
Issue published March 1, 1999 Previous issue | Next issue



-
Research Articles
×
Abstract
The transporter associated with antigen processing (TAP), which is composed of two subunits (TAP1 and TAP2) that have different biochemical and functional properties, plays a key role in peptide loading and the cell surface expression of HLA class I molecules. Three cases of HLA class I deficiency have previously been shown to result from the absence of a functional TAP2 subunit. In the present study, we analyzed two cases displaying not only the typical lung syndrome of HLA class I deficiency but also skin lesions, and found these patients to be TAP1-deficient. This defect leads to unstable HLA class I molecules and their retention in the endoplasmic reticulum. However, the absence of TAP1 is compatible with life and does not seem to result in higher susceptibility to viral infections than TAP2 deficiency. This work also reveals that vasculitis is often observed in HLA class I–deficient patients.
Authors
Henri de la Salle, Jacques Zimmer, Dominique Fricker, Catherine Angenieux, Jean-Pierre Cazenave, Mitsuo Okubo, Hiroo Maeda, Alessandro Plebani, Marie-Marthe Tongio, Anne Dormoy, Daniel Hanau
×Abstract
Proliferation and apoptosis are increased in many types of inflammatory diseases. A role for the cyclin kinase inhibitor p27Kip1 (p27) in limiting proliferation has been shown. In this study, we show that p27–/– mesangial cells and fibroblasts have strikingly elevated rates of apoptosis, not proliferation, when deprived of growth factors. Apoptosis was rescued by restoration of p27 expression. Cyclin A–cyclin-dependent kinase 2 (CDK2) activity, but not cyclin E–CDK2 activity, was increased in serum-starved p27–/– cells, and decreasing CDK2 activity, either pharmacologically (Roscovitine) or by a dominant–negative mutant, inhibited apoptosis. Our results show that a new biological function for the CDK inhibitor p27 is protection of cells from apoptosis by constraining CDK2 activity. These results suggest that CDK inhibitors are necessary for coordinating the cell cycle and cell-death programs so that cell viability is maintained during exit from the cell cycle.
Authors
Keiju Hiromura, Jeffrey W. Pippin, Matthew L. Fero, James M. Roberts, Stuart J. Shankland
×Abstract
In vivo confocal imaging of the mucosal surface of rat stomach was used to measure pH noninvasively under the mucus gel layer while simultaneously imaging mucus gel thickness and tissue architecture. When tissue was superfused at pH 3, the 25 μm adjacent to the epithelial surface was relatively alkaline (pH 4.1 ± 0.1), and surface alkalinity was enhanced by topical dimethyl prostaglandin E2 (pH 4.8 ± 0.2). Luminal pH was changed from pH 3 to pH 5 to mimic the fasted-to-fed transition in intragastric pH in rats. Under pH 5 superfusion, surface pH was relatively acidic (pH 4.2 ± 0.2). This surface acidity was enhanced by pentagastrin (pH 3.5 ± 0.2) and eliminated by omeprazole, implicating parietal cell H,K-ATPase as the dominant regulator of surface pH under pH 5 superfusion. With either pH 5 or pH 3 superfusion (a) gastric pit lumens had the most divergent pH from luminal superfusates; (b) qualitatively similar results were observed with and without superfusion flow; (c) local mucus gel thickness was a poor predictor of surface pH values; and (d) no channels carrying primary gastric gland fluid through the mucus were observed. The model of gastric defense that includes an alkaline mucus gel and viscous fingering of secreted acid through the mucus may be appropriate at the intragastric pH of the fasted, but not fed, animal.
Authors
Shaoyou Chu, Shin Tanaka, Jonathan D. Kaunitz, Marshall H. Montrose
×Abstract
We have shown previously that treatment of human aortic endothelial cells (HAECs) with minimally modified low-density lipoprotein (MM-LDL) induces monocyte but not neutrophil binding. This monocyte binding was not mediated by endothelial E-selectin, P-selectin, vascular cell adhesion molecule-I, or intercellular adhesion molecule-I, suggesting an alternative monocyte-specific adhesion molecule. We now show that moncytic α4β1 integrins mediate binding to MM-LDL-treated endothelial cells. We present data suggesting that the expression of the connecting segment-1 (CS-1) domain of fibronectin (FN) is induced on the apical surface of HAEC by MM-LDL and is the endothelial α4β1 ligand in MM-LDL-treated cells. Although the levels of CS-1 mRNA and protein were not increased, we show that MM-LDL treatment causes deposition of FN on the apical surface by activation of β1integrins, particularly those associated with α5 integrins.Activation of β1 by antibody 8A2 also induced CS-1-mediated monocyte binding. Confocal microscopy demonstrated the activated β1 and CS-1colocalize in concentrated filamentous patches on the apical surface of HAEC. Both anti-CS-1 and an antibody to activated β1 showed increased staining on the luminal endothelium of human coronary lesions with active monocyte entry. These results suggest the importance of these integrin ligand interactions in human atherosclerosis.
Authors
Peggy T. Shih, Mariano J. Elices, Zhuang T. Fang, Tatiana P. Ugarova, Dana Strahl, Mary C. Territo, Joy S. Frank, Nicholas L. Kovach, Carlos Cabanas, Judith A. Berliner, Devendra K. Vora
×Abstract
Several lines of evidence show the importance of angiotensin II (AII) in renal injuries, especially when hemodynamic abnormalities are involved. To elucidate the role of AII in immune-mediated renal injury, we studied anti–glomerular basement membrane (GBM) nephritis in AII type 1a receptor (AT1a)–deficient homozygous (AT1a–/–) and wild-type (AT1a+/+) mice. A transient activation of the renin–angiotensin system (RAS) was observed in both groups of mice at around day 1. A renal expression of monocyte chemoattractant protein-1 (MCP-1) was transiently induced at six hours in both groups, which was then downregulated at day 1. In the AT1a+/+ mice, after RAS activation, the glomerular expression of MCP-1 was exacerbated at days 7 and 14. Thereafter, severe proteinuria developed, and the renal expressions of transforming growth factor-β1 (TGF-β1) and collagen type I increased, resulting in severe glomerulosclerosis and interstitial fibrosis. In contrast, glomerular expression of MCP-1, proteinuria, and tissue damage were markedly ameliorated in the AT1a–/– mice. Because this amelioration is likely due to the lack of AT1a, we can conclude that AII action, mediated by AT1a, plays a pathogenic role in anti-GBM nephritis, in which AII may contribute to the exacerbation of glomerular MCP-1 expression. These results suggest the involvement of AII in immune-mediated renal injuries.
Authors
Yutaka Hisada, Takeshi Sugaya, Masaya Yamanouchi, Hiromi Uchida, Hisako Fujimura, Hiroaki Sakurai, Akiyoshi Fukamizu, Kazuo Murakami
×Abstract
It is well established that maternal smoking during pregnancy is a leading preventable cause of low birth weight and prematurity. Less appreciated is that maternal smoking during pregnancy is also associated with alterations in pulmonary function at birth and greater incidence of respiratory illnesses after birth. To determine if this is the direct result of nicotine interacting with nicotinic cholinergic receptors (nAChRs) during lung development, rhesus monkeys were treated with 1 mg/kg/day of nicotine from days 26 to 134 of pregnancy. Nicotine administration caused lung hypoplasia and reduced surface complexity of developing alveoli. Immunohistochemistry and in situ α-bungarotoxin (αBGT) binding showed that α7 nAChRs are present in the developing lung in airway epithelial cells, cells surrounding large airways and blood vessels, alveolar type II cells, free alveolar macrophages, and pulmonary neuroendocrine cells (PNEC). As detected both by immunohistochemistry and by αBGT binding, nicotine administration markedly increased α7 receptor subunit expression and binding in the fetal lung. Correlating with areas of increased α7 expression, collagen expression surrounding large airways and vessels was significantly increased. Nicotine also significantly increased numbers of type II cells and neuroendocrine cells in neuroepithelial bodies. These findings demonstrate that nicotine can alter fetal monkey lung development by crossing the placenta to interact directly with nicotinic receptors on non-neuronal cells in the developing lung, and that similar effects likely occur in human infants whose mothers smoke during pregnancy.
Authors
Harmanjatinder S. Sekhon, Yibing Jia, Renee Raab, Alexander Kuryatov, James F. Pankow, Jeffrey A. Whitsett, Jon Lindstrom, Eliot R. Spindel
×Abstract
P-selectin is a leukocyte adhesion receptor present in endothelial cells and platelets. We examined the role of P-selectin in the autologous phase of an accelerated model of anti-glomerular basement membrane (GBM) glomerulonephritis using P-selectin–deficient mice and chimeric mice expressing P-selectin only in platelets or endothelial cells. P-selectin–deficient mice exhibited more severe glomerular damage with increased interstitial mononuclear leukocytic infiltrates, and had significantly increased proteinuria and mortality when compared to wild-type mice. P-selectin on the endothelium was predominantly responsible for protection from the exacerbated disease, because chimeric mice with endothelial P-selectin, and not mice with platelet P-selectin, showed glomerular injury similar to that in wild-type animals. Levels of soluble circulating P-selectin were increased in nephritic wild-type mice and in chimeric mice with endothelial P-selectin, but not platelet P-selectin. Levels of soluble P-selectin, which has been shown to be anti-inflammatory in vitro, were inversely associated with the severity of disease. P-selectin was not expressed in the endothelium of the glomerulus or interstitium. Thus, the protective effect in wild-type mice may be accounted for, in part by soluble P-selectin shed by non-renal endothelial cells, although other endothelial P-selectin–dependent mechanisms cannot be ruled out.
Authors
Alexander R. Rosenkranz, Donna L. Mendrick, Ramzi S. Cotran, Tanya N. Mayadas
×Abstract
Excitation–contraction coupling in cardiac muscle of familial hypertrophic cardiomyopathy (FHC) remains poorly understood, despite the fact that the genetic alterations are well defined. We characterized calcium cycling and contractile activation in trabeculae from a mutant mouse model of FHC (Arg403Gln knockin, α-myosin heavy chain). Wild-type mice of the same strain and age (∼20 weeks old) served as controls. During twitch contractions, peak intracellular Ca2+ ([Ca2+]i) was higher in mutant muscles than in the wild-type (P < 0.05), but force development was equivalent in the two groups. Ca2+ transient amplitude increased dramatically in both groups as stimulation rate increased from 0.2 to 4 Hz. Nevertheless, developed force fell at the higher stimulation rates in the mutants but not in controls (P < 0.05). The steady-state force–[Ca2+]i relationship was less steep in mutants (Hill coefficient, 2.94 ± 0.27 vs. 5.28 ± 0.64; P > 0.003), with no changes in the [Ca2+]i required for 50% activation or maximal Ca2+-activated force. Thus, calcium cycling and myofilament properties are both altered in FHC mutant mice: more Ca2+ is mobilized to generate force, but this does not suffice to maintain contractility at high stimulation rates.
Authors
Wei Dong Gao, Nestor Gustavo Pérez, Christine E. Seidman, Jonathan G. Seidman, Eduardo Marbán
×Abstract
Liddle's syndrome is an inherited form of hypertension linked to mutations in the epithelial Na+ channel (ENaC). ENaC is composed of three subunits (α, β, γ), each containing a COOH-terminal PY motif (xPPxY). Mutations causing Liddle's syndrome alter or delete the PY motifs of β- or γ-ENaC. We recently demonstrated that the ubiquitin–protein ligase Nedd4 binds these PY motifs and that ENaC is regulated by ubiquitination. Here, we investigate, using the Xenopus oocyte system, whether Nedd4 affects ENaC function. Overexpression of wild-type Nedd4, together with ENaC, inhibited channel activity, whereas a catalytically inactive Nedd4 stimulated it, likely by acting as a competitive antagonist to endogenous Nedd4. These effects were dependant on the PY motifs, because no Nedd4-mediated changes in channel activity were observed in ENaC lacking them. The effect of Nedd4 on ENaC missing only one PY motif (of β-ENaC), as originally described in patients with Liddle's syndrome, was intermediate. Changes were due entirely to alterations in ENaC numbers at the plasma membrane, as determined by surface binding and immunofluorescence. Our results demonstrate that Nedd4 is a negative regulator of ENaC and suggest that the loss of Nedd4 binding sites in ENaC observed in Liddle's syndrome may explain the increase in channel number at the cell surface, increased Na+ reabsorption by the distal nephron, and hence the hypertension.
Authors
Hugues Abriel, Johannes Loffing, John F. Rebhun, J. Howard Pratt, Laurent Schild, Jean-Daniel Horisberger, Daniela Rotin, Olivier Staub
×Abstract
The adenosine triphosphate (ATP)–sensitive K+ (KATP) channel is the most abundant K+ channel active in the skeletal muscle fibers of humans and animals. In the present work, we demonstrate the involvement of the muscular KATP channel in a skeletal muscle disorder known as hypokalemic periodic paralysis (HOPP), which is caused by mutations of the dihydropyridine receptor of the Ca2+ channel. Muscle biopsies excised from three patients with HOPP carrying the R528H mutation of the dihydropyridine receptor showed a reduced sarcolemma KATP current that was not stimulated by magnesium adenosine diphosphate (MgADP; 50–100 μM) and was partially restored by cromakalim. In contrast, large KATP currents stimulated by MgADP were recorded in the healthy subjects. At channel level, an abnormal KATP channel showing several subconductance states was detected in the patients with HOPP. None of these were surveyed in the healthy subjects. Transitions of the KATP channel between subconductance states were also observed after in vitro incubation of the rat muscle with low-K+ solution. The lack of the sarcolemma KATP current observed in these patients explains the symptoms of the disease, i.e., hypokalemia, depolarization of the fibers, and possibly the paralysis following insulin administration.
Authors
Domenico Tricarico, Serenella Servidei, Pietro Tonali, Karin Jurkat-Rott, Diana Conte Camerino
×Abstract
Deficiency of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) in humans leads to the syndrome of apparent mineralocorticoid excess (SAME), in which cortisol illicitly occupies mineralocorticoid receptors, causing sodium retention, hypokalemia, and hypertension. However, the disorder is usually incompletely corrected by suppression of cortisol, suggesting additional and irreversible changes, perhaps in the kidney. To examine this further, we produced mice with targeted disruption of the 11β-HSD2 gene. Homozygous mutant mice (11β-HSD2–/–) appear normal at birth, but ∼50% show motor weakness and die within 48 hours. Both male and female survivors are fertile but exhibit hypokalemia, hypotonic polyuria, and apparent mineralocorticoid activity of corticosterone. Young adult 11β-HSD2–/– mice are markedly hypertensive, with a mean arterial blood pressure of 146 ± 2 mmHg, compared with 121 ± 2 mmHg in wild-type controls and 114 ± 4 mmHg in heterozygotes. The epithelium of the distal tubule of the nephron shows striking hypertrophy and hyperplasia. These histological changes do not readily reverse with mineralocorticoid receptor antagonism in adulthood. Thus, 11β-HSD2–/– mice demonstrate the major features of SAME, providing a unique rodent model to study the molecular mechanisms of kidney resetting leading to hypertension.
Authors
Yuri Kotelevtsev, Roger W. Brown, Stewart Fleming, Christopher Kenyon, Christopher R.W. Edwards, Jonathan R. Seckl, John J. Mullins
×Abstract
Chronic hypoxia induces polycythemia, pulmonary hypertension, right ventricular hypertrophy, and weight loss. Hypoxia-inducible factor 1 (HIF-1) activates transcription of genes encoding proteins that mediate adaptive responses to hypoxia, including erythropoietin, vascular endothelial growth factor, and glycolytic enzymes. Expression of the HIF-1α subunit increases exponentially as O2 concentration is decreased. Hif1a–/– mouse embryos with complete deficiency of HIF-1α due to homozygosity for a null allele at the Hif1a locus die at midgestation, with multiple cardiovascular malformations and mesenchymal cell death. Hif1a+/– heterozygotes develop normally and are indistinguishable from Hif1a+/+ wild-type littermates when maintained under normoxic conditions. In this study, the physiological responses of Hif1a+/– and Hif1a+/+ mice exposed to 10% O2 for one to six weeks were analyzed. Hif1a+/– mice demonstrated significantly delayed development of polycythemia, right ventricular hypertrophy, pulmonary hypertension, and pulmonary vascular remodeling and significantly greater weight loss compared with wild-type littermates. These results indicate that partial HIF-1α deficiency has significant effects on multiple systemic responses to chronic hypoxia.
Authors
Aimee Y. Yu, Larissa A. Shimoda, Narayan V. Iyer, David L. Huso, Xing Sun, Rita McWilliams, Terri Beaty, James S.K. Sham, Charles M. Wiener, J.T. Sylvester, Gregg L. Semenza
×Abstract
We have isolated a cardiomyogenic cell line (CMG) from murine bone marrow stromal cells. Stromal cells were immortalized, treated with 5-azacytidine, and spontaneously beating cells were repeatedly screened. The cells showed a fibroblast-like morphology, but the morphology changed after 5-azacytidine treatment in ∼30% of the cells; they connected with adjoining cells after one week, formed myotube-like structures, began spontaneously beating after two weeks, and beat synchronously after three weeks. They expressed atrial natriuretic peptide and brain natriuretic peptide and were stained with anti-myosin, anti-desmin, and anti-actinin antibodies. Electron microscopy revealed a cardiomyocyte-like ultrastructure, including typical sarcomeres, a centrally positioned nucleus, and atrial granules. These cells had several types of action potentials, such as sinus node–like and ventricular cell–like action potentials. All cells had a long action potential duration or plateau, a relatively shallow resting membrane potential, and a pacemaker-like late diastolic slow depolarization. Analysis of the isoform of contractile protein genes, such as myosin heavy chain, myosin light chain, and α-actin, indicated that their muscle phenotype was similar to that of fetal ventricular cardiomyocytes. These cells expressed Nkx2.5/Csx, GATA4, TEF-1, and MEF-2C mRNA before 5-azacytidine treatment and expressed MEF-2A and MEF-2D after treatment. This new cell line provides a powerful model for the study of cardiomyocyte differentiation.
Authors
Shinji Makino, Keiichi Fukuda, Shunichirou Miyoshi, Fusako Konishi, Hiroaki Kodama, Jing Pan, Motoaki Sano, Toshiyuki Takahashi, Shingo Hori, Hitoshi Abe, Jun-ichi Hata, Akihiro Umezawa, Satoshi Ogawa
×Abstract
Alcoholic beverages produced by fermentation (e.g., beer and wine) are powerful stimulants of gastric acid output and gastrin release in humans. The aim of this study was to separate and specify the gastric acid stimulatory ingredients in alcoholic beverages produced by fermentation. Yeast-fermented glucose was used as a simple model of fermented alcoholic beverages; it was stepwise separated by different methods of liquid chromatography, and each separated solution was tested in human volunteers for its stimulatory action on gastric acid output and gastrin release. Five substances were detected by high-performance liquid chromatography and were analyzed by mass spectrometry and 1H-13C nuclear magnetic resonance spectroscopy. At the end of the separation process of the five identified substances, only the two dicarboxylic acids, maleic acid and succinic acid, had a significant (P < 0.05) stimulatory action on gastric acid output (76% and 70% of fermented glucose, respectively), but not on gastrin release. When given together, they increased gastric acid output by 100% of fermented glucose and by 95% of maximal acid output. We therefore conclude that maleic acid and succinic acid are the powerful stimulants of gastric acid output in fermented glucose and alcoholic beverages produced by fermentation, and that gastrin is not their mediator of action.
Authors
Stephan Teyssen, Gloria González-Calero, Michael Schimiczek, Manfred V. Singer
×Abstract
We have shown previously that a group IIA phospholipase A2 (PLA2) is responsible for the potent bactericidal activity of inflammatory fluids against many Gram-positive bacteria. To exert its antibacterial activity, this PLA2 must first bind and traverse the bacterial cell wall to produce the extensive degradation of membrane phospholipids (PL) required for bacterial killing. In this study, we have examined the properties of the cell-wall that may determine the potency of group IIA PLA2 action. Inhibition of bacterial growth by nutrient deprivation or a bacteriostatic antibiotic reversibly increased bacterial resistance to PLA2-triggered PL degradation and killing. Conversely, pretreatment of Staphylococcus aureus or Enterococcus faecium with subinhibitory doses of β-lactam antibiotics increased the rate and extent of PL degradation and/or bacterial killing after addition of PLA2. Isogenic wild-type (lyt+) and autolysis-deficient (lyt–) strains of S. aureus were equally sensitive to the phospholipolytic action of PLA2, but killing and lysis was much greater in the lyt+ strain. Thus, changes in cell-wall cross-linking and/or autolytic activity can modulate PLA2 action either by affecting enzyme access to membrane PL or by the coupling of massive PL degradation to autolysin-dependent killing and bacterial lysis or both. Taken together, these findings suggest that the bacterial envelope sites engaged in cell growth may represent preferential sites for the action and cytotoxic consequences of group IIA PLA2 attack against Gram-positive bacteria.
Authors
Amy K. Foreman-Wykert, Yvette Weinrauch, Peter Elsbach, Jerrold Weiss
×Abstract
We have investigated the cellular pathology of the syndrome called thiamine-responsive megaloblastic anemia (TRMA) with diabetes and deafness. Cultured diploid fibroblasts were grown in thiamine-free medium and dialyzed serum. Normal fibroblasts survived indefinitely without supplemental thiamine, whereas patient cells died in 5–14 days (mean 9.5 days), and heterozygous cells survived for more than 30 days. TRMA fibroblasts were rescued from death with 10–30 nM thiamine (in the range of normal plasma thiamine concentrations). Positive terminal deoxynucleotide transferase–mediated dUTP nick end-labeling (TUNEL) staining suggested that cell death was due to apoptosis. We assessed cellular uptake of [3H]thiamine at submicromolar concentrations. Normal fibroblasts exhibited saturable, high-affinity thiamine uptake (Km 400–550 nM; Vmax 11 pmol/min/106 cells) in addition to a low-affinity unsaturable component. Mutant cells lacked detectable high-affinity uptake. At 30 nM thiamine, the rate of uptake of thiamine by TRMA fibroblasts was 10-fold less than that of wild-type, and cells from obligate heterozygotes had an intermediate phenotype. Transfection of TRMA fibroblasts with the yeast thiamine transporter gene THI10 prevented cell death when cells were grown in the absence of supplemental thiamine. We therefore propose that the primary abnormality in TRMA is absence of a high-affinity thiamine transporter and that low intracellular thiamine concentrations in the mutant cells cause biochemical abnormalities that lead to apoptotic cell death.
Authors
Amy R. Stagg, Judith C. Fleming, Meghan A. Baker, Massayuki Sakamoto, Nadine Cohen, Ellis J. Neufeld
×Abstract
Cathepsin K, a lysosomal cysteine protease critical for bone remodeling by osteoclasts, was recently identified as the deficient enzyme causing pycnodysostosis, an autosomal recessive osteosclerotic skeletal dysplasia. To investigate the nature of molecular lesions causing this disease, mutations in the cathepsin K gene from eight families were determined, identifying seven novel mutations (K52X, G79E, Q190X, Y212C, A277E, A277V, and R312G). Expression of the first pro region missense mutation in a cysteine protease, G79E, in Pichia pastoris resulted in an unstable precursor protein, consistent with misfolding of the proenzyme. Expression of five mature region missense defects revealed that G146R, A277E, A277V, and R312G precursors were unstable, and no mature proteins or protease activity were detected. The Y212C precursor was activated to its mature form in a manner similar to that of the wild-type cathepsin K. The mature Y212C enzyme retained its dipeptide substrate specificity and gelatinolytic activity, but it had markedly decreased activity toward type I collagen and a cathepsin K–specific tripeptide substrate, indicating that it was unable to bind collagen triple helix. These studies demonstrated the molecular heterogeneity of mutations causing pycnodysostosis, indicated that pro region conformation directs proper folding of the proenzyme, and suggested that the cathepsin K active site contains a critical collagen-binding domain.
Authors
Wu-Shiun Hou, Dieter Brömme, Yingming Zhao, Ernest Mehler, Craig Dushey, Harel Weinstein, Clara Sa Miranda, Claudia Fraga, Fenella Greig, John Carey, David L. Rimoin, Robert J. Desnick, Bruce D. Gelb
×Abstract
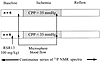
Conventional approaches for the treatment of myocardial ischemia increase coronary blood flow or reduce myocardial demand. To determine whether a rightward shift in the hemoglobin–oxygen saturation curve would reduce the metabolic and contractile effects of a myocardial oxygen-supply imbalance, we studied the impact of a potent synthetic allosteric modifier of hemoglobin–oxygen affinity, a 2-[4-[[(3,5-disubstituted anilino)carbonyl]methyl] phenoxy] -2-methylproprionic acid derivative (RSR13), during low-flow ischemia. Changes in myocardial high-energy phosphate levels and pH were studied by 31P nuclear magnetic resonance (NMR) spectroscopy in 12 open-chest dogs randomized to receive RSR13 or vehicle control during a reversible reduction of left anterior descending (LAD) coronary artery blood flow. Changes in cardiac metabolites and regional ventricular function studied by pressure segment–length relations were also investigated in additional animals before and after RSR13 administration during low-flow LAD ischemia. The intravenous administration of RSR13 before ischemia resulted in a substantial increase in the mean hemoglobin p50 and attenuated the decline in cardiac creatine phosphate/adenosine triphosphate (PCr/ATP), percent PCr, and pH during ischemia without a change in regional myocardial blood flow, heart rate, or systolic blood pressure. RSR13 given after the onset of low-flow ischemia also improved cardiac PCr/ATP ratios and regional function as measured by fractional shortening and regional work. Thus, synthetic allosteric reduction in hemoglobin–oxygen affinity may be a new and important therapeutic strategy to ameliorate the metabolic and functional consequences of cardiac ischemia.
Authors
Robert G. Weiss, Marco A. Mejia, David A. Kass, Anthony F. DiPaula, Lewis C. Becker, Gary Gerstenblith, V.P. Chacko
×Abstract
Epidemiological investigations have linked Chlamydia pneumoniae infection to atherosclerosis. It is not clear, however, whether C. pneumoniae infection plays a causal role in the development of atherosclerosis. Mice with low-density lipoprotein receptor deficiency were induced to develop atherosclerotic lesions in aorta with a cholesterol-enriched diet that increased serum cholesterol by two- to threefold. Using this mouse model, we found that the chlamydial infection alone with either the C. pneumoniae AR39 or the C. trachomatis MoPn strain failed to induce any significant atherosclerotic lesions in aorta over a period of nine months. However, in the presence of a high-cholesterol diet, infection with the C. pneumoniae AR39 strain significantly exacerbated the hypercholesterolemia-induced atherosclerosis, demonstrating that a hypercholesterolemic condition is required for the C. pneumoniae to aggravate the development of atherosclerosis. Although both AR39 and MoPn antigens were detected in aorta of mice infected with the corresponding strains, only mice infected with the C. pneumoniae strain AR39 displayed enhanced atherosclerotic lesions, suggesting that the C. pneumoniae species may possess a unique atherogenic property. This study may provide a model for further understanding the mechanisms of C. pneumoniae atherogenesis and evaluating chlamydial intervention strategies for preventing the advancement of atherosclerotic lesions enhanced by C. pneumoniae infection.
Authors
He Hu, Grant N. Pierce, Guangming Zhong
×Abstract
Expression of histocompatibility leukocyte antigen (HLA) class I molecules on the cell surface depends on the heterodimer of the transporter associated with antigen processing 1 and 2 (TAP1 and TAP2), which transport peptides cleaved by proteasome to the class I molecules. Defects in the TAP2 protein have been reported in two families with HLA class I deficiency, the so-called bare lymphocyte syndrome (BLS) type I. We have, to our knowledge, identified for the first time a splice site mutation in the TAP1 gene of another BLS patient. In addition, class I heavy chains (HCs) did not form the normal complex with tapasin in the endoplasmic reticulum (ER) of the cells of our patient.
Authors
Hiroshi Furukawa, Shigeo Murata, Toshio Yabe, Naoki Shimbara, Naoto Keicho, Kouichi Kashiwase, Kaoru Watanabe, Yoshihide Ishikawa, Tatsuya Akaza, Kenji Tadokoro, Shigeto Tohma, Tetsufumi Inoue, Katsushi Tokunaga, Kazuhiko Yamamoto, Keiji Tanaka, Takeo Juji









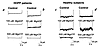












Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)