CN111039919A - L-malic acid dimer impurity and preparation method thereof - Google Patents
L-malic acid dimer impurity and preparation method thereof Download PDFInfo
- Publication number
- CN111039919A CN111039919A CN201911350262.XA CN201911350262A CN111039919A CN 111039919 A CN111039919 A CN 111039919A CN 201911350262 A CN201911350262 A CN 201911350262A CN 111039919 A CN111039919 A CN 111039919A
- Authority
- CN
- China
- Prior art keywords
- impurity
- compound
- malic acid
- dimer
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 title claims abstract description 102
- 239000012535 impurity Substances 0.000 title claims abstract description 92
- 238000002360 preparation method Methods 0.000 title abstract description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims abstract description 42
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims abstract description 42
- 235000011090 malic acid Nutrition 0.000 claims abstract description 42
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims abstract description 39
- 150000001875 compounds Chemical class 0.000 claims abstract description 30
- 229940116298 l- malic acid Drugs 0.000 claims abstract description 30
- 229940126214 compound 3 Drugs 0.000 claims abstract description 24
- 229940125782 compound 2 Drugs 0.000 claims abstract description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 23
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims abstract description 21
- 229940125904 compound 1 Drugs 0.000 claims abstract description 18
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 6
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims abstract description 5
- 230000003301 hydrolyzing effect Effects 0.000 claims abstract description 3
- 238000006243 chemical reaction Methods 0.000 claims description 64
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 40
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 30
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 16
- 229960000583 acetic acid Drugs 0.000 claims description 15
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 14
- 238000010992 reflux Methods 0.000 claims description 14
- 239000000539 dimer Substances 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 239000001257 hydrogen Substances 0.000 claims description 10
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 claims description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 9
- 239000012362 glacial acetic acid Substances 0.000 claims description 9
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 239000003054 catalyst Substances 0.000 claims description 8
- 238000000034 method Methods 0.000 claims description 8
- 239000007810 chemical reaction solvent Substances 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 238000006555 catalytic reaction Methods 0.000 claims description 4
- 239000002253 acid Substances 0.000 claims description 2
- 239000011230 binding agent Substances 0.000 claims description 2
- 238000005984 hydrogenation reaction Methods 0.000 claims description 2
- 239000002904 solvent Substances 0.000 claims description 2
- 125000006278 bromobenzyl group Chemical group 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- 229940099690 malic acid Drugs 0.000 abstract description 12
- 239000001630 malic acid Substances 0.000 abstract description 12
- 238000011160 research Methods 0.000 abstract description 9
- 238000001228 spectrum Methods 0.000 abstract description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 45
- 238000005303 weighing Methods 0.000 description 28
- 238000001914 filtration Methods 0.000 description 27
- 238000003756 stirring Methods 0.000 description 21
- 239000000126 substance Substances 0.000 description 21
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- 238000010438 heat treatment Methods 0.000 description 17
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- 238000001035 drying Methods 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- 238000005406 washing Methods 0.000 description 12
- 239000007864 aqueous solution Substances 0.000 description 7
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- 150000002431 hydrogen Chemical class 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 description 6
- 239000003814 drug Substances 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 238000003908 quality control method Methods 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- -1 2,2' - ((2S, 5S) -3, 6-dioxo-1, 4-dioxane-2, 5-diyl) diacetic acid Chemical compound 0.000 description 1
- 208000037157 Azotemia Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010019663 Hepatic failure Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001793 charged compounds Chemical class 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000007903 liver failure Diseases 0.000 description 1
- 231100000835 liver failure Toxicity 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 208000009852 uremia Diseases 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/12—1,4-Dioxanes; Hydrogenated 1,4-dioxanes not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention provides an L-malic acid dimer impurity and a preparation method thereof, wherein the L-malic acid dimer impurity (impurity X) is discovered and separated for the first time and has the following structure:
Description
Technical Field
The invention belongs to the technical field of medicines, and particularly relates to an L-malic acid dimer impurity and a preparation method thereof.
Background
The malic acid is a molecule containing a chiral center, the L-malic acid is a levorotatory isomer of the malic acid, and documents show that the L-malic acid has important physiological functions, is an important intermediate product of internal circulation of a human body, is easy to be absorbed by the human body, can be used for treating various diseases such as liver diseases, anemia, low immunity, uremia, hypertension, liver failure and the like, and can relieve the toxic effect of an anticancer medicament on normal cells.
L-malic acid has been widely applied in the field of medicine, the literature research on impurities of the L-malic acid is mainly focused on fumaric acid, maleic acid and tartaric acid, and the research on related impurities of the L-malic acid is still incomplete.
In the process of the stability research of the L-malic acid, the technical personnel of the invention find a degradation impurity which is polymerized by the L-malic acid. At present, the impurity is not reported in the literature.
Disclosure of Invention
The invention aims to provide an L-malic acid dimer impurity which is discovered and separated for the first time, and a preparation method of the impurity.
The molecular formula of the L-malic acid dimer impurity (impurity X) is C8H8O8The structure is as follows:
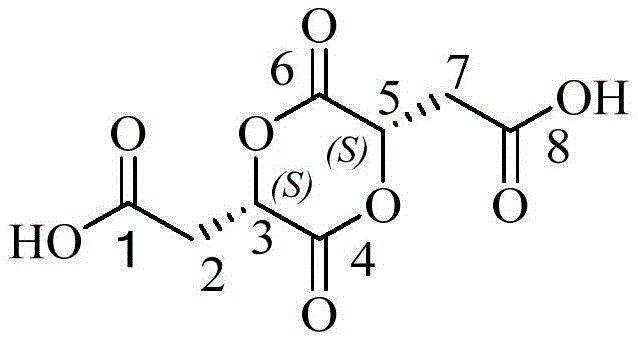
the chiral centers of the 3-position carbon and the 5-position carbon of the L-malic acid dimer impurity (impurity X) are in S configuration.
This L-malic acid dimer impurity (impurity X) was chemically named 2,2' - ((2S, 5S) -3, 6-dioxo-1, 4-dioxane-2, 5-diyl) diacetic acid, which is1H-NMR(DMSO-d,500MHz)、13C-NMR(DMSO-d, 500MHz) and mass spectral structure confirmation characteristic peak data were as follows:
nuclear magnetic resonance hydrogen spectrum data of L-malic acid dimer impurity (impurity X)
| Proton type | Chemical shift (PPM) | Home H number | Number of protons |
| O-H | 12.747 | C1-H,C8- |
2 |
| C-H | 5.723-5.746 | C3-H,C5- |
2 |
| C-H | 2.499-3.025 | C2-H,C7- |
4 |
Nuclear magnetic resonance hydrogen spectrum data of L-malic acid dimer impurity (impurity X)
| Chemical shift/ppm | Type of carbon atom | Home C number | Number of carbons |
| 170.16 | Quaternary carbon | C1,C8 | 2 |
| 166.47 | Quaternary carbon | C4,C6 | 2 |
| 72.63 | Tertiary carbon | C3,C5 | 2 |
| 35.06 | Secondary carbon | C2,C7 | 4 |
Mass spectral data of L-malic acid dimer impurity (impurity X)
| Peak of molecular ion | Theoretical nuclear to proton ratio | The result of the detection |
| [M+H] | 233.14 | 233.1 |
| [M+H2O+H] | 251.16 | 251.1 |
| [M+2H2O+H] | 268.17 | 268.1 |
| [SM2+H] | 135.1 | 135.1 |
The L-malic acid dimer impurity (impurity X) provided by the invention is unstable under acidic or alkaline conditions, and is easy to be converted into malic acid especially in an acidic or alkaline aqueous solution; while malic acid is easily degraded into L-malic acid dimer impurity (impurity X) at high temperature.
The discovery of the L-malic acid dimer impurity (impurity X) fills the defects in the impurity research, and makes the impurity research more complete and sufficient. Meanwhile, in the research on the stability of the malic acid, the explanation characteristic of the impurity is relatively large under the influence of temperature, and a more comprehensive evidence is provided for the selection basis of storage conditions.
The invention also provides a preparation method of the L-malic acid dimer impurity (impurity X), which comprises the following steps:
(1) reacting L-malic acid with 2, 2-dimethoxypropane at room temperature under the catalysis of p-toluenesulfonic acid to protect carboxyl on one side of the L-malic acid to obtain a compound 1;
(2) under the conditions that acetone is used as a solvent and triethylamine is used as an acid-binding agent, protecting carboxyl on the other side with benzyl to obtain a compound 2;
(3) hydrolyzing the compound 2 under the condition of acetic acid/water/tetrahydrofuran to obtain a compound 3;
(4) under the catalysis of p-toluenesulfonic acid, refluxing toluene, and self-dehydrating and condensing to obtain a compound 4;
(5) adding the compound 4 into a reaction solvent, and hydrogenating to remove a benzyl protecting group under the action of palladium carbon and hydrogen to obtain an impurity X.
The synthetic route of the dimer L-malic acid impurity (impurity X) is as follows:
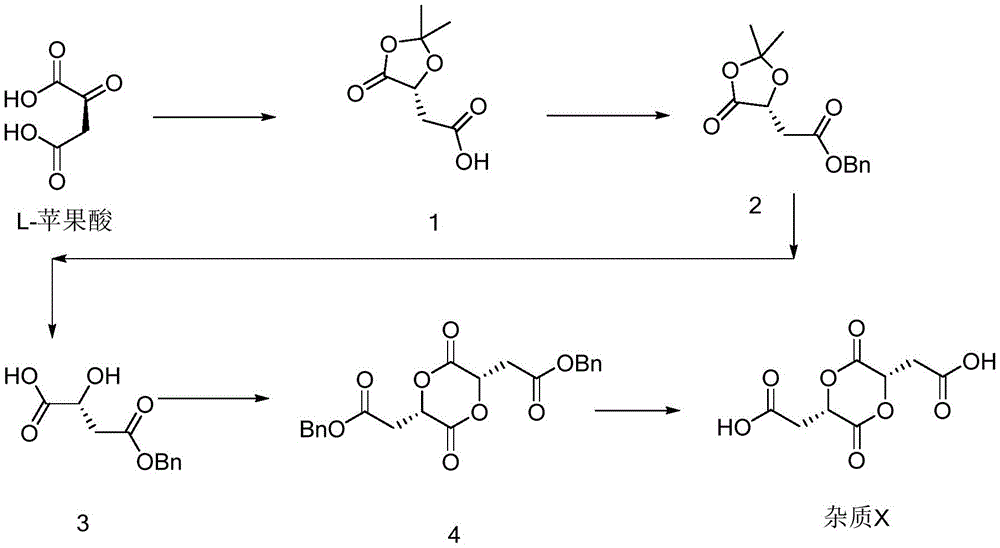
wherein the feeding ratio of the L-malic acid, the p-toluenesulfonic acid and the 2, 2-dimethoxypropane in the step (1) is 1:0.015: 3-1: 0.03: 5, preferably 1:0.015: 3-1: 0.02: 4; the reaction temperature is 25-35 ℃, preferably 25-30 ℃.
In the step (2), the feeding ratio of the compound 1, triethylamine and benzyl bromide is 1:2: 2-1: 3:3, preferably 1:2.5:2 in terms of weight-volume ratio (g: ml: ml).
In the step (3), the feeding ratio of the compound 2 to acetic acid/water/tetrahydrofuran is 1: 15-1: 20, preferably 1:15, in terms of weight-volume ratio (g: ml); the feeding ratio of the acetic acid, the water and the tetrahydrofuran is 1:1:1 according to the volume.
In the step (4), the feeding ratio of the compound 3, toluene and p-toluenesulfonic acid is 1:60: 0.05-1: 75:0.1, preferably 1:65:0.1 according to the weight-volume ratio (g: ml: g).
In the step (5), the feeding ratio of the compound 4, the reaction solvent and the palladium-carbon catalyst is 1:15: 0.1-1: 25:0.3, preferably 1:20:0.2 according to the weight-volume ratio (g: ml: g); the reaction solvent is selected from tetrahydrofuran, methanol and glacial acetic acid, and tetrahydrofuran is preferred; the pressure of the hydrogenation reaction was atmospheric pressure.
The L-malic acid dimer impurity and the preparation method thereof provided by the invention have important significance for the quality control of malic acid:
(1) the L-malic acid dimer impurity is a new discovery, and a document report is not seen before, so that the quality control of malic acid is more complete, and the related contents of domestic and foreign pharmacopoeias are expected to be supplemented, and the quality standard of malic acid is promoted to be improved;
(2) the invention researches the physical and chemical properties of the compound of the L-malic acid dimer impurity, and has important guiding significance for the selection of the storage condition of the malic acid.
Drawings
FIG. 1 NMR spectrum of dimer L-malic acid impurity (impurity X)
FIG. 2 NMR spectrum of dimer impurity L-malic acid (impurity X)
FIG. 3 Mass Spectrometry of L-malic acid dimer impurity (impurity X)
Detailed Description
The following examples are intended to illustrate the invention in detail, but are not intended to limit the invention.
Example 1
Preparation method of L-malic acid dimer impurity (impurity X)
(1) Weighing 50.0g of L-malic acid and 0.80g of p-toluenesulfonic acid, adding into a 500ml three-mouth reaction bottle, adding 180ml of 2, 2-dimethoxypropane, reacting for 4-5h at 25-30 ℃ under the protection of nitrogen, washing and extracting with 300ml of sodium bicarbonate aqueous solution and 300ml of dichloromethane after the reaction is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating the organic phase to obtain an oily substance, adding 50ml of diethyl ether, stirring and crystallizing, and filtering to obtain 28.56g of compound 1, wherein the yield is 41.5%.
(2) Adding the compound 1 into a 1L three-necked bottle, adding 450ml of acetone, stirring for dissolving, adding 75ml of triethylamine and 60ml of benzyl bromide, heating to reflux for 5-6h, filtering after the reaction is finished, concentrating the filtrate under reduced pressure, adding 400ml of ethyl acetate and 200ml of water for washing and extracting after the concentration is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating, adding 50ml of diethyl ether after the concentration is finished, stirring for crystallization, and filtering to obtain 32.15g of the compound 2 with the yield of 74.6%.
(3) Weighing 32.00g of the compound 2, adding the compound 2 into a 1L reaction bottle, adding 160ml of tetrahydrofuran, 160ml of water and 160ml of glacial acetic acid, heating to 40 ℃, reacting for 18-20h, concentrating under reduced pressure to obtain yellow oily matter, weighing 25.18g of the compound 3, and obtaining the yield of 89.2%.
(4) Weighing 25.00g of compound 3, adding the compound 3 into a 2L three-mouth reaction bottle, adding 1.65L of toluene and 2.6g of p-toluenesulfonic acid, heating to reflux, carrying out water-splitting reaction for 48-50h, after the reaction is finished, carrying out reduced pressure concentration to obtain an oily substance, separating a main product point through a chromatographic column, concentrating to obtain the oily substance, adding diethyl ether, stirring, crystallizing, and filtering to obtain 1.84g of compound 4, wherein the yield is 3.86%.
(5) Weighing 1.8g of compound 4, adding the compound 4 into a 100ml three-mouth reaction bottle, adding 36ml of tetrahydrofuran and 0.37g of palladium-carbon catalyst (the palladium content is 10 percent, the moisture content is 43 percent), introducing hydrogen, hydrogenating at normal pressure, reacting for 4-5h at the temperature of 30-35 ℃, filtering after the reaction is finished, and concentrating the filtrate to be dry to obtain 0.86g of impurity X, wherein the yield is 88.1 percent.
1H-NMR(DMSO-d,500MHz)δ:12.761(s,2H),5.723-5.746(s,2H),2.502-3.028(d,4H)。ESI-MS(m/z):233.1[M+H]+.
Example 2
Preparation method of L-malic acid dimer impurity (impurity X)
(1) Weighing 50.0g of L-malic acid and 1.0g of p-toluenesulfonic acid, adding into a 500ml three-mouth reaction bottle, adding 200ml of 2, 2-dimethoxypropane, reacting for 4-5h at 25-30 ℃ under the protection of nitrogen, washing and extracting with 300ml of sodium bicarbonate aqueous solution and 300ml of dichloromethane after the reaction is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating the organic phase to obtain an oily substance, adding 50ml of diethyl ether, stirring and crystallizing, and filtering to obtain 28.5g of compound 1, wherein the yield is 43.2%.
(2) Adding the compound 1 into a 1L three-necked bottle, adding 450ml of acetone, stirring for dissolving, adding 72ml of triethylamine and 57ml of benzyl chloride, heating to reflux for reaction for 5-6h, filtering after the reaction is finished, concentrating the filtrate under reduced pressure, adding 400ml of ethyl acetate and 200ml of water for washing and extracting after the concentration is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating, adding 50ml of diethyl ether after the concentration is finished, stirring for crystallization, and filtering to obtain 30.26g of the compound 2 with the yield of 69.98%.
(3) Weighing 30.0g of the compound 2, adding the compound into a 1L reaction bottle, adding 150ml of tetrahydrofuran, 150ml of water and 150ml of glacial acetic acid, heating to 40 ℃, reacting for 18-20h, after the reaction is finished, concentrating under reduced pressure to obtain yellow oily matter, weighing 23.36g of the compound 3, and obtaining the yield of 91.7%.
(4) Weighing 23.00g of compound 3, adding the compound 3 into a 2L three-mouth reaction bottle, adding 1.5L of toluene and 2.33g of p-toluenesulfonic acid, heating to reflux, carrying out water-splitting reaction for 48-50h, after the reaction is finished, carrying out reduced pressure concentration to obtain an oily substance, separating a main product point through a chromatographic column, adding diethyl ether into the oily substance obtained by concentration, stirring and crystallizing, and filtering to obtain 2.12g of compound 4 with the yield of 5%.
(5) Weighing 2.0g of compound 4, adding the compound 4 into a 100ml three-mouth reaction bottle, adding 40ml of tetrahydrofuran and 0.4g of palladium-carbon catalyst (the palladium content is 10 percent, the moisture content is 43 percent), introducing hydrogen, hydrogenating at normal pressure, reacting for 4-5h at the temperature of 30-35 ℃, filtering after the reaction is finished, and concentrating the filtrate to be dry to obtain 1.05g of impurity X, wherein the yield is 93.7 percent.
Example 3
Preparation method of L-malic acid dimer impurity (impurity X)
(1) Weighing 50.0g of L-malic acid and 0.90g of p-toluenesulfonic acid, adding into a 500ml three-mouth reaction bottle, adding 150ml of 2, 2-dimethoxypropane, reacting for 4-5h at 25-30 ℃ under the protection of nitrogen, washing and extracting with 300ml of sodium bicarbonate aqueous solution and 300ml of dichloromethane after the reaction is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating the organic phase to obtain an oily substance, adding 50ml of diethyl ether, stirring and crystallizing, and filtering to obtain 27.6g of compound 1, wherein the yield is 41.8%.
(2) Adding the compound 1 into a 1L three-necked bottle, adding 450ml of acetone, stirring for dissolving, adding 70ml of triethylamine and 60ml of benzyl bromide, heating to reflux for 5-6h, filtering after the reaction is finished, concentrating the filtrate under reduced pressure, adding 400ml of ethyl acetate and 200ml of water for washing and extracting after the concentration is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating, adding 50ml of diethyl ether after the concentration is finished, stirring for crystallization, and filtering to obtain 31.2g of the compound 2 with the yield of 74.5%.
(3) 31.0g of the compound 2 is weighed and added into a 1L reaction bottle, 155ml of tetrahydrofuran, 155ml of water and 155ml of glacial acetic acid are added, the temperature is raised to 40 ℃, the reaction is carried out for 18 to 20 hours, after the reaction is finished, yellow oily matter is obtained by decompression and concentration, 21.36g of the compound 3 is weighed, and the yield is 81.2 percent.
(4) Weighing 21.0g of compound 3, adding the compound 3 into a 2L three-mouth reaction bottle, adding 1.35L of toluene and 2.1g of p-toluenesulfonic acid, heating to reflux, carrying out water-splitting reaction for 48-50h, after the reaction is finished, carrying out reduced pressure concentration to obtain an oily substance, separating a main product point through a chromatographic column, adding diethyl ether into the oily substance obtained by concentration, stirring and crystallizing, and filtering to obtain 2.32g of compound 4 with the yield of 6%.
(5) Weighing 2.2g of the compound 4, adding the compound 4 into a 100ml three-mouth reaction bottle, adding 44ml of tetrahydrofuran and 0.44g of palladium-carbon catalyst (the palladium content is 10 percent, the moisture content is 43 percent), introducing hydrogen, hydrogenating at normal pressure, reacting for 4-5h at the temperature of 30-35 ℃, filtering after the reaction is finished, and concentrating the filtrate to be dry to obtain 1.08g of impurity X, wherein the yield is 91.8 percent.
Example 4
Preparation method of L-malic acid dimer impurity (impurity X)
(1) Weighing 50.0g of L-malic acid and 0.75g of p-toluenesulfonic acid, adding into a 500ml three-mouth reaction bottle, adding 150ml of 2, 2-dimethoxypropane, reacting for 4-5h at 30-35 ℃ under the protection of nitrogen, washing and extracting with 300ml of sodium bicarbonate aqueous solution and 300ml of dichloromethane after the reaction is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating the organic phase to obtain an oily substance, adding 50ml of diethyl ether, stirring and crystallizing, and filtering to obtain 26.6g of compound 1 with the yield of 40.3%.
(2) Adding the compound 1 into a 1L three-necked bottle, adding 450ml of acetone, stirring for dissolving, adding 80ml of triethylamine and 80ml of benzyl bromide, heating to reflux for reaction for 5-6h, filtering after the reaction is finished, concentrating the filtrate under reduced pressure, adding 400ml of ethyl acetate and 200ml of water for washing and extracting after the concentration is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating, adding 50ml of diethyl ether after the concentration is finished, stirring for crystallization, and filtering to obtain 30.2g of the compound 2 with the yield of 74.8%.
(3) Weighing 30.0g of the compound 2, adding the compound into a 1L reaction bottle, adding 150ml of tetrahydrofuran, 150ml of water and 150ml of glacial acetic acid, heating to 40 ℃, reacting for 18-20h, after the reaction is finished, concentrating under reduced pressure to obtain yellow oily matter, weighing 23.28g of the compound 3, and obtaining the yield of 91.5%.
(4) Weighing 23.0g of compound 3, adding the compound 3 into a 2L three-mouth reaction bottle, adding 1.7L of toluene and 2.3g of p-toluenesulfonic acid, heating to reflux and carrying out water separation reaction for 48-50h, after the reaction is finished, carrying out reduced pressure concentration to obtain an oily substance, separating a main product point through a chromatographic column, and concentrating to obtain an oily substance, and freeze-drying to obtain 6.56g of compound 4, wherein the yield is 15.5%.
(5) Weighing 6.0g of compound 4, adding the compound 4 into a 250ml three-mouth reaction bottle, adding 120ml of tetrahydrofuran and 1.2g of palladium-carbon catalyst (the palladium content is 10 percent, the moisture content is 43 percent), introducing hydrogen, hydrogenating at normal pressure, reacting for 4-5h at the temperature of 30-35 ℃, filtering after the reaction is finished, and concentrating the filtrate to be dry to obtain 2.83g of impurity X, wherein the yield is 83.9 percent.
Example 5
Preparation method of L-malic acid dimer impurity (impurity X)
(1) Weighing 50.0g of L-malic acid and 1.2g of p-toluenesulfonic acid, adding into a 500ml three-mouth reaction bottle, adding 200ml of 2, 2-dimethoxypropane, reacting for 4-5h at 25-30 ℃ under the protection of nitrogen, washing and extracting with 300ml of sodium bicarbonate aqueous solution and 300ml of dichloromethane after the reaction is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating the organic phase to obtain an oily substance, adding 50ml of diethyl ether, stirring and crystallizing, and filtering to obtain 25.6g of compound 1, wherein the yield is 38.8%.
(2) Adding the compound 1 into a 1L three-necked bottle, adding 450ml of acetone, stirring for dissolving, adding 65ml of triethylamine and 65ml of benzyl bromide, heating to reflux for 5-6h, filtering after the reaction is finished, concentrating the filtrate under reduced pressure, adding 400ml of ethyl acetate and 200ml of water for washing and extracting after the concentration is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating, adding 50ml of diethyl ether after the concentration is finished, stirring for crystallization, and filtering to obtain 30.5g of a compound 2 with the yield of 76.8%.
(3) Weighing 30.0g of the compound 2, adding the compound 2 into a 1L reaction bottle, adding 200ml of tetrahydrofuran, 200ml of water and 20ml of glacial acetic acid, heating to 40 ℃, reacting for 18-20h, concentrating under reduced pressure after the reaction is finished to obtain yellow oily matter, weighing 23.1g of the compound 3, and obtaining the yield of 90.2%.
(4) Weighing 23.0g of compound 3, adding the compound 3 into a 2L three-mouth reaction bottle, adding 1.4L of toluene and 1.15g of p-toluenesulfonic acid, heating to reflux and carrying out water separation reaction for 48-50h, after the reaction is finished, carrying out reduced pressure concentration to obtain an oily substance, separating a main product point through a chromatographic column, and concentrating to obtain an oily substance, and freeze-drying to obtain 6.36g of compound 4, wherein the yield is 13.7%.
(5) Weighing 6.0g of compound 4, adding the compound 4 into a 250ml three-mouth reaction bottle, adding 90ml of methanol and 0.6g of palladium-carbon catalyst (the palladium content is 10 percent, and the moisture content is 43 percent), introducing hydrogen, hydrogenating at normal pressure, reacting for 4-5h at the temperature of 30-35 ℃, filtering after the reaction is finished, and concentrating the filtrate to be dry to obtain 2.73g of impurity X, wherein the yield is 81.5 percent.
Example 6
Preparation method of L-malic acid dimer impurity (impurity X)
(1) Weighing 50.0g of L-malic acid and 1.5g of p-toluenesulfonic acid, adding into a 500ml three-mouth reaction bottle, adding 250ml of 2, 2-dimethoxypropane, reacting for 4-5h at 30-35 ℃ under the protection of nitrogen, washing and extracting with 300ml of sodium bicarbonate aqueous solution and 300ml of dichloromethane after the reaction is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating the organic phase to obtain an oily substance, adding 50ml of diethyl ether, stirring and crystallizing, and filtering to obtain 26.4g of compound 1, wherein the yield is 39.7%.
(2) Adding the compound 1 into a 1L three-necked bottle, adding 450ml of acetone, stirring for dissolving, adding 52ml of triethylamine and 52ml of benzyl bromide, heating to reflux for reaction for 5-6h, filtering after the reaction is finished, concentrating the filtrate under reduced pressure, adding 400ml of ethyl acetate and 200ml of water for washing and extracting after the concentration is finished, collecting an organic phase, drying with anhydrous sodium sulfate, concentrating, adding 50ml of diethyl ether after the concentration is finished, stirring for crystallization, and filtering to obtain 30.9g of a compound 2 with the yield of 75.8%.
(3) Weighing 30.0g of the compound 2, adding the compound 2 into a 1L reaction bottle, adding 180ml of tetrahydrofuran, 180ml of water and 180ml of glacial acetic acid, heating to 40 ℃, reacting for 18-20h, concentrating under reduced pressure to obtain yellow oily matter, weighing 23.8g of the compound 3, and obtaining the yield of 91.2%.
(4) Weighing 23.0g of compound 3, adding the compound 3 into a 2L three-mouth reaction bottle, adding 1.55L of toluene and 1.85g of p-toluenesulfonic acid, heating to reflux and carrying out water separation reaction for 48-50h, after the reaction is finished, carrying out reduced pressure concentration to obtain an oily substance, separating a main product point through a chromatographic column, and concentrating to obtain an oily substance, and freeze-drying to obtain 6.26g of compound 4 with the yield of 12.9%.
(5) Weighing 6.0g of compound 4, adding the compound 4 into a 250ml three-mouth reaction bottle, adding 180ml of glacial acetic acid and 1.8g of palladium-carbon catalyst (the palladium content is 10 percent, and the water content is 43 percent), introducing hydrogen, hydrogenating at normal pressure, reacting for 4-5h at the temperature of 30-35 ℃, filtering after the reaction is finished, and concentrating the filtrate to be dry to obtain 2.53g of impurity X, wherein the yield is 76.5 percent.
Claims (10)
1. An L-malic acid dimer impurity (impurity X), characterized in that the molecular formula of the impurity X is C8H8O8。
2. The dimer L-malic acid impurity (impurity X) according to claim 1, wherein the impurity X has the structure:
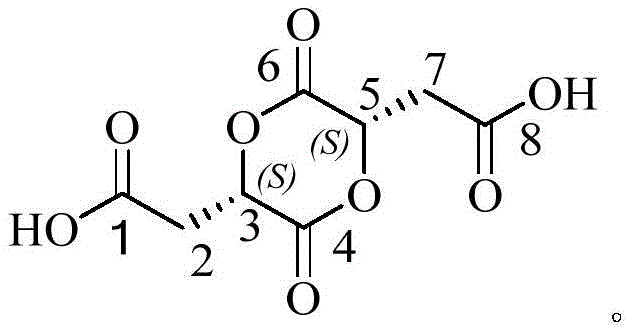
3. the dimer L-malic acid impurity (impurity X) according to claim 1, wherein the chiral centers of the 3-and 5-carbon of impurity X are in S-configuration.
4. The method of claim 1, wherein the method comprises the steps of:
(1) reacting L-malic acid with 2, 2-dimethoxypropane at room temperature under the catalysis of p-toluenesulfonic acid to protect carboxyl on one side of the L-malic acid to obtain a compound 1;
(2) under the conditions that acetone is used as a solvent and triethylamine is used as an acid-binding agent, protecting carboxyl on the other side with benzyl to obtain a compound 2;
(3) hydrolyzing the compound 2 under the condition of acetic acid/water/tetrahydrofuran to obtain a compound 3;
(4) under the catalysis of p-toluenesulfonic acid, refluxing toluene, and self-dehydrating and condensing to obtain a compound 4;
(5) adding the compound 4 into a reaction solvent, and hydrogenating to remove a benzyl protecting group under the action of palladium carbon and hydrogen to obtain an impurity X.
5. The method for preparing L-malic acid dimer impurity (impurity X) according to claim 4, wherein the charging ratio of L-malic acid, p-toluenesulfonic acid, 2-dimethoxypropane in step (1) is 1:0.015: 3-1: 0.03: 5, preferably 1:0.015: 3-1: 0.02: 4; the reaction temperature is 25-35 ℃, preferably 25-30 ℃.
6. The method for preparing dimer L-malic acid impurity (impurity X) according to claim 4, wherein the compound 1, triethylamine and bromobenzyl are fed in a weight/volume ratio (g: ml: ml) of 1:2:2 to 1:3:3, preferably 1:2.5:2 in step (2).
7. The method for preparing dimer L-malic acid impurity (impurity X) according to claim 4, wherein the charge ratio of compound 2, acetic acid/water/tetrahydrofuran in step (3) is 1:15 to 1:20, preferably 1:15, in terms of weight to volume ratio (g: ml); the feeding ratio of the acetic acid, the water and the tetrahydrofuran is 1:1:1 according to the volume.
8. The method for preparing dimer L-malic acid impurity (impurity X) according to claim 4, wherein the charge ratio of compound 3, toluene and p-toluenesulfonic acid in step (4) is 1:60:0.05 to 1:75:0.1, preferably 1:65:0.1, in terms of weight to volume ratio (g: ml: g).
9. The method for preparing dimer L-malic acid impurity (impurity X) according to claim 4, wherein the charge ratio of the compound 4, the reaction solvent and the palladium-on-carbon catalyst in the step (5) is 1:15:0.1 to 1:25:0.3, preferably 1:20:0.2, in terms of weight to volume ratio (g: ml: g); the reaction solvent is selected from tetrahydrofuran, methanol, glacial acetic acid, and preferably tetrahydrofuran.
10. The method for producing dimer L-malic acid impurity (impurity X) according to claim 4, wherein the pressure of the hydrogenation in the step (5) is normal pressure.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911350262.XA CN111039919A (en) | 2019-12-24 | 2019-12-24 | L-malic acid dimer impurity and preparation method thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911350262.XA CN111039919A (en) | 2019-12-24 | 2019-12-24 | L-malic acid dimer impurity and preparation method thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111039919A true CN111039919A (en) | 2020-04-21 |
Family
ID=70239308
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201911350262.XA Pending CN111039919A (en) | 2019-12-24 | 2019-12-24 | L-malic acid dimer impurity and preparation method thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111039919A (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112730637A (en) * | 2020-11-18 | 2021-04-30 | 石家庄四药有限公司 | HPLC detection method of L-malic acid related substances |
| WO2025134071A1 (en) | 2023-12-22 | 2025-06-26 | Sanofi | Malic and glutaric acid based ionizable lipids |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3162619A (en) * | 1961-06-14 | 1964-12-22 | Dow Chemical Co | New polyamides from malide and diamines |
| WO2005077882A1 (en) * | 2004-02-18 | 2005-08-25 | Tokai Education Instruments Co., Ltd. | Oligolactate having substituent in side chain |
-
2019
- 2019-12-24 CN CN201911350262.XA patent/CN111039919A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3162619A (en) * | 1961-06-14 | 1964-12-22 | Dow Chemical Co | New polyamides from malide and diamines |
| WO2005077882A1 (en) * | 2004-02-18 | 2005-08-25 | Tokai Education Instruments Co., Ltd. | Oligolactate having substituent in side chain |
Non-Patent Citations (4)
| Title |
|---|
| NIKOLAUS G. TURRINI等: "Biocatalytic access to nonracemic γ-oxo esters via stereoselective reduction using ene-reductases", 《GREEN CHEMISTRY》 * |
| OUCHI, TATSURO等: "Synthesis of poly(α-malic acid) and its hydrolysis behavior in vitro", 《MAKROMOLEKULARE CHEMIE》 * |
| POUNDER, RYAN J等: "Stereocomplexation in novel degradable amphiphilic block copolymer micelles of poly(ethylene oxide) and poly(benzyl a-malate)", 《SOFT MATTER》 * |
| RYAN J. POUNDER等: "Synthesis and Organocatalytic Ring-Opening Polymerization of Cyclic Esters Derived from L-Malic Acid", 《BIOMACROMOLECULES》 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112730637A (en) * | 2020-11-18 | 2021-04-30 | 石家庄四药有限公司 | HPLC detection method of L-malic acid related substances |
| CN112730637B (en) * | 2020-11-18 | 2023-02-28 | 石家庄四药有限公司 | HPLC detection method for related substances of L-malic acid |
| WO2025134071A1 (en) | 2023-12-22 | 2025-06-26 | Sanofi | Malic and glutaric acid based ionizable lipids |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111039919A (en) | L-malic acid dimer impurity and preparation method thereof | |
| CN101863948B (en) | High-purity (2 beta, 3 alpha, 5 alpha, 16 beta, 17 beta)-2-(4-morpholinyl)-16-(1-pyrrolidinyl)-androstane-3,17-diol or composition thereof and preparation method thereof | |
| CN111925293A (en) | Preparation method of dopamine hydrochloride | |
| CN104761555A (en) | Tofacitinib intermediate preparation method and method for preparing tofacitinib or its salt by using tofacitinib intermediate preparation method | |
| CN104557845B (en) | A kind of preparation method of lubiprostone compound | |
| US9447067B2 (en) | Method of preparing intermediate of salmeterol | |
| CN114685390A (en) | Preparation method of gadobutrol derivative | |
| CN117105884B (en) | Preparation method of 1- (2-aminoethyl) pyrrolidine | |
| EP4112622A1 (en) | Improved processes for making opioids including 14-hydroxycodeinone and 14-hydroxymorphinone | |
| CN113480481B (en) | A kind of preparation method of degradation impurity in ivabradine hydrochloride | |
| CN111100042B (en) | Preparation method of 2-methoxy-5-sulfonamide benzoic acid | |
| CN111285876A (en) | Linagliptin intermediate isomer impurity, preparation method and application thereof | |
| CN1023121C (en) | Preparation method of etopodophylloside-2-dimethylamino compound hydrochloride dihydrate crystal | |
| CN111978188B (en) | Preparation method of mexiletine hydrochloride impurity C | |
| CN113234076A (en) | Preparation method of doxofylline | |
| CN113563285A (en) | Preparation method of a new type of major depressive disorder drug vortioxetine | |
| CN112679363A (en) | Method for preparing pentazocine intermediate | |
| CN113956239A (en) | Azelastine hydrochloride, and preparation method and application thereof | |
| CN117050035B (en) | A kind of preparation method of vortioxetine hydrobromide | |
| CN116554191B (en) | A method for synthesizing bergamot lactone | |
| CN115521317B (en) | Preparation of nafurala method of forming intermediates for use in the preparation of pharmaceutical compositions | |
| CN113636980B (en) | Preparation method of dexrazoxane | |
| CN109761835A (en) | The preparation method of tetracaine hydrochloride | |
| CN112409338B (en) | Midazolam hydrochloride syrup impurity C and impurity D and application thereof | |
| CN110317142B (en) | Preparation method of voglibose |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20200421 |