CN101657462B - The preparation method and intermediates of capecitabine - Google Patents
The preparation method and intermediates of capecitabine Download PDFInfo
- Publication number
- CN101657462B CN101657462B CN200780052717.9A CN200780052717A CN101657462B CN 101657462 B CN101657462 B CN 101657462B CN 200780052717 A CN200780052717 A CN 200780052717A CN 101657462 B CN101657462 B CN 101657462B
- Authority
- CN
- China
- Prior art keywords
- formula
- deoxidation
- grams
- compound
- capecitabine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 C[C@]1OC(C2)(C2*(C=C(C(*[U]C)=*2)N)C2=O)[C@@]2OC(c3cccc(N)c3)O[C@]12 Chemical compound C[C@]1OC(C2)(C2*(C=C(C(*[U]C)=*2)N)C2=O)[C@@]2OC(c3cccc(N)c3)O[C@]12 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/073—Pyrimidine radicals with 2-deoxyribosyl as the saccharide radical
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to the preparation method and intermediates of capecitabine. The present invention provides a new synthesis route for capecitabine, in which doxifluridine is used as the starting material and capecitabine is obtained through three steps of reaction. The present invention also provides the intermediates in the said synthesis route. The synthesis has short route, which can avoid producing stereoisomers. The method has a high yield, the technical process is easy to control and the quality of the products is stable.
Description
Technical field
The present invention relates to the Preparation Method And Their Intermediate of capecitabine.
Background technology
Capecitabine (Capecitabine) is the prodrug of 5 FU 5 fluorouracil, and tumour cell is had selectively acting, can be used as oral cytotoxicity preparation.
Capecitabine itself is cytotoxicity not, has Cytotoxic 5 FU 5 fluorouracil but be converted into through three steps under the effect of enzyme in vivo.High in concentration compared with normal tissue with capecitabine metabolism involved enzyme in tumor tissues, thus make it have selecting cell toxicity to tumour cell.Its structural formula is as follows:

The synthetic method of the capecitabine of report mainly comprises following several at present:
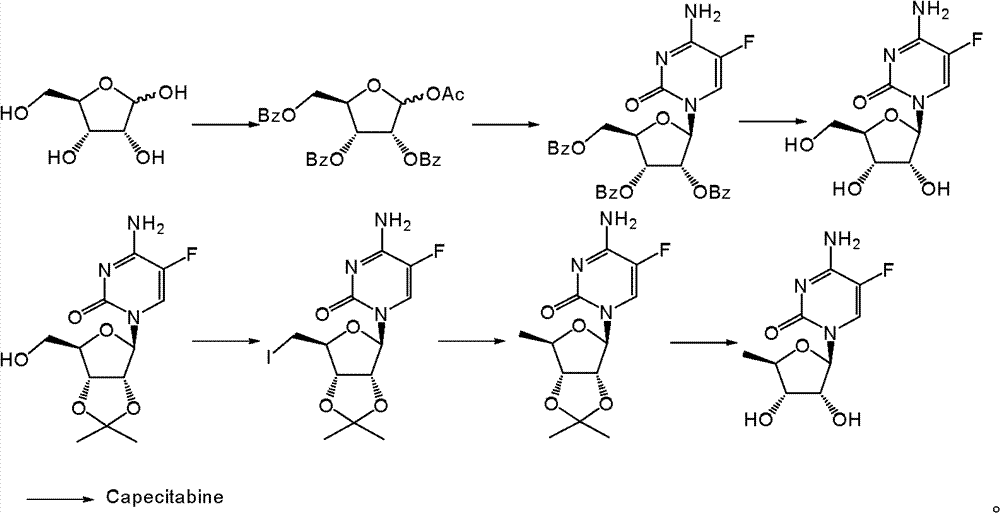
1. use the triacetoxyl group ribofuranose of racemization to dock with 5-flurocytosine, then obtain acylate with acyl chloride reaction, then be hydrolyzed and obtain capecitabine (Bioorganic﹠amp; MedicinalChemistry 8,2000,1697-1706)

2. use 5 '-deoxidation-5-fluoro-cytidine as starting raw material through two acidylate steps, then hydrolysis obtain product (Drug of the Future 21,1996,358-360).

3. use the pentyloxy formyl chloride as acylating reagent, hydroxyl and amino are carried out acidylate, then selective hydrolysis obtains the finished product (US005476932).

4. acylated 5-flurocytosine is as raw material in use, and docking reaction then hydrolysis obtains the finished product (CN166089).

5. use ribose to be raw material, transform through seven steps and obtain final product (Chinese pharmaceutical chemistry magazine, 15,2005,173).
Summary of the invention
The invention provides a kind of new capecitabine synthetic route, prepare capecitabine take deoxidation fluorouracil glucoside as raw material.
Technical scheme of the present invention is as follows:
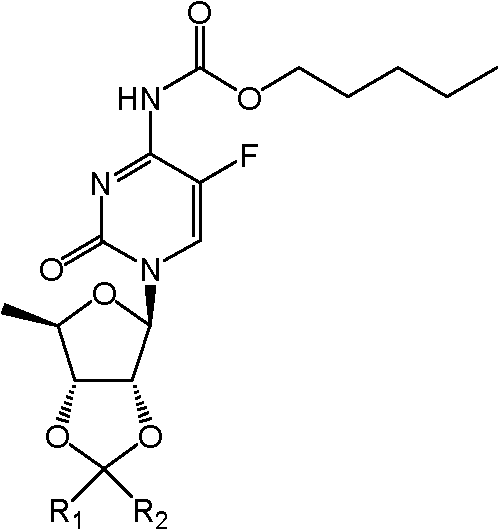
The invention provides a kind of compound, the deoxidation fluorouracil glycoside derivates, shown in (I):
(I), wherein, R
1Can be selected from hydrogen atom, contain the phenyl ring of straight or branched alkyl, phenyl ring or the replacement of 1~8 carbon atom; R
2Can be selected from hydrogen atom, contain the phenyl ring of straight or branched alkyl, phenyl ring or the replacement of 1~8 carbon atom.
The present invention also provides the preparation method of deoxidation fluorouracil glycoside derivates shown in formula (I), and the method is carried out condensation reaction with deoxidation fluorouracil glucoside and aldehydes or ketones and obtained deoxidation fluorouracil glycoside derivates shown in formula (I) in the presence of an acidic catalyst.
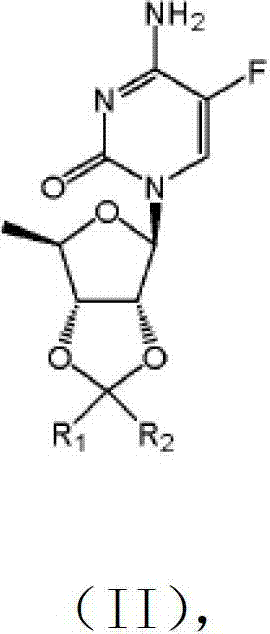
The invention provides a kind of compound, the deoxidation fluorcytidines, shown in (II):

(II), wherein, R
1Can be selected from the phenyl ring of hydrogen atom, alkyl, phenyl ring or replacement; R
2Can be selected from the phenyl ring of hydrogen atom, alkyl, phenyl ring or replacement.
The present invention also provides the preparation method of deoxidation fluorcytidines shown in formula (II), and the method obtains deoxidation fluorcytidines shown in formula (II) with deoxidation fluorouracil glycoside derivates shown in formula (I) and phosphorus oxychloride, organic bases, ammoniacal liquor effect.
The invention provides a kind of compound, the deoxidation fluorcytidines, shown in (III):
(III), wherein, R
1Can be selected from the phenyl ring of hydrogen atom, alkyl, phenyl ring or replacement; R
2Can be selected from the phenyl ring of hydrogen atom, alkyl, phenyl ring or replacement.
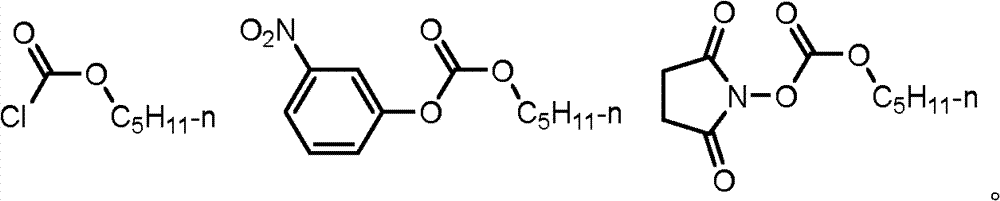
The present invention also provides the preparation method of deoxidation fluorcytidines shown in formula (III), and the method is reacted deoxidation fluorcytidines shown in the formula of obtaining (III) with compound shown in deoxidation fluorcytidines shown in formula (II) and formula (IV).

The present invention also provides the preparation method of capecitabine, and the method is sloughed hydroxy-protective group with deoxidation fluorcytidines shown in formula (III) and obtained capecitabine under acidic conditions.
Deoxidation fluorcytidines shown in described formula (III) reacts deoxidation fluorcytidines shown in the formula of obtaining (III) with compound shown in deoxidation fluorcytidines shown in formula (II) and formula (IV).
Deoxidation fluorcytidines shown in described formula (II) obtains deoxidation fluorcytidines shown in formula (II) with deoxidation fluorouracil glycoside derivates shown in formula (I) and phosphorus oxychloride, organic bases, ammoniacal liquor effect.
Deoxidation fluorouracil glycoside derivates shown in described formula (I) carries out condensation reaction with deoxidation fluorouracil glucoside and aldehydes or ketones and obtains deoxidation fluorouracil glycoside derivates shown in formula (I) in the presence of an acidic catalyst.
Above method concrete steps are as follows:
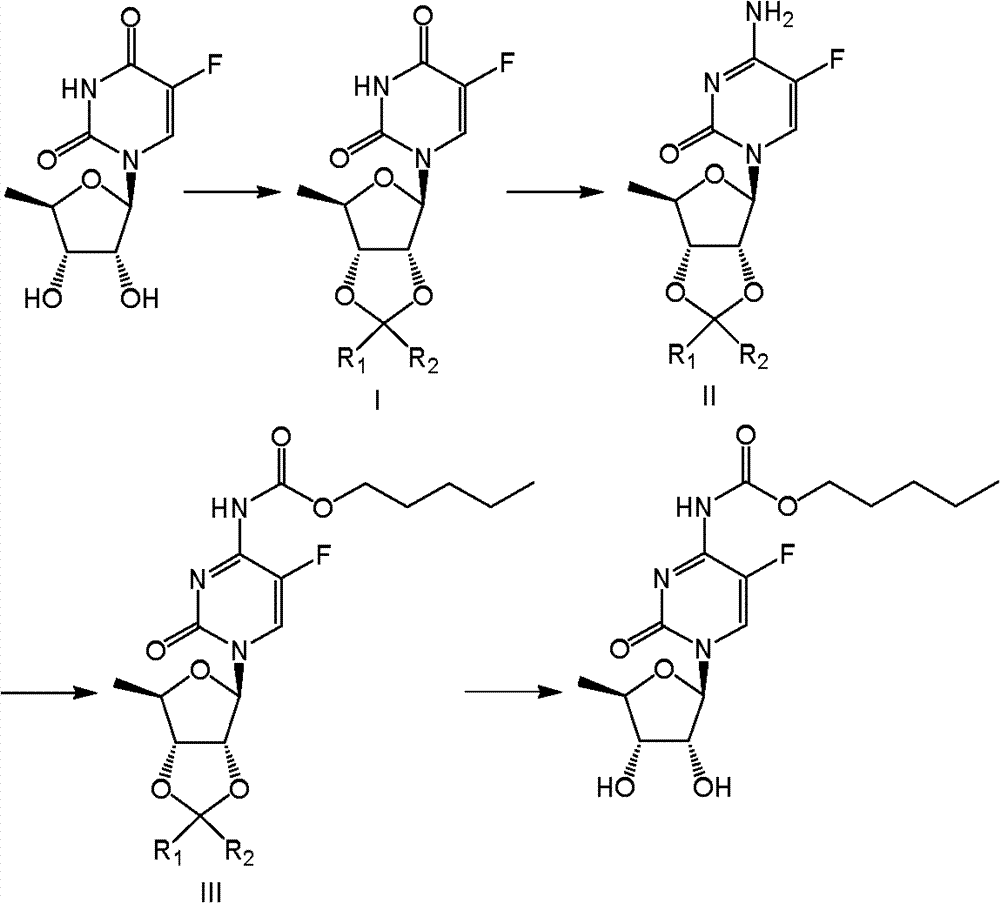
Deoxidation fluorouracil glucoside carries out condensation reaction from different aldehydes or ketones and obtains deoxidation fluorouracil glycoside derivates shown in formula (I); then pass through and phosphorus oxychloride; organic bases; the effect of ammoniacal liquor obtains deoxidation fluorcytidines shown in formula (II); obtain deoxidation fluorcytidines shown in formula (III) with acylating reagent generation acylation reaction shown in formula (IV) again, slough hydroxy-protective group at last under acidic conditions and obtain capecitabine.
In above reaction, the condensation reaction that deoxidation fluorouracil glucoside and aldehydes or ketones carry out can be carried out in arbitrarily than mixed solvent at a kind of of the toluene, benzene, acetone, tetrahydrofuran (THF), methylene dichloride or the ethylene dichloride that add an acidic catalyst or its.Described an acidic catalyst can be selected from tosic acid, zinc chloride, tin chloride.Temperature of reaction can change in the larger context, is generally-20 ℃-120 ℃, and preferred 80 ℃-120 ℃, the molar ratio of deoxidation fluorouracil glucoside and aldehydes or ketones is 1: 1-1: 2.
The reaction of deoxidation fluorouracil glycoside derivates and phosphorus oxychloride, organic bases, ammoniacal liquor shown in formula (I) can be carried out in the aprotic solvent mixed solvent that acetonitrile or other and water dissolve each other.Temperature of reaction is-10 ℃-30 ℃, preferably carries out at-5 ℃-20 ℃.
Acylating reagent shown in deoxidation fluorcytidines shown in formula (II) and formula (IV) can carry out in the acetonitrile that adds basic catalyst or other non-protonic solvents.Described basic catalyst can be mineral alkali or organic bases, specifically can be selected from salt of wormwood, triethylamine, pyridine.Temperature of reaction is-10 ℃-50 ℃, preferably carries out at 0 ℃-20 ℃.Shown in deoxidation fluorcytidines shown in formula (II) and formula (IV), the molar ratio of acylating reagent is 1: 1.1-1: 3, be preferably 1: 1.1-1: 2.
Shown in formula (III), the deoxidation fluorcytidines is sloughed blocking group, obtains the reaction of capecitabine, can carry out in the proton aqueous acid, can carry out in the alcoholic solution of protonic acid or in ethereal solution, also can carry out in the solution of aprotic acid.Preferably carry out in the alcoholic solution of protonic acid.
The technique effect that the present invention realizes is as follows:
The method uses the definite deoxidation fluorouracil glucoside of configuration as raw material, has obtained capecitabine through three-step reaction, and synthetic route is short, has avoided the generation of steric isomer.Through evidence, the yield of the method is high, and technological process is easily controlled, and product quality is stable.
Specific implementation method:
The present invention further illustrates by following examples, and following examples only limit technical scheme of the present invention for the preferred embodiment of the present invention more specifically being described, being not used in.The technical scheme of the invention described above is the technical scheme that can realize the object of the invention.Be temperature that following examples adopt and reagent, all available relevant temperature mentioned above and reagent substitute to realize the present invention's purpose.
Embodiment 1:

0.26 gram (1.5mmol) tosic acid is dissolved in 20ml acetone, add 3.69 grams (15mmol) deoxidation fluorouracil glucoside, stirring at room 24 hours adds solid carbonic acid potassium in reaction system, regulating the pH value is 7, filter, filtrate concentrating removed acetone and got white solid, and dissolving adds methylene chloride, wash with water, anhydrous sodium sulfate drying, removal of solvent under reduced pressure obtain white solid (Ia) 3.83 grams, yield 89.2%.Ia:
1H?NMR(300MHz,CDCl
3):δ7.36(d,1H,J=7.6Hz),5.67(s,1H),4.67(dd,1H,J=8.0,3.2Hz),4.50(dd,1H,J=4.5,3.6Hz),4.24(m,1H),1.56(s,3H),1.40(d,3H,J=6.3Hz),1.34(s,3H);ESI-MS?m/z(M+1
+)287。
Embodiment 2:
0.2 gram (1.5mmol) zinc chloride is dissolved in 20ml acetone, add 3.69 grams (15mmol) deoxidation fluorouracil glucoside,-20 ℃ were stirred 24 hours, added salt of wormwood in reaction system, and regulating the pH value is 7, filter, filtrate concentrating removed acetone and got white solid, and the dissolving that adds methylene chloride washes with water, anhydrous sodium sulfate drying, removal of solvent under reduced pressure get white solid Ia.
Embodiment 3:
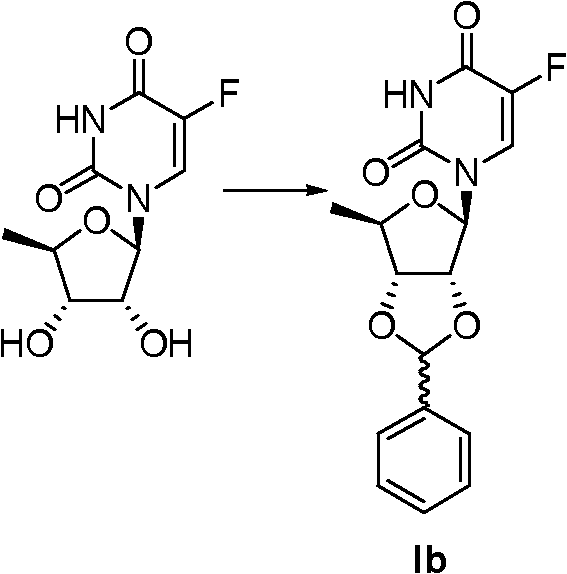
0.35 gram (2mmol) tosic acid is dissolved in 100ml toluene, add 4.92 grams (20mmol) deoxidation fluorouracil glucoside and 6.87 grams (65.0mmol) phenyl aldehyde to reflux, stirred 4 hours, cooling, filter, filter residue is raw material 2.48 grams, and filtrate is concentrated, resistates obtains solid (Ib) 2.80 grams, yield 91.0% with ethyl acetate and normal hexane recrystallization.Ib:
1H?NMR(300MHz,CDCl
3):δ9.17(s,1H),7.32-7.50(m,6H),6.09(s,1H),5.74(d,1H,J=2.7Hz),4.96(dd,1H,J=3.0,3.9Hz),4.69(m,1H),4.30(m,1H),1.47(d,3H,J=6.3Hz);EI-MS?m/z(M+1
+)334。
Embodiment 4:

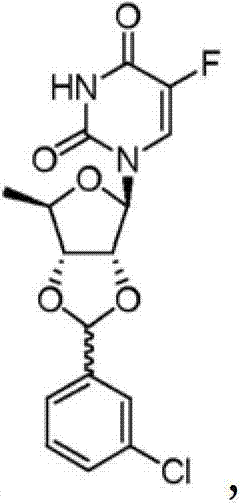
0.2 gram (1.2mmol) tosic acid is dissolved in 40ml toluene, add 3.0 grams (12.2mmol) deoxidation fluorouracil glucoside and 2.01 grams (14.3mmol) m chlorobenzaldehyde, reflux, stirred 4 hours, cooling, filter, filter residue is raw material 0.7 gram, filtrate is concentrated, and resistates obtains solid (Ic) 3.15 grams, yield 91.0% with ethyl acetate and normal hexane recrystallization.Ic:
1H?NMR(300MHz,CDCl
3):δ9.17(s,1H),7.32-7.50(m,6H),6.09(s,1H),5.74(d,1H,J=2.7Hz),4.96(dd,1H,J=3.0,3.9Hz),4.69(m,1H),4.30(m,1H),1.47(d,3H,J=6.3Hz);EI-MS?m/z(M+1
+)368。
Embodiment 5:

0.2 gram (1.2mmol) tosic acid is dissolved in 40 acetonitriles, add 3.0 grams (12.2mmol) deoxidation fluorouracil glucoside and 3.26 grams (14.3mmol) methoxyl group ditan, reflux, stirred 4 hours, cooling, to filter, filtrate is concentrated, resistates obtains solid (Id) 2.3 grams, yield 46.0% with ethyl acetate and normal hexane recrystallization.Id:
1H?NMR(300MHz,CDCl
3):δ9.22(brs,1H),7.31-7.52(m,6H),5.82(d,1H,J=2.1Hz),4.88(dd,1H,J=6.9,2.4Hz),4.55(dd,1H,J=6.6,4.2Hz),4.42(dd,1H,J=6.6,4.5Hz),1.42(d,3H,J=6.6Hz);EI-MS?m/z(M
+)410。
Embodiment 6:
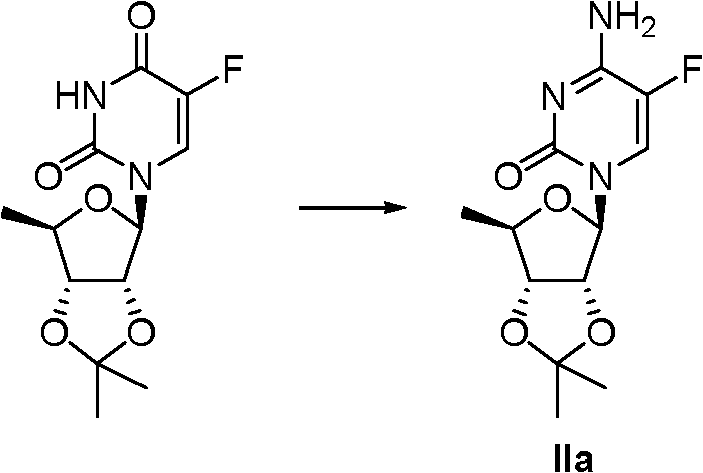
3.83 grams (13.4mmol) Ia is dissolved in the 35ml anhydrous acetonitrile, adds 3.17 grams (40.0mmol) pyridine and 4.88 grams (40.0mmol) N, the N-Dimethylamino pyridine, be cooled to 0 ℃, drip 6.14 grams (40.0mmol) phosphorus oxychloride, stirred 6 hours, reaction solution is poured in 0 ℃ of ammoniacal liquor, stirred 0.5 hour, separatory, the organic phase washed with dichloromethane merges organic phase, anhydrous sodium sulfate drying, removal of solvent under reduced pressure obtain crude product (IIa) 6.0 grams.IIa:
1H?NMR(300MHz,CDCl
3):δ7.42(d,1H,J=5.7Hz),5.57(d,1H,J=1.2Hz),4.93(dd,1H,J=6.6,1.8Hz),4.49(dd,1H,J=6.6,1.5Hz),4.27(dd,1H,J=6.3,4.2Hz),1.55(s,3H),1.38(d,1H,J=6.6Hz),1.32(s,3H);EI-MS?m/z(M
+)285。
Embodiment 7:
3.83 grams (13.4mmol) Ia is dissolved in the 35ml anhydrous acetonitrile, adds 3.17 grams (40.0mmol) pyridine and 4.88 grams (40.0mmol) N, the N-Dimethylamino pyridine, 30 ℃, drip 6.14 grams (40.0mmol) phosphorus oxychloride, stirred 6 hours, reaction solution is poured in 0 ℃ of ammoniacal liquor, stirred 0.5 hour, separatory, the organic phase washed with dichloromethane merges organic phase, anhydrous sodium sulfate drying, removal of solvent under reduced pressure obtain crude product (IIa) 6.0 grams.
Embodiment 8-10:
Take Ib, Ic or Id as raw material, the method according to identical with embodiment 7 has obtained Compound I Ib, IIc and IId respectively.
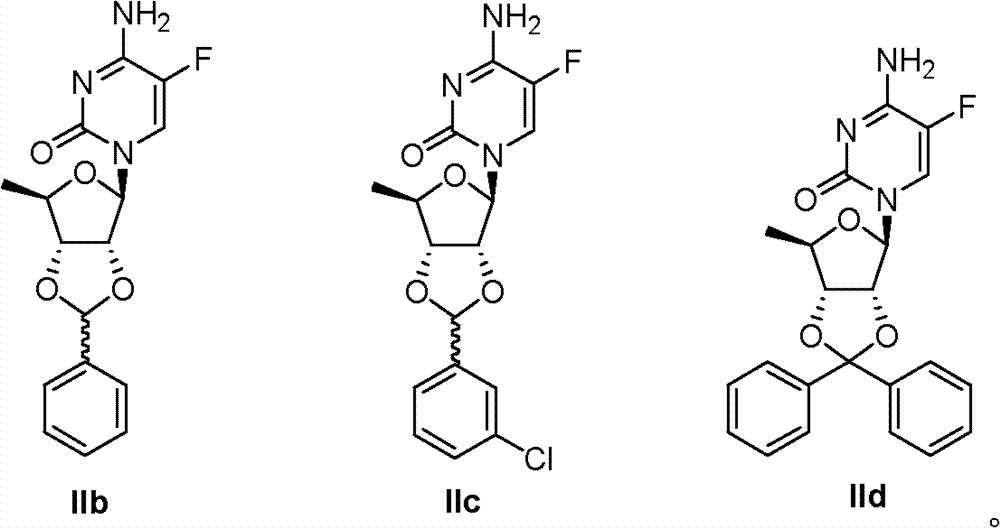
IIb:
1H?NMR(300MHz,CDCl
3):δ8.00(d,1H,J=8.6Hz),7.40-7.54(m,5H),6.11(s,1H),5.88(d,1H,J=4.4Hz),4.94(dd,1H,J=8.8,4.0Hz),4.69(m,1H),4.20(m,1H),1.36(d,3H,J=7.6Hz);EI-MSm/z?(M
+)333。
IIc:EI-MS?m/z(M
+)367。
IId:
1H?NMR(300MHz,CDCl
3):δ8.00(d,1H,J=8.6Hz),7.26-7.52(m,10H),6.11(s,1H),5.73(d,1H,J=2.4Hz),4.96(dd,1H,J=6.8,2.4Hz),4.59(dd,1H,J=6.6,4.2Hz),4.46(m,1H),1.42(d,3H,J=6.6Hz).ESI-MS?m/z(M+Na
+)432。
Embodiment 11:
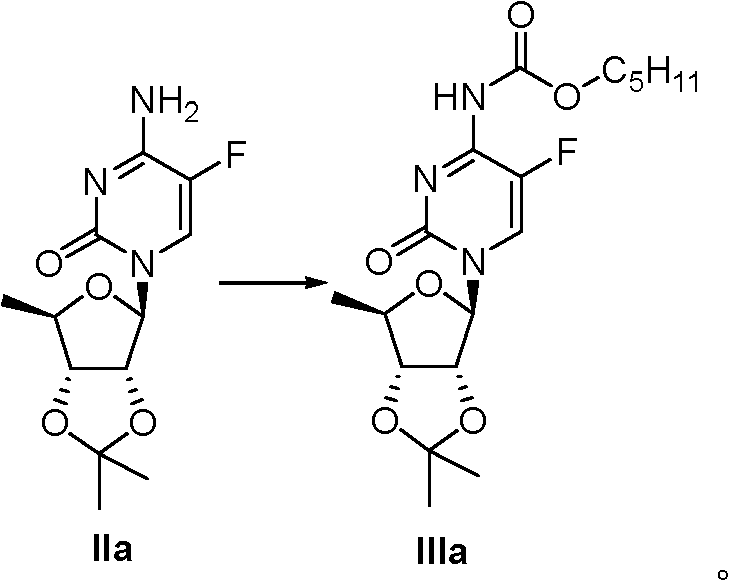
6.0 gram IIa are dissolved in the 40ml acetonitrile, add 7.6 grams (26.8mmol) N-, penta oxygen ketonic oxygen base succimide and 3.7 grams (26.8mmol) salt of wormwood, stirring at room 24 hours is filtered, removal of solvent under reduced pressure, resistates is dissolved in methylene dichloride, and with 1N hydrochloric acid washed twice, the saturated common salt water washing once, anhydrous sodium sulfate drying, removal of solvent under reduced pressure gets resistates, gets product (IIIa) 3.43 grams through column chromatography, and two step yields are 64%.IIIa:
1H?NMR(300MHz,CDCl
3):δ12.05(brs,1H),7.41(d,1H,J=4.8Hz),5.65(s,1H),4.87(d,1H,J=5.4Hz),4.49(dd,1H,J=4.2,6.3Hz),4.14-4.28(m,1H),1.70(m,2H),1.56(s,3H),1.28-1.42(m,11H,J=6.3Hz),0.89(t,3H,J=7.2Hz);ESI-MS?m/z(M+Na
+)422。
Embodiment 12:
6.0 gram IIa are dissolved in the 40ml acetonitrile, add 6.78 grams (26.8mmol) m-nitro base n-pentyl carbonic ether and 3.7 grams (26.8mmol) salt of wormwood, stirring at room 24 hours is filtered, removal of solvent under reduced pressure, resistates is dissolved in methylene dichloride, and with 1N hydrochloric acid washed twice, the saturated common salt water washing once, anhydrous sodium sulfate drying, removal of solvent under reduced pressure gets resistates, must produce (IIIa) 3.33 grams through column chromatography, and two step yields are 62%.
Embodiment 13:
6.0 gram IIa are dissolved in the 40ml acetonitrile, add 4.02 grams (26.8mmol) n-amyl chlorocarbonate and 2.1 grams (26.8mmol) salt of wormwood, 0 ℃ was stirred 2 hours, and removal of solvent under reduced pressure is dissolved in methylene dichloride with resistates, with 1N hydrochloric acid washed twice, the saturated common salt water washing once, anhydrous sodium sulfate drying, removal of solvent under reduced pressure gets resistates, must produce (IIIa) 3.63 grams through column chromatography, two step yields are 66%.
Embodiment 14~16:
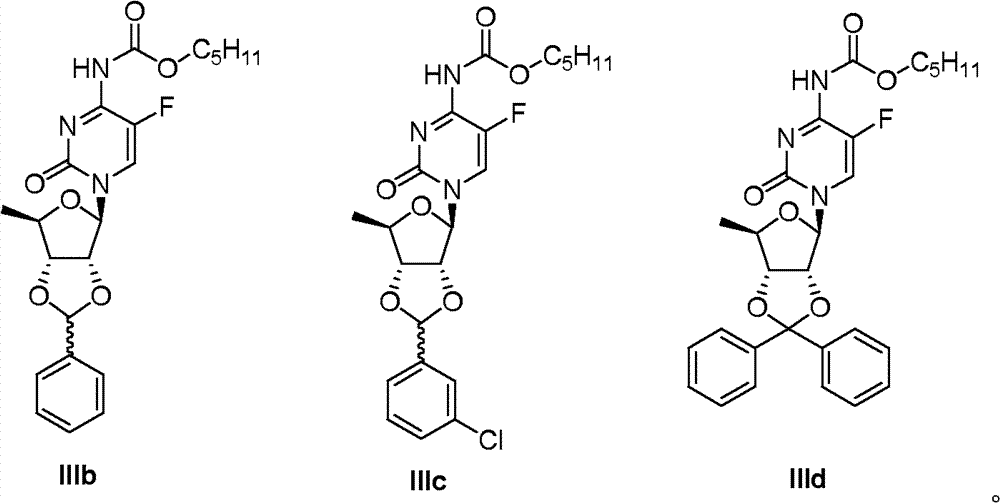
Take IIb and IIc as raw material, the method according to identical with embodiment 11 has obtained compound III b, IIIc and IIId respectively.
IIIb:
1H?NMR(300MHz,CDCl
3):δ12.09(brs,1H),7.42-7.58(m,6H),6.13(s,1H),5.75(d,1H,J=2.7Hz),5.03(m,1H),4.74(m,1H),4.19-4.40(m,3H),1.74(m,2H),1.48(d,3H,J=8.4Hz),1.29-1.43(m,5H,J=6.3Hz),0.90(t,3H,J=7.2Hz);ESI-MS?m/z(M+Na
+)470。
IIIc:
1H?NMR(300MHz,CDCl
3):δ12.05(brs,1H),7.30-7.50(m,5H),6.07(s,1H),5.68(m,1H),4.97-5.11(m,1H),4.66(m,1H),4.17-4.44(m,3H),1.70(m,2H),1.48(d,3H,J=8.4Hz),1.20-1.37(m,5H),0.85(t,3H,J=7.2Hz);ESI-MS?m/z(M+Na
+)504。
IIId:
1H?NMR(300MHz,CDCl
3):δ12.03(brs,1H),7.31-7.51(m,11H),5.80(d,1H,J=2.1Hz),4.90(m,1H),4.55(dd,1H,J=6.9,4.2Hz),4.45(dd,1H,J=5.4,4.8Hz),4.01-4.18(m,2H),1.72(m,2H),1.40(d,3H,J=8.4Hz),1.25-1.37(m,5H,J=6.3Hz),0.90(t,3H,J=7.2Hz)。
Embodiment 17:
1.3 gram IIIa are dissolved in 10ml ethanol, are cooled to 0 ℃, stir, slowly drip the 10mlHCl/ ethanolic soln, keeping temperature of reaction is 0 ℃, and removal of solvent under reduced pressure after 4 hours is dissolved in methylene dichloride with resistates, uses saturated NaHCO
3Solution washing, anhydrous sodium sulfate drying filters, and removal of solvent under reduced pressure gets 1.0 gram products (capecitabine).
1H?NMR(300MHz,d-DMSO):δ8.03(brs,1H),5.67(d,1H,J=4.8Hz),4.08(m,3H),3.90(m,1H),3.68(q,1H,J=6.0Hz),1.60(m,2H),1.22-1.31(m,7H),0.88(t,3H,J=6.4Hz);ESI-MS?m/z(M
+)358。
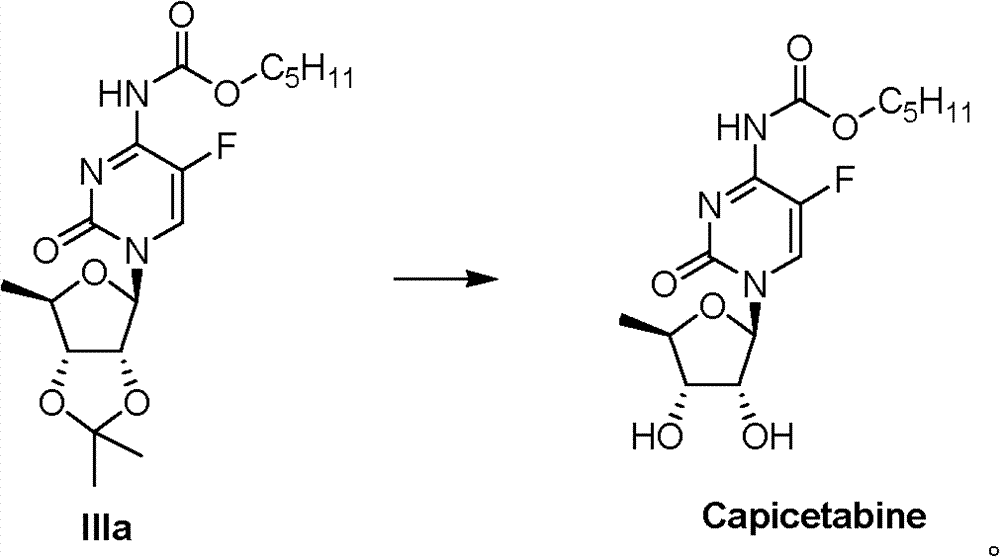
Embodiment 18~20:
Take IIIb, IIIc or IIId as raw material, the method according to identical with embodiment 17 has obtained capecitabine respectively.
Claims (4)


Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2007/070051 WO2008144980A1 (en) | 2007-05-25 | 2007-05-25 | The preparation method and intermediates of capecitabine |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101657462A CN101657462A (en) | 2010-02-24 |
| CN101657462B true CN101657462B (en) | 2013-06-05 |
Family
ID=40316812
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200780052717.9A Expired - Fee Related CN101657462B (en) | 2007-05-25 | 2007-05-25 | The preparation method and intermediates of capecitabine |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101657462B (en) |
| WO (1) | WO2008144980A1 (en) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009094847A1 (en) * | 2007-12-28 | 2009-08-06 | Topharman Shanghai Co., Ltd. | A capecitabine hydroxyl-derivative, its preparation processes and uses for preparing capecitabine |
| CN101469008B (en) * | 2007-12-29 | 2013-08-07 | 上海特化医药科技有限公司 | Capecitabine hydroxy derivatives, preparation thereof and use in capecitabine preparation |
| WO2011067588A1 (en) | 2009-12-04 | 2011-06-09 | Generics [Uk] Limited | Cyclic sulphinyl esters of cytidine |
| WO2011104540A1 (en) | 2010-02-24 | 2011-09-01 | Generics [Uk] Limited | One step process for the preparation of capecitabine |
| CN101845070B (en) * | 2010-05-25 | 2012-05-16 | 郑州大学 | Synthesis method of antineoplastic medicine capecitabine |
| CN103897005B (en) * | 2012-12-27 | 2017-07-28 | 鲁南制药集团股份有限公司 | Method for continuously synthesizing capecitabine |
| CN103113441A (en) * | 2013-03-13 | 2013-05-22 | 上海龙翔生物医药开发有限公司 | Method for preparing capecitabine |
| US10308597B2 (en) | 2014-04-30 | 2019-06-04 | Rgenix, Inc. | Inhibitors of creatine transport and uses thereof |
| CN104478975A (en) * | 2014-11-24 | 2015-04-01 | 苏州乔纳森新材料科技有限公司 | Synthesis method of capecitabine |
| CN106699825A (en) * | 2016-12-01 | 2017-05-24 | 齐鲁天和惠世制药有限公司 | Method for preparing capecitabine from capecitabine waste water extract |
| US10435429B2 (en) | 2017-10-03 | 2019-10-08 | Nucorion Pharmaceuticals, Inc. | 5-fluorouridine monophosphate cyclic triester compounds |
| CN117402189A (en) | 2018-01-10 | 2024-01-16 | 广东集宝医药技术有限公司 | Aminophosphorus (phosphine) acid acetal and phosphorus (phosphine) acid acetal compounds |
| CN111801339B (en) * | 2018-01-19 | 2025-06-24 | 纽科利制药公司 | 5-Fluorouracil compounds |
| US11427550B2 (en) | 2018-01-19 | 2022-08-30 | Nucorion Pharmaceuticals, Inc. | 5-fluorouracil compounds |
| US12110311B2 (en) | 2019-07-17 | 2024-10-08 | Nucorion Pharmaceuticals, Inc. | Cyclic deoxyribonucleotide compounds |
| WO2021216427A1 (en) | 2020-04-21 | 2021-10-28 | Ligand Pharmaceuticals, Inc. | Nucleotide prodrug compounds |
| CN114644666A (en) * | 2020-12-18 | 2022-06-21 | 上海特化医药科技有限公司 | Process for the preparation of 5' -nucleoside prodrugs and intermediates |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6310039B1 (en) * | 1996-09-11 | 2001-10-30 | Felix Kratz | Antineoplastic conjugates of transferrin, albumin and polyethylene glycol |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU664948B2 (en) * | 1993-02-23 | 1995-12-07 | City Of Hope | 4-ethoxy 5-fluoro 2'deoxyuridine |
| CN100425617C (en) * | 2006-10-31 | 2008-10-15 | 浙江海正药业股份有限公司 | Fluoropyrimidine compound carbalkoxylation method |
-
2007
- 2007-05-25 CN CN200780052717.9A patent/CN101657462B/en not_active Expired - Fee Related
- 2007-05-25 WO PCT/CN2007/070051 patent/WO2008144980A1/en not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6310039B1 (en) * | 1996-09-11 | 2001-10-30 | Felix Kratz | Antineoplastic conjugates of transferrin, albumin and polyethylene glycol |
Non-Patent Citations (2)
| Title |
|---|
| 余建鑫等.抗肿瘤药物卡培他滨的合成工艺改进.《中国药物化学杂志》.2005,第15卷(第3期),173-175,187. * |
| 董辉等.去氧氟尿苷合成工艺改进.《中国医药工业杂志》.2002,第33卷(第3期),1-4,13. * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101657462A (en) | 2010-02-24 |
| WO2008144980A1 (en) | 2008-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101657462B (en) | The preparation method and intermediates of capecitabine | |
| ES2398934T3 (en) | Production process of 1- (3- (2- (1-benzothiophene-5-yl) ethoxy) propyl) acetidinol or salts thereof | |
| EP3712130A1 (en) | Method for synthesis of roxadustat and intermediate compounds thereof | |
| CN110590635A (en) | Preparation method of levetiracetam and intermediate thereof | |
| CN107963996A (en) | Method for preparing 3-trifluoromethyl isoxazole compound by one-pot method | |
| WO2015012110A1 (en) | Method for manufacturing c-glycoside derivative | |
| WO2021020998A1 (en) | Method for producing roxadustat | |
| CN101575298B (en) | Method for preparing chiral medicinal intermediate 2-amido-1-phenylethylalcohol | |
| CN101952242B (en) | Convergent synthesis of renin inhibitors and intermediates useful therein | |
| CN101759716B (en) | Preparation method and intermediate of water-soluble pleuromutilin compound | |
| CN101469008A (en) | Capecitabine hydroxy derivatives, preparation thereof and use in capecitabine preparation | |
| CN102757365B (en) | Method for preparing peramivir key intermediate | |
| CN113943240A (en) | Novel preparation method of brivaracetam | |
| CN107382867B (en) | 4-isothiocyanatopyrazolones | |
| CN117362142B (en) | Asymmetric catalytic hydrogenation method for cyclohexene derivatives | |
| EP3026047A1 (en) | Method for producing heterocyclic compound | |
| CN113773294B (en) | Preparation method and application of flavonoids and isoflavones | |
| CN117417272B (en) | Preparation method of Boc-D-isoleucine | |
| CN113004161B (en) | Preparation method of (2R, 3R) -3-methyl-3-phenylalanine | |
| CN114539044B (en) | A dehydrated icariin intermediate compound | |
| AU2006325622B2 (en) | A manufacturing process of 2',2'-difluoronucleoside and intermediate | |
| CN111662233B (en) | Method for synthesizing 4-chloro-1H-imidazole-2-carboxylic acid ethyl ester by one-step method | |
| CN109776612A (en) | A kind of synthetic method of phospha chromogen ketone derivatives | |
| CN108558968A (en) | Maleimide derivatives of the triazole structure containing glucose and the preparation method and application thereof | |
| CN120987896A (en) | A synthetic method for 8-bromobenzodihydropyran-3-carboxylic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20130605 Termination date: 20150525 |
|
| EXPY | Termination of patent right or utility model |