CN103872348A - catalyst, electrode and preparation method thereof - Google Patents
catalyst, electrode and preparation method thereof Download PDFInfo
- Publication number
- CN103872348A CN103872348A CN201210580508.4A CN201210580508A CN103872348A CN 103872348 A CN103872348 A CN 103872348A CN 201210580508 A CN201210580508 A CN 201210580508A CN 103872348 A CN103872348 A CN 103872348A
- Authority
- CN
- China
- Prior art keywords
- metal
- catalyst
- solution
- catalyst powder
- powder
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 247
- 238000002360 preparation method Methods 0.000 title claims abstract description 28
- 229910052751 metal Inorganic materials 0.000 claims abstract description 121
- 239000002184 metal Substances 0.000 claims abstract description 121
- 229910044991 metal oxide Inorganic materials 0.000 claims abstract description 20
- 150000004706 metal oxides Chemical class 0.000 claims abstract description 20
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 55
- 229910052759 nickel Inorganic materials 0.000 claims description 29
- 229910052746 lanthanum Inorganic materials 0.000 claims description 19
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 10
- 239000011148 porous material Substances 0.000 claims description 10
- 229920000428 triblock copolymer Polymers 0.000 claims description 10
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 claims description 6
- 229910052742 iron Inorganic materials 0.000 claims description 5
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 claims description 5
- 229910017052 cobalt Inorganic materials 0.000 claims 6
- 239000010941 cobalt Substances 0.000 claims 6
- 239000003795 chemical substances by application Substances 0.000 claims 5
- 229920001577 copolymer Polymers 0.000 claims 4
- 239000004094 surface-active agent Substances 0.000 abstract description 46
- 238000010438 heat treatment Methods 0.000 abstract description 40
- 229920001400 block copolymer Polymers 0.000 abstract description 35
- 239000000843 powder Substances 0.000 description 169
- 239000000243 solution Substances 0.000 description 108
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 78
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 72
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 62
- 230000000052 comparative effect Effects 0.000 description 54
- 239000008367 deionised water Substances 0.000 description 50
- 229910021641 deionized water Inorganic materials 0.000 description 50
- UFMZWBIQTDUYBN-UHFFFAOYSA-N cobalt dinitrate Chemical compound [Co+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O UFMZWBIQTDUYBN-UHFFFAOYSA-N 0.000 description 44
- 239000007864 aqueous solution Substances 0.000 description 43
- 238000003756 stirring Methods 0.000 description 39
- 229960000583 acetic acid Drugs 0.000 description 36
- 239000012362 glacial acetic acid Substances 0.000 description 36
- 238000002441 X-ray diffraction Methods 0.000 description 29
- -1 ethyleneoxy-propyleneoxy-ethyleneoxy Chemical group 0.000 description 29
- FYDKNKUEBJQCCN-UHFFFAOYSA-N lanthanum(3+);trinitrate Chemical compound [La+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O FYDKNKUEBJQCCN-UHFFFAOYSA-N 0.000 description 28
- 238000004458 analytical method Methods 0.000 description 24
- 229940045032 cobaltous nitrate Drugs 0.000 description 22
- 239000006261 foam material Substances 0.000 description 18
- 230000003197 catalytic effect Effects 0.000 description 14
- CZMAIROVPAYCMU-UHFFFAOYSA-N lanthanum(3+) Chemical compound [La+3] CZMAIROVPAYCMU-UHFFFAOYSA-N 0.000 description 13
- 238000000034 method Methods 0.000 description 13
- XLJKHNWPARRRJB-UHFFFAOYSA-N cobalt(2+) Chemical compound [Co+2] XLJKHNWPARRRJB-UHFFFAOYSA-N 0.000 description 11
- 229960004106 citric acid Drugs 0.000 description 10
- 239000012535 impurity Substances 0.000 description 10
- 229910020599 Co 3 O 4 Inorganic materials 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 238000001354 calcination Methods 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- KBJMLQFLOWQJNF-UHFFFAOYSA-N nickel(ii) nitrate Chemical compound [Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O KBJMLQFLOWQJNF-UHFFFAOYSA-N 0.000 description 6
- 241000877463 Lanio Species 0.000 description 5
- 239000003792 electrolyte Substances 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 229910021193 La 2 O 3 Inorganic materials 0.000 description 3
- 239000000853 adhesive Substances 0.000 description 3
- 230000001070 adhesive effect Effects 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 230000002209 hydrophobic effect Effects 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 229910002254 LaCoO3 Inorganic materials 0.000 description 2
- VEQPNABPJHWNSG-UHFFFAOYSA-N Nickel(2+) Chemical compound [Ni+2] VEQPNABPJHWNSG-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- NNBFNNNWANBMTI-UHFFFAOYSA-M brilliant green Chemical compound OS([O-])(=O)=O.C1=CC(N(CC)CC)=CC=C1C(C=1C=CC=CC=1)=C1C=CC(=[N+](CC)CC)C=C1 NNBFNNNWANBMTI-UHFFFAOYSA-M 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- MPMSMUBQXQALQI-UHFFFAOYSA-N cobalt phthalocyanine Chemical compound [Co+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 MPMSMUBQXQALQI-UHFFFAOYSA-N 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- LVIYYTJTOKJJOC-UHFFFAOYSA-N nickel phthalocyanine Chemical compound [Ni+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 LVIYYTJTOKJJOC-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- UBEWDCMIDFGDOO-UHFFFAOYSA-N cobalt(II,III) oxide Inorganic materials [O-2].[O-2].[O-2].[O-2].[Co+2].[Co+3].[Co+3] UBEWDCMIDFGDOO-UHFFFAOYSA-N 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000004146 energy storage Methods 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000004626 scanning electron microscopy Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000007784 solid electrolyte Substances 0.000 description 1
Images






















Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
- H01M4/9016—Oxides, hydroxides or oxygenated metallic salts
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/88—Processes of manufacture
- H01M4/8825—Methods for deposition of the catalytic active composition
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Catalysts (AREA)
Abstract
一种触媒、电极及其制备方法,触媒包括一金属氧化物,包含一第一金属及一第二金属,金属氧化物具有单一相的钙钛矿结构。电极包含一金属网及一触媒。触媒负载于金属网,并且触媒包含一单一相的钙钛矿结构的金属氧化物。电极的制备方法包括下列步骤。提供一第一金属以及一第二金属。混合第一金属与第二金属于一溶液,溶液包含一三区块共聚物界面活性剂。提供一金属网。将金属网放置于溶液。加热溶液与金属网使第一金属与第二金属于金属网反应成一触媒,以使触媒与金属网组成一电极。
A catalyst, an electrode and a preparation method thereof. The catalyst includes a metal oxide, including a first metal and a second metal. The metal oxide has a single-phase perovskite structure. The electrode includes a metal mesh and a catalyst. The catalyst is loaded on the metal mesh, and the catalyst includes a single-phase metal oxide with a perovskite structure. The electrode preparation method includes the following steps. A first metal and a second metal are provided. The first metal and the second metal are mixed in a solution containing a three-block copolymer surfactant. A metal mesh is provided. Place the metal mesh into the solution. Heating the solution and the metal mesh causes the first metal and the second metal to react with the metal mesh to form a catalyst, so that the catalyst and the metal mesh form an electrode.
Description
技术领域technical field
本发明涉及一种触媒,特别涉及一种触媒制成的电极及其制备方法。The invention relates to a catalyst, in particular to an electrode made of the catalyst and a preparation method thereof.
背景技术Background technique
近年来随着电动车的发展以及为了满足未来电力网路以及电能储存的需求,二次电池已成为热门的研究课题。一般而言,为了提升二次电池的效能,常见的方法是针对二次电池的触媒进行改进。举例来说,提高触媒的比表面积是研究人员需要解决的问题。除此之外,在制备触媒的过程中,常会生成不具有触媒效能的其他杂相,这些杂相会减少触媒可以进行反应的表面积,进而降低触媒的效能,而造成二次电池的供电电压、供电电流的下降。目前主要是利用控制触媒的成分比例来减少杂相的生成,然而这样的操作方法较为繁琐,并且效果不甚理想。In recent years, with the development of electric vehicles and in order to meet the needs of future power grids and electric energy storage, secondary batteries have become a hot research topic. Generally speaking, in order to improve the efficiency of the secondary battery, a common method is to improve the catalyst of the secondary battery. For example, increasing the specific surface area of the catalyst is a problem that researchers need to solve. In addition, in the process of preparing the catalyst, other impurity phases that do not have catalytic performance are often formed. These impurity phases will reduce the surface area where the catalyst can react, thereby reducing the efficiency of the catalyst, resulting in secondary battery power supply voltage, drop in supply current. At present, the main method is to reduce the formation of impurity phases by controlling the composition ratio of the catalyst, but such an operation method is cumbersome and the effect is not ideal.
因此,如何设计一种触媒及电极,以减少在制备触媒的过程中,会产生杂相而降低所制成电池的效能,就成为研究人员需要解决的问题。Therefore, how to design a catalyst and electrode to reduce the generation of impurity phases during the preparation of the catalyst and reduce the performance of the battery has become a problem that researchers need to solve.
发明内容Contents of the invention
鉴于以上的问题,本发明的目的在于提供一种触媒、电极及其制备方法,藉以减少在制备触媒的过程中,会产生杂相而降低所制成电池的效能的问题。In view of the above problems, the object of the present invention is to provide a catalyst, an electrode and a preparation method thereof, so as to reduce the problem of generating impurity phases during the preparation of the catalyst and lowering the efficiency of the resulting battery.
本发明一实施例所揭露的触媒,包括一金属氧化物,金属氧化物包含一第一金属以及一第二金属,金属氧化物具有单一相的钙钛矿结构。The catalyst disclosed in an embodiment of the present invention includes a metal oxide, the metal oxide includes a first metal and a second metal, and the metal oxide has a single-phase perovskite structure.
本发明一实施例所揭露的电极的制备方法,包括下列步骤。提供一第一金属以及一第二金属。混合第一金属与第二金属于一溶液,溶液包含一三区块共聚物界面活性剂。提供一金属网。将金属网放置于溶液。加热溶液与金属网使第一金属与第二金属于金属网反应成一触媒,以使触媒与金属网组成一电极。A method for preparing an electrode disclosed in an embodiment of the present invention includes the following steps. A first metal and a second metal are provided. The first metal and the second metal are mixed in a solution containing a triblock copolymer surfactant. A metal mesh is provided. A metal mesh is placed in the solution. Heating the solution and the metal net makes the first metal and the second metal react in the metal net to form a catalyst, so that the catalyst and the metal net form an electrode.
本发明一实施例所揭露的电极,包含一金属网以及一触媒。触媒负载于金属网。触媒包含一单一相的钙钛矿结构的金属氧化物,金属氧化物包含一第一金属以及一第二金属。An electrode disclosed in an embodiment of the present invention includes a metal mesh and a catalyst. The catalyst is loaded on the metal mesh. The catalyst includes a single-phase perovskite metal oxide, and the metal oxide includes a first metal and a second metal.
根据本发明实施例所揭露的触媒、电极及其制备方法,由于在制备电极的过程中加入了三区块共聚物界面活性剂,使得所制备的触媒具有单一相的钙钛矿结构,因此解决了制备触媒过程中,会产生杂相的问题。因而可避免杂相降低触媒可进行反应的表面积,进而降低以该触媒制成的电池的效能。According to the catalyst, electrode and preparation method disclosed in the embodiments of the present invention, since the three-block copolymer surfactant is added in the process of preparing the electrode, the prepared catalyst has a single-phase perovskite structure, thus solving the problem of In the process of preparing the catalyst, the problem of impurity will be generated. Therefore, it is avoided that the impurity phase reduces the surface area available for the catalyst to react, thereby reducing the efficiency of the battery made with the catalyst.
以下结合附图和具体实施例对本发明进行详细描述,但不作为对本发明的限定。The present invention will be described in detail below in conjunction with the accompanying drawings and specific embodiments, but not as a limitation of the present invention.
附图说明Description of drawings
图1为本发明一实施例所揭露的触媒的制备方法的流程图;FIG. 1 is a flow chart of a method for preparing a catalyst disclosed in an embodiment of the present invention;
图2A为比较例一的触媒粉末的X光绕射分析结果;Fig. 2A is the X-ray diffraction analysis result of the catalyst powder of comparative example one;
图2B为实施例二、实施例五、比较例一、比较例二的触媒粉末的X光绕射分析结果;Fig. 2B is the X-ray diffraction analysis result of the catalyst powder of embodiment two, embodiment five, comparative example one, comparative example two;
图2C为实施例一至实施例四的触媒粉末的X光绕射分析结果;Fig. 2 C is the X-ray diffraction analysis result of the catalyst powder of embodiment one to embodiment four;
图2D为实施例二的触媒粉末、实施例六的触媒粉末以及实施例七的触媒粉末的X光绕射分析结果;Fig. 2D is the X-ray diffraction analysis result of the catalyst powder of embodiment two, the catalyst powder of embodiment six and the catalyst powder of embodiment seven;
图3A为比较例一的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3 A is the analysis result of the scanning electron microscope of the catalytic powder of comparative example 1;
图3B为实施例一的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3B is the analysis result of the scanning electron microscope of the catalytic powder of embodiment one;
图3C为实施例二的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3 C is the analysis result of the scanning electron microscope of the catalytic powder of embodiment two;
图3D为实施例三的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3D is the analysis result of the scanning electron microscope of the catalytic powder of embodiment three;
图3E为实施例四的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3 E is the analysis result of the scanning electron microscope of the catalytic powder of embodiment four;
图3F为实施例五的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3 F is the analysis result of the scanning electron microscope of the catalytic powder of embodiment five;
图3G为实施例六的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3G is the analysis result of the scanning electron microscope of the catalytic powder of embodiment six;
图3H为实施例七的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3H is the analysis result of the scanning electron microscope of the catalytic powder of embodiment seven;
图3I为比较例二的触媒粉末的扫描式电子显微镜的分析结果;Fig. 3I is the analysis result of the scanning electron microscope of the catalytic powder of comparative example two;
图4为比较例三的触媒粉末、实施例八的触媒粉末以及实施例九的触媒粉末的X光绕射分析结果;Fig. 4 is the X-ray diffraction analysis result of the catalyst powder of comparative example three, the catalyst powder of embodiment eight and the catalyst powder of embodiment nine;
图5A为比较例三的触媒粉末的扫描式电子显微镜的分析结果;Fig. 5A is the analysis result of the scanning electron microscope of the catalytic powder of comparative example 3;
图5B为实施例八的触媒粉末的扫描式电子显微镜的分析结果;Fig. 5B is the analysis result of the scanning electron microscope of the catalytic powder of embodiment eight;
图5C为实施例九的触媒粉末的扫描式电子显微镜的分析结果;Fig. 5 C is the analysis result of the scanning electron microscope of the catalytic powder of embodiment 9;
图6为锌空气电池放电的电压与时间的关系图;Fig. 6 is the relationship diagram of the voltage and time of zinc-air battery discharge;
图7为本发明一实施例所揭露的电极的制备方法的流程图;FIG. 7 is a flowchart of a method for preparing an electrode disclosed in an embodiment of the present invention;
图8A为镍发泡材的X光绕射分析结果;Fig. 8 A is the X-ray diffraction analysis result of nickel foam material;
图8B为实施例十一的电极的X光绕射分析结果;Fig. 8B is the X-ray diffraction analysis result of the electrode of the eleventh embodiment;
图9A为镍发泡材的扫描式电子显微镜的分析结果;Fig. 9 A is the analysis result of the scanning electron microscope of nickel foam;
图9B为实施例十的电极的扫描式电子显微镜的分析结果;Fig. 9B is the analysis result of the scanning electron microscope of the electrode of embodiment ten;
图9C为实施例十一的电极的扫描式电子显微镜的分析结果;Fig. 9C is the analysis result of the scanning electron microscope of the electrode of the eleventh embodiment;
图10为本发明一实施例所揭露的改进空气电池的示意图。FIG. 10 is a schematic diagram of an improved air battery disclosed by an embodiment of the present invention.
其中,附图标记Among them, reference signs
100 电池100 batteries
110 疏水层110 hydrophobic layer
120 触媒层120 catalyst layer
130 电解质层130 electrolyte layer
140 金属层140 metal layers
具体实施方式Detailed ways
以下在实施方式中详细叙述本发明的详细特征以及优点,其内容足以使任何本领域技术人员了解本发明的技术内容并据以实施,且根据本说明书所揭露的内容、权利要求范围及附图,任何本领域技术人员可轻易地理解本发明相关的目的及优点。以下的实施例进一步详细说明本发明的观点,但非以任何观点限制本发明的范畴。The detailed features and advantages of the present invention are described in detail below in the embodiments, the content of which is sufficient to enable any person skilled in the art to understand the technical content of the present invention and implement it accordingly, and according to the content disclosed in this specification, the scope of claims and the accompanying drawings , any person skilled in the art can easily understand the related objects and advantages of the present invention. The following examples further illustrate the concept of the present invention in detail, but do not limit the scope of the present invention in any way.
首先,请参阅图1,图1为本发明一实施例所揭露的触媒的制备方法的流程图。First, please refer to FIG. 1 . FIG. 1 is a flowchart of a catalyst preparation method disclosed in an embodiment of the present invention.
首先,提供一三区块共聚物界面活性剂(步骤S101)。在本发明实施例中,使用乙烯氧基-丙烯氧基-乙烯氧基的三区块共聚物界面活性剂,例如P123,分子式为HO(CH2CH2O)20(CH2CH(CH3)O)70(CH2CH2O)20H的三区块共聚物界面活性剂,或者是F127,分子式为HO(CH2CH2O)106(CH2CH(CH3)O)70(CH2CH2O)106H的三区块共聚物界面活性剂。另,使用例如搅拌子进行搅拌可使三区块共聚物界面活性剂均匀溶解于溶液中。First, a three-block copolymer surfactant is provided (step S101 ). In the embodiment of the present invention, a three-block copolymer surfactant of ethyleneoxy-propyleneoxy-ethyleneoxy is used, such as P123, the molecular formula is HO(CH 2 CH 2 O) 20 (CH 2 CH(CH 3 )O) 70 (CH 2 CH 2 O) 20 H triblock copolymer surfactant, or F127, the molecular formula is HO(CH 2 CH 2 O) 106 (CH 2 CH(CH 3 )O) 70 ( Triblock copolymer surfactant of CH 2 CH 2 O) 106 H. In addition, the three-block copolymer surfactant can be uniformly dissolved in the solution by stirring with a stirring bar, for example.
接着,提供一第一金属以及一第二金属(步骤S102),以作为触媒中用于进行催化的金属。在本发明实施例中,第一金属为镧,第二金属为锰、铁、亚钴或亚镍。其中,为了使第一金属与第二金属形成一具有钙钛矿结构的触媒,第一金属与第二金属的含量比大致为1比1的比例,藉以满足钙钛矿结构的简式ABO3(A为第一金属,B为第二金属)中第一金属与第二金属的比例。Next, a first metal and a second metal are provided (step S102 ) as metals used in the catalyst for catalysis. In the embodiment of the present invention, the first metal is lanthanum, and the second metal is manganese, iron, cobaltous or nickelous. Wherein, in order to make the first metal and the second metal form a catalyst with a perovskite structure, the content ratio of the first metal and the second metal is approximately 1 to 1, so as to satisfy the simple formula ABO of the perovskite structure (A is the first metal, B is the second metal) in the ratio of the first metal to the second metal.
其中,第一金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至10:1之间,而第二金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至10:1之间。在一实施例中,第一金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至25:1之间,而第二金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至25:1之间。Wherein, the molar ratio of the first metal to the three-block copolymer surfactant is between 100:1 and 10:1, and the molar ratio of the second metal to the three-block copolymer surfactant It is between 100:1 and 10:1. In one embodiment, the molar ratio of the first metal to the triblock copolymer surfactant is between 100:1 and 25:1, and the molar ratio of the second metal to the triblock copolymer surfactant is The molar ratio is between 100:1 and 25:1.
然后,将第一金属以及第二金属加入该三区块共聚物界面活性剂中,并混合均匀形成一溶液(步骤S103)。Then, the first metal and the second metal are added into the three-block copolymer surfactant, and mixed uniformly to form a solution (step S103 ).
最后,将该溶液加热,以使第一金属、第二金属与三区块共聚物界面活性剂反应成一触媒(步骤S104)。详细来说,是先将溶液加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,去除三区块共聚物界面活性剂,最后再将溶液加热至600℃并维持2小时,使溶液反应成本发明实施例所揭露的触媒粉末,并且触媒粉末具有单一相钙钛矿的结构。其中,单一相是指触媒粉末具有单一种的结晶相。在本发明实施例中,触媒的型态为粉末状,在以下叙述中,将以触媒粉末作为触媒的举例说明。然,粉末状的触媒并非用以限定本发明。Finally, the solution is heated to make the first metal, the second metal and the three-block copolymer surfactant react to form a catalyst (step S104 ). Specifically, the solution was first heated to 100°C and maintained for 30 minutes, then heated to 450°C and maintained for 1 hour to remove the three-block copolymer surfactant, and finally the solution was heated to 600°C and maintained for 2 hours, the solution is reacted with the catalyst powder disclosed in the embodiment of the present invention, and the catalyst powder has a single-phase perovskite structure. Here, a single phase means that the catalyst powder has a single crystal phase. In the embodiment of the present invention, the form of the catalyst is powder, and in the following description, catalyst powder will be used as an example for illustration. However, the powdered catalyst is not intended to limit the present invention.
触媒的主要成分包含第一金属与第二金属的金属氧化物。由于在制备触媒的过程中加入了三区块共聚物界面活性剂,因此金属氧化物具有单一相的钙钛矿结构并且具有高比表面积。The main components of the catalyst include metal oxides of the first metal and the second metal. The metal oxide has a single-phase perovskite structure and a high specific surface area due to the addition of a three-block copolymer surfactant during the preparation of the catalyst.
以下将以多个比较例及实施例进行本发明所制成的触媒的举例说明及实验验证,其中所列举的%是代表界面活性剂与第一金属(例如为镧)的比值。In the following, several comparative examples and examples will be used to illustrate and verify the catalyst prepared by the present invention, wherein the listed % represents the ratio of the surfactant to the first metal (such as lanthanum).
比较例一(LaCoO3/未使用三区块共聚物界面活性剂)Comparative example 1 (LaCoO 3 / no three-block copolymer surfactant)
量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O],并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O], and add deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
将溶液倒入坩锅中并置于高温炉进行加热,以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为比较例一的触媒粉末。Pour the solution into a crucible and place it in a high-temperature furnace for heating, heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour, and then heating the solution to 600°C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder of Comparative Example 1.
实施例一(LaCoO3/1%P123)Example 1 (LaCoO 3 /1%P123)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.0035M的P123水溶液(步骤S101)。其中,P123、去离子水以及冰醋酸莫耳数比为0.01:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.0035 M aqueous solution of P123 (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.01:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.01,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.01, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例一所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in
实施例二(LaCoO3/2%P123)Example 2 (LaCoO 3 /2%P123)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.007M的P123溶液(步骤S101)。其中,P123、去离子水以及冰醋酸莫耳数比为0.02:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.007 M P123 solution (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.02, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例二所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in the second embodiment of the present invention.
实施例三(LaCoO3/4%P123)Example 3 (LaCoO 3 /4%P123)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.014M的P123溶液(步骤S101)。其中,P123、去离子水以及冰醋酸的莫耳数为0.04:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir with a stirring bar for 1 hour, so that P123 is uniformly dissolved in water to form a 0.014 M P123 solution (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.04:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.04,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.04, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例三所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in the third embodiment of the present invention.
实施例四(LaCoO3/10%P123)Example 4 (LaCoO 3 /10%P123)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.035M的P123溶液(步骤S101)。其中,P123、去离子水以及冰醋酸的莫耳数0.1:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir with a stirring bar for 1 hour, so that P123 is uniformly dissolved in water to form a 0.035 M P123 solution (step S101). Among them, the molar numbers of P123, deionized water and glacial acetic acid are 0.1:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.1,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.1, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例四所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in Example 4 of the present invention.
实施例五(LaCoO3/2%F127)Example 5 (LaCoO 3 /2%F127)
首先,将F127溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使F127均匀溶解于水中,形成0.035M的F127溶液(步骤S101)。其中,F127、去离子水以及冰醋酸的莫耳数比为0.02:320:1。First, dissolve F127 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that F127 is uniformly dissolved in water to form a 0.035 M F127 solution (step S101). Among them, the molar ratio of F127, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有F127的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及F127的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing F127 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and F127 is 1:1:1:1:320:0.02, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例五所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in Example 5 of the present invention.
实施例六(LaCoO3/2%P123/1小时)Example 6 (LaCoO 3 /2%P123/1 hour)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.007M的P123溶液(步骤S101)。其中,P123、去离子水以及冰醋酸的莫耳数比以0.02:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.007 M P123 solution (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.02, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持1小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例六所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then heated the solution to 600° C. for 1 hour, and finally cooled the solution to room temperature. The solution reacts to become the catalyst powder disclosed in Example 6 of the present invention.
实施例七(LaCoO3/2%P123/4小时)Example 7 (LaCoO 3 /2%P123/4 hours)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.007M的P123溶液(步骤S101)。其中,P123、去离子水以及冰醋酸的莫耳数比0.02:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.007 M P123 solution (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.02, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持4小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例七所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600° C. and maintained for 4 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in Example 7 of the present invention.
比较例二(LaCoO3/2%PEG)Comparative Example 2 (LaCoO 3 /2%PEG)
首先,将PEG(PolyehyleneGlycol,HO(C2H4O)nH)溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使PEG均匀溶解于水中,形成0.007M的PEG溶液(步骤S101)。其中,PEG、去离子水以及冰醋酸的莫耳数比0.02:320:1。First, PEG (PolyehyleneGlycol, HO(C 2 H 4 O) n H) was dissolved in 20 mL of deionized water, and 0.417 g of glacial acetic acid was added as a catalyst, and stirred with a stirring bar for 1 hour, so that PEG was uniformly dissolved in water, A 0.007 M PEG solution is formed (step S101 ). Among them, the molar ratio of PEG, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有PEG的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及PEG的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing PEG (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and PEG is 1:1:1:1:320:0.02, and the solution is a clear pink solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明比较例二所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in Comparative Example 2 of the present invention.
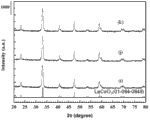
将比较例一、比较例二的触媒粉末、实施例一至实施例七的触媒粉末进行X光绕射分析(X-Ray Diffraction,XRD),结果如图2A至图2D所示,图2A为比较例一的触媒粉末的X光绕射分析结果,图2B为实施例二、实施例五、比较例一及比较例二的触媒粉末的X光绕射分析结果,图2C为实施例一至实施例四的触媒粉末的X光绕射分析结果,图2D为实施例二的触媒粉末、实施例六的触媒粉末以及实施例七的触媒粉末的X光绕射分析结果。The catalyst powders of Comparative Example 1 and Comparative Example 2, and the catalyst powders of Examples 1 to 7 were subjected to X-ray diffraction analysis (X-Ray Diffraction, XRD). The results are shown in Figure 2A to Figure 2D, and Figure 2A is a comparison The X-ray diffraction analysis result of the catalyst powder of Example 1, Fig. 2B is the X-ray diffraction analysis result of the catalyst powder of Embodiment 2, Embodiment 5, Comparative Example 1 and Comparative Example 2, and Fig. 2C is
首先请参阅图2A,将比较例一的触媒粉末的X光绕射分析结果与Co3O4及La2O3比较,比较例一的触媒粉末的2θ于31度及37度具有与Co3O4相同的信号,代表比较例一的触媒粉末还存在Co3O4晶相,并且比较例一的触媒粉末的2θ于32度及54度具有与La2O3相同的信号,代表比较例一的触媒粉末还存在La2O3晶相。Please refer to Fig. 2A first, compare the X-ray diffraction analysis results of the catalyst powder of Comparative Example 1 with Co 3 O 4 and La 2 O 3 , the 2θ of the catalyst powder of Comparative Example 1 has the same properties as Co 3 at 31 degrees and 37 degrees The same signal of O 4 means that the catalyst powder of Comparative Example 1 still has Co 3 O 4 crystal phase, and the 2θ of the catalyst powder of Comparative Example 1 has the same signal as La 2 O 3 at 32 degrees and 54 degrees, which represents the comparative example One catalyst powder still has La 2 O 3 crystal phase.
接着请参阅图2B,其中(a)为实施例二的触媒粉末(2%P123)、(b)为实施例五的触媒粉末(2%F127)、(c)为比较例二的触媒粉末(2%PEG)、(d)为比较例一的触媒粉末(未加入三区块共聚物界面活性剂)。其中,P123以及F127为三区块共聚物界面活性剂,而PEG并非三区块共聚物界面活性剂。Then please refer to Fig. 2B, wherein (a) is the catalyst powder (2% P123) of embodiment two, (b) is the catalyst powder (2% F127) of embodiment five, (c) is the catalyst powder of comparative example two ( 2% PEG), (d) is the catalyst powder of Comparative Example 1 (no three-block copolymer surfactant was added). Among them, P123 and F127 are three-block copolymer surfactants, but PEG is not a three-block copolymer surfactant.
将这些触媒粉末的X光绕射分析结果与Co3O4比较,比较例一及比较例二的触媒粉末在2θ于31度及37度具有与Co3O4相同的信号,代表比较例一及比较例二的触媒粉末包含有Co3O4。接着,再将实施例二及实施例五的触媒粉末的X光绕射分析结果与LaCoO3比较,其信号结果相吻合,显示实施例二及实施例五的触媒粉末应为LaCoO3。因此,添加了三区块共聚物界面活性剂所制成的触媒粉末,确实具有单一相的钙钛矿结构。Comparing the X-ray diffraction analysis results of these catalyst powders with Co 3 O 4 , the catalyst powders of Comparative Example 1 and Comparative Example 2 have the same signals as Co 3 O 4 at 2θ at 31 degrees and 37 degrees, representing Comparative Example 1 and the catalyst powder of Comparative Example 2 contained Co 3 O 4 . Next, the X-ray diffraction analysis results of the catalyst powders of Examples 2 and 5 were compared with LaCoO 3 , and the signal results were consistent, indicating that the catalyst powders of Examples 2 and 5 should be LaCoO 3 . Therefore, the catalyst powder made by adding the three-block copolymer surfactant does have a single-phase perovskite structure.
接着请参阅图2C,其中(e)为实施例一的触媒粉末(1%P123)、(f)为实施例二的触媒粉末(2%P123)、(g)为实施例三的触媒粉末(4%P123)、(h)为实施例四的触媒粉末(10%P123)。Then please refer to Fig. 2C, wherein (e) is the catalyst powder (1% P123) of embodiment one, (f) is the catalyst powder (2% P123) of embodiment two, (g) is the catalyst powder of embodiment three ( 4% P123), (h) is the catalyst powder (10% P123) of Example 4.
将实施例一至实施例四的触媒粉末的X光绕射分析结果与Co3O4比较,实施例一至实施例四的触媒粉末在31度及37度并无明显的信号,显示实施例一至实施例四的触媒粉末不具有Co3O4。接着,再将实施例一至实施例四的触媒粉末的X光绕射分析结果与LaCoO3比较,实施例一至实施例四的触媒粉末的信号与LaCoO3的信号相吻合,显示实施例一至实施例四的触媒粉末应为LaCoO3。因此,即使改变了P123的浓度,所制成的触媒粉末仍然具有单一相的钙钛矿结构。Comparing the X-ray diffraction analysis results of the catalyst powders in Examples 1 to 4 with Co 3 O 4 , the catalyst powders in Examples 1 to 4 have no obvious signals at 31 degrees and 37 degrees, which shows that the catalyst powders in Examples 1 to 4 have no obvious signal. The catalyst powder of Example 4 does not have Co 3 O 4 . Then, compare the X-ray diffraction analysis results of the catalyst powders of Examples 1 to 4 with LaCoO 3 , the signals of the catalyst powders of Examples 1 to 4 are consistent with the signals of LaCoO 3 , showing that the signals of Examples 1 to 4 are The catalyst powder of IV should be LaCoO 3 . Therefore, even if the concentration of P123 is changed, the prepared catalyst powder still has a single-phase perovskite structure.
接着请参阅图2D,其中(i)为实施例六的触媒粉末(2%P123/1小时)、(j)为实施例二的触媒粉末(2%P123/2小时)、(k)为实施例七的触媒粉末(2%P123/4小时)。Then please refer to Fig. 2D, wherein (i) is the catalyst powder (2%P123/1 hour) of embodiment six, (j) is the catalyst powder (2%P123/2 hour) of embodiment two, (k) is the implementation Catalyst powder (2% P123/4 hours) of example seven.
将实施例二、实施例六以及实施例七的触媒粉末的X光绕射分析结果与Co3O4比较,实施例二、实施例六以及实施例七的触媒粉末在31度及37度并无明显的信号,显示实施例二、实施例六以及实施例七的触媒粉末不具有Co3O4。接着,再将实施例二、实施例六以及实施例七的触媒粉末的X光绕射分析结果与LaCoO3比较,实施例二、实施例六以及实施例七的触媒粉末的信号与LaCoO3的信号相吻合,显示实施例二、实施例六以及实施例七的触媒粉末应为LaCoO3。因此,即使改变触媒粉末的煅烧时间,所制成的触媒粉末仍然具有单一相的钙钛矿结构。By comparing the X-ray diffraction analysis results of the catalyst powders of Example 2, Example 6, and Example 7 with Co3O4, the catalyst powders of Example 2, Example 6, and Example 7 have no obvious changes at 31 degrees and 37 degrees. The signal shows that the catalyst powders of Example 2, Example 6 and Example 7 do not contain Co 3 O 4 . Then, compare the X-ray diffraction analysis results of the catalyst powders of Embodiment 2, Embodiment 6, and Embodiment 7 with LaCoO 3 , the signal of the catalyst powders of Embodiment 2, Embodiment 6, and Embodiment 7 is comparable to that of LaCoO 3 . The signals are consistent, showing that the catalyst powders in Example 2, Example 6 and Example 7 should be LaCoO 3 . Therefore, even if the calcination time of the catalyst powder is changed, the prepared catalyst powder still has a single-phase perovskite structure.
接着请参阅图3A至图3G,图3A为比较例一的触媒粉末的扫描式电子显微镜(ScanningElectronMicroscope,SEM)的分析结果,图3B至图3H分别为实施例一至实施例七的触媒粉末的扫描式电子显微镜的分析结果,图3I为比较例二的触媒粉末的扫描式电子显微镜的分析结果。比较例一的触媒粉末主要是破碎片状的组成,并且会团聚成大聚集块体。而添加界面活性剂所制成的触媒粉末(实施例一至实施例七以及比较例二),由于在制备触媒粉末时P123、F127、PEG会形成微胞,使得触媒粉末主要是形成小颗粒的组成,并且使触媒粉末具有孔洞及孔穴而提升了触媒粉末的比表面积。Please refer to Fig. 3A to Fig. 3G. Fig. 3A is the scanning electron microscope (Scanning Electron Microscope, SEM) analysis result of the catalyst powder of Comparative Example 1, and Fig. 3B to Fig. 3H are the scans of the catalyst powder of Example 1 to Example 7 respectively. 3I is the analysis result of the scanning electron microscope of the catalytic powder of Comparative Example 2. The catalyst powder in Comparative Example 1 is mainly composed of broken flakes and will agglomerate into large aggregates. The catalyst powders made by adding surfactants (Example 1 to Example 7 and Comparative Example 2), because P123, F127, and PEG will form microcells when preparing the catalyst powder, the catalyst powder is mainly composed of small particles , and make the catalyst powder have holes and holes to increase the specific surface area of the catalyst powder.
接着,将比较例一、实施例一、实施例二的触媒粉末进行比表面积分析(Brunauer-Emmett-Teller analysis,BET analysis),结果如下表。Next, the specific surface area analysis (Brunauer-Emmett-Teller analysis, BET analysis) of the catalyst powders of Comparative Example 1, Example 1, and Example 2 was carried out, and the results are shown in the following table.
实施例一的触媒粉末以及实施例二的触媒粉末与未添加P123的比较例一的触媒粉末相比,实施例一的触媒粉末以及实施例二的触媒粉末的比表面积分别为未添加P123的触媒粉末的比表面积的3倍及4倍。实施例一的触媒粉末以及实施例二的触媒粉末的孔洞体积分别为未添加P123的触媒粉末的孔洞体积的3倍及4倍。实施例一的触媒粉末以及实施例二的触媒粉末的平均孔径分别为未添加P123的触媒粉末的平均孔径的1.2倍及1.1倍。因此,P123确实可提升制成的触媒粉末的比表面积。The catalyst powder of embodiment one and the catalyst powder of embodiment two are compared with the catalyst powder of the comparative example one that does not add P123, and the specific surface area of the catalyst powder of embodiment one and the catalyst powder of embodiment two is respectively the catalyst powder that does not add P123 3 times and 4 times the specific surface area of the powder. The pore volumes of the catalyst powder of Example 1 and the catalyst powder of Example 2 are 3 times and 4 times of the pore volume of the catalyst powder without adding P123, respectively. The average pore diameters of the catalyst powder of Example 1 and the catalyst powder of Example 2 are 1.2 times and 1.1 times of the average pore diameter of the catalyst powder without adding P123, respectively. Therefore, P123 can indeed increase the specific surface area of the catalyst powder produced.
接着,将实施例一、实施例二及实施例三的触媒粉末进行比表面积分析,结果如下表。Next, the catalyst powders of Example 1, Example 2, and Example 3 were analyzed for specific surface area, and the results are shown in the following table.
实施例一至实施例三的触媒粉末的比表面积均较比较例一的触媒粉末的比表面积大。The specific surface areas of the catalyst powders of Examples 1 to 3 are larger than those of the catalyst powders of Comparative Example 1.
接着,由X光绕射分析结果计算比较例一、比较例二、实施例一至实施例五的触媒粉末的粒径,结果如下表。Next, the particle diameters of the catalyst powders of Comparative Example 1, Comparative Example 2, and Examples 1 to 5 were calculated from the X-ray diffraction analysis results, and the results are shown in the following table.
比较例一、比较例二、实施例一至实施例五的触媒粉末的粒径均为纳米等级。The particle diameters of the catalyst powders in Comparative Example 1, Comparative Example 2, and Examples 1 to 5 are all in the nanometer range.
然后,由X光绕射分析结果计算实施例二、实施例六以及实施例七的触媒粉末的粒径,结果如下表。其中,实施例二、实施例六以及实施例七在600℃的反应时间分别为1小时、2小时以及4小时。Then, the particle diameters of the catalyst powders in Example 2, Example 6, and Example 7 were calculated from the X-ray diffraction analysis results, and the results are shown in the following table. Wherein, the reaction times of Example 2, Example 6, and Example 7 at 600° C. are 1 hour, 2 hours, and 4 hours, respectively.
实施例二、实施例六以及实施例七的触媒粉末的粒径均为纳米等级。The particle diameters of the catalyst powders in Embodiment 2, Embodiment 6, and Embodiment 7 are all in the nanometer range.
比较例三(LaNiO3/未使用三区块共聚物界面活性剂)Comparative example 3 (LaNiO 3 / no three-block copolymer surfactant)
量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚镍[Ni(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O],并加入去离子水,使水溶液中的镧离子以及亚镍离子的浓度为0.35M。Lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], nickel nitrate [Ni(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O], and add deionized water to make the concentration of lanthanum ions and nickelous ions in the aqueous solution be 0.35M.
将溶液倒入坩锅中并置于高温炉进行加热,以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为比较例三的触媒粉末。Pour the solution into a crucible and place it in a high-temperature furnace for heating, heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour, and then heating the solution to 600°C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder of Comparative Example 3.
实施例八(LaNiO3/1%P123)Example 8 (LaNiO 3 /1%P123)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.0035M的P123水溶液(步骤S101)。其中,P123、去离子水以及冰醋酸莫耳数比为0.01:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.0035 M aqueous solution of P123 (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.01:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚镍[Ni(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚镍离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], nickel nitrate [Ni(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and nickelous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚镍离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚镍、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.01,并且溶液为澄清的翠绿色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and nickelous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, nickel nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.01, and the solution is a clear emerald green solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例八所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution reacts to become the catalyst powder disclosed in the eighth embodiment of the present invention.
实施例九(LaNiO3/2%P123)Example 9 (LaNiO 3 /2%P123)
首先,将P123溶于20mL去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.007M的P123水溶液(步骤S101)。其中,P123、去离子水以及冰醋酸莫耳数比为0.02:320:1。First, dissolve P123 in 20 mL of deionized water, add 0.417 g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.007 M aqueous solution of P123 (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚镍[Ni(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S102),并加入去离子水,使水溶液中的镧离子以及亚镍离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], nickel nitrate [Ni(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S102 ), and adding deionized water, so that the concentration of lanthanum ion and nickelous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚镍离子的水溶液缓慢滴入含有P123的水溶液(步骤S103),并持续搅拌2小时。其中,硝酸镧、硝酸亚镍、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的翠绿色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and nickelous ions into the aqueous solution containing P123 (step S103 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, nickel nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.02, and the solution is a clear emerald green solution.
最后,将溶液倒入坩锅中并置于高温炉进行加热(步骤S104),以每分钟3℃的速率由室温加热至100℃并维持30分钟,再将溶液加热至450℃并维持1小时,再将溶液加热至600℃并维持2小时,最后将溶液冷却至室温。溶液即反应成为本发明实施例九所揭露的触媒粉末。Finally, pour the solution into a crucible and place it in a high-temperature furnace for heating (step S104), heating from room temperature to 100°C at a rate of 3°C per minute and maintaining it for 30 minutes, then heating the solution to 450°C and maintaining it for 1 hour , and then the solution was heated to 600 ° C and maintained for 2 hours, and finally the solution was cooled to room temperature. The solution immediately reacts to become the catalyst powder disclosed in Example 9 of the present invention.
将比较例三的触媒粉末、实施例八的触媒粉末以及实施例九的触媒粉末进行X光绕射分析(X-Ray Diffraction,XRD),结果如图4所示,其中(l)为比较例三的触媒粉末、(m)为实施例八的触媒粉末、(n)为实施例九的触媒粉末。将三触媒粉末的X光绕射分析结果与La2NiO4比较,比较例三的触媒粉末在31度至32度之间具有与La2NiO4相同的信号,代表未添加三区块共聚物界面活性剂制备而成的触媒粉末包含有La2NiO4,而实施例八的触媒粉末以及实施例九的触媒粉末在31度至32度之间则无明显的信号,显示实施例八的触媒粉末以及实施例九的触媒粉末不具有La2NiO4。接着,再将实施例八的触媒粉末以及实施例九的触媒粉末的X光绕射分析结果与LaNiO3比较,其信号结果相吻合,显示实施例八的触媒粉末以及实施例九的触媒粉末应为LaNiO3。因此,添加了三区块共聚物界面活性剂所制成的触媒粉末,确实具有单一相的钙钛矿结构。The catalyst powder of Comparative Example 3, the catalyst powder of Example 8 and the catalyst powder of Example 9 were subjected to X-ray diffraction analysis (X-Ray Diffraction, XRD), and the results are shown in Figure 4, wherein (l) is a comparative example The catalyst powder of three, (m) is the catalyst powder of embodiment eight, (n) is the catalyst powder of embodiment nine. Comparing the X-ray diffraction analysis results of the three catalyst powders with those of La 2 NiO 4 , the catalyst powder of Comparative Example 3 has the same signal as La 2 NiO 4 between 31°C and 32°C, indicating that no three-block copolymer was added The catalyst powder prepared by the surfactant contains La 2 NiO 4 , while the catalyst powder of Example 8 and the catalyst powder of Example 9 have no obvious signal between 31 degrees and 32 degrees, showing that the catalyst powder of Example 8 The powder and the catalyst powder of Example 9 do not have La 2 NiO 4 . Then, the X-ray diffraction analysis results of the catalyst powder of Example 8 and the catalyst powder of Example 9 are compared with LaNiO 3 , and the signal results are consistent, showing that the catalyst powder of Example 8 and the catalyst powder of Example 9 should be is LaNiO 3 . Therefore, the catalyst powder made by adding the three-block copolymer surfactant does have a single-phase perovskite structure.
接着请参阅图5A至图5C,图5A为比较例三的触媒粉末的扫描式电子显微镜的分析结果,图5B为实施例八的触媒粉末的扫描式电子显微镜的分析结果,图5C为实施例九的触媒粉末的扫描式电子显微镜的分析结果。未添加三区块共聚物界面活性剂制备而成的触媒粉末主要是破碎片状的组成,并且会团聚成大聚集块体。而添加三区块共聚物界面活性剂(P123)的触媒粉末(实施例八、九),由于在制备触媒时P123会形成微胞,使得触媒粉末主要是形成小颗粒的组成,并且使触媒粉末具有孔洞而提升了触媒粉末的比表面积。Then please refer to Fig. 5A to Fig. 5C, Fig. 5A is the analysis result of the scanning electron microscope of the catalyst powder of comparative example three, Fig. 5B is the analysis result of the scanning electron microscope of the catalyst powder of embodiment eight, Fig. 5C is embodiment 9. SEM analysis results of catalyst powders. The catalyst powder prepared without the addition of three-block copolymer surfactant is mainly composed of broken flakes and will agglomerate into large aggregates. And add the catalyst powder (embodiment eight, nine) of three-block copolymer surfactant (P123), because P123 can form microcell when preparing catalyst, make catalyst powder mainly form the composition of small particle, and make catalyst powder Having holes increases the specific surface area of the catalyst powder.
接着,进行比表面积分析,结果如下表。Then, the specific surface area analysis is carried out, and the results are shown in the following table.

实施例八的触媒粉末以及实施例九的触媒粉末与比较例三的触媒粉末相比,实施例八的触媒粉末以及实施例九的触媒粉末的比表面积分别为比较例三的触媒粉末的比表面积的1.4倍及1.3倍。实施例八的触媒粉末以及实施例九的触媒粉末的孔洞体积分别为比较例三的触媒粉末的孔洞体积的1.9倍及2倍。实施例八的触媒粉末以及实施例九的触媒粉末的平均孔径分别为比较例三的触媒粉末的平均孔径的1.2倍及1.4倍。实施例八的触媒粉末以及实施例九的触媒粉末的微孔表面积分别为比较例三的触媒粉末的微孔表面积的3倍及4.2倍。实施例八的触媒粉末以及实施例九的触媒粉末的微孔体积分别为比较例三的触媒粉末的微孔体积的6倍及9倍。因此,三区块共聚物界面活性剂确实可提升制成的触媒粉末的比表面积。The catalyst powder of embodiment eight and the catalyst powder of embodiment nine are compared with the catalyst powder of comparative example three, and the specific surface area of the catalyst powder of embodiment eight and the catalyst powder of embodiment nine is respectively the specific surface area of the catalyst powder of comparative example three 1.4 times and 1.3 times. The pore volumes of the catalyst powder of Example 8 and the catalyst powder of Example 9 are 1.9 times and 2 times of the pore volume of the catalyst powder of Comparative Example 3, respectively. The average pore diameters of the catalyst powder of Example 8 and the catalyst powder of Example 9 are 1.2 times and 1.4 times of the average pore diameter of the catalyst powder of Comparative Example 3, respectively. The micropore surface areas of the catalyst powder of Example 8 and the catalyst powder of Example 9 are 3 times and 4.2 times of the micropore surface area of the catalyst powder of Comparative Example 3, respectively. The micropore volumes of the catalyst powder of Example 8 and the catalyst powder of Example 9 are 6 times and 9 times of the micropore volume of the catalyst powder of Comparative Example 3, respectively. Therefore, the three-block copolymer surfactant can indeed increase the specific surface area of the prepared catalyst powder.
综上所述,无论是使用硝酸镧及硝酸亚钴来制备触媒或者是使用硝酸镧及硝酸亚镍来制备触媒粉末,只要在制备的过程中添加了三区块共聚物界面活性剂,均能增加所制成的触媒粉末的比表面积并且可抑制杂相而生成具有单一相钙钛矿结构的触媒粉末。这些触媒粉末可应用于电极,例如是燃料电池的电极或者是空气电池的电极等。In summary, no matter whether lanthanum nitrate and cobaltous nitrate are used to prepare the catalyst or lanthanum nitrate and nickel nitrate are used to prepare the catalyst powder, as long as the three-block copolymer surfactant is added during the preparation process, all can The specific surface area of the prepared catalyst powder is increased and the impurity phase can be suppressed to generate a catalyst powder with a single-phase perovskite structure. These catalyst powders can be applied to electrodes, such as electrodes of fuel cells or electrodes of air batteries.
接着,混合实施例二的触媒粉末、碳粉以及质子交换膜(Nafion),并且涂布在碳纸上,即完成一简单的空气电极的制备。Next, the catalyst powder, carbon powder and proton exchange membrane (Nafion) of Example 2 were mixed, and coated on carbon paper, that is, the preparation of a simple air electrode was completed.
将上述空气电极与锌片组成一锌空气电池,并且锌空气电池放电的电压与时间的关系如图6所示,图6为锌空气电池放电的电压与时间的关系图。其中,(o)为
首先,请参阅图7,图7为本发明一实施例所揭露的电极的制备方法的流程图。First, please refer to FIG. 7 . FIG. 7 is a flowchart of a method for preparing an electrode disclosed in an embodiment of the present invention.
首先,提供一三区块共聚物界面活性剂(步骤S701)。在本发明实施例中,是使用乙烯氧基-丙烯氧基-乙烯氧基的三区块共聚物界面活性剂,例如P123,分子式为HO(CH2CH2O)20(CH2CH(CH3)O)70(CH2CH2O)20H(P123)的三区块共聚物界面活性剂,或者是F127,分子式为HO(CH2CH2O)106(CH2CH(CH3)O)70(CH2CH2O)106H的三区块共聚物界面活性剂。另,使用例如搅拌子进行搅拌可使三区块共聚物界面活性剂均匀溶解于溶液中。以下将以P123作为三区块共聚物界面活性剂的举例说明。First, a three-block copolymer surfactant is provided (step S701 ). In the embodiment of the present invention, a three-block copolymer surfactant of ethyleneoxy-propyleneoxy-ethyleneoxy is used, such as P123, the molecular formula is HO(CH 2 CH 2 O) 20 (CH 2 CH(CH 3 )O) 70 (CH 2 CH 2 O) 20 H (P123) triblock copolymer surfactant, or F127 with the formula HO(CH 2 CH 2 O) 106 (CH 2 CH(CH 3 ) O) 70 (CH 2 CH 2 O) 106 H triblock copolymer surfactant. In addition, the three-block copolymer surfactant can be uniformly dissolved in the solution by stirring with a stirring bar, for example. The following will use P123 as an example of a three-block copolymer surfactant.
接着,提供一第一金属以及一第二金属(步骤S702),以作为触媒中用于进行催化的金属。在本发明实施例中,第一金属为镧,第二金属为锰、铁、亚钴或亚镍。其中,为了使第一金属与第二金属形成一具有钙钛矿结构的触媒,第一金属与第二金属的含量比大致为1比1的比例,藉以满足钙钛矿结构的简式ABO3(A为第一金属,B为第二金属)中第一金属与第二金属的比例。Next, a first metal and a second metal are provided (step S702 ) as metals used in the catalyst for catalysis. In the embodiment of the present invention, the first metal is lanthanum, and the second metal is manganese, iron, cobaltous or nickelous. Wherein, in order to make the first metal and the second metal form a catalyst with a perovskite structure, the content ratio of the first metal and the second metal is approximately 1 to 1, so as to satisfy the simple formula ABO3 of the perovskite structure ( A is the first metal, B is the ratio of the first metal to the second metal in the second metal).
其中,第一金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至10:1之间,而第二金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至10:1之间。在一实施例中,第一金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至25:1之间,而第二金属与三区块共聚物界面活性剂的莫耳数比是介于100:1至25:1之间。Wherein, the molar ratio of the first metal to the three-block copolymer surfactant is between 100:1 and 10:1, and the molar ratio of the second metal to the three-block copolymer surfactant It is between 100:1 and 10:1. In one embodiment, the molar ratio of the first metal to the triblock copolymer surfactant is between 100:1 and 25:1, and the molar ratio of the second metal to the triblock copolymer surfactant is The molar ratio is between 100:1 and 25:1.
然后,将第一金属以及第二金属加入该三区块共聚物界面活性剂中,并混合均匀形成一溶液(步骤S703)。Then, the first metal and the second metal are added into the three-block copolymer surfactant, and mixed uniformly to form a solution (step S703 ).
接着,提供一金属网(步骤S704),金属网例如但不限于镍发泡材,利用金属网以承载触媒,并且金属网还可提供作为电极的集电网。在其他实施例中,金属网可以是金网、钯网或是铂网,并不以镍发泡材为限。Next, a metal mesh is provided (step S704 ), the metal mesh is such as but not limited to nickel foam material, the metal mesh is used to carry the catalyst, and the metal mesh can also provide a collector network as an electrode. In other embodiments, the metal mesh can be gold mesh, palladium mesh or platinum mesh, and is not limited to nickel foam.
然后,将金属网浸泡于溶液中(步骤S705)。Then, soak the metal mesh in the solution (step S705 ).
最后,将金属网与溶液在惰性气体下进行煅烧(步骤S706),以使触媒担载在金属网上,即完成了电极的制备。其中,惰性气体例如为氮气、氩气或者是氦气。Finally, the metal mesh and the solution are calcined under inert gas (step S706 ), so that the catalyst is loaded on the metal mesh, and the preparation of the electrode is completed. Wherein, the inert gas is, for example, nitrogen, argon or helium.
经由本实施例所揭露的电极的制备方法,由于在制备电极的过程中加入了三区块共聚物界面活性剂,因此在制作过程中无须使用接着剂仍可将触媒与金属网接合,而使得触媒直接接触金属网。此外,三区块共聚物界面活性剂还可使触媒具有单一相的钙钛矿结构并且具有高比表面积。Through the preparation method of the electrode disclosed in this embodiment, since the three-block copolymer surfactant is added during the preparation of the electrode, the catalyst and the metal mesh can still be bonded without using an adhesive during the production process, so that The catalyst directly contacts the metal mesh. In addition, the three-block copolymer surfactant can also make the catalyst have a single-phase perovskite structure and a high specific surface area.
以下将以多个实施例进行本发明所制成的电极的举例说明及实验验证。In the following, several examples will be used to illustrate and verify the electrodes made by the present invention.
实施例十(镍发泡材+LaCoO3/1%P123)Example 10 (nickel foam + LaCoO 3 /1%P123)
首先,将P123溶于20ml去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.0035M的P123水溶液(步骤S101)。其中,P123、去离子水以及冰醋酸莫耳数比为0.01:320:1。First, dissolve P123 in 20ml of deionized water, add 0.417g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.0035M aqueous solution of P123 (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.01:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S702),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S702 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S703),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.01,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S703 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.01, and the solution is a clear pink solution.
接着,提供一镍发泡材(步骤S704),并将镍发泡材浸泡于溶液中(步骤S705)。其中,镍发泡材为一具有多孔性的金属网,因此可具有较高的表面积以担载触媒。Next, a nickel foam material is provided (step S704 ), and the nickel foam material is soaked in the solution (step S705 ). Wherein, the nickel foam material is a porous metal mesh, so it can have a relatively high surface area to support the catalyst.
最后,将镍发泡材与溶液在1大气压的氮气下进行煅烧,分为两阶段煅烧。首先是在450℃下煅烧1小时,接着在600℃下煅烧2小时(步骤S706),以使触媒担载在金属网上,即完成了实施例十的电极的制备。Finally, the nickel foam material and the solution are calcined under 1 atmosphere of nitrogen, which is divided into two stages of calcination. Firstly, calcining at 450° C. for 1 hour, and then calcining at 600° C. for 2 hours (step S706 ), so that the catalyst is loaded on the metal mesh, and the preparation of the electrode of Example 10 is completed.
实施例十一(镍发泡材+LaCoO3/2%P123)Example eleven (nickel foam + LaCoO3/2% P123)
首先,将P123溶于20ml去离子水,并加入0.417g冰醋酸当触媒,并且使用搅拌子进行搅拌1小时,使P123均匀溶解于水中,形成0.007M的P123水溶液(步骤S101)。其中,P123、去离子水以及冰醋酸莫耳数比为0.02:320:1。First, dissolve P123 in 20ml of deionized water, add 0.417g of glacial acetic acid as a catalyst, and stir for 1 hour with a stirring bar, so that P123 is uniformly dissolved in water to form a 0.007M aqueous solution of P123 (step S101). Among them, the molar ratio of P123, deionized water and glacial acetic acid is 0.02:320:1.
接着,量秤莫耳数比为1:1:1的硝酸镧[La(NO3)3˙6H2O]、硝酸亚钴[Co(NO3)2˙6H2O]以及柠檬酸[C6H8H7˙6H2O](步骤S702),并加入去离子水,使水溶液中的镧离子以及亚钴离子的浓度为0.35M。Next, lanthanum nitrate [La(NO 3 ) 3 ˙6H 2 O], cobaltous nitrate [Co(NO 3 ) 2 ˙6H 2 O] and citric acid [C 6 H 8 H 7 ˙6H 2 O] (step S702 ), and adding deionized water, so that the concentration of lanthanum ion and cobaltous ion in the aqueous solution is 0.35M.
然后,将含有镧离子以及亚钴离子的水溶液缓慢滴入含有P123的水溶液(步骤S703),并持续搅拌2小时。其中,硝酸镧、硝酸亚钴、柠檬酸、冰醋酸、去离子水、以及P123的莫耳数比为1:1:1:1:320:0.02,并且溶液为澄清的粉红色溶液。Then, slowly drop the aqueous solution containing lanthanum ions and cobaltous ions into the aqueous solution containing P123 (step S703 ), and keep stirring for 2 hours. Among them, the molar ratio of lanthanum nitrate, cobaltous nitrate, citric acid, glacial acetic acid, deionized water, and P123 is 1:1:1:1:320:0.02, and the solution is a clear pink solution.
接着,提供一镍发泡材(步骤S704),并将镍发泡材浸泡于溶液中(步骤S705)。其中,镍发泡材为一具有多孔性的金属网,因此可具有较高的表面积以担载触媒。Next, a nickel foam material is provided (step S704 ), and the nickel foam material is soaked in the solution (step S705 ). Wherein, the nickel foam material is a porous metal mesh, so it can have a relatively high surface area to support the catalyst.
最后,将镍发泡材与溶液在1大气压的氮气下进行煅烧,分为两阶段煅烧。首先是在450℃下煅烧1小时,接着在600℃下煅烧2小时(步骤S706),以使触媒担载在金属网上,即完成了实施例十一的电极的制备。Finally, the nickel foam material and the solution are calcined under 1 atmosphere of nitrogen, which is divided into two stages of calcination. Firstly, calcining at 450° C. for 1 hour, and then calcining at 600° C. for 2 hours (step S706 ), so that the catalyst is loaded on the metal mesh, and the preparation of the electrode of Example 11 is completed.
将镍发泡材以及实施例十一(2%P123)的电极进行X光绕射分析,结果如图8A以及图8B所示,其中图8A为镍发泡材的X光绕射分析结果,图8B为实施例十一的电极的X光绕射分析结果。其中,图8A在2θ为44.50度、51.84度、76.37度的处出现信号,将这些信号与数据库进行比对,可得知这些信号为镍金属相(ICDD code:00-004-0850)的特性峰。另一方面,图8B在2θ为32.88度、33.29度、47.50度、58.9度的处出现信号,将这些信号与数据库进行比对,可得知这些信号是为LaCoO3钙钛矿相(ICDD code:01-084-0848)的特性峰。因此,依据图8A以及图8B可得知,本发明实施例的镍发泡材的表面确实担载了LaCoO3的触媒。The nickel foam material and the electrode of Example 11 (2%P123) are subjected to X-ray diffraction analysis, and the results are shown in Figure 8A and Figure 8B, wherein Figure 8A is the X-ray diffraction analysis result of the nickel foam material, Fig. 8B is the X-ray diffraction analysis result of the electrode of the eleventh embodiment. Among them, in Figure 8A, signals appear at 2θ of 44.50 degrees, 51.84 degrees, and 76.37 degrees. Comparing these signals with the database, it can be known that these signals are characteristics of the nickel metal phase (ICDD code: 00-004-0850) peak. On the other hand, in Figure 8B, signals appear at 2θ of 32.88 degrees, 33.29 degrees, 47.50 degrees, and 58.9 degrees. By comparing these signals with the database, it can be known that these signals are LaCoO3 perovskite phases (ICDD code: 01-084-0848) characteristic peak. Therefore, according to FIG. 8A and FIG. 8B , it can be seen that the surface of the nickel foam material according to the embodiment of the present invention is indeed loaded with the catalyst of LaCoO 3 .
接着请参阅图9A至图9C,图9A至图9C分别为镍发泡材、实施例十的电极以及实施例十一的电极的扫描式电子显微镜(Scanning ElectronMicroscope,SEM)的分析结果。如图9B及图9C所示,镍发泡材上确实担载了LaCoO3的触媒并且均匀地分布在镍发泡材表面,且无明显改变镍发泡材的表面形貌。藉此,本发明的电极的制备方法无须使用接着剂仍可将触媒与载体接合,在制备上具有简单、快速、便宜等优点。Next, please refer to FIG. 9A to FIG. 9C . FIG. 9A to FIG. 9C are the analysis results of the nickel foam material, the electrode of Example 10, and the electrode of Example 11, respectively, by a Scanning Electron Microscope (SEM). As shown in Figure 9B and Figure 9C, the LaCoO 3 catalyst is indeed loaded on the nickel foam material and evenly distributed on the surface of the nickel foam material, and the surface morphology of the nickel foam material does not change significantly. Thereby, the preparation method of the electrode of the present invention can still join the catalyst and the carrier without using an adhesive, and has the advantages of simplicity, speed, and cheapness in preparation.
最后,请参阅图10,图10为本发明一实施例所揭露的改进空气电池的示意图。电池100包含疏水层110、触媒层120、电解质层130以及金属层140。其中,疏水层110包含镍发泡材以及铁氟龙,触媒层120包含如实施例十的电极,电解质层130包含碱性溶液、胶态电解质以及固态电解质,140金属层例如可包含锂、钠或锌。Finally, please refer to FIG. 10 , which is a schematic diagram of an improved air battery disclosed in an embodiment of the present invention. The
根据本发明实施例所揭露的触媒、电极及其制备方法,由于在制备触媒及电极的过程中加入了三区块共聚物界面活性剂,使得所制备的触媒具有单一相的钙钛矿结构,因此解决了在制备触媒的过程中,会产生杂相并且杂相会降低触媒的表面积并进而降低所制成电池的效能的问题。According to the catalyst, electrode and preparation method thereof disclosed in the embodiments of the present invention, since a three-block copolymer surfactant is added in the process of preparing the catalyst and electrode, the prepared catalyst has a single-phase perovskite structure, Therefore, the problem that impurity phases will be generated during the preparation of the catalyst and the surface area of the catalyst will be reduced by the impurity phases will further reduce the efficiency of the fabricated battery.
此外,在制备触媒及电极的过程中加入三区块共聚物界面活性剂,还可提升所制成的触媒及电池的比表面积,进而可提高所制成的电池的放电电压及电容密度。此外,三区块共聚物界面活性剂还可使触媒无须接着剂即可与金属网接合而使触媒直接接触金属网,因而在制备上还具有简单、快速、便宜等优点。In addition, adding the three-block copolymer surfactant in the process of preparing the catalyst and the electrode can also increase the specific surface area of the prepared catalyst and battery, thereby increasing the discharge voltage and capacitance density of the prepared battery. In addition, the three-block copolymer surfactant can also enable the catalyst to be bonded to the metal mesh without an adhesive, so that the catalyst directly contacts the metal mesh, and thus has the advantages of simplicity, speed, and cheapness in preparation.
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员当可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明所附的权利要求的保护范围。Certainly, the present invention also can have other multiple embodiments, without departing from the spirit and essence of the present invention, those skilled in the art can make various corresponding changes and deformations according to the present invention, but these corresponding Changes and deformations should belong to the scope of protection of the appended claims of the present invention.
Claims (17)
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| TW101146634A TWI482660B (en) | 2012-12-11 | 2012-12-11 | Electrode, and manufacturing method thereof |
| TW101146634 | 2012-12-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN103872348A true CN103872348A (en) | 2014-06-18 |
| CN103872348B CN103872348B (en) | 2016-08-24 |
Family
ID=50910654
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210580508.4A Active CN103872348B (en) | 2012-12-11 | 2012-12-27 | catalyst, electrode and preparation method thereof |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN103872348B (en) |
| TW (1) | TWI482660B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111668377A (en) * | 2020-06-08 | 2020-09-15 | 河南大学 | A kind of perovskite solar cell with Mo-tin dioxide as electron transport layer and preparation method thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1336016A (en) * | 1999-10-08 | 2002-02-13 | 全球热电公司 | Composite electrodes for solid state electrochemical devices |
| CN101083334A (en) * | 2007-07-13 | 2007-12-05 | 西安交通大学 | Borohydride alkaline dry cell |
| CN101383418A (en) * | 2007-08-31 | 2009-03-11 | 丹麦技术大学 | Electrodes based on ceria and stainless steel |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013534697A (en) * | 2010-06-22 | 2013-09-05 | ビーエーエスエフ ソシエタス・ヨーロピア | Electrodes and their manufacture and use |
-
2012
- 2012-12-11 TW TW101146634A patent/TWI482660B/en active
- 2012-12-27 CN CN201210580508.4A patent/CN103872348B/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1336016A (en) * | 1999-10-08 | 2002-02-13 | 全球热电公司 | Composite electrodes for solid state electrochemical devices |
| CN101083334A (en) * | 2007-07-13 | 2007-12-05 | 西安交通大学 | Borohydride alkaline dry cell |
| CN101383418A (en) * | 2007-08-31 | 2009-03-11 | 丹麦技术大学 | Electrodes based on ceria and stainless steel |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111668377A (en) * | 2020-06-08 | 2020-09-15 | 河南大学 | A kind of perovskite solar cell with Mo-tin dioxide as electron transport layer and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI482660B (en) | 2015-05-01 |
| TW201422302A (en) | 2014-06-16 |
| CN103872348B (en) | 2016-08-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Yu et al. | Advances in porous perovskites: synthesis and electrocatalytic performance in fuel cells and metal–air batteries | |
| CN100435392C (en) | Catalyst for fuel cell and its preparation, membrane-electrode assembly and fuel cell system | |
| CN107159297B (en) | Double-function oxygen catalyst cobalt/cobaltosic oxide/nitrogen carbon composite material and preparation method thereof | |
| CN103427094B (en) | Oxide of perovskite structure and its production and use | |
| CN104538647B (en) | A kind of preparation method of lithium-air battery catalyst | |
| CN111054411B (en) | Preparation method of multi-metal carbide electrocatalyst | |
| Wang et al. | recent advance in self-supported electrocatalysts for rechargeable Zinc-air batteries | |
| US11901566B2 (en) | Bifunctional oxygen electrocatalyst, preparation method and use thereof | |
| CN113955728B (en) | Preparation of cobalt phosphide/cobalt manganese phosphide with hollow grade structure and application of electrolytic water | |
| CN110280288A (en) | The preparation method of bifunctional catalyst is precipitated in a kind of nitrogen co-doped carbon material hydrogen reduction of novel transition metal -/oxygen | |
| CN111430734A (en) | (Pr0.5Sr0.5)xFe1-yRuyO3-Perovskite material and preparation method and application thereof | |
| CN113964331B (en) | Nano-micron multilevel structure strontium-cobalt-based perovskite composite cathode and preparation method thereof | |
| KR101308020B1 (en) | Composite powders having core-shell structure and methods for fabricating the same | |
| CN102364737A (en) | Preparation method of a flat SOFC anti-carbon composite anode membrane material | |
| CN114620772A (en) | Doped transition metal oxide and preparation method and application thereof | |
| CN110729489B (en) | Alkaline fuel cell and preparation method of molybdenum-nickel alloy nano material | |
| CN113555569B (en) | Catalyst precursor, metal carbon-based catalyst, and preparation methods and applications thereof | |
| CN114792815B (en) | A single-atom iron catalyst for electrocatalytic oxygen reduction to water and its preparation method and application | |
| CN103872348B (en) | catalyst, electrode and preparation method thereof | |
| Li et al. | In situ construction of Co3O4 nanoarray catalysts on (La0. 8Sr0. 2) 0.95 MnO3–δ cathode for high-efficiency intermediate-temperature solid oxide fuel cells | |
| CN115224293B (en) | An ORR and OER dual-functional catalyst and its preparation method and application | |
| Wu et al. | Active sites derived from heteroatom doping in carbon materials for oxygen reduction reaction | |
| JPWO2019177060A1 (en) | Electrode catalyst for fuel cells and fuel cells using them | |
| CN111744496B (en) | Self-supporting composite electrode material and preparation method and application thereof | |
| CN101306842A (en) | A preparation method of ceramic cathode nanopowder for solid oxide fuel cell |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |