CN110797080A - Prediction of synthetic lethal genes based on cross-species transfer learning - Google Patents
Prediction of synthetic lethal genes based on cross-species transfer learning Download PDFInfo
- Publication number
- CN110797080A CN110797080A CN201910991037.8A CN201910991037A CN110797080A CN 110797080 A CN110797080 A CN 110797080A CN 201910991037 A CN201910991037 A CN 201910991037A CN 110797080 A CN110797080 A CN 110797080A
- Authority
- CN
- China
- Prior art keywords
- species
- synthetic lethal
- distribution
- cross
- transfer learning
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 108700005090 Lethal Genes Proteins 0.000 title claims abstract description 24
- 238000013526 transfer learning Methods 0.000 title claims abstract description 17
- 238000009826 distribution Methods 0.000 claims abstract description 39
- 238000000034 method Methods 0.000 claims abstract description 32
- 241000894007 species Species 0.000 claims abstract description 31
- 240000004808 Saccharomyces cerevisiae Species 0.000 claims abstract description 11
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 claims abstract description 11
- 241000282412 Homo Species 0.000 claims abstract description 9
- 239000011159 matrix material Substances 0.000 claims description 23
- 230000006870 function Effects 0.000 claims description 11
- 241000235347 Schizosaccharomyces pombe Species 0.000 claims description 8
- 230000006916 protein interaction Effects 0.000 claims description 7
- 241000235346 Schizosaccharomyces Species 0.000 claims description 3
- 230000003044 adaptive effect Effects 0.000 claims description 3
- 238000013480 data collection Methods 0.000 claims description 3
- 238000007781 pre-processing Methods 0.000 claims description 3
- 238000004422 calculation algorithm Methods 0.000 claims description 2
- 230000001131 transforming effect Effects 0.000 claims description 2
- 102000003839 Human Proteins Human genes 0.000 claims 1
- 108090000144 Human Proteins Proteins 0.000 claims 1
- 108010031271 Saccharomyces cerevisiae Proteins Proteins 0.000 claims 1
- 108010058778 Schizosaccharomyces pombe Proteins Proteins 0.000 claims 1
- 238000011524 similarity measure Methods 0.000 claims 1
- 108090000623 proteins and genes Proteins 0.000 description 35
- 231100000225 lethality Toxicity 0.000 description 12
- 230000001665 lethal effect Effects 0.000 description 8
- 238000012216 screening Methods 0.000 description 7
- 108020004459 Small interfering RNA Proteins 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 231100000518 lethal Toxicity 0.000 description 4
- 108091033409 CRISPR Proteins 0.000 description 3
- 238000010354 CRISPR gene editing Methods 0.000 description 3
- 230000031018 biological processes and functions Effects 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 238000012226 gene silencing method Methods 0.000 description 3
- 230000004879 molecular function Effects 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 238000007418 data mining Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 238000000054 nanosphere lithography Methods 0.000 description 2
- 238000007637 random forest analysis Methods 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 101100126955 Arabidopsis thaliana KCS2 gene Proteins 0.000 description 1
- 244000260524 Chrysanthemum balsamita Species 0.000 description 1
- 235000005633 Chrysanthemum balsamita Nutrition 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 108700019961 Neoplasm Genes Proteins 0.000 description 1
- 102000048850 Neoplasm Genes Human genes 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 238000000205 computational method Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 230000009643 growth defect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000010399 physical interaction Effects 0.000 description 1
- 238000013179 statistical model Methods 0.000 description 1
- 238000012706 support-vector machine Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
Images




Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B15/00—ICT specially adapted for analysing two-dimensional or three-dimensional molecular structures, e.g. structural or functional relations or structure alignment
- G16B15/20—Protein or domain folding
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Physics & Mathematics (AREA)
- Health & Medical Sciences (AREA)
- Medical Informatics (AREA)
- Engineering & Computer Science (AREA)
- Spectroscopy & Molecular Physics (AREA)
- General Health & Medical Sciences (AREA)
- Theoretical Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Bioinformatics & Computational Biology (AREA)
- Biotechnology (AREA)
- Evolutionary Biology (AREA)
- Biophysics (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Epidemiology (AREA)
- Artificial Intelligence (AREA)
- Bioethics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Data Mining & Analysis (AREA)
- Databases & Information Systems (AREA)
- Chemical & Material Sciences (AREA)
- Evolutionary Computation (AREA)
- Public Health (AREA)
- Software Systems (AREA)
- Management, Administration, Business Operations System, And Electronic Commerce (AREA)
- Information Retrieval, Db Structures And Fs Structures Therefor (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description
技术领域technical field
本发明属于生物信息学领域,特别是涉及一种基于跨物种迁移学习预测合成致死基因方法。The invention belongs to the field of bioinformatics, in particular to a method for predicting synthetic lethal genes based on cross-species transfer learning.
背景技术Background technique
目前潜在合成致死(syntheticlethality,SL)基因对的筛选方法可以归纳为三类。The current screening methods for potential synthetic lethality (SL) gene pairs can be classified into three categories.
第一种是基于模型生物的方法。它们的基因组很小,很容易突变和匹配;因此,基因沉默技术更容易在模型生物中进行。然而,与所有模型生物的同源推断方法一样,模型生物SL基因对中的大部分基因在人类基因组中没有同源基因。尽管在人类基因组中可以找到同源基因,但它们的功能却发生了巨大的变化,不能直接转化为SL基因。The first is a model organism-based approach. Their genomes are small and easily mutated and matched; thus, gene silencing techniques are easier to perform in model organisms. However, as with the homology inference method for all model organisms, most of the genes in the model organism SL gene pairs have no homologous genes in the human genome. Although homologous genes can be found in the human genome, their functions have changed dramatically and cannot be directly translated into SL genes.
第二种筛选方法是哺乳动物的基因沉默方法,目前已发展出两种基因沉默方法。一种是基于先验知识的推测。潜在的SL基因对包含两种基因,即突变的癌症基因和SL伴侣基因。因此,SL伴侣基因应直接敲除并逐个检测。另一种是基于高通量实验技术对整个基因组进行无偏筛选。最终,siRNA和CRISPR筛选被证明是检测SL基因对s15最可靠的方法。然而,与模型遗传系统相比,人类细胞系统在全基因组siRNA或CRISPR筛选面临更大的挑战。而且,这些方法要昂贵得多,耗费大量劳动和时间,因此发现的许多基本基因要么局限于这些细胞系模型,要么常常在癌症中过度表达。The second screening method is the mammalian gene silencing method, and two gene silencing methods have been developed. One is speculation based on prior knowledge. Potential SL gene pairs contained two genes, the mutated cancer gene and the SL partner gene. Therefore, SL partner genes should be directly knocked out and tested one by one. The other is based on high-throughput experimental techniques for unbiased screening of the entire genome. Ultimately, siRNA and CRISPR screens proved to be the most reliable methods to detect SL genes against s15. However, compared with model genetic systems, human cell systems face greater challenges in genome-wide siRNA or CRISPR screening. Also, these methods are much more expensive, labor-intensive and time-intensive, so many of the essential genes discovered are either limited to these cell line models or are often overexpressed in cancer.
第三种是基于大数据和数据挖掘的计算方法。这种数据驱动的方法又包括生物网络拓扑的方法、数据挖掘方法和统计筛选的方法。与全基因组sirna或基于CRISPR的人细胞系筛选方法相比,计算方法是一种有吸引力的替代方法,它可以帮助识别并优先排序潜在SL基因,以便进行进一步的实验验证。这些方法包括从酵母SL基因中推断人的同源SL基因;利用肿瘤PPI网络的鲁棒性特征评价基因对的重要性;利用基因突变/转录表达数据的统计模型进行互斥性计算;结合体细胞拷贝数改变、siRNA筛选、细胞存活和基因共表达信息的SL(DAISY)数据驱动检测在数据驱动SL基因,并取得了良好的效果;以及基于学习的训练和预测管道,将突变覆盖、驱动突变概率和网络信息中心性这三个特征组合成流形排序模型,生成潜在SL对的排序列表。The third is the computing method based on big data and data mining. This data-driven approach includes biological network topology, data mining and statistical screening. Computational approaches are an attractive alternative to genome-wide siRNA or CRISPR-based human cell line screening methods that can help identify and prioritize potential SL genes for further experimental validation. These methods include inferring human homologous SL genes from yeast SL genes; evaluating the importance of gene pairs using robust features of tumor PPI networks; using statistical models of gene mutation/transcription expression data for mutual exclusion calculations; SL (DAISY) data-driven detection of cell copy number alterations, siRNA screening, cell survival and gene co-expression information has achieved good results in data-driven SL genes; and a learning-based training and prediction pipeline that covers mutation coverage, drives The three features, mutation probability and network information centrality, are combined into a manifold ranking model that generates a ranked list of potential SL pairs.
综上所述,现有的方法预测人类合成致死基因成本较高,需要耗费大量劳动和时间。To sum up, the existing methods for predicting human synthetic lethal genes are costly and require a lot of labor and time.
发明内容SUMMARY OF THE INVENTION
本发明针对现有的监督学习方法的效用受到限制,人类的合成致死基因数据量少的问题,我们提出了基于跨物种迁移学习预测合成致死基因。从酵母、小鼠等模型有机体获得丰富的、经过实验验证的合成致死性作用预测人类的合成致死基因。所叙述方法步骤包括:Aiming at the problem that the utility of the existing supervised learning methods is limited and the amount of synthetic lethal gene data for humans is small, the present invention proposes to predict synthetic lethal genes based on cross-species transfer learning. Obtain abundant, experimentally validated synthetic lethal effects from model organisms such as yeast and mice to predict synthetic lethal genes in humans. The described method steps include:
1.数据收集阶段1. Data collection stage
我们从BioGrid蛋白质相互作用数据库收集的数据生成PPI网络,每个节点代表一种蛋白质,而每条边代表蛋白质之间的相互作用。然后从PPI网络中获取的源物种和目标物种基因使用训练分类器进行分类,具有合成致死性的基因对为阳性数据集,不具有合成致死性的基因对为阴性数据集。两个基因之间已知的合成致死性用二元矩阵Ys,Yt表示,用1表示具有合成致死性,0表示不具有合成致死性。We generated PPI networks from data collected from the BioGrid protein interaction database, with each node representing a protein and each edge representing interactions between proteins. The source and target species genes obtained from the PPI network were then classified using a trained classifier, with gene pairs with synthetic lethality being the positive dataset, and gene pairs not having synthetic lethality being the negative dataset. The known synthetic lethality between two genes is represented by a binary matrix Ys,Yt, with 1 for synthetic lethality and 0 for not synthetic lethality.
2.数据预处理阶段2. Data preprocessing stage
对源物种和目标物种进行PPI网络拓扑相似性度量得到拓扑相似度矩阵Ns∈Rn×k,Nt∈Rm×k,其中k是基因对的网络参数。对源物种和目标物种进行GO语义相似性度量得到语义相似度矩阵Gs∈Rn×d,Gt∈Rm×d,其中d是计算GO相似性的方法数。然后基于PPI网络拓扑相似度矩阵和基于GO方法的语义相似度矩阵的线性组合得到了源物种和目标物种的特征矩阵Xs,Xt,如下:The PPI network topological similarity measurement is performed on the source species and the target species to obtain the topological similarity matrix Ns∈Rn×k, Nt∈Rm×k, where k is the network parameter of the gene pair. Measure the GO semantic similarity between the source species and the target species to obtain the semantic similarity matrix Gs∈Rn×d, Gt∈Rm×d, where d is the number of methods to calculate the GO similarity. Then, based on the linear combination of the topological similarity matrix of the PPI network and the semantic similarity matrix based on the GO method, the characteristic matrices Xs, Xt of the source species and the target species are obtained, as follows:
Xs=[Ns Gs]X s =[N s G s ]
Xt=[Nt Gt]X t =[N t G t ]
跨物种迁移学习方法由两个基本步骤组成。首先,进行流形特征学习,学习两个物种的新特征表示。其次,采用动态分布对齐的方法,定量评价了边缘分布和条件分布的相对重要性,并自适应地最小化了两个物种之间的边缘分布和条件分布差异。最后,可以通过总结这两个步骤来学习域不变的合成致死分类器f。形式上,流形特征学习函数用g(·)表示,目标函数表述如下:The cross-species transfer learning method consists of two basic steps. First, manifold feature learning is performed to learn new feature representations for both species. Second, using a dynamic distribution alignment approach, the relative importance of marginal and conditional distributions was quantitatively evaluated, and the marginal and conditional distribution differences between the two species were adaptively minimized. Finally, a domain-invariant synthetic lethal classifier f can be learned by summarizing these two steps. Formally, the manifold feature learning function is represented by g( ), and the objective function is expressed as follows:
其中第一项表示数据样本的损失。是f的平方范数。Df(·,·)表示动态分布对齐。Rf(·,·)为拉普拉斯正则化,η,λ和ρ是相应的正则化参数。where the first term represents the loss of the data sample. is the square norm of f. Df(·,·) denotes dynamic distribution alignment. Rf(·,·) is the Laplace regularization, and η, λ and ρ are the corresponding regularization parameters.
3.流形特征学习阶段3. Manifold feature learning stage
流形特征学习的目的是确定一个新的特征空间,使源物种和目标物种表现出共同的特征。共同特征的新特征表示是域不变的,因此能够将分类器从源物种迁移到目标物种。我们将源数据集和目标数据集嵌入到Grassmann流形方法G(d)中,它可以看作是所有d维子空间{Φ(T):0≤t≤1}的集合。对于两个原始的基因对xi和xj的D维特征向量,我们计算了Φ(T)tx,它是一个特征向量x在这个子空间中的投影,对于从0到1的连续t,并将所有投影串联到无限维特征向量zi和zj中。将特征向量zi和zj内积产生了一个正半定测地线流核函数为:The goal of manifold feature learning is to determine a new feature space in which the source and target species exhibit common features. The new feature representation of common features is domain-invariant, thus enabling the transfer of classifiers from source species to target species. We embed the source and target datasets into the Grassmann manifold method G(d), which can be viewed as the set of all d-dimensional subspaces {Φ(T): 0≤t≤1}. For the D-dimensional eigenvectors of the two original gene pairs xi and xj, we compute Φ(T)tx, which is the projection of an eigenvector x in this subspace, for a continuous t from 0 to 1, and assign All projections are concatenated into infinite-dimensional eigenvectors zi and zj. The inner product of the eigenvectors zi and zj produces a positive semidefinite geodesic flow kernel function as:
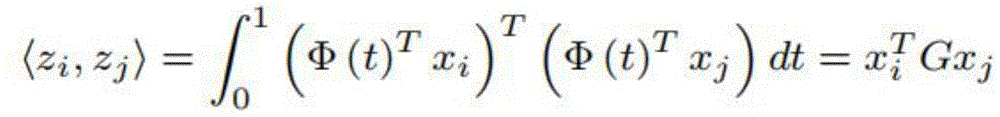
因此,将源特征空间转化为z=g(X)=√gx的Grassmann流形特征空间,通过奇异值分解可有效地计算G,目标函数可表示为:Therefore, by transforming the source feature space into a Grassmann manifold feature space with z=g(X)=√gx, G can be efficiently calculated by singular value decomposition, and the objective function can be expressed as:

然后将Ds的结构最小化:Then the structure of Ds is minimized:
其中是Frobenius范数。K∈r(Nm)×(Nm)是核矩阵,Kij=k(zi,ZJ),A∈r(Nm)×(Nm)是一个对角矩阵,如果i∈Ds,则Aii=1,否则Aii=0。y=y1,y2,.,y(Nm)是酿酒酵母和目标物种种的标签矩阵。tr(·)为跟踪操作。where is the Frobenius norm. K∈r(Nm)×(Nm) is the kernel matrix, Kij=k(zi, ZJ), A∈r(Nm)×(Nm) is a diagonal matrix, if i∈Ds, then Aii=1, otherwise Aii=0. y=y1, y2, ., y(Nm) is the label matrix of S. cerevisiae and target species. tr( ) is a trace operation.
4.动态分布对齐阶段4. Dynamic distribution alignment stage
动态分布对齐主要目的是分布自适应,以最小化域之间的分布差异。采用动态分布对齐的方法,定量地评价了两个物种之间的边缘分布(P)和条件分布分布(Q)的重要性。为此引入自适应因子μ,将动态分布对齐函数定义为:The main purpose of dynamic distribution alignment is distribution adaptation to minimize distribution differences between domains. Using dynamic distribution alignment, the importance of marginal distribution (P) and conditional distribution (Q) between two species was quantitatively evaluated. To this end, an adaptive factor μ is introduced, and the dynamic distribution alignment function is defined as:

(1)分布散度测量(1) Measurement of distribution divergence
边缘分布P和条件分布Q之间的最大平均偏差MMD定义如下:The maximum mean deviation MMD between the marginal distribution P and the conditional distribution Q is defined as:

因此动态分布对齐函数可表示为:Therefore, the dynamic distribution alignment function can be expressed as:

其中第一项表示物种之间的边缘分布偏差,第二项表示条件分布偏差。通过进一步利用具象定理和核技巧,可以将上式中的动态分布对齐函数转化为:where the first term represents the marginal distribution bias between species and the second term represents the conditional distribution bias. By further utilizing the concrete theorem and the kernel trick, the dynamic distribution alignment function in the above equation can be transformed into:

(2)自适应因子μ(2) Adaptive factor μ
A-distance作为一种基本的测量方法被用来获得自适应因子。将A-distance定义为建立线性分类器来区分两个域的误差。ε(H)表示线性分类器h判别两个区域Ds和Dt的误差。A-distance定义如下:A-distance is used as a basic measure to obtain the adaptation factor. Define A-distance as the error in building a linear classifier to distinguish two domains. ε(H) represents the error of the linear classifier h discriminating the two regions Ds and Dt. A-distance is defined as follows:
dA(Ds,Dt)=2(1-2ε(h))d A (D s , D t )=2(1-2ε(h))
然后μ可估计为:Then μ can be estimated as:

其中dM表示第c类A-distance的边缘分布,dC表示A-distance的条件分布。where dM represents the marginal distribution of the c-th class A-distance, and dC represents the conditional distribution of A-distance.
5.拉普拉斯正则化引入拉普拉斯正则化来进一步利用流形方法G中邻近点的相似几何性质,pair-wise affinity矩阵如下:5. Laplacian regularization Laplacian regularization is introduced to further exploit the similar geometric properties of adjacent points in the manifold method G. The pair-wise affinity matrix is as follows:

其中sim(·,·)是度量两点间距离的相似函数(如余弦距离)。Np(Zi)表示点Zi的最近邻集。P是一个自由参数,必须在该方法中设置。通过引入对角矩阵的拉普拉斯矩阵L=D-W,得到了方程的最终拉普拉斯正则化项。where sim(·,·) is a similarity function (such as cosine distance) that measures the distance between two points. Np(Zi) represents the nearest neighbor set of point Zi. P is a free parameter that must be set in this method. By introducing the Laplacian matrix L=D-W of the diagonal matrix, the final Laplacian regularization term of the equation is obtained.

最终目标函数表示为:The final objective function is expressed as:
设置导数
β*=((A+λW+ρL)K+ηI)-1AYT β * = ((A+λW+ρL)K+ηI) -1 AY T
附图说明Description of drawings
图1:基因对的相似性度量Figure 1: Similarity Metrics for Gene Pairs
图2:流形特征矩阵转换Figure 2: Manifold feature matrix transformation
图3:动态分布对齐Figure 3: Dynamic Distribution Alignment
图4:两个不同目标域Figure 4: Two different target domains
具体实施方式Detailed ways
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实验,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。In order to make the objectives, technical solutions and advantages of the present invention clearer, the present invention will be further described in detail below in conjunction with experiments. It should be understood that the specific embodiments described herein are only used to explain the present invention, but not to limit the present invention.
1.数据收集1. Data collection
我们从BioGrid数据库中获得了物种特异性的PPI网络,其中包括酿酒酵母中的740000多种蛋白质相互作用,裂殖酵母中的74000多种蛋白质相互作用,以及人类在的470000多种蛋白质相互作用。BioGrid数据库还提供了实验验证的基因之间的合成致死性,包括酿酒酵母中14000多个具有合成致死作用的基因,裂殖酵母中900多个具有合成致死作用的基因,人类中800多个具有合成致死作用的基因。GO有三种亚本体,即生物过程(BP)、分子功能(MF)和细胞成分(CC)。BP为29660项,MF为11120项,CC为4115项。在各种基于GO语义相似度的计算方法中,我们使用了Mazandu等人所提出的蛋白质语义相似度工具。合成致死预测算法如下:We obtained species-specific PPI networks from the BioGrid database, which included more than 740,000 protein interactions in Saccharomyces cerevisiae, more than 74,000 protein interactions in Schizosaccharomyces cerevisiae, and more than 470,000 protein interactions in humans. The BioGrid database also provides synthetic lethality among experimentally validated genes, including more than 14,000 genes with synthetic lethal effects in Saccharomyces cerevisiae, more than 900 genes with synthetic lethal effects in fission yeast, and more than 800 genes with synthetic lethal effects in humans Synthetic lethal genes. GO has three sub-ontologies, namely biological process (BP), molecular function (MF) and cellular component (CC). BP has 29660 items, MF has 11120 items, and CC has 4115 items. Among various computational methods based on GO semantic similarity, we used the protein semantic similarity tool proposed by Mazandu et al. The synthetic lethal prediction algorithm is as follows:

2.裂殖酵母合成致死基因预测2. Prediction of synthetic lethal genes in fission yeast
我们将TLSL模型应用于酿酒酵母和酿酒酵母,以酿酒酵母(S.cerevisiae)为源物种,以裂殖酵母为目标物种。在酿酒酵母中,我们构建了PPI网络,其中包括9000种实验得到的合成致死性。PPI网络由904种合成致死性、50种剂量致死性、200种负遗传、200种综合生长缺陷和200种正遗传相互作用五种类型组成。在酿酒酵母中,我们考虑了8500对合成致死基因对作为阳性数据集,在一个连通分量图中生成了18000个随机对作为阴性数据集。在裂殖酵母中,906个SLs为阳性数据集,8237个NSLs为阴性数据集。其次,分别计算了基于拓扑结构的PPI相似度矩阵和基于GO的语义相似度矩阵。最后,去除功能相似性缺失的基因对,通过线性组合得到酿酒酵母和裂殖酵母的特征矩阵Xs∈R25039×35,Xt∈R8463×35。利用特征矩阵作为迁移学习模型的输入,得到了裂殖酵母的合成致死预测结果Yt。为了评估所提出的方法的性能,我们采用了一系列的性能评估程序来评估我们的模型来预测SLS,包括准确度(ACC)、灵敏度(Se)、特异性(Sp)、精密度(Pr)、F1-测量(F1)、G-均值(GM)、Matthews相关系数(MCC)。TLSL识别出裂殖酵母中缺少的SL,我们希望找到177个SL对,但只找到了65个。表1显示,本方法的灵敏度为95.9%~80.5%,特异性为91.6%~89.7%,准确度为88.6%~85.1%。We applied the TLSL model to S. cerevisiae and S. cerevisiae, with S. cerevisiae as the source species and Schizosaccharomyces cerevisiae as the target species. In Saccharomyces cerevisiae, we constructed a PPI network that includes 9000 experimentally derived synthetic lethalities. The PPI network consisted of five types of 904 synthetic lethality, 50 dose lethality, 200 negative genetics, 200 comprehensive growth defects, and 200 positive genetic interactions. In Saccharomyces cerevisiae, we considered 8500 pairs of synthetic lethal genes as the positive dataset and 18000 random pairs were generated in a connected component graph as the negative dataset. In fission yeast, 906 SLs were positive datasets and 8237 NSLs were negative datasets. Second, the topology-based PPI similarity matrix and the GO-based semantic similarity matrix are calculated respectively. Finally, the gene pairs with missing functional similarity were removed, and the feature matrices Xs ∈ R 25039×35 and Xt ∈ R 8463×35 were obtained by linear combination. Using the feature matrix as the input of the transfer learning model, the synthetic lethal prediction result Yt of fission yeast was obtained. To evaluate the performance of the proposed method, we employ a series of performance evaluation procedures to evaluate our model to predict SLS, including accuracy (ACC), sensitivity (Se), specificity (Sp), precision (Pr) , F1-measure (F1), G-mean (GM), Matthews correlation coefficient (MCC). TLSL identifies SLs that are missing in fission yeast, and we expected to find 177 SL pairs, but only 65 were found. Table 1 shows that the sensitivity of this method is 95.9%-80.5%, the specificity is 91.6%-89.7%, and the accuracy is 88.6%-85.1%.
表1.裂殖酵母合成致死预测模型的性能比较Table 1. Performance comparison of fission yeast synthetic lethality prediction models

3.人类合成致死基因预测3. Human synthetic lethal gene prediction
我们将酿酒酵母为标记的源物种,人类为未标记的目标物种。我们使用了在裂殖酵母合成致死基因预测中的源数据集。利用BiorGrid数据库构建了人类PPI网络,包括6645个基因和17083个物理相互作用。随机选择803个SLs作为阳性数据集,6000个NSLs为阴性数据集。其次,分别计算了基于拓扑结构的PPI相似度矩阵和基于GO的语义相似度矩阵。最后,去除功能相似性缺失的基因对,通过线性组合得到人类的特征矩阵Xt∈R8463×35。利用特征矩阵作为迁移学习模型的输入,得到了人类的合成致死预测结果Yt。为了评价TLSL方法的预测性能,将其结果与SINaTRA方法进行了比较。结果(表1)表明,TLSL对人类SL基因对分类的所有指标表现最佳,每个指标都有明显的改善。We used Saccharomyces cerevisiae as the tagged source species and humans as the untagged target species. We used the source dataset in fission yeast synthetic lethal gene prediction. A human PPI network was constructed using the BiorGrid database, including 6645 genes and 17083 physical interactions. 803 SLs were randomly selected as the positive dataset and 6000 NSLs as the negative dataset. Second, the topology-based PPI similarity matrix and the GO-based semantic similarity matrix are calculated respectively. Finally, the gene pairs with missing functional similarity are removed, and the human feature matrix Xt∈R 8463×35 is obtained by linear combination. Using the feature matrix as the input to the transfer learning model, the synthetic lethal prediction result Yt for humans is obtained. To evaluate the predictive performance of the TLSL method, its results were compared with the SINaTRA method. The results (Table 1) show that TLSL performs best on all metrics for human SL gene pair classification, with significant improvements in each.
表2.人类合成致死预测模型的性能比较Table 2. Performance comparison of human synthetic lethality prediction models

4.实验及结果分析4. Experiment and result analysis
实验结果表明,迁移学习模型在酿酒酵母的合成致死基因迁移到人类的合成致死基因这一跨物种学习任务中优于当代最先进的分类器。迁移学习模型的经验成功可以归因于以下优点。首先,迁移学习模型的流形特征学习能够学习对两个物种不变的共同特征的新特征表示。因此,浅层模型只关注观测变量的协方差,如随机森林和支持向量机,将很难捕捉到两个域之间的这种共同特征。其次,迁移学习模型中的动态分布对齐考虑了物种间的边缘分布和条件分布,并自适应地利用了每种分布的重要性。而传统的分类器,如随机森林,通常不能捕捉到域间的分布差异,从而限制了它们在跨物种任务上的性能,从而导致性能较差。Experimental results demonstrate that the transfer learning model outperforms contemporary state-of-the-art classifiers in the cross-species learning task of translocating synthetic lethal genes from Saccharomyces cerevisiae to synthetic lethal genes in humans. The empirical success of transfer learning models can be attributed to the following advantages. First, manifold feature learning of transfer learning models is able to learn new feature representations that are invariant to common features of both species. Therefore, shallow models that only focus on the covariance of observed variables, such as random forests and support vector machines, will have a hard time capturing this common feature between the two domains. Second, dynamic distribution alignment in transfer learning models takes into account marginal and conditional distributions across species and adaptively exploits the importance of each distribution. While traditional classifiers, such as random forests, often fail to capture distribution differences between domains, which limits their performance on cross-species tasks, resulting in poor performance.
Claims (6)






Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910991037.8A CN110797080A (en) | 2019-10-18 | 2019-10-18 | Prediction of synthetic lethal genes based on cross-species transfer learning |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910991037.8A CN110797080A (en) | 2019-10-18 | 2019-10-18 | Prediction of synthetic lethal genes based on cross-species transfer learning |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN110797080A true CN110797080A (en) | 2020-02-14 |
Family
ID=69440465
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201910991037.8A Pending CN110797080A (en) | 2019-10-18 | 2019-10-18 | Prediction of synthetic lethal genes based on cross-species transfer learning |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN110797080A (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111863123A (en) * | 2020-06-08 | 2020-10-30 | 深圳大学 | A gene synthesis lethal association prediction method |
| CN113436729A (en) * | 2021-07-08 | 2021-09-24 | 湖南大学 | Synthetic lethal interaction prediction method based on heterogeneous graph convolution neural network |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150045369A1 (en) * | 2012-04-10 | 2015-02-12 | Vib Vzw | Novel markers for detecting microsatellite instability in cancer and determining synthetic lethality with inhibition of the dna base excision repair pathway |
| US20160283650A1 (en) * | 2015-02-26 | 2016-09-29 | The Trustees Of Columbia University In The City Of New York | Method for identifying synthetic lethality |
| WO2017083716A2 (en) * | 2015-11-13 | 2017-05-18 | The Board Of Trustees Of The Leland Stanford Junior University | Determination of synthetic lethal partners of cancer-specific alterations and methods of use thereof |
| CN106778070A (en) * | 2017-03-31 | 2017-05-31 | 上海交通大学 | A kind of human protein's subcellular location Forecasting Methodology |
| CN107133496A (en) * | 2017-05-19 | 2017-09-05 | 浙江工业大学 | Gene expression characteristicses extracting method based on manifold learning Yu closed loop depth convolution dual network model |
| CN108197430A (en) * | 2018-01-22 | 2018-06-22 | 哈尔滨工程大学 | Functional form microexon recognition methods based on transfer learning |
| CN109906486A (en) * | 2016-10-03 | 2019-06-18 | 伊鲁米那股份有限公司 | Use phenotype/disease specific gene order of common recognition gene pool and network-based data structure |
-
2019
- 2019-10-18 CN CN201910991037.8A patent/CN110797080A/en active Pending
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150045369A1 (en) * | 2012-04-10 | 2015-02-12 | Vib Vzw | Novel markers for detecting microsatellite instability in cancer and determining synthetic lethality with inhibition of the dna base excision repair pathway |
| US20160283650A1 (en) * | 2015-02-26 | 2016-09-29 | The Trustees Of Columbia University In The City Of New York | Method for identifying synthetic lethality |
| WO2017083716A2 (en) * | 2015-11-13 | 2017-05-18 | The Board Of Trustees Of The Leland Stanford Junior University | Determination of synthetic lethal partners of cancer-specific alterations and methods of use thereof |
| CN109906486A (en) * | 2016-10-03 | 2019-06-18 | 伊鲁米那股份有限公司 | Use phenotype/disease specific gene order of common recognition gene pool and network-based data structure |
| CN106778070A (en) * | 2017-03-31 | 2017-05-31 | 上海交通大学 | A kind of human protein's subcellular location Forecasting Methodology |
| CN107133496A (en) * | 2017-05-19 | 2017-09-05 | 浙江工业大学 | Gene expression characteristicses extracting method based on manifold learning Yu closed loop depth convolution dual network model |
| CN108197430A (en) * | 2018-01-22 | 2018-06-22 | 哈尔滨工程大学 | Functional form microexon recognition methods based on transfer learning |
Non-Patent Citations (4)
| Title |
|---|
| JACUNSKI A ET AL.: "Connectivity homology enables inter-species network models of synthetic lethality" * |
| JASON FAN ET AL.: "Functional protein representations from biological networks enable diverse cross-species inference" * |
| JINDONG WANG ET AL.: "Visual Domain Adaptation with Manifold Embedded Distribution Alignment" * |
| 王建明: "基于基因表达谱芯片确定前列腺癌致病有关基因及其功能" * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111863123A (en) * | 2020-06-08 | 2020-10-30 | 深圳大学 | A gene synthesis lethal association prediction method |
| CN111863123B (en) * | 2020-06-08 | 2023-07-28 | 深圳大学 | Gene synthesis death association prediction method |
| CN113436729A (en) * | 2021-07-08 | 2021-09-24 | 湖南大学 | Synthetic lethal interaction prediction method based on heterogeneous graph convolution neural network |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Maulik et al. | Combining pareto-optimal clusters using supervised learning for identifying co-expressed genes | |
| Hopfensitz et al. | Multiscale binarization of gene expression data for reconstructing Boolean networks | |
| Zhu et al. | Collaborative decision-reinforced self-supervision for attributed graph clustering | |
| CN114420201B (en) | A method for predicting drug target interactions through efficient fusion of multi-source data | |
| CN115394348B (en) | Method, equipment and medium for predicting lncRNA subcellular localization based on graph rolling network | |
| Hong et al. | The entropy and PCA based anomaly prediction in data streams | |
| CN108564009A (en) | A kind of improvement characteristic evaluation method based on mutual information | |
| CN110797080A (en) | Prediction of synthetic lethal genes based on cross-species transfer learning | |
| Nath et al. | Inferring biological basis about psychrophilicity by interpreting the rules generated from the correctly classified input instances by a classifier | |
| Zhu et al. | Multiobjective evolutionary algorithm-based soft subspace clustering | |
| Susanty et al. | Leveraging protein language model embeddings and logistic regression for efficient and accurate in-silico acidophilic proteins classification | |
| Tamee et al. | Towards clustering with XCS | |
| Green et al. | MACEst: The reliable and trustworthy model agnostic confidence estimator | |
| WO2021208993A1 (en) | Information processing method and apparatus for predicting drug target | |
| Tahir et al. | Protein subcellular localization in human and hamster cell lines: employing local ternary patterns of fluorescence microscopy images | |
| Cui et al. | An improved method for K-means clustering | |
| Chen et al. | A novel selective ensemble classification of microarray data based on teaching-learning-based optimization | |
| CN116108892A (en) | A community detection method and system based on network representation learning | |
| Dhyaram et al. | RANDOM SUBSET FEATURE SELECTION FOR CLASSIFICATION. | |
| CN109215741A (en) | Oncogene based on double hypergraph regularizations expresses modal data double focusing class method | |
| Wang et al. | Anfis-based fuzzy systems for searching dna-protein binding sites | |
| Ning et al. | Intrusion detection research based on improved PSO and SVM | |
| Gamage et al. | A robust ensemble regression model for reconstructing genetic networks | |
| Tamee et al. | A learning classifier system approach to clustering | |
| CN119323695B (en) | Parameter fine adjustment method for interpretability of medicine molecular diagram structure |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20200214 |