CN101043893A - Fluorinated pyrrolo[2,3-d]pyrimidine nucleosides for the treatment of rna-dependent rna viral infection - Google Patents
Fluorinated pyrrolo[2,3-d]pyrimidine nucleosides for the treatment of rna-dependent rna viral infection Download PDFInfo
- Publication number
- CN101043893A CN101043893A CN 200580036083 CN200580036083A CN101043893A CN 101043893 A CN101043893 A CN 101043893A CN 200580036083 CN200580036083 CN 200580036083 CN 200580036083 A CN200580036083 A CN 200580036083A CN 101043893 A CN101043893 A CN 101043893A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- rna
- amino
- hydrogen
- carbonyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
发明领域field of invention
本发明涉及作为RNA-依赖性RNA病毒聚合酶抑制剂的氟化吡咯并[2,3-d]嘧啶核苷化合物及其某些衍生物,它们的合成和它们的用途。本发明的化合物是RNA-依赖性RNA病毒复制的抑制剂并且可用于治疗RNA-依赖性RNA病毒病毒感染。特别地,它们可用作丙型肝炎病毒(HCV)NS5B聚合酶的抑制剂,用作HCV复制的抑制剂和用于治疗丙型肝炎病毒感染。The present invention relates to fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds and certain derivatives thereof, their synthesis and their use as inhibitors of RNA-dependent RNA viral polymerases. The compounds of the present invention are inhibitors of RNA-dependent RNA virus replication and are useful in the treatment of RNA-dependent RNA virus viral infections. In particular, they are useful as inhibitors of hepatitis C virus (HCV) NS5B polymerase, as inhibitors of HCV replication and for the treatment of hepatitis C virus infection.
本发明的背景Background of the invention
综述治疗HCV感染的现有技术水平在很多受感染的个体中丙型肝炎病毒(HCV)是导致慢性肝病,例如肝硬化和肝细胞癌的主要健康问题,估计受感染的个体为世界人口的2-15%。根据美国疾病控制中心的估计单在美国就有3900000人受到感染,大约是感染人类免疫缺陷性病毒(HIV)的五倍。根据世界卫生组织的估计,全世界有超过1700,000,000的人受到感染,每年有3百万到4百万人受到感染。一旦被感染,大约20%的人可以清除病毒,而其余的人此后将终身携带HCV。10%至20%的慢性感染个体最终会发展成肝脏破坏的肝硬化或癌症。该病毒病可通过被污染的血液和血液制品,被污染的针经肠胃外途径传染,或经性传染和由受感染的母亲或为携带者的母亲垂直传染给她们的后代。当前的HCV感染治疗,仅限于单独使用重组干扰素-α或将其与核苷类似物利巴韦林联合的免疫治疗,其临床益处有限。而且,还没有确定的HCV疫苗。因此,急需能够有效对抗慢性HCV感染的改善的治疗剂。已经对治疗HCV感染的技术状态进行了综述,可参考以下出版物:B.Dymock,等.,″Novel approaches to the treatment ofhepatitis C virus infection,″ Antiviral Chemistry & Chemotherapy.11:79-96(2000);H.Rosen,等.,″Hepatitis C virus:current understandingand prospects for future therapies,″ Molecular Medicine Today.5:393-399(1999);D.Moradpour,等.,″Current and evolving therapies forhepatitis C,″ European J.Gastroenterol. Hepatol.,11:1189-1202(1999);R.Bartenschlager,″Candidate Targets for Hepatitis C Virus-SpecificAntiviral Therapy,″ Intervirology.40:378-393(1997);G.M.Lauer和B.D.Walker,″Hepatitis C Virus Infection,″ N.Engl.J.Med..345:41-52(2001);B.W.Dymock,″Emerging therapies for hepatitis C Virusinfection,″ Emerging Drugs.6:13-42(2001);和C.Crabb,″Hard-WonAdvances Spark Excitement about Hepatitis C,″ Science:506-507(2001);将其全部内容完整引入本文作为参考。Review of the State of the Art in the Treatment of HCV Infection Hepatitis C virus (HCV) is a major health problem leading to chronic liver diseases such as cirrhosis and hepatocellular carcinoma in many infected individuals, estimated to be 2% of the world's population. -15%. The US Centers for Disease Control estimates that 3,900,000 people are infected in the US alone, roughly five times the number infected with the human immunodeficiency virus (HIV). According to World Health Organization estimates, more than 1700,000,000 people are infected worldwide, and 3 to 4 million people are infected every year. Once infected, about 20% of people clear the virus, while the remainder carry HCV for life thereafter. Ten to 20 percent of chronically infected individuals eventually develop cirrhosis or cancer, which destroys the liver. The viral disease can be transmitted parenterally through contaminated blood and blood products, contaminated needles, or sexually and vertically from infected or carrier mothers to their offspring. Current treatment of HCV infection, limited to immunotherapy using recombinant interferon-α alone or in combination with the nucleoside analog ribavirin, has limited clinical benefit. Furthermore, there is no definitive HCV vaccine. Therefore, there is an urgent need for improved therapeutic agents that are effective against chronic HCV infection. The state of the art in the treatment of HCV infection has been reviewed, with reference to the following publications: B. Dymock, et al., "Novel approaches to the treatment of hepatitis C virus infection," Antiviral Chemistry & Chemotherapy . 11:79-96 (2000) ; H. Rosen, et al., "Hepatitis C virus: current understanding and prospects for future therapies," Molecular Medicine Today .5: 393-399 (1999); D. Moradpour, et al., "Current and evolving therapies for hepatitis C," European J. Gastroenterol . Hepatol. , 11:1189-1202 (1999); R. Bartenschlager, "Candidate Targets for Hepatitis C Virus-Specific Antiviral Therapy," Intervirology . 40:378-393 (1997); GMLauer and BD Walker, "Hepatitis C Virus Infection, " N. Engl. J. Med. .345:41-52 (2001); BW Dymock, "Emerging therapies for hepatitis C Virus infection, " Emerging Drugs . 6:13-42 (2001); and C. Crabb , "Hard-Won Advances Spark Excitement about Hepatitis C," Science : 506-507 (2001); incorporated herein by reference in its entirety.
已经采用不同的途径治疗HCV,包括抑制病毒的丝氨酸蛋白酶(NS3蛋白酶),解旋酶,和RNA-依赖性RNA聚合酶(NS5B),和开发疫苗。Different approaches have been taken to treat HCV, including inhibition of the viral serine protease (NS3 protease), helicase, and RNA-dependent RNA polymerase (NS5B), and the development of vaccines.
HCV病毒体是有包膜的正链RNA病毒,所具有的单个寡核糖核苷酸基因组序列有大约9600个碱基,能够编码具有大约3,010氨基酸的多蛋白。HCV基因的蛋白产物由结构蛋白质C,E1和E2与非结构蛋白NS2,NS3,NS4,NS4B,NS5a和NS5B组成。认为非结构(NS)蛋白质为病毒复制提供催化手段(catalytic machinery)。NS3蛋白酶由多蛋白链释放NS5B,其为RNA-依赖性RNA聚合酶。由在HCV复制循环中作为模板的单链病毒RNA合成双链RNA需要HCV NS5B聚合酶。因此在HCV复制复合体中认为NS5B聚合酶是必要组分。[参见K.Ishi,等.,″Expression of Hepatitis C Virus NS5B Protein:Characterization of Its RNA Polymerase Activity and RNA Binding,″Hepatology,29:1227-1235(1999)和V.Lohmann,等.,″Biochemical andKinetic Analyses of NS5B RNA-Dependent RNA Polymerase of theHepatitis C Virus,″ Viroloey.249:108-118(1998)].抑制HCV NS5B聚合酶可防止形成双链HCV RNA,因此,构成开发HCV特异性抗病毒治疗的颇具吸引力的途径。The HCV virion is an enveloped, positive-strand RNA virus with a single oligoribonucleotide genome sequence of approximately 9600 bases, capable of encoding a polyprotein of approximately 3,010 amino acids. The protein products of HCV genes consist of structural proteins C, E1 and E2 and nonstructural proteins NS2, NS3, NS4, NS4B, NS5a and NS5B. Nonstructural (NS) proteins are thought to provide the catalytic machinery for viral replication. The NS3 protease releases NS5B, an RNA-dependent RNA polymerase, from the polyprotein chain. HCV NS5B polymerase is required for the synthesis of double-stranded RNA from single-stranded viral RNA that serves as a template in the HCV replication cycle. The NS5B polymerase is therefore considered an essential component in the HCV replication complex. [See K.Ishi, etc., "Expression of Hepatitis C Virus NS5B Protein: Characterization of Its RNA Polymerase Activity and RNA Binding," Hepatology , 29:1227-1235 (1999) and V.Lohmann, etc., "Biochemical and Kinetic Analyzes of NS5B RNA-Dependent RNA Polymerase of the Hepatitis C Virus," Viroloey .249: 108-118 (1998)]. Inhibition of HCV NS5B polymerase prevents the formation of double-stranded HCV RNA and, therefore, constitutes a key to the development of HCV-specific antiviral therapies. attractive avenue.
已经对潜在可治疗HCV感染的HCV NS5B抑制剂的发展进行了综述,参见M.P.Walker等.″Promising candidates for the treatment ofchronic hepatitis C,″ Expert Opin.Invest.Drugs.12:1269-1280(2003);P.Hoffmann等.,″Recent patents on experimental therapy for hepatitisC virus infection(1999-2002),″ Expert Opin.Ther.Patents,″13:1707-1723(2003);和V.Brass,等,″Recent developments in targetidentification against HCV,″ Expert Opin.Ther.Targets,″8:295-307(2004).A.E.Eldrup等在″Structure-Activity Relationship of PurineRibonucleosides for Inhibition of HCV RNA-Dependent RNAPolymerase,″J.Med.Chem..47:2283-2295(2004)中报道了通过嘌呤核糖核苷抑制HCV复制。持续需要作为HCV聚合酶抑制剂的结构不同的核苷衍生物作为HCV治疗的治疗途径The development of HCV NS5B inhibitors for the potential treatment of HCV infection has been reviewed, see MP Walker et al. "Promising candidates for the treatment of chronic hepatitis C," Expert Opin. Invest. Drugs. 12:1269-1280 (2003); P .Hoffmann et al., "Recent patents on experimental therapy for hepatitis C virus infection (1999-2002), " Expert Opin. Ther. Patents , "13:1707-1723 (2003); and V.Brass, et al., "Recent developments in target identification against HCV, " Expert Opin. Ther. Targets, "8: 295-307 (2004). Inhibition of HCV replication by purine ribonucleosides was reported in : 2283-2295 (2004). There is a continuing need for structurally distinct nucleoside derivatives that are HCV polymerase inhibitors as therapeutic approaches for HCV treatment
美国专利第6,777,396(2004年8月17日出版)公开了作为HCVNS5B聚合酶抑制剂用于治疗HCV感染的一系列新结构的吡咯并[2,3-d]嘧啶核苷衍生物。D.B.Olsen等在″A 7-Deaza-Adenosine Analog is aPotent and Selective Inhibitor of HCV Replication with ExcellentPharmacokinetic Properties,″ Antimicrob.Agents Chemother..48:3944-3953(2004)中和A.E.Eldrup,等在″Structure-ActivityRelationship of Heterobase-Modified 2′-C-Methyl Ribonucleosides asInhibitors of Hepatitis C Virus RNA Replication,″ J.Med.Chem.,47:5284-5297(2004).对此类化合物的生物学性质进行了描述。现在发现在吡咯并[2,3-d]嘧啶核的C-5的位置引入氟提供的核苷衍生物是具有优良药动学性质,例如更好分布于肝脏的更为有效的HCV RNA复制抑制剂。US Patent No. 6,777,396 (published August 17, 2004) discloses a series of new structural pyrrolo[2,3-d]pyrimidine nucleoside derivatives as HCV NS5B polymerase inhibitors for the treatment of HCV infection. DBOlsen etc. in "A 7-Deaza-Adenosine Analog is a Potent and Selective Inhibitor of HCV Replication with Excellent Pharmacokinetic Properties," Antimicrob.Agents Chemother .. 48:3944-3953 (2004) and AEEldrup, etc. in "Structure-Activity Relationship of Heterobase -Modified 2′-C-Methyl Ribonucleosides as Inhibitors of Hepatitis C Virus RNA Replication,” J. Med. Chem ., 47: 5284-5297 (2004). Biological properties of such compounds are described. It is now found that the introduction of fluorine at the C-5 position of the pyrrolo[2,3-d]pyrimidine nucleus provides nucleoside derivatives with excellent pharmacokinetic properties, such as more efficient HCV RNA replication with better distribution in the liver Inhibitors.
因此,本发明的一个目的是提供用作RNA-依赖性RNA病毒聚合酶抑制剂,特别是HCV NS5B聚合酶抑制剂的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物。Accordingly, it is an object of the present invention to provide fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds and certain derivatives thereof useful as inhibitors of RNA-dependent RNA viral polymerases, particularly HCV NS5B polymerase inhibitors. biology.
本发明的另一个目的是提供用作RNA-依赖性RNA病毒聚合酶抑制剂,特别是丙型肝炎病毒复制抑制剂的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物。Another object of the present invention is to provide fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds and certain derivatives thereof useful as RNA-dependent RNA viral polymerase inhibitors, especially hepatitis C virus replication inhibitors. biology.
本发明的另一个目的是提供用于治疗RNA-依赖性RNA病毒感染,特别是HCV感染的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物。Another object of the present invention is to provide fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds and certain derivatives thereof for use in the treatment of RNA-dependent RNA viral infections, particularly HCV infections.
本发明的另一个目的是提供包括与药学上可接受的载体联合的氟化吡咯并[2,3-d]嘧啶核苷化合物的药物组合物。Another object of the present invention is to provide a pharmaceutical composition comprising a fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compound in association with a pharmaceutically acceptable carrier.
本发明的另一个目的是提供包括用作RNA-依赖性RNA病毒聚合酶抑制剂,特别是HCV NS5B聚合酶抑制剂的本发明的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物的药物组合物。Another object of the present invention is to provide the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention which are useful as RNA-dependent RNA viral polymerase inhibitors, especially HCV NS5B polymerase inhibitors and Pharmaceutical compositions of certain derivatives thereof.
本发明的另一个目的是提供包括用作RNA-依赖性RNA病毒复制的抑制剂,特别是HCV复制抑制剂的本发明的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物的药物组合物。Another object of the present invention is to provide the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention and certain compounds thereof which are useful as inhibitors of RNA-dependent RNA virus replication, especially HCV replication inhibitors. Pharmaceutical compositions of these derivatives.
本发明的另一个目的是提供包括用于治疗RNA-依赖性RNA病毒感染,特别是HCV感染的本发明的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物的药物组合物。Another object of the present invention is to provide a medicament comprising the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention and certain derivatives thereof for use in the treatment of RNA-dependent RNA viral infections, particularly HCV infections combination.
本发明的另一个目的是包括与其它抗RNA-依赖性RNA病毒,特别是抗HCV的活性剂联合的本发明的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物的药物组合物。Another object of the present invention is to include the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention and certain derivatives thereof in combination with other active agents against RNA-dependent RNA viruses, especially against HCV pharmaceutical composition.
本发明的另一个目的是提供抑制RNA-依赖性RNA病毒聚合酶,特别是HCV NS5B聚合酶的方法。Another object of the present invention is to provide methods for inhibiting RNA-dependent RNA viral polymerase, particularly HCV NS5B polymerase.
本发明的另一个目的是提供抑制RNA-依赖性RNA病毒复制,特别是抑制HCV复制的方法。Another object of the present invention is to provide methods for inhibiting the replication of RNA-dependent RNA viruses, especially HCV.
本发明的另一个目的是提供治疗RNA-依赖性RNA病毒感染,特别是治疗HCV感染的方法。Another object of the present invention is to provide a method of treating RNA-dependent RNA virus infection, especially HCV infection.
本发明的另一个目的是提供与其它抗RNA-依赖性RNA病毒联合治疗RNA-依赖性RNA病毒,感染,特别是与其它抗HCV的活性剂联合治疗HCV感染的方法。Another object of the present invention is to provide a method for treating RNA-dependent RNA virus infections in combination with other anti-RNA-dependent RNA viruses, especially in combination with other anti-HCV active agents for HCV infection.
本发明的另一个目的是用作抑制RNA-依赖性RNA病毒复制和/或治疗RNA-依赖性RNA病毒感染,特别是抑制HCV复制和/或治疗HCV感染的药物的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物及其药物组合物。Another object of the present invention is a fluorinated pyrrolo[2, 3-d] Pyrimidine nucleoside compounds and certain derivatives thereof and pharmaceutical compositions thereof.
本发明的另一个目的是提供本发明的氟化吡咯并[2,3-d]嘧啶核苷化合物和其某些衍生物及其药物组合物制备抑制RNA-依赖性RNA病毒复制和/或治疗RNA-依赖性RNA病毒感染,特别是抑制HCV复制和/或治疗HCV感染的药物的用途。Another object of the present invention is to provide the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention and certain derivatives thereof and pharmaceutical compositions thereof for the preparation of inhibitory RNA-dependent RNA virus replication and/or therapeutic RNA - Dependent RNA virus infection, in particular the use of a drug that inhibits HCV replication and/or treats HCV infection.
根据以下的详细描述这些和其它目的将变得显而易见。These and other objects will become apparent from the detailed description below.
发明概述Summary of the invention
本发明涉及标明立体化学的构型的结构式I的核苷化合物The present invention relates to the nucleoside compound of structural formula I indicating the configuration of stereochemistry
及其药学上可接受的盐;其中and their pharmaceutically acceptable salts; where
R1是氢或者氟;R 1 is hydrogen or fluorine;
R2是氟或者羟基;R 2 is fluorine or hydroxyl;
R3是氢,C1-16烷基羰基,C2-18烯基羰基,C1-10烷氧基羰基,C3-6环烷基羰基,C3-6环烷氧基羰基,或以下结构式的氨基酰基残基;R 3 is hydrogen, C 1-16 alkylcarbonyl, C 2-18 alkenylcarbonyl, C 1-10 alkoxycarbonyl, C 3-6 cycloalkylcarbonyl, C 3-6 cycloalkoxycarbonyl, or An aminoacyl residue of the following structural formula;
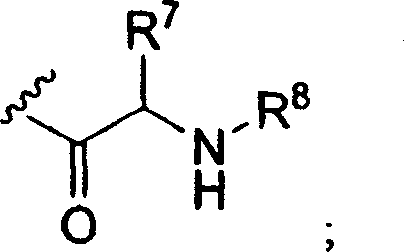
R4是氢,C1-10烷基羰基,磷酰基或其环状前药酯,二磷酰基,三磷酰基,C2-18烯基羰基,C1-10烷氧基羰基,C3-6环烷基羰基,C3-6环烷氧基羰基,CH2O(C=O)C1-4烷基,CH(C1-4烷基)O(C=O)C1-4烷基或以下结构式的氨基酰基残基;R 4 is hydrogen, C 1-10 alkylcarbonyl, phosphoryl or its cyclic prodrug ester, diphosphoryl, triphosphoryl, C 2-18 alkenylcarbonyl, C 1-10 alkoxycarbonyl, C 3 -6 cycloalkylcarbonyl, C 3-6 cycloalkoxycarbonyl, CH 2 O(C=O)C 1-4 alkyl, CH(C 1-4 alkyl)O(C=O)C 1- 4 alkyl or aminoacyl residues of the following structural formula;
以下结构式的残基Residues of the formula
R5是氨基或者羟基;R 5 is amino or hydroxyl;
R6是氢,氨基或者氟;R 6 is hydrogen, amino or fluoro;
R7是氢,C1-5烷基或苯基C0-2烷基;和R 7 is hydrogen, C 1-5 alkyl or phenyl C 0-2 alkyl; and
R8是氢,C1-4烷基,C1-4酰基,苯甲酰基。C1-4烷基氨基羰基,苯基C0-2烷基氨基羰基,C1-4烷基磺酰基或苯基C0-2烷基磺酰基;R 8 is hydrogen, C 1-4 alkyl, C 1-4 acyl, benzoyl. C 1-4 alkylaminocarbonyl, phenyl C 0-2 alkylaminocarbonyl, C 1-4 alkylsulfonyl or phenyl C 0-2 alkylsulfonyl;
R9是氢,C1-5烷基,苯基或苯甲基,其中烷基未取代或被一个选自羟基,甲氧基,氨基,羧基,氨甲酰基,胍基,巯基,甲硫基,1H-咪唑啉基和1H-吲哚-3-基的取代基取代并且其中苯基和苯甲基未取代或被一个或两个独立选自卤素,羟基和甲氧基的取代基取代;R 9 is hydrogen, C 1-5 alkyl, phenyl or benzyl, wherein the alkyl is unsubstituted or replaced by one selected from hydroxyl, methoxy, amino, carboxyl, carbamoyl, guanidino, mercapto, methylthio substituted by substituents of 1H-imidazolinyl and 1H-indol-3-yl and wherein phenyl and benzyl are unsubstituted or substituted by one or two substituents independently selected from halogen, hydroxy and methoxy ;
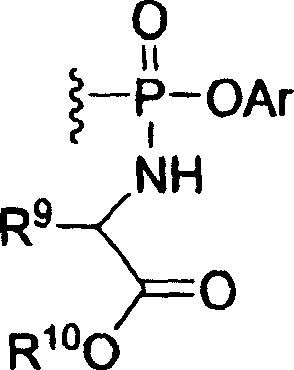
R10是氢,C1-6烷基,C3-6环烷基,苯基或苯甲基,其中烷基和环烷基未取代或被一个至3个独立选自卤素,羟基,羧基,C1-4烷氧基的取代基取代并且其中苯基和苯甲基未取代或被一个至三个独立选自卤素,羟基,氰基,C1-4烷氧基和三氟甲基的取代基取代;Ar是未取代的苯基或被一个至3个独立地选自卤素,C1-4烷基,C1-4烷氧基,C1-4烷硫基,氰基,硝基,氨基,羧基,三氟甲基,C1-4烷基氨基,二(C1-4烷基)氨基,C1-4烷基羰基,C1-4烷基羰基氧基和C1-4烷氧基羰基;R 10 is hydrogen, C 1-6 alkyl, C 3-6 cycloalkyl, phenyl or benzyl, wherein alkyl and cycloalkyl are unsubstituted or are independently selected from halogen, hydroxy, carboxyl by one to three , a substituent of C 1-4 alkoxy and wherein phenyl and benzyl are unsubstituted or are independently selected from one to three halogen, hydroxyl, cyano, C 1-4 alkoxy and trifluoromethyl Substituents; Ar is unsubstituted phenyl or by one to three independently selected from halogen, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 alkylthio, cyano, Nitro, amino, carboxyl, trifluoromethyl, C 1-4 alkylamino, di(C 1-4 alkyl)amino, C 1-4 alkylcarbonyl, C 1-4 alkylcarbonyloxy and C 1-4 alkoxycarbonyl;
条件是当R1,R3,R4,和R6是氢并且R2是羟基时,R5不是氨基。with the proviso that when R1 , R3 , R4 , and R6 are hydrogen and R2 is hydroxy, R5 is not amino.
式I的化合物可用作RNA-依赖性RNA病毒聚合酶,特别是HCVNS5B聚合酶的抑制剂。它们也是RNA-依赖性RNA病毒复制,特别是HCV复制的抑制剂并可用于治疗RNA-依赖性RNA病毒感染,特别是治疗HCV感染。The compounds of formula I are useful as inhibitors of RNA-dependent RNA viral polymerases, in particular HCV NS5B polymerase. They are also inhibitors of RNA-dependent RNA virus replication, especially HCV replication and are useful in the treatment of RNA-dependent RNA virus infections, especially HCV infection.
本发明还包括单独含有该化合物的药物组合物或与其它抗RNA-依赖性RNA病毒,特别是抗HCV的其它活性剂联合的该化合物的药物组合物,以及抑制RNA-依赖性RNA病毒复制和治疗RNA-依赖性RNA病毒感染的方法。The present invention also includes the pharmaceutical composition containing this compound alone or the pharmaceutical composition of this compound combined with other anti-RNA-dependent RNA viruses, particularly other active agents against HCV, and inhibiting RNA-dependent RNA virus replication and Methods of treating RNA-dependent RNA viral infections.
发明详述Detailed description of the invention
本发明涉及标明立体化学的构型的结构式I的核苷化合物The present invention relates to the nucleoside compound of structural formula I indicating the configuration of stereochemistry

及其药学上可接受的盐;其中and their pharmaceutically acceptable salts; where
R1是氢或者氟;R 1 is hydrogen or fluorine;
R2是氟或者羟基;R 2 is fluorine or hydroxyl;
R3是氢,C1-16烷基羰基,C2-18烯基羰基,C1-10烷氧基羰基,C3-6环烷基羰基,C3-6环烷氧基羰基,或以下结构式的氨基酰基残基;R 3 is hydrogen, C 1-16 alkylcarbonyl, C 2-18 alkenylcarbonyl, C 1-10 alkoxycarbonyl, C 3-6 cycloalkylcarbonyl, C 3-6 cycloalkoxycarbonyl, or An aminoacyl residue of the following structural formula;
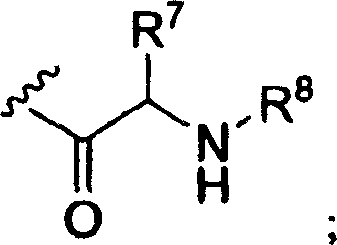
R4是氢,C1-10烷基羰基,磷酰基或其环状前药酯,二磷酰基,三磷酰基,C2-18烯基羰基,C1-10烷氧基羰基,C3-6环烷基羰基,C3-6环烷氧基羰基,CH2O(C=O)C1-4烷基,CH(C1-4烷基)O(C=O)C1-4烷基或以下结构式的氨基酰基残基;R 4 is hydrogen, C 1-10 alkylcarbonyl, phosphoryl or its cyclic prodrug ester, diphosphoryl, triphosphoryl, C 2-18 alkenylcarbonyl, C 1-10 alkoxycarbonyl, C 3 -6 cycloalkylcarbonyl, C 3-6 cycloalkoxycarbonyl, CH 2 O(C=O)C 1-4 alkyl, CH(C 1-4 alkyl)O(C=O)C 1- 4 alkyl or aminoacyl residues of the following structural formula;

以下结构式的残基Residues of the formula

R5是氨基或者羟基;R 5 is amino or hydroxyl;
R6是氢,氨基或者氟;R 6 is hydrogen, amino or fluoro;
R7是氢,C1-5烷基或苯基C0-2烷基;和R 7 is hydrogen, C 1-5 alkyl or phenyl C 0-2 alkyl; and
R8是氢,C1-4烷基氨基羰基,苯基C0-2烷基氨基羰基,C1-4烷基磺酰基或苯基C0-2烷基磺酰基;R 8 is hydrogen, C 1-4 alkylaminocarbonyl, phenyl C 0-2 alkylaminocarbonyl, C 1-4 alkylsulfonyl or phenyl C 0-2 alkylsulfonyl;
R9是氢,C1-5烷基,苯基或苯甲基,其中烷基未取代或被一个选自羟基,甲氧基,氨基,羧基,氨甲酰基,胍基,巯基,甲硫基,1H-咪唑啉基和1H-吲哚-3-基的取代基取代并且其中苯基和苯甲基未取代或被一个或两个独立选自卤素,羟基和甲氧基的取代基取代;R 9 is hydrogen, C 1-5 alkyl, phenyl or benzyl, wherein the alkyl is unsubstituted or replaced by one selected from hydroxyl, methoxy, amino, carboxyl, carbamoyl, guanidino, mercapto, methylthio substituted by substituents of 1H-imidazolinyl and 1H-indol-3-yl and wherein phenyl and benzyl are unsubstituted or substituted by one or two substituents independently selected from halogen, hydroxy and methoxy ;
R10是氢,C1-6烷基,C3-6环烷基,苯基或苯甲基,其中烷基和环烷基未取代或被一个至3个独立选自卤素,羟基,羧基,C1-4烷氧基的取代基取代并且其中苯基和苯甲基未取代或被一个或两个独立选自卤素,羟基和,氰基,C1-4烷氧基和三氟甲基的取代基取代;Ar是未取代的苯基或被一个至3个独立地选自卤素,C1-4烷基,C1-4烷氧基,C1-4烷硫基,氰基,硝基,氨基,羧基,三氟甲基,C1-4烷基氨基,二(C1-4烷基)氨基,C1-4烷基羰基,C1-4烷基羰基氧基和C1-4烷氧基羰基的基团取代;R 10 is hydrogen, C 1-6 alkyl, C 3-6 cycloalkyl, phenyl or benzyl, wherein alkyl and cycloalkyl are unsubstituted or are independently selected from halogen, hydroxy, carboxyl by one to three , a substituent of C 1-4 alkoxy and wherein phenyl and benzyl are unsubstituted or are independently selected from one or two of halogen, hydroxy and, cyano, C 1-4 alkoxy and trifluoromethane Substituents of substituents; Ar is unsubstituted phenyl or by one to three independently selected from halogen, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 alkylthio, cyano , nitro, amino, carboxyl, trifluoromethyl, C 1-4 alkylamino, di(C 1-4 alkyl)amino, C 1-4 alkylcarbonyl, C 1-4 alkylcarbonyloxy and C 1-4 alkoxycarbonyl group substitution;
条件是当R1,R3,R4,和R6是氢并且R2是羟基时,R5不是氨基。with the proviso that when R1 , R3 , R4 , and R6 are hydrogen and R2 is hydroxy, R5 is not amino.
式I化合物可用作RNA依赖性RNA病毒聚合酶的抑制剂。它们也是RNA依赖性RNA病毒复制的抑制剂,可用于治疗RNA依赖性RNA病毒感染。Compounds of formula I are useful as inhibitors of RNA-dependent RNA viral polymerases. They are also inhibitors of RNA-dependent RNA virus replication and are useful in the treatment of RNA-dependent RNA virus infections.
在本发明化合物的一个实施方案中,R1是氢;R2是羟基;R3和R4是氢。In one embodiment of the compounds of the invention, R1 is hydrogen; R2 is hydroxyl; R3 and R4 are hydrogen.
在本发明化合物的第二个实施方案中,R1是氢;R2是氟;R3和R4是氢;条件是当R1,R3,R4,和R6是氢并且R2是羟基时,R5不是氨基。In a second embodiment of the compounds of this invention, R 1 is hydrogen; R 2 is fluoro; R 3 and R 4 are hydrogen; provided that when R 1 , R 3 , R 4 , and R 6 are hydrogen and R 2 When is a hydroxyl group, R 5 is not an amino group.
在本发明化合物的第三个实施方案中,Ar是未取代的苯基。In a third embodiment of the compounds of this invention Ar is unsubstituted phenyl.
在本发明化合物的第四个实施方案中,R9选自氢,甲基,乙基,正丙基,异丙基,异丁基,2-甲基-1-丙基,羟甲基,巯基甲基,羧甲基,氨基甲酰甲基,1-羟乙基,2-羧乙基,2-氨基甲酰甲基,2-甲硫乙基,4-氨基的-1-丁基,3-氨基-1-丙基,3-胍基-1-丙基,1H-咪唑-4-基甲基,苯基,4-羟基苯甲基,1H-吲哚-3-基甲基。在一类这种实施方案中,R9是甲基或苯甲基。In a fourth embodiment of the compounds of the invention, R is selected from hydrogen, methyl, ethyl, n-propyl, isopropyl, isobutyl, 2-methyl-1-propyl, hydroxymethyl, Mercaptomethyl, carboxymethyl, carbamoylmethyl, 1-hydroxyethyl, 2-carboxyethyl, 2-carbamoylmethyl, 2-methylthioethyl, 4-amino-1-butyl , 3-amino-1-propyl, 3-guanidino-1-propyl, 1H-imidazol-4-ylmethyl, phenyl, 4-hydroxybenzyl, 1H-indol-3-ylmethyl . In a class of such embodiments, R9 is methyl or benzyl.
在本发明化合物的第五个实施方案中,R10是C1-6烷基,环己基,苯基或者苯甲基。在一类这种实施方案中,R10是甲基。In a fifth embodiment of the compounds of this invention, R 10 is C 1-6 alkyl, cyclohexyl, phenyl or benzyl. In a class of such embodiments, R 10 is methyl.
在本发明化合物的第六个实施方案中,Ar是未取代的苯基,R9是甲基或苯甲基,R10是甲基。In a sixth embodiment of the compounds of this invention, Ar is unsubstituted phenyl, R9 is methyl or benzyl, R10 is methyl.
用作RNA依赖性RNA病毒聚合酶的抑制剂的结构式I的本发明化合物的例举性而非限制性的实例如下:Illustrative, non-limiting examples of compounds of the invention of structural formula I useful as inhibitors of RNA-dependent RNA viral polymerases are as follows:
2,4-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶;2,4-Amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine;
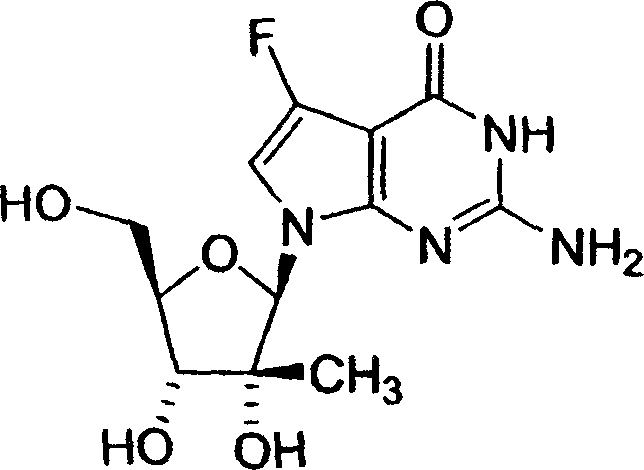
2-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶--4(3H)-酮;2-Amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin--4(3H)-one;

2,4-二氨基-5-氟-7-(2-氟-2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶;2,4-Diamino-5-fluoro-7-(2-fluoro-2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine;
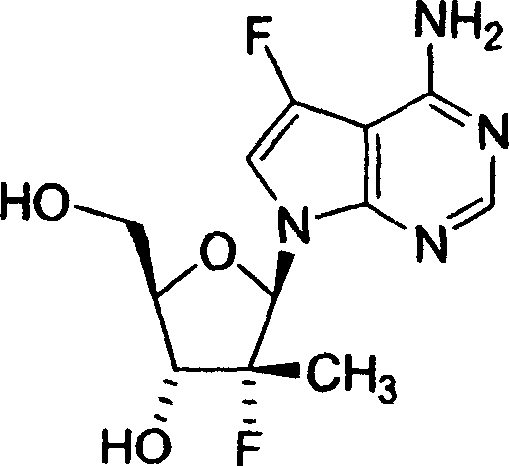
4-氨基-5-氟-7-(2-氟-2-C-甲基-β-D-呋喃核糖基)-7H-;4-Amino-5-fluoro-7-(2-fluoro-2-C-methyl-β-D-ribofuranosyl)-7H-;
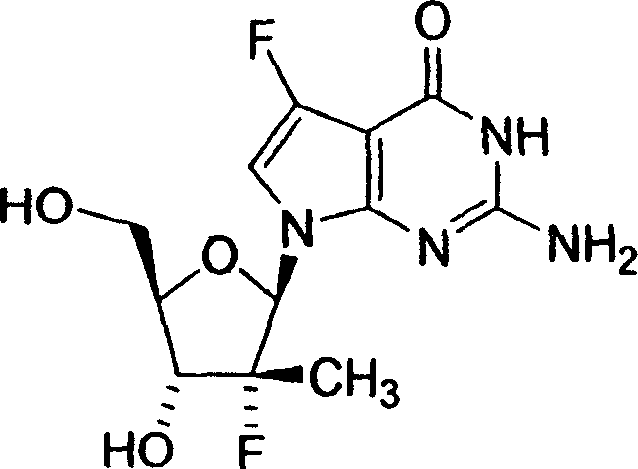
2-氨基-5-氟-7-(2-氟-2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶--4(3H)-酮;2-Amino-5-fluoro-7-(2-fluoro-2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine--4(3H)- ketone;
及其药学上可接受的盐。and pharmaceutically acceptable salts thereof.
在本发明的一个实施方案中,本发明的氟化吡咯并[2,3-d]嘧啶核苷化合物可用作正义单链RNA依赖性RNA病毒聚合酶的抑制剂,正义单链RNA依赖性RNA病毒复制的抑制剂,和/或用于治疗正义单链RNA依赖性的RNA病毒感染。在一类这种实施方案中,正义单链RNA依赖性RNA病毒是黄病毒科(Flaviviridae)病毒或小核糖核酸病毒科(Picornaviridae)病毒。在该类的亚类中,小核糖核酸病毒科病毒是鼻病毒、脊髓灰质炎病毒或甲型肝炎病毒。在该类的第二亚类中,黄病毒科病毒选自丙型肝炎病毒、黄热病病毒、登革病毒、西尼罗病毒、日本脑炎病毒、班齐病毒和牛病毒性腹泻病毒(BVDV)。在该亚类的子集中,黄病毒科病毒为丙型肝炎病毒。In one embodiment of the invention, the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the invention are useful as inhibitors of positive-sense single-stranded RNA-dependent RNA viral polymerases, positive-sense single-stranded RNA-dependent Inhibitors of RNA virus replication, and/or for the treatment of positive-sense single-stranded RNA-dependent RNA virus infections. In one class of such embodiments, the positive-sense single-stranded RNA-dependent RNA virus is a Flaviviridae virus or a Picornaviridae virus. In a subclass of this class, the picornaviridae viruses are rhinoviruses, polioviruses or hepatitis A viruses. In the second subgroup of this class, the Flaviviridae viruses are selected from the group consisting of hepatitis C virus, yellow fever virus, dengue virus, West Nile virus, Japanese encephalitis virus, Banzi virus and bovine viral diarrhea virus (BVDV ). In a subset of this subclass, the Flaviviridae virus is the hepatitis C virus.
本发明的另一方面涉及抑制RNA依赖性RNA病毒聚合酶的方法,抑制RNA依赖性RNA病毒复制的方法,和/或治疗需要这种治疗的哺乳动物的RNA依赖性RNA病毒感染的方法,该方法包括给哺乳动物施用治疗有效量的结构式I的化合物。Another aspect of the invention relates to methods of inhibiting RNA-dependent RNA virus polymerase, methods of inhibiting replication of RNA-dependent RNA viruses, and/or methods of treating RNA-dependent RNA virus infections in mammals in need of such treatment, the The method comprises administering to a mammal a therapeutically effective amount of a compound of formula I.
在本发明的这方面的一个实施方案中,RNA依赖性RNA病毒聚合酶是正义单链RNA依赖性RNA病毒聚合酶。在一类这种实施方案中,正义单链RNA依赖性RNA病毒聚合酶是黄病毒科病毒聚合酶或小核糖核酸病毒科病毒聚合酶。在该类的亚类中,小核糖核酸病毒科病毒聚合酶是鼻病毒聚合酶、脊髓灰质炎病毒聚合酶或甲型肝炎病毒聚合酶。在该类的第二亚类中,黄病毒科病毒聚合酶选自丙型肝炎病毒聚合酶、黄热病毒聚合酶、登革病毒聚合酶、西尼罗病毒聚合酶、日本脑炎病毒聚合酶、班齐病毒聚合酶和牛病毒性腹泻病毒(BVDV)聚合酶。在该亚类的子集中,黄病毒科病毒聚合酶为丙型肝炎病毒聚合酶。In one embodiment of this aspect of the invention, the RNA-dependent RNA viral polymerase is a positive-sense single-stranded RNA-dependent RNA viral polymerase. In one class of such embodiments, the positive-sense single-stranded RNA-dependent RNA viral polymerase is a Flaviviridae viral polymerase or a Picornaviridae viral polymerase. In a subclass of this class, the picornaviridae viral polymerase is a rhinovirus polymerase, a poliovirus polymerase, or a hepatitis A virus polymerase. In the second subclass of this class, the Flaviviridae viral polymerase is selected from the group consisting of hepatitis C virus polymerase, yellow fever virus polymerase, dengue virus polymerase, West Nile virus polymerase, Japanese encephalitis virus polymerase , Banzi virus polymerase and bovine viral diarrhea virus (BVDV) polymerase. In a subset of this subclass, the Flaviviridae viral polymerase is the hepatitis C virus polymerase.
在本发明的这方面的第二个实施方案中,RNA依赖性RNA病毒复制是正义单链RNA依赖性RNA病毒复制。在一类这种实施方案中,正义单链RNA依赖性RNA病毒复制是黄病毒科病毒复制或小核糖核酸病毒科病毒复制。在该类的亚类中,小核糖核酸病毒科病毒复制是鼻病毒复制、脊髓灰质炎病毒复制或甲型肝炎病毒复制。在该类的第二亚类中,黄病毒科病毒复制选自丙型肝炎病毒复制、黄热病毒复制、登革病毒复制、西尼罗病毒复制、日本脑炎病毒复制、班齐病毒复制和牛病毒复制性腹泻病毒复制。在该亚类的子集中,黄病毒科病毒复制为丙型肝炎病毒复制。In a second embodiment of this aspect of the invention, the RNA-dependent RNA viral replication is positive-sense single-stranded RNA-dependent RNA viral replication. In one class of such embodiments, the positive-sense single-stranded RNA-dependent RNA viral replication is Flaviviridae viral replication or Picornaviridae viral replication. In a subclass of this class, picornaviridae viral replication is rhinovirus replication, poliovirus replication, or hepatitis A virus replication. In the second subgroup of this class, the Flaviviridae virus replication is selected from the group consisting of hepatitis C virus replication, yellow fever virus replication, dengue virus replication, West Nile virus replication, Japanese encephalitis virus replication, Banzi virus replication and bovine virus replication. Viral replicative diarrhea virus replication. In a subset of this subclass, Flaviviridae viruses replicate as Hepatitis C viruses.
在本发明的这方面的第三个实施方案中,RNA依赖性RNA病毒感染是正义单链RNA依赖性RNA病毒感染。在一类这种实施方案中,正义单链RNA依赖性RNA病毒感染是黄病毒科病毒感染或小核糖核酸病毒科病毒感染。在该类的亚类中,小核糖核酸病毒科病毒感染是鼻病毒感染、脊髓灰质炎病毒感染或甲型肝炎病毒感染。在该类的第二亚类中,黄病毒科病毒感染选自丙型肝炎病毒感染、黄热病毒感染、登革病毒感染、西尼罗病毒感染、日本脑炎病毒感染、班齐病毒感染和牛病毒性腹泻病毒感染。在该亚类的子集中,黄病毒科病毒感染为丙型肝炎病毒感染。In a third embodiment of this aspect of the invention, the RNA-dependent RNA viral infection is a positive-sense single-stranded RNA-dependent RNA viral infection. In one class of such embodiments, the positive-sense single-stranded RNA-dependent RNA viral infection is a Flaviviridae viral infection or a Picornaviridae viral infection. In a subclass of this class, the picornaviridae virus infection is a rhinovirus infection, a poliovirus infection, or a hepatitis A virus infection. In the second subclass of this class, the Flaviviridae viral infection is selected from the group consisting of hepatitis C virus infection, yellow fever virus infection, dengue virus infection, West Nile virus infection, Japanese encephalitis virus infection, Banzi virus infection and bovine Viral diarrhea virus infection. In a subset of this subclass, the Flaviviridae virus infection is the hepatitis C virus infection.
贯穿本申请始终,以下术语具有指出的含意:Throughout this application, the following terms have the indicated meanings:
“烷基”,以及具有前缀“alk”的其他基团,例如烷氧基或烷硫基指直链或支链的碳链,或其组合,除非另外对碳链进行定义。烷基的实例有甲基、乙基、丙基、异丙基、丁基、仲丁基、叔丁基、戊基、异戊基、己基、庚基、辛基、壬基等。在规定碳原子数目的情况下,例如C3-10,术语“烷基”还包括环烷基,以及与环烷基结构结合的直链或支链烷基链的组合。"Alkyl", as well as other groups having the prefix "alk", such as alkoxy or alkylthio, refers to straight or branched carbon chains, or combinations thereof, unless the carbon chain is defined otherwise. Examples of alkyl groups are methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert-butyl, pentyl, isopentyl, hexyl, heptyl, octyl, nonyl and the like. Where the number of carbon atoms is specified, eg C3-10 , the term "alkyl" also includes cycloalkyl, and combinations of straight or branched alkyl chains combined with a cycloalkyl structure.
术语“环烷基”指一组烷基并且指具有规定数目碳原子的烷基饱和碳环。环烷基的实例包括环丙基、环丁基、环戊基、环己基、环庚基或环辛基等。除非另外指明,环烷基通常为单环。除非另外指明,环烷基是饱和的。The term "cycloalkyl" refers to a group of alkyl groups and refers to an alkyl saturated carbocyclic ring having the specified number of carbon atoms. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl and the like. Unless otherwise specified, cycloalkyl groups are generally monocyclic. Cycloalkyl groups are saturated unless otherwise indicated.
术语“链烯基”指总碳原子数为2-6或该范围内任何数字的直链或支链烯烃(例如乙烯基、丙烯基、丁烯基、戊烯基等)。The term "alkenyl" refers to a straight or branched chain alkene (eg vinyl, propenyl, butenyl, pentenyl, etc.) having a total carbon number of 2-6 or any number within the range.
术语“炔基”指总碳原子数为2-6或者该范围内任何数字的直链或支链炔烃(例如乙炔基、丙炔基、丁炔基、戊炔基等)。The term "alkynyl" refers to a straight or branched chain alkyne (eg, ethynyl, propynyl, butynyl, pentynyl, etc.) with a total carbon number of 2-6 or any number within the range.
术语“烷氧基”指碳原子数为规定数目(例如C1-4烷氧基)或者该范围内任何数目的直链或支链烷基氧[即甲氧基(MeO-)、乙氧基、异丙氧基等]。The term "alkoxy" refers to a specified number of carbon atoms (such as C 1-4 alkoxy) or any number of linear or branched alkyl oxygen within the range [ie methoxy (MeO-), ethoxy base, isopropoxy, etc.].
术语“烷硫基”指碳原子数为规定数目(例如C1-4烷硫基)或者该范围内任何数目的直链或支链烷基硫[即甲硫基(MeS-)、乙硫基、异丙硫基等]。The term "alkylthio" refers to a specified number of carbon atoms (such as C 1-4 alkylthio) or any number of straight-chain or branched alkylthio within the range [ie methylthio (MeS-), ethylthio group, isopropylthio group, etc.].
术语“烷基氨基”指碳原子数为规定数目(例如C1-4烷基氨基)或者该范围内任何数目的直链或支链烷基胺[即甲基氨基、乙基氨基、异丙基氨基、叔丁基氨基等]。The term "alkylamino" refers to a specified number of carbon atoms (such as C 1-4 alkylamino) or any number of linear or branched alkylamines within this range [i.e. methylamino, ethylamino, isopropylamino, baseamino, tert-butylamino, etc.].
术语“烷基磺酰基”指碳原子数为规定数目(例如C1-6烷基磺酰基)或者该范围内任何数目的直链或支链烷基砜[即甲基磺酰基(MeSO2-)、乙基磺酰基、异丙基磺酰基等]。The term "alkylsulfonyl" refers to a specified number of carbon atoms (such as C 1-6 alkylsulfonyl) or any number of straight-chain or branched alkylsulfones within this range [ie methylsulfonyl (MeSO 2 - ), ethylsulfonyl, isopropylsulfonyl, etc.].
术语“烷氧基羰基”指碳原子数为规定数目(例如C1-4烷氧基羰基)或该范围内任何数目的本发明羧酸衍生物的直链或支链酯[即甲氧基羰基(MeOCO-)、乙氧基羰基或丁氧基羰基]。The term "alkoxycarbonyl" refers to a straight-chain or branched ester of a carboxylic acid derivative of the present invention having a specified number of carbon atoms (e.g. C1-4 alkoxycarbonyl) or any number within this range [i.e. methoxy carbonyl (MeOCO-), ethoxycarbonyl or butoxycarbonyl].
术语“烷基羰基”指碳原子数为规定数目(例如C1-4烷基羰基)或该范围内任何数目的直链或支链烷基酰基[即甲基羰基(MeCO-)、乙基羰基或丁基羰基]。The term "alkylcarbonyl" refers to a straight - chain or branched chain alkylacyl group [i.e. methylcarbonyl (MeCO-), ethyl, or methylcarbonyl (MeCO-), ethyl carbonyl or butylcarbonyl].
术语“卤素”意欲包括卤素原子氟、氯、溴和碘。The term "halogen" is intended to include the halogen atoms fluorine, chlorine, bromine and iodine.
术语“磷酰基”指-P(O)(OH)2-The term "phosphoryl" refers to -P(O)(OH) 2 -
术语“二磷酰基”指具有以下结构的基:The term "diphosphoryl" refers to a radical having the following structure:
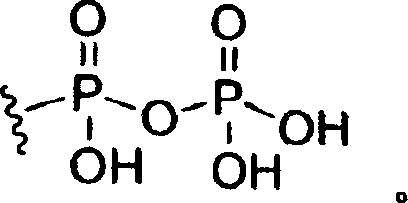
术语“三磷酰基”指具有以下结构的基:The term "triphosphoryl" refers to a group having the following structure:

术语“取代的”应被认为包括被指定的取代基多重取代。当多重取代部分被公开了或者被要求保护时,取代化合物可以独立被一个或多个公开的或者被要求保护的取代基部分单一或者多处取代。The term "substituted" shall be considered to include multiple substitutions by the indicated substituents. When multiple substituted moieties are disclosed or claimed, the substituted compounds may independently be single or multiple substituted with one or more disclosed or claimed substituent moieties.
当R3和R4的氨基酰基残基实施方案中的R7在下式中不是氢时,When R in the aminoacyl residue embodiment of R and R is not hydrogen in the following formulae,
氨基酰基残基含有不对称中心并且意欲包括单独的R-和S-立体异构体以及RS-非对映异构混合物。Aminoacyl residues contain asymmetric centers and are intended to include individual R- and S-stereoisomers as well as RS-diastereomeric mixtures.
术语“5’-三磷酸酯”指具有下述通用结构式II的本发明氟化吡咯并[2,3-d]嘧啶核苷化合物的5’-羟基的三磷酸酯衍生物:The term "5'-triphosphate" refers to a triphosphate derivative of the 5'-hydroxyl group of the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention having the following general structural formula II:

其中R1-R3,R5和R6如上定义。本发明化合物还意欲包括三磷酸酯的药学上可接受的盐和分别具有结构式III和IV的5’-一磷酸酯和5’-二磷酸酯衍生物的药学上可接受的盐。wherein R 1 -R 3 , R 5 and R 6 are as defined above. The compounds of the present invention are also intended to include pharmaceutically acceptable salts of triphosphates and pharmaceutically acceptable salts of 5'-monophosphate and 5'-bisphosphate derivatives having structural formulas III and IV, respectively.
“药物组合物”中的术语“组合物”意欲包括包含活性成分和形成载体的惰性成分的产物,以及由任何两种或多种成分混合、复合或聚集,或者由一种或多种成分离解,或者由一种或多种成分的其它类型反应或相互作用而直接或间接得到的任何产物。相应地,本发明药物组合物包括通过混合本发明化合物与药学上可接受的载体制成的任何组合物。The term "composition" in "pharmaceutical composition" is intended to include products comprising the active ingredient and an inert ingredient forming a carrier, as well as mixtures, complexes, or aggregates of any two or more ingredients, or separation of one or more ingredients. , or any product resulting directly or indirectly from any other type of reaction or interaction of one or more components. Accordingly, the pharmaceutical compositions of the present invention include any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
术语“施与”和“施用”化合物应理解为是指向有需要的个体提供本发明化合物或本发明化合物的前药。The terms "administering" and "administering" a compound are understood to mean providing a compound of the invention or a prodrug of a compound of the invention to a subject in need thereof.
本发明的另一方面涉及抑制HCV NS5B聚合酶,抑制HCV复制的方法,或者用本发明化合物与一种或多种用于治疗HCV感染的试剂结合治疗HCV感染的方法。这样的可有效对抗HCV的试剂包括但不限于利巴韦林、左旋韦林(levovirin)、viramidine、胸腺素α-1、干扰素β、干扰素α、聚乙二醇化(pegylated)干扰素α(聚乙二醇干扰素-α)、干扰素-α与利巴韦林的联合、聚乙二醇干扰素-α与利巴韦林的联合、干扰素α与左旋韦林的联合、聚乙二醇干扰素-α与左旋韦林的联合。干扰素α包括但不限于重组干扰素α2a(如Hoffmann-LaRoche,Nutley,NJ获得的Roferon interferon)、聚乙二醇化干扰素α2a(PegasysTM)、干扰素α2b(如Schering Corp.,Kenilworth,NJ获得的Intron-Ainterferon)、聚乙二醇化干扰素α2b(PegIntronTM)、重组体共有干扰素(如干扰素alphacon-1)和纯化的干扰素α产品。Amgen′s重组体共有干扰素具有商品名Infergen。左旋韦林是利巴韦林的L-对映异构体,其显示出近似于利巴韦林的免疫调节活性。Viramidine表示WO01/60379(转让给ICN Pharmaceuticals)中所公开的利巴韦林类似物。根据本发明的方法,可将联合的各成分在治疗期间内的不同时间分别施用,或者以分开的或单一组合形式同时施用。因此,应将本发明理解为包括所有这些同时或交替治疗的疗法,术语“施用”也作相应解释。应当理解本发明化合物与其它用于治疗HCV感染的试剂联合的范围原则上包括与任何用于治疗HCV感染的药物组合物的任何联合。当将本发明化合物或其药学上可接受的盐与可有效对抗HCV的第二种治疗试剂联合时,各化合物的剂量可以与单独使用化合物时的剂量相同或不同。Another aspect of the invention pertains to methods of inhibiting HCV NS5B polymerase, inhibiting HCV replication, or treating HCV infection using a compound of the invention in combination with one or more agents useful in the treatment of HCV infection. Such agents effective against HCV include, but are not limited to, ribavirin, levovirin, viramidine, thymosin alpha-1, interferon beta, interferon alpha, pegylated interferon alpha (pegylated interferon-α), combination of interferon-α and ribavirin, combination of pegylated interferon-α and ribavirin, combination of interferon-α and levovirin, poly The combination of ethylene glycol interferon-alpha and levovirin. Interferon alpha includes, but is not limited to, recombinant interferon alpha 2a (such as Roferon interferon available from Hoffmann-LaRoche, Nutley, NJ), pegylated interferon alpha 2a (Pegasys ™ ), interferon alpha 2b (such as Schering Corp., Kenilworth, NJ Intron-Ainterferon), pegylated interferon alpha 2b (PegIntron ™ ), recombinant consensus interferon (such as interferon alphacon-1) and purified interferon alpha products. Amgen's recombinant consensus interferon has the trade name Infergen(R). Levovirin is the L-enantiomer of ribavirin, which exhibits immunomodulatory activity similar to that of ribavirin. Viramidine means the ribavirin analog disclosed in WO 01/60379 (assigned to ICN Pharmaceuticals). According to the methods of the invention, the components of the combination may be administered separately at different times during the treatment period, or administered simultaneously in separate or single combinations. Accordingly, the invention is to be understood as including all such therapies of simultaneous or alternating treatment, and the term "administration" is to be construed accordingly. It is to be understood that the scope of combinations of the compounds of the present invention with other agents useful in the treatment of HCV infection includes in principle any combination with any pharmaceutical composition useful in the treatment of HCV infection. When a compound of the present invention, or a pharmaceutically acceptable salt thereof, is combined with a second therapeutic agent effective against HCV, the dosage of each compound may be the same or different than when the compound is used alone.
为治疗HCV感染,也可将本发明化合物与HCV NS3丝氨酸蛋白酶抑制剂的试剂联合施用。HCV NS3丝氨酸蛋白酶是必需的病毒酶,并被描述为是抑制HCV复制的优良靶标。HCV NS3蛋白酶抑制剂的底物和非底物基抑制剂都公开在WO98/22496、WO98/46630、WO99/07733、WO99/07734、WO99/38888、WO99/50230、WO99/64442、WO00/09543、WO00/59929、GB2337262、WO02/48116、WO02/48172和美国专利第6,323,180号中。B.W.Dymock,″Emergingtherapies for hepatitis C virus infection,″Emerging Drugs,6:13-42(2001)中讨论了作为开发HCV复制抑制剂和治疗HCV感染的靶标的HCV NS3蛋白酶。For the treatment of HCV infection, the compounds of the invention may also be administered in combination with agents that are HCV NS3 serine protease inhibitors. The HCV NS3 serine protease is an essential viral enzyme and has been described as an excellent target for inhibition of HCV replication. Both substrate and non-substrate-based inhibitors of HCV NS3 protease inhibitors are disclosed in WO98/22496, WO98/46630, WO99/07733, WO99/07734, WO99/38888, WO99/50230, WO99/64442, WO00/09543, WO00/59929, GB2337262, WO02/48116, WO02/48172 and US Patent No. 6,323,180. The HCV NS3 protease as a target for developing HCV replication inhibitors and treating HCV infection is discussed in B.W. Dymock, "Emerging therapies for hepatitis C virus infection," Emerging Drugs, 6:13-42 (2001).
利巴韦林、左旋韦林和viramidine可通过经由抑制胞内酶肌苷一磷酸脱氢酶(IMPDH)调节鸟嘌呤核苷酸细胞内池(pool)发挥其抗HCV作用。IMPDH是从头鸟嘌呤核苷酸生物合成中生物合成途径的限速酶。利巴韦林容易被细胞内磷酰化,一磷酸衍生物是IMPDH的抑制剂。因此,IMPDH的抑制代表发现HCV复制抑制剂的另一有效靶标。从而,本发明化合物也可与下述物质联合施用:IMPDH抑制剂,如WO 97/41211和WO 01/00622(转让给Vertex)中公开的VX-497;另一IMPDH抑制剂,如WO 00/25780(转让给Bristol-Myers Squibb)中所公开的物质;或者麦考酚酸吗乙酯[参见A.C.Allison和E.M Eugui,Agents Action,44(Suppl.):165(1993)]。Ribavirin, levovirin and viramidine can exert their anti-HCV effects by regulating the intracellular pool of guanine nucleotides by inhibiting the intracellular enzyme inosine monophosphate dehydrogenase (IMPDH). IMPDH is the rate-limiting enzyme of the biosynthetic pathway in de novo guanine nucleotide biosynthesis. Ribavirin is readily phosphorylated intracellularly, and its monophosphate derivatives are inhibitors of IMPDH. Therefore, inhibition of IMPDH represents another effective target for the discovery of inhibitors of HCV replication. Thus, the compounds of the present invention may also be administered in combination with an IMPDH inhibitor such as VX-497 disclosed in WO 97/41211 and WO 01/00622 (assigned to Vertex); another IMPDH inhibitor such as WO 00/00622 25780 (assigned to Bristol-Myers Squibb); or mycophenolate mofetil [see A.C. Allison and E.M Eugui, Agents Action, 44 (Suppl.): 165 (1993)].
为治疗HCV感染,可将本发明化合物与抗病毒剂金刚烷胺(1-氨基金刚烷)联合施用[该药剂的综述参见J.Kirschbaum,Anal.Profiles-DrugSubs.12:1-36(1983)]。For the treatment of HCV infection, the compounds of the present invention may be administered in combination with the antiviral agent amantadine (1-aminoadamantane) [for a review of this agent, see J. Kirschbaum, Anal. Profiles-Drug Subs. 12: 1-36 (1983) ].
本发明的化合物还可以与2′-C-支链核糖核苷联合用于治疗HCV感染,2′-C-支链核糖核苷公开于R.E.Harry-O′kuru等.,J.Org.Chem..62:1754-1759(1997);M.S.Wolfe等.,Tetrahedron Lett..36:7611-7614(1995);美国专利第3,480,613号(1969年11月25日);国际出版号WO01/90121(2001年11月29);国际出版号WO 01/92282(2001年12月6日);国际出版号WO 02/32920(2002年4月25日);国际出版号WO04/002999(2004年1月8日);国际出版号WO 04/003000(2004年1月8日);国际出版号WO 04/002422(2004年1月8日);将其内容完整引入作为参考。这样的2′-C-支链核糖核苷包括但是不限于2′-C-甲基胞苷,2′-C-甲基尿苷,2′-C-甲基腺苷,2′-C-甲基鸟苷,9-(2-C-甲基-β-D-呋喃核糖基)-2,6-二氨基嘌呤,核糖C-2′,C-3和C-5′羟基相应的氨基酸酯(例如,3′-O-(L-缬氨酰基)-2′-C-甲基胞苷)和相应的任选取代的5′-磷酸脂衍生物的环1,3-丙二醇酯。The compounds of the present invention can also be used in the treatment of HCV infection in combination with 2'-C-branched ribonucleosides disclosed in R.E.Harry-O'kuru et al., J.Org.Chem .. 62: 1754-1759 (1997); M.S. Wolfe et al., Tetrahedron Lett.. 36: 7611-7614 (1995); U.S. Patent No. 3,480,613 (November 25, 1969); International Publication No. WO 01/90121 ( November 29, 2001); International Publication No. WO 01/92282 (December 6, 2001); International Publication No. WO 02/32920 (April 25, 2002); International Publication No. WO 04/002999 (January 2004 8); International Publication No. WO 04/003000 (January 8, 2004); International Publication No. WO 04/002422 (January 8, 2004); the contents of which are incorporated by reference in their entirety. Such 2'-C-branched ribonucleosides include, but are not limited to, 2'-C-methylcytidine, 2'-C-methyluridine, 2'-C-methyladenosine, 2'-C- -Methylguanosine, 9-(2-C-methyl-β-D-ribofuranosyl)-2,6-diaminopurine, ribose C-2′, C-3 and C-5′ hydroxyl corresponding Cyclic 1,3-propanediol esters of amino acid esters (e.g., 3′-O-(L-valyl)-2′-C-methylcytidine) and the corresponding optionally substituted 5′-phospholipid derivatives .
本发明的化合物还可与其它具有抗HCV性质的其它核苷联合用于治疗HCV感染,其它核苷公开于WO02/51425(2002年7月4日),转让给Mitsubishi Pharma Corp.;WO01/79246,WO02/32920,WO02/48165(200年6月20日),转让给Pharmasset,Ltd.;WO01/686632001年9月20日),转让给ICN Pharmaceuticals;WO99/43691(1999年9月2日);WO02/18404(2002年3月7日),转让给Hoffmann-LaRoche;U.S2002/0019363(2002年2月14日);WO02/100415(2002年12月19日);WO03/026589(2003年4月3日);WO03/026675(2003年4月3日);WO03/093290(2003年11月13日):US2003/0236216(2003年12月25日);US2004/0006007(2004年1月8日);WO04/011478(2004年2月5日);WO04/013300(2004年2月12日);US 2004/0063658(2004年4月1日);WO04/028481(2004年4月8日)。Compounds of the present invention can also be used in combination with other nucleosides having anti-HCV properties for the treatment of HCV infection, other nucleosides disclosed in WO02/51425 (July 4, 2002), assigned to Mitsubishi Pharma Corp.; WO01/79246 , WO02/32920, WO02/48165 (June 20, 200), assigned to Pharmasset, Ltd.; WO01/68663, September 20, 2001), assigned to ICN Pharmaceuticals; WO99/43691 (September 2, 1999) ; WO02/18404 (7 March 2002), assigned to Hoffmann-LaRoche; U.S2002/0019363 (14 February 2002); WO02/100415 (19 December 2002); WO03/026589 (2003 April 3, 2003); WO03/026675 (April 3, 2003); WO03/093290 (November 13, 2003): US2003/0236216 (December 25, 2003); US2004/0006007 (November 2004 February 8); WO04/011478 (February 5, 2004); WO04/013300 (February 12, 2004); US 2004/0063658 (April 1, 2004); WO04/028481 (April 2004 8).
本发明的化合物还可与HCV聚合酶抑制剂联合用于治疗HCV感染,HCV聚合酶抑制剂公开于WO01/77091(2001年10月18日),转让给Tularik,Inc.;WO01/47883(2001年7月5日),转让给JapanTobacco,Inc.;WO02/04425(2002年1月17日),转让给BoehringerIngelheim;WO02/06246(2002年1月24日),转让给Istituto di Ricerchedi Biologia Moleculare P.Angeletti S.P.A.;WO02/20497(2002年3月3日)。Compounds of the present invention can also be used in combination with HCV polymerase inhibitors for the treatment of HCV infection. HCV polymerase inhibitors are disclosed in WO01/77091 (October 18, 2001), assigned to Tularik, Inc.; WO01/47883 (2001 Jul. 5, 2002), assigned to Japan Tobacco, Inc.; WO02/04425 (Jan. 17, 2002), assigned to Boehringer Ingelheim; WO02/06246 (Jan. 24, 2002), assigned to Istituto di Ricerchedi Biologia Moleculare P . Angeletti S.P.A.; WO02/20497 (March 3, 2002).
“药学上可接受的”意指载体、稀释剂或赋形剂必须与其它制剂成分相容,并且对其接受者无害。"Pharmaceutically acceptable" means that the carrier, diluent or excipient must be compatible with the other formulation ingredients and not deleterious to the recipient thereof.
也包括在本发明范围内的是包含本发明氟化吡咯并[2,3-d]嘧啶核苷化合物及其衍生物和药学上可接受的载体的药物组合物。本发明的另一个实例是通过组合任何上述化合物和药学上可接受的载体而制成的药物组合物。本发明的另一示例制备药物组合物的方法,该方法包括组合任何上述化合物和药学上可接受的载体。Also included within the scope of the invention are pharmaceutical compositions comprising the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds and derivatives thereof of the invention and a pharmaceutically acceptable carrier. Another example of the present invention is a pharmaceutical composition prepared by combining any of the above compounds and a pharmaceutically acceptable carrier. Another example of the present invention is a method for preparing a pharmaceutical composition, the method comprising combining any of the above-mentioned compounds and a pharmaceutically acceptable carrier.
还包括在本发明范围内的是可用于抑制RNA依赖性RNA病毒聚合酶,特别是HCV NS5B聚合酶的药物组合物,该组合物包含有效量的本发明化合物和药学上可接受的载体。可用于治疗RNA依赖性RNA病毒感染,特别是HCV感染的药物组合物也包括在本发明范围内,还有抑制RNA依赖性RNA病毒聚合酶,特别是HCVNS5B聚合酶的方法,以及治疗RNA依赖性RNA病毒复制,特别是HCV复制的方法也都包括在本发明范围内。此外,本发明涉及一种药物组合物,该组合物包含治疗有效量的本发明化合物和治疗有效量的可有效对抗RNA依赖性RNA病毒,特别是对抗HCV的另一药剂。有效对抗HCV的药剂包括但不限于利巴韦林、左旋韦林、viramidine、胸腺素α-1、HCV NS3丝氨酸蛋白酶抑制剂、干扰素α、聚乙二醇化干扰素α(聚乙二醇干扰素-α)、干扰素-α与利巴韦林的联合、聚乙二醇干扰素-α与利巴韦林的联合、干扰素α与左旋韦林的联合、聚乙二醇干扰素-α与左旋韦林的联合。干扰素α包括但不限于重组干扰素α2a(如Hoffmann-LaRoche,Nutley,NJ获得的Roferon interferon)、聚乙二醇化干扰素α2a(PegasysTM)、干扰素α2b(如Schering Corp.,Kenilworth,NJ获得的Intron-Ainterferon)、聚乙二醇化干扰素α2b(PegIntronTM)、重组体共有干扰素(如干扰素alphacon-1)和纯化的干扰素α产品。对利巴韦林及其对抗HCV的活性的讨论参见J.O.Saunders和S.A.Raybuck,″Inosine Monophosphate Dehydrogenase:Cohsideration of Stmcture,Kinetics,and Therapeutic Potential,″Ann.Rep.Med.Chem,35:201-210(2000)。Also included within the scope of the present invention are pharmaceutical compositions useful for inhibiting RNA-dependent RNA viral polymerase, particularly HCV NS5B polymerase, comprising an effective amount of a compound of the present invention and a pharmaceutically acceptable carrier. Pharmaceutical compositions useful for treating RNA-dependent RNA viral infections, particularly HCV infections, are also included within the scope of the invention, as are methods of inhibiting RNA-dependent RNA viral polymerases, particularly HCV NS5B polymerases, and the treatment of RNA-dependent Methods for the replication of RNA viruses, particularly HCV, are also within the scope of the invention. Furthermore, the present invention relates to a pharmaceutical composition comprising a therapeutically effective amount of a compound of the present invention and a therapeutically effective amount of another agent effective against RNA-dependent RNA viruses, in particular against HCV. Agents effective against HCV include but are not limited to ribavirin, levovirin, viramidine, thymosin alpha-1, HCV NS3 serine protease inhibitors, interferon alpha, pegylated interferon alpha (peginterferon interferon-α), the combination of interferon-α and ribavirin, the combination of pegylated interferon-α and ribavirin, the combination of interferon-α and levovirin, the combination of pegylated interferon-α Alpha in combination with levovirin. Interferon alpha includes, but is not limited to, recombinant interferon alpha 2a (such as Roferon interferon available from Hoffmann-LaRoche, Nutley, NJ), pegylated interferon alpha 2a (Pegasys ™ ), interferon alpha 2b (such as Schering Corp., Kenilworth, NJ Intron-Ainterferon), pegylated interferon alpha 2b (PegIntron ™ ), recombinant consensus interferon (such as interferon alphacon-1) and purified interferon alpha products. For a discussion of ribavirin and its activity against HCV see JOSaunders and SA Raybuck, "Inosine Monophosphate Dehydrogenase: Cohsideration of Structure, Kinetics, and Therapeutic Potential," Ann. Rep. Med. Chem, 35: 201-210 (2000) .
本发明的另一方面提供氟化吡咯并[2,3-d]嘧啶核苷化合物及其衍生物和它们的药物组合物在制造药物中的用途,所述药物用于RNA依赖性RNA病毒复制,特别是HCV复制,和/或治疗RNA依赖性RNA病毒感染,特别是HCV感染。本发明的再一方面提供可用作药物的氟化吡咯并[2,3-d]嘧啶化合物及其衍生物和它们的药物组合物,所述药物用于抑制RNA依赖性RNA病毒复制,特别是HCV复制,和/或治疗RNA依赖性RNA病毒感染,特别是HCV感染。Another aspect of the present invention provides the use of fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds and their derivatives and their pharmaceutical compositions in the manufacture of a medicament for the replication of RNA-dependent RNA viruses , particularly HCV replication, and/or treatment of RNA-dependent RNA virus infections, particularly HCV infections. Still another aspect of the present invention provides fluorinated pyrrolo[2,3-d]pyrimidine compounds and their derivatives and their pharmaceutical compositions useful as medicaments for inhibiting the replication of RNA-dependent RNA viruses, in particular is HCV replication, and/or treatment of RNA-dependent RNA viral infections, particularly HCV infections.
本发明的药物组合物包含作为有效成分的结构式I的化合物或其药学上可接受的盐,还可包含药学上可接受的载体和任选的其它治疗成分。The pharmaceutical composition of the present invention contains the compound of structural formula I or a pharmaceutically acceptable salt thereof as an active ingredient, and may also contain a pharmaceutically acceptable carrier and optional other therapeutic ingredients.
组合物包括适于经口、直肠、局部、肠胃外(包括皮下、肌内和静脉内)、眼(眼用)、肺(经鼻或口腔吸入)或者经鼻施用的组合物,尽管在任何情况下最合适的给药途径将取决于所治疗病症的性质和严重程度以及活性成分的性质。它们可方便地以单元剂量的形式存在,可通过药剂学领域内已知的任何方法制备。Compositions include those suitable for oral, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ophthalmic (ophthalmic), pulmonary (nasal or buccal inhalation) or nasal administration, although in any The most suitable route of administration in a case will depend upon the nature and severity of the condition being treated and the nature of the active ingredient. They may conveniently be presented in unit dosage form, prepared by any of the methods known in the art of pharmacy.
在实际应用中,可将结构式I的化合物作为活性成分与药用载体通过常规制药混合技术紧密掺混而组合。根据施用所需制剂,例如经口或肠胃外(包括静脉内)施用所需制剂的形式,所述载体可以采用各种形式。制备经口剂型用组合物时,在口服液制剂例如混悬液、酏剂和溶液的情况下,可以使用任何常用药物介质,例如水、二醇、油、醇、调味剂、防腐剂、着色剂等;或者在口服固体制剂例如粉末、硬和软胶囊剂以及片剂的情况下,可以使用载体如淀粉、糖、微晶纤维素、稀释剂、粒化剂、润滑剂、粘结剂、崩解剂等,固体口服制剂比液体制剂更优选。In practical application, the compound of structural formula I can be combined as an active ingredient and a pharmaceutical carrier by intimate mixing through conventional pharmaceutical mixing techniques. The carrier can take a variety of forms depending on the form of preparation desired for administration, eg, oral or parenteral (including intravenous). In the preparation of compositions for oral dosage forms, in the case of oral liquid preparations such as suspensions, elixirs and solutions, any usual pharmaceutical media such as water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents can be used. or in the case of oral solid preparations such as powders, hard and soft capsules, and tablets, carriers such as starch, sugar, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, Disintegrants and the like, solid oral preparations are more preferable than liquid preparations.
由于易于施用,因而片剂和胶囊是最好的口服剂量单元形式,这时明显是采用固体制药载体。如果需要,可以用常规水性或非水性工艺对片剂进行包衣。这样的组合物和制剂应含有至少0.1%活性化合物。当然,这些组合物中活性化合物的百分比可以改变,适宜在该单元的约2%至约60%重量范围内。活性化合物在这类可用于治疗的组合物中的量是可以获得有效剂量的量。所述活性化合物也可以经鼻内施用,例如作为液滴或喷雾施用。Because of their ease of administration, tablets and capsules are the most preferred oral dosage unit forms when obviously solid pharmaceutical carriers are employed. Tablets may be coated, if desired, by conventional aqueous or non-aqueous techniques. Such compositions and preparations should contain at least 0.1% active compound. The percentage of active compound in these compositions may, of course, be varied and suitably ranges from about 2% to about 60% by weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained. The active compounds can also be administered intranasally, eg, as liquid drops or spray.
片剂、丸剂、胶囊等也可包含粘合剂如西黄蓍胶、阿拉伯胶、玉米淀粉或明胶;赋形剂如磷酸二钙;崩解剂如玉米淀粉、马铃薯淀粉、海藻酸;润滑剂如硬脂酸镁;和甜味剂如蔗糖、乳糖或糖精。当单元剂型为胶囊时,它除了上述类型的材料外,还可以包含液体载体如脂肪油。Tablets, pills, capsules, etc. may also contain binders such as tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; disintegrants such as corn starch, potato starch, alginic acid; lubricants such as magnesium stearate; and sweeteners such as sucrose, lactose, or saccharin. When the unit dosage form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
可以存在各种其它材料作为包衣剂或用于改变剂量单元的物理形态。例如,可以用虫胶、糖或两种一起对片剂进行包衣。糖浆剂或酏剂除了活性成分外,还可以包含蔗糖作为甜味剂、包含对羟基苯甲酸甲酯和对羟基苯甲酸丙酯作为防腐剂、包含染料和调味剂作为樱桃味或橙味香料。Various other materials may be present as coatings or to modify the physical form of the dosage unit. For example, tablets may be coated with shellac, sugar or both. A syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavoring as cherry or orange flavor.
结构式I的化合物也可以肠胃外施用。这些活性化合物的溶液或混悬液可以通过在水中与表面活性剂如羟丙基纤维素适当混合来制备。分散体也可以在乙二醇、液态聚乙二醇及其油中混合物中制备。在通常的储存和使用条件下,这些制剂含有防腐剂以防止微生物生长。Compounds of formula I may also be administered parenterally. Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxypropylcellulose. Dispersions can also be prepared in glycols, liquid polyethylene glycols, and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
适于注射用途的药用剂型包括灭菌水溶液或分散体以及即时制备灭菌注射溶液或分散体的灭菌粉末。在各种情况下,所述剂型必须是灭菌的,并且必须是易于注射程度上的液态。在制造和储存条件下必须是稳定的,并且必须防止被微生物如细菌和真菌污染。载体可以是溶剂或含有例如水、乙醇、多元醇(例如丙三醇、丙二醇和液态聚乙二醇)、其稳定的混合物和植物油的分散介质。The pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersion. In each case, the dosage form must be sterile and must be liquid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be protected against the contamination by microorganisms such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (eg, glycerol, propylene glycol, and liquid polyethylene glycol), stable mixtures thereof, and vegetable oils.
可以采用任何适合的施用途径以向哺乳动物,特别是人提供有效剂量的本发明化合物。例如,可以采用经口、直肠、局部、肠胃外、眼、肺、鼻等。剂型包括片剂、锭剂、分散体、混悬液、溶液、胶囊、乳膏、软膏、气雾剂等。优选经口施用结构式I的化合物。Any suitable route of administration may be employed to provide an effective dose of a compound of the invention to a mammal, especially a human. For example, oral, rectal, topical, parenteral, ophthalmic, pulmonary, nasal, etc. can be used. Dosage forms include tablets, lozenges, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like. The compounds of formula I are preferably administered orally.
对人经口施用时,分剂量的剂量范围为0.01-1000mg/kg体重。在一个实施方案中,分剂量的剂量范围为0.1-100mg/kg体重。在另一个实施方案中,分剂量的剂量范围为0.5-20mg/kg体重。对于经口施用,优选将组合物以含1.0-1000mg活性成分,特别是1、5、10、15、20、25、50、75、100、150、200、250、300、400、500、600、750、800、900和1000mg活性成分的片剂或胶囊的形式施用以调节所治疗患者的症状。For oral administration to humans, the dose ranges in divided doses from 0.01 to 1000 mg/kg body weight. In one embodiment, the dose range in divided doses is 0.1-100 mg/kg body weight. In another embodiment, the dose in divided doses ranges from 0.5-20 mg/kg body weight. For oral administration, the composition is preferably formulated to contain 1.0-1000 mg of active ingredient, especially 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600 , 750, 800, 900 and 1000 mg of active ingredient in the form of tablets or capsules to regulate the symptoms of the treated patients.
所用活性成分的有效剂量可以随所采用的具体化合物、施用方式、所治疗病症和所治疗病症的严重程度而变化。本领域技术人员可以容易地确定这样的剂量。可以调节该剂量方案以提供最佳治疗反应。The effective dosage of the active ingredient employed may vary with the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosages can be readily determined by those skilled in the art. The dosage regimen can be adjusted to provide the optimum therapeutic response.
本发明化合物包含一个或多个非对称中心,因此可以作为外消旋体和外消旋混合物、单一的对映体、非对映体混合物和单独的非对映体存在。本发明包括具有下述结构式的五元呋喃糖环为β-D立体化学构型的氟化吡咯并[2,3-d]嘧啶核苷化合物,即,其中五元呋喃糖环的C-1和C-4上的取代基具有β-立体化学构型的氟化吡咯并[2,3-d]嘧啶核苷化合物(以粗线表示的“向上”方向)。The compounds of the present invention contain one or more asymmetric centers and can therefore exist as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. The present invention includes fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds having a five-membered furanose ring with a β-D stereochemical configuration, that is, wherein C-1 of the five-membered furanose ring is Fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds with substituents on C-4 having a β-stereochemical configuration ("up" direction indicated by bold lines).

本文所述的一些化合物包含烯属双键,除非另外指明,否则指包括E和Z两种几何异构体。Some of the compounds described herein contain olefinic double bonds and, unless otherwise indicated, are meant to include both E and Z geometric isomers.
本文所述的一些化合物可以作为互变异构体如酮-烯醇互变异构体存在。各种互变异构体及其混合物包括在结构式I的化合物中。包括在本发明化合物范围内的酮-烯醇和亚胺-烯胺互变异构体的例子如下:Some of the compounds described herein may exist as tautomers such as keto-enol tautomers. Included within the compounds of formula I are the various tautomers and mixtures thereof. Examples of keto-enol and imine-enamine tautomers included within the scope of the compounds of the present invention are as follows:
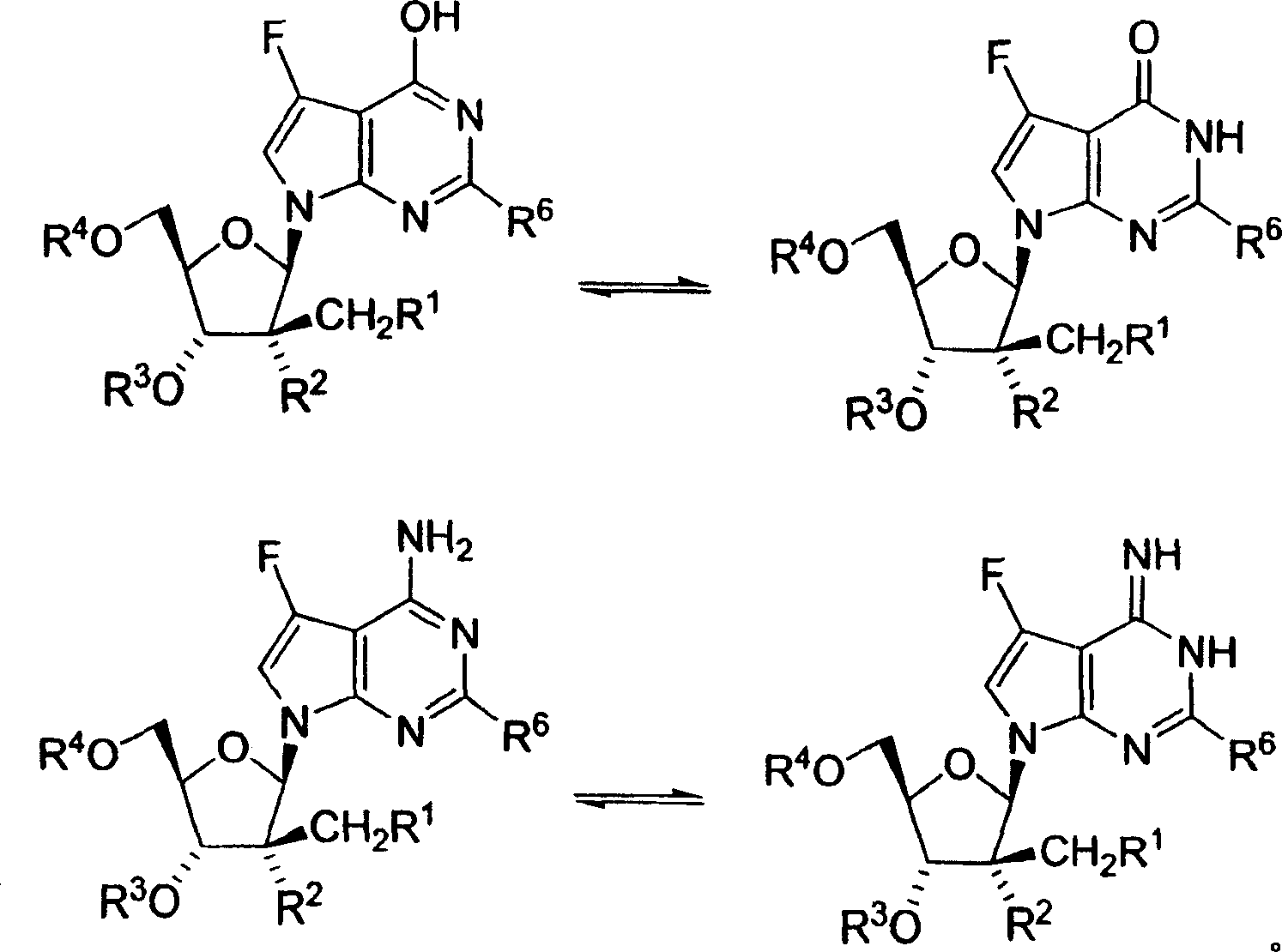
可以通过例如分级结晶从适当的溶剂例如甲醇或乙酸乙酯或其混合物中或者经由使用旋光活性固定相的手性色谱将结构式I的化合物分离为它们的单独的非对映体。The compounds of formula I can be separated into their individual diastereomers by eg fractional crystallization from a suitable solvent such as methanol or ethyl acetate or mixtures thereof or via chiral chromatography using optically active stationary phases.
可选地,可以通过使用已知构型的光学纯起始原料或试剂进行立体有择合成得到结构式I的化合物的任何立体异构体。Alternatively, any stereoisomer of a compound of formula I may be obtained by stereospecific synthesis using optically pure starting materials or reagents of known configuration.
本发明化合物可以以药学上可接受的盐的形式施用。术语“药学上可接受的盐”指由药学上可接受的无毒碱或酸包括无机或有机碱和无机或有机酸所制成的盐。包括在术语“药学上可接受的盐”范围内的碱性化合物的盐指本发明化合物的无毒盐,它通常通过游离碱与适当的有机或无机酸的反应制备。本发明碱性化合物的具有代表性的盐包括但不限于下列盐:乙酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、酒石酸氢盐、硼酸盐、溴化物、樟磺酸盐、碳酸盐、氯化物、克拉维酸盐、柠檬酸盐、二盐酸盐、依地酸盐、乙二磺酸盐、依托酸盐、乙磺酸盐、富马酸盐、葡庚糖酸盐、葡糖酸盐、谷氨酸盐、乙醇酰对氨苯基胂酸盐、己基间苯二酚盐(hexylresorcinate)、海巴明、氢溴酸盐、盐酸盐、羟萘甲酸盐、碘化物、异硫代硫酸盐(isothionate)、乳酸盐、乳糖酸盐、月桂酸盐、苹果酸盐、马来酸盐、扁桃酸盐、甲磺酸盐、甲基溴化物、甲基硝酸盐、甲基硫酸盐、粘酸盐、萘磺酸盐、硝酸盐、N-甲基葡糖胺铵盐、油酸盐、草酸盐、巴莫酸盐(双羟萘酸盐)、棕榈酸盐、泛酸盐、磷酸盐/二磷酸盐、聚半乳糖醛酸盐、水杨酸盐、硬脂酸盐、硫酸盐、碱式乙酸盐、琥珀酸盐、单宁酸盐、酒石酸盐、茶氯酸盐、甲苯磺酸盐、三乙基碘和戊酸盐。此外,当本发明化合物带有酸性部分时,其适当的药学上可接受的盐包括但不限于衍生自无机碱的盐,包括铝、铵、钙、铜、铁、亚铁、锂、镁、锰、mangamous、钾、钠、锌等。特别优选的是铵、钙、镁、钾和钠盐。衍生自药学上可接受的有机无毒碱的盐包括伯、仲和叔胺、环胺的盐以及碱性离子交换树脂,如精氨酸、甜菜碱、咖啡碱、胆碱、N,N-二苄基乙二胺、二乙胺、2-二乙基氨基乙醇、2-二甲基氨基乙醇、乙醇胺、乙二胺、N-乙基吗啉、N-乙基哌啶、葡糖胺、壳糖胺、组氨酸、海巴明、异丙基胺、赖氨酸、甲葡糖胺、吗啉、哌嗪、哌啶、聚胺树脂、普鲁卡因、嘌呤、可可碱、三乙胺、三甲胺、三丙胺、氨丁三醇等。The compounds of the present invention may be administered in the form of pharmaceutically acceptable salts. The term "pharmaceutically acceptable salt" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids, including inorganic or organic bases and inorganic or organic acids. Salts of basic compounds encompassed within the term "pharmaceutically acceptable salt" refer to non-toxic salts of the compounds of this invention which are usually prepared by reacting the free base with a suitable organic or inorganic acid. Representative salts of basic compounds of the invention include, but are not limited to, the following salts: acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide , camphorate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, ettoate, ethanesulfonate, fumaric acid salt, glucoheptonate, gluconate, glutamate, glycolyl p-aminophenylarsinate, hexylresorcinate (hexylresorcinate), hepamine, hydrobromide, hydrochloride , xinafoate, iodide, isothiosulfate (isothionate), lactate, lactobionate, laurate, malate, maleate, mandelate, methanesulfonate, formazan bromide, methyl nitrate, methyl sulfate, mucate, naphthalene sulfonate, nitrate, N-methylglucamine ammonium salt, oleate, oxalate, balm salt (bis moxamate), palmitate, pantothenate, phosphate/diphosphate, polygalacturonate, salicylate, stearate, sulfate, hydroxyacetate, succinate , tannin, tartrate, theanate, tosylate, triethyl iodide and valerate. Additionally, when compounds of the present invention bear an acidic moiety, suitable pharmaceutically acceptable salts thereof include, but are not limited to, salts derived from inorganic bases, including aluminium, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, Manganese, mangamous, potassium, sodium, zinc, etc. Particularly preferred are the ammonium, calcium, magnesium, potassium and sodium salts. Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary and tertiary amines, cyclic amines, and basic ion exchange resins such as arginine, betaine, caffeine, choline, N,N-di Benzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucosamine, Glucosamine, histidine, hepamine, isopropylamine, lysine, methyl glucosamine, morpholine, piperazine, piperidine, polyamine resin, procaine, purine, theobromine, three Ethylamine, trimethylamine, tripropylamine, tromethamine, etc.
还有,在本发明化合物中存在羧酸(-COOH),磷酸[-OP(O)(OH)2]或醇基的情况下,可以使用羧酸衍生物的药学上可接受的酯,如甲基、乙基或新戊酰氧基甲基酯;氟化吡咯并[2,3-d]嘧啶核苷的5′-磷酸衍生物(包括5′-一磷酸酯,5′-二磷酸酯和5′-三磷酸酯)的药学上可接受的前药酯;或核糖C-2′,C-3′,和C-5′羟基的前药酰基衍生物,如O-乙酸酯或O-马来酸酯或O-氨酰基。包括在内的有本领域已知用于改进用作持续释放或前药制剂的生物利用度,组织分布,溶解性或水解特性的那些酯和酰基。还包括C-2′和C-3′的羟基的五元环碳酸酯衍生物。预期的衍生物在体外容易地转化成所需的化合物。因此,在本发明的治疗方法中,术语“施与”和“施用”指包括使用明确公开的化合物或使用没有明确公开的但在给哺乳动物包括人类患者施用后在体内转化为指定化合物的化合物治疗所描述的病毒感染。常规的选择和制备适宜的前药衍生物的方法例如在″Design of Prodrugs,″H.Bundgaard编辑,Elsevier,1985中有描述,将其完整引入本文作为参考。Also, where carboxylic acid (-COOH), phosphate [-OP(O)(OH) 2 ] or alcohol groups are present in the compounds of the present invention, pharmaceutically acceptable esters of carboxylic acid derivatives such as Methyl, ethyl or pivaloyloxymethyl esters; 5′-phosphate derivatives of fluorinated pyrrolo[2,3-d]pyrimidine nucleosides (including 5′-monophosphate, 5′-diphosphate esters and 5'-triphosphate) to pharmaceutically acceptable prodrug esters; or prodrug acyl derivatives of ribose C-2', C-3', and C-5' hydroxyl groups, such as O-acetate Or O-maleate or O-aminoacyl. Included are those esters and acyl groups known in the art to modify bioavailability, tissue distribution, solubility or hydrolysis properties for use as sustained release or prodrug formulations. Also included are five-membered ring carbonate derivatives of the C-2' and C-3' hydroxyl groups. The expected derivatives are readily converted to the desired compounds in vitro. Therefore, in the method of treatment of the present invention, the terms "administering" and "administering" are meant to include the use of a specifically disclosed compound or the use of a compound that is not specifically disclosed but is converted in vivo to the specified compound after administration to a mammal, including a human patient. Treat viral infections as described. Routine methods for selecting and preparing suitable prodrug derivatives are described, for example, in "Design of Prodrugs," edited by H. Bundgaard, Elsevier, 1985, which is incorporated herein by reference in its entirety.
本发明化合物的制备:Preparation of compounds of the present invention:
用于制备本发明化合物的起始物料为4-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶 (1-9),流程图1描述了其合成。The starting material for the preparation of the compounds of the present invention is 4-amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (1-9) , the synthesis of which is described in Scheme 1.
流程图1Flowchart 1

5-氟-4-氯-7H-吡咯并[2,3-d]嘧啶的制备(1-4):Preparation of 5-fluoro-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1-4):
步骤A:5-溴-4氯-7H-吡咯并[2,3-d]嘧啶的制备(1-2)Step A: Preparation of 5-bromo-4chloro-7H-pyrrolo[2,3-d]pyrimidine (1-2)
在0℃向在DMF(20 mL)中的4氯-7H-吡咯并[2,3-d]嘧啶( 1-1)(1.53g,10.0mmol)的溶液中滴加在(10mL)中的N-溴代琥珀酰亚胺(1.78g,10.0mmol)的溶液。将反应混合物在0℃搅拌30分钟然后在室温搅拌1小时。加入甲醇(25mL),然后将反应混合物搅拌另外1小时。蒸发溶剂并由甲醇重结晶残留物得到为白色固体的标题化合物。To a solution of 4chloro-7H-pyrrolo[2,3-d]pyrimidine ( 1-1 ) (1.53 g, 10.0 mmol) in DMF (20 mL) was added dropwise in (10 mL) at 0 °C Solution of N-bromosuccinimide (1.78 g, 10.0 mmol). The reaction mixture was stirred at 0 °C for 30 minutes then at room temperature for 1 hour. Methanol (25 mL) was added and the reaction mixture was stirred for an additional 1 hour. Evaporation of the solvent and recrystallization of the residue from methanol gave the title compound as a white solid.
步骤B:5-(三甲基甲锡烷基)-4氯-7H-吡咯并[2,3-d]嘧啶(1-3)的制备Step B: Preparation of 5-(trimethylstannyl)-4chloro-7H-pyrrolo[2,3-d]pyrimidine (1-3)
在-78℃向在THF(25mL)中的由步骤A获得的化合物(0.92g,4mmol)的溶液中滴加n-BuLi(2.5M己烷溶液,3.48mL)。加完后,将反应混合物在-78℃搅拌另外30分钟。在10分钟内向该溶液中滴加在THF(8mL)中的三甲基氯化锡(0.88g,4.4mmol)。将反应混合物缓慢恢复至室温并在室温搅拌过夜。加入饱和的氯化铵溶液(60mL)并用乙酸乙酯进行萃取(3×70mL)。将合并的有机萃取物用盐水洗涤,Na2SO4干燥,蒸发至变干。在硅胶上将残留物纯化得到为无色固体的标题化合物。To a solution of the compound obtained from step A (0.92 g, 4 mmol) in THF (25 mL) was added n-BuLi (2.5M in hexane, 3.48 mL) dropwise at -78°C. After the addition was complete, the reaction mixture was stirred at -78°C for an additional 30 minutes. To this solution was added trimethyltin chloride (0.88 g, 4.4 mmol) in THF (8 mL) dropwise over 10 minutes. The reaction mixture was slowly brought to room temperature and stirred overnight at room temperature. Saturated ammonium chloride solution (60 mL) was added and extracted with ethyl acetate (3 x 70 mL). The combined organic extracts were washed with brine, dried over Na2SO4 and evaporated to dryness. The residue was purified on silica gel to give the title compound as a colorless solid.
步骤C:5-氟-4-氯-7H-吡咯并[2,3-d]嘧啶(1-4)Step C: 5-fluoro-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1-4)
向在CH3CN(60mL)中的由步骤B获得的化合物的溶液中加入一份[1-(氯甲基)-4-氟-1,4-二氮杂双环[2.2.2]辛烷双(四氟硼酸酯)](SELECTFLUOR氟化试剂)(2.40g,6.5mmol)并将反应混合物在室温搅拌7小时。滤出白色沉淀,将滤液蒸发至变干。在硅胶上使用乙酸乙酯/己烷(3∶7)作为洗脱液将残留物纯化。将含有产物的级分集中并真空蒸发得到为无色固体的标题化合物。To a solution of the compound obtained from step B in CH3CN (60 mL) was added one portion of [1-(chloromethyl)-4-fluoro-1,4-diazabicyclo[2.2.2]octane Bis(tetrafluoroborate)] (SELECTFLUOR(R) fluorinating reagent) (2.40 g, 6.5 mmol) and the reaction mixture was stirred at room temperature for 7 hours. The white precipitate was filtered off and the filtrate was evaporated to dryness. The residue was purified on silica gel using ethyl acetate/hexane (3:7) as eluent. Fractions containing product were pooled and evaporated in vacuo to give the title compound as a colorless solid.
1H-NMR(500MHz,MeOH-d4):δ8.53(s,1H),7.37(d,J=2.8Hz);19F-NMR(DMSO-d6):δ-171.5. 1 H-NMR (500MHz, MeOH-d 4 ): δ8.53 (s, 1H), 7.37 (d, J=2.8Hz); 19 F-NMR (DMSO-d 6 ): δ-171.5.
2-C-甲基-3.5-二-O-(对-甲苯酰基)-D-呋喃核糖(1-6)的制备:Preparation of 2-C-methyl-3.5-di-O-(p-toluoyl)-D-ribofuranose (1-6):
向在35l乙腈中的3-O-苯甲基-1,2-O-异丙叉基-3-C-甲基-α-D-呋喃葡萄糖(1-5)(制备参见Carbohydr.Res..44:275-283(1975)(5.0kg,15.4mol)和吡啶(3.7kg,46.2mol)的溶液中加入对-苯甲酰氯(5.2kg,33.9mol),将反应在50-55℃加热12小时。在50-55℃加入在9L水中的48wt%的HBF4(四氟硼酸)的溶液6.0L(46.2mol)。2小时后,蒸馏除去10L乙腈,加入10L乙腈。在转化97%时,蒸馏除去10L乙腈,将反应溶液冷却至0-5℃。加入在10L水中的高碘酸溶液(4.2kg,18.5mol)。在将反应陈化30分钟后,加入35L乙酸异丙酯和10L水。用25L水,随后使用20L NaHCO3水溶液,15L在水中的5%硫代硫酸钠,15L水洗涤有机相。将乙酸异丙酯溶液浓缩至10-15L,加入40L甲醇。将溶液冷却至0℃并加入二异丙胺(0.78kg,7.7mol)。在0℃两天后,在0-5℃加入HCl水溶液(1N,7.7L),随后加入30L乙酸异丙酯和40L水。使用1N HCl,NaHCO3和盐水洗涤有机相。通过共沸蒸馏干燥有机相并有活性炭进行处理。通过过滤除去碳并使用乙酸异丙酯将所得溶液稀释至75L并氢化(45psi,50℃,1.5kg 10%Pd/C)24小时。将滤液浓缩至15L并在50℃加入60L庚烷。用10L的在庚烷中20%乙酸异丙酯洗涤过滤分离得到晶物产物。干燥得到4.03kg需要的二醇1-6。To 3-O-benzyl-1,2-O-isopropylidene-3-C-methyl-α-D-glucofuranose (1-5) in 35 l of acetonitrile (for preparation see Carbohydr. Res. .44:275-283 (1975) (5.0kg, 15.4mol) and pyridine (3.7kg, 46.2mol) were added to a solution of p-benzoyl chloride (5.2kg, 33.9mol), and the reaction was heated at 50-55°C 12 hours. Add 48wt% HBF 4 (tetrafluoroboric acid) solution 6.0L (46.2mol) in 9L water at 50-55 ° C. After 2 hours, distill off 10L acetonitrile, add 10L acetonitrile. When converting 97% , 10L of acetonitrile was distilled off, and the reaction solution was cooled to 0-5°C. Periodic acid solution (4.2kg, 18.5mol) in 10L of water was added. After aging the reaction for 30 minutes, 35L of isopropyl acetate and 10L of Water. Wash the organic phase with 25 L of water, followed by 20 L of NaHCO aqueous solution, 15 L of 5% sodium thiosulfate in water, 15 L of water. Concentrate the isopropyl acetate solution to 10-15 L and add 40 L of methanol. Cool the solution to 0° C. and diisopropylamine (0.78 kg, 7.7 mol) was added. After two days at 0° C., aqueous HCl (1 N, 7.7 L) was added at 0-5° C., followed by 30 L isopropyl acetate and 40 L water. 1 N HCl was used , NaHCO3 and brine. The organic phase was dried by azeotropic distillation and treated with activated carbon. The carbon was removed by filtration and the resulting solution was diluted to 75 L using isopropyl acetate and hydrogenated (45 psi, 50 °C, 1.5 kg 10% Pd /c) 24 hours. The filtrate was concentrated to 15 L and 60 L of heptane was added at 50° C. The crystalline product was isolated by washing with 10 L of 20% isopropyl acetate in heptane by filtration. Drying gave 4.03 kg of the required diol 1 -6.
1H NMR(CDCl3,400MHz):CDCl3中α∶β异构体的比例为大约5∶1.对于主要的异构体:δ7.95-7.90(m,4H),7.26(d,J=8.0Hz,2H),7.17(d,J=8.0Hz,2H),5.53(d,J=7.2Hz,1H),5.22(d,J=2.8Hz,1H),4.65-4.49(m,3H),3.08(d,J=3.2Hz,1H),2.44(s,3H),2.38(s,3H),2.26(s,1H),1.44(s,3H)ppm;对于次要的异构体:δ7.95-7.90(m,4H),7.27(d,J=8.0Hz,2H),7.22(d,J=8.0Hz,2H),5.16(d,J=5.6Hz,1H),5.12(d,J=5.6Hz,1H),4.66-4.49(m,3H),3.54(d,J=5.6Hz,1H),2.91(s,1H),2.43(s,3H),2.40(s,3H),1.44(s,3H)ppm.。 1 H NMR (CDCl 3 , 400 MHz): The ratio of α:β isomer in CDCl 3 is about 5:1. For the major isomer: δ 7.95-7.90 (m, 4H), 7.26 (d, J =8.0Hz, 2H), 7.17(d, J=8.0Hz, 2H), 5.53(d, J=7.2Hz, 1H), 5.22(d, J=2.8Hz, 1H), 4.65-4.49(m, 3H ), 3.08 (d, J = 3.2Hz, 1H), 2.44 (s, 3H), 2.38 (s, 3H), 2.26 (s, 1H), 1.44 (s, 3H) ppm; for minor isomers : δ7.95-7.90(m, 4H), 7.27(d, J=8.0Hz, 2H), 7.22(d, J=8.0Hz, 2H), 5.16(d, J=5.6Hz, 1H), 5.12( d, J=5.6Hz, 1H), 4.66-4.49(m, 3H), 3.54(d, J=5.6Hz, 1H), 2.91(s, 1H), 2.43(s, 3H), 2.40(s, 3H ), 1.44 (s, 3H) ppm.
4-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(1-9)的制备Preparation of 4-amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (1-9)
步骤A:1.2-脱水-3.5-二-O-(对-甲苯酰基)-2-C-甲基-α-D-呋喃核糖(1-7)Step A: 1.2-Anhydro-3.5-di-O-(p-toluoyl)-2-C-methyl-α-D-ribofuranose (1-7)
向72L的容器中装入干燥的二氯甲烷(32L),三乙胺(3.0L),和二醇(3.44kg,90wt%纯)。将混合物加热至30℃,然后在40分钟内加入甲磺酰氯(0.79L)。1小时后,将该批产物(batch)在pH7的缓冲液(20L)和甲基叔丁基醚(44L)之间分配。使用1M NaCl水溶液(38L)洗涤有机相,然后转换到真空蒸馏甲苯,随后浓缩至大约9L。所得环氧化物溶液直接步骤B中使用。A 72 L vessel was charged with dry dichloromethane (32 L), triethylamine (3.0 L), and diol (3.44 kg, 90 wt% pure). The mixture was heated to 30°C, then methanesulfonyl chloride (0.79 L) was added over 40 minutes. After 1 hour, the batch was partitioned between pH 7 buffer (20 L) and methyl tert-butyl ether (44 L). The organic phase was washed with 1M aqueous NaCl (38 L), then switched to vacuum distillation of toluene, followed by concentration to approximately 9 L. The resulting epoxide solution was used directly in Step B.
步骤B:4-氯-5-氟-7-(2-C-甲基-3,5-二-O-(对-甲苯酰基)-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(1-8)Step B: 4-Chloro-5-fluoro-7-(2-C-methyl-3,5-di-O-(p-toluoyl)-β-D-ribofuranosyl)-7H-pyrrolo[ 2,3-d] pyrimidine (1-8)
在室温向在N,N-二甲乙酰胺(300mL)的4-氯-5-氟-7H-吡咯并[2,3-d]嘧啶(1-4)(28.4g,0.165mol)的溶液中portionwise加入60%氢化钠(6.6g,0.165mol)。加完后,将反应混合物在60℃搅拌1小时。向反应混合物中加入在THF(200mL)的1.2-脱水-3.5-二-O-(对-甲苯酰基)-2-C-甲基-α-D-呋喃核糖(1-7)的溶液(63.4g,0.166mol),将反应混合物在60℃加热18小时。将反应混合物冷却至室温,倒入水(1L)和乙酸乙酯(2L)中。有机萃取物用水(500mL)洗涤,MgSO4干燥,蒸发至变干。将残留物在硅胶上使用10-40%乙酸乙酯/己烷作为洗脱液进行纯化。将含有产物的级分合并浓缩得到泡沫状物,直接在以下的步骤C中使用。To a solution of 4-chloro-5-fluoro-7H-pyrrolo[2,3-d]pyrimidine (1-4) (28.4 g, 0.165 mol) in N,N-dimethylacetamide (300 mL) at room temperature Portionwise 60% sodium hydride (6.6 g, 0.165 mol) was added. After the addition was complete, the reaction mixture was stirred at 60°C for 1 hour. To the reaction mixture was added a solution (63.4 g, 0.166 mol), the reaction mixture was heated at 60°C for 18 hours. The reaction mixture was cooled to room temperature and poured into water (1 L) and ethyl acetate (2 L). The organic extracts were washed with water (500 mL), dried over MgSO 4 and evaporated to dryness. The residue was purified on silica gel using 10-40% ethyl acetate/hexanes as eluent. Fractions containing product were pooled and concentrated to give a foam which was used directly in Step C below.
步骤C:4-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(1-9)Step C: 4-Amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (1-9)
在密封的容器中将在无水氨(300mL)中的4-氯-5-氟-7-(2-C-甲基-3,5-二-O-(对-甲苯酰基)-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶的溶液(29.4g,0.053mol)在85℃加热48小时。将反应混合物升温至室温,将残留物在甲醇(200mL)中成浆,过滤,将滤液吸附到硅胶(200g)上,然后通过色谱使用0-30%甲醇/二氯甲烷作为洗脱液进行纯化,将含有产物的级分合并,蒸发得到为固体的标题化合物 1-9。4-Chloro-5-fluoro-7-(2-C-methyl-3,5-di-O-(p-toluoyl)-β- A solution of D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (29.4 g, 0.053 mol) was heated at 85°C for 48 hours. The reaction mixture was warmed to room temperature, the residue was slurried in methanol (200 mL), filtered, the filtrate was absorbed onto silica gel (200 g) and purified by chromatography using 0-30% methanol/dichloromethane as eluent , fractions containing product were combined and evaporated to give the title compound 1-9 as a solid.
1H-NMR(500MHz,MeOH-d4):δ8.07(s,1H),7.41(d,J=2.2Hz,1H),6.25(d,J=1.8Hz),4.09-3.95(m,3H),3.82(dd,J=2.7,12.5Hz,1H);19F-NMR(MeOH-d4):δ-170.4;质谱:321(M+Na)+。 1 H-NMR (500MHz, MeOH-d 4 ): δ8.07(s, 1H), 7.41(d, J=2.2Hz, 1H), 6.25(d, J=1.8Hz), 4.09-3.95(m, 3H), 3.82 (dd, J=2.7, 12.5Hz, 1H); 19 F-NMR (MeOH-d 4 ): δ-170.4; Mass Spectrum: 321 (M+Na) + .
在中间体 1-9的6-氨基-7-氮杂嘌呤(7-deazaadenine)环的C-2位上引入氨基可按Zhao等,在J.Org.Chem.,62:7832-7835(1997)中所述合成方法进行,如流程图2所例示。2,6-二氨基-7-氮杂嘌呤环转化为7-deazaguanine系统可按K.Alarcon等人在Tetrahedron Lett,41:7211-7215(2000)中所述方法进行,如流程图3所示。The introduction of the amino group at the C-2 position of the 6-amino-7-azapurine (7-deazaadenine) ring of the intermediate 1-9 can be carried out according to Zhao et al., in J.Org.Chem., 62:7832-7835 (1997 ) The synthetic method described in ) was carried out, as illustrated in Scheme 2. The conversion of 2,6-diamino-7-azapurine ring into 7-deazaguanine system can be carried out according to the method described in K.Alarcon et al. in Tetrahedron Lett, 41:7211-7215 (2000), as shown in flow chart 3 .
下面的实施例对在制备本发明化合物中使用的条件进行了举例说明。实施例无意以任何方式限制本发明的范围,也不应对其作如是解释。核苷和核苷酸合成领域的技术人员将容易地意识到可对下述制备过程的条件和方法作众所周知的变更,以制备本发明的这些和其它化合物。除非另有说明,否则所有的温度都是摄氏度。The following examples illustrate the conditions used in the preparation of compounds of the invention. The examples are not intended to limit the scope of the invention in any way, nor should they be construed as such. Those skilled in the art of nucleoside and nucleotide synthesis will readily recognize that well-known modifications to the conditions and methods of the preparative procedures described below can be made to prepare these and other compounds of the invention. All temperatures are in degrees Celsius unless otherwise stated.
流程图2Flowchart 2
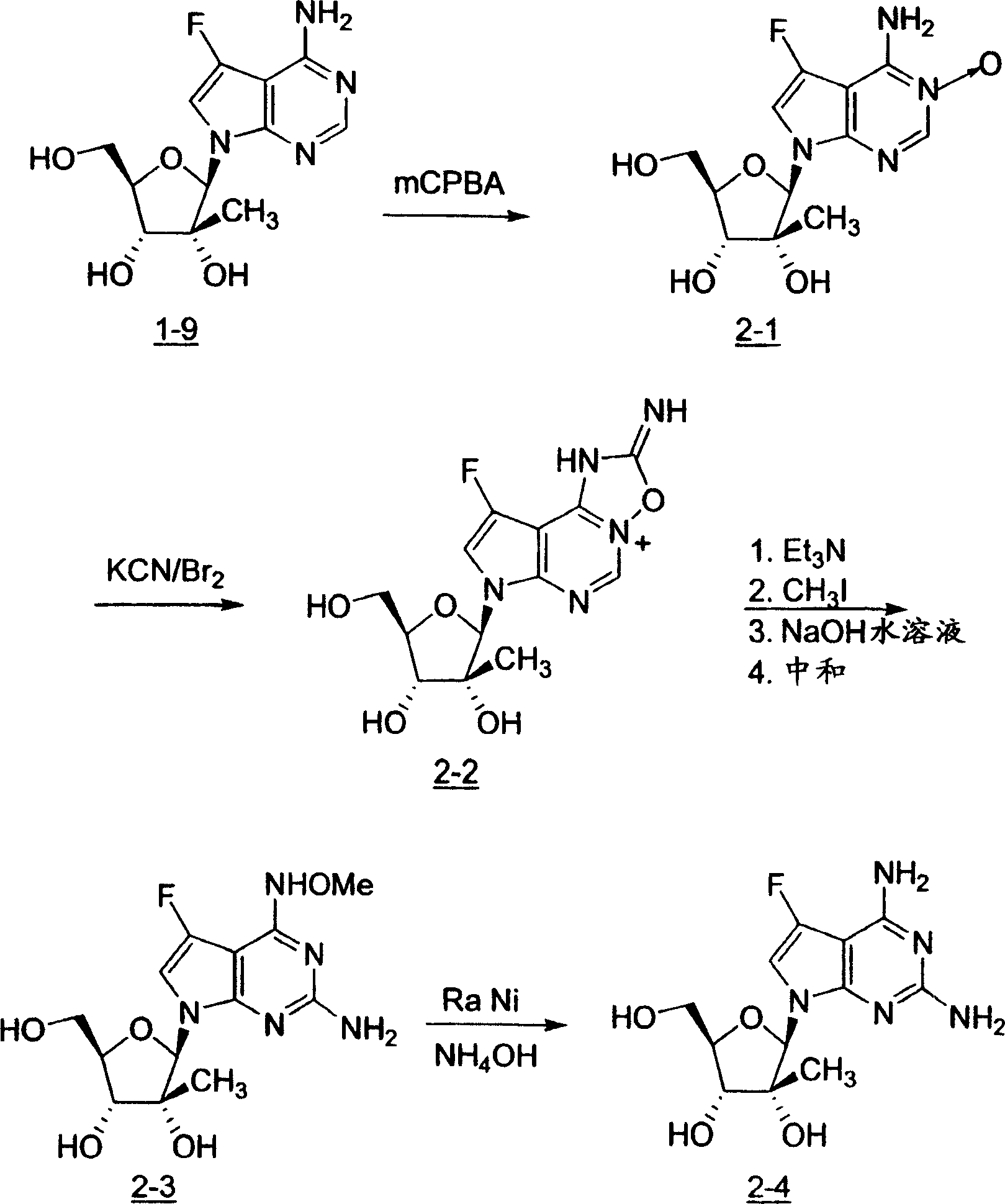
实施例1Example 1
2,4-二氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(2-4)2,4-Diamino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (2-4)
步骤A:4-氨基-5-氟-3-N-氧-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(2-1)Step A: 4-Amino-5-fluoro-3-N-oxo-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (2 -1)
向在50%甲醇/水(20mL)的4-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶的的溶液(268mg,0.899mmol)中加入m-氯过氧苯甲酸(444mg,1.80mmol)。将反应混合物在室温搅拌18小时。将溶剂蒸发并将残留物与甲苯共沸两次得到为米色固体的标题化合物。To 4-amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine in 50% methanol/water (20 mL) (268mg, 0.899mmol) was added m-chloroperoxybenzoic acid (444mg, 1.80mmol). The reaction mixture was stirred at room temperature for 18 hours. The solvent was evaporated and the residue was azeotroped twice with toluene to give the title compound as a beige solid.
步骤B:2,4-二氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(2-4)Step B: 2,4-Diamino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (2-4)
在0℃向在水中(3mL)的溴化氰生物溶液中加入在水中(3mL)的4-氨基-5-氟-3-N-氧-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶(2-1)(160mg,0.509mmol)的溶液。将所得溶液在0℃搅拌1.5小时。蒸发溶剂并与甲苯共沸。向参礼物中加入N,N-二甲基酰胺(3.5mL)和三乙胺(0.25mL,1.79mol),将所得溶液在室温搅拌45分钟。分批加入碘甲烷(0.25mL,4.0mmol),在室温避光情况下将反应混合物搅拌1.5小时。蒸发溶剂并向残留物中加入0.25M氢氧化钠水溶液(10mL),将其在室温下搅拌30分钟。用1M HCl中和反应混合物,用乙醇(10mL)稀释,在60℃加热8小时。在冷却至室温后,加入浓缩的氢氧化铵(12mL),将反应混合物加热至90℃因此加入拉尼镍。15分钟后,通过将热反应混合物过滤,将溶剂蒸发,在硅胶上使用甲醇/二氯甲烷作为洗脱液将残留物纯化。将含有产物的级分蒸发得到为固体的标题化合物 2-4。To a biological solution of cyanogen bromide in water (3 mL) was added 4-amino-5-fluoro-3-N-oxygen-7-(2-C-methyl-β-D A solution of -ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (2-1 ) (160 mg, 0.509 mmol). The resulting solution was stirred at 0 °C for 1.5 hours. The solvent was evaporated and azeotroped with toluene. N,N-Dimethylamide (3.5 mL) and triethylamine (0.25 mL, 1.79 mol) were added to the solution, and the resulting solution was stirred at room temperature for 45 minutes. Iodomethane (0.25 mL, 4.0 mmol) was added in portions, and the reaction mixture was stirred at room temperature for 1.5 hours in the dark. The solvent was evaporated and 0.25M aqueous sodium hydroxide solution (10 mL) was added to the residue, which was stirred at room temperature for 30 minutes. The reaction mixture was neutralized with 1M HCl, diluted with ethanol (10 mL), and heated at 60° C. for 8 hours. After cooling to room temperature, concentrated ammonium hydroxide (12 mL) was added and the reaction mixture was heated to 90° C. whereby Raney nickel was added. After 15 minutes, the residue was purified on silica gel using methanol/dichloromethane as eluent by filtering the hot reaction mixture, evaporating the solvent. Fractions containing product were evaporated to give the title compound 2-4 as a solid.
1H-NMR(500MHz,MeOH-d4):δ 7.3(s,1H),6.1(s,1H),4.0(m,3H),3.8(d,1H),0.9(s,3H);质谱:314(M+1). 1 H-NMR (500MHz, MeOH-d 4 ): δ 7.3(s, 1H), 6.1(s, 1H), 4.0(m, 3H), 3.8(d, 1H), 0.9(s, 3H); mass spectrum : 314(M+1).
流程图3Flowchart 3
实施例2Example 2
2-氨基-5-氟-7-(2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶-4(3H)-酮(3-2)2-Amino-5-fluoro-7-(2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-4(3H)-one (3-2 )
该化合物通过使用在DMF中的1,2-双[(二甲基氨基)-亚甲基]肼处理化合物 2-4以提供化合物 3-1,将其按K.Alarcon等人在TetrahedronLett,41:7211-7215(2000)中所述条件下使用在DMSO中的1N NaOH含水溶液水解来进行制备。This compound was treated by treatment of compound 2-4 with 1,2-bis[(dimethylamino)-methylene]hydrazine in DMF to provide compound 3-1 , which was described by K. Alarcon et al. in Tetrahedron Lett, 41 : 7211-7215 (2000) using hydrolysis of aqueous 1 N NaOH in DMSO solution under the conditions described in DMSO.
本发明的2-氟-2-C-甲基核糖核苷(结构式I中R2=F)可根据核苷和核苷酸化学实践中行之有效的合成方法进行制备。提供了化合物 6-3的合成作为示例,如流程图4-6中所描述。D-核糖( 4-1)首先被保护。在这种情况下,酯,例如乙酸酯和苯甲酸酯提供了合适的保护基,但是也可使用替代性的保护基。任选在溶剂,例如二乙醚,二恶烷,四氢呋喃和二氯甲烷存在的情况下使D-核糖与适当的酰基卤或酐反应实现酯化。这样的转化在化学文献文献中是众所周知的,在Greene,T.W.,Wuts,P.G.M.,″ProtectiVe Groups in Organic Synthesis,″,John Wiley & Sons,,Inc.,第3版,1999中可发现实例。The 2-fluoro-2-C-methyl ribonucleoside (R 2 =F in the formula I) of the present invention can be prepared according to the effective synthetic methods in nucleoside and nucleotide chemistry practice. The synthesis of compound 6-3 is provided as an example, as described in Schemes 4-6. D-ribose ( 4-1 ) was first protected. In this case, esters such as acetate and benzoate provide suitable protecting groups, but alternative protecting groups may also be used. Esterification is achieved by reacting D-ribose with an appropriate acid halide or anhydride, optionally in the presence of a solvent such as diethyl ether, dioxane, tetrahydrofuran and methylene chloride. Such transformations are well known in the chemical literature and examples can be found in Greene, TW, Wuts, PGM, "ProtectiVe Groups in Organic Synthesis," John Wiley & Sons, Inc., 3rd Ed., 1999.
流程图4Flowchart 4
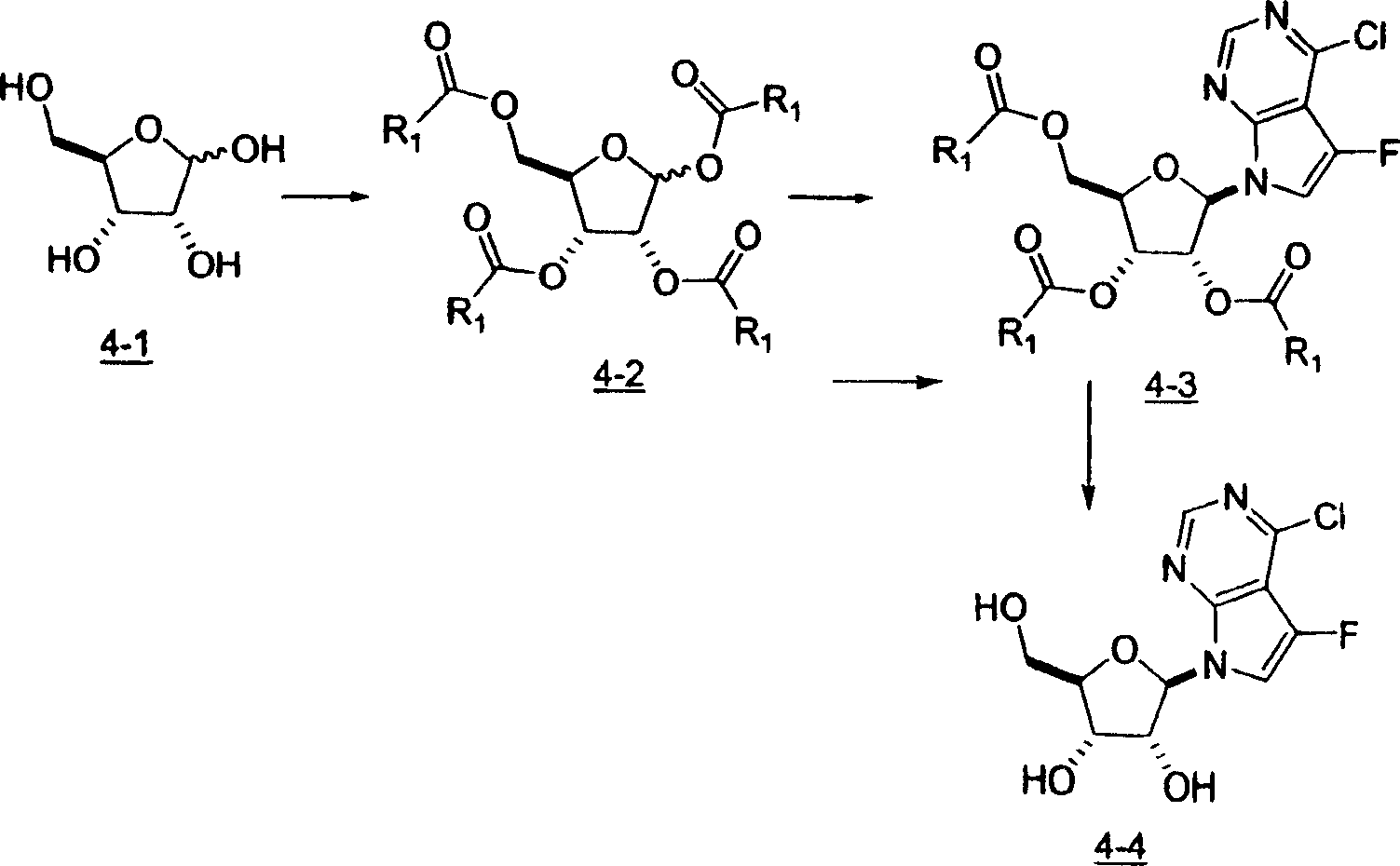
中间体 4-3可以使用很多方法进行制造。在流程图4中使用Vorbruggen反应(″Synthesis of Nucleosides″,H.Vorbruggen和C.Ruh-Pohlenz,在Organic Reactions,vol.55,pp 1-630,2000中)连接nucleobase5-氟-4-氯-7H-吡咯并[2,3-d]嘧啶( 1-4)形成被保护的核苷4-3。然后通过任何的适当的方法,例如酸或碱催化水解和酯交换(例如使用在甲醇中的甲醇钠)除去酯保护基以提供 4-4。Intermediate 4-3 can be made using a number of methods. In Scheme 4, the Vorbruggen reaction ("Synthesis of Nucleosides", H.Vorbruggen and C.Ruh-Pohlenz, in Organic Reactions, vol.55, pp 1-630, 2000) was used to link the nucleobase 5-fluoro-4-chloro- 7H-Pyrrolo[2,3-d]pyrimidine ( 1-4 ) forms protected nucleoside 4-3. The ester protecting group is then removed by any suitable method, such as acid or base catalyzed hydrolysis and transesterification (eg using sodium methoxide in methanol) to provide 4-4 .
接下来如流程图5所述引入C-2′甲基。为了能够正交操控(orthogonal manipulation)C-2′羟基,在5′和3′位的羟基首先被保护。这可以通过很多方法实现,流程图5对其中之一进行了描述。前述参考文献,Greene,T.W.,Wuts,P.G.M.,″Protective Groups in OrganicSynthesis,″,John Wiley & Sons,,Inc.,第3版,1999包括很多实例;特别适合的是四异丙基二硅恶烷基亚基(tetraisopropyldisiloxanylidene)环醚 5-1。The C-2' methyl group is then introduced as described in Scheme 5. To enable orthogonal manipulation of the C-2' hydroxyl group, the hydroxyl groups at the 5' and 3' positions were first protected. This can be accomplished in a number of ways, one of which is depicted in Flowchart 5. The aforementioned reference, Greene, TW, Wuts, PGM, "Protective Groups in Organic Synthesis," John Wiley & Sons, Inc., 3rd Ed., 1999, contains many examples; especially suitable is tetraisopropyldisiloxane Subunit (tetraisopropyldisiloxanylidene) cyclic ether 5-1 .
流程图5Flowchart 5
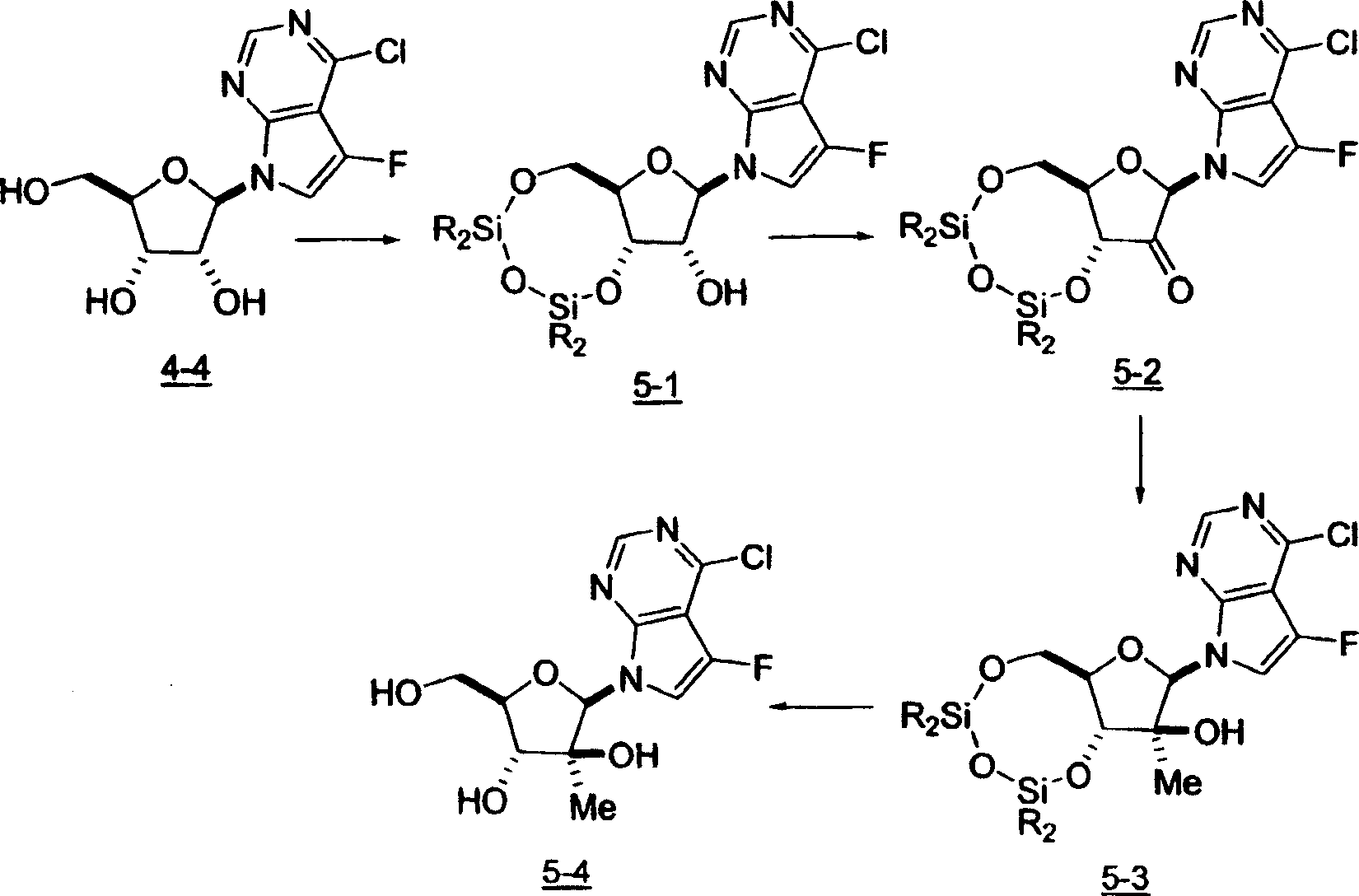
然后通过使用适宜的氧化方法,例如,Swern或Moffatt氧化或应用Dess-Martin过碘烷(periodinane)将剩余游离羟基氧化成酮 5-2。这样的方法的实例可以在相关化学文献,例如,在作者为Richard C.Larock,VCH Publishers1989年出版的“Comprehensive Organic Transfor-mations”中找到。然后使 5-2与适宜的有机金属的试剂,例如,甲基锂和卤化甲基镁反应。这样的反应优选在低温在适当的溶剂,例如四氢呋喃和二乙醚中进行。在这种情况下,由阻碍更少的面引入甲基,这种立体控制主要产生一种异构体。这样的立体控制的实例可以在相关化学文献,例如,在作者为Ernest L.Eliel和Samuel H.Wilen由Wiley-IntersciencePublications在1994年出版的“Stereochemistry of Organic Compounds”中找到。然后如流程图6所述在C-2′位引入氟基。The remaining free hydroxyl group is then oxidized to the ketone 5-2 by using a suitable oxidation method, eg, Swern or Moffatt oxidation or using Dess-Martin periodinane. Examples of such methods can be found in the relevant chemical literature, eg, in "Comprehensive Organic Transformations" by Richard C. Larock, VCH Publishers, 1989. 5-2 is then reacted with a suitable organometallic reagent, eg methyllithium and methylmagnesium halide. Such reactions are preferably carried out at low temperatures in suitable solvents such as tetrahydrofuran and diethyl ether. In this case, the methyl group is introduced from the less hindered face, and this stereocontrol produces mainly one isomer. Examples of such stereocontrol can be found in the relevant chemical literature, eg, in "Stereochemistry of Organic Compounds" by Ernest L. Eliel and Samuel H. Wilen, Wiley-Interscience Publications, 1994. A fluorine group is then introduced at the C-2' position as described in Scheme 6.
流程图6Flowchart 6
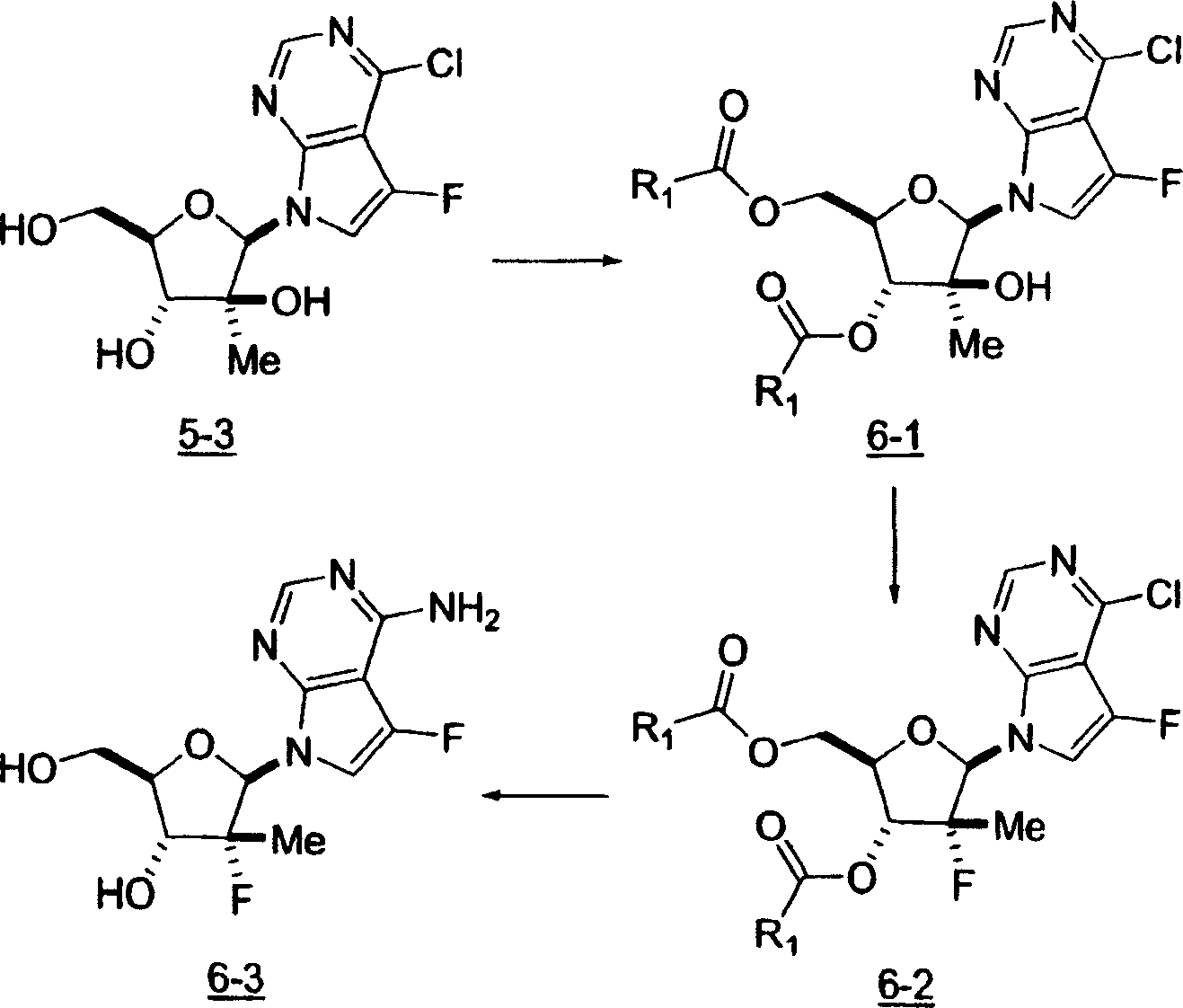
如上所述可选择性地保护在3′和5′位存在的羟基。使用低级烷基或简单芳香酯,例如乙酸酯和苯甲酸酯是有利的。如此选择反应条件以使空间位阻的C-2′羟基保持不受影响。任选在溶剂,例如芳烃,四氢呋喃,氯仿存在的情况下在低温,室温或升温下通过使用三氟化二乙氨基硫(DAST)或其他的适宜的氟化试剂来实现引入氟。在美国专利公布第2005/0009737号(2005年1月13日出版)中描述了这样的转化的实例。然后通过水解除去酯保护基,随后用氨置换在nucleobase中存在的氯原子得到需要的核苷。但是在一个釜中通过在适当的溶剂,例如甲醇的氨在室温或升温下进行最后两步操作是有利的,需要的话,可使用高压。The hydroxyl groups present at the 3' and 5' positions can be optionally protected as described above. It is advantageous to use lower alkyl or simple aromatic esters such as acetates and benzoates. The reaction conditions are chosen such that the sterically hindered C-2' hydroxyl group remains unaffected. The introduction of fluorine is accomplished by using diethylaminosulfur trifluoride (DAST) or other suitable fluorinating reagents optionally in the presence of solvents such as aromatic hydrocarbons, tetrahydrofuran, chloroform at low temperature, room temperature or elevated temperature. Examples of such transformations are described in US Patent Publication No. 2005/0009737 (published January 13, 2005). The ester protecting group is then removed by hydrolysis followed by replacement of the chlorine atom present in the nucleobase with ammonia to yield the desired nucleoside. However, it is advantageous to carry out the last two steps in one kettle by ammonia in a suitable solvent, eg methanol, at room temperature or elevated temperature, using high pressure if necessary.
实施例3Example 3

4-氨基-5-氟-7-(2-氟-2-C-甲基-β-D-呋喃核糖基)-7H-吡咯并[2,3-d]嘧啶4-Amino-5-fluoro-7-(2-fluoro-2-C-methyl-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine
步骤A:Step A:

使用对甲苯酰氯(22mL,165mmol)逐滴处理在无水DMF(80mL)中的D-核糖(5.00g,33mmol),三乙胺(46mL,330mmol)和DMAP(810mg,6.6mmol)的溶液并在室温继续搅拌3小时。通过倾倒在300g冰上使反应淬灭。在冰融化后二氯甲烷萃取粗制产物(3×100mL),用无水硫酸镁干燥,过滤并真空除去溶剂。由丙酮粉碎剩余的油状残留物。将溶剂倾析,将固体残留物由异丙醇(200mL)重结晶。在下一步中使用该产物。A solution of D-ribose (5.00 g, 33 mmol), triethylamine (46 mL, 330 mmol) and DMAP (810 mg, 6.6 mmol) in anhydrous DMF (80 mL) was treated dropwise with p-toluoyl chloride (22 mL, 165 mmol) and Stirring was continued for 3 hours at room temperature. The reaction was quenched by pouring onto 300 g of ice. The crude product (3 x 100 mL) was extracted with dichloromethane after the ice had melted, dried over anhydrous magnesium sulfate, filtered and the solvent was removed in vacuo. The remaining oily residue was triturated with acetone. The solvent was decanted and the solid residue was recrystallized from isopropanol (200 mL). This product was used in the next step.
步骤B:Step B:

使用如H.Vorbruggen和U.Niedballa在 J.Ore.Chem.,39:3654-3660(1974)所述方法合成该化合物。按A.B.Eldrup等人,在 J.Med. Chem.,47:5284(2004)中所述方法制备在该转化中使用的4-氯-5-氟-7H-吡咯并[2,3-d]嘧啶( 1-4)。This compound was synthesized using the method described by H. Vorbruggen and U. Niedballa in J. Ore. Chem ., 39:3654-3660 (1974). The 4-chloro-5-fluoro-7H-pyrrolo[2,3-d]pyrimidine used in this transformation was prepared as described by AB Eldrup et al., in J.Med.Chem . , 47:5284 (2004) ( 1-4 ).
步骤C:Step C:

由步骤B的产物按K.L.Smith等人,在 Bioorg.Med.Chem.Lett,14:3517-3520(2004)中所述方法合成该化合物。This compound was synthesized from the product of step B as described in KL Smith et al., Bioorg. Med. Chem. Lett , 14:3517-3520 (2004).
步骤D:Step D:
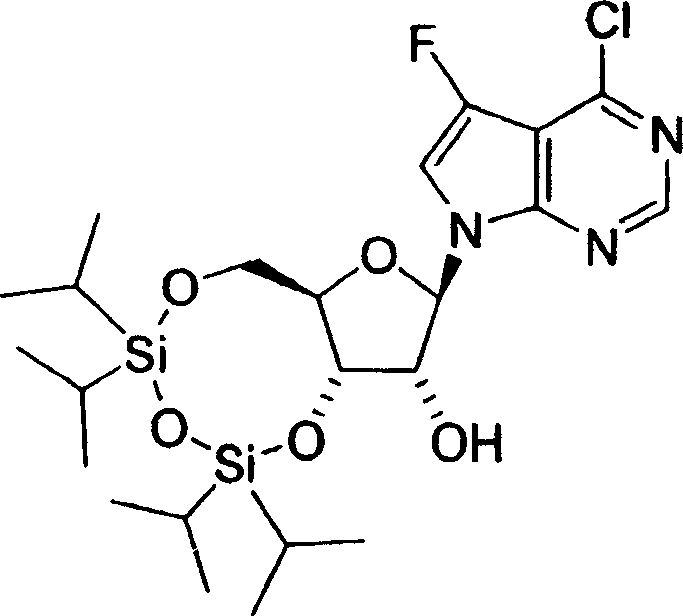
由步骤C的产物按G.Gaubert等人,在 Tetrahedron Lett,45:5629-5632(2004)中所述方法合成该化合物。The compound was synthesized from the product of step C according to the method described in G. Gaubert et al., Tetrahedron Lett , 45:5629-5632 (2004).
步骤E:Step E:

由步骤D的产物按V.L.Moore等人,在 Biochemistry,41:14066-14075(2002)中所述方法合成该化合物。This compound was synthesized from the product of step D according to the method described in VL Moore et al., Biochemistry , 41:14066-14075 (2002).
步骤F:Step F:

由步骤E的产物按V.L.Moore等人,在 Biochemistry,4:14066-14075(2002)中所述方法合成该化合物。This compound was synthesized from the product of step E as described in VL Moore et al., Biochemistry , 4: 14066-14075 (2002).
步骤G:Step G:

由步骤F的产物使用由M.Gallo等人,在 Tetrahedron.57:5707-5713(2001).中公布的方法合成该化合物。This compound was synthesized from the product of step F using the method published by M. Gallo et al., in Tetrahedron . 57:5707-5713 (2001).
步骤H:Step H:
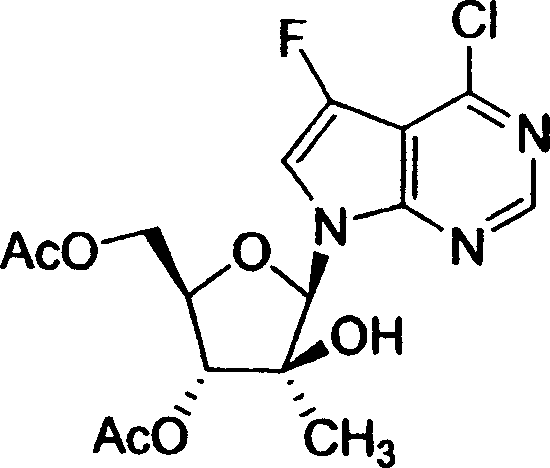
由步骤G的产物使用由M.Akira等人,在 Chem.Pharm.Bull.,35:3967-3970(1987)。中公布的方法合成该二酯。The product from step G was used by M. Akira et al., in Chem. Pharm. Bull ., 35:3967-3970 (1987). The diester was synthesized by the method published in.
步骤I:Step I:
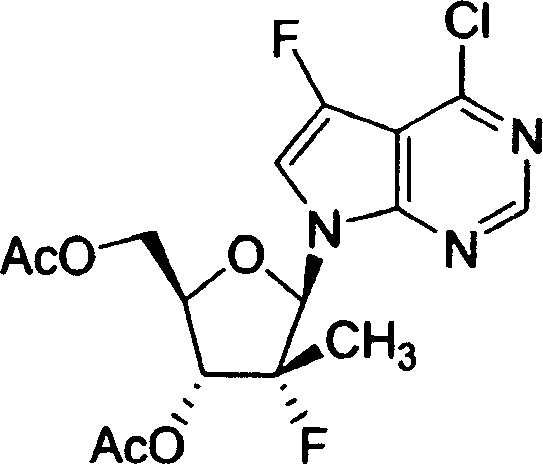
按照美国专利公布第2005/0009737号所述方法使用DAST处理步骤H的产物来制备该化合物。This compound was prepared using DAST treatment of the product of Step H as described in US Patent Publication No. 2005/0009737.
步骤J:Step J:

通过按美国专利第6,777,395号实施例62,步骤F的条件使用甲醇氨处理步骤I的产物来合成实施例3,将其内容完整引入作为参考。Example 3 was synthesized by treating the product of step I with methanolic ammonia according to the conditions of US Pat. No. 6,777,395, Example 62, Step F, which is incorporated by reference in its entirety.
实施例4Example 4
核苷5′-三磷酸酯nucleoside 5′-triphosphate
按照Chem.Rev.100:2047(2000)中所述的通用步骤制备本发明的核苷5′-三磷酸酯。The nucleoside 5'-triphosphates of the invention were prepared following the general procedure described in Chem. Rev. 100:2047 (2000).
实施例5Example 5
核苷5′-三磷酸酯衍生物的纯化和纯度分析Purification and Purity Analysis of Nucleoside 5′-triphosphate Derivatives
将三磷酸酯过用具有pH8的50mM Tris缓冲系统的30×100mmMono Q柱(Pharmacia)进行阴离子交换(AX)色谱提纯。洗脱梯度通常在两柱体积内为40mM NaCl至0.8M NaCl,6.5mL/分钟。收集阴离子交换色谱得到的适当的级分,通过使用Luna C18 250×21mm柱(Phenomenex),流速为10mL/分钟的反相(RP)色谱除去盐分。洗脱梯度通常为1%-95%甲醇,14分钟,5mM乙酸三乙铵(TEAA)的恒定浓度。Triphosphate was purified by anion exchange (AX) chromatography over a 30×100 mm Mono Q column (Pharmacia) with a 50 mM Tris buffer system at pH 8. The elution gradient is typically 40mM NaCl to 0.8M NaCl in two column volumes at 6.5mL/min. Appropriate fractions from anion exchange chromatography were collected and desalted by reverse phase (RP) chromatography using a Luna C18 250 x 21 mm column (Phenomenex) at a flow rate of 10 mL/min. The elution gradient was typically 1% to 95% methanol over 14 minutes with a constant concentration of 5 mM triethylammonium acetate (TEAA).
在Hewlett-Packard(Palo Alto,CA)MSD 1100上用联机HPLC质谱测定纯化三磷酸酯的质谱。RP HPLC用Phenomenex Luna(C18(2)),150×2mm,加30×2mm保护柱,3-μm粒度。在负离子化模型中实施乙腈的20mM TEAA(乙酸三乙铵)pH7溶液的0-50%线性梯度(15分钟)。用氮气和气动喷雾器产生电喷射。以150-900的分子量范围为样本。用HP Chemstation分析程序包测定分子量。Mass spectra of the purified triphosphates were determined by on-line HPLC mass spectrometry on a Hewlett-Packard (Palo Alto, CA) MSD 1100. RP HPLC uses Phenomenex Luna (C18(2)), 150×2mm, plus 30×2mm guard column, 3-μm particle size. A 0-50% linear gradient (15 minutes) of acetonitrile in 20 mM TEAA (triethylammonium acetate) pH 7 was implemented in negative ionization mode. Electrospray was generated with nitrogen and a pneumatic nebulizer. Take the molecular weight range of 150-900 as the sample. Molecular weights were determined using the HP Chemstation analysis package.
纯化三磷酸酯的纯度通过分析用RP和AX HPLC测定,具有Phenomonex Luna或Jupiter柱(250×4.6mm),5μm粒度通常用在100mM TEAA,pH7中的15分钟内2-70%乙腈梯度运转。AX HPLC在1.6×5mm Mono Q柱(Pharmacia)上进行。以50mM Tris,pH8的恒定浓度,0-0.4M NaCl梯度洗脱三磷酸酯。三磷酸酯的纯度一般>80%。Purity of purified triphosphates was determined by analytical RP and AX HPLC with Phenomonex Luna or Jupiter columns (250 x 4.6 mm), 5 μm particle size typically run with a 2-70% acetonitrile gradient in 100 mM TEAA, pH 7, over 15 minutes. AX HPLC was performed on a 1.6 x 5mm Mono Q column (Pharmacia). Triphosphate was eluted with a gradient of 0-0.4M NaCl at a constant concentration of 50mM Tris at pH 8. The purity of the triphosphate is generally >80%.
生物学试验biological test
下面描述用于测定抑制HCV NS5B聚合酶和HCV复制的试验。The assays used to determine inhibition of HCV NS5B polymerase and HCV replication are described below.
在下面的试验中测定本发明化合物作为HCV NS5B RNA依赖性RNA聚合酶(RdRp)的抑制剂的有效性。The effectiveness of the compounds of the invention as inhibitors of HCV NS5B RNA-dependent RNA polymerase (RdRp) was determined in the following assay.
A.抑制HCV NS5B聚合酶试验:A. Inhibition of HCV NS5B polymerase test:
本试验用于测定本发明的氟化吡咯并[2,3-d]嘧啶核苷三磷酸酯抑制丙型肝炎病毒(HCV)的RNA依赖性RNA聚合酶(NS5B)在异数(heteromeroc)RNA模板上的酶活性的能力。This test is used for determining that fluorinated pyrrolo [2,3-d] pyrimidine nucleoside triphosphate of the present invention inhibits the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus (HCV) in different numbers (heteromeroc) RNA Capacity for enzymatic activity on the template.
步骤:step:
试验缓冲条件:(50μL-总量/反应物)Test buffer conditions: (50μL-total amount/reactant)
20mM Tris,pH7.520mM Tris, pH7.5
50μM EDTA50 μM EDTA
5mM DTT5mM DTT
2mM MgCl2 2mM MgCl2
80mM KCl80mM KCl
0.4U/μL RNAsin(Promega,储备液为40单位/μL)0.4U/μL RNAsin (Promega, stock solution is 40 units/μL)
0.75μgt500(一个用T7 runoff转录制成的500-nt RNA,具有来自丙型肝炎基因组的NS2/3区域的序列)0.75 μgt500 (a 500-nt RNA made by T7 runoff transcription with sequences from the NS2/3 region of the hepatitis C genome)
1.6μg纯化丙型肝炎NS5B(由21个C-末端截短的氨基酸形成)1.6 μg purified hepatitis C NS5B (formed from 21 C-terminal truncated amino acids)
1μM A,C,U,GTP(核苷三磷酸酯混合物)1 μM A, C, U, GTP (mixture of nucleoside triphosphates)
[α-32P]-GTP或[α-33P]-GTP[α- 32P ]-GTP or [α- 33P ]-GTP
在高达100μM最终浓度的各种浓度下测试核苷三磷酸酯。Nucleoside triphosphates were tested at various concentrations up to a final concentration of 100 μM.
制备包括酶和模板t500的适当体积的反应缓冲液。将本发明的核苷三磷酸酯吸移到96孔板的孔中。制备包括放射性标记GTP的核苷三磷酸酯(NTP’s)混合物,并将其吸移到96孔板的孔中。加入酶-模板反应溶液引发反应,并使反应在室温下进行1-2小时。Prepare an appropriate volume of reaction buffer including enzyme and template T500. The nucleoside triphosphates of the invention were pipetted into wells of a 96-well plate. A mixture of nucleoside triphosphates (NTP's) including radiolabeled GTP was prepared and pipetted into wells of a 96-well plate. The reaction was initiated by adding the enzyme-template reaction solution and allowed to proceed at room temperature for 1-2 hours.
加入20μL 0.5M EDTA,pH8.0淬灭反应。包括空白试验,空白试验中将淬灭溶液在加入反应缓冲液之前加入到NTPs中。The reaction was quenched by adding 20 μL of 0.5M EDTA, pH 8.0. A blank experiment was included in which the quenching solution was added to the NTPs prior to the addition of the reaction buffer.
将50μL淬灭后的反应物点滴到DE8l滤片(Whatman)上,并使其干燥30分钟。用0.3M甲酸铵,pH8(每次洗涤用150mL,直到1mL洗液中的cpm小于100,通常洗6次)洗涤滤片。在闪烁计数器中在5mL闪烁液中对滤片计数。50 [mu]L of the quenched reaction was spotted onto a DE81 filter disc (Whatman) and allowed to dry for 30 minutes. Wash the filter disc with 0.3M ammonium formate, pH 8 (150 mL for each wash, until the cpm in 1 mL of the wash solution is less than 100, usually 6 washes). Filters were counted in 5 mL of scintillation fluid in a scintillation counter.
通过下述公式计算抑制百分比:抑制%=[1-(测试反应中的cpm-空白反应中的cpm)/(对照反应中的cpm-空白反应中的cpm)]×100。Percent inhibition was calculated by the following formula: % inhibition = [1 - (cpm in test reaction - cpm in blank reaction)/(cpm in control reaction - cpm in blank reaction)] x 100.
在HCV NS5B聚合酶试验中测试的代表性的化合物显示出小于50微摩尔的IC50值。Representative compounds tested in the HCV NS5B polymerase assay showed IC50 values of less than 50 micromolar.
B.抑制HCV RNA复制试验:B. Inhibition of HCV RNA replication test:
还评价本发明化合物在含有亚基因组HCV复制子的培养肝癌(HuH-7)细胞中对丙型肝炎病毒RNA复制的影响。试验的细节如下所述。本复制子试验在V.Lohmann,F.Komer,J-O.Koch,U.Herian,L.Theilmann和R.Bartenschlager,″Replication of a Sub-genomicHepatitis C Virus RNAs in a Hepatoma Cell Line,″ Science 28 5:110(1999)中所述内容的基础上进行了修改。Compounds of the invention were also evaluated for their effect on hepatitis C virus RNA replication in cultured hepatocarcinoma (HuH-7) cells containing a subgenomic HCV replicon. The details of the test are described below. This replicon test is in V.Lohmann, F.Komer, JO.Koch, U.Herian, L.Theilmann and R.Bartenschlager, "Replication of a Sub-genomic Hepatitis C Virus RNAs in a Hepatoma Cell Line," Science 28 5: 110 (1999), modified from what was stated in 110 (1999).
实验方案:Experimental program:
本试验是原位核糖核酸酶保护、基于亲和闪烁的平板测定(SPA)。将10,000-40,000个细胞加入到96孔cytostar板(Amersham)中的100-200μL含有0.8mg/mL G418的培养基中。在0-18小时,以在1%DMSO中的高达100μM的各种浓度将化合物加入到细胞中,然后培养24-96小时。将细胞固定(20分钟,10%福尔马林),透化(20分钟,0.25%TritonX-100/PBS)并与单链33P RNA探针杂交(过夜,50℃),所述单链33PRNA与RNA病毒基因组中所含的(+)链NS5B(或其它基因)互补。将细胞洗涤,用RNAse处理,洗涤,加热至65℃,并在Top-Count中计数。读取每分钟计数(cpm)的减少作为对复制的抑制。The assay is an in situ ribonuclease protected, affinity scintillation based plate assay (SPA). 10,000-40,000 cells were added to 100-200 μL of medium containing 0.8 mg/mL G418 in 96-well cytostar plates (Amersham). Compounds were added to cells at various concentrations up to 100 [mu]M in 1% DMSO at 0-18 hours and then incubated for 24-96 hours. Cells were fixed (20 min, 10% formalin), permeabilized (20 min, 0.25% TritonX-100/PBS) and hybridized (overnight, 50°C) with a single-stranded 33 P RNA probe, which 33 PRNA is complementary to the (+) strand NS5B (or other genes) contained in the RNA virus genome. Cells were washed, treated with RNAse, washed, heated to 65°C, and counted in a Top-Count. A decrease in counts per minute (cpm) was read as inhibition of replication.
被选择来含有亚基因组复制子的人HuH-7肝癌细胞带有细胞质RNA,该细胞质RNA由HCV 5’非翻译区(NTR)、新霉素选择性标记、EMCV IRES(内部核糖体进入位点)和HCV非结构蛋白NS3-NS5B以及跟随其后的3’NTR构成。Human HuH-7 hepatoma cells selected to contain a subgenomic replicon harbor cytoplasmic RNA consisting of HCV 5' untranslated region (NTR), neomycin selectable marker, EMCV IRES (internal ribosome entry site ) and HCV nonstructural protein NS3-NS5B followed by 3'NTR.
在本复制试验中测试的代表性的化合物显示出小于100微摩尔的EC50值。Representative compounds tested in this replicate assay showed EC50 values of less than 100 micromolar.
C.细胞内代谢试验:C. Intracellular metabolism test:
还评价了本发明化合物进入人肝癌细胞株以及在细胞内被转化为相应的核苷5′-单-,二-和三磷酸酯的能力The compounds of the present invention were also evaluated for their ability to enter human liver cancer cell lines and be converted into the corresponding nucleoside 5'-mono-, di- and triphosphates in the cells
将两种细胞株,HuH-7和HBIlOA用于本发明化合物的细胞内代谢研究。HuH-7是人肝癌细胞株,HBIlOA表示由存在HCV双顺反子复制子的HuH-7细胞获得的克隆株。以每60-毫米培养皿1.5×106细胞将HuH-7细胞涂布到含有10%胎牛血清的完全Dulbecco′s改良Eagle′s培养基,将HBIlOA细胞涂布到含有G418(0.8mg/mL)的同样的培养基中,以使在加入化合物的时候有80%的细胞融合。将氚标记化合物以2μM在细胞培养基中孵育3或23小时。收集细胞,用磷酸盐-缓冲盐水洗涤,计数。然后将细胞用70%甲醇,20m氚标记化合物EDTA,20mm EGTA提取并离心。将溶胞产物干燥,使用离子对反相(C-18)HPLC在与在线β-RAM闪烁检测器连接的Waters Millenium系统上(IN/USSystems)测定放射性标记的核苷酸。HPLC流动相(a)具有2mm氢氧化四丁铵的10mm磷酸钾和(b)包含具有2mm氢氧化四丁铵的10mm磷酸钾的50%甲醇。通过比较相对于标准的保留时间来进行峰鉴别。活性表示为在106HuH-7或HBIlOA细胞中检测到的皮摩尔的核苷酸。Two cell lines, HuH-7 and HBI10A, were used for intracellular metabolism studies of compounds of the present invention. HuH-7 is a human liver cancer cell line, and HBI10A represents a clone obtained from HuH-7 cells in which the HCV bicistronic replicon exists. Spread HuH-7 cells with 1.5×10 6 cells per 60-mm culture dish to the complete Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, and spread HBI10A cells to the medium containing G418 (0.8mg/ mL) such that 80% of the cells were confluent at the time of compound addition. Tritiated compounds were incubated at 2 μM in cell culture medium for 3 or 23 hours. Cells were harvested, washed with phosphate-buffered saline, and counted. Cells were then extracted with 70% methanol, 20 mM tritiated compound EDTA, 20 mM EGTA and centrifuged. Lysates were dried and radiolabeled nucleotides were assayed using ion-pair reversed-phase (C-18) HPLC on a Waters Millenium system (IN/US Systems) coupled to an on-line β-RAM scintillation detector. HPLC mobile phase (a) 10 mm potassium phosphate with 2 mm tetrabutylammonium hydroxide and (b) 50% methanol containing 10 mm potassium phosphate with 2 mm tetrabutylammonium hydroxide. Peak identification was performed by comparing retention times relative to standards. Activity is expressed as picomoles of nucleotides detected in 106 HuH-7 or HBI10A cells.
在以下所述反过滤(counterscreens)中还评价本发明氟化吡咯并[2,3-d]嘧啶核苷化合物的细胞毒性和抗病毒特异性。The cytotoxicity and antiviral specificity of the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the invention were also evaluated in counterscreens as described below.
C.反过滤:C. Anti-filtering:
在下面的试验中测试了本发明氟化吡咯并[2,3-d]嘧啶核苷化合物对人DNA聚合酶的抑制能力。The inhibitory ability of the fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the present invention on human DNA polymerase was tested in the following assay.
a.对人DNA聚合酶α和β的抑制:a. Inhibition of human DNA polymerase α and β:
反应条件:Reaction conditions:
50μL反应体积50μL reaction volume
反应缓冲组分:Reaction Buffer Components:
20mM Tris-HCl,pH7.520mM Tris-HCl, pH7.5
200μg/mL牛血清清蛋白200μg/mL bovine serum albumin
100mM KCl100mM KCl
2mM β-巯基乙醇2mM β-mercaptoethanol
10mM MgCl2 10mM MgCl2
1.6μM dA,dG,dC,dTTP1.6 μM dA, dG, dC, dTTP
α-33P-dATPα- 33 P-dATP
酶和模板:Enzymes and templates:
0.05mg/mL缺口鱼精子DNA模板0.05mg/mL Gap Fish Sperm DNA Template
0.01U/μL DNA聚合酶α或β0.01U/μL DNA polymerase α or β
缺口鱼精子DNA模板的制备:Preparation of gapped fish sperm DNA template:
将5μL 1M MgCl2加入到500μL活化鱼精子DNA(USB 70076)中;加热至37℃,并加入30μL 65U/μL的外切核酸酶III(GibcoBRL18013-011);Add 5 μL 1M MgCl 2 to 500 μL activated fish sperm DNA (USB 70076); heat to 37°C, and add 30 μL 65U/μL exonuclease III (GibcoBRL18013-011);
在37℃孵育5分钟;Incubate at 37°C for 5 minutes;
通过加热至65℃10分钟终止反应;The reaction was terminated by heating to 65 °C for 10 minutes;
将50-100μL等分试样装到Bio-spin 6色谱柱(Bio-Rad 732-6002)中,该色谱柱已用20mM Tris-HCl,pH7.5进行平衡;Load a 50-100 μL aliquot onto a Bio-spin 6 column (Bio-Rad 732-6002) equilibrated with 20 mM Tris-HCl, pH 7.5;
通过以1,000Xg离心4分钟进行洗脱;Elution was performed by centrifugation at 1,000×g for 4 minutes;
收集洗出液,并在260nm测定吸光度,以确定浓度。The eluate was collected and the absorbance was measured at 260nm to determine the concentration.
将DNA模板稀释至20mM Tris-HCl,pH7.5的适当体积,将酶稀释至20mM Tris-HCl的适当体积,含有2mM β-巯基乙醇和100mMKCl。将模板和酶吸移至微量离心管或96孔板中。还分别用酶稀释缓冲液和测试化合物溶剂制备了排除酶的空白反应物和排除测试化合物的对照反应物。用具有上述组分的反应缓冲液引发反应。将反应物在37℃孵育1小时。加入20μL 0.5M EDTA淬灭反应。将50μL淬灭后的反应物点滴到Whatman DE81滤片上并风干。用150mL 0.3M pH8的甲酸铵反复洗涤滤片,直到1mL洗液<100cpm。将滤片用150mL无水乙醇洗涤两次,用150mL无水乙醚洗涤一次,干燥并在5mL闪烁液中计数。Dilute the DNA template to an appropriate volume of 20mM Tris-HCl, pH 7.5, and dilute the enzyme to an appropriate volume of 20mM Tris-HCl containing 2mM β-mercaptoethanol and 100mM KCl. Pipette template and enzyme into microcentrifuge tubes or 96-well plates. Blank reactions excluding enzymes and control reactions excluding test compounds were also prepared in enzyme dilution buffer and test compound solvent, respectively. The reaction was initiated with a reaction buffer having the above composition. Reactions were incubated at 37°C for 1 hour. Add 20 μL of 0.5M EDTA to quench the reaction. 50 μL of the quenched reaction was spotted onto Whatman DE81 filter discs and air dried. Wash the filter piece repeatedly with 150mL of 0.3M ammonium formate pH8 until 1mL of the washing solution is <100cpm. The filter was washed twice with 150 mL of absolute ethanol and once with 150 mL of absolute ether, dried and counted in 5 mL of scintillation fluid.
通过下述公式计算抑制的百分比:抑制%=[1-(测试反应中的cpm-空白反应中的cpm)/(对照反应中的cpm-空白反应中的cpm)]×100。Percent inhibition was calculated by the following formula: % inhibition = [1 - (cpm in test reaction - cpm in blank reaction)/(cpm in control reaction - cpm in blank reaction)] x 100.
b.对人DNA聚合酶γ的抑制:b. Inhibition of human DNA polymerase gamma:
在包含下述成分的反应中测试了对人DNA聚合酶γ的抑制能力:0.5ng/μL酶;10μm dATP,dGTP,dCTP和TTP;2μCi/反应[a-33P]-dATP和0.4μg/μL活化鱼精子DNA(购自US Biochemical),在含有20mM Tris pH8,2mM β-巯基乙醇、50mM KCl、10mM MgCl2和0.1μg/μL BSA的缓冲液中。使反应在37℃进行1小时,然后通过加入0.5M EDTA至142mM的最终浓度淬灭反应。通过阴离子交换过滤结合和闪烁计数对所形成的产物定量。在高达50μM的浓度测试化合物。Inhibition of human DNA polymerase gamma was tested in reactions containing the following components: 0.5 ng/μL enzyme; 10 μM dATP, dGTP, dCTP and TTP; 2 μCi/reaction [a- 33 P]-dATP and 0.4 μg/ μL of activated fish sperm DNA (purchased from US Biochemical) in a buffer containing 20 mM Tris pH 8, 2 mM β-mercaptoethanol, 50 mM KCl, 10 mM MgCl 2 and 0.1 μg/μL BSA. The reaction was allowed to proceed for 1 hour at 37°C and then quenched by adding 0.5M EDTA to a final concentration of 142mM. The product formed was quantified by anion exchange filter binding and scintillation counting. Compounds were tested at concentrations up to 50 μM.
通过下述公式计算抑制的百分比:抑制%=[1-(测试反应中的cpm-空白反应中的cpm)/(对照反应中的cpm-空白反应中的cpm)]×100。Percent inhibition was calculated by the following formula: % inhibition = [1 - (cpm in test reaction - cpm in blank reaction)/(cpm in control reaction - cpm in blank reaction)] x 100.
在下述试验中,测定了本发明氟化吡咯并[2,3-d]嘧啶核苷化合物抑制HIV感染和HIV扩散的能力。The ability of fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the invention to inhibit HIV infection and HIV spread was determined in the assay described below.
c.HIV感染性试验:c. HIV infectivity test:
用表达CXCR4和CCR5的HeLa Magi细胞的变体进行本试验,根据低背景β-半乳糖苷酶(β-gal)表达进行选择。使细胞感染48小时,用化学发光底物(Galactolight Plus,Tropix,Bedford,MA)对得自整合(integrated)HIV-1 LTR启动子的β-gal产物进行定量。在开始于100μm的两倍连续稀释液中滴定(在重复试验中)抑制物;计算相对于对照感染的各浓度下的抑制百分比。The assay was performed with a variant of HeLa Magi cells expressing CXCR4 and CCR5, selected for low background β-galactosidase (β-gal) expression. Cells were infected for 48 hours and β-gal product from the integrated HIV-1 LTR promoter was quantified using a chemiluminescent substrate (Galactolight Plus, Tropix, Bedford, MA). Inhibitors were titrated (in replicates) in two-fold serial dilutions starting at 100 [mu]M; percent inhibition at each concentration was calculated relative to control infection.
d.对HIV扩散的抑制:d. Inhibition of HIV spread:
通过美国专利第5,413,999号(1995年5月9日)和J.P.Vacca等的Proc.Natl.Acad.Sci.,91:4096-4100(1994)中所述的方法测试本发明化合物抑制人类免疫缺陷性病毒(HIV)扩散的能力,将所述文献的内容完整引入作为参考。Compounds of the invention were tested for inhibition of human immunodeficiency virus by the methods described in U.S. Patent No. 5,413,999 (May 9, 1995) and JPVacca et al. , Proc. (HIV) diffusion capacity, the contents of which are incorporated by reference in their entirety.
在下列试验中所述的基于MTS细胞的试验中,筛选了本发明氟化吡咯并[2,3-d]嘧啶核苷化合物对含有亚基因组HCV复制子的培养肝癌(HuH-7)细胞的细胞毒性。在H.Nakabayashi等的 Cancer Res.,42:3858(1982)中描述了HuH-7细胞株。Fluorinated pyrrolo[2,3-d]pyrimidine nucleoside compounds of the invention were screened for their activity on cultured hepatoma (HuH-7) cells containing a subgenomic HCV replicon in the MTS cell-based assay described in the following assay. Cytotoxicity. The HuH-7 cell line is described in H. Nakabayashi et al., Cancer Res ., 42:3858 (1982).
e.细胞毒性试验:e. Cytotoxicity test:
在适当的培养基中制备细胞培养物,浓度为大约1.5×105个细胞/mL用于3天孵育中的悬浮培养,浓度为5.0×104个细胞/mL用于3天孵育中的贴壁培养。将99μL细胞培养物转移至96孔组织培养处理板的孔中,加入1μL在DMSO中的100倍最终浓度的测试化合物。将平板在37℃和5%CO2条件下孵育一段特定时间。培育期之后,向各孔中加入20μL CellTiter 96 Aqueous One Solution Cell Proliferation试验试剂(MTS)(Promega),再将平板在37℃和5%CO2条件下培育最长为3小时的一段时间。振荡平板以使各孔混合,用平板读数器读取490nm下的吸光度。用临添加MTS试剂前的已知细胞数制作悬浮培养细胞的标准曲线。代谢活性细胞将MTS分解为甲月替,甲月替在490nm吸收。将化合物存在下490nm的吸光度与未添加任何化合物的细胞的吸光度相比。Prepare cell cultures in appropriate media at a concentration of approximately 1.5 x 10 cells/mL for suspension cultures in 3-day incubations and 5.0 x 10 cells/mL for stickers in 3-day incubations wall culture. 99 μL of cell culture was transferred to wells of a 96-well tissue culture treated plate and 1 μL of test compound at 100-fold final concentration in DMSO was added. Incubate the plate at 37 °C and 5% CO2 for a specific period of time. After the incubation period, 20 μL of CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS) (Promega) was added to each well and the plates were incubated at 37° C. and 5% CO 2 for a period of up to 3 hours. The plate was shaken to mix the wells and the absorbance at 490 nm was read with a plate reader. A standard curve of suspension cultured cells was prepared using the known number of cells immediately before the addition of MTS reagent. Metabolically active cells break down MTS to formazan, which absorbs at 490nm. Absorbance at 490 nm in the presence of compounds was compared to the absorbance of cells without any added compound.
参考:Cory,A.H等,“Use of an aqueous soluble tetrazolium/formazanassay for cell growth assays inculture,” Cancer Commun.3:207(1991)。Reference: Cory, AH et al., "Use of an aqueous soluble tetrazolium/formazana assay for cell growth assays inculture," Cancer Commun . 3: 207 (1991).
将下面的试验用于测定本发明化合物对其它RNA依赖性RNA病毒的活性。The following assay was used to determine the activity of compounds of the invention against other RNA-dependent RNA viruses.
a.测定化合物对抗鼻病毒的体外抗病毒活性(致细胞病变效应抑制试验):a. Determination of the in vitro antiviral activity of the compound against rhinovirus (cytopathic effect inhibition test):
试验条件描述在Sidwell and Huffman,″Use of disposablemicrotissue culture plates for antiviral and interferon inductionstudies,″ Appl.Microbiol.22:797-801(1971)的文章中。Experimental conditions are described in the article by Sidwell and Huffman, "Use of disposable microtissue culture plates for antiviral and interferon induction studies," Appl. Microbiol . 22:797-801 (1971).
病毒:Virus:
如Sidwell and Huffman参考中所述,使用2型鼻病毒(RV-2)HGP毒株与KB细胞和培养基(0.1%NaHCO3,无抗生素)。由ATCC获得的病毒取自具有轻微的急性发热上呼吸道疾病的成年男性的咽喉棉棒。9型鼻病毒(RV-9)211毒株和14型鼻病毒(RV-14)Tow毒株也由Rockville,MD的American Type Culture Collection(ATCC)获得。RV-9取自人咽喉洗液,RV-14取自具有上呼吸道疾病的年轻成人的咽喉棉棒。将这两种病毒与人子宫颈上皮样癌细胞HeLa Ohio-1细胞(Dr.FredHayden,UniV.of VA)一起使用。用具有5%胎牛血清(FBS)和0.1%NaHCO3的MEM(Eagle’s最低基础培养基)作为生长培养基。全部三种病毒类型的抗病毒测试培养基都是具有5%FBS、0.1%NaHCO3、50μg庆大霉素/mL和10mM MgCl2的MEM。Rhinovirus type 2 (RV-2) HGP strain was used with KB cells and medium (0.1% NaHCO3 , no antibiotics) as described in the Sidwell and Huffman reference. Viruses obtained by ATCC were obtained from throat swabs of adult males with mild acute febrile upper respiratory illness. Rhinovirus type 9 (RV-9) 211 strain and rhinovirus type 14 (RV-14) Tow strain were also obtained from the American Type Culture Collection (ATCC) in Rockville, MD. RV-9 was obtained from human throat washes, and RV-14 was obtained from throat swabs of young adults with upper respiratory disease. These two viruses were used together with human cervical epithelioid carcinoma cells HeLa Ohio-1 cells (Dr. Fred Hayden, UniV. of VA). MEM (Eagle's minimal basal medium) with 5% fetal bovine serum (FBS) and 0.1% NaHCO3 was used as growth medium. The antiviral test medium for all three virus types was MEM with 5% FBS, 0.1% NaHCO 3 , 50 μg gentamicin/mL and 10 mM MgCl 2 .
2000μg/mL是用于测试本发明化合物的最高浓度。在加入受试化合物之后大约5分钟,将病毒加入到试验板中。同时制作适当的对照试样。将试验板在潮湿空气、5%CO2、37℃条件下进行培养。通过显微镜观察对照细胞的形态变化来监测细胞毒性。病毒CPE数据和毒性对照数据的回归分析给出了ED50(50%有效剂量)和CC50(50%细胞毒性浓度)。通过式子:SI=CC50÷ED50计算选择性指数(SI)。2000 μg/mL was the highest concentration used to test the compounds of the invention. Virus was added to the assay plate approximately 5 minutes after the addition of the test compound. At the same time, appropriate control samples were prepared. The test plate was cultured under the conditions of humid air, 5% CO 2 , and 37°C. Cytotoxicity was monitored by microscopic observation of morphological changes in control cells. Regression analysis of virus CPE data and toxicity control data gave ED50 (50% effective dose) and CC50 (50% cytotoxic concentration). The selectivity index (SI) is calculated by the formula: SI=CC50÷ED50.
b.测定化合物对抗登革热 班齐和黄热病的体外抗病毒活性(CPE抑制试验):b. measure the in vitro antiviral activity (CPE inhibition test) of compound against dengue fever Banzi and yellow fever:
试验详细内容在上述Sidwell and Huffman参考文献中给出。Experimental details are given in the aforementioned Sidwell and Huffman reference.
病毒:Virus:
从疾病控制中心获取登革热2型病毒New Guinea毒株。用两株非洲绿猴肾细胞培养病毒(Vero)并进行抗病毒测试(MA-104)。从ATCC获得自被感染的鼠脑制取的黄热病病毒17D毒株和分离自南非发热男孩的血清的班齐病毒H336毒株。这两种病毒和试验都使用Vero细胞。Obtain the New Guinea strain of dengue type 2 virus from the Centers for Disease Control. The virus (Vero) was cultured with two lines of African green monkey kidney cells and tested for anti-virus (MA-104). The yellow fever virus 17D strain prepared from infected mouse brains and the Banzi virus H336 strain isolated from the sera of febrile boys in South Africa were obtained from the ATCC. Both viruses and assays use Vero cells.
细胞和培养基:Cells and media:
在具有5%FBS和0.1%NaHCO3、没有抗生素的199培养基中使用MA-104细胞(Bio Whittaker,Inc.,Walkersville,MD)和Vero细胞(ATCC)。MA-104 cells (Bio Whittaker, Inc., Walkersville, MD) and Vero cells (ATCC) were used in 199 medium with 5% FBS and 0.1% NaHCO3 , without antibiotics.
用于登革热、黄热病和班齐病毒的测试培养基是MEM、2%FBS、0.18%NaHCO3和50μg庆大霉素/mL。The test medium for dengue, yellow fever and Banzi virus was MEM, 2% FBS, 0.18% NaHCO 3 and 50 μg gentamicin/mL.
本发明化合物的抗病毒测试按照Sidwell and Huffman参考文献中所述进行,类似于上述鼻病毒抗病毒测试。对于各种病毒,在5-6天后获得适当的致细胞病变效应(CPE)记录。Antiviral testing of compounds of the invention was performed as described in the Sidwell and Huffman reference, similar to the rhinovirus antiviral testing described above. Appropriate cytopathic effect (CPE) records were obtained after 5-6 days for each virus.
c.测定化合物对西尼罗病毒的体外抗病毒活性(CPE抑制试验):c. measure the in vitro antiviral activity (CPE inhibition test) of compound to West Nile virus:
试验详细内容在上述引用的Sidwell and Huffman参考文献中给出。从疾病控制中心获得取自鸦脑的西尼罗病毒、New York分离物。使Vero细胞生长并如上使用。测试培养基为MEM、1%FBS、0.1%NaHCO3和50μg庆大霉素/mL。Experimental details are given in the Sidwell and Huffman reference cited above. West Nile virus, New York isolates from crow brain were obtained from the Centers for Disease Control. Vero cells were grown and used as above. The test medium was MEM, 1% FBS, 0.1% NaHCO 3 and 50 μg gentamicin/mL.
本发明化合物的抗病毒测试按照Sidwell and Huffman参考文献中所述方法进行,类似于用于鼻病毒活性测试的那些方法。在5-6天后获得适当的致细胞病变效应(CPE)记录。Antiviral testing of the compounds of the present invention was performed according to the methods described in the Sidwell and Huffman reference, similar to those used for rhinovirus activity testing. Appropriate cytopathic effect (CPE) records were obtained after 5-6 days.
d.测定化合物对抗鼻病毒、黄热病、登革热、班齐和西尼罗病毒的体外抗病毒活性(中心红摄取试验):d. Determination of in vitro antiviral activity of compounds against rhinovirus, yellow fever, dengue fever, Banzi and West Nile virus (center red uptake test):
如上进行CPE抑制测试后,再使用“Microtiter Assay forInterferon:Microspectrophotometric Quantitation of CytopathicEffect,” Appl.Environ.Microbiol.31:35-38(1976)中所述的细胞病变检测方法。用EL309型小平板读出器(Bio-Tek Instruments Inc.)对试验平板读数。如上计算出ED50值和CD50值。After the CPE inhibition assay was performed as above, the cytopathic assay described in "Microtiter Assay for Interferon: Microspectrophotometric Quantitation of Cytopathic Effect," Appl. Environ. Microbiol . 31:35-38 (1976) was used. Assay plates were read with a model EL309 miniplate reader (Bio-Tek Instruments Inc.). ED50 and CD50 values were calculated as above.
药物制剂的实施例Examples of Pharmaceutical Formulations
作为本发明化合物的口服组合物的具体实施方案,用50mg实施例1或实施例2的化合物与充分粉碎后的乳糖一起配制制剂,以提供580-590mg的总量,将其装进0号硬质明胶胶囊中。As a specific embodiment of the oral composition of the compound of the present invention, 50 mg of the compound of Example 1 or Example 2 is used to prepare the preparation together with fully pulverized lactose to provide a total amount of 580-590 mg, which is packed into a No. 0 hard in gelatin capsules.
虽然对本发明进行了描述并参照其具体实施方案进行了举例说明,但本领域技术人员都将意识到在不背离本发明的精神和范围的前提下,可以对其进行各种变化、改进和替换。例如,根据需要治疗HCV严重传染的人的反应的各种变化的推断,前文中列出的优选剂量以外的有效剂量也是适用的。同样,所观察到的药理学反应也可根据并依赖所选择的具体活性化合物或者是否存在药学载体以及制剂类型和所用施用方式而变化,根据本发明的目的和实践可预料这些预期的变化或结果差异。因此,本发明仅由所附权利要求的范围限定,这些权利要求适当地保持较宽的范围。Although the invention has been described and illustrated with reference to specific embodiments thereof, those skilled in the art will appreciate that various changes, modifications and substitutions can be made thereto without departing from the spirit and scope of the invention. . For example, effective doses other than the preferred doses listed above may be suitable, as inferred from the variable response of humans in need of treatment for severe HCV infection. Likewise, the observed pharmacological responses may also vary according to and depend upon the particular active compound chosen or the presence or absence of a pharmaceutical carrier as well as the type of formulation and mode of administration used, and such expected changes or results may be expected in light of the objectives and practice of the present invention difference. Accordingly, the invention is to be limited only by the scope of the appended claims, which claims are afforded the broadest.
Claims (7)

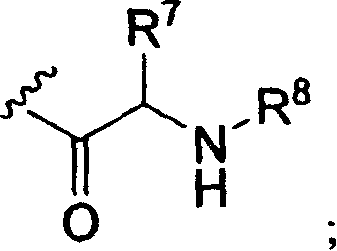
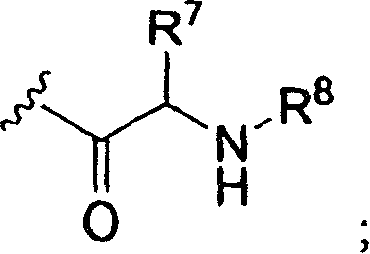




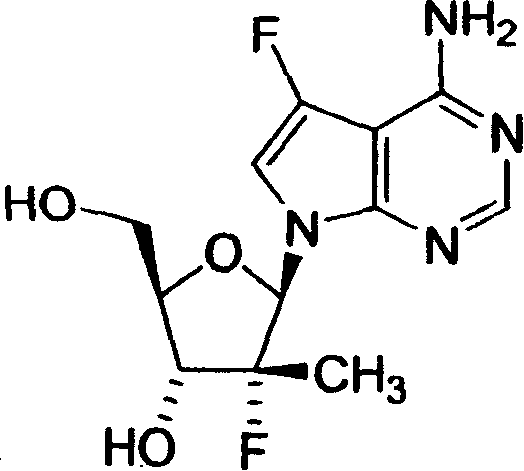
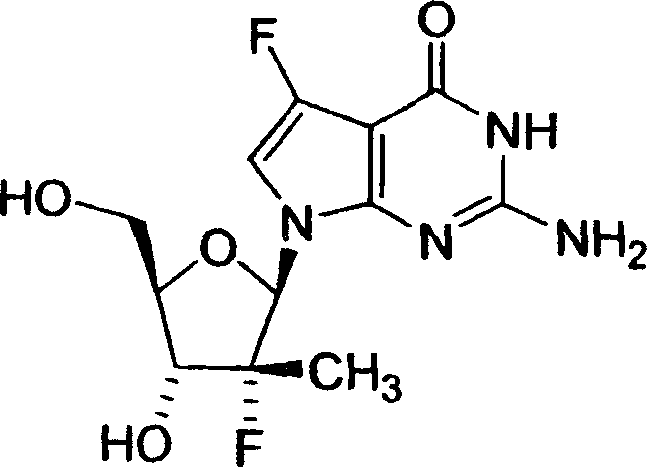
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62074304P | 2004-10-21 | 2004-10-21 | |
| US60/620,743 | 2004-10-21 | ||
| US60/651,366 | 2005-02-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101043893A true CN101043893A (en) | 2007-09-26 |
Family
ID=38808923
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 200580036083 Pending CN101043893A (en) | 2004-10-21 | 2005-10-17 | Fluorinated pyrrolo[2,3-d]pyrimidine nucleosides for the treatment of rna-dependent rna viral infection |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101043893A (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102596958A (en) * | 2009-09-21 | 2012-07-18 | 吉里德科学公司 | 2'-fluoro CARBA-nucleoside analogues for antiviral therapy |
| CN103108876A (en) * | 2010-09-20 | 2013-05-15 | 吉里德科学公司 | 2 -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US8853171B2 (en) | 2008-04-23 | 2014-10-07 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| CN101977923B (en) * | 2008-01-18 | 2014-10-08 | 捷克有机化学和生物化学研究院 | Novel cytostatic 7-deazapurine nucleosides |
| US9090642B2 (en) | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US9724360B2 (en) | 2014-10-29 | 2017-08-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10065958B2 (en) | 2010-07-22 | 2018-09-04 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US10251904B2 (en) | 2015-09-16 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US10675296B2 (en) | 2017-07-11 | 2020-06-09 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| US10988498B2 (en) | 2009-09-21 | 2021-04-27 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
-
2005
- 2005-10-17 CN CN 200580036083 patent/CN101043893A/en active Pending
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101977923B (en) * | 2008-01-18 | 2014-10-08 | 捷克有机化学和生物化学研究院 | Novel cytostatic 7-deazapurine nucleosides |
| USRE46762E1 (en) | 2008-04-23 | 2018-03-27 | Gilead Sciences, Inc | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8853171B2 (en) | 2008-04-23 | 2014-10-07 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US10988498B2 (en) | 2009-09-21 | 2021-04-27 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| CN102596958A (en) * | 2009-09-21 | 2012-07-18 | 吉里德科学公司 | 2'-fluoro CARBA-nucleoside analogues for antiviral therapy |
| US9090642B2 (en) | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US10065958B2 (en) | 2010-07-22 | 2018-09-04 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US10696679B2 (en) | 2010-07-22 | 2020-06-30 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
| CN103108876A (en) * | 2010-09-20 | 2013-05-15 | 吉里德科学公司 | 2 -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US9949994B2 (en) | 2014-10-29 | 2018-04-24 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US9724360B2 (en) | 2014-10-29 | 2017-08-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10251898B2 (en) | 2014-10-29 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10695357B2 (en) | 2014-10-29 | 2020-06-30 | Gilead Sciences, Inc. | Methods for treating filoviridae virus infections |
| US11344565B2 (en) | 2014-10-29 | 2022-05-31 | Gilead Sciences, Inc. | Methods for the preparation of ribosides |
| US10251904B2 (en) | 2015-09-16 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| US10675296B2 (en) | 2017-07-11 | 2020-06-09 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1267446C (en) | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerases | |
| CN1816558A (en) | Modified Fluorinated Nucleoside Analogs | |
| JP5055564B2 (en) | C-purine nucleoside analogues as inhibitors of RNA-dependent RNA viral polymerase | |
| KR101682494B1 (en) | Nucleoside cyclicphosphates | |
| CN101044151A (en) | Antiviral 4'-azido-nucleosides | |
| EP2195326B1 (en) | Nucleoside derivatives as inhibitors of viral polymerases | |
| CN1852915A (en) | Purine nucleoside analogs for the treatment of diseases caused by viruses of the Flaviviridae family including hepatitis C | |
| JP2008517912A (en) | Fluorinated pyrrolo [2,3-d] pyrimidine nucleosides for the treatment of RNA-dependent RNA viral infections | |
| JP2005533777A (en) | Carbocyclic nucleoside analogs as RNA antiviral agents | |
| CN1527836A (en) | Antiviral agents for the treatment of Flaviviridae viral infections | |
| CN1653077A (en) | Nucleoside derivatives for treating hepatitis C virus infection | |
| US20060264389A1 (en) | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase | |
| CN1871250A (en) | Nucleoside compounds for the treatment of viral infections | |
| CN1863813A (en) | Novel tricyclic nucleosides or nucleotides as therapeutic agents | |
| CN1678326A (en) | 2'-C-methyl-3'-O-L-valine ester ribofuranocytidine for the treatment of flavivirus infection | |
| JP2006512288A (en) | Nucleoside derivatives as RNA-dependent RNA viral polymerase inhibitors | |
| CN1466591A (en) | nucleoside derivatives | |
| JP2005530843A (en) | Nucleoside derivatives as RNA-dependent RNA viral polymerase inhibitors | |
| CN1777432A (en) | Antiviral nucleoside analogues and methods for treating viral infections, especially HIV infections | |
| CN1646141A (en) | Modified nucleoside compounds for the treatment of viral infections and abnormal cell proliferation | |
| JP2004534830A (en) | Nucleoside compounds in HCV | |
| CN1646534A (en) | Modified fluorinated nucleoside analogues | |
| CN101043893A (en) | Fluorinated pyrrolo[2,3-d]pyrimidine nucleosides for the treatment of rna-dependent rna viral infection | |
| CN1023561C (en) | Process for preparing cryptoclindamycin/adenosine derivatives | |
| CN1968605A (en) | C-purine nucleoside analogs as inhibitors of RNA-dependent RNA viral polymerase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Open date: 20070926 |