Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma and to Provide New Insights into Intrinsic and Acquired Mechanism of Resistance

 ,
,  , , , and
, , , and {kind=link}
<p>Serine/threonine and tyrosine kinase activities of seven pre-treatment melanoma biopsies harbouring the BRAF V600E mutation. (<b>A</b>) Serine Threonine kinase (STK, top) and tyrosine kinase (PTK, bottom) activity profiles in clinical responders and non-responders to vemurafenib. Log<sub>2</sub> transformed signals represented as a heat map. Rows represent peptides, sorted on overall mean signal intensity. Columns represent the samples; (<b>B</b>) Upstream kinase analysis of basal kinase activity profiles of pre-treatment melanoma tumour samples to identify kinases that show higher activity in the non-responder groups compared to the responders. A brighter colour corresponds with more significant differences. Illustration reproduced courtesy of Cell Signaling Technology, Inc. (<a href="http://www.cellsignal.com" target="_blank">www.cellsignal.com</a>) using the Kinmap.</p> "> Figure 1 Cont.
{kind=link}
<p>Serine/threonine and tyrosine kinase activities of seven pre-treatment melanoma biopsies harbouring the BRAF V600E mutation. (<b>A</b>) Serine Threonine kinase (STK, top) and tyrosine kinase (PTK, bottom) activity profiles in clinical responders and non-responders to vemurafenib. Log<sub>2</sub> transformed signals represented as a heat map. Rows represent peptides, sorted on overall mean signal intensity. Columns represent the samples; (<b>B</b>) Upstream kinase analysis of basal kinase activity profiles of pre-treatment melanoma tumour samples to identify kinases that show higher activity in the non-responder groups compared to the responders. A brighter colour corresponds with more significant differences. Illustration reproduced courtesy of Cell Signaling Technology, Inc. (<a href="http://www.cellsignal.com" target="_blank">www.cellsignal.com</a>) using the Kinmap.</p> "> Figure 2
{kind=link}
<p>Effect of dabrafenib on kinase activity of the melanoma tissues. (<b>A</b>) Clinical responders and non-responders to vemurafenib: Median signal intensity of all peptides for serine/threonine and tyrosine kinase activity as a function of dabrafenib concentration; (<b>B</b>,<b>C</b>) Log<sub>2</sub> Fold Change of kinase activity with dabrafenib concentration. Only peptides that show significant (<span class="html-italic">p</span> < 0.05) difference in inhibition between responder and non-responders are shown. For STK (<b>B</b>), lysates were incubated with 0, 1, 10 or 25 µM dabrafenib (<span class="html-italic">n</span> = 3). For PTK (<b>C</b>), lysates were incubated with 0, 0.1, 0.2 or 0.5 µM dabrafenib (<span class="html-italic">n</span> = 3); (<b>D</b>) Network of parental protein for peptides that differentiated clinical responders from non-responders based on inhibition by dabrafenib. Network was made in STRING, using peptides with <span class="html-italic">p</span> < 0.05 for STK with 1 and 10 µM and for PTK with 0.1, 0.2 and 0.5 µM dabrafenib. The BRAF identity was added to illustrate its position in the network.</p> "> Figure 2 Cont.
{kind=link}
<p>Effect of dabrafenib on kinase activity of the melanoma tissues. (<b>A</b>) Clinical responders and non-responders to vemurafenib: Median signal intensity of all peptides for serine/threonine and tyrosine kinase activity as a function of dabrafenib concentration; (<b>B</b>,<b>C</b>) Log<sub>2</sub> Fold Change of kinase activity with dabrafenib concentration. Only peptides that show significant (<span class="html-italic">p</span> < 0.05) difference in inhibition between responder and non-responders are shown. For STK (<b>B</b>), lysates were incubated with 0, 1, 10 or 25 µM dabrafenib (<span class="html-italic">n</span> = 3). For PTK (<b>C</b>), lysates were incubated with 0, 0.1, 0.2 or 0.5 µM dabrafenib (<span class="html-italic">n</span> = 3); (<b>D</b>) Network of parental protein for peptides that differentiated clinical responders from non-responders based on inhibition by dabrafenib. Network was made in STRING, using peptides with <span class="html-italic">p</span> < 0.05 for STK with 1 and 10 µM and for PTK with 0.1, 0.2 and 0.5 µM dabrafenib. The BRAF identity was added to illustrate its position in the network.</p> "> Figure 2 Cont.
{kind=link}
<p>Effect of dabrafenib on kinase activity of the melanoma tissues. (<b>A</b>) Clinical responders and non-responders to vemurafenib: Median signal intensity of all peptides for serine/threonine and tyrosine kinase activity as a function of dabrafenib concentration; (<b>B</b>,<b>C</b>) Log<sub>2</sub> Fold Change of kinase activity with dabrafenib concentration. Only peptides that show significant (<span class="html-italic">p</span> < 0.05) difference in inhibition between responder and non-responders are shown. For STK (<b>B</b>), lysates were incubated with 0, 1, 10 or 25 µM dabrafenib (<span class="html-italic">n</span> = 3). For PTK (<b>C</b>), lysates were incubated with 0, 0.1, 0.2 or 0.5 µM dabrafenib (<span class="html-italic">n</span> = 3); (<b>D</b>) Network of parental protein for peptides that differentiated clinical responders from non-responders based on inhibition by dabrafenib. Network was made in STRING, using peptides with <span class="html-italic">p</span> < 0.05 for STK with 1 and 10 µM and for PTK with 0.1, 0.2 and 0.5 µM dabrafenib. The BRAF identity was added to illustrate its position in the network.</p> "> Figure 3
{kind=link}
<p>Comparative analyses of kinome profiles between BRAFi sensitive and resistant melanoma cell lines. (<b>A</b>) Serine Threonine kinase (STK, top) and tyrosine kinase (PTK, bottom) activity profiles cell lines sensitive (S), with acquired resistance (AR) or intrinsically resistant (IR) to vemurafenib; (<b>B</b>) Peptides that show significant (<span class="html-italic">p</span> < 0.05) difference in inhibition between cell lines with acquired resistance to vemurafenib: effects per cell line. <sup>2</sup>Log Fold Change vs. parental cell line. Red—increase, blue—decrease in resistant cell line. Asterisks indicate significance; (<b>C</b>) Western Blot analysis of key targeted protein in MAPK, PI3K/AKT and SRC signalling pathways in four BRAFi sensitive, and six cell lines with intrinsic (MM043 and MM054) and acquired resistance to vemurafenib, (R refers to acquired resistant cells compared to parental sensitive cells).</p> "> Figure 3 Cont.
{kind=link}
<p>Comparative analyses of kinome profiles between BRAFi sensitive and resistant melanoma cell lines. (<b>A</b>) Serine Threonine kinase (STK, top) and tyrosine kinase (PTK, bottom) activity profiles cell lines sensitive (S), with acquired resistance (AR) or intrinsically resistant (IR) to vemurafenib; (<b>B</b>) Peptides that show significant (<span class="html-italic">p</span> < 0.05) difference in inhibition between cell lines with acquired resistance to vemurafenib: effects per cell line. <sup>2</sup>Log Fold Change vs. parental cell line. Red—increase, blue—decrease in resistant cell line. Asterisks indicate significance; (<b>C</b>) Western Blot analysis of key targeted protein in MAPK, PI3K/AKT and SRC signalling pathways in four BRAFi sensitive, and six cell lines with intrinsic (MM043 and MM054) and acquired resistance to vemurafenib, (R refers to acquired resistant cells compared to parental sensitive cells).</p> "> Figure 4
{kind=link}
<p>Common effects in cells lines with acquired resistance. (<b>A</b>) Common effects in the four cell line pairs determined with mixed model analysis. Peptides that were significantly differently (<span class="html-italic">p</span> < 0.01) phosphorylated in sensitive and resistant cell line pairs (<sup>2</sup>log fold change vs. sensitive cell lines). Red—increase, blue—decrease in resistant cell line; (<b>B</b>) STRING network analysis of all proteins with <span class="html-italic">p</span> < 0.01 in PTK or STK analysis.</p> "> Figure 4 Cont.
{kind=link}
<p>Common effects in cells lines with acquired resistance. (<b>A</b>) Common effects in the four cell line pairs determined with mixed model analysis. Peptides that were significantly differently (<span class="html-italic">p</span> < 0.01) phosphorylated in sensitive and resistant cell line pairs (<sup>2</sup>log fold change vs. sensitive cell lines). Red—increase, blue—decrease in resistant cell line; (<b>B</b>) STRING network analysis of all proteins with <span class="html-italic">p</span> < 0.01 in PTK or STK analysis.</p> "> Figure 5
{kind=link}
<p>Effect of dabrafenib, trametinib and their combination on STK and PTK kinase activities. (<b>A</b>) Median Log Fold Change of all peptides (median of LFC for all peptides of replicate experiments) of serine/threonine or tyrosine kinase activity upon addition of dabrafenib, trametinib or the combination to the kinase assay. Cell lines are coloured by resistance type; (<b>B–D</b>) Network representation (STRING) of peptides that show a significant interaction between dabrafenib and trametinib in sensitive cell lines (<b>B</b>), cell lines with intrinsic (<b>C</b>) or acquired resistance (<b>D</b>).</p> "> Figure 5 Cont.
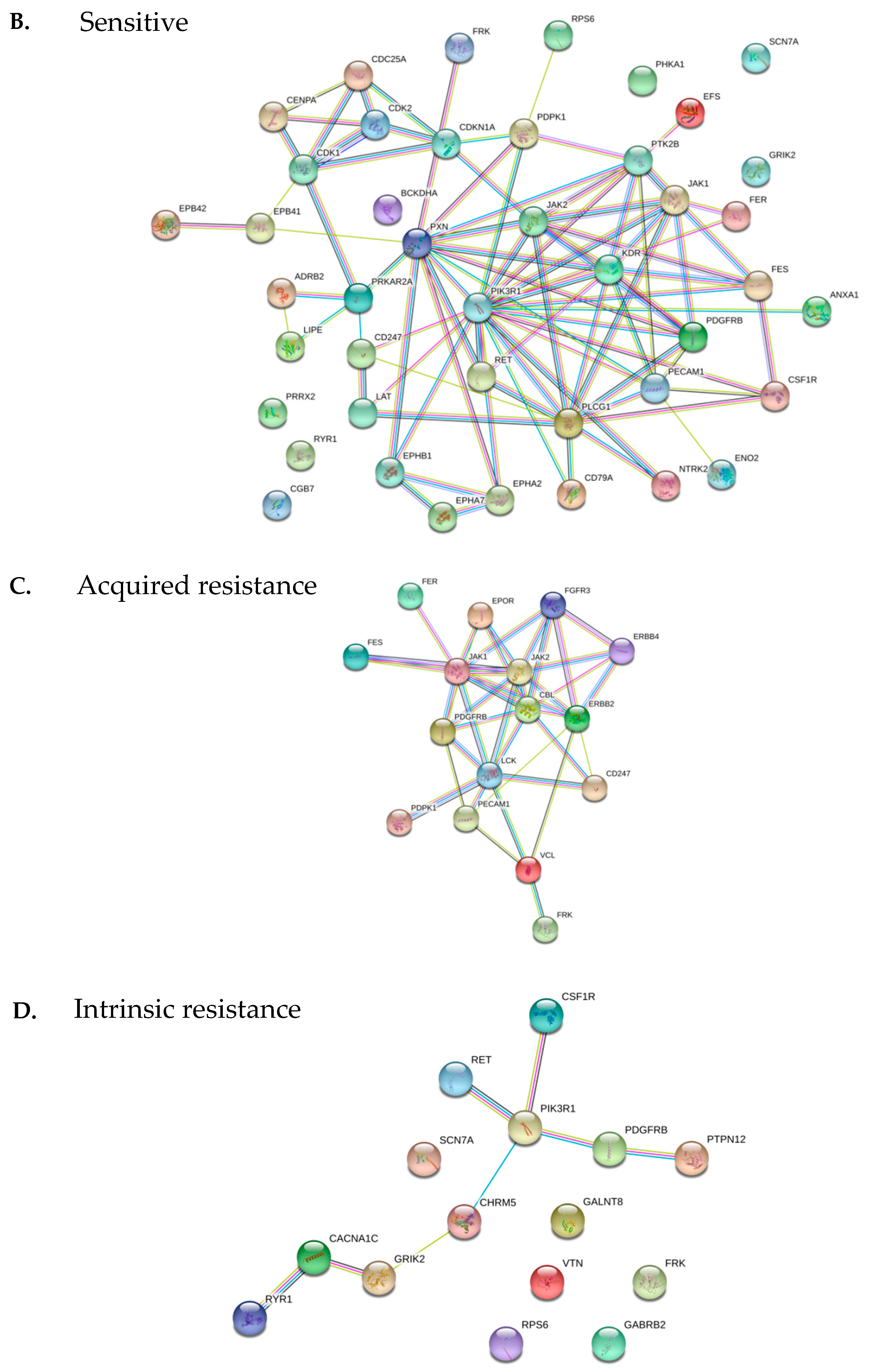{kind=link}
<p>Effect of dabrafenib, trametinib and their combination on STK and PTK kinase activities. (<b>A</b>) Median Log Fold Change of all peptides (median of LFC for all peptides of replicate experiments) of serine/threonine or tyrosine kinase activity upon addition of dabrafenib, trametinib or the combination to the kinase assay. Cell lines are coloured by resistance type; (<b>B–D</b>) Network representation (STRING) of peptides that show a significant interaction between dabrafenib and trametinib in sensitive cell lines (<b>B</b>), cell lines with intrinsic (<b>C</b>) or acquired resistance (<b>D</b>).</p> ">
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Melanoma Cell Lines
2.3. Cell Culture Conditions
2.4. Patients and Tissue Collection
2.5. Lysis of Tissues and Cells
2.6. Kinase Activity Profiling on PamChip® Peptide Microarrays
2.7. Data Analysis and Quality Control of the PamChip® Peptide Microarrays
2.8. Western Blot Analysis
3. Results
3.1. Kinome Profiling Reveals Differential Activity between Responder and Non-Responder Patients to BRAF Inhibition
3.2. Effect of a V600EBRAF Inhibitor on Kinase Activity in Melanoma Tissue Lysates
3.3. Comparative Analysis of Kinome Profiles between Melanoma Cell Lines Sensitive and Resistant to a V600EBRAF Inhibitor
3.4. BRAFi and MEKi Combination Effect on STK and PTK Activities in Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Miller, A.J.; Mihm, M.C.J. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Berwick, M.; Erdei, E.; Hay, J. Melanoma epidemiology and public health. Dermatol. Clin. 2009, 27, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet Lond. Engl. 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Buscà, R.; Abbe, P.; Mantoux, F.; Aberdam, E.; Peyssonnaux, C.; Eychène, A.; Ortonne, J.P.; Ballotti, R. Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000, 19, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Zavala-Pompa, A.; Sequeira, J.H.; Shoji, M.; Sexton, D.G.; Cotsonis, G.; Cerimele, F.; Govindarajan, B.; Macaron, N.; Arbiser, J.L. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3728–3733. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004, 64, 2338–2342. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Weber, B.L.; Herlyn, M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell 2003, 4, 95–98. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet Lond. Engl. 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet Lond. Engl. 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Alcalá, A.M.; Flaherty, K.T. BRAF inhibitors for the treatment of metastatic melanoma: Clinical trials and mechanisms of resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 33–39. [Google Scholar]
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired resistance to BRAFi reverses senescence-like phenotype in mutant BRAF melanoma. Oncotarget 2018, 9, 31888–31903. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur. J. Cancer Oxf. Engl. 1990 2016, 55, 98–110. [Google Scholar]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.L.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef]
- Krayem, M.; Sabbah, M.; Najem, A.; Wouters, A.; Lardon, F.; Simon, S.; Sales, F.; Journe, F.; Awada, A.; Ghanem, G.E.; et al. The Benefit of Reactivating p53 under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma. Cancers 2019, 11, 1093. [Google Scholar] [CrossRef]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G. Prominent role of cyclic adenosine monophosphate signalling pathway in the sensitivity of (WT)BRAF/(WT)NRAS melanoma cells to vemurafenib. Eur. J. Cancer Oxf. Engl. 1990 2014, 50, 1310–1320. [Google Scholar]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer Oxf. Engl. 1990 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Wahl, R.L.; Jacene, H.; Kasamon, Y.; Lodge, M.A. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2009, 50 (Suppl. 1), 122S–150S. [Google Scholar] [CrossRef]
- Alonzo, T.A. Standards for reporting prognostic tumor marker studies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 9053–9054. [Google Scholar] [CrossRef]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics REporting recommendations for tumor MARKer prognostic studies (REMARK). Nat. Clin. Pract. Urol. 2005, 2, 416–422. [Google Scholar]
- Rosenberger, A.F.N.; Hilhorst, R.; Coart, E.; García Barrado, L.; Naji, F.; Rozemuller, A.J.M.; van der Flier, W.M.; Scheltens, P.; Hoozemans, J.J.M.; van der Vies, S.M. Protein Kinase Activity Decreases with Higher Braak Stages of Alzheimer’s Disease Pathology. J. Alzheimers Dis. JAD 2016, 49, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Chirumamilla, C.S.; Fazil, M.H.U.T.; Perez-Novo, C.; Rangarajan, S.; de Wijn, R.; Ramireddy, P.; Verma, N.K.; Vanden Berghe, W. Profiling Activity of Cellular Kinases in Migrating T-Cells. Methods Mol. Biol. Clifton NJ 2019, 1930, 99–113. [Google Scholar]
- Lezcano, C.; Shoushtari, A.N.; Ariyan, C.; Hollmann, T.J.; Busam, K.J. Primary and Metastatic Melanoma with NTRK Fusions. Am. J. Surg. Pathol. 2018, 42, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.M.; de Roos, J.A.D.M.; van Doornmalen, A.M.; Prinsen, M.B.W.; de Man, J.; Tanizawa, Y.; Kawase, Y.; Yoshino, K.; Buijsman, R.C.; Zaman, G.J.R. Comparison of the cancer gene targeting and biochemical selectivities of all targeted kinase inhibitors approved for clinical use. PLoS ONE 2014, 9, e92146. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355. [Google Scholar] [CrossRef] [PubMed]
- Ruijtenbeek, R.; Kerkhoff, L.H.; Hilhorst, M.; Mulders, P.; Oosterwijk-Wakka, J.; Kiemeney, L.; Oosterwijk, E. Abstract 2419: Predicting clinical response based on ex vivo drug response in renal cell carcinoma using kinase activity profiling. Cancer Res. 2015, 75, 2419. [Google Scholar]
- Mohr, S.E.; Smith, J.A.; Shamu, C.E.; Neumüller, R.A.; Perrimon, N. RNAi screening comes of age: Improved techniques and complementary approaches. Nat. Rev. Mol. Cell Biol. 2014, 15, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Stranger, B.E.; Stahl, E.A.; Raj, T. Progress and Promise of Genome-Wide Association Studies for Human Complex Trait Genetics. Genetics 2011, 187, 367–383. [Google Scholar] [CrossRef]
- López Villar, E.; Wu, D.; Cho, W.C.; Madero, L.; Wang, X. Proteomics-based discovery of biomarkers for paediatric acute lymphoblastic leukaemia: Challenges and opportunities. J. Cell. Mol. Med. 2014, 18, 1239–1246. [Google Scholar]
- Jarboe, J.S.; Jaboin, J.J.; Anderson, J.C.; Nowsheen, S.; Stanley, J.A.; Naji, F.; Ruijtenbeek, R.; Tu, T.; Hallahan, D.E.; Yang, E.S.; et al. Kinomic profiling approach identifies Trk as a novel radiation modulator. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2012, 103, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Tahiri, A.; Røe, K.; Ree, A.H.; de Wijn, R.; Risberg, K.; Busch, C.; Lønning, P.E.; Kristensen, V.; Geisler, J. Differential inhibition of ex-vivo tumor kinase activity by vemurafenib in BRAF(V600E) and BRAF wild-type metastatic malignant melanoma. PLoS ONE 2013, 8, e72692. [Google Scholar] [CrossRef] [PubMed]
- Lassen, A.; Atefi, M.; Robert, L.; Wong, D.J.; Cerniglia, M.; Comin-Anduix, B.; Ribas, A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer 2014, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Kumar, S.M.; Zhang, G.; Bastian, B.C.; Arcasoy, M.O.; Karande, P.; Pushparajan, A.; Acs, G.; Xu, X. Erythropoietin receptor contributes to melanoma cell survival in vivo. Oncogene 2012, 31, 1649–1660. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Marini, A.; Gustrau, A.; Delia, D.; Nambiar, S.; Hassan, M.; Hengge, U.R. Role of erythropoietin receptor expression in malignant melanoma. J. Investig. Dermatol. 2010, 130, 201–210. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef]
- Gough, N.R. Resistance through cAMP Signaling. Sci. Signal. 2013, 6, ec305. [Google Scholar] [CrossRef]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res. 2006, 66, 9483–9491. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; McCormick, F. Inhibition by cAMP of Ras-dependent activation of Raf. Science 1993, 262, 1069–1072. [Google Scholar] [CrossRef] [PubMed]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krayem, M.; Aftimos, P.; Najem, A.; van den Hooven, T.; van den Berg, A.; Hovestad-Bijl, L.; de Wijn, R.; Hilhorst, R.; Ruijtenbeek, R.; Sabbah, M.; et al. Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma and to Provide New Insights into Intrinsic and Acquired Mechanism of Resistance. Cancers 2020, 12, 512. https://doi.org/10.3390/cancers12020512
Krayem M, Aftimos P, Najem A, van den Hooven T, van den Berg A, Hovestad-Bijl L, de Wijn R, Hilhorst R, Ruijtenbeek R, Sabbah M, et al. Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma and to Provide New Insights into Intrinsic and Acquired Mechanism of Resistance. Cancers. 2020; 12(2):512. https://doi.org/10.3390/cancers12020512
Chicago/Turabian StyleKrayem, Mohammad, Philippe Aftimos, Ahmad Najem, Tim van den Hooven, Adriënne van den Berg, Liesbeth Hovestad-Bijl, Rik de Wijn, Riet Hilhorst, Rob Ruijtenbeek, Malak Sabbah, and et al. 2020. "Kinome Profiling to Predict Sensitivity to MAPK Inhibition in Melanoma and to Provide New Insights into Intrinsic and Acquired Mechanism of Resistance" Cancers 12, no. 2: 512. https://doi.org/10.3390/cancers12020512