Hémoglobine
Pour les articles homonymes, voir Hémoglobine (homonymie), Hb et Hgb.
L'hémoglobine, couramment symbolisée Hb, parfois Hgb, est un pigment respiratoire (de la famille moléculaire des métalloprotéines, ici contenant du fer) présent essentiellement dans le sang des vertébrés, au sein de leurs globules rouges, ainsi que dans les tissus de certains invertébrés. Elle a pour fonction de transporter l'oxygène O2 depuis l'appareil respiratoire (poumons, branchies) vers le reste de l'organisme. La quantité d'hémoglobine est un paramètre mesuré lors d'un hémogramme.
| Hémoglobine | ||
 Représentation d'une hémoglobine A humaine montrant les quatre hèmes en vert, avec le cation de fer en orange (tétramère α2β2, PDB 3HHB[1]) | ||
| Caractéristiques générales | ||
|---|---|---|
| Gène HBA1 – Chaînes α | ||
| Homo sapiens | ||
| Locus | 16p13.3 | |
| Masse moléculaire | 15 258 Da[2] | |
| Nombre de résidus | 142 acides aminés[2] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
| Gène HBA2 – Chaînes α | ||
| Homo sapiens | ||
| Locus | 16p13.3 | |
| Masse moléculaire | 15 258 Da[2] | |
| Nombre de résidus | 142 acides aminés[2] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
| Gène HBB – Chaînes β | ||
| Homo sapiens | ||
| Locus | 11p15.4 | |
| Masse moléculaire | 15 998 Da[2] | |
| Nombre de résidus | 147 acides aminés[2] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
| Gène HBD – Chaînes δ | ||
| Homo sapiens | ||
| Locus | 11p15.4 | |
| Masse moléculaire | 16 055 Da[2] | |
| Nombre de résidus | 147 acides aminés[2] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
| Gène HBG1 – Chaînes γ | ||
| Homo sapiens | ||
| Locus | 11p15.4 | |
| Masse moléculaire | 16 140 Da[2] | |
| Nombre de résidus | 147 acides aminés[2] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
| Gène HBG2 – Chaînes γ | ||
| Homo sapiens | ||
| Locus | 11p15.4 | |
| Masse moléculaire | 16 126 Da[2] | |
| Nombre de résidus | 147 acides aminés[2] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
Rôle
modifierL'hémoglobine libère l'oxygène dans les tissus afin d'y permettre la respiration cellulaire aérobie, laquelle, à travers le métabolisme, fournit l'énergie des processus biologiques essentiels à la vie.
Chez l'humain, l'hémoglobine est une protéine hétéro-tétramérique formée de chaînes peptidiques identiques deux à deux. L'hémoglobine A (HbA) représente environ 95 % des molécules d'hémoglobines chez l'adulte, constituée de deux chaînes α et de deux chaînes β ; il existe également une hémoglobine A2 (HbA2) de formule α2δ2, et une hémoglobine F (HbF, fœtale) de formule α2γ2. Chacune des quatre chaînes est associée à un groupe prosthétique appelé hème et constitué d'un cation de fer complexé avec une porphyrine. L'hémoglobine est donc une hémoprotéine.
Chez les mammifères, l'hémoglobine constitue près de 96 % de la masse de matière sèche des globules rouges, et environ 35 % de leur contenu total en incluant l'eau[3]. Chaque molécule d'hémoglobine peut fixer jusqu'à quatre molécules d'oxygène O2, et l'hémoglobine du sang peut transporter 1,34 mL d'O2 par gramme de protéine[4], ce qui lui permet de transporter 70 fois plus d'oxygène que la quantité d'O2 dissoute dans le sang. L'hémoglobine intervient aussi dans le transport d'autres gaz que l'oxygène. Elle assure notamment le transport d'une partie du dioxyde de carbone CO2 produit par la respiration cellulaire, et transporte également du monoxyde d'azote NO, qui joue un rôle significatif dans la signalisation cellulaire de certains processus physiologiques, et qui est libéré en même temps que l'oxygène après avoir été transporté sur un groupe thiol de l'apoprotéine[5].
La majeure partie de l'hémoglobine se trouve dans les globules rouges, eux-mêmes produits par la moelle osseuse. Cependant, toute l'hémoglobine n'est pas concentrée dans les globules rouges. On en trouve ainsi par exemple dans les neurones dopaminergiques du groupe A9 de la substantia nigra, dans les macrophages, dans les cellules alvéolaires et, au niveau des reins, dans les cellules du mésangium. Dans ces tissus, l'hémoglobine joue un rôle d'antioxydant et de régulateur du métabolisme du fer[6].
L'hémoglobine et diverses molécules apparentées sont également présentes chez un grand nombre d'invertébrés, de champignons et de plantes[7]. Chez ces organismes, l'hémoglobine peut avoir pour fonction de transporter l'oxygène O2, mais peut aussi intervenir comme transporteur et régulateur d'autres espèces chimiques telles que le dioxyde de carbone CO2, le monoxyde d'azote NO, le sulfure d'hydrogène HS et l'anion sulfure S2−. Une variante de l'hémoglobine, appelée léghémoglobine, assure l'élimination de l'oxygène des systèmes anaérobies, par exemple des nodules de Rhizobium chez les fabacées, avant que celui-ci ne les inactive.
Structure et fonctionnement
modifierSous-unités
modifierL'hémoglobine possède une structure quaternaire caractéristique de nombreuses protéines à sous-unités globulaires. La plupart de ses résidus d'acides aminés sont engagés dans des hélices α reliées entre elles par des segments non hélicoïdaux. Les sections hélicoïdales sont stabilisées par des liaisons hydrogène qui confèrent à la protéine sa structure tridimensionnelle caractéristique, appelée repliement globine car on le retrouve également dans d'autres globines à groupe prosthétique héminique telles que la myoglobine[8]. Ce repliement caractéristique présente une cavité dans laquelle est étroitement insérée une molécule d'hème constituant le groupe prosthétique de la protéine. L'hémoglobine contient donc une molécule d'hème par sous-unité.
-
 Représentation générique d'une molécule d'hémoglobine, montrant les quatre sous unités, identiques deux à deux, avec chacune une molécule d'hème insérée dans des cavités à l'intérieur des sous-unités.
Représentation générique d'une molécule d'hémoglobine, montrant les quatre sous unités, identiques deux à deux, avec chacune une molécule d'hème insérée dans des cavités à l'intérieur des sous-unités.

Chez la plupart des vertébrés, la molécule d'hémoglobine est un assemblage de quatre sous-unités globulaires selon un arrangement grossièrement tétraédrique. Ces sous-unités sont maintenues ensemble par des liaisons hydrogène, par des liaisons ioniques et par effet hydrophobe. Chez l'Homme adulte, le type d'hémoglobine le plus courant est l'hémoglobine A, constituée de deux sous-unités α et deux sous-unités β, formées chacune de 141 et 146 résidus d'acides aminés respectivement. Cette structure est symbolisée par α2β2. Ces sous-unités sont structurellement très semblables et ont sensiblement la même taille. Chacune a une masse moléculaire d'environ 16 kDa, soit 64 kDa (64 458 g·mol-1) pour la protéine complète[9]. Chez l'enfant, l'hémoglobine principale est dite hémoglobine F (fœtale), de formule α2γ2, les chaînes γ étant progressivement remplacée par des chaînes β au cours de la croissance.
Hème
modifierL'hème est constitué d'un cation de fer(II) coordonné à quatre atomes d'azote d'une porphyrine, un tétrapyrrole dont la molécule est plane. Ce cation Fe2+ est également lié par covalence au résidu d'histidine F8 de la globine dans laquelle l'hème est inséré ; ce résidu, appelé histidine proximale, est situé sous le plan de l'hème. Fe2+ peut également se lier de manière réversible par une liaison covalente de coordination à une molécule d'oxygène O2 au-dessus du plan de l'hème, à l'opposé de l'histidine proximale, ce qui complète la géométrie de coordination octaédrique à six ligands du cation de fer(II) dans l'oxyhémoglobine ; en l'absence d'oxygène, dans la désoxyhémoglobine, ce sixième site est occupé par une molécule d'eau très faiblement liée.
Le fer ferreux de la désoxyhémoglobine est dans un état haut spin, c'est-à-dire que ses cinq orbitales d sont occupées, essentiellement par des électrons célibataires, d'où un rayon ionique de l'ordre de 92 pm[10], tandis que, dans l'oxyhémoglobine, le fer ferreux est dans un état bas spin, c'est-à-dire que ses orbitales d sont occupées par six électrons appariés qui se limitent aux trois orbitales de plus basse énergie, d'où un rayon ionique de seulement 75 pm. Pour cette raison, l'ion Fe2+ est décalé d'environ 40 pm par rapport au plan de l'hème dans la désoxyhémoglobine, mais de seulement 10 pm dans l'oxyhémoglobine. Cette variation est à la base du basculement entre forme tendue et forme relâché de l'hémoglobine.
-
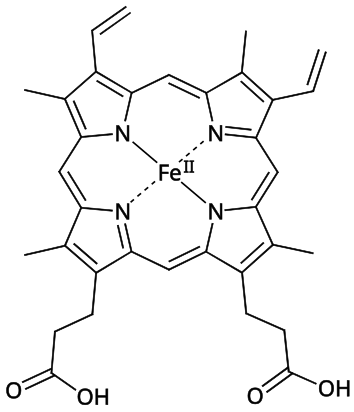 Structure de l'hème b, groupe prosthétique de l'hémoglobine.
Structure de l'hème b, groupe prosthétique de l'hémoglobine. -
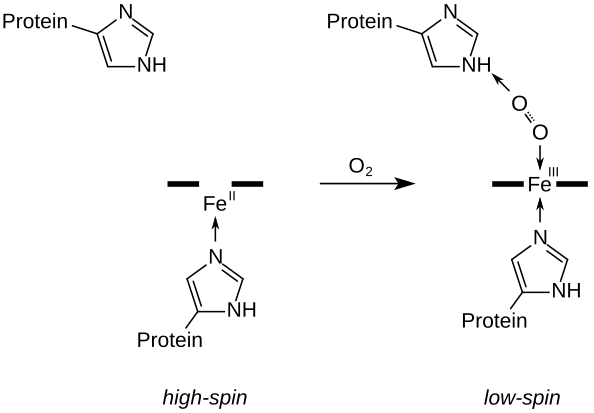 (en) Schéma de principe de la liaison d'une molécule d'oxygène O2 sur l'hème, symbolysé ici par un trait épais. L'ion superoxyde O2•− résultant est lié d'un côté au cation de fer(III) par une liaison covalente de coordination et de l'autre côté à l'histidine distale. Le cation Fe(II) de la désoxyhémoglobine est à l'état haut spin et décalé hors du plan de l'hème vers l'histidine proximale, mais est ramené dans ce plan en passant à l'état bas spin par la liaison à l'oxygène, ce qui déplace l'histidine proximale vers l'hème et favorise le basculement du reste de la protéine de la forme tendue (T) vers la forme relâchée (R).
(en) Schéma de principe de la liaison d'une molécule d'oxygène O2 sur l'hème, symbolysé ici par un trait épais. L'ion superoxyde O2•− résultant est lié d'un côté au cation de fer(III) par une liaison covalente de coordination et de l'autre côté à l'histidine distale. Le cation Fe(II) de la désoxyhémoglobine est à l'état haut spin et décalé hors du plan de l'hème vers l'histidine proximale, mais est ramené dans ce plan en passant à l'état bas spin par la liaison à l'oxygène, ce qui déplace l'histidine proximale vers l'hème et favorise le basculement du reste de la protéine de la forme tendue (T) vers la forme relâchée (R).


Le cation de fer peut être à l'état d'oxydation +2 ou +3 : dans ce dernier cas, on a affaire à de la méthémoglobine, qui se lie à l'oxygène de manière moins réversible que l'hémoglobine, et avec une affinité inférieure. En effet, lorsqu'elle se lie à l'hème ferreux, la molécule d'oxygène O2 tend à être réduite en ion superoxyde O2•− tandis que le cation Fe2+ tend à être oxydé en Fe3+, mécanisme qui s'inverse lors de la libération de l'oxygène ; en revanche, la liaison de l'oxygène à l'hème ferrique est essentiellement irréversible et tend à bloquer la protéine en forme R, ce qui empêche la libération de l'oxygène et inhibe sa fonctionnalité de transporteur d'oxygène. La cytochrome b5 réductase, ou méthémoglobine réductase, est l'enzyme qui assure la réduction de la méthémoglobine en hémoglobine fonctionnelle par réduction du cation Fe3+ en Fe2+, ce qui en fait une enzyme essentielle au maintien des propriétés du sang.
Forme tendue (T) et forme relâchée (R)
modifierL'hémoglobine désoxygénée (désoxyhémoglobine) présente une conformation dite T, ou tendue, tandis que l'hémoglobine oxygénée (oxyhémoglobine) présente une conformation dite R, ou relâchée. La forme T a une faible affinité pour l'oxygène et tend par conséquent à le libérer, tandis que la forme R a une forte affinité pour l'oxygène et tend à le fixer. Plusieurs facteurs favorisent l'une ou l'autre de ces conformations. Ainsi, la forme T est favorisée par un pH faible (acide), une forte concentration en CO2 et un taux élevé en 2,3-bisphosphoglycérate (2,3-BPG), ce qui favorise la libération de l'oxygène lorsque le sang circule à travers les tissus, tandis que la forme R est favorisée par un pH élevé, une faible pression partielle de CO2 et un faible taux de 2,3-BPG, ce qui favorise la fixation de l'oxygène lorsque le sang circule au niveau des alvéoles pulmonaires.
-
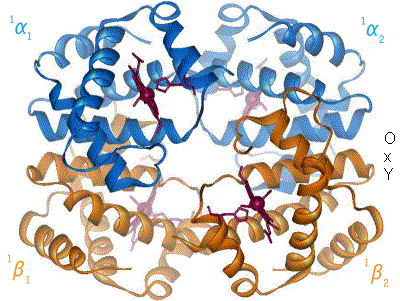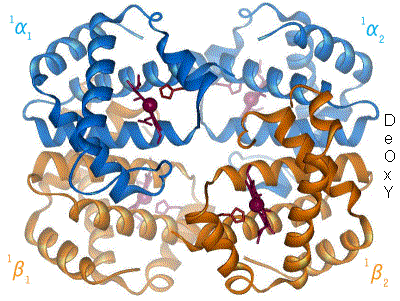 Représentation schématique du basculement de l'hémoglobine entre les formes T (désoxy) et R (oxy). Les déplacements de l'hème et de l'histidine proximale sont bien visibles dans les sous-unités α1 et β2.
Représentation schématique du basculement de l'hémoglobine entre les formes T (désoxy) et R (oxy). Les déplacements de l'hème et de l'histidine proximale sont bien visibles dans les sous-unités α1 et β2. -
 Taux de saturation en O2 de l'hémoglobine en fonction de la pression partielle d'O2 ; parfois appelée courbe de Barcroft, elle est sigmoïde en raison de l'effet coopératif accompagnant la liaison de l'oxygène à l'hémoglobine.
Taux de saturation en O2 de l'hémoglobine en fonction de la pression partielle d'O2 ; parfois appelée courbe de Barcroft, elle est sigmoïde en raison de l'effet coopératif accompagnant la liaison de l'oxygène à l'hémoglobine.


Le basculement entre la forme T et la forme R de l'hémoglobine est un mécanisme dit coopératif, c'est-à-dire allostérique, car la liaison d'une molécule d'oxygène à la forme T induit un changement conformationnel qui se propage partiellement aux sous-unités adjacentes, dont l'affinité pour l'oxygène augmente progressivement au fur et à mesure que d'autres molécules d'oxygène se lient à l'hémoglobine, jusqu'à ce que toute la protéine adopte la conformation R ; à l'inverse, la libération d'une molécule d'oxygène par la forme R induit un changement conformationnel qui se propage partiellement aux sous-unités adjacentes, dont l'affinité pour l'oxygène décroît graduellement au fur et à mesure que l'hémoglobine libère de l'oxygène, jusqu'à ce que toute la protéine adopte la conformation T[11]. C'est la raison pour laquelle la courbe de liaison de l'oxygène à l'hémoglobine en fonction de la pression partielle d'oxygène présente une forme sigmoïde, alors qu'elle serait hyperbolique en l'absence d'allostérie.
Courbe de saturation de l'hémoglobine en oxygène
modifier
On a coutume de tracer le taux de saturation de l'hémoglobine en oxygène O2 représentée en ordonnée en fonction de la pression partielle de l'oxygène O2, donnée en abscisse. Dans cette représentation, la courbe est sigmoïde et tend à glisser vers la gauche lorsque l'affinité de l'hémoglobine pour l'oxygène augmente, et vers la droite lorsqu'elle diminue. La pression partielle d'oxygène à laquelle l'hémoglobine est saturée d'oxygène à 50 % est appelée p50 : plus sa valeur est faible et plus l'affinité de l'hémoglobine pour l'oxygène est élevée. À titre indicatif, la p50 de l'hémoglobine d'un adulte sain vaut typiquement 3,5 kPa, souvent écrite 26,6 mmHg, tandis que celle de la myoglobine vaut typiquement 130 Pa.
Plusieurs facteurs augmentent la p50 et font donc glisser cette courbe vers la droite :
- une baisse du pH, qui devient acide : c'est l'effet Bohr ;
- une augmentation du taux de dioxyde de carbone CO2 : c'est l'effet Haldane ;
- une augmentation du taux de 2,3-bisphosphoglycérate (2,3-BPG) ;
- une élévation de la température, avec cependant un effet relativement faible.
Ces effets sont réversibles, et l'inversion du sens de variation de ces facteurs fait glisser la courbe vers la gauche.
Autres ligands transportés par l'hémoglobine
modifierOutre l'oxygène O2, qui se lie à l'hémoglobine selon un mécanisme dit coopératif, cette protéine transporte également d'autres ligands dont certains sont des inhibiteurs compétitifs, comme le monoxyde de carbone CO, et d'autres sont des ligands allostériques tels que le dioxyde de carbone CO2 et le monoxyde d'azote NO. Le CO2 se lie de manière réversible à des groupes amine de l'apoprotéine pour former de la carbaminohémoglobine, dont on pense qu'elle assure environ 10 % du transport de CO2 chez les mammifères, le reste étant transporté essentiellement sous forme d'ions bicarbonate HCO3−. Le monoxyde d'azote se lie de manière réversible à des groupes thiol de l'apoprotéine pour former un S-nitrosothiol. Il est possible que le transport du monoxyde d'azote intervienne indirectement pour favoriser le transport de l'oxygène par l'hémoglobine en agissant comme vasodilatateur dans les tissus où la pression partielle d'oxygène est faible[12].
Inhibiteurs par compétition avec l'oxygène
modifier


La liaison de l'oxygène à l'hémoglobine est efficacement bloquée par le monoxyde de carbone CO, provenant par exemple de la fumée de cigarettes, de pots d'échappement ou d'une combustion incomplète par une chaudière. Le monoxyde de carbone entre en compétition avec l'oxygène au niveau du site de liaison de ce dernier sur l'hème. L'affinité de l'hémoglobine pour le monoxyde de carbone est environ 230 fois supérieure à celle de l'hémoglobine pour l'oxygène[13],[14],[15], de sorte que de faibles quantités de monoxyde de carbone suffisent pour significativement réduire l'oxygénation de l'hémoglobine lors de l'hématose, et donc la capacité du sang à oxygéner l'organisme. L'hypoxie qui résulte ainsi d'une exposition continue à 0,16 % de CO dans l'air provoque vertige, nausée, céphalée et tachycardie en 20 minutes, et conduit à la mort en moins de deux heures ; un taux de 1,28 % de CO dans l'air provoque une perte de connaissance après seulement deux à trois inspirations, et la mort en moins de trois minutes[16],[17]. Lorsqu'elle se combine au monoxyde de carbone, l'hémoglobine est une protéine appelée carboxyhémoglobine dont la couleur rouge très vif est susceptible de colorer en rose la peau des victimes mortes d'une intoxication au monoxyde de carbone, qui auraient sans cela le teint pâle ou bleui.
De manière semblable, l'hémoglobine présente, sur son site de liaison à l'oxygène, une affinité compétitive pour l'ion cyanure CN−, le monoxyde de soufre SO et l'ion sulfure S2−, comme avec le sulfure d'hydrogène H2S. Ceux-ci se lient au cation de fer de l'hème sans modifier son état d'oxydation, mais ils inhibent cependant la liaison de l'oxygène à l'hème, d'où leur grande toxicité.
Ligands allostériques de l'hémoglobine
modifierLe dioxyde de carbone CO2 se lie plus facilement à la désoxyhémoglobine, ce qui facilite son élimination de l'organisme. C'est ce qu'on appelle l'effet Haldane.
Par ailleurs, le CO2 dissous dans le sang est converti en anion bicarbonate HCO3− par l'anhydrase carbonique, selon la réaction :
Il s'ensuit que le sang riche en CO2 est également plus acide, c'est-à-dire que son pH est abaissé sous l'effet de l'acide carbonique. La liaison de protons H+ et de molécules de CO2 à l'hémoglobine induit un changement conformationnel qui en favorise la forme T, et donc la libération de l'oxygène. Les protons se lient sur différents sites de l'hémoglobine, tandis que le dioxyde de carbone se lie aux groupes amine α pour former de la carbaminohémoglobine. La baisse d'affinité de l'hémoglobine pour l'oxygène en présence de CO2 et de pH acide est appelée effet Bohr.
Les personnes acclimatées aux altitudes élevée présentent un taux sanguin accru de 2,3-bisphosphoglycérate (2,3-BPG). Ce dernier est un effecteur hétéroallostérique qui a pour effet de réduire l'affinité de l'hémoglobine pour l'oxygène en stabilisant la forme T : sous une pression partielle d'oxygène plus faible qu'au niveau de la mer, une baisse d'affinité de l'hémoglobine pour l'oxygène a pour effet d'accroître l'efficacité globale du transport de l'oxygène par l'hémoglobine. On observe plus généralement une augmentation du taux de 2,3-BPG lorsque la pression partielle en oxygène décroît dans les tissus périphériques, par exemple en cas d'hypoxémie, de maladie respiratoire chronique, d'anémie, ou encore d'insuffisance cardiaque. A contrario, le taux de 2,3-BPG décroît en cas de choc septique et d'hypophosphatémie (en).
Biosynthèse et dégradation
modifier

La biosynthèse de l'hémoglobine fait intervenir un ensemble complexe d'étapes. L'hème est issu d'une suite de réactions qui commencent dans les mitochondries et se poursuivent dans le cytosol d'érythrocytes immatures, tandis que l'apoprotéine est produite au niveau de ribosomes du cytosol. La production d'hémoglobine se produit aux premiers stades de l'érythropoïèse, depuis le stade proérythroblaste jusqu'au stade réticulocyte dans la moelle osseuse. C'est à ce niveau que les érythrocytes des mammifères perdent leur noyau, tandis que ce dernier demeure dans les érythrocytes chez les oiseaux et de nombreuses autres espèces. La biosynthèse de l'apoprotéine se poursuit cependant après la perte du noyau car il subsiste de l'ARN messager dans la cellule, qui peut être traduit par les ribosomes du cytosol jusqu'à la mise en fonction de l'érythrocyte dans l'appareil cardiovasculaire[18].
Chez les vertébrés, les érythrocytes parvenus en fin de vie du fait de leur sénescence ou de leur altération sont retirés du sang par phagocytose par des macrophages dans la rate et dans le foie. En cas d'hémolyse dans la circulation sanguine, l'hémoglobine se lie à l'haptoglobine, tandis que l'hème libre est fixé par l'hémopexine, ce qui limite l'effet oxydant. L'hémoglobine incomplètement dégradée ou libérée en trop grande quantité à partir de globules rouges endommagés est susceptible d'obstruer les vaisseaux sanguins, notamment les capillaires des reins, ce qui peut provoquer des néphropathies. L'hémoglobine libérée est éliminée du sang par la protéine CD163, exclusivement exprimée dans les monocytes et les macrophages. L'hémoglobine est dégradée dans ces cellules et le fer de l'hème est recyclé, tandis qu'une molécule de monoxyde de carbone est libérée par molécule d'hème dégradée[19] : la dégradation de l'hème est l'un des rares processus naturels produisant du monoxyde de carbone dans le corps humain et est responsable de la présence de CO dans le sang d'individus respirant même l'air le plus pur. Ce processus forme de la biliverdine, puis de la bilirubine, de couleur jaune. Insoluble, elle est libérée par les macrophages dans le plasma sanguin, où elle se lie à la sérum albumine, qui la transporte jusqu'aux hépatocytes. Ces derniers la solubilisent par conjugaison avec l'acide glucuronique et la sécrètent dans les intestins avec la bile. Les intestins métabolisent la bilirubine en urobilinogène, qui est excrété dans les fèces sous forme de stercobiline ainsi que dans les urines. Lorsque la bilirubine ne peut être excrétée, sa concentration sanguine augmente et elle est éliminée essentiellement par les urines, qui deviennent foncées tandis que les fèces sont décolorées.
Le fer issu de la dégradation de l'hème est stocké dans les ferritines des tissus et véhiculé dans le plasma sanguin par des β-globulines telles que les transferrines.
Génétique
modifierLes molécules d'hémoglobine sont constituées de sous-unités de type globine dont la séquence diffère selon les espèces. Il existe également des variantes d'hémoglobines au sein d'une même espèce, bien que l'une de ces variantes soit généralement largement prépondérante sur les autres. Chez l'Homme, la forme prépondérante d'hémoglobine est appelée hémoglobine A ; elle est codée par les gènes HBA1, HBA2 et HBB situés sur le chromosome 16 pour les deux premiers et sur le chromosome 11 pour le dernier[20].
Évolution
modifierIl est généralement admis que la divergence entre hémoglobine et myoglobine est postérieure à la séparation des gnathostomes (vertébrés à mâchoire) d'avec les lamproies[21]. La myoglobine a été orientée vers le stockage de l'oxygène tandis que l'hémoglobine a été spécialisée dans le transport de l'oxygène[22]. Les sous-unités de la protéine sont codées par des gènes de type globines α et β[20]. Les prédécesseurs de ces gènes sont apparus lors d'une duplication survenue après l'apparition des gnathostomes, il y a environ 450 à 500 millions d'années[21]. L'apparition de gènes α et β a ouvert la voie à la polymérisation de ces globines, et donc à la formation d'une protéine plus grosse constituée de sous-unités distinctes. Le fait que l'hémoglobine soit une protéine polymérique est à la base du mécanisme allostérique qui sous-tend notamment le caractère coopératif de la liaison de l'oxygène à l'hémoglobine[22]. Le gène α a par la suite subi une seconde duplication qui conduit à la formation des gènes HBA1 et HBA2[23]. Ces multiples duplications et divergences ont créé tout un ensemble de gènes apparentés aux globines α et β dont la régulation conduit à les exprimer à différents stades de développement[22].
-
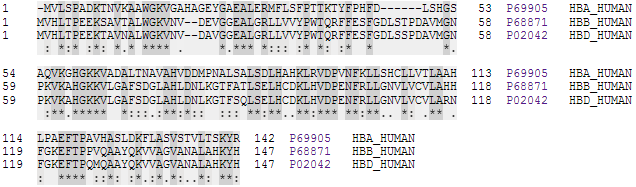 Alignement de séquences de chaînes α, β et δ d'hémoglobine humaine (source UniProt).
Alignement de séquences de chaînes α, β et δ d'hémoglobine humaine (source UniProt).

Mutations
modifierLes mutations sur les gènes de l'hémoglobine peuvent conduire à des variantes d'hémoglobine. La plupart de ces variantes sont fonctionnelles et sans effet sur la santé. Certaines mutations de l'hémoglobine, en revanche, sont susceptibles de provoquer des maladies génétiques appelées hémoglobinopathies. La mieux connue de ces affections est la drépanocytose, qui a été la première maladie humaine dont le mécanisme a été élucidé au niveau moléculaire. Les thalassémies forment un autre groupe d'hémoglobinopathies impliquant une altération de la régulation des gènes des globines constituant l'hémoglobine. Toutes ces maladies ont pour conséquence une anémie.
La modification de la séquence en acides aminés de l'hémoglobine peut être adaptative. On a pu ainsi montrer que l'hémoglobine s'adapte à la baisse de la pression partielle en oxygène observée en haute altitude. L'hémoglobine doit alors être en mesure de se lier à l'oxygène à une pression plus faible, ce qui peut se manifester par une modification de la séquence au niveau des acides aminés intervenant dans l'affinité de l'hémoglobine pour l'oxygène, comme cela a été observé par exemple chez des colibris de la cordillère des Andes : ainsi, chez les espèces du genre Oreotrochilus, chez le colibri de Castelnau, l'inca violifère ou encore le colibri géant, ces mutations réduisent l'affinité de l'hémoglobine pour l'acide phytique, qui joue chez ces oiseaux le même rôle que le 2,3-bisphospoglycérate chez l'homme ; cette baisse d'affinité a pour effet d'accroître l'efficacité du transport de l'oxygène lorsque la pression partielle de ce dernier est réduite[24].
L'adaptation de l'hémoglobine aux altitudes élevées touche également les humains. On ainsi identifié un groupe de femmes tibétaines dont le génotype code une hémoglobine dont l'affinité pour l'oxygène est accrue à faible pression partielle[25]. Ceci a pour effet de réduire la mortalité infantile dans ces conditions extrêmes, ce qui offre un avantage sélectif favorisant les individus porteurs de ces mutations de l'hémoglobine.
Variantes humaines de l'hémoglobine
modifier
Chez l'adulte, la principale variante d'hémoglobine est l'hémoglobine A, ou HbA, de formule α2β2, qui représente plus de 97 % de l'hémoglobine totale d'un adulte sain. L'autre variante d'hémoglobine adulte est l'hémoglobine A2, ou HbA2, de formule α2δ2, qui représente entre 1,5 % et 3,1 % de l'hémoglobine totale d'un adulte sain, mais dont la proportion augmente chez les patients drépanocytaires. Outre ces variantes adultes saines, il existe une douzaine d'autres variantes d'hémoglobine humaine, qu'on rencontre chez l'embryon, le fœtus, ou les patients atteints d'une ou plusieurs formes d'hémoglobinopathies.
Hémoglobines embryonnaires
modifierOn connaît quatre types d'hémoglobine embryonnaire chez l'homme :
- Hb Gower-1, de formule ζ2ε2, est relativement instable et se décompose facilement[26] ;
- Hb Gower-2, de formule α2ε2, plus stable que la variante Gower-1, existe en petites quantités au cours de la vie embryonnaire et fœtale ; elle a été proposée comme traitement par réactivation du gène chez les patients souffrant d'hémoglobinopathies telles qu'une thalassémie β chez lesquels une réactivation de l'hémoglobine F est contre-indiquée pour des raisons de toxicité[26] ;
- Hb Portland-1, de formule ζ2γ2, est présente en faibles quantités au cours de la vie embryonnaire et fœtale[26] ;
- Hb Portland-2, de formule ζ2β2, est encore plus instable que la variante Gower-1 mais a été proposée comme traitement par réactivation du gène chez les patients souffrant de thalassémie α[26],[27].
L'hémoglobine embryonnaire est parfois symbolisée par Hbε, qui ne doit pas être confondue avec l'hémoglobine E, notée HbE, laquelle est une variante pathologique d'HbA présentant une mutation délétère sur les sous-unités β, notées βE (le « E » fait dans ce cas référence au résidu de glutamate modifié par mutation).
Hémoglobine fœtale
modifier
L'hémoglobine fœtale HbF, de formule α2γ2, remplace l'hémoglobine embryonnaire après 10 à 12 semaines de développement. Elle constitue jusqu'à 95 % du sang du nouveau-né, et est progressivement remplacée par l'hémoglobine adulte HbA à partir du sixième mois suivant la naissance ; elle demeure cependant présente à l'état de traces chez l'adulte, où elle n'excède pas 1 % de toutes les variantes d'hémoglobine détectables. Elle demeure produite chez l'enfant lors de certaines thalassémies particulières, parfois jusqu'à l'âge de cinq ans, et une maladie rare, dite syndrome de persistance héréditaire de l'hémoglobine fœtale (en) (HPFH), se traduit par la production d'HbF au lieu d'HbA au-delà de la période normale. Par ailleurs, la production d'HbF peut être réactivée chez l'adulte dans un cadre thérapeutique pour traiter la drépanocytose[28].
L'hémoglobine fœtale est caractérisée par une plus grande affinité pour l'oxygène que l'hémoglobine adulte, ce qui permet au fœtus de s'oxygéner à partir du sang de sa mère : en effet, la p50 d'HbF vaut environ 19 mmHg (2,6 kPa), contre 26,8 mmHg (3,6 kPa) pour HbA. Cette différence d'affinité pour l'oxygène résulte d'une différence d'affinité pour l'un des effecteurs allostériques de l'hémoglobine : le 2,3-bisphosphoglycérate (2,3-BPG), dont la liaison avec l'hémoglobine a pour effet de stabiliser la forme T de cette protéine, laquelle correspond à la désoxyhémoglobine, ce qui réduit l'affinité de l'hémoglobine pour l'oxygène. Dans le cas de l'hémoglobine fœtale, la sous-unité γ présente un résidu de sérine en position 143, là où une sous-unité β d'HbA présente un résidu d'histidine : cette position se trouve au niveau du site de liaison au 2,3-BPG, et le remplacement d'une histidine, dont la chaîne latérale porte une charge électrique positive, par une sérine, électriquement neutre, affaiblit l'interaction du 2,3-BPG avec l'hémoglobine, car le 2,3-BPG est une petite molécule porteuse de cinq charges électriques négatives.
Hémoglobinopathies
modifierLes thalassémies sont caractérisées par l'insuffisance de production d'un des deux types de sous-unités de l'hémoglobine adulte. On distingue ainsi la thalassémie α, plutôt rare, dans laquelle les sous-unités α sont insuffisamment produites, et la thalassémie β, la plus courante, dans laquelle ce sont les sous-unités β qui sont insuffisamment produites. La première conduit à la formation de tétramères de β-globine dits hémoglobine H, de formule β4, qui sont assez instables. Les homozygotes α0 ne survivent généralement pas longtemps après la naissance en raison d'une altération profonde de l'hémoglobine fœtale HbF, donnant dans ces conditions de l'hémoglobine Barts, de formule γ4.
Les principales mutations de l'hémoglobine sont :
- l'hémoglobine C, de formule α2βC2, qui correspond à une substitution E6K, remplacement du résidu de glutamate en position 6 par un résidu de lysine. Le premier possède une chaîne latérale courte et chargée négativement, tandis que le second possède une chaîne latérale longue chargée positivement, ce qui affecte la plasticité générale des érythrocytes. Les hétérozygotes comptent de 28 % à 44 % d'hémoglobine C, ce qui demeure asymptomatique, tandis que les homozygotes comptent 100 % d'hémoglobine C, ce qui provoque une légère anémie hémolytique. Le gène de l'hémoglobine C est surtout présent en Afrique de l'Ouest, où il peut présenter un avantage préventif contre le paludisme à l'instar d'autres hémoglobinopathies, ainsi qu'en Europe du Sud, en Amérique latine et dans les Caraïbes ;
- l'hémoglobine E', de formule α2βE2, qui correspond à une substitution E26K, remplacement du résidu de glutamate en position 26 par un résidu de lysine. Cette mutation touche environ 1 million de personnes dans le monde, essentiellement en Asie du Sud-Est. Elle affecte l'expression de la β-globine en induisant un épissage alternatif de l'ARN messager au niveau des codons 25-27, d'où un déficit de production de β-globine normale, ce qui conduit à une thalassémie β. De plus, les sous-unités βE interagissent moins fortement avec les sous-unités α, ce qui rend les molécules d'hémoglobine E moins stables en présence d'oxydants[29] ;
- l'hémoglobine S, de formule α2βS2, qui correspond à une substitution E6V, remplacement du résidu de glutamate en position 6 par un résidu de valine. Cette substitution, qui place un résidu d'acide aminé hydrophobe à la surface de la protéine, crée une zone d'adhérence qui favorise la précipitation de l'hémoglobine S en longs filaments qui allongent les érythrocytes en leur donnant une forme de faucille (sickle en anglais, d'où le « S » de cette variante), d'où le nom d'anémie falciforme également donnée à cette maladie, également appelée drépanocytose. Le gène S est présent essentiellement en Afrique subsaharienne, au Moyen-Orient et en Inde centrale : on estimait en 2013 à 3,2 millions le nombre d'homozygotes souffrant de drépanocytose, et à 43 millions le nombre d'hétérozygotes ayant le trait drépanocytaire[30]. L'anémie de ces derniers les protège du paludisme, d'où un avantage sélectif qui favorise la prévalence du gène drépanocytaire dans les régions impaludées.
Molécules analogues
modifier
Il existe, chez les plantes et les animaux, une grande diversité de protéines qui se lient à l'oxygène pour en assurer le stockage ou le transport. Les bactéries, les protozoaires et les champignons possèdent tous également des protéines apparentées à l'hémoglobine qui, par leur fonction connue ou prédite, se lient à des ligands gazeux de manière réversible. Outre le transport et la détection de l'oxygène, ces protéines peuvent intervenir pour éliminer l'oxygène des milieux qui sont censés demeurer anaérobies[32], comme c'est par ailleurs le cas de la léghémoglobine.
Dans la mesure où de nombreuses protéines de ce type sont formées de globines et d'hème, elles sont souvent appelées « hémoglobine » même si leur structure générale est très différente de l'hémoglobine des vertébrés. En particulier, la distinction entre myoglobine et hémoglobine est souvent impossible chez les animaux les plus simples en l'absence de muscles chez ces derniers, tandis que le système circulatoire de la plupart des insectes n'intervient pas dans la diffusion de l'oxygène à travers l'organisme. Un certain nombre d'arthropodes (araignées, scorpions, certains crustacés) ont recours à l'hémocyanine, qui est une métalloprotéine dépourvue d'hème mais utilisant des cations de cuivre directement coordonnés à des résidus d'histidine, mais cette protéine n'est pas homologue de l'hémoglobine.
La structure des hémoglobines est très variable selon les espèces considérées. Elle est souvent mono-globine chez les bactéries, les protozoaires, les algues et les plantes, tandis que de nombreux nématodes, mollusques et crustacés possèdent de très grandes protéines contenant un nombre de sous-unités bien plus élevé que chez les vertébrés. Les champignons et les annélides possèdent en particulier des hémoglobines chimériques contenant à la fois des globines et d'autres types de protéines[7]. Ainsi, le ver tubicole géant des monts hydrothermaux contient une variété d'hémoglobine comprenant pas moins de 144 sous-unités globine, associées chacune à un groupe héminique, dont le rôle est de capter l'oxygène O2 et le sulfure d'hydrogène H2S nécessaires aux bactéries qui vivent en symbiose avec lui, ainsi que le dioxyde de carbone CO2 nécessaire à l'anabolisme du ver. Ces structures sont remarquables en ce qu'elles peuvent transporter l'oxygène en présence d'ions sulfure et transporter ces ions eux-mêmes sans être empoisonnées par eux comme le sont les hémoglobines des autres espèces[33],[34].
Parmi les protéines autres que l'hémoglobine capables de se lier à l'oxygène, on peut retenir les molécules suivantes :
- Myoglobine — Présente dans les muscles de la plupart des vertébrés, y compris chez les humains, elle donne à ces tissus une teinte rouge ou gris foncé. Sa structure est très semblable aux sous-unités globine de l'hémoglobine, mais est monomérique, et ne présente donc pas d'effet coopératif en se liant à l'oxygène. Elle intervient plutôt dans le stockage de l'oxygène que dans son transport.
- Hémocyanine — Deuxième transporteur d'oxygène le plus courant dans la nature après l'hémoglobine, on la trouve chez de nombreux arthropodes et mollusques. Elle utilise un groupe prosthétique constitué de cuivre et non de fer héminique, et présente une couleur bleue lorsqu'elle est oxygénée.
- Hémérythrine — Certains invertébrés marins et quelques espèces d'annélides utilisent cette protéine à fer non héminique pour transporter l'oxygène. Elle présente une couleur rose ou violette lorsqu'elle est oxygénée, et est claire lorsqu'elle n'est pas oxygénée.
- Chlorocruorine — Présente chez de nombreux annélides, elle est très semblable à l'érythrocruorine mais son groupe héminique présente une structure sensiblement différente. Elle est de couleur rouge lorsqu'elle est oxygénée, et verte lorsqu'elle est désoxygénée (d'où son nom).
- Érythrocruorine — Présente chez de nombreux annélides, y compris les vers de terre, il s'agit d'une très grosse protéine pouvant contenir plus d'une centaine de sous-unités protéiques et d'unités héminiques, l'ensemble ayant une masse moléculaire pouvant atteindre 3 600 kDa.
- Léghémoglobine — Présente dans les gousses, telles que la luzerne cultivée et le soja, elle a pour fonction de protéger les bactéries fixant l'azote de l'oxygène, afin de permettre à la nitrogénase de réduire l'azote, ce qu'elle ne peut faire en présence d'oxygène.
Utilisation clinique
modifierEn médecine, plusieurs termes se rapportent à l'hémoglobine :
- Le taux d'hémoglobine est exprimé en g/100 mL. Les valeurs normales du taux d'hémoglobine dépendent du sexe et de l'âge du sujet. Un taux d'hémoglobine inférieur à la norme définit une anémie. Les valeurs de références sont plus élevées chez les hommes que chez les femmes. Une étude remet en question les valeurs de références de l'hémoglobine, avançant qu'avoir des valeurs de référence différentes concernant l'hémoglobine pour les hommes et les femmes n'est pas justifié[35].
- La saturation SaO2 définie en % est calculée par la quantité d'oxyhémoglobine divisé par la quantité totale d'hémoglobine du sang. La saturation SaO2 peut être mesurée sur du sang veineux ou du sang artériel. La saturation en oxygène du sang est un des paramètres d'un examen appelé gaz du sang. La valeur de la saturation est considérée comme dangereuse si elle est inférieure à 90 % pour du sang artériel. La valeur normale est d'environ 96-100 % pour des conditions atmosphériques normales. À cette valeur, on parle de capacité en O2 du sang.
- La cyanose est un signe clinique. Il s'agit de la coloration bleutée des téguments. Elle apparaît lorsque la concentration d'hémoglobine réduite dépasse les 5 g/100 ml de sang capillaire. Elle peut être masquée par une anémie.
Maladies génétiques de l'hémoglobine
modifierComme de nombreuses protéines, les chaines d'hémoglobine présentent diverses mutations qui n'ont le plus souvent aucune incidence clinique. Plus de 500 hémoglobines anormales ont été répertoriées[36]. Certaines mutations (Hb Köln, Indianapolis, etc.) entraînent une instabilité du tétramère précipitant en corps de Heinz, ou une méthémoglobinémie (hémoglobines M).
Parfois cette mutation entraîne une affinité anormale pour l'oxygène, soit, telle l'Hb Hope, une diminution d'affinité avec une P50 élevée donnant une anémie bien tolérée et une cyanose au repos, l'effort et l'altitude étant mal supportés, soit, telle l'Hb Chesapeake, Malmö, ou Olympia, une augmentation d'affinité avec une P50 diminuée et une polyglobulie compensatrice entraînant des manifestations cliniques à partir d'un certain âge.
D'autres peuvent être responsables d'une hémolyse chronique, HbS (par mutation de glutamine en valine ce qui va provoquer la polymérisation d'Hb), HbC, ou aggraver à l'état hétérozygote une autre hémoglobinopathie, HbO Arabe, HbD Punjab ou Hb Lepore, ou une β-thalassémie, HbE.
Enfin, l'atteinte génétique peut porter non sur la structure primaire de la protéine, mais sur un défaut quantitatif de sa synthèse, ou une persistance anormalement élevée de l'hémoglobine fœtale HbF.
Les défauts de synthèse, ou l'anomalie moléculaire sont décrits sous les noms de :
Galerie
modifier-
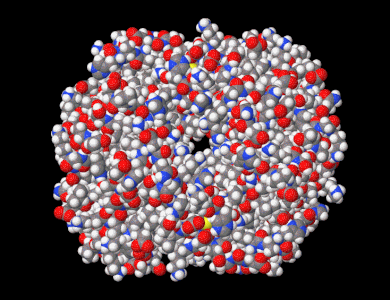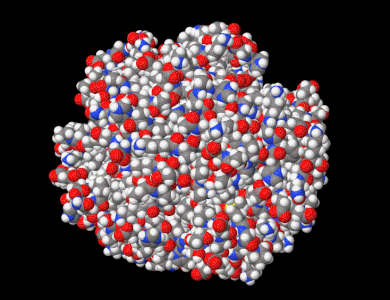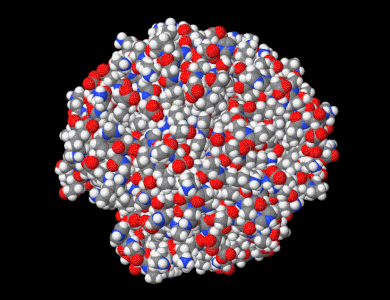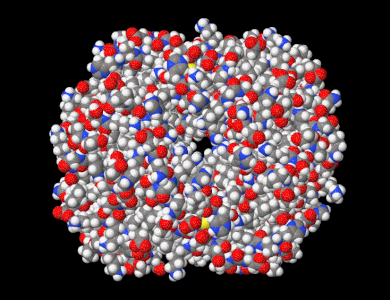 Représentation de l'hémoglobine avec tous les atomes. (7dy3).
Représentation de l'hémoglobine avec tous les atomes. (7dy3). -
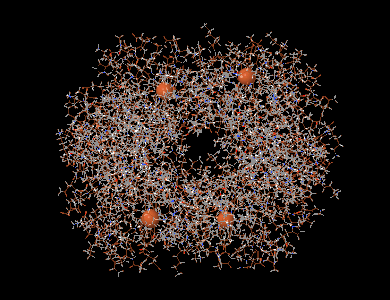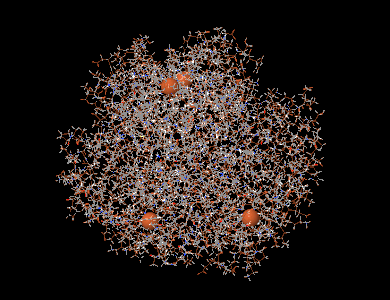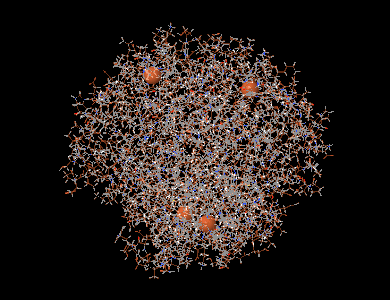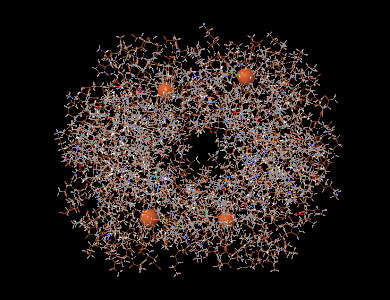 La même molécule avec les atomes de fer mis en évidence.
La même molécule avec les atomes de fer mis en évidence.


Historique
modifierLes premières études sur l'hémoglobine ont été conduites au XIXe siècle en Allemagne. Découverte en 1840 par Hünefeld, l'hémoglobine a été cristallisée en 1851 par Otto Funke (en), et c'est Felix Hoppe-Seyler qui mit en évidence la fixation réversible de l'oxygène sur cette protéine en 1866[37]. La nature tétramérique et la masse moléculaire de l'hémoglobine furent établies par Gilbert Smithson Adair (en) en 1925 par mesure de la pression osmotique de solutions d'hémoglobine[38], qui identifia également les bases de l'effet coopératif de la liaison de l'oxygène à cette protéine par allostérie.
La structure tridimensionnelle de l'hémoglobine fut établie par Max Perutz en 1959 par cristallographie aux rayons X[39],[40], ce qui lui valut de partager le prix Nobel de chimie 1962 avec John Kendrew[41], qui avait conduit des travaux semblables sur la myoglobine.
L'hémoglobine dans les arts
modifier
En 2005, l’artiste Julian Voss-Andreae a réalisé la sculpture Heart of Steel (Hemoglobin), ayant pour modèle l’épine dorsale de la protéine. La sculpture est faite de verre et d’acier Corten. L’aspect rouillé de l’œuvre est intentionnel et évoque la réaction chimique fondamentale de l’oxygène se liant au fer contenu dans l’hémoglobine[42],[43].
L'artiste montréalais Nicolas Baier a réalisé la sculpture Lustre (hémoglobine), une sculpture en acier inoxydable poli qui montre la structure de la molécule d'hémoglobine. La sculpture se trouve à l'atrium du centre de recherches du Centre universitaire de santé McGill à Montréal. La taille de la sculpture est d'environ 10 mètres par 10 mètres par 10 mètres[44],[45],[46].
Notes et références
modifier- (en) G. Fermi, M.F. Perutz et B. Shaanan, « The crystal structure of human deoxyhaemoglobin at 1.74 A resolution », Journal of Molecular Biology, vol. 175, no 2, , p. 159-174 (PMID 6726807, DOI 10.1016/0022-2836(84)90472-8, lire en ligne)
- Les valeurs de la masse et du nombre de résidus indiquées ici sont celles du précurseur protéique issu de la traduction du gène, avant modifications post-traductionnelles, et peuvent différer significativement des valeurs correspondantes pour la protéine fonctionnelle.
- (en) Robert I. Weed, Claude F. Reed et George Berg, « Is Hemoglobin an Essential Structural Component of Human Erythrocyte Membranes? », Journal of Clinical Investigation, vol. 42, , p. 581-588 (PMID 13999462, PMCID 289318, DOI 10.1172/JCI104747, lire en ligne)
- (en) E. Domínguez de Villota, M. T. García Carmona, J. J. Rubio, et S. Ruiz de Andrés, « Equality of the in vivo and in vitro oxygen-binding capacity of haemoglobin in patients with severe respiratory disease », British Journal of Anaesthesia, vol. 53, no 12, , p. 1325-1328 (PMID 7317251, PMCID 289318, DOI 10.1093/bja/53.12.1325, lire en ligne)
- (en) Connie C. W. Hsia, « Respiratory Function of Hemoglobin », The New England Journal of Medicine, vol. 338, no 4, , p. 239-247 (PMID 9435331, DOI 10.1056/NEJM199801223380407, lire en ligne)
- (en) M. Biagioli, M. Pinto, D. Cesselli et al., « Unexpected expression of α- and β-globin in mesencephalic dopaminergic neurons and glial cells », Proceedings of the National Academy of Sciences of the United States of America, vol. 106, no 36, , p. 15454-15459 (PMID 19717439, PMCID 2732704, DOI 10.1073/pnas.0813216106, lire en ligne [PDF])
- (en) Roy E. Weber et Serge N. Vinogradov, « Nonvertebrate hemoglobins: functions and molecular adaptations », Physiological Review, vol. 81, no 2, , p. 569-628 (PMID 11274340, lire en ligne)
- (en) Ross C. Hardison, « A brief history of hemoglobins: plant, animal, protist, and bacteria », Proceedings of the National Academy of Sciences of the United States of America, vol. 93, no 12, , p. 5675-5679 (PMID 8650150, PMCID 39118, DOI 10.1073/pnas.93.12.5675, JSTOR 39604, Bibcode 1996PNAS...93.5675H, lire en ligne)
- (en) Mireille C. P. Van Beekvelt, Willy N. J. M. Colier, Ron A. Wevers et Baziel G. M. Van Engelen, « Performance of near-infrared spectroscopy in measuring local O2 consumption and blood flow in skeletal muscle », Journal of Applied Physiology, vol. 90, no 2, , p. 511-519 (PMID 11160049)
- (en) R. D. Shannon, « Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides », Acta Crystallographica Section A: Crystal Physics, Diffraction, Theoretical and General Crystallography, vol. 32, no 5, septembre, p. 1976 (DOI 10.1107/S0567739476001551, Bibcode 1976AcCrA..32..751S, lire en ligne)
- (en) Wray H. Huestis et Michael A. Raftery, « Conformation and cooperativity in hemoglobin », Biochemistry, vol. 14, no 9, , p. 1886-1892 (PMID 235969, DOI 10.1021/bi00680a013, lire en ligne)
- (en) Frank B. Jensen, « The dual roles of red blood cells in tissue oxygen delivery: oxygen carriers and regulators of local blood flow », Journal of Experimental Biology, vol. 212, no Pt 21, , p. 3387-3393 (PMID 19837879, DOI 10.1242/jeb.023697, lire en ligne)
- (en) D. Nicholas Bateman, « Carbon Monoxide », Medicine, vol. 31, no 10, , p. 41-42 (DOI 10.1383/medc.31.10.41.27810, lire en ligne)
- (en) C. L. Townsend et R. L. Maynard, « Effects on health of prolonged exposure to low concentrations of carbon monoxide », Occupational & Environmental Medicine, vol. 59, no 10, , p. 708-711 (PMID 12356933, PMCID 1740215, DOI 10.1136/oem.59.10.708, JSTOR 27731796, lire en ligne)
- (en) John Haldane, « The Action of Carbonic Oxide on Man », The Journal of Physiology, vol. 18, nos 5-6, , p. 430-462 (PMID 16992272, PMCID 1514663, DOI 10.1113/jphysiol.1895.sp000578, lire en ligne)
- (en) Mark Goldstein, « Carbon Monoxide Poisoning », Journal of Emergency Nursing, vol. 34, no 6, , p. 538-542 (PMID 19022078, DOI 10.1016/j.jen.2007.11.014, lire en ligne)
- (en) Tim Struttmann, Amy Scheerer, T. Scott Prince et Linda A. Goldstein, « Unintentional carbon monoxide poisoning from an unlikely source », Journal of the American Board of Family Medicine, vol. 11, no 6, , p. 481-484 (PMID 9876005, DOI 10.3122/jabfm.11.6.481, lire en ligne)
- (en) Edward R. Burka, « Characteristics of RNA degradation in the erythroid cell », Journal of Clinical Investigation, vol. 48, no 7, , p. 1266-1272 (PMID 5794250, PMCID 322349, DOI 10.1172/JCI106092, lire en ligne)
- (en) Goro Kikuchi, Tadashi Yoshida et Masato Noguchi, « Heme oxygenase and heme degradation », Biochemical and Biophysical Research Communications, vol. 338, no 1, , p. 558-567 (PMID 16115609, DOI 10.1016/j.bbrc.2005.08.020, lire en ligne)
- (en) Ross C. Hardison, « Evolution of Hemoglobin and Its Genes », Cold Spring Harbor Perspectives in Medicine, vol. 2, no 12, , a011627 (PMID 23209182, PMCID 3543078, DOI 10.1101/cshperspect.a011627, lire en ligne)
- (en) Morris Goodman, G. William Moore et Genji Matsuda, « Darwinian evolution in the genealogy of haemoglobin », Nature, vol. 253, no 5493, , p. 603-608 (PMID 1089897, DOI 10.1038/253603a0, Bibcode 1975Natur.253..603G, lire en ligne)
- (en) Jay F. Storz, Juan C. Opazo et Federico G. Hoffmann, « Gene duplication, genome duplication, and the functional diversification of vertebrate globins », Molecular Phylogenetics and Evolution, vol. 66, no 2, , p. 469-478 (PMID 22846683, PMCID 4306229, DOI 10.1016/j.ympev.2012.07.013, lire en ligne)
- (en) E. A. Zimmer, S. L. Martin, S. M. Beverley, Y. W. Kan et A. C. Wilson, « Rapid duplication and loss of genes coding for the alpha chains of hemoglobin », Proceedings of the National Academy of Sciences of the United States, vol. 77, no 4, , p. 2158-2162 (PMID 6929543, PMCID 348671, DOI 10.1073/pnas.77.4.2158, lire en ligne)
- (en) Joana Projecto-Garcia, Chandrasekhar Natarajan, Hideaki Moriyama, Roy E. Weber, Angela Fago, Zachary A. Cheviron, Robert Dudley, Jimmy A. McGuire, Christopher C. Witt et Jay F. Storz, « Repeated elevational transitions in hemoglobin function during the evolution of Andean hummingbirds », Proceedings of the National Academy of Sciences of the United States of America, vol. 11à, no 51, , p. 20669-20674 (PMID 24297909, PMCID 3870697, DOI 10.1073/pnas.1315456110, lire en ligne)
- (en) Cynthia M. Beall, Kijoung Song, Robert C. Elston et Melvyn C. Goldstein, « Higher offspring survival among Tibetan women with high oxygen saturation genotypes residing at 4,000 m », Proceedings of the National Academy of Sciences of the United States of America, vol. 101, no 39, , p. 14300–14304 (PMID 15353580, PMCID 521103, DOI 10.1073/pnas.0405949101, lire en ligne)
- (en) Zhenning He et J. Eric Russell, « Expression, purification, and characterization of human hemoglobins Gower-1 (ζ2ε2), Gower-2 (α2ε2), and Portland-2 (ζ2β2) assembled in complex transgenic–knockout mice », Blood, vol. 97, no 4, , p. 1099-1105 (PMID 11159543, lire en ligne)
- (en) J. Eric Russell et Stephen A. Liebhaber, « Reversal of Lethal α- and β-Thalassemias in Mice by Expression of Human Embryonic Globins », Blood, vol. 92, no 9, , p. 3057-3063 (PMID 9787139, lire en ligne)
- (en) Sophie Lanzkron, John J. Strouse, Renee Wilson, Mary Catherine Beach, Carlton Haywood, HaeSong Park, Catherine Witkop, Eric B. Bass et Jodi B. Segal, « Systematic Review: Hydroxyurea for the Treatment of Adults with Sickle Cell Disease », Annals of Internal Medicine, vol. 148, no 12, , p. 939-955 (PMID 18458272, PMCID 3256736, DOI 10.7326/0003-4819-148-12-200806170-00221, lire en ligne)
- (en) Amoz I. Chernoff, Virginia Minnich, Supa Na-Nakorn, Soodsarkorn Tuchinda, Channivat Kashemsant et Renate R. Chernoff, « Studies on hemoglobin E – I. The clinical, hematologic, and genetic characteristics of the hemoglobin E syndromes », Journal of Laboratory and Clinical Medicine, vol. 47, no 3, , p. 455-489 (PMID 13353880, lire en ligne)
- (en) « Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013 », Lancet, vol. 386, no 9995, , p. 743-800 (PMID 26063472, DOI 10.1016/S0140-6736(15)60692-4, lire en ligne)
- (en) William E. Royer Jr., Hitesh Sharma, Kristen Strand, James E. Knapp et Balaji Bhyravbhatla, « Lumbricus Erythrocruorin at 3.5 Å Resolution: Architecture of a Megadalton Respiratory Complex », Structure, vol. 14, no 7, , p. 1167-1177 (PMID 16843898, DOI 10.1016/j.str.2006.05.011, lire en ligne)
- (en) Luc Int Panis, Boudewijn Goddeeris et Rudolf Verheyen, « The hemoglobin concentration of Chironomus cf. Plumosus l. (Diptera: Chironomidae) larvae from two lentic habitats », Netherland Journal of Aquatic Ecology, vol. 29, no 1, , p. 1-4 (DOI 10.1007/BF02061785, lire en ligne)
- (en) Franck Zal, François H. Lallier, Brian N. Green, Serge N. Vinogradov et André Toulmond, « The multi-hemoglobin system of the hydrothermal vent tube worm Riftia pachyptila. II. Complete polypeptide chain composition investigated by maximum entropy analysis of mass spectra », Journal of Biological Chemistry, vol. 271, no 15, , p. 8875-8881 (PMID 8621529, DOI 10.1074/jbc.271.15.8875, lire en ligne)
- (en) Zoran Minic et Guy Hervé, « iochemical and enzymological aspects of the symbiosis between the deep-sea tubeworm Riftia pachyptila and its bacterial endosymbiont », The FEBS Journal, vol. 271, no 15, , p. 3093-3102 (PMID 15265029, DOI 10.1111/j.1432-1033.2004.04248.x, lire en ligne)
- « What is the evidence for gender differences in ferritin and haemoglobin? » (consulté le )
- l'Hématologie de Bernard Dreyfus, Médecine-Sciences- Flammarion1992
- (de) Felix Hoppe-Seyler, « Über die oxydation in lebendem blute », Med-chem Untersuch Lab, , p. 133–140
- (en) Gilbert S. Adair, « The Osmotic Pressure of Haemoglobin in the Absence of Salts », Proceedings of the Royal Society of London. Series B, Containing Papers of a Biological Character, vol. 98, no 692, , p. 524 (DOI 10.1098/rspa.1925.0126, JSTOR 94515, Bibcode 1925RSPSB..98..524A, lire en ligne)
- (en) M. F. Perutz, M. G. Rossmann, Ann F. Cullis, Hilary MUIRHEAD, GEORG WILL & A. C. T. NORTH, « Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis », Nature, vol. 185, no 4711, , p. 416-422 (PMID 18990801, DOI 10.1038/185416a0, lire en ligne)
- (en) Max F. Perutz, « Structure of hemoglobin », Brookhaven Symposia in Biology, vol. 13, , p. 165-183 (PMID 13734651)
-
(en) « The Nobel Prize in Chemistry 1962 » (consulté le ) :
« The Nobel Prize in Chemistry 1962 was awarded jointly to Max Ferdinand Perutz and John Cowdery Kendrew "for their studies of the structures of globular proteins". »
- (en) Constance Holden, « Blood and Steel », Science, vol. 309, no 5744, , p. 2160 (DOI 10.1126/science.309.5744.2160d, lire en ligne [PDF])
- (en) Moran L, Horton RA, Scrimgeour G, Perry M, Principles of Biochemistry, Boston, MA, Pearson, , 786 p. (ISBN 978-0-321-70733-8 et 0-321-70733-8), p. 127
- (en) Sean Henry, « Take a sneak peek at the MUHC's art collection », sur CBC News, (consulté le )
- « Lustre (Hémoglobine) 2014 », sur Art Public Montréal (consulté le )
- « Nicolas Baier », sur Centre universitaire de santé McGill (consulté le )
Voir aussi
modifierBibliographie
modifier- Banerjee, R., & Sagaert, L. (1967). Dissociation de l'hémoglobine humaine en milieu acide. Biochimica et Biophysica Acta (BBA)-Protein Structure, 140(2), 266-273 (résumé).
- Blum, N., Maleknia, M., & Schapira, G. (1970). α-et β-globines libres et biosynthese de l'hemoglobine. Biochimica et Biophysica Acta (BBA)-Nucleic Acids and Protein Synthesis, 199(1), 236-247.
- Breton-Gorius, J. (1970). Utilisation de la diaminobenzidine pour la mise en évidence au microscope électronique de l'hémoglobine intracellulaire. Nouv. Rev. Fr. Hematologie, 10, 243-256.
- Caffin, J. P., Chauvet, J. P., & Acher, R. (1969). Les hémoglobines des amphibiens: Separation et caracterisation préliminaire des chaînes d'une hémoglobine du crapaud Bufo bufo. FEBS letters, 5(3), 196-198 (résumé).
- Bardakdjian-Michau, J., Dhondt, J. L., Ducrocq, R., Galactéros, F., Guyard, A., Huchet, F. X., ... & Wajcman, H. (2003, July). Bonnes pratiques de l’étude de l’hémoglobine. In Annales de Biologie Clinique (Vol. 61, No. 4, pp. 401-409).
- Bernard, M., Bordas-Fonfrède, M., Grimaldi, A., Guillemin, C., Stahl, A., Leutenegger, M., & Gillery, P. (1995). Intérêts respectifs des dosages d'hémoglobine glyquée et de fructosamines dans la surveillance du diabète sucré. In Annales de biologie clinique (Vol. 53, No. 6, pp. 321-327). John Libbey Eurotext.
- Bert, P. (1882). Sur la richesse en hemoglobine du sang des animaux vivant sur les hauts lieux. CR Acad Sci Paris, 94, 805-807.
- Bloch-Raphaël, C. (1939). Localisation, formation et destruction de l'hémoglobine chez les Annélides polychètes (Doctoral dissertation).
- Eaton, W. A., & Hofrichter, J. (1990). Sickle cell hemoglobin polymerization. Advances in protein chemistry, 40, 263-279.
- Foettinger, A. (1880). Sur l’existence de l’hémoglobine chez les échinodermes. Arch. Biol. Paris, 1, 405-415.
- Kruh, J., Dreyfus, J. C., & Schapira, G. (1964). Activation de la synthèse acellulaire de l'hémoglobine par l'acide ribonucléique: III. Action de l'acide ribonucléique total de foie. Biochimica et Biophysica Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects, 91(3), 494-505 (résumé).
- Kruh, J., Dreyfus, J. C., Rosa, J., & Schapira, G. (1962). Synthèse de l'hémoglobine par des systèmes acellulaires de réticulocytes. Biochimica et Biophysica Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects, 55(5), 690-703.
- Lambert, P. P., Grégoire, F., & Royers, E. (1955). Hémodynamique glomérulaire et excrétion de l'hémoglobine. Archives Of Physiology And Biochemistry, 63(1), 7-34 (résumé).
- Lena-Russo, D., North, M. L., & Girot, R. (1992). Epidémiologie des maladies génétiques de l'hémoglobine en France métropolitaine. La Revue du praticien, 42(15), 1867-1872.
- Robert, M. (1975). Affinité de l'hémoglobine pour l'oxygène. Hôpital cantonal, département de médecine, clinique médicale thérapeutique.
- Uriel, J. (1958). Détection des activités catalasiques et péroxydasiques de l'hémoglobine apres électrophorèse en gélose. Bulletin de la Société de chimie biologique, 40, 277-280.
- White, C. T., Murray, A. J., Smith, D. J., Greene, J. R., & Bolin, R. B. (1986). Synergistic toxicity of endotoxin and hemoglobin. J Lab Clin Med, 108(2), 132-137.