


Cells, Volume 11, Issue 2 (January-2 2022) – 131 articles
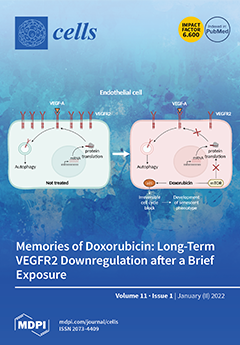
Cover Story (view full-size image):
Doxorubicin (Dox) remains an essential drug in several anticancer regimens even though its use is associated with severe cardiotoxic side effects that persist after drug withdrawal and can lead to heart failure. In addition to cardiomyocytes, damaged cardiac endothelial cells are a culprit of Dox-induced cardiotoxicity. We show that a brief exposure of endothelial cells to low Dox concentrations leads to long-lasting effects that include cellular senescence and downregulation of VEGFR2, crucial for endothelial cell activation. Mechanistically, Dox represses global protein synthesis by inhibiting mTOR. Senescence and downregulation of VEGFR2 in endothelial cells might participate in Dox-induced cardiotoxicity. View this paper
- Issues are regarded as officially published after their release is announced to the table of contents alert mailing list.
- You may sign up for e-mail alerts to receive table of contents of newly released issues.
- PDF is the official format for papers published in both, html and pdf forms. To view the papers in pdf format, click on the "PDF Full-text" link, and use the free Adobe Reader to open them.
Previous Issue
Next Issue