CN108440526B - 一种手性巴比妥螺四氢喹啉类化合物及制备方法 - Google Patents
一种手性巴比妥螺四氢喹啉类化合物及制备方法 Download PDFInfo
- Publication number
- CN108440526B CN108440526B CN201810449570.7A CN201810449570A CN108440526B CN 108440526 B CN108440526 B CN 108440526B CN 201810449570 A CN201810449570 A CN 201810449570A CN 108440526 B CN108440526 B CN 108440526B
- Authority
- CN
- China
- Prior art keywords
- compound
- barbiturate
- chiral
- reaction
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
一种手性巴比妥螺四氢喹啉类化合物及制备方法,属于化合物的制备技术领域。具体为以乙烯基苯并噁嗪酮化合物和巴比妥烯烃化合物为反应物,并加入金属钯催化剂以及含磷手性配体,在室温下反应得到产物。此制备方法反应条件温和,反应速度快,后处理简单,适用底物范围广,并且合成的绝大多数目标物具有较高的产率、优秀的对映选择性和非对映选择性。这是一种全新的高效合成具有潜在药用价值的手性巴比妥螺四氢喹啉类化合物的方法。
Description
技术领域
本发明涉及一种手性巴比妥螺四氢喹啉类化合物及制备方法,属于化合物的制备技术领域。
背景技术
巴比妥螺四氢喹啉类化合物是一类结构复杂的含氮杂环,据文献报道,许多药效化合物含有其结构片段,具有抗菌和抗肿瘤活性,在生物学和医学上有重要作用。四氢喹啉类化合物可以用于治疗和预防多种疾病。许多具有生物活性的天然产物以及药物分子中都含有四氢喹啉的骨架结构。巴比妥类化合物是普遍性中枢抑制药。巴比妥螺四氢喹啉类化合物含有巴比妥和四氢喹啉结构单元,作为药效化合物的片段具有重要意义。
在过去几年中,大多文献报道的是关于巴比妥螺四氢喹啉类化合物外消旋体的合成方法。然而,很多天然产物和药物中间体都是手性分子。也有一些文献关于手性巴比妥螺四氢喹啉类化合物的对映选择性的合成,但其合成方法主要涉及手性底物和手性拆分。其对仪器和技术要求较高,而且适用底物范围窄。因此寻找一种反应条件温和、操作简单、收率高和对映选择性优秀的催化体系制备手性巴比妥螺四氢喹啉类化合物是很有必要的。本发明的手性巴比妥螺四氢喹啉类化合物类药性结构显著,在抗菌和抗肿瘤作用方面具有潜在的应用价值。
发明内容
本发明目的在于提供一种手性巴比妥螺四氢喹啉类化合物的制备方法。
为达到上述发明目的,本发明采取的技术方案是:
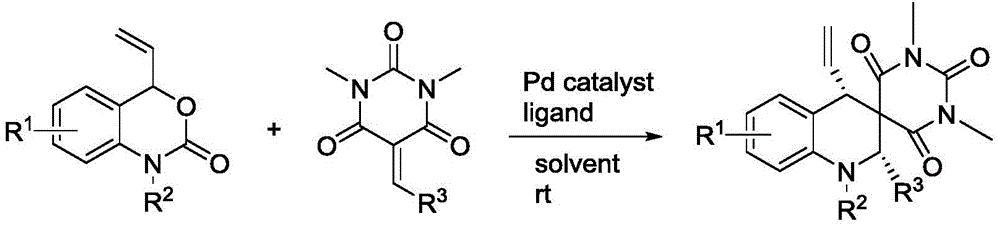
一种手性巴比妥螺四氢喹啉类化合物的制备方法,所述制备方法为:以乙烯基苯并噁嗪酮化合物和巴比妥烯烃化合物为反应物,并加入金属钯催化剂以及含磷手性配体,在极性为2~6的有机溶剂中,室温下反应得到产物手性巴比妥螺四氢喹啉类化合物;优选所述乙烯基苯并噁嗪酮化合物和巴比妥酸盐烯烃化合物的摩尔比为1:1;
所述手性巴比妥螺四氢喹啉类化合物的结构式为:
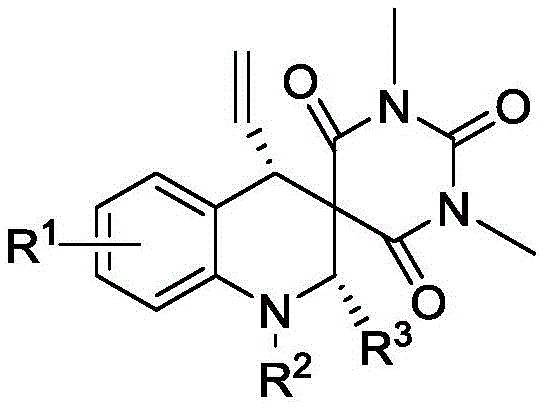
其中,R1为氟、氯、溴、甲基中的一种;R2为氢或对芳基磺酰基;R3为芳基或杂环基。
上述的芳基是指萘基、苯基或具有1~3个取代基的苯基。例如:一取代苯基、二取代苯基、三取代苯基。
上述苯基上的取代基选自:甲基、甲氧基、氟、氯、溴或三氟甲基。
上述的杂环基是指无取代的包含1~4个杂原子的五元环或六元杂环基,所述的杂原子选自N、S、O,例如:吡啶基、呋喃基、哌啶基、嘧啶基、噻唑基、噻吩基。
上述技术方案中,所述有机溶剂为二氯甲烷、四氢呋喃、1,2-二氯乙烷、甲苯、乙腈或三氯甲烷。
上述技术方案中,所述的钯催化剂为四(三苯基膦)钯、醋酸钯、三(二亚苄基丙酮)二钯或三(二亚苄基丙酮)二钯-氯仿加合物。
上述技术方案中,所述含磷手性配体为:基于联二萘酚(BINOL)为骨架的手性磷酸配体或手性联萘型双磷配体(BINAP),例如:R-(+)-1,1'-联萘-2,2'-双二苯膦、(R)-(-)-(3,5-二氧杂-4-磷杂环庚二烯并[2,1-A;3,4-A']二萘-4-基)二甲胺。
上述技术方案中,所述反应时间为3小时~48小时。
上述技术方案中,所述钯催化剂的用量为乙烯基苯并噁嗪酮化合物摩尔量的2.5%~10%。
上述技术方案中,所述含磷手性配体的用量为乙烯基苯并噁嗪酮化合物摩尔量的10%~20%。
上述技术方案中,反应过程包括向反应瓶中加入乙烯基苯并噁嗪酮化合物、巴比妥烯烃化合物、金属钯催化剂以及含磷手性配体,再加入有机溶剂在室温下进行反应,用TLC检测反应进程,反应结束后,粗产物通过简单的柱层析(洗脱剂选为体积比1:5~6的乙酸乙酯/石油醚混合溶液)即可得到目标产物。
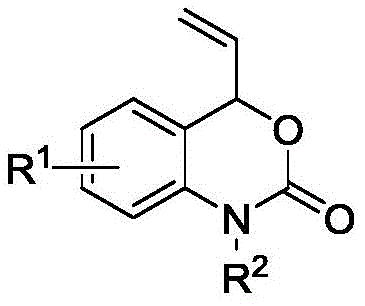
本发明中,乙烯基苯并噁嗪酮化合物的制备方法属于现有技术,其结构式如下所示:
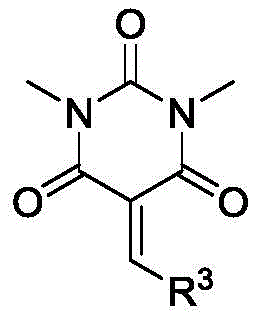
本发明中,巴比妥烯烃化合物的制备方法属于现有技术,其结构式如下所示:
本发明公开的反应过程如下所示:
由于上述技术方案运用,本发明与现有技术相比具有下列优点:
1.本发明首次提供了以乙烯基苯并噁嗪酮化合物、巴比妥烯烃化合物为反应物,并加入金属钯催化剂以及含磷手性配体制备手性巴比妥螺四氢喹啉类化合物的方法;该方法操作简单,反应条件温和,时间短。
2.本发明所公开的制备方法中后处理简单,反应不存在手性拆分。
3.本发明所公开的方法适用底物范围广,合成的绝大多数目标物对映选择性和非对映选择性优秀,收率较高。
具体实施方式
下面结合实施例对本发明作进一步描述,但本发明并不限于以下实施例。
实施例1:
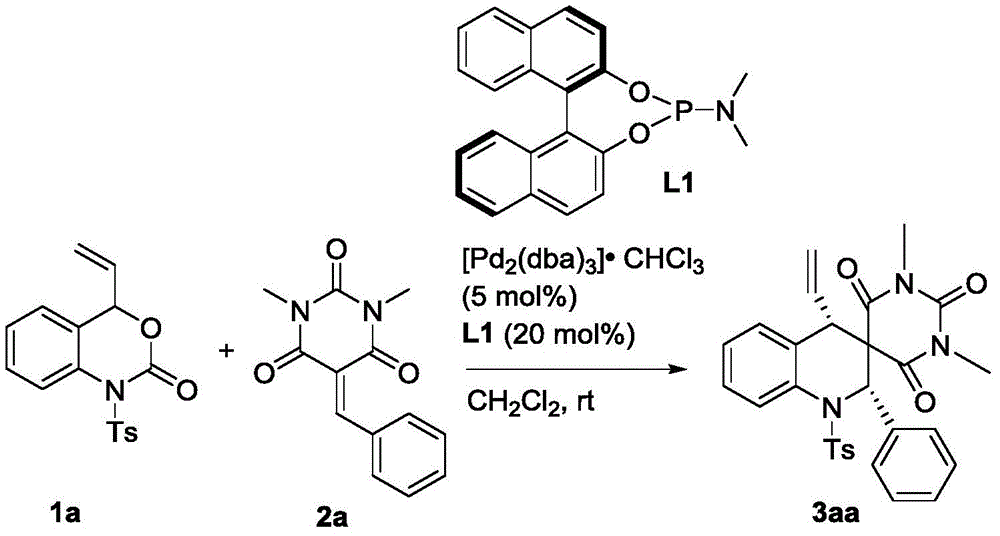
称取1a(32.9mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3aa(40mg),得率为76%。
目标物的表征及分析:白色固体,dr>99:1;ee=96%;[α]1 D 8:160(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ8.01(d,J=7.6Hz,1H),7.52–7.47(m,3H),7.29–7.24(m,5H),7.21–7.18(m,3H),6.89(d,J=7.6Hz,1H),6.31(s,1H),5.46–5.36(m,1H),5.22(dd,J=10.0,1.2Hz,1H),4.80(d,J=16.4Hz,1H),3.32(s,3H),2.80(d,J=10.0Hz,1H),2.64(s,3H),2.42(s,3H)ppm;HRMS(ESI)m/z:C29H28N3O5S[M+H]+理论计算值530.1744,实测值530.1751。
实施例2:
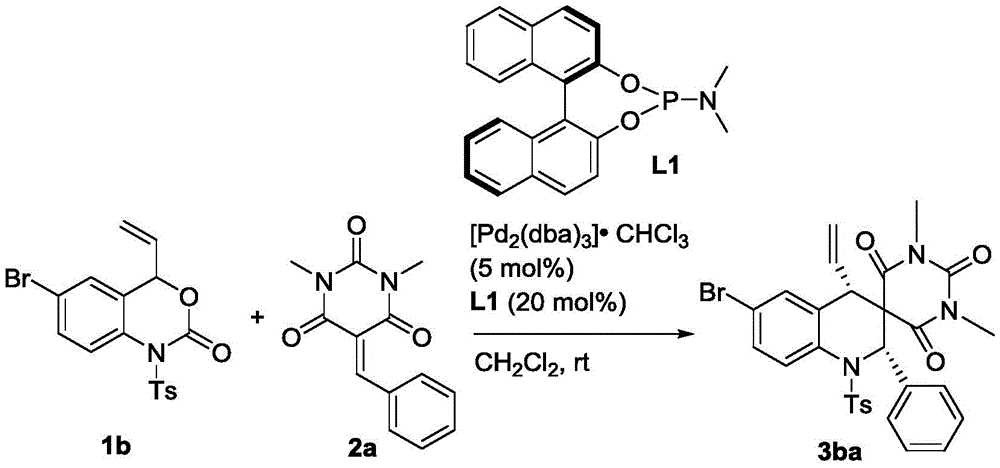
称取1b(40.8mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ba(40mg),得率为66%。
目标物的表征及分析:白色固体,dr>99:1;ee=58%;[α]1 D 8:94(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.87(d,J=8.8Hz,1H),7.60(dd,J=8.4,1.6Hz,1H),7.54(d,J=8.0Hz,2H),7.30–7.21(m,5H),7.14(d,J=6.8Hz,2H),7.02(s,1H),6.22(s,1H),5.40–5.31(m,1H),5.26(dd,J=10.0,1.2Hz1H),4.85–4.80(m,1H),3.32(s,3H),2.81(d,J=9.6Hz,1H),2.64(s,3H),2.44(s,3H)ppm;HRMS(ESI)m/z:C29H27BrN3O5S[M+H]+理论计算值608.0849,实测值608.0833。
实施例3:
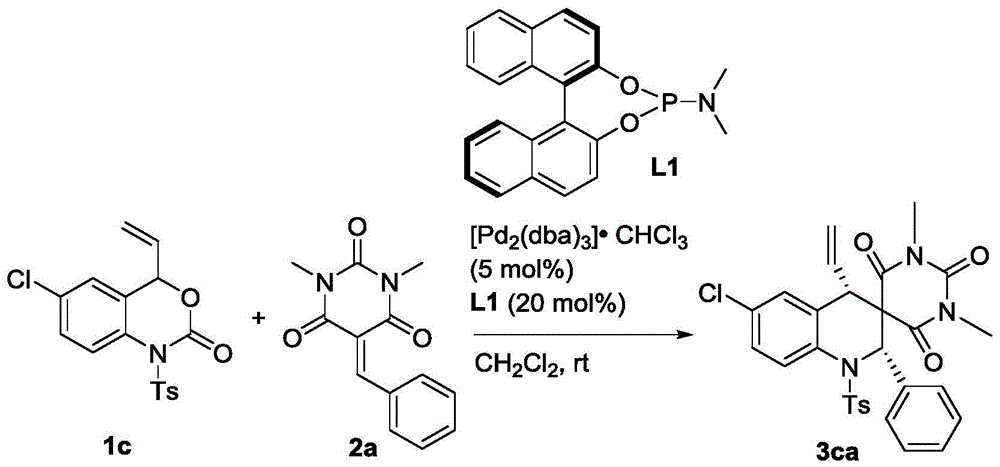
称取1c(36.3mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌5小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ca(37mg),得率为66%。
目标物的表征及分析:白色固体,dr>99:1;ee=66%;[α]1 D 8:84(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.93(d,J=8.4Hz,1H),7.53(d,J=8.0Hz,2H),7.45(d,J=7.2Hz,1H),7.30–7.21(m,5H),7.14(d,J=6.8Hz,2H),6.87(s,1H),6.23(s,1H),5.40–5.31(m,1H)5.26(d,J=9.6Hz,1H),4.82(d,J=16.4Hz1H),3.32(s,3H),2.78(d,J=9.6Hz,1H),2.64(s,3H),2.44(s,3H)ppm;HRMS(ESI)m/z:C29H27ClN3O5S[M+H]+理论计算值564.1354,实测值564.1342。
实施例4:
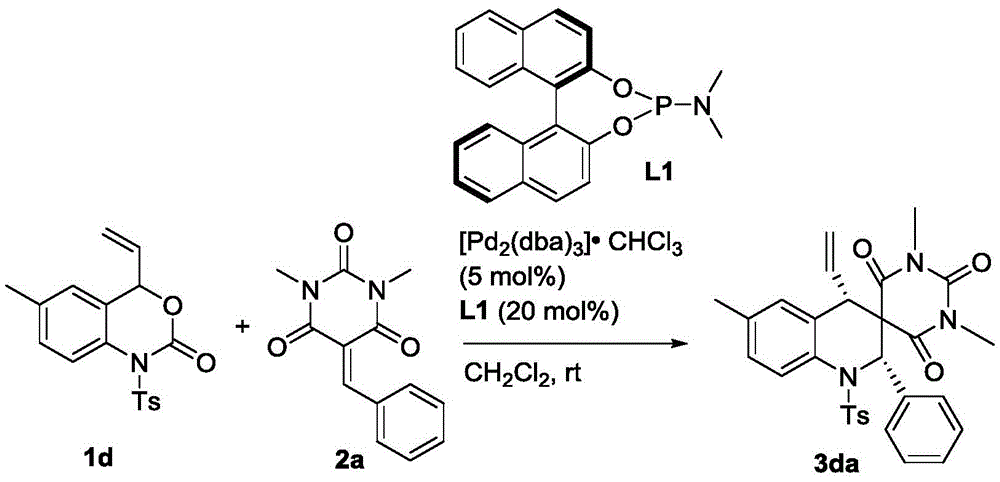
称取1d(34.3mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌12小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3da(40mg),得率为74%。
目标物的表征及分析:白色固体,dr>99:1;ee=80%;[α]1 D 8:116(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.87(d,J=8.0Hz,1H),7.52(d,J=8.0Hz,2H),7.30–7.20(m,5H),7.19–7.16(m,3H),6.68(s,1H),6.28(s,1H),5.44–5.35(m,1H),5.21(d,J=10.0Hz,1H),4.78(d,J=16.8Hz,1H),3.32(s,3H),2.76(d,J=10.0Hz,1H),2.64(s,3H),2.42(s,3H),2.36(s,3H)ppm;HRMS(ESI)m/z:C30H30N3O5S[M+H]+理论计算值544.1901,实测值544.1905。
实施例5:
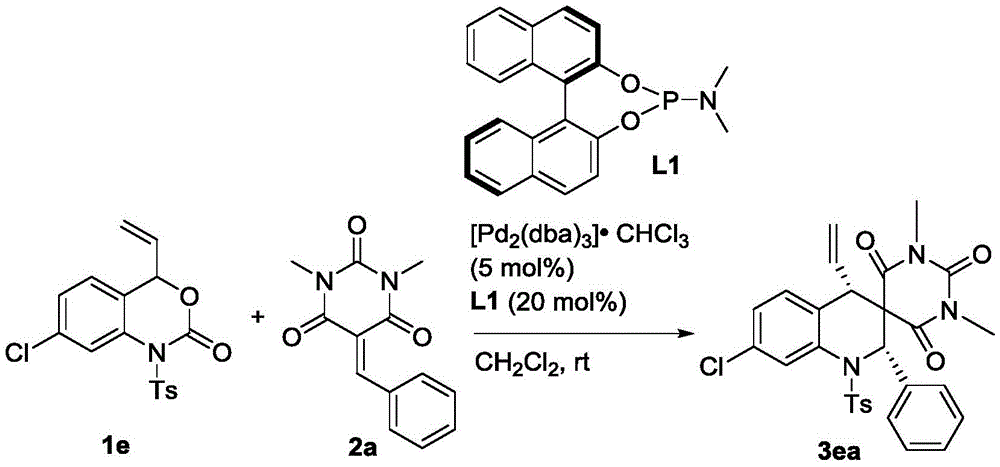
称取1e(36.3mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ea(48mg),得率为85%。
目标物的表征及分析:白色固体,dr>99:1;ee=92%;[α]1 D 8:110(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ8.02(s,1H),7.55(d,J=8.0Hz,2H),7.28–7.21(m,6H),7.15(d,J=7.6Hz,2H),6.81(d,J=8.0Hz,1H),6.26(s,1H),5.41–5.32(m,1H),5.24(dd,J=10.0,1.2Hz,1H),4.83(d,J=16.8Hz,1H),3.32(s,3H),2.80(d,J=9.6Hz,1H),2.64(s,3H),2.42(s,3H)ppm;HRMS(ESI)m/z:C29H27ClN3O5S[M+H]+理论计算值564.1354,实测值564.1355。
实施例6:
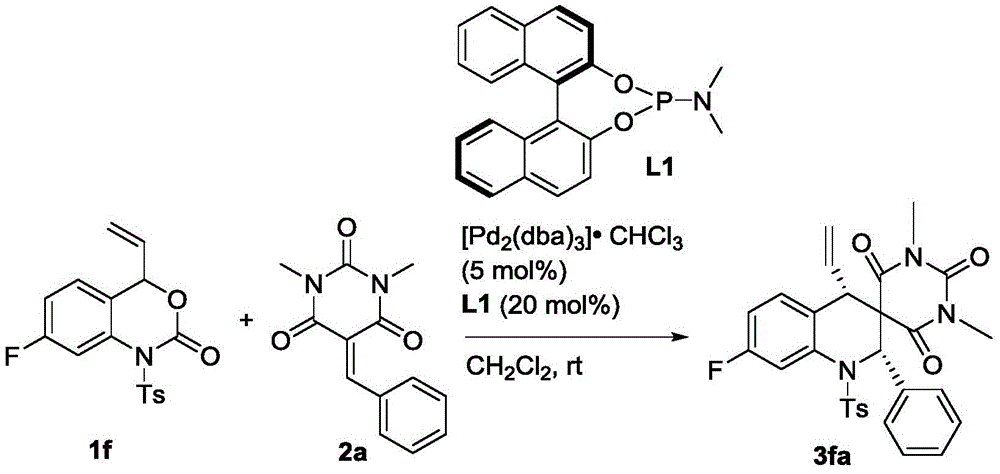
称取1f(34.7mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌5小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3fa(45mg),得率为82%。
目标物的表征及分析:白色固体,dr>99:1;ee=96%;[α]1 D8:148(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.79–7.77(m,1H),7.55(d,J=7.6Hz,2H),7.28–7.16(m,7H),6.97–6.94(m,1H),6.85–6.81(m,1H),6.28(s,1H),5.42–5.33(m,1H),5.24(d,J=10.0Hz,1H),4.84(d,J=16.8Hz,1H),3.32(s,3H),2.80(d,J=10.0Hz,1H),2.65(s,3H),2.43(s,3H)ppm;HRMS(ESI)m/z:C29H27FN3O5S[M+H]+理论计算值548.1650,实测值548.1650。
实施例7:
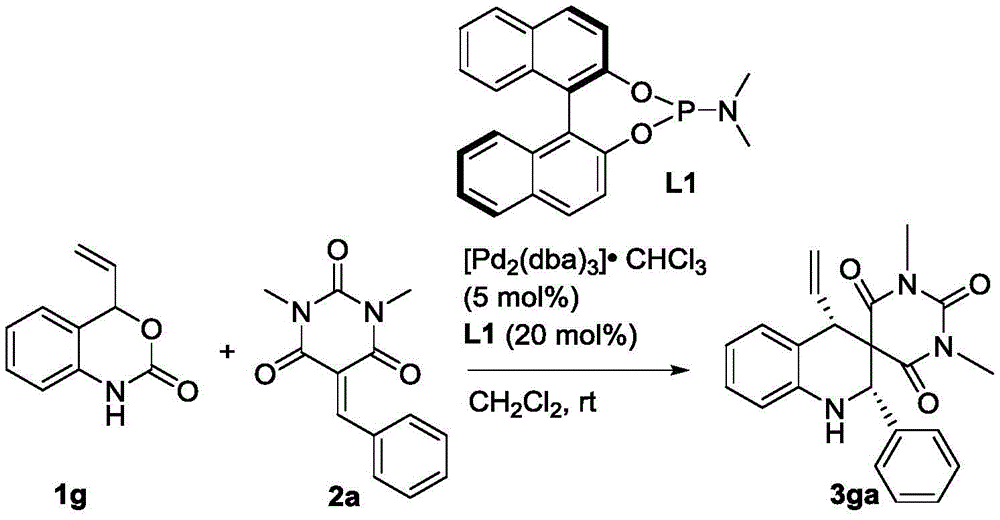
称取1g(17.5mg,0.1mmol)、2a(24.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌12小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ga(36mg),得率为96%。
目标物的表征及分析:白色固体,dr>99:1;ee=35%;[α]1D8:28(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.37–7.31(m,3H),7.21–7.12(m,4H),6.84–6.81(m,1H),6.71(d,J=7.6Hz,1H),5.70–5.61(m,1H),5.39–5.29(m,2H),4.81(s,1H),4.56(d,J=9.6Hz,1H),4.44(s,1H),3.08(s,3H),3.03(s,3H)ppm;HRMS(ESI)m/z:C22H22N3O3[M+H]+理论计算值376.1656,实测值376.1656。
实施例8:
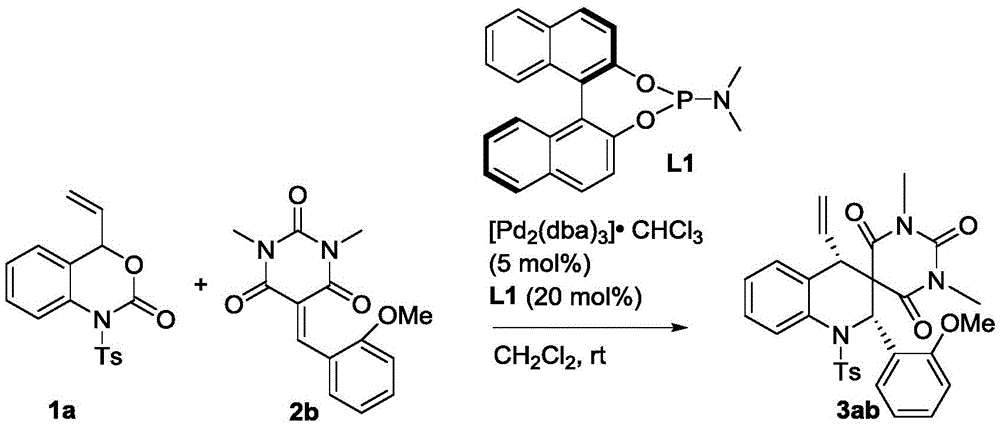
称取1a(32.9mg,0.1mmol)、2b(27.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ab(38mg),得率为68%。
目标物的表征及分析:白色固体,dr>99:1;ee=91%;[α]1D8:240(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ8.06(d,J=8.0Hz,1H),7.62–7.56(m,3H),7.48(t,J=7.6Hz,1H),7.28–7.22(m,3H),7.17(t,J=7.6Hz,1H),6.97–6.93(m,1H),6.88(d,J=7.6Hz,1H),6.69(d,J=8.0Hz,1H),6.63(s,1H),5.48–5.39(m,1H),5.19(d,J=9.6Hz,1H),4.72(d,J=16.8Hz,1H),3.64(s,3H),3.34(s,3H),2.81(d,J=10.0Hz,1H),2.66(s,3H),2.40(s,3H)ppm;HRMS(ESI)m/z:C30H30N3O6S[M+H]+理论计算值560.1850,实测值560.1847。
实施例9:
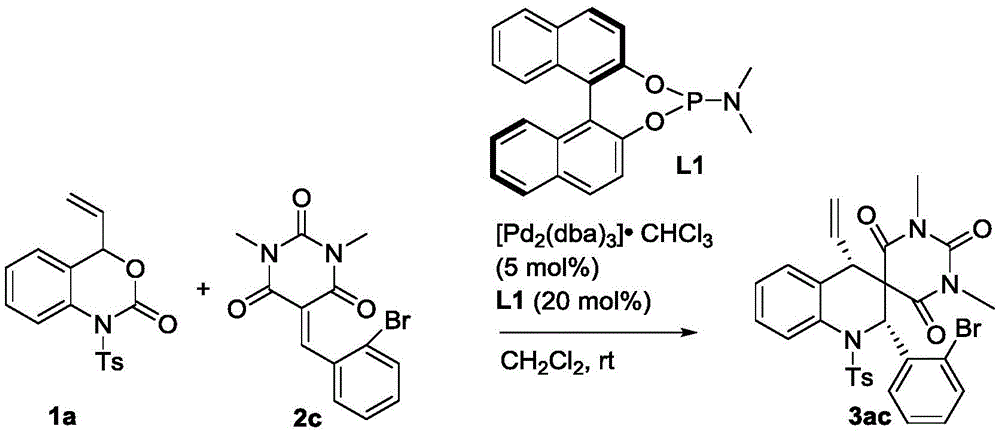
称取1a(32.9mg,0.1mmol)、2b(32.2mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌5小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ac(41mg),得率为68%。
目标物的表征及分析:白色固体,dr>99:1;ee=94%;[α]1D8:188(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.94(d,J=7.6Hz,1H),7.58(d,J=8.4Hz,2H),7.51–7.44(m,2H),7.35–7.26(m,4H),7.22–7.19(m,1H),7.10–7.06(m,1H),7.00(d,J=7.6Hz,1H),6.58(s,1H),5.50–5.40(m,1H),5.24(dd,J=10.0,1.2Hz,1H),4.83(dd,J=16.8,1.2Hz,1H),3.28(s,3H),3.02(d,J=10.0Hz,1H),2.58(s,3H),2.43(s,3H)ppm;HRMS(ESI)m/z:C29H27BrN3O5S[M+H]+理论计算值608.0849,实测值608.0857。
实施例10:
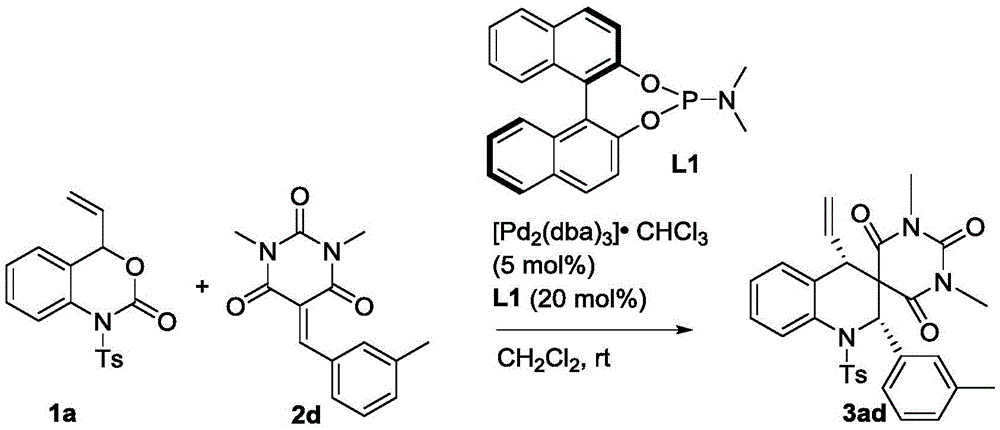
称取1a(32.9mg,0.1mmol)、2d(25.8mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ad(49mg),得率为90%。
目标物的表征及分析:白色固体,dr>99:1;ee=88%;[α]1D8:142(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ8.00(d,J=8.0Hz,1H),7.49(t,J=8.0Hz,3H),7.28–7.24(m,3H),7.13(t,J=7.6Hz,1H),7.02–6.94(m,3H),6.89(d,J=7.6Hz,1H),6.22(s,1H),5.45–5.36(m,1H),5.22(dd,J=10.0,1.2Hz,1H),4.81(d,J=16.8Hz,1H),3.32(s,3H),2.82(d,J=10.0Hz,1H),2.63(s,3H),2.42(s,3H),2.28(s,3H)ppm;HRMS(ESI)m/z:C30H30N3O5S[M+H]+理论计算值544.1901,实测值544.1899。
实施例11:
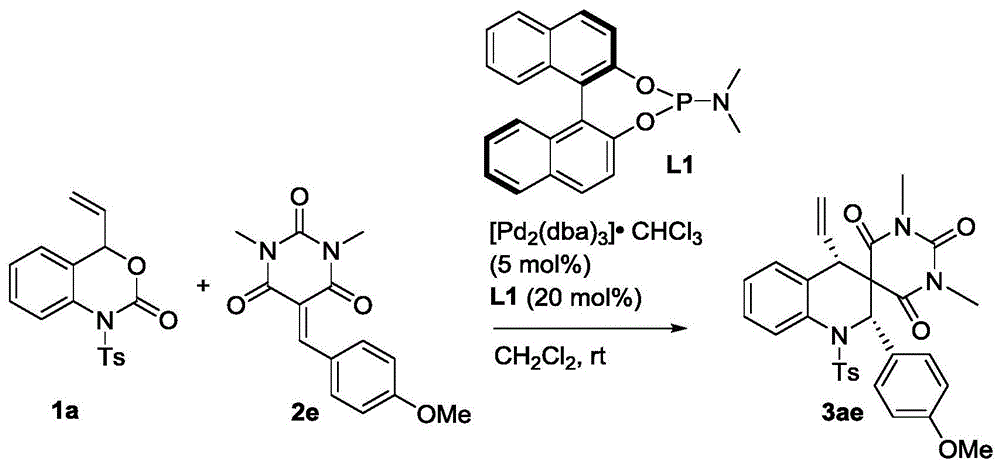
称取1a(32.9mg,0.1mmol)、2e(27.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6~1/6)即可得到目标产物3ae(45mg),得率为81%。
目标物的表征及分析:白色固体,dr>99:1;ee=92%;[α]1 D 8:130(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.98(d,J=8.0Hz,1H),7.50–7.46(m,3H),7.28–7.24(m,3H),7.11(d,J=8.4Hz,2H),6.88(d,J=7.6Hz,1H),6.78(d,J=8.8Hz,2H),6.23(s,1H),5.45–5.35(m,1H),5.22(d,J=10Hz,1H),4.79(d,J=16.8Hz,1H),3.76(s,3H),3.31(s,3H),2.79(d,J=10.0Hz,1H),2.68(s,3H),2.41(s,3H)ppm;HRMS(ESI)m/z:C30H30N3O6S[M+H]+理论计算值560.1850,实测值560.1854。
实施例12:
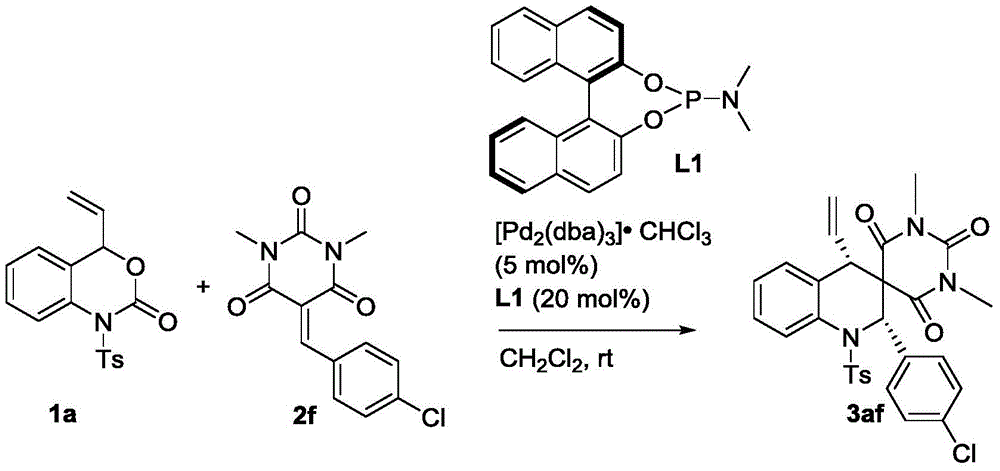
称取1a(32.9mg,0.1mmol)、2f(27.8mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌5小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6~1/6)即可得到目标产物3af(44mg),得率为78%。
目标物的表征及分析:白色固体,dr>99:1;ee=89%;[α]1 D 8:126(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.99(d,J=8.0Hz,1H),7.49(d,J=7.6Hz,3H),7.28–7.23(m,5H),7.16(d,J=8.0Hz,2H),6.86(d,J=7.6Hz,1H),6.32(s,1H),5.44–5.35(m,1H),5.24(d,J=9.6Hz,1H),4.78(d,J=16.4Hz,1H),3.31(s,3H),2.72(d,J=10.0Hz,4H),2.42(s,3H)ppm;1HRMS(ESI)m/z:C29H27ClN3O5S[M+H]+理论计算值564.1354,实测值564.1350。
实施例13:

称取1a(32.9mg,0.1mmol)、2g(33.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ag(52mg),得率为84%。
目标物的表征及分析:白色固体,dr>99:1;ee=89%;[α]1 D 8:166(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.97(d,J=8.0Hz,1H),7.51–7.48(m,3H),7.28–7.25(m,3H),6.89(d,J=8.0Hz,1H),6.40(s,2H),6.20(s,1H),5.44–5.35(m,1H),5.23(d,J=10.0Hz,1H),4.81(d,J=16.8Hz,1H),3.78(d,J=8.8Hz,9H),3.32(s,3H),2.81(d,J=10.0Hz,1H),2.71(s,3H),2.42(s,3H)ppm;HRMS(ESI)m/z:C32H34N3O8S[M+H]+理论计算值620.2061实测值620.2068。
实施例14:
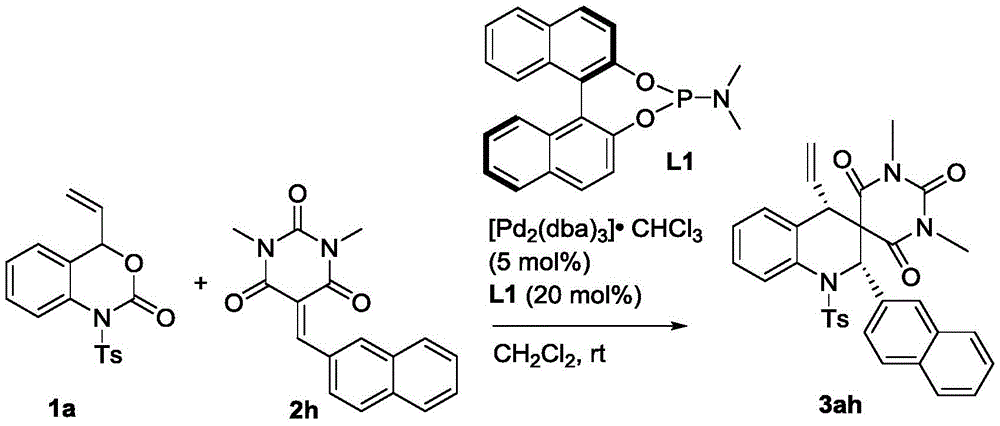
称取1a(32.9mg,0.1mmol)、2h(29.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ah(44mg),得率为76%。
目标物的表征及分析:白色固体,dr>99:1;ee=90%;[α]1 D 8:126(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ8.11(d,J=8.0Hz,1H),7.78–7.70(m,4H),7.55(d,J=8.0Hz,3H),7.47–7.45(m,2H),7.33–7.26(m,4H),6.91(d,J=7.6Hz,1H),6.51(s,1H),5.48–5.39(m,1H),5.25(d,J=10.0Hz,1H),4.83(d,J=16.8Hz,1H),3.35(s,3H),2.85(d,J=10.0Hz,1H),2.55(s,3H),2.43(s,3H)ppm;HRMS(ESI)m/z:C33H30N3O5S[M+H]+理论计算值580.1901,实测值580.1912。
实施例15:

称取1a(32.9mg,0.1mmol)、2i(23.4mg,0.1mmol)、钯催化剂(5.2mg,0.005mmol)以及手型配体L1(7.2mg,0.02mmol)溶于1mL的二氯甲烷,在室温下搅拌3小时(用TLC检测反应),待反应完全后,粗产物经过柱层析(洗脱剂为乙酸乙酯/石油醚=1/5~1/6)即可得到目标产物3ai(46mg),得率为89%。
目标物的表征及分析:白色固体,dr>99:1;ee=90%;[α]1 D 8:120(c=0.10,CHCl3),1H NMR(400MHz,CDCl3):δ7.88(d,J=7.6Hz,1H),7.51–7.42(m,3H),7.28–7.22(m,3H),7.13(s,1H),6.83(d,J=7.6Hz,1H),6.55(s,1H),6.38(s,1H),6.30(s,1H),5.48–5.39(m,1H),5.23(d,J=10.0Hz,1H),4.71(d,J=16.8Hz,1H),3.34(s,3H),2.92(s,3H),2.56(d,J=10.0Hz,1H),2.41(s,3H)ppm;HRMS(ESI)m/z:C27H26N3O6S[M+H]+理论计算值520.1537,实测值520.1536。
以上结果可以看出,本发明公开的制备方法反应条件温和,反应速度快,后处理简单,并且合成的绝大多数目标物具有较高的产率、优秀的对映选择性和非对映选择性。
Claims (7)
1.一种手性巴比妥螺四氢喹啉类化合物的制备方法,其特征在于,所述手性巴比妥螺四氢喹啉类化合物的结构式为:

其中,R1为氟、氯、溴、甲基中的一种;R2为氢或芳基磺酰基;R3为芳基或杂环基;
上述的芳基是指萘基、苯基或具有1~3个取代基的苯基;上述苯基上的取代基选自:甲基、甲氧基、氟、氯、溴或三氟甲基;
上述的杂环基是指无取代的包含1~4个杂原子的五元环或六元杂环基,所述的杂原子选自N、S、O;
所述手性巴比妥螺四氢喹啉类化合物的制备方法,包括向反应瓶中加入乙烯基苯并噁嗪酮化合物、巴比妥烯烃化合物、金属钯催化剂以及含磷手性配体,再加入有机溶剂在室温下进行反应,用TLC检测反应进程,反应结束后,粗产物通过简单的柱层析即可得到目标产物;洗脱剂选为体积比1:5~6的乙酸乙酯/石油醚混合溶液;
所述含磷手性配体的结构式为:
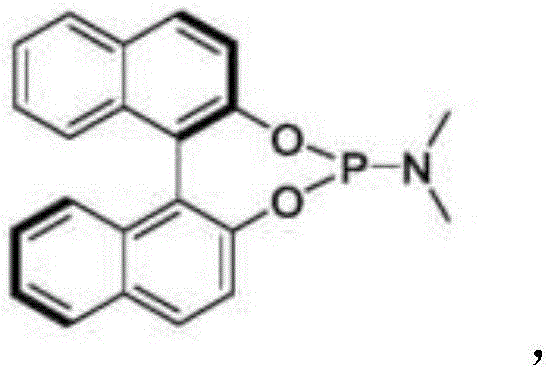
2.按照权利要求1所述的方法,其特征在于,所述乙烯基苯并噁嗪酮化合物和巴比妥烯烃化合物的摩尔比为1:1。
3.按照权利要求1所述的方法,其特征在于,有机溶剂为二氯甲烷、四氢呋喃、1,2-二氯乙烷、甲苯、乙腈或三氯甲烷。
4.按照权利要求1所述的方法,其特征在于,所述的钯催化剂为四(三苯基膦)钯、醋酸钯、三(二亚苄基丙酮)二钯或三(二亚苄基丙酮)二钯-氯仿加合物。
5.按照权利要求1所述的方法,其特征在于,所述反应时间为3小时~48小时。
6.按照权利要求1所述的方法,其特征在于,所述钯催化剂的用量为乙烯基苯并噁嗪酮化合物摩尔量的2.5%~10%。
7.按照权利要求1所述的方法,其特征在于,乙烯基苯并噁嗪酮化合物结构式如下所示:

R1为氟、氯、溴、甲基中的一种;R2为氢或芳基磺酰基;
巴比妥烯烃化合物结构式如下所示:

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810449570.7A CN108440526B (zh) | 2018-05-11 | 2018-05-11 | 一种手性巴比妥螺四氢喹啉类化合物及制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810449570.7A CN108440526B (zh) | 2018-05-11 | 2018-05-11 | 一种手性巴比妥螺四氢喹啉类化合物及制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108440526A CN108440526A (zh) | 2018-08-24 |
| CN108440526B true CN108440526B (zh) | 2020-10-16 |
Family
ID=63203530
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201810449570.7A Active CN108440526B (zh) | 2018-05-11 | 2018-05-11 | 一种手性巴比妥螺四氢喹啉类化合物及制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN108440526B (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109354583B (zh) * | 2018-12-21 | 2020-07-17 | 北京工业大学 | 一种手性3,4-二氢-2(1h)-喹啉酮类化合物及制备方法 |
| CN113880775A (zh) * | 2021-10-20 | 2022-01-04 | 北京工业大学 | 一种巴比妥类化合物及制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104387310A (zh) * | 2014-11-12 | 2015-03-04 | 华中师范大学 | 具有光学活性的3-乙烯基吲哚啉类衍生物及其不对称合成方法 |
| CN105732495A (zh) * | 2015-12-16 | 2016-07-06 | 华中师范大学 | 具有光学活性的四氢喹啉类化合物及其制备方法 |
| CN107417615A (zh) * | 2017-09-13 | 2017-12-01 | 华中师范大学 | 可见光促进的手性喹啉酮类衍生物制备新方法 |
| CN107573285A (zh) * | 2017-09-13 | 2018-01-12 | 华中师范大学 | 钯催化的不对称合成含有季碳中心的喹啉酮类及其衍生物的新方法 |
-
2018
- 2018-05-11 CN CN201810449570.7A patent/CN108440526B/zh active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104387310A (zh) * | 2014-11-12 | 2015-03-04 | 华中师范大学 | 具有光学活性的3-乙烯基吲哚啉类衍生物及其不对称合成方法 |
| CN105732495A (zh) * | 2015-12-16 | 2016-07-06 | 华中师范大学 | 具有光学活性的四氢喹啉类化合物及其制备方法 |
| CN107417615A (zh) * | 2017-09-13 | 2017-12-01 | 华中师范大学 | 可见光促进的手性喹啉酮类衍生物制备新方法 |
| CN107573285A (zh) * | 2017-09-13 | 2018-01-12 | 华中师范大学 | 钯催化的不对称合成含有季碳中心的喹啉酮类及其衍生物的新方法 |
Non-Patent Citations (3)
| Title |
|---|
| A catalytic asymmetric construction of a tetrahydroquinoline-based spirooxindole framework via a diastereo- and enantioselective decarboxylative [4+2] cycloaddition;Guang-Jian Mei et al.;《Chem. Commun.》;20170817;第53卷;10030-10033 * |
| Interaction of barbituric acids with o-dialkylaminobenzaldehydes;Konstantin A. Krasnov et al.;《Mendeleev Commun.》;20061231;第16卷(第1期);52-54 * |
| Konstantin A. Krasnov et al..Interaction of barbituric acids with o-dialkylaminobenzaldehydes.《Mendeleev Commun.》.2006,第16卷(第1期),52-54. * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN108440526A (zh) | 2018-08-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Mu et al. | Access to phosphine-containing quinazolinones enabled by photo-induced radical phosphorylation/cyclization of unactivated alkenes | |
| CN104276994B (zh) | 3,3′‑双取代氧化吲哚与3‑烯键氧化吲哚拼接衍生物及其制备方法及应用 | |
| Sun et al. | Pyridine-phosphinimine ligand-accelerated Cu (i)-catalyzed azide–alkyne cycloaddition for preparation of 1-(pyridin-2-yl)-1, 2, 3-triazole derivatives | |
| CN109354583B (zh) | 一种手性3,4-二氢-2(1h)-喹啉酮类化合物及制备方法 | |
| Song et al. | Asymmetric synthesis of highly functionalized spirothiazolidinone tetrahydroquinolines via a squaramide-catalyzed cascade reaction | |
| CN103992334A (zh) | 吲哚酮螺四氢硫代吡喃类抗肿瘤衍生物及其制备方法 | |
| CN106496222A (zh) | 一种咪唑并[1,2‑a]吡啶化合物及其制备方法和应用 | |
| CN111763148B (zh) | 一种含三氟甲基的炔基环戊烯衍生物及其制备方法和应用 | |
| Ren et al. | C (sp2)–H Functionalization of Imidazole at the C2-and C4-Position via Palladium-Catalyzed Isocyanide Insertion Leading to Indeno [1, 2-d] imidazole and Imidazo [1, 2-a] indole Derivatives | |
| CN108440526B (zh) | 一种手性巴比妥螺四氢喹啉类化合物及制备方法 | |
| Liu et al. | Transition-metal-free synthesis of indolizines via [3+ 2]-annulation from α-bromoenals and 2-substituted azaarenes | |
| CN108864147B (zh) | 一种八元氮氧杂环螺吲哚酮化合物及制备方法 | |
| CN101792438A (zh) | 一种1-取代-1,2,3-三氮唑的合成方法 | |
| CN112645958B (zh) | 一种手性螺环吡唑啉酮化合物及制备方法 | |
| CN111303028B (zh) | 一种4-氰基-2-二氟甲基取代的喹啉类化合物及合成方法 | |
| CN108440483B (zh) | 一种3,4-二氢氧基-2(7h)-酮及制备方法 | |
| CN115613060A (zh) | 一种直接电化学氧化合成c-3磷酸化2h-吲唑类化合物的方法 | |
| Jiang et al. | 1, 3-Dipolar cycloaddition of uracil derivatives with nitrile oxides: Synthesis of [1, 2, 4] oxadiazolo [4, 5-c] pyrimidine-5, 7 (6H)-dione derivatives | |
| CN107602559A (zh) | 一种通过迈克尔加成引发的不对称环丙化合成手性三元碳环核苷的方法 | |
| CN107445914B (zh) | 一种2,2,5-三取代1,3,4噁二唑衍生物及其合成方法 | |
| CN114478388A (zh) | 一种多官能团化的吡唑酮类化合物及制备方法 | |
| CN111393437A (zh) | 三取代吲嗪类化合物及其制备方法 | |
| CN112174958B (zh) | 一种吡啶并[2,3-d]嘧啶类化合物及其制备方法和用途 | |
| CN106632374B (zh) | 异甘露糖醇—双苯并咪唑盐类化合物及其制备方法 | |
| WO2024012425A1 (zh) | 作为ripk1抑制剂的杂环化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |