CN105793424B - 孢囊菌酰胺 - Google Patents
孢囊菌酰胺 Download PDFInfo
- Publication number
- CN105793424B CN105793424B CN201480050439.3A CN201480050439A CN105793424B CN 105793424 B CN105793424 B CN 105793424B CN 201480050439 A CN201480050439 A CN 201480050439A CN 105793424 B CN105793424 B CN 105793424B
- Authority
- CN
- China
- Prior art keywords
- group
- mmol
- cystobactamide
- nmr
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/44—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having carbon atoms of carboxamide groups, amino groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/02—Amides, e.g. chloramphenicol or polyamides; Imides or polyimides; Urethanes, i.e. compounds comprising N-C=O structural element or polyurethanes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyrrole Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Pyridine Compounds (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明提供了一种如式(I)所示的孢囊菌酰胺,以及其用于细菌感染的治疗或预防的用途。R1‑Ar1‑L1‑Ar2‑L2‑Ar3‑L3‑Ar4‑L4‑Ar5‑R2(I)。
Description
孢囊菌酰胺是一种从粘细菌深棕色孢囊杆菌(Cystobac ter vela tus)(MCy8071;内部名称:深棕色孢囊杆菌(Cystobacter ferrugineus))中 分离出的新天然产物。孢囊菌酰胺表现出良好的抗菌活性(特别是对于选定的革 兰氏阴性菌例如大肠杆菌、绿脓杆菌和鲍曼不动杆菌),且对革兰氏阳性菌具有 广谱的活性。
本发明提供了一种式(I)化合物,或其药学上可接受的盐、溶剂化物或水合 物,或其药学上可接受的制剂
R1-Ar1-L1-Ar2-L2-Ar3-L3-Ar4-L4-Ar5-R2
(I)
其中,
Ar1为任选取代的亚苯基,或任选取代的具有5至6个环原子(其中包括1、 2、3或4个选自下组的杂原子:氧、硫和氮)的杂亚芳基;
Ar2为任选取代的亚苯基,或任选取代的具有5至6个环原子(其中包括1、 2、3或4个选自下组的杂原子:氧、硫和氮)的杂亚芳基;
Ar3为任选取代的亚苯基,或任选取代的具有5至6个环原子(其中包括1、 2、3或4个选自下组的杂原子:氧、硫和氮)的杂亚芳基;
Ar4为无,或任选取代的亚苯基,或任选取代的具有5至6个环原子(其中包 括1、2、3或4个选自下组的杂原子:氧、硫和氮)的杂亚芳基;
Ar5为无,或任选取代的亚苯基,或任选取代的具有5至6个环原子(其中包 括1、2、3或4个选自下组的杂原子:氧、硫和氮)的杂亚芳基;
L1为键、氧原子、硫原子或如式NH、CONH、NHCO、COO、OCO、CONR3、 NR3CO、OCONH、NHCOO、NHCONH、OCONR3、NR3COO、NR3CONR4、NR3、-CNR3-、-CO- 、-SO-、-SO2-、-SO2NH-、-NHSO2-、-SO2NR3-、-NR3SO2-、-COCH2-、-CH2CO-、- COCR3R4-、-CR3R4CO-、-NHCSNH-、-NR3CSNR4、-CH=CH-、-CR3=CR4-所示的基团, 或任选取代的具有5至6个环原子(其中包括1、2或3个选自下组的杂原子: 氧、硫和氮)的杂亚芳基,或杂亚烃基;
L2为键、氧原子、硫原子或如式NH、CONH、NHCO、COO、OCO、CONR3、 NR3CO、OCONH、NHCOO、NHCONH、OCONR3、NR3COO、NR3CONR4、NR3、-CNR3-、-CO- 、-SO-、-SO2-、-SO2NH-、-NHSO2-、-SO2NR3-、-NR3SO2-、-COCH2-、-CH2CO-、- COCR3R4-、-CR3R4CO-、-NHCSNH-、-NR3CSNR4、-CH=CH-、-CR3=CR4-所示的基团,或 任选取代的具有5至6个环原子(其中包括1、2或3个选自下组的杂原子:氧、 硫和氮)的杂亚芳基,或杂亚烃基;
L3为无,或键、氧原子、硫原子或如式NH、CONH、NHCO、COO、OCO、 CONR3、NR3CO、OCONH、NHCOO、NHCONH、OCONR3、NR3COO、NR3CONR4、NR3、-CNR3- 、-CO-、-SO-、-SO2-、-SO2NH-、-NHSO2-、-SO2NR3-、-NR3SO2-、-COCH2-、-CH2CO- 、-COCR3R4-、-CR3R4CO-、-NHCSNH-、-NR3CSNR4、-CH=CH-、-CR3=CR4-所示的基 团,或任选取代的具有5至6个环原子(其中包括1、2或3个选自下组的杂原 子:氧、硫和氮)的杂亚芳基,或杂亚烃基;
L4为无,或键、氧原子、硫原子或如式NH、CONH、NHCO、COO、OCO、 CONR3、NR3CO、OCONH、NHCOO、NHCONH、OCONR3、NR3COO、NR3CONR4、NR3、-CNR3- 、-CO-、-SO-、-SO2-、-SO2NH-、-NHSO2-、-SO2NR3-、-NR3SO2-、-COCH2-、-CH2CO- 、-COCR3R4-、-CR3R4CO-、-NHCSNH-、-NR3CSNR4、-CH=CH-、-CR3=CR4-所示的基 团,或任选取代的具有5至6个环原子(其中包括1、2或3个选自下组的杂原 子:氧、硫和氮)的杂亚芳基,或杂亚烃基;
R1为氢原子,卤素原子,羟基,氨基,硫醇基,硝基,如式-COOH、- SO2NH2、-CONH2、-NO2或-CN所示的基团,烷基,烯基,炔基,杂烷基,环烷基, 杂环烷基,烷基环烷基,杂烷基环烷基,芳基,杂芳基,芳烷基或杂芳基;
R2为氢原子,卤素原子,羟基,氨基,硫醇基,硝基,如式-COOH、- SO2NH2、-CONH2、-NO2或-CN所示的基团,烷基,烯基,炔基,杂烷基,环烷基, 杂环烷基,烷基环烷基,杂烷基环烷基,芳基,杂芳基,芳烷基或杂芳基;
R3基团各自独立地为氢原子或C1-6烷基基团;和
R4基团各自独立地为氢原子或C1-6烷基基团。
表述烷基是指饱和的直链或支链烃基,它包含1至20个碳原子,优选1至 15个碳原子,特别是1至10个(例如1、2、3或4)个碳原子,例如甲基、乙 基、丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、正己 基、2,2-二甲基丁基或正辛基。
表述烯基和炔基指至少部分不饱和的直链或支链烃基,包含2至20个碳原 子,优选2至15个碳原子,特别是2至10(例如2、3或4)个碳原子,例如乙烯 基(乙烯基)、丙烯基(烯丙基)、异丙烯基、丁烯基、乙炔基、丙炔基、丁炔基、 乙炔基、炔丙基、异戊二烯基或己-2-烯基。优选地,烯基团具有一个或两个(特 别优选一个)双键,且炔基基团具有一个或两个(特别优选一个)三键。
此外,术语烷基、烯基和炔基指其中一个或多个氢原子已被替换为卤素原子 (优选F或Cl)的基团,诸如,例如,2,2,2-三氯乙基或三氟甲基。
表述杂烷基是指烷基、烯基或炔基基团,所述基团中一个或多个(优选1至8 个,特别优选为1、2、3或4个)碳原子已被氧、氮、磷、硼、硒、硅或硫原子 (优选由氧、硫或氮原子)取代,或被SO或SO2基取代。表述杂烷基进一步是指羧 酸或由羧酸衍生的基团,如,例如,酰基、酰基烷基、烷氧基羰基、酰氧基、酰 氧基烷基、羧基烷基酰胺、或烷氧基羰基氧基。
优选地,杂烷基基团含有1至12个碳原子和1至8个选自氧、氮和硫(特别 是氧和氮)的杂原子。特别优选地,杂烷基基团含有1至6个(例如,1、2、3或 4)个碳原子和1、2、3或4个(特别是1、2或3)选自氧、氮和硫的杂原子(特别 是氧和氮)。术语C1-C6杂烷基指具有1至6个碳原子和1、2或3个选自O,S和/ 或N(特别是O和/或N)的杂原子的杂烷基基团。术语C1-C4杂烷基指具有1至4个 碳原子和1、2或3个选自O,S和/或N(特别是O和/或N)的杂原子的杂烷基基 团。此外,术语杂烷基是指其中一个或多个氢原子已被替换为卤素原子(优选F 或Cl)的基团。
特别优选的是,表述杂烷基是指如上所定义的(直链或支链)的烷基基团,其 中一个或多个(优选1至6;特别优选1、2、3或4)个碳原子已被氧、硫或氮原 子所替换。上述基团优选含有1至6个(例如,1、2、3或4)个碳原子和1、2、3 或4(特别是1、2或3)选自氧、氮和硫(特别是氧和氮)的杂原子;所述的基团可 以优选被一个或多个(优选为1至6;特别优选1、2、3或4)个氟、氯、溴或碘 原子、或OH、=O、SH、=S、NH2、=NH、N3、CN或NO2基团所取代。
表述亚杂烷基基团指二价的杂烷基基团。
杂烷基基团的例子是下式所示的基团:Ra-O-Ya-、Ra-S-Ya-、Ra-SO-Ya-、Ra- SO2-Ya-、Ra-N(Rb)-Ya-、Ra-CO-Ya-、Ra-O-CO-Ya-、Ra-CO-O-Ya-、Ra-CO-N(Rb)-Ya-、 Ra-N(Rb)-CO-Ya-、Ra-O-CO-N(Rb)-Ya-、Ra-N(Rb)-CO-O-Ya-、Ra-N(Rb)-CO-N(Rc)-Ya- 、Ra-O-CO-O-Ya-、Ra-N(Rb)-C(=NRd)-N(Rc)-Ya-、Ra-CS-Ya-、Ra-O-CS-Ya-、Ra-CS- O-Ya-、Ra-CS-N(Rb)-Ya-、Ra-N(Rb)-CS-Ya-、Ra-O-CS-N(Rb)-Ya-、Ra-N(Rb)-CS-O-Ya- 、Ra-N(Rb)-CS-N(Rc)-Ya-、Ra-O-CS-O-Ya-、Ra-S-CO-Ya-、Ra-CO-S-Ya-、Ra-S-CO- N(Rb)-Ya-、Ra-N(Rb)-CO-S-Ya-、Ra-S-CO-O-Ya-、Ra-O-CO-S-Ya-、Ra-S-CO-S-Ya-、 Ra-S-CS-Ya-、Ra-CS-S-Ya-、Ra-S-CS-N(Rb)-Ya-、Ra-N(Rb)-CS-S-Ya-、Ra-S-CS-O- Ya-、Ra-O-CS-S-Ya-,其中Ra为氢原子、C1-C6烷基、C2-C6烯基或C2-C6炔基基团; Rb为氢原子、C1-C6烷基、C2-C6烯基或C2-C6炔基基团;Rc为氢原子、C1-C6烷基、 C2-C6烯基或C2-C6炔基基团;Rd为氢原子、C1-C6烷基、C2-C6烯基或C2-C6炔基基 团,且Ya为键、C1-C6亚烷基、C2-C6亚烯基或C2-C6亚炔基基团,其中各个杂烷基 基团含有至少一个碳原子,并且一个或多个氢原子可被氟或溴原子取代。
杂烷基基团的具体例子是甲氧基、三氟甲氧基、乙氧基、正丙氧基、异丙氧 基、丁氧基、叔丁氧基、甲氧基甲基、乙氧基甲基、-CH2CH2OH、-CH2OH、- SO2Me、甲氧基乙基、1-甲氧基乙基、1-乙氧基乙基、2-甲氧基乙基或2-乙氧基 乙基、甲氨基、乙氨基、丙氨基、异丙氨基、二甲氨基、二乙氨基、异丙基乙基 氨基、甲氨基甲基、乙基氨基甲基、二异丙基氨基乙基、甲硫基、乙硫基、异丙 硫基、烯醇醚、二甲基氨基甲基、二甲基氨基乙基、乙酰基、丙酰基、丁酰氧 基、乙酰氧基、甲氧羰基、乙氧羰基、丙酰氧基、乙酰氨基或丙酰胺基、羧甲 基、羧乙基或羧丙基、N-乙基-N-甲基氨基甲酰基或N-甲基氨基甲酰基。杂烷基 基团的进一步例子为腈、异腈、氰酸酯、硫代氰酸酯、异氰酸酯、异硫氰酸酯和 烷基腈基。
表述环烷基指的是饱和或部分不饱和的(例如,环烯基基团)环基团,它包含 一个或多个(优选1或2)环,且含有3至14个环碳原子,优选为3至10(特别为 3、4、5、6或7)个环碳原子。表述环烷基进一步指一个或多个氢原子已被氟、 氯、溴或碘原子,或由OH、=O、SH、=S、NH2、=NH、N3或NO2基团所取代的基 团,因此,例如,环酮,例如,环己酮、2-环己烯酮或环戊酮。环烷基团进一步 具体例子为环丙基、环丁基、环戊基、螺[4,5]癸基、降冰片基、环己基、环戊 烯基、环己二烯基、十氢萘基、双环[4.3.0]壬基、四氢化萘、环戊基环己基、 氟代环己基或环己-2-烯基。
表述杂环烷基是指如上所述的环烷基基团,且所述基团中一个或多个(优选 为1、2或3)碳原子已被氧、氮、硅、硒、磷或硫原子(优选为氧、硫或氮原 子),或被SO或SO2基取代。杂环烷基基团具有优选1或2个环,各环含有3至 10个(特别是3、4、5、6或7个)环原子(优选选自C、O、N和S)。表述杂环烷 基进一步是指被氟、氯、溴或碘原子或被OH、=O、SH、=S、NH2、=NH、N3或NO2基团取代的基团。例子为哌啶、脯氨基(prolinyl)、咪唑烷基、哌嗪基、吗啉 基、乌洛托品基(urotropinyl)、吡咯烷基、四氢噻吩基、四氢吡喃基、四氢呋 喃基或2-吡唑啉基团,以及内酰胺、内酯、环状酰亚胺和环状酸酐。
表述烷基环烷基指同时含有如上文所述的环烷基以及烷基、烯基或炔基的基 团,例如烷基环烷基、环烷基烷基、烷基环烯基、烯基环烷基和炔基环烷基基 团。烷基环烷基团优选含有一个环烷基基团(其中包括一个或两个环,各环具有3 至10个(特别是3、4、5、6或7)个环碳原子)和一个或两个具有1或2至6个碳 原子的烷基、烯基或炔基基团(特别是烷基基团)。
表述杂烷基环烷基是指如上所述的,所述基团中一个或多个(优选为1、2或 3)碳原子已被氧、氮、硅、硒、磷或硫原子(优选为氧、硫或氮原子),或SO或 SO2基取代。杂烷基环烷基基团优选含有1或2个环,各环包括3至10个(特别是 3、4、5、6或7)个环原子,并含有一个或两个具有1或2至6个碳原子的烷 基、烯基、炔基或杂烷基基团(特别是烷基或杂烷基基团)。所述基团的例子为烷 基杂环烷基、烷基杂环烯基、烯基杂环烷基、炔基杂环烷基、杂烷基环烷基、杂 烷基杂环烷基和杂烷基杂环烯基,所述的环状基团可以是饱和的或单-、双-或 三-不饱和的。
表述芳基指包含一个或多个环的芳香基团,各环具有6至14个环碳原子, 优选6至10(特别是6)个环碳原子。表述芳基进一步是指被氟、氯、溴或碘原子 或被OH、SH、NH2、N3或NO2基团所取代的基团。例子为苯基、萘基、联苯基、2- 氟苯基、苯胺基(anilinyl)、3-硝基苯基或4-羟基苯基。
表述“杂芳基”指包含一个或多个环的芳香基团,各环含有5至14个环原 子,优选为5至10(特别是5或6或9或10)个环原子,且包含一个或多个(优选 1、2、3或4)氧、氮、磷或硫环原子(优选O、S或N)。表述“杂芳基”进一步是 指被氟、氯、溴或碘原子或被OH、SH、NH2、N3或NO2基团取代的基团。例子为吡 啶基(例如4-吡啶基)、咪唑基(例如2-咪唑基)、苯基吡咯基(例如3-苯基吡咯 基)、噻唑基、异噻唑基、1,2,3-三唑基、1,2,4-三唑基、噁二唑基、噻二唑 基、吲哚基、吲唑基、四唑基、吡嗪基、嘧啶基、哒嗪基、噁唑基、异噁唑基、 三唑基、四唑基、异噁唑基、吲唑基、吲哚基、苯并咪唑基、苯并噁唑基、苯并 异噁唑基、苯并噻唑基、哒嗪基、喹啉基、异喹啉基、吡咯基、嘌呤基、咔唑 基、吖啶基、嘧啶基、2,3’-二呋喃基、吡唑基(例如3-吡唑基)和异喹啉基。
表述芳烷基是指同时含有如上述的定义的芳基以及烷基、烯基、炔基和/或 环烷基基团的基团,如,例如,芳基烷基、芳基烯基、芳基炔基、芳基环烷基、 芳基环烯基、烷基芳基环烷基和烷基芳基环烯基团。芳烷基的具体例子是甲苯、 二甲苯、均三甲苯、苯乙烯、苄基氯、邻-氟甲苯、1H-茚、四氢化萘、二氢化 萘、茚满酮、苯基环戊基、枯烯、环己基苯基、芴和二氢化茚。芳烷基基团优选 含有一个或两个芳香的环体系(特别是1或2个环),每一个含有6至10个碳原 子和一个或两个含有1或2到6个碳原子的烷基、烯基和/或炔基基团,和/或含 5或6个环碳原子的环烷基基团。
表述杂芳烷基指同时分别具有如上述定义的芳基或杂芳基,并具有烷基、烯 基、炔基和/或杂烷基和/或环烷基和/或杂环烷基基团的基团。杂芳烷基基团优 选含有一个或两个芳香的环体系(特别是1或2个环),每一个环体系含有5或6 至9或10个环碳原子以及一个或两个含有1或2至6个碳原子的烷基、烯基和/ 或炔基基团,和/或一个或两个含有1至6个碳原子以及1、2或3个选自O、S 和N的杂原子的杂烷基基团,和/或一个或两个分别含有5或6个环碳原子的环 烷基基团,和/或一个或两个各含有5或6个环原子(其中包括1、2、3或4个 氧、硫或氮原子)的杂环烷基基团。
例子有芳杂烷基、芳杂环烷基、芳杂环烯基、芳基烷基杂环烷基、芳烯基杂 环烷基、芳炔基杂环烷基、芳基烷基杂环烯基、杂芳基烷基、杂芳烯基、杂芳炔 基、杂芳杂烷基、杂芳环烷基、杂芳环烯基、杂芳杂环烷基、杂芳杂环烯基、杂 芳基烷基环烷基、杂芳基烷基杂环烯基、杂芳杂烷基环烷基、杂芳杂烷基环烯基 和杂芳杂烷基杂环烷基基团,上述的环基团可以为饱和的或单-、二-或三-不饱 和的。具体例子是四氢异喹啉、苯甲酰基、2-或3-乙基吲哚基、4-甲基吡啶、2- ,3-或4-甲氧基苯基、4-乙氧基苯基、2-,3-或4-羧基苯基烷基基团。
如前所述,表述环烷基、杂环烷基、烷基环烷基、杂烷基环烷基、芳基、杂 芳基、芳烷基和杂芳烷基也指被氟、氯、溴或碘原子或被OH、=O、SH、=S、 NH2、=NH、N3或NO2基团取代的基团。
表述“任选取代”指基团可以被氟、氯、溴或碘原子或被OH、=O、SH、=S、 NH2、=NH、N3或NO2基团任选地取代。上述表述进一步指可被一个、两个、三个或 更多个未取代的C1-C10烷基、C2-C10烯基、C2-C10炔基、C1-C10杂烷基、C3-C18环烷 基、C2-C17杂环烷基、C4-C20烷基环烷基、C2-C19杂烷基环烷基、C6-C18芳基、C1-C17杂芳基、C7-C20芳烷基或C2-C19杂芳烷基基团所取代的基团。上述表述进一步指被 一个、两个、三个或更多个未取代的C1-C6烷基、C2-C6烯基、C2-C6炔基、C1-C6杂 烷基、C3-C10环烷基、C2-C9杂环烷基、C7-C12烷基环烷基、C2-C11杂烷基环烷基、 C6-C10芳基、C1-C9杂芳基、C7-C12芳烷基或C2-C11杂芳烷基基团所取代的基团。
特别优选地,在基团Ar1、Ar2、Ar3、Ar4和Ar5中,表述“任选取代的”指基 团被一个、两个、三个独立选自下组的基团所取代:卤素原子、羟基、如式-O- 烷基(例如,-O-C1-6烷基如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O- tBu)所示的基团、-NH2、-NR5aR6a(其中R5a和R6a各自独立地为氢原子或烷基基团如 C1-6烷基基团)、-SO2NH2、-CONH2、-CN、-烷基(例如-C1-6烷基、-CF3)、-SH、-S-烷 基(例如,-S-C1-6烷基)。
最优选地,在基团Ar1、Ar2、Ar3、Ar4和Ar5中,表述“任选取代的”指基团 被一个、两个、三个独立选自下组的基团所取代:F、Cl、羟基、如式-O-C1-6烷基 (特别是-O-C1-4烷基如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O-tBu) 所示的基团、和-C1-6烷基(例如-C1-4烷基如-CH3或-CF3)。
特别优选地,在基团Ar6中,表述“任选取代的”指基团被一个、两个、三 个独立选自下组的基团所取代:卤素原子、羟基、如式-O-烷基(例如,-O-C1-6烷 基如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O-tBu)所示的基团、- NH2、-NR5aR6a(其中R5a和R6a各自独立地为氢原子或烷基基团如C1-6烷基基团)、- SO2NH2、-CONH2、-CN、-烷基(例如-C1-6烷基、-CF3)、-SH、-S-烷基(例如,-S-C1-6烷基),和NO2。
最优选地,在基团Ar6中,表述“任选取代的”指基团被一个、两个、三个 选自下组的基团所取代:F、Cl、羟基、-NH2、-NO2、如式-O-C1-6烷基(特别是-O- C1-4烷基如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O-tBu)所示的基 团、和-C1-6烷基(例如-C1-4烷基如-CH3或-CF3)。
术语卤素指氟、氯、溴或碘。
根据一个优选的实施例,所有本文所述的烷基、烯基、炔基、杂烷基、芳 基、杂芳基、环烷基、杂环烷基、烷基环烷基、杂烷基环烷基、芳烷基和杂芳烷 基基团可以各自独立地为任选取代的。
当芳基、杂芳基、环烷基、烷基环烷基、杂烷基环烷基、杂环烷基、芳烷基 或杂芳烷基基团含有多于一个环时,所述的环可以是通过单键或双键互相连接 的,或所述的环可以是环合的。
由于它们的取代,式(I)的化合物可含有一个或多个手性中心。因此,本发 明既包括所有的纯的对映异构体,也包括所有的纯的非对映异构,及其以任意混 合比例混合的混合物。此外,本发明还包括通式(I)的化合物的所有顺式/反式异 构体及其混合物。本发明还包括式(I)的化合物的所有互变异构形式。
优选地,当Ar4为无,L3同样为无。
更优选地,当Ar5为无,L4同样为无。
优选地,Ar1为任选取代的1,4-亚苯基,或任选取代的具有5个环原子(其中 包括1、2或3个选自下组的杂原子:氧、硫和氮)的1,3-亚杂芳基。
更优选地,Ar1为任选取代的1,4-亚苯基。
优选地,Ar2为任选取代的1,4-亚苯基,或任选取代的具有5个环原子(其中 包括1、2或3个选自下组的杂原子:氧、硫和氮)的1,3-亚杂芳基;
更优选地,Ar2为任选取代的1,4-亚苯基。
优选地,Ar2为任选取代的1,4-亚苯基,或任选取代的具有5个环原子(其中 包括1、2或3个选自下组的杂原子:氧、硫和氮)的1,3-亚杂芳基。
更优选地,Ar3为任选取代的1,4-亚苯基。
优选地,Ar4为任选取代的1,4-亚苯基,或任选取代的具有5个环原子(其中 包括1、2或3个选自下组的杂原子:氧、硫和氮)的1,3-亚杂芳基。
更优选地,Ar4为任选取代的1,4-亚苯基。
优选地,Ar5为任选取代的1,4-亚苯基,或任选取代的具有5个环原子(其中 包括1、2或3个选自下组的杂原子:氧、硫和氮)的1,3-亚杂芳基;
更优选地,Ar5为任选取代的1,4-亚苯基。
更优选地,Ar4为无。
更优选地,Ar5为无。
术语“具有5个环原子(其中包括1、2或3个选自下组的杂原子:氧、硫和 氮)的1,3-亚杂芳基”特别优选地指以下的基团之一:

其中A为O、S或NH;U为N或CH;V为N或CH;W为N或CH;且X为N或 CH。
更优选地,L1为如式-CONH-、-NHCO-、-SO2NH-、-NHSO2-、-CH=CH-、- CR3=CR4-所示的基团,或任选取代的具有5个环原子(其中包括1、2或3个选自 下组的杂原子:氧、硫和氮)的亚杂芳基,其中R3和R4各自独立地为C1-6烷基基 团。
更优选地,L2为如式-CONH-、-NHCO-、-SO2NH-、-NHSO2-、-CH=CH-、- CR3=CR4-所示的基团,或任选取代的具有5个环原子(其中包括1、2或3个选自 下组的杂原子:氧、硫和氮)的亚杂芳基,其中R3和R4各自独立地为C1-6烷基基 团。
更优选地,L3为无,或如式-CONH-、-NHCO-、-SO2NH-、-NHSO2-、-CH=CH- 、-CR3=CR4-所示的基团,或任选取代的具有5个环原子(其中包括1、2或3个选 自下组的杂原子:氧、硫和氮)的亚杂芳基,其中R3和R4各自独立地为C1-6烷基基 团。
更优选地,L4为无,或如式-CONH-、-NHCO-、-SO2NH-、-NHSO2-、-CH=CH- 、-CR3=CR4-所示的基团,或任选取代的具有5个环原子(其中包括1、2或3个选 自下组的杂原子:氧、硫和氮)的亚杂芳基,其中R3和R4各自独立地为C1-6烷基基 团。
更优选地,L1为NHCO(其中氮原子与Ar2相连接),或如下式所示的基团:

(其中NH基团与Ar1连接),其中R30为氢原子或C1-3烷基基团。
特别优选地,L1为NHCO(其中氮原子与Ar1相连接)。
更为优选地,L2为NHCO(其中氮原子与Ar2相连接),或如下式所示的基团:

(其中NH基团与Ar2连接),其中R30为氢原子或C1-3烷基基团。
特别优选地,L2为NHCO(其中氮原子与Ar1相连接)。
更加优选地,L3为无或如下式所示的基团:

(其中NH基团与Ar3连接),其中R30为氢原子或C1-3烷基基团。
更优选地,L4为无或NHCO(其中氮原子与Ar4相连接)。
更为优选地,R30为氢原子。
更优选地,R1为氢原子、卤素原子,或如下式-OH、-NH2、-COOH、-SO2NH2、- CONH2、-NO2、-CN、-烷基(例如-CF3)、-O-烷基、-O-CO-烷基、-NH-烷基、-NH- CO-烷基所示的基团,或任选取代的具有5个环原子(其中包括1、2、3或4个选 自下组的杂原子:氧、硫和氮)的杂芳基,或任选取代的具有5个环原子(其中包 括1、2、3或4个选自下组的杂原子:氧、硫和氮)的杂环烷基基团。
更优选地,R2为氢原子、卤素原子,或如下式-OH、-NH2、-COOH、-SO2NH2、- CONH2、-NO2、-CN、-烷基(例如-CF3)、-O-烷基、-O-CO-烷基、-NH-烷基、-NH- CO-烷基所示的基团,或任选取代的具有5个环原子(其中包括1、2、3或4个选 自下组的杂原子:氧、硫和氮)的杂芳基,或任选取代的具有5个环原子(其中包 括1、2、3或4个选自下组的杂原子:氧、硫和氮)的杂环烷基基团。
作为基团R1和R2的、优选的任选取代的具有5个环原子(其中包括1、2、3 或4个选自下组的杂原子:氧、硫和氮)的杂芳基和任选取代的具有5个环原子 (其中包括1、2、3或4个选自下组的杂原子:氧、硫和氮)的杂环烷基基团的例 子,是羧酸的电子等排体,例如下式基团:

所有上述基团可以任选地被进一步取代。
特别优选地,R1为如下式-NH2、-NO2、COOR11、或-CONR12R13所示的基团;其中 R11、R12和R13各自独立地为氢原子或C1-6烷基基团;更优选地,R1为如式-COOH所 示的基团。
进一步特别优选地,R2为如下式-NH2、-NO2、COOR11a、或-CONR12aR13a所示的基 团;其中R11a、R12a和R13a各自独立地为氢原子或C1-6烷基基团;更优选地,R2为如 式-NH2或-NO2所示的基团。
进一步特别优选地,R1为被羟基取代的、具有5个环原子(其中包括1、2、3 或4个选自下组的杂原子:氧、硫和氮)的亚杂芳基。
进一步特别优选地,R2为被羟基取代的、具有5个环原子(其中包括1、2、3 或4个选自下组的杂原子:氧、硫和氮)的亚杂芳基。
特别优选的是如式(I)所示的化合物,或其药学上可接受的盐、溶剂化物或 水合物,或其药学上可接受的制剂
R1-Ar1-L1-Ar2-L2-Ar3-L3-Ar4-L4-Ar5-R2
(I)
其中,
Ar1为任选取代的1,4-亚苯基;
Ar2为任选取代的1,4-亚苯基;
Ar3为任选取代的1,4-亚苯基;
Ar4为无或任选取代的1,4-亚苯基;
Ar5为无或任选取代的1,4-亚苯基;
L1为如式-CONH-、-NHCO-、-SO2NH-或-NHSO2-所示的基团,或如下式所示的 基团:

(其中NH基团与Ar1相连);
L2为如式-CONH-、-NHCO-、-SO2NH-或-NHSO2-所示的基团;
L3为无,或如式-CONH-、-NHCO-、-SO2NH-或-NHSO2-所示的基团,或如下式 所示的基团:

(其中NH基团与Ar3相连);
L4为无,或如式-CONH-、-NHCO-、-SO2NH-或-NHSO2-所示的基团;
R30为氢原子,或C1-3烷基基团(特别优选地为氢原子);
R1为如下式-NH2、-NO2、COOR11、或-CONR12R13所示的基团;其中R11、R12和R13各自独立地为氢原子或C1-6烷基基团(特别优选地,R1为如式-COOH所示的基团); 和
R2为如下式-NH2、-NO2、COOR11a、或-CONR12aR13a所示的基团;其中R11a、R12a和 R13a各自独立地为氢原子或C1-6烷基基团(特别选地,R2为如式-NH2或-NO2所示的基 团)。
其中,优选地,L1为如式-CONH-、-NHCO-、-SO2NH-或-NHSO2-所示的基团, 且L3为无,或如下式所示的基团:

(其中NH基团与Ar3相连)。
更加优选的为如式(II)所示的化合物
R1-Ar1-L1-Ar2-L2-Ar3-R2
(II)
其中Ar1、Ar2、Ar3、L1、L2、R1以及R2如上文中所述。
更加优选的为如式(III)所示的化合物

其中,
n为0、1、2、3或4;
m为0、1、2、3或4;
p为0、1、2、3或4;
基团R21各自独立地选自卤素原子、羟基、如式-O-烷基(例如,-O-C1-6烷基 如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O-tBu)所示的基团、- NH2、-NR5aR6a(其中R5a和R6a各自独立地为氢原子或烷基基团如C1-6烷基基团)、- SO2NH2、-CONH2、-CN、-烷基(例如-C1-6烷基、-CF3)、-SH、-S-烷基(例如,-S-C1-6烷基);
基团R22各自独立地选自卤素原子、羟基、如式-O-烷基(例如,-O-C1-6烷基 如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O-tBu)所示的基团、- NH2、-NR5aR6a(其中R5a和R6a各自独立地为氢原子或烷基基团如C1-6烷基基团)、- SO2NH2、-CONH2、-CN、-烷基(例如-C1-6烷基、-CF3)、-SH、-S-烷基(例如,-S-C1-6烷基);
基团R23各自独立地选自卤素原子、羟基、如式-O-烷基(例如,-O-C1-6烷基 如-OMe、-OEt、-O-nPr、-O-iPr、-O-nBu、-O-iBu或-O-tBu)所示的基团、- NH2、-NR5aR6a(其中R5a和R6a各自独立地为氢原子或烷基基团如C1-6烷基基团)、- SO2NH2、-CONH2、-CN、-烷基(例如-C1-6烷基、-CF3)、-SH、-S-烷基(例如,-S-C1-6烷基);和
R1、R2、L1和L2如上所述。
更加优选的为如式(IV)所示的化合物

其中,
R5为如式-O-C1-6烷基所示的基团;
R6为羟基基团;
R7为如式-O-C1--6烷基所示的基团;和
R8为氢原子、烷基、烯基、炔基、杂烷基、环烷基、杂环烷基、烷基环烷 基、杂烷基环烷基、芳基、杂芳基、芳烷基或杂芳烷基基团。
更加优选地,R8为氢原子或如下式所示的基团:

其中R9为COOH或CONH2,且R10为COOH或CONH2。
更加优选地,R5为如式-O-C1-4烷基所示的基团,且R7为如式-O-C1-4烷基所示 的基团。
更加优选的为如式(V)所示的化合物,或其药学上可接受的盐、溶剂化物或 水合物,或其药学上可接受的制剂:

其中,
R51为氢原子,或C1-6烷基基团;
R52为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R53为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R54为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R55为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
D为N或CR56;
E为N或CR57;
G为N或CR58;
M为N或CR59;
R56为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R57为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R58为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R59为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;和
Ar6为(被一个、两个或更多取代基如R2、R8或NHR8)任选取代的苯基、或(被 一个、两个或更多取代基如R2、R8或NHR8)任选取代的具有5个或6个环原子的杂 芳基,其中,所述的环原子中具有1、2、3或4个选自氧、硫和氮的杂原子。
特别优选的为如式(V)所示的化合物,其中:
R51为氢原子,或C1-4烷基基团;
R52为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R53为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R54为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R55为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
D为N或CR56;
E为N或CR57;
G为N或CR58;
M为N或CR59;
R56为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R57为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R58为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;和
R59为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-4烷基的基团。
特别优选地,D,E,G和M中仅有一个或两个(特别是仅有一个)为N。
更加优选的为如式(VI)所示的化合物,或其药学上可接受的盐、溶剂化物或 水合物,或其药学上可接受的制剂:

其中,
R51为氢原子,或C1-6烷基基团;
R53为F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团(特别优选地 为如式-O-C1-6烷基的基团);
D为N或CR56;
R56为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R57为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R58为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R59为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;和
Ar6为(被一个、两个或更多取代基如R2、R8或NHR8)任选取代的苯基,或(被 一个、两个或更多取代基如R2、R8或NHR8)任选取代的具有5个或6个环原子的杂 芳基,其中,所述的环原子中具有1、2、3或4个选自氧、硫和氮的杂原子。
特别优选的为如式(VI)所示的化合物,其中:
R51为氢原子,或C1-4烷基基团;
R53为F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团(特别优选地 为如式-O-C1-4烷基的基团);
D为N或CR56;
R56为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R57为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R58为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;和
R59为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团。
更加优选的为如式(VII)所示的化合物,或其药学上可接受的盐、溶剂化物 或水合物,或其药学上可接受的制剂:

其中,
R51为氢原子,或C1-6烷基基团;
R53为F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团(特别优选地 为如式-O-C1-6烷基的基团);
D为N或CR56;
R56为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R57为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R58为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R59为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R60为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;
R61为氢原子、F、Cl、羟基基团、C1-6烷基基团或如式-O-C1-6烷基的基团;和
R8为氢原子、烷基、烯基、炔基、杂烷基、环烷基、杂环烷基、烷基环烷 基、杂烷基环烷基、芳基、杂芳基、芳烷基或杂芳烷基基团。
特别优选的为如式(VII)所示的化合物,其中:
R51为氢原子,或C1-4烷基基团;
R53为F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团(特别优选地 为如式-O-C1-4烷基的基团);
D为N或CR56;
R56为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R57为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R58为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R59为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;
R60为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团;和
R61为氢原子、F、Cl、羟基基团、C1-4烷基基团或如式-O-C1-4烷基的基团。
更加优选地,R8为氢原子或如下式所示的基团:

其中R9为COOH或CONH2,且R10为COOH或CONH2。
特别优选的为以下化合物:

孢囊菌酰胺A(1);

孢囊菌酰胺B(2);

孢囊菌酰胺C(3);
孢囊菌酰胺D(4);和

孢囊菌酰胺E(5)(R’为NH2或OH且R”为NH2或OH)。
此外,特别优选的为以下化合物:

孢囊菌酰胺F(6),孢囊菌酰胺G(7);和

孢囊菌酰胺H(8)。
此外,特别优选地为以下化合物:



本发明还提供了含有本发明所述的一种或多种化合物或其药学上可接受的 盐、溶剂合物或水合物,以及任选的一种或多种载体物质和/或一种或多种佐剂 的药物组合物。
本发明还提供了一种所述的化合物或药物组合物用于治疗和/或预防细菌感 染的用途,特别是由于大肠杆菌、绿脓杆菌、鲍曼不动杆菌或其它革兰氏阴性细 菌,以及革兰氏阳性细菌导致的感染。
更加优选地,本发明提供了化合物用于治疗和/或预防细菌感染,特别是由 绿脓杆菌和其他革兰氏阴性菌引起的感染。
本发明的另一目的是提供一种如本文所述的化合物或药物组合物用于制备治 疗和/或预防细菌感染的药物组合物的用途,特别是由选定的革兰氏阴性细菌和 革兰氏阳性细菌所引起的感染。
具有足够碱性的化合物的药理学上可接受的盐的实例,为生理学上可接受的 无机酸如盐酸、氢溴酸、硫酸和磷酸的盐;或有机酸如甲磺酸、对甲苯磺酸、乳 酸、乙酸、三氟乙酸、柠檬酸、琥珀酸、富马酸、马来酸和水杨酸的盐。此外, 足够酸性的化合物可形成碱金属盐或碱土金属盐,例如钠、钾、锂、钙或镁的 盐;铵盐;或有机碱盐,例如甲胺、二甲胺、三甲胺、三乙胺、乙二胺、乙醇 胺、胆碱氢氧化物、甲基葡胺、哌啶、吗啉,三(2-羟乙基)胺、赖氨酸或精氨酸 的盐;所有这些也都是本文所述的化合物的盐的进一步实例。本发明所述的化合物可以是溶剂合物,优选水合物。在生产的过程中,或由于最初无水的化合物的 吸湿性质导致的结果,可能会发生水合/水化。其溶剂合物和/或水合物可以以例 如固体或液体形式存在。
本文所述的化合物,其药理学上可接受的盐,溶剂合物和水合物,以及制剂 和药物组合物的各自治疗用途,也属于本发明的范围之内。
如本发明所述的药物组合物包含至少一种本发明所述化合物和任选的一种或 多种载体物质和/或佐剂。
如上所述,包含本发明所述的化合物、其溶剂合物,盐或制剂的治疗上有用 的药剂,也包括在本发明的范围内。在一般情况下,可通过使用本领域已知的公 知的和可接受的方式,将本发明所述的化合物单独施用地或与任何其它治疗剂组 合施用。
对于口服给药,所述的治疗上有用的药剂可以通过下列途径之一施用:口 服,例如作为片剂、锭剂、包衣片剂、丸剂、半固体、软或硬胶囊(例如软或硬 明胶胶囊)、水性或油性溶液剂、乳剂、混悬剂或糖浆剂,肠胃外给药包括静脉 内、肌内和皮下注射,例如作为可注射溶液或悬浮液,直肠中作为栓剂,通过吸 入或吹入(例如作为粉末制剂,如微晶或作为喷雾剂(例如液体气雾剂)),经皮, 例如通过透皮递送系统(TDS),例如包含活性成分的膏药,或鼻内。为了生产这 样的片剂、丸剂、半固体、包衣片剂、糖衣丸和硬胶囊例如明胶胶囊,治疗有用 的产物可以与药学惰性的无机或有机的赋形剂混合,例如,乳糖、蔗糖、葡萄糖、明胶、麦芽、硅胶、淀粉或其衍生物、滑石、硬脂(stearinic)酸或其盐、 脱脂奶粉等。为生产软胶囊,可以使用赋形剂,例如,植物油、石油、动物油或 合成油、蜡、脂肪和多元醇。用于生产液体溶液、乳剂或混悬剂或糖浆剂,可以 使用以下物质作为赋形剂,例如水、醇、含水盐水、葡萄糖水溶液、多元醇、甘 油、脂质、磷脂、环糊精、植物油、石油、动物油或合成油。特别优选的为脂 质,而更优选的是磷脂(优选天然来源的;特别优选具有300至350nm之间的粒 径),优选在磷酸盐缓冲盐水中(pH值=7-8,优选7.4)。用于栓剂,可以使用的 赋形剂是例如,植物油、石油、动物油或合成油、蜡、脂肪和多元醇。用于气雾 剂制剂,可以使用对于此目的合适的压缩气体,例如,氧、氮和二氧化碳。该药 学上有用的药剂也可以含有添加剂用于保存、稳定,如UV稳定剂、乳化剂、甜 味剂、芳香剂、盐类(用于改变渗透压)、缓冲剂、包衣添加剂和抗氧化剂。
通常,口服或肠胃外给药于体重约80公斤的成人的情况,合适的日剂量为 约1mg至约10,000mg,优选为约5mg至约1,000mg,尽管在有提示时其上限 可超标。日剂量可以为单剂量或分剂量,或对于肠胃外给药,它可以通过连续输 注或皮下注射给药。
本发明的化合物可以通过发酵(例如,通过菌株MCy8071DSM27004的发酵) 或通过本领域技术人员已知的化学合成应用程序来制备。
例如,本发明的化合物可以通过以下流程制备:
从各个被任选取代的构建单元构建单元(例如Ar1、Ar2、Ar3、Ar4和Ar5)起 始,这些构建单元构建单元可使用本领域技术人员所知的酰氯或偶联试剂互相连 接,例如按照下列反应方案:
R1-Ar1-NH2+HOOC-Ar2-L2-Ar3-L3-Ar4-L4-Ar5-R2
R1-Ar1-NH2+HO3SC-Ar2-L2-Ar3-L3-Ar4-L4-Ar5-R2
如果L1、L2、L3和/或L4为式-CH=CH-基团(或其他烯烃基团),各个被任选 取代的构建单元构建单元(例如Ar1、Ar2、Ar3、Ar4和Ar5)可以通过Wittig或 Horner反应互相连接,例如:按照下列反应方案:
R1-Ar1-CHO+BrPh3P-CH2-Ar2-L2-Ar3-L3-Ar4-L4-Ar5-R2
R1-Ar1-CHO+(EtO)2OPCH2-Ar2-L2-Ar3-L3-Ar4-L4-Ar5-R2
如果L1、L2、L3和/或L4为杂环烷基或杂芳基,各个被任选取代的构建单元构 建单元(例如Ar1、Ar2、Ar3、Ar4和Ar5)可以使用类似的反应条件互相连接。
孢囊菌酰胺生物合成基因簇的鉴定:
孢囊菌酰胺生产菌的基因组用鸟枪法测序法进行测序。作为孢囊菌酰胺的主 要构建单元构建单元是非蛋白原性的氨基酸对-氨基苯甲酸(PABA),对氨基苯甲 酸合酶(查询号NP_415614)被用作查询项,以识别在Cbv34基因组中推定的孢囊 菌酰胺生物合成簇。重要的是,对-氨基苯甲酸合酶的同源物可以被鉴定出 (CysD,图12和表A),它在计算机预测的约48KB大的NRPS基因簇(图12,分 配:表A)中与非核糖体肽合成酶(CysG,H和K)一起构成一个操纵子。在此NRPS 簇中的基因用pfam、NCBI BLAST和phyre2进行分析。除了对氨基苯甲酸合酶的 同源物,在该基因簇中还可以发现两个其他的PABA生物合成酶:氨基脱氧分支 酸裂合酶(CysI)和3-脱氧-d-阿拉伯-庚酮糖-7-磷酸(DAHP)合成酶(CysN)。DAHP 合成酶(CysN)是用于生产莽草酸和分支酸的关键酶。在莽草酸途径的主干线,D- 赤藓糖-4-磷酸和磷酸烯醇丙酮酸盐(DAHP合成酶)经由莽草酸转化为分支酸。 CysI和CysD使得能够从分支酸直接生物合成PABA。此外,所述的簇包含对氨基 苯甲酸N-氧合酶的同源物(CysR)。
图12显示了本发明的孢囊菌酰胺的生物合成簇。
能够合成选自下组的孢囊菌酰胺:孢囊菌酰胺A、B、C、D、E、F、G和H的 重组生物合成簇,其中所述的簇包括如SEQ ID NO:40至73的所有多肽,或其 功能性变体。
如本文所用的术语“功能性变体”是指具有与本文所述的多肽序列有至少 85%,90%,95%或99%序列相同的多肽。一个多肽的“功能性变体”可保留被确认 为天然多肽中保守的氨基酸残基,和/或可具有非保守氨基酸残基。氨基酸可以 是,相对于天然多肽而言的取代(不同氨基酸),插入或缺失,但是变体相比本文 中所描述的多肽具有大致相似的(酶)活性或功能。“功能性变体”可以在自然界 中找到,或为其工程化的突变体(重组体)。
术语“同源性”指用于衡量序列的相似性或关系的一种序列特性。同源性是 通过相同残基的数目除以残基的总数,并将结果乘以100测得的。
如本文所用,术语“蛋白质”,“多肽”,“肽”用于定义排成直链的,两 个或多个氨基酸残基构成的有机化合物,其中在所述有机化合物的单个氨基酸之 间通过肽键(即相邻氨基酸残基之间的酰胺键)连接。按照惯例,一种蛋白质的一 级结构的描述是从氨基端(N)开始并朝向羧基端(C)。
如本文所用,“包含”,“包括”,“含有”,“特征在于”,以及它们的 语法等同词是包含性的或开放式的术语,不排除另外的、未陈述的要素或方法步 骤。例如,“包括”等可被解释为包括更具限制性的术语“由......组成”。
如本文所用,“由......组成”不包括在权利要求中没有指定的任何元素 (元件)、步骤或成分。
当商品名在本文中被使用,则独立地包括了商品名的产品配方,仿制药,和 商品名产品的活性药物成分。
一般而言,除非另外定义,本文所用的技术和科学术语具有通常本发明所属 领域的普通技术人员理解的相同的含义,并与一般的教科书和字典一致。
优选地,本发明的NRPS酶是非天然存在的NRPS。本发明的NRPS也可以是杂 合型NRPS,它包括来自于两个或更多的NRPS或来自于一个或多个聚酮化合物合 成酶(PKSs)的模块、结构域和/或其部分,或其功能性变体。
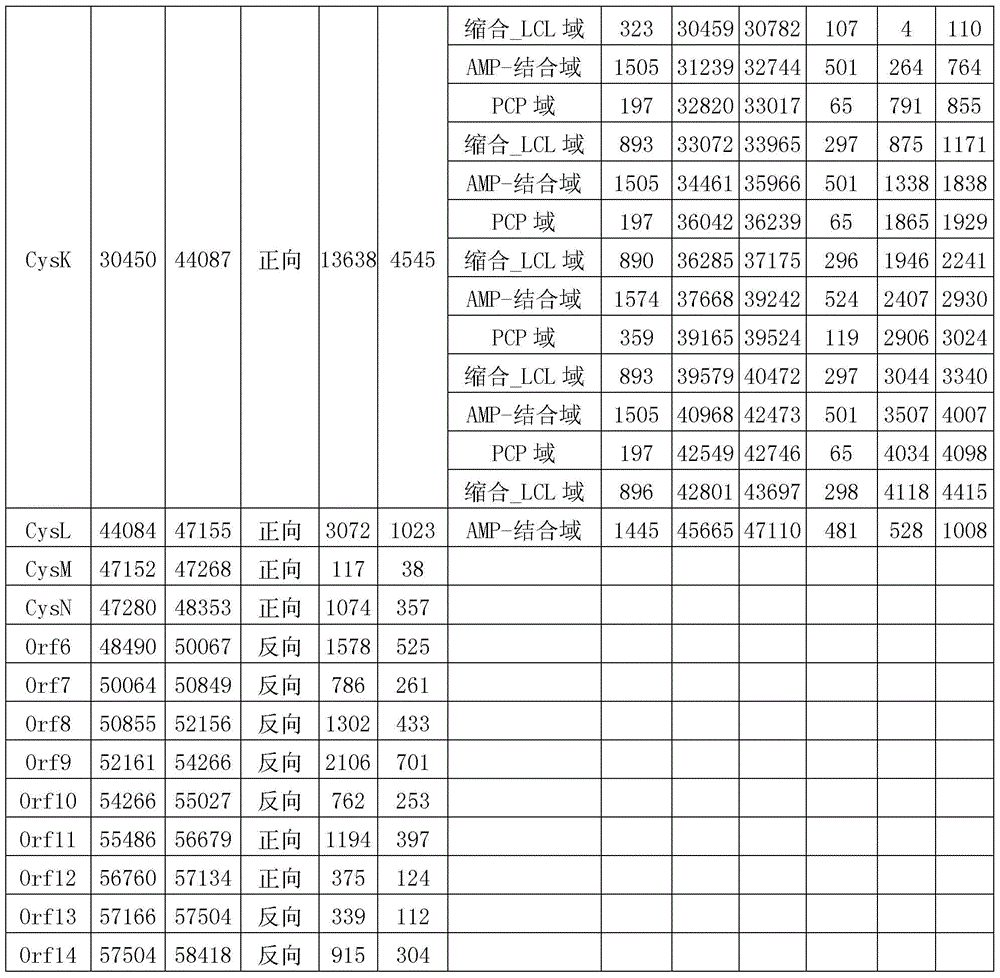
本发明的所述孢囊菌酰胺的生物合成簇,优选包括表A中的元件。
表A:本发明的孢囊菌酰胺基因簇。基因和NRPS域的注解对应于SEQ ID NO. 1的基因簇序列。

本发明还提供了分离的、合成的或重组的用于编码本发明的NRPS的核酸。 所述核酸包括包括本发明的NRPS的一部分或全部的核酸,进一步包括调控序列 (如启动子和翻译的起始序列和终止序列)的核酸,以及可以进一步包括促进维持 在宿主细胞中稳定的序列,即提供复制起点的功能的序列或通过同源重组促进整 合入宿主细胞染色体或其它DNA的序列。这些NRPSs可以用作研究工具或作为重 组NRPS或PKS簇的模块。
优选地,本发明涉及分离的,合成的或重组的核酸,其中包括:
(i)编码孢囊菌酰胺生物合成簇的序列,其中所述的序列与SEQ ID NO.1 全长序列具有至少85%、90%、95%、96%、97%、98%、98.5%、99%、或99.5%至 100%的同源性;
(ii)编码NRPS的序列,其中所述的序列与SEQ ID NO.8、9、12或13具 有至少85%、90%、95%、96%、97%、98%、98.5%、99%,或99.5%至100%的同源 性;
(iii)与(i)或(ii)中的任何核酸的全长序列完全互补的序列;或
(iv)编码如SEQ ID NO.46,47,50或51中任一多肽的序列。
如本文所用,词语“核酸”或“核酸序列”是指寡核苷酸、核苷酸、多核苷 酸、或任何其片段,也指基因组或合成来源的DNA,它可以是单链或双链的,并 且可以代表有义链或反义链,可以是天然的或合成来源的。“寡核苷酸”包括单 链多脱氧核苷酸,或者两条互补的多脱氧核苷酸链,它们可以是化学合成的。这 种合成的寡核苷酸没有5’磷酸,并因此在激酶的存在下不通过ATP添加一个磷酸 根,将不会连接于另一个寡核苷酸。合成的寡核苷酸可以连接于尚未去磷酸化的 片段。编码特定多肽或蛋白质的“编码序列”或“核苷酸序列”是这样的核酸序 列,它被置于适当调控序列的控制下,并被转录和翻译成多肽或蛋白质。用于实 施本发明的核酸可以分离自多种不同来源,被遗传改造,被扩增和/或被表达/被重组。用于核酸操作的技术,如,例如,亚克隆,标记探针(例如,使用Klenow 聚合酶的随机引物标记、切口平移、扩增),测序,杂交等,在科学和专利文献 中有很好的描述,参见例如,Sambrook编,《分子克隆:实验手册(第二版)》, VoI.1-3,冷泉港实验室,(1989);《分子生物学实验指南(CURRENT PROTOCOLS IN MOLECULAR BIOLOGY)》,Ausubel编辑,John Wiley&Sons,Inc.,纽约 (1997年);《生物化学与分子生物学实验室技术:核酸探针杂交》,第一部分, 理论和核酸合成,Tijssen编辑,Elsevier,纽约(1993)。编码本发明多肽的核 酸,与能够指导被翻译的多肽或其片段的分泌的前导序列按合适的读框进行组 装。
如本文所用,术语“分离的”一词是指材料(例如,核酸、多肽、载体、细 胞)与其原始环境例如自然环境(如果它是天然存在的)相分开。例如,天然存在 的、位于活动物中的多核苷酸或多肽是未被分离的,而同样的多核苷酸或多肽, 当与在自然系统中共存的一些或全部的物质分开时,即为分离的。此类多核苷酸 可以是载体的一部分,和/或此类多核苷酸或多肽可以是组合物的一部分,并且 仍然是分离的,只要此类载体或组合物不是其天然环境中的一部分。
如本文所用,术语“合成的”是指该材料,例如核酸,在体外通过公知的化 学合成技术被合成,如描述于例如,Adams(1983)J.Am.Chem.Soc. 105:661;Belousov(1997)Nucleic Acids Res.25:3440-3444;Frenkel(1995) Free Radic.Biol.Med.19:373-380;Blommers(1994)Biochemistry 33:7886-7896;Narang(1979)Meth.Enzymol.68:90;Brown(1979)Meth. Enzymol.68:109;Beaucage(1981)Tetra.Lett.22:1859中。
术语“重组的”是指该核酸邻接于在天然环境下并不相邻的“骨架”核酸。 本发明的骨架分子包括核酸,如克隆载体和表达载体、自我复制的核酸、病毒、 整合核酸和用于维持或操作感兴趣核酸插入序列的其他载体或核酸。从这些核酸 产生的本发明重组多肽可被单独分离或克隆,并测试其期望活性。可以使用任何 重组表达系统,包括细菌、哺乳动物、酵母、昆虫或植物细胞表达系统。
还提供了包含至少一种本发明核酸的载体。所述载体可以是克隆载体、表达 载体或人工染色体。
如本文所用,术语“载体”是指能够将与其连接的另一核酸分子进行转运的 核酸。载体(包括克隆载体和表达载体)包含了本发明的核酸或其功能等同物。本 发明的核酸可以整合入重组的可复制载体中,例如克隆载体或表达载体中。该载 体可用于在相容的宿主细胞中复制该核酸。因此,本发明还提供了制备本发明的 多核苷酸的方法,其中通过将本发明的多核苷酸引入可复制载体,将该载体导入 相容的宿主细胞,并在能够复制载体的条件下培养所述宿主细胞。载体可以从宿 主细胞中回收。合适的宿主细胞在下文中进行了描述。用于插入表达盒或本发明 的核酸的载体,可以是可便利地进行重组DNA操作的任何载体,并且载体的选择 常常取决于待被引入的宿主细胞。多种用于与原核和真核宿主一起使用的克隆载 体和表达载体如Sambrook等人,分子克隆:实验室手册,第二版,冷泉港,纽 约(1989)所述。
如本发明所述的载体可以是自主复制载体,即,其中作为染色体外的实体存 在的载体,其复制是独立于染色体的复制,例如,质粒。或者,所述的载体可以 是当导入宿主细胞时,被整合到宿主细胞基因组中并与其整合的染色体一起复制 的载体。
一种类型的载体是“质粒”,它指环状双链DNA环,在该环中可连入另外的 DNA片段。另一类型的载体是病毒载体,其中额外的DNA片段可以连接入病毒基 因组中。某些载体能够在它们所导入的宿主细胞中自主复制(例如,具有细菌复 制起点的细菌载体,和游离型(episomal)哺乳动物载体)。其他载体(例如,非游 离型哺乳动物载体)被引入宿主细胞后被整合入宿主细胞的基因组中,并由此随 着宿主基因组复制。此外,特定的载体能够指导与它们可操作地连接的基因的表 达。此类载体在本文中称为“表达载体”。一般而言,用于重组DNA技术的表达 载体通常是质粒的形式。术语“质粒”和“载体”在本文中可以互换使用,因为 质粒是最常用的载体形式。然而,本发明旨在包括此类其它形式的表达载体,如粘粒,病毒载体(例如,复制缺陷型逆转录病毒,腺病毒和腺相关病毒)及与其发 挥等同功能的噬菌体载体。
本发明的载体可以体外使用,例如用于生产RNA或用于转染或转化宿主细 胞。
本发明的载体可包含两种或更多,例如三,四或五种本发明的核酸,例如用 于过表达。
本发明的重组表达载体包含本发明核酸,其形式适合于核酸在宿主细胞中表 达所述核酸,这意味着该重组表达载体包括一个或多个调控序列,所述调控序列 可基于用于表达的宿主细胞进行选择,并可操作地连接于待表达的核酸序列。
在载体(如表达载体)中,“可操作地连接”是用于指感兴趣的核苷酸序列以 允许所述核苷酸序列表达的方式连接于调控序列(例如,在体外转录/翻译系统 中,或当载体被引入宿主细胞时在宿主细胞中)。即,术语“可操作地连接”是 指其中所述元件处于允许它们以其预期方式发挥功能的并列关系。“可操作地连 接”于编码序列的调控序列如启动子、增强子或其它表达调控信号,是用以下方 式进行定位:在与控制序列相容的条件下实现该编码序列的表达,或者各序列被 被排列从而使得它们共同起作用以达到预期目的,例如转录在启动子处开始并持 续进行并通过编码多肽的DNA序列。
术语“调控序列”或“控制序列”旨在包括启动子、操纵子、增强子、减弱 子(attenuator)和其他表达控制元件(例如,多腺苷酸化信号)。此类调节序列描 述于例如在Goeddel;基因表达技术:酶学方法,185,学术出版社,圣地亚哥, 加利福尼亚(1990)中。
术语调控或控制序列,包括在许多类型宿主细胞中指导核苷酸序列组成型表 达的序列,以及仅在特定的宿主细胞中指导核苷酸序列表达的序列(如组织特异 性的调控序列)。
对于给定的宿主细胞的载体或表达构建,可因此包含可操作地依次互相连接 的下列元件,按相对于编码本发明多肽的编码链序列的从5’端至3’端方向表 示:(i)能够指导编码多肽的核苷酸序列在给定的宿主细胞中的转录的启动子序 列;(ii)任选的,能够指导多肽从给定的宿主细胞分泌到培养基中的信号序 列;(iii)任选的,编码C-末端、N-末端或内部的,用于纯化、检测或标记多肽 的表位标签序列,或上述序列的组合;(iv)编码本发明多肽的本发明的核酸序 列;以及还优选地包括(v)转录终止区(终止子),其能够终止位于编码多肽的核 苷酸序列下游的转录。特定的被命名的细菌启动子包括lad、lacZ、T3、T7、 SP6、K1F、tac、tet、gpt、λ(lambda)PR、PL和trp。真核启动子包括CMV即时 早期启动,HSV胸苷激酶,早期和晚期SV40,来自逆转录病毒的LTR和小鼠金属 硫蛋白-I等启动子。选择合适的载体和启动子是在普通技术人员的技术水平范围 内的。本发明的核苷酸序列的下游可以存在含有一个或多个转录终止位点(例如 终止子)的3’非翻译区。终止子的来源是不太关键的。终止子可以例如,是对于 编码多肽的DNA序列而言天然的终止子。优选地,终止子是宿主细胞内源性的(在所述宿主细胞中,编码多肽的核苷酸序列是待表达的)。在被转录的区域中, 可以存在用于翻译的核糖体结合位点。由构建物表达的成熟转录本的编码部分, 包括位于待翻译的多肽的起始处的用于翻译起始的AUG(或在原核生物内为TUG或 GUG)以及位于末端的终止密码子。
本发明的多核苷酸的增强表达也可以通过异源调控区的选择实现,例如启动 子、分泌前导和/或终止子区域,其可用于增加感兴趣蛋白质的表达和(如果需要 的话)从表达宿主分泌的水平和/或提供本发明多肽表达的诱导型控制。本领域的 技术人员可以理解,该表达载体的设计可取决于这些因素:如待转化的宿主细胞 的选择,所需的蛋白质表达水平等。如本文所述,载体例如本发明的表达载体可 以被引入到宿主细胞中从而产生核酸所编码的蛋白质或肽。
本发明的载体,如重组表达载体,可以被设计用于在原核或真核细胞的表达 部分或全部的本发明的NRPS。例如,部分或全部的本发明的NRPS可以在细菌细 胞如大肠杆菌、芽孢杆菌菌株、昆虫细胞(使用杆状病毒表达载体)、丝状真菌、 酵母细胞或哺乳动物细胞中表达。合适的宿主细胞在Goeddel,基因表达技术: 酶学方法,185,学术出版社,圣地亚哥,加利福尼亚(1990)中有进一步描述。 合适宿主的代表性实例在下文中说明。上述宿主细胞的适当培养基和培养条件是 现有技术公知的。
如上所述,术语“控制序列”或“调控序列”在本文中定义为包括至少任何 一种对于多肽的表达可能是必要的和/或有利的元件。任何控制序列可以是对于 编码多肽的本发明的核酸序列而言天然的或外源的。这样的控制序列可包括,但 不限于:启动子、前导序列、优化翻译起始序列(如Kozak,1991,J.Biol. Chem.266:19867-19870中所述)、分泌信号序列、原肽序列、多聚腺苷酸化序 列、转录终止子。至少,该控制序列通常包括启动子和转录和翻译的终止信号。 稳定转化的微生物是指已引入一个或多个DNA片段,从而使得所引入的分子在生 长培养过程中被保留、复制和分开。稳定转化可能是由于多个或单个染色体整 合,或由于染色体外的元件,例如质粒载体。质粒载体能够指导由特定DNA片段 编码的多肽表达。表达可以是组成型或是由诱导型(或阻抑型)启动子所调节,这 些启动子使得编码特定多肽的功能性相关的DNA片段被高水平转录。
本发明的表达载体还可以包括选择性标记基因,以便选择出被转化的细菌菌 株,例如,导致赋予细菌针对药物(例如氯霉素、红霉素、卡那霉素、新霉素、 四环素、以及氨苄青霉素和其他青霉素衍生物如羧苄青霉素)抗性的基因。可供 选择的标记也可包括生物合成基因,例如在组氨酸、色氨酸和亮氨酸生物合成途 径中的基因。
合适的多核苷酸序列可以通过各种不同程序插入载体中。在一般情况下,多 核苷酸序列可以在用适当的限制性内切酶对插入片段和载体进行酶切后,连接到 载体的所需位置。或者,可以对插入元件和载体的平末端进行连接。多种克隆技 术在Ausubel等人,《目前的分子生物学方法》,John Wiley 503Sons,Inc. 1997以及Sambrook等,《分子克隆实验手册》,第二版,冷泉港实验室出版社 (1989)中有公开。多核苷酸序列也可通过同源重组的技术(包括在体外以及在体 内重组)进行克隆。这样的方法和其它方法被认为是本领域技术人员的能力范围 之内的。所述载体可以是,例如,质粒、病毒颗粒或噬菌体的形式。其它载体包 括染色体的、非染色体的和合成的多核苷酸序列、SV40的衍生物;细菌质粒、噬 菌体DNA、杆状病毒、酵母质粒、基于质粒和噬菌体DNA组合而衍生的载体,病 毒DNA如牛痘、腺病毒、禽痘病毒和伪狂犬病的组合。
本发明还提供了一种工程或重组宿主细胞,即包含本发明的多核苷酸序列作 为异源或非天然多核苷酸(例如,编码孢囊菌酰胺生物合成簇或本发明的NRPS), 或包含本发明的载体的转化细胞。所述宿主细胞可以是本领域技术人员熟悉的任 何宿主细胞,包括原核细胞和真核细胞,例如细菌细胞、真菌细胞、酵母细胞、 哺乳动物细胞、昆虫细胞、或植物细胞。
优选的哺乳动物细胞包括例如中国仓鼠卵巢(CHO)细胞、COS细胞、293细 胞、PerC6细胞、杂交瘤细胞、Bowes黑素瘤细胞或任何小鼠或任何人细胞系。 示例性昆虫细胞包括夜蛾属或果蝇属的任何物种,包括果蝇S2和草地夜蛾Sf- 9。示例性的真菌细胞包括任何的曲霉种类。优选的酵母细胞包括,例如,念珠 菌属、汉逊酵母属、克鲁维酵母菌属、毕赤酵母属、酿酒酵母属、裂殖酵母属、 或耶氏酵母属菌株的细胞。更优选为乳酸克鲁维酵母、酿酒酵母、多形汉逊酵 母、解脂耶氏酵母、或毕赤酵母。根据本发明,宿主细胞可以是原核细胞。优选 地,所述的原核宿主细胞是细菌细胞。术语“细菌细胞”包括革兰氏阴性和革兰氏阳性以及古细菌的微生物。合适的细菌可选自例如埃希氏杆菌属、鱼腥藻属、 柄杆菌属、葡糖杆菌属、光合细菌属、假单胞菌属、副球菌属、芽孢杆菌属、短 杆菌属、棒状杆菌属、根瘤菌属(中华根瘤菌)、黄杆菌属、克雷伯氏菌属、肠杆 菌属、乳杆菌属、乳球菌属、甲基杆菌属、葡萄球菌或链霉菌属。优选地,所述 细菌细胞是选自枯草芽孢杆菌(B.subtilis)、解淀粉芽孢杆菌(B. amyloliquefaciens)、地衣芽孢杆菌(B.licheniformis)、普氏芽孢杆菌(B. puntis)、巨大芽孢杆菌(B.megaterium)、耐盐芽孢杆菌(B.halodurans)、短 小芽孢杆菌(B.pumilus)、氧化葡糖杆菌(G.oxydans)、新月柄杆菌 CB15(Caulobactercrescentus)、扭脱原单胞菌(Methylobacterium extorquens)、球形红细菌(Rhodobactersphaeroides)、恶臭假单胞菌 (Pseudomonas putida)、zeaxanthinifaciens副球菌(Paracoccus zeaxanthinifaciens)、脱氮副球菌(Paracoccus denitrificans)、大肠杆菌、 谷氨酸棒杆菌(C.glutamicum)、肉葡萄球菌(Staphylococcus carnosus)、变青 链霉菌(Streptomyces lividans)、草木樨中华根瘤菌(Sinorhizobium melioti) 和放射根瘤菌(Rhizobium radiobacter)。合适的宿主的选择是在本领域技术人 员的能力范围内的。
所述载体可被使用任何各种不同技术引入到宿主细胞,包括转化、转染、转 导、病毒感染、基因枪或Ti介导的基因转移。具体的方法包括磷酸钙转染、 DEAE-葡聚糖介导的转染、脂转染、或电穿孔(Davis,L.,Dibner,M.,Battey, I.,分子生物学基本方法,(1986))。本发明核酸或载体可被引入到细胞中,以 便由此筛选核酸是否以适合随后表达核酸的方式进入细胞。引入的方法在很大程 度上取决于靶细胞类型。示例性的方法包括CaPO4沉淀、脂质体融合、脂质转染 (例如,LIPOFECTINTM)、电穿孔、病毒感染等。候选的核酸可以稳定地整合到宿 主细胞的基因组(例如,逆转录病毒引入),或者可以瞬时或稳定存在于细胞质(即,通过使用传统的质粒,利用标准的调控序列、选择标记、等等)。因为许多 药学上重要的筛选需要人或模型哺乳动物的靶细胞,可使用能够转染这些靶细胞 的逆转录病毒载体。
在适当情况下,工程宿主细胞可以在常规营养培养基中进行培养,该培养基 可改良以便适合激活启动子、选择转化子或扩增本发明的核酸。在转化合适的宿 主菌株并宿主菌株生长至适当的细胞密度之后,可通过适当的手段(例如,温度 变化或化学诱导)来诱导选定的启动子,而细胞可以被额外培养一段时间以允许 它们以产生其所需的多肽或其片段。细胞可以通过离心收获,通过物理或化学方 法破碎细胞,保留得到的粗提取物以用于进一步纯化。用于表达蛋白质的微生物 细胞可以通过任何常规方法来破碎,包括冻融循环、超声处理、机械破碎或使用 细胞裂解剂。这样的方法是本领域技术人员所熟知的。表达的多肽或其片段可被 通过以下方法从重组细胞培养物中回收并纯化,其中包括硫酸铵或乙醇沉淀、酸 提取、阴离子或阳离子交换层析、磷酸纤维素层析、疏水相互作用层析、亲和层 析、羟基磷灰石层析和凝集素层析。如必要,可采用蛋白质重折叠步骤,以完成 所述多肽的构型。如果需要的话,高效液相色谱(HPLC)可用于最终纯化步骤。在 宿主细胞中的构建物可用于以常规的方式产生重组序列所编码的基因产物。取决 于在重组生产方法中使用的宿主,由含有载体的宿主细胞产生的多肽可以是糖基 化或是未糖基化的。本发明的多肽还可以或可以不包括起始甲硫氨酸的氨基酸残 基。无细胞翻译系统也可以用来生产本发明的多肽。无细胞翻译系统可以使用从 DNA构建物转录出的mRNA,该构建物包括可操作连接于编码多肽或其片段的核酸 的启动子。在一些方面,该DNA构建物在进行体外转录反应之前,可以被线性 化。转录的mRNA然后与合适的无细胞翻译提取物(如兔网状细胞提取物)一同温 育,以产生所需的多肽或其片段。
含有感兴趣的多核苷酸(例如,本发明的核酸)的宿主细胞,可以在常规营养 培养基中培养,该培养基可经改良而适合激活启动子、选择转化子或扩增基因。 培养条件如温度、pH等,就是先前该选定的用于表达的宿主细胞所用的条件,并 且对于普通技术人员是显而易见的。被识别为具有特定酶活性的克隆随后被测 序,以确定编码本发明的一部分或全部的NRPS的多核苷酸序列。
重组DNA可通过任何手段被引入到宿主细胞中,包括但不限于,质粒、粘 粒、噬菌体、酵母人工染色体或其它介导遗传元件转移至宿主细胞中的载体。这 些载体可包括复制起点,以及控制该载体和由该载体所携带的遗传因子的复制的 顺式作用控制元件。选择性标记可以存在于载体上,以协助识别在其中已引入遗 传元件的宿主细胞。用于将遗传元件引入到宿主细胞的方法(例如,克隆)是本领 域技术人员公知的。其它克隆方法包括但不限于,遗传物质直接整合入染色体。 这可以通过各种手段实现,包括在侧接宿主染色体的同源DNA序列的非复制质粒 上克隆本文中描述的遗传元件;一旦将所述重组质粒转入宿主,遗传元件可以通 过DNA重组被引入到染色体。如果整合的DNA片段含有可选择的标记,如抗生素 抗性,则这样的重组菌株可以回收。或者,该遗传元件可以被直接导入到宿主细 胞的染色体中,而不使用非复制型质粒。这可以通过合成产生本发明的遗传元件 的DNA片段来实现,其中该DNA片段还包含宿主染色体的同源DNA序列。同样, 如果这些合成的DNA片段也包含可选择标记,那么该遗传元件可以被插入到宿主 染色体中。
所述孢囊菌酰胺生物合成簇或本发明的NRPS在任何上述宿主细胞中可以有 利地被表达。因此,本发明提供各种不同的宿主细胞,它们包含一种或多种分离 的、合成的或重组的核酸和/或本发明的NRPSs。所述的宿主细胞在合适的条件下 培养时,能够产生选自孢囊菌酰胺A、B、C、D、E、F、G和H的孢囊菌酰胺,而 在不存在本发明的核酸的情况下这些孢囊菌酰胺原本是不产生的或产生的水平较 低。
本发明还涉及分离的、合成的或重组的具有SEQ ID NO.40至73任一的氨 基酸序列的多肽,或由本发明核酸所编码的氨基酸序列。
本发明进一步提供了制备选自下组的孢囊菌酰胺的方法:孢囊菌酰胺A、B、 C、D、E、F、G和H,所述方法一般包括:提供本发明的宿主细胞,并在合适的 培养基中在合适的条件下培养所述宿主细胞,使得至少一种选自下组的孢囊菌酰 胺被生产:孢囊菌酰胺A、B、C、D、E、F、G和H。该方法还可以包括分离选自 孢囊菌酰胺A、B、C、D、E、F、G和H中的孢囊菌酰胺的步骤,即从培养液中分 离和保留化合物。所述分离步骤可以利用亲和层析、阴离子交换层析、或反相色 谱法进行。
实施例
制备方法
生产制备用菌株
菌株深棕色孢囊杆菌(Cystobacter velatus)MCy8071属于粘细菌目(粘细 菌)、孢囊杆菌亚目、孢囊杆菌科、孢囊杆菌属。部分16S rRNA基因序列与公共 数据库的序列比较(BLAST,由NCBI(美国国家生物技术信息中心)所提供的“基本 的局部比对搜索工具”)显示,与深棕色孢囊杆菌菌株DSM14718相似度100%。
MCy8071是从1982年采集的中国的土壤样品中,在亥姆霍兹感染研究中心 (HZI,前身为GBF)分离出。该菌株于2013年3月保藏在布伦瑞克(Braunschweig) 的德国微生物保藏中心(DSM),保藏号为DSM27004。
培养
菌株MCy8071在酵母琼脂(VY/2:0.5%酿酒酵母,0.14%CaCl2x 2H2O, 0.5μg维生素B12/l,1.5%琼脂,pH 7.4),CY-琼脂(酪胨0.3%,酵母提取物 0.1%,CaCl2x 2H2O0.1%,琼脂1.5%,pH 7.2)和P-琼脂(马考尔蛋白胨 (peptone Marcor)0.2%,淀粉0.8%,单细胞蛋白Probione 0.4%,酵母提取物 0.2%,CaCl2x 2H2O 0.1%,MgSO4 0.1%,Fe-EDTA 8mg/l,1.5%琼脂,pH 7.5) 上充分生长。工作培养物培育于液体介质CY/H(50%CY-培养基+50毫摩尔的 Hepes缓冲液,50%的H-培养基:大豆粉0.2%,葡萄糖0.8%,淀粉0.2%,酵母提 取物0.2%,CaCl2x 2H2O 0.1%,MgSO4 0.1%,Fe-EDTA 8mg/l,Hepes 50mMpH 7.4)中。液体培养物以180rpm在30℃下摇动。为了保存,将2毫升每份的三 天的培养物保存在-80℃下。即使经过好几年,在上述的琼脂平板上或在20毫升 CY/H介质(于具有栓塞和铝盖的100ml锥形瓶中)中激活也没有问题。一两天 后,20毫升培养物可以升级至100毫升。
形态描述
在液态介质CY/H中两天后,上述菌株MCy8071的杆状细胞具有9.0–14.5μm 的长度,以及0.8–1.0μm的宽度。在上述琼脂平板上,菌落是圆形的。在VY/2- 琼脂中,菌落是薄而透明的。在VY/2-琼脂中,酵母的退化是可见的。在CY-琼 脂中,培养物看起来是透明的橙色。在P-琼脂上,细胞群的产生是独特的,且菌 落聚集行为减少。菌落是橙棕色的。P-琼脂中的淀粉被分解。
MCy8071对以下抗生素耐药:氨苄青霉素、庆大霉素、潮霉素、多粘菌素(polymycin)、杆菌肽、大观霉素、新霉素、和夫西地酸(fusidinic acid)。在 头孢菌素和春雷霉素存在下可能有微弱生长,在硫链丝菌素、甲氧苄啶、卡那霉 素、和土霉素存在下生长是不可能的(所有抗生素的终浓度调节至50μg ml-1)。
孢囊菌酰胺A、B、C、D、E、F、G和H的生产
菌株在复合介质中生产。它更喜欢在含氮的营养素,如单细胞蛋白(Probion) 和蛋白质分解产物如蛋白胨、胰蛋白胨、酵母提取物、大豆粉和肉提取物等。在 此,多个所述的蛋白混合物存在下的生产情况优于使用单一的蛋白质。
孢囊菌酰胺是在生长对数期至稳定期之间产生的。经过100升发酵(培养基E) 两天后,产物的量不再增加。
孢囊菌酰胺被递送到培养基,并与XAD-吸附树脂结合。XAD用金属筛进行筛 分,并在丙酮中洗脱。在不同的生产温度下进行了测试(21℃,30℃,37℃和 42℃),而在42℃下是不可能进行生产的。在最大通气量下,最适温度为30℃。
MCy8071的发酵是在150升发酵罐中进行,其中采用100升培养基E(脱脂奶 0.4%,大豆粉0.4%,酵母提取物0.2%,淀粉1.0%,MgSO4 0.1%,Fe-EDTA 8mg/l,甘油0.5%;pH为7.4)进行发酵,以及在100升发酵罐中进行,其中用 70升培养基M(大豆蛋白胨1.0%,麦芽糖1.0%,CaCl2x 2H2O 0.1%,MgSO4 0.1 %,Fe-EDTA 8mg/l;pH值7.2),并在30℃下发酵四天。用氢氧化钾(2.5%)和硫 酸将pH调节至7.2到7.4之间。搅拌器速度为100–400rpm,用0.05vvm压 缩空气进行通气。发酵液中的溶解氧的含量通过搅拌器速度调控至pO2 40%。为 结合孢囊菌酰胺,1%的吸附树脂被加入到发酵液中。将5升的三天预培养物(分 别为E或M-培养基),接种于发酵罐。在发酵过程中的产物经HPLC-MS-分析和针 对大肠杆菌的甲醇提取物的系列稀释试验进行检查。该菌株生产孢囊菌酰胺A、 B、C、D、E、F、G和H。
基因敲除实验
为了确认孢囊菌酰胺生物合成基因簇是负责生产孢囊菌酰胺的,进行了敲除(KO)实验,其中CysK(NRPS)和CysL(苯甲酰辅酶A连接酶)被分别敲除。具体而 言,使用Taq聚合酶,根据MCy8071基因组DNA,产生CysK和CysL基因的 1000bp片段PCR产物。该引物设计成在PCR产物的末端添加3个终止密码子。
CysL敲除,正向
TGATTGATTGATCGGCGCGATTCGGCCTCTGG
CysL敲除,反向
TCAATCAATCATCGGGTCGCGGTCTCAGGCTC
CysK敲除,正向
TGATTGATTGAAAAACAGTCGGAGGAGTTTCTTGTCC
CysK敲除,反向
TCAATCAATCAACTCCCAGTGCCCTCAGCCTC
将PCR产物进行凝胶纯化,其中使用 凝胶和马谢雷-内格尔(Macherey-Nagel)PCR清理(Clean up)试剂盒,并克隆至pCR2.1-TOPO载体中。 该构建物通过热休克整合成到化学感受态大肠杆菌HS996,并在添加有卡那霉素 的LB琼脂平板上完成挑选。通过碱裂解质粒制备和EcoRI限制性消化,来筛选 具有正确构建物的单菌落。然后对构建物进行测序,以确保序列同源性。
凝胶和马谢雷-内格尔(Macherey-Nagel)PCR清理(Clean up)试剂盒,并克隆至pCR2.1-TOPO载体中。 该构建物通过热休克整合成到化学感受态大肠杆菌HS996,并在添加有卡那霉素 的LB琼脂平板上完成挑选。通过碱裂解质粒制备和EcoRI限制性消化,来筛选 具有正确构建物的单菌落。然后对构建物进行测序,以确保序列同源性。
用于各KO的正确的构建物被转入非甲基化的化学感受态大肠杆菌SCS110。 质粒通过使用Thermo Scientific公司的GeneJET质粒小量制备试剂盒制备,并 通过电穿孔导入MCy8071。转化的复制体的选择在添加有卡那霉素的CTT琼脂平 板上进行。KO突变体和野生型培养物在吸附树脂(XAD-16)存在下进行平行生长, 并对培养物的粗提取物样品进行分析。
结果表明,在KO突变体中完全没有孢囊菌酰胺生产,这提示CysK和CysL 对于孢囊菌酰胺的生产是必不可少的。此外,该结果提示了孢囊菌酰胺生物合成 基因簇用于生产孢囊菌酰胺的基本性质。
结构分析:
孢囊菌酰胺A(1)的HRESI(+)MS分析返回了准分子式离子(M+H)+,其符合分 子式C46H45N7O14,需要28个双键当量(DBE)。13C NMR(DMSO-d6)数据显示7酯/酰胺 羰基(δC 163.7至169.6)以及进一步的30sp2共振态(δC 114.2至150.8),对应 于22DBE。对一维和二维NMR数据(表1)的考虑,显示了一套五芳香自旋系统, 其中三个对应于对位取代,1,3,4-三取代和1,2,3,4-四取代的苯环。一组HMBC 相关性:从H-6,6′(δH 7.96)和NH(δH 8.92)到酰胺羰基C-4(δc 166.5);从NH(δH 10.82)到C-7/7′(δc 119.8)和第二个酰胺羰基C-10(δc164.6);从H-12/12(δH 8.20)到C-10,建立了两个对位取代的芳香环体系的连接关系(图1)。对于1H和 COSY NMR数据的进一步检验,建立了酰胺NH(δH 8.92)延伸至次甲基H-2(δH4.96) 和H-1(δH 4.70)的连接关系。低磁场特征H-1(δC 79.4)提示了氧取代,上述特征 被HMBC的从H-1到1-OMe(δH 3.53,δC 59.6)相关性所证实。还观察到HMBC中从 H-1和H-2到酯/酰胺羰基(δC 169.6)的相关性,这导致指向亚基A的结构(图 1)。
对于1,3,4-三取代的苯环,观察到HMBC中从H-17(δH 7.58)到酯/酰胺羰基 C-15(δC 167.3)、氧季碳C-18(δC 146.8)、C-19(δC 133.6)和C-21(δC 122.9)的 相关性。1,2,3,4-四取代苯环的独立的自旋系统,显示了HMBC中以下相关性:i) 从H-25(δH 7.82,d,8.7)到酯/酰胺羰基C-23(δC 163.7)、C-27(δC 136.2)以及氧 季碳C-29(δC 150.8);i i)从H-26(δH 7.42)到C-24(δC 117.3)和C-28(δC 139.5) 的相关性,以及酚羟基(δH 11.25)与C-24和C-28的相关性。三和四取代的苯环 互为对位连接,这可由HMBC中酰胺NH(δH 10.98)与C-20(δC 119.8)、C-18(δC 146.7)以及C-23(δC 163.7)的相关性得出(图1)。最后对位取代的芳族自旋系统 的H-33/33′(δH 8.11,d,8.3)和H-34/34′(δH 7.44,d,8.3),显示了连接于1,2,3- 三取代苯环,这可由HMBC中酰胺NH(δH 9.88)和H-33/33′与酰胺羰基C-31(δC 164.3)的相关性得出。对COSY数据的额外解读,显示了两个异丙氧基残留(H3- 39(δH 1.38)-H-38(δH 4.76)-H-40(δH 1.38))和(H3-42(δH 1.25)-H-41(δH 4.30)- H3-43(δH 1.25)。两个异丙氧基残留确认与氧季碳C-18(δc 146.7)和C-28(δc 139.5)相连,基于ROESY中H-38/H-39与H-17/NH,以及H-42/43与NH/29- OH/H-33/33′的相关性(图1)。亚基A和B的关系未被建立,不过基于孢囊菌酰胺 B的结构相似性,亚基A和B的连接点被推测出。在解释了大部分共振之后, N2O3H2和1DBE被留下来用于进一步解释。化合物的UV光谱显示出λmax为301和320nm,这暗示一个偶联系统,该偶联系统只可能是由硝基官能团对位连接于亚 基A的芳基上所产生的。剩余的MF被调节为生成位于亚基B的1,2,3-取代的芳 环上的羧酸残基(C-15),产生了4-氨基-3-异丙氧基苯甲酸部分,从而导致构成 了孢囊菌酰胺A的平面结构。
孢囊菌酰胺B(2)的HRESI(+)MS分析返回了准分子式离子(M+H)+,其符合分 子式C46H44N6O15,需要28个双键当量(DBE)。2的NMR数据(表2)与(1)高度相似, 并且现在NH(δH 10.19)和氧次甲基H-1(δH 4.32)看到了羰基C-37(δC 168.6),这 确认了亚基A和B的连接位点。除了这个,唯一的变化是羰酰胺现在调整为羧 酸,这随后被孢囊菌酰胺B二甲酯的生成所证实。
孢囊菌酰胺C(3)的HRESI(+)MS分析返回了准分子式离子(M+H)+,其符合分 子式C27H29N3O7,需要15个双键当量(DBE)。孢囊菌酰胺C的1H NMR数据显示了芳 香信号,这使人想起孢囊菌酰胺A和B,但它缺乏两组对位取代的芳族单元的芳 族共振。COSY数据揭示了存在两组异丙氧基残基连同一组对位取代的芳环体系。 基于与,对1D和2D NMR数据的解释(表3,图2)鉴定了孢囊菌酰胺C(3)与孢囊 菌酰胺A和B的右侧部分相似,该部分由3-异丙氧基苯甲酸,2-羟基-3-异丙氧 基苯甲酰胺与对氨基苯甲酰胺单元组成。
表1.孢囊菌酰胺A(1)的NMR(700MHz,DMSO-d6)数据


a重叠信号,*通过HSQC和HMBC实验支持的分配。
表2.孢囊菌酰胺B(2)的NMR(700MHz,DMSO-d6)数据


表3.孢囊菌酰胺C(3)的NMR(500MHz,DMSO-d6)数据


无-未观察到,*通过HSQC和HMBC实验支持的分配。
孢囊菌酰胺D(4)的HRESI(+)MS分析返回了准分子式离子([M+H]+),表示 分子式(C42H37O14N7),需要28个双键当量。NMR(DMSO-d6)数据(表4)的解释揭示了 磁性相当的芳香质子H-12′/12(δH 8.17,d,8.0)和H-13/13′(δH 8.36,d,8.0),它 们对应于第一个对位取代的苯环。1H-1H COSY数据的进一步解释反映了两个额外 的对位取代苯环的存在,(H-35/35′)(δH 7.80,d,8.1)和H-36/36′(δH 7.94, d,8.1);第二组芳基高度重叠(H-6/6′)和(H-7/7′(δH 7.88)。芳香质子的(H- 12/12′)与酰胺羰基C-10(δC 165.1)的HMBC相关性判断以及可交换的(NH)(δH 10.82)与C-10,C-7/7′,建立了两个对位取代的芳环之间的连接(图3),这进一步 被NH/H-12和NH/H-7之间ROESY的相关性所证实。COSY数据反映了一个额外的自旋系统,这基于氧次甲基H-1(δH 4.08,d,8.0)和α-质子H-2(δH 4.91,dd,8.0, 7.7)到可交换质子(NH)(δH 8.47)。以下HMBC相关性:(i)从H-2到三个酰胺羰基 C-4(δC 166.4)、C-15(δC 171.8)和C-32(δC 169.2);(ii)从NH(δH 8.48)到C-4; (iii)从NH(δH 10.54)到C-35/35′(δC 119.5),(iv)从H-6/6′到C-4,进一步拓展 了孢囊菌酰胺D(4)的部分结构。对于1-D和2-D NMR数据的考虑,揭示了额外的 1,3,4-三取代和1,2,3,4-四取代的苯环。观察到从芳香质子H-27(δH 7.55)和H- 29(δH 7.60)到羰基C-31(δC 167.8)以及季碳C-25(δC133.0)的HMBC相关性,同 时H-30(δH 8.47,d,7.0)和甲氧基信号(δH 3.96)被连于与负载有氧的碳C-26(δC 149.1),因此揭示了4-氨基-3-甲氧基苯甲酸部分,其之后被酯化所证实。除此 之外,观察到从可交换质子(NH)(δH 7.46)到负载有氧的碳C-1(δC 80.8),C- 18(δC141.0)和芳香碳原子C-22(δC 116.2)的HMBC相关性,其中H-22(δH 7.48,d,8.8)和甲氧基显示了与连接于酰胺羰基C-23(δC 164.8)的C-18和H- 21(δH 7.77,d,8.8)之间的连接。羟基官能的邻位的甲氧基的存在,后来被酯化证 实(4a)(图4),反映了4-氨基-2-羟基-3-甲氧基苯甲酰胺的存在。4-氨基-3-甲 氧基苯甲酸和4-氨基-2-羟基-3-甲氧基苯甲酰胺取代基的连接,被ROESY和从孢 囊菌酰胺D二甲酯(4a)中观察到的可交换的NH的HMBC相关性所证实。所缺乏的 取代基被指定为位于C-14(δC 150.0)和羰基C-38。λmax(320nm)和C-14的低场化 学位移揭示了在C-14有硝基取代基,并且伯胺连接于羰基C-38,这产生了4的 平面结构。
表4.孢囊菌酰胺D(4)的NMR(700MHz,DMSO-d6)数据


a,b,c重叠信号,通过2D HSQC和HMBC实验得到13C位移。无-未观察
表5.孢囊菌酰胺D二甲酯(4a)的NMR(700MHz,DMSO-d6)数据


a,b重叠信号,通过2D HSQC和HMBC实验得到13C位移。无-未观察
孢囊菌酰胺E(5)的HRESI(+)MS分析返回了准分子式离子([M+H]+),其提 示分子式(C26H23O9N5),需要18个双键当量。1H NMR谱图与孢囊菌酰胺D类似,其 主要区别是缺乏对应于4-氨基-3-甲氧基苯甲酸和4-氨基-2-羟基-3-甲氧基苯甲 酰基部分的信号。1-D和2-DNMR数据(表6)的具体分析,导致了孢囊菌酰胺E(5) 的平面结构。
表6.孢囊菌酰胺E(5)的NMR(700MHz,DMSO-d6)数据

a重叠信号,通过2D HSQC和HMBC实验得到13C位移。
孢囊菌酰胺F(6)的HRESI(+)MS分析返回了准分子式离子(M+H)+,其符合分 子式C43H39N7O13,需要28个双键当量(DBE)。NMR(DMSO-d6)数据的解释(表7)揭示 三组磁性相当的芳族质子,它们可能通过COSY(6/6’和7/7’,12/12’和13/13’, 33/33’和34/34’)被关联;此外,与所有其他孢囊菌酰胺不同,还揭示了一组磁 性相当的芳香质子(26/26’和27/27’),它们也可以通过COSY被关联。上述四组 在分子中占有四个对位取代的苯环,而非在所有其他孢囊菌酰胺中发现的三个。 仅有一个1,2,3,4-四取代的苯环可以被检测到,其中在芳香质子H-22(dH 7.22) 与C-18(dC 137.1)和C-20(dH 114.0)之间的,以及芳香质子H-21(dH7.51)与C- 23(dC 167.3)之间的HMBC相关性被观察到。质子H-21和H-22可以通过COSY相关性被关联。由于碳C-17、C-19和C-22不能被观察到,所有的肽-片段的HR- MS/MS质量已经建立并且揭示了在各自的片段中存在7个碳、11个质子、1个氮 和3个氧,这证实了在这个位置上存在1,2,3,4-取代的对-氨基苯部分(见图 1)。HMBC数据进一步证实了H-37(dH 4.93)和C-18(dc 137.1)的联系。HMBC和 COSY数据证实了在分子的两个芳族部分之间相同的连接结构,正如孢囊菌酰胺D 中发现的。可交换质子H-9(dH 10.82)和C-10(dC 163.9)以及C-7/7’(dC 119.4) 之间,H-3(dH 8.49)和C-4(dC 165.1)之间,H-31(dH 10.56)和C-30(dC168.3)以 及C-32(dC 141.5)之间,和H-16(dH 8.91)与C-36(dC 163.1)和C-18(dC 137.1)之间的HMBC相关性,以及H-2(dH 4.92)与可交换质子H-3(dH 8.49)之间的COSY 相关性,以及HRMS片段数据,建立了各个片段之间的系列相关性。硝基基团的 位置和芳族链之间的连接基中自由酰胺基的存在,是通过用HR-MS/MS碎片以产 生相应片段的总式而建立的。
表7:孢囊菌酰胺F(6)的NMR(700MHz,DMSO-d6)数据


a重叠信号,无-未观察,*通过HSQC和HMBC实验支持的分配。
孢囊菌酰胺G(7)的HRESI(+)MS分析返回了准分子式离子(M+H)+,其符合分 子式C44H41N7O14,需要28个双键当量(DBE)。由于在DMSO-d6中芳香信号重叠,在 甲醇-d4中获得的NMR数据被用于建立芳香和连接片段的局部结构(表8)。对位取 代的苯环可以经由COSY、HSQC和HMBC相关性来确定。通过HSQC、COSY和HMBC 相关性建立了1,3,4-三取代苯环(4-氨基-3-甲氧基-苯甲酰胺)和甲氧基取代基 (1-OMe,(dC 55.2,dH 3.50)的结构。由于并非所有1,2,3,4-取代的苯部分的信 号均可以在甲醇-d4中被检测到,对DMSO-d6中测定的NMR数据进行解读,以建立 4-氨基-3-异丙基-2-羟基-苯甲酰胺和在孢囊菌酰胺D中所鉴定的芳香部分之间 相同的连接结构。C-39(dC 74.4)和碳原子C-40/40’(dC 22.7)之间的联系通过H- 39(dH 4.82)和H-40/40’(dH 1.31)之间的COSY相关性建立,且1,2,3,4-取代的 苯环和H-39(dH 4.82)之间的连接,是通过H-39和C-18(dC 137.3,在DMSO-d6中) 之间的HMBC相关性建立的。这个苯部分的结构被DMSO-d6中H-22(dH 7.04)与C- 18(dC 137.3)和C-20(dC116.1)之间的HMBC相关性,以及(dH 7.45)与C-23(dC 165.4)之间的HMBC相关性,以及H-21和H-22之间的COSY相关性进一步证实。 整体序列,所述硝基基团的位置和芳香族链之间的连接结构中自由酰胺基团的 存,是在通过HR-MS/MS碎片以产生相应片段的总式而建立的。
表8:孢囊菌酰胺G(7)的NMR(700MHz,甲醇-d4)数据,包括位置17-23和 39-40/40’(700MHz,DMSO-d6)的数据.


a重叠信号,无-未观察,*通过HSQC和HMBC实验支持的分配。
孢囊菌酰胺H(8)的HRESI(+)MS分析返回了准分子式离子(M+H)+,其符合分 子式C43H39N7O14,需要28个双键当量(DBE)。据发现,芳香链之间的连接结构与孢 囊菌酰胺D中所发现的一致。在DMSO-d6中得到的HSQC、HMBC和COSY数据显示 出,三个对位取代苯单元,这与在孢囊菌酰胺A、B、D、F和G中所发现的相 同。对COSY、HSGC和HMBC数据的进一步解释,显示了相同的1,3,4-三取代苯部 分,它显示出与甲氧基的HMBC相关性,这与在除了孢囊菌酰胺F之外的所有其 他孢囊菌酰胺中发现的情况相同(这通过1-OMe(dH 3.27)和C-26(dC147.4)之间 的HMBC相关性得以确认)。NMR数据的分析显示了1,2,3,4-取代的苯部分,这与其他孢囊菌酰胺相一致。显著的改变源自于乙氧基单元的建立,这是通过亚甲基 质子H-39(dH 4.17)和甲基基团H-40(dH 1.27)之间的COSY相关性,以及亚甲基 基团H-39(dH 4.17)和C-18(dc 139.5)之间的HMBC相关性得出的,从而将4-氨 基-2-羟基-3-X-苯甲酰胺结构部分的取代方式扩展至:位置3上X=甲氧基、异丙 基或乙氧基。孢囊菌酰胺H的连续序列通过可交换质子H-9(dH 10.93)与C-10(dc 163.9)和C-7/7’(dC 119.6)之间,H-33(dH 10.85)与C-32(dC 168.7)和C- 35/35’(dC 118.8)之间,H-16(dH 8.91)与C-38(dC 163.1)之间,C-18(dC 139.5) 和C-22(dC 100.4)和H-24(dH 14.71)与C-20(dC 116.1)之间,C-25(dC131.0)、 C-26(dC 147.4)和C-30(dC 118.5)以及H-2(dH 4.85)与C-4(dc 165.5)之间的HMBC相关性,以及HR-MS2碎片数据得以建立,HR-MS2碎片数据还使得硝基基团 被定位以及确定了连接部分中的自由酰胺基。
表9:孢囊菌酰胺H(8)的NMR(700MHz,DMSO-d6)


a重叠信号,无-未观察,*通过HSQC和HMBC实验支持的分配。
附图说明:
图1:孢囊菌酰胺A(1)关键的2D NMR相关性(700MHz,DMSO-d6)
图2:孢囊菌酰胺C(3)关键的2D NMR相关性(500MHz,DMSO-d6)
图3:孢囊菌酰胺D(4)关键的2D NMR相关性(700MHz,DMSO-d6)
图4:孢囊菌酰胺D二甲酯(4a)关键的2D NMR相关性
图5:孢囊菌酰胺E(5)关键的2D NMR相关性
图6:孢囊菌酰胺F(6)关键的2D NMR相关性(700MHz,DMSO-d6)
图7:孢囊菌酰胺G(7)关键的2D NMR相关性(700MHz,MeOH-d4)
图8:孢囊菌酰胺H(8)关键的2D NMR相关性(700MHz,DMSO-d6)
孢囊菌酰胺的生物学评价
如在表10a/b中所概括,评价了孢囊菌酰胺对于若干微生物和细胞系的作 用。所有的衍生物表现出对各种不同大肠杆菌菌株(包括对抗萘啶酸(NALR)的分离 株)的有效抑制作用。测试衍生物的整体效力按照以下顺序增加(平均MIC值): CysA1,CysC<CysB<CysA,CysG<CysF。重要的是,在低μg/ml范围内,病原 性革兰氏阴性菌株鲍曼不动杆菌和绿脓杆菌也为活性最好的衍生物CysA、CysB、 CysG和CysF所抑制,以MIC值表示,仅比参考药物环丙沙星高一个数量级。
对革兰氏阳性菌,如粪肠球菌、金黄色葡萄球菌和肺炎链球菌的平均MIC值 部分地在低于μg/ml范围,且CysA和CysB的平均效力超过了环丙沙星的效力。
另外,它表明孢囊菌酰胺即不抑制酵母,也不抑制哺乳动物细胞的生长。因 此,该孢囊菌酰胺没有造成明显的细胞毒性。
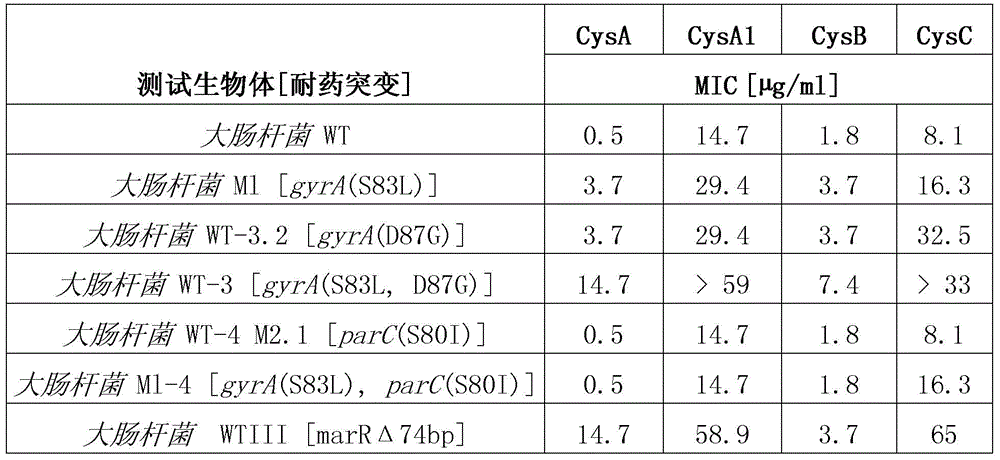
突变的大肠杆菌菌株对于孢囊菌酰胺的敏感性
喹诺酮类药物是一类广泛使用的,针对II型拓扑异构酶、DNA促旋酶和拓扑 异构酶IV的抗生素。由此,喹诺酮类耐药性常常是由导致药物靶标改变的染色 体基因突变所介导。在GyrA中,喹诺酮类耐药决定区(QRDR)位于氨基酸67和 106之间,而氨基酸83(Ser)和87(Asp)是最经常参与的[1,2]。在ParC类似的区域 中(ParC是喹诺酮类的次要目标),已发现,氨基酸80(Ser)的变化赋予喹诺酮类 药物耐药性[3,4]。
用在gyrA和parC基因具有典型突变的大肠杆菌菌株平板,对孢囊菌酰胺进 行筛选(表11)。使用环丙沙星时,MIC值对于单个的gyrA突变提高了大约30个 因子(倍)(菌株MI和WT-3.2)。然而,这两种gyrA突变的组合(菌株WT-3)导致 了接近临床耐药性(1mg/L)。parC突变(菌株WT-4M2.1)导致环丙沙星的MIC增 加了两倍。然而,孢囊菌酰胺的MIC值对于gyrA和parC突变的大肠杆菌菌株没 有增加或仅略微增加,这表明相比于环丙沙星中观察到的情况,孢囊菌酰胺可能 与gyrA的氨基酸87和83以及ParC的氨基酸80之间的干扰程度较低。
高浓度的喹诺酮类药物耐药性,通常是由多个目标位点突变和改变的外排机 制相结合所导致的。在体外选定的突变株WT III(marR Δ74bp)不会产生功能性 MarR,而MarR的作用是作为marA表达的阻遏子。这反过来,导致MarA和AcrAB 的生产过剩,而AcrAB外排泵的过表达是与MAR(多重耐药性)表型相关联的[5]。 大肠杆菌菌株WT III对于环丙沙星治疗更不敏感,其因子为约4(与大肠杆菌 WT相比)。相比之下,孢囊菌酰胺B、F和G的MIC值仍处于μg/ml范围内。值得 注意的是,CysF对大肠杆菌菌株WT III的MIC仅较野生型大肠杆菌DSM-1116增 加了2个因子(倍),而环丙沙星的MIC增加约10倍。
表10a:孢囊菌酰胺(Cys)的抗菌活性




CIP对照抗生素环丙沙星
-未测定
表10b:孢囊菌酰胺(Cys)的细胞毒性


-未测定
表11:孢囊菌酰胺(Cys)对大肠杆菌突变菌株的抗菌活性

CIP对照抗生素环丙沙星
-未测定
基于细胞的测定的实验步骤
细胞系和原代细胞。人HCT-116结肠癌细胞(CCL-247)来自于美国典型培养 物保藏中心(ATCC),中国仓鼠卵巢CHO-K1细胞(ACC-110)来自于德国微生物和细 胞培养物保藏中心(DSMZ)。两种细胞系都在各自保藏者所推荐的条件下进行培 养。原代HUVEC(人脐静脉内皮细胞;单个供体)购自PromoCell(海德堡,德 国),并在含有如下补充剂的内皮细胞生长培养基(PromoCell)中培养:2%FCS, 0.4%ECGS,0.1ng/ml EGF,1ng/ml bFGF,90μg/ml肝素,1μg/ml氢化可的 松。
细菌菌株。在敏感性试验中使用的细菌的野生型菌株,或者是我们菌株收集 的一部分,或是从德国微生物和细胞培养保藏中心(DSMZ)或从美国典型培养物保 藏中心(ATCC)购得。大肠杆菌菌株WT[6]以及大肠杆菌突变株为汉堡大学医药生物 和微生物学的P.Heisig教授(博士)惠赠。
细胞毒性检测。细胞以6x 103个细胞/每孔接种于96-孔板(Corning  )上的完全培养基(180μl)中,并直接用系列稀释的溶解于甲醇的孢 囊菌酰胺进行处理。化合物以及内部溶剂对照,一式两份测试5天。在5天孵育 后,5mg/ml MTT的PBS(20μL)溶液被加入到每孔中,并在37℃下继续孵育2 小时[7]。弃去培养基,将细胞用PBS(100μl)洗涤,然后加入2-丙醇/10N盐酸 (250:1,v/v;100μl)以溶解甲瓒颗粒。在570nm处的吸光度用酶标仪测定 (EL808,Bio-Tek仪器公司)。
)上的完全培养基(180μl)中,并直接用系列稀释的溶解于甲醇的孢 囊菌酰胺进行处理。化合物以及内部溶剂对照,一式两份测试5天。在5天孵育 后,5mg/ml MTT的PBS(20μL)溶液被加入到每孔中,并在37℃下继续孵育2 小时[7]。弃去培养基,将细胞用PBS(100μl)洗涤,然后加入2-丙醇/10N盐酸 (250:1,v/v;100μl)以溶解甲瓒颗粒。在570nm处的吸光度用酶标仪测定 (EL808,Bio-Tek仪器公司)。
敏感性测试。MIC值通过微量稀释试验测定。过夜培养物在适当的生长培养 基中稀释,以获得104-106cfu/mL的接种物。酵母生长于NYC培养基(1%植物蛋 白胨,1%葡萄糖,50mM HEPES,pH7.0),肺炎链球菌和粪肠球菌在胰蛋白酶大豆 培养基中(TSB:1.7%蛋白胨酪蛋白,0.3%蛋白胨豆粕,0.25%葡萄糖,0.5%的 NaCl,0.25%K2HPO4;pH7.3);耻垢分枝杆菌在补充了10%的Middlebrook ADC富 集物和2ml/l甘油的Middlebrook 7H9培养基中。列出的所有其他细菌在米勒 -辛顿培养基(0.2%牛肉输注固体,1.75%酪蛋白水解物,0.15%的淀粉,pH 7.4) 中培养。孢囊菌酰胺和对照药物被直接加入到在无菌96孔板中的培养物中,一 式两份,并制备系列稀释物。微生物生长在微孔板摇床上(750rpm,30-37℃,18-48h)中,除了肺炎链球菌,其生长在非振荡条件下(37℃,5%CO2,18小 时)。生长抑制通过目测评估,且MIC被定义为抑制可见生长的化合物的最低浓 度。
靶标识别
为了测试孢囊菌酰胺的抑制促旋酶的活性,使用商业大肠杆菌旋转酶超螺旋 试剂盒(Inspiralis)。孢囊菌酰胺A抑制大肠杆菌促旋酶(20.5nM=1单位)的表 观IC50为6μM。孢囊菌酰胺A1抑制大肠杆菌促旋酶(20.5nM=1单位)的表观IC50为2.5μM。孢囊菌酰胺D抑制大肠杆菌促旋酶(20.5nM=1单位)的表观IC50为1 μM。孢囊菌酰胺C抑制大肠杆菌促旋酶(20.5nM=1单位)的表观IC50为7.7μM。 因此,孢囊菌酰胺为细菌DNA促旋酶的新型抑制剂。
孢囊菌酰胺A-D在促旋酶抑制活性实验中的IC50值:


图9a显示了促旋酶抑制测定的结果。促旋酶反应用不同浓度孢囊菌酰胺A、 A1、C和D进行滴定,并通过琼脂糖凝胶电泳进行解析。对于IC50测定,超螺旋 质粒的带强度使用Adobe Photoshop来确定,对[孢囊菌酰胺]进行作图,并用 Hill’s方程拟合。
原核DNA促旋酶和拓扑异构酶IV具有高度的同源性,而促旋酶抑制剂通常 显示出拓扑异构酶IV抑制活性8。为测试孢囊菌酰胺对于拓扑异构酶IV的影 响,使用了市售的大肠杆菌拓扑异构酶IV试剂盒(Inspiralis)。
孢囊菌酰胺A只在815μM的最高测试浓度下抑制大肠杆菌拓扑异构酶IV的 活性。孢囊菌酰胺A1抑制大肠杆菌拓扑异构酶IV,显示出6.4±1.8μM的IC50值。孢囊菌酰胺C只在300μM的最高测试浓度下抑制大肠杆菌拓扑异构酶IV的 活性。孢囊菌酰胺D抑制大肠杆菌拓扑异构酶IV,显示出10±3μM的IC50值。
孢囊菌酰胺A-D对大肠杆菌拓扑异构酶IV的IC50值抑制试验:
| 化合物 | IC<sub>50</sub>/μM |
| 孢囊菌酰胺A | >160 |
| 孢囊菌酰胺A1 | 6.4±1.8 |
| 孢囊菌酰胺C | >60 |
| 孢囊菌酰胺D | 10±3 |
图9b显示了拓扑异构酶IV抑制测定的结果。促旋酶反应用不同浓度A-D 进行滴定,并通过琼脂糖凝胶电泳解析。对于IC50测定,超螺旋质粒的带强度使 用AdobePhotoshop来确定,对[孢囊菌酰胺]进行作图,并用Hill’s方程拟 合。
原核DNA拓扑异构酶IV和真核拓扑异构酶II具有高度的同源性(IIa型拓扑 异构酶),而原核酶抑制剂通常也抑制真核中的对应物8。为测试孢囊菌酰胺对于 真核拓扑异构酶IV的影响,使用了市售的人拓扑异构酶II试剂盒 (Inspiralis)。
孢囊菌酰胺A只在815μM的最高测试浓度抑制人拓扑异构酶II的活性。孢 囊菌酰胺A1抑制人拓扑异构酶II,显示出9±0.03μM的IC50值。孢囊菌酰胺 C只在300μM最高测试浓度抑制人拓扑异构酶II的活性。孢囊菌酰胺D抑制人 拓扑异构酶II显示出41.2±3μM的IC50值。
孢囊菌酰胺A-D对人拓扑异构酶II的IC50值抑制试验:
| 化合物 | IC<sub>50</sub>/μM |
| 孢囊菌酰胺A | >160 |
| 孢囊菌酰胺A1 | 9±0.03 |
| 孢囊菌酰胺C | >60 |
| 孢囊菌酰胺D | 41.2±3 |
图9c显示了拓扑异构酶II抑制测定的结果。拓扑异构酶II的反应用不同 浓度A-D进行滴定,并通过琼脂糖凝胶电泳解析。对于IC50测定,超螺旋质粒的 带强度使用AdobePhotoshop来确定,对[孢囊菌酰胺]进行作图,并用Hill’s 方程拟合。
除了ATP依赖型IIa型拓扑异构酶如大肠杆菌促旋酶,拓扑酶IV和人拓扑 酶II之外,孢囊菌酰胺对于ATP非依赖型人拓扑异构酶I的活性同样被测试。
孢囊菌酰胺A-D对人拓扑异构酶I的IC50值试验:
| 化合物 | IC<sub>50</sub>/μM |
| 孢囊菌酰胺A | ~10 |
| 孢囊菌酰胺A1 | ~0.7 |
| 孢囊菌酰胺C | ~6 |
| 孢囊菌酰胺D | ~33.6 |
图9d显示了拓扑异构酶I抑制测定的结果。拓扑I反应用不同浓度A-D进 行滴定,并通过琼脂糖凝胶电泳解析。对于IC50测定,超螺旋质粒的带强度使用 Adobe Photoshop来确定,对[孢囊菌酰胺]进行作图,并用Hill’s方程拟合。
孢囊菌酰胺A-D的IC50(促旋酶)对IC50(拓扑异构酶IV)的值的比较:

孢囊菌酰胺A和C显示了对于促旋酶分子靶点的强烈倾向(对促旋酶有40- 100倍更强的偏好)。A1和D几乎同样好地都靶向螺旋酶和拓扑异构酶IV(对促旋 酶有2.6-10倍更强的偏好)。
通常,对促旋酶抑制剂有两种描述的抑制模式/结合位点:
1.如氟喹诺酮类化合物结合到GyrA DNA复合物,并避免切口的双链DNA的 再连接(促旋酶中毒);和
2.另一方面氨基香豆素结合于GyrB上的ATP结合口袋(竞争性抑制)8。
为测试孢囊菌酰胺是否按照这两个抑制模式中任一种,用孢囊菌酰胺D进行 DNA/促旋酶复合物的线性化测定(A)和ATP竞争测定(B)。(A)在此,DNA与促旋酶 的复合物使用SDS进行捕获,而促旋酶使用蛋白酶K进行消化。如果促旋酶/DNA 复合体是由1型的促旋酶抑制剂捕获,这将导致线性化的质粒的形成(因为再连 接被抑制)。2型抑制剂-结合或无化合物的样品将不显示出线性化质粒的形成。 评估的结果在图10a中显示。环丙沙星(一种已知的促旋酶/DNA稳定剂)和孢囊 菌酰胺D显示出,在经蛋白酶K处理后,形成了线性质粒。在未处理的对照组中 未显示上述效果。因此,可能似乎孢囊菌酰胺以类似于氟喹诺酮类化合物的方式 稳定了共价的GyrA-DNA复合体。(B)在此,标准促旋酶反应被用一恒定量的孢囊菌酰胺D抑制,并用数量增加的ATP进行滴定。如果ATP和孢囊菌酰胺D都竞争 结合于促旋酶GyrB亚基上的ATP结合口袋,那么ATP的量增加将导致超螺旋质 粒在测定中形成。图10b显示了实验结果。甚至在10mM的最高的ATP浓度下(孢 囊菌酰胺浓度的2000倍),促旋酶的活性也没有恢复,这表明ATP结合口袋并不 是孢囊菌酰胺的结合位点。这一结果是与线性化的测定结果一致。
图11示出了DNA/促旋酶复合线性测定的结果。
实验步骤
促旋酶超螺旋法
为了测试孢囊菌酰胺,使用了市售大肠杆菌旋转酶超螺旋试剂盒 (Inspiralis,诺维奇,英国)。3对于标准反应,将0.5μg松弛型质粒与1单位 (~20.5nM)的大肠杆菌促旋酶混合于1×反应缓冲液中(30μl最终体积,参见试 剂盒手册),并在37℃下温育30分钟。该反应通过加入含有10%(w/v)SDS的DNA 凝胶上样缓冲液进行淬灭。将样品在0.8%(w/v)琼脂糖凝胶进行分离,并使用 Roti-凝胶染色(Carl Roth)观察DNA。
所有天然产物原液和稀释液在100%DMSO中制备,并加入到超螺旋反应中, 得到DMSO终浓度为5%(v/v)。环丙沙星原液和稀释液制备于10mM HCl和50%的 DMSO中,并在最后的分析中按1:10使用。
在测定中使用以下的天然产物浓度:
孢囊菌酰胺A:815.8μM;163μM;80μM,16μM;8μM;1.6μM;0.8μM;0.16μM; 0.08μM;0.016μM
孢囊菌酰胺A1:543.5μM;108.7μM;54μM;10.8μM;5.4μM;1.087μM; 0.54μM;0.108μM;0.054μM;0.0108μM
孢囊菌酰胺C:300μM;60μM;30μM;6μM;3μM;0.6μM;0.3μM;0.06μM; 0.03μM;0.006μM
孢囊菌酰胺D:347μM;173.5μM;86.75μM;43.38μM;21.69μM;10.84μM; 5.42μM;2.71μM;1.36μM;0.68μM;0.34μM;0.17μM;0.085μM;0.042μM; 0.021μM;0.0106μM;0.0053μM
对照组反应为:在5%(v/v)DMSO存在下进行标准反应,无酶。
在室温,DNA不存在情况下,所有反应样品平衡10分钟。然后加入松弛型质 粒开始反应。
蛋白酶K线性化分析
为测试孢囊菌酰胺是否稳定DNA促旋酶和切口的DNA底物之间的共价复合 物,进行蛋白酶K线性化测定(见a)。
在孢囊菌酰胺D(18μM;1.8μM)存在下,进行标准的促旋酶超螺旋实验。对照 反应不含促旋酶,没有抑制或已知的促旋酶/DNA复合物稳定剂环丙沙星(1μM)。 反应通过加入1/10体积的10%SDS进行淬灭。为了线性化带切口的DNA促旋酶 复合物,将50μg/ml蛋白酶K加入到该反应中,并在37℃下孵育30分钟。将样 品用0.8%(w/v)琼脂糖凝胶进行分离,并使用Roti-凝胶染色(Carl Roth)观察 DNA。为了检测线性化质粒带,松弛型质粒用单切割型限制性内切酶Nde I消化。
用浓度变化ATP进行解旋酶超螺旋测试
为测试孢囊菌酰胺是否与ATP竞争结合于GyrB的ATP结合口袋,进行了不 同浓度ATP的标准促旋酶超螺旋测定(见a)。标准反应混合物(1mM ATP)中分别 补加ATP(0.5M ATP储备溶液,ATP购自Sigma-Aldrich公司)至ATP终浓度为 2.5;5和10mM。所有反应一式三份进行。
拓扑异构酶IV的松弛测试
为了测试孢囊菌酰胺的抗拓扑异构酶IV活性,使用了市售的大肠杆菌拓扑 异构酶IV松弛试剂盒(Inspiralis,诺维奇,英国)。4对于标准反应,将0.5μg 松弛型质粒与1单位(~20.5nM)的大肠杆菌拓扑异构酶IV混合于1×反应缓冲 液中(参见试剂盒手册),并在37℃下温育30分钟。该反应通过加入含有 10%(w/v)SDS的DNA凝胶上样缓冲液进行淬灭。将样品用0.8%(w/v)琼脂糖凝胶 分离,并使用Roti-凝胶染色(Carl Roth)观察DNA。
在测定中使用以下的天然产物浓度:
孢囊菌酰胺A:815.8μM;163μM;80μM,16μM;8μM;1.6μM;0.8μM;0.16μM; 0.08μM;0.016μM
孢囊菌酰胺A1:543.5μM;108.7μM;54μM;10.8μM;5.4μM;1.087μM; 0.54μM;0.108μM;0.054μM;0.0108μM
孢囊菌酰胺C:300μM;60μM;30μM;6μM;3μM;0.6μM;0.3μM;0.06μM; 0.03μM;0.006μM
孢囊菌酰胺D:347μM;173.5μM;86.75μM;43.38μM;21.69μM;10.84μM; 5.42μM;2.71μM;1.36μM;0.68μM;0.34μM;0.17μM;0.085μM;0.042μM; 0.021μM;0.0106μM;0.0053μM
对照组反应为:在5%(v/v)DMSO存在下进行标准反应,无酶。在室温,DNA 不存在情况下,所有反应样品平衡10分钟。然后加入松弛型质粒开始反应。
拓扑异构酶II的松弛测试
为了测试孢囊菌酰胺的抗拓扑异构酶II活性,使用了市售的人拓扑异构酶 IV松弛试剂盒(Inspiralis,诺维奇,英国)。4对于标准反应,将0.5μg超螺旋 质粒与1单位(~20.5nM)的大肠杆菌拓扑异构酶II混合于1×反应缓冲液中(参 见试剂盒手册),并在37℃下温育30分钟。该反应通过加入含有10%(w/v)SDS的 DNA凝胶上样缓冲液进行淬灭。将样品用0.8%(w/v)琼脂糖凝胶分离,并使用 Roti-凝胶染色(Carl Roth)观察DNA。
在测定中使用以下的天然产物浓度:
孢囊菌酰胺A:815.8μM;163μM;80μM,16μM;8μM;1.6μM;0.8μM;0.16μM; 0.08μM;0.016μM
孢囊菌酰胺A1:543.5μM;108.7μM;54μM;10.8μM;5.4μM;1.087μM; 0.54μM;0.108μM;0.054μM;0.0108μM
孢囊菌酰胺C:300μM;60μM;30μM;6μM;3μM;0.6μM;0.3μM;0.06μM; 0.03μM;0.006μM
孢囊菌酰胺D:347μM;173.5μM;86.75μM;43.38μM;21.69μM;10.84μM; 5.42μM;2.71μM;1.36μM;0.68μM;0.34μM;0.17μM;0.085μM;0.042μM; 0.021μM;0.0106μM;0.0053μM
对照组反应为:无酶,在5%(v/v)DMSO存在下进行标准反应。在室温, DNA不存在情况下,所有反应样品平衡10分钟。然后加入松弛型质粒开始反应。
拓扑异构酶I的松弛测试
为了测试孢囊菌酰胺的抗拓扑异构酶II活性,使用了市售的人的拓扑异构 酶I松弛试剂盒(Inspiralis,诺维奇,英国)。4对于标准反应,将0.5μg松弛 型质粒与1单位(~20.5nM)的智人的拓扑异构酶I混合于1×反应缓冲液中(参 见试剂盒手册),并在37℃下温育30分钟。该反应通过加入含有10%(w/v)SDS的 DNA凝胶上样缓冲液进行淬灭。将样品用0.8%(w/v)的琼脂糖凝胶进行分离,并 使用Roti-凝胶染色(Carl Roth)观察DNA。
在测定中使用以下的天然产物浓度:
孢囊菌酰胺A:815μM;81.5μM;8.15μM
孢囊菌酰胺A1:543μM;54.3μM;5.43μM
孢囊菌酰胺C:300μM;30μM;3μM
孢囊菌酰胺D:277μM;27.2μM;2.77μM
对照组反应为:无酶,在5%(v/v)DMSO存在下进行标准反应。在室温,DNA 不存在情况下,所有反应样品平衡10分钟。然后加入松弛型质粒开始反应。
定量和分析
为了确定IC50值,超螺旋的(螺旋酶)或宽松型(拓扑异构酶I,II,IV)质粒 的形成使用Adobe Photoshop(直方图模式)量化。这些值相对于化合物浓度的作 图,产生了S形曲线的形状,这是用Hill’s方程拟合的(Origin Pro 8.5)。所 有确定的IC50值是三次独立实验的平均值。
参考文献:
[1]T.Gruger,J.L.Nitiss,A.Maxwell,E.L.Zechiedrich,P. Heisig,S.Seeber,Y.Pommier,D.Strumberg,抗微生物剂化疗 (Antimicrob.Agents Chemother.)48,2004,4495-4504.
[2]H.Schedletzky,B.Wiedemann,P.Heisig,抗微生物化学治疗杂 志(J.Antimicrob.Chemother.)43,1999,31-37.
[3]A.B.Khodursky,E.L.Zechiedrich,N.R.Cozzarelli,美国 科学院院报(Proc.Natl.Acad.Sci.USA)92,1995,11801-11805.
[4]A.Schulte,P.Heisig,抗微生物化学治疗杂志(J.Antimicrob. Chemother.)46,2000,1037-1046.
[5]D.Keeney,A.Ruzin,F.McAleese,E.Murphy,P.A.Bradford, 抗微生物化学治疗杂志(J.Antimicrob.Chemother.)61,2008,46-53.
[6]P.Heisig,H.Schedletzky,H.Falkenstein-Paul,抗微生物剂 化疗(Antimicrob.Agents Chemother.)37,1993,669-701.
[7]T.Mosmann,免疫学方法杂志(J.Immunol.Meth.)65,1983,55- 63.
[8]Pommier,Y.;Leo,E.;Zhang,H.;Marchand,C.化学和生物学 (Chemistry&Biology)2010,17,421.
孢囊菌酰胺A和C的合成
首先,对于孢囊菌酰胺C的合成的描述,可进一步阐述其他的孢囊菌酰胺。
1.1.孢囊菌酰胺C
以下方案1和2提供了合成单个芳香构建单元,随后通过组装这些构建单元 生成孢囊菌酰胺C的概述。
可选地,方案1中的步骤e)可以通过使用其他的醇(R’OH)替代iPrOH进行改 变。如果,例如使用了EtOH,则可以制备得到孢囊菌酰胺H的构建单元。同样的 也适用于方案1中的第二个反应顺序中的步骤b)。在此,iPrOH同样也可以用其 他的醇(R’OH)替代。如果,例如使用了MeOH,则可以制备得到孢囊菌酰胺C、G 和H的构建单元。对于孢囊菌酰胺F的制备,对氨基苯甲酸衍生物如对氨基苯甲 酸或相应的N-保护的氨基苯甲酸衍生物,以及对-硝基苯甲酸被使用以代替构建 单元B。
方案1:合成芳烃A和B然后酰胺偶合。
(中央芳香部分)

a)BBr3,CH2Cl2,-40℃-rt,17h(95%);b)NaBH4,THF,-40℃-rt,30min(91%);c)PhCH(OMe)2, pTSA.H2O,THF,rt,5天(56%);d)Ni(NO3)2.5H2O,pTsOH.H2O,丙酮,rt,2.5h(74%);e)iPrOH,DEAD, PPh3,THF,rt,17h(85%);f)Pd2(dba)3,(PhO)3P,iPrOH,二氧六环,80℃,1.5h(70%);g)崁酮-1莰酮磺酸, CH2Cl2/MeOH(1:2),0℃-rt,17h(90%);h)MnO2,CH2Cl2,rt,17h(81%);i)2-甲基-2-丁烯, NaClO2/NaH2PO4,tBuOH,rt,17h(75%);j)TMSCHN2,MeOH/PhMe,0℃-rt,30min(57%);k)BnOH, DEAD,PPh3,THF,rt,17h(90%);l)LiOH,THF/H2O(1:1),rt,17h(99%).
(末端三取代芳香部分)

a)TMSCHN2,MeOH/PhMe,0℃-rt,30min(90%);b)/PrOH,DEAD,PPh3,THF,rt,17h(定量);c) Pd/C,MeOH,H2atm.,rt,17h(定量).
(耦合芳香部分A和B)

a)I.A,Goshez‘s试剂,CH2Cl2,40℃,3h;II.B,DIPEA,CH2Cl2,rt,10min,然后I.,40℃,2天(68%)
方案2:最终合成孢囊菌酰胺C。
孢囊菌酰胺C(最终合成)

a)Pd/C,MeOH,H2atm.,rt,3h(96%);b)I.4-Boc氨基苯甲酸,Goshez’试剂,CH2Cl2,rt,1h;II.B, DIPEA,CH2Cl2;然后I.,rt,1day(7天);c)TFA/CH2Cl2(10:1),rt,17h(定量);d)LiOH,THF/H2O(1:1), rt,17h(99%).
1.2孢囊菌酰胺A
更复杂的孢囊菌酰胺由双酰胺(表示为孢囊菌酰胺C),二芳基酰胺(片段C) 和手性接头元件组成。本节中,首先报道片段C和手性接头元件,随后组装所有 三个元件从而提供孢囊菌酰胺A。
1.2.1二芳烃C的合成。
方案3:活化片段C的合成。
片段C

a)P(OMe)3,I2,THF,3天(75%);b)LiOH,THF/H2O(1:1),rt,17h(80%);
c)氯甲酸乙酯,Et3N,CH3CN,0℃30min(67%)。
1.2.2合成连接有二芳烃C的手性构建单元D
该合成从肉桂酸甲酯起始,手性通过Sharpless不对称二羟基化引入。该苯 环作为第二羧酸的保护基,其不会被氧化释放。最后,构建单元C与自由氨基基 团相连接。相应的对映体片段(ent)-D通过使用AD混合物α替代AD混合物β 进行制备。
方案4:从肉桂酸甲酯起始合成羧酸D。

a)AD混合物β,MeSO2NH2,tBuOH/H2O(1:1),0℃,12h,然后25 ℃,12h,(79%,ee>99%);b)33%HBr/HOAc,45℃,30min.,(71%);c) NaN3,DMF,25℃,3h,然后40℃,2h,(89%);d)KOH,THF/H2O;e)2. MeI,Ag2O,CaSO4(两步骤74%);f)KOH,THF/H2O;g)Me2N-CH(OtBu)2,甲苯, 80℃,(两步骤87%);h)RuCl3·H2O,NaIO4,CHCl3/CH3CN/H2O,70℃;i)MeI,Ag2O,CaSO4;j)Ph3P,THF/H2O,50℃,k)DMF(四步骤16%);l)CF3CO2H, CH2Cl2,(定量).
方案5:最终合成孢囊菌酰胺A。

a)HOAt,EDC.HCl,DIPEA,CH2Cl2,rt,17h(75%);b)LiOH, THF/H2O(1/1),rt,(95%).
2.实验
2.1常规实验信息
除非另有说明,所有反应均在烘箱干燥的玻璃器皿中,在氮气气氛下进行。 1H-NMR谱用Bruker AVS-400,在400MHz下记录,或用Bruker DRX-500,在500 MHz下记录。13C-NMR谱用Bruker AVS-400,在100MHz下记录,或用Bruker DRX-500,在125MHz下记录。多重性使用以下缩写描述:s=单峰,d=双峰, t=三重峰,q=四重峰,m=多重峰,b=宽峰。1H和13C NMR谱的化学位移值通常报 告为相对于作为内标的残留溶剂信号的ppm值。重数指在偏共振解耦光谱的共 振。这些通过使用无畸变极化转移增强(DEPT)光谱编辑技术,次级脉冲在90°和 135°阐明。多重性使用以下缩写报导:s=单重峰(由于季碳),d=双重峰(次甲基),t=三重峰(亚甲基),q=四重峰(甲基)。质谱(EI)在70eV下,将VG Autospec型分光计(质谱仪)、LCT型(ESI)(质谱仪)或Q-TOF型(质谱仪)光谱仪 与Waters Aquity超高效液相色谱系统组合使用获得。使用预涂硅胶60F254板 (Merck,达姆施塔特Darmstadt)进行薄层色谱法分析,在254nm紫外光下显示 斑点,或可选地用高锰酸钾、磷钼酸、2,4-二硝基苯酚或对-茴香醛溶液进行染 色。四氢呋喃(THF)在氮气下从钠/苯甲酮蒸馏得到。二氯甲烷(CH2Cl2)使用溶剂 纯化系统(SPS)进行干燥。使用供应的市售可得的试剂。制备高效液相色谱法使用 Merck Hitachi LaChrom系统(泵L-7150,接口D-7000,二极管阵列检测器L-7450(λ=220-400nm,优选在监测λ=230nm))和柱(在括号中给出的实验部分提到的 缩写):Trentec Reprosil-PUR120C18AQ 5μm,250×8mm,保护柱,40×8 mm(C18-SP)。用Merck硅胶60(230-400目)进行快速柱色谱。用于快速色谱法的 洗脱剂在使用前蒸馏。使用的SRSOptiMelt装置测量熔点。旋光度[α]用旋光计 341(Perkin Elmer公司),在波长为589nm下测量,以10-1度cm2g-1示出。
2.2具体步骤
4-氨基甲基苯甲酸

在烧瓶中加入甲醇(200ml)溶液,并缓慢加入乙酰氯(2.6mL,36.5mmol,1 eq)。然后加入4-氨基苯甲酸(5.00g,36.5mmol),并在室温下将该溶液搅拌7天。 减压下除去溶剂,得到米色固体4-氨基甲基苯甲酸酯(5.38g,35.59mmol,定 量)。
达到熔点前,题述化合物被分解。
ATR-IR(净值): 2015,1724,1612,1558,1508,1430,1316,1280,1181,1109,1072,1022,984,959,853,786,757,686,653cm-1.
2015,1724,1612,1558,1508,1430,1316,1280,1181,1109,1072,1022,984,959,853,786,757,686,653cm-1.
1H-NMR(400MHz,CD3OD):δ8.19-8.13(m,2H),7.53-7.48(m,2H),3.93(s,3H) ppm.
13C-NMR(100MHz,CD3OD):δ167.2 137.0,132.4,131.7,124.2,53.0ppm
HRMS(ESI):C8H10NO2(M+H)+计算值:152.0712,测定值:152.0706.
4-(4-硝基苯甲酰氨基)苯甲酸甲酯

P(OMe)3(3.5mL,29.8mmol)的CH2Cl2(100mL)溶液在冰浴中冷却,然后加 入I2(7.56g,29.8mmol)。固体碘完全溶解后,相继加入对硝基苯甲酸(5.52g,29.8 mmol)和Et3N(4.70mL,33.7mmol),溶液在冰浴中搅拌10分钟。加入4-氨基苯 甲酸甲酯(3.00gr,19.9mmol),并将该混合物搅拌10分钟。除去冷却浴后,在室 温下,将反应混合物搅拌3天,然后用饱和NaHCO3水溶液稀释,并用二氯甲烷 (3x)萃取。合并的有机相依次用H2O,1M HCl,H2O和盐水洗涤。合并的有机层用 无水MgSO4干燥,溶剂在真空下浓缩,得到米色固体题述化合物(4.4g,14.65 mmol,75%)。
mp:245–246℃
1H NMR(400MHz,DMSO)δ10.87(s,1HNH),8.39(d,J=8.8Hz,2H),8.20(d,J =8.8Hz,2H),7.99(d,J=8.8Hz,2H),7.95(d,J=8.8Hz,2H),3.84(s,3HOMe)ppm.
13C NMR(100MHz,DMSO)δ166.2,164.9,149.77,143.6,140.7,130.7,129.8,125.3,124.2,120.2,52.4ppm.
HRMS(ESI):C15H13N2O2Na(M+H)+计算值:301.0824,测定值:301.0828.
4-(4-硝基苯甲酰氨基)苯甲酸

4-(4-硝基苯甲酰氨基)苯甲酸甲酯(4.32g,14.38mmol)溶于THF/H2O(77/77 mL)的1/1混合物中。然后,加入固体LiOH(5.16g,215.66mmol),并将该体系在 室温下搅拌17小时。加入1M HCl直到pH~1,过滤所得的固体,真空下干燥。 得到浅黄色固体题述化合物(3.3g,11.54mmol,80%)。
mp:322–324℃
1H NMR(400MHz,C6D6)δ10.83(s,1HCO2H),8.34(d,J=8.6Hz,1H),8.29(d,J =8.6Hz,1H),8.13(d,J=8.6Hz,1H),8.06(d,J=8.6Hz,1H),7.75(s,1HNH)ppm.
13C NMR(100MHz,C6D6)δ168.2,164.6,162.2,149.7,143.9,141.1,131.1, 129.8,123.5,120.4ppm.
HRMS(ESI):C14H9N2O5(M-H)-计算值:285.0511,测定值:285.0506.
(乙基碳酸)4-(4-硝基苯甲酰氨基)苯甲酸酐

搅拌下,在0℃下向4-氨基苯甲酸(1.5g,10.9mmol)和N,N-二甲基苯胺(2.0g,10.9mmol)的丙酮溶液中加入4-硝基苯甲酰氯。然后,将反应混合物温热至室温 并再搅拌一小时。将所得的固体过滤并在DMF中通过重结晶纯化,得到4-(4-硝 基-苯甲酰基氨基)-苯甲酸(2.75g,88%)。
4-(4-硝基-苯甲酰基氨基)-苯甲酸(0.6g,2.1mmol)溶解于14ml CH3CN中。然 后在0℃下将Et3N(0.31ml,2.2mmol)加入。在所得的溶液中加入氯甲酸乙酯。在 0℃下搅拌30分钟后,将白色沉淀物过滤并用冷CH3CN洗涤,然后在室温下高 真空干燥,得到题述酐0.5克,67%。
1H-NMR(400MHz,DMSO,DMSO=2.50ppm):δ=1.33(dd,J=7.2Hz,3H), 4.37(q,J=7.2Hz,2H),8.02-8.09(m,4H),8.21(d,J=8.8Hz,2H),8.40(d,J=8.8Hz, 2H),11.01(s,1H).
3-羟基-4-硝基苯甲酸甲酯

0℃下,TMSCHN2(2.0M的Et2O溶液,13.20mL,26.48mmol)加入3-羟基- 2-硝基苯甲酸(2.50g,13.65mmol)的甲苯/甲醇混合物(81/36mL)溶液中。在0℃ 下搅拌30分钟后,将溶剂在真空中蒸发,得到油状残余物,将其通过快速色谱 纯化(石油醚/乙酸乙酯=9:1),得到标题化合物(2.43g,12.33mmol,90%),为黄色 固体。
mp:91–92℃
1H NMR(400MHz,CDCl3)δ10.49(s,1H-OH),8.17(d,J=8.8Hz,1H),7.83(d,J =1.8Hz,1H),7.61(dd,J=8.8,1.8Hz,1H),3.96(s,3H)ppm.
13C NMR(100MHz,CDCl3)δ165.0,154.8,138.1,125.4,121.8,120.74,53.1 ppm.
HRMS(ESI):C8H6NO5(M-H)-计算值:196.0246,测定值:196.0249.
3-异丙氧基-4-硝基苯甲酸甲酯

3-羟基-4-硝基苯甲酸甲酯(2.30g,10.89mmol)溶解于THF(100mL)中。加入 iPrOH(1.10mL,14.16mmol)和PPh3(3.90g,14.70mmol),混合物搅拌直至所有成 份被溶解。然后加入DEAD(2.2M的甲苯溶液,14.16mmol,6.50mL),并将该混 合物在室温下搅拌17小时。真空蒸发溶剂,得到油状残余物,将其通过快速色 谱纯化(石油醚/乙酸乙酯=95:5),得到标题化合物(2.61g,10.91mmol,定量),为 黄色油状物。
1H NMR(400MHz,CDCl3)δ7.75(d,J=8.4Hz,2H),7.64(dd,J=8.3,1.6Hz, 1H),4.77(hept,J=6.1Hz,1H),3.95(s,3H),1.41(s,3H),1.40(s,3H)ppm.
13C NMR(100MHz,CDCl3)δ165.5,150.9,134.6,125.2,121.2,117.1,73.2,52.9,21.9ppm.
HRMS(Qtof):C8H6NO5(M+Na)+计算值:262.0691,测定值:262.0700.
3-异丙氧基-4-氨基苯甲酸甲酯
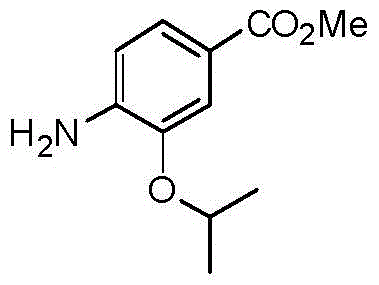
3-异丙氧基-4-硝基苯甲酸甲酯(2.60g,10.87mmol)溶于MeOH(91.0mL)中并 脱气。加入Pd/C(10%wt.,0.58g,0.54mmol),冷却下抽气除去空气。烧瓶用H2冲洗,悬浮液在室温下搅拌17小时。催化剂用硅藻土过滤,用MeOH洗涤并减 压除去溶剂。将粗产物通过快速色谱法(石油醚/EtOAc=7/3)纯化。得到3-异丙基- 4-氨基苯甲酸甲酯(2.27g,10.85mmol,定量),为浅橙色固体。
mp:55–57℃
1H NMR(400MHz,CDCl3)δ7.51(dd,J=8.2,1.7Hz,1H),7.46(d,J=1.7Hz, 1H),6.66(dd,J=8.2,5.1Hz,1H),4.63(sept,J=5.1Hz,1H),3.85(s,3H),1.36(s, 3H),1.35(s,3H)ppm.
13C NMR(100MHz,CDCl3)δ167.5,144.24,142.3,124.0,119.5,114.1,113.5,70.9,51.8,22.3ppm.
HRMS(ESI):C11H16NO3(M+H)+计算值:210.1130,测定值:210.1126.
6-溴-2,3-二羟基苯甲醛

在-30℃下,往6-溴-2-羟基-3-甲氧基苯甲醛(25.0g,108.2mmol)的CH2Cl2(270mL)溶液中,通过附加的漏斗在45分钟内缓慢加入BBr3(1M的CH2Cl2溶 液,200.0mL,200.0mmol)。溶液被升温至室温,搅拌17小时。加入H2O,反应 混合物继续搅拌30分钟。溶液用EtOAc(3x)萃取,并用H2O洗涤。合并的有机 层用无水MgSO4干燥,过滤,并在真空下浓缩,得到题述化合物(22.16g,102.11 mmol,95%),为黄色固体。
mp:135–136℃
1H NMR(400MHz,CDCl3)δ12.13(d,J=0.5Hz,1H-OH),10.27(s,1H-CHO), 7.07(d,J=8.5Hz,1H),7.02(dd,J=8.5,0.5Hz,1H),5.67(s,1H-OH)ppm.
13C NMR(100MHz,CDCl3)δ198.4,151.2,145.0,124.4,122.0,117.5,116.1 ppm.
HRMS(ESI):C7H4BrO3(M-H)-计算值:214.3943,测定值:214.9344.
4-溴-3-羟基甲基苯-1,2-二醇

在-40℃下,6-溴-2,3-二羟基苯甲醛(22.16g,102.10mmol)的THF(650mL) 溶液用NaBH4(3.86g,102.10mmol)分批(3x)处理。所得的混合物在室温下搅拌30 分钟。加入NH4Cl的饱和水溶液,混合物再度搅拌10分钟,最后用1M HCl处 理。再度搅拌10分钟后,水相用EtOAc(3x)萃取。合并的有机层用无水MgSO4干燥并过滤。减压去除溶剂,得到题述化合物(20.27g,92.53mmol,91%),为无色 固体。
mp:90–92℃
1H NMR(400MHz,MeOD)δ6.88(d,J=8.5Hz,1H),6.64(d,J=8.5Hz,1H), 4.82(s,2H)ppm.
13C NMR(100MHz,MeOD)δ147.1,146.1,126.9,123.9,116.6,114.4,61.1ppm.
HRMS(ESI):C7H6BrO3(M-H)-计算值:216.9500,测定值:216.9505.
5-溴-2-苯基-4H-苯并-[1,3]二氧杂环己烯-8-醇

4-溴-3-羟基甲基苯-1,2-二醇(20.27g,92.53mmol)的THF(550mL)溶液用 PhCH(OMe)2(20.8mL,138.8mmol)和pTSA.H2O(0.19g,1.02mmol)处理。混合物 在室温下搅拌5天。加入CH2Cl2,然后相继用5%NaHCO3水溶液和盐水洗涤。 水相用EtOAc(3x)萃取。合并的有机萃取物用无水MgSO4干燥,过滤并减压除去 溶剂。通过快速色谱法(石油醚/EtOAc=95/5)纯化,得到5-溴-2-苯基-4H-苯并-[1,3] 二氧杂环己烯-8-醇(16.02g,52.16mmol,56%),为无色固体。
mp:89–91℃
1H NMR(400MHz,CDCl3)δ7.62–7.55(m,2H),7.50–7.43(m,3H),7.07(d,J =8.6Hz,1H),6.78(d,J=8.6Hz,1H),5.97(s,1H),5.40(s,1H-OH),4.99(s,2H)ppm.
13C NMR(100MHz,CDCl3)δ144.0,141.8,136.1,130.1,128.8,126.7,124.9,121.0,115.0,109.4,100.0,67.8ppm.
HRMS(ESI):C14H10BrO3(M-H)-计算值:304.9813,测定值:304.9813.
5-溴-7-硝基-2-苯基-4H-苯并-[1,3]二氧杂环己烯-8-醇

5-溴-2-苯基-4H-苯并[1,3]二氧杂环己烯-8-醇(6.00g,19.54mmol;最大量)溶解于丙酮(250mL)。然后加入Ni(NO3)2.5H2O(5.68g,19.54mmol)和pTSA.H2O (3.72g,19.54mmol)。将混合物在室温下搅拌2.5小时。将反应混合物通过硅藻土 过滤,用CH2Cl2洗涤,真空下浓缩。通过快速色谱法(干燥负载:SiO2+ CH2Cl2;石油醚/乙酸乙酯=9:1),得到题述化合物(5.08g,14.43mmol,74%),为亮 黄色固体。
mp:154–156℃
1H NMR(400MHz,CDCl3)δ10.60(s,1H-OH),7.96(s,1H),7.65–7.57(m,2H), 7.48–7.42(m,3H),6.02(s,1H),4.99(s,2H)ppm.
13C NMR(100MHz,CDCl3)δ144.9,135.5,133.2,130.2,129.0,128.9,126.7,119.2,109.2,99.9,67.4ppm.
HRMS(ESI):C14H9BrNO5(M-H)-计算值:359.9664,测定值:349.9660.
5-溴-8-异丙氧基-7-硝基-2-苯基-4H-苯并-[1,3]二氧杂环己烯

5-溴-7-硝基-2-苯基-4H-苯并-[1,3]-二氧杂环己烯-8-醇(13.79g,39.16mmol)溶 解于THF(429mL)中。加入iPrOH(4.00mL,50.91mmol)和PPh3(13.87g,52.87 mmol),搅拌混合物直至所有成份被溶解。缓慢(通过注射器泵)加入DEAD(2.2M 的甲苯溶液,23.1mL,50.91mmol),并将该混合物在室温下搅拌17小时。真空 蒸发溶剂,得到油状残余物,将其通过快速色谱纯化(石油醚/乙酸乙酯=96:4), 得到标题化合物(13.08g,33.18mmol,85%),为无色固体。
mp:87–89℃
1H NMR(400MHz,CDCl3)δ7.59(s,1H),7.59–7.54(m,2H),7.50–7.43(m, 3H),5.97(s,1H),5.00(s,2H),4.69(hept,J=6.2Hz,1H),1.31(d,J=6.2Hz,3H), 1.28(d,J=6.2Hz,3H)ppm.
13C NMR(100MHz,CDCl3)δ216.8,149.0,144.5,139.9,135.7,130.1,128.8,126.4,126.2,119.8,112.7,99.7,78.1,67.6,22.6,22.4ppm.
HRMS(Qtof):C14H9BrNO5(M+Na)+计算值:416.0110,测定值:416.0101.
8-异丙氧基-7-硝基-2-苯基-4H-苯并-[1,3]-二氧杂环己烯,73

5-溴-8-异丙氧基-7-硝基-2-苯基-4H-苯并-[1,3]二氧杂环己烯72(4.00g,10.15mmol),Pd2(dba)3(0.93g,1.01mmol),(PhO)3P(0.53mL,2.03mmol),Cs2CO3(4.30g,13.19mmol)和iPrOH(4.7mL,60.88mmol)溶解于1,4-二氧六环(28mL)中。油浴 预热至60℃,混合物在80℃下搅拌1.5小时。反应混合物通过硅藻土过滤并用 EtOAc洗涤。合并的有机提取物用无水MgSO4干燥,并在真空下浓缩。粗产物用 快速色谱纯化(石油醚/乙酸乙酯=96:4),得到题述化合物(2.24g,7.10mmol, 70%),为浅黄色固体。
mp:80–82℃
1H NMR(400MHz,CDCl3)δ7.65–7.55(m,2H),7.51–7.41(m,3H),7.37(d,J =8.5Hz,1H),6.81(d,J=8.5Hz,1H),6.01(s,1H),5.19(d,J=15.5Hz,1H),5.03(d, J=15.5Hz,1H),4.71(hept,J=6.2Hz,1H),1.32(d,J=6.2Hz,3H),1.28(d,J=6.2 Hz,3H)ppm.
13C NMR(100MHz,CDCl3)δ147.67,144.27,140.55,136.26,129.85,128.72,126.54,126.34,118.82,116.69,99.61,77.71,66.44,22.65,22.41ppm.
HRMS(QTof):C17H17NO5Na(M+Na)+计算值:338.1004.测定值:338.1003.
6-羟甲基-2-异丙氧基-3-硝基苯酚

0℃下,向8-异丙氧基-7-硝基-2-苯基-4H-苯并-[1,3]-二氧杂环己烯(4.24g,13.43mmol)在MeOH(102mL)和CH2Cl2(42mL)的混合物中,加入樟脑磺酸(3.12 g,13.43mmol)。混合物在室温下搅拌17小时。反应混合物用Et3N淬灭至pH~ 8,真空下浓缩并用快速色谱纯化(石油醚/乙酸乙酯=7:3),得到题述化合物(2.75 g,12.09mmol,90%),为褐色固体。
mp:39–41℃
1H NMR(400MHz,CDCl3)δ7.46(d,J=7.4Hz,1H),7.12(d,J=7.4Hz,1H), 6.61(s,1H-OH),4.81(d,J=3.5Hz,2H),4.39(hept,J=7.4Hz,1H),1.36(s,3H),1.35 (s,3H)ppm.
13C NMR(100MHz,CDCl3)δ148.9,138.5,132.4,122.1,116.5,79.2,61.3,22.5ppm.
HRMS(ESI):C10H12NO5(M-H)-计算值:226.0715,测定值:226.0717.
2-羟基-3-异丙氧基-4-硝基苯甲醛

6-羟甲基-2-异丙氧基-3-硝基苯酚(2.97g,13.05mmol)溶于CH2Cl2(58mL) 中。然后加入MnO2(11.35g,130.53mmol),混合物在室温下搅拌17h。混合物 用硅藻土过滤,并用CH2Cl2洗涤。将溶剂浓缩,得到标题化合物(2.38g,10.57 mmol,81%),为棕色油状物。
1H NMR(400MHz,CDCl3)δ11.44(s,1H-CHO),9.97(s,1H-OH),7.39(d,J=8.4 Hz,1H),7.23(d,J=8.4Hz,1H),4.88(hept,J=6.2Hz,1H),1.33(s,3H),1.32(s,3H) ppm.
13C NMR(100MHz,CDCl3)δ196.39,156.53,149.36,139.74,127.28,122.57,114.32,77.42,77.16,22.51.ppm.
HRMS(ESI):C10H10NO5(M-H)-计算值:224.0559.测定值:224.0535.
2-羟基-3-异丙氧基-4-硝基苯甲酸

2-羟甲基-3-异丙氧基-4-硝基苯甲醛(2.36g,10.49mmol)溶于叔丁醇(71mL) 中。相继加入2-甲基-2-丁烯(2M的THF溶液,36.7mL,73.45mmol)以及NaClO2 (2.85g,31.48mmol)和NaH2PO4(6.32g,47.22mmol)的H2O溶液(51mL)。反应混 合物在室温下搅拌17小时。加入6M NaOH,直到ph~10并在真空中浓缩溶剂。 加入H2O,有机层用石油醚(2x)提取。水层用6M HCl酸化直至pH~1,并用乙酸 乙酯(3x)萃取。合并有机萃取物,用MgSO4干燥并过滤。在真空下将溶剂浓缩, 得到标题化合物(1.90g,7.87mmol,75%),为黑色蜡状。
1H NMR(400MHz,MeOD)δ7.72(d,J=8.7Hz,1H),7.15(d,J=8.7Hz,1H), 4.86–4.82(m,1H),1.28(s,3H),1.26(s,3H)ppm.
13C NMR(100MHz,MeOD)δ172.7,158.0,140.0,125.8,117.4,113.8,77.5,22.6ppm.
HRMS(ESI):C10H10NO6(M-H)-计算值:240.0508,测定值:240.0510.
2-羟基-3-异丙氧基-4-硝基苯甲酸酯

0℃下,将TMSCHN2(2.0M的Et2O溶液,0.87mL,1.75mmol)加入2-羟基- 3-异丙氧基-4-硝基苯甲酸(0.32g,1.35mmol)的甲苯/甲醇混合物(10.4/2mL)溶液 中。在0℃下搅拌30分钟后,在真空中蒸发溶剂,得到油状残余物,将其通过 快速色谱纯化(SiO2.Et3N;石油醚/乙酸乙酯=95:5),得到标题化合物(0.24g,0.94 mmol,57%),为黄色油状物。
1H NMR(400MHz,CDCl3)δ11.29(s,1H-OH),7.63(d,J=8.8Hz,1H),7.12(d,J =8.8Hz,1H),4.84(hept,J=6.2Hz,1H),4.00(s,3H),1.32(s,3H),1.31(s,3H)ppm.
13C NMR(100MHz,CDCl3)δ198.2,188.9,176.1,170.0,157.0,149.2,139.8,123.9,115.7,113.4,77.4,53.2,22.5ppm.
HRMS(ESI):C11H12NO6(M-H)-计算值:254.0665,测定值:254.0666.
2-苄氧基-3-异丙氧基-4-硝基苯甲酸酯

2-羟基-3-异丙氧基-4-硝基苯甲酸酯(0.17g,0.69mmol)溶于THF(7.5mL)中。 加入BnOH(92.6μL,0.89mmol)和PPh3(0.24g,0.93mmol),搅拌混合物直至所有 的成份溶解。缓慢(通过注射器泵)加入DEAD(2.2M的甲苯溶液,0.41mL,0.89 mmol),并将该混合物在室温下搅拌17小时。在真空中蒸发溶剂,得到油状残余 物,将其通过快速色谱纯化(石油醚/乙酸乙酯=95:5),得到标题化合物(0.20g, 0.58mmol,85%),为无色油状物。
1H NMR(400MHz,CDCl3)δ7.53(d,J=8.6Hz,1H),7.50(d,J=8.6Hz,1H), 7.48–7.44(m,2H),7.42–7.35(m,3H),5.14(s,2H),4.74(hept,J=6.2Hz,1H),3.86 (s,3H),1.28(s,3H),1.26(s,3H)ppm.
13C NMR(100MHz,CDCl3)δ165.3,153.4,148.4,145.7,136.4,130.9,128.7,128.7,128.7,125.1,119.3,78.2,76.4,52.8,22.5ppm.
HRMS(QTof):C18H19NO6Na(M+Na)+计算值:368.1110,测定值:368.1112.
2-苄氧基-3-异丙氧基-4-硝基苯甲酸

2-苄氧基-3-异丙氧基-4-硝基苯甲酸酯(0.23g,0.67mmol)溶于THF/H2O (3.5/3.5mL)的1/1混合物中。然后,加入固体LiOH(0.16g,6.67mmol),并将该体 系在室温下搅拌17小时。水层用1M HCl酸化直至pH~1,并用EtOAc(3x)萃 取。合并有机萃取物,用MgSO4干燥并过滤。在真空下浓缩溶剂,得到标题化合 物(0.21g,0.63mmol,95%),为黄色蜡状。
1H NMR(400MHz,CDCl3)δ7.91(d,J=8.7Hz,1H),7.58(d,J=8.7Hz,1H), 7.41(s,5H),5.35(s,2H),4.71–4.62(m,1H),1.36(s,3H),1.35(s,3H)ppm.
13C NMR(100MHz,CDCl3)δ164.3,152.8,149.7,144.7,134.1,129.8,129.4,129.2,126.98,120.0,79.1,77.7,22.5ppm.
HRMS(ESI):C17H16NO6(M-H)-计算值:330.0978,测定值:330.0976.
4-(2-(苄氧基)-3-异丙氧基-4-硝基苯甲酰胺)-3-异丙基苯甲酸酯

40℃下,2-苄氧基-3-异丙氧基-4-硝基苯甲酸(51.5mg,0.16mmol)溶解于 CH2Cl2(8mL)并与Ghosez's试剂(66.0μL,0.50mmol)预活化3小时。3-异丙氧基- 4-氨基苯甲酸甲酯(0.12g,0.55mmol)溶解于CH2Cl2(8mL)中,并加入N,N-二异丙 基乙胺(DIPEA)(0.20mL,1.12mmol)。然后加入含酰氯的溶液,并在40℃下将反 应混合物搅拌2天。然后将溶剂除去并且将粗产物通过制备型HPLC纯化(RP- 18;运行时间100分钟;H2O/MeCN=100:0→0:100;tr=80分钟),得到标题 化合物(56.9mg,0.11mmol,68%),为浅黄色油状物。
1H NMR(400MHz,CDCl3)δ10.33(s,1H-NH),8.55(d,J=8.5Hz,1H),7.85(d,J =8.7Hz,1H),7.70(dd,J=8.5,1.7Hz,1H),7.59(d,J=8.7Hz,1H),7.57(d,J=1.7 Hz,1H),7.25–7.12(m,5H),5.25(s,2H),4.75–4.67(m,1H),4.67–4.59(m,1H), 3.93(s,3H),1.40(d,J=6.2Hz,6H),1.28(d,J=6.0Hz,6H)ppm.
13C NMR(100MHz,CDCl3)δ167.0,161.4,151.1,147.9,146.1,145.2,134.1,132.9,132.9,130.0,129.4,128.7,125.79,125.6,123.3,120.1,119.5,113.3,78.9,77.4,71.7,52.3,22.6,22.1ppm.
HRMS(ESI):C28H31N2O8(M+H)+计算值:523.2080,测定值:523.2075.
4-(4-氨基-2-羟基-3-异丙氧基苯甲酰氨基)-3-异丙氧基苯甲酸酯

4-[2-(苄氧基)-3-异丙氧基-4-硝基苯甲酰基]-3-异丙氧基苯甲酸酯(7.9mg,0.015mmol)溶于MeOH(0.5mL)中并脱气。加入Pd/C(10%wt.,2mg, 0.0014mmol),冷却下抽气除去空气。烧瓶用H2冲洗,悬浮液在室温下搅拌3小 时。催化剂用硅藻土过滤,用MeOH洗涤并在减压下除去溶剂。粗产物用快速色 谱纯化(石油醚/乙酸乙酯=7:3),得到题述化合物(5.8g,0.014mmol,96%),为黄 色固体。
1H NMR(400MHz,CDCl3)δ12.21(s,1H-OH),8.81(s,1H-NH),8.49(d,J=8.5 Hz,1H),7.69(dd,J=8.5,1.8Hz,1H),7.58(d,J=1.7Hz,1H),7.07(d,J=8.8Hz, 1H),6.28(d,J=8.7Hz,1H),4.80–4.72(m,1H),4.72–4.63(m,1H),4.28(s,2H-NH2), 3.91(s,3H),1.44(d,J=6.1Hz,6H),1.34(d,J=6.2Hz,7H)ppm.
13C NMR(100MHz,CDCl3)δ168.5,166.9,156.4,146.5,146.0,132.7,132.0,125.1,123.40,121.5,119.1,113.4,106.5,106.3,77.4,74.4,72.0,52.3,22.9,22.4ppm.
HRMS(ESI):C21H25N2O6(M-H)-计算值:401.1713,测定值:401.1716.
4-(叔丁氧基羰基氨基)苯甲酸

4-氨基苯甲酸(1.00g,7.29mmol)溶解于1,4-二氧六环(15mL)和H2O(7mL) 中。Et3N(2.0mL,14.58mmol)加入到该溶液中,在室温下将反应混合物搅拌5分 钟。然后一次性将二叔丁基二碳酸酯(3.18g,14.58m mol)加入到该溶液中并将反 应混合物搅拌24小时。在真空中除去溶剂,然后将3M盐酸加入到残余物中,得 到白色沉淀。然后过滤该浆体,并用H2O洗涤,然后在高真空下干燥。用热甲醇 重结晶,得到标题化合物,为无色固体(1.63g,6.85mmol,94%产率)。
mp:192–194℃.
1H NMR(400MHz,DMSO)δ9.73(s,1H-CO2H),7.83(d,2H,J=8.9Hz),7.55(d, 2H,J=8.9Hz),1.47(s,9H)ppm.
13C NMR(100MHz,CDCl3)δ167.1,152.6,143.8,130.4,124.0,117.2,79.7,28.1ppm.
HRMS(ESI):C12H15NnaO4(M+Na)+计算值:260.0893,测定值:260.0897.
光谱数据与文献报道一致(J.Am.Chem.Soc.2012,134,7406-7413)。
甲基-4-(4-(4-(叔丁氧羰基)氨基)苯甲酰氨基)-2-羟基-3-异丙氧基苯甲酰氨基)- 3-异丙氧基苯甲酸酯

4-(叔丁氧羰基氨基)苯甲酸(40.0mg,0.17mmol)溶解于CH2Cl2(8.4mL)中, 并在室温下与Ghosez's试剂(22.5μL,0.17mmol)预活化2个小时。4-(4-氨基-2-羟 基-3-异丙氧基苯甲酰氨基)-3-异丙氧基苯甲酸酯(68.4mg,0.17mmol)溶解在 CH2Cl2(8.4mL)溶液中,并加入N,N-二异丙基乙胺(DIPEA)(59.2μL,0.34 mmol)。然后加入含酰氯的溶液,在室温下将反应混合物搅拌1天。然后将溶剂 除去,粗产物通过制备型HPLC纯化(RP-18;运行时间100分钟;H2O/MeCN= 100:0→0:100;tr=70分钟)得到浅黄色油状标题化合物(47.3mg,0.076mmol, 72%)。
1H NMR(400MHz,CDCl3)δ7.98(d,J=7.5Hz,2H),7.78(d,J=1.4Hz,1H), 7.72(dd,J=7.5,1.4Hz,1H),7.69(s,1H-NH),7.68(d,J=7.3Hz,3H),7.56(d,J=7.5 Hz,1H),7.17(d,J=7.5Hz,1H),5.72(s,1H-NH),5.49(s,1H-NH),4.02–3.96(m,2H), 3.95(d,J=3.7Hz,3H),1.49(s,9H),1.46(d,J=5.6Hz,6H),1.41(d,J=5.5Hz,6H) ppm.
13C NMR(100MHz,CDCl3)δ166.89,166.67,166.61,158.88,154.93,146.90,141.47,135.07,134.68,131.70,130.38,130.38,127.26,127.17,123.25,121.40,120.63,120.63,115.87,114.85,113.39,106.06,80.65,75.89,74.13,52.08,28.41,28.41,28.41,21.80,21.80,21.80,21.80ppm.
HRMS(ESI):C33H38N3O9(M-H)-计算值:620.2687,测定值:620.2689.
甲基-4-(4-(4-氨基苯甲酰氨基)-2-羟基-3-异丙氧基苯甲酰氨基)-3-异丙氧基苯 甲酸酯

甲基-4-(4-(4-(叔丁氧基羰基)氨基)苯甲酰氨基)-2-羟基-3-异丙氧基苯甲酰氨基)-3-异丙氧基苯甲酸酯(40.0mg,0.064mmol)溶解于二氯甲烷/三氟乙酸的10/1混 合物(1mL)中,并在室温下搅拌17小时。减压下除去溶剂,并在高真空下除去残 留的酸,得到标题化合物(33.4mg,0.064mmol,定量),为黄色油状物。
1H NMR(400MHz,CDCl3)δ7.86(d,J=1.4Hz,1H),7.83(s,1H-NH),7.79(dd,J =7.5,1.4Hz,1H),7.75(d,J=7.5Hz,1H),7.70(d,J=7.5Hz,2H),7.65(d,J=7.5Hz, 1H),7.05(d,J=7.5Hz,1H),6.94(s,1H-NH),6.75(d,J=7.5Hz,2H),6.09(s,1H-OH), 4.02–3.97(m,1H),3.95–3.89(s,3H),3.92(m,1H),3.85(s,2H-NH),1.47(d,J=5.7 Hz,6H),1.40(d,J=5.5Hz,6H)ppm.
13C NMR(100MHz,CDCl3)δ166.89,166.67,166.61,158.88,152.59,146.90,135.07,134.68,131.70,130.93,130.93,127.17,123.25,122.42,121.40,115.87,114.85,114.35,114.35,113.39,106.06,75.89,74.13,52.08,21.80,21.80,21.80,21.80ppm.
HRMS(ESI):C28H32N3O7(M+H)+计算值:522.2162,测定值:522.2160.
孢囊菌酰胺C

甲基-4-[4-(4-氨基苯甲酰氨基)-2-羟基-3-异丙氧基苯甲酰氨基]-3-异丙氧基苯 甲酸酯(30.0mg,0.058mmol)溶于THF/H2O(0.3/0.3mL)的1/1混合物中。然后, 加入固体LiOH(13.9mg,0.58mmol),并将该体系在室温下搅拌17小时。水层用 1M HCl酸化直至pH~1,并用乙酸乙酯(3x)萃取。合并有机萃取物,用MgSO4干燥并过滤。在真空下将溶剂浓缩,得到标题化合物(27.4mg,0.054mmol, 93%),为黄色油状。
1H NMR(400MHz,CDCl3)δ7.91(d,J=1.4Hz,1H),7.87(dd,J=7.5,1.4Hz, 1H),7.70(d,J=7.5Hz,2H),7.65(d,J=7.5Hz,1H),7.53(d,J=7.5Hz,1H),7.05(d, J=7.5Hz,1H),6.95(s,1H-NH),6.77(s,1H-NH),6.75(d,J=7.5Hz,2H),6.12(s,1H- OH),3.97–3.89(m,2H),3.85(s,2H-NH),1.40(d,J=5.5Hz,6H),1.39(d,J=5.5Hz, 6H)ppm.
13C NMR(100MHz,CDCl3)δ167.79,166.67,166.61,158.88,152.59,149.81,136.38,135.07,134.68,130.93,130.93,125.08,123.25,122.80,122.42,120.37,114.35,114.35,113.76,113.39,106.06,75.89,74.13,21.80,21.80,21.80,21.80ppm.
HRMS(ESI):C28H32N3O7(M+H)+计算值:508.2006,测定值:508.2008.
(2S,3R)-2,3-二羟基-3-苯基丙酸甲酯
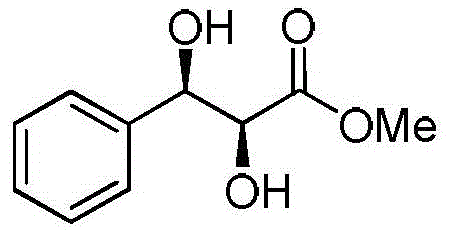
25℃下,AD混合物β(20.0g)溶解于tBuOH/H2O(1:1;142mL)混合物中。 然后将CH3SO2NH2(1.36g,14.3mmol,1.0eq.)加入,反应混合物冷却至0℃。然 后加入肉桂酸甲酯(2.31g,14.3mmol,1.0eq.),所得的混合物在0℃下猛烈搅拌 16h。在25℃下继续进行搅拌6h。反应混合物通过加入Na2SO3水溶液(21.4g, 170mmol,12.0eq.)水解,并继续搅拌2.5h。反应混合物用乙酸乙酯稀释并分层。 水相用EtOAc(3x)萃取。合并的有机相用H2O(1x)洗涤,并用Na2SO4干燥,过滤 并减压浓缩。通过快速色谱法(石油醚/乙酸乙酯=1:1)纯化,得到所需的二醇 (2.21g,11.3mmol,79%),为无色固体。光谱数据与文献报道一致。
Rf=0.38(PE/EtOAc 1:1);m.p.=84–85℃(lit:m.p.=80–81℃);[α]D 20=- 9.8°(c 1.28,CHCl3){文献:[α]D 26=-9.8°(c 1.07,CHCl3)};
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=7.42-7.29(5H,m,ArH), 5.03(1H,dd,J=2.7,7.2Hz,H-3),4.38(1H,dd,J=2.7,6.0Hz,H-2),3.82(3H,s,H- 8),3.12(1H,d,J=6.0Hz,OH-α),2.76(1H,d,J=7.2Hz,OH-β)ppm;
13C-NMR(100MHz,CDCl3,CHCl3=77.16ppm):δ=173.3(q,C-1),140.1(q, C-4),128.6(2C,t,C-6),128.3(t,C-7),126.3(2C,t,C-5),74.8(t,C-2),74.6(t,C-3), 53.1(p,C-8)ppm;HRMS(ESI):m/z C10H12O4Na[M+Na]+计算值:219.0633;测 量值219.0633.
(2R,3S)-2-乙酰氧基-3-溴-3-苯基丙酸甲酯(3)

在25℃下,向(2S,3R)-2,3-二羟基-3-苯基丙酸甲酯(2.15g,10.9mmol,1.0eq.)中逐滴加入HBr/HOAc(33%;16.9mL)。所得的混合物加热至45℃并搅拌30分 钟。然后反应混合物冷却至25℃,倒入冰冷的NaHCO3溶液(40mL)中。水相用 Et2O(3x)萃取。合并的有机相用H2O(1x)和盐水洗涤。然后,合并的有机相用 Na2SO4干燥,过滤并减压浓缩。通过快速色谱法(石油醚/乙酸乙酯=12.5:1)纯 化,得到题述化合物(2.32g,7.71mmol,71%),为无色固体。光谱数据与文献报道 一致。
Rf=0.79(PE/EtOAc 1:1);m.p.=78–82℃(lit:m.p.=78–79℃);[α]D20= +89.9°(c 1.74,CHCl3){文献:[α]D26=+100.3°(c 1.36,CHCl3)};
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=7.46-7.44(2H,m,H-6), 7.36-7.30(3H,m,H-5,H-7),5.65(1H,d,J=6.3Hz,H-3),5.35(1H,d,J=6.3Hz,H- 2),3.71(3H,s,H-9),2.11(3H,s,H-10)ppm;
13C-NMR(100MHz,CDCl3,CHCl3=77.16ppm):δ=169.7(q,C-1),167.5(q, C-8),136.8(q,C-4),129.3(t,C-7),128.7(4C,t,C-5,C-6),75.4(t,C-3),52.9(p,C-9), 49.3(t,C-2),20.6(p,C-10)ppm;
HRMS(ESI):m/z C12H13O4BrNa[M+Na]+计算值:322.9895;测量值 322.9891.
(2S,3R)-2-乙酰氧基-3-叠氮基-3-苯基丙酸甲酯

在25℃下,(2S,3R)-2-乙酰氧基-3-叠氮基-3-苯基丙酸甲酯(2.27g,7.55 mmol,1.0eq.)溶解于DMF(27.0mL)。然后加入NaN3(1.96g,30.2mmol,4.0 eq.),所得的混合物40℃加热3h。冷却后,反应混合物冷却至25℃,并加入 EtOAc。有机层用H2O(2x)洗涤,然后用盐水(1x)洗涤。合并的有机相用Na2SO4干燥,过滤并减压浓缩。通过快速色谱法(石油醚/乙酸乙酯=10:1)纯化,得到题 述化合物(1.77g,6.71mmol,89%),为黄色油状物。光谱数据与文献报道一致。
Rf=0.24(PE/EtOAc=10:1);[α]D 20=-97.8°(c 2.3,CHCl3);{文献:[α]D 26=-104.2°(c 2.33,CHCl3)};
IR: 2103(s,azide),1747(s,C=O),1495(w),1454(m),1437(m),1373(m),1210(s),1099(m),1030(m),910(m),751(m),701(s)cm-1;
2103(s,azide),1747(s,C=O),1495(w),1454(m),1437(m),1373(m),1210(s),1099(m),1030(m),910(m),751(m),701(s)cm-1;
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=7.42-7.33(5H,m,ArH), 5.24(1H,d,J=4.8Hz,H-2),5.07(1H,d,J=4.8Hz,H-3),3.69(3H,s,H-9),2.14(3H, s,H-10)ppm;
13C-NMR(100MHz,CDCl3,CHCl3=77.16ppm):δ=169.9(q,C-1),168.0(q, C-8),134.6(q,C-4),129.3(t,C-7),129.0(2C,t,C-6),127.6(2C,t,C-5),74.9(t,C-2), 65.4(t,C-3),52.8(p,C-9),20.5(p,C-10)ppm;
HRMS(ESI):m/z C12H13N3O4Na[M+Na]+计算值:286.0804;测量值 286.0805.
(2S,3R)-3-叠氮基-2-甲氧基-3-苯基丙酸甲酯

0℃下,(2S,3R)-2-乙酰氧基-3-叠氮基-3-苯基丙酸甲酯(2.5g,1.0eq)溶解 于190ml THF中。然后滴加加入KOH(0.5M,10.0eq)溶液,反应混合物在0℃下 搅拌5h。然后将2N HCl水溶液加入到反应混合物中,水相用乙酸乙酯萃取。将 有机相合并,用硫酸钠干燥,过滤,减压浓缩得到粗酸,将其直接用于下一步 骤,无需进一步纯化。粗产物(0.5g,1.0eq)溶解于17ml碘甲烷中。然后加入 CaSO4(2.6g,8.0eq)和Ag2O(1.7g,3.0eq),室温下在暗处搅拌所述悬浮液22h。 然后过滤该粗混合物并在真空下浓缩得到题述化合物(70%产率),可以直接用于 下一步骤而无需纯化。
[α]D 20=-143.7°(c 1.1,CHCl3);
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=3.44(s,3H),3.61(s,3H), 3.94(d,J=6.4Hz,1H),4.79(d,J=6.4Hz,1H),7.35-7.36(m,5H);
13C-NMR(100MHz,CDCl3,CHCl3=77.0ppm):δ=52.2,59.1,66.9,84.7,127.7,128.7,128.9,135.1,170.0;
HRMS(ESI):m/z C11H13N3O3Na[M+Na]+计算值:258.0855;测量值 258.0852.
(2S,3S)-3-叠氮基-2-甲氧基-3-苯基丙酸叔丁酯

边搅拌,边往(2S,3R)-3-叠氮基-2-甲氧基-3-苯基丙酸甲酯(1.2g,1.0eq)的100ml THF溶液中逐滴加入KOH(0.5M,10.0eq)水溶液。反应混合物在室温下搅拌 5h并通过加入2N HCl水解。水相用乙酸乙酯萃取,合并的有机相经硫酸钠干燥 并减压浓缩,得到羧酸(1.2g,产率98%),其不经过进一步纯化而用于下一反应。 在室温下,粗酸(0.3g,1.0eq)和3.9ml二甲基甲酰胺二叔丁基缩醛(3.9ml,12eq)溶 解于8ml甲苯中。所得的反应混合物加热至80℃并搅拌7h。减压下除去溶剂, 粗产物通过快速柱色谱纯化(石油醚/乙酸乙酯=30:1),得到题述化合物(0.34g, 89%收率)。
[α]D 20=-113.3°(c 1.0,CHCl3);
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=1.26(s,9H),3.45(s,3H), 3.85(d,J=7.2Hz,1H),4.70(d,J=7.2Hz,1H),7.34-7.35(m,5H);
13C-NMR(100MHz,CDCl3,CHCl3=77.0ppm):δ=27.7,58.6,67.2,82.3,85.1,128.2,128.6,128.9,135.2,168.5;
HRMS(ESI):m/z C14H19O3N3Na[M+Na]+计算值:300.1324;测量值 300.1332.
(2S,3S)-4-叔丁基-1-甲基-2-叠氮基-3-甲氧基琥珀酸酯

在室温下,边搅拌边向(2S,3S)-3-叠氮基-2-甲氧基-3-苯基丙酸叔丁酯(310 mg,1.0eq)的3ml CHCl3,13ml CH3CN和26ml H2O混合溶液中逐滴加入NaIO4 (7.2g,30eq)和RuCl3(0.3eq,69mg)。在回流条件下,将反应混合物加热3小时。 冷却至室温后形成白色沉淀。过滤得到固体,滤液用二乙醚萃取。合并的有机相 在减压下浓缩,得到粗产物。所述的物料溶解于9ml碘甲烷中。然后加入CaSO4 (1.2g,8.0eq)和Ag2O(778mg,3.0eq),室温下在暗处搅拌所述反应混合物22h。 过滤,滤液在减压下浓缩得到纯品形式的题述化合物,其可以直接用于下一步骤 而无需进一步纯化。
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=1.51(s,3H),3.48(s,3H), 4.15(d,J=3.6Hz,1H),4.21(d,J=4.0Hz,1H);
13C-NMR(100MHz,CDCl3,CHCl3=77.0ppm):δ=28.1,53.0,59.5,63.4,81.2,83.0,167.7,168.3.
(2S,3R)-1-叔丁基-4-甲基-2-甲氧基-3-[4-(4-硝基苯甲酰氨基)苯甲酰氨基]琥珀 酸酯

粗品混合物(2S,3S)-4-叔丁基-1-甲基-2-叠氮基-3-甲氧基琥珀酯溶解于12mlTHF,然后加入0.5ml水和PPh3(881mg,3.0eq)。所得的反应混合物加热至50℃ 并继续进行搅拌12h。然后在减压下除去溶剂,得到粗产物,其足够纯可直接用 于下一步骤。粗产物溶解于5ml DMF,在室温下加入(乙基碳酸)4-(4-硝基苯甲酰 氨基)苯甲酸酐(481mg,1.2eq)。搅拌20小时后,加入水,水溶液用乙酸乙酯萃 取。减压浓缩合并的有机相。通过快速色谱法(石油醚/乙酸乙酯=2:1)纯化,得 到题述化合物(81mg,四步收率16%)。
[α]D 20=-11.8°(c 1.1,CHCl3);
1H-NMR(400MHz,CDCl3,CHCl3=7.26ppm):δ=1.41(s,9H),3.45(s,3H), 3.78(s,3H),4.34(d,J=2.4Hz,1H),5.29(dd,J=2.4,9.6Hz,1H),6.76(d,J= 9.6Hz,1H),7.27-7.35(m,4H),8.07(d,J=8.8Hz,2H),8.26(2,J=8.8Hz,2H), 8.83(s,1H);
13C-NMR(100MHz,CDCl3,CHCl3=77.0ppm):δ=27.9,52.9,54.8,59.1,79.8,83.2,120.1,123.8,128.3,128.7,129.6,140.3,141.1,149.7,164.1,166.9,168.0,169.7.
HRMS(ESI):m/z C24H27O9N3Na[M+Na]+计算值:524.1645;测量值 524.1647.

在室温下,边搅拌边向(2S,3R)-1-叔丁基-4-甲基-2-甲氧基-3-[4-(4-硝基苯甲酰氨基)苯甲酰氨基]琥珀酸酯(74.3mg,0.15mmol)的2.5ml二氯甲烷溶液中加入 1.5mL的TFA。搅拌5小时后,向反应混合物中加入水,并用乙酸乙酯萃取。合 并的有机相用水洗涤(三次),经硫酸钠干燥,减压下浓缩得到定量收率的标题化 合物(65.9mg,定量)。
[α]D 20=-16.4°(c 1.1,EtOAc);
1H-NMR(400MHz,DMSO,DMSO=2.50ppm):δ=3.37(s,3H),3.69(s,J= 3H),4.34(d,J=4.4Hz,1H),5.09(dd,J=4.8,8.8Hz,1H),7.89-7.90(m,4H),8.21 (dd,J=2,6.8Hz,1H),8.39(dd,J=2,6.8Hz,1H),8.55(d,J=8.8Hz,1H),10.8(s, 1H).
13C-NMR(100MHz,DMSO,DMSO=40.0ppm):δ=52.9,54.8,58.7,79.5, 120.0,124.1,129.0,129.2,129.8,140.8,142.2,149.8,164.7,166.6,170.2,170.9.
HRMS(ESI):m/z C20H19O9N3Na[M+Na]+计算值:468.1019;测量值 468.1016.
另一对映体旋光度:

[α]D 20=+13.9°(c 1.1,EtOAc);
4-(4-(4-((2S,3S)-2,4-二甲氧基-3-(4-(4-硝基苯甲酰氨基)苯甲酰氨基)-4-氧代 丁酰胺)苯甲酰氨基)-2-羟基-3-异丙氧基苯甲酰氨基)-3-异丙氧基苯甲酸甲酯

将4-[4-(4-氨基苯甲酰氨基)-2-羟基-3-异丙氧基苯甲酰氨基]-3-异丙氧基苯甲酸甲酯(15.3mg,0.029mmol)和(2S,3R)-2,4-二甲氧基-3-[4-(4-硝基苯甲酰氨基)苯 甲酰氨基]琥珀酸酯(14.2mg,0.032mmol)溶解于CH2Cl2(3.4mL)中,并冷却至 0℃。然后,加入HOAt(5.9mg,0.044mmol),DIPEA(7.7μL,0.044mmol)和 EDC.HCl(6.9mg,0.036mmol)。混合物在0℃至室温下搅拌17小时。在真空中 浓缩溶剂,得到油状残余物,将其通过快速色谱纯化(石油醚/乙酸乙酯=94:6), 得到标题化合物(20.1mg,0.021mmol,73%),为无色油状物。
1H NMR(400MHz,CDCl3)δ9.07(s,1H-OH),8.37(d,J=7.5Hz,2H),8.20(d,J =7.5Hz,2H),8.11(s,1H-NH),8.02(s,1H-NH),8.01(d,J=1.4Hz,2H),7.98(d,J=7.5 Hz,2H),7.90(d,J=1.3Hz,1H),7.81(dd,J=7.5,1.4Hz,1H),7.78(d,J=7.4Hz, 1H),7.69(d,J=7.5Hz,1H),7.61(d,J=7.5Hz,2H),7.55(s,1H),7.54(s,1H-NH), 7.53(s,1H),7.41(d,J=7.5Hz,1H),5.72(s,1H-NH),5.63(s,1H-NH),5.10(d,J=3.8 Hz,1H),4.76(d,J=3.8Hz,1H),4.04–3.98(m,2H),3.97(s,J=3.1Hz,3H),3.74(s, 3H),3.32(s,3H),1.47(d,J=5.7Hz,6H),1.39(d,J=5.7Hz,6H)ppm.
13C NMR(100MHz,CDCl3)δ173.30,168.15,168.07,167.77,166.93,166.88,166.82,158.83,151.01,146.97,140.78,139.42,138.71,134.97,134.55,131.57,130.00,130.00,129.41,129.41,129.39,129.39,128.12,127.53,127.24,124.17,124.17,123.28,122.61,122.61,121.78,121.78,121.44,115.94,114.88,113.30,106.09,78.00,75.89,74.13,58.51,56.50,52.17,52.08,21.80,21.80,21.80,21.80ppm.
HRMS(ESI):C48H47N6O15(M-H)-计算值:947.3178,测定值:947.3175.
孢囊菌酰胺B
4-4-[4-((2S,3S)-2,4-二甲氧基-3-(4-(4-硝基苯甲酰氨基)苯甲酰氨基)-4-氧代丁 酰胺]苯甲酰氨基}-2-羟基-3-异丙氧基苯甲酰氨基)-3-异丙氧基苯甲酸甲酯(15.2mg, 0.016mmol)溶于THF/H2O(0.2/0.2mL)的1/1混合物中。然后,加入固体 LiOH(3.8mg,0.16mmol),并将该反应混合物在室温下搅拌17小时。水层用1M HCl酸化直至pH~1,并用乙酸乙酯(3x)萃取。合并有机萃取物,用MgSO4干燥 并过滤。在真空下将溶剂浓缩,得到标题化合物(13.3mg,0.014mmol,90%),为 黄色蜡状。
[α]D 20=-19.1°(c 1.1,EtOAc)
1H NMR(400MHz,CDCl3)δ8.35(d,J=7.5Hz,2H),8.15(d,J=7.5Hz,2H), 8.00(d,J=1.8Hz,2H),7.98(d,J=1.8Hz,2H),7.90(d,J=1.8Hz,1H),7.86(dd,J= 7.5,1.8Hz,1H),7.78(d,J=7.5Hz,1H),7.65(s,1H),7.63(d,J=7.5Hz,2H),7.58(s, 1H-NH),7.54(d,J=7.5Hz,2H),7.51(s,1H-NH),7.10(s,1H-NH),7.03(d,J=7.5Hz, 1H),6.35(s,1H-NH),5.57(s,1H-NH),5.42(s,1H-OH),4.93(s,1H),4.70(s,1H),4.01 (hept,J=5.6Hz,1H),3.95(hept,J=5.6Hz,1H),3.38(s,3H),1.48(s,6H),1.47(s, 6H)ppm.
13C NMR(100MHz CDCl3)δ173.30,169.54,168.18,168.07,167.77,166.88,166.82,158.83,151.01,149.88,140.78,139.42,138.71,136.26,134.97,134.55,130.00,130.00,129.41,129.41,129.39,129.39,128.12,127.53,125.15,124.17,124.17,123.28,122.84,122.61,122.61,121.78,121.78,120.41,113.82,113.30,106.09,77.86,75.89,74.13,58.51,54.58,21.80,21.80,21.80,21.80ppm.
HRMS(ESI):C46H43N6O15(M-H)-计算值:920.2865,测定值:920.2866.
孢囊菌酰胺C衍生物的合成



1.1.所用的不同单环的合成
此处对所述孢囊菌酰胺C衍生物的合成过程中使用的不同的单个环的制备进 行说明。
环C的制备
a)BrCH(CH3)2,K2CO3,DMF,90℃,过夜;b)SO2(OMe)2,K2CO3,DMF,90℃,过夜;c)Fe,NH4Cl, EtOH/H2O,回流,2小时
环B的制备

a)BrCH(CH3)2,K2CO3,DMF,90℃,过夜;b)SO2(OMe)2,K2CO3,DMF,90℃,过夜;d)NaOH/MeOH,45℃, 过夜;e)KOH,MeOH/H2O;f)i-PrOH/NaH;g)H2C=CHSn(Bu)3,Pd[(Ph)3P]4;h)KMnO4;i)AcCl/吡啶;j) KNO3/TFAA,k)NaOH,l)AgNO3/NaOH
环A的制备

a)BrCH(CH3)2,K2CO3,DMF,90℃,过夜;d)NaOH/MeOH,45℃,过夜
1.2.环B和C的偶联以提供不同的制备的BC片段


c)Fe,NH4Cl,EtOH/H2O,回流,2小时;m)PCl3,CH2Cl2,二甲苯,145℃,2小时;n)Cl2PPh3,CHCl3;o)
EDC,HOBT
1.3.环A和BC片段的偶联
1.3.1.环A和BC片段的偶联(BC1、BC2、BC3、BC5、BC6、BC7)从而合 成孢囊菌酰胺C衍生物(1a)-(23a)

c)Fe,NH4Cl,EtOH/H2O,回流,2小时;d)NaOH/MeOH,45℃,过夜;m)PCl3,CH2Cl2,二甲苯,145℃,2小 时;n)Cl2PPh3,CHCl3


1.3.2.环A和BC1片段的偶联从而合成孢囊菌酰胺C衍生物(24a)-(31a)

d)NaOH/MeOH,45℃,过夜;m)PCl3,CH2Cl2,二甲苯,145℃,2小时

1.3.3.环A和BC4片段的偶联从而合成孢囊菌酰胺C衍生物(32a)-(33a)

d)NaOH/MeOH,45℃,过夜;p)CH2Cl2,吡啶,rt,过夜
2.实验
2.1常规实验信息
起始材料和溶剂从供应商处市售购买,且不进行进一步纯化即被使用。所有 化学产率是指纯化的化合物,并没有进行优化。反应过程通过TLC硅胶60F254铝片,并通过254nmUV进行可视化。使用硅胶 (40-63μm)进行快速色谱 法。制备RP-HPLC通过包含2767样品管理器、2545二元梯度模块、2998PDA 检测器和3100电子喷雾质谱仪的Waters公司设置进行。使用WatersXBridge柱 (C18,150×19mm,5μm)进行纯化,二元溶剂系统A和B(A=含0.1%甲酸的水; B=含0.1%甲酸的MeCN)作为洗脱剂,流量速率为20mL/min,并在8分钟施加 60%至95%B的梯度。熔点是在斯图尔特科学熔点测定装置SMP3(Bibby Sterilin,英国)上测定,并且未校正。NMR光谱是于300K在Bruker DRX-500(1H, 500MHz;13C,126MHz),或布鲁克傅立叶(Bruker Fourier)300(1H,300MHz;13C, 75MHz)光谱仪上记录。化学位移记录为δ值,单位为ppm,参照氘代试剂的卤 代残留物为内标(CDCl3:δ=7.26,77.02;DMSO-d6:δ=2.50,39.99)。分裂模式描 述为表观多重性,并被指定为s(单峰),br s(宽单峰),d(二重峰),dd(二重二重 峰),t(三重峰),q(四重峰),m(多重峰)。偶合常数(J)通过赫兹(Hz)表示。在生物 测定中使用的所有化合物的纯度为≥95%,通过LC/MS Finnigan SurveyorMSQ Plue(Thermo Fisher Scientific公司,Dreieich,德国)测定。该系统由LC泵,自动进样器,PDA检测器,和单四极杆质谱仪,以及标准的Xcalibur软件工作。RP C18 Nucleodur100-5(125×3mm)柱(Macherey-Nagel GmbH公司,Dühren,德国)用作 固定相,而二元溶剂系统A和B(A=含0.1%TFA的水;B=含0.1%TFA的 MeCN)作为流动相。在梯度运行中,B的百分比的从初始0分钟的浓度0%,在 15分钟内增加至100%,并在100%保持5分钟。注射体积为10μL,并且流动速 率设定为800μL/min。MS(ESI)分析在3800伏的喷射电压,350℃的毛细管温度和 10V的源CID下进行。光谱通过100至1000m/z的正离子模式获得,并在254nm 下紫外光追踪。
(40-63μm)进行快速色谱 法。制备RP-HPLC通过包含2767样品管理器、2545二元梯度模块、2998PDA 检测器和3100电子喷雾质谱仪的Waters公司设置进行。使用WatersXBridge柱 (C18,150×19mm,5μm)进行纯化,二元溶剂系统A和B(A=含0.1%甲酸的水; B=含0.1%甲酸的MeCN)作为洗脱剂,流量速率为20mL/min,并在8分钟施加 60%至95%B的梯度。熔点是在斯图尔特科学熔点测定装置SMP3(Bibby Sterilin,英国)上测定,并且未校正。NMR光谱是于300K在Bruker DRX-500(1H, 500MHz;13C,126MHz),或布鲁克傅立叶(Bruker Fourier)300(1H,300MHz;13C, 75MHz)光谱仪上记录。化学位移记录为δ值,单位为ppm,参照氘代试剂的卤 代残留物为内标(CDCl3:δ=7.26,77.02;DMSO-d6:δ=2.50,39.99)。分裂模式描 述为表观多重性,并被指定为s(单峰),br s(宽单峰),d(二重峰),dd(二重二重 峰),t(三重峰),q(四重峰),m(多重峰)。偶合常数(J)通过赫兹(Hz)表示。在生物 测定中使用的所有化合物的纯度为≥95%,通过LC/MS Finnigan SurveyorMSQ Plue(Thermo Fisher Scientific公司,Dreieich,德国)测定。该系统由LC泵,自动进样器,PDA检测器,和单四极杆质谱仪,以及标准的Xcalibur软件工作。RP C18 Nucleodur100-5(125×3mm)柱(Macherey-Nagel GmbH公司,Dühren,德国)用作 固定相,而二元溶剂系统A和B(A=含0.1%TFA的水;B=含0.1%TFA的 MeCN)作为流动相。在梯度运行中,B的百分比的从初始0分钟的浓度0%,在 15分钟内增加至100%,并在100%保持5分钟。注射体积为10μL,并且流动速 率设定为800μL/min。MS(ESI)分析在3800伏的喷射电压,350℃的毛细管温度和 10V的源CID下进行。光谱通过100至1000m/z的正离子模式获得,并在254nm 下紫外光追踪。
2.2.三芳基衍生物的LC/MS数据


2.3常规合成步骤:
a)酸(25mmol)、异丙基溴(52mmol)和碳酸钾(52mmol)的100ml DMF溶液在 90℃下加热过夜。减压下去除多余的DMF,剩余的残留物在水和乙酸乙酯之间 分配。将有机层用硫酸钠干燥,然后在减压下除去过量的溶剂,得到纯的产物。
c)55℃下,边搅拌边向硝基衍生物(10mmol)的EtOH(60mL)溶液中加入铁 粉(2.80g,50mmol),然后加入NH4Cl(266mg,5mmol)的水(30mL)溶液。将反应 混合物回流1-2小时,然后趁热过滤铁,将滤液在真空下浓缩至干。将残余物用 水(30mL)稀释并用NaHCO3(饱和水溶液)碱化至pH 7-8。混合物用EtOAc萃取。 合并的有机相用盐水洗涤,干燥(MgSO4),真空蒸发除去溶剂。将得到的粗物质 物用正己烷研磨,并通过过滤收集。
d)根据以下报道的步骤1进行酯水解。酯(0.1mmol),氢氧化钠1M(3mL)和 无水甲醇在45℃下加热过夜。冷却后,将反应混合物酸化至pH 1(3mL,盐酸1 M)并用二氯甲烷(3×150mL)萃取。将有机层用硫酸钠干燥,然后在减压下除去溶 剂,得到纯的产物。
m)根据以下报道的步骤2进行氨基化。煮沸的酸(1mmol)和胺(1mmol)的2.5 ml二甲苯溶液用2M PCl3的CH2Cl2溶液(0.4mmol)进行处理。2小时后,蒸发除 去多余溶剂,残留物用柱色谱纯化。
n)氮气气氛下,边搅拌边向酸(2mmol),胺(2.4mmol)的无水CHCl3(50mL) 溶液中加入二氯三苯基膦(3.0g,9mmol)。反应在80℃下加热5h。溶剂通过真 空蒸馏除去。残留物通过快速色谱纯化。
2.4具体合成步骤:
3-甲氧基-4-硝基苯甲酸甲酯

边搅拌边往3-羟基-4-硝基苯甲酸(9.16g,50mmol)和K2CO3(15.2g,110 mmol)在DMF(150mL)中的混合物中分批加入硫酸二甲酯(25.2g,200mmol),然 后在90℃下将反应搅拌过夜。冷却后,将混合物倒入冰水(400mL),将沉淀物过 滤,先后用冷水和正己烷洗涤。
产率95%(浅黄色固体),m/z(ESI+)212[M+H]+。
3-甲氧基-4-硝基苯甲酸

边搅拌边往3-甲氧基-4-硝基苯甲酸甲酯(2.11g,10mmol)的MeOH(30mL)溶 液中加入KOH(1.68g,30mmol)的水(30mL)溶液。将反应物回流2小时,然后通 过真空蒸馏将MeOH蒸发。残留物用水稀释(20mL)。将溶液在冰浴中冷却,用 KHSO4(饱和水溶液)酸化至pH3-4。析出的固体通过过滤收集,先后用冷水和正 己烷洗涤。
产率96%(灰白色固体),m/z(ESI+)198[M+H]+。
6-氯-2-异丙氧基-3-硝基吡啶

边搅拌边往2.6-二氯-3-硝基吡啶(3.86g,20mmol)的甲苯(30mL)溶液中加入 异丙醇(1.44g,24mmol)。将混合物在0℃下搅拌15分钟。然后在氮气气氛下分 批加入NaH(50-60%在矿物油中,1.22g,28mmol),并将该反应物在室温下搅拌 过夜。该反应用盐水淬灭,然后用水稀释并用EtOAc萃取。合并的有机萃取物用 盐水洗涤,干燥(MgSO4),真空蒸发除去溶剂。将残余物溶于甲苯,并用快速色 谱纯化(SiO2,n-己烷–EtOAc=5:1)。
产率70%(微黄色的白色固体),m/z(ESI+)217[M+H]+。
2-异丙氧基-3-硝基-6-乙烯基吡啶

在氮气气氛中,边搅拌边向6-氯-2-异丙氧基-3-硝基吡啶(650mg,3mmol)和 三丁基(乙烯基)锡(1.0g,3.15mmol)的甲苯溶液(20mL)中加入四(三苯基膦)钯(0) (180mg,5%eq.)。反应回流过夜。加入盐水后,将该反应用EtOAc萃取。合并的 有机萃取物用盐水洗涤,干燥(MgSO4),真空蒸发除去溶剂。粗产物不经纯化直 接用于下一步反应。产率90%(黄色液体),m/z(ESI+)208[M]+。
6-异丙氧基-5-硝基吡啶-2-羧酸

边搅拌边向2-异丙氧基-3-硝基-6-乙烯基吡啶(625mg,3mmol)的丙酮(10mL) 溶液中加入KMnO4(1.9g,12mmol)的50%的丙酮(50mL)水溶液。反应在室温下 搅拌24h。加入NaOH 0.5M(5mL),然后混合物过滤,滤液在真空下浓缩。残 留物在冰浴中冷却,并小心地用KHSO4(饱和水溶液)酸化至pH 4–5,然后用 EtOAc萃取。合并的有机萃取物用盐水洗涤,干燥(MgSO4),真空蒸发除去溶 剂。将得到的粗物质物用正己烷研磨,并通过过滤收集。
产率75%(米黄色固体),m/z(ESI+)227[M+H]+。
3-异丙氧基-4-{[(6-异丙氧基-5-硝基吡啶-2-基)羰基]氨基}苯甲酸异丙酯

0℃下,在氮气气氛中,边搅拌边往6-异丙氧基-5-硝基吡啶-2-羧酸(226mg,1mmol)和4-氨基-3-异丙氧基苯甲酸异丙酯(237mg,1mmol)的无水CHCl3(5o mL) 和DMF(1mL)混合溶液中加入HOBt(676mg,5mmol),然后加入EDC.HCl(958 mg,5mmol)。反应物在0℃下搅拌2h,然后在室温下过夜。溶剂通过真空蒸馏 除去。将残余物溶于甲苯,并使用快速色谱纯化(SiO2,n-己烷–EtOAc=2:1)。产 率70%(浅黄色固体),m/z(ESI+)446[M+H]+。
2-甲酰基-6-甲氧基苯基乙酸酯

边搅拌边向3-甲氧基水杨醛的(4.56g,30mmol)和吡啶(2.43mL,30mmol)的 DCM(40mL)溶液中滴加加入乙酰氯(2.36g,30mmol)。反应在室温下搅拌过夜, 然后溶剂通过真空蒸馏除去。将残余物在冷的稀HCl溶液中研磨,并过滤,先后 用冷水和正己烷洗涤。
产率94%(灰白色色固体),m/z(ESI+)195[M+H]+。
6-甲酰基-2-甲氧基-3-硝基苯基乙酸酯

边搅拌边向冰冷的2-甲酰基-6-甲氧基苯基乙酸酯(1.94g,10mmol)和KNO3(1.01g,10mmol)的CHCl3(15mL)悬浮液中加入三氟乙酸酐(12mL)。反应在冰浴 中搅拌2h,然后在室温下过夜。非常小心地用水(50mL)稀释反应物,并用 CHCl3萃取。干燥(MgSO4)合并的有机萃取物,真空蒸发除去溶剂。将残余物溶 于甲苯,并用快速色谱纯化(SiO2,n-己烷–EtOAc=3:1)。产率45%(黄色半固体), m/z(ESI+)239[M]+。
2-羟基-3-甲氧基-4-硝基苯甲醛

边搅拌边往6-甲酰基-2-甲氧基-3-硝基苯基乙酸酯(957mg,4mmol)的水(30 mL)悬浊液中加入NaOH(0.8g,20mmol)。反应回流2h,然后在室温下搅拌过 夜。将溶液在冰浴中冷却,用2M HCl酸化至pH3-4。析出的固体通过过滤收 集,先后用冷水和正己烷洗涤。
产率90%(黄棕色固体),m/z(ESI+)197[M]+。
2-羟基-3-甲氧基-4-硝基苯甲酸

边搅拌边向2-羟基-3-甲氧基-4-硝基苯甲醛(788mg,4mmol)和NaOH(0.8g,20mmol)的水(50mL)溶液中分批加入AgNO3(3.4g,20mmol)。反应回流过夜, 然后冷却并通过硅藻土过滤。溶液在冰浴中冷却,并用37%HCl酸化至pH 3-4。 析出的固体通过过滤收集,先后用冷水和正己烷洗涤。产率65%(米黄色固体), m/z(ESI+)213[M]+。
3-异丙氧基-4-[({6-异丙氧基-5-[(4-硝基苯甲酰基)氨基]吡啶-2-基}羰基)氨基] 苯甲酸异丙酯

边搅拌边向4-{[(5-氨基-6-异丙氧基吡啶-2-基)羰基]氨基}-3-异丙氧基苯甲酸异丙酯(207mg,0.5mmol)和吡啶(0.1mL)的DCM(20mL)溶液中加入4-硝基苯甲 酰氯(185mg,1mmol)。反应在室温下搅拌过夜,然后加入2M HCl(20mL)。混 合物依次用DCM和EtOAc萃取。干燥(MgSO4)合并的有机萃取物,真空蒸发除 去溶剂。将残余物溶于甲苯,并用快速色谱纯化(SiO2,n-己烷–EtOAc=1:1)。产 率80%(黄色晶体),m/z(ESI+)565[M+H]+。
5.参考文献
1)Valeria Azzarito,Panchami Prabhakaran,Alice I.Bartlett,NatashaMurphy, Michaele J.Hardie,Colin A.Kilner,Thomas A.Edwards,Stuart L.Warriner,Andrew J. Wilson.2-O-烷基化的对苯酰胺α-螺旋模拟物:骨架曲率的作用(2-O-Alkylated Para-Benzamideα-Helix Mimetics:The Role of Scaffold Curvature.)有机生物分子化 学(Org.Biomol.Chem.),2012,10,6469.
2)Alina Fomovska,Richard D.Wood,Ernest Mui,Jitenter P.Dubey,LeandraR. Ferreira,Mark R.Hickman,Patricia J.Lee,Susan E.Leed,Jennifer M.Auschwitz,William J.Welsh,Caroline Sommerville,Stuart Woods,Craig Roberts,和RimaMcLeod.弓形虫的水杨酰苯胺抑制剂(Salicylanilide Inhibitors of Toxoplasmagondii.),医学化学杂志(J.Med.Chem.),2012,55(19),pp 8375–8391.
6.这些化合物的活性
根据上述方法测试了这些化合物中的几个对大肠杆菌菌株(TolC-缺陷型)的活性。大多数测试的化合物体现出1到320μM的活性(MIC)。








































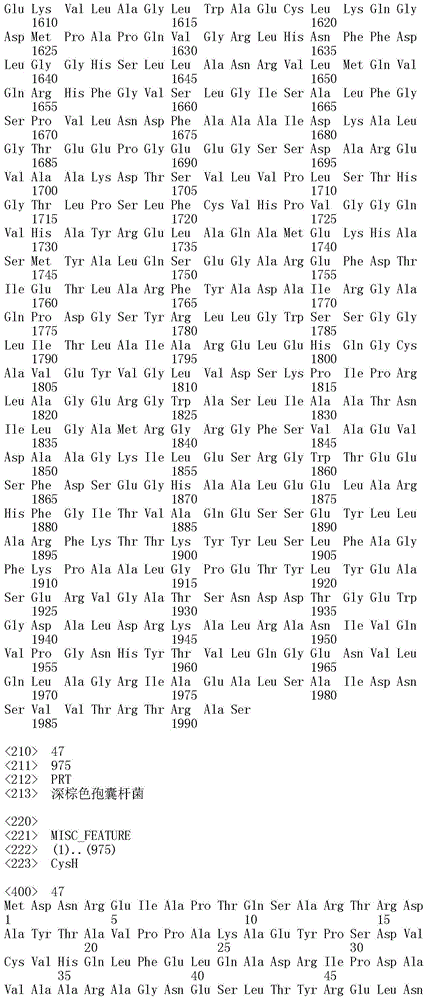









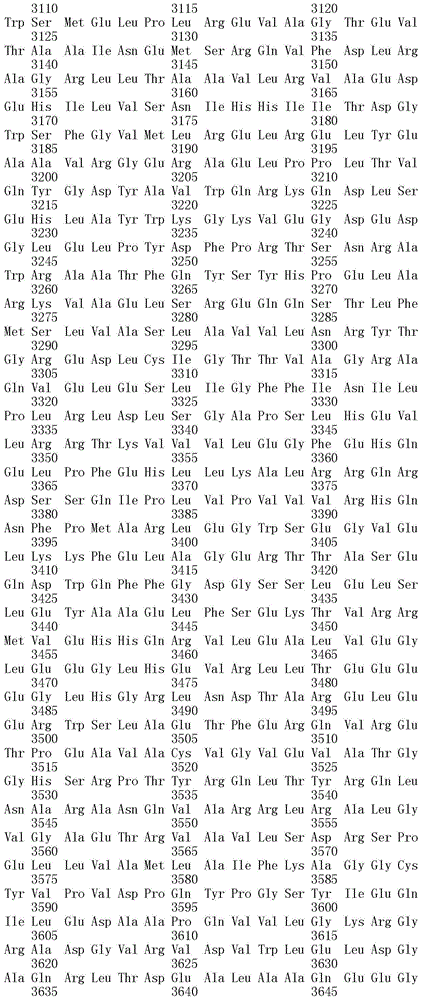





















Claims (5)
1.一种选自下组的化合物:




2.如权利要求1所述的化合物,其特征在于,所述的化合物选自下组:



3.一种药物组合物,包括根据前述任一权利要求所述的化合物,以及任选的一种或多种载体物质和/或一种或多种佐剂。
4.如权利要求1-2任一所述的化合物的用途,其特征在于,用于制备治疗或预防细菌感染的药物组合物。
5.一种制备如权利要求1所述的化合物的方法,其特征在于,所述方法包括以下步骤:
(a)培养保藏号为DSM27004的深棕色孢囊杆菌菌株;和
(b)从培养液中分离并保留所述化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13003539 | 2013-07-12 | ||
| EP13003539.7 | 2013-07-12 | ||
| PCT/EP2014/001925 WO2015003816A2 (en) | 2013-07-12 | 2014-07-14 | Cystobactamides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN105793424A CN105793424A (zh) | 2016-07-20 |
| CN105793424B true CN105793424B (zh) | 2021-02-19 |
Family
ID=48792948
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201480050439.3A Active CN105793424B (zh) | 2013-07-12 | 2014-07-14 | 孢囊菌酰胺 |
Country Status (7)
| Country | Link |
|---|---|
| US (3) | US20160145304A1 (zh) |
| EP (1) | EP3019615B1 (zh) |
| JP (1) | JP6730183B2 (zh) |
| CN (1) | CN105793424B (zh) |
| AU (1) | AU2014289663B2 (zh) |
| CA (1) | CA2917767C (zh) |
| WO (1) | WO2015003816A2 (zh) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105793424B (zh) | 2013-07-12 | 2021-02-19 | 亥姆霍兹感染研究中心有限公司 | 孢囊菌酰胺 |
| GB201402277D0 (en) | 2014-02-10 | 2014-03-26 | Sentinel Oncology Ltd | Pharmaceutical compounds |
| KR101747702B1 (ko) * | 2014-05-15 | 2017-06-19 | 한국생명공학연구원 | 신규한 항균성 화합물 및 이의 용도 |
| CN107001241B (zh) * | 2014-11-26 | 2020-03-27 | 亥姆霍兹感染研究中心有限公司 | 新型孢囊菌酰胺 |
| CN105712894A (zh) * | 2016-03-23 | 2016-06-29 | 叶芳 | 一种3-甲氧基-4-氨基苯甲酸及其制备方法 |
| WO2018062422A1 (ja) * | 2016-09-28 | 2018-04-05 | 日産化学工業株式会社 | ジアミンおよびその利用 |
| TW201920081A (zh) | 2017-07-11 | 2019-06-01 | 美商維泰克斯製藥公司 | 作為鈉通道調節劑的羧醯胺 |
| CA3070240A1 (en) | 2017-07-18 | 2019-01-24 | Technische Universitat Berlin | Novel albicidin derivatives, their use and synthesis |
| EP3672937A1 (en) * | 2017-08-23 | 2020-07-01 | Helmholtz-Zentrum für Infektionsforschung GmbH | Novel cystobactamide derivatives |
| CN111148743B (zh) | 2017-10-06 | 2023-12-15 | 福马治疗有限公司 | 抑制泛素特异性肽酶30 |
| SG11202005785XA (en) | 2018-01-10 | 2020-07-29 | Recurium Ip Holdings Llc | Benzamide compounds |
| CR20200347A (es) | 2018-02-13 | 2020-09-23 | Gilead Sciences Inc | Inhibidores pd-1/pd-l1 |
| KR102591947B1 (ko) | 2018-04-19 | 2023-10-25 | 길리애드 사이언시즈, 인코포레이티드 | Pd-1/pd-l1 억제제 |
| IL278291B2 (en) | 2018-05-17 | 2023-10-01 | Forma Therapeutics Inc | Fused bicyclic compounds useful as ubiquitin-specific peptidase 30 inhibitors |
| PT3820572T (pt) | 2018-07-13 | 2023-11-10 | Gilead Sciences Inc | Inibidores pd-1/pd-l1 |
| SG11202102815SA (en) | 2018-10-05 | 2021-04-29 | Forma Therapeutics Inc | Fused pyrrolines which act as ubiquitin-specific protease 30 (usp30) inhibitors |
| JP7158577B2 (ja) | 2018-10-24 | 2022-10-21 | ギリアード サイエンシーズ, インコーポレイテッド | Pd-1/pd-l1阻害剤 |
| WO2020146612A1 (en) | 2019-01-10 | 2020-07-16 | Vertex Pharmaceuticals Incorporated | Esters and carbamates as modulators of sodium channels |
| US12441703B2 (en) | 2019-01-10 | 2025-10-14 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| JP2022539208A (ja) | 2019-07-03 | 2022-09-07 | スミトモ ファーマ オンコロジー, インコーポレイテッド | チロシンキナーゼ非受容体1(tnk1)阻害剤およびその使用 |
| UY38979A (es) | 2019-12-06 | 2021-07-30 | Vertex Pharma | Tetrahidrofuranos sustituidos como moduladores de canales de sodio |
| WO2022212080A1 (en) * | 2021-03-31 | 2022-10-06 | Medibeacon Inc. | Purification of substituted diaminopyrazine dicarboxylic acids |
| TW202313593A (zh) | 2021-06-04 | 2023-04-01 | 美商維泰克斯製藥公司 | 作為鈉通道調節劑之n—(羥基烷基(雜)芳基)四氫呋喃甲醯胺 |
| CN113563220B (zh) * | 2021-06-25 | 2023-08-29 | 华中农业大学 | 一种抗菌化合物及其应用 |
| WO2025003147A1 (en) | 2023-06-26 | 2025-01-02 | Helmholtz-Zentrum für Infektionsforschung GmbH | Cystobactamid derivatives |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001080846A8 (en) * | 2000-04-26 | 2003-03-20 | Linkshare Corp | Improved transaction tracking, managing, assessment, and auditing data processing system and network |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4525354A (en) * | 1983-09-14 | 1985-06-25 | University Of Hawaii | Antibiotic and process for the production thereof |
| FR2772025B1 (fr) | 1997-12-10 | 2000-03-03 | Guerbet Sa | Chelates metalliques de macrocycles polyaminocarboxyliques et leur application a l'imagerie par resonance magnetique |
| NZ546497A (en) * | 2000-04-28 | 2008-04-30 | Kosan Biosciences Inc | Production of polyketides |
| KR20030017569A (ko) * | 2000-06-28 | 2003-03-03 | 테바 파마슈티컬 인더스트리즈 리미티드 | 카르베딜올 |
| WO2004035760A2 (en) * | 2002-10-18 | 2004-04-29 | University Of Florida | Complete biosynthetic gene set for biosynthesis of albicidin, resistance genes, and uses thereof |
| JPWO2006109846A1 (ja) | 2005-04-06 | 2008-11-20 | 武田薬品工業株式会社 | トリアゾール誘導体およびその用途 |
| JP5379965B2 (ja) * | 2006-09-26 | 2013-12-25 | 株式会社半導体エネルギー研究所 | スチルベン誘導体、発光素子および発光装置 |
| US7758972B2 (en) * | 2006-09-26 | 2010-07-20 | Semiconductor Energy Laboratory Co., Ltd. | Stilbene derivative, light emitting element, light emitting device, and electronic appliance |
| US8236983B2 (en) * | 2008-03-13 | 2012-08-07 | Board Of Regents, The University Of Texas System | Composition and method for the treatment of diseases affected by apoptosis |
| US7816324B2 (en) * | 2007-03-13 | 2010-10-19 | Board Of Regents, The University Of Texas System | Composition and method for the treatment of diseases affected by a peptide receptor |
| WO2013078277A1 (en) * | 2011-11-23 | 2013-05-30 | The Board Of Regents Of The University Of Texas System | Oligo-benzamide compounds and their use in treating cancers |
| US10308595B2 (en) * | 2013-02-15 | 2019-06-04 | Technische Universität Berlin | Albicidin derivatives, their use and synthesis |
| CN105793424B (zh) | 2013-07-12 | 2021-02-19 | 亥姆霍兹感染研究中心有限公司 | 孢囊菌酰胺 |
| KR101747702B1 (ko) | 2014-05-15 | 2017-06-19 | 한국생명공학연구원 | 신규한 항균성 화합물 및 이의 용도 |
| CN107001241B (zh) * | 2014-11-26 | 2020-03-27 | 亥姆霍兹感染研究中心有限公司 | 新型孢囊菌酰胺 |
-
2014
- 2014-07-14 CN CN201480050439.3A patent/CN105793424B/zh active Active
- 2014-07-14 JP JP2016524705A patent/JP6730183B2/ja active Active
- 2014-07-14 EP EP14747296.3A patent/EP3019615B1/en active Active
- 2014-07-14 AU AU2014289663A patent/AU2014289663B2/en active Active
- 2014-07-14 US US14/904,654 patent/US20160145304A1/en not_active Abandoned
- 2014-07-14 WO PCT/EP2014/001925 patent/WO2015003816A2/en not_active Ceased
- 2014-07-14 CA CA2917767A patent/CA2917767C/en active Active
-
2018
- 2018-07-23 US US16/042,753 patent/US10793600B2/en active Active
-
2020
- 2020-06-10 US US16/897,497 patent/US11225503B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001080846A8 (en) * | 2000-04-26 | 2003-03-20 | Linkshare Corp | Improved transaction tracking, managing, assessment, and auditing data processing system and network |
Non-Patent Citations (2)
| Title |
|---|
| EBI accession no. UniprotKB-Q70C52,"Submitted name:Non-ribosomal peptide synthase";无;《EMBL-EBI》;20040705;第3-5页sequence部分 * |
| Jean Euze'by.List of new names and new combinations previously effectively, but not validly, published.《International Journal of Systematic and Evolutionary Microbiology》.2007,第57卷第893–897页. * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20190185514A1 (en) | 2019-06-20 |
| AU2014289663A1 (en) | 2016-02-04 |
| US11225503B2 (en) | 2022-01-18 |
| CN105793424A (zh) | 2016-07-20 |
| AU2014289663B2 (en) | 2019-03-07 |
| WO2015003816A3 (en) | 2015-03-12 |
| US20160145304A1 (en) | 2016-05-26 |
| CA2917767A1 (en) | 2015-01-15 |
| EP3019615B1 (en) | 2021-04-07 |
| EP3019615A2 (en) | 2016-05-18 |
| US20200331964A1 (en) | 2020-10-22 |
| US10793600B2 (en) | 2020-10-06 |
| JP2016527215A (ja) | 2016-09-08 |
| CA2917767C (en) | 2022-05-03 |
| JP6730183B2 (ja) | 2020-07-29 |
| WO2015003816A2 (en) | 2015-01-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105793424B (zh) | 孢囊菌酰胺 | |
| Degrassi et al. | Plant growth-promoting Pseudomonas putida WCS358 produces and secretes four cyclic dipeptides: cross-talk with quorum sensing bacterial sensors | |
| CN110325545B (zh) | 基于减毒细菌的蛋白递送 | |
| WO2019117399A1 (ko) | 신규 폴리펩타이드 및 이를 이용한 imp 생산방법 | |
| CN107001241B (zh) | 新型孢囊菌酰胺 | |
| JP5854515B2 (ja) | リベロマイシンaまたはその合成中間体の製造法、スピロケタール環含有化合物の製造方法、並びに新規抗癌剤、抗真菌剤および骨疾患治療剤 | |
| JP2014530609A (ja) | グリセリマイシン及びメチルグリセリマイシンの生合成のための遺伝子クラスター | |
| Fu et al. | Biosynthesis of 3-hydroxy-5-methyl-O-methyltyrosine in the saframycin/safracin biosynthetic pathway | |
| Gromyko et al. | Generation of Streptomyces globisporus SMY622 strain with increased landomycin E production and it's initial characterization | |
| JP5227219B2 (ja) | クルクミノイド合成酵素およびクルクミノイド製造方法 | |
| EP2154150B1 (en) | Genes for biosynthesis of tetracycline compounds and uses thereof | |
| KR101774940B1 (ko) | 아스트린진 제조용 신규한 조성물 | |
| HK1229840B (zh) | 孢囊菌酰胺 | |
| Jiang et al. | The importance of start codon of nosM in nosiheptide production | |
| CN105755076B (zh) | 利用突变合成获得sansanmycin结构类似物的方法 | |
| US9499825B2 (en) | Dual inducible system for the condensed single protein production system | |
| KR101936975B1 (ko) | 활성이 개선된 메틸로시누스 트리코스포리움 유래의 돌연변이 가용성 메탄 일원자산소화효소 수산화효소 및 그 용도 | |
| CN117642417A (zh) | 新型达罗巴汀衍生物 | |
| CN112980756B (zh) | 一种提高嘌呤操纵子pur基因表达量的突变体及其构建方法与应用 | |
| Kraxner | Novel insights into the transcriptional regulation of cell division in Corynebacterium glutamicum | |
| CN121079081A (zh) | 转录因子HilD抑制剂 | |
| JP5596271B2 (ja) | 複素環含有ペプチド化合物の製造方法及びゴードスポリンの類縁体 | |
| Bell | Analysis and manipulation of “actagardine” gene clusters from Actinoplanes | |
| JP2014166185A (ja) | 複素環含有ペプチド化合物の製造方法及びゴードスポリンの類縁体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |