CN109096218B - Oxydterol hydrochloride crystal form A and preparation method thereof - Google Patents
Oxydterol hydrochloride crystal form A and preparation method thereof Download PDFInfo
- Publication number
- CN109096218B CN109096218B CN201810884531.XA CN201810884531A CN109096218B CN 109096218 B CN109096218 B CN 109096218B CN 201810884531 A CN201810884531 A CN 201810884531A CN 109096218 B CN109096218 B CN 109096218B
- Authority
- CN
- China
- Prior art keywords
- stirring
- hydrochloride
- temperature
- preparation
- isopropanol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000013078 crystal Substances 0.000 title claims abstract description 74
- 238000002360 preparation method Methods 0.000 title claims abstract description 38
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 title claims abstract description 37
- 229960001733 olodaterol hydrochloride Drugs 0.000 claims abstract description 52
- 238000002441 X-ray diffraction Methods 0.000 claims abstract description 16
- KCEHVJZZIGJAAW-FERBBOLQSA-N olodaterol hydrochloride Chemical compound Cl.C1=CC(OC)=CC=C1CC(C)(C)NC[C@H](O)C1=CC(O)=CC2=C1OCC(=O)N2 KCEHVJZZIGJAAW-FERBBOLQSA-N 0.000 claims abstract description 16
- 238000010521 absorption reaction Methods 0.000 claims abstract description 13
- 238000000034 method Methods 0.000 claims abstract description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 73
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 60
- 238000003756 stirring Methods 0.000 claims description 50
- 239000012043 crude product Substances 0.000 claims description 24
- 238000006243 chemical reaction Methods 0.000 claims description 21
- 239000012046 mixed solvent Substances 0.000 claims description 16
- 239000000047 product Substances 0.000 claims description 16
- 238000001914 filtration Methods 0.000 claims description 15
- 238000001816 cooling Methods 0.000 claims description 14
- 238000001035 drying Methods 0.000 claims description 14
- 238000010438 heat treatment Methods 0.000 claims description 14
- 238000004321 preservation Methods 0.000 claims description 7
- 239000003054 catalyst Substances 0.000 claims description 5
- 230000005855 radiation Effects 0.000 claims description 5
- 239000000843 powder Substances 0.000 claims description 4
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 3
- 239000000443 aerosol Substances 0.000 claims description 2
- ANFZRGMDGDYNGA-UHFFFAOYSA-N ethyl acetate;propan-2-ol Chemical group CC(C)O.CCOC(C)=O ANFZRGMDGDYNGA-UHFFFAOYSA-N 0.000 claims description 2
- 229940041682 inhalant solution Drugs 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 239000003814 drug Substances 0.000 abstract description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 abstract description 8
- 230000009286 beneficial effect Effects 0.000 abstract description 5
- 229940079593 drug Drugs 0.000 abstract description 5
- 238000003908 quality control method Methods 0.000 abstract description 5
- 238000009776 industrial production Methods 0.000 abstract description 4
- 230000000857 drug effect Effects 0.000 abstract description 2
- 239000000048 adrenergic agonist Substances 0.000 abstract 1
- COUYJEVMBVSIHV-SFHVURJKSA-N olodaterol Chemical compound C1=CC(OC)=CC=C1CC(C)(C)NC[C@H](O)C1=CC(O)=CC2=C1OCC(=O)N2 COUYJEVMBVSIHV-SFHVURJKSA-N 0.000 description 39
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 239000007787 solid Substances 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 9
- 239000012065 filter cake Substances 0.000 description 9
- 238000012360 testing method Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 5
- DKLVIQDZNAPVFJ-VWLOTQADSA-N 8-[(1r)-1-hydroxy-2-[[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino]ethyl]-6-phenylmethoxy-4h-1,4-benzoxazin-3-one Chemical compound C1=CC(OC)=CC=C1CC(C)(C)NC[C@H](O)C1=CC(OCC=2C=CC=CC=2)=CC2=C1OCC(=O)N2 DKLVIQDZNAPVFJ-VWLOTQADSA-N 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- YBFVFTWCEOCKBE-NHSZFOGYSA-N (2R)-2-(2-chloro-1-hydroxyethyl)-4H-1,4-benzoxazin-3-one Chemical compound ClCC(O)[C@H]1OC2=C(NC1=O)C=CC=C2 YBFVFTWCEOCKBE-NHSZFOGYSA-N 0.000 description 2
- DUMKDWRRTLFHTA-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-methylpropan-2-amine Chemical compound COC1=CC=C(CC(C)(C)N)C=C1 DUMKDWRRTLFHTA-UHFFFAOYSA-N 0.000 description 2
- QWELSVCCXLINHO-UHFFFAOYSA-N 3-[1-hydroxy-2-(propan-2-ylamino)ethyl]phenol;hydrochloride Chemical compound [Cl-].CC(C)[NH2+]CC(O)C1=CC=CC(O)=C1 QWELSVCCXLINHO-UHFFFAOYSA-N 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- FBXGQDUVJBKEAJ-UHFFFAOYSA-N 4h-oxazin-3-one Chemical class O=C1CC=CON1 FBXGQDUVJBKEAJ-UHFFFAOYSA-N 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- 229910017488 Cu K Inorganic materials 0.000 description 2
- 229910017541 Cu-K Inorganic materials 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000002447 crystallographic data Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000000967 suction filtration Methods 0.000 description 2
- BXOCBNXPOIPDTL-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-methylpropan-2-amine;hydrochloride Chemical compound Cl.COC1=CC=C(CC(C)(C)N)C=C1 BXOCBNXPOIPDTL-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- COUYJEVMBVSIHV-UHFFFAOYSA-N olodaterol Chemical compound C1=CC(OC)=CC=C1CC(C)(C)NCC(O)C1=CC(O)=CC2=C1OCC(=O)N2 COUYJEVMBVSIHV-UHFFFAOYSA-N 0.000 description 1
- 238000000634 powder X-ray diffraction Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000004537 pulping Methods 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- REFMEZARFCPESH-UHFFFAOYSA-M sodium;heptane-1-sulfonate Chemical compound [Na+].CCCCCCCS([O-])(=O)=O REFMEZARFCPESH-UHFFFAOYSA-M 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/34—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings
- C07D265/36—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention relates to a long-acting beta2The crystal form A of the adrenergic agonist drug of the odaterol hydrochloride, a preparation method thereof and a pharmaceutical composition containing the crystal form A are characterized by an X-ray diffraction pattern characteristic absorption peak of the crystal form A. Compared with the prior art, the crystal form A of the olodaterol hydrochloride provided by the invention is not easy to absorb moisture, has obviously improved stability and is convenient for quality control of products; and the solubility of the crystal in water is improved, which is beneficial to the exertion of drug effect. The preparation method provided by the invention is simple in process, is beneficial to cost control in industrial production and has higher economic value.
Description
Technical Field
The invention belongs to the field of medicines, and particularly relates to a novel crystal form of olodaterol hydrochloride and a preparation method thereof.
Background
Oxdaterol hydrochloride, systematic name: 6-hydroxy-8- { (1R) -1-hydroxy-2- { [2- (4-methoxyphenyl) -1, 1-dimethylethyl]Amino } ethyl } -4H-benzo [1,4]]Oxazin-3-one hydrochloride (6-Hydroxy-8- { (1R) -1-Hydroxy-2- { [2- (4-methoxyphenyl) -1,1-dimethyl-ethyl]amino}ethyl}-4H-[1,4]-benzoxazin-3-one, hydrochloride), molecular formula C21H27ClN2O5Molecular weight of 386.44, CAS registry number of 868049-49-4, structural formula shown in 1

The olodaterol hydrochloride is long-acting beta2A receptor agonist, suitable for use in patients with Chronic Obstructive Pulmonary Disease (COPD).
The specification of the chinese invention patent application publication No. CN101817800A (published 2010, 9/1) describes 6-hydroxy-8- { 1-hydroxy-2- { [2- (4-methoxyphenyl) -1, 1-dimethylethyl ] amino } ethyl } -4H-benzo [1,4] oxazin-3-one without enantiomer discrimination, and its preparation method and use. Chinese patent application publication No. CN102827097 a (publication No. 2012, 12, 19) discloses a pure enantiomer represented by structural formula 1, and discloses the following preparation methods:
"5.25 g (17.7 mmol) 6-benzyloxy-8- (R) -oxiranyl-4H-benzo [1,4] oxazin-3-one and 6.30 g (35.1 mmol) 2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamine are mixed with 21 ml isopropanol and stirred in a closed reaction vessel under microwave irradiation at 135 ℃ for 30 minutes. The solvent was evaporated off and the residue was chromatographed (alumina; ethyl acetate/methanol gradient). The product thus obtained was further purified "by recrystallization from a diethyl ether/diisopropyl ether mixture to give 6-benzyloxy-8- { (R) -1-hydroxy-2- [2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamino ] -ethyl } -4H-benzo [1,4] oxazin-3-one.
Then "a suspension of 5.33 g (11.2 mmol) 6-benzyloxy-8- { (R) -1-hydroxy-2- [2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamino ] -ethyl } -4H-benzo [1,4] oxazin-3-one in 120 ml methanol was mixed with 0.8g palladium on charcoal (10%), heated to 50 ℃ and hydrogenated under 3 bar hydrogen pressure. The catalyst was then filtered off with suction and the filtrate was evaporated to dryness. The residue was dissolved in 20 ml of isopropanol and 2.5 ml of 5 mol hydrochloric acid in isopropanol were added. This product was precipitated with 200 ml of diethyl ether, filtered off with suction and dried to give 6-hydroxy-8- { (R) -1-hydroxy-2- [2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamino ] -ethyl } -4H-benzo [1,4] oxazin-3-one-hydrochloride (olodaterol hydrochloride).
However, the inventor finds that the olodaterol hydrochloride is extremely easy to absorb moisture and is converted into oily matter during suction filtration according to the method, and cannot obtain a stable crystalline product, thereby causing great difficulty in subsequent quality control and preparation work.
As is well known, the physical and chemical properties of the same drug in various crystal forms such as solubility, stability, fluidity, compressibility, bioavailability and the like may be greatly different, thereby affecting the curative effect of the drug. Therefore, there is a need to develop a new crystal form of odaterol hydrochloride having superior physiochemical properties, thereby facilitating formulation processing and clinical use.
Disclosure of Invention
In order to overcome the defects of the prior art, the invention provides a novel crystal form of the oxdaterol hydrochloride, namely a crystal form A of the oxdaterol hydrochloride. The crystal form is not easy to absorb moisture, the stability is obviously improved, and the quality control of the product is convenient; and the preparation process is simple and is beneficial to cost control in industrial production.
In order to achieve the purpose of the invention, the invention adopts the following technical scheme:
a crystal form a of olodaterol hydrochloride shown in structural formula 1, wherein an X-ray diffraction pattern expressed by an angle of 2 theta by using Cu-K alpha radiation has characteristic absorption peaks at 8.01 ° ± 0.2 °, 16.06 ° ± 0.2 °, 21.67 ° ± 0.2 °, 25.32 ° ± 0.2 ° and 25.78 ° ± 0.2 °.
Preferably, the crystal form A of the olodaterol hydrochloride has characteristic absorption peaks at 8.01 degrees +/-0.2 degrees, 16.06 degrees +/-0.2 degrees, 17.64 degrees +/-0.2 degrees, 19.16 degrees +/-0.2 degrees, 20.17 degrees +/-0.2 degrees, 21.67 degrees +/-0.2 degrees, 25.32 degrees +/-0.2 degrees and 25.78 degrees +/-0.2 degrees by using Cu-Kalpha radiation and an X-ray diffraction pattern expressed by an angle of 2 theta.
Preferably, the form a of the olodaterol hydrochloride has an X-ray diffraction pattern substantially in accordance with fig. 1 expressed in 2 theta angles using Cu-ka radiation.
The invention also aims to provide a preparation method of the crystal form A of the metaterol hydrochloride, which comprises the following steps:
I. putting the crude product of the olodaterol hydrochloride and the mixed solvent into a reaction container, stirring, heating to ensure that the solution is clear, and keeping the temperature and continuing stirring;
and II, reducing the temperature, adding seed crystals, stirring, crystallizing, filtering and drying to obtain the crystal.
Preferably, the mixed solvent is selected from two or more of isopropanol, ethyl acetate, dichloromethane, toluene and n-propanol.
More preferably, the mixed solvent is isopropanol-ethyl acetate.
More preferably, the volume ratio of isopropanol to ethyl acetate is:
isopropyl alcohol: and ethyl acetate is 7: 1-15: 1.
Most preferably, the volume ratio of isopropanol to ethyl acetate is:
isopropyl alcohol: ethyl acetate 10: 1.
Preferably, the weight-to-volume ratio of the crude olodaterol hydrochloride to the mixed solvent is as follows:
crude product of olodaterol hydrochloride: 8-1 g of mixed solvent and 15ml of mixed solvent;
more preferably, the weight-to-volume ratio of the crude olodaterol hydrochloride to the mixed solvent is as follows:
crude product of olodaterol hydrochloride: the mixed solvent (1 g) and (13 ml).
Preferably, in the step I, the temperature is 50-55 ℃, and the mixture is stirred for 20 min-1 h, preferably 0.5h, after being dissolved and cleaned.
Preferably, in the step II, the temperature is reduced to 25-30 ℃, and the seed crystal is added and stirred for 1-3 h, preferably 2h at 25-30 ℃.
In the above preparation method, the seed crystal is prepared by the following steps:
I. putting the crude product of the oldacterol hydrochloride, isopropanol and ethyl acetate into a reaction container, stirring, heating to 50-55 ℃, dissolving to be clear, keeping the temperature and continuously stirring for half an hour; stirring for 2 hours at the temperature of 25-30 ℃. Wherein the weight volume ratio of the olodaterol hydrochloride to the isopropanol is as follows:
crude product of olodaterol hydrochloride: isopropyl alcohol: 1g of ethyl acetate, 8-15 ml: 3-8 ml;
II, reducing the temperature to 25-30 ℃, preferably 30 ℃, and adding seed crystal B;
III, keeping the temperature in the reaction container at 25-30 ℃, keeping the temperature and stirring for 0.5-3 h, preferably 2h, filtering and drying to obtain the catalyst;
wherein the seed crystal B is prepared by the following method:
i. putting the crude product of the olodaterol hydrochloride and isopropanol into a reaction container, stirring, heating to 50-55 ℃, dissolving to be clear, keeping the temperature, and continuing stirring for half an hour; wherein the weight volume ratio of the olodaterol hydrochloride to the isopropanol is as follows: crude product of olodaterol hydrochloride: 8 ml-1 g of isopropanol and 15ml of isopropanol;
reducing the temperature to 25-30 ℃, and keeping the temperature for 30 min-1 h;
iii, raising the temperature to 50-55 ℃ again, and stirring for 0.5-3 h under the condition of heat preservation;
and iv, cooling to 25-30 ℃ again, keeping the temperature, stirring for 1-3 h, filtering and drying to obtain the product.
The crude product of the olodaterol hydrochloride can be prepared by the method described in the following steps 1-3 of example 1.
The third objective of the present invention is to provide a pharmaceutical composition, which includes the crystal form a of the olodaterol hydrochloride or the crystal form a of the olodaterol hydrochloride prepared by the preparation method, and pharmaceutically acceptable excipients.
Preferably, the pharmaceutical composition is an inhalation aerosol, an inhalation powder or an inhalation solution.
In addition, the invention also aims to provide the crystal form A of the aodalterol hydrochloride, the crystal form A of the aodalterol hydrochloride prepared by the preparation method, or the application of the pharmaceutical composition in preparing a medicament for treating COPD.
The crystal form A of the olodaterol hydrochloride provided by the invention is not easy to absorb moisture, the stability is obviously improved compared with the prior art, and the quality control of a product and the subsequent preparation processing are facilitated. In addition, the solubility of the crystal form A of the odaterol in water is obviously improved compared with the original ground crystal form, and the bioavailability of the crystal form A of the odaterol can be improved, so that the drug effect is greatly improved. Meanwhile, the preparation process of the crystal form provided by the invention is simple, is beneficial to cost control in industrial production, and has high economic value.
Drawings
The invention will be further described with reference to the accompanying drawings.
FIG. 1 is an X-ray diffraction pattern of an metaterol hydrochloride seed crystal B prepared in step 4) of example 1.
Fig. 2 is an X-ray diffraction pattern of the form a of the olodaterol hydrochloride prepared in example 1.
Detailed Description
The invention is illustrated below with reference to specific examples. It will be understood by those skilled in the art that these examples are for illustrative purposes only and are not intended to limit the scope of the present invention in any way. Further, it should be understood that various changes or modifications of the present invention may be made by those skilled in the art after reading the teachings herein, and such equivalents may fall within the scope of the invention as defined in the appended claims.
The experimental procedures in the following examples are conventional unless otherwise specified. The raw materials and reagent materials used in the following examples are all commercially available products unless otherwise specified. Wherein, the purchase conditions of partial reagents, raw materials and instruments are as follows:
6-benzyloxy-8- ((R) -2-chloro-1-hydroxy-ethyl-4H-benzo [1,4] -oxazin-3-one, CAS869478-11-5, 98% pure and 2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamine hydrochloride CAS56490-93-8, 99% pure, all purchased from Gekko Ji pharmaceutical science, Inc. in Shanghai.
Nuclear magnetic resonance apparatus: bruker AVANCE III HD 500;
mass spectrometry: LTQ Orbitrap Elite;
liquid chromatograph: agilent 1260.
In the following examples and comparative examples, HPLC measurements were carried out as follows:
a chromatographic column: waters XBridge 5 μm (4.6 x 250 mm);
mobile phase: a0.1% aqueous solution of sodium heptanesulfonate (pH3.2) and B acetonitrile, the gradient elution procedure is shown in Table 1:
TABLE 1 HPLC gradient elution procedure
| Time(min) | A% | B% |
| 0 | 73 | 27 |
| 3 | 69 | 27 |
| 23 | 49 | 51 |
| 43 | 35 | 65 |
| 55 | 32 | 68 |
| 55.1 | 73 | 27 |
| 60 | 73 | 27 |
Flow rate: 1.0 ml/min;
column temperature: 30 ℃;
detection wavelength: 220 nm;
sample introduction volume: 10 mu L of the solution;
test sample diluent: and (3) acetonitrile.
In the following examples and comparative examples, the detection conditions for X-ray diffraction were:
the instrument model is as follows: bruker D8 Advance
And (3) testing conditions are as follows: x-ray source: Cu-K
Working current: 40mA
Working voltage: 40KV
A detector: PSD
Initial angle: 4 ° (2- θ)
End angle: 40 ° (2- θ)
Increment: 0.05 °/step
Scanning speed: 1sec/step
Example 1 preparation of form a of olodaterol hydrochloride
1)6-benzyloxy-8- (R) -oxiranyl-4H-benzo [1,4]]Preparation of oxazin-3-ones
Adding 100.6g (0.3mol) of 6-benzyloxy-8- ((R) -2-chloro-1-hydroxy-ethyl-4H-benzo [1,4] -oxazine-3-one and 2L of DMF into a reaction bottle, stirring, cooling in an ice bath, cooling to 0 ℃, adding 400ml of 2N sodium hydroxide aqueous solution, reacting for four hours at 0-5 ℃, pouring the reaction solution into ice water, stirring for 1 hour at 0-5 ℃, filtering, and drying a filter cake in vacuum at 50 ℃ to obtain 86g of white solid, the yield is 96%, and the purity HPLC is 96.5%.
2)6-benzyloxy-8- { (R) -1-hydroxy-2- [2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamino]-B Radical } -4H-benzo [1,4]]Preparation of oxazin-3-ones
Adding 52.5g (0.178mol) of 6-benzyloxy-8- (R) -oxiranyl-4H-benzo [1,4] oxazine-3-one prepared in the step 1), 63g (0.351mol) of 2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamine and 500ml of isopropanol into a reaction bottle, carrying out reflux reaction for 24 hours, cooling to 20-30 ℃, then adding 30g (0.3mol) of concentrated hydrochloric acid, stirring at 0-5 ℃ for 1 hour, filtering, and pulping a filter cake with 500ml of anhydrous methanol to obtain 60g of off-white solid, wherein the yield is as follows: 66.2 percent and the purity is 95.8 percent
3)Preparation of crude Oldhuterol hydrochloride
60g (0.117mol) of 6-benzyloxy-8- { (R) -1-hydroxy-2- [2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamino ] -ethyl } -4H-benzo [1,4] oxazin-3-one prepared in step 2), 600ml of methanol and 8g of 10% palladium on charcoal were added to a reaction flask, heated to 50 ℃ and hydrogenated under 3 bar of hydrogen pressure. Then the catalyst was removed by suction filtration, and the filtrate was evaporated to dryness in vacuo at 50 ℃ to give 49.1g of a white foamy solid, yield: 99.27%, purity: 95.3 percent.
4)Preparation of seed B
Adding 1g of the crude product of the olodaterol hydrochloride prepared in the step 3) and 9ml of isopropanol into a reaction bottle, stirring, heating to 50-55 ℃, dissolving to be clear, and stirring for half an hour under the condition of heat preservation; cooling to 30 deg.C, separating out solid to obtain suspension, and keeping the temperature for half an hour; then heating to 50-55 ℃, stirring for 1 hour under heat preservation, then cooling to 25-30 ℃, stirring for 2 hours under heat preservation, filtering, and drying a filter cake in vacuum at 50 ℃ to obtain 0.8g of off-white solid, wherein the yield is as follows: 80%, purity: 99.8 percent. The X-ray diffraction pattern of the crystal is shown in figure 1, and the X-ray diffraction data is shown in table 2.
Table 2X-ray powder diffraction data of the obtained olodaterol hydrochloride seed crystal B prepared in step 4) of the method


5)Preparation of crystal seed with crystal form of Aodaterol hydrochloride as A
Adding 5g of the crude product of the olodaterol hydrochloride prepared in the step 2), 50ml of isopropanol and 5ml of ethyl acetate into a reaction bottle, stirring, heating to 50-55 ℃, dissolving, keeping the temperature and stirring for half an hour, cooling to 30 ℃, adding a crystal seed with the crystal form B, keeping the temperature and stirring for 2 hours at 25-30 ℃, filtering, and drying a filter cake at 50 ℃ in vacuum to obtain 4.3g of a white-like solid with the yield: 90%, purity: 99.8 percent. The crystalline X-ray powder diffraction data are in accordance with those shown in table 3.
6)Preparation of crystal form A of olodaterol hydrochloride
Adding 5g of the crude product of the oldamterol hydrochloride prepared in the step 2), 50ml of isopropanol and 5ml of ethyl acetate into a reaction bottle, stirring, heating to 50-55 ℃, dissolving, stirring while keeping the temperature for half an hour, cooling to 30 ℃, adding the crystal seed of the crystal form A of the oldamterol hydrochloride prepared in the step 5), stirring while keeping the temperature for 2 hours at 25-30 ℃, filtering, and drying the filter cake at 50 ℃ in vacuum to obtain 4.3g of white-like solid with the yield: 90%, purity: 99.8 percent.
1HNMR(DMSO-d6):ppm 1.19(d,6H),2.90(m,3H),3.14(m,1H),3.74(s,3H),4.60(m,2H),5.15(m,1H),6.10(d,1H),6.37(d,1H),6.58(d,1H),6.90(d,2H),7.15(d,2H),8.58(m,1H),8.89(m,1H),9.33(s,1H),10.65(s,1H).
MS(ESI)387m/z(M-HCl+H)+。
The X-ray diffraction pattern of the crystal form a of the olodaterol hydrochloride obtained in the example is shown in fig. 2, and the specific results are shown in table 3.
Table 3X-ray powder diffraction data for crystal form a of odaterol hydrochloride
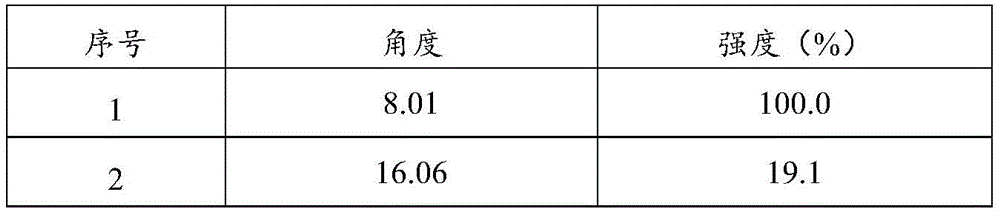

The crystal form a of the odaterol hydrochloride prepared in the example was tested according to the guiding principle of moisture absorption test of XIX J medicament in the appendix of the second part of the chinese pharmacopoeia (2005 edition), and the moisture absorption increased by 0.25% as a result. The solubility of the crystals in water, measured at room temperature, was 950mg/10 ml.
Example 2 preparation of form a of olodaterol hydrochloride
Adding 6g of the crude product of the olodaterol hydrochloride obtained in the example 1, 45ml of isopropanol and 3ml of ethyl acetate into a reaction bottle, stirring, heating to 50-55 ℃ to dissolve, keeping the temperature and stirring for half an hour, cooling to 30 ℃, adding the seed crystal prepared in the example 1, keeping the temperature and stirring for 2 hours at 25-30 ℃, filtering, and drying a filter cake in vacuum at 50 ℃ to obtain 4.9g of an off-white solid with the yield: 82%, purity: 99.2 percent.
Using a Bruker D8 Advance X-ray diffractometer, the product of this example gave an X-ray diffraction pattern (map-less) substantially in accordance with FIG. 2.
The crystal form a of the odaterol hydrochloride prepared in the example was tested according to the guiding principle of moisture absorption test of XIX J medicament in the appendix of the second part of the chinese pharmacopoeia (2005 edition), and the moisture absorption increased by 0.27% as a result. The solubility of the crystals in water, measured at room temperature, was 950mg/10 ml.
Example 3 preparation of form a of olodaterol hydrochloride
Adding 5.3g of the crude product of the olodaterol hydrochloride obtained in the example 1, 70ml of isopropanol and 10ml of ethyl acetate into a reaction bottle, stirring, heating to 50-55 ℃ to dissolve the crude product clear, keeping the temperature and stirring for half an hour, cooling to 30 ℃, adding seed crystals, keeping the temperature and stirring for 2 hours at 25-30 ℃, filtering, and drying a filter cake at 50 ℃ in vacuum to obtain 4.3g of an off-white solid with the yield: 81%, purity: 99.3 percent.
Using a Bruker D8 Advance X-ray diffractometer, the product of this example gave an X-ray diffraction pattern (map-less) substantially in accordance with FIG. 2.
The crystal form a of the odaterol hydrochloride prepared in the example was tested according to the guiding principle of moisture absorption test of XIX J medicament in the appendix of the second part of the chinese pharmacopoeia (2005 edition), and the moisture absorption increased by 0.30% as a result. The solubility of the crystals in water, measured at room temperature, was 950mg/10 ml.
Example 4 preparation of form a of olodaterol hydrochloride
Adding 5g of the crude product of the olodaterol hydrochloride obtained in the example 1, 60ml of isopropanol and 5ml of ethyl acetate into a reaction bottle, stirring, heating to 50-55 ℃, dissolving to clear, keeping the temperature and stirring for half an hour, cooling to 30 ℃, adding seed crystals, keeping the temperature and stirring for 2 hours at 25-30 ℃, filtering, and drying a filter cake in vacuum at 50 ℃ to obtain 4.4g of off-white solid with the yield: 88%, purity: 99.5 percent. Using a Bruker D8 Advance X-ray diffractometer, the product of this example gave an X-ray diffraction pattern (map-less) substantially in accordance with FIG. 2.
The crystal form a of the odaterol hydrochloride prepared in the example was tested according to the guiding principle of moisture absorption test of XIX J medicament in the appendix of the second part of the chinese pharmacopoeia (2005 edition), and the moisture absorption increased by 0.28%. The solubility of the crystals in water, measured at room temperature, was 950mg/10 ml.
Example 5Preparation of crystal form A of olodaterol hydrochloride
Adding 5g of the crude product of the olodaterol hydrochloride obtained in the example 1, 60ml of isopropanol and 5ml of dichloromethane into a reaction bottle, stirring, heating to 50-55 ℃, dissolving to clear, keeping the temperature and stirring for half an hour, cooling to 30 ℃, adding seed crystals, keeping the temperature and stirring for 2 hours at 25-30 ℃, filtering, and drying a filter cake in vacuum at 50 ℃ to obtain 4.5g of off-white solid with the yield: 90%, purity: 99.4 percent.
Using a Bruker D8 Advance X-ray diffractometer, the product of this example gave an X-ray diffraction pattern (map-less) substantially in accordance with FIG. 2.
The crystal form B of the ondansterol hydrochloride prepared in this example was tested according to the guidelines of the hygroscopicity test of the drug, XIX J, appendix ii of the chinese pharmacopoeia (2005 edition), and the hygroscopicity increased by 0.34%. The solubility of the crystals in water, measured at room temperature, was 950mg/10 ml.
Comparative example 1 preparation of Oldaterol hydrochloride
The preparation of odaterol hydrochloride was carried out according to the method described in the chinese invention patent application publication No. CN102827097 (example 1) and the procedure was as follows:
a suspension of 5.33 g (11.2 mmol) 6-benzyloxy-8- { (R) -1-hydroxy-2- [2- (4-methoxy-phenyl) -1, 1-dimethyl-ethylamino ] -ethyl } -4H-benzo [1,4] oxazin-3-one in 120 ml methanol was mixed with 0.8g palladium on charcoal (10%), heated to 50 ℃ and hydrogenated under 3 bar of hydrogen pressure. The catalyst was then filtered off with suction and the filtrate was evaporated to dryness. The residue is dissolved in 20 ml of isopropanol and 2.5 ml of 5 mol hydrochloric acid in isopropanol are added, the product is precipitated with 200 ml of diethyl ether, filtered off with suction and dried.
However, when the solution is filtered, the oldacterol hydrochloride is found to be extremely easy to absorb moisture and change into oily substance, and a stable crystal product cannot be obtained.
Comparative example 2 preparation of Oldaterol hydrochloride
The preparation of the odaterol hydrochloride was carried out according to the method described in example 1 of the chinese patent application publication No. CN 101208316A. The yield is 63-70%, and the purity is 98.5%.
The crystal form of the olodaterol hydrochloride prepared by the comparative example uses Cu-Kalpha radiation, and an X-ray diffraction pattern expressed by a 2 theta angle has characteristic absorption peaks at 11.91 degrees +/-0.2 degrees, 15.19 degrees +/-0.2 degrees, 16.88 degrees +/-0.2 degrees, 17.34 degrees +/-0.2 degrees, 19.04 degrees +/-0.2 degrees, 19.71 degrees +/-0.2 degrees, 21.02 degrees +/-0.2 degrees, 21.35 degrees +/-0.2 degrees, 22.15 degrees +/-0.2 degrees, 23.21 degrees +/-0.2 degrees, 28.41 degrees +/-0.2 degrees, 29.79 degrees +/-0.2 degrees and 30.71 degrees +/-0.2 degrees. The crystal had a solubility in water of 900mg/10ml at room temperature.
In a word, the invention provides a new crystal form of the aodalterol hydrochloride, namely the crystal form A of the aodalterol hydrochloride, the crystal form is not easy to absorb moisture, the stability is obviously improved, and the quality control of a product and the subsequent preparation processing are convenient. Meanwhile, the preparation process of the crystal form provided by the invention is simple, is beneficial to cost control in industrial production, and has high economic value.
Claims (8)
1. A crystal form a of olodaterol hydrochloride, characterized in that the crystal form a of olodaterol hydrochloride has characteristic absorption peaks at 8.01 ° ± 0.2 °, 16.06 ° ± 0.2 °, 17.64 ° ± 0.2 °, 19.16 ° ± 0.2 °, 20.17 ° ± 0.2 °, 21.67 ° ± 0.2 °, 25.32 ° ± 0.2 ° and 25.78 ° ± 0.2 ° in an X-ray diffraction pattern expressed by 2 θ angles using Cu-ka radiation.
2. A process for the preparation of form a of olodaterol hydrochloride according to claim 1, comprising the steps of:
I. putting the crude product of the olodaterol hydrochloride and the mixed solvent into a reaction container, stirring, heating to 50-55 ℃, and keeping the temperature and stirring for 20 min-1 h after dissolving;
wherein the weight volume ratio of the crude product of the olodaterol hydrochloride to the mixed solvent is as follows: crude product of olodaterol hydrochloride: 8-1 g of mixed solvent and 15ml of mixed solvent; the mixed solvent is isopropanol-ethyl acetate, and the volume ratio of the isopropanol to the ethyl acetate is as follows: isopropyl alcohol: ethyl acetate is 7: 1-15: 1;
II, cooling to 25-30 ℃, adding seed crystals, stirring for 1-3 h at 25-30 ℃, crystallizing, filtering, and drying to obtain the product;
the seed crystal is prepared by the following steps:
I. putting the crude product of the oldacterol hydrochloride, isopropanol and ethyl acetate into a reaction container, stirring, heating to 50-55 ℃, dissolving to be clear, keeping the temperature and continuously stirring for half an hour; stirring for 2 hours at the temperature of 25-30 ℃; wherein the weight volume ratio of the olodaterol hydrochloride to the isopropanol to the ethyl acetate is as follows:
crude product of olodaterol hydrochloride: isopropyl alcohol: 1g of ethyl acetate, 8-15 ml: 3-8 ml;
II, reducing the temperature to 25-30 ℃, and adding a seed crystal B;
III, keeping the temperature in the reaction container at 25-30 ℃, keeping the temperature, stirring for 0.5-3 h, filtering and drying to obtain the catalyst;
wherein the seed crystal B is prepared by the following method:
i. putting the crude product of the olodaterol hydrochloride and isopropanol into a reaction container, stirring, heating to 50-55 ℃, dissolving to be clear, keeping the temperature, and continuing stirring for half an hour; wherein the weight volume ratio of the olodaterol hydrochloride to the isopropanol is as follows: 8 ml-1 g of isopropanol as a crude product of the oldamterol hydrochloride, namely 15 ml;
reducing the temperature to 25-30 ℃, and keeping the temperature for 30 min-1 h;
iii, raising the temperature to 50-55 ℃ again, and stirring for 0.5-3 h under the condition of heat preservation;
and iv, cooling to 25-30 ℃ again, keeping the temperature, stirring for 1-3 h, filtering and drying to obtain the product.
3. The preparation method according to claim 2, wherein the volume ratio of the mixed solvent, isopropanol and ethyl acetate is as follows: isopropyl alcohol: ethyl acetate 10: 1.
4. The preparation method according to claim 2 or 3, wherein the weight-to-volume ratio of the crude olodaterol hydrochloride to the mixed solvent is as follows:
crude product of olodaterol hydrochloride: the mixed solvent (1 g) and (13 ml).
5. The preparation method according to claim 2, wherein in the step I, stirring is carried out for 0.5h under heat preservation after the solution is clear; and in the step II, adding seed crystals and stirring for 2 hours at 25-30 ℃.
6. The preparation method according to claim 2, wherein the seed crystal is prepared by, in the step II, reducing the temperature to 30 ℃, and adding the seed crystal B; and in the step III, stirring for 2 hours under the condition of heat preservation.
7. A pharmaceutical composition comprising the form a of olodaterol hydrochloride according to claim 1 or the form a of olodaterol hydrochloride prepared by the preparation method according to any one of claims 2 to 6, and pharmaceutically acceptable excipients;
8. the pharmaceutical composition of claim 7, wherein the pharmaceutical composition is an inhalation aerosol, an inhalation powder or an inhalation solution.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810884531.XA CN109096218B (en) | 2018-08-06 | 2018-08-06 | Oxydterol hydrochloride crystal form A and preparation method thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810884531.XA CN109096218B (en) | 2018-08-06 | 2018-08-06 | Oxydterol hydrochloride crystal form A and preparation method thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109096218A CN109096218A (en) | 2018-12-28 |
| CN109096218B true CN109096218B (en) | 2020-10-27 |
Family
ID=64848853
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201810884531.XA Active CN109096218B (en) | 2018-08-06 | 2018-08-06 | Oxydterol hydrochloride crystal form A and preparation method thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN109096218B (en) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101133039A (en) * | 2004-05-14 | 2008-02-27 | 贝林格尔·英格海姆国际有限公司 | Novel enantiomerically pure beta agonists, method for the production thereof and use thereof in the form of medicaments |
| CN101208316A (en) * | 2005-08-15 | 2008-06-25 | 贝林格尔·英格海姆国际有限公司 | Method for preparing beta-mimetics |
| CN101578274A (en) * | 2007-01-25 | 2009-11-11 | 贝林格尔.英格海姆国际有限公司 | Method for producing betamimetics |
| CN101735166A (en) * | 2002-11-15 | 2010-06-16 | 贝林格尔英格海姆法玛两合公司 | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| WO2016038628A2 (en) * | 2014-09-09 | 2016-03-17 | Reddy G Pratap | A process for preparing olodaterol and intermediates thereof |
| WO2018114887A1 (en) * | 2016-12-20 | 2018-06-28 | Laboratorios Lesvi, S.L. | Improved process for the manufacture of r-6-hydroxy-8-[1-hydroxy-2-[2-(4-methoxyphenyl)-1,1-dimethylethylaminoethyl]-2h-1,4-benzoxazin-3(4h)-one hydrochloride |
-
2018
- 2018-08-06 CN CN201810884531.XA patent/CN109096218B/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101735166A (en) * | 2002-11-15 | 2010-06-16 | 贝林格尔英格海姆法玛两合公司 | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| CN101133039A (en) * | 2004-05-14 | 2008-02-27 | 贝林格尔·英格海姆国际有限公司 | Novel enantiomerically pure beta agonists, method for the production thereof and use thereof in the form of medicaments |
| CN101208316A (en) * | 2005-08-15 | 2008-06-25 | 贝林格尔·英格海姆国际有限公司 | Method for preparing beta-mimetics |
| CN101578274A (en) * | 2007-01-25 | 2009-11-11 | 贝林格尔.英格海姆国际有限公司 | Method for producing betamimetics |
| WO2016038628A2 (en) * | 2014-09-09 | 2016-03-17 | Reddy G Pratap | A process for preparing olodaterol and intermediates thereof |
| WO2018114887A1 (en) * | 2016-12-20 | 2018-06-28 | Laboratorios Lesvi, S.L. | Improved process for the manufacture of r-6-hydroxy-8-[1-hydroxy-2-[2-(4-methoxyphenyl)-1,1-dimethylethylaminoethyl]-2h-1,4-benzoxazin-3(4h)-one hydrochloride |
Non-Patent Citations (1)
| Title |
|---|
| "奥达特罗的合成工艺优化";祝淳君 等;《中国医药工业杂志》;20171231;第48卷(第11期);第1582-1586页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109096218A (en) | 2018-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6239977B2 (en) | Pharmaceutical composition for the treatment of obesity | |
| CN101891738B (en) | Dasatinib polymorph, its preparation method and pharmaceutical composition | |
| CN108794481A (en) | Polymorphic, the preparation method and use of compound | |
| EP3565819B1 (en) | Solid forms of [(1s)-1-[(2s,4r,5r)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-tetrahydrofuran-2-yl]propyl] acetate | |
| CN109438370B (en) | Methylpyrazine derivative anhydrous crystal form | |
| CN108822054B (en) | Oxydterol hydrochloride crystal form C and preparation method thereof | |
| CN109096218B (en) | Oxydterol hydrochloride crystal form A and preparation method thereof | |
| TWI773987B (en) | Solid form of diaminopyrimidine compound or its hydrate and its preparation method and use | |
| CN116041351B (en) | New crystal form of midazolam hydrochloride and preparation method thereof | |
| US20250361226A1 (en) | Pharmaceutically acceptable salt of benzo[c]chroman compound and polymorphic form and use of pharmaceutically acceptable salt | |
| CN108997248B (en) | Crystal form B of ondarot hydrochloride and preparation method thereof | |
| EP4289826A1 (en) | Salt and crystal form of ha inhibitor compound | |
| US11434226B2 (en) | Salt and polymorph of benzopyrimidinone compound and pharmaceutical composition and use thereof | |
| US20220119415A1 (en) | Solid forms of [(1s)-1-[(2s,4r,5r)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-tetrahydrofuran-2-yl]propyl] acetate | |
| CN1603324A (en) | Levohalogenated sculiline salt and its preparation method and use | |
| CN116323593A (en) | A kind of crystal form of pyrimidine derivative and its preparation method | |
| CN111732586A (en) | Crystal form of alkynyl-containing compound salt, preparation method and application | |
| TWI857250B (en) | Treprostinil monohydrate crystals and methods for preparation thereof | |
| CN115583952B (en) | Polycrystal of phosphodiesterase 5 inhibitor, preparation method and application thereof | |
| EP4282860A1 (en) | Crystal form of anti-influenza virus compound, preparation method for crystal form, and use of crystal form | |
| CN110627777B (en) | Maleate of benzothiophene compound, crystal form and application thereof | |
| CN110218209A (en) | It is a kind of according to crystal form A, preparation method and an application for piperazine azoles laurate | |
| CA3048771C (en) | Solid forms of [(1s)-1-[(2s,4r,5r)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-tetrahydrofuran-2-yl]propyl] acetate | |
| EP3960742A1 (en) | Crystals of alkynyl-containing compound, salt and solvate thereof, preparation method, and applications | |
| CN117466914A (en) | A new crystal form of baloxavir distil and its method for product purification |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |