims description 2
- 239000004327 boric acid Substances 0.000 claims description 2
- 238000006555 catalytic reaction Methods 0.000 claims description 2
- 239000012024 dehydrating agents Substances 0.000 claims description 2
- UQSQSQZYBQSBJZ-UHFFFAOYSA-N fluorosulfonic acid Chemical compound OS(F)(=O)=O UQSQSQZYBQSBJZ-UHFFFAOYSA-N 0.000 claims description 2
- 229910017053 inorganic salt Inorganic materials 0.000 claims description 2
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 claims description 2
- 239000001301 oxygen Substances 0.000 claims description 2
- 125000006505 p-cyanobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C#N)C([H])([H])* 0.000 claims description 2
- 229960003424 phenylacetic acid Drugs 0.000 claims description 2
- 239000003279 phenylacetic acid Substances 0.000 claims description 2
- 239000010452 phosphate Substances 0.000 claims description 2
- 229910052711 selenium Inorganic materials 0.000 claims description 2
- 239000011592 zinc chloride Substances 0.000 claims description 2
- 235000005074 zinc chloride Nutrition 0.000 claims description 2
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims 1
- 150000008065 acid anhydrides Chemical group 0.000 claims 1
- 238000003786 synthesis reaction Methods 0.000 abstract description 64
- 230000015572 biosynthetic process Effects 0.000 abstract description 58
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical class S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 abstract description 19
- 238000010521 absorption reaction Methods 0.000 abstract description 16
- 238000009776 industrial production Methods 0.000 abstract description 5
- 125000002755 pyrazolinyl group Chemical group 0.000 abstract description 4
- 239000002994 raw material Substances 0.000 abstract description 2
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 102
- 239000000047 product Substances 0.000 description 71
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 27
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 15
- 239000000243 solution Substances 0.000 description 15
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 14
- 239000011248 coating agent Substances 0.000 description 12
- 238000000576 coating method Methods 0.000 description 12
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 11
- 238000001914 filtration Methods 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 238000000862 absorption spectrum Methods 0.000 description 9
- 239000003822 epoxy resin Substances 0.000 description 9
- 229920000647 polyepoxide Polymers 0.000 description 9
- 230000035484 reaction time Effects 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 8
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical compound CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 7
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 7
- 150000003839 salts Chemical class 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 6
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 6
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 6
- GNKZMNRKLCTJAY-UHFFFAOYSA-N 4'-Methylacetophenone Chemical compound CC(=O)C1=CC=C(C)C=C1 GNKZMNRKLCTJAY-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 6
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 5
- GYZLOYUZLJXAJU-UHFFFAOYSA-N diglycidyl ether Chemical class C1OC1COCC1CO1 GYZLOYUZLJXAJU-UHFFFAOYSA-N 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 230000005284 excitation Effects 0.000 description 5
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- IGJQUJNPMOYEJY-UHFFFAOYSA-N 2-acetylpyrrole Chemical compound CC(=O)C1=CC=CN1 IGJQUJNPMOYEJY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 238000012512 characterization method Methods 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 238000001723 curing Methods 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 238000012544 monitoring process Methods 0.000 description 4
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 4
- 229940043267 rhodamine b Drugs 0.000 description 4
- JPJALAQPGMAKDF-UHFFFAOYSA-N selenium dioxide Chemical compound O=[Se]=O JPJALAQPGMAKDF-UHFFFAOYSA-N 0.000 description 4
- 230000003595 spectral effect Effects 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 3
- 235000019441 ethanol Nutrition 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 238000003825 pressing Methods 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000007790 scraping Methods 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- WIEXMPDBTYDSQF-UHFFFAOYSA-N 1,3-bis(furan-2-yl)propan-2-one Chemical compound C=1C=COC=1CC(=O)CC1=CC=CO1 WIEXMPDBTYDSQF-UHFFFAOYSA-N 0.000 description 2
- FBNAYEYTRHHEOB-UHFFFAOYSA-N 2,3,5-triphenyl-1,3-dihydropyrazole Chemical compound N1N(C=2C=CC=CC=2)C(C=2C=CC=CC=2)C=C1C1=CC=CC=C1 FBNAYEYTRHHEOB-UHFFFAOYSA-N 0.000 description 2
- UBUGJNBXPJDIJY-UHFFFAOYSA-N 2,3-diphenyl-5-thiophen-2-yl-1,3-dihydropyrazole Chemical compound C1(=CC=CC=C1)N1NC(=CC1C1=CC=CC=C1)C=1SC=CC=1 UBUGJNBXPJDIJY-UHFFFAOYSA-N 0.000 description 2
- WYJOVVXUZNRJQY-UHFFFAOYSA-N 2-Acetylthiophene Chemical compound CC(=O)C1=CC=CS1 WYJOVVXUZNRJQY-UHFFFAOYSA-N 0.000 description 2
- AOBIOSPNXBMOAT-UHFFFAOYSA-N 2-[2-(oxiran-2-ylmethoxy)ethoxymethyl]oxirane Chemical compound C1OC1COCCOCC1CO1 AOBIOSPNXBMOAT-UHFFFAOYSA-N 0.000 description 2
- DGUJJOYLOCXENZ-UHFFFAOYSA-N 4-[2-[4-(oxiran-2-ylmethoxy)phenyl]propan-2-yl]phenol Chemical compound C=1C=C(OCC2OC2)C=CC=1C(C)(C)C1=CC=C(O)C=C1 DGUJJOYLOCXENZ-UHFFFAOYSA-N 0.000 description 2
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 2
- 102100025403 Epoxide hydrolase 1 Human genes 0.000 description 2
- 239000004593 Epoxy Substances 0.000 description 2
- 101100451963 Homo sapiens EPHX1 gene Proteins 0.000 description 2
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- GGRQLKPIJPFWEZ-UHFFFAOYSA-N cycloprop-2-en-1-one Chemical class O=C1C=C1 GGRQLKPIJPFWEZ-UHFFFAOYSA-N 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- OIRDBPQYVWXNSJ-UHFFFAOYSA-N methyl trifluoromethansulfonate Chemical compound COS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-UHFFFAOYSA-N 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- FXLOVSHXALFLKQ-UHFFFAOYSA-N p-tolualdehyde Chemical compound CC1=CC=C(C=O)C=C1 FXLOVSHXALFLKQ-UHFFFAOYSA-N 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000004321 preservation Methods 0.000 description 2
- 238000007142 ring opening reaction Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910001495 sodium tetrafluoroborate Inorganic materials 0.000 description 2
- KVCGISUBCHHTDD-UHFFFAOYSA-M sodium;4-methylbenzenesulfonate Chemical compound [Na+].CC1=CC=C(S([O-])(=O)=O)C=C1 KVCGISUBCHHTDD-UHFFFAOYSA-M 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 235000011149 sulphuric acid Nutrition 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229930192474 thiophene Natural products 0.000 description 2
- JNBRDOQHPXUXLY-UHFFFAOYSA-N (2,5-difluorophenyl)hydrazine Chemical compound NNC1=CC(F)=CC=C1F JNBRDOQHPXUXLY-UHFFFAOYSA-N 0.000 description 1
- SCZGZDLUGUYLRV-UHFFFAOYSA-N (2-methylphenyl)hydrazine Chemical compound CC1=CC=CC=C1NN SCZGZDLUGUYLRV-UHFFFAOYSA-N 0.000 description 1
- XEVQXKKKAVVSMW-CPCISQLKSA-N (6s,7as)-6-hydroxy-4,4,7a-trimethyl-6,7-dihydro-5h-1-benzofuran-2-one Chemical compound C1[C@@H](O)CC(C)(C)C2=CC(=O)O[C@]21C XEVQXKKKAVVSMW-CPCISQLKSA-N 0.000 description 1
- UWFRVQVNYNPBEF-UHFFFAOYSA-N 1-(2,4-dimethylphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=C(C)C=C1C UWFRVQVNYNPBEF-UHFFFAOYSA-N 0.000 description 1
- IQQRAVYLUAZUGX-UHFFFAOYSA-N 1-butyl-3-methylimidazolium Chemical compound CCCCN1C=C[N+](C)=C1 IQQRAVYLUAZUGX-UHFFFAOYSA-N 0.000 description 1
- OSSNTDFYBPYIEC-UHFFFAOYSA-N 1-ethenylimidazole Chemical compound C=CN1C=CN=C1 OSSNTDFYBPYIEC-UHFFFAOYSA-N 0.000 description 1
- YWDALJMLXXWXFP-UHFFFAOYSA-N 1-ethyl-4-[2-(4-methoxyphenyl)ethynyl]benzene Chemical compound C1=CC(CC)=CC=C1C#CC1=CC=C(OC)C=C1 YWDALJMLXXWXFP-UHFFFAOYSA-N 0.000 description 1
- XKQQFGSSJLXNRD-UHFFFAOYSA-N 1-methyl-4-[3-[3-(6-methyl-7-oxabicyclo[4.1.0]heptan-3-yl)prop-2-enoxy]prop-1-enyl]-7-oxabicyclo[4.1.0]heptane Chemical compound CC12C(CC(CC1)C=CCOCC=CC1CC3C(CC1)(O3)C)O2 XKQQFGSSJLXNRD-UHFFFAOYSA-N 0.000 description 1
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical compound C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 description 1
- ZFOCMWZKYXXLBD-UHFFFAOYSA-N 2,4-dimethylfuro[3,2-c]quinoline Chemical compound C1=CC=CC2=C(OC(C)=C3)C3=C(C)N=C21 ZFOCMWZKYXXLBD-UHFFFAOYSA-N 0.000 description 1
- VMSDHCPTIMUSRH-UHFFFAOYSA-N 2,5-diphenyl-3-thiophen-2-yl-1,3-dihydropyrazole Chemical compound C1(=CC=CC=C1)N1NC(=CC1C=1SC=CC1)C1=CC=CC=C1 VMSDHCPTIMUSRH-UHFFFAOYSA-N 0.000 description 1
- STMDPCBYJCIZOD-UHFFFAOYSA-N 2-(2,4-dinitroanilino)-4-methylpentanoic acid Chemical compound CC(C)CC(C(O)=O)NC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O STMDPCBYJCIZOD-UHFFFAOYSA-N 0.000 description 1
- BBBUAWSVILPJLL-UHFFFAOYSA-N 2-(2-ethylhexoxymethyl)oxirane Chemical compound CCCCC(CC)COCC1CO1 BBBUAWSVILPJLL-UHFFFAOYSA-N 0.000 description 1
- SNGZGCFWZHOVOS-UHFFFAOYSA-N 2-(2-methyloctoxymethyl)oxirane Chemical compound CCCCCCC(C)COCC1CO1 SNGZGCFWZHOVOS-UHFFFAOYSA-N 0.000 description 1
- PDAIFTPWQFGJGD-UHFFFAOYSA-N 2-(4-methoxyphenyl)pyrimidine Chemical compound C1=CC(OC)=CC=C1C1=NC=CC=N1 PDAIFTPWQFGJGD-UHFFFAOYSA-N 0.000 description 1
- NUUPCWDAIXYCAG-UHFFFAOYSA-N 2-(4-methylphenyl)-1,3-dihydropyrazole Chemical compound C1=CC(C)=CC=C1N1NC=CC1 NUUPCWDAIXYCAG-UHFFFAOYSA-N 0.000 description 1
- YSUQLAYJZDEMOT-UHFFFAOYSA-N 2-(butoxymethyl)oxirane Chemical compound CCCCOCC1CO1 YSUQLAYJZDEMOT-UHFFFAOYSA-N 0.000 description 1
- WAPRZVXVTPSWEB-UHFFFAOYSA-N 2-[(2-butan-2-ylphenoxy)methyl]oxirane Chemical compound CCC(C)C1=CC=CC=C1OCC1OC1 WAPRZVXVTPSWEB-UHFFFAOYSA-N 0.000 description 1
- HHRACYLRBOUBKM-UHFFFAOYSA-N 2-[(4-tert-butylphenoxy)methyl]oxirane Chemical compound C1=CC(C(C)(C)C)=CC=C1OCC1OC1 HHRACYLRBOUBKM-UHFFFAOYSA-N 0.000 description 1
- SYEWHONLFGZGLK-UHFFFAOYSA-N 2-[1,3-bis(oxiran-2-ylmethoxy)propan-2-yloxymethyl]oxirane Chemical compound C1OC1COCC(OCC1OC1)COCC1CO1 SYEWHONLFGZGLK-UHFFFAOYSA-N 0.000 description 1
- DOQQIEJZBMOSKR-UHFFFAOYSA-N 2-[2,3,4,5-tetramethyl-6-(oxiran-2-ylmethoxy)phenyl]phenol Chemical compound CC=1C(C)=C(C)C(C)=C(C=2C(=CC=CC=2)O)C=1OCC1CO1 DOQQIEJZBMOSKR-UHFFFAOYSA-N 0.000 description 1
- KUAUJXBLDYVELT-UHFFFAOYSA-N 2-[[2,2-dimethyl-3-(oxiran-2-ylmethoxy)propoxy]methyl]oxirane Chemical compound C1OC1COCC(C)(C)COCC1CO1 KUAUJXBLDYVELT-UHFFFAOYSA-N 0.000 description 1
- IEMMBWWQXVXBEU-UHFFFAOYSA-N 2-acetylfuran Chemical compound CC(=O)C1=CC=CO1 IEMMBWWQXVXBEU-UHFFFAOYSA-N 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- VYSRZETUSAOIMP-UHFFFAOYSA-N 2-furanacetic acid Chemical compound OC(=O)CC1=CC=CO1 VYSRZETUSAOIMP-UHFFFAOYSA-N 0.000 description 1
- 239000001431 2-methylbenzaldehyde Substances 0.000 description 1
- FCUOBLMMCCBYKO-UHFFFAOYSA-N 2-oxochromene-4-carbaldehyde Chemical compound C1=CC=CC2=C1OC(=O)C=C2C=O FCUOBLMMCCBYKO-UHFFFAOYSA-N 0.000 description 1
- MRXCTUHFMJGHPM-UHFFFAOYSA-N 3-(furan-2-yl)-2,5-diphenyl-1,3-dihydropyrazole Chemical compound N1N(C=2C=CC=CC=2)C(C=2OC=CC=2)C=C1C1=CC=CC=C1 MRXCTUHFMJGHPM-UHFFFAOYSA-N 0.000 description 1
- MECNWXGGNCJFQJ-UHFFFAOYSA-N 3-piperidin-1-ylpropane-1,2-diol Chemical compound OCC(O)CN1CCCCC1 MECNWXGGNCJFQJ-UHFFFAOYSA-N 0.000 description 1
- PSGQCCSGKGJLRL-UHFFFAOYSA-N 4-methyl-2h-chromen-2-one Chemical compound C1=CC=CC2=C1OC(=O)C=C2C PSGQCCSGKGJLRL-UHFFFAOYSA-N 0.000 description 1
- PIMQQGJMDMAZGT-UHFFFAOYSA-N 4-methylthiobenzaldehyde Chemical compound CC1=CC=C(C=S)C=C1 PIMQQGJMDMAZGT-UHFFFAOYSA-N 0.000 description 1
- GRACMMPOKSUSPN-UHFFFAOYSA-N 5-(4-methylphenyl)-2-phenyl-3-thiophen-2-yl-1,3-dihydropyrazole Chemical compound C1(=CC=CC=C1)N1NC(=CC1C=1SC=CC=1)C1=CC=C(C=C1)C GRACMMPOKSUSPN-UHFFFAOYSA-N 0.000 description 1
- ORTNTAAZJSNACP-UHFFFAOYSA-N 6-(oxiran-2-ylmethoxy)hexan-1-ol Chemical compound OCCCCCCOCC1CO1 ORTNTAAZJSNACP-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- OGFDPHNOHNFRDB-UHFFFAOYSA-N C(=O)OC1(CC2C(CC1)O2)CCC2CC1C(CC2)O1 Chemical compound C(=O)OC1(CC2C(CC1)O2)CCC2CC1C(CC2)O1 OGFDPHNOHNFRDB-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- WPYCRFCQABTEKC-UHFFFAOYSA-N Diglycidyl resorcinol ether Chemical compound C1OC1COC(C=1)=CC=CC=1OCC1CO1 WPYCRFCQABTEKC-UHFFFAOYSA-N 0.000 description 1
- JJHHIJFTHRNPIK-UHFFFAOYSA-N Diphenyl sulfoxide Chemical compound C=1C=CC=CC=1S(=O)C1=CC=CC=C1 JJHHIJFTHRNPIK-UHFFFAOYSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-L PdCl2(PPh3)2 Substances [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 1
- FQYUMYWMJTYZTK-UHFFFAOYSA-N Phenyl glycidyl ether Chemical compound C1OC1COC1=CC=CC=C1 FQYUMYWMJTYZTK-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- AIGAZQPHXLWMOJ-UHFFFAOYSA-N Tanshinone I Chemical compound C1=CC2=C(C)C=CC=C2C(C(=O)C2=O)=C1C1=C2C(C)=CO1 AIGAZQPHXLWMOJ-UHFFFAOYSA-N 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- ZXUJYIQZKNWLQN-UHFFFAOYSA-N [3-(7-oxabicyclo[4.1.0]heptan-3-ylmethyl)-7-oxabicyclo[4.1.0]heptan-3-yl] formate Chemical compound C(=O)OC1(CC2C(CC1)O2)CC1CC2C(CC1)O2 ZXUJYIQZKNWLQN-UHFFFAOYSA-N 0.000 description 1
- 229920006243 acrylic copolymer Polymers 0.000 description 1
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 150000003934 aromatic aldehydes Chemical class 0.000 description 1
- XTKDAFGWCDAMPY-UHFFFAOYSA-N azaperone Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCN(C=2N=CC=CC=2)CC1 XTKDAFGWCDAMPY-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- JRPRCOLKIYRSNH-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) benzene-1,2-dicarboxylate Chemical compound C=1C=CC=C(C(=O)OCC2OC2)C=1C(=O)OCC1CO1 JRPRCOLKIYRSNH-UHFFFAOYSA-N 0.000 description 1
- XFUOBHWPTSIEOV-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) cyclohexane-1,2-dicarboxylate Chemical compound C1CCCC(C(=O)OCC2OC2)C1C(=O)OCC1CO1 XFUOBHWPTSIEOV-UHFFFAOYSA-N 0.000 description 1
- 239000004841 bisphenol A epoxy resin Substances 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000010538 cationic polymerization reaction Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000001793 charged compounds Chemical class 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- ZZTCPWRAHWXWCH-UHFFFAOYSA-N diphenylmethanediamine Chemical compound C=1C=CC=CC=1C(N)(N)C1=CC=CC=C1 ZZTCPWRAHWXWCH-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 238000005286 illumination Methods 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000012263 liquid product Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 125000006431 methyl cyclopropyl group Chemical group 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229920003986 novolac Polymers 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- RPQRDASANLAFCM-UHFFFAOYSA-N oxiran-2-ylmethyl prop-2-enoate Chemical compound C=CC(=O)OCC1CO1 RPQRDASANLAFCM-UHFFFAOYSA-N 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- KRIOVPPHQSLHCZ-UHFFFAOYSA-N propiophenone Chemical compound CCC(=O)C1=CC=CC=C1 KRIOVPPHQSLHCZ-UHFFFAOYSA-N 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000001725 pyrenyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000027756 respiratory electron transport chain Effects 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 150000003385 sodium Chemical class 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000010025 steaming Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- JIPKHPRMLCLPSP-UHFFFAOYSA-N sulfanium;methanesulfonate Chemical compound [SH3+].CS([O-])(=O)=O JIPKHPRMLCLPSP-UHFFFAOYSA-N 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229920006305 unsaturated polyester Polymers 0.000 description 1
- 238000007738 vacuum evaporation Methods 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/06—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/46—Polymerisation initiated by wave energy or particle radiation
- C08F2/48—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light
- C08F2/50—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light with sensitising agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/68—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used
- C08G59/687—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used containing sulfur
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The present application relates to N-para-sulfonium salt substituted pyrazoline derivatives represented by the following formula (I), photocurable compositions and processes for producing the sameAnd a preparation method of the pyrazoline derivative substituted by the N-para-sulfonium salt shown in the formula (I). The N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I) has good absorption at the wavelength of more than 350nm, and compared with 5-substituted sulfonium salt, the N-para-sulfonium salt substituted pyrazoline derivative has the advantages of simpler and simpler molecular synthesis steps and reduced raw material cost, and is more suitable for industrial production and application.
Description
Technical Field
The invention belongs to the field of new material organic chemistry, and particularly relates to preparation of an N-para-sulfonium salt substituted pyrazoline derivative.
Background
In the beginning of the 20 th century, IBM ita first proposed a chemically amplified resist (h.ito, c.g.wilson, j.m.s.frechet, Process of 1982 Symposium on V L SI Technology,1982, 1: 86-87). after more than thirty years of development, the chemically amplified resist has gained wide acceptance and application in the field of imaging materials.
Among various photoacid generators, sulfonium salts have been widely regarded and applied in industrial production and scientific research due to their good thermal stability, molecular designability, and excellent photogenerated acidity. However, the conjugated system of sulfonium salt which has been commercially used at present is small, so that the absorption wavelength of the main absorption peak thereof is relatively small, and the application thereof in the long wavelength field cannot be satisfied. The sulfonium salt with a larger wavelength absorption peak which is designed and synthesized at present mainly comprises a sulfonium salt (X.Y.Wu, M.jin, J.C.Xie, J.P.Malverl, D.C.Wan, Chemistry-A European Journal,2017,23(62):15783-15789) with a D-pi-A (D is an electron donor and A is an electron acceptor) structure, and in order to enable the sulfonium salt to have good absorption at a long wavelength (>350nm), the sulfonium salt needs to have a larger conjugated structure, and the synthesis of the larger conjugated structure is difficult, the steps are more, the yield is lower, and the industrial production is difficult.
Therefore, how to obtain sulfonium salts with a large wavelength absorption peak as photoacid generators by a simple synthesis method is a problem to be solved urgently.
Disclosure of Invention
The present inventors have conducted extensive studies with respect to the deficiencies of the prior art and, as a result, have found that an N-para-sulfonium salt-substituted pyrazoline derivative represented by formula (I) of the present invention obtained by introducing a pyrazoline group into a sulfonium salt exhibits good absorption at wavelengths of 350nm or more by causing an absorption peak thereof to red-shift to the near ultraviolet-visible region by intramolecular electron transfer, and thus the N-para-sulfonium salt-substituted pyrazoline derivative represented by formula (I) of the present invention further overlaps with the emission wavelength of a currently commercially available L ED light source, thereby improving the excitation efficiency.
Furthermore, the preparation method of the N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I) is simple and convenient, high in yield, low in cost and suitable for industrial production and application. The N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I) has good application prospect as a photoacid generator.
Specifically, the present invention provides the following scheme:
in a first aspect, the present invention provides an N-p-sulfonium salt-substituted pyrazoline derivative represented by the following formula (I),

wherein:
R1、R2each independently selected from C1-12Alkyl of (A), CSubstituted or substituted by 1-5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, or unsubstituted or substituted by 1-8R7Substituted benzoaromatic heterocyclic groups;
R3selected from H, C1-6Alkyl radical, C3-6Cycloalkyl, unsubstituted or substituted by 1-5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, unsubstituted or substituted by 1-8R7Substituted benzoaromatic heterocyclic groups;
R4selected from unsubstituted or substituted by 1-4RaSubstituted C1-6Alkyl, -F, -Cl, -Br, -I, -CN, -CF2CF3、-CF3、-NO2、-NRbRb、-ORb、-SRb、-C(=O)Rb、-CO2Rb、-OC(=O)Rb、 -NRbC(=O)Rb、-S(=O)Rb、-S(=O)2Rb;
y is 0, 1, 2,3 or 4;
R5、R6each independently selected from C1-12Unsubstituted or substituted by 1 to 5R7Substituted benzyl, unsubstituted or substituted by 1 to 5R7Substituted phenyl;
R7each independently selected from unsubstituted or substituted by 1-5RaSubstituted C1-6Alkyl, -F, -Cl, -Br, -I, -CN, -CF2CF3、-CF3、-NO2、-NRbRb、-ORb、-SRb、-C(=O)Rb、-CO2Rb、 -OC(=O)Rb、-NRbC(=O)Rb、-S(=O)Rb、-S(=O)2RbUnsubstituted or substituted by 1 to 5RcSubstituted carbocyclic ring, unsubstituted or substituted by 1 to 5RdSubstituted heterocycle, OR, P (═ O) (OR)b)2;
RaEach independently selected from C1-6Alkyl group, (CH)2)rC3-6Cycloalkyl or- (CH)2)rA phenyl group;
Rbeach independently selected from H, unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted- (CH)2)rPh;
RcEach independently selected from unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted (CH)2)rPh;
RdEach independently selected from unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted (CH)2)rPh;
ReEach independently selected from-F, -Cl, -Br, -I, -OH, -NO2、-CN,-CF3、-CF2CF3、 C1-4Alkyl radical, C1-4Alkoxy radical, C3-7Cycloalkyl, phenyl, benzyl, phenethyl, naphthyl, heterocyclic aryl, or, keto;
each r is independently 0, 1, 2,3, or 4;
X-is an anion.
In a second aspect, the present invention provides a photocurable composition comprising an N-para-sulfonium salt-substituted pyrazoline derivative represented by formula (I) of the present invention and a polymerizable component comprising a monomer or polymer having an ethylenic bond or an epoxy group.
In a third aspect, the present invention provides a process for producing an N-p-sulfonium salt-substituted pyrazoline derivative represented by the formula (I), which comprises the following step (c):

in the above step (c), the compound represented by the formula (I) -b and R5、R6Substituted sulfoxide by Metal+X-The compound containing the metal element reacts to obtain the N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I),
the R is1、R2、R3、R4、R5、R6、n、y、X-The definition of (A) is the same as that of the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I).
The N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I) has good absorption at the wavelength of more than 350nm, has good application prospect as a photoacid generator, can be used as a photoinitiator to be applied to a photocuring composition, and also can be used as an intermediate for synthesizing other compounds.
The photocurable composition of the present invention contains the N-para-sulfonium salt-substituted pyrazoline derivative represented by formula (I) of the present invention, and thus has good absorption at a wavelength of 350nm or more.
Compared with 5-substituted sulfonium salt, the preparation method of the N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I) has the advantages of simpler molecular synthesis steps, reduced raw material cost and suitability for industrial production and application.
Drawings
FIG. 1: the anion is PF6 -(I) -1((I) -1-PF) of6 -) And (I) -2((I) -2-PF)6 -) Ultraviolet and visible absorption spectrum of (1).
FIG. 2: (I) -1-PF6 -After rhodamine B is added, the mixture is exposed for different time under L ED light, and then an ultraviolet-visible absorption spectrum is scanned to indicate an ultraviolet absorption spectrogram of the generated acid.
FIG. 3: (I) -2-PF6 -After rhodamine B is added, the mixture is exposed for different time under L ED light, and then ultraviolet-visible absorption spectrum is scanned to indicate the ultraviolet absorption of generated acidAnd (6) spectrum collection.
Detailed Description
[ N-para-sulfonium salt-substituted pyrazoline derivative ]
The N-para-sulfonium salt substituted pyrazoline derivative is shown in the following formula (I),

wherein R is1、R2Each independently selected from C1-12Unsubstituted or substituted by 1 to 5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, or unsubstituted or substituted by 1-8R7Substituted benzoheteroaromatic group.
R3Selected from H, C1-6Alkyl radical, C3-6Cycloalkyl, unsubstituted or substituted by 1-5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, unsubstituted or substituted by 1-8R7Substituted benzoaromatic heterocyclic groups;
R4selected from unsubstituted or substituted by 1-4RaSubstituted C1-6Alkyl, -F, -Cl, -Br, -I, -CN, -CF2CF3、-CF3、-NO2、-NRbRb、-ORb、-SRb、-C(=O)Rb、-CO2Rb、-OC(=O)Rb、 -NRbC(=O)Rb、-S(=O)Rb、-S(=O)2Rb;
y is 0, 1, 2,3 or 4;
R5、R6each independently selected from C1-12Unsubstituted or substituted by 1 to 5R7Substituted benzyl, unsubstituted or substituted by 1 to 5R7Substituted phenyl;
R7each independently selected from unsubstituted or substituted by 1-5RaSubstituted C1-6Alkyl, -F, -Cl, -Br, -I, -CN, -CF2CF3、-CF3、-NO2、-NRbRb、-ORb、-SRb、-C(=O)Rb、-CO2Rb、 -OC(=O)Rb、-NRbC(=O)Rb、-S(=O)Rb、-S(=O)2RbUnsubstituted or substituted by 1 to 5RcSubstituted carbocyclic ring, unsubstituted or substituted by 1 to 5RdSubstituted heterocycle, OR, P (═ O) (OR)b)2;
RaEach independently selected from C1-6Alkyl group, (CH)2)rC3-6Cycloalkyl or- (CH)2)rA phenyl group;
Rbeach independently selected from H, unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted- (CH)2)rPh;
RcEach independently selected from unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted (CH)2)rPh;
RdEach independently selected from unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted (CH)2)rPh;
ReEach independently selected from-F, -Cl, -Br, -I, -OH, -NO2、-CN,-CF3、-CF2CF3、 C1-4Alkyl radical, C1-4Alkoxy radical, C3-7Cycloalkyl, phenyl, benzyl, phenethyl, naphthyl, heterocyclic aryl, or, keto;
each r is independently 0, 1, 2,3, or 4;
X-is an anion.
The foregoing term "C1-12The "alkyl group" of (1) is an alkyl group having 1 to 12 carbon atoms, and may be a linear or branched alkyl group, and is not particularly limited. As "C1-12Examples of the "alkyl group" include, for example, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, tert-pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl and the like.
The foregoing term "C1-6The "alkyl group" of (a) is an alkyl group having 1 to 6 carbon atoms, and may be a linear or branched alkyl group, and is not particularly limited. As "C1-6Examples of the "alkyl group" include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, tert-pentyl and hexyl groups.
The aforementioned term "condensed ring aryl group" means a polycyclic aryl group in which two or more benzene rings are constituted by sharing a ring edge, and examples of the condensed ring aryl group include, for example, naphthyl, anthryl, phenanthryl, pyrenyl, and the like;
the term "aromatic heterocyclic group" refers to a heterocyclic group having aromatic characteristics, and examples of the aromatic heterocyclic group include furyl group, imidazolyl group, pyridyl group and the like.
The term "benzoaromatic heterocyclic group" as used herein means an aromatic heterocyclic group in which a benzene ring is fused with a heterocyclic ring, and examples of the "benzoaromatic heterocyclic group" include quinolyl, indolyl, purinyl and the like.
The foregoing term "C3-6The "cycloalkyl group" is a cycloalkyl group having 3 to 6 carbon atoms, and the term "C" is3-6Examples of cycloalkyl "are, for example, cyclopropyl, methylcyclopropyl, cyclobutyl, cyclopentyl, methylcyclobutyl, dimethylcyclobutyl, cyclohexyl and the like.
The foregoing term "C1-4The "alkyl group" is an alkyl group having 1 to 4 carbon atoms, and is "C1-4Examples of the "alkyl group" include, for example, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl and tert-butyl.
The foregoing "Ph" represents a phenyl group.
The N-para-sulfonium salt shown in formula (I) of the invention is used for substituting pyrazolineIn (A), preferably, R5、 R6Each independently selected from methyl, cyclopropyl, phenyl, benzyl, 4-cyanobenzyl, or 4-trifluoromethylbenzyl. Thus, the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I) can be produced at low cost, and the sulfonium salt can generate a radical or cation after photolysis to initiate radical polymerization or cation polymerization.
In the N-para-sulfonium salt-substituted pyrazoline derivative of the formula (I) of the present invention, R is preferably1、 R2Each independently an aromatic heterocyclic group selected from the following (A) and (B):
(A) a 6-membered aromatic heterocyclic group containing 1 to 3 heteroatoms selected from the group consisting of O, N, S and Se on the heterocyclic ring;
(B) a 5-membered aromatic heterocyclic group containing a hetero atom of any one of the following groups in the heterocyclic ring,
1) 1O, 1N, or, 1S;
2) 1S and 1N, 1O and 1N, or, 2N; or
3) 3N, 1O and 2N, or, 1S and 2N.
In addition, by introducing hybridization into the molecular structure, a new electronic structure can be further formed by the pyrazoline group and the lone pair of electrons of the heteroatom, and the absorption of the molecule is further influenced.
In the N-para-sulfonium salt-substituted pyrazoline derivative of the formula (I) of the present invention, R is preferably1、 R2Each independently selected from the group consisting of the following structural formulae:


wherein R is7The definitions of (a) are the same as those described above.
In the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I) of the present invention, it is preferably selected from the group consisting of compounds represented by the following structural formulae,





wherein, X-Is an anion.
Among the N-p-sulfonium salt-substituted pyrazoline derivatives of the formula (I) of the present invention, X is preferably selected from-Selected from the group consisting of halogens, oxygen-containing acid radicals, borate radicals, phosphate radicals, antimonate radicals, or aluminate radicals. Further preferably, X-Selected from Cl-、CF3SO3 -、CH3SO3 -、p-MePhSO3 -、BF4 -、B(Ph)4 -、B(PhF5)4 -、 PF6 -、SbF6 -Or, Al (t-Bu)4 -。
[ Photocurable composition ]
The photocurable composition of the present invention contains the aforementioned N-para-sulfonium salt-substituted pyrazoline derivative of the present invention and a polymerizable component, and the aforementioned polymerizable component contains a monomer or polymer having an ethylenic bond or an epoxy group.
In the photocurable composition of the present invention, the content of the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I) is preferably 0.1 to 15 parts by weight relative to 100 parts by weight of the total amount of the polymerizable components. More preferably, the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I) is contained in an amount of 0.5 to 10% by weight.
Examples of the monomer having an ethylenic bond include (meth) acrylates, acrolein, olefins, conjugated dienes, styrene, maleic anhydride, fumaric anhydride, vinyl acetate, vinylpyrrolidone, vinylimidazole, (meth) acrylic acid, and (meth) acrylic acid derivatives such as (meth) acrylamide, vinyl halides, vinylidene halides, and the like.
Examples of the monomer having an epoxy group include monofunctional glycidyl ethers, polyfunctional aliphatic glycidyl ethers, polyfunctional aromatic glycidyl ethers, glycidyl esters, and aliphatic epoxy compounds.
Examples of the monofunctional glycidyl ether include allyl glycidyl ether, butyl glycidyl ether, phenyl glycidyl ether, 2-ethylhexyl glycidyl ether, sec-butylphenyl glycidyl ether, tert-butylphenyl glycidyl ether, and 2-methyloctyl glycidyl ether.
Examples of the polyfunctional aliphatic glycidyl ether include 1, 6-hexanediol glycidyl ether, trimethylolpropane triglycidyl ether, neopentyl glycol diglycidyl ether, glycerol triglycidyl ether, ethylene glycol diglycidyl ether, polyethylene glycol diglycidyl ether, and polypropylene glycol diglycidyl ether.
Examples of the polyfunctional aromatic glycidyl ethers include bisphenol a glycidyl ether, bisphenol F glycidyl ether, brominated bisphenol a glycidyl ether, biphenol glycidyl ether, tetramethylbiphenol glycidyl ether, and resorcinol glycidyl ether.
Examples of the glycidyl esters include glycidyl acrylate, glycidyl methacrylate, diglycidyl phthalate, and diglycidyl hexahydrophthalate.
Examples of the aliphatic epoxy compound include 3, 4-epoxycyclohexylmethyl-3, 4-epoxycyclohexyl formate, 3, 4-epoxycyclohexylethyl-3, 4-epoxycyclohexyl formate, ethylene cyclohexenyl dioxide, propylene cyclohexenyl dioxide, and 3, 4-epoxy-4-methylcyclohexyl-2-propenyl oxide.
In the photocurable composition of the present invention, the polymerizable component may be in the form of a polymer such as an oligomer or a prepolymer, or a copolymer formed from at least one of a monomer, an oligomer, and a prepolymer. In addition, it may be in the form of an aqueous dispersion.
Examples of such an ethylenic bond-containing polymer include (meth) acrylic copolymers having a (meth) acryloyl functional group, urethane (meth) acrylates, polyester (meth) acrylates, unsaturated polyesters, polyether (meth) acrylates, silicone (meth) acrylates, epoxy resin (meth) acrylates, and the like which are water-soluble or water-dispersible.
As the aforementioned epoxy group-containing polymer, for example, an epoxy group-containing polymer or resin such as bisphenol a epoxy resin, dicyclopentadiene type epoxy resin, diaminodiphenylmethane type epoxy resin, aminophenol type epoxy resin, naphthalene type epoxy resin, novolak type epoxy resin, biphenyl type epoxy resin, hydrogenated biphenyl type epoxy resin, aliphatic type epoxy resin, and the like can be cited.
[ preparation method of N-para-sulfonium salt substituted pyrazoline derivative ]
The preparation method of the N-para-sulfonium salt substituted pyrazoline derivative comprises the following steps (c):

in the above step (c), the compound represented by the formula (I) -b and R5、R6Substituted sulfoxide by Metal+X-The compound containing the metal element reacts to obtain the N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I),
the R is1、R2、R3、R4、R5、R6、n、y、X-The definition of (A) is the same as that of the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I).
The aforementioned "Metal element-containing compound" is Metal+X-Is represented by a monovalent metal cation with X-The anion composition indicated.
As an example of the aforementioned step (c), for example, a method of reacting a compound represented by the formula (I) -b with R5、R6Substitution of sulfoxides in methanesulfonic acid and phosphorus pentoxide (P)2O5) Generating methylsulfonyl sulfonium salt in the formed system, and then adding the reaction product into Metal+X-The precipitate separated out is recrystallized to obtain the N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I), namely the target product.
Preferably, the aforementioned methanesulfonic acid and phosphorus pentoxide (P)2O5) In the formed system, P2O5The mass fraction in the methanesulfonic acid is 7.5-15%.
Preferably, the aforementioned Metal+X-X in the aqueous solution of (1)-The molar concentration of (b) is about 0.8 to 1.2, and more preferably about equimolar amount to the molar concentration of the sulfonium methanesulfonate salt.
The step (c) may be optionally carried out in a system comprising an organic solvent, an acid catalyst and an inorganic salt under the action of a water absorbent, preferably, the acid catalyst is selected from protonic acids such as hydrochloric acid, sulfuric acid, phosphoric acid, perfluorosulfonic acid and benzenesulfonic acid, or L ewis acids such as aluminum trichloride, ferric trichloride and zinc dichloride.
As an embodiment of the aforementioned process for producing an N-p-sulfonium salt-substituted pyrazoline derivative, R3Is H or C1-6N of the aforementioned formula (I) for alkylA process for producing a pyrazoline derivative substituted with a para-sulfonium salt, wherein the process for producing the compound represented by the formula (I) -b comprises the steps (a) and (b),

in the above step (a), R1、R3Substituted alkyl ketones with R2The substituted formaldehyde reacts in absolute ethyl alcohol by taking alkali as a catalyst to obtain a compound shown as (I) -a. The reaction temperature in the step (a) is preferably room temperature to 60 ℃. The reaction time of the step (a) may be, for example, 1 to 6 hours, preferably 2 to 4 hours;
in the step (b), the compound represented by the formula (I) -a obtained in the step (a) is reacted with a compound containing n R in the presence of a base as a catalyst in anhydrous ethanol4And refluxing phenyl hydrazine of a substituent group, and reacting to generate a compound shown as (I) -b, wherein n is 0-4. The reaction time of the step (b) may be, for example, 1 to 10 hours, preferably 4 to 8 hours;
in the above step (c), the compound represented by the above (I) -b obtained in the above step (b) and R5、R6The substituted sulfoxide is reacted in a system containing methanesulfonic acid and phosphorus pentoxide to generate methylsulfonyl sulfonium salt, and then the reaction system is added into a system containing Metal+X-Separating the precipitate from the aqueous solution of the metal element-containing compound, and recrystallizing the precipitate to obtain the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I).
The base in the step (a) is not particularly limited, and is preferably sodium hydroxide, potassium hydroxide or potassium carbonate.
The base in the step (b) is not particularly limited, and is preferably sodium hydroxide, potassium hydroxide or potassium carbonate.
As another embodiment of the aforementioned process for producing an N-p-sulfonium salt-substituted pyrazoline derivative, there is provided R3Is C1-6A process for producing an N-p-sulfonium salt-substituted pyrazoline derivative represented by the following general formula (I) wherein the compound is represented by the following general formula (I) -bThe preparation method comprises the following steps (a '), (b ') and (c '):

in the step (a'), R is a catalyst of alkali in absolute ethanol1Substituted ethanones and R2Substituting formaldehyde to generate the compound shown in the formula (I) -a'. The reaction time in the step (a') is not particularly limited, but is preferably 2 to 4 hours;
in the above step (b '), the compound represented by the formula (I) -a ' produced in the above step (a ') is reacted with a compound containing n R4And (2) carrying out reflux reaction on phenyl hydrazine of a substituent group in absolute ethyl alcohol by using alkali as a catalyst to generate a compound shown as (I) -b', wherein n is 0-4. The reaction time in the step (b') is not particularly limited, but is preferably 4 to 8 hours;
in the above step (c '), the products (I) -b ' and R of the above step (b ') are3The substituted iodoalkane is reacted in tetrahydrofuran by taking alkali as a catalyst to generate a compound shown as a formula (I) -b, wherein n is 0-4.
The base in the aforementioned step (a') is not particularly limited, and is preferably sodium hydroxide, potassium hydroxide or potassium carbonate.
The base in the aforementioned step (b') is not particularly limited, and is preferably sodium hydroxide, potassium hydroxide or potassium carbonate.
The base in the step (c') is not particularly limited, and lithium diisopropylamide is preferable.
As another embodiment of the aforementioned process for producing an N-p-sulfonium salt-substituted pyrazoline derivative, there is provided R3Is unsubstituted or substituted by 1 to 5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, unsubstituted or substituted by 1-8R7Substituted benzoheteroaromatic radical (R)7The same definition as in the aforementioned N-p-sulfonium salt-substituted pyrazoline derivative represented by the formula (I) and an N-p-sulfonium salt-substituted pyrazoline represented by the formula (I)A process for producing a derivative, wherein the process for producing a compound represented by the formula (I) -b comprises the following steps (a "), (b"), and (c "):

in the aforementioned step (a'), R1Substituted formaldehydes and R2Substituted formaldehyde in 1-butyl-3-methylimidazole ionic liquid [ bmim]OH reacts in the presence of benzimidazole catalysts to generate compounds shown in the formula (I) -a';
in the step (b '), the compound shown in the formula (I) -a' generated in the step (a ') is converted into the compound shown in the formula (I) -b' in benzene by using phosphorus pentoxide as a water absorbent;
in the step (c ″), the compound represented by the formula (I) -b ″ produced in the step (b ") is reacted with phenylhydrazine having n substituents in anhydrous ethanol using a base as a catalyst to produce a compound represented by the formula (I) -b, wherein n is 0 to 4.
Preferably, in the step (a ″), the benzimidazole catalyst is selected from 1, 3-dimethyl-1-H-benzimidazole-3- And (4) iodide.
And (4) iodide.
Preferably, in the aforementioned step (a ″), R1≠R2In this case, the two substituted formaldehydes are not added simultaneously. That is, R is added first1CHO、R2One of CHO and R3CH2And (4) reacting the X, and adding another aldehyde to react after the reaction is finished.
The base in the step (c ") is not particularly limited, and lithium diisopropylamide is preferable.
In another embodiment of the method for producing an N-p-sulfonium salt-substituted pyrazoline derivative, the steps (a ") and (b") may be replaced with the following steps (a '"), (b'"), step (c '"), and step (d'").

In the aforementioned step (a'), R1The substituted phenylacetic acid reacts in tetrahydrofuran by using Dicyclohexylcarbodiimide (DCC) as a dehydrating agent and 4-Dimethylaminopyridine (DMAP) as a catalyst to generate a compound shown as a formula (I) -a'. The reaction time of the step (a' ") can be, for example, 0.1 to 5 hours, preferably 0.5 to 2 hours;
in the step (b '), the compound of formula (I) -a', which is formed in the step (a '), is reacted with bromine in acetic acid to form the compound of formula (I) -b'. The reaction time of the step (b' ") can be, for example, 0.5 to 6 hours, preferably 1 to 4 hours;
in the step (c '), the compound represented by the formula (I) -b ', which is generated in the step (b '), is generated in anhydrous dichloromethane by using a base as a catalyst. The reaction time of the step (c') may be, for example, 0.5 to 4 hours, preferably 1 to 2 hours;
in the step (d '), the compound represented by the formula (I) -c ', which is formed in the step (c '), is reacted with R in 1, 4-dioxane under the action of a palladium catalyst3And (3) reacting the substituted boric acid compound to generate the compound shown as (I) -b'. The reaction time in the step (d') may be, for example, 0.5 to 8 hours, preferably 1 to 5 hours.
The base in the step (c' ") is not particularly limited, and is preferably an organic strong base such as triethylamine;
preferably, the palladium-based catalyst in the step (d') is not particularly limited, and is preferably Pd (OAc)2、 PdCl2(PPh3)2Or, Pd (PPh)3)4。
In another embodiment of the method for producing an N-p-sulfonium salt-substituted pyrazoline derivative, the steps (a ") and (b") are replaced with the following steps (a ""), (b ""), and (c "").
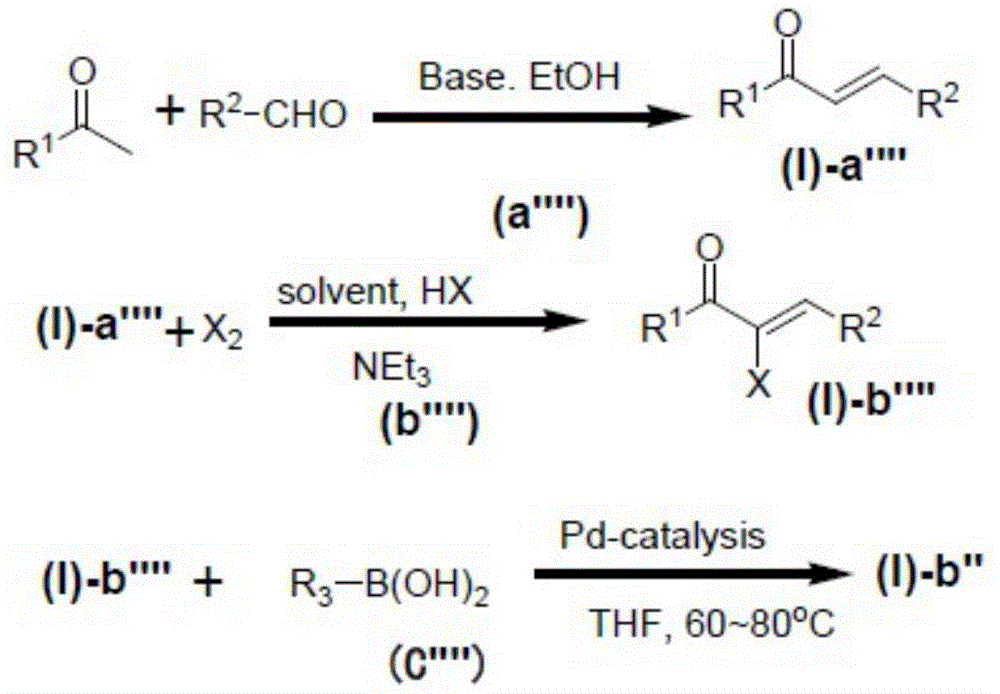
In the above step (a'), R1Substituted ethanones and R2The substituted formaldehyde reacts in absolute ethyl alcohol by taking alkali as a catalyst to generate a compound shown as a formula (I) -a'. The reaction time of the step (a') can be, for example, 1 to 6 hours, preferably 2 to 4 hours;
in the step (b '), the compound shown in the formula (I) -a' generated in the step (a ') reacts with monohalogen and corresponding halogen acid in an organic solvent, and then triethylamine is added to continue the reaction to generate the compound shown in the formula (I) -b';
in the above step (c '), the compound represented by the formula (I) -b ' produced in the above step (b ') and R3-B (OH)2The compound shown in the formula (I) -b' is generated by reaction in tetrahydrofuran under the catalysis of a palladium catalyst. The reaction temperature in the step (c') is preferably 60-80 ℃.
The base in the aforementioned step (a "") is not particularly limited, and is preferably sodium hydroxide, potassium hydroxide or potassium carbonate;
the simple substance of halogen in the step (b') is not particularly limited, and is preferably a simple substance of bromine or iodine. The amount of the halogen acid is 0 to 2 equivalents;
the palladium catalyst in the step (c') is not particularly limited, and is preferably tetrakis (triphenylphosphine) palladium or bis (triphenylphosphine) palladium dichloride.
Examples
In order to more clearly illustrate the disclosure, the disclosure is further described below in connection with preferred embodiments. It is to be understood by persons skilled in the art that the following detailed description is illustrative and not restrictive, and is not to be taken as limiting the scope of the present disclosure.
Example 1: the anion PF is synthesized according to the following route6 -Target molecule (I) -1

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) methanesulfonic acid, phosphorus pentoxide and dark at room temperature for 6 hours;
(d) potassium hexafluorophosphate in water at room temperature.
1. Synthesis of 1, 3-diphenyl-2-en-1-one
Adding acetophenone (24.00g, 0.20mol), benzaldehyde (21.20g, 0.20mol) and solvent absolute ethyl alcohol (60m L) into a 500m L three-neck flask containing a magnetic rotor, stirring at room temperature, then preparing an aqueous solution of sodium hydroxide (16g, 0.40mol, 20m L), dropwise adding the aqueous solution into a reaction system through a constant-pressure dropping funnel, reacting for 2 hours after the addition is finished, monitoring the reaction process through a silica gel chromatography plate, filtering after the reaction is finished, concentrating the filtrate, filtering again, washing the solid obtained by filtering twice with water, washing twice with absolute ethyl alcohol, drying, recrystallizing with absolute ethyl alcohol to obtain yellow crystals, wherein the yield is 84.3% and the yield is HRMSfor C15H12O: 208.0941 (calculated), 208.0923 (actual).
2. Synthesis of 1,3, 5-triphenylpyrazoline
Adding sodium hydroxide (4.4g, 0.11mol) and anhydrous ethanol (200m L) serving as a solvent into a 500m L three-neck flask containing a magnetic rotor, refluxing and stirring at 80 ℃ to dissolve, then adding phenylhydrazine (12.43g, 0.11mol), adding 1, 3-diphenyl-2-en-1-one (20.83g, 0.10mol) after 15 minutes, carrying out heat preservation reaction, monitoring the reaction process through a silica gel chromatography plate, cooling to room temperature after the reaction is finished, filtering, washing the obtained solid twice with 95% ethanol, and recrystallizing through an anhydrous ethanol/ethyl acetate (10/1, v/v) mixed solvent to obtain a yellow crystal product with the yield of 86.2%. HRMS for C21H18N2: 298.1534 (calculated), 298.1542 (actual).
3. Synthesis of target molecule (I) -1-PF6 -
1,3, 5-Triphenylpyrazoline (3.25g, 10.89mmol), phosphorus pentoxide (1.67g) and the solvent methanesulfonic acid (15m L) were charged in a 100m L three-necked flask containing a magnetic rotor at room temperatureStirring, then dropwise adding dimethyl sulfoxide (0.93g, 12.00mmol), keeping the temperature away from the sun and stirring for reaction for 6h, pouring the product into 500m L deionized water after the reaction is finished, then adding potassium hexafluorophosphate solution to obtain yellow precipitate, filtering, passing through a silica gel column by taking a dichloromethane/methanol mixed solvent (v: v ═ 10:1) as a developing agent to obtain a crude product, dissolving the crude product into a small amount of acetone, slowly dropwise adding the crude product into 10 volumes of stirred saturated potassium hexafluorophosphate aqueous solution, separating out the precipitate, filtering, and drying in vacuum to obtain white powder, namely the target product with hexafluorophosphate, wherein the total yield is 68.5%23H23N2S+: 359.5084 (calculated), 359.5096 (actual).
The solid line in FIG. 1 shows the target product (I) -1-PF obtained in example 16 -Ultraviolet and visible absorption spectrum of (1). As can be seen from FIG. 1, the target product (I) -1-PF6 -The maximum ultraviolet-visible absorption peak of (2) is located at 359 nm.
In addition, in the target product (I) -1-PF6 -After adding rhodamine, the UV-visible absorption spectrum of the acid is scanned after exposure for different time under L ED light to indicate that the UV absorption spectrum of the acid is shown in figure 2, the wavelength of L ED light is 365nm, and the light intensity is 2mW/cm2Sulfonium salt concentration 6.96 × 10-5mol L-1. As can be seen from FIG. 2, the absorption peak at 555nm, which is generated by ring opening of rhodamine B due to the generated proton, gradually rises and linearly changes with time as the light irradiation progresses. Indicating that the photo-generated acid has good properties.
Example 2: the anion PF is synthesized according to the following route6 -Synthesis of target molecule (I) -5

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) acetic anhydride, acetonitrile, potassium hexafluorophosphate, 98% H2SO4,5-10℃,3h
1. Synthesis of 3-phenyl-1- (2-furyl) -2-en-1-one
The product is obtained by reacting 2-acetylfuran with benzaldehyde, and the yield is 89.7% in the same way as in example one. HRMSfor C13H10O2: 198.0731 (calculated), 198.0728 (actual).
2. Synthesis of 1-phenyl-3- (2-furyl) -5-phenylpyrazoline
The product is obtained by reacting 3-phenyl-1- (2-furyl) -2-en-1-one with phenylhydrazine, the method is the same as the first embodiment, and the yield is 88.4%. HRMS for C19H16N2O: 288.1313 (calculated), 288.1318 (actual).
3. Synthesis of target molecule (I) -5-PF6 -
Sequentially adding 2.88g (0.01mol) of 1-phenyl-3- (2-furyl) -5-phenylpyrazoline, 2.20g (0.012mol) of potassium hexafluorophosphate, 0.82g (0.0105mol) of dimethyl sulfoxide and 2.55g (0.025mol) of acetic anhydride into a 100ml three-necked flask at room temperature, using 16g of acetonitrile as a solvent, cooling to 5 ℃ by using an ice water bath, slowly dropwise adding 1.2g (0.012mol) of 98% sulfuric acid, keeping the reaction system at below 10 ℃, reacting at 5-10 ℃ for 3h after the addition, sampling T L C, detecting the reaction, adding 40ml of water and 40ml of dichloromethane after the reaction is finished, layering, washing the organic layer with an aqueous solution of sodium bicarbonate until the pH is 7-8, separating the organic layer, washing with water once, separating the organic layer, spin-drying to obtain 6.5g, wherein the yield is 94.8% in HRMS for C21H21N2OS+: 349.1437 (calculated), 349.1420 (actual).
Example 3: the anion PF is synthesized according to the following route6 -Synthesis of target molecule (I) -9

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) methanesulfonic acid, phosphorus pentoxide and dark at room temperature for 6 hours;
(d) potassium hexafluorophosphate in water at room temperature.
1. Synthesis of 3-phenyl-1- (2-thienyl) -2-en-1-one
The product is obtained by reacting 2-acetylthiophene with benzaldehyde, and the method is the same as example one, and the yield is 89.7%. HRMSfor C13H10And OS: 214.0531 (calculated), 214.0528 (actual).
2. Synthesis of 1-phenyl-3- (2-thienyl) -5-phenylpyrazoline
The product is obtained by reacting 3-phenyl-1- (2-thienyl) -2-en-1-one with phenylhydrazine, the method is the same as the first embodiment, and the yield is 88.4%. HRMS for C19H16N2S: 304.1013 (calculated), 304.1018 (actual).
3. Synthesis of target molecule (I) -9-PF6 -
Using 1-phenyl-3- (2-thienyl) -5-phenylpyrazoline reacted with dimethyl sulfoxide and potassium hexafluorophosphate, the same procedure as in example one was carried out, giving a yield of 78.5%. HRMS for C21H21N2S2 +: 365.1074 (calculated), 365.1080 (actual).
Example 4: the anion PF is synthesized according to the following route6 -Synthesis of target molecule (I) -13

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) acetic anhydride, acetonitrile, potassium hexafluorophosphate, 98% H2SO4,5-10℃,3h
1. Synthesis of 3-phenyl-1- (2-pyrrolyl) -2-en-1-one
The product is obtained by reacting 2-acetyl pyrrole with benzaldehyde, and the method is the same as example one, and the yield is 89.7%. HRMSfor C13H11NO: 197.0821 (calculated), 197.0828 (actual).
2. Synthesis of 1-phenyl-3- (2-pyrrolyl) -5-phenylpyrazoline
The product is obtained by reacting 3-phenyl-1- (2-pyrrolyl) -2-en-1-one with phenylhydrazine, the method is the same as the first embodiment, and the yield is 88.4%. HRMS for C19H17N3: 287.1413 (calculated), 287.1418 (actual).
3. Synthesis of target molecule (I) -13-PF6 -
Using 1-phenyl-3- (2-pyrrolyl) -5-phenylpyrazoline reacted with dimethyl sulfoxide and potassium hexafluorophosphate in acetonitrile/sulfuric acid, the same procedure as in example two was carried out, giving a yield of 91.5%. HRMS for C21H22N3S+: 348.1503 (calculated), 348.1510 (actual).
Example 5: synthesizing (I) -1, (I) -5, (I) -9, (I) -13 containing other anions, and target products (I) -2 to (I) -28 (except for (I) -5, (I) -9, (I) -13) and target products (I) -5 'to (I) -16'.
The preparation method of the sulfonium salt is basically the same as the above embodiment, and can be realized by changing the substituent of acetophenone and different MX.
Specific yields and mass spectral characterization results are as follows. (taking the product of hexafluorophosphate as an anion for example, the molecular weight of the corresponding anion cannot be shown by mass spectrum, the molecular ion peak of the sulfonium salt is not changed by the anion; the other various salts show similar results and are not described again)




The dotted line in FIG. 2 shows the target product (I) -2 obtained in example 5-PF6 -Ultraviolet and visible absorption spectrum of (1). In addition, the target product (I) -2-PF6 -After adding rhodamine, the acid is exposed to L ED light for different time, and the ultraviolet absorption spectrum of the acid is indicated by scanning ultraviolet-visible absorption spectrum and shown in figure 3, wherein the wavelength of L ED light is 365nm, and the light intensity is 2mW/cm2Sulfonium salt concentration 6.71 × 10-5mol L-1. As can be seen from FIG. 3, the absorption peak at 555nm, which is generated by ring opening of rhodamine B due to the generated proton, gradually rises and linearly changes with time as the illumination progresses. Indicating that the photo-generated acid has good properties.
Example 6: containing SbF6 -Synthesis of the target product (I) -29

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) methanesulfonic acid, phosphorus pentoxide and dark at room temperature for 6 hours;
(d) aqueous sodium hexafluoroantimonate solution at room temperature.
1. Synthesis of 3- (4-methylphenyl) -1- (2-furyl) -2-en-1-one
The product is obtained by reacting 2-aldehyde furan with 4-methylacetophenone, the method is the same as the first example, and the yield is 86.7%. HRMS for C14H12O2: 212.0841 (calculated), 212.0838 (actual).
2. Synthesis of 1-phenyl-3- (4-methylphenyl) -5- (2-furyl) -pyrazoline
The product was obtained by reacting 3- (4-methylphenyl) -1- (2-furyl) -2-en-1-one with phenylhydrazine in the same manner as in example one, 81.3% yield. HRMS for C20H18N2O: 302.1401 (calculated), 302.1420 (actual).
3. Synthesis of target molecule (I) -29-SbF6 -
Obtained by reacting 1-phenyl-3-phenyl-5- (2-furyl) -pyrazoline with dimethyl sulfoxideThe methylsulfonate product was salt exchanged with sodium hexafluoroantimonate in the same manner as example one, 74.2% yield. HRMS for C22H23N2OS+: 363.1511 (calculated), 363.1530 (actual).
Example 7: synthesis of target product (I) -33 containing SbF6-

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) acetic anhydride, acetonitrile, sodium hexafluoroantimonate, 98% H2SO4,5-10℃,3h
1. Synthesis of 3- (4-methylphenyl) -1- (2-thienyl) -2-en-1-one
The product is obtained by reacting 2-aldehyde thiophene with 4-methylacetophenone, and the yield is 86.7% by the same method as in example one. HRMS for C14H12And OS: 228.0641 (calculated), 228.0638 (actual).
2. Synthesis of 1-phenyl-3- (4-methylphenyl) -5- (2-thienyl) -pyrazoline
The product was obtained by reacting 3- (4-methylphenyl) -1- (2-thienyl) -2-en-1-one with phenylhydrazine in the same manner as in example one, in 81.3% yield. HRMS for C20H18N2S: 318.1201 (calculated), 318.1220 (actual).
3. Synthesis of target molecule (I) -33-SbF6 -
The reaction of 1-phenyl-3-phenyl-5- (2-thienyl) -pyrazoline with dimethyl sulfoxide and sodium hexafluoroantimonate in acetonitrile/sulfuric acid is carried out in 74.2% yield as in example two. HRMS for C22H23N2S2 +: 379.1211 (calculated), 379.1330 (actual).
Example 8: containing SbF6 -Synthesis of the target product (I) -37

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) methanesulfonic acid, phosphorus pentoxide and dark at room temperature for 6 hours;
(d) aqueous sodium hexafluoroantimonate solution at room temperature.
1. Synthesis of 3- (4-methylphenyl) -1- (2-pyrrolyl) -2-en-1-one
The product is obtained by reacting 2-aldehyde pyrrole with 4-methylacetophenone, and the method is the same as the first example, and the yield is 86.7%. HRMS for C14H13NO: 211.1041 (calculated), 211.1038 (actual).
2. Synthesis of 1-phenyl-3- (4-methylphenyl) -5- (2-pyrrolyl) -pyrazoline
The product was obtained by reacting 3- (4-methylphenyl) -1- (2-pyrrolyl) -2-en-1-one with phenylhydrazine in 81.3% yield as in example one. HRMS for C20H19N3: 301.1601 (calculated), 301.1620 (actual).
3. Synthesis of target molecule (I) -37-SbF6 -
The method is the same as the first example and the yield is 74.2 percent by utilizing the reaction of 1-phenyl-3-phenyl-5- (2-pyrrolyl) -pyrazoline and dimethyl sulfoxide to obtain a methylsulfonate product and using sodium hexafluoroantimonate to carry out salt exchange. HRMS for C22H24N3S+: 362.1731 (calculated), 362.1730 (actual).
Example 9: (I) -29, (I) -33, (I) -37, the target products (I) -30 to (I) -52 (except for (I) -33, (I) -37) and the target products (I) -29 'to (I) -40' containing other anions are synthesized.
The preparation method of the sulfonium salt is basically the same as that of the six to eight examples, and can be realized by changing the substituent of benzaldehyde and different MX.
Specific yields and mass spectral characterization results are as follows. (taking the product in which hexafluoroantimonate is an anion as an example)



Example 10: the anion being BF4 -Synthesis of target molecule (I) -53

(a) Sodium hydroxide, ethanol/water, 60 ℃,2 h;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) methanesulfonic acid, phosphorus pentoxide and dark at room temperature for 6 hours;
(d) sodium tetrafluoroborate aqueous solution, room temperature.
1. Synthesis of 2-methyl-1, 3-diphenyl-2-one-1-ene
Adding propiophenone (4.03g, 30.0mmol), benzaldehyde (3.18g, 30.0mmol) and 95% EtOH (40.0 m L) into a 100m L round bottom flask with a magnetic stirrer, stirring, slowly heating to 60 ℃, then dropwise adding an ethanol-water solution of NaOH (1.4g, 35.0mmol, 15m L, 1/1, v/v ethanol/water), keeping the temperature for reaction for 2h, monitoring the reaction process by a silica gel chromatography plate, after the reaction is finished, extracting and separating the product by dichloromethane, suspending and steaming an organic phase solvent, washing by petroleum ether and absolute ethanol to obtain a colorless liquid with the yield of 84.3%, HRMS for C16H14O: 222.1041 (calculated), 222.1023 (actual).
2. Synthesis of 1- (2-methylphenyl) -3, 5-diphenyl-4-methyl-pyrazoline
The product is obtained by reacting 2-methyl-1, 3-diphenyl-2-en-1-one with 2-methylphenylhydrazine, the method is the same as the first embodiment, and the yield is 88.6%. HRMS for C23H22N2: 326.1821 (calculated), 326.1823 (actual).
3. Synthesis of target molecule (I) -53-BF4 -
1- (2-methylphenyl) -3, 5-diphenyl-4-methyl-pyrazoline is reacted with dimethyl sulfoxide to obtain a methylsulfonate product, and sodium tetrafluoroborate is used for salt exchange, wherein the yield is 74.2 percent. HRMS for C25H27N2S+: 387.1931 (calculated), 387.1930 (actual).
Example 11: (I) -53, the target products (I) -54 to (I) -57 and the target products (I) -53 'to (I) -56' containing other anions were synthesized.
The preparation method of the sulfonium salt is basically the same as that of the ninth embodiment, and the modification of the molecular structure of the aryl acetone and the modification of the molecular structure of different MX can be realized.
Specific yields and mass spectral characterization results are as follows. (products wherein tetrafluoroborate is an anion are exemplified)


Example 12: synthesis of (I) -58 containing tetraphenylborate.

The synthetic route is shown as the following formula
(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) drying tetrahydrofuran and lithium diisopropylamide at-78 ℃ for 3h at room temperature for 20 h;
(d) acetic anhydride, acetonitrile, 4-potassium phenylboronate, 98% H2SO4, 5-10 ℃ for 3H.
1. Synthesis of 1, 3-bis (4-methylphenyl) -2-en-1-one
By using 4-methylbenzaldehyde and4-methylacetophenone was reacted to give the product in the same manner as in example one, 90.3% yield. HRMS for C17H16O: 236.1241 (calculated), 236.1223 (actual).
2. Synthesis of 1- (2, 5-difluoro) -3, 5-bis (4-methylphenyl-) pyrazoline
The product was obtained by reacting 1, 3-bis (4-methylphenyl) -2-ene 1-one with 2, 5-difluorophenylhydrazine in the same manner as in example one, 85.0% yield. HRMS for C23H20F2N2: 362.1641 (calculated), 362.1633 (actual).
3. Synthesis of 1- (2, 5-difluoro) -3, 5-bis (4-methylphenyl) -4-ethylpyrazoline
A dry three-necked flask with a nitrogen blanket of 100m L was charged with a dry tetrahydrofuran solution of diisopropylamine (0.50g, 5.0mmol, 10m L), then cooled to-78 deg.C and n-butyllithium (1.6mol L) was added dropwise-13.1m L, 5mmol), incubation for 1h, then dropwise addition of a dry tetrahydrofuran solution of 1- (2, 5-difluoro) -3, 5-bis (4-methylphenyl) -pyrazoline (1.09g, 3.0mmol, 10m L), stirring with incubation for 1h, then dropwise addition of iodoethane (0.94g, 6.0mmol), incubation for 1h, then reaction at room temperature for 20h, dilution of the product with saturated brine and dichloromethane, separation of the aqueous layer after stirring for 20 min, extraction with dichloromethane (10m L× 3), combination of the organic layers, drying over anhydrous magnesium sulfate, evaporation of the organic solvent, separation by silica gel chromatography (ethyl acetate/n-hexane 1/20, v/v) to give 0.82g of a yellow liquid product, 70% yield, hrformms c25H24F2N2: 390.1881 (calculated), 390.1893 (actual).
4. Synthesis of the target molecule (I) -58-B (Ph)4 -
The product was obtained by reacting 1- (2, 5-difluoro) -3, 5-bis (4-methylphenyl) -4-ethylpyrazoline with diphenyl sulfoxide, potassium tetraphenylborate and acetic anhydride in the same manner as in example two, giving a yield of 95.2%. HRMS for C27H29F2N2S+: 451.2039 (calculated), 451.2024 (actual).
Example 13: synthesis of target molecule (I) -59 containing methylsulfonate

(a) Dicyclohexylcarbodiimide (DCC), 4-Dimethylaminopyridine (DMAP), tetrahydrofuran, room temperature, 1 h;
(b) bromine, acetic acid, room temperature, 3 h;
(c) triethylamine, dichloromethane, room temperature, 1 h;
(d) tetrakis (triphenylphosphine) palladium ((PPh3)4Pd), ethyl acetate, nitrogen, 0 ℃,1 h;
(e) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(f) methanesulfonic acid, phosphorus pentoxide, away from light, room temperature, 6 h.
1. Synthesis of 1, 3-bis (2-furyl) -2-propanone
DCC (4.3g, 21.0mmol), DMAP (0.73g, 6.0mmol) and THF (15m L) were added to a 50m L three-necked flask containing a magnetic rotor followed by 2- (2-furyl) -acetic acid (2.5g, 20.0mmol), and after 60 minutes of incubation the reaction was filtered through celite.
2. Synthesis of 1, 3-dibromo-1, 3-di (2-furyl) -2-propanone
In a 50m L three-necked flask containing a magnetic rotor, a solution of 1, 3-bis (2-furyl) -2-propanone in acetic acid (1.90g, 10.0mmol, 10m L) was added followed by dropwise addition of a solution of bromine in acetic acid (4.0g, 25.0mmol, 10m L) and after 3 hours of reaction, the product was poured into water, collected by filtration and dried for further use.
3. Synthesis of difuranyl cyclopropenones
1, 3-dibromo-1, 3-bis (2-furyl) -2-propanone (3.48g, 10.0mmol, 5.0m L) was dissolved in anhydrous dichloromethane (10m L), the solution was added to a 50m L three-necked flask with a dichloromethane solution of triethylamine (2.53g, 25.0mmol, 10m L), and stirred at room temperature for 1 hour, then the reaction mixture was stirred with 1mol L-1Washed with HCl, then brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The product was purified by silica gel column chromatography. The yield was 70.2%. HRMS for C11H16O3: 186.0281 (calculated), 186.0293 (actual).
4. Synthesis of 2-phenyl-1, 3-di (2-furyl) -2-one-1-ene
A50 m L three-necked flask containing a magnetic rotor was charged with difuryl cyclopropenone (1.86g, 10.0mmol) and phenylboronic acid (1.22g, 10.0mmol), sealed, deoxygenated, and then the catalyst (PPh) was added under nitrogen atmosphere (PPh)3)4Pd (0.23g, 0.2mmol) and ethyl acetate (15m L), cooling to 0 ℃, starting stirring, reacting for 1h, adding saturated salt water, extracting, drying the organic phase with anhydrous sodium sulfate, evaporating the solvent, and recrystallizing with anhydrous ethanol to obtain the product, wherein the yield is 84.3%17H12O3: 264.2781 (calculated), 264.2793 (actual).
5. Synthesis of 1-phenyl-3, 5-bis- (2-furyl) 4-phenylpyrazoline
The product was obtained by reacting 2-phenyl-1, 3-bis (2-furyl) -2-keto-1-ene with phenylhydrazine in 80.3% yield as in example one. HRMS for C23H16N2O2: 352.1201 (calculated), 352.1213 (actual).
6. Synthesis of target molecule (I) -59-CH3SO3 -
The 1-phenyl-3, 5-di- (2-furyl) 4-phenylpyrazoline is reacted with dimethyl sulfoxide, the method is the same as the first embodiment, and the product after the reaction is 76.1%. HRMS for C25H23N2O2S+: 415.1501 (calculated), 415.1513 (actual).
Example 14: synthesis of chloride ion-containing target molecule (I) -60 (Synthesis method shown below)
(a) 1-butyl-3-methylimidazole ionic liquid ([ bmim ] OH), N, N-dimethylbenzimidazole iodide for 14 h.
(b) Phosphorus pentoxide and benzene at 85 ℃ for 16 h;
(c) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(d) acetic anhydride, acetonitrile, potassium hexafluorophosphate, 98% H2SO4,5-10℃,3h

1. Synthesis of 3-phenyl-1- (2-thienyl) -3-hydroxy-2-phenylpropan-1-one
To [ bmim ] containing N, N-dimethylbenzimidazole iodide (1.37g, 5mmol) at room temperature was added]Adding 2-aldehyde thiophene (0.56g, 5.0mmol) and benzyl bromide (3.42, 20.0mmol) to a stirred solution of OH (15ml), maintaining the temperature for 20 minutes, adding benzaldehyde (0.53g, 5.0mmol), maintaining the temperature for 14 hours, detecting the progress of the reaction by a silica gel chromatography plate, extracting the reaction mixture with ethyl acetate (3 × 30ml) after the reaction is finished, drying the combined organic layers with anhydrous sodium sulfate, removing the solvent by vacuum evaporation, purifying the residue by a silica gel chromatography column (the developing agent ethyl acetate/n-hexane is 1/10, v/v) to obtain a light yellow target product with a yield of 74.2%. HRMS for C19H16O2S: 308.0911 (calculated), 308.0923 (actual).
2. Synthesis of 2, 3-diphenyl-1- (2-thienyl) -2-en-1-one
3-phenyl-1- (2-thienyl) -3-hydroxy-2-phenylpropan-1-one (0.92g, 3mmol) was added to P with stirring2O5(0.4g, 2.8mmol) in a suspension of benzene (10m L.) the reaction mixture was then heated at 90 ℃ for 9h after the reaction was complete, the product was poured into ice water, the organic layer was separated, the aqueous layer was extracted with ether (3 × 10m L), the combined organic layers were extracted with saturated NaHCO3The solution (20m L) and saturated NaCl solution (20m L) were washed, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the product in 89% yield, HRMS for C19H14And OS: 290.0841 (calculated), 290.0823 (actual).
3. Synthesis of 1,4, 5-triphenyl-3- (2-thienyl) pyrazoline
The synthesized 2, 3-diphenyl-1- (2-thienyl) -2-alkene-1-ketone reacts with phenylhydrazine to obtain a product, and the yield is 80.2 percent by the same method as the first embodiment. HRMS for C25H20N2S: 380.1341 (calculated), 380.1323 (actual).
4. Synthesis of target molecule (I) -59-CH3SO3 -
1,4, 5-triphenyl-3- (2-thienyl) pyrazoline is reacted with dimethyl sulfoxide and sodium chloride in an acetonitrile/concentrated sulfuric acid system, and the method is the same as the second embodiment, and the reacted product is 76.1 percent. HRMS for C27H25N2S2 +: 441.1501 (calculated), 441.1513 (actual).
Example 15: synthesis of target molecule (I) -61 containing p-toluenesulfonate

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) acetic anhydride, acetonitrile, sodium p-toluenesulfonate, 98% H2SO4, 5-10 deg.C, 3H
1. Synthesis of 1, 3-di (2-pyrrolyl) -2-en-1-one
The product is obtained by reacting 2-acetyl pyrrole with 2-aldehyde pyrrole, and the method is the same as the first embodiment, and the yield is 89.7%. HRMS for C11H10N2O: 186.0821 (calculated), 186.0828 (actual).
2. Synthesis of 1-phenyl-3, 5-bis (2-pyrrolyl) -pyrazoline
The product is obtained by reacting 1, 3-di (2-pyrrolyl) -2-en-1-one with phenylhydrazine, the method is the same as the first embodiment, and the yield is 88.4%. HRMS for C17H16N4: 276.1413 (calculated), 276.1418 (actual).
3. Synthesis of target molecule (I) -61-p-MePhSO3 -
The 1-phenyl-3, 5-bis (2-pyrrolyl) -pyrazoline is reacted with dimethyl sulfoxide and sodium p-toluenesulfonate in acetonitrile/sulfuric acid in the same manner as in example II in a yield of 91.5%. HRMS for C19H21N4S+: 337.1503 (calculated), 337.1510 (actual).
Example 16: synthesis of (I) -61, the target products (I) -62 to (I) -66 and the target molecules (I) -62 'to (I) -64' containing other anions
The preparation method of the sulfonium salt is basically the same as the above embodiment, and the change of aromatic aldehyde and aromatic methyl ketone substituted aromatic ring and different MX can be realized.
Specific yields and mass spectral characterization results are as follows. (products with tetraphenylborate as anion are exemplified)


Example 17: target product (I) -67 containing methylsulfonate

(a) Selenium dioxide, xylene, 100 ℃, 140 DEG C
(b) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(c) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(d) methanesulfonic acid, phosphorus pentoxide and dark at room temperature for 6 hours;
1. synthesis of 4-formyl coumarin
Adding 4-methylcoumarin (4.39g, 27.4mmol) and xylene (40m L) as a solvent into a 250m L three-neck flask containing a magnetic rotor, heating to 100 ℃, then adding selenium dioxide (4.55g, 41.1mol), heating to 140 ℃, carrying out heat preservation after xylene refluxes, carrying out reaction for 2 hours, monitoring the reaction process by a silica gel chromatography plate, directly carrying out heat filtration after the reaction is finished, washing the obtained filter residue with ethyl acetate, evaporating all organic phases, and separating the product by a silica gel chromatography column (petroleum ether/ethyl acetate 3/1, v/v) to obtain the product with the yield of 64.3% by HRMS for C10H6O3: 174.0341 (calculated), 174.0323 (actual).
2. Synthesis of 3- (4-coumarinyl) -1-phenyl-2-en-1-one
The product is synthesized by using 4-aldehyde coumarin and acetophenone, the method is the same as the first embodiment, and the yield is 85.9%. HRMS for C18H12O3: 276.0791 (calculated), 276.0783 (actual).
3. Synthesis of 1, 3-diphenyl-5- (4-coumarinyl) -pyrazoline
The product was synthesized from 3- (4-coumarinyl) -1-phenyl-2-en-1-one and phenylhydrazine in 88.1% yield as in example one. HRMS for C24H18N2O2: 366.1421 (calculated), 366.1433 (actual).
4. Synthesizing the target product (I) -67-CH3SO3 -
The product is obtained by reacting 1, 3-diphenyl-5- (4-coumarinyl) -pyrazoline with dimethyl sulfoxide in methanesulfonic acid and phosphorus pentoxide, the method is the same as the first embodiment, and the yield is 76.1%. HRMS for C26H23N2O2S+: 427.1511 (calculated), 427.1523 (actual).
Example eighteen: synthesis of a catalyst containing Al (t-Bu)4 -Target product (I) -68 of (1)

(a) Sodium hydroxide and absolute ethyl alcohol are used at normal temperature for 2 hours;
(b) sodium hydroxide and absolute ethyl alcohol at 80 ℃ for 2 h;
(c) acetic anhydride, acetonitrile, potassium hexafluorophosphate, 98% sulfuric acid, and keeping away from light at 5-10 ℃ for 3 h;
(d) methyl trifluoromethanesulfonate, dichloromethane, cesium carbonate for 24 h;
(e) potassium hexafluorophosphate in water at room temperature.
1. Synthesis of 3- (4-methylmercaptophenyl) -1-phenyl-2-en-1-one
The product was synthesized from acetophenone and 4-methylthiobenzaldehyde in the same manner as in example one, yield 90.2%. HRMSfor C16H14And OS: 254.0811 (calculated), 254.0823 (actual).
2. Synthesis of 1, 3-dimethyl-5- (4-methylmercaptophenyl) -pyrazoline
The product was synthesized from 3- (4-methylmercaptophenyl) -1-phenyl-2-en-1-one and phenylhydrazine in 88.2% yield as in example one. HRMS for C22H20N2S: 344.1311 (calculated), 344.1323 (actual).
3. Synthesis of 1- (4-dimethylsulfonium salt-based phenyl) -3-phenyl-5- (4-methylmercaptophenyl) -pyrazoline
The product is synthesized by the reaction of 1, 3-dimethyl-5- (4-methylmercaptophenyl) -pyrazoline and dimethyl sulfoxide, the method is the same as the third embodiment, and the yield is 90.0%. HRMS for C24H25N2S2 +: 405.1491 (calculated), 405.1483 (actual).
4. Synthesis of the target product (I) -68-Al (t-Bu)4 -
1- (4-Dimethylthio-onium-phenyl) -3-phenyl-5- (4-methylmercaptophenyl) -pyrazoline (7.5g, 14.51mmol) and cesium carbonate (0.95g, 2.90mmol) were charged in a 100m L three-necked flask containing a magnetic rotor, and the system was evacuated and N was charged2Cooling to room temperature after three times, injecting anhydrous dichloromethane (20m L) by using an injector, placing a reaction system at minus 20 ℃ in a dark place after organic matters are dissolved, dropwise adding methyl trifluoromethanesulfonate (2.95g, 18.00mmol) by using the injector, after the dropwise addition is finished, reacting at room temperature in the dark place for 24 hours, filtering to remove inorganic salts, concentrating filtrate, passing through a silica gel column by using pure dichloromethane and dichloromethane/methanol (10/1, v/v) as developing agents to obtain a product, dissolving sulfonium salts in a small amount of acetone, slowly dropwise adding the solution into a saturated sodium tetra-tert-butyl aluminate solution with 5 times volume of stirring, precipitating, filtering, and drying in vacuum to obtain yellow powder, namely a target product with hexafluorophosphate, wherein the total yield is 65.1 percent25H28N2S2 2+: 210.0811 (calculated), 210.0823 (actual).
Example 19: (I) -1-PF6 -L ED photocuring experiments and coating Property testing
Epoxy group-containing sample systems were prepared according to the following formulation (in weight percent)
Dual functional group resin (EPOX): 97 percent
Photoinitiator ((I) -1-PF6 -):3%
The mixture of the preparation examples is coated on a cardboard to form a coating with the thickness of about 25-30 microns, and the unit power produced by Shenzhen Shenri science and technology company is 64mW/cm2L ED light source with an emission wavelength of 365nm (3L ED area light source with the width of cm and the length of 80 cm) as an excitation light source, and placing the area light source on a variable-speed conveyor belt.
The results show that the compounds of this example all contained more than2Curing was effected at a speed of 5 m/min.
The coating obtained by photocuring was subjected to hardness test by a hand-operated pencil hardness tester, and the hardness was measured to be 4H.
Examples20:(I)-44-SbF6 -L ED photocuring experiments and coating Property testing
Epoxy group-containing sample systems were prepared according to the following formulation (in weight percent)
Monofunctional resin (CHO): 98.5 percent
Photoinitiator ((I) -44-SbF)6 -):1.5%
The mixture of the preparation examples is coated on a cardboard to form a coating of about 30-35 microns, and the unit power produced by Shenzhen Shenri science and technology company is 64mW/cm2The emission wavelength of the light source is 365nm, L ED light source (3 cm wide and 80 cm long L ED light source) is an excitation light source, the light source is placed on a variable speed conveyor belt, and repeated pressing and scraping of nails are used for avoiding imprinting as a criterion for completing photopolymerization curing.
The results show that the compounds containing this example all cured efficiently at a rate of greater than 30 m/min.
The coating obtained by photocuring was subjected to hardness test by a hand-operated pencil hardness tester, and the hardness was measured to be 4H.
Example 21: (I) -78-BF4 -L ED photocuring experiments and coating Property testing
Epoxy group-containing sample systems were prepared according to the following formulation (in weight percent)
Dual functional group resin (EPOX): 97 percent
Photoinitiator ((I) -78-BF)4 -):1%
The mixture of the preparation examples is coated on a cardboard to form a coating with the thickness of about 25-30 microns, and the unit power produced by Shenzhen Shenri science and technology company is 64mW/cm2The emission wavelength of the light source is 365nm, L ED light source (3 cm wide and 80 cm long L ED light source) is an excitation light source, the light source is placed on a variable speed conveyor belt, and repeated pressing and scraping of nails are used for avoiding imprinting as a criterion for completing photopolymerization curing.
The results show that the compounds containing this example all cure efficiently at a rate of greater than 25 m/min.
The coating obtained by photocuring was subjected to hardness test by a hand-operated pencil hardness tester, and the hardness was measured to be 4H.
Example 22: (I) -88-B (Ph)4 -L ED photocuring experiments and coating Property testing
Epoxy group-containing sample systems were prepared according to the following formulation (in weight percent)
Monofunctional resin (CHO): 98.5 percent
Photoinitiator ((I) -88-B (Ph)4 -):0.5%
The mixture of the preparation examples is coated on a cardboard to form a coating of about 30-35 microns, and the unit power produced by Shenzhen Shenri science and technology company is 64mW/cm2The emission wavelength of the light source is 365nm, L ED light source (3 cm wide and 80 cm long L ED light source) is an excitation light source, the light source is placed on a variable speed conveyor belt, and repeated pressing and scraping of nails are used for avoiding imprinting as a criterion for completing photopolymerization curing.
The results show that the compounds containing this example all cured efficiently at a rate of greater than 30 m/min.
The coating obtained by photocuring was subjected to hardness test by a hand-operated pencil hardness tester, and the hardness was measured to be 4H.
It should be understood that the above-mentioned examples are for illustrative purposes only and are not intended to limit the embodiments of the present disclosure, and that various other modifications and changes in light thereof will be suggested to persons skilled in the art and are not intended to be exhaustive or to limit the present disclosure to the precise embodiments disclosed herein.
Claims (16)
1. An N-para-sulfonium salt substituted pyrazoline derivative represented by the following formula (I),

wherein:
R1、R2each independently selected from C1-12Unsubstituted or substituted by 1 to 5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, or unsubstituted or substituted by 1-8R7Substituted benzoaromatic heterocyclic groups;
R3selected from H, C1-6Alkyl radical, C3-6Cycloalkyl, unsubstituted or substituted by 1-5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, unsubstituted or substituted by 1-8R7Substituted benzoaromatic heterocyclic groups;
R4selected from unsubstituted or substituted by 1-4RaSubstituted C1-6Alkyl, -F, -Cl, -Br, -I, -CN, -CF2CF3、-CF3、-NO2、-NRbRb、-ORb、-SRb、-C(=O)Rb、-CO2Rb、-OC(=O)Rb、-NRbC(=O)Rb、-S(=O)Rb、-S(=O)2Rb;
y is 0, 1, 2,3 or 4;
R5、R6each independently selected from C1-12Unsubstituted or substituted by 1 to 5R7Substituted benzyl, unsubstituted or substituted by 1 to 5R7Substituted phenyl;
R7each independently selected from unsubstituted or substituted by 1-5RaSubstituted C1-6Alkyl, -F, -Cl, -Br, -I, -CN, -CF2CF3、-CF3、-NO2、-NRbRb、-ORb、-SRb、-C(=O)Rb、-CO2Rb、-OC(=O)Rb、-NRbC(=O)Rb、-S(=O)Rb、-S(=O)2RbUnsubstituted or substituted by 1 to 5RcSubstituted carbocyclic ring, unsubstituted or substituted by 1 to 5RdSubstituted heterocycle, OR, P (═ O) (OR)b)2;
RaEach independently selected from C1-6Alkyl group, (CH)2)rC3-6Cycloalkyl or- (CH)2)rA phenyl group;
Rbeach independently selected from H, unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted- (CH)2)rPh;
RcEach independently selected from unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted (CH)2)rPh;
RdEach independently selected from unsubstituted or substituted by 1-5ReSubstituted C1-6Alkyl, unsubstituted or substituted by 1-5ReSubstituted (CH)2)rPh;
ReEach independently selected from-F, -Cl, -Br, -I, -OH, -NO2、-CN,-CF3、-CF2CF3、C1-4Alkyl radical, C1-4Alkoxy radical, C3-7Cycloalkyl, phenyl, benzyl, phenethyl, naphthyl, heterocyclic aryl, or, keto;
each r is independently 0, 1, 2,3, or 4;
X-is an anion.
2. The N-p-sulfonium salt-substituted pyrazoline derivative according to claim 1, wherein R is5、R6Each independently selected from methyl, cyclopropyl, phenyl, benzyl, 4-cyanobenzyl, or 4-trifluoromethylbenzyl.
3. The N-p-sulfonium salt-substituted pyrazoline derivative according to claim 1, wherein R is1、R2Each independently an aromatic heterocyclic group selected from the following (A) and (B):
(A) a 6-membered aromatic heterocyclic group containing 1 to 3 heteroatoms selected from the group consisting of O, N, S and Se on the heterocyclic ring;
(B) a 5-membered aromatic heterocyclic group containing a hetero atom of any one of the following groups in the heterocyclic ring,
1) 1O, 1N, or, 1S;
2) 1S and 1N, 1O and 1N, or, 2N; or
3) 3N, 1O and 2N, or, 1S and 2N.
4. The N-p-sulfonium salt-substituted pyrazoline derivative according to claim 1, wherein R is1、R2Each independently selected from the group consisting of the following structural formulae:


wherein R is7Is as defined in claim 1.
5. The N-para-sulfonium salt-substituted pyrazoline derivative according to claim 1, which is selected from the group consisting of compounds represented by the following structural formulae,

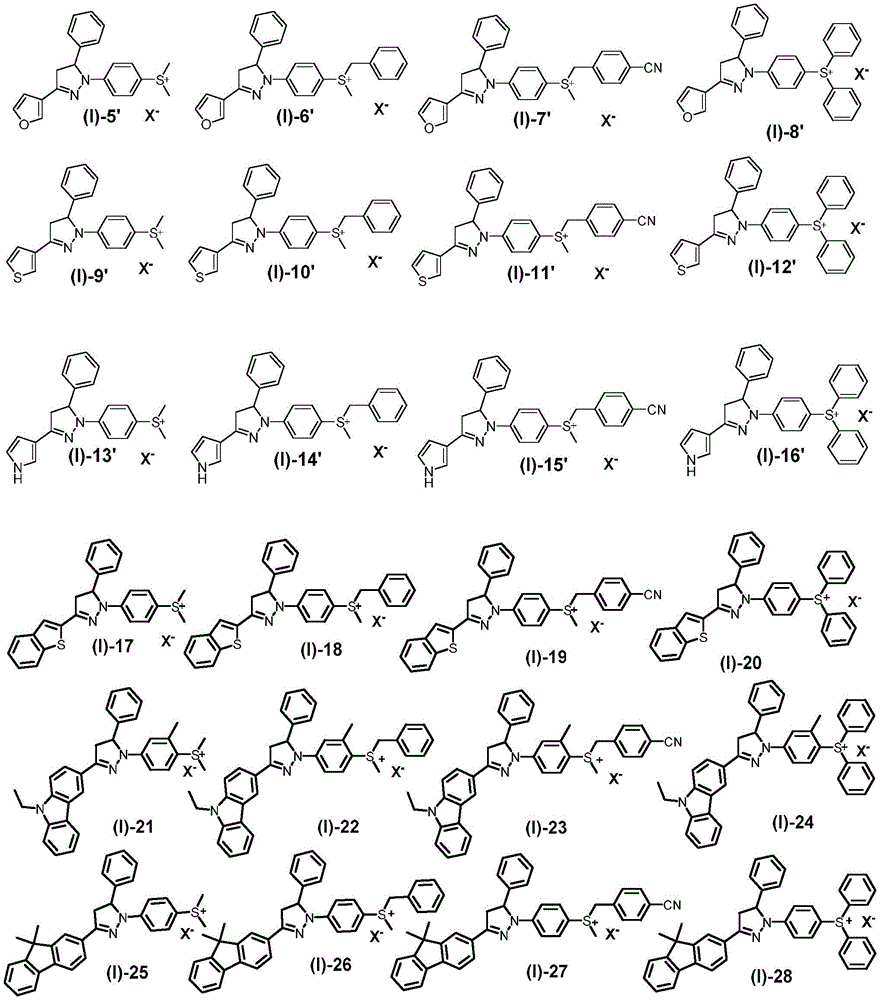



wherein, X-Is an anion.
6. The N-para-sulfonium salt-substituted pyrazoline derivative according to any one of claims 1 to 5, wherein X is-Selected from the group consisting of halogens, oxygen-containing acid radicals, borate radicals, phosphate radicals, antimonate radicals, or aluminate radicals.
7. The N-p-sulfonium salt-substituted pyrazoline derivative according to claim 6, wherein X is-Selected from Cl-、CF3SO3 -、CH3SO3 -、p-MePhSO3 -、BF4 -、B(Ph)4 -、B(PhF5)4 -、PF6 -、SbF6 -Or, Al (t-Bu)4 -。
8. A photocurable composition comprising the N-para-sulfonium salt-substituted pyrazoline derivative in accordance with any one of claims 1 to 7 and a polymerizable component comprising a monomer or polymer having an ethylenic bond or an epoxy group.
9. The photocurable composition according to claim 8, wherein the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I) is contained in an amount of 0.1 to 15 parts by weight relative to 100 parts by weight of the total amount of the polymerizable components.
10. The process for producing an N-p-sulfonium salt-substituted pyrazoline derivative in accordance with any one of claims 1 to 7, which comprises the following step (c):

in the step (c), the compound shown as the formula (I) -b and R5、R6Substituted sulfoxide by Metal+X-The compound containing the metal element reacts to obtain the N-para-sulfonium salt substituted pyrazoline derivative shown in the formula (I),
the R is1、R2、R3、R4、R5、R6、n、y、X-Is as defined in claim 1.
11. The process for producing an N-p-sulfonium salt-substituted pyrazoline derivative according to claim 10, wherein in the compound represented by the formula (I), R is3Is H or C1-6An alkyl group;
the preparation method of the compound shown in the formula (I) -b comprises the following steps (a) and (b):
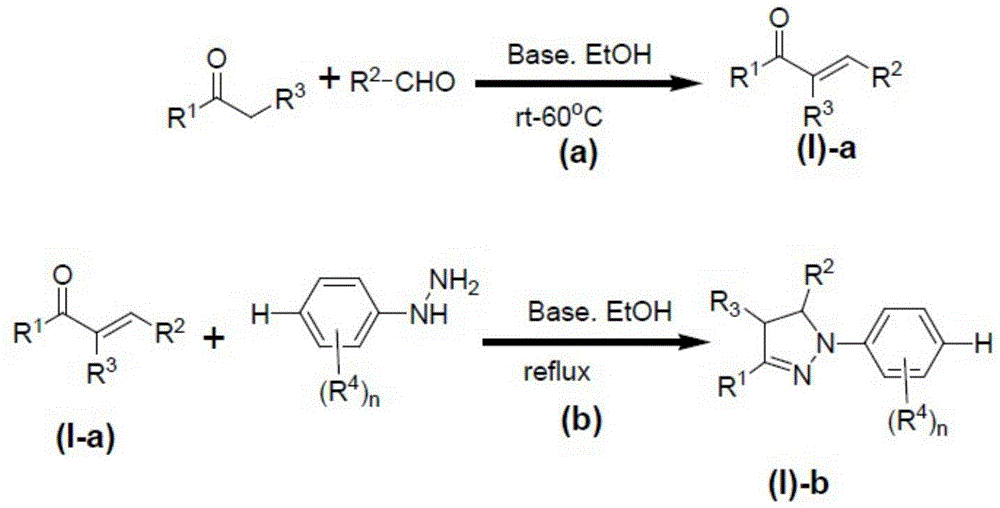
in the step (a), R1、R3Substituted alkyl ketones with R2The substituted formaldehyde reacts in absolute ethyl alcohol by taking alkali as a catalyst to obtain a compound shown as (I) -a, and the reaction temperature is between room temperature and 60 ℃;
in the step (b), the compound shown in the (I) -a obtained in the step (a) is reacted with a compound containing n R in absolute ethyl alcohol by taking alkali as a catalyst4Performing reflux reaction on phenyl hydrazine of a substituent group to generate a compound shown as (I) -b, wherein n is 0-4;
in the step (c), the compound represented by the formula (I) -b obtained in the step (b) is reacted with R5、R6The substituted sulfoxide is reacted in a system containing methanesulfonic acid and phosphorus pentoxide to generate methylsulfonyl sulfonium salt, and then the reaction system is added into a system containing Metal+X-Separating the precipitated precipitate from the aqueous solution of the metal element-containing compound represented by the formula (I), and recrystallizing the precipitate to obtain the N-para-sulfonium salt-substituted pyrazoline derivative represented by the formula (I).
12. The process for producing an N-p-sulfonium salt-substituted pyrazoline derivative according to claim 10, wherein in the compound represented by the formula (I), R is3Is C1-6An alkyl group;
the preparation method of the compound shown in the formula (I) -b comprises the following steps (a '), step (b ') and step (c '):

in the step (a'), in absolute ethyl alcohol, alkali is used as a catalyst, and R1Substituted ethanones and R2Substituting formaldehyde to generate a compound shown as a formula (I) -a';
in the step (b '), the compound of formula (I) -a ' produced in the step (a ') is reacted with a compound containing n R4Carrying out reflux reaction on phenyl hydrazine of a substituent group in absolute ethyl alcohol by taking alkali as a catalyst to generate a compound shown as (I) -b', wherein n is 0-4;
in the step (c '), the compound represented by the formula (I) -b ' produced in the step (b ') is reacted with R3The substituted iodoalkane is reacted in tetrahydrofuran by taking alkali as a catalyst to generate a compound shown as a formula (I) -b, wherein n is 0-4.
13. The process for producing an N-p-sulfonium salt-substituted pyrazoline derivative according to claim 10, wherein in the compound represented by the formula (I), R is3Is unsubstituted or substituted by 1 to 5R7Substituted phenyl, unsubstituted or substituted by 1 to 9R7Substituted condensed ring aryl, unsubstituted or substituted by 1 to 4R7Substituted aromatic heterocyclic radical, unsubstituted or substituted by 1-8R7Substituted benzoheteroaromatic radical, wherein R7Is as defined in claim 1, wherein,
the preparation method of the compound represented by the formula (I) -b comprises the following steps (a '), step (b '), and step (c '):

in said step (a'), R1Substituted formaldehydes and R2Substituted formaldehyde in 1-butyl-3-methylimidazole ionic liquid [ bmim]OH reacts in the presence of benzimidazole catalysts to generate compounds shown in the formula (I) -a';
in the step (b '), the compound shown in the formula (I) -a' generated in the step (a ') is converted into the compound shown in the formula (I) -b' in benzene by using phosphorus pentoxide as a water absorbent;
in the step (c ″), the compound represented by the formula (I) -b ″ generated in the step (b ") is reacted with phenylhydrazine containing n substituents in absolute ethyl alcohol by using a base as a catalyst to generate the compound represented by the formula (I) -b, wherein n is 0-4.
14. The process for the preparation of N-para-sulfonium salt-substituted pyrazoline derivatives in accordance with claim 13, wherein the steps (a '), step (b'), step (c '), step (d') are replaced by the following steps (a '), step (b'),

the step (a') In, R1Substituted phenylacetic acid reacts in tetrahydrofuran by using dicyclohexylcarbodiimide as a dehydrating agent and 4-dimethylamino pyridine as a catalyst to generate a compound shown as a formula (I) -a';
in step (b '"), reacting the compound of formula (I) -a'" produced in step (a '") with bromine in acetic acid to produce a compound of formula (I) -b'";
in the step (c '), the compound shown in the formula (I) -b ', which is generated in the step (b '), is generated in anhydrous dichloromethane by taking a base as a catalyst;
in the step (d '), the compound represented by the formula (I) -c ', which is generated in the step (c '), is reacted with R in 1, 4-dioxane under the action of a palladium catalyst3And (3) reacting the substituted boric acid compound to generate the compound shown as (I) -b'.
15. The process for producing an N-p-sulfonium salt-substituted pyrazoline derivative in accordance with claim 13, wherein the step (a ") and the step (b") are replaced with the following step (a ""), step (b ""), step (c ""),

in said step (a'), R1Substituted ethanones and R2Substituted formaldehyde reacts in absolute ethyl alcohol by taking alkali as a catalyst to generate a compound shown as a formula (I) -a';
in the step (b '), the compound shown in the formula (I) -a' generated in the step (a ') reacts with a halogen simple substance and corresponding halogen acid in an organic solvent, and then triethylamine is added for continuous reaction to generate the compound shown in the formula (I) -b';
in the step (c '), the compound represented by the formula (I) -b ' produced in the step (b ') and R3-B(OH)2The compound shown in the formula (I) -b' is generated by reaction in tetrahydrofuran under the catalysis of a palladium catalyst, and the reaction temperature is 60-80 ℃.
16. The process for producing an N-p-sulfonium salt-substituted pyrazoline derivative in accordance with claim 10, wherein the step (c) is carried out in a system comprising an organic solvent, an acid catalyst and an inorganic salt under the action of a water absorbent,
the acid catalyst is selected from the group consisting of hydrochloric acid, sulfuric acid, phosphoric acid, perfluorosulfonic acid, benzenesulfonic acid, aluminum trichloride, ferric trichloride, and zinc dichloride;
the organic solvent is selected from the group consisting of acetonitrile, DMF, DMAc, dichloromethane;
the water absorbent is selected from acid anhydride and phosphorus pentoxide.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010354947.8A CN111393367A (en) | 2020-04-29 | 2020-04-29 | N-para-sulfonium salt substituted pyrazoline derivative, photocuring composition and preparation method |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010354947.8A CN111393367A (en) | 2020-04-29 | 2020-04-29 | N-para-sulfonium salt substituted pyrazoline derivative, photocuring composition and preparation method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111393367A true CN111393367A (en) | 2020-07-10 |
Family
ID=71426718
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010354947.8A Pending CN111393367A (en) | 2020-04-29 | 2020-04-29 | N-para-sulfonium salt substituted pyrazoline derivative, photocuring composition and preparation method |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111393367A (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109776419A (en) * | 2019-03-07 | 2019-05-21 | 同济大学 | Sulfonium salt containing pyrazoline group and preparation method and application thereof |
| CN112300042A (en) * | 2020-09-27 | 2021-02-02 | 同济大学 | 4-Benzene five-membered ring-phenylsulfonium salt compound and preparation method and application thereof |
| CN112574110A (en) * | 2020-12-25 | 2021-03-30 | 同济大学 | Preparation and application of acyl-substituted pyrazoline sulfonium salt derivative |
| CN112574184A (en) * | 2020-12-25 | 2021-03-30 | 同济大学 | Epoxide substituted pyrazoline derivative, light-cured composition and preparation method |
| CN112707863A (en) * | 2020-12-25 | 2021-04-27 | 同济大学 | Acrylate-substituted pyrazoline derivative, photocuring composition and preparation method |
| CN114702458A (en) * | 2022-04-10 | 2022-07-05 | 同济大学 | Benzo five-membered ring-phenylalkynyl sulfonium salt derivative and preparation method and application thereof |
| CN119569697A (en) * | 2024-12-23 | 2025-03-07 | 佛山市新量子环保材料有限公司 | Cationic photoinitiator intermediate and preparation method thereof, water-soluble cationic photoinitiator and preparation method thereof |
| CN120192301A (en) * | 2025-05-26 | 2025-06-24 | 浙江扬帆新材料股份有限公司 | A kind of arylsulfonium salt photoinitiator, preparation method and application thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109776419A (en) * | 2019-03-07 | 2019-05-21 | 同济大学 | Sulfonium salt containing pyrazoline group and preparation method and application thereof |
| CN112300042A (en) * | 2020-09-27 | 2021-02-02 | 同济大学 | 4-Benzene five-membered ring-phenylsulfonium salt compound and preparation method and application thereof |
| CN112574110A (en) * | 2020-12-25 | 2021-03-30 | 同济大学 | Preparation and application of acyl-substituted pyrazoline sulfonium salt derivative |
-
2020
- 2020-04-29 CN CN202010354947.8A patent/CN111393367A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109776419A (en) * | 2019-03-07 | 2019-05-21 | 同济大学 | Sulfonium salt containing pyrazoline group and preparation method and application thereof |
| CN112300042A (en) * | 2020-09-27 | 2021-02-02 | 同济大学 | 4-Benzene five-membered ring-phenylsulfonium salt compound and preparation method and application thereof |
| CN112574110A (en) * | 2020-12-25 | 2021-03-30 | 同济大学 | Preparation and application of acyl-substituted pyrazoline sulfonium salt derivative |
Non-Patent Citations (1)
| Title |
|---|
| NIRMALENDU DATTA-GUPTA 等: ""Synthesis and Properties of Three Sulfur-Containing Porphyrins, One Water-Soluble"", 《BULL. CHEM. SOC. JPN.》 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109776419A (en) * | 2019-03-07 | 2019-05-21 | 同济大学 | Sulfonium salt containing pyrazoline group and preparation method and application thereof |
| CN109776419B (en) * | 2019-03-07 | 2021-06-08 | 同济大学 | Pyrazoline group-containing sulfonium salt and preparation method and application thereof |
| CN112300042A (en) * | 2020-09-27 | 2021-02-02 | 同济大学 | 4-Benzene five-membered ring-phenylsulfonium salt compound and preparation method and application thereof |
| CN112574110A (en) * | 2020-12-25 | 2021-03-30 | 同济大学 | Preparation and application of acyl-substituted pyrazoline sulfonium salt derivative |
| CN112574184A (en) * | 2020-12-25 | 2021-03-30 | 同济大学 | Epoxide substituted pyrazoline derivative, light-cured composition and preparation method |
| CN112707863A (en) * | 2020-12-25 | 2021-04-27 | 同济大学 | Acrylate-substituted pyrazoline derivative, photocuring composition and preparation method |
| CN114702458A (en) * | 2022-04-10 | 2022-07-05 | 同济大学 | Benzo five-membered ring-phenylalkynyl sulfonium salt derivative and preparation method and application thereof |
| CN114702458B (en) * | 2022-04-10 | 2023-06-02 | 同济大学 | Benzo five-membered ring-phenylynylsulfonium salt derivative and its preparation method and application |
| CN119569697A (en) * | 2024-12-23 | 2025-03-07 | 佛山市新量子环保材料有限公司 | Cationic photoinitiator intermediate and preparation method thereof, water-soluble cationic photoinitiator and preparation method thereof |
| CN120192301A (en) * | 2025-05-26 | 2025-06-24 | 浙江扬帆新材料股份有限公司 | A kind of arylsulfonium salt photoinitiator, preparation method and application thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111393367A (en) | N-para-sulfonium salt substituted pyrazoline derivative, photocuring composition and preparation method | |
| CN104991418B (en) | A kind of sensitizer and its preparation method and application for UV-LED photocuring | |
| WO2011024774A1 (en) | Second-order non-linear optical compound and non-linear optical element comprising same | |
| CN108084450B (en) | Polymerization complex containing vinylphenyl and p-vinylphenol methylamine derivative cadmium and preparation method and application thereof | |
| JP2016509099A (en) | Carbazole ketoxime ester based photoinitiator with high photosensitivity | |
| CN112574110B (en) | Preparation and application of acyl-substituted pyrazoline sulfonium salt derivative | |
| Wua et al. | Effects of conjugated systems on UV-visible light-sensitive D-π-A type sulfonium salt photoacid generators | |
| CN104559286B (en) | Triphenylamine-boron fluoride complex dimethyl pyrrole methine derivative organic dye and preparation method thereof | |
| CN112300042B (en) | 4-Benzene five-membered ring-phenylsulfonium salt compound and preparation method and application thereof | |
| CN104030974A (en) | Aryl-substituted terpyridyl compound and preparation method and application thereof | |
| CN108976252B (en) | Preparation method of trimeric indenyl BODIPY-coumarin star-shaped compound | |
| CN112250686A (en) | A kind of mouth-shaped lattice hole transport material and its preparation method and application | |
| CN107033121A (en) | The aryl ethylene compound of heterozygosis four, polymer and preparation method and application | |
| CN101624408A (en) | Pendant naphthalene group-containing benzene ligand ferrocenium salt cationic photoinitiators and preparation method thereof | |
| CN104447824A (en) | Fluoro-boron diisoindole compounds and preparation method thereof | |
| CN112574184B (en) | Epoxide-substituted pyrazoline derivatives, photocurable composition and preparation method | |
| CN112707863A (en) | Acrylate-substituted pyrazoline derivative, photocuring composition and preparation method | |
| CN112079856A (en) | 4-Iodophenyl substituted carborane derivatives and preparation method thereof | |
| CN112300026B (en) | 4-benzo five-membered ring-phenyl ketoxime ester compound and preparation method and application thereof | |
| CN104710481B (en) | The molysite class two-photon absorbing material of butylcyclopentadiene containing tolans and preparation method | |
| CN115850993A (en) | Chiral amphiphilic near-infrared aza-BODIPY dye and preparation method thereof | |
| CN111807977B (en) | 9-aniline fluorene-9-carboxylate compound and preparation method thereof | |
| CN116283917A (en) | Preparation method of carbazole free radical-cation coupling type visible light photoinitiator | |
| CN114957151A (en) | Benzo five-membered ring-phenylethynyl ketoxime ester compound and preparation and application thereof | |
| CN107522643A (en) | A kind of novel preparation method using talan as the sulfosalt derivative of conjugated system |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20200710 |