CN110452165B - Preparation method of 2-methylpyridine-4-formic acid - Google Patents
Preparation method of 2-methylpyridine-4-formic acid Download PDFInfo
- Publication number
- CN110452165B CN110452165B CN201810427394.7A CN201810427394A CN110452165B CN 110452165 B CN110452165 B CN 110452165B CN 201810427394 A CN201810427394 A CN 201810427394A CN 110452165 B CN110452165 B CN 110452165B
- Authority
- CN
- China
- Prior art keywords
- formula
- compound
- methylpyridine
- reaction
- compound shown
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- PMDHIMMPXRSDML-UHFFFAOYSA-N 2-methylpyridine-4-carboxylic acid Chemical compound CC1=CC(C(O)=O)=CC=N1 PMDHIMMPXRSDML-UHFFFAOYSA-N 0.000 title claims abstract description 66
- 238000002360 preparation method Methods 0.000 title claims abstract description 21
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims abstract description 30
- 238000000034 method Methods 0.000 claims abstract description 29
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims abstract description 26
- 238000007254 oxidation reaction Methods 0.000 claims abstract description 21
- 238000006243 chemical reaction Methods 0.000 claims abstract description 20
- 229910021529 ammonia Inorganic materials 0.000 claims abstract description 12
- 238000007259 addition reaction Methods 0.000 claims abstract description 11
- 150000003863 ammonium salts Chemical class 0.000 claims abstract description 11
- 238000007142 ring opening reaction Methods 0.000 claims abstract description 11
- 239000002253 acid Substances 0.000 claims abstract description 10
- 238000006467 substitution reaction Methods 0.000 claims abstract description 10
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 claims abstract description 9
- BULLHNJGPPOUOX-UHFFFAOYSA-N chloroacetone Chemical compound CC(=O)CCl BULLHNJGPPOUOX-UHFFFAOYSA-N 0.000 claims abstract description 9
- 238000006460 hydrolysis reaction Methods 0.000 claims abstract description 9
- 238000007363 ring formation reaction Methods 0.000 claims abstract description 9
- 230000020477 pH reduction Effects 0.000 claims abstract description 5
- 150000001875 compounds Chemical class 0.000 claims description 65
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 30
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 24
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 22
- 239000002904 solvent Substances 0.000 claims description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 18
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical group [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 claims description 16
- 230000001590 oxidative effect Effects 0.000 claims description 13
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 12
- 239000007800 oxidant agent Substances 0.000 claims description 12
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 12
- 235000011121 sodium hydroxide Nutrition 0.000 claims description 10
- 239000000243 solution Substances 0.000 claims description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- 235000019270 ammonium chloride Nutrition 0.000 claims description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 8
- 239000011230 binding agent Substances 0.000 claims description 7
- 239000003054 catalyst Substances 0.000 claims description 7
- 239000003153 chemical reaction reagent Substances 0.000 claims description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 6
- 230000037361 pathway Effects 0.000 claims description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 claims description 5
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 claims description 5
- 238000005580 one pot reaction Methods 0.000 claims description 5
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 claims description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 claims description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 claims description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 4
- 239000003513 alkali Substances 0.000 claims description 4
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 claims description 4
- 150000007529 inorganic bases Chemical group 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 150000007530 organic bases Chemical class 0.000 claims description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 claims description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 4
- 235000011181 potassium carbonates Nutrition 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- 239000002585 base Substances 0.000 claims description 3
- 239000000543 intermediate Substances 0.000 claims description 3
- 239000002994 raw material Substances 0.000 claims description 3
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 claims description 2
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 claims description 2
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 claims description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 2
- 239000004254 Ammonium phosphate Substances 0.000 claims description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 claims description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 claims description 2
- 229910000148 ammonium phosphate Inorganic materials 0.000 claims description 2
- 235000019289 ammonium phosphates Nutrition 0.000 claims description 2
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 claims description 2
- 229910052921 ammonium sulfate Inorganic materials 0.000 claims description 2
- 235000011130 ammonium sulphate Nutrition 0.000 claims description 2
- 239000007864 aqueous solution Substances 0.000 claims description 2
- ZXVOCOLRQJZVBW-UHFFFAOYSA-N azane;ethanol Chemical compound N.CCO ZXVOCOLRQJZVBW-UHFFFAOYSA-N 0.000 claims description 2
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 claims description 2
- 239000001639 calcium acetate Substances 0.000 claims description 2
- 235000011092 calcium acetate Nutrition 0.000 claims description 2
- 229960005147 calcium acetate Drugs 0.000 claims description 2
- NKWPZUCBCARRDP-UHFFFAOYSA-L calcium bicarbonate Chemical compound [Ca+2].OC([O-])=O.OC([O-])=O NKWPZUCBCARRDP-UHFFFAOYSA-L 0.000 claims description 2
- 229910000020 calcium bicarbonate Inorganic materials 0.000 claims description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 claims description 2
- 235000010216 calcium carbonate Nutrition 0.000 claims description 2
- MNNHAPBLZZVQHP-UHFFFAOYSA-N diammonium hydrogen phosphate Chemical compound [NH4+].[NH4+].OP([O-])([O-])=O MNNHAPBLZZVQHP-UHFFFAOYSA-N 0.000 claims description 2
- 230000003301 hydrolyzing effect Effects 0.000 claims description 2
- 238000002955 isolation Methods 0.000 claims description 2
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 claims description 2
- 235000011056 potassium acetate Nutrition 0.000 claims description 2
- 239000011736 potassium bicarbonate Substances 0.000 claims description 2
- 235000015497 potassium bicarbonate Nutrition 0.000 claims description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 claims description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 claims description 2
- 239000001632 sodium acetate Substances 0.000 claims description 2
- 235000017281 sodium acetate Nutrition 0.000 claims description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 claims description 2
- 239000007858 starting material Substances 0.000 claims description 2
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 claims description 2
- 230000009286 beneficial effect Effects 0.000 abstract description 4
- 230000007062 hydrolysis Effects 0.000 abstract description 4
- 238000009776 industrial production Methods 0.000 abstract description 3
- 239000002699 waste material Substances 0.000 abstract description 3
- 239000002351 wastewater Substances 0.000 abstract description 3
- KVNRLNFWIYMESJ-UHFFFAOYSA-N butyronitrile Chemical compound CCCC#N KVNRLNFWIYMESJ-UHFFFAOYSA-N 0.000 abstract description 2
- 238000003756 stirring Methods 0.000 description 25
- 238000001816 cooling Methods 0.000 description 20
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- 230000035484 reaction time Effects 0.000 description 11
- 239000000047 product Substances 0.000 description 9
- 238000001914 filtration Methods 0.000 description 8
- 239000012295 chemical reaction liquid Substances 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 238000001035 drying Methods 0.000 description 6
- 239000007791 liquid phase Substances 0.000 description 6
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 238000004821 distillation Methods 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 230000000052 comparative effect Effects 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 229950000908 nazartinib Drugs 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 238000007086 side reaction Methods 0.000 description 4
- -1 2-methyl-1, 4-dihydro-4-pyridinecarboxylate Chemical compound 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- ZEKUVSJJRLBURC-UHFFFAOYSA-N 2-methyl-1,4-dihydropyridine-4-carbonitrile Chemical compound CC1=CC(C=CN1)C#N ZEKUVSJJRLBURC-UHFFFAOYSA-N 0.000 description 2
- XJHNCGVHPLDMRK-UHFFFAOYSA-N 4,4-dimethoxybutanenitrile Chemical compound COC(OC)CCC#N XJHNCGVHPLDMRK-UHFFFAOYSA-N 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- MMRXJPOAPCEDLP-UHFFFAOYSA-N methyl 4,4-dimethoxybutanoate Chemical compound COC(OC)CCC(=O)OC MMRXJPOAPCEDLP-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 238000006359 acetalization reaction Methods 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000007037 hydroformylation reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 229940127285 new chemical entity Drugs 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000000575 pesticide Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- DKGAVHZHDRPRBM-UHFFFAOYSA-N tert-butyl alcohol Substances CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/803—Processes of preparation
- C07D213/807—Processes of preparation by oxidation of pyridines or condensed pyridines
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention provides a preparation method of 2-methylpyridine-4-formic acid, which is characterized in that 4, 4-dialkoxy n-butyronitrile or 4, 4-dialkoxy n-butyric ester (II) and chloroacetone are subjected to substitution reaction, or ring-opening addition reaction and oxidation reaction with epoxypropane are subjected to ring-opening addition reaction to prepare 6, 6-dialkoxy-4-cyano n-hexyl-2-ketone or 6, 6-dialkoxy-4-alkoxycarbonyl n-hexyl-2-ketone (III), then the cyclization reaction is carried out on the 6, 6-dialkoxy-4-alkoxycarbonyl n-hexyl-2-ketone and ammonia and ammonium salt, and finally the 2-methylpyridine-4-formic acid (I) is prepared through oxidation reaction, hydrolysis and hydrochloric acid acidification. The method has mild conditions, simple process flow and safe and simple operation; the waste acid and waste water yield is low, and the environment is protected; good reaction selectivity, low product cost, high product yield and purity, and is beneficial to the green industrial production of the 2-methylpyridine-4-formic acid and the popularization of downstream products thereof.
Description
Technical Field
The invention relates to a preparation method of 2-methylpyridine-4-formic acid, belonging to the technical field of medical chemistry.
Background
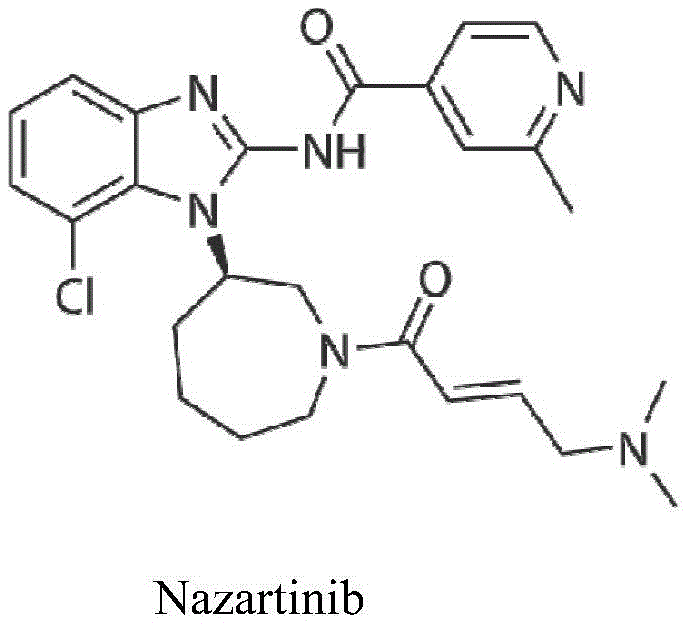
2-methylpyridine-4-formic acid or 2-methyl-4-picolinic acid is an important pyridine derivative and can be used for preparing medicine and pesticide active compounds such as Nazartinib and the like. The Nazartinib, abbreviated as EGF816 or NVS-816, is an oral non-small cell lung cancer new chemical entity drug developed by Novartis company, is currently in clinical stage III research and shows good treatment effect; the 2-methylpyridine-4-formic acid is an intermediate for preparing the Nazartinib, and the optimized preparation method of the 2-methylpyridine-4-formic acid has important significance for clinical research and drug production of the Nazartinib.
Disclosure of Invention
Aiming at the defects in the prior art, the invention provides a preparation method of 2-methylpyridine-4-formic acid. The method has mild conditions, simple process flow and safe and simple operation; the waste acid and waste water yield is low, and the environment is protected; good reaction selectivity, low product cost, high product yield and purity, and is beneficial to the green industrial production of the 2-methylpyridine-4-formic acid and the popularization of downstream products thereof.
Description of terms:
a compound of formula II: 4, 4-dialkoxy n-butyronitrile or 4, 4-dialkoxy n-butyrate;
a compound of formula III: 6, 6-dialkoxy-4-cyano-n-hexyl-2-one or 6, 6-dialkoxy-4-alkoxycarbonyl-n-hexyl-2-one;
a compound of formula IV: 2-methyl-1, 4-dihydro-4-pyridinecarboxylate or 2-methyl-1, 4-dihydro-4-pyridinecarbonitrile;
a compound of formula I: 2-methylpyridine-4-carboxylic acid.
The technical scheme of the invention is as follows:
a preparation method of 2-methylpyridine-4-formic acid comprises the following steps:
(1) preparing a compound shown in a formula III by using a compound shown in a formula II as an initial raw material;
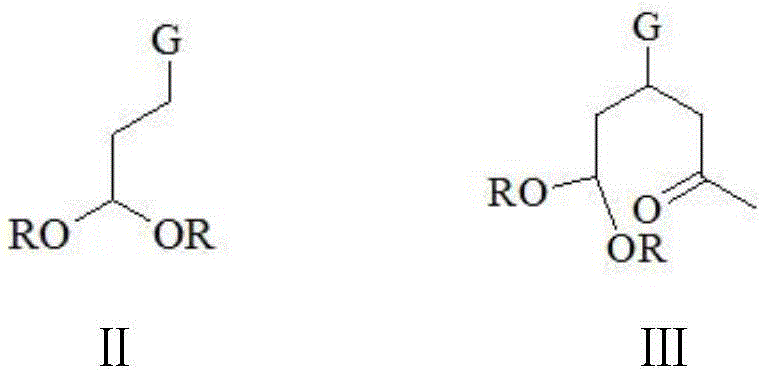
in the compounds shown in the formulas II and III, R is methyl, ethyl, isopropyl, n-butyl or tert-butyl; g is-CN or-COOR ', R' in the-COOR 'is methyl, ethyl, isopropyl, n-butyl or tert-butyl, and R' and R are the same or different;
(2) in a solvent B, in the presence of ammonia and ammonium salt, carrying out cyclization reaction on a compound shown in a formula III to prepare a compound shown in a formula IV, then carrying out oxidation reaction under the action of an oxidant, hydrolyzing under the action of an alkaline reagent, and acidifying with hydrochloric acid to obtain 2-methylpyridine-4-formic acid (I);

wherein G in the compound of formula IV has the same meaning as G in the compound of formula II.
According to the invention, the compounds of the formula II are commercially available or can be prepared from acrylonitrile or acrylic esters by hydroformylation and acetalization.
According to the present invention, the method for preparing the compound of formula III in step (1) using the compound of formula II as a starting material is route 1 or route 2, respectively; wherein,
route 1 comprises the steps of:
in a solvent A, in the presence of an acid binding agent, performing substitution reaction on a compound shown in a formula II and chloroacetone to obtain a compound shown in a formula III;
route 2 comprises the steps of:
in the presence of an alkali catalyst, carrying out ring-opening addition reaction on a compound shown in a formula II and propylene oxide; then under the action of an oxidant, carrying out oxidation reaction to obtain a compound shown in a formula III.
In the route 1 of the step (1), the solvent A is one or a combination of tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, N-dimethylformamide and toluene; the mass ratio of the solvent A to the compound shown in the formula II is (3-15) to 1; preferably, the mass ratio of the solvent A to the compound of the formula II is (4.5-10): 1.
In the way 1 of the step (1), the acid-binding agent is an inorganic base or an organic base, the inorganic base is selected from one or a combination of potassium carbonate, sodium methoxide, sodium ethoxide, calcium carbonate, sodium hydroxide, potassium bicarbonate, sodium bicarbonate, calcium bicarbonate, potassium acetate, sodium acetate and calcium acetate, and the organic base is selected from one or a combination of triethylamine and tri-n-butylamine; the molar ratio of the acid-binding agent to the compound shown in the formula II is (1.0-2.0): 1; preferably, the molar ratio of the acid-binding agent to the compound of the formula II is (1.0-1.5): 1.
In route 1 of step (1), the molar ratio of the chloroacetone to the compound of formula II is (1.0-1.5): 1.
In route 1 of step (1), the temperature of the substitution reaction is 30 to 100 ℃; preferably, the temperature of the substitution reaction is 60 to 80 ℃. The substitution reaction time is 2-8 hours; preferably, the substitution reaction time is 3 to 5 hours.
In pathway 2 of step (1), the base catalyst is one or a combination of 1, 8-diazabicyclo [5.4.0] -7-undecene (DBU), 1, 5-diazabicyclo [4.3.0] -5-nonene (DBN), piperidine, and 4-dimethylaminopyridine; the mass of the alkali catalyst is 1-5% of that of the compound shown in the formula II; preferably, the mass of the base catalyst is 1.5-4% of the mass of the compound of formula II.
In route 2 of step (1), the molar ratio of propylene oxide to the compound of formula II is (1.0-1.5): 1.
In the route 2 of the step (1), the temperature of the ring-opening addition reaction is 20-70 ℃; preferably, the temperature of the ring-opening addition reaction is 40 to 55 ℃. The ring-opening addition reaction time is 2-10 hours; preferably, the ring-opening addition reaction time is 4 to 6 hours.
In the path 2 of the step (1), the oxidant is one or a combination of hydrogen peroxide, tert-butyl peroxide and m-chloroperoxybenzoic acid; the molar ratio of the oxidant to the compound of formula II is (1.0-1.3): 1.
In the route 2 of the step (1), the temperature of the oxidation reaction is 40-100 ℃; preferably, the temperature of the oxidation reaction is 60 to 80 ℃. The oxidation reaction time is 2-7 hours; preferably, the oxidation reaction time is 3 to 5 hours.
The reaction in pathway 2 of step (1) is carried out as a "one-pot" process without isolation of the intermediate product.
According to the present invention, preferably, the solvent B in step (2) is one or a combination of two or more of water, acetonitrile, methanol, ethanol, isopropanol or toluene; the mass ratio of the solvent B to the compound shown in the formula III is (2-20) to 1; preferably, the mass ratio of the solvent B to the compound of the formula III is (4-10): 1.
According to the present invention, it is preferable that the ammonia in step (2) is ammonia gas, ammonia water, an ammonia methanol solution or an ammonia ethanol solution; the ammonium salt is ammonium chloride, ammonium sulfate, ammonium nitrate or ammonium phosphate; the molar ratio of ammonia, ammonium salt and compound of formula III is (2.0-5.0): (0.5-1.5): 1.
according to the present invention, it is preferable that the cyclization reaction temperature in step (2) is 20 to 100 ℃; preferably, the cyclization reaction temperature is 50-70 ℃. The cyclization reaction time is 2 to 8 hours, preferably 3 to 5 hours.
According to the present invention, it is preferred that in step (2), the process for preparing the compound of formula iv from the compound of formula iii comprises the steps of: dissolving ammonia and ammonium salt in solvent B, dripping the compound of formula III at 20-100 deg.C, and reacting at 20-100 deg.C for 2-8 hr.
According to the invention, preferably, the oxidant in the step (2) is one of hydrogen peroxide, tert-butyl peroxide and m-chloroperoxybenzoic acid or a combination thereof; the molar ratio of the oxidant to the compound of formula III is (1.0-1.5): 1.
According to the present invention, it is preferable that the temperature of the oxidation reaction in the step (2) is 20 to 100 ℃; preferably, the temperature of the oxidation reaction is 50 to 80 ℃. The oxidation reaction time is 2-7 hours; preferably, the oxidation reaction time is 3 to 5 hours.
According to the present invention, preferably, the alkaline reagent in step (2) is one or a combination of sodium hydroxide, potassium hydroxide and lithium hydroxide; the molar ratio of the alkaline reagent to the compound of formula III is (1.0-1.5): 1.
According to the present invention, it is preferable that the temperature of the hydrolysis reaction in step (2) is 20 to 100 ℃; preferably, the temperature of the hydrolysis reaction is 30 to 90 ℃. The hydrolysis reaction time is 0.5-5 hours; preferably, the hydrolysis reaction time is 1 to 3 hours.
According to the invention, preferably, the hydrochloric acid acidification in the step (2) is carried out by using 20-30% by mass of hydrochloric acid aqueous solution to adjust the pH value of the system to 3.0-3.5.
According to the invention, step (2) is preferably a "one-pot" reaction, the intermediate product not being isolated.
The process of the present invention is depicted as scheme 1 below:

in the compounds shown in the formulas II and III, R is methyl, ethyl, isopropyl, n-butyl or tert-butyl; g is-CN or-COOR ', R' in the-COOR 'is methyl, ethyl, isopropyl, n-butyl or tert-butyl, and R' and R are the same or different; the meaning of G in the compounds of the formula IV is identical to that of G in the compounds of the formula II.
The invention has the technical characteristics and beneficial effects that:
1. the invention provides two ways for preparing a compound shown in a formula III, and a method for preparing 2-methylpyridine-4-formic acid from the compound shown in the formula III by a one-pot method. The invention uses a compound of formula II and chloroacetone to prepare a compound of formula III through substitution reaction or ring-opening addition reaction and oxidation reaction of the compound of formula II and epoxypropane, the obtained compound of formula III and ammonia and ammonium salt are subjected to cyclization reaction to prepare 2-methyl-1, 4-dihydro-4-picolinate or 2-methyl-1, 4-dihydro-4-pyridinecarbonitrile, and then the 2-methylpyridine-4-formic acid (I) is prepared through oxidation reaction, hydrolysis and hydrochloric acid acidification.
2. The invention has reasonable route design and high related reaction selectivity, and utilizes the high stability and the only active methylene position of the compound shown in the formula II to react with the chloroacetone or the epoxypropane under the alkaline condition to obtain carbonyl or obtain a carbonyl compound, namely the compound shown in the formula III by oxidizing hydroxyl, the compound shown in the formula III undergoes acetal hydrolysis under an ammonia and ammonium salt system to generate aldehyde group and then cyclizes to prepare the compound shown in the formula IV, and then the compound shown in the formula IV is subjected to oxidation reaction, hydrolysis and hydrochloric acid acidification to prepare the 2-methylpyridine-4-formic acid (I). Reaction sites in each step are single, and the low concentration effect of the compound of the formula III is controlled by combining and preferably dripping to reduce intermolecular side reactions, so that the high purity and the high yield of the product are ensured.
3. The method has mild conditions and simple process flow, can be prepared by only two steps, and has safe and simple operation and low product cost; the waste acid and waste water yield is low, and the environment is protected; the reaction selectivity is good, the product yield and purity are high, the total yield can reach 84.2%, and the green industrial production of the 2-methylpyridine-4-formic acid and the popularization of downstream products thereof are facilitated.
Detailed Description
The present invention is described in detail below with reference to examples, but the present invention is not limited thereto.
The 4, 4-dialkoxy-n-butyronitrile and 4, 4-dialkoxy-n-butyrate used in the examples are commercially available from Shandong Ruihui pharmaceutical Co., Ltd. The rest raw materials and reagents are all commercial products.
In the examples, "%" is given by weight unless otherwise specified. The yields in the examples are all molar yields.
Example 1: preparation of 6, 6-dimethoxy-4-cyano-n-hexyl-2-one (III 1) (route 1 method)
400 g of tetrahydrofuran, 64.5 g (0.5 mol) of 4, 4-dimethoxy n-butyronitrile (II 1), 55.5 g (0.6 mol) of chloroacetone and 83.0 g (0.6 mol) of potassium carbonate are added into a reaction flask with a stirring, thermometer and reflux condenser, stirred and reacted for 4 hours at the temperature of 60-65 ℃, then cooled to 20-25 ℃, filtered, filter cakes are washed twice with tetrahydrofuran, 20 g for each time, the filtrate is combined, the solvent is recovered by distillation, and 84.6 g of 6, 6-dimethoxy-4-cyano n-hexyl-2-ketone (III 1) is obtained by reduced pressure distillation (85-105 ℃/2-3mmHg), the yield is 91.5 percent, and the gas phase purity is 99.6 percent.
Example 2: preparation of 6, 6-dimethoxy-4-cyano-n-hexyl-2-one (III 1) (route 2 method)
Adding 64.5 g (0.5 mol) of 4, 4-dimethoxy n-butyronitrile (II 1), 35.0 g (0.6 mol) of propylene oxide, 1.5 g of DBU, stirring and reacting at 50-55 ℃ for 5 hours, then cooling to 20-25 ℃, transferring the obtained reaction liquid into a reaction flask with a stirring thermometer, adding 68.0 g (0.6 mol) of 30 wt% hydrogen peroxide, stirring and reacting at 75-80 ℃ for 4 hours, then cooling to 20-25 ℃, adding 200 g of dichloromethane, layering, extracting a water layer with dichloromethane for 2 times, 50 g each time, combining organic phases, distilling and recovering dichloromethane, then carrying out reduced pressure distillation to collect a fraction (85-105 ℃/2-3mmHg) to obtain 81.2 g of 6, 6-dimethoxy-4-cyano n-hexyl-2-ketone (III 1), the yield was 87.8%, and the gas phase purity was 99.2%.
Example 3: preparation of 6, 6-dimethoxy-4-methoxycarbonyl n-hexyl-2-one (III 2) (route 1 method)
Into a reaction flask equipped with a stirrer, a thermometer, and a reflux condenser, 400 g of N, N-dimethylformamide, 81.0 g (0.5 mol) of methyl 4, 4-dimethoxybutyrate (II 2), 55.5 g (0.6 mol) of chloroacetone, 83.0 g (0.6 mol) of potassium carbonate were added, and stirred at 75-80 ℃ for reaction for 3 hours, followed by cooling to 20-25 ℃, filtration, washing the cake twice with N, N-dimethylformamide, 20 g each time, combining the filtrates, recovering the solvent by distillation, and distillation under reduced pressure (90-115 ℃/2-3mmHg) to obtain 100.8 g of 6, 6-dimethoxy-4-methoxycarbonyln-hex-2-one (III 2), yield 92.5%, and gas phase purity 99.3%.
Example 4: preparation of 6, 6-dimethoxy-4-methoxycarbonyl n-hexyl-2-one (III 2) (route 2 method)
Adding 81.0 g (0.5 mol) of 4, 4-dimethoxy n-butyric acid methyl ester (II 2), 35.0 g (0.6 mol) of propylene oxide, 1.5 g of DBU, stirring and reacting at 45-50 ℃ for 6 hours, cooling to 20-25 ℃, transferring the obtained reaction liquid into a reaction flask with a stirring thermometer, adding 68.0 g (0.6 mol) of 30 wt% hydrogen peroxide, stirring and reacting at 75-80 ℃ for 4 hours, cooling to 20-25 ℃, adding 200 g of dichloromethane, demixing, extracting a water layer with dichloromethane for 2 times, combining dichloromethane organic phases, distilling and recovering a solvent, distilling under reduced pressure (90-115 ℃/2-3mmHg) to obtain 96.8 g of 6, 6-dimethoxy-4-methoxycarbonyl n-2-ketone (III 2), wherein the yield is 88.8%, the gas phase purity was 99.2%.
Example 5: preparation of 2-methylpyridine-4-carboxylic acid (I)
Adding 100 g of water, 40.0 g (0.4 mol) of 17 wt% ammonia water, 3.0 g of ammonium chloride, 18.5 g (0.1 mol) of 6, 6-dimethoxy-4-cyano n-hexyl-2-ketone (III 1) obtained in example 2 dropwise at 60-65 ℃ into a reaction flask with a stirring device, a thermometer, a reflux condenser and a constant pressure dropping funnel, finishing dropwise adding for about 1 hour, then stirring and reacting at 65-70 ℃ for 3 hours, cooling to 20-25 ℃, adding 17.0 g (0.15 mol) of 30 wt% hydrogen peroxide, stirring and reacting at 65-70 ℃ for 4 hours, cooling to 20-25 ℃, adding 4.8 g (0.12 mol) of sodium hydroxide, stirring and reacting at 85-90 ℃ for 2 hours, cooling to 0-5 ℃, adjusting the pH value of the reaction liquid to 3.0-3.5 by using 30 wt% hydrochloric acid, filtering, washing by 10 g of ice water, drying, 11.8 g of 2-methylpyridine-4-carboxylic acid (I) were obtained as a white solid in 86.1% yield and 99.7% purity in the liquid phase.
The nuclear magnetic data of the product obtained are as follows:
1HNMR(DMSO-D6,,ppm):2.32(s,3H),7.76(s,1H),7.82(d,1H),8.62(d,1H),10.19(s,1H)。
example 6: preparation of 2-methylpyridine-4-carboxylic acid (I)
Adding 50 g of water, 40.0 g (0.4 mol) of a 17 wt% ammonia methanol solution, 3.5 g of ammonium chloride, dropwise adding 18.5 g (0.1 mol) of a solution of 6, 6-dimethoxy-4-cyano n-hexyl-2-one (III 1) and 50 g of methanol obtained in example 1 at 60-65 ℃, after dropwise adding, stirring at 65-70 ℃ for 3 hours, cooling to 20-25 ℃, adding 17.0 g (0.15 mol) of 30 wt% hydrogen peroxide, stirring at 65-70 ℃ for 4 hours, cooling to 20-25 ℃, adding 4.8 g (0.12 mol) of sodium hydroxide, stirring at 80-85 ℃ for 2 hours, recovering the distilled methanol, cooling to 0-5 ℃, adjusting the pH value of the reaction liquid to 3.0-3.5 by using 30 wt% hydrochloric acid, filtration, washing with 10 g of ice water and drying gave 12.6 g of 2-methylpyridine-4-carboxylic acid (I) as a white solid in 92.0% yield and 99.6% purity in the liquid phase.
Example 7: preparation of 2-methylpyridine-4-carboxylic acid (I)
120 g of water, 40.0 g (0.4 mol) of 17 wt% ammonia water, 5.0 g of ammonium chloride, 21.8 g (0.1 mol) of 6, 6-dimethoxy-4-methoxycarbonyl n-2-one (III 2) obtained in example 3 was added dropwise at 65 to 70 ℃ for about 1 hour, after which the reaction was stirred at 65 to 70 ℃ for 3 hours, cooled to 20 to 25 ℃, 14.0 g (0.11 mol) of 70 wt% t-butanol peroxide was added, stirred at 50 to 55 ℃ for 3 hours, cooled to 20 to 25 ℃, 4.8 g (0.12 mol) of sodium hydroxide was added, stirred at 40 to 45 ℃ for 2 hours, cooled to 0 to 5 ℃, the pH of the reaction solution was adjusted to 3.0 to 3.5 with 30 wt% hydrochloric acid, filtered, and washed with 10 g of ice water, drying gave 12.1 g of 2-methylpyridine-4-carboxylic acid (I) as a white solid in 88.3% yield and 99.8% purity in the liquid phase.
Example 8: preparation of 2-methylpyridine-4-carboxylic acid (I)
Adding 50 g of water, 40.0 g (0.4 mol) of a 17 wt% ammonia methanol solution, 4.0 g of ammonium chloride, and a solution of 21.8 g (0.1 mol) of the 6, 6-dimethoxy-4-methoxycarbonyl n-2-one (III 2) and 50 g of methanol obtained in example 4 dropwise at 60-65 ℃ into a reaction flask with a stirring device, a thermometer, a condenser and a constant pressure dropping funnel, after dropwise addition over about 2 hours, stirring and reacting at 65-70 ℃ for 3 hours, cooling to 20-25 ℃, adding 17.0 g (0.15 mol) of 30 wt% hydrogen peroxide, stirring and reacting at 65-70 ℃ for 4 hours, recovering the distilled methanol, cooling to 20-25 ℃, adding 4.8 g (0.12 mol) of sodium hydroxide, stirring and reacting at 35-40 ℃ for 2 hours, cooling to 0-5 ℃, adjusting the pH of the reaction liquid to 3.0-3.5 with 30 wt% hydrochloric acid, filtration, washing with 10 g of ice water and drying gave 12.3 g of 2-methylpyridine-4-carboxylic acid (I) as a white solid in 89.8% yield and 99.5% purity in the liquid phase.
Comparative example 1: preparation of 2-methylpyridine-4-carboxylic acid (I)
Adding 100 g of water, 40.0 g (0.4 mol) of 17 wt% ammonia water, 3.0 g of ammonium chloride and 18.5 g (0.1 mol) of 6, 6-dimethoxy-4-cyano n-hexyl-2-ketone (III 1) obtained in example 1 into a reaction flask with a stirring thermometer and a reflux condenser, stirring and reacting at 65-70 ℃ for 4 hours, cooling to 20-25 ℃, adding 17.0 g (0.15 mol) of 30 wt% hydrogen peroxide, stirring and reacting at 65-70 ℃ for 4 hours, cooling to 20-25 ℃, adding 4.8 g (0.12 mol) of sodium hydroxide, stirring and reacting at 85-90 ℃ for 2 hours, cooling to 0-5 ℃, adjusting the pH value of the reaction liquid to 3.0-3.5 by using 30 wt% hydrochloric acid, filtering, washing by 10 g of ice water, drying to obtain 12.1 g of viscous white solid, heating and dissolving by 100 g of acetonitrile for 1 hour, filtering to remove insoluble substances while the solid is hot, acetonitrile was recovered to obtain 8.3 g of 2-methylpyridine-4-carboxylic acid (I) in 60.6% yield and 99.1% liquid phase purity.
As can be seen from the comparative example, the dropwise addition of the 6, 6-dimethoxy-4-cyano n-hexyl-2-one (III 1) material is beneficial to reducing intermolecular side reactions by controlling the concentration of the material, thereby improving the yield and the purity of the final product.
Comparative example 2: preparation of 2-methylpyridine-4-carboxylic acid (I)
Adding 50 g of water, 50 g of methanol, 40.0 g (0.4 mol) of 17 wt% ammonia methanol solution, 4.0 g of ammonium chloride and 21.8 g (0.1 mol) of 6, 6-dimethoxy-4-methoxycarbonyl n-2-one (III 2) obtained in example 3 into a reaction flask with a stirrer, a thermometer and a condenser, stirring and reacting at 65-70 ℃ for 4 hours, cooling to 20-25 ℃, adding 17.0 g (0.15 mol) of 30 wt% hydrogen peroxide, stirring and reacting at 65-70 ℃ for 4 hours, simultaneously recovering the evaporated methanol, cooling to 20-25 ℃, adding 4.8 g (0.12 mol) of sodium hydroxide, stirring and reacting at 35-40 ℃ for 2 hours, cooling to 0-5 ℃, adjusting the pH value of the reaction liquid to 3.0-3.5 by using 30 wt% hydrochloric acid, filtering, washing by 10 g, drying to obtain 12.3 g of viscous white solid, the resulting mixture was dissolved in 100 g of acetonitrile under heating for 1 hour, and the insoluble matter was removed by hot filtration, and acetonitrile was recovered to obtain 7.6 g of 2-methylpyridine-4-carboxylic acid (I), yield 55.5%, and liquid-phase purity 98.6%.
As can be seen from the comparative example, the one-time material addition mode results in more side reactions between molecules and ammonia, and a large amount of polymer is generated; 6, 6-dimethoxy-4-methoxycarbonyl n-hexyl-2-ketone (III 2) is dropwise added, so that the intermolecular side reaction is reduced by controlling the system concentration, and the yield and the purity of the final product are improved.
Claims (14)
1. A preparation method of 2-methylpyridine-4-formic acid comprises the following steps:
(1) preparing a compound shown in a formula III by using a compound shown in a formula II as an initial raw material;
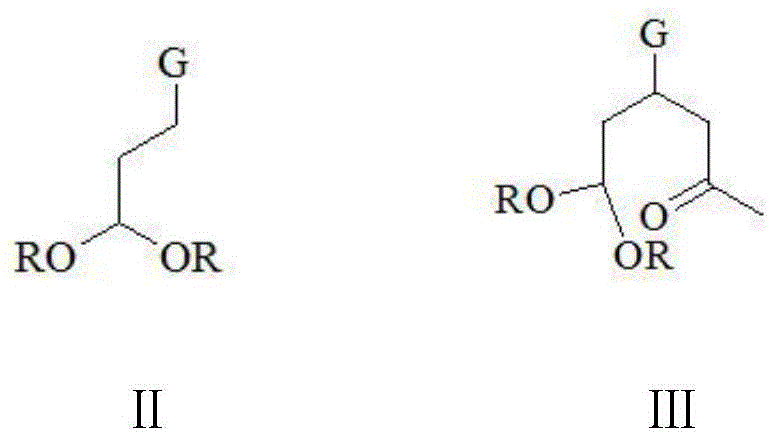
in the compounds shown in the formulas II and III, R is methyl, ethyl, isopropyl, n-butyl or tert-butyl; g is-CN or-COOR ', R' in the-COOR 'is methyl, ethyl, isopropyl, n-butyl or tert-butyl, and R' and R are the same or different;
(2) in a solvent B, in the presence of ammonia and ammonium salt, carrying out cyclization reaction on a compound shown in a formula III to prepare a compound shown in a formula IV, then carrying out oxidation reaction under the action of an oxidant, hydrolyzing under the action of an alkaline reagent, and acidifying with hydrochloric acid to obtain 2-methylpyridine-4-formic acid (I);
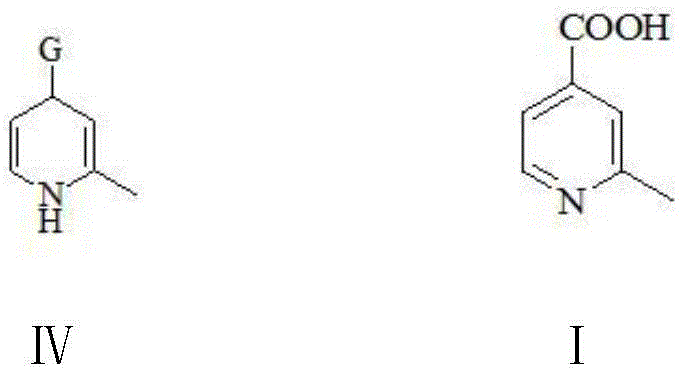
wherein G in the compound of formula IV has the same meaning as G in the compound of formula II.
2. The process for preparing 2-methylpyridine-4-carboxylic acid according to claim 1, wherein the process for preparing the compound of formula iii in step (1) using the compound of formula ii as a starting material is pathway 1 or pathway 2; wherein,
route 1 comprises the steps of:
in a solvent A, in the presence of an acid binding agent, performing substitution reaction on a compound shown in a formula II and chloroacetone to obtain a compound shown in a formula III;
route 2 comprises the steps of:
in the presence of an alkali catalyst, carrying out ring-opening addition reaction on a compound shown in a formula II and propylene oxide; then under the action of an oxidant, carrying out oxidation reaction to obtain a compound shown in a formula III.
3. The process for the preparation of 2-methylpyridine-4-carboxylic acid according to claim 2, wherein pathway 1 of step (1) comprises one or more of the following conditions:
a. the solvent A is one or a combination of tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentyl methyl ether, N-dimethylformamide and toluene; the mass ratio of the solvent A to the compound shown in the formula II is (3-15) to 1;
b. the acid-binding agent is inorganic base or organic base, the inorganic base is selected from one or a combination of potassium carbonate, sodium methoxide, sodium ethoxide, calcium carbonate, sodium hydroxide, potassium bicarbonate, sodium bicarbonate, calcium bicarbonate, potassium acetate, sodium acetate and calcium acetate, and the organic base is selected from one or a combination of triethylamine and tri-n-butylamine; the molar ratio of the acid-binding agent to the compound shown in the formula II is (1.0-2.0): 1;
c. the molar ratio of the chloroacetone to the compound of the formula II is (1.0-1.5) to 1;
d. the temperature of the substitution reaction is 30-100 ℃.
4. The process for producing 2-methylpyridine-4-carboxylic acid according to claim 2, wherein the temperature of the substitution reaction in route 1 of step (1) is 60 to 80 ℃.
5. The process for the preparation of 2-methylpyridine-4-carboxylic acid according to claim 2, wherein route 2 of step (1) comprises one or more of the following conditions:
a. the base catalyst is one or a combination of 1, 8-diazabicyclo [5.4.0] -7-undecene (DBU), 1, 5-diazabicyclo [4.3.0] -5-nonene (DBN), piperidine and 4-dimethylaminopyridine; the mass of the alkali catalyst is 1-5% of that of the compound shown in the formula II;
b. the molar ratio of the propylene oxide to the compound shown in the formula II is (1.0-1.5) to 1;
c. the temperature of the ring-opening addition reaction is 20-70 ℃;
d. the oxidant is one or a combination of hydrogen peroxide, tert-butyl peroxide and m-chloroperoxybenzoic acid; the molar ratio of the oxidant to the compound of formula II is (1.0-1.3) to 1;
e. the temperature of the oxidation reaction is 40-100 ℃.
6. The process for producing 2-methylpyridine-4-carboxylic acid according to claim 2, wherein the temperature of the ring-opening addition reaction in route 2 of step (1) is 40 to 55 ℃; the temperature of the oxidation reaction is 60-80 ℃.
7. The process for the preparation of 2-methylpyridine-4-carboxylic acid according to claim 2, wherein the reaction in pathway 2 of step (1) is carried out in a "one-pot" process without isolation of intermediates.
8. The process for producing 2-methylpyridine-4-carboxylic acid according to claim 1, wherein the step (2) comprises one or more of the following conditions:
a. the solvent B is one or the combination of more than two of water, acetonitrile, methanol, ethanol, isopropanol or toluene; the mass ratio of the solvent B to the compound shown in the formula III is (2-20) to 1;
b. the ammonia is ammonia gas, ammonia water, ammonia methanol solution or ammonia ethanol solution; the ammonium salt is ammonium chloride, ammonium sulfate, ammonium nitrate or ammonium phosphate; the molar ratio of ammonia, ammonium salt and compound of formula III is (2.0-5.0): (0.5-1.5): 1;
c. the cyclization reaction temperature is 20-100 ℃.
9. The process according to claim 1, wherein the cyclization reaction temperature in the step (2) is 50 to 70 ℃.
10. The process for producing 2-methylpyridine-4-carboxylic acid according to claim 1, wherein the process for producing the compound of formula IV from the compound of formula III in the step (2) comprises the steps of: dissolving ammonia and ammonium salt in solvent B, dripping the compound of formula III at 20-100 deg.C, and reacting at 20-100 deg.C for 2-8 hr.
11. The process for producing 2-methylpyridine-4-carboxylic acid according to claim 1, wherein the step (2) comprises one or more of the following conditions:
a. the oxidant is one or the combination of hydrogen peroxide, tert-butyl peroxide and m-chloroperoxybenzoic acid; the molar ratio of the oxidant to the compound of formula III is (1.0-1.5): 1;
b. the temperature of the oxidation reaction is 20-100 ℃.
12. The process according to claim 1, wherein the temperature of the oxidation reaction in the step (2) is 50 to 80 ℃.
13. The process for producing 2-methylpyridine-4-carboxylic acid according to claim 1, wherein the step (2) comprises one or more of the following conditions:
a. the alkaline reagent is one or a combination of sodium hydroxide, potassium hydroxide and lithium hydroxide; the molar ratio of the alkaline reagent to the compound of formula III is (1.0-1.5): 1;
b. the temperature of the hydrolysis reaction is 20-100 ℃;
c. the hydrochloric acid acidification is to use 20-30% hydrochloric acid aqueous solution to acidify the system until the pH value is 3.0-3.5.
14. The process according to claim 1, wherein step (2) is a "one-pot" reaction and the intermediate is not isolated.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810427394.7A CN110452165B (en) | 2018-05-07 | 2018-05-07 | Preparation method of 2-methylpyridine-4-formic acid |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810427394.7A CN110452165B (en) | 2018-05-07 | 2018-05-07 | Preparation method of 2-methylpyridine-4-formic acid |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN110452165A CN110452165A (en) | 2019-11-15 |
| CN110452165B true CN110452165B (en) | 2020-09-25 |
Family
ID=68472016
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201810427394.7A Active CN110452165B (en) | 2018-05-07 | 2018-05-07 | Preparation method of 2-methylpyridine-4-formic acid |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN110452165B (en) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070179060A1 (en) * | 2006-01-13 | 2007-08-02 | Balko Terry W | 6-(Poly-substituted aryl)-4-aminopicolinates and their use as herbicides |
| CN101056857A (en) * | 2004-11-05 | 2007-10-17 | 弗·哈夫曼-拉罗切有限公司 | Process for preparation of isonicotinic acid derivatives |
| CN104945308A (en) * | 2015-07-14 | 2015-09-30 | 武汉康贝德生物科技有限公司 | Method for reducing pyridine ring to piperidine in 2-picoline-4-formic acid |
| WO2016125192A2 (en) * | 2015-02-06 | 2016-08-11 | Mylan Laboratories Limited | Process for the preparation of dolutegravir |
-
2018
- 2018-05-07 CN CN201810427394.7A patent/CN110452165B/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101056857A (en) * | 2004-11-05 | 2007-10-17 | 弗·哈夫曼-拉罗切有限公司 | Process for preparation of isonicotinic acid derivatives |
| US20070179060A1 (en) * | 2006-01-13 | 2007-08-02 | Balko Terry W | 6-(Poly-substituted aryl)-4-aminopicolinates and their use as herbicides |
| WO2016125192A2 (en) * | 2015-02-06 | 2016-08-11 | Mylan Laboratories Limited | Process for the preparation of dolutegravir |
| CN104945308A (en) * | 2015-07-14 | 2015-09-30 | 武汉康贝德生物科技有限公司 | Method for reducing pyridine ring to piperidine in 2-picoline-4-formic acid |
Non-Patent Citations (2)
| Title |
|---|
| "吡啶甲酸的制备研究进展";刘小华等;《化学研究与应用》;20030428;第15卷(第2期);第161-164页 * |
| "吡啶甲酸的合成及其用途";温飞鹏等;《应用化工》;20101028;第39卷(第10期);第1552-1556页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110452165A (en) | 2019-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3476838B1 (en) | Method for preparing azoxystrobin | |
| JP2023532362A (en) | Method for producing phenylisoxazoline compound | |
| CN110878084A (en) | Preparation method of nicosulfuron original drug | |
| RU2470919C2 (en) | Method of producing toluidine compound | |
| US10053420B2 (en) | Processes for the preparation of compounds, such as 3-arylbutanals, useful in the synthesis of medetomidine | |
| CN110452165B (en) | Preparation method of 2-methylpyridine-4-formic acid | |
| JP4049534B2 (en) | Process for producing O-alkyl-N-cyanoacetimidate | |
| CN116082181B (en) | A method for preparing 3-amino-5-ethoxy-benzoic acid | |
| CN114539085B (en) | Preparation of ureido derivatives | |
| CN100497309C (en) | Method for synthesizing heteroaryl thiosemicarbazone antineoplastic | |
| CN115710213A (en) | Preparation method of cis-chiral 3-fluoro-4-hydroxypiperidine and derivatives thereof | |
| JP6284942B2 (en) | Improved method for preparing 2-amino-5,8-dimethoxy [1,2,4] triazolo [1,5-c] pyrimidine from 4-amino-2,5-dimethoxypyrimidine | |
| CN114702425A (en) | Preparation method of (S) -2-amino- (S) -3- [ pyrrolidone-2' ] alanine derivative and intermediate | |
| CN114591226B (en) | Preparation method of nicotinaldehyde derivative | |
| CN116003271B (en) | A method for synthesizing quaternary ammonium lipids | |
| JPH03271273A (en) | Production of 2-chloro-5-(aminomethyl)pyridine | |
| WO2025022637A1 (en) | Method for producing cyclized product by cyclization reaction involving dehydration condensation of carboxylic acid ester derivative, and method for producing 1,3,4-substituted-pyrazole-5-carboxylic acid ester | |
| JP3272340B2 (en) | Method for producing 1-[(cyclopent-3-en-1-yl) methyl] -5-ethyl-6- (3,5-dimethylbenzoyl) -2,4-pyrimidinedione | |
| KR102157528B1 (en) | Method for producing 2-aminonicotinic acid benzyl ester derivative | |
| JPS5840939B2 (en) | Method for producing cyclohexanedione derivatives | |
| JP3592747B2 (en) | N-tert-butyl-2,3-pyrazinedicarboxamide and method for producing the same | |
| RU2455301C2 (en) | Method of producing n-(6-methyl-2,4-dioxo-1,2,3,4-tetrahydro-5-pyrimidine-sulphone)-n′-isonictinoyl hydrazide | |
| JPS58103379A (en) | Preparation of alpha-acetyl lactone | |
| JPS6183168A (en) | 2-Mercapto-4-amino-5-formylpyrimidine and its production method | |
| JPS62267267A (en) | Pyrazole derivative and production thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| PE01 | Entry into force of the registration of the contract for pledge of patent right | ||
| PE01 | Entry into force of the registration of the contract for pledge of patent right |
Denomination of invention: A preparation method of 2-methylpyridine-4-formic acid Effective date of registration: 20211130 Granted publication date: 20200925 Pledgee: Zhejiang Commercial Bank Co.,Ltd. Dongying Branch Pledgor: Xinfa pharmaceutical Co.,Ltd. Registration number: Y2021980013546 |