CN111187200A - Synthesis method of lomitapide-D8 - Google Patents
Synthesis method of lomitapide-D8 Download PDFInfo
- Publication number
- CN111187200A CN111187200A CN202010271711.8A CN202010271711A CN111187200A CN 111187200 A CN111187200 A CN 111187200A CN 202010271711 A CN202010271711 A CN 202010271711A CN 111187200 A CN111187200 A CN 111187200A
- Authority
- CN
- China
- Prior art keywords
- compound
- reaction
- hours
- room temperature
- carrying
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000001308 synthesis method Methods 0.000 title abstract description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 52
- 238000006243 chemical reaction Methods 0.000 claims abstract description 41
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims abstract description 20
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims abstract description 18
- 229940125782 compound 2 Drugs 0.000 claims abstract description 16
- 229940126214 compound 3 Drugs 0.000 claims abstract description 14
- 229940125898 compound 5 Drugs 0.000 claims abstract description 14
- 229940125904 compound 1 Drugs 0.000 claims abstract description 10
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims abstract description 8
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 claims abstract description 8
- 238000006467 substitution reaction Methods 0.000 claims abstract description 7
- 238000006482 condensation reaction Methods 0.000 claims abstract description 6
- 238000006356 dehydrogenation reaction Methods 0.000 claims abstract description 3
- 238000005658 halogenation reaction Methods 0.000 claims abstract description 3
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims abstract description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 30
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical group CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 30
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 18
- -1 benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate Chemical compound 0.000 claims description 17
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 claims description 15
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 14
- 229910052757 nitrogen Inorganic materials 0.000 claims description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 10
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 claims description 10
- 239000007810 chemical reaction solvent Substances 0.000 claims description 10
- 230000035484 reaction time Effects 0.000 claims description 10
- 238000000034 method Methods 0.000 claims description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- 238000010438 heat treatment Methods 0.000 claims description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 6
- 239000007864 aqueous solution Substances 0.000 claims description 5
- 238000003756 stirring Methods 0.000 claims description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 5
- 230000002194 synthesizing effect Effects 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 2
- 230000002378 acidificating effect Effects 0.000 claims description 2
- WYURNTSHIVDZCO-SVYQBANQSA-N oxolane-d8 Chemical compound [2H]C1([2H])OC([2H])([2H])C([2H])([2H])C1([2H])[2H] WYURNTSHIVDZCO-SVYQBANQSA-N 0.000 abstract description 8
- 230000000694 effects Effects 0.000 abstract description 3
- 239000002994 raw material Substances 0.000 abstract description 3
- ULTHEAFYOOPTTB-SVYQBANQSA-N 1,4-dibromo-1,1,2,2,3,3,4,4-octadeuteriobutane Chemical class [2H]C([2H])(Br)C([2H])([2H])C([2H])([2H])C([2H])([2H])Br ULTHEAFYOOPTTB-SVYQBANQSA-N 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 239000012043 crude product Substances 0.000 description 5
- 102100031545 Microsomal triglyceride transfer protein large subunit Human genes 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 108010038232 microsomal triglyceride transfer protein Proteins 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 3
- ZHNUHDYFZUAESO-UHFFFAOYSA-N formamide Substances NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 3
- MBBCVAKAJPKAKM-UHFFFAOYSA-N lomitapide Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1(C(=O)NCC(F)(F)F)CCCCN(CC1)CCC1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 MBBCVAKAJPKAKM-UHFFFAOYSA-N 0.000 description 3
- 229960003566 lomitapide Drugs 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 208000035150 Hypercholesterolemia Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- GIMDPFBLSKQRNP-UHFFFAOYSA-N 1,1-diphenylethanol Chemical compound C=1C=CC=CC=1C(O)(C)C1=CC=CC=C1 GIMDPFBLSKQRNP-UHFFFAOYSA-N 0.000 description 1
- IQOMYCGTGFGDFN-UHFFFAOYSA-N 2-[4-(trifluoromethyl)phenyl]benzoic acid Chemical compound OC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 IQOMYCGTGFGDFN-UHFFFAOYSA-N 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 108010004103 Chylomicrons Proteins 0.000 description 1
- 238000005727 Friedel-Crafts reaction Methods 0.000 description 1
- 208000000563 Hyperlipoproteinemia Type II Diseases 0.000 description 1
- 108010028554 LDL Cholesterol Proteins 0.000 description 1
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 1
- 108010062497 VLDL Lipoproteins Proteins 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 238000007098 aminolysis reaction Methods 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 201000001386 familial hypercholesterolemia Diseases 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- LWMPFIOTEAXAGV-UHFFFAOYSA-N piperidin-1-amine Chemical compound NN1CCCCC1 LWMPFIOTEAXAGV-UHFFFAOYSA-N 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000002639 sodium chloride Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention provides a synthesis method of lomitapide-D8, which comprises the following steps: 1) performing halogenation reaction on the compound 2a and hydrobromic acid to obtain a compound 2; 2) firstly, carrying out hydrogen abstraction reaction on the compound 1 and n-butyllithium, and then adding the compound 2 for substitution reaction to prepare a compound 3; 3) carrying out condensation reaction on the compound 3 and the compound 4 to obtain a compound 5; 4) carrying out condensation reaction on the compound 6 and the compound 7 to obtain a compound 8; 5) removing tert-butyloxycarbonyl protection from the compound 8 and trifluoroacetic acid to obtain a compound 9; 6) and carrying out substitution reaction on the compound 5 and the compound 9 to obtain the lomitapide-D8. The invention firstly uses the relatively low-cost deuterated tetrahydrofuran as the raw material to synthesize the deuterated 1, 4-dibromobutane-D8, successfully synthesizes the deuterated lomitapide-D8, changes the conventional synthetic route, and enables the difficultly obtained deuterated atoms to achieve the maximum atom economic effect.
Description
Technical Field
The invention relates to a synthesis method of lomitapide-D8, belonging to the technical field of organic chemical synthesis.
Background
Lomitapide is an oral small molecule microsomal triglyceride transfer protein (MTP) inhibitor that was approved by the FDA in the united states for marketing in 2012 for the treatment of hypercholesterolemia, including primary hypercholesterolemia and familial hypercholesterolemia. The action mechanism of the lometasapine is that the lometasapine can be retained in endoplasmic reticulum, is directly combined with MTP and has inhibition effect on the MTP, prevents the assembly and secretion of apolipoprotein in intestinal epithelial cells and liver cells, inhibits the synthesis of chylomicron and very low density lipoprotein, and reduces the level of plasma low density lipoprotein cholesterol.
Lomitapide-D8 can be used for clinical pharmacology and toxicology research. No report on the synthesis of Lomitapide-D8 exists at present.
The conventional synthetic route of lomitapide takes diphenylethanol as a raw material, N- (2, 2, 2-trifluoroethyl) -9- (4-bromobutyl) -9H-fluorenyl-9-formamide is obtained through Friedel-Crafts reaction, nucleophilic substitution and aminolysis, the N- (2, 2, 2-trifluoroethyl) -9- [4- (4-aminopiperidine-1-yl) -butyl ] -9H-fluorenyl-9-formamide is subjected to N-substitution reaction with amino piperidine protected by Boc, deprotection is carried out to obtain N- (2, 2, 2-trifluoroethyl) -9- [4- (4-aminopiperidine-1-yl) -butyl ] -9H-fluorenyl-9-formamide, and acyl chloride prepared from 4' -trifluoromethyl biphenyl-2-carboxylic acid is condensed to obtain the product.
Lomitapide-D8 cannot be synthesized by adopting the conventional synthetic route of Lomitapide.
Disclosure of Invention
The invention aims to overcome the problems in the prior art and provides a synthesis method of lomitapide-D8.
The technical scheme of the invention is as follows:
a method for synthesizing lometasai-D8, the structure of lometasai-D8 is shown as formula I,
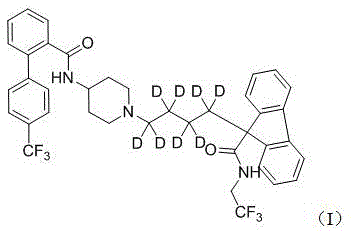
the synthetic route is as follows:

the method comprises the following steps:
1) under the acidic condition, carrying out halogenation reaction on the compound 2a and hydrobromic acid to obtain a compound 2;
2) under the protection of nitrogen, firstly, carrying out hydrogen abstraction reaction on the compound 1 and n-butyllithium, and then adding the compound 2 for substitution reaction to prepare a compound 3;
3) carrying out condensation reaction on the compound 3 and the compound 4 in the presence of 1-hydroxybenzotriazole, benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and N, N-diisopropylethylamine to prepare a compound 5;
4) carrying out condensation reaction on a compound 6 and a compound 7 in the presence of 1-hydroxybenzotriazole, benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and N, N-diisopropylethylamine to prepare a compound 8;
5) removing tert-butyloxycarbonyl protection from the compound 8 and trifluoroacetic acid to obtain a compound 9;
6) carrying out substitution reaction on the compound 5 and the compound 9 in the presence of potassium carbonate to obtain lomitapide-D8;
in the step 1), the acid is concentrated sulfuric acid, the reaction temperature is 110 ℃, and the reaction time is 6 hours;
in the step 2), the reaction solvent is anhydrous tetrahydrofuran, the reaction condition of the compound 1 and n-butyllithium is 0 ℃ for 1 hour, and the reaction condition of adding the compound 2 is room temperature reaction for 24 hours;
in the step 3), the reaction solvent is N, N-dimethylformamide, the reaction temperature is room temperature, and the reaction time is 24 hours;
in the step 4), the reaction solvent is N, N-dimethylformamide, the reaction temperature is room temperature, and the reaction time is 24 hours;
in the step 5), the reaction solvent is dichloromethane, the reaction temperature is room temperature, and the reaction time is 1 hour;
in the step 6), the reaction solvent is N, N-methylformamide, the reaction temperature is 50 ℃, and the reaction time is 24 hours.
Preferably, the first and second electrodes are formed of a metal,
the method comprises the following steps:
1) under the ice bath condition, slowly adding concentrated sulfuric acid into an aqueous solution of hydrobromic acid, then slowly adding a compound 2a, and after completion, heating to 110 ℃ for reacting for 6 hours to obtain a compound 2;
2) dissolving compound 1 in anhydrous tetrahydrofuran under nitrogen protection at 0oSlowly adding n-butyllithium under the condition of C, preserving heat, reacting for 1 hour, then adding the compound 2, heating to room temperature, and reacting for 24 hours to obtain a compound 3;
3) adding the compound 3, the compound 4, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and the N, N-diisopropylethylamine into the N, N-dimethylformamide in sequence, and reacting at room temperature for 24 hours to obtain a compound 5;
4) adding the compound 6, the compound 7, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and the N, N-diisopropylethylamine into the N, N-dimethylformamide in sequence, and stirring at room temperature for reacting for 24 hours to obtain a compound 8;
5) dissolving the compound 8 in dichloromethane, slowly dropwise adding trifluoroacetic acid while stirring at room temperature, and reacting at room temperature for 1 hour to obtain a compound 9;
6) and sequentially adding the compound 5, the compound 9 and potassium carbonate into N, N-methylformamide, heating to 50 ℃ and reacting for 24 hours to obtain the lomitapide-D8.
More preferably still, the first and second liquid crystal compositions are,
in the step 1), the molar ratio of the compound 2a to the hydrobromic acid is 1.0: 2.85;
in the step 2), the mol ratio of the compound 1, the n-butyllithium and the compound 2 is 1.0: 2.25: 1.20;
in the step 3), the molar ratio of the compound 3, the compound 4, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate to the N, N-diisopropylethylamine is 1.21: 1: 1.51: 1.51: 2.96;
in the step 4), the molar ratio of the compound 6, the compound 7, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate to the N, N-diisopropylethylamine is 1.19: 1: 1.51: 1.51: 1.98 of;
in the step 5), the molar ratio of the compound 8 to the trifluoroacetic acid is 1: 2.51;
in the step 6), the molar ratio of the compound 5 to the compound 9 to the potassium carbonate is 1: 1.13: 2.52.
the invention has the following technical effects: the invention firstly uses the low-price deuterated tetrahydrofuran as the raw material to synthesize the deuterated 1, 4-dibromobutane-D8, uses the deuterated tetrahydrofuran as a deuterated intermediate for synthesizing the deuterated lomitapide-D8, successfully synthesizes the deuterated lomitapide-D8, changes the conventional synthetic route, and enables the difficultly obtained deuterated atoms to achieve the maximum atom economic benefit.
Drawings
FIG. 1 is a HNMR map of lometaside-D8 prepared in example 1.
Detailed Description
Deuterated tetrahydrofuran was purchased from Cambridge Isotope Laboratories, inc.
Example 1
The synthesis method of the lomitapide-D8 comprises the following steps:
step 1)
The synthetic route is as follows:

under the ice bath condition, 22 ml of concentrated sulfuric acid is slowly added into 60 g of 48% wt hydrobromic acid aqueous solution, then compound 2a (10.0 g, 124.8 mmol) is slowly added, after the reaction is completed, the temperature is raised to 110 ℃ for reaction for 6 hours, the reaction is monitored, after the reaction is completed, the reaction is cooled to room temperature, the lower layer is separated, the lower layer is dissolved by 150 ml of n-hexane, washed by water for 2 times, washed by saturated sodium bicarbonate aqueous solution for 1 time, dried by anhydrous sodium sulfate, and distilled under reduced pressure to obtain 26.2 g of colorless liquid, compound 2, and the yield is 94%.
Step 2)
The synthetic route is as follows:

compound 1 (4.2 g, 20.0 mmol) was dissolved in 100 ml of anhydrous tetrahydroIn furan under nitrogen protection at 0oSlowly adding n-butyllithium (2.5M, 18mL) at the temperature of C, keeping the temperature for reaction for 1 hour, then adding the compound 2 (5.4 g, 24.1 mmol), heating to room temperature after the reaction is finished, reacting for 24 hours, monitoring the reaction, adding 100 mL of ice water in an ice bath after the reaction is completed, extracting the reaction liquid by using ethyl acetate (80 mL x 3), combining organic phases, washing the organic phases once by using saturated salt solution, drying by using anhydrous sodium sulfate, spin-drying, and carrying out column chromatography on a crude product to obtain 4.8g of the compound 3 with the yield of 68 percent.
Step 3)
The synthetic route is as follows:

compound 3 (2.0 g, 5.7 mmol), compound 4 (0.64 g, 4.7 mmol), 1-hydroxybenzotriazole (HOBT, 0.96g, 7.1 mmol), benzotriazole-N, N' -tetramethyluronium hexafluorophosphate (HBTU, 2.7g, 7.1 mmol), N-diisopropylethylamine (DIPEA, 1.8g, 13.9 mmol) were added to 20 ml of N, N-dimethylformamide in this order to react at room temperature for 24 hours, the reaction was monitored, after completion of the reaction, 100 ml of water was added, the reaction solution was extracted with ethyl acetate (60 ml × 3), the organic layers were combined and washed with water once, brine once, dried over anhydrous sodium sulfate, spin-dried, and crude product column chromatography gave 1.4g of compound 5 in 68% yield.
Step 4)
The synthetic route is as follows:

compound 6 (1.5 g, 5.6 mmol), compound 7 (0.94 g, 4.7 mmol), 1-Hydroxybenzotriazole (HOBT) (0.96 g, 7.1 mmol), benzotriazole-N, N' -tetramethyluronium hexafluorophosphate (HBTU, 2.7g, 7.1 mmol), N-diisopropylethylamine (DIPEA, 1.2g, 9.3 mmol) were added to 15 ml of N, N-dimethylformamide in this order, after completion, the reaction was stirred at room temperature for 24 hours, the reaction was monitored, after completion, 60ml of water was added, the reaction solution was extracted with ethyl acetate (50 ml × 3), the organic layers were combined and washed with water once, saturated salt once, dried over anhydrous sodium sulfate, spin-dried, and the crude product of column chromatography gave 1.7g of compound 8, yield 81%.
Step 5)
The synthetic route is as follows:

dissolving a compound 8 (1.5 g and 3.3 mmol) in 15 ml of dichloromethane, slowly dropwise adding trifluoroacetic acid (0.95 g and 8.3 mmol) under stirring at room temperature, reacting at room temperature for 1 hour, monitoring the reaction, removing the trifluoroacetic acid after the reaction is completed, adjusting a crude product to be alkalescent by using a saturated sodium bicarbonate aqueous solution, extracting the dichloromethane (50 ml x 4), combining organic layers, washing once by using saturated salt solution, drying by using anhydrous sodium sulfate, and spin-drying to obtain 1.1g of a compound 9 which is directly used for the next reaction.
Step 6)
The synthetic route is as follows:

adding compound 5 (1.0 g, 2.3 mmol), compound 9 (0.9 g, 2.6 mmol) and potassium carbonate (0.8 g, 5.8 mmol) into 20 ml of N, N-methylformamide in sequence, heating the reaction solution to 50 ℃ after the reaction is finished, reacting for 24 hours, monitoring the reaction, adding 60ml of water after the reaction is finished, extracting with ethyl acetate (30 ml x 3), combining organic layers and washing with water once, washing with saturated common salt water once, drying with anhydrous sodium sulfate, spin-drying, performing column chromatography on a crude product to obtain 1.1g of a final product with the yield of 68%, M/z, M +1=702.4
The detection result of the obtained product is shown in FIG. 1, which shows that the product is Lomitapide-D8.
Claims (3)
1. A method for synthesizing lometasai-D8, the structure of lometasai-D8 is shown as formula I,

the method is characterized in that the synthetic route is as follows:

the method comprises the following steps:
1) under the acidic condition, carrying out halogenation reaction on the compound 2a and hydrobromic acid to obtain a compound 2;
2) under the protection of nitrogen, firstly, carrying out hydrogen abstraction reaction on the compound 1 and n-butyllithium, and then adding the compound 2 for substitution reaction to prepare a compound 3;
3) carrying out condensation reaction on the compound 3 and the compound 4 in the presence of 1-hydroxybenzotriazole, benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and N, N-diisopropylethylamine to prepare a compound 5;
4) carrying out condensation reaction on a compound 6 and a compound 7 in the presence of 1-hydroxybenzotriazole, benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and N, N-diisopropylethylamine to prepare a compound 8;
5) removing tert-butyloxycarbonyl protection from the compound 8 and trifluoroacetic acid to obtain a compound 9;
6) carrying out substitution reaction on the compound 5 and the compound 9 in the presence of potassium carbonate to obtain lomitapide-D8;
in the step 1), the acid is concentrated sulfuric acid, the reaction temperature is 110 ℃, and the reaction time is 6 hours;
in the step 2), the reaction solvent is anhydrous tetrahydrofuran, the reaction condition of the compound 1 and n-butyllithium is 0 ℃ for 1 hour, and the reaction condition of adding the compound 2 is room temperature reaction for 24 hours;
in the step 3), the reaction solvent is N, N-dimethylformamide, the reaction temperature is room temperature, and the reaction time is 24 hours;
in the step 4), the reaction solvent is N, N-dimethylformamide, the reaction temperature is room temperature, and the reaction time is 24 hours;
in the step 5), the reaction solvent is dichloromethane, the reaction temperature is room temperature, and the reaction time is 1 hour;
in the step 6), the reaction solvent is N, N-methylformamide, the reaction temperature is 50 ℃, and the reaction time is 24 hours.
2. The method of claim 1, wherein the method comprises the steps of:
1) under the ice bath condition, slowly adding concentrated sulfuric acid into an aqueous solution of hydrobromic acid, then slowly adding a compound 2a, and after completion, heating to 110 ℃ for reacting for 6 hours to obtain a compound 2;
2) dissolving compound 1 in anhydrous tetrahydrofuran under nitrogen protection at 0oSlowly adding n-butyllithium under the condition of C, preserving heat, reacting for 1 hour, then adding the compound 2, heating to room temperature, and reacting for 24 hours to obtain a compound 3;
3) adding the compound 3, the compound 4, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and the N, N-diisopropylethylamine into the N, N-dimethylformamide in sequence, and reacting at room temperature for 24 hours to obtain a compound 5;
4) adding the compound 6, the compound 7, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate and the N, N-diisopropylethylamine into the N, N-dimethylformamide in sequence, and stirring at room temperature for reacting for 24 hours to obtain a compound 8;
5) dissolving the compound 8 in dichloromethane, slowly dropwise adding trifluoroacetic acid while stirring at room temperature, and reacting at room temperature for 1 hour to obtain a compound 9;
6) and sequentially adding the compound 5, the compound 9 and potassium carbonate into N, N-methylformamide, heating to 50 ℃ and reacting for 24 hours to obtain the lomitapide-D8.
3. The method according to claim 1 or 2,
in the step 1), the molar ratio of the compound 2a to the hydrobromic acid is 1.0: 2.85;
in the step 2), the mol ratio of the compound 1, the n-butyllithium and the compound 2 is 1.0: 2.25: 1.20;
in the step 3), the molar ratio of the compound 3, the compound 4, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate to the N, N-diisopropylethylamine is 1.21: 1: 1.51: 1.51: 2.96;
in the step 4), the molar ratio of the compound 6, the compound 7, the 1-hydroxybenzotriazole, the benzotriazole-N, N, N ', N' -tetramethylurea hexafluorophosphate to the N, N-diisopropylethylamine is 1.19: 1: 1.51: 1.51: 1.98 of;
in the step 5), the molar ratio of the compound 8 to the trifluoroacetic acid is 1: 2.51;
in the step 6), the molar ratio of the compound 5 to the compound 9 to the potassium carbonate is 1: 1.13: 2.52.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010271711.8A CN111187200A (en) | 2020-04-09 | 2020-04-09 | Synthesis method of lomitapide-D8 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010271711.8A CN111187200A (en) | 2020-04-09 | 2020-04-09 | Synthesis method of lomitapide-D8 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111187200A true CN111187200A (en) | 2020-05-22 |
Family
ID=70704996
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010271711.8A Pending CN111187200A (en) | 2020-04-09 | 2020-04-09 | Synthesis method of lomitapide-D8 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111187200A (en) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5712279A (en) * | 1995-02-21 | 1998-01-27 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| WO1998031366A1 (en) * | 1997-01-17 | 1998-07-23 | Bristol-Myers Squibb Company | Method for treating atherosclerosis with an mpt inhibitor and cholesterol lowering drugs |
| WO1998031367A1 (en) * | 1997-01-17 | 1998-07-23 | Bristol-Myers Squibb Company | Method for treating acid lipase deficiency diseases with an mtp inhibitor and cholesterol lowering drugs |
| US20160083345A1 (en) * | 2014-09-19 | 2016-03-24 | Cadila Healthcare Limited | Polymorphic forms of lomitapide and its salts and processes for their preparation |
| CN105481758A (en) * | 2016-01-13 | 2016-04-13 | 天津药物研究院有限公司 | Lomitapide crystal form I as well as preparation method and application thereof |
| WO2016055934A1 (en) * | 2014-10-09 | 2016-04-14 | Glenmark Pharmaceuticals Limited | Amorphous form of lomitapide mesylate |
| CN105523994A (en) * | 2016-02-03 | 2016-04-27 | 南京华威医药科技开发有限公司 | Crystal form III of lomitapide mesylate |
| WO2016071849A1 (en) * | 2014-11-05 | 2016-05-12 | Hetero Research Foundation | Process for the preparation of lomitapide |
| CN106146385A (en) * | 2015-04-03 | 2016-11-23 | 天津药物研究院有限公司 | A kind of synthetic method of triglyceride transfer protein enzyme inhibitor |
| CN105461618B (en) * | 2016-01-26 | 2018-09-25 | 南京华威医药科技集团有限公司 | Methanesulfonic acid Lome Tapai novel crystal forms and preparation method thereof |
-
2020
- 2020-04-09 CN CN202010271711.8A patent/CN111187200A/en active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5712279A (en) * | 1995-02-21 | 1998-01-27 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| WO1998031366A1 (en) * | 1997-01-17 | 1998-07-23 | Bristol-Myers Squibb Company | Method for treating atherosclerosis with an mpt inhibitor and cholesterol lowering drugs |
| WO1998031367A1 (en) * | 1997-01-17 | 1998-07-23 | Bristol-Myers Squibb Company | Method for treating acid lipase deficiency diseases with an mtp inhibitor and cholesterol lowering drugs |
| US20160083345A1 (en) * | 2014-09-19 | 2016-03-24 | Cadila Healthcare Limited | Polymorphic forms of lomitapide and its salts and processes for their preparation |
| WO2016055934A1 (en) * | 2014-10-09 | 2016-04-14 | Glenmark Pharmaceuticals Limited | Amorphous form of lomitapide mesylate |
| WO2016071849A1 (en) * | 2014-11-05 | 2016-05-12 | Hetero Research Foundation | Process for the preparation of lomitapide |
| CN106146385A (en) * | 2015-04-03 | 2016-11-23 | 天津药物研究院有限公司 | A kind of synthetic method of triglyceride transfer protein enzyme inhibitor |
| CN105481758A (en) * | 2016-01-13 | 2016-04-13 | 天津药物研究院有限公司 | Lomitapide crystal form I as well as preparation method and application thereof |
| CN105461618B (en) * | 2016-01-26 | 2018-09-25 | 南京华威医药科技集团有限公司 | Methanesulfonic acid Lome Tapai novel crystal forms and preparation method thereof |
| CN105523994A (en) * | 2016-02-03 | 2016-04-27 | 南京华威医药科技开发有限公司 | Crystal form III of lomitapide mesylate |
Non-Patent Citations (3)
| Title |
|---|
| KEN KAWAMOTO,等: "Loops versus Branch Functionality in Model Click Hydrogels", 《MACROMOLECULES》 * |
| 王世真: "《分子核医学 第2版》", 30 April 2004 * |
| 王德心: "《组合化学原理》", 31 January 2005 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5768712B2 (en) | Diphenylmethane compounds | |
| Tamiaki et al. | A novel protecting group for constructing combinatorial peptide libraries | |
| CN111253287A (en) | Method for synthesizing side chain of Somalutide in liquid phase convergence manner | |
| JPH07304770A (en) | New benzazepinone derivative | |
| CN107810189B (en) | Method for preparing nitrogen mustard derivatives | |
| CN111187200A (en) | Synthesis method of lomitapide-D8 | |
| US20240262805A1 (en) | Method for producing compound or pharmaceutically acceptable salt thereof | |
| EP3398933A1 (en) | Method for preparing long-chain compound | |
| Isowa et al. | Synthesis of rhodotorulic acid | |
| CN113024637A (en) | Method for preparing carfilzomib by using water-soluble alkynylamide as condensing agent | |
| CN109369779B (en) | Synthetic method of taltirelin | |
| CN111848546B (en) | A kind of 2-(aminomethyl)thiazole-5-carbonitrile and its synthetic method | |
| US6225480B1 (en) | Sulphonyl compounds for use as linkers in solid phase and combinatorial synthesis | |
| CN109574860B (en) | A kind of method for preparing vilanterol | |
| CN106167457A (en) | Novel aspartame, its preparation method and the purposes in Solid phase peptide synthssis thereof | |
| CN111393426A (en) | Rivaroxaban thiophene carboxylate impurity reference substance and preparation method thereof | |
| JP3207018B2 (en) | Method for producing benzylsuccinic acid derivative and intermediate for producing the same | |
| CN112375015A (en) | Preparation method of di-tert-butyloxycarbonylaminoacetic acid | |
| CN105949279A (en) | Method for preparing proteasome inhibitor Oprozomib and analogs thereof | |
| KR100543980B1 (en) | 2- (4-nitrophenyl) sulfonylethoxycarbonyl substituted amino acid derivative and preparation method thereof | |
| CN119708127B (en) | Method for synthesizing connector-MMAF | |
| JPH078855B2 (en) | Sulfonium compound | |
| CN120025261A (en) | A method for synthesizing 2-aminoacyl-substituted L-phenylalanine | |
| CN116693428A (en) | Synthesis method of tri (p-toluenesulfonyloxymethyl) nitromethane | |
| CA2596419A1 (en) | Use of functionalized onium salts for peptide synthesis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20200522 |
|
| RJ01 | Rejection of invention patent application after publication |