Abstract
Aim: This study aims to assess the microbiome variations related to intraoral halitosis and its relationship with volatile sulfur compounds (VSCs) among periodontally healthy Chinese adults. Material and methods: Tongue coating samples were collected from 28 periodontally healthy subjects (16 subjects with halitosis and 12 subjects without halitosis) who fulfilled the selection criteria. The organoleptic score (OS) was used to evaluate the halitosis status. The characterization of associated microbial communities was performed using 16S rRNA gene pyrosequencing and metagenomics methods. Results: A wide range of microbial communities, including 13 phyla, 23 classes, 37 orders, 134 genera, 266 species and 349 operational taxonomic units (OTUs), were detected. The Shannon index values were significantly higher in the halitosis group. Genera, such as Prevotella, Alloprevotella, Leptotrichia, Peptostreptococcus and Stomatobaculum, exhibited significantly higher relative percentages in halitosis samples, when compared to healthy samples. Peptostreptococcus, Alloprevotella, Eubacterium nodatum and Stomatobaculum exhibited significantly positive correlations with the total number of VSCs. Prevotella, Peptostreptococcus, Eubacterium nodatum and Alloprevotella were correlated with increased H2S and CH3SH concentration values. Bergeyella was correlated with decreased total VSC, H2S and CH3SH concentration values. Conclusion: Microbial diversity was higher in the halitosis group than in the control group, and several bacteria were significantly correlated to halitosis. Furthermore, there were correlations between tongue bacterial composition structure and VSC gases. Tongue coating microbiota can offer important clues in the investigation of the pathogenesis and treatment of halitosis.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Halitosis or bad breath is a widespread disorder that leads to social embarrassment and personal discomfort [1, 2]. Although the aetiology of halitosis is multifactorial, which involves various body organs (oral cavity, lungs and stomach), approximately 90% of all types of halitosis can be classified as intraoral halitosis, which originates from oral cavity-associated pathological conditions (periodontitis and gingivitis) and physiological traits, particularly tongue microbial coating [1]. The tongue coating comprises of a wide range of bacteria, and secretions from the postnasal area and gingiva, saliva and dead epithelial cells [1, 3]. The microorganisms present in tongue coating produce various metabolites, which include volatile sulfur compounds (VSCs), such as hydrogen sulfide (H2S), methyl mercaptan (CH3SH) and dimethyl sulfide (CH3SCH3). The production of VSC metabolites has been considered as the leading cause of intraoral halitosis [4–6]. Furthermore, short-chain fatty acids (SCFAs), such as propanoic acid and butyric acid, cadaverine, indole and skatole, may also contribute to the development of halitosis [7, 8].
The sulfur-containing amino acids (cysteine and methionine) are degraded by both Gram-negative and Gram-positive anaerobic bacteria to produce VSCs [5, 9, 10]. A wide range of oral bacteria, including Porphyromonas gingivalis (P. gingivalis), Fusobacterium nucleatum (F. nucleatum), Prevotella intermedia (P. intermedia), Treponema denticola (T. denticola) and Tannerella forsythia (T. forsythia), have been regarded to be closely associated with intraoral halitosis [5, 9]. In the developed tongue coatings, microorganisms, especially anaerobic bacteria, rapidly grow due to the low oxygen potential facilitated by favourable morphological features (such as roughness, papillae, fissures and crypts) present on the tongue surface [11]. The deep fissures present on the tongue surface facilitate bacterial colonization by providing low oxygen conditions for halitosis-related anaerobic bacteria. The tongue coating acts as a reservoir for periodontal pathogenic bacteria in both periodontally healthy and inflammatory environments [12]. Due to the large amount of microorganisms and exfoliated cells from these large papillary surfaces, the tongue dorsum has been considered as a major source of VSC production.
At present, the capabilities of next-generation sequencing (NGS) [13] technology have enabled the analysis of halitosis-related microbial communities, including previously uncultured microorganisms [14], providing a deeper understanding of the micro-ecological changes associated with intraoral halitosis. In previous studies, tongue coating has been widely explored [15, 16]. Furthermore, oral microbiome differences can be observed in oral environments in patients with and without halitosis [15]. Similarly, Ren et al [16] used 16S rRNA gene pyrosequencing and metagenomic sequencing, and found that Leptotrichia wadei (L. wadei), Peptostreptococcus stomatis (P. stomatis) and Prevotella shahii (P. shahii) had different abundances and prevalence in tongue coating samples obtained from children with or without halitosis. Based on the existing information regarding tongue microbial composition, the relationship between the microbiome and intraoral halitosis remains insufficiently clear. Furthermore, the microbial basis of intraoral halitosis in the oral environment remains largely unknown. Therefore, the aim of the present study was to investigate the microbial communities related to intraoral halitosis, and determine the relationships with VSCs in periodontally healthy environments among Chinese adults.
Methods
The present study was conducted in the Department of Preventive Dentistry, the Ninth People's Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, China. The Ethics Committee of the Ninth People's Hospital of Shanghai Jiao Tong University School of Medicine reviewed and approved the present study (No. 201609). All procedures were performed in accordance to the principles of the Declaration of Helsinki, and a written informed consent was obtained from all recruited patients prior to participation into the study.
Study population and inclusion criteria
Prior to the clinical examination, all participants received a letter with written instructions. At two days before the appointment, all participants were prohibited from smoking and consuming any spicy food, including garlic and onions. In the morning of the examination, the participants were instructed not to use any perfume, cosmetics, or fragrances. Chewing gums, mints, drops, scents and mouth rinses were also not allowed on the morning of the appointment. In order to distinguish between halitosis and morning bad breath, participants were allowed to brush their teeth using water at least two hours before the assessment. After monitoring the inclusion and exclusion criteria (table 1), all participants underwent a comprehensive clinical oral examination. All participants were in good state of general and periodontal health, and maintained good oral hygiene. Following the clinical examination, each participant was requested to complete a detailed halitosis and medical questionnaire, and received a thorough halitosis examination.
Table 1. Criteria used to recruit participants in the present study.
| Inclusion criteria | Exclusion criteria |
|---|---|
|
|
The organoleptic score (OS) was determined by a qualified and calibrated examiner (W.Y.), who has been trained in distinguishing odours using smell identification tests (Sensonics Inc., Haddon Heights, NJ, USA). The examiner was instructed to detect olfactory sensitivity using a series of gradient concentrations of the following substances: skatole, putrescine, isovaleric acid and dimethyl disulphide. All participants were reexamined to evaluate for intra-examiner reliability, and the Cohen's kappa value (κ) for the OS was 0.778. In this method, the participants were instructed to close their mouth for three minutes (nasal breathing) and slowly blow out air through the mouth through a paper tube with a 10-cm distance from the examiner's nose. The '0–5 Rosenberg scale' was used to determine the scores: '0' represented the absence of odour; '1' represented barely noticeable odour; '2' represented slight malodour; '3' represented moderate malodour; '4' represented strong malodour; '5' represented severe malodour. All subjects were instructed to measure their oral VSCs using a Halimeter for three times, according to manufacturer's instructions. The average value (in parts per billion (ppb)) displayed by the Halimeter was recorded. Oral ChromaTM (CHM-1, Abimedical Corporation, Osaka, Japan) VSC measurements were performed for the quantitative analysis of H2S, CH3SH and CH3SCH3 (the VSC gas concentration values were also displayed in ppb).
The halitosis participants were selected based on an OS ≥ 2 [17] and a VSC value ≥ 110 ppb, according to manufacturer's instructions (http://halimeter.com/calibration-procedure). Participants with OS = 0 and a VSC value of <110 ppb were selected as the control group. The sample size was referred to previous studies [16, 18]. Patients within 18–55 years old in the case group (n = 16) and control group (n = 12) were included in the present study. The subjects with halitosis were patients from the halitosis clinic in this department. The controls were patients who primary requested for a periodic oral health examination without complaints of discomfort. Tongue coating was evaluated before sampling, which included the tongue coating area (0 = none, 1 ≤ 1/3 of the tongue, 2 = 1/3–2/3 of the tongue, and 3 ≥ 2/3 of the tongue) and tongue coating thickness (0 = none; 1 = thin, tongue papillae visible; 2 = thick, tongue papillae invisible) [19].
Sample collection and DNA extraction
All tongue coating samples were collected from participants in both groups in the morning, from March 2018 to April 2018. In order to minimize the saliva contamination, saliva was removed with a stream of air from the tongue dorsum, and the subjects were instructed to keep their tongue up from the mouth floor during sampling. The tongue coating was scraped from the dorsal to ventral areas using a tongue cleaning device, and transferred to a 1-ml Tris-EDTA buffer. Then, the samples were stored at −80 °C for no more than two months until further analysis. The microbiome of samples and variations among the communities were analyzed using 16S rRNA gene pyrosequencing and metagenomics methods.
According to manufacturer's protocols, the E.Z.N.A.® soil DNA kit (Omega Bio-tek, Norcross, GA, USA) was used to extract the microbial DNA from the tongue coating. A UV-vis spectrophotometer (NanoDrop 2000, Thermo Scientific, Wilmington, USA) was used to determine the final DNA concentration and purification. The quality of the DNA was assessed using 1% agarose gel electrophoresis.
PCR amplification and Illumina MiSeq sequencing
The V3–V4 hypervariable regions of the bacteria 16S rRNA gene were amplified with primers 338F (5'-ACTCCTACGGGAGGCAGCAG-3') and 806R (5'-GGACTACHVGGGTWTCTAAT-3') using the thermocycler PCR system (GeneAmp 9700, ABI, USA). The following steps were used to perform the PCR reactions: three minutes of denaturation at 95 °C, 27 cycles for 30 s at 95 °C, 30 s of annealing at 55 °C, 45 s of elongation at 72 °C, and a final extension at 72 °C for 10 min [20]. The PCR reactions were performed in triplicate using an aliquot of 20 μl of the mixture, containing 4 μl of 5 × FastPfu Buffer, 2 μl of 2.5 mM dNTPs, 0.8 μl of each primer (5 μM), 0.4 μl of FastPfu Polymerase, and 10 ng of the template DNA. According to manufacturer's protocol, the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) was used to extract and purify the resulting PCR products from the 2% agarose gel. Then, further quantification was performed using QuantiFluor™-ST (Promega, USA).
The purified amplicons were pooled in equimolar amounts, and paired-end sequencing (2 × 300) was performed on the Illumina MiSeq platform (Illumina, San Diego, USA), according to the standard protocols provided by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). All data extracted from the present study were submitted to the SRA database (SRA accession: PRJNA504320).
Processing of sequencing data
Raw fastq files were demultiplexed, quality-filtered by Trimmomatic, and merged using the FLASH software (http://cbcb.umd.edu/software/flash) [21, 22]. Operational taxonomic units (OTUs) were clustered with 97% similarity cut-off using UPARSE (version 7.1, http://drive5.com/uparse/) [23], and the chimeric sequences were identified and removed using UCHIME (version 4.1). The taxonomy of each 16S rRNA gene sequence was analyzed using the RDP Classifier algorithm (http://rdp.cme.msu.edu/) against the Silva (SSU123) and 16S rRNA databases at a confidence threshold of 70%. Then, the data were analyzed using the free online Majorbio I-Sanger Cloud Platform (www.i-sanger.com).
The representative sequences were assigned from phylum to species levels. The alpha (α) Shannon index was used to assess the tongue bacterial richness and diversity across samples [24]. Venn diagrams were implemented by Venn Diagram (version 1.6.20). The beta diversity analysis was performed using the weighted UniFrac and Bray-Curtis distance matrices to compare the results of the principal coordination analysis (PCoA). The community ecology package R-forge (Vegan 2.0 package) was used to generate the PCoA and heat map figures, and perform the redundancy analysis (RDA) with Monte Carlo permutations (permu = 999). The variance inflation factor (VIF) was measured to assess the collinearity among the various environmental factors (VSC gas concentration values). Environmental factors with VIF >10 were removed after the Spearman's correlation analysis. The relationship between the tongue microbial community structure and each associated factor was analyzed using RDA and variation partitioning.
Statistical analysis
Statistical analysis was conducted using the SPSS software (ver. 20.0; IBM, Armonk, NY, USA). The significance level was set at P < 0.05. The socio-demographic characteristics and clinical measurement results were obtained by descriptive statistical analysis. The comparisons of continuous variables between the halitosis and control groups were analyzed using Student's t-test. Tongue coating microbiota composition comparisons were analyzed using the Mann-Whitney U-test. The correlations between the relative abundance of genera and volatile sulfur compound values were evaluated using Spearman's correlation analysis.
Results
Participant characteristics and overall sequencing data
A total of 28 participants were included in the present study. These participants were divided into two groups according to their OS: participants with an OS ≥ 2 were assigned to the halitosis group (n = 16, 11 males and five females, mean age: 37.7 ± 6.89 years old), while participants with an OS < 2 were assigned to the control group (n = 12, seven males and five females, mean age: 33.3 ± 9.48 years old). There were no significant differences in age (P = 0.262) and gender (P = 0.397) distribution between these two groups. Furthermore, these two groups were matched in terms of socio-demographic background, oral health-related behaviours, and oral conditions (table S1 is available online at stacks.iop.org/JBR/14/016005/mmedia). The concentrations of all compounds (total VSC, H2S, CH3SH and CH3SCH3) were significantly higher in the halitosis group, when compared to the control group (448.69 ± 147.68 versus 75.00 ± 60.62, P = 0.04; 1070.63 ± 481.18 versus 72.67 ± 53.06, P < 0.01; 379.19 ± 208.11 versus 26.08 ± 36.25, P = 0.013; 59.44 ± 58.85 versus 11.42 ± 19.65, P < 0.01; respectively).
A total of 28 tongue coatings specimens were collected from each subject after clinical measurement, and sequenced. Finally, a total of 1 290 271 quality-filtered reads were obtained. The number of reads ranged from 30 595 to 68 515 per sample, with an average of 46 081 reads per sample (table S2). Sequence OTU clustering and notation were performed on the quality sequences at a 3% divergence level, and 13 phyla, 23 classes, 37 orders, 134 genera, 266 species and 349 OTUs were detected. The rarefaction curves determined by the Shannon index revealed an adequate sequencing depth (figure S1(A)). The Venn diagram revealed the OTU distributions, indicating that the majority of OTUs (287) were preserved and shared in both groups (figure S1(B)).
Microbial richness and diversity
The alpha (α) diversity (observed at the OTU level, Shannon index) was calculated. The Shannon index was significantly higher in the halitosis group than in the control group (3.19 ± 0.22 versus 2.94 ± 0.24, P = 0.009; figure 1(A)), indicating that the microbial diversity was remarkably greater in the halitosis group than in the control group.
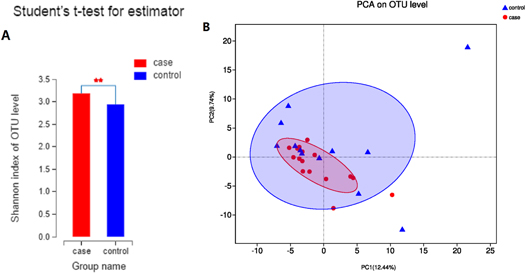
Figure 1. Microbial richness and diversity in the halitosis and control groups: (A) Comparison of α diversity by the Shannon index in healthy and halitosis tongue coating samples. **P < 0.01. (B) Weighted UniFrac principal coordination analysis at the OTU level.
Download figure:
Standard image High-resolution imageIn order to analyze the similarity and difference of the overall microbial composition and structure among the different samples obtained from the two groups, PCoA was applied. Based on the weighted UniFrac and Bray-Curtis distance matrices, no significant difference was found in the microbial community composition between the halitosis and control samples (figure 1(B)).
Characterization of tongue microbiota and microorganisms associated with halitosis
The bacterial taxa from the phylum to the genus level were quantitatively identified through taxonomic assignment against reference databases. Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria and Fusobacteria comprised more than 90% of all sequences, while Firmicutes was the most abundant phyla in both groups. Phyla with average relative abundances of less than 1% were referred to as 'others' (figure 2(A)). At the genus level, genera with mean relative abundances of >1% were assessed, and the most frequently detected genera in both groups included Neisseria (12.52% versus 18.61%), Streptococcus (11.88% versus 13.60%), Prevotella_7 (10.88% versus 10.82%), Veillonella (9.64% versus 8.97%), Haemophilus (5.88% versus 8.44%), Rothia (5.71% versus 7.76%), Actinomyces (7.08% versus 5.03%), Fusobacterium (5.28% versus 4.63%), Porphyromonas (2.90% versus 3.31%), Prevotella (3.36% versus 1.70%), and Leptotrichia (3.46% versus 1.59%), which together constitutes approximately 50% of the tongue bacterial composition (figure 2(B)). The bacterial average relative abundances at other levels in the two groups are presented in supplementary figure S2.
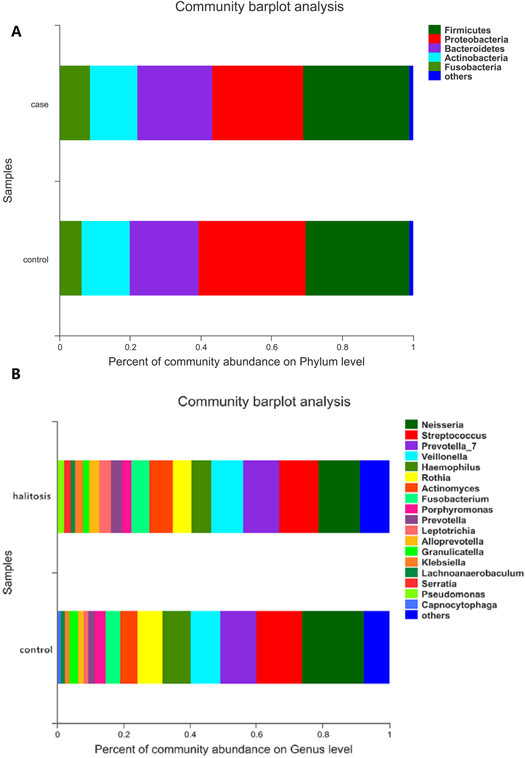
Figure 2. The composition of tongue coating bacterial communities across samples at the phylum and genus level. (A) The community bar-plot analysis reveals the relative abundance of bacteria at the phylum level in both the halitosis and control groups. (B) The community bar-plot analysis reveals the relative abundance of bacteria at the genus level in the halitosis and control groups.
Download figure:
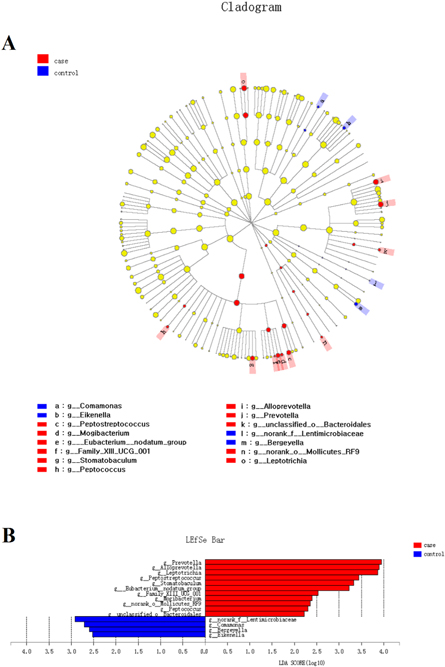
Standard image High-resolution imageThe various bacterial compositional structures were presented through a cladogram using the linear discriminant analysis effect size (LEfSe, figure 3(A)). The differences were analyzed according to the linear discriminant analysis (LDA) score (figure 3(B)). The analysis results revealed that there were significant bacterial differences in tongue bacterial composition at the genus level in the halitosis and control groups. The relative abundances of 11 taxa, including Prevotella, Alloprevotella, Leptotrichia, Peptostreptococcus and Stomatobaculum, were significantly higher in the halitosis tongue coating samples, when compared to control samples (P < 0.05). Four taxa, including Comamonas and Bergeyella, revealed significantly higher relative percentages in the control group (table S3).
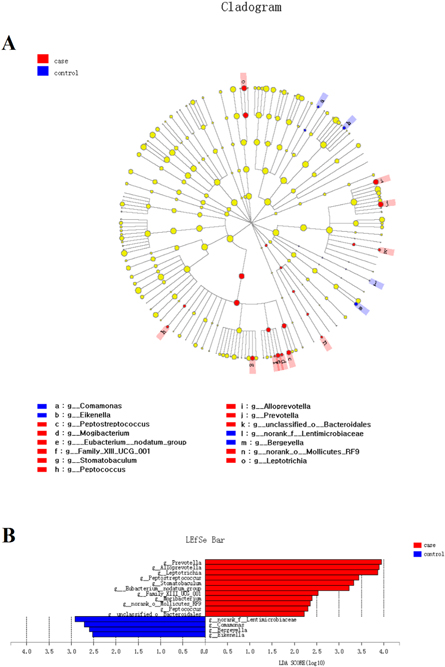
Figure 3. The LEfSe analysis of distinct taxa identified in the halitosis and control groups. (A) The cladogram based on the LEfSe method depicts the different microbiota structures in the halitosis and control groups. The diameter of each circle is proportional to the taxon's abundance. (B) The linear discriminant analysis scores show that there are significant bacterial differences at the genus level in the halitosis and control groups.
Download figure:
Standard image High-resolution imageThe correlation between tongue microbiota structure and volatile sulfur compounds
The VIF was measured to assess the collinearity among various environmental factors, such as VSC gas concentration values. Factors with a VIF >10 were removed from the analysis. The VIF value for total VSC, H2S, CH3SH and CH3SCH3 was 2.07, 4.37, 4.29 and 1.57, respectively. The RDA indicated a significant correlation between the value of VSCs and the tongue bacterial community composition (r2 = 0.47, P = 0.001; figure 4).
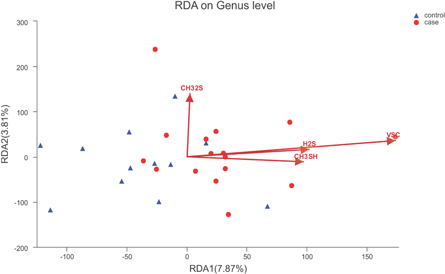
Figure 4. Environmental factors related to the tongue coating microbiota based on the redundancy analysis at the genus level.
Download figure:
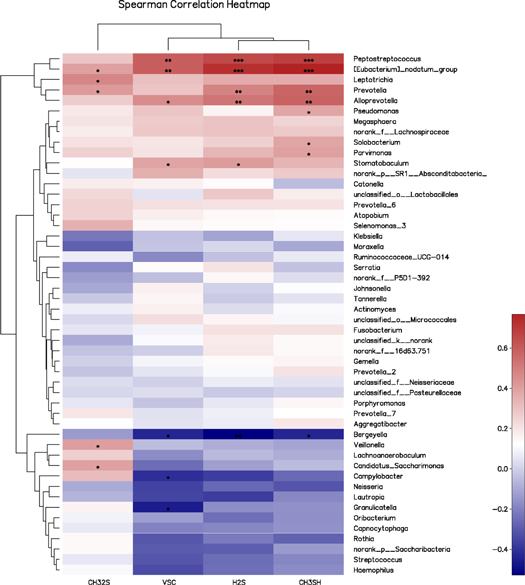
Standard image High-resolution imageIn order to identify the specific bacteria potentially associated with certain VSC gases, further correlation analyses between the relative abundance (%) of the top 50 genus and the values of VSCs were performed using Spearman's correlation analysis (figure 5). Four bacterial genera, including Peptostreptococcus (r = 0.591), Alloprevotella (r = 0.458), Eubacterium nodatum (r = 0.593) and Stomatobaculum (r = 0.386), were found to be significantly and positively correlated with total VSC, when detected by a Halimeter (P < 0.05), while three bacterial genera, including Granulicatella (r = −0.458), Bergeyella (r = −0.434) and Campylobacter (r = −0.412), were significantly and negatively correlated with the total VSC values (P < 0.05). Prevotella (r = 0.484), Peptostreptococcus (r = 0.654), Eubacterium nodatum (r = 0.726), Alloprevotella (r = 0.524) and Stomatobaculum (r = 0.411) were correlated with elevated H2S concentration values (P < 0.05), while Bergeyella (r = −0.528) was correlated with decreased H2S concentration values (P < 0.05). Regarding the CH3SH concentration values, Peptostreptococcus (r = 0.680), Eubacterium nodatum (r = 0.768), Alloprevotella (r = 0.580), Prevotella (r = 0.573), Parvimonas (r = 0.400), Solobacterium (r = 0.384) and Pseudomonas (r = 0.383) were positively correlated, while Bergeyella (r =−0.434) was negatively correlated. A total of 22 genera, including Prevotella (r = 0.415), Eubacterium nodatum (r = 0.399), Leptotrichia (r = 0.475), Veillonella (r = 0.404) and Candidatus_Saccharimonas (r = 0.396), were significantly and positively correlated with the CH3SCH3 value. The genus level Spearman correlation coefficients are presented in supplementary table S4.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. The correlation analysis between the relative abundance (%) of the 50 most abundant genera in the tongue bacterial community and volatile sulfur compounds using Spearman's correlation analysis. The colour of the spots in the right panel represents the relative abundance. *P < 0.05, **P < 0.01, ***P < 0.001.
Download figure:
Standard image High-resolution image{kind=link}
{kind=link}
Discussion
The present study investigated the microbiome variations related to intraoral halitosis and its association with VSCs among periodontally healthy Chinese adults. Microbial diversity was higher in the halitosis group than in the control group, and several bacteria were significantly associated with halitosis. Furthermore, there were correlations between tongue bacterial composition structure and VSC gases. The present study provides in-depth information about VSC-producing bacteria associated with intraoral halitosis.
Most studies [1, 18, 25] have not explicitly excluded the effects of periodontitis in investigating intraoral halitosis. The study conducted by Yaegaki et al [3, 26] revealed that the amount of tongue coating in participants with periodontal disease was three times greater than that in participants with a healthy periodontal environment. In the present study, all subjects were periodontally healthy to eliminate the influence of periodontal factors. Furthermore, there were no significant differences in tongue coating area and thickness between the halitosis and control groups. The consistent tongue coating suggests that the bacterial composition, rather than the amount of tongue coating, may be responsible for the oral halitosis. Therefore, characterizing the bacterial composition of the tongue microbiome may provide some insight regarding the key organisms involved in the development of oral halitosis. The α-diversity (measured by the Shannon index) was significantly higher in the halitosis group, when compared to the control group (P = 0.009), suggesting that the increase in microbial diversity favoured the halitosis status in the present study. These findings are in agreement with previous studies, showing a greater species diversity in patients with halitosis, when compared to controls [16, 25, 27]. In contrast, few studies reported no differences in microbial diversity in patients with halitosis [15, 18]. There were a number of possible reasons for these inconsistent results, such as the use of various assessment techniques, inclusion criteria, race of participants, sequencing segments, and database selection. Therefore, further studies are needed to verify these findings.
In terms of RDA data (figure 4), the total VSC concentration value was considered as the most important environmental factor related to the composition and nature of the tongue microbiome, followed by H2S and CH3SH. In addition, these environmental factors revealed a positive correlation. Halitosis is mainly attributed to VSCs, particularly H2S and CH3SH, which are considered to account for 90% of the VSCs in the oral cavity [4], while CH3SCH3 is the main cause of extraoral halitosis. In few halitosis cases, CH3SH may turn into CH3SCH3 in the oral environment [28]. Sulfur-containing substrates, such as cysteine and methionine, can also degrade into H2S and CH3SH through the Gram-negative and anaerobic tongue bacteria present in the oral cavity [4].
These present results reveal the unique OTUs associated with the control (19 of 306 OTUs) and halitosis (26 of 313 OTUs) groups. However, these unique OTUs had a low relative abundance level (<0.001), and may not play an important contribution in developing halitosis. According to relative abundance, the most relative abundant bacteria were shared by both groups. Overall, Neisseria, Streptococcus, Veillonella, Haemophilus, Rothia, Actinomyces, Fusobacterium, Porphyromonas, Prevotella and Leptotrichia were frequently detected in both groups. The most abundant genera found were consistent with those in previous studies [29, 30]. Veillonella, Fusobacterium, Prevotella, Streptococcus and Actinomyces have been widely reported as predominant bacterial genera in both halitosis and healthy tongue coating microbiota [27, 31, 32]. The PCoA also indicated that the overall bacterial composition and structure of tongue biofilms were similar in both groups in the present study. Considering the similarities in tongue bacterial composition in these groups, more attention should be given to the differences in flora abundance. Furthermore, the relationship between the tongue microbial community and each VSC gas should be further analyzed.
At the genus level, Prevotella was significantly abundant in the halitosis group, when compared to the control group (P < 0.01), which is consistent with previous studies [15, 33]. Further analysis revealed the significantly positive correlation of Prevotella with VSC gas parameters (total VSC, H2S, CH3SH and CH3SCH3). H2S producing bacteria are considered pathogenic and predominant contributors for the development of halitosis [6, 10, 34]. For Prevotella, cysteine undergoes enzymatic degradation through cystathionine γ-lyase (cysteine desulfhydrase) to pyruvate, producing H2S and ammonia. Then, methionine is degraded by methionine γ-lyase (methioninase) into 2-oxobutyrate, producing methyl mercaptan (methanetiol) and ammonia [35–38].
In addition, Leptotrichia, Alloprevotella, Peptostreptococcus and Stomatobaculum also exhibited a significantly higher relative abundance in tongue coatings obtained from the halitosis group. The total VSC and H2S concentration values revealed the significantly positive correlation of Peptostreptococcus, Eubacterium nodatum and Alloprevotella (P < 0.05). However, Bergeyella was observed to be significantly and negatively correlated with the total VSC, H2S and CH3SH values (P < 0.05). Similarly, Ren et al [16] reported that Peptostreptococcus was significantly more prevalent in the tongue coating of halitosis samples, when compared to healthy samples. A number of studies have reported the increased abundance of Leptotrichia in subjects with halitosis [15, 16, 18, 39], and that this exhibited a positive correlation with H2S concentrations [15]. The present study infers that Leptotrichia and Peptostreptococcu may play a role in halitosis.
The analysis of subgingival bacterial samples confirms Eubacterium as an abundant producer of VSCs [40, 41]. Kazor et al [42] reported that Eubacterium is associated with halitosis. These findings are in agreement with the present results. In addition, Prevotella and Eubacterium were also found to be positively correlated to the values of CH3SCH3. Haraszthy et al [31, 32] isolated different bacterial species in halitosis groups, and revealed that Gram-positive Solobacterium moorei (S. moorei) only existed in halitosis samples. This species was considered a key halitosis-associated bacterium [43], and are capable of generating VSCs [44, 45]. In the present study, Solobacterium had a significant and positive correlation with CH3SH (r = 0.384). However, this was detected in both the halitosis and control groups.
The halitosis and control groups did not have any significant differences in terms of the presence or relative abundance of some other VSC-producing pathogens, including P. gingivalis and T. forsythia. In addition, some differences were found, such as the presence of Alloprevotella, which has not been previously described, suggesting the need for further research on tongue biofilms. Furthermore, there were no remarkable differences between the halitosis and control groups, in terms of the relative abundance of certain periodontitis pathogens associated with intraoral halitosis, such as P. gingivalis and F. nucleatum [46, 47]. Yang et al [15] reported that Haemophilus and Gemella decreased the H2S levels. However, the present study did not observe such negative correlations.
It was observed that Bergeyella was correlated with decreased total VSC, H2S and CH3SH concentration values, while Alloprevotella was correlated with increased total VSC, H2S and CH3SH concentration values. These two bacteria have not been previously reported, and Alloprevotella particularly exhibited a relative abundance in the sequenced samples, thereby advocating their role in intraoral halitosis. Such inconsistencies may have been caused by the different selection criteria used for subjects, and the application of different molecular analytical techniques. Furthermore, the present study focused on periodontally healthy adults, which may provide a better understanding of the halitosis-associated microbiota.
At present, research results regarding predominant microorganisms associated with halitosis and healthy tongue dorsum surfaces are not in agreement. Once the bacteria involved in intraoral halitosis are confirmed, the roles of specific organisms, and their interactions, functional activities and associated mechanism in inducing halitosis can be disclosed. Due to the complexity of the tongue coating microbiome, the interplay among factors, such as bacteria, tongue coating, saliva, oral pH value and low oxygen environment, requires more extensive investigations. Further correlation analyses among tongue bacterial community composition, specific tongue organisms and sulfur-containing substrates, such as cysteine and methionine, needs to be conducted. The tongue coating microbiome can be investigated by combining transcriptomics and metabolomics analyses, in order to identify the microbial community expression in halitosis.
Conclusion
In conclusion, the present study described the microbial composition and characteristics of tongue coating in halitosis and healthy participants with a healthy periodontium. Participants had a higher microbial diversity in the halitosis group, when compared to the control group, and several bacteria were found to be significantly correlated to halitosis. Furthermore, the present study explored the correlations between tongue bacterial composition structure and VSC gases. Tongue coating microbiota can offer important clues in the investigation of the pathogenesis and treatment of halitosis.
Acknowledgments
The authors declare that they have no conflict of interest. This research was funded by the National Natural Science Foundation of China (Grant numbers: 81800967 and 81470737).